63
HYDROCEPHALUS HYDROCEPHALUS W & W 495-503 W & W 495-503
| Date post: | 16-Dec-2015 |
| Category: |
Documents |
| Upload: | maria-montgomery |
| View: | 221 times |
| Download: | 7 times |
HYDROCEPHALUSHYDROCEPHALUSHYDROCEPHALUSHYDROCEPHALUS
W & W 495-503W & W 495-503

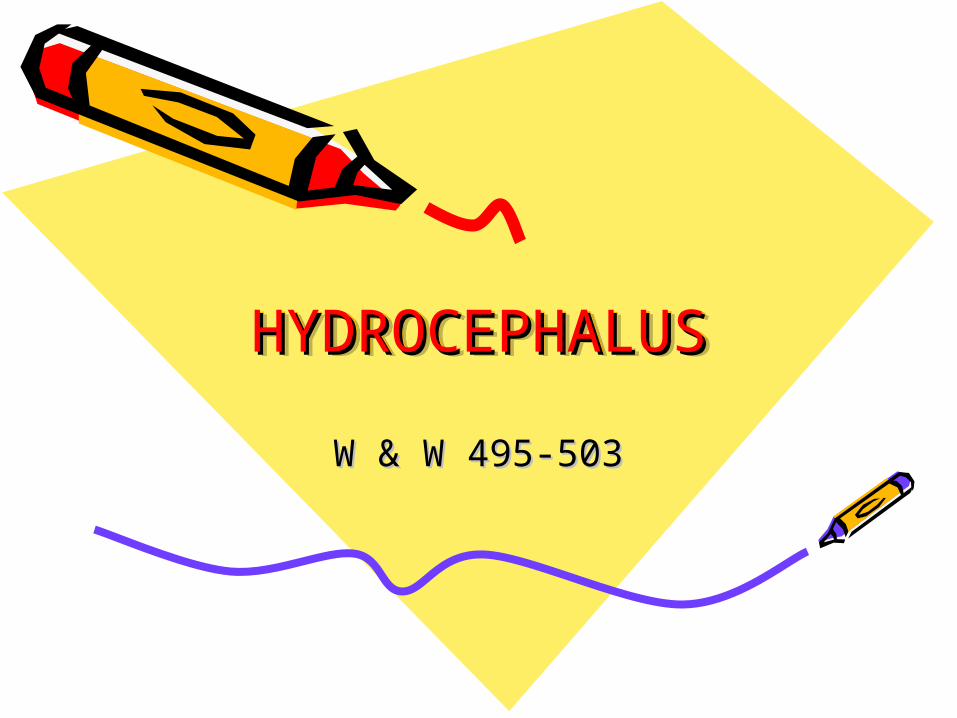
Hydrocephalus• A syndrome, or sign, resulting
from disturbances in the dynamics of cerebrospinal fluid (CSF), which may be caused by several diseases.


Incidence• Occurs in 3-4 of every 1000 births.• Cause may be congenital or acquired.• Congenital- may be due to
maldevelopment or intrauterine infection
• Acquired- may be due to infection, neoplasm or hemorrhage.

Pathophysiology• CSF is formed by two mechanisms:
– Secretion by the choroid plexus,– Lymphatic-like drainage by the
extracellular fluid in brain.
CSF circulates thru ventricular system and is absorbed within subarachnoid spaces by unknown mechanism.

Mechanisms of Fluid Imbalance
• Hydrocephalus results from:• 1. Impaired absorption of CSF within
the subarachnoid space (communicating hydrocephalus), or
• 2. Obstruction to the flow of CSF through the ventricular system (non-communicating hydrocephalus)

Mechanisms of fluid imbalance
• Both lead to increase accumulation of CSF in the ventricles!
• Ventricles become dilated and compress the brain.
• When this happens before cranial sutures are closed, skull enlarges.
• In children <10-12, previously closed sutures may open.

Hydrocephalus• Most cases of non-communicating
(obstructive) hydrocephalus are a result of developmental malformations.
• Other causes: neoplasms, intrauterine infections, trauma.
• Developmental defects account for most causes of hydrocephalus from birth to 2 years of age. (Table 11-3, page 497- sites and types of hydrocephalus)

Common Defects• Arnold-Chiari Malformation (ACM)
– Type 2 malformation of brain seen most exclusively with myelomeningocele, is characterized by herniation of a small cerebellum, medulla, pons, and fourth ventricle into the cervical spinal canal through an enlarged foramen magnum.


Clinical manifestations• Clinical picture depends on acuity
of onset and presence of preexisting structural lesions.

Infancy• Head grows at alarming rate with
hydrocephalus.– First signs- bulging of fontanels without head
enlargement.– Tense, bulging, non-pulsatile anterior
fontanel– Dilated scalp veins, esp. when crying– Thin skull bones with separated sutures
(cracked pot sounds on percussion)




Infancy• Protruding forehead or bossing.• Depressed eyes or setting-sun eyes (eyes
rotating or downward with sclera visible above pupil)
• Pupils sluggish with unequal response to light• Irritability, lethargy, feeds poorly, changes in
LOC, arching of back (opisthotonos), lower extremity spasticity.
• May cry when picked up or rocked; quiets when allowed to lay still.

Infancy• Swallowing difficulties, stridor, apnea,
aspiration, respiratory difficulties and arm weakness may indicate brain stem compression.
• If hydrocephalus progresses, difficulty sucking and feeding, and a high-pitched shrill cry results. (lower brain stem dysfunction)

Infancy• Emesis, somnolence, seizures, and
cardiopulmonary distress ensues and hydrocephalus progresses.
• Severely affected infants may not survive neonatal period.


Childhood• Signs and symptoms caused by
increased ICP.• Manifestations caused by posterior
neoplasms and aqueduct stenosis, manifestations associated with space-occupying lesions.

Childhood
• Headache on awakening with improvement following emesis or sitting up.
• Papilledema (swelling of optic disc DT obstruction), strabismus, and extrapyramidal tract signs such as ataxia
• Irritability, lethargy, apathy, confusion, and often incoherent


Childhood• Dandy-Walker syndrome- congenital
defect-late onset.– Obstruction of foramen of Lushka and
Magendie– Bulging occiput, nystagmus, ataxia,
cranial nerve palsies– Female predominance (3:1)– Absence or occlusion of ventricles

Diagnostic Evaluation• Antenatal- fetal ultrasound as early as
14 weeks• Infancy- based on head circumference
crosses one or more grid lines on the infant growth chart within a 4 week period and there are progressive neuro signs.
• CT and MRI to localize site of obstruction; reveal large ventricles

Therapeutic management
• Goals:• Relieve hydrocephaly• Treat complications• Manage problem resulting from
effects of disorder on psychomotor development
• USUALLY SURGICAL!

Surgical Treatment• Therapy of choice!• Direct removal of source of obstruction
(neoplasm, cyst, or hematoma)• Most require shunt procedure to drain
CSF from ventricles to extracranial area; usually peritoneum(VP shunt), or right atrium (VA shunt) for absorption.


VP shunt• Used in neonates and young
infants• Greater allowance for excess
tubing; which minimizes number of revisions needed as child grows


VA shunt• Reserved for older children who
have attained most of somatic growth, or children with abdominal pathology.
• Contraindicated in children with cardiopulmonary disease or with elevated CSF protein.



Major Complications• Shunt infection is most serious
complication!• Period of greatest risk is 1 to 2
months following placement.• Staph and strep most common
organisms

Complications• Mechanical difficulties
kinking, plugging, migration of tubing.
• Malfunction is most often by mechanical obstruction!
• Look for signs of increased ICP; fever, inflammation and abdominal pain.

Post-op care• In addition to routine post-op care:
– 1. Place on unoperated side to prevent pressure on shunt valve
– 2. Keep HOB flat; rapid decrease in IC fluid may cause subdural hematoma due to small vein rupture in cerebral cortex.
– 3. Do not pump shunt without specific direction from doctor (too many different pump devices)

Post-op care• 4. Observe for signs of Increased ICP!
May indicate obstruction of shunt!– Assess pupil size; as pressure on
oculomotor nerve may cause dilation on same side as pressure.
– Blood pressure may be variable due to hypoxia to brainstem
– Abdominal distention- due to CSF peritonitis or post-op ileus due to catheter placement.

Post-op• 5. Monitor I and O- may be on fluid
restriction or NPO for 24 hours to prevent fluid overload.
• 6. Monitor VS- increased temp may indicate infection.
• 7. Give good skin care to prevent tissue damage, etc.

Family support• Fear• Communication of procedures• Prepare for discharge.

SPINA BIFIDA• Neural Tube defects
are largest group of congenital anomalies.
• Failure of neural tube to close produces defects of either entire neural tube or small areas.

Etiology• Anacephaly and spina bifida occur
together very often.• Higher in females than males• 50% occur due to nutritional
deficiency (folic acid)

Spina Bifida• Defined as midline defects involving failure of
the bony spine to close.• Spina bifida occulta- defect not visible
externally.– Occurs most often in lumbosacral area.– Not apparent unless there are gait disturbances,
foot deformities, sphincter dysfunction or other neuromuscular manifestations.
– Many people with occulta will never have any deficits and may not know they have it.

Spina Bifida• Spina Bifida cystica- visible defect
with external saclike protrusion.– A. meningocele- encases meninges and
spinal fluid, but no neurological deficits.– B. meningomyelocele-contains
meninges, spinal fluid, and nerves. Neuromotor deficits depend on anatomic level of protrusion and nerves involved.


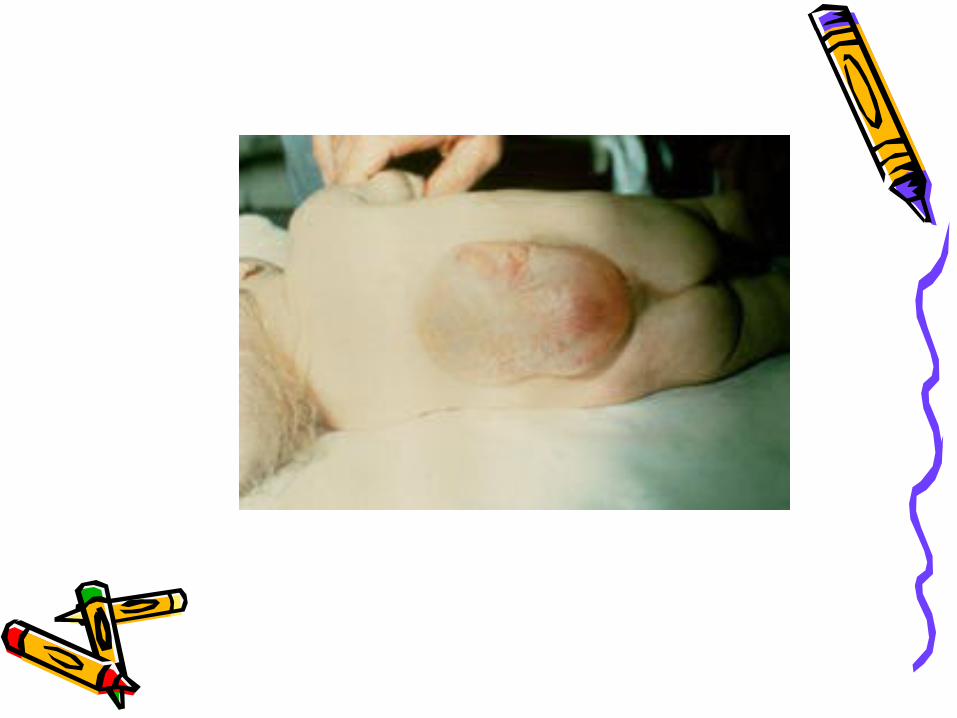

Meningomyelocele• AKA spina bifida• Develops during first 28 days of
pregnancy when neural tube fails to close and fuse.
• 90% of spinal cord lesions, and may occur at any point along spine.
• Sac usually enclosed in fine membrane that is prone to tears.

meningomyelocele• Largest number in lumbar or lumbosacral
area• 90-95% of children have hydrocephalus• Careful monitoring of head size important• Chiari malformation may be present:
observe infant for stridor, hoarse cry from vocal cord paralysis; feeding difficulties, deteriorating upper extremity function.

Clinical manifestations• S/S vary according to degree of spinal
defect.• Readily apparent on inspection!• Loss of sensation below lesion• Poor urinary and bladder control• Joint deformities in lower extremities• Scoliosis or kyphosis• Hip dislocations

Diagnostic evaluation• Examination of meningeal sac and clinical
manifestations• MRI, CT to assess condition of brain and spinal
cord. Other defects may be present.• Prenatal- fetal ultrasound or amniotic fluid
sample for (alpha-fetal protein (AFP).• Test should be done between 16 and 18
weeks of gestation. Afterwards AFP level drops, making detection of SB difficult. Also, therapeutic abortion not option after this time.


Therapeutic management
• Multi-disciplinary approach– Neurology, neuro-surgery, pediatrics,
urology, orthopedics, rehabilitation, PT, OT, social services, intensive nursing in many areas.

Goals• 1. prevent infection• 2. early closure of lesion, within 72
hours (prevents infection and trauma to exposed tissues, and prevents further motor impairment). Goal is satisfactory skin coverage of lesion!
• 3. PT for specific deformity

Goals• Physical therapy to prevent joint
contractures.– Correct deformity, prevent skin
breakdown, obtain best ambulatory functioning

Goals• Management of genitourinary
function:– Mylemeningocele is a common cause
of neurogenic bladder which leads to urinary system distress (frequent UTI’s, ureterohydronephrosis, vesicoureteral reflux, renal insufficiency.
– Urinary incontinence is common

Goals• Clean, intermittent catherization
as a conservative treatment.• Vesicostomy (anterior wall of
bladder brought through abdominal wall to create stoma) may be done for bladder control.
• Meticulous skin care is needed!

Nursing care• Pre-op
– Positioning to keep off sac (use diaper rolls, pads or sandbags)
– Keep in prone position– Can be challenging!– More difficult to keep clean, pressure
areas a threat, feeding a problem

Nursing care• Pre-op
– Turn head on side for feeding– Diapering may be contraindicated until
repair and healing has taken place.– Constant stooling due to affected
bowel sphincter (not diarrhea).– Keep skin clean.

Post-op care• Prone position until healing takes
place!• May allow side-lying- depends on
doctor.• Feeding resumed after anesthesia
wears off• Comfort measure/vital signs/I & O

Latex allergy• 70% of children and adolescents
with SB are sensitive to latex. – Cause unknown-probably continued
exposure to latex.
Avoid latex, balloons, condoms, catheters, or anything with latex!

Post-op• Use touch for stimulation, since
can’t hold• Observe for increased ICP, such as
bulging fontanels• Assess for infection:
– Increased or decreased temperature, irritability, nuchal rigidity,

Home Care• Involve parents in care
– Positioning/feeding/skin care/range of motion exercises/ clean catheterization when prescribed, complications.
– Help with assistive devices (if child is paraplegic use hands/arms, etc.)
– Long range planning– Spina Bifida Association of America








![Management of subdural effusion and hydrocephalus ......hydrocephalus in patients with DC for TBI is 10 to 40% [10, 12, 13, 19]. SDE is defined as cerebrospinal fluid (CSF) accumula-tion](https://static.documents.pub/doc/80x56/60f77946a97a3c60fd2cc41f/management-of-subdural-effusion-and-hydrocephalus-hydrocephalus-in-patients.jpg)









