45
Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations İnanç Birol 13 October 2011

Haploid Assembly of Diploid Genomes
Challenges, Trials, Tribulations
İnanç Birol
13 October 2011

IEEE InfoVis 2009
Assembly By Short Sequencing
2

3

in Literature
• ~40 citations on tool comparisons
• ~20 citations on using ABySS for a biology study
• Crowded field – 17 teams in Assemblathon 1
4
Overlap-Overlay-Consensus
ARACHNE
CAP3
Celera assembler
MIRA
Newbler
Phred/Phrap
SGA
De Bruijn Graph
Euler
Velvet
ABySS
SOAPdenovo
ALLPATHS
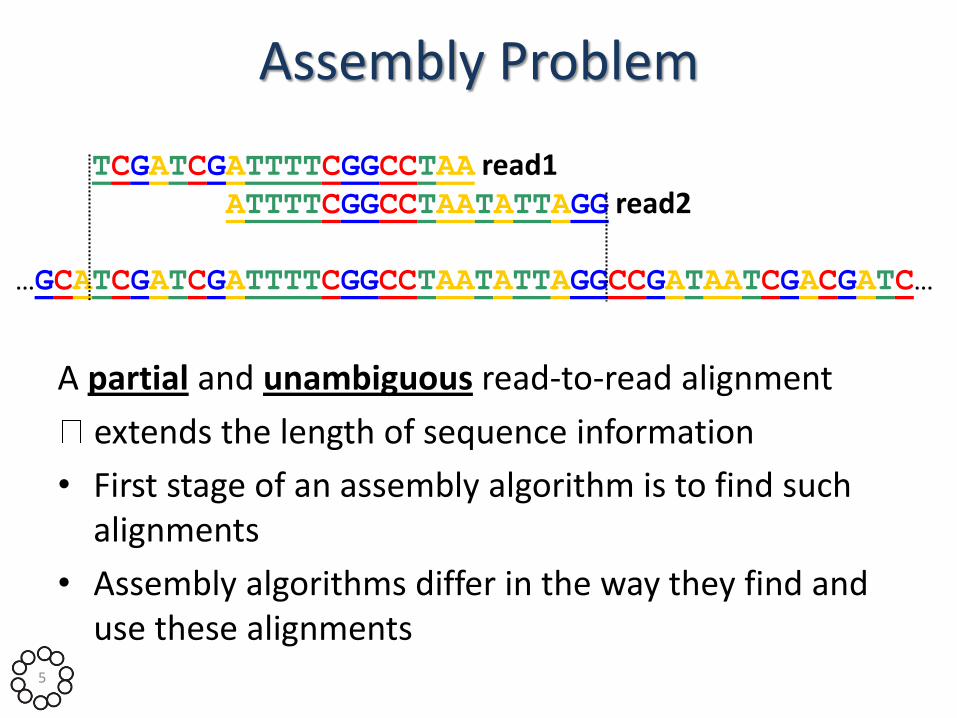
Assembly Problem
A partial and unambiguous read-to-read alignment
extends the length of sequence information
• First stage of an assembly algorithm is to find such alignments
• Assembly algorithms differ in the way they find and use these alignments
5
TCGATCGATTTTCGGCCTAA read1 ATTTTCGGCCTAATATTAGG read2
…GCATCGATCGATTTTCGGCCTAATATTAGGCCGATAATCGACGATC…

Algorithm
• SE Assembly:
• PE Assembly:
• Scaffolding:
k-mer extension on a de Bruijn graph
search for unambiguous contig merging along paths
search for unambiguous linkage across distant contigs
6
d=6±5
d=5±4
d=26±9
d=12±5
Software
7

De Bruijn Graph
• Description of read-to-read overlaps
– 2x4 possible extension of every k-mer
• Provides and O(n) algorithm for SE assembly
8
…GACATTGC… seq1 …GACATTAT… seq2
GACAT ACATT
ATTAT CATTA
CATTG ATTGC
…
…
…
k = 5

Adjacency Graph
• Description of contig overlaps
– Built during SE assembly
• Overlap = k-1 bp
– Generalized during PE assembly
• Arbitrary overlap
9

Linkage Graph
• Built through read pairs aligned to different contigs
– PE reads from a tight fragment length distribution
• Reliable distance estimates
– MP reads from broader insert length distribution
• Noisy data
• Used in PE assembly (PE) and scaffolding (PE and MP) stages
10

Anchor
• Scrubbing “homozygous” variations
Indel SNPs
11
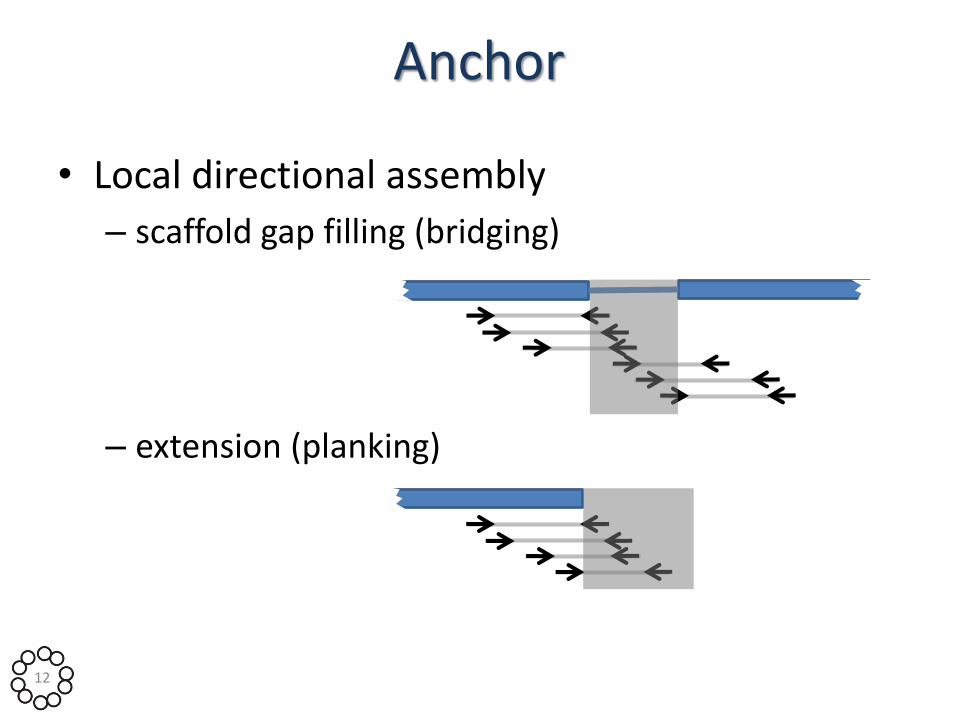
Anchor
• Local directional assembly
– scaffold gap filling (bridging)
– extension (planking)
12

Case Study
Mountain Pine Beetle Genome Assembly
13
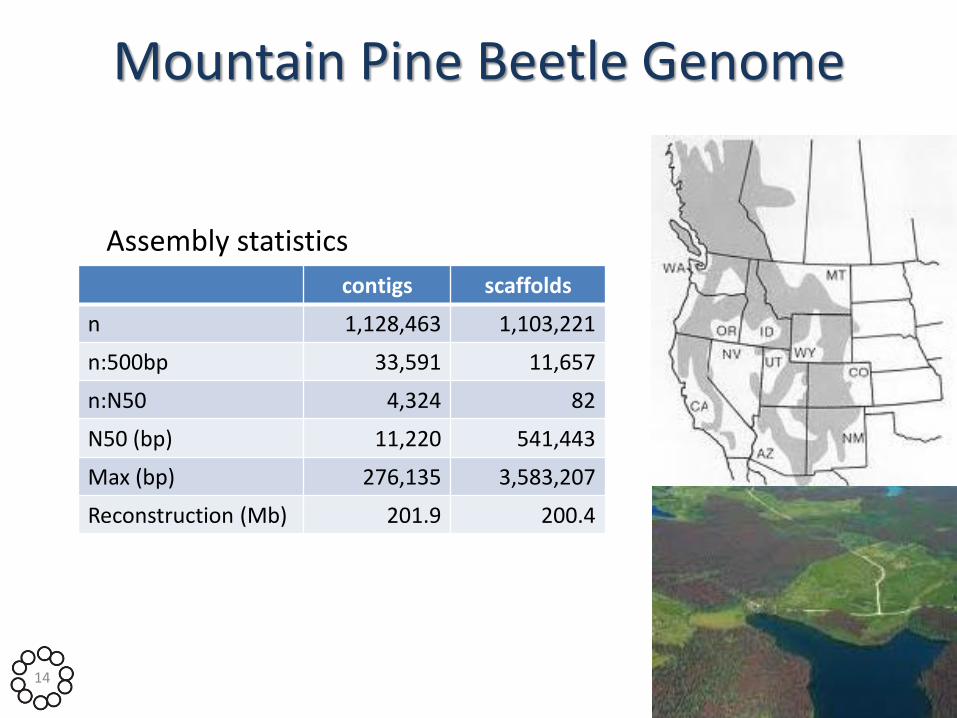
Mountain Pine Beetle Genome
Assembly statistics
contigs scaffolds
n 1,128,463 1,103,221
n:500bp 33,591 11,657
n:N50 4,324 82
N50 (bp) 11,220 541,443
Max (bp) 276,135 3,583,207
Reconstruction (Mb) 201.9 200.4
14

Assembly As a Hairball
• ABySS v1.2.7
– PE/MP information disambiguates short contig extensions
1 2 3 4 5 6+ 1 15822 7354 1882 530 109 1
2 7354 9814 1817 456 72 3
3 1882 1817 1074 238 31 1
4 530 456 238 126 13 1 5 109 72 31 13 10 0 6+ 1 3 1 1 0 0
Node connectivity*
out in
* For contigs 2 kb
15

Scaffolding
16

Quality Assessment
Alignment of 81,047,980 reads
Gene alignments
17
Before Anchor After Anchor Change
Mapped 65,624,456 (80.97%)
66,949,341 (82.60%)
+ 1,324,885
Paired 43,207,118 (53.31%)
44,732,320 (55.19%)
+ 1,525,202
Single-end 9,536,178 (11.77%)
8,846,977 (10.92%)
-689,201
2,180 ESTs 248 Conserved Genes
Complete Partial Complete Partial
Contigs 968 1169 212 18
Scaffolds 1,481 619 228 5
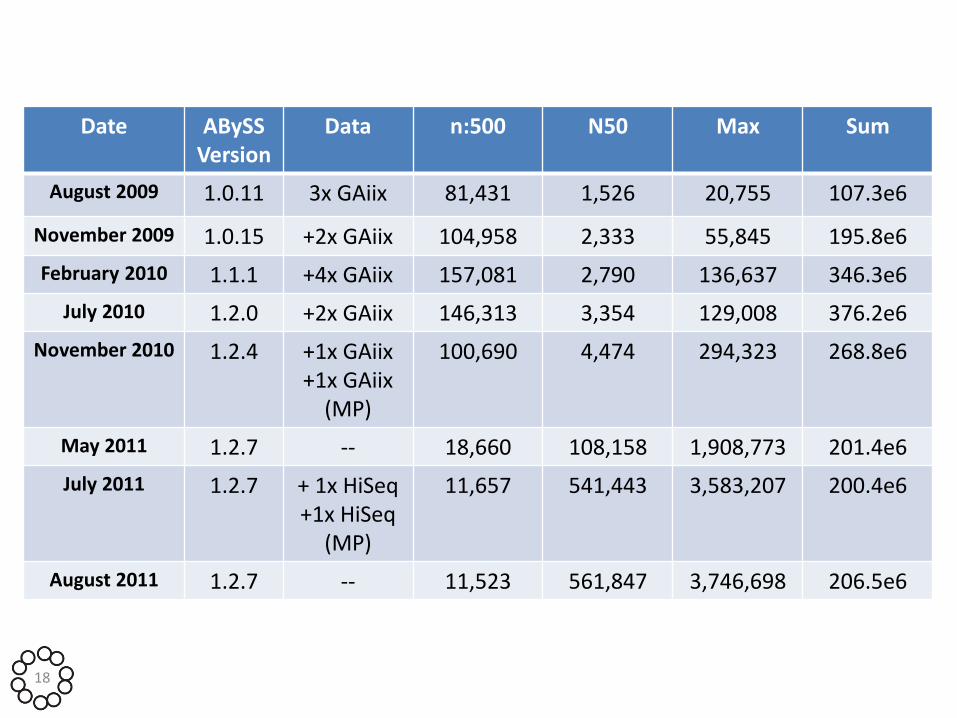
Date ABySS Version
Data n:500 N50 Max Sum
August 2009 1.0.11 3x GAiix 81,431 1,526 20,755 107.3e6
November 2009 1.0.15 +2x GAiix 104,958 2,333 55,845 195.8e6
February 2010 1.1.1 +4x GAiix 157,081 2,790 136,637 346.3e6
July 2010 1.2.0 +2x GAiix 146,313 3,354 129,008 376.2e6
November 2010 1.2.4 +1x GAiix +1x GAiix
(MP)
100,690 4,474 294,323 268.8e6
May 2011 1.2.7 -- 18,660 108,158 1,908,773 201.4e6
July 2011 1.2.7 + 1x HiSeq +1x HiSeq
(MP)
11,657 541,443 3,583,207 200.4e6
August 2011 1.2.7 -- 11,523 561,847 3,746,698 206.5e6
18

Transcriptome Assembly
19

Transcriptome Sequencing
• RNA-seq protocol
• Brings information on how a genome “acts”
– Expression levels
• Allelic expression
– Present isoforms
– Gene fusions
– Other transcriptional events
– Post-transcriptional RNA editing Rodrigo Goya
20

Transcript models
Transcriptome Assembly
Transcriptome assembly is different from genome assembly
– varying coverage levels ⇒ varying expression levels
– split assembly paths ⇒ isoforms/splice variants
– small contig sizes ⇒ small product sizes
21

What Overlap to Choose?
22

Selection of k
23
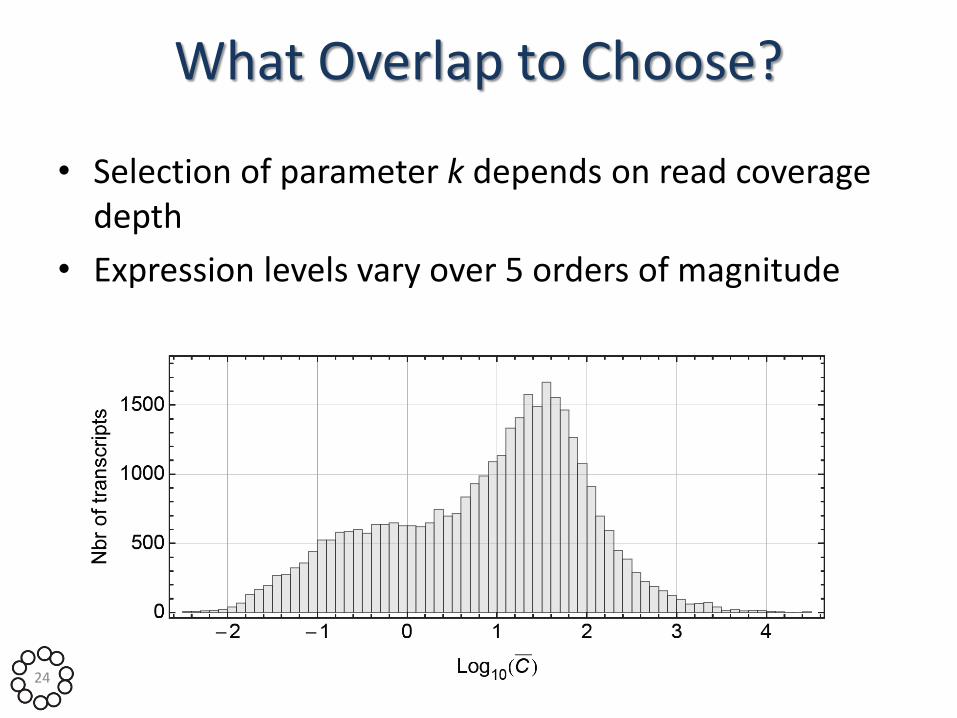
What Overlap to Choose?
• Selection of parameter k depends on read coverage depth
• Expression levels vary over 5 orders of magnitude
24

Assembly Merging
25
buried parent untouched

Multi-k Assembly
We capture a wide range of expression levels
• Gray: all transcripts with a read alignment
• Blue: at least 80% of a transcript in a single contig
• Red: at least 80% of a transcript is reconstructed
26

Trans-ABySS
A versatile tool for
• Transcript reconstruction
• Gene identification
• InDel and SNV discovery
• Chimeric transcript discovery
– Gene fusions
– Trans-splicing
• Expression analysis
27

Trans-ABySS
Cufflinks 0.8.3
Scripture
28
Transcriptome Assembly
De novo assembly based on ABySS
Reference-based assembly based on TopHat alignments [Trapnell et al., 2010; Guttman et al., 2010; Trapnell et al., 2009]

Events
29 + chimeric transcripts
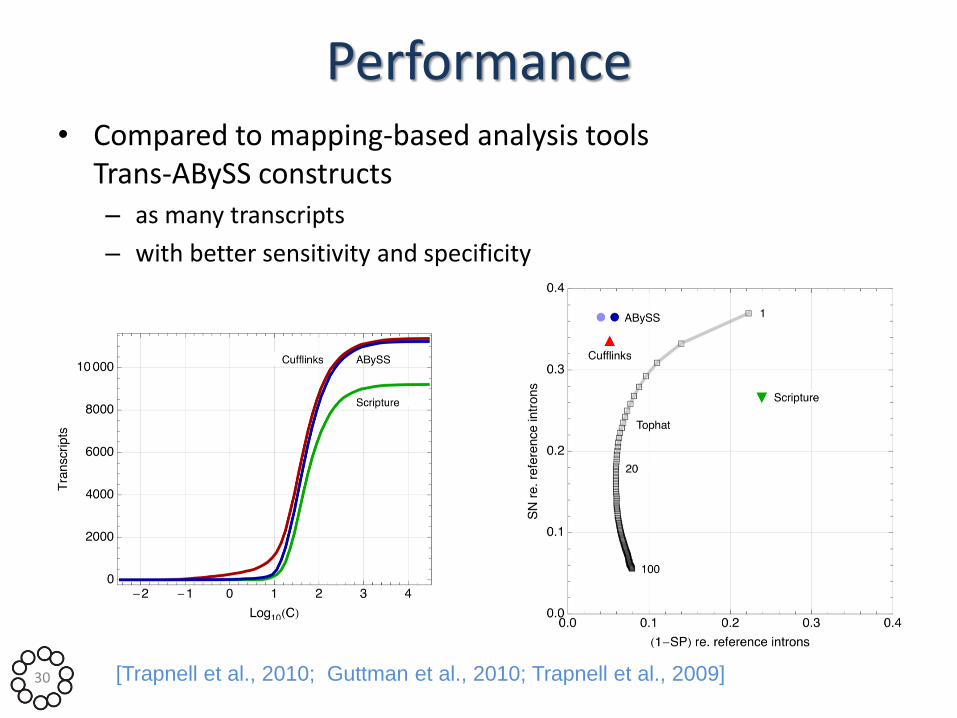
Performance • Compared to mapping-based analysis tools
Trans-ABySS constructs – as many transcripts
– with better sensitivity and specificity
30 [Trapnell et al., 2010; Guttman et al., 2010; Trapnell et al., 2009]

Case Study
Acute Myeloid Leukemia Transcriptome Assembly
31

Fusions • Assembled transcriptome
contigs span multiple genes
• Break point (usually) corresponds to exon boundaries
• Break point is supported by – Spanning reads – Read pairs linking regions
• Gene fusions are often drivers in AML and define subtypes (e.g. PML/RARα and M3 subtype)
1 2
4 5 6
Lucas Swanson, Readman Chiu and Gordon Robertson
32

AML Gene Fusions
0
2
4
6
8
10
12
14
16
Nu
mb
er
of
pat
ien
ts
Candidate fusion events
9%
5%
4% MLL fusions
Known AML fusion events (12) Known polymorphism (1) Novel fusion event (17)
Low frequency (<1%)
71 events in 65/173 (38%) patients 30 different gene fusions identified ≥94% validation by RT-PCR sequencing
Karen Mungall 33
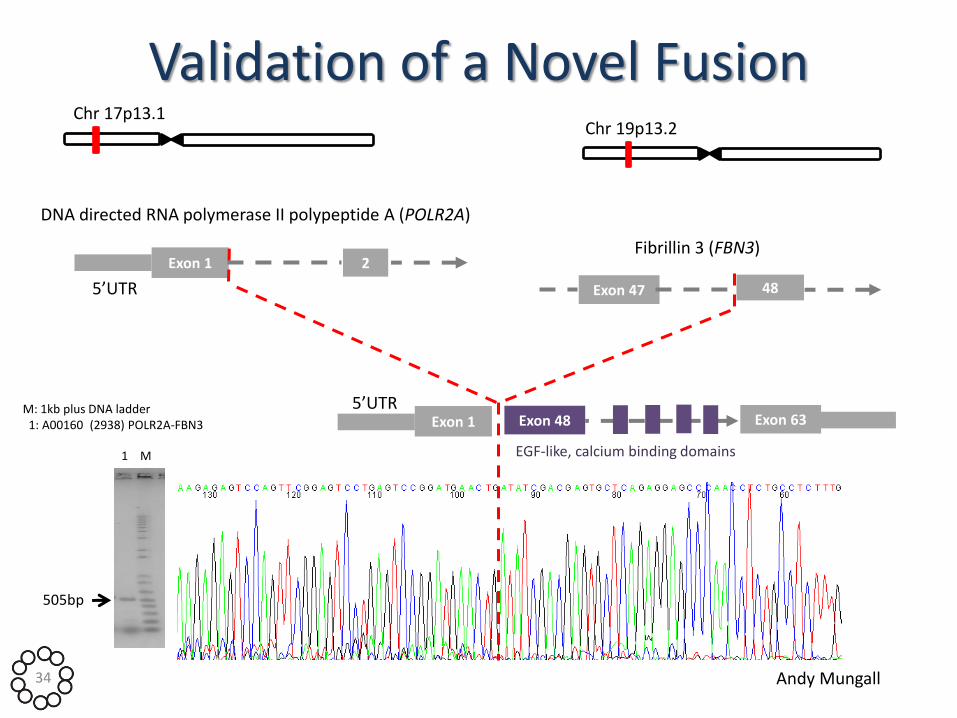
Validation of a Novel Fusion
M: 1kb plus DNA ladder 1: A00160 (2938) POLR2A-FBN3
505bp
Chr 17p13.1
DNA directed RNA polymerase II polypeptide A (POLR2A)
Exon 1 2
5’UTR
Fibrillin 3 (FBN3)
Chr 19p13.2
Exon 47 48
Exon 1 5’UTR
Exon 48 Exon 63
EGF-like, calcium binding domains 1 M
Andy Mungall 34
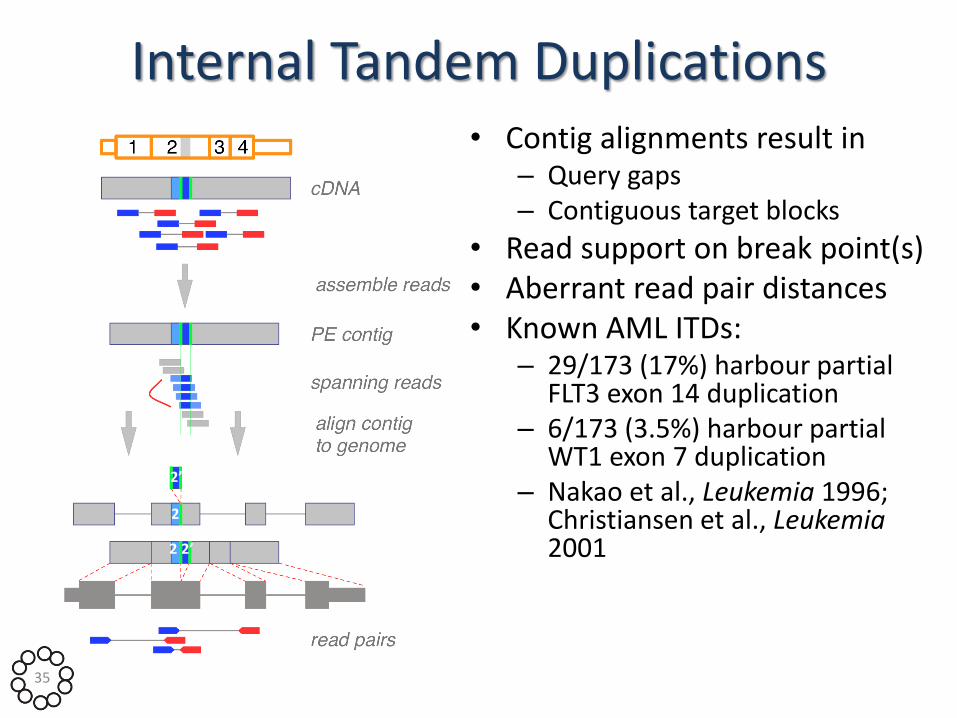
Internal Tandem Duplications • Contig alignments result in
– Query gaps – Contiguous target blocks
• Read support on break point(s) • Aberrant read pair distances • Known AML ITDs:
– 29/173 (17%) harbour partial FLT3 exon 14 duplication
– 6/173 (3.5%) harbour partial WT1 exon 7 duplication
– Nakao et al., Leukemia 1996; Christiansen et al., Leukemia 2001 2 2’
2’
2
35

Known ITD in FLT3
• A 33 bp duplication in exon 14 CTCCCATttgagatcatattcatattctctgaaatcaacgTTGAGATCATATTCATATTCTCTGAAATCAACGTAGAA
Karen Mungall 36
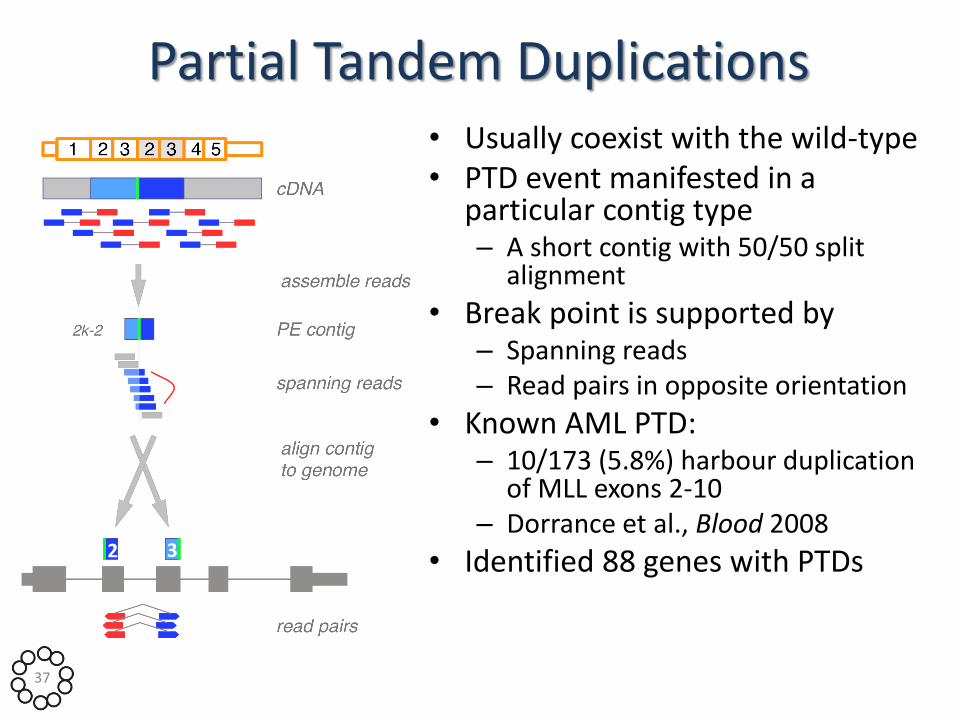
Partial Tandem Duplications • Usually coexist with the wild-type • PTD event manifested in a
particular contig type – A short contig with 50/50 split
alignment
• Break point is supported by – Spanning reads – Read pairs in opposite orientation
• Known AML PTD: – 10/173 (5.8%) harbour duplication
of MLL exons 2-10 – Dorrance et al., Blood 2008
• Identified 88 genes with PTDs 2 3
37

Novel PTD in Arid1a
• Exons 2-4 tandemly repeated in 5 AML libraries
• Recurrent across tissues and species
WT CT
Source Observations
AML 5/173 Libraries
LBC 5/54 Libraries
Normal mouse 3/7 Libraries
NCBI EST colon_ins , placenta_normal
38

Summary
39

ABySS Team: Shaun Jackman Tony Raymond Rod Docking Beetle Project: Joerg Bohlmann Chris Keeling Nancy Liao Greg Taylor Simon Chan Diana Palmquist
Trans-ABySS Team: Readman Chiu Karen Mungall Gordon Robertson Ka Ming Nip Jenny Qian Rong She Lucas Swanson AML Project: Richard Moore Yongjun Zhao Andy Mungall Aly Karsan
GSC: Sequencing Team Library Core Systems Team Steven Jones Marco Marra

Final Hairball
• ABySS v1.2.7
– Read pairs and inferred distances allow for scaffolding
41
contigs scaffolds
n 1,128,463 1,103,221
n:500bp 33,591 11,657
n:N50 4,324 82
N50 (bp) 11,220 541,443
Max (bp) 276,135 3,583,207
Reconstruction (Gb) 201.9 200.4

Biotin Read-Through
circularized insert
42

43

Triage of MP Reads
Challenge: A B
A B
Which
one?
Information:
• Distances from contig ends
• Base mismatches on read ends
• Inferred contig orientations 44
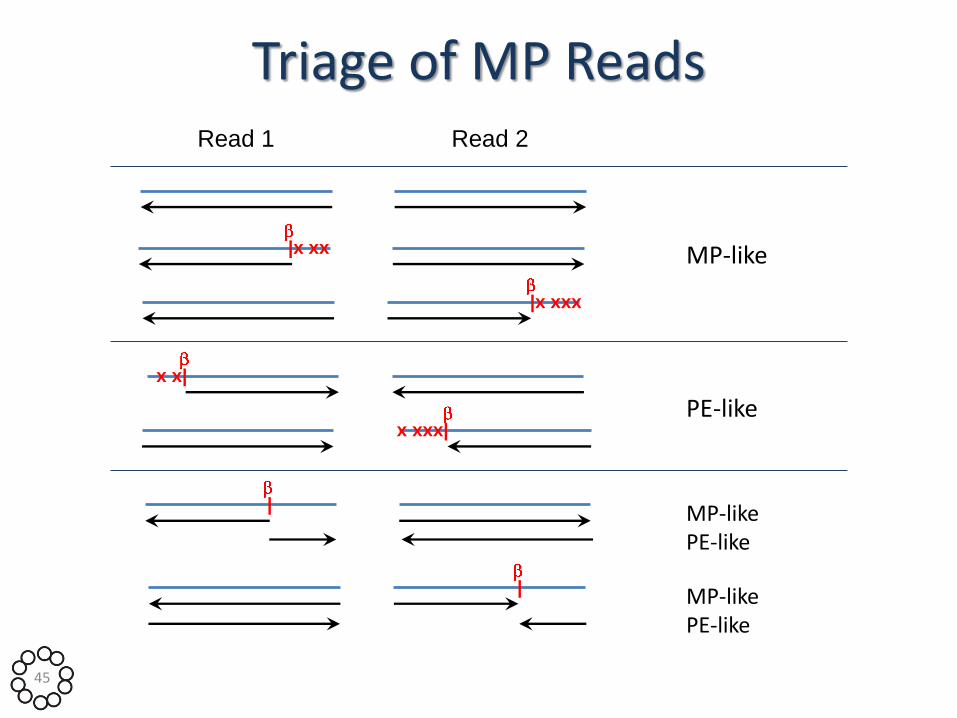
Triage of MP Reads Read 1 Read 2
MP-like
PE-like
MP-like PE-like
MP-like PE-like
|x xx
|x xxx
x x|
x xxx|
|
|
45

















