30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom An agency of the European Union Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5520 Send a question via our website www.ema.europa.eu/contact 5 March 2018 EMA/261438/2018 Committee for Medicinal Products for Human Use (CHMP) Assessment report Invented name: Hizentra International non-proprietary name: human normal immunoglobulin Procedure No. EMEA/H/C/002127/II/0087 Note Variation assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted.
Transcript
30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom
An agency of the European Union
Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5520 Send a question via our website www.ema.europa.eu/contact
5 March 2018 EMA/261438/2018 Committee for Medicinal Products for Human Use (CHMP)
Assessment report
Invented name: Hizentra
International non-proprietary name: human normal immunoglobulin
Procedure No. EMEA/H/C/002127/II/0087
Note
Variation assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted.
Assessment report EMA/261438/2018 Page 2/55
Table of contents
1. Background information on the procedure .............................................. 4 1.1. Type II variation .................................................................................................. 4 1.2. Steps taken for the assessment of the product ........................................................ 4
2. Scientific discussion ................................................................................ 5 2.1. Introduction ........................................................................................................ 5 2.2. Non-clinical aspects .............................................................................................. 7 2.3. Clinical aspects .................................................................................................... 7 2.3.1. Introduction...................................................................................................... 7 2.3.2. Pharmacokinetics .............................................................................................. 8 2.3.3. Pharmacokinetic/Pharmacodynamic modelling .................................................... 18 2.3.4. Discussion on clinical pharmacology ................................................................... 21 2.3.5. Conclusions on clinical pharmacology ................................................................. 23 2.4. Clinical efficacy .................................................................................................. 23 2.4.1. Main study ..................................................................................................... 23 2.4.2. Discussion on clinical efficacy ............................................................................ 42 2.4.3. Conclusions on the clinical efficacy .................................................................... 44 2.5. Clinical safety .................................................................................................... 44 2.5.1. Discussion on clinical safety .............................................................................. 48 2.5.2. Conclusions on clinical safety ............................................................................ 49 2.5.3. PSUR cycle ..................................................................................................... 49 2.6. Risk management plan ....................................................................................... 49 2.7. Update of the Product information ........................................................................ 51
3. Benefit-Risk Balance ............................................................................. 51 3.1. Favourable effects .............................................................................................. 51 3.2. Uncertainties and limitations about favourable effects ............................................. 51 3.3. Unfavourable effects ........................................................................................... 51 3.4. Uncertainties and limitations about unfavourable effects ......................................... 52 3.5. Effects Table ...................................................................................................... 52 3.6. Benefit-risk assessment and discussion ................................................................. 54 3.6.1. Importance of favourable and unfavourable effects .............................................. 54 3.6.2. Balance of benefits and risks ............................................................................ 55 3.7. Conclusions ....................................................................................................... 55
List of abbreviations AE Adverse Event AESI Adverse Event of Special Interest BRDM Blinded Data Review Meetings bw Bodyweight CI Confidence Interval CIDP Chronic Inflammatory Demyelinating Polyneuropathy CMAP Compound Muscle Action Potential DAT Direct Antiglobulin Test ECG Electrocardiogram EFNS European Federation of Neurological Societies EMA European Medicines Agency IDMC Independent Data Monitoring Committee INCAT Inflammatory Neuropathy Cause and Treatment ITTS Intention-to-Treat Set IV / i.v. Intravenous IVIG Intravenous Immunoglobulin MAH Marketing Authorisation Holder MedDRA Medical Dictionary for Regulatory Activities MRC Medical Research Council N.A. / n.a. Not Applicable PID Primary Immunodeficiency PK Pharmacokinetic(s) PNS Peripheral Nerve Society PP-PSDS Per Protocol-Pre Randomization Safety Data Set PPS Per Protocol Set PSDS Pre Randomization Safety Data Set PT Preferred Term RMP Risk Management Plan R-ODS Rasch-built Overall Disability Scale RSDS Rescue Medication Safety Data Set SA Scientific Advice SAE Serious Adverse Event SC / s.c. Subcutaneous SCIG Subcutaneous Immunoglobulin SE Standard Error SID Secondary Immunodeficiency SOC System Organ Class SDS Safety Data Set SmPC Summary of Product Characteristics
Assessment report EMA/261438/2018 Page 4/55
1. Background information on the procedure
1.1. Type II variation
Pursuant to Article 16 of Commission Regulation (EC) No 1234/2008, CSL Behring GmbH submitted to the European Medicines Agency on 15 June 2017 an application for a variation.
The following variation was requested:
Variation requested Type Annexes affected
C.I.6.a C.I.6.a - Change(s) to therapeutic indication(s) - Addition of a new therapeutic indication or modification of an approved one
Type II I and IIIB
Extension of Indication to include immunomodulatory therapy for the treatment of patients with chronic inflammatory demyelinating polyneuropathy (CIDP) as maintenance therapy to prevent relapse of neuromuscular disability and impairment. As a consequence, sections 4.1, 4.2, 4.8, 5.1 and 5.2 of the SmPC are updated. The Package Leaflet is updated in accordance. The RMP is updated (v. 4.0)
The requested variation proposed amendments to the Summary of Product Characteristics and Package Leaflet and to the Risk Management Plan (RMP).
Information on paediatric requirements
Not applicable
Information relating to orphan market exclusivity
Similarity
Pursuant to Article 8 of Regulation (EC) No. 141/2000 and Article 3 of Commission Regulation (EC) No 847/2000, the applicant did not submit a critical report addressing the possible similarity with authorised orphan medicinal products because there is no authorised orphan medicinal product for a condition related to the proposed indication.
Scientific advice
The applicant did not seek Scientific Advice at the CHMP.
1.2. Steps taken for the assessment of the product
The Rapporteur and Co-Rapporteur appointed by the CHMP were:
Rapporteur: Jan Mueller-Berghaus Co-Rapporteur: N/A
Assessment report EMA/261438/2018 Page 5/55
Timetable Planned dates Actual dates
Start of procedure: 15 July 2017 15 July 2017
CHMP Rapporteur Assessment Report 8 September 2017 8 September 2017
PRAC Rapporteur Assessment Report 15 September 2017 13 September 2017
PRAC members comments 20 September 2017 n/a
Updated PRAC Rapporteur Assessment Report 21 September 2017 n/a
Request for Supplementary Information (RSI) 12 October 2017 12 October 2017
Responses to RSI: 14 November 2017 13 November 2017
PRAC Rapporteur Assessment Report 20 November 2017 16 November 2017
PRAC members comments 22 November 2017 23 November 2017
Updated PRAC Rapporteur Assessment Report 23 November 2017 23 November 2017
CHMP Rapporteur Assessment Report 29 November 2017 29 November 2017
PRAC Outcome 30 November 2017 30 November 2017
CHMP members comments 4 December 2017 5 December 2017
Updated CHMP Rapporteur’s Assessment Report 7 December 2017 7 December 2017
2nd Request for Supplementary Information (RSI) 14 December 2017 14 December 2017
Responses to 2nd RSI 19 December 2017 20 December 2017
PRAC Rapporteur Assessment Report 10 January 2018
n/a
PRAC members comments 15 January 2018 15 January 2018
Updated PRAC Rapporteur Assessment Report 18 January 2018 n/a
CHMP Rapporteur Assessment Report 10 January 2018 15 January 2018
CHMP members comments 15 January 2018 15 January 2018
Updated CHMP Rapporteur’s Assessment Report 18 January 2018 n/a
Opinion 25 January 2018 25 January 2018
2. Scientific discussion
2.1. Introduction
IgPro20 is a ready-to-use 20% protein liquid formulation of a polyvalent human immunoglobulin G (IgG) preparation for subcutaneous administration. The protein moiety of IgPro20 is highly purified IgG (≥ 98% purity); more than 90% of the IgG consists of monomers and dimers. IgG function (Fc and Fab mediated activity) is retained. The sterile 20% IgG solution is formulated with 250 mmol/L L-proline and 20 mg/L polysorbate 80 at pH 4.8. IgPro20 contains no preservative. Wherever possible, specifications and analytical methods have been selected in compliance with both the USP and the Ph. Eur. The manufacturing process of the subcutaneous immunoglobulin (SCIG) solution Hizentra is based on the IgPro10 (Privigen: EMEA/H/C/831) process except for formulation and final protein concentration. Filling sizes include 5 mL (1 g), 10 mL (2 g), 15 mL (3 g) and 20 mL (4 g).
Assessment report EMA/261438/2018 Page 6/55
IgPro20 is approved in the US, EU, Switzerland, Latin America, Eastern Europe, Canada, Japan, and Australia under the trade name of Hizentra for s.c. application in the treatment of Primary Immunodeficiency (PID). In the EU, IgPro20 is also approved for replacement therapy in myeloma or chronic lymphocytic leukemia with severe secondary hypogammaglobulinemia and recurrent infections.
In general, IV immunoglobulins (IVIG) targets various cellular (such as dendritic cells, macrophages, monocytes, B and T cells) and soluble compartments (cytokines, complements, auto-antibodies, and auto-antigens) of the immune system that are involved in the pathogenesis of autoimmune disease. These mechanisms are non-exclusive and work synergistically to provide their therapeutic effects, which is essentially neutralization of the activated complement, inactivation of pro-inflammatory cytokines, downregulation of Fc receptors, adhesion to molecules on macrophage, and modulation of B-cells.
The exact pharmacotherapeutic mechanisms in auto-immune disorders are unclear; however, there is some evidence that IGs exert their action in part through up-regulation of the inhibitory FcγIIB cell receptor on effector cells, whose function is to balance the activity of activating FcγRs, dismissing inflammatory response by delivering inhibitory signals. This mechanism was shown for CIDP patients when compared to healthy subjects. In addition, number of circulating CD4+ CD25+ T- regulatory cells was shown to be reduced in CIDP patients. Increased frequency of genotype GA13-16 of the SH2D2A gene encoding for a T-cell-specific adapter protein in CIDP patients may result in a defective control and elimination of autoreactive T cells. IVlG treatment has been shown to increase numbers and function of peripheral CD4+ CD25+ T-regulatory cells in a mouse model.
CIDP is an acquired polyneuropathy within the peripheral nerve system with an assumed autoimmune-mediated pathogenesis. Its presentation is heterogeneous, and the clinical, serological, and electrophysiologic diagnostic procedures have limitations. The probable autoimmune nature of the condition is most strongly suggested by response to various immunotherapies. This assumption is further supported by the fact that the histology of active lesions is characterized by endoneurally located inflammatory mediators, deposits of complement and infiltrates of T-cells, and macrophage-associated demyelination. Patients with CIDP have symmetrical weakness in both proximal and distal muscles that worsens progressively. The condition is usually associated with impaired sensation, absent or diminished tendon reflexes, an elevated cerebrospinal fluid protein level, and changes in electrophysiology parameters. Nerve biopsy specimens are characterized by signs of demyelination. The clinical course can be relapsing or chronic and progressive, the former being much more common in young adults.
CIDP is a rare disease with an estimated prevalence of about 1.6 to 8.9 per 100,000 adults and about 0.5 per 100,000 children.
Primary treatment modalities for CIDP include intravenous immunoglobulins (IVIGs) and plasma exchange, for which there is randomized, double-blind, placebo-controlled evidence. In addition, despite less definitive published evidence of efficacy, corticosteroids are also considered as first-line therapy because of their long history of use. Studies have failed to demonstrate a difference in efficacy among these 3 treatments; consequently, the choice is usually based on availability, cost, and side-effect profile. Another therapy option for CIDP is the subcutaneous (s.c.) administration of IgG.
In the last years, possible prognostic biomarkers for CIDP have been discussed: Antibodies against NF155 (Neurofascin-155) were found in subgroups of CIDP patients and correlated with a more severe phenotype of disease, younger age at onset, ataxia, CNS demyelination and poor response to IVIG treatment. The presence of antibodies against CNTN1 (Contactin-1) characterized subgroups of CIDP patients with an acute and aggressive symptom onset, poor response to IVIG treatment but positive response to corticosteroids. Both autoantibodies could possibly serve as biomarkers to guide treatment option and therapy decision, if they were fully validated.
Rationale for the proposed change:
According to international guidelines [eg, Joint Task Force of the European Federation of Neurological Societies (EFNS) and the Peripheral Nerve Society (PNS), 2010], IVIG products have become established in the treatment of sensory and motor CIDP (recommendation level A). SCIG is an alternative treatment option for CIDP that allows patients to self-administer the product in the home setting. As demonstrated for SCIG treatment of primary immune deficiency, many patients prefer treatment at home to IVIG
Assessment report EMA/261438/2018 Page 7/55
treatment in the hospital. SCIG treatment increases autonomy, quality of life, and may reduce costs by less hospitalization. In addition, and in contrast to IVIG, serum IgG peak levels are lower and troughs are higher with SCIG; thus, a more constant IgG level is achieved, leading to a reduction in the wearing-off effect at the end of an IV treatment cycle. SCIG also results in an improved side-effect profile, with a lower rate of systemic reaction observed in SCIG studies in PID and does not require venous access that can be associated with complications. Several published trials and cases indicate the benefits of SCIG as a treatment of CIDP.
Scientific advice
CSL Behring obtained initial and follow-up Scientific Advice from EMA in February 2010 and April 2011. The main aspects discussed included:
• Study design as randomized, double-blind, placebo-controlled phase III trial
• Primary endpoint dependent on INCAT score
• Acceptability of study for type II variation
• Sample size, inclusion criteria, dose selection
• Planned extension study for evaluation of long-term safety and efficacy (IgPro20_3004)
The EMA endorsed the proposed development program for IgPro20 and the design of Study 3003, including the dosing rationale and considerations for dose selection [Advice letters EMEA/H/SA/1468/1/2009/II of 18 Feb 2010 and EMEA/H/SA/1468/1/FU/1/2011/II of 18 Apr 2011].
2.2. Non-clinical aspects
No new clinical data have been submitted in this application, which was considered acceptable by the CHMP.
2.3. Clinical aspects
2.3.1. Introduction
GCP
The Clinical trial was performed in accordance with GCP as claimed by the applicant.
The applicant has provided a statement to the effect that clinical trials conducted outside the community were carried out in accordance with the ethical standards of Directive 2001/20/EC.
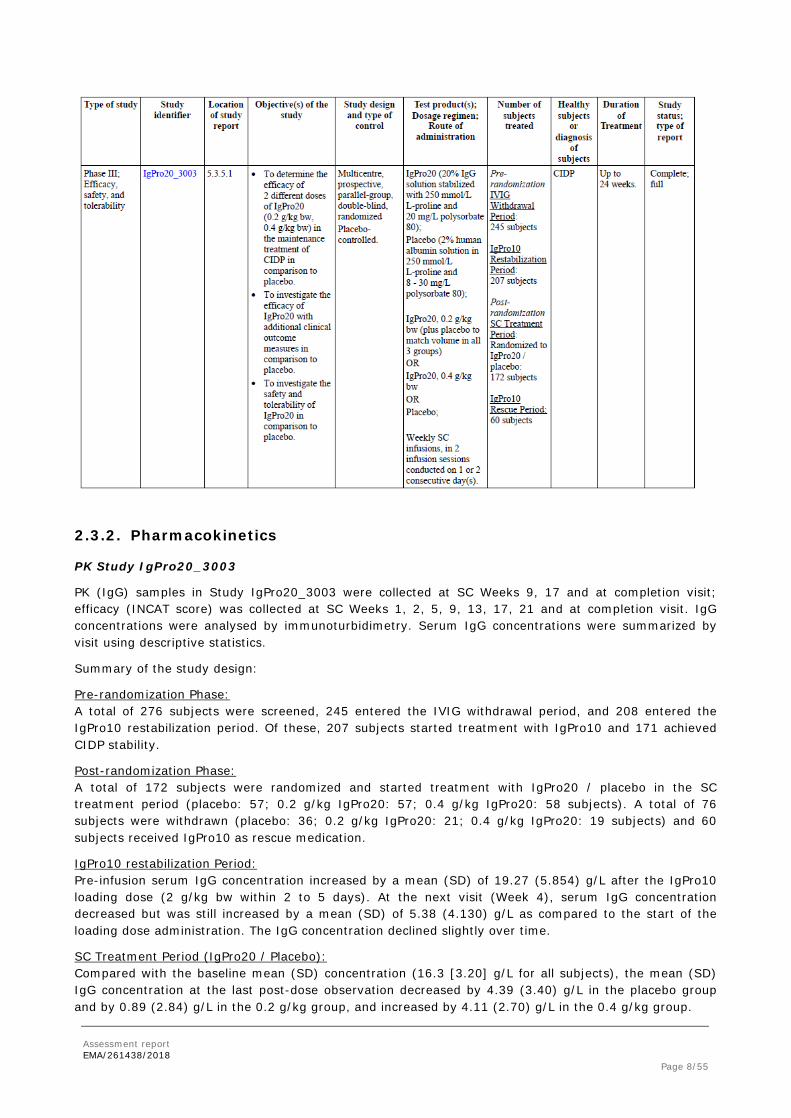
• Tabular overview of clinical study
Assessment report EMA/261438/2018 Page 8/55
2.3.2. Pharmacokinetics
PK Study IgPro20_3003
PK (IgG) samples in Study IgPro20_3003 were collected at SC Weeks 9, 17 and at completion visit; efficacy (INCAT score) was collected at SC Weeks 1, 2, 5, 9, 13, 17, 21 and at completion visit. IgG concentrations were analysed by immunoturbidimetry. Serum IgG concentrations were summarized by visit using descriptive statistics.
Summary of the study design:
Pre-randomization Phase: A total of 276 subjects were screened, 245 entered the IVIG withdrawal period, and 208 entered the IgPro10 restabilization period. Of these, 207 subjects started treatment with IgPro10 and 171 achieved CIDP stability.
Post-randomization Phase: A total of 172 subjects were randomized and started treatment with IgPro20 / placebo in the SC treatment period (placebo: 57; 0.2 g/kg IgPro20: 57; 0.4 g/kg IgPro20: 58 subjects). A total of 76 subjects were withdrawn (placebo: 36; 0.2 g/kg IgPro20: 21; 0.4 g/kg IgPro20: 19 subjects) and 60 subjects received IgPro10 as rescue medication.
IgPro10 restabilization Period: Pre-infusion serum IgG concentration increased by a mean (SD) of 19.27 (5.854) g/L after the IgPro10 loading dose (2 g/kg bw within 2 to 5 days). At the next visit (Week 4), serum IgG concentration decreased but was still increased by a mean (SD) of 5.38 (4.130) g/L as compared to the start of the loading dose administration. The IgG concentration declined slightly over time.
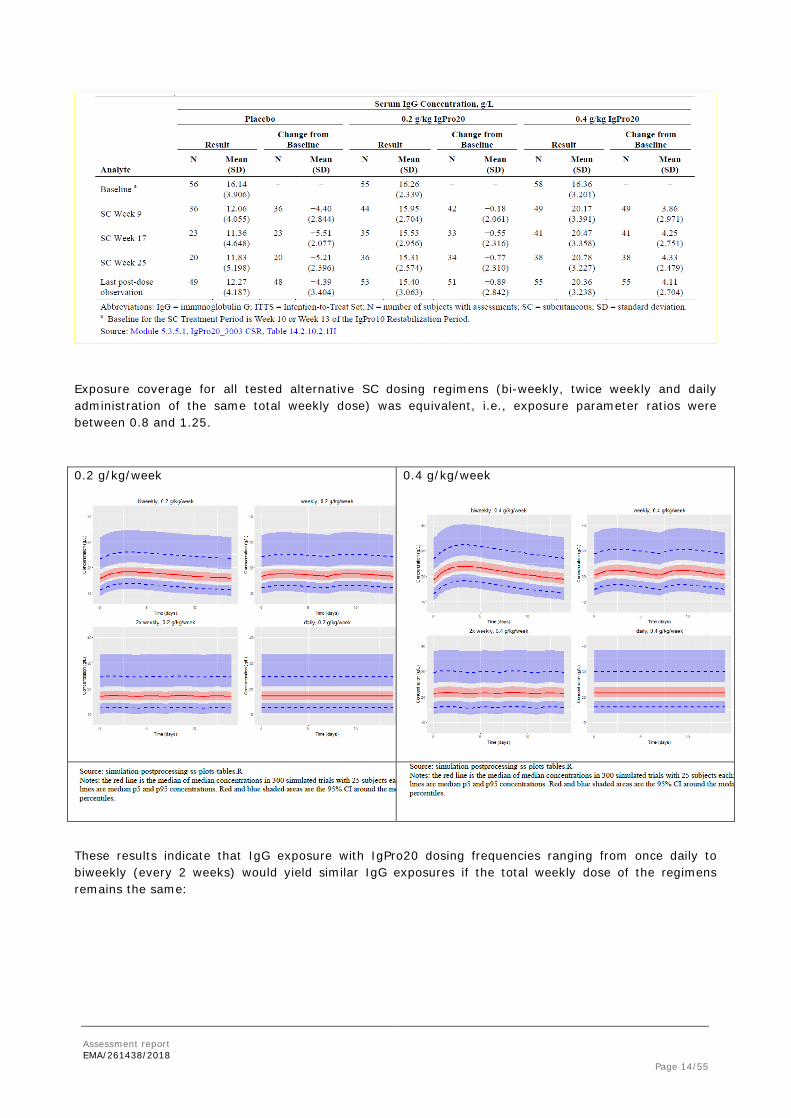
SC Treatment Period (IgPro20 / Placebo): Compared with the baseline mean (SD) concentration (16.3 [3.20] g/L for all subjects), the mean (SD) IgG concentration at the last post-dose observation decreased by 4.39 (3.40) g/L in the placebo group and by 0.89 (2.84) g/L in the 0.2 g/kg group, and increased by 4.11 (2.70) g/L in the 0.4 g/kg group.
Assessment report EMA/261438/2018 Page 9/55
Population PK Model and Model Evaluation
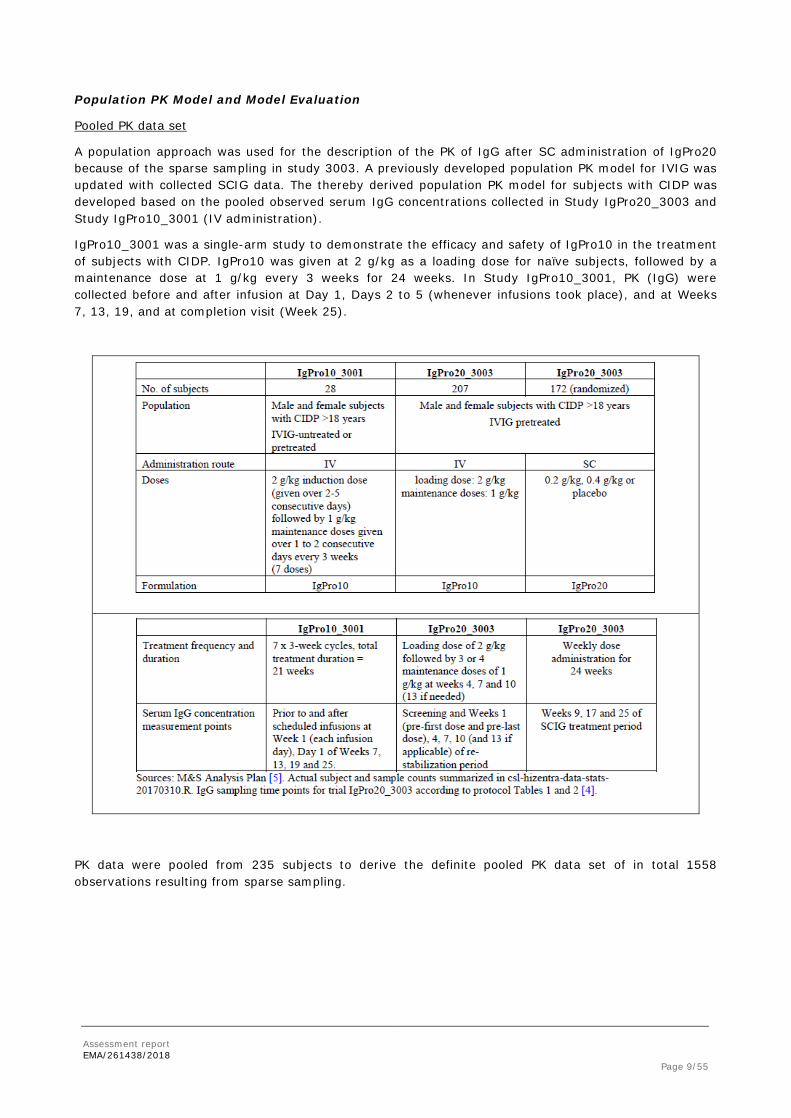
Pooled PK data set
A population approach was used for the description of the PK of IgG after SC administration of IgPro20 because of the sparse sampling in study 3003. A previously developed population PK model for IVIG was updated with collected SCIG data. The thereby derived population PK model for subjects with CIDP was developed based on the pooled observed serum IgG concentrations collected in Study IgPro20_3003 and Study IgPro10_3001 (IV administration).
IgPro10_3001 was a single-arm study to demonstrate the efficacy and safety of IgPro10 in the treatment of subjects with CIDP. IgPro10 was given at 2 g/kg as a loading dose for naïve subjects, followed by a maintenance dose at 1 g/kg every 3 weeks for 24 weeks. In Study IgPro10_3001, PK (IgG) were collected before and after infusion at Day 1, Days 2 to 5 (whenever infusions took place), and at Weeks 7, 13, 19, and at completion visit (Week 25).
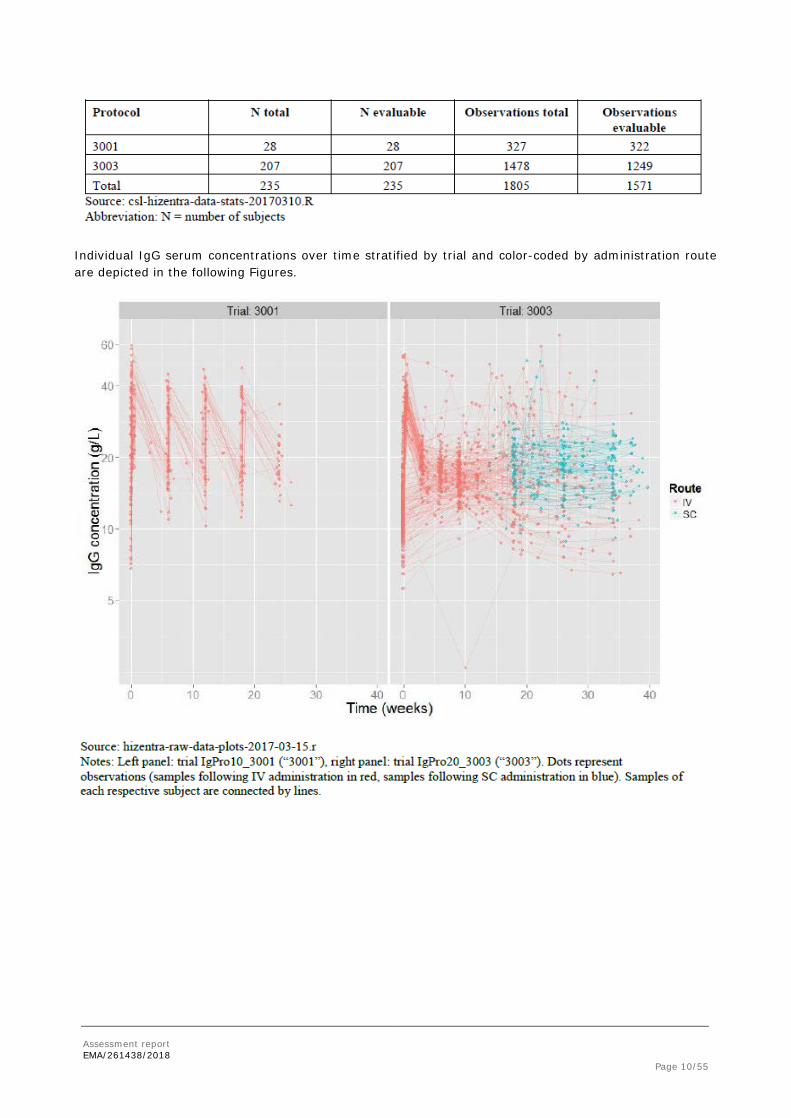
PK data were pooled from 235 subjects to derive the definite pooled PK data set of in total 1558 observations resulting from sparse sampling.
Assessment report EMA/261438/2018 Page 10/55
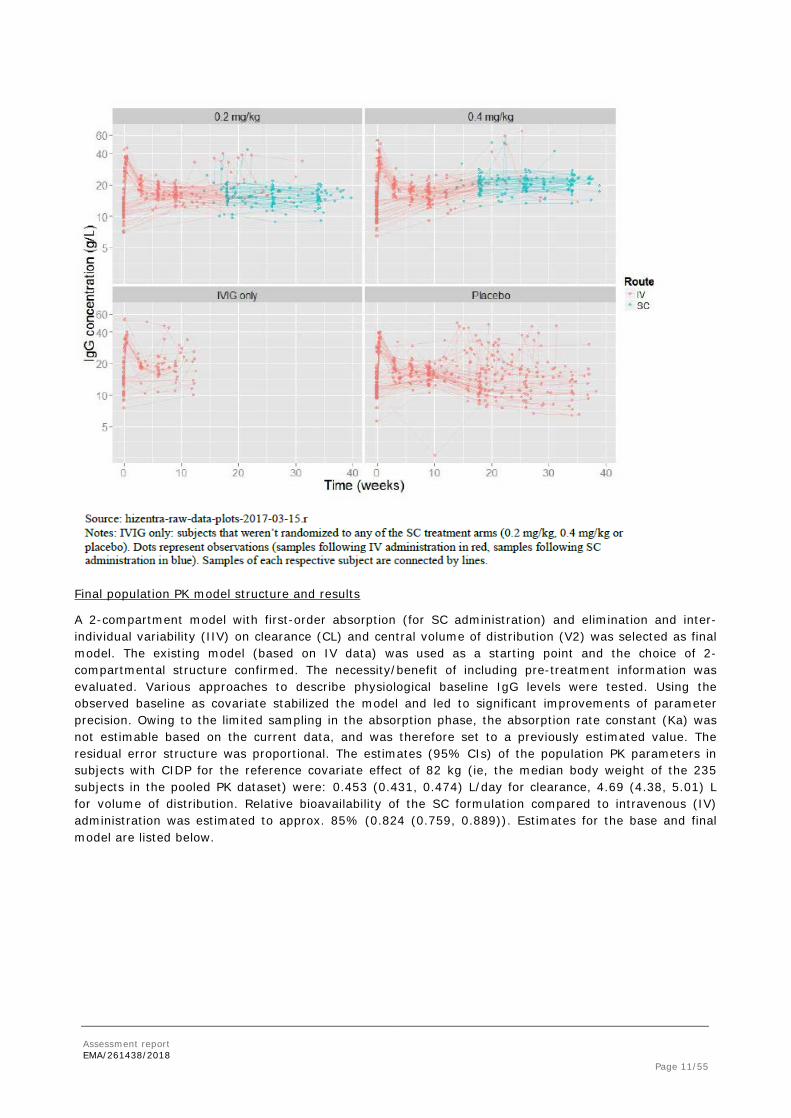
Individual IgG serum concentrations over time stratified by trial and color-coded by administration route are depicted in the following Figures.
Assessment report EMA/261438/2018 Page 11/55
Final population PK model structure and results
A 2-compartment model with first-order absorption (for SC administration) and elimination and inter-individual variability (IIV) on clearance (CL) and central volume of distribution (V2) was selected as final model. The existing model (based on IV data) was used as a starting point and the choice of 2-compartmental structure confirmed. The necessity/benefit of including pre-treatment information was evaluated. Various approaches to describe physiological baseline IgG levels were tested. Using the observed baseline as covariate stabilized the model and led to significant improvements of parameter precision. Owing to the limited sampling in the absorption phase, the absorption rate constant (Ka) was not estimable based on the current data, and was therefore set to a previously estimated value. The residual error structure was proportional. The estimates (95% CIs) of the population PK parameters in subjects with CIDP for the reference covariate effect of 82 kg (ie, the median body weight of the 235 subjects in the pooled PK dataset) were: 0.453 (0.431, 0.474) L/day for clearance, 4.69 (4.38, 5.01) L for volume of distribution. Relative bioavailability of the SC formulation compared to intravenous (IV) administration was estimated to approx. 85% (0.824 (0.759, 0.889)). Estimates for the base and final model are listed below.
Assessment report EMA/261438/2018 Page 12/55
Base population PK Model
Final population PK Model
Model evaluation
Prognostic plots of the final population PK model are shown below.
Assessment report EMA/261438/2018 Page 13/55
Model simulations
Based on these estimates, the following steady-state PK parameters for serum IgG after administration of IgPro20 (SC weekly dose of 0.2 g/kg or 0.4 g/kg) during maintenance therapy were derived in a simulated population of subjects with CIDP.
In comparison, trough serum IgG concentrations and changes from baseline associated with SC administration of IgPro20 or placebo are listed in the table below.
Assessment report EMA/261438/2018 Page 14/55
Exposure coverage for all tested alternative SC dosing regimens (bi-weekly, twice weekly and daily administration of the same total weekly dose) was equivalent, i.e., exposure parameter ratios were between 0.8 and 1.25.
0.2 g/kg/week
0.4 g/kg/week
These results indicate that IgG exposure with IgPro20 dosing frequencies ranging from once daily to biweekly (every 2 weeks) would yield similar IgG exposures if the total weekly dose of the regimens remains the same:
Assessment report EMA/261438/2018 Page 15/55
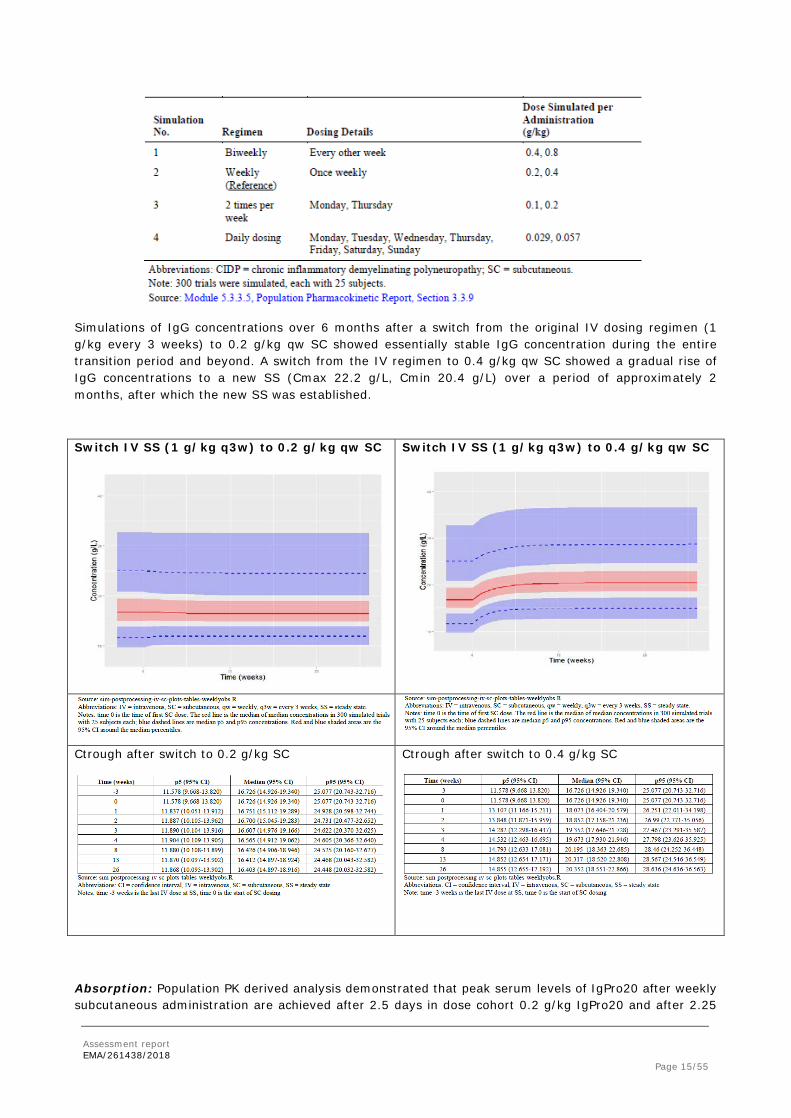
Simulations of IgG concentrations over 6 months after a switch from the original IV dosing regimen (1 g/kg every 3 weeks) to 0.2 g/kg qw SC showed essentially stable IgG concentration during the entire transition period and beyond. A switch from the IV regimen to 0.4 g/kg qw SC showed a gradual rise of IgG concentrations to a new SS (Cmax 22.2 g/L, Cmin 20.4 g/L) over a period of approximately 2 months, after which the new SS was established.
Switch IV SS (1 g/kg q3w) to 0.2 g/kg qw SC
Switch IV SS (1 g/kg q3w) to 0.4 g/kg qw SC
Ctrough after switch to 0.2 g/kg SC
Ctrough after switch to 0.4 g/kg SC
Absorption: Population PK derived analysis demonstrated that peak serum levels of IgPro20 after weekly subcutaneous administration are achieved after 2.5 days in dose cohort 0.2 g/kg IgPro20 and after 2.25
Assessment report EMA/261438/2018 Page 16/55
days in dose cohort 0.4 g/kg IgPro20, respectively. Analogously, the mean (95% CI) absolute bioavailability of SC administered IgPro20 relative to IV administered IgPro10 was estimated to be 82% (76%, 89%). The absorption constant was fixed to the value of 0.439 1/day and has been estimated in the base model to 0.143 (0.008 – 0.277) 1/day.
Distribution: The Population-PK derived estimated mean (95% CI) central volume of distribution of IgPro20 in study IgPro20_3003 was 4.69 (4.38 - 5.01) L. Peripheral volume of distribution was estimated to 1.87 (1.29 – 2.45) L.
Elimination: The elimination of IgG occurs mostly via intracellular catabolism, after fluid-phase or receptor-mediated endocytosis. Because neonatal Fc receptor (FcRn) expression in vascular endothelium and in various other organs and tissues is limited, FcRn-mediated recycling is capacity-limited. The mean (95% CI) clearance of IgPro20 was estimated to be 0.453 (0.431, 0.474) L/day.
Special populations
Covariate analysis
The following table provides a statistical summary of continuous covariates in the population PK data set. No correlations between evaluated continuous covariates were observed.
Descriptive statistics of categorical covariates are presented below. Japanese subjects had a lower median body weight and age, and a higher median baseline IgG value than non-Japanese subjects. Female subjects had a lower median body weight than males, and Baseline IgG values were slightly lower in treatment-naïve subjects than in pre-treated subjects.
Assessment report EMA/261438/2018 Page 17/55
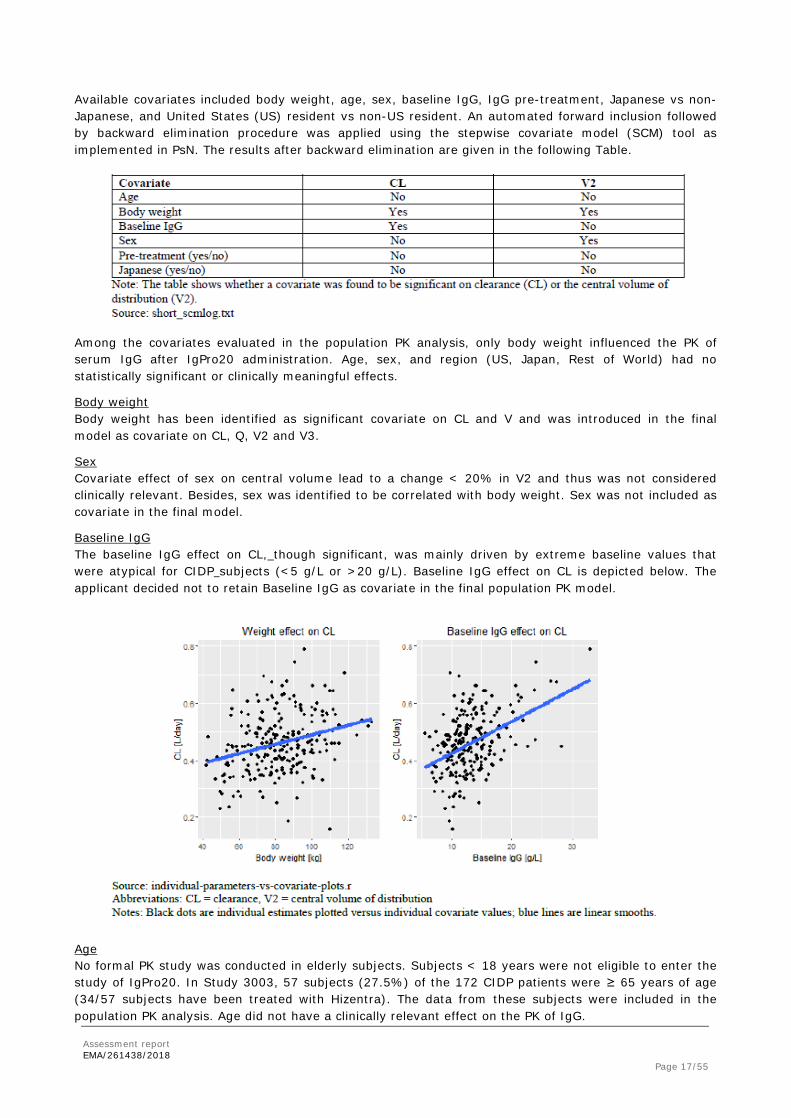
Available covariates included body weight, age, sex, baseline IgG, IgG pre-treatment, Japanese vs non-Japanese, and United States (US) resident vs non-US resident. An automated forward inclusion followed by backward elimination procedure was applied using the stepwise covariate model (SCM) tool as implemented in PsN. The results after backward elimination are given in the following Table.
Among the covariates evaluated in the population PK analysis, only body weight influenced the PK of serum IgG after IgPro20 administration. Age, sex, and region (US, Japan, Rest of World) had no statistically significant or clinically meaningful effects.
Body weight Body weight has been identified as significant covariate on CL and V and was introduced in the final model as covariate on CL, Q, V2 and V3.
Sex Covariate effect of sex on central volume lead to a change < 20% in V2 and thus was not considered clinically relevant. Besides, sex was identified to be correlated with body weight. Sex was not included as covariate in the final model.
Baseline IgG The baseline IgG effect on CL, though significant, was mainly driven by extreme baseline values that were atypical for CIDP subjects (<5 g/L or >20 g/L). Baseline IgG effect on CL is depicted below. The applicant decided not to retain Baseline IgG as covariate in the final population PK model.
Age No formal PK study was conducted in elderly subjects. Subjects < 18 years were not eligible to enter the study of IgPro20. In Study 3003, 57 subjects (27.5%) of the 172 CIDP patients were ≥ 65 years of age (34/57 subjects have been treated with Hizentra). The data from these subjects were included in the population PK analysis. Age did not have a clinically relevant effect on the PK of IgG.
Assessment report EMA/261438/2018 Page 18/55
The covariate categories “treatment-naïve” and “Japanese” contained only 15 subjects each, limiting the ability to interpret these effects. However, both were not found to be significant on clearance and central volume of distribution.
By incorporation of covariate effects on CL and V (only BW-related effects), unexplained IIV (%CV) for CL was reduced by 7% for CL and 6% for V2 compared to the base model.
2.3.3. Pharmacokinetic/Pharmacodynamic modelling
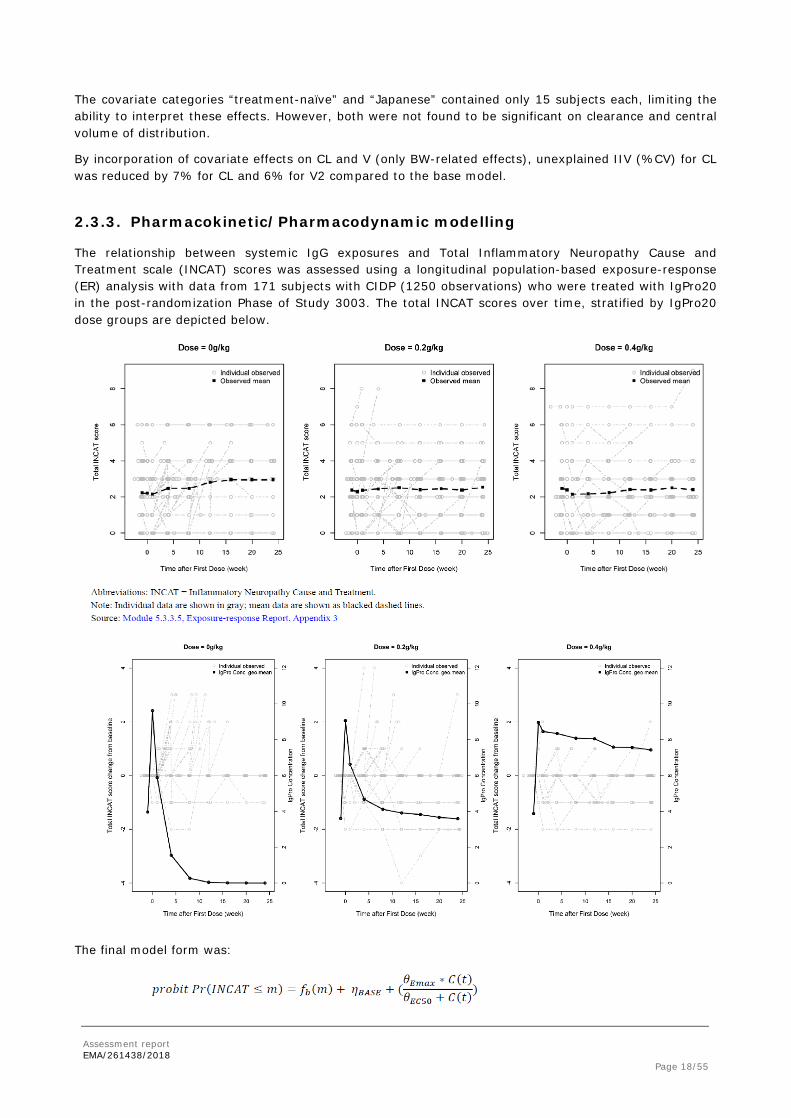
The relationship between systemic IgG exposures and Total Inflammatory Neuropathy Cause and Treatment scale (INCAT) scores was assessed using a longitudinal population-based exposure-response (ER) analysis with data from 171 subjects with CIDP (1250 observations) who were treated with IgPro20 in the post-randomization Phase of Study 3003. The total INCAT scores over time, stratified by IgPro20 dose groups are depicted below.
The final model form was:
Assessment report EMA/261438/2018 Page 19/55
With the final model estimates listed below:
The key parameter estimates from the final model included an exposure Emax of 2.27, corresponding to an infinite dose, and a half-maximal effective concentration (EC50) for exogenous IgG of 5.37 g/L. Observed and predicted total INCAT scores over time and stratified by dose groups are depicted below as visual predictive check plot.
Assessment report EMA/261438/2018 Page 20/55
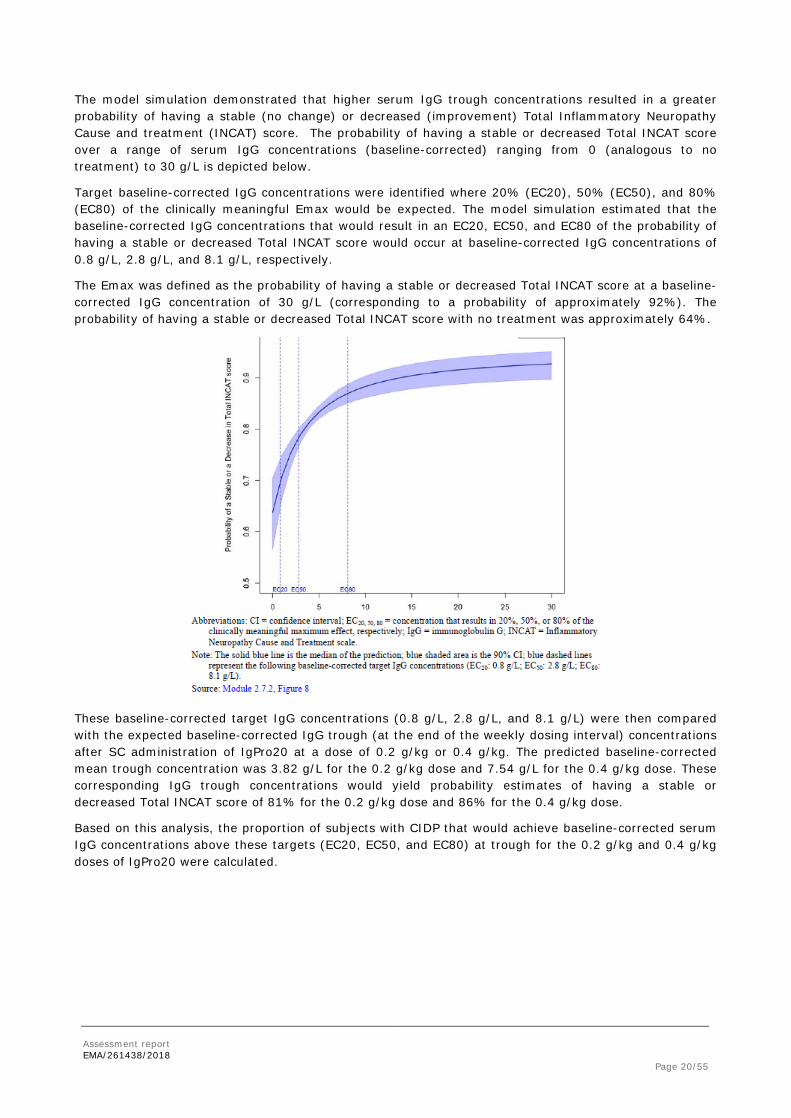
The model simulation demonstrated that higher serum IgG trough concentrations resulted in a greater probability of having a stable (no change) or decreased (improvement) Total Inflammatory Neuropathy Cause and treatment (INCAT) score. The probability of having a stable or decreased Total INCAT score over a range of serum IgG concentrations (baseline-corrected) ranging from 0 (analogous to no treatment) to 30 g/L is depicted below.
Target baseline-corrected IgG concentrations were identified where 20% (EC20), 50% (EC50), and 80% (EC80) of the clinically meaningful Emax would be expected. The model simulation estimated that the baseline-corrected IgG concentrations that would result in an EC20, EC50, and EC80 of the probability of having a stable or decreased Total INCAT score would occur at baseline-corrected IgG concentrations of 0.8 g/L, 2.8 g/L, and 8.1 g/L, respectively.
The Emax was defined as the probability of having a stable or decreased Total INCAT score at a baseline-corrected IgG concentration of 30 g/L (corresponding to a probability of approximately 92%). The probability of having a stable or decreased Total INCAT score with no treatment was approximately 64%.
These baseline-corrected target IgG concentrations (0.8 g/L, 2.8 g/L, and 8.1 g/L) were then compared with the expected baseline-corrected IgG trough (at the end of the weekly dosing interval) concentrations after SC administration of IgPro20 at a dose of 0.2 g/kg or 0.4 g/kg. The predicted baseline-corrected mean trough concentration was 3.82 g/L for the 0.2 g/kg dose and 7.54 g/L for the 0.4 g/kg dose. These corresponding IgG trough concentrations would yield probability estimates of having a stable or decreased Total INCAT score of 81% for the 0.2 g/kg dose and 86% for the 0.4 g/kg dose.
Based on this analysis, the proportion of subjects with CIDP that would achieve baseline-corrected serum IgG concentrations above these targets (EC20, EC50, and EC80) at trough for the 0.2 g/kg and 0.4 g/kg doses of IgPro20 were calculated.
Assessment report EMA/261438/2018 Page 21/55
The simulations indicate that both doses of IgPro20 SC would achieve serum IgG concentrations that exceed the EC50 (2.8 g/L) at trough, specifically 96% of subjects with CIDP would have minimum (trough) concentrations at steady-state (Ctrough) above the EC50 after receiving IgPro20 at a weekly SC dose of 0.4 g/kg, and 72% of subjects with CIDP would have Ctrough concentrations above the EC50 after receiving IgPro20 at a weekly SC dose of 0.2 g/kg. In addition, the ER model was used to compare the probability of having a stable or decreased Total INCAT score after SCIG treatment with IgPro20 (at the weekly doses of 0.2 g/kg and 0.4 g/kg investigated in Study 3003) with the probability of having a stable or decreased Total INCAT score after IVIG treatment with IgPro10 every 3 weeks at a dose of 1 g/kg, representative of an approved IVIG treatment regimen for CIDP:
A higher probability (86%) of having a stable of decreased Total INCAT score was predicted for the 0.4 g/kg SC dose of IgPro20 as a result of the higher IgG trough concentrations obtained relative to the low SC dose of IgPro20 (81%) and the IV dose of IgPro10.
2.3.4. Discussion on clinical pharmacology
PK modelling and simulation have previously been performed and successfully concluded for Hizentra in the past and this allowed for two-weekly doing and frequent dosing of more than once a week. These variations applied to the PID/SID population, in whom dosing is generally lower than in autoimmune disorders such as CIDP.
The PK of IgG after SC administration of IgPro20 was described using a population approach because of the sparse sampling in Study IgPro20_3003 (0.2 g/kg, 0.4 g/kg SC QW).This was considered acceptable by CHMP. A previously developed population PK model for IV administered IgG was updated with SC data. PK data were pooled from 235 subjects to derive the definite pooled PK data set of in total 1558 observations resulting from both studies, IgPro10_3001 (IV, N =28) and IgPro20_3003 (IV & SC, N =207).
A 2-compartment model with first-order absorption (for SC administration) and elimination and inter-individual variability (IIV) on clearance (CL) and central volume of distribution (V2) was selected to describe SC PK data. Due to limited sampling in the absorption phase, the absorption rate constant (Ka) could not properly be estimated based on the current data and instead, literature references were used.
Assessment report EMA/261438/2018 Page 22/55
As requested by the CHMP, a sensitivity analysis was provided showing that varying ka values over the range 0.05 – 0.6 has only minor influence on the objective function values and consequently also on trough concentrations in the steady state. This mirrors the poor identifiability of ka and in turn the low quantitative predictive power. Thus, the choice of the more robust literature-based value is acceptable. It is agreed that differences in ka between PID and CIDP patients are not expected. Due to the above mentioned reasons the model is not qualified to detect possible differences in ka.
Diagnostic plots and VPC plots showed that the PK of IgG following IV and SC administration was in general well characterized. Relating observed and predicted values showed an over-estimation of small values, high values are slightly over-predicted on the individual level, however, these are not considered substantial.
The estimates (95% CIs) of the population PK parameters were: 0.453 (0.431, 0.474) L/day for clearance, 4.69 (4.38, 5.01) L for the central volume and 1.87 (1.29, 2.45) L for the peripheral volume of distribution. Relative bioavailability of the SC formulation compared to intravenous administration was estimated to approx. 85% (0.824 (0.759, 0.889)).
Body weight had a significant impact on both IgG CL and V2. By incorporation of covariate effects (only BW-related effects), unexplained IIV (%CV) for CL (28 %CV) was reduced by 7% and by 6% for V2 (23 %CV) compared to the base model.
Based on the final model estimates, the following steady-state PK parameters for serum IgG after administration of IgPro20 (SC weekly dose of 0.2 g/kg or 0.4 g/kg, 6 months) during maintenance therapy were derived after switching from IV regimen to SC in subjects with CIDP.
While the 0.2 g/kg SC qw dose level (trough levels) stayed almost unchanged at the SS after standard IV administration (Cmax 17.4 g/L, Cmin 16.5 g/L), simulations of 0.4 g/kg SC qw dosing resulted in a gradual rise of IgG concentrations to a slightly higher SS level over approximately 2 months (Cmax 22.2 g/L, Cmin 20.4 g/L). This is in accordance with the data collected from SC treatment period: Compared with the Baseline mean (SD) concentration (16.3 [3.20] g/L for all subjects), the mean (SD) IgG concentration at the last post-dose observation decreased by 4.39 (3.40) g/L in the placebo group and by 0.89 (2.84) g/L in the 0.2 g/kg group, and increased by 4.11 (2.70) g/L in the 0.4 g/kg group. In the placebo group IgG Ctrough values decline after approx. Week 10. Thus, the carry-over effect of IVIG to IgPro 20 could also be estimated to last this length of time.
Model-based simulations indicate that flexible SC dosing scenarios (bi-weekly, weekly, twice-weekly and daily administration) would lead to equivalent exposure. A switch from the established IV regimen to one of proposed SC weekly dose levels (0.2 g/kg, 0.4 g/kg) would achieve comparable trough IgG concentration levels with slightly higher level in the steady state regarding the 0.4 g/kg SC dosing regimen.
In conclusion, the derived population PK model described the PK of IgPro20 for SC use acceptably well.
PK/PD
The pharmacodynamics IgPro20 effect was described by an Emax model that relates exogenous IgG concentration with INCAT score changes observed. Data, population based PK analysis and simulation indicate that there is a dose-response relations with regard to efficacy. Weekly dosing of 0.4 g/kg SC
Assessment report EMA/261438/2018 Page 23/55
resulted in a more pronounced change from baseline and higher Ctrough IgG values as compared to 0.2 g/kg dosing. Exposure-efficacy response was described by an Emax model; however the EC50 value could not be estimated precisely.
Model-based simulations indicate that both doses of IgPro20 achieve serum IgG concentrations that exceed the EC50 (2.8 g/L). Specifically 96% of subjects with CIDP would have minimum (trough) concentrations at steady-state above the EC50 after receiving IgPro20 at a weekly SC dose of 0.4 g/kg, and 72% of subjects with CIDP would have Ctrough concentrations above the EC50 after receiving IgPro20 at a weekly SC dose of 0.2 g/kg. Only 4% of the patients would achieve Ctrough above the EC80 (8.1 g/L) in the 0.2 g/kg group, but 44% of the patients that have been dosed with 0.4 g/kg SC. Analogously, a higher probability (86%) of having a stable of decreased Total INCAT score is expected for the 0.4 g/kg SC dose of IgPro20 as a result of the higher IgG trough concentrations obtained relative to the low SC dose of IgPro20 (81%) and the IV dose of IgPro10.
In conclusion, as diagnostic plots regarding the population PK as well as for the PK-PD model indicate that precise quantitative predictions based on these models might be biased, nevertheless, data and model indicate a relationship between dose/exposure and efficacy that is in favour of the 0.4 g/kg SC dosing regimen.
2.3.5. Conclusions on clinical pharmacology
Overall. the clinical pharmacology data provided for this new indication of Hizentra for use in CIDP patients is considered adequate by CHMP.
2.4. Clinical efficacy
2.4.1. Main study
Title of Study
Study 3003 (PATH Study) This was a phase 3, prospective, multi-center, international, randomized, double-blind, placebo-controlled, parallel-group, 3-arm study with 2 study phases: a Pre-randomization Phase (consisting of an IVIG Withdrawal Period up to 12 weeks and an IgPro10 Restabilization Period with IgPro10 of 10 or 13 weeks) and a Post-randomization Phase (consisting of a randomized placebo-controlled s.c. Treatment Period with 2 doses of IgPro20 for 24 weeks and an IgPro10 Rescue Period).
Methods
Study Design
Assessment report EMA/261438/2018 Page 24/55
Deterioration: A clinically meaningful deterioration is defined as a total INCAT disability score increase by≥1 point, I-RODS deterioration by≥4 points (using the centile metric), or a mean grip strength deterioration by≥8 kiloPascal (kPa) in one hand using the handheld vigorimeter. Restabilisation: Only patients whose INCAT total score improves to at least the INCAT total score recorded at the screening visit (i.e., ≥ INCAT score at screening) and who maintain a stable INCAT total score at weeks 7 and 10 (or at weeks 10 and 13) are eligible for randomization. Relapse: Relapse is defined as an increase of ≥ 1 point in adjusted INCAT score compared with Baseline (for full definition see 1° endpoint). Visit periods:
Study participants
Inclusion criteria
1. Definite or probable CIDP according to the EFNS / PNS criteria 2010 2. Age ≥ 18 years. 3. Male or female. 4. Written informed consent for study participation obtained before undergoing any study-specific
procedures. Additional Inclusion Criterion to Enter IgPro10 Restabilization Period: All subjects were required to experience CIDP deterioration (i.e. before amendment 3, an increase in adjusted INCAT score by ≥ 1 point. After amendment 3, an increase in adjusted INCAT score by ≥ 1 point, a decrease in R-ODS total
Assessment report EMA/261438/2018 Page 25/55
score by ≥ 4 points, or a decrease in mean grip strength by ≥ 8 kPa) before entering the IgPro10 Restabilization Period.
Exclusion criteria (summarized)
There was a number of exclusion criteria, amongst these were:
1. Any polyneuropathy of other causes 2. Any other disease (mainly neurological or chronic orthopedic) that has caused neurological symptoms or may interfere with treatment or outcome assessments 3. Severe diseases and conditions that are likely to interfere with evaluation of the study product or satisfactory conduct of the study (e.g. current malignancy or history of allogeneic bone marrow / stem cell transplant, cardiac insufficiency, cardiomyopathy, significant cardiac arrhythmia requiring treatment, unstable or advanced ischemic heart disease, congestive heart failure or severe hypertension, chronic kidney disease stage IV and V, etc) 4. History of thrombotic episodes within the 2 years before enrolment 5. Known allergic or other severe reactions to blood products including intolerability to previous IVIG And other criteria.
Treatments
IVIG during pre-study/screening: Subjects received their last IVIG during Pre-study / Screening (before amendment 3) or after Screening eligibility determination (after amendment 3) before the start of the IVIG Withdrawal Period. Subjects received their regular / required non-study IVIG. Any locally available IVIG was used. The dosage was the same dosage the subject usually received or the dose the subject required by judgment of the Treating Physician. IVIG withdrawal period: No IVIG was administered during the IVIG Withdrawal Period (for other medication see “Concomitant Medication” below). IgPro10 during IgPro10 restabilization period: Eligible subjects were treated with the IVIG product IgPro10, administered as follows: • 1 loading dose of 2 g/kg bw, administered over 2 to 5 consecutive days (in Japan: dose was given
over 5 days), with a maximum of 1 g/kg bw on a single day, followed by • 3 or 4 maintenance doses (depending on the time needed for restabilization) of 1 g/kg bw given
every 3 weeks over 1 or 2 consecutive days. Maintenance treatment was given at Weeks 4, 7, and 10 (and Week 13, if needed).
Subjects who did not achieve CIDP stability (i.e. CIDP status did not show a clinically meaningful difference during the last 2 visits. In addition, to be considered CIDP stable, the CIDP status had to recover back to at least the status at Screening, as assessed by adjusted INCAT score) during the last 2 visits (either Weeks 7 and 10 or Weeks 10 and 13) were discontinued and not randomized.
SC treatment period with IgPro20 or placebo: The SC treatment dose with IgPro20 or placebo was based on body weight, and eligible subjects were randomized to weekly SC infusions for 24 weeks to 1 of the following 3 treatment groups: • IgPro20 at 0.2 g/kg bw. • IgPro20 at 0.4 g/kg bw. • Placebo (2% human albumin solution).
The dose was administered once a week in 2 infusion sessions conducted on 1 or 2 consecutive day(s).
Rescue medication with IgPro10: Subjects who experienced CIDP relapse (i.e. an increase of ≥ 1 point in adjusted INCAT score compared to Baseline) during the SC Treatment Period were withdrawn from further SC treatment and were offered IgPro10 as rescue treatment within 1 week of CIDP relapse determination. Before any rescue therapy was administered, all assessments were performed. Rescue treatment with IgPro10 included:
Assessment report EMA/261438/2018 Page 26/55
• 1 loading dose of 2 g/kg bw, administered over 2 to 5 consecutive days (in Japan: dose was given over 5 days), with a maximum of 1 g/kg bw on a single day, followed by:
• A maximum of 4 maintenance doses of 1 g/kg bw given every 3 weeks over 1 or 2 consecutive days, depending on the time needed to return to Baseline INCAT score.
INCAT score was assessed before the subsequent IgPro10 maintenance dose. If the INCAT score had improved (i.e., the INCAT score returned back to or below the Baseline score), IgPro10 was administered and Completion Visit tasks were performed.
If INCAT score had not improved, another maintenance dose was administered and INCAT was assessed according to above-mentioned procedure at the next visit scheduled 3 weeks later.
If INCAT score had not improved after 4 maintenance doses of IgPro10, Completion Visit tasks were performed and further treatment was at the discretion of the Treating Physician. Concomitant medication: Concomitant CIDP treatments eg, methotrexate, azathioprine, mycophenolate corticosteroids (maintenance dose ≤ 20 mg), topical and inhaled corticosteroids, or topical immunosuppressants were permitted, provided that their dose and frequency were kept stable during the whole study and were stable during the 3 months before enrolment.
Objectives
Primary objective To determine the efficacy of 2 different doses of IgPro20 (0.2 g/kg bw and 0.4 g/kg bw) in the maintenance treatment of CIDP in comparison to placebo.
Secondary objectives • To investigate the efficacy of IgPro20 with additional clinical outcome measures in comparison to
placebo. • To investigate the safety and tolerability of IgPro20 in comparison to placebo. • To investigate the safety and efficacy of IgPro10 restabilization therapy. • To investigate the safety and efficacy of IgPro10 rescue therapy.
Exploratory objectives • To investigate health-related quality of life (HRQL) following treatment with IgPro20. • To investigate exploratory safety and efficacy endpoints. • To investigate serum IgG concentrations. • To investigate the effect of IgPro20 on electrophysiology parameters.
Outcomes/endpoints
Primary Endpoint The primary efficacy endpoint was the percentage of subjects who had CIDP relapse during the SC Treatment Period or were withdrawn from SC treatment for any reason. CIDP relapse was defined as follows: An increase of ≥ 1 point in adjusted INCAT score compared with Baseline, excluding an increase in INCAT score of 1 point if this is only due to an increase of the arm score from 0 to 1 (not clinically meaningful worsening) or an unchanged adjusted INCAT score compared with Baseline where the arm score decreased from 1 to 0 (not clinically meaningful improvement) and the leg score increased by 1 point (clinically meaningful worsening). Secondary Endpoints Efficacy:
• Changes in means during SC Treatment Period between groups in (i) INCAT score, (ii) maximum grip strength (dominant/non-dominant hand), (iii) MRC sum score, and (iv) R-ODS.
• Difference in “time to CIDP relapse” using a Kaplan-Meier estimation comparing both IgPro20 groups with placebo as well as the 2 IgPro20 groups pair-wise.
Assessment report EMA/261438/2018 Page 27/55
Safety: • Rate of AEs per infusion during the SC Treatment Period, grouped by Medical Dictionary for Regulatory
Activities (MedDRA) system organ class (SOC) and preferred term (PT).
• Number and percentage of subjects with AEs during the SC Treatment Period, grouped by MedDRA, SOC and PT.
Efficacy of IgPro10 and IgPro20 was assessed on the basis of the following variables: • INCAT score. • R-ODS score. • Mean grip strength. • MRC sum score (8 muscle groups). • Electrophysiology parameters: distal and proximal latencies, Compound action potential (CMAP)
amplitudes, nerve conduction velocities, and conduction block in 3 motor nerves (SC Treatment Period only)
Safety was assessed on the basis of the following variables recorded during the study: • AEs. • Laboratory safety parameters (haematology and serum chemistry). • Vital signs. • Physical examination. • 12-lead electrocardiogram (ECG) (Japan only)
During the double-blind SC treatment period, randomization to 0.2 g/kg bw IgPro20, 0.4 g/kg bw IgPro20, or placebo was controlled centrally by the Interactive Voice Response System (IVRS) / Interactive Web Response System (IWRS). Randomization was stratified by region (Japan versus non-Japan) to ensure that the treatments were evenly distributed among the subgroup of Japanese patients.
Blinding (masking)
All subjects and study personnel were blinded to IgPro20/placebo treatment. Standard measures were taken for the 2 doses of IgPro20 and placebo to ensure adequate blinding of the investigational product. In addition, the investigators and subjects were blinded to the randomized treatment assignment. In addition, the blind was preserved by administration of the same volume for all 3 treatment groups (subjects randomized to 0.2 g/kg bw IgPro20 received 1 session of IgPro20 and 1 session of placebo, randomly selected, each week.) To minimize potential unblinding, a 2-physician approach (Treating Physician / Evaluating Physician) was used. The results of immunoglobulin concentration assessments remained blinded until post-database lock. For the planned interim analysis and ongoing risk-benefit evaluations, members of the IDMC were unblinded. Access to study documents containing information on IgG concentrations and treatment groups was restricted to ensure that no person involved in operations, analysis, or management of the study was unblinded.
Assessment report EMA/261438/2018 Page 28/55
Statistical methods
Study IgPro20_3003 is a randomized, multicenter, double-blind, placebo-controlled phase III study. The study has three arms; a low-dose, a high-dose, and a placebo arm with 57, 58, and 57 subjects randomized respectively (planned with the sample size calculation according to study protocol were 58 subjects each).
The study was powered to demonstrate that at least the high dose of IgPro20 is superior to placebo with regard to the primary efficacy endpoint. A monotonic dose-response was expected for the primary endpoint, with placebo ≥ IgPro20 low dose ≥ IgPro20 high dose and with at least 1 strict inequality among the 2 non-strict inequalities. This means that if the null hypothesis was rejected, the primary efficacy endpoint would have a statistically significant higher result for placebo than for at least 1 of the investigated doses. Based on the (recalculated) results from the ICE study extension period [Hughes et al, 2008; Hughes, 2009], it was assumed that the percentages of subjects who would relapse during SC treatment were 35% for the IgPro20 high dose, 52% for the IgPro20 low dose, and 65% for placebo. Further it was assumed that 60% of subjects were randomized under protocol amendment 3 or later. The proportion of IVIG-independent subjects included in the study was assumed to be 15%. It was further assumed that these subjects would have a relapse rate of 10% regardless of the treatment. A discontinuation rate of 15% due to other reasons than CIDP relapse in the placebo group and 10% in the IgPro20 treatment groups after implementation of amendment 3 was included in the calculation of the relapse rates. The exact CA trend test with equally spaced scores was used for the purpose of sample size calculation. With a 1-sided significance level of 2.5%, a sample size of 58 was needed in each treatment group to achieve a power of approximately 90% in the ITT analysis based on the above assumptions. Thus, the overall planned sample size was 174 subjects treated either with IgPro20 or placebo. Approximately 350 subjects were planned to be enrolled to ensure that 174 subjects would be treated with IgPro20 or placebo.
Results
Participant flow
Assessment report EMA/261438/2018 Page 29/55
Recruitment
A total of 172 subjects were randomized and started treatment with IgPro20 / placebo in the SC Treatment Period of the Post-randomization Phase. A total of 57 subjects received placebo; of these, 36 subjects were withdrawn (CIDP relapse: 32 subjects, withdrawal by subject: 3 subjects, physician decision: 1 subject). A total of 57 subjects received 0.2 g/kg bw IgPro20; of these, 21 subjects were withdrawn (CIDP relapse: 18 subjects, withdrawal by subject: 2 subjects, adverse event [AE]: 1 subject). A total of 58 subjects received 0.4 g/kg bw IgPro20; of these, 19 subjects were withdrawn (CIDP relapse: 10 subjects, withdrawal by subject: 8 subjects, AE: 1 subject). The demographic and primary disease characteristics of the ITTS were balanced across the treatment groups, except for sex: there were more male subjects in the 0.2 g/kg IgPro20 group (42 subjects [73.7%]) than in the 0.4 g/kg IgPro20 group (31 subjects [53.4%]). In the placebo group, 37 subjects (64.9%) were male. All subjects received prior treatment with immunoglobulins before enrolment. There was no dose-related pattern in the use of concomitant medication.
Conduct of the study
Protocol amendments
Assessment report EMA/261438/2018 Page 30/55
The following two important changes were implemented:
(1) The IVIg withdrawal phase was modified to an IgG dependency test and additional deterioration criteria were implemented as described above. Fulfillment on one of these criteria, in the event of an unchanged INCAT score, qualified the patient to move to the next study phase (Amendment 3). Relapse rates in the IgPro20 groups were anticipated to increase after this change due to the fact that significant decrease in grip strength (i.e., 8-point deterioration) is not always accompanied by a corresponding worsening in adjusted INCAT score by 1 point. To correct for the new assumptions for relapse percentages underlying the power calculation, the sample size was increased from 150 to 174, and the screening numbers were increased from 250 to 350. The underlying assumptions were that 90 % of subjects would be recruited after Amendment 3 and the dropout rates for placebo subjects would increase to 15 % (while being around 10 % in the active treatment groups).
(2) The length of time required for prestudy IVIg has been reduced to 8 weeks. The change in this requirement is not expected to adversely affect the outcomes of the SC treatment period because all patients must show IgG dependency (up to 12 weeks) and IVIg restabilization (up to 13 weeks) before randomization and start of SC treatment.
Assessment report EMA/261438/2018 Page 31/55
Protocol violations
Baseline data
Demographic and Baseline characteristics
Primary Disease Characteristics at Screening
Assessment report EMA/261438/2018 Page 32/55
In the PSDS, the median time since CIDP diagnosis was 3.0 years (0.1 to 33.5 years). A total of 185 subjects (89.4%) had a definite CIDP diagnosis per the EFNS / PNS diagnostic criteria. The mean (SD) INCAT score at Screening was 2.7 (1.67) points. The minimum (0 points) and maximum (8 points) INCAT scores show that subjects were affected by the disease to very different degrees. The ITTS and the RSDS had similar primary disease characteristics as the PSDS. The primary disease characteristics were balanced across treatment groups in the ITTS. In the PSDS, the most frequent SOC recorded in medical history was Vascular Disorders (94 subjects, 45.4%), followed by Musculoskeletal and Connective Tissue Disorders (78 subjects, 37.7%) and Metabolism and Nutrition Disorders (72 subjects, 34.8%). The most frequent PT recorded in medical history was Hypertension (85 subjects, 41.1%), followed by Hypercholesterolaemia (25 subjects, 12.1%).
Prior Treatment With Immunoglobulins: All subjects in the PSDS received prior treatment with immunoglobulins before enrollment. A total of 56 subjects (27.1%) in the PSDS received at least 1 dose of Privigen before enrollment. The mean (SD) IVIG dose in the 3 months before Screening in the PSDS was 2.6 (1.52) g/kg bw. A similar result was observed for the ITTS, the mean IVIG dose was balanced across the treatment groups.
Prior Medication for CIDP (Excluding Treatment With Immunoglobulins): During the IVIG Withdrawal Period, 21 (10.1%) of the subjects in the PSDS received Glucocorticoids and 24 (11.6%) subjects received Other Analgesics and Antipyretics that were reported as CIDP medication. A small proportion of subjects (10 subjects, 4.8%) in the PSDS received prior medication, mainly Glucocorticoids (6 subjects, 2.9%), for CIDP other than IgG within a maximum of 6 months before enrollment. Prior medication was not analyzed for the ITTS.
Prior Medication (Excluding Treatment of CIDP): The most frequently used medications in the PSDS during the IVIG Withdrawal Period were Proton Pump Inhibitors (43 subjects, 20.8%), ACE Inhibitors (33 subjects, 15.9%), and 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG CoA) Reductase Inhibitors (30 subjects, 14.5%). The most frequently used prior medications in the PSDS were Proton Pump Inhibitors (5 subjects, 2.4%) and Contact Laxatives (3 subjects, 1.4%). Prior medication was not analyzed for the ITTS.
Numbers analysed
A total of 172 subjects were randomized and started treatment with IgPro20 / placebo in the SC Treatment Period of the Post-randomization Phase. A total of 57 subjects received placebo; of these, 36
Assessment report EMA/261438/2018 Page 33/55
subjects were withdrawn (CIDP relapse: 32 subjects, withdrawal by subject: 3 subjects, physician decision: 1 subject). A total of 57 subjects received 0.2 g/kg bw IgPro20; of these, 21 subjects were withdrawn (CIDP relapse: 18 subjects, withdrawal by subject: 2 subjects, adverse event [AE]: 1 subject). A total of 58 subjects received 0.4 g/kg bw IgPro20; of these, 19 subjects were withdrawn (CIDP relapse: 10 subjects, withdrawal by subject: 8 subjects, AE: 1 subject).
Outcomes and estimation
Efficacy results are given on the ITTS group. The per-protocol analyses of the efficacy data supported the overall efficacy results obtained from the ITTS. Study 3003 was considered successful if a superiority of at least 1 dose of IgPro20 over placebo was shown.
Primary Efficacy Endpoint
CIDP relapse
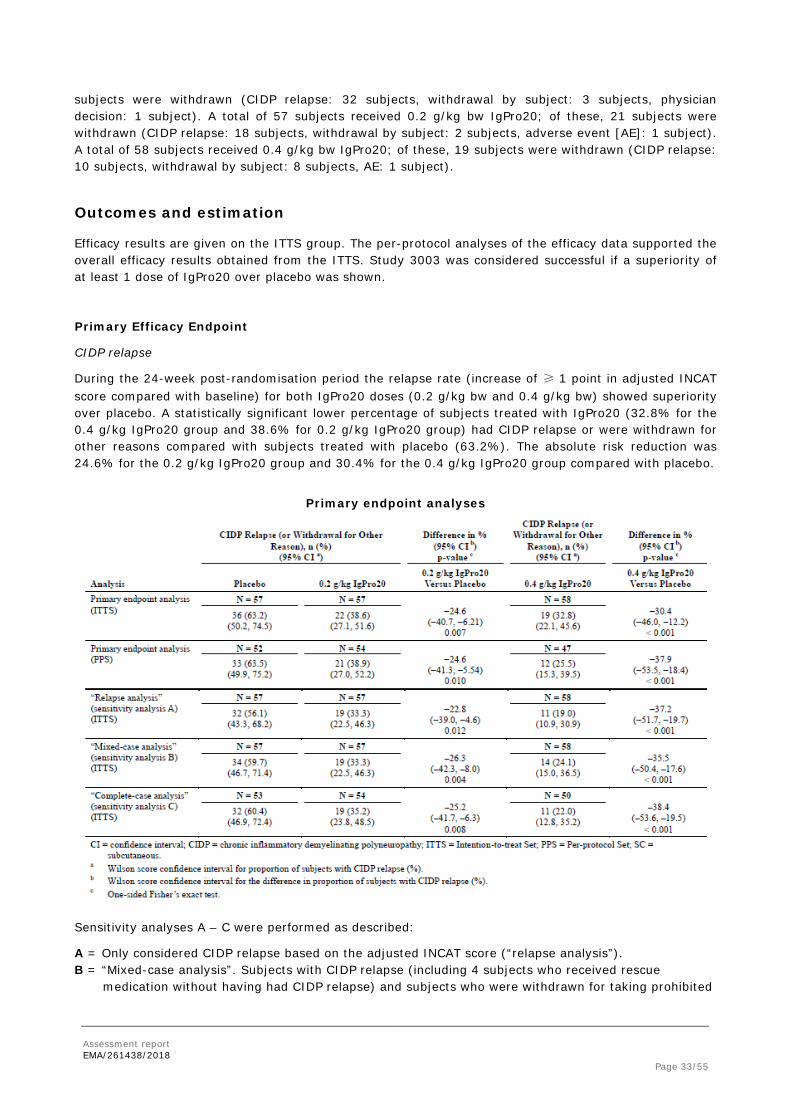
During the 24-week post-randomisation period the relapse rate (increase of ≥ 1 point in adjusted INCAT score compared with baseline) for both IgPro20 doses (0.2 g/kg bw and 0.4 g/kg bw) showed superiority over placebo. A statistically significant lower percentage of subjects treated with IgPro20 (32.8% for the 0.4 g/kg IgPro20 group and 38.6% for 0.2 g/kg IgPro20 group) had CIDP relapse or were withdrawn for other reasons compared with subjects treated with placebo (63.2%). The absolute risk reduction was 24.6% for the 0.2 g/kg IgPro20 group and 30.4% for the 0.4 g/kg IgPro20 group compared with placebo.
Primary endpoint analyses
Sensitivity analyses A – C were performed as described:
A = Only considered CIDP relapse based on the adjusted INCAT score (“relapse analysis”). B = “Mixed-case analysis”. Subjects with CIDP relapse (including 4 subjects who received rescue
medication without having had CIDP relapse) and subjects who were withdrawn for taking prohibited
Assessment report EMA/261438/2018 Page 34/55
medication and subjects who were withdrawn due to physician’s decision were considered relapsers. All other subjects were considered non-relapsers.
C = “Complete-case analysis”. All subjects who were withdrawn for any other reason than CIDP relapse were excluded from the analysis; thus, the population included in this analysis was smaller than that in the primary endpoint analysis.
Secondary Efficacy Endpoints
Time to CIDP Relapse (or Withdrawal for any Other Reason)
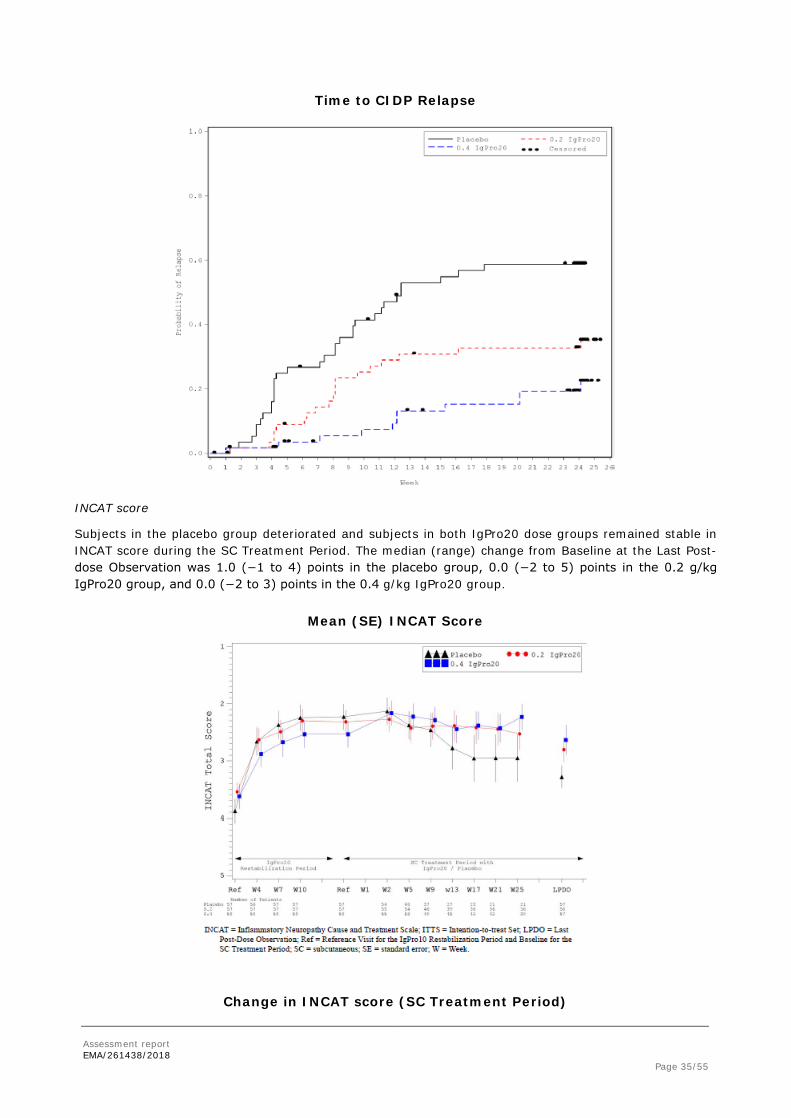
After switching from the IgPro10 Restabilization Period to the randomized SC Treatment Period with IgPro20 / placebo, subjects in the placebo group had a rapidly increasing probability of CIDP relapse or withdrawal from SC Week 3 onwards. In the 0.2 g/kg IgPro20 group, the probability of relapse or withdrawal increased above the level of the 0.4 g/kg IgPro20 group at SC Week 6 and remained higher until the end of the SC Treatment Period (Figure E 3). In the 0.4 g/kg IgPro20 group, a gradual increase was observed. At Week 25, the probability of CIDP relapse or withdrawal for any other reason was 0.63 for placebo, 0.39 for 0.2 g/kg IgPro20, and 0.34 for 0.4 g/kg IgPro20.
Time to CIDP Relapse or Withdrawal for any Other Reason
• Censored: Subjects who neither relapsed nor withdrew for any other reason were censored at the date of their Completion Visit.
Time to CIDP Relapse
The probability of CIDP relapse alone increased in the 0.2 g/kg IgPro20 group above the level of the 0.4 g/kg IgPro20 group at SC Week 4 and remained higher until the end of the SC Treatment Period (Figure E 4). At Week 25, the probability of CIDP relapse was 0.59 for placebo, 0.35 for 0.2 g/kg IgPro20, and 0.22 for 0.4 g/kg IgPro20.
Assessment report EMA/261438/2018 Page 35/55
Time to CIDP Relapse
INCAT score
Subjects in the placebo group deteriorated and subjects in both IgPro20 dose groups remained stable in INCAT score during the SC Treatment Period. The median (range) change from Baseline at the Last Post-dose Observation was 1.0 (−1 to 4) points in the placebo group, 0.0 (−2 to 5) points in the 0.2 g/kg IgPro20 group, and 0.0 (−2 to 3) points in the 0.4 g/kg IgPro20 group.
Mean (SE) INCAT Score
Change in INCAT score (SC Treatment Period)
Assessment report EMA/261438/2018 Page 36/55
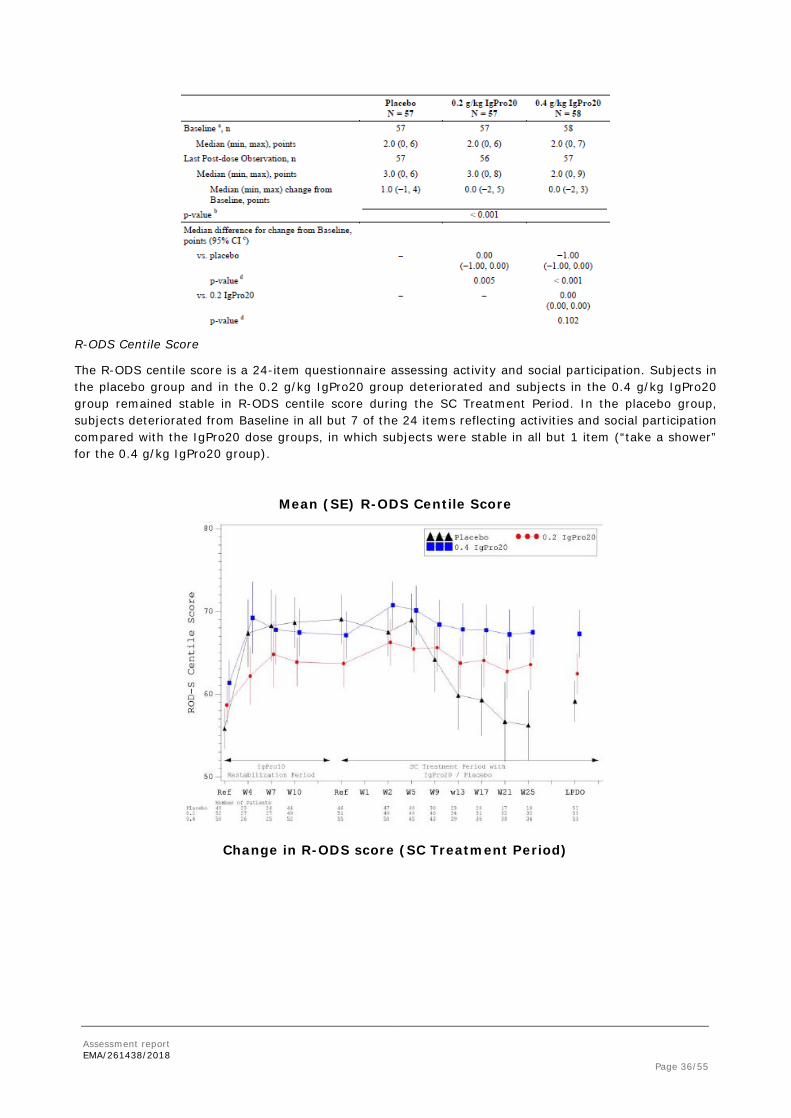
R-ODS Centile Score
The R-ODS centile score is a 24-item questionnaire assessing activity and social participation. Subjects in the placebo group and in the 0.2 g/kg IgPro20 group deteriorated and subjects in the 0.4 g/kg IgPro20 group remained stable in R-ODS centile score during the SC Treatment Period. In the placebo group, subjects deteriorated from Baseline in all but 7 of the 24 items reflecting activities and social participation compared with the IgPro20 dose groups, in which subjects were stable in all but 1 item (“take a shower” for the 0.4 g/kg IgPro20 group).
Mean (SE) R-ODS Centile Score
Change in R-ODS score (SC Treatment Period)
Assessment report EMA/261438/2018 Page 37/55
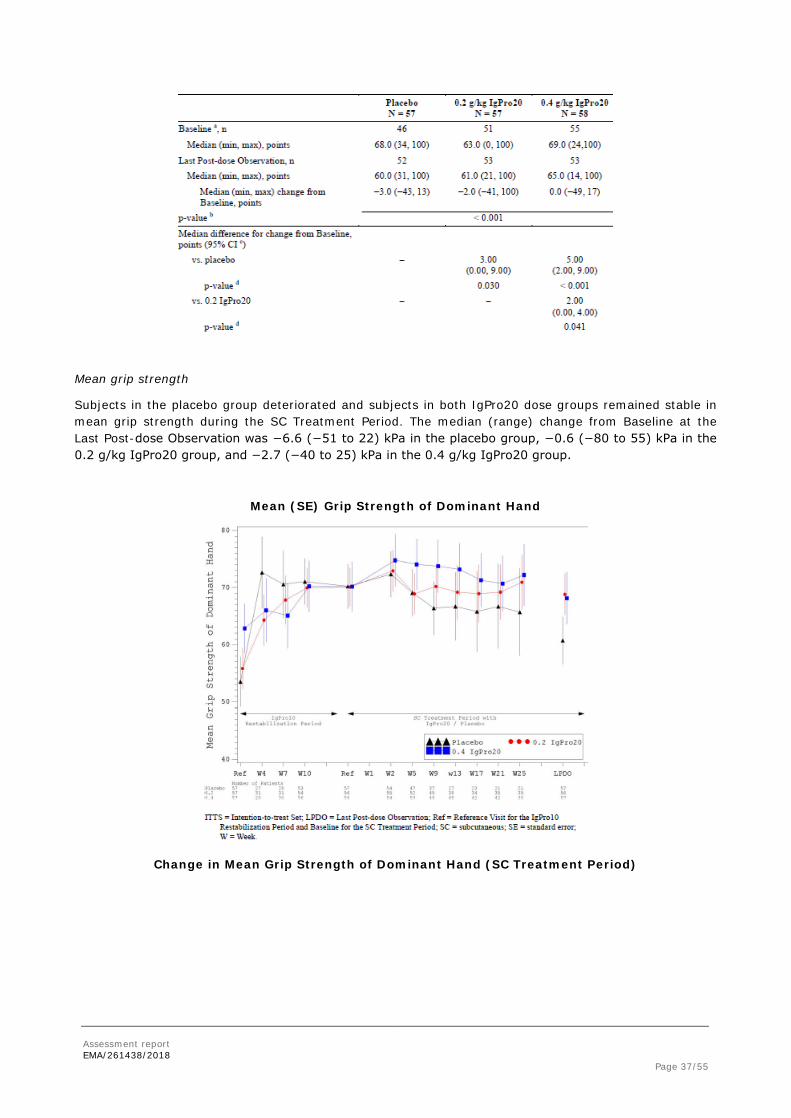
Mean grip strength
Subjects in the placebo group deteriorated and subjects in both IgPro20 dose groups remained stable in mean grip strength during the SC Treatment Period. The median (range) change from Baseline at the Last Post-dose Observation was −6.6 (−51 to 22) kPa in the placebo group, −0.6 (−80 to 55) kPa in the 0.2 g/kg IgPro20 group, and −2.7 (−40 to 25) kPa in the 0.4 g/kg IgPro20 group.
Mean (SE) Grip Strength of Dominant Hand
Change in Mean Grip Strength of Dominant Hand (SC Treatment Period)
Assessment report EMA/261438/2018 Page 38/55
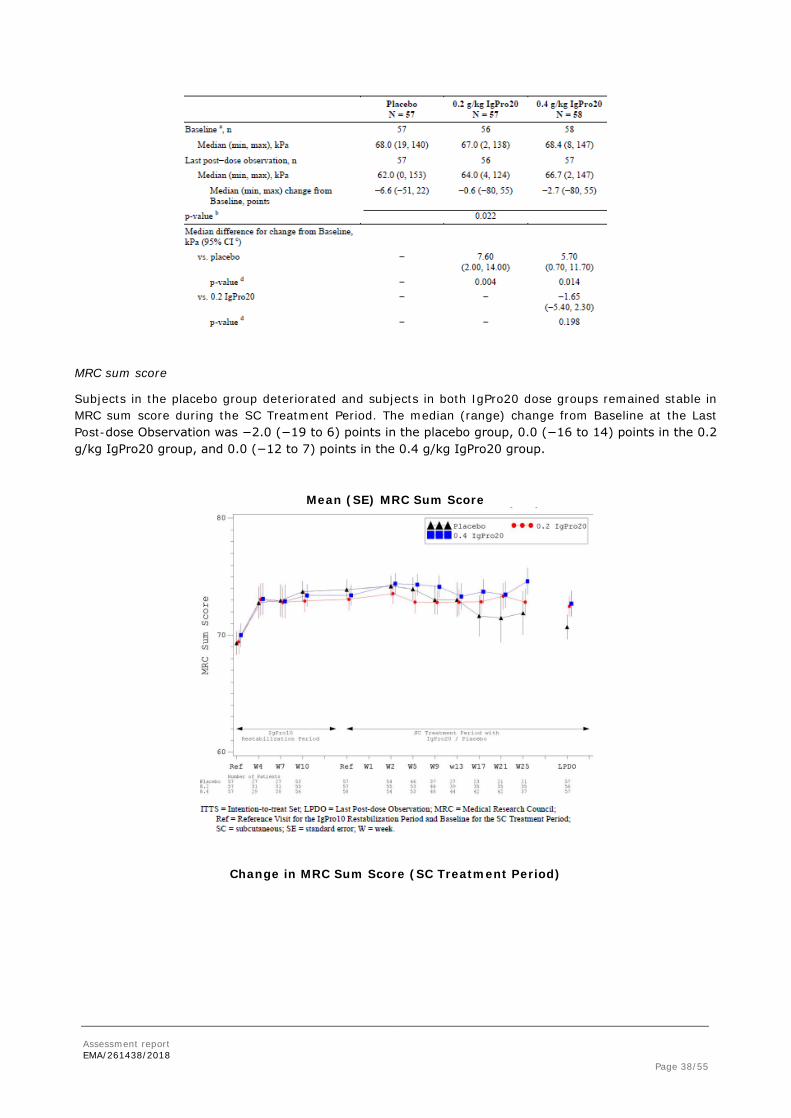
MRC sum score
Subjects in the placebo group deteriorated and subjects in both IgPro20 dose groups remained stable in MRC sum score during the SC Treatment Period. The median (range) change from Baseline at the Last Post-dose Observation was −2.0 (−19 to 6) points in the placebo group, 0.0 (−16 to 14) points in the 0.2 g/kg IgPro20 group, and 0.0 (−12 to 7) points in the 0.4 g/kg IgPro20 group.
Mean (SE) MRC Sum Score
Change in MRC Sum Score (SC Treatment Period)
Assessment report EMA/261438/2018 Page 39/55
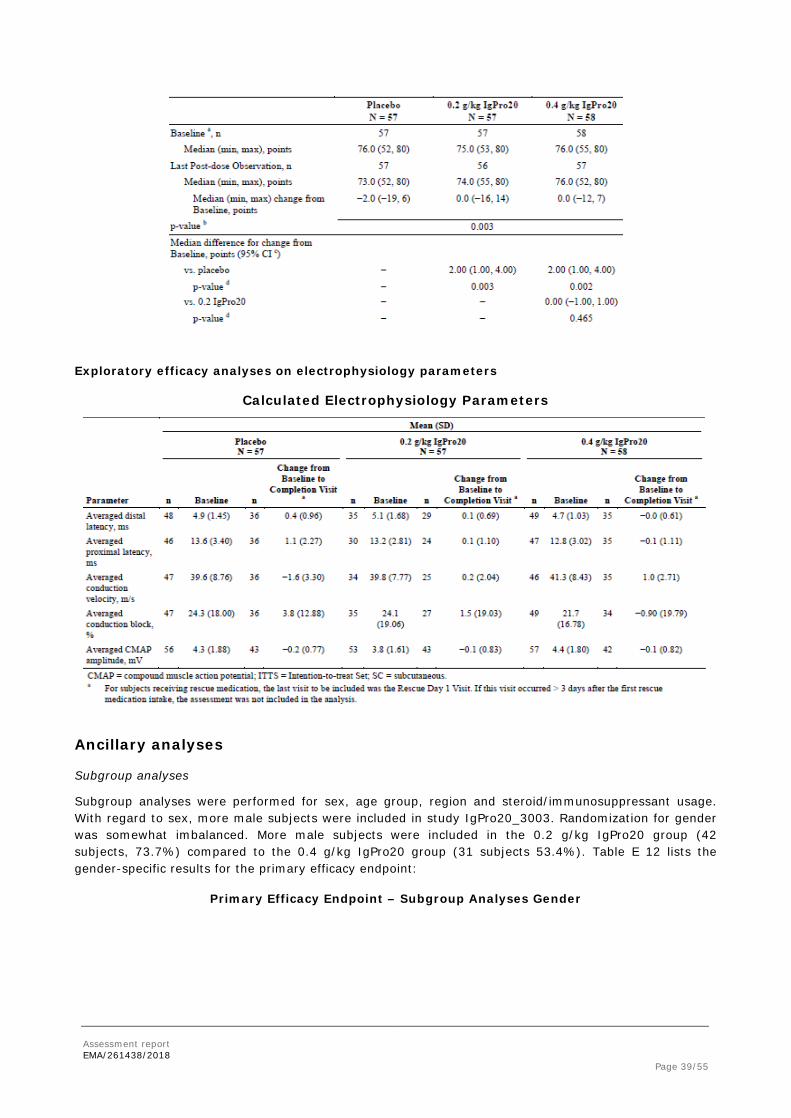
Exploratory efficacy analyses on electrophysiology parameters
Calculated Electrophysiology Parameters
Ancillary analyses
Subgroup analyses
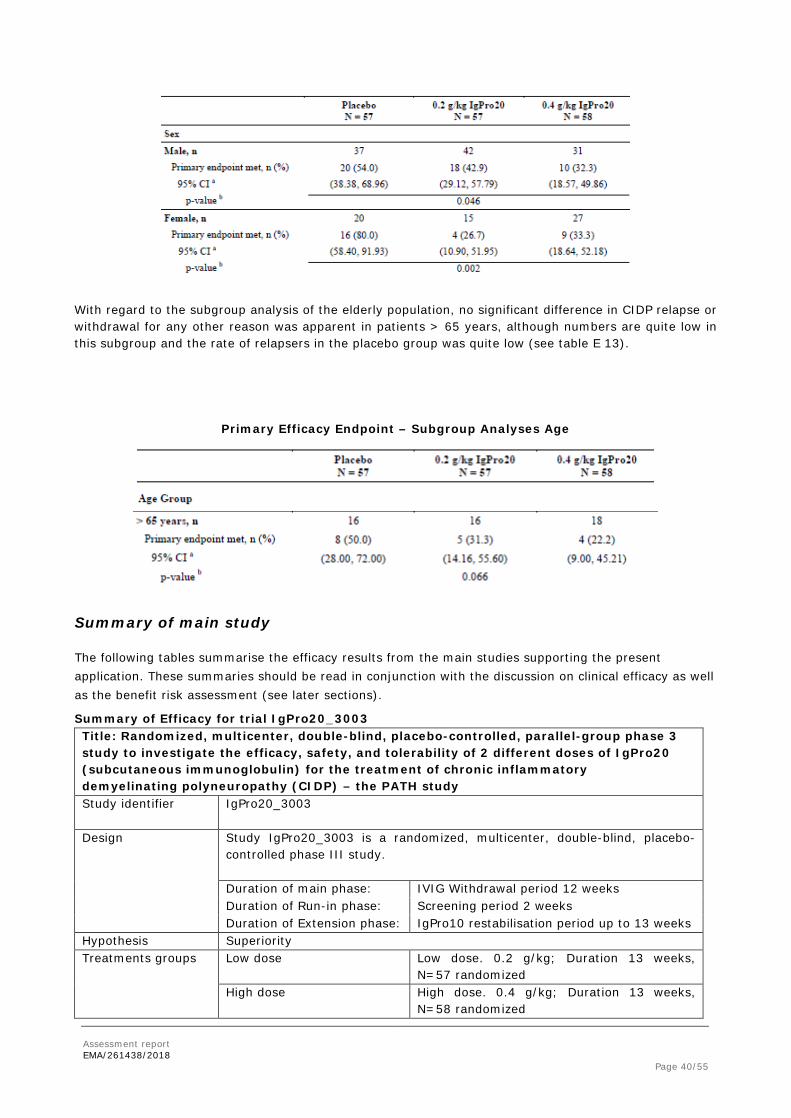
Subgroup analyses were performed for sex, age group, region and steroid/immunosuppressant usage. With regard to sex, more male subjects were included in study IgPro20_3003. Randomization for gender was somewhat imbalanced. More male subjects were included in the 0.2 g/kg IgPro20 group (42 subjects, 73.7%) compared to the 0.4 g/kg IgPro20 group (31 subjects 53.4%). Table E 12 lists the gender-specific results for the primary efficacy endpoint:
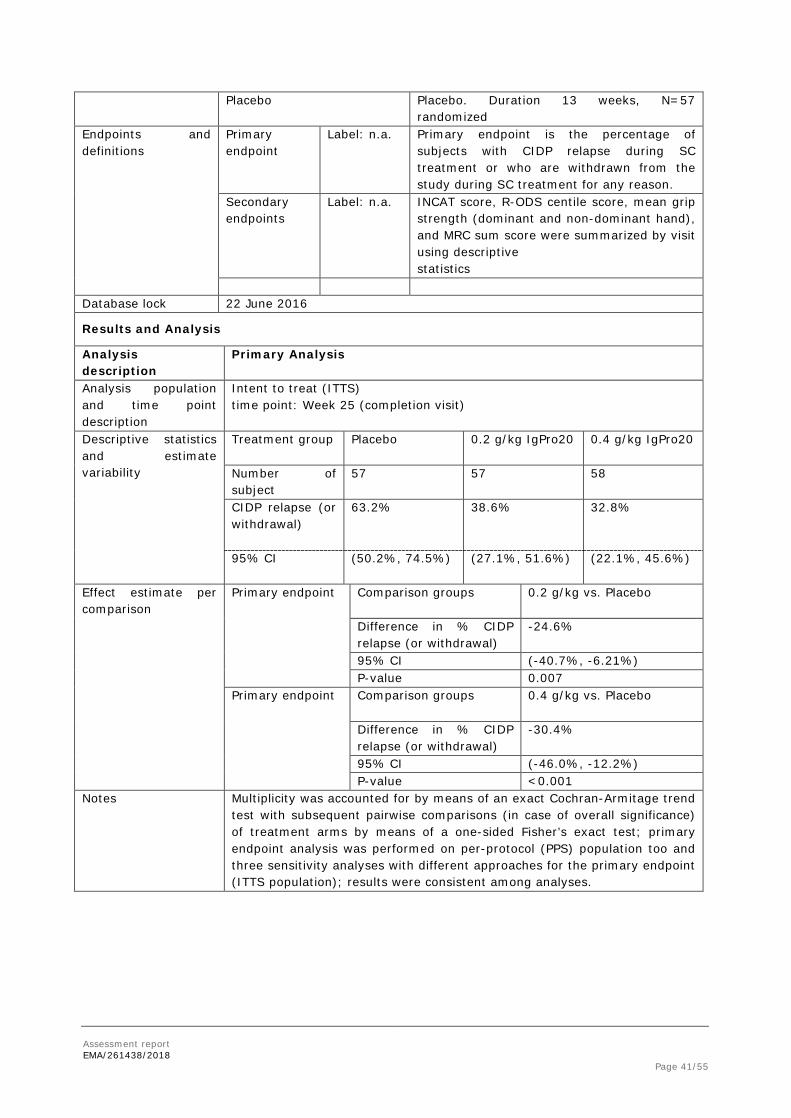
With regard to the subgroup analysis of the elderly population, no significant difference in CIDP relapse or withdrawal for any other reason was apparent in patients > 65 years, although numbers are quite low in this subgroup and the rate of relapsers in the placebo group was quite low (see table E 13).
Primary Efficacy Endpoint – Subgroup Analyses Age
Summary of main study
The following tables summarise the efficacy results from the main studies supporting the present application. These summaries should be read in conjunction with the discussion on clinical efficacy as well as the benefit risk assessment (see later sections).
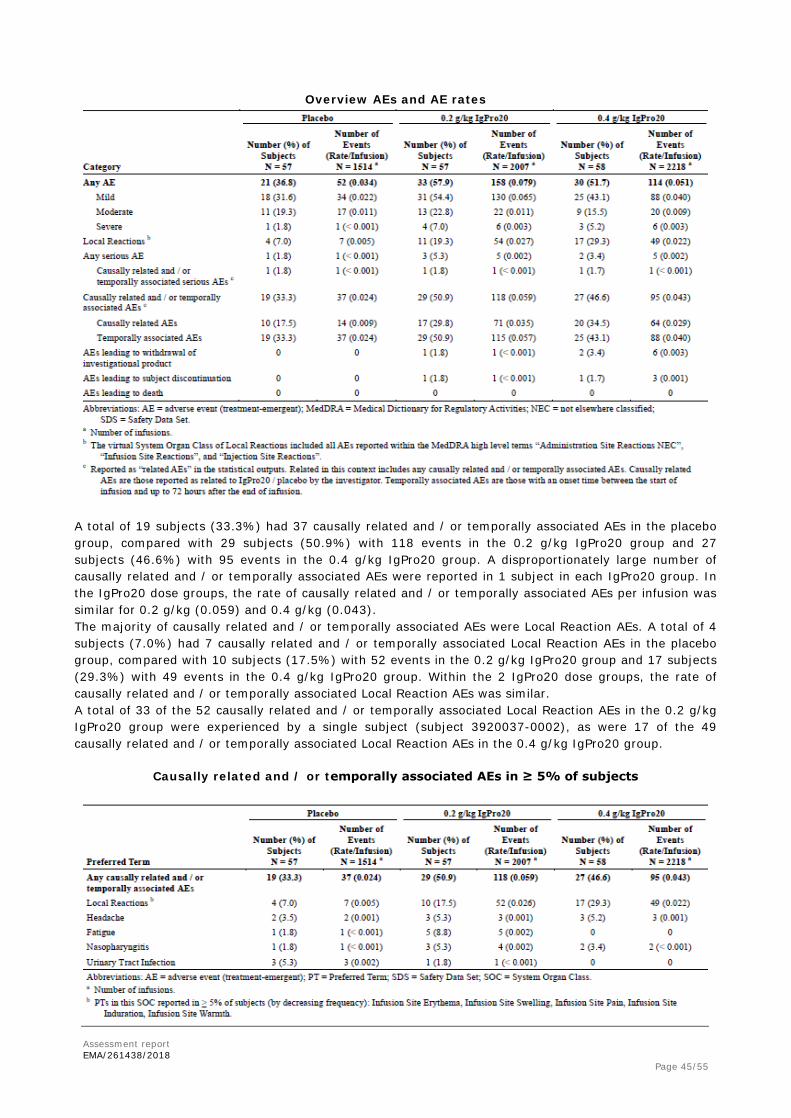
Summary of Efficacy for trial IgPro20_3003 Title: Randomized, multicenter, double-blind, placebo-controlled, parallel-group phase 3 study to investigate the efficacy, safety, and tolerability of 2 different doses of IgPro20 (subcutaneous immunoglobulin) for the treatment of chronic inflammatory demyelinating polyneuropathy (CIDP) – the PATH study Study identifier IgPro20_3003
Design Study IgPro20_3003 is a randomized, multicenter, double-blind, placebo-
controlled phase III study. Duration of main phase: IVIG Withdrawal period 12 weeks Duration of Run-in phase: Screening period 2 weeks Duration of Extension phase: IgPro10 restabilisation period up to 13 weeks
Primary endpoint is the percentage of subjects with CIDP relapse during SC treatment or who are withdrawn from the study during SC treatment for any reason.
Secondary endpoints
Label: n.a.
INCAT score, R-ODS centile score, mean grip strength (dominant and non-dominant hand), and MRC sum score were summarized by visit using descriptive statistics
Database lock 22 June 2016
Results and Analysis
Analysis description
Primary Analysis
Analysis population and time point description
Intent to treat (ITTS) time point: Week 25 (completion visit)
Descriptive statistics and estimate variability
Treatment group Placebo
0.2 g/kg IgPro20
0.4 g/kg IgPro20
Number of subject
57 57 58
CIDP relapse (or withdrawal)
63.2% 38.6% 32.8%
95% CI
(50.2%, 74.5%) (27.1%, 51.6%) (22.1%, 45.6%)
Effect estimate per comparison
Primary endpoint Comparison groups 0.2 g/kg vs. Placebo
Difference in % CIDP relapse (or withdrawal)
-24.6%
95% CI (-40.7%, -6.21%) P-value 0.007
Primary endpoint Comparison groups 0.4 g/kg vs. Placebo
Difference in % CIDP relapse (or withdrawal)
-30.4%
95% CI (-46.0%, -12.2%) P-value <0.001
Notes Multiplicity was accounted for by means of an exact Cochran-Armitage trend test with subsequent pairwise comparisons (in case of overall significance) of treatment arms by means of a one-sided Fisher’s exact test; primary endpoint analysis was performed on per-protocol (PPS) population too and three sensitivity analyses with different approaches for the primary endpoint (ITTS population); results were consistent among analyses.
Assessment report EMA/261438/2018 Page 42/55
2.4.2. Discussion on clinical efficacy
Design and conduct of clinical studies
Study IgPro20_3003 was a phase 3, prospective, multicenter, randomized, double-blind, placebo-controlled, parallel-group, 3-arm study in adult CIDP patients with 2 study phases: a Pre-randomization Phase (consisting of an IVIG Withdrawal Period and an IgPro10 (Privigen) Restabilization Period) and a Post-randomization Phase (consisting of a randomized placebo-controlled SC Treatment Period with 2 doses of IgPro20 (Hizentra) and an IgPro10 Rescue Period). The study is in compliance with the preceding initial and follow-up scientific advice.
Of 276 subjects screened, 172 were randomized to IgPro20/placebo, 96 subjects completed the study. Subjects were mostly withdrawn due to no CIDP deterioration during Withdrawal Period (n=28), no restabilization during IgPro10 Restabilization Period (n=22) or CIDP relapse during Post-randomization Phase (n=60). In total, there were 28 withdrawals by the subject concerned and 3 withdrawals due to physician decision. During the Pre-randomization Phase 4 subjects and during Post-randomization Phase 2 subjects were withdrawn due to adverse events. Protocol violations were recorded for 2 subjects during Pre-randomization phase which led to subject withdrawal.
The demographic and baseline characteristics were balanced across the 3 treatment groups in the ITTS, except for sex: there were more male subjects in the 0.2 g/kg IgPro20 group (42 subjects, 73.7%) than in the 0.4 g/kg IgPro20 group (31 subjects 53.4%).
The primary efficacy endpoint was the rate of CIDP relapse or withdrawal for any other reason; relapse was defined as an increase of ≥ 1 point in adjusted INCAT score compared to baseline. The higher SC dose of IgPro20 investigated in Study 3003 (0.4 g/kg bw) prevented CIDP relapse or withdrawal for any other reason in 67% of subjects, and the lower dose (0.2 g/kg bw) prevented relapse in 61% of subjects, whereas only 37% of subjects on placebo remained relapse-free.
In general, analysis of the secondary efficacy endpoints (time to CIDP relapse, INCAT score, R-ODS score, mean grip strength, MRC sum score) and exploratory efficacy endpoints (electrophysiologic parameters) revealed efficacy of maintenance treatment with IgPro20 and therefore support the primary efficacy endpoint.
Efficacy data and additional analyses
Study IgPro20_3003 proved that maintenance treatment with IgPro20 in both dosing groups significantly reduced CIDP relapse rate when compared to placebo (CIDP relapse or withdrawal for any other reason: 63.2% for placebo vs. 38.6% for 0.2 g/kg IgPro20 vs. 32.8% for 0.4 g/kg IgPro20). However, the carry-over effect of the preceding IVIG treatment with IgPro10 may have exerted an effect for ~ 10 weeks as seen by the PK data of the placebo group. This would imply that during the post-randomization treatment with IgPro20 (=24 weeks), residual effects of the IgPro10 pre-treatment cannot be excluded. Thus, results from expansion study IgPro20_3004 are important and could offer valuable information on long-term efficacy. Therefore the company was requested to provide these data as soon as available. A restriction if the indication was requested by the CHMP, because of this carry-over effect from IVIG and because SCIG alone or stabilisation with some other therapy haven’t been studied. Hizentra is indicated for the treatment of patients with CIDP as maintenance therapy after stabilization with IVIg. In addition, the CHMP also considered that the age range of the CIDP patients for whom Hizentra is indicated should be stated in the indication and this was agreed by the applicant.
No significant difference between the high and low dose of IgPro20 was observed in study IgPro20_3003. However, for the primary efficacy endpoint, no statistical analysis comparing the 0.2 g/kg IgPro20 group with the 0.4 g/kg IgPro20 group has been provided. The difference in CIDP relapse (excluding withdrawal for any other reason) appears to be high enough to reveal a significant difference and patients could thus have a greater benefit from treatment with 0.4 g/kg IgPro20. This is further supported by the analysis of R-ODS centile score, where the outcome in the 0.4 g/kg IgPro20 group was significantly better compared to the 0.2 g/kg IgPro20 group. Nevertheless, the posology range given in the PI is supported by the
Assessment report EMA/261438/2018 Page 43/55
CHMP, since both low and high dose (0.2 g/kg and 0.4 g/kg) will be made available to patients and the treatment with Hizentra will be adjusted in each patient according to their individual response.
Although subgroup analysis (gender, age, region [United States, Japan], steroid / immunosuppressant usage) revealed no clinically relevant differences for the primary endpoint, the CIDP relapse rate in female subjects on placebo (80%) was much higher compared to male subjects (54%) and treatment with 0.2 g/kg IgPro20 appeared to be more effective in women (26.7% CIDP relapse) compared to men (42.9% CIDP relapse) (see table E 12). Randomization for gender and dosing group is somewhat imbalanced and may explain some of the differences observed, however, the numbers are too small to actually draw any meaningful conclusions
With regard to the subgroup analysis of the elderly population (> 65 years), although no significant difference in CIDP relapse or withdrawal for any other reason was apparent comparing treatment with IgPro20 vs. placebo (p=0.066), the same trend is seen for the primary efficacy endpoint of IgPro20 as compared to the total set.
Although any non-head-to-head comparison should be viewed with caution due to differing study designs, patient populations, and concomitant medications etc. a general overview of the efficacy of Ig in CIDP is provided here. IVIG is considered to be established in this indication. From a Cochrane Review analyzing IVIG treatment in CIDP, 8 randomized controlled trials including 332 participants were eligible for evaluation. Five randomized trials prove that intravenous immunoglobulin improves disability more than placebo. In the trials comparing IVIg with placebo a significantly higher proportion of participants improved in disability within six weeks after the onset of treatment with IVIg compared with placebo, risk ratio (RR) 2.40 (95% CI 1.72 to 3.36) and a number needed to treat for an additional beneficial outcome (NNTB) of 3.03 (95% CI 2.33 to 4.55). Three other small trials showed no significant difference between intravenous immunoglobulin and plasma exchange, corticosteroids or methylpredisolone. No new trials were found for this 2013 update. In this review, mild and transient side effects were reported in approximately half of treated participants; serious side effects were reported in six per cent of the treated participants. This did not differ significantly from plasma exchange or corticosteroids treated participants.
Whether the improvements are equally clinically relevant cannot be deduced from this analysis because each trial used a different disability scale with a unique definition of a significant improvement. Only one study included in this review had a long-term follow-up. These results suggest that intravenous immunoglobulin improves disability more than placebo over 24 and 48 weeks. The authors conclude that further research is needed to compare the long-term benefits as well as side effects of intravenous immunoglobulin with other treatments.
Study IgPro20_3003 was the first randomized, controlled trial evaluating SCIG in CIDP therapy. For this reason, and since this study was designed to show eligibility for maintenance instead of primary treatment, comparison to other studies on IVIGs in CIDP is difficult. Due to the restabilization criteria, subjects generally not responding to IVIG treatment have already been excluded. Only stable subjects were eligible for treatment with IgPro20 and study IgPro20_3003 aimed at showing disease stability instead of improvement. In other studies, e.g. the ICE study or study IgPro10_3001, subjects had to be IVIG-free for at least 3 months or a prior IVIG washout was performed.
The primary efficacy endpoint of study IgPro20_3003 was CIDP relapse based on the INCAT score, which corresponds to the primary endpoint of other IVIG studies (e.g. study IgPro10_3001 (Privigen) or the ICE-study (Gamunex)), which analyzed the responder rate based on INCAT score points. Secondary efficacy endpoints of study IgPro20_3003 (grip strength, time to relapse, MRC sum score) have also been used in other studies on IVIGs in CIDP.
Study IgPro20_3003 also analyzed subject’s preference for treatment comparing SC to IV treatment. At the Last Post-dose Observation, a larger percentage of subjects preferred current SC treatment over pre-study IV treatment (placebo group: 38.6% versus 24.6%; 0.2 g/kg IgPro20 group: 52.6% versus 17.5%; 0.4 g/kg IgPro20 group: 53.4% versus 19.0%), mainly due to feeling that SC treatment offered participating subjects greater independence.
Overall, the efficacy data demonstrated that IgPro20, administered subcutaneously either as low (0.2 g/kg) or high (0.4 g/kg) dose for maintenance treatment of CIDP, effectively prevented relapse of
Assessment report EMA/261438/2018 Page 44/55
neuromuscular disability and impairment in 61.6% and 67.2% of cases, respectively. Results for CIDP relapse rate based on the adjusted INCAT score were further supported by the secondary efficacy outcome measures (time to relapse, INCAT score, R-ODS centile score, mean grip strength, MRC sum score) showing CIDP stability in IgPro20 treated subjects. The beneficial outcomes of study IgPro20_3003 therefore justify a transition from IVIG treatment to SCIG treatment in CIDP.
2.4.3. Conclusions on the clinical efficacy
Results for the primary endpoint show a significant lower CIDP relapse rate (ITTS and PPS population) for both 0.2 g/kg and 0.4 g/kg IgPro20 maintenance therapy compared to placebo. These results were also confirmed for three sensitivity analyses performed on ITTS population. Secondary efficacy endpoints of both IgPro20 study arms confirmed the primary endpoint and revealed stable disease for subjects who did not relapse. The originally proposed indication was requested to be amended in order to describe the age group of the treated population (adults, children and adolescents (0-18 years)). The indication was also narrowed to reflect the correct population suitable for treatment, i.e. maintenance therapy of patients with CIDP already stabilised with IVIg.
With respect to the possible overlapping effect of the preceding IgPro10 treatment it would be beneficial to present long-term efficacy results from the expansion study IgPro20_3004 and the applicant was requested to provide these data as soon as possible in the post marketing setting.
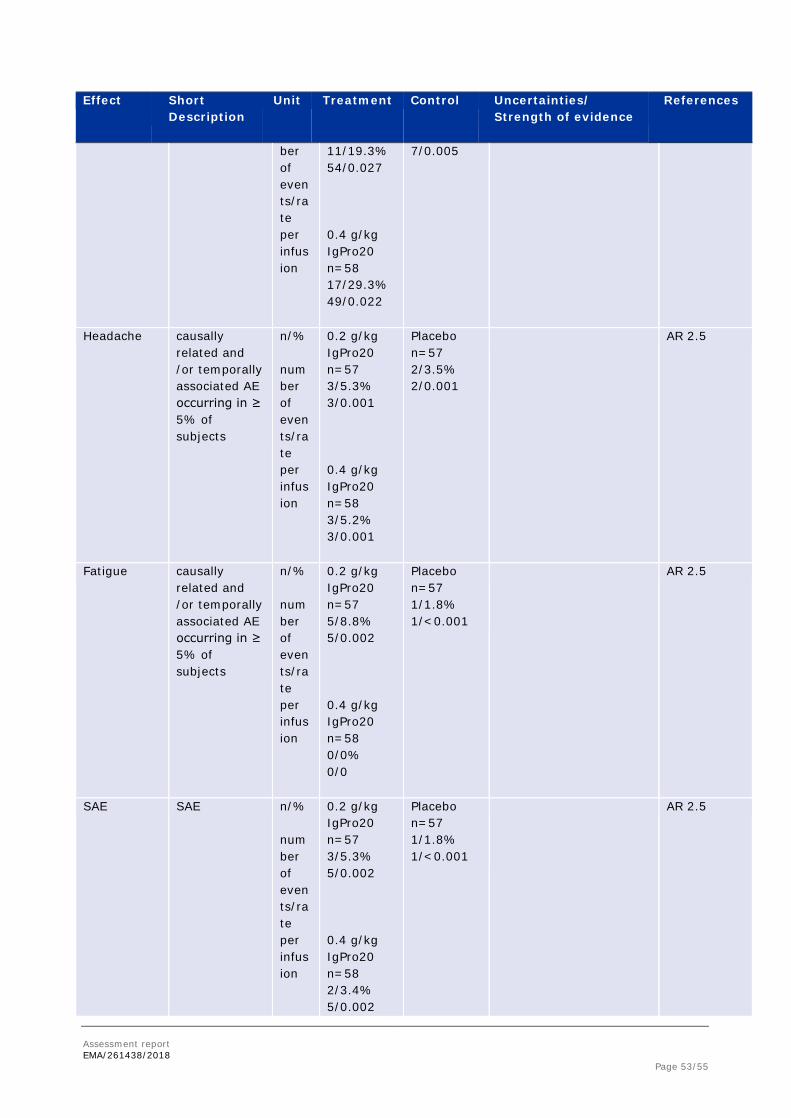
2.5. Clinical safety
Patient exposure
In the post-randomization phase, 57 subjects were administered a total of 1514 infusions with placebo, 57 subjects were administered a total of 2007 infusions with 0.2 g/kg IgPro20, and 58 subjects were administered a total of 2218 infusions with 0.4 g/kg IgPro20. Subjects generally used 4 injections sites (maximum: 9 sites) and infused an average of 20 mL per site (maximum: 50 mL), with an infusion rate of 20 mL/h (maximum: 50 mL/h). The infusion time was approximately 1 hour and the maximum infusion volume was 140 mL per infusion session.
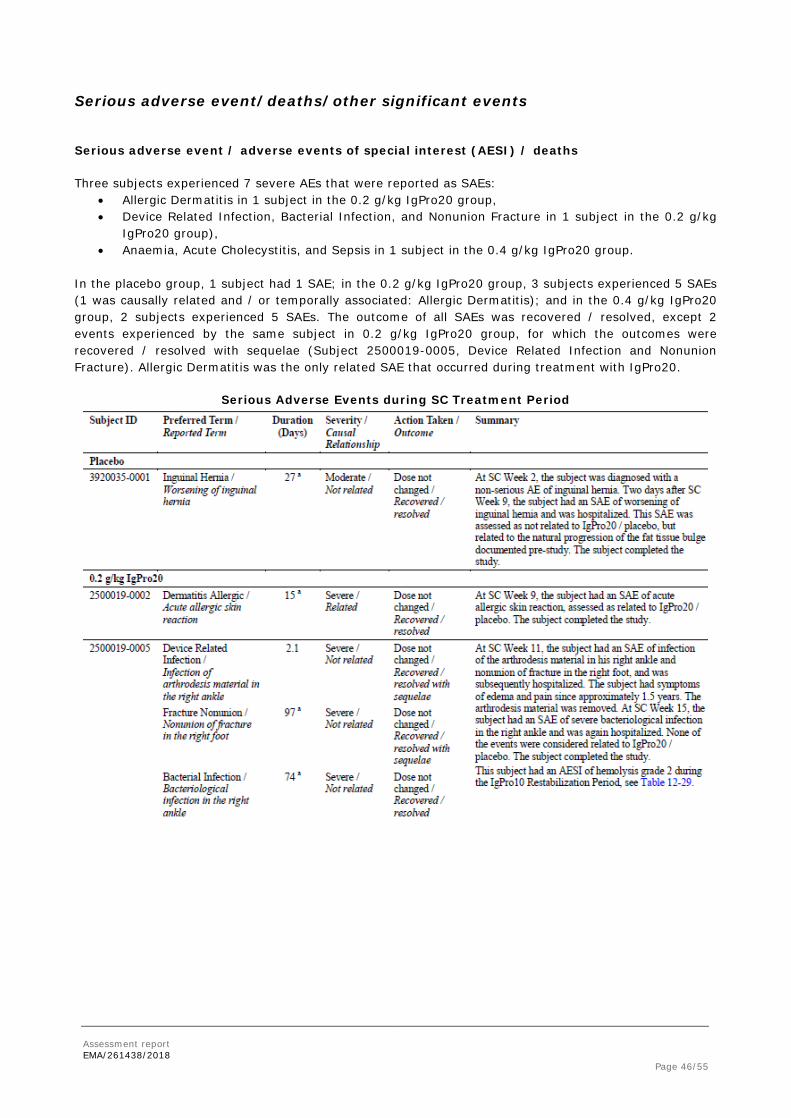
Adverse events
A total of 21 subjects (36.8%) in the placebo group had 52 AEs, compared with 33 subjects (57.9%) in the 0.2 g/kg IgPro20 group with 158 AEs and 30 subjects (51.7%) in the 0.4 g/kg IgPro20 group with 114 AEs. Most of these AEs were Local Reaction AEs. In the 0.2 g/kg IgPro20 group, 11 subjects (19.3%) had 54 Local Reaction AEs (1 subject had 30 Local Reaction AEs), and in the 0.4 g/kg IgPro20 group, 17 subjects (29.3%) had 49 Local Reaction AEs (1 subject had 17 Local Reaction AEs). All Local Reaction AEs were either mild (94.5%) or moderate (5.5%) intensity, and the frequency decreased over time. Most AEs reported in the 3 treatment groups were mild or moderate.
Assessment report EMA/261438/2018 Page 45/55
Overview AEs and AE rates
A total of 19 subjects (33.3%) had 37 causally related and / or temporally associated AEs in the placebo group, compared with 29 subjects (50.9%) with 118 events in the 0.2 g/kg IgPro20 group and 27 subjects (46.6%) with 95 events in the 0.4 g/kg IgPro20 group. A disproportionately large number of causally related and / or temporally associated AEs were reported in 1 subject in each IgPro20 group. In the IgPro20 dose groups, the rate of causally related and / or temporally associated AEs per infusion was similar for 0.2 g/kg (0.059) and 0.4 g/kg (0.043). The majority of causally related and / or temporally associated AEs were Local Reaction AEs. A total of 4 subjects (7.0%) had 7 causally related and / or temporally associated Local Reaction AEs in the placebo group, compared with 10 subjects (17.5%) with 52 events in the 0.2 g/kg IgPro20 group and 17 subjects (29.3%) with 49 events in the 0.4 g/kg IgPro20 group. Within the 2 IgPro20 dose groups, the rate of causally related and / or temporally associated Local Reaction AEs was similar. A total of 33 of the 52 causally related and / or temporally associated Local Reaction AEs in the 0.2 g/kg IgPro20 group were experienced by a single subject (subject 3920037-0002), as were 17 of the 49 causally related and / or temporally associated Local Reaction AEs in the 0.4 g/kg IgPro20 group.
Causally related and / or temporally associated AEs in ≥ 5% of subjects
Serious adverse event / adverse events of special interest (AESI) / deaths
Three subjects experienced 7 severe AEs that were reported as SAEs: • Allergic Dermatitis in 1 subject in the 0.2 g/kg IgPro20 group, • Device Related Infection, Bacterial Infection, and Nonunion Fracture in 1 subject in the 0.2 g/kg
IgPro20 group), • Anaemia, Acute Cholecystitis, and Sepsis in 1 subject in the 0.4 g/kg IgPro20 group.
In the placebo group, 1 subject had 1 SAE; in the 0.2 g/kg IgPro20 group, 3 subjects experienced 5 SAEs (1 was causally related and / or temporally associated: Allergic Dermatitis); and in the 0.4 g/kg IgPro20 group, 2 subjects experienced 5 SAEs. The outcome of all SAEs was recovered / resolved, except 2 events experienced by the same subject in 0.2 g/kg IgPro20 group, for which the outcomes were recovered / resolved with sequelae (Subject 2500019-0005, Device Related Infection and Nonunion Fracture). Allergic Dermatitis was the only related SAE that occurred during treatment with IgPro20.
Serious Adverse Events during SC Treatment Period
Assessment report EMA/261438/2018 Page 47/55
Adverse Events of special interest (AESI): The following AEs related to class effects for IgG products were considered as AESIs: Acute Systemic Hypersensitivity Reactions, Aseptic Meningitis Syndrome, Haemolysis, and Thrombotic Events. The only AESI reported during the SC Treatment Period was 1 SAE of Allergic Dermatitis. The event was experienced by Subject 2500019-0002 in the 0.2 g/kg IgPro20 group and was assessed as causally related and / or temporally associated with IgPro20 / placebo. The outcome was recovered / resolved. No subject experienced AEs of Haemolysis, Aseptic Meningitis Syndrome, or Thrombotic Events during the SC Treatment Period. There were no deaths during the SC Treatment Period with IgPro20.
Laboratory findings
During the SC Treatment Period, 4 subjects (2.3%) had 5 laboratory abnormalities reported as AEs and potentially related to hemolysis; however, none of these subjects fulfilled the hemolysis criteria.
Laboratory Abnormalities Reported as AEs and potentially related to hemolysis
Assessment report EMA/261438/2018 Page 48/55
All of these AEs were mild to moderate in intensity; the majority was considered to be not related to IgPro20 by the investigator and was resolved at the final follow-up. None of these led to discontinuation or changed doses of study product, to any other intervention, or to discontinuation from the study.
At Last Post-dose Observation there was a relevant shift in leukocytes compared to Baseline: 5 subjects (8.6%) had a shift from normal to low in the 0.4 g/kg IgPro20 group, whereas no subjects had a shift from normal to low in the 0.2 g/kg IgPro20 group or in the placebo group. In group 0.2 g/kg IgPro20, 7 subjects revealed a shift in reticulocytes from normal to high and in group 0.4 g/kg IgPro20, 4 subjects had a shift in reticulocytes from normal to high. There was also a relevant shift recorded for LDH: overall, 7 subjects had shifts from normal to high; 4 subjects (6.9%) in the 0.4 g/kg IgPro20 group, 2 subjects (3.5%) in the 0.2 g/kg IgPro20 group, and 1 subject (1.8%) in the placebo group. In group 0.2 g/kg IgPro20, 4 subjects revealed a shift in haptoglobin from normal to high and in group 0.4 g/kg IgPro20, 3 subjects had a shift in haptoglobin from normal to high.
Safety in special populations
There were no regional effects in the pattern of AEs (US versus non-US and Japan versus non-Japan. No PT was experienced by > 2 US subjects or > 1 Japanese subject in any treatment group.
Discontinuation due to adverse events
One subject in the 0.2 g/kg IgPro20 group experienced 1 non-serious, causally related and / or temporally associated AE of Fatigue that led to withdrawal of the investigational product and subject discontinuation. In the 0.4 g/kg IgPro20 group, 1 subject experienced 3 events leading to withdrawal of the investigational product and subject discontinuation (anaemia, acute cholecystitis, and sepsis). All 3 events were also serious. None of them were assessed as causally related and / or temporally associated. These 3 SAEs had an outcome of recovered / resolved.
Post marketing experience
The post-marketing safety profile of Hizentra for the previously approved indications has been characterized during the 7-year post-marketing period. During this period, 38,330,927 g of Hizentra, corresponding to 3,833,093 estimated standard doses of 10 g, were distributed. As of 31 May 2016 (Data Lock Point [DLP] of last periodic report), a worldwide cumulative total of 9277 post-marketing case reports of suspected adverse drug reactions (ADRs) were reported to CSL Behring’s Pharmacovigilance database. Based on these case reports of suspected ADRs and the class effects of Hizentra, the identified risks of Hizentra have been established as local reactions including ulceration-like infusion site reactions (UL-ISRs), anaphylactic reactions, aseptic meningitis syndrome, and thromboembolic events. The potential risks of Hizentra are increased or unknown risks in the home-based SC (self-) administration, exacerbation of existing hyperprolinemia (product specific), hemolysis, and transmission of infectious agents.
2.5.1. Discussion on clinical safety
The safety profile of IgPro20 in maintenance treatment of CIDP was evaluated in 57 subjects treated with 0.2 g/kg IgPro20 and 58 subjects treated with 0.4 g/kg IgPro20. 57 subjects were administered a total of 2007 infusions with 0.2 g/kg IgPro20, and 58 subjects were administered a total of 2218 infusions with 0.4 g/kg IgPro20, whereby 33 subjects (58%) experienced at least 1 AE in the 0.2 g /kg IgPro20 group and 30 subjects (52%) experienced at least 1 AE in the 0.4 g/kg IgPro20 group. AEs were causally related to study drug in 17 subjects (30%, 0.2 g/kg IgPro20) and 20 subjects (34.5% 0.4 g/kg IgPro20).
Assessment report EMA/261438/2018 Page 49/55
Temporally associated AEs (within 72 hours) were observed in 29 subjects (51%, 0.2 g/kg IgPro20) and 25 subjects (43%, 0.4 g/kg IgPro20). Based on the amount of infusions administered, the overall AE rate per infusion was 0.079 (0.2 g/kg IgPro20) and 0.051 (0.4 g/kg IgPro20), the rate of causally related AEs per infusion was 0.035 (0.2 g/kg IgPro20) and 0.029 (0.4 g/kg IgPro20) and the rate of temporally associated AEs per infusion was 0.057 (0.2 g/kg IgPro20) and 0.040 (0.4 g/kg IgPro20).
Causally related and/or temporally associated AEs experienced by ≥ 5% of subjects were local reactions, headache, fatigue, nasopharyngitis and 1 urinary tract infection. Almost all AEs were mild or moderate in intensity. The laboratory abnormalities reported as AEs were mild to moderate in intensity; the majority was considered to be not related or unlikely related to study drug, and were resolved at the final follow-up. There were no clinically relevant changes in vital signs or physical examinations.
No deaths occurred in this study. In the 0.2 g/kg IgPro20 group, 3 subjects experienced 5 SAEs. Of these, 1 was causally related and / or temporally associated to IgPro20 (Allergic Dermatitis), 4 were considered not related (infection of arthrodesis material in the right ankle, nonunion of fracture in the right foot, bacteriological infection in the right ankle, arthralgia). In the 0.4 g/kg IgPro20 group, 2 subjects had 5 SAEs. None of them were considered related to the study drug (2x Acute Cholecystitis, anaemia, sepsis, arthropathy). Except for the nonunion of the fracture in the right foot (resolved with sequelae), all SAEs were recovered/resolved.
In total, 2 subjects had AEs leading to study discontinuation; 1 subject with 1 event in the 0.2 g/kg IgPro20 group, and 1 subject with 4 events in the 0.4 g/kg IgPro20 group.
The only AESI reported during the SC treatment period was 1 event of allergic dermatitis. There was no hemolysis, aseptic meningitis syndrome, or thrombotic events during the SC Treatment Period.
Safety data indicate that both low and high dose IgPro20 were safe and reasonably well tolerated when administered s.c. as maintenance treatment to subjects with CIDP.
2.5.2. Conclusions on clinical safety
The CHMP considered that the safety profile of Hizentra when used in the treatment of CIDP is sufficiently characterised and all adverse events have been included in the Product Information.
2.5.3. PSUR cycle
The requirements for submission of periodic safety update reports for this medicinal product are set out in the list of Union reference dates (EURD list) provided for under Article 107c(7) of Directive 2001/83/EC and any subsequent updates published on the European medicines web-portal.
2.6. Risk management plan
The CHMP received the following PRAC Advice on the submitted Risk Management Plan:
The PRAC considered that the risk management plan version 4.2 is acceptable.
The MAH implemented the changes in the RMP as requested by PRAC and CHMP.
The CHMP endorsed the Risk Management Plan version 4.2 with the following content:
Safety concerns
Summary of safety concerns
Important identified risks • Local Reactions including ulceration like-infusion site reactions (UL-ISRs)
Assessment report EMA/261438/2018 Page 50/55
• Anaphylactic reactions
• Aseptic Meningitis Syndrome (AMS)
• Thromboembolic events (TEE)
Important potential risks • Increased or unknown risks in the home-based SC (self-) administration
• Exacerbation of existing hyperprolinaemia (product specific)
SC = subcutaneous, TEE = thromboembolic event; UL-ISR = ulceration like-infusion site reaction.
Assessment report EMA/261438/2018 Page 51/55
2.7. Update of the Product information
As a consequence of this new indication, sections 4.1, 4.2, 4.8, 5.1 and 5.2 of the SmPC have been updated.
In addition, section 4.7 of the SmPC has been updated in accordance with the latest QRD template.
The Package Leaflet is updated in accordance
3. Benefit-Risk Balance
3.1. Favourable effects
Results of the main clinical study show a significant lower CIDP relapse rate (ITTS population) for both active arms compared to placebo (32.8% for 0.4 g/kg IgPro20, 38.6% for 0.2 g/kg IgPro20, and 63.2% for placebo). These results were also confirmed for the PPS population and three sensitivity analyses performed on ITTS population. Positive differences between active arms and placebo could also be demonstrated for the SC treatment phase for secondary endpoints “Time to CIDP relapse”, “INCAT Score”, “R-ODS Centile Score” (Questionnaire), “mean grip strength”, and “MRC Sum Score”.
Study IgPro20_3003 also showed that a larger percentage of subjects preferred SC treatment over IV treatment because SC treatment offered greater independence.
3.2. Uncertainties and limitations about favourable effects
Long-term beneficial effects of maintenance treatment with IgPro20 are not foreseen yet, especially when considering the overlapping effects of prior IVIG treatment. In the placebo group IgG Ctrough values decline after approx. Week 10. Thus, the carry-over effect of IVIG to IgPro 20 could also be estimated to last this length of time. Thus, results from expansion study IgPro20_3004 are important and could offer valuable long-term data. Therefore the company is requested to provide these data as soon as possible. Since the study did not investigate the paediatric population, there is some uncertainty as to the extent of beneficial effects of s.c. treatment with IgPro20 in maintenance treatment of children with CIDP. Nevertheless, the extrapolation to the paediatric population was accepted by the CHMP and Hizentra is indicated also in children and adolescents.
The CIDP relapse rate (excluding withdrawal for any other reason) appears to be lower in subjects treated with 0.4 g/kg IgPro20 vs. 0.2 g/kg IgPro20.
Study IgPro20_3003 did not include enough subjects for showing beneficial effects in the elderly population (> 65 years), however, a comparable beneficial outcome as observed in the age group 18 – 65 years might have become apparent if more subjects > 65 years had been included in the study.
3.3. Unfavourable effects
The unfavourable effects of IgPro20 seen in the study mainly include local reactions (most frequent), fatigue and headache. The overall frequency of AEs was low (36.8% of placebo subjects, 57.9% of subjects on 0.2 g/kg, and 51.7% of subjects on 0.4 g/kg), with very few systemic AEs. Most AEs reported in the 3 treatment groups were mild or moderate. The only AESI reported during the SC treatment period was 1 event of allergic dermatitis. There were no proven cases of haemolysis, aseptic meningitis syndrome, or thrombotic events during the s.c. treatment period.
Assessment report EMA/261438/2018 Page 52/55
3.4. Uncertainties and limitations about unfavourable effects
During the s.c. treatment period with IgPro20, 4 subjects in the 0.2 g/kg IgPro20 group and 2 subjects of the 0.4 g/kg IgPro20 group yielded insufficient data to evaluate haemolysis cases. In these subjects either baseline data for haemoglobin were missing + at least 1 post-baseline DAT was positive + at least 1 of the criteria B was met or no post-baseline laboratory data were available. Thus, the occurrence of hemolysis cases due to IgPro20 treatment in these patients cannot be entirely ruled out. In addition, the MAH plans to increase the infusion volume and infusion rate of IgPro20. However, based on the sparse data obtained and provided, safety of the increased infusion volume and infusion rate is uncertain and cannot be adequately assessed for acceptability at the present time.
3.5. Effects Table
Effects Table for IgPro20 (indication CIDP) (data cut-off: 22 June 2016)
Effect Short Description
Unit Treatment Control Uncertainties/ Strength of evidence
References
Favourable Effects CIDP relapse or withdrawal for any other reason
Percentage of subjects who had CIDP relapse during the SC Treatment Period or were withdrawn from SC treatment for any reason
The difference in the percentage of relapse between the 0.2 g/kg IgPro20 dose group and placebo was -24.6% (95% CI: -40.7, -6.21) p=0.007 The difference in the percentage of relapse between the 0.4 g/kg IgPro20 dose group and placebo was -30.4% (95% CI: -46.0, -12.2) p<0.001
AR 2.4.2.
CIDP relapse
Percentage of subjects who had CIDP relapse during the SC Treatment Period
The difference in the percentage of relapse between the 0.2 g/kg IgPro20 dose group and placebo was -22.8% (95% CI: -39.0, -4.6) p=0.012 The difference in the percentage of relapse between the 0.4 g/kg IgPro20 dose group and placebo was -37.2% (95% CI: -51.7, -19.7) p<0.001
AR 2.4.2.
Unfavourable Effects Local Reactions
AE n/% num
0.2 g/kg IgPro20 n=57
Placebo n=57 4/7.0%
AR 2.5
Assessment report EMA/261438/2018 Page 53/55
Effect Short Description
Unit Treatment Control Uncertainties/ Strength of evidence
3.6.1. Importance of favourable and unfavourable effects
Treatment of both low and high dose IgPro20 led to a significant reduction of CIDP relapse (or withdrawal for any other reason) compared to placebo. CIDP relapse was based on the adjusted INCAT score, which was the basis for calculation of CIDP response rates in various other studies on IVIGs in CIDP. The 10-point INCAT score is a globally accepted, validated, and reliable scale of disability. A change of 1 INCAT score point is considered clinically meaningful, (excluding an increase in INCAT score of 1 point if this is only due to an increase of the arm score from 0 to 1 or an unchanged adjusted INCAT score compared with the Reference Visit (IgPro10 Restabilization Period) or to Baseline (SC Treatment Period), where the arm score decreased from 1 to 0 (not clinically meaningful improvement) and the leg score increased by 1 point (clinically meaningful worsening)). Secondary efficacy endpoints analysed in this study also revealed positive differences between both IgPro20 treatment arms and placebo. Thus, s.c. treatment with IgPro20 was shown to be effective in CIDP maintenance treatment.
Marginal uncertainty exists with respect to the overlapping effect of prior IVIG treatment and potential increase of CIDP relapse rates after complete washout of Privigen. With respect to long-term efficacy and safety, results from the extension study IgPro20_3004 which enrolled subjects who completed Study 3003 or were successfully rescued from CIDP relapse in Study 3003 could provide further information.
In general, IgPro20 revealed a reasonable safety profile in the maintenance treatment of CIDP. Except for 1 acute systemic hypersensitivity reaction (allergic dermatitis), no other adverse events of special interest (hemolysis, aseptic meningitis, thrombotic events) occurred during the study. However, there is uncertainty with regard to haemolytic cases, since 6 subjects yielded insufficient data to evaluate hemolysis. The most frequent AEs were local reactions, causally related AEs included headache and fatigue. The majority of AEs was mild to moderate in severity and manageable. There was 1 case of non-serious, causally related and / or temporally associated AE of fatigue that led to subject discontinuation from the study. Overall, the safety profile was rather similar as it is known from other indications and no new safety signals were identified.
Assessment report EMA/261438/2018 Page 55/55
3.6.2. Balance of benefits and risks
IgPro20_3003 analysed SCIG in the maintenance treatment of CIDP and showed a clinically and statistically relevant reduction in CIDP relapse rate based on the adjusted INCAT score. The safety profile of IgPro20 was consistent with that previously reported. The benefit-risk balance is positive.
The difference in CIDP relapse comparing both high and low dose IgPro20 with placebo is statistically significant and clinically relevant. Since only IVIG-stabilized subjects were included, the extension of indication solely affects maintenance treatment of CIDP after prior IVIG treatment. Results from secondary endpoints are generally consistent with the primary endpoint. The acceptable safety profile also indicates a positive benefit-risk balance.
3.7. Conclusions
The overall B/R of Hizentra is positive.
4. Recommendations
Outcome
Based on the review of the submitted data, the CHMP considers the following variation acceptable and therefore recommends the variation to the terms of the Marketing Authorisation, concerning the following change:
Variation accepted Type Annexes affected
C.I.6.a C.I.6.a - Change(s) to therapeutic indication(s) - Addition of a new therapeutic indication or modification of an approved one
Type II I and IIIB
Extension of indication to include immunomodulatory therapy in adults, children and adolescents (0-18 years), for the treatment of patients with chronic inflammatory demyelinating polyneuropathy (CIDP) as maintenance therapy after stabilization with IVIg; as a consequence, sections 4.1, 4.2, 4.8, 5.1 and 5.2 of the SmPC are updated. Section 4.7 of the SmPC was updated to bring it in line with the latest QRD template. The Package Leaflet is updated in accordance. The RMP is updated (finally agreed version 4.2).
The variation leads to amendments to the Summary of Product Characteristics and Package Leaflet and to the Risk Management Plan (RMP).