1Division of Hematology, Stanford University School of Medicine / Stanford Cancer Institute, Stanford, CA Keywords: Atypical CML; MDS/MPN; BCR-ABL1 negative; SETBP1 Corresponding Author Jason Gotlib, MD, MS Professor of Medicine (Hematology) Stanford Cancer Institute 875 Blake Wilbur Drive, Room 2324 Stanford, CA 94305-5821 TEL: 650-736-1253 FAX: 650-724-5203 Email: [email protected] Word Count: Abstract: 140 Text: 4336 Tables: 1 Figures: 2
Blood First Edition Paper, prepublished online November 29, 2016; DOI 10.1182/blood-2016-08-693630
1. Arber DA, Orazi A, Hasserjian R, Thiele J, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
myeloid leukaemia, BCR-ABL1 negative. In: Swerdlow S, Harris NL, Stein H, Jaffe ES, Theile J, Vardiman JW, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2008. pp. 80–81.
3. Dao KH, Tyner JW. What’s different about atypical CML and chronic
neutrophilic leukemia? Hematology Am Soc Hematol Educ Program. 2015;2015:264-71
4. Mughal TI, Cross NCP, Padron E, et al. An International MDS/MPN Working
Group's perspective and recommendations on molecular pathogenesis, diagnosis and clinical characterization of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2015;100(9):1117-1130.
5. Tiu RV, Sekeres MA. Making sense of the myelodysplastic/
10. Martiat P, Michaux JL, Rodhain J. Philadelphia- negative (Ph-) chronic myeloid leukemia (CML): comparison with Ph1 CML and chronic myelomonocytic leukemia. The Groupe Français de Cytogénétique Hématologique. Blood. 1991;78(1):205-211.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
11. Hernández JM, del Cañizo MC, Cuneo A, et al. Clinical, hematological and cytogenetic characteristics of atypical chronic myeloid leukemia. Ann Oncol. 2000;11(4):441-444.
12. Zoi K, Cross NCP. Molecular pathogenesis of atypical CML, CMML and MDS/MPN-unclassifiable. Int J Hematol. 2015;101(3):229-242.
13. Piazza R, Valletta S, Winkelmann N, et al. Recurrent SETBP1 mutations in
atypical chronic myeloid leukemia. Nat Genet. 2013;45(1):18-24. 14. Meggendorfer M, Bacher U, Alpermann T, et al. SETBP1 mutations occur in
9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27(9):1852-1860.
15. Meggendorfer M, Haferlach T, Alpermann T, et al. Specific molecular
16. Minakuchi M, Kakazu N, Gorrin-Rivas MJ, et al. Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia- associated protein SET. Eur J Biochem. 2001; 268(5):1340-1351.
17. Cristóbal I, Blanco FJ, Garcia-Orti L, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 2010;115(3):615-625.
18. Hoischen A, van Bon BW, Gilissen C, et al.�De novo mutations of SETBP1 cause Schinzel- Giedion syndrome. Nat Genet. 2010;42(6):483-485.
19. Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942-946.
20. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in
chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013. 368(19):1781-1790.
21. Pardanani A, Lasho TL, Laborde RR, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013; 27(9):1870-1873.
22. Maxson JE, Luty SB, MacManiman JD, et al. The colony-stimulating factor 3 receptor T640N mutation Is oncogenic, sensitive to JAK inhibition, and mimics T618I. Clin Cancer Res. 2016;22(3):757-764.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
23. Bartels S, Lehmann U, Busche G, et al. De novo CSF3R mutation associated with transformation of myeloproliferative neoplasm to atypical CML. Ann Hematol. 2015;94(7):1255-1256.
24. Gambacorti-Passerini CB, Donadoni C, Parmiani A, et al. Recurrent ETNK1 mutations in atypical chronic myeloid leukemia. Blood. 2015;125(3):499-503.
25. Lasho TL, Finke CM, Zblewski D, et al. Novel recurrent mutations in ethanolamine kinase 1 (ETNK1) gene in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia. Blood Cancer J. 2015;5:e275.
26. Bousquet M, Quelen C, De Mas V, et al. The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene. 2005;24(48):7248-7252.
27. Reiter A, Walz C, Watmore A, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65(7):2662-2667.
28. Baxter EJ, Hochhaus A, Bolufer P, et al. The t(4;22)(q12;q11) in atypical chronic myeloid leukaemia fuses BCR to PDGFRA. Hum Mol Genet. 2002;11(12):1391-1397.
29. Safley AM, Sebastian S, Collins TS, et al. Molecular and cytogenetic characterization of a novel translocation t(4;22) involving the breakpoint cluster regiona and platelet-derived growth factor receptor-alpha genes in a patient with atypical chronic myeloid leukemia. Genes Chromosomes Cancer. 2004;40(1):44-50.
30. Wittman B, Horan J, Goldberg J, et al. A 2-year-old with atypical CML with a t(5;12)(q33;p13) treated successfully with imatinib mesylate. Leuk Res. 2004;28 (Suppl1):S65-S69.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
31. Grand FH, Iqbal S, Zhang L, et al. A constitutively active SPTBN1-FLT3 fusion in atypical chronic myeloid leukemia is sensitive to tyrosine kinase inhibitors and immunotherapy. Exp Hematol. 2007;35(11):1723-1727.
32. Fu Y, Schroeder T, Zabelina T, et al. Postallogeneic monitoring with molecular markers detected by pretransplant next-generation or Sanger sequencing predicts clinical relapse in patients with myelodysplastic/ Mmyeloproliferative neoplasms. Eur J Haematol. 2014;92(3):189-194. �
33. Lim SN, Lee JH, Lee JH, et al. Allogeneic hematopoietic cell transplantation in adult patients with myelodysplastic/myeloproliferative neoplasms. Blood Res. 2013;48(3):178-184. �
34. Koldehoff M, Beelen DW, Trenschel R, et al. Outcome of hematopoietic stem
cell transplantation in patients with atypical chronic myeloid leukemia. Bone Marrow Transplant. 2004;34(12):1047-1050.
35. Koldehoff M, Steckel NK, Hegerfeldt Y, et al. Clinical course and molecular features in 21 patients with atypical chronic myeloid leukemia. Int J Lab Hematol. 2012;34(1):e3-5.
36. Mittal P, Saliba RM, Giralt SA, et al. Allogeneic transplantation: a therapeutic option for myelofibrosis, chronic myelomonocytic leukemia and Philadelphia-negative/BCR-ABL-negative chronic myelogenous leukemia. Bone Marrow Transplant. 2004;33(10):1005-1009.
37. Langabeer SE, McCarron SL, Haslam K, O'Donovan MT, Conneally E. The CSF3R T618I mutation as a disease-specific marker of atypical CML post allo-SCT. Bone Marrow Transplant. 2014;49(6):843-844.
38. Patnaik MM, Tefferi A. Chronic myelomonocytic leukemia: 2016 update on diagnosis, risk stratification, and management. Am J Hematol. 2016;91:631-642.
39. Ades L, Sekeres MA, Wolfromm A, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37:609-613.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
40. Mao L, You L, Yang M, et al. The first case of decitabine successfully in treatment of atypical chronic myeloid leukemia with CEPBPA double mutation. Chemotherapy. 2013;2:114
41. Tong, Li J, Zhou Z, et al. Efficacy and side-effects of decitabine in treatment of atypical chronic myeloid leukemia. Leuk Lymphoma. 2015;56(6):1911-1913.
43. Jiang H, Wu Z, Ren LI, Tao D, Tong H. Decitabine for the treatment of atypical chronic myeloid leukemia: A report of two cases. Oncol Lett. 2016;11(1):689-692.
44. Fleischman AG, Maxson JE, Luty SB, The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood. 2013;122(22):3628-3631.
45. Shanavas M, Popat U, Michaelis LC, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with myelofibrosis with prior exposure to Janus kinase 1/2 inhibitors. Biol Blood Marrow Transplant. 2016;22(3):432-440.
46. Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67-69.
47. Freedman JL, Desai AV, Bailey LC, et al. Atypical Chronic Myeloid Leukemia in Two Pediatric Patients. Pediatr Blood Cancer. 2016;63(1):156-159.
48. Ammatuna E, Eefting M, van Lom K, et al. Atypical chronic myeloid leukemia with concomitant CSF3R T618I and SETBP1 mutations unresponsive to JAK inhibitor ruxolitinib. Ann Hematol. 2015;94:879-880.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
49. Lierman E, Selleslag D, Smits S, Billiet J, Vandenberghe P. Ruxolitinib inhibits transforming JAK2 fusion proteins in vitro and induces complete cytogenetic remission in t(8;9)(p22;p24)/PCM1-JAK2-positive chronic eosinophilic leukemia. Blood. 2012;120(7):1529-31
50. Schwaab J, Knut M, Haferlach C, et al. Limited duration of complete remission on ruxolitinib in myeloid neoplasms with PCM1-JAK2 and BCR-JAK2 fusion genes. Ann Hematol. 2015;94(2):233-238.
51. Rumi E, Milosevic JD, Selleslag D, et al. Efficacy of ruxolitinib in myeloid neoplasms with PCM1-JAK2 fusion gene. Ann Hematol. 2015;94(11):1927-1928.
52. Kurzrock R, Kantarjian H, Shtalrid M, Gutterman JU, Talpaz M. Philadelphia chromosome-negative chronic myelogenous leukemia without breakpoint cluster region rearrangement: a chronic myeloid leukemia with a distinct clinical course. Blood. 1990;75(2):445-452.
53. Montefusco E, Alimena G, Lo Coco F, et al. Ph-negative and bcr-negative atypical chronic myelogenous leukemia: biological features and clinical outcome. Ann Hematol. 1992;65(1):17-21.
54. Costello R, Lafage M, Toiron Y, et al. Philadelphia chromosome-negative chronic myeloid leukaemia: a report of 14 new cases. Br J Haematol. 1995;90(2):346-352.
55. Jabbour E, Kantarjian H, Cortes J. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer. 2007;110(9):2012-2018.
56. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111(2):574-582.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
57. Khanna V, Pierce ST, Dao KH, et al. Durable Disease Control with MEK Inhibition in a Patient with NRAS-mutated Atypical Chronic Myeloid Leukemia. Cureus. 2015;7(12):e414.
58. Borthakur G, Popplewell L, Boyiadzis M, et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer. 2016;122(12):1871-1879
59. Burgess MR, Hwang E, Firestone AJ, et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014;124(26):3947-3955.
60. Cristóbal I, Garcia-Orti L, Cirauqui C, et al. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forksolin has a potent anti-leukemic effect. Leukemia. 2011;24(4):606-614.
61. Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14(6):e229-238.
62. Lee SC, Dvinge H, Kim E, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22(6):672-678.
63. Savona MR, Malcovati L, Komrokji R, et al. MDS/MPN International Working Group. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125(12):1857-1865.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
Table 1. World Health Organization Diagnostic Criteria for Atypical Chronic Myeloid Leukemia, BCR-ABL1-negative
PV: polycythemia vera; ET: essential thrombocythemia; PMF: primary myelofibrosis * Cases of MPN, particularly those in accelerated phase and/or in post-polycythemic or post-essential thrombocythemic myelofibrosis, if neutrophilic, may simulate aCML. A previous history of MPN, the presence of MPN features in the BM and/or MPN-associated mutations (in JAK2, CALR, or MPL) tend to exclude a diagnosis of aCML. Conversely, a diagnosis of aCML is supported by the presence of SETBP1 and/or ETNK1 mutations. The presence of a CSF3R mutation is uncommon in aCML and if detected should prompt a careful morphologic review to exclude an alternative diagnosis of CNL or other myeloid neoplasm.1
• Peripheral blood leukocytosis (WBC > 13x109/L) due to increased numbers of neutrophils and their precursors with prominent dysgranulopoiesis
• Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) > 10% of leukocytes • No Ph chromosome or BCR-ABL1 fusion gene and not meeting criteria for PV, ET, or
PMF* • No evidence of PDGFRA, PDGFRB, FGFR1 rearrangement, or PCM1-JAK2 • Minimal absolute basophilia; basophils usually <2% of leukocytes • No or minimal absolute monocytosis; monocytes usually < 10% of leukocytes • Hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia,
with or without dysplasia in the erythroid and megakaryocytic lineages • Less than 20% blasts in the blood and bone marrow
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
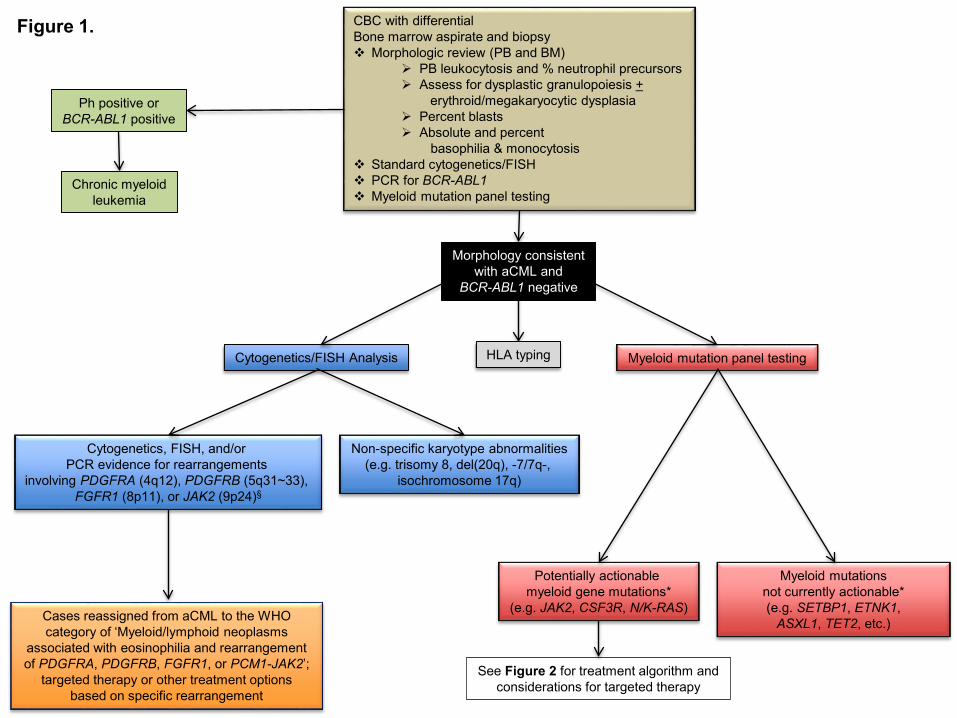
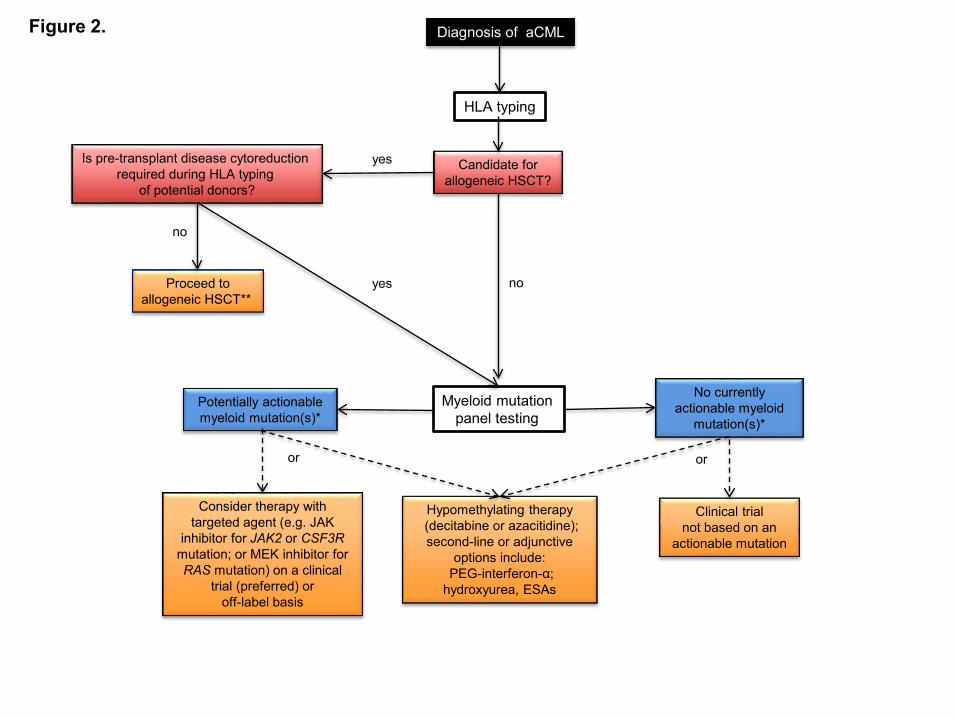
Figure Legends Figure 1. Diagnostic evaluation for atypical CML and identification of opportunities for targeted therapy. If a morphologic diagnosis of aCML is rendered, cytogenetic, FISH, and myeloid mutation panel testing are critical as they can unmask karyotypic or molecular abnormalities that have potential implications for use of targeted therapy approaches. *The ability to target certain genes is expected to change over time as new therapeutics are developed § Additional JAK2 rearrangements besides the PCM1-JAK2 fusion may present with morphologic features of aCML Figure 2. Treatment algorithm for atypical CML. Please refer to the text section ‘Treatment’ for a discussion of this treatment scheme for aCML. This algorithm is based on several decision nodes, including: 1) potential candidacy for allogeneic hematopoietic stem cell transplantation (HSCT); 2) the results of myeloid mutation panel testing; 3) eligibility for enrollment in clinical trials; and 4) opportunities to adopt strategies used for MDS or MPN (e.g. hypomethylating agents or second line/adjunctive therapies). HSCT: hematopoietic stem cell transplantation; ESAs: erythropoiesis-stimulating agents. *The ability to target certain genes is expected to change over time as new therapeutics are developed ** Myeloid mutation panel testing may also be performed prior to patients proceeding directing to allogeneic HSCT who do not require pre-transplant disease cytoreduction.
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom
doi:10.1182/blood-2016-08-693630Prepublished online November 29, 2016;
Jason Gotlib How I treat atypical chronic myeloid leukemia
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. indexed by PubMed from initial publication. Citations to Advance online articles must include final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of
For personal use only.on May 31, 2018. by guest www.bloodjournal.orgFrom