Homework Assignments due next session 1. Find a entry of interest in OMIM ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM ) 2. Find a Gene associated with that entry 1. Click on the “links” link on the right and choose “Gene”
Transcript
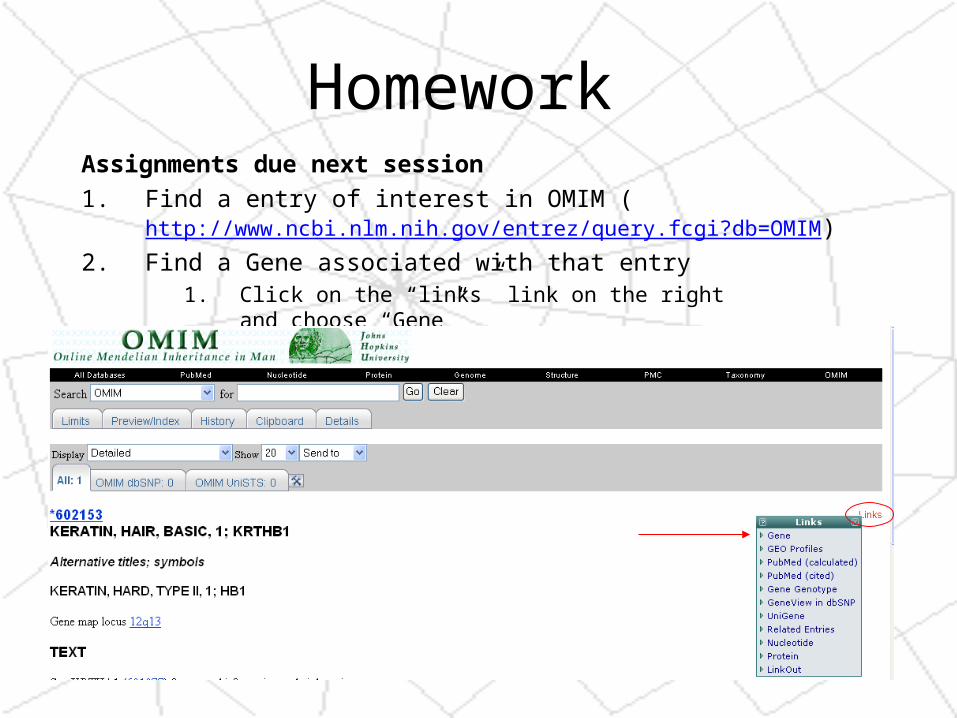
Homework Assignments due next session
1. Find a entry of interest in OMIM (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM)
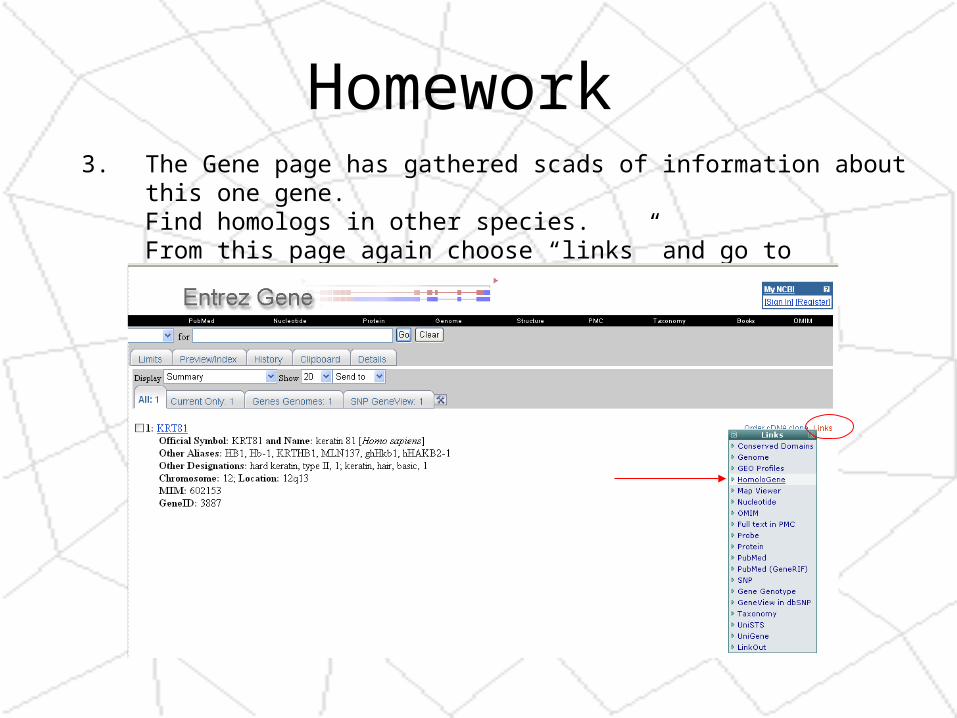
2. Find a Gene associated with that entry1. Click on the “links” link on the right
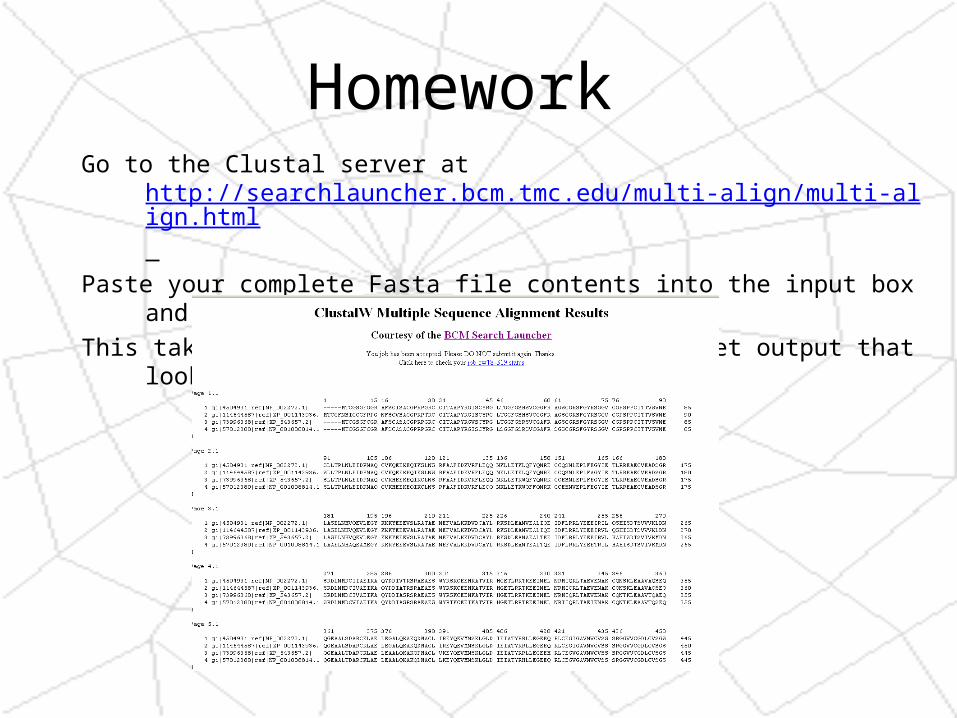
Homework At the bottom of the alignment file is the same results in “Fasta” format. Copy the
complete Fasta results and paste it into the input box at a BoxShade server (http://bioweb.pasteur.fr/seqanal/interfaces/boxshade.html)
Homework Depending on the parameters chosen for BoxShade, you will see something like this.
Regions which are the same in all species are likely involved in function in some way.
HomeworkAfter all that work, your boss comes to you ands says that sequence comparison is
obsolete! He wants you do structural alignments of these proteins. Figure out what a structural alignment is, find two different tools to find conserved 3D structures and choose which one you would use for this. Describe why this tool is preferable to the other.
NOTE: You do not need to actually do any structural alignments. Just find out how you would go about doing on if you had to.