Review How to write a surgical clinical research protocol: literature review and practical guide Rachel Rosenthal, M.D., M.Sc. a, *, Juliane Scha ¨fer, Ph.D. a,b , Matthias Briel, M.D., M.Sc. b,c , Heiner C. Bucher, M.D., M.P.H. b , Daniel Oertli, M.D. a , Salome Dell-Kuster, M.D., M.Sc. a,b a Department of Surgery, Basel University Hospital, Spitalstrasse 26, CH-4031 Basel, Switzerland; b Basel Institute for Clinical Epidemiology and Biostatistics, Basel University Hospital, Basel, Switzerland; c Department of Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, Ontario, Canada KEYWORDS: Study protocol; Surgery; Design; Clinical research Abstract BACKGROUND: The study protocol is the core document of every clinical research project. Clinical research in studies involving surgical interventions presents some specific challenges, which need to be accounted for and described in the study protocol. The aim of this review is to provide a practical guide for developing a clinical study protocol for surgical interventions with a focus on methodologic issues. DATA SOURCES: On the basis of an in-depth literature search of methodologic literature and on some cardinal published surgical trials and observational studies, the authors provides a 10-step guide for developing a clinical study protocol in surgery. CONCLUSIONS: This practical guide outlines key methodologic issues important when planning an ethically and scientifically sound research project involving surgical interventions, with the ultimate goal of providing high-level evidence relevant for health care decision making in surgery. Ó 2014 Elsevier Inc. All rights reserved. The study protocol as a core document in clinical research The study protocol is the central document of a clinical research project and takes into account scientific, ethical, and regulatory considerations. It provides detailed infor- mation on all aspects of the planning and conduct of a research project and is the main document for evaluation of the planned research (eg, by an independent ethics com- mittee [IEC] and regulatory authorities). It guides study investigators to conduct the study according to standardized criteria, and it allows replication in subsequent studies. The protocol includes the justification for the planned research, the objectives, details on the intervention and the study population, information on data management, quality as- surance, statistical analyses, and ethical considerations. Importantly, the protocol should be developed in an inter- disciplinary setting, including clinicians, scientists, statis- ticians, and other involved parties. Study protocols need to be approved by an IEC and by the regulatory authorities according to local guidelines. Types and phases of surgical research Surgical clinical research may involve pharmaceuticals, medical devices, surgical procedures, and other interventions The authors declare no conflicts of interest. * Corresponding author. Tel.: 141-61-556-53-63; fax: 141-61-265- 88-81. E-mail address: [email protected]Manuscript received April 24, 2013; revised manuscript July 15, 2013 0002-9610/$ - see front matter Ó 2014 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.amjsurg.2013.07.039 The American Journal of Surgery (2014) 207, 299-312
Transcript
The American Journal of Surgery (2014) 207, 299-312
Review
How to write a surgical clinical research protocol:literature review and practical guide
aDepartment of Surgery, Basel University Hospital, Spitalst
rasse 26, CH-4031 Basel, Switzerland; bBasel Institute forClinical Epidemiology and Biostatistics, Basel University Hospital, Basel, Switzerland; cDepartment of ClinicalEpidemiology and Biostatistics, McMaster University, Hamilton, Ontario, Canada
KEYWORDS:Study protocol;Surgery;Design;Clinical research
The authors declare no conflicts of i
* Corresponding author. Tel.: 141-
88-81.
E-mail address: rachel.rosenthal@us
Manuscript received April 24, 2013;
0002-9610/$ - see front matter � 2014
http://dx.doi.org/10.1016/j.amjsurg.20
AbstractBACKGROUND: The study protocol is the core document of every clinical research project. Clinical
research in studies involving surgical interventions presents some specific challenges, which need to beaccounted for and described in the study protocol. The aim of this review is to provide a practical guidefor developing a clinical study protocol for surgical interventions with a focus on methodologic issues.
DATA SOURCES: On the basis of an in-depth literature search of methodologic literature and onsome cardinal published surgical trials and observational studies, the authors provides a 10-step guidefor developing a clinical study protocol in surgery.
CONCLUSIONS: This practical guide outlines key methodologic issues important when planning anethically and scientifically sound research project involving surgical interventions, with the ultimategoal of providing high-level evidence relevant for health care decision making in surgery.� 2014 Elsevier Inc. All rights reserved.
The study protocol as a core document inclinical research
The study protocol is the central document of a clinicalresearch project and takes into account scientific, ethical,and regulatory considerations. It provides detailed infor-mation on all aspects of the planning and conduct of aresearch project and is the main document for evaluation ofthe planned research (eg, by an independent ethics com-mittee [IEC] and regulatory authorities). It guides study
nterest.
61-556-53-63; fax: 141-61-265-
b.ch
revised manuscript July 15, 2013
Elsevier Inc. All rights reserved.
13.07.039
investigators to conduct the study according to standardizedcriteria, and it allows replication in subsequent studies. Theprotocol includes the justification for the planned research,the objectives, details on the intervention and the studypopulation, information on data management, quality as-surance, statistical analyses, and ethical considerations.Importantly, the protocol should be developed in an inter-disciplinary setting, including clinicians, scientists, statis-ticians, and other involved parties. Study protocols need tobe approved by an IEC and by the regulatory authoritiesaccording to local guidelines.
Types and phases of surgical research
Surgical clinical research may involve pharmaceuticals,medical devices, surgical procedures, and other interventions
300 The American Journal of Surgery, Vol 207, No 2, February 2014
concerning prevention, diagnostics, treatment, and rehabil-itation. In drug development, the phases of investigation havebeen well defined, and most typically range from humanpharmacologic studies (phase 1) through therapeutic explor-atory (phase 2) to therapeutic confirmatory studies (phase 3),followed by postmarketing studies (phase 4).1 Similar phasesranging from pilot, pivotal, through to postmarketing surveil-lance have been described for medical devices, for which re-quirements for demonstrating safety and efficacy depend onthe risk associated with the devices.2 In the example of re-search involving surgical interventions, the IDEAL frame-work has been proposed, the acronym standing for thestages (1) idea (including proof of concept); (2a) develop-ment, (2b) exploration; (3) assessment; and (4) long-termstudy.3,4 An overview of the IDEAL framework with exam-ples is provided in Table 1.
In this review article, we focus on surgical interventions,although some of the concepts may be extrapolated to otherinterventions.
Challenges in clinical research of surgicalinterventions
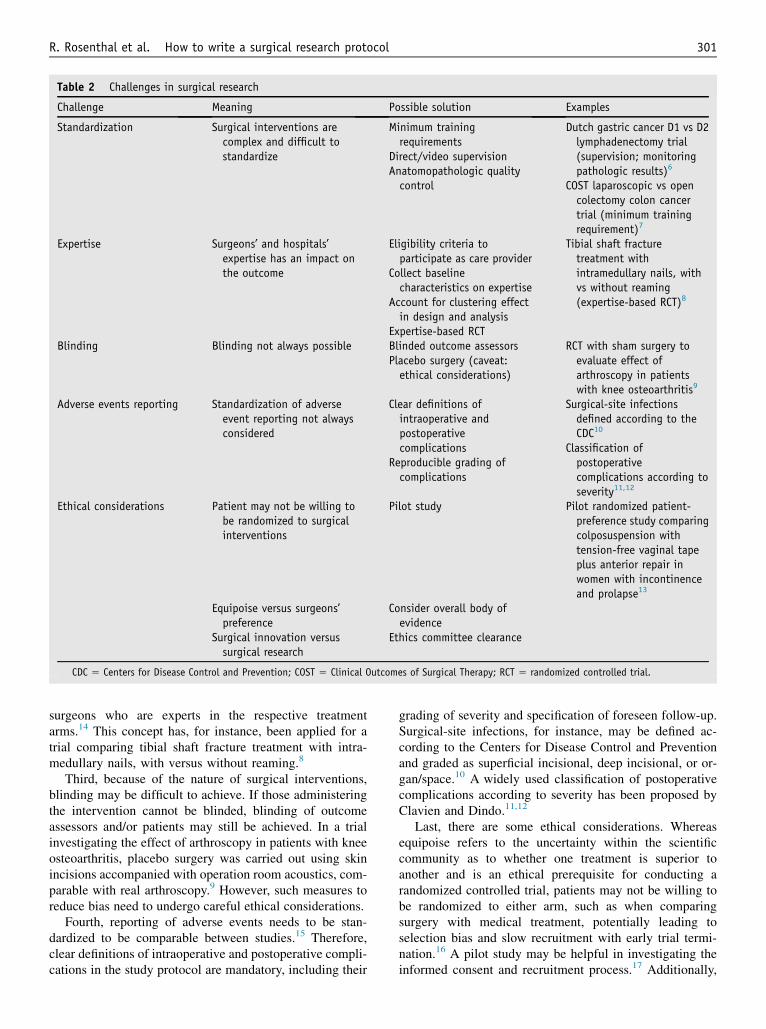
When planning surgical research involving surgicalinterventions, some specific challenges need to be ad-dressed (for an overview, possible solutions, and examples,refer to Table 2).5–13
Table 1 Stages of surgical innovation
IDEAL stage Purpose Study design
1. Idea Proof of concept Case report/case seri
2a. Development Development Prospective cohort
2b. Exploration
3. Assessment
Learning
Assessment
Research database (pFeasibility/explanatoRCTAlternative designs iapplicable� Matched case-c� Interrupted tim
observations oted by interven
� Controlled befo(observation btervention in icontrol group)
� Step-wedged deder of introduction in a prosp
4. Long-term study Surveillance Routine database/regcohort)
First, compared with pharmacologic trials, surgicalinterventions are more complex and may thus be moredifficult to standardize. Standardization may be enhancedand controlled by specific surgeon selection (ie, minimumtraining requirements) and training, direct and video-recorded supervision, as well as anatomopathologic qualitycontrol, as for instance in the Dutch gastric cancer D1versus D2 lymphadenectomy trial6 and the Clinical Out-comes of Surgical Therapy laparoscopic versus open colec-tomy colon cancer trial.7 If applicable, details should beprovided in the protocol for how interventions are tailoredto individual patients.5
Second, surgeons’ expertise or hospital standards mayhave an impact on clinical outcomes and treatment effects,which needs to be accounted for in the design and analysisphase of the study.5,14 This may, for instance, be addressedby defining eligibility criteria to participate as a care pro-vider and center in a trial and further be enhanced by fore-seeing baseline data on the care providers’ and centers’case volumes, expertise, and qualifications, as well as bytaking into account the clustering effect of care providersand centers in sample-size calculations, statistical analyses,and reporting.5 However, surgeons may tend to be most ex-perienced in one surgical approach, which potentially leadsto differential expertise bias, even if they meet minimumcriteria for participation in a trial.14 This problem may beaddressed by surgical expertise–based randomized con-trolled trials, in which patients are randomized to different
Example: INRA after ulcerative colitisand familial adenomatous polyposis4
es Based on animal experiments (pigmodel), pilot study in 11 patients
Confirmation of pilot study findings andrefining of technique in 26 patients
rospective cohort)ry RCT
f RCT not
Prospective cohort with extendedinclusion criteria
ontrol studye series (multiplever time, interrup-tion)re-and-after studyefore and after in-ntervention and
sign (random or-tion of interven-ective cohort)
Matched case-control study (INRA vsIPAA as gold standard) with long-term results showing comparablemorbidity and functional results, noadvantage from the patients’ point ofview, and a disadvantage from thesurgeons’ point of view (technicallydemanding and long surgery)
/ Decision not to offer INRA anymore,use of IPAA as standard
istry (prospective
nts)
RCT 5 randomized controlled trial. Adapted from McCulloch et al.3
Table 2 Challenges in surgical research
Challenge Meaning Possible solution Examples
Standardization Surgical interventions arecomplex and difficult tostandardize
Surgical-site infectionsdefined according to theCDC10
Classification ofpostoperativecomplications according toseverity11,12
Ethical considerations Patient may not be willing tobe randomized to surgicalinterventions
Pilot study Pilot randomized patient-preference study comparingcolposuspension withtension-free vaginal tapeplus anterior repair inwomen with incontinenceand prolapse13
Equipoise versus surgeons’preference
Consider overall body ofevidence
Surgical innovation versussurgical research
Ethics committee clearance
CDC 5 Centers for Disease Control and Prevention; COST 5 Clinical Outcomes of Surgical Therapy; RCT 5 randomized controlled trial.
R. Rosenthal et al. How to write a surgical research protocol 301
surgeons who are experts in the respective treatmentarms.14 This concept has, for instance, been applied for atrial comparing tibial shaft fracture treatment with intra-medullary nails, with versus without reaming.8
Third, because of the nature of surgical interventions,blinding may be difficult to achieve. If those administeringthe intervention cannot be blinded, blinding of outcomeassessors and/or patients may still be achieved. In a trialinvestigating the effect of arthroscopy in patients with kneeosteoarthritis, placebo surgery was carried out using skinincisions accompanied with operation room acoustics, com-parable with real arthroscopy.9 However, such measures toreduce bias need to undergo careful ethical considerations.
Fourth, reporting of adverse events needs to be stan-dardized to be comparable between studies.15 Therefore,clear definitions of intraoperative and postoperative compli-cations in the study protocol are mandatory, including their
grading of severity and specification of foreseen follow-up.Surgical-site infections, for instance, may be defined ac-cording to the Centers for Disease Control and Preventionand graded as superficial incisional, deep incisional, or or-gan/space.10 A widely used classification of postoperativecomplications according to severity has been proposed byClavien and Dindo.11,12
Last, there are some ethical considerations. Whereasequipoise refers to the uncertainty within the scientificcommunity as to whether one treatment is superior toanother and is an ethical prerequisite for conducting arandomized controlled trial, patients may not be willing tobe randomized to either arm, such as when comparingsurgery with medical treatment, potentially leading toselection bias and slow recruitment with early trial termi-nation.16 A pilot study may be helpful in investigating theinformed consent and recruitment process.17 Additionally,
302 The American Journal of Surgery, Vol 207, No 2, February 2014
surgeons should be well aware of their potentially conflict-ing roles as clinicians versus investigators. Even if clinicalequipoise is established within the expert clinical commu-nity, an individual surgeon may still have a preference forone treatment. This dilemma may be addressed by recog-nizing that the overall body of evidence does not suggestany treatment to be superior.18 Moreover, sometimes regu-lar practice, surgical innovation, and surgical research maybe difficult to discriminate. In such circumstances, ethicscommittees should be liberally consulted.18
The purpose of this report is to provide a guide fordeveloping a study protocol while focusing on the keymethodologic issues to consider when investigating asurgical intervention, be it in an observational or interven-tional setting.
Guide for Developing a clinical studyProtocol for surgical Interventions
This report provides a 10-step practical guide for devel-oping a clinical study protocol investigating a surgicalintervention using observational or interventional data. Itfocuses on methodologic issues and may be used as adjunctto existing international guidelines,19 local regulations, andthe recommendations of the Standard Protocol Items: Rec-ommendations for Interventional Trials initiative.20,21 Theinformed-consent process is beyond the scope of this article.
Step 1: defining the research question
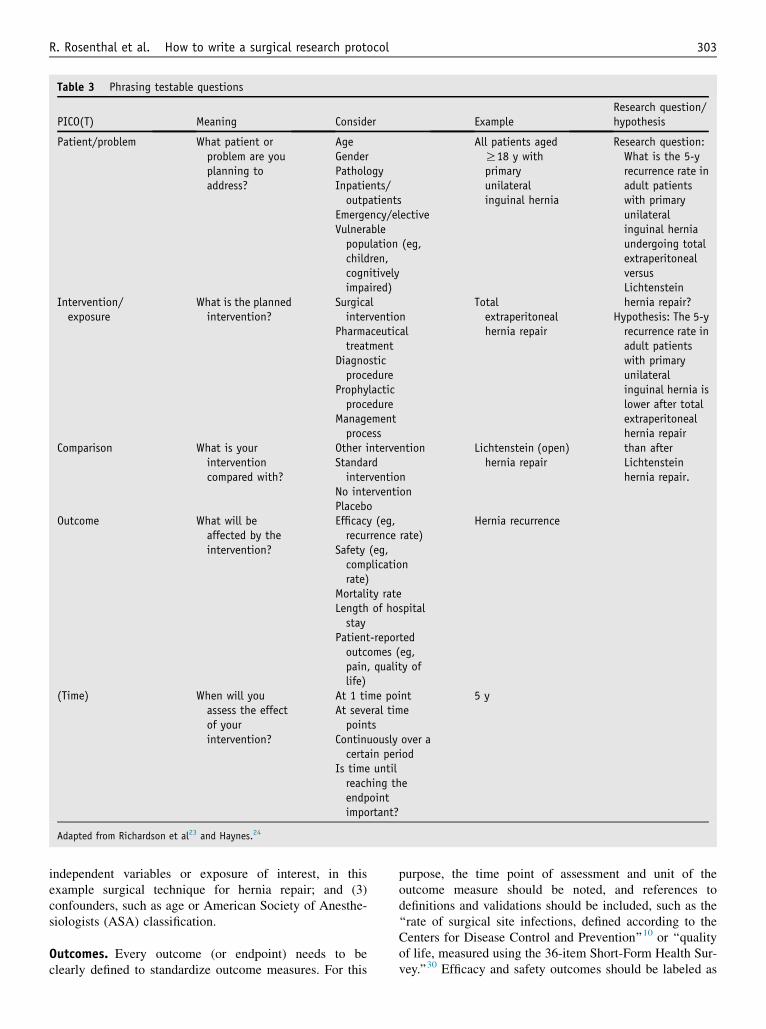
The heart of every protocol is the research question. Itdefines the knowledge gap that will be filled with theplanned research. Characteristics of a good research ques-tion are easily described using the mnemonic ‘‘FINER,’’ asproposed by Cummings et al,22 standing for feasible interms of scope, expertise, resources, and recruitment; inter-esting to the investigator and the scientific community;novel, targeting new findings or the extension, confirma-tion, or rejection of previous findings; ethical, with fair sub-ject selection and a favorable risk-benefit ratio; and relevantto scientific knowledge, daily practice, health policy, andfuture research. It is crucial to precisely formulate the re-search question. This allows the development of a statisticalanalysis plan and the determination of the sample size nec-essary to attain a targeted power. When formulating a re-search question, the acronym ‘‘PICO’’23 may be helpfulfor phrasing testable questions. ‘‘PICO’’ stands for the pa-tient/problem, the intervention or exposure, the compari-son, and the outcome. Some add a T (PICOT) as a 5thelement, which stands for time (time frame of outcome as-sessment),24 whereas in review questions, it may stand forstudy type (eg, randomized controlled trial, cohort study).The relevant points to consider when formulating the re-search question and examples are presented in Table 3.
There may be several research questions, but in general,the most important one should be labeled as the primary
research question and the other(s) as secondary researchquestion(s). For each research question, a hypothesis shouldbe formulated to prespecify what results are expected.Because the sample-size calculation is based on the primaryoutcome, secondary research questions may not necessarilybe answered with sufficient power. They are thus often moreexploratory in nature. In an inguinal hernia trial comparing 2surgical techniques, a secondary research question could be,for instance, to compare the postoperative quality of lifebetween the 2 techniques, whereas the primary researchquestion may be the comparison of recurrence rates.
It is important to define these research questions andoutcomes in advance. Post hoc specification with the risk fordata-driven selection may first introduce considerable out-come reporting bias (ie, significant results being more likelyto be reported than negative results) and, second, lead to theerror-prone acceptance of an association on the basis ofmultiple post hoc tests.25 Trial registries havebeen introducedto enhance transparency and to address the problem of publi-cation bias and outcome reporting bias.26 Trial registration in-cludes information on the choice of primary and secondaryoutcomes.27 The International Committee ofMedical JournalEditors28 and subsequently the Surgical Journal EditorsGroup29 have published guidelines for mandatory trial regis-tration for all trials as a prerequisite for considering a scien-tific report for publication in the respective member journals.
Step 2: justification of the planned research
It is important for the reader to understand why thisresearch is planned. This involves an overview of thecurrent knowledge in the field (‘‘What has been done?’’)and a presentation of the knowledge gap that will beaddressed with the planned research (‘‘What needs to bedone?’’). The ultimate purpose is to justify from scientific,ethical, and economic points of view the conduct of theresearch. The presentation of the current state of the art andknowledge in the field implies a systematic review of theliterature, including published literature and the gray liter-ature, along with consulting trial registries to obtaininformation about ongoing trials or past unpublished trials.The presented literature should be critically commented on,indicating eventual discrepancies in study results or limi-tations of study design, methodologic quality componentssuch as blinding or extent of follow-up, and the number ofincluded participants. The key information from the citedstudies may be presented within a table. This part of theprotocol directly leads the reader to the aim of the plannedresearch, which represents the logical consequence of thelack of knowledge previously described.
Step 3: deciding on outcomes and confounders
Variables may be divided into (1) outcome or dependentvariables, such as the recurrence rate in a study comparing2 different surgical techniques for hernia repair; (2)
Table 3 Phrasing testable questions
PICO(T) Meaning Consider ExampleResearch question/hypothesis
Patient/problem What patient orproblem are youplanning toaddress?
All patients agedR18 y withprimaryunilateralinguinal hernia
Research question:What is the 5-yrecurrence rate inadult patientswith primaryunilateralinguinal herniaundergoing totalextraperitonealversusLichtensteinhernia repair?
Hypothesis: The 5-yrecurrence rate inadult patientswith primaryunilateralinguinal hernia islower after totalextraperitonealhernia repairthan afterLichtensteinhernia repair.
(Time) When will youassess the effectof yourintervention?
At 1 time pointAt several timepoints
Continuously over acertain period
Is time untilreaching theendpointimportant?
5 y
Adapted from Richardson et al23 and Haynes.24
R. Rosenthal et al. How to write a surgical research protocol 303
independent variables or exposure of interest, in thisexample surgical technique for hernia repair; and (3)confounders, such as age or American Society of Anesthe-siologists (ASA) classification.
Outcomes. Every outcome (or endpoint) needs to beclearly defined to standardize outcome measures. For this
purpose, the time point of assessment and unit of theoutcome measure should be noted, and references todefinitions and validations should be included, such as the‘‘rate of surgical site infections, defined according to theCenters for Disease Control and Prevention’’10 or ‘‘qualityof life, measured using the 36-item Short-Form Health Sur-vey.’’30 Efficacy and safety outcomes should be labeled as
304 The American Journal of Surgery, Vol 207, No 2, February 2014
such, and standard procedures for reporting and patientfollow-up of adverse events need to be described.
When choosing outcomes, it is important to be aware ofseveral points that affect the statistical analysis plan andsample-size calculation: (1) the types of variables that arecollected31 (eg, categorical, metric, time-to-event data); (2)in the case of continuous variables, whether they can be ex-pected to be normally distributed or not; and (3) whetherpaired or unpaired data are collected. Categorical (binaryin the case of 2 categories) or qualitative variables have nounits and may be divided into nominal variables in thecase of unordered categories (eg, blood group) and ordinalvariables in the case of ordered categories (eg, ASA class).They are most often displayed in frequency tables and barcharts. Metric or quantitative variables are referred to eitheras discrete variables with integer values and counted units(eg, number of episodes of angina pectoris per week) or ascontinuous variables with stepless values andmeasured units(eg, blood pressure). They are typically displayed reportingtheir central values and variation (ie, mean and standard de-viation in case of normal distribution, else median and rangeor interquartile range). In graphs, box plots and histogramsare used for displaying metric variables. Typical examplesof time-to-event data are overall or progression-free sur-vival. Categorization of continuous variables should beavoided, because this is associated with a loss of informationand therefore a loss of power and precision. Right-skeweddata (ie, most of the data are concentrated on the left, withrelatively few high values) are relatively frequent; examplesare many laboratory findings and the duration of surgery orof hospitalization. Skewed data may be transformed for sta-tistical analysis to achieve better approximation to normalityby, for example, logarithmic transformation. Paired or clus-tered data are generated if the same measurement is repeatedin the same patient over time (eg, repeated measurements ofpain in the same patient after hernia repair) or twice in thesame patient at 1 point in time, such as evaluation of herniarecurrence on the right and the left sides after bilateral ingui-nal hernia repair. Paired and clustered data will have an im-pact on the choice of methods for statistical analysis,because the variability of several measurements within 1 pa-tient is smaller than the variability of measurements betweenseveral independent patients.
For some outcome variables and settings, it is a prereq-uisite to obtain baseline information, such as evaluating thequality of life before and after hernia repair.
Outcomes may be objective, such as mortality or sub-jective, such as pain. In any case, patient-important out-comes should be considered. For subjective outcomes,blinding is especially relevant.
In the case of rare events with insufficient power toevaluatemultiple single outcomes, orwhenno single outcomeoptimally represents the outcome of interest, a compositeendpointmay be chosen.32 To enhance feasibility and compa-rability of randomized controlled trials, this has, for instance,been proposed for liver surgery with a composite endpoint in-volving ‘‘ascites, postresectional liver failure, bile leakage,
intra-abdominal haemorrhage, intra-abdominal abscess andoperative mortality.’’33 Hereby, the individual componentsof the composite endpoint should be of similar importanceto patients, they should occurwith similar frequency, and sim-ilar treatment effects (eg, relative risk reductions) should beexpected,34 whereas components that are redundant or mar-ginally related to the intervention should be avoided.32
Surrogate endpoints and surrogate biomarkers are fre-quently used, because they may be easier and faster toassess compared with the patient-important outcome.35 Anexample is the surrogate endpoint of lipid profile instead ofmajor cardiovascular events. A surrogate endpoint can bedefined as ‘‘a laboratory measurement or a physical signused as a substitute for a clinically meaningful endpointthat measures directly how a patient feels, functions or sur-vives.’’36 The effect of the intervention on the surrogateendpoint should predict the effect on the clinically relevantoutcome.37 Thus, the use of surrogate endpoints needs to becarefully evaluated.38,39
Independent and confounding variables. In a random-ized controlled trial, confounders should be equally dis-tributed in the different treatment arms through the processof randomization, if randomization has been correctlyconducted and if the number of randomized individuals issufficiently large. This is not the case in observational data.In a cohort study including patients having undergonelaparoscopic or open left colectomy, for example, not onlythe surgical technique but also age or ASA classificationmay have an impact on the length of hospital stay and willprobably not be equally distributed between groups. To beregarded as a confounder, these factors need to have animpact not only on the outcome but also on the choice ofintervention (ie, independent variable), meaning an olderpatient may be more likely to be assigned to one treatmentoption than to the other. Which factors qualify to beconfounders (ie, are associated with the outcome as well aswith the exposure of interest) should be prespecified in thestudy protocol according to expert opinion and informationgathered accordingly.40
Confounding may be controlled for in the design as wellas in the analysis of a study. In the design, randomizationshould lead to equally distributed known and unknownconfounders in the groups. Stratification with or withoutrandomization aims at balancing the groups for specificprognostic patient characteristics.41 In the Clinical Out-comes of Surgical Therapy trial comparing laparoscopi-cally assisted with open colectomy for colon cancer,randomization stratified for the site of primary tumor,ASA classification, and surgeon was undertaken.7 If a ran-domized study is not possible, matching may be a strategyto reduce confounding. In the statistical analysis, potentialconfounding may be addressed by adjusting for these vari-ables using multivariate regression analysis. Other ways tocontrol for confounding are using propensity scores (theprobability of an individual to be treated with an interven-tion given all available baseline information on the patient)
R. Rosenthal et al. How to write a surgical research protocol 305
or inverse probability weighting (the reciprocal of an indi-vidual’s probability of receiving the treatment that he or sheactually received).42,43
Superiority, equivalence, and noninferiority. An a prioristatement of the overall goal of comparison needs to beprovided in the protocol. The reader should know whetherthe goal is to show that treatment A is superior, equivalent, ornoninferior to treatment B. From a superiority study withnonsignificant results, one may not conclude that the inter-ventions are equivalent.44 In an equivalence (2-sided hypoth-esis) or noninferiority (1-sided hypothesis) setting, themargin of noninferiority, respectively the 2 margins of equiv-alence, needs to be prespecified, that is, respectively the larg-est or smallest values representing clinically irrelevantdifferences need to be defined in advance. An aid when de-fining the margin(s) is the question of whether the investiga-tional intervention is equivalent if its efficacy or safetyoutcome is within the chosen boundaries. This margin hasimplications for the sample size (ie, the smaller the margin,the larger the sample size). As a rule of thumb, the requiredsample size is higher with equivalence and noninferiority de-signs than in superiority trials. Examples of noninferioritytrials are trials in surgical oncology with the ultimate goalof assessing whether a new intervention with potential ben-efits such as lower invasiveness, lower toxicity, or reducedcost is equivalent or not inferior to an established therapyconcerning efficacy (ie, cancer control).45 Examples arethe aforementioned gastric lymphadenectomy trial6 and theClinical Outcomes of Surgical Therapy trial.7
Multiple comparisons, multiple testing, and interimanalyses. Multiple comparisons between different groupsneed to be carefully justified; the same is true for multipletesting because of multiple outcomes or multiple timepoints in the case of interim analyses.
If there is a possibility that a treatment effect might bedifferent in different subgroups of patients, this should beexamined through an additional interaction term in theregression model46 rather than multiple testing of each sub-group.47 To give an example, in a long-term comparison ofendovascular versus open aortic aneurysm repair, a signifi-cant interaction between age and type of treatment wasfound with a better survival in patients ,70 years of age af-ter endovascular repair versus borderline better survival inpatients R70 years of age after open repair.48 Subgroupsshould be prespecified in the study protocol, and post hocsubgroup analyses should be declared as such and thusare more explorative in nature. All subgroup analysesshould be reported to avoid the risk for selective data-driven reporting.47 In case of a continuous covariate, whichmay influence response to treatment, advanced methods formodeling treatment-covariate interactions using fractionalpolynomials should be considered.49
Because multiple comparisons and multiple testingincrease the chance of committing an a error (type I error,ie, concluding that there is a difference when in fact there is
no difference), this needs to be accounted for with a morestringent P value considered as significant. To give an ex-ample, if 20 independent outcomes are compared between2 groups using hypothesis tests, the global type I errorrate will increase to 64%. Various procedures have been de-scribed to control for the multiple type I error rate, amongwhich the Bonferroni (a divided by the number of tests)and Bonferroni-Holm procedures are quite common, asthey strictly control the multiple type I error rate.50 Thus,in the case of 20 outcomes, to be statistically significant,the P value must be ,.0025 (.05/20) after correction formultiple testing according to the Bonferroni procedure.
In an interim analysis, trial data are analyzed by treatmentgroup for study monitoring purposes before the final anal-ysis. Reasons may be monitoring for superiority, harm, orfutility. On the basis of the results of interim analyses, trialsmay be stopped early, typically as evaluated by an indepen-dent data safety and monitoring board. The number ofinterim analyses and the definition of stopping rules ac-counting for multiple testing (eg, according to O’Brien-Fleming, Peto, or Pocock) needs to be prespecified in thestudy protocol.47 These rules define P values for consideringstopping a trial early depending on the overall number ofplanned interim analyses and preserving the overall type I er-ror rate.47 In a trial with 2 interim analyses and 1 final anal-ysis, for instance, the P values for the interim stopping levelwould be, for the first interim, second interim, and final anal-ysis, .0005, .014, and .045 according to the rule of O’Brien-Fleming and .001, .001, and .05 according to the rule of Peto,the latter applying constant stopping levels until the finalanalysis.47 To give an example, a study investigating surgeryfollowed by radiotherapy versus radiotherapy alone for met-astatic cancer spinal cord compression was stopped early bythe data safety and monitoring board at a planned interimanalysis after recruitment of half of the foreseen patientsfor superiority of the surgical intervention arm at a P valueof .001 according to the rule of O’Brien-Fleming.51 Stop-ping a trial early for superiority or futility, however, mustbe carefully evaluated. Empirical evidence indicates thattrials having been stopped early for benefit tend to overesti-mate the underlying true treatment effect. Therefore, interimanalysis should be well justified and if possible not be con-ducted for detecting an early benefit.52
Step 4: choosing the appropriate design
Types of study design and potential biases. Dependingon the research question, the appropriate study design needsto be chosen and described in the protocol. An overview ofstudy designs53 with examples54–59 is provided in Table 4.A more detailed description of the pros and cons of differ-ent study designs is beyond the scope of the present article.
Bias (ie, a systematic error) jeopardizes the internal (relia-bility and accuracy) and external (generalizability) validity ofstudies. Therefore,methods tominimize the risk for bias need tobe outlined in the protocol.33,34 Many different types of biases
Table 4 Design types
Study type
Descriptive Analytic
Population Individual ObservationalInterventional(experimental)
Ecologic (correla-tional) study Case report/case series
Attrition bias The loss of participants from thestudy (ie, dropout, withdrawalfor example because ofdeviation from the protocol) isdifferent in the interventionand control group
A higher dropout rate in theintervention group than in thecontrol group mayunderestimate herniarecurrences in a trialcomparing 2 hernia repairtechniques
Measures to reduce dropoutsDocumentation of patient flowincluding dropouts
Intention-to-treat analysisDeclaration of strategies to dealwith missing data (eg, lastobserved value carriedforward, best-case/worst-casescenario assumption, multipleimputation)
Adapted from Akobeng61 and Bornhoft et al.62
R. Rosenthal et al. How to write a surgical research protocol 307
have been described, and they may be classified in differentways, such as by the direction of resulting change in the estimateor by the stage of research, in which they occur.60 Examples arerandomization or matched pairs to address selection bias, blind-ing to address performance and detection bias, and measures toreduce loss to follow-up to address attrition bias. An outline ofbiases and possible solutions to address them is provided inTable 5.61,62
Table 6 Hypothesis tests and multivariate analysis
Outcome Parametric No
ContinuousUnpaired measurements(comparison of 2 groups)
Unpaired t test W
Paired measurements(comparison of 2 groups)
Paired t test W
Comparison of .2 groups Analysis of variance KrCategoricalUnpaired measurements Chi-square test FiPaired measurements McNemar’s chi-square test M
Time to event Log-rank test
*Applicable if the expected value (under the null) of any of the cells in th
Randomization. The process of randomization comprisesthe allocation sequence generation, allocation concealment(ie, neither the participants nor the investigators are able topredict group assignment), and allocation sequenceimplementation.61
The protocol needs to specify how the randomizationsequence is generated; examples are variable–block sizerandomization, stratified randomization, and cluster
nparametricMultivariate analysis(examples)
ilcoxon’s rank-sum test(Mann-Whitney U test)
Continuous: linear regressionDiscrete: Poisson regression
ilcoxon’s signed-rank test,sign testuskal-Wallis test
sher’s exact test* Binary: logistic regressionethods based on exactprobabilities
Regression analysis ofsurvival/time-to-eventdata (eg, Cox or Poissonregression or parametricmodels)
e table is ,5. Adapted from Kirkwood and Sterne.66
Example binaryoutcome variable:complication yes/no
Example quasi-continuous outcomevariable (normallydistributed):quality-of-life score
Univariable Assumptions Significance level/ type I error(studyerroneouslyrejects the null,ie, claims adifferencealthough there isnone)
Most typically.05.01
.05 .05
Power / type IIerror (studyerroneouslyaccepts the null,ie, finds nodifferencealthough there isone)
Most typically80%90%
80% 90%
Effect control group LiteraturePilot study
10% Mean, 35.5
Effect interventiongroup
Clinically relevantdifference
5% Mean, 45.5
Standarddeviation/variance
Check assumptionof equal SD inintervention/control group
Not necessary; willbe derived fromthe choseneffects
SD, 9.0 in controland interventiongroups
1 sided/2 sided Superiority designmost typically 2sided
2 sided 2 sided
Sample size Calculated samplesize*
Lack of normalityfor continuousoutcome /
Inflate sample sizeby 15%68
Estimate samplesize withsimulation69
434 per group or868 in total
18 per group or 36in total
Final sample sizeaccounting fordropouts
LiteraturePilot study
10% dropout,resulting in 478per group or 956in total
20% dropout,resulting in 22per group or 44in total
Sample-sizestatement
Assuming a 10%dropout rate, asample size of478 per group isnecessary to havean 80% chance ofdetecting, assignificant at the5% level, adecrease incomplication ratefrom 10% in thecontrol group to5% in theinterventiongroup
Assuming a dropoutrate of 20%, 44patients arerequired to have a90% chance ofdetecting, assignificant at the5% level, anincrease in theprimary outcomemeasure from35.5(SD 9.0) in thecontrol group to45.5 (SD 9.0) inthe experimentalgroup
(continued on next page)
308 The American Journal of Surgery, Vol 207, No 2, February 2014
Avoid overfitting(do not includetoo manyvariables)
To adjust for age,surgeonexperience (highvs low), and ASAclass (R3 vs ,3)(ie,1 independentvariable and 3confounders), aminimum of 40patients with/withoutcomplication(whichever is thesmallerpercentage) needto be observed
To adjust for age,gender, and ASAclass (R3 vs ,3)(ie,1 independentvariable and 3confounders), aminimum of 40patients need tobe observed
*Using R version 2.14.2 (R Foundation for Statistical Computing, Vienna, Austria).
R. Rosenthal et al. How to write a surgical research protocol 309
randomization. Pseudo-randomization or quasi-randomization (eg, according to date of birth, date of entry,patient ID, or alternation) should be avoided, becauseallocation will be easily predictable.
Additionally, the method of information transfer needs tobe described (eg, central Web-based randomization, centraltelephone randomization, or serially numbered opaquesealed envelopes). Central randomization is preferred be-cause it is more reliable to ensure allocation concealment.
Because the surgeons’ and institutions’ expertise mayhave an impact on the clinical outcome and treatment effect(ie, performance bias), an expertise-based design may beappropriate in certain circumstances.14
Blinding. Whenever possible, blinding should be consideredand outlined in the study protocol. Studies may be unblinded(open label), single blind (ie, patient blind), or double blind(patient and caregiver blind).63 Other terms have been used,such as ‘‘triple blind,’’ referring to the patient, caregiver, andassessor. Because there are other persons involved in a trial(data collector, outcome adjudicator, data analyst), it is bestto describe in detail who is blinded and to what.
In surgery, blinding may be a challenge. To limit detectionbias, it is advisable to use blinded outcome assessment (eg,through a separate team of assessors not involved in surgery).Sham or placebo surgery has been previously conducted,9 butthis approach requires special ethical justification.
Step 5: description of the study procedures
A detailed description of all study procedures should beprovided. Importantly, it should be made clear what is part
of clinical routine and what is study specific. Becausesurgical interventions are complex, they need to be stan-dardized as outlined above to be able to draw generalizableconclusions from the study. Moreover, the surgeons’ ex-pertise or specific hospital standards may have an impact onthe outcome, which needs to be accounted for.5 If applica-ble, measures to ensure compliance should be described.
An activity plan in the format of a table describing allactivities during the study periods of enrollment, allocation,postallocation, and close-out, such as screening procedures,intervention, and different types of outcome assessmentswith corresponding timelines and allowed deviance fromthe foreseen date, could be helpful in providing an over-view of all involved procedures.22 Discontinuation criteriafor study participants, parts of the trial, or the trial as awhole should be described as well.
Step 6: description of the study population
There are medical, methodologic, and ethical criteria todefine a study population. The choice of participant inclu-sion and exclusion criteria will have an impact on theinternal (reliability and accuracy) and external (generaliz-ability) validity of a study61,62 and depend on the goal ofthe study (ie, an explanatory trial testing efficacy with rig-orous control of internal validity vs a pragmatic trial eval-uating effectiveness under clinical real-life conditions).64
The inclusion of a vulnerable population, such aschildren or cognitively impaired adults (eg, in emergencysettings), requires special justification and measures ofparticipant protection.65
310 The American Journal of Surgery, Vol 207, No 2, February 2014
Step 7: development of a statistical analysisplan
The goal of a quantitative assessment entails that thescientific research question be translated into a statisticalproblem. In the respective protocol section, the statisticalmethods should be described in sufficient detail, includingthe statistical software to be used, the analysis population(eg, intention to treat [ITT] or per protocol [PP]), descrip-tive or exploratory statistics, hypothesis testing indicatingthe level of significance and taking into account the type ofoutcome, effect measures (with confidence intervals), typeof sample (paired vs unpaired), assumption of data distri-bution (normally vs not normally distributed data) andmodeling if applicable. Table 6 summarizes the most com-mon hypothesis tests and examples of regression modelsdepending on the type of outcome variable.66
Confidence intervals are preferred over P values becausethey provide information not only regarding statistical sig-nificance but also about the smallest and largest plausiblevalues of the effect measure of interest. Importantly, ‘‘ab-sence of evidence’’ commonly does not equal ‘‘evidenceof absence,’’ and statistical significance is not to be consid-ered equivalent to clinical relevance.44
According to the design, the analysis population (ITT vsPP) needs to be prespecified. ITT refers to the population asrandomized, regardless of factors such as compliance,crossover, or loss to follow-up, whereas PP refers to thepatients actually treated and followed as foreseen in theprotocol. In a superiority trial, the ITT analysis is preferred.It is conservative, because noncompliant participants gen-erally reduce the treatment effect. In contrast, in anequivalence or noninferiority design, because of the poten-tially reduced treatment effect, ITT is no longer conserva-tive. Therefore, the PP analysis is the conservative andpreferred primary analysis in the noninferiority setting,complemented by an ITT analysis.67
Procedures for handling missing data (eg, last observa-tion carried forward, best-case or worst-case scenarioimputation, multiple imputation, censoring) should beoutlined, as well as any planned interim analyses indicatingthe number, time point, and definition of stopping rules asoutlined above. Subgroup investigations and interactionanalyses should be determined in advance.
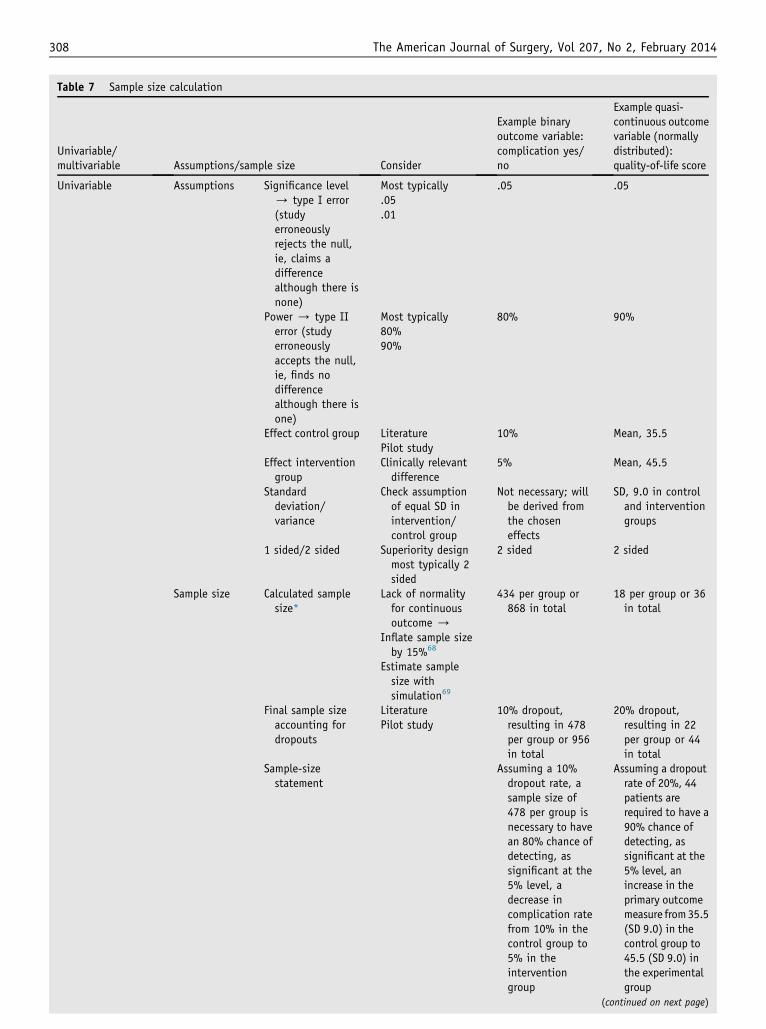
Step 8: sample-size calculation
The sample size is chosen to ensure that the study willhave sufficient power to allow conclusive inferences re-garding the primary outcome, given the assumptions for thesample-size calculation happen to be realistic. In a supe-riority trial, a sample-size statement should include the alevel, power (equals 12 b or type II error), the event rate orvalue in the control group, the expected (or clinicallyrelevant) effect in the experimental group, 1-sided versus 2-sided testing, and the expected rate of loss to follow-up. In
case of binary outcomes, the effects are estimated asproportions and, in case of continuous normally distributedoutcome measures, as means and standard deviations, thelatter as a measure of variability in the 2 groups. Table 7summarizes the assumptions required for sample-size cal-culation depending on the type of outcome variable. Thesource of information for the assumed treatment effects(eg, literature, pilot study) should be indicated.
In equivalence and noninferiority trials, the boundariesof equivalence and the noninferiority margin, respectively,need to be prespecified instead of the expected effect in theexperimental group.
In case of non-normally distributed data, as a rule ofthumb, the sample size may be computed for a 2-sample ttest and then inflated by 15%.68 Alternatively, the samplesize may be determined using computer simulations or, incase of availability of pilot or historical data, using boot-strap methods.69
In multivariate analysis, particularly relevant when an-alyzing observational data, as a rule of thumb, a minimumof 10 events, and a corresponding number of nonevents, pervariable in the model are necessary to achieve reliableestimates from logistic regression (binary outcome)70 and10 to 15 observations in multiple linear regression (contin-uous outcome).40 For example in a study evaluating riskfactors for the development of surgical-site infection afterhernia repair, a minimum of 10 surgical-site infectionsand of 10 non-surgical-site infections are necessary to eval-uate 1 candidate risk factor, whereas to evaluate predictorsof length of hospital stay, 10 patients per predictor shouldbe included (Table 7).
In retrospective studies and pilot studies, there is gen-erally no formal sample-size calculation, but a plausiblerationale for the choice of sample size should be provided.
Step 9: description of data management andquality assurance
The process of data entry, data management, monitoring,quality control, and quality assurance should be described.A statement of permitting access to source data for thepurpose of audits and inspections by the IEC and regulatoryauthorities should be included. The process of privacyprotection (eg, reversible anonymization) and the durationof data storage should be described. A detailed descriptionof data management and quality assurance options isbeyond the scope of this article.
Step 10: ethical considerations
Under ethical considerations, a risk-benefit assessmentshould be presented. Potential benefits, risks, and inconve-niences should be mentioned. These should refer to theindividual study participants but may also include potentialbenefits for future patients. The inclusion of a vulnerablepopulation should be further elaborated and justified in this
R. Rosenthal et al. How to write a surgical research protocol 311
section. Other ethical aspects should be mentioned here, suchas participation being entirely voluntary and withdrawalbeing possible at any time without giving any reason andwithout any impact on patient management. The handling ofincidental findings and genetic information as well as thejustification of placebo procedures, if applicable, should beincluded. Additionally, a statement that the study will beconducted according to the study protocol and Good ClinicalPractice should be included, as well as that the study protocoland any potential amendments will be submitted to an IECand potential regulatory authorities. A funding statement, adescriptionof any potential conflicts of interest, and insuranceissues should be provided. Any clinical study with ethicalapproval should disseminate its results through publication;the agreed publication policy can complete this section.
Conclusions
The study protocol is the core document when planningand conducting clinical research. It should be created in aninterdisciplinary setting, approved and strictly followed.Any changes require an amendment approved by an IECand the regulatory authorities. The ultimate goal of theprotocol is to support the conduct of scientifically andethically sound research providing high-level evidencerelevant for health care decision making.
References
1. The International Conference on Harmonisation of Technical Require-
ments for Registration of Pharmaceuticals for Human Use. Ich
Harmonised Tripartite Guideline: General Considerations for Clinical
Trials: E8. Available at: http://www.ich.org/. Accessed January 30,
2013.
2. Kaplan AV, Baim DS, Smith JJ, et al. Medical device development:
from prototype to regulatory approval. Circulation 2004;109:3068–72.
3. McCulloch P, Altman DG, Campbell WB, et al. No surgical innovation
without evaluation: the IDEAL recommendations. Lancet 2009;374:
1105–12.
4. Heikens JT, Gooszen HG, Rovers MM, et al. Stages and evaluation of
surgical innovation: a clinical example of the ileo neorectal anastomo-
sis after ulcerative colitis and familial adenomatous polyposis. Surg In-
nov 2013;20:459–65.
5. Boutron I, Moher D, Altman DG, et al, CONSORT Group. Extending
the CONSORT statement to randomized trials of nonpharmacologic
treatment: explanation and elaboration. Ann Int Med 2008;148:
295–309.
6. Bonenkamp JJ, Hermans J, Sasako M, et al, Dutch Gastric Cancer
Group. Extended lymph-node dissection for gastric cancer. N Engl J
Med 1999;340:908–14.
7. Clinical Outcomes of Surgical Therapy Study Group. A comparison of
laparoscopically assisted and open colectomy for colon cancer. N Engl
J Med 2004;350:2050–9.
8. Finkemeier CG, Schmidt AH, Kyle RF, et al. A prospective, random-
ized study of intramedullary nails inserted with and without reaming
for the treatment of open and closed fractures of the tibial shaft.
J Orthop Trauma 2000;14:187–93.
9. Moseley JB, O’Malley K, Petersen NJ, et al. A controlled trial of ar-
throscopic surgery for osteoarthritis of the knee. N Engl J Med
2002;347:81–8.
10. Mangram AJ, Horan TC, Pearson ML, et al, Centers for Disease Con-
trol and Prevention (CDC) Hospital Infection Control Practices Advi-
sory Committee. Guideline for prevention of surgical site infection
1999. Am J Infect Control 1999;27:97–132.
11. Dindo D, Demartines N, Clavien PA. Classification of surgical compli-
cations: a new proposal with evaluation in a cohort of 6336 patients
and results of a survey. Ann Surg 2004;240:205–13.
12. Clavien PA, Barkun J, de Oliveira ML, et al. The Clavien-Dindo clas-
sification of surgical complications: five-year experience. Ann Surg
2009;250:187–96.
13. Tincello DG, Kenyon S, Slack M, et al. Colposuspension or TVT with
anterior repair for urinary incontinence and prolapse: results of and
lessons from a pilot randomised patient-preference study (CARPET
1). BJOG 2009;116:1809–14.
14. Devereaux PJ, Bhandari M, Clarke M, et al. Need for expertise based
randomised controlled trials. BMJ 2005;330:88.
15. Martin II RC, Brennan MF, Jaques DP. Quality of complication report-
ing in the surgical literature. Ann Surg 2002;235:803–13.
16. McCulloch P, Taylor I, Sasako M, et al. Randomised trials in surgery:
problems and possible solutions. BMJ 2002;324:1448–51.
17. Lancaster GA, Dodd S,Williamson PR. Design and analysis of pilot stud-
ies: recommendations for good practice. J EvalClinPrac 2004;10:307–12.
18. McDonald PJ, Kulkarni AV, Farrokhyar F, et al. Ethical issues in sur-
gical research. Can J Surg 2010;53:133–6.
19. The International Conference on Harmonisation of Technical Require-
ments for Registration of Pharmaceuticals for HumanUse. Ich Harmon-
ised Tripartite Guideline: Guideline for Good Clinical Practice: E6(R1).
Available at: http://www.ich.org/. Accessed April 4, 2012.
20. Chan AW, Tetzlaff JM, Gøtzsche PC, et al. SPIRIT 2013 explanation and
elaboration: guidance for protocols of clinical trials. BMJ2013;346:e7586.