ORIGINAL PAPER Hydrophilic properties as a new contribution for computer-aided identification of short peptides in complex mixtures Christelle Harscoat-Schiavo & Claudia Nioi & Evelyne Ronat-Heit & Cédric Paris & Régis Vanderesse & Frantz Fournier & Ivan Marc Received: 14 February 2012 / Revised: 20 March 2012 / Accepted: 27 March 2012 / Published online: 28 April 2012 # Springer-Verlag 2012 Abstract A new method to predict elementary amino acid (AA) composition of peptides (molar mass <1,000 g/mol) is described. This procedure is based on a computer-aided method using three combined analyses—reversed phase liquid chromatography (RPLC), hydrophilic interaction chromatography (HILIC) and capillary electrophoresis cou- pled with mass spectrometry—and using a software calcu- lating all possible amino acid combinations from the mass of any given peptide. The complementarity between HILIC and RPLC was demonstrated. Peptide retention prediction in HILIC was successfully modelled, and the achieved pre- diction accuracy was as high as r² 0 0.97. This mathematical model, based on amino acid retention contributions and peptide length, provided the information about peptide hydrophilicity that was not redundant with its hydrophobic- ity. Correlations between respectively the hydrophobicity coefficients and RPLC retention time, hydrophilicity and HILIC retention time, and electrophoretic mobility and migration time were used for ranking all potential AA combinations corresponding to the given mass. The essen- tial contribution of HILIC in this identification strategy and the need to combine the three models to significantly in- crease identification capabilities were both shown. Applied to an 18-standard peptide mixture, the identification proce- dure enabled the actual AA combination determination of the 14 di- to pentapeptides, in addition to an over 98 % reduction of possible combination numbers for the four hexapeptides. This procedure was then applied to the iden- tification of 24 unknown peptides in a rapeseed protein hydrolysate. The effective AA composition was found for ten peptides, whereas for the 14 other peptides, the number of possible combinations was reduced by over 95 % thanks to the association of the three analyses. Finally, as a result of the information provided by the analytical techniques about peptides present in the mixture, the proposed method could become a highly valuable tool to recover bioactive peptides from undefined protein hydrolysates. Keywords HILIC-MS . Identification strategy . Peptides . Prediction method Introduction Peptides, especially short peptides (molar mass (MM) <1,000 g/mol), obtained from enzymatic protein hydrolysis are of interest to various industries with respect to their numerous bioactivities. For instance, many studies reported the presence of anti-hypertensive [1–4], opioid [5, 6] and antimicrobial [7–9] peptides in various protein hydrolysates. Furthermore, a growth-promoting [ 10–12] or growth- inhibiting [13] effect in cell cultures has been reported for C. Harscoat-Schiavo (*) : C. Nioi : E. Ronat-Heit : F. Fournier : I. Marc Laboratoire Réactions et Génie des Procédés—U.P.R. 3349 C.N.R.S, Université de Lorraine, Plate forme SVS, 13 rue du bois de la Champelle, 54500 Vandœuvre-lès-Nancy, France e-mail: [email protected]C. Paris Université de Lorraine, Laboratoire d’Ingénierie des BIOmolécules, 2 avenue de la forêt de Haye, 54505 Vandœuvre-lès-Nancy, France R. Vanderesse Université de Lorraine, Laboratoire de Chimie Physique Macromoléculaire—U.M.R. 7568 C.N.R.S, 1 rue Grandville, B.P. 451, 54001 Nancy Cedex, France Anal Bioanal Chem (2012) 403:1939–1949 DOI 10.1007/s00216-012-5987-6
Transcript
ORIGINAL PAPER
Hydrophilic properties as a new contributionfor computer-aided identification of shortpeptides in complex mixtures
Received: 14 February 2012 /Revised: 20 March 2012 /Accepted: 27 March 2012 /Published online: 28 April 2012# Springer-Verlag 2012
Abstract A new method to predict elementary amino acid(AA) composition of peptides (molar mass <1,000 g/mol) isdescribed. This procedure is based on a computer-aidedmethod using three combined analyses—reversed phaseliquid chromatography (RPLC), hydrophilic interactionchromatography (HILIC) and capillary electrophoresis cou-pled with mass spectrometry—and using a software calcu-lating all possible amino acid combinations from the mass ofany given peptide. The complementarity between HILICand RPLC was demonstrated. Peptide retention predictionin HILIC was successfully modelled, and the achieved pre-diction accuracy was as high as r²00.97. This mathematicalmodel, based on amino acid retention contributions andpeptide length, provided the information about peptidehydrophilicity that was not redundant with its hydrophobic-ity. Correlations between respectively the hydrophobicitycoefficients and RPLC retention time, hydrophilicity andHILIC retention time, and electrophoretic mobility and
migration time were used for ranking all potential AAcombinations corresponding to the given mass. The essen-tial contribution of HILIC in this identification strategy andthe need to combine the three models to significantly in-crease identification capabilities were both shown. Appliedto an 18-standard peptide mixture, the identification proce-dure enabled the actual AA combination determination ofthe 14 di- to pentapeptides, in addition to an over 98 %reduction of possible combination numbers for the fourhexapeptides. This procedure was then applied to the iden-tification of 24 unknown peptides in a rapeseed proteinhydrolysate. The effective AA composition was found forten peptides, whereas for the 14 other peptides, the numberof possible combinations was reduced by over 95 % thanksto the association of the three analyses. Finally, as a result ofthe information provided by the analytical techniques aboutpeptides present in the mixture, the proposed method couldbecome a highly valuable tool to recover bioactive peptidesfrom undefined protein hydrolysates.
Peptides, especially short peptides (molar mass (MM)<1,000 g/mol), obtained from enzymatic protein hydrolysisare of interest to various industries with respect to theirnumerous bioactivities. For instance, many studies reportedthe presence of anti-hypertensive [1–4], opioid [5, 6] andantimicrobial [7–9] peptides in various protein hydrolysates.Furthermore, a growth-promoting [10–12] or growth-inhibiting [13] effect in cell cultures has been reported for
C. Harscoat-Schiavo (*) :C. Nioi : E. Ronat-Heit : F. Fournier :I. MarcLaboratoire Réactions et Génie desProcédés—U.P.R. 3349 C.N.R.S, Université de Lorraine,Plate forme SVS, 13 rue du bois de la Champelle,54500 Vandœuvre-lès-Nancy, Francee-mail: [email protected]
C. ParisUniversité de Lorraine, Laboratoire d’Ingénierie des BIOmolécules,2 avenue de la forêt de Haye,54505 Vandœuvre-lès-Nancy, France
R. VanderesseUniversité de Lorraine, Laboratoire de Chimie PhysiqueMacromoléculaire—U.M.R. 7568 C.N.R.S,1 rue Grandville, B.P. 451, 54001 Nancy Cedex, France
some protein hydrolysates. Various additional functional-ities and bioactivities have been described [9, 14], and theliterature on this subject is expanding.
However, depending on further use, protein hydrolysates,which are complex mixtures of peptides (up to severalhundreds) varying in sizes and properties, should be wellcharacterized through accurate identification of the aminoacid composition of the released peptides. Peptide sequenc-ing by the Edman method is the most accurate tool todetermine their amino acid composition, but it is costlyand highly time-consuming as it requires peptide isolation.It also has low sensitivity. An alternative to this method isliquid chromatography hyphenated with tandem mass spec-trometry, which has become the most powerful analyticaltechnique for analysing peptides [15, 16] or monitoring thetime-dependent release of a particular bioactive peptideduring protein hydrolysis [17]. Unfortunately, mass spec-trometry performance can be limited in cases of ionizationsuppression, especially occurring when a complex mixtureof molecules is analysed. Actually, molecular species withhigh ionization efficiencies dominate the spectrum, thussuppressing species in similar abundance but with lowionization efficiencies [18].
Furthermore, tandem mass spectrometry (MS) analysesrely on computer-aided database searching, whilst databasesare sometimes unavailable. Therefore, the analysis of non-standard species requires direct peptide sequencing, i.e.without any use of databases, called de novo sequencing.This newly developed procedure is fast and reliable in somecases, but the whole sequence can be obtained only whenthe spectrum contains the complete series of fragments andalso when only an ideal fragmentation process occurs.Incomplete fragment information is often obtained, andrearrangement products formed in collision-induced disso-ciation (CID), as well as different types of cleavage, leadingto a highly complex fragmentation spectra which may bevery difficult to interpret unequivocally and thus causesequencing failure [19, 20]. Several authors have describedattempts to improve de novo sequencing procedures.Bertsch et al. [21] used electron transfer dissociation com-plementary to common CID to significantly increase thesequence coverage. Spengler et al. [20, 22] elaborated atwo-step database-independent sequencing strategy calledcomposition-based sequencing. After possible amino acid(AA) compositions had been determined from the accuratemass value measured in tandem MS, the resulting expectedfragment ion signals of permuted sequence propositionswere compared with the observed fragment ion information.Bruni et al. [19, 23] developed a computational analysisprocedure obtained by building a mathematical model ofpeptide sequence determination and by searching for allpossible sequences of given components compatible withparticular constraints.
These alternative procedures clearly require sufficientfragment information; otherwise, they possibly lead to thenon-uniqueness of the achieved peptide sequence. One wayto discriminate among various sequences would be to com-pare between the expected and the observed chromatographicbehaviours.
In this context, predictive models were established torelate the behaviour of peptides under specific separationconditions with their physicochemical properties. Acomputer-aided strategy based on the simultaneous use ofa list of all possible AA compositions corresponding to amass value and retention predictive models was developed.The first retention model was based on the hydrophobiccoefficient (HC) of peptides, from reversed phase liquidchromatography (RPLC)-MS analysis. The association oftwo data (HC and mass) for the prediction of possible AAcompositions of small peptides enhanced identification byeliminating proposed compositions that did not satisfy thehydrophobicity criterion [24]. Similarly, a second modelwas established from capillary electrophoresis (CE)-MSanalysis as a function of the charge-to-mass ratio (electro-phoretic mobility, μ) of peptides [25]. These two modelswere successfully combined and led to a significantimprovement in the AA combination prediction [26].However, the hydrophobicity model is only valid forhydrophobic enough peptides. Furthermore, in this strategy,the more information is available, the more accurate thisidentification is.
Hydrophilic interaction chromatography (HILIC) is be-coming increasingly popular [27–30] and has been success-fully used for the separation of peptides [31, 32] and ofamino acids [33, 34]. The growing interest in HILIC ispartly due to its high solvent compatibility with MS in theelectrospray ionization (ESI) mode [35, 36]. One of themajor fields currently under investigation is the use ofHILIC as a separation tool for proteomic applications [33,36–38], but it is also studied for use in mixed-mode [39–41]and two-dimensional liquid chromatography (2D LC)[42–45].
The analysis of peptides in complex mixtures has alreadybeen performed successfully by 2D LC-MS, couplingHILIC and RPLC, in plasma [45] and milk hydrolysates[44]. However, studies regarding complex hydrolysatesfrom non-defined proteins are still limited, and very fewretention prediction models have been established for HILIC[46–51].
The present work describes the addition of HILIC hy-phenated with MS to the already available analytical tech-niques, RPLC-MS and CE-MS. Parameters influencingpeptide retention behaviour are identified and an accuratepredictive model of peptide retention in HILIC is provided.Finally, the added value for identification is illustrated by itsuse in the characterization of both a synthetic model peptide
1940 C. Harscoat-Schiavo et al.
mixture and peptides standing in a natural complex mixture,i.e. a mixed protein enzymatic hydrolysate.
Experimental
Chemicals and reagents
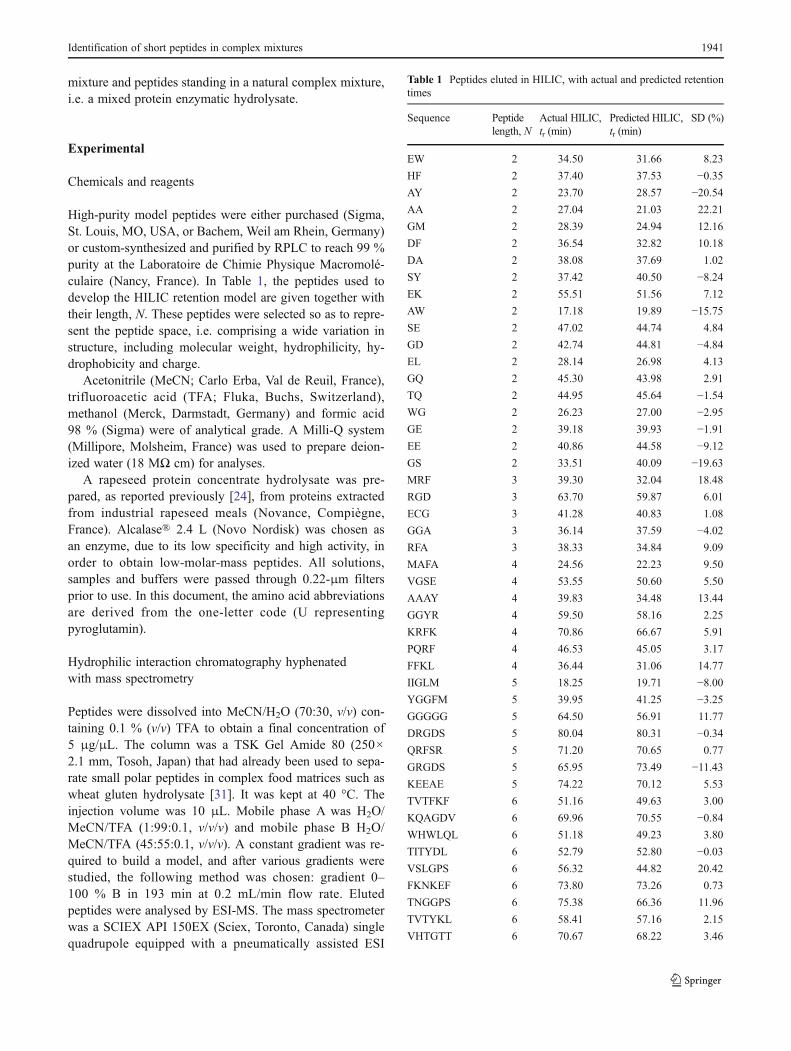
High-purity model peptides were either purchased (Sigma,St. Louis, MO, USA, or Bachem, Weil am Rhein, Germany)or custom-synthesized and purified by RPLC to reach 99 %purity at the Laboratoire de Chimie Physique Macromolé-culaire (Nancy, France). In Table 1, the peptides used todevelop the HILIC retention model are given together withtheir length, N. These peptides were selected so as to repre-sent the peptide space, i.e. comprising a wide variation instructure, including molecular weight, hydrophilicity, hy-drophobicity and charge.
Acetonitrile (MeCN; Carlo Erba, Val de Reuil, France),trifluoroacetic acid (TFA; Fluka, Buchs, Switzerland),methanol (Merck, Darmstadt, Germany) and formic acid98 % (Sigma) were of analytical grade. A Milli-Q system(Millipore, Molsheim, France) was used to prepare deion-ized water (18 MΩ cm) for analyses.
A rapeseed protein concentrate hydrolysate was pre-pared, as reported previously [24], from proteins extractedfrom industrial rapeseed meals (Novance, Compiègne,France). Alcalase® 2.4 L (Novo Nordisk) was chosen asan enzyme, due to its low specificity and high activity, inorder to obtain low-molar-mass peptides. All solutions,samples and buffers were passed through 0.22-μm filtersprior to use. In this document, the amino acid abbreviationsare derived from the one-letter code (U representingpyroglutamin).
Hydrophilic interaction chromatography hyphenatedwith mass spectrometry
Peptides were dissolved into MeCN/H2O (70:30, v/v) con-taining 0.1 % (v/v) TFA to obtain a final concentration of5 μg/μL. The column was a TSK Gel Amide 80 (250×2.1 mm, Tosoh, Japan) that had already been used to sepa-rate small polar peptides in complex food matrices such aswheat gluten hydrolysate [31]. It was kept at 40 °C. Theinjection volume was 10 μL. Mobile phase A was H2O/MeCN/TFA (1:99:0.1, v/v/v) and mobile phase B H2O/MeCN/TFA (45:55:0.1, v/v/v). A constant gradient was re-quired to build a model, and after various gradients werestudied, the following method was chosen: gradient 0–100 % B in 193 min at 0.2 mL/min flow rate. Elutedpeptides were analysed by ESI-MS. The mass spectrometerwas a SCIEX API 150EX (Sciex, Toronto, Canada) singlequadrupole equipped with a pneumatically assisted ESI
Table 1 Peptides eluted in HILIC, with actual and predicted retentiontimes
Sequence Peptidelength, N
Actual HILIC,tr (min)
Predicted HILIC,tr (min)
SD (%)
EW 2 34.50 31.66 8.23
HF 2 37.40 37.53 −0.35
AY 2 23.70 28.57 −20.54
AA 2 27.04 21.03 22.21
GM 2 28.39 24.94 12.16
DF 2 36.54 32.82 10.18
DA 2 38.08 37.69 1.02
SY 2 37.42 40.50 −8.24
EK 2 55.51 51.56 7.12
AW 2 17.18 19.89 −15.75
SE 2 47.02 44.74 4.84
GD 2 42.74 44.81 −4.84
EL 2 28.14 26.98 4.13
GQ 2 45.30 43.98 2.91
TQ 2 44.95 45.64 −1.54
WG 2 26.23 27.00 −2.95
GE 2 39.18 39.93 −1.91
EE 2 40.86 44.58 −9.12
GS 2 33.51 40.09 −19.63
MRF 3 39.30 32.04 18.48
RGD 3 63.70 59.87 6.01
ECG 3 41.28 40.83 1.08
GGA 3 36.14 37.59 −4.02
RFA 3 38.33 34.84 9.09
MAFA 4 24.56 22.23 9.50
VGSE 4 53.55 50.60 5.50
AAAY 4 39.83 34.48 13.44
GGYR 4 59.50 58.16 2.25
KRFK 4 70.86 66.67 5.91
PQRF 4 46.53 45.05 3.17
FFKL 4 36.44 31.06 14.77
IIGLM 5 18.25 19.71 −8.00
YGGFM 5 39.95 41.25 −3.25
GGGGG 5 64.50 56.91 11.77
DRGDS 5 80.04 80.31 −0.34
QRFSR 5 71.20 70.65 0.77
GRGDS 5 65.95 73.49 −11.43
KEEAE 5 74.22 70.12 5.53
TVTFKF 6 51.16 49.63 3.00
KQAGDV 6 69.96 70.55 −0.84
WHWLQL 6 51.18 49.23 3.80
TITYDL 6 52.79 52.80 −0.03
VSLGPS 6 56.32 44.82 20.42
FKNKEF 6 73.80 73.26 0.73
TNGGPS 6 75.38 66.36 11.96
TVTYKL 6 58.41 57.16 2.15
VHTGTT 6 70.67 68.22 3.46
Identification of short peptides in complex mixtures 1941
(ionspray) interface. Data acquisition and processing wereperformed with a computer running the Masschrom® appli-cation. Retention time standard deviation was 0.1 min. Allanalyses were carried out in the positive ionization mode.The operating parameters were tuned as follows: massscan0100–1,500 mass units (m/z), ion spray voltage05.5 kV, scan time01.4 s, step size00.2 mass unit (mu) anddwell time00.1 ms.
Reversed phase chromatography and capillaryelectrophoresis coupled with mass spectrometry:determination of the corresponding peptidephysicochemical properties
Reversed phase chromatography and capillary electrophore-sis were performed as described previously [26]. The oper-ating conditions of the mass spectrometer were the same asthose used for HILIC-MS.
The HCs of peptides were calculated from the AA hy-drophobic coefficients described by Schweizer et al. [24].The net charge of the peptides at pH 2.75 was calculated byconsidering any dissociable group separately using theHenderson–Hasselbach equation [52]. The experimentalelectrophoretic mobility of each peptide (μexp(pep)) wasmeasured as described by Janini et al. [53].
Software and data processing
Matlab for Windows (The Math Works Inc.) was used toperform (1) multilinear regression (MLR), (2) hydrophilicindex multi-parametric identification using a genetic–evolutionary algorithm [54, 55] and (3) statistical analysisof the obtained regression model.
The MLR method provides an equation that relates thestructural features to the retention time (tr) of the peptides.
MLR models usually take the form “tr0a0+a1x1+…+anxn”,where the intercept (a0) and the regression coefficients ofthe predictors (ai) are determined using the least-squaresmethod. The predictors (xi) included in the equation areused to describe the molecular structure of the components,where n is the number of predictors. The physicochemicaldescriptors used to produce the prediction models wereeither only peptide hydrophilicity or peptide hydrophilicityand peptide length. The stepwise regression technique wasused to establish a correlation between the HILIC retentiontime, tr, of peptides and respectively either only theirhydrophilic property or their hydrophilic property andlength. The model used a training set of retention timesof peptides of known sequence for the multi-parametricoptimization of retention coefficients for each amino acidresidue to achieve the best fit between predicted andobserved retention.
The model accuracy, predictive ability and robustnesswere evaluated by cross-validation.
Various cross-validation approaches were performed:
– The leave-one-out approach consists of an m timesprocedure where each molecule is used in turn as a testfor the model built from the remaining (m−1) mole-cules, with m as the number of peptides in data.
– The tenfold cross-validation method consists in a ten-time procedure where the data are partitioned into tengroups; each group in turn is used for testing the modelbuilt from the peptides of the nine remaining groups.
The root mean squared error (RMSE), the root meansquared error of calibration (RMSEC) and the root meansquared error of cross-validation (RMSECV) were calculatedand compared with one another. In the results, RMSECV1
corresponds to the leave-one-out approach and RMSECV10 tothe tenfold cross-validation. From a qualitative point of view,comparable RMSE, RMSEC and RMSECV values indicatethat both interpolations and extrapolations of the modelshould be adequate, whereas large differences point out a lackof robustness.
The coefficient of determination (r2), the r2 statisticadjusted for degrees of freedom (radj
2), the significance(p value), the Fisher ratio (F) and the Durbin–Watsonvalue (DW) were also evaluated. Autocorrelation ofresidues is usually considered acceptable when DWremains in the range of 1.5–2.5.
Data bank of rapeseed proteins
The sequences of the main rapeseed proteins (cruciferinsand napins) have been obtained from a protein database(PubMed: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi).The software tool FindPept (http://www.expasy.org/tools/findpept.html) has been used to determine the amino acid
sequences corresponding to a precise mass into theseproteins.
Results and discussion
HILIC-MS separation
In order to identify peptides in complex mixtures, the moreinformation by various analyses is obtained, the more accu-rate their identification may be. After the capacity of HILICto separate peptides that were unretained in RPLC had beenverified, peptide retention was modelled.
The retention of 63 independent peptides was studied inHILIC, coupled with mass spectrometry. Table 1 gives onlypeptides with different AA compositions. With the aim ofproposing a retention model allowing for the retention pre-diction of peptides, the suitability of a simple additiveretention law was first checked. Therefore, a possible impactof amino acid sequence on peptide retention was examinedthrough the observation of elution behaviour in some pairsof homologous standard peptides: AP/PA, GW/WG, AGG/GGA and FKNKEF/KNFFKE. For each pair, the retentiontimes were similar, whatever the sequence. It was thusadmitted that the retention of a peptide resulted from thesummation of the retention contributions of the amino acidscomprising it. These retention contributions were namedhydrophilic coefficients or hydrophilic indices (IHI).
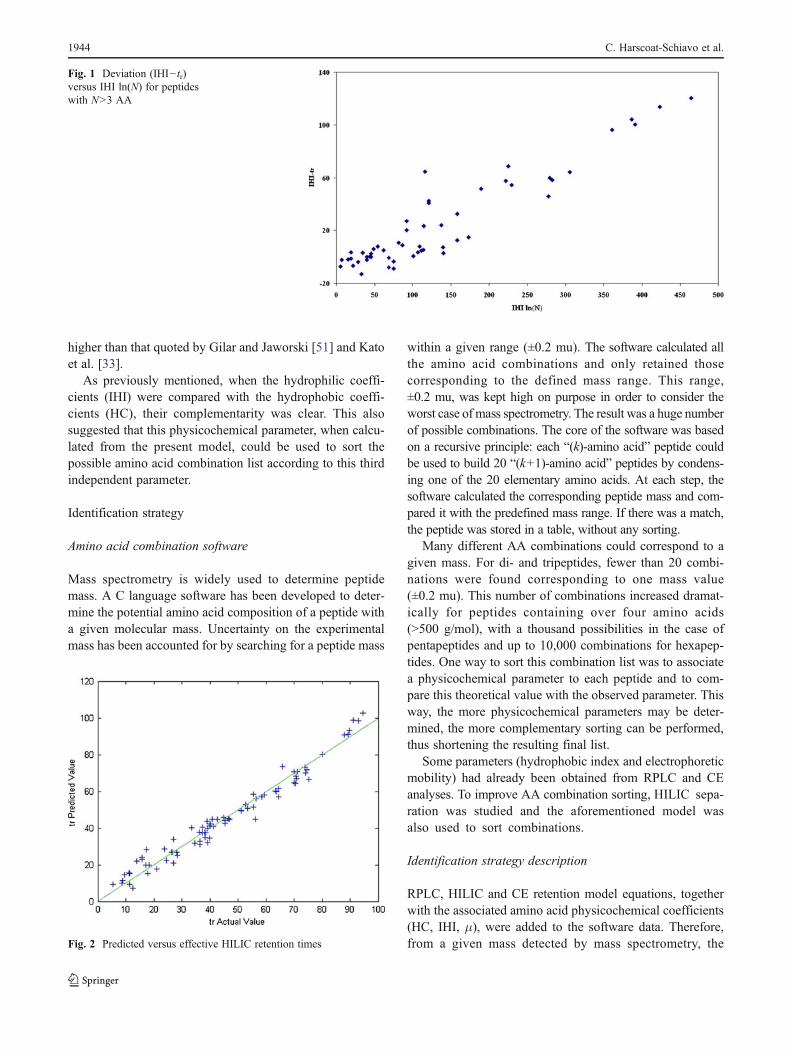
An attempt to simply model retention from peptide hy-drophilic indices was made. The first approach consideredthat hydrophilic coefficients IHI could be determined foreach amino acid in order that the resulting hydrophiliccoefficient of a peptide (being the sum of AA coefficients)should correspond to its retention time. This model consid-ered only the contribution of the hydrophilic characteristicsof peptides. IHI values for amino acids were determinedfrom the AA retention times as initial values and by multi-parametric optimization to reach the best fit (data notshown). The obtained model showed deviation from thelinear relation for peptides with more than six residues andfor shorter but highly hydrophilic peptides. Additionally,this deviation from the linear model increased with peptidehydrophilicity, namely when IHI increased. When the devia-tion (IHI−tr) was plotted versus IHI ln(N), with N the numberof residues (AA), a correlation was made clear (Fig. 1). As aconsequence, the proposed model took into account both theaddition of constitutive amino acid retention contributions anda linearization by the logarithmic function of the peptide chainlength. The model equation was as follows:
tr ¼ a IHI� b IHI lnðNÞ þ c;
where a and b are iteratively optimized coefficients, c a linearintercept of the model and IHI the sum of amino acid IHI
coefficients determined by multi-parametric identificationusing a genetic–evolutionary algorithm.
Finally, the following model equation was established:
tr ¼ 1:05IHI� 0:27IHI lnðNÞ þ 7:78
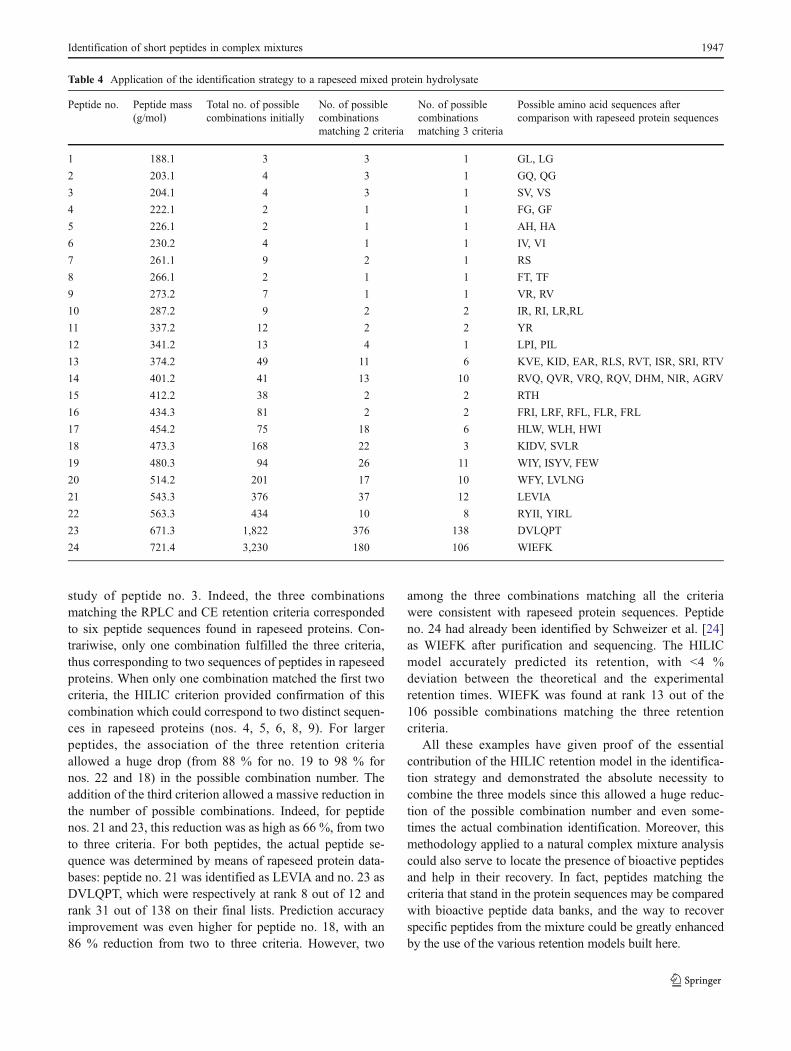
The predicted retention time values were in good agree-ment with the observed values (Fig. 2), thus validating theuse of IHI ln(N) as a correcting factor in the HILIC separa-tion model. Linearization by ln(N) was in accordance withthe model recently described by Gilar and Jaworski [51],and the prediction accuracy of the present model (r200.97)was as high as that reported by the authors.
The robustness of this model, together with its predictiveability, has been investigated by cross-validation and statis-tical analysis. The standard errors of coefficients were 0.04and 0.02 for IHI and IHI ln(N), respectively. For both termsof the equation, p values were 0, thus validating their fullsignificance. The very close values of the root mean squarederror (RMSE04.42), the root mean squared error of calibration(RMSEC04.3) and the root mean squared error of cross-validation (RMSECV1004.35, RMSECV103.6) indicatedgood robustness and showed that interpolations of the modelshould create nomajor obstacle. The Fisher ratio (F01142) andthe Durbin–Watson value (DW01.76) also validated the model.
Hydrophilic indices (IHI) obtained for amino acidsthrough parametric identification using a genetic–evolutionaryalgorithm are collected in Table 2. Hydrophobic coefficientsfrom Schweizer et al. [24] and hydrophilic retention coeffi-cients for amide columns reported by Yoshida et al. [46] andGilar and Jaworski [51] are also listed in this table. First, itappears clearly that the basic amino acids, amino acids withamide or hydroxyl residues and the acid amino acids arecontributing the most to retention in HILIC. Coefficientsarising from the present study are consistent with what hadpreviously been described in the literature. However, the orderof contribution is not exactly similar to that described byYoshida [47], i.e. basic amino acids>amino acids with amideresidues>amino acids with hydroxyl residues>acid aminoacids. Moreover, neither coefficients reported by Gilar et al.actually followed this exact order. In their study, contributionsdecreased as follows: basic amino acids>acid amino acids>amino acids with amide residues>amino acids with hydroxylresidues.
All these IHI values could be compared with the elutionorder of amino acids separated by ZIC-HILIC reported byKato et al. [33]:
F < M < L < I < Y < P < V < A < S < G < H < R
In the present study, the contribution of P to peptidehydrophilic characteristics was found lower than that quotedby Yoshida et al. [46], Gilar and Jaworski [51] and Kato etal. [33]. Conversely, the retention contribution of Y wasconsistent with that reported by Yoshida et al. [46], i.e.
Identification of short peptides in complex mixtures 1943
higher than that quoted by Gilar and Jaworski [51] and Katoet al. [33].
As previously mentioned, when the hydrophilic coeffi-cients (IHI) were compared with the hydrophobic coeffi-cients (HC), their complementarity was clear. This alsosuggested that this physicochemical parameter, when calcu-lated from the present model, could be used to sort thepossible amino acid combination list according to this thirdindependent parameter.
Identification strategy
Amino acid combination software
Mass spectrometry is widely used to determine peptidemass. A C language software has been developed to deter-mine the potential amino acid composition of a peptide witha given molecular mass. Uncertainty on the experimentalmass has been accounted for by searching for a peptide mass
within a given range (±0.2 mu). The software calculated allthe amino acid combinations and only retained thosecorresponding to the defined mass range. This range,±0.2 mu, was kept high on purpose in order to consider theworst case of mass spectrometry. The result was a huge numberof possible combinations. The core of the software was basedon a recursive principle: each “(k)-amino acid” peptide couldbe used to build 20 “(k+1)-amino acid” peptides by condens-ing one of the 20 elementary amino acids. At each step, thesoftware calculated the corresponding peptide mass and com-pared it with the predefined mass range. If there was a match,the peptide was stored in a table, without any sorting.
Many different AA combinations could correspond to agiven mass. For di- and tripeptides, fewer than 20 combi-nations were found corresponding to one mass value(±0.2 mu). This number of combinations increased dramat-ically for peptides containing over four amino acids(>500 g/mol), with a thousand possibilities in the case ofpentapeptides and up to 10,000 combinations for hexapep-tides. One way to sort this combination list was to associatea physicochemical parameter to each peptide and to com-pare this theoretical value with the observed parameter. Thisway, the more physicochemical parameters may be deter-mined, the more complementary sorting can be performed,thus shortening the resulting final list.
Some parameters (hydrophobic index and electrophoreticmobility) had already been obtained from RPLC and CEanalyses. To improve AA combination sorting, HILIC sepa-ration was studied and the aforementioned model wasalso used to sort combinations.
Identification strategy description
RPLC, HILIC and CE retention model equations, togetherwith the associated amino acid physicochemical coefficients(HC, IHI, μ), were added to the software data. Therefore,from a given mass detected by mass spectrometry, the
Fig. 1 Deviation (IHI− tr)versus IHI ln(N) for peptideswith N>3 AA
Fig. 2 Predicted versus effective HILIC retention times
1944 C. Harscoat-Schiavo et al.
software provided an unsorted list of all possible amino acidcombinations with, for each combination, its theoreticalRPLC, HILIC and CE retention times. Analyses of peptidemixtures by RPLC-MS, HILIC-MS and CE-MS provided,for each detected peptide, its molar mass and experimentalRPLC, HILIC and CE retention times.
Theoretical and experimental retention time values werethen compared so that possible AA combinations could besorted according to the smallest difference between them.Since both experimental and theoretical retention timevalues carried uncertainties, a maximum overall uncertaintyrate of 20 % has been considered for RPLC and HILIC(Table 1) and of 15 % for CE, respectively. The threeanalytical techniques led to three independent sortings thatwere combined to improve peptide identification. Onlycombinations that matched the three criteria simultaneouslywere retained in the final combination list.
Validation: application to a synthetic peptide mixturecharacterization and to the identification of peptidesin a protein enzymatic hydrolysate
First, the identification strategy was implemented to charac-terize an 18-synthetic peptide mixture. Then, this strategy
was applied to a protein concentrate hydrolysate to validatethe presented original methodology.
Synthetic peptide mixture characterization
The 18 peptides in mixture were representative of thepeptide space, with a wide range of charge, hydropho-bic and hydrophilic properties. The peptides were com-posed of two to six AAs which can be described asfollows: some were either hydrophobic AA or not,some either hydrophilic AA or not, and some eithercharged or not.
The mixture was analysed by RPLC-MS, HILIC-MS andCE-MS. From the mass provided by MS analysis, and the0.2-mu range, an unsorted list of possible AA combinationswas built. From the three models, three sorted tables wereestablished and the position of the actual combination of theanalysed peptide was located in each table. Then, the threecriteria were combined to improve prediction accuracy andreduce the list of possible combinations. All these data aregiven in Table 3.
For short peptides, up to 300 g/mol, the number ofpossible amino acid combinations was rather limited,between 2 and 4. Nevertheless, these combinations had
Table 2 IHI values obtained in this study compared with HC coefficients [24] and previous IHI values [46, 51]
Identification of short peptides in complex mixtures 1945
to be sorted to find the effective one. When these pep-tides were not hydrophobic enough, no sorting could beperformed through RPLC analysis. Although the actualpeptide was often in first position on both CE and HILIClists, which allowed confirmation, it is interesting topoint out that HILIC seemed more accurate than CEsince the actual peptide was found to be in first positionon HILIC lists for all of the five peptides, although thiswas true only for three peptides among five on the CElists. In the case of small hydrophobic peptides, theactual combination usually stood in the first position onthe three lists.
When the peptide molar mass (around 300–450 g/mol)corresponded to a moderate number of AA combinations,namely below 50 combinations, the three rankings showedvaluable complementarity, and their association alloweddetermining the effective combination.
Then, with higher molar masses, the number of totalpossible combinations increased strongly, from 1,197 forKKKKK to 10,183 for TITYEY. Among these peptides,the effective combination of KKKKK, UHWSY andTITYDL was determined by this method. Although theirmasses corresponded to a high number of possiblecombinations, up to 3,000, the association of the threeanalytical data successfully led to the determination ofthe actual one. Finally, for TVTFKF, KNFFKE andTITYEY, for which the total number of possible combinationsranged from 3,875 to 10,183, the association of the threeanalytical techniques led to an over 98 % reduction of thisnumber.
Application of the identification strategy to a rapeseedprotein hydrolysate
The present strategy was applied to 24 peptides (numbered 1to 24) randomly extracted from about 150 peptides found ina mixed protein hydrolysate that was subjected to RPLC-MS, HILIC-MS and CE-MS analyses. For each peptidemass, the total number of possible combinations was calcu-lated and the number of remaining possible combinationsthat fulfilled simultaneously the RPLC and CE retentioncriteria was determined. In order to investigate the predic-tion accuracy improvement attributable to the addition of theHILIC retention model, the number of remaining possiblecombinations that matched simultaneously the three reten-tion criteria was also determined and compared with theformer. Finally, the sorted list was compared with thesequences of rapeseed proteins (cruciferins and napins) us-ing available protein databases and a software able to locatea specific peptide (from the mass) within a given protein. Allthese data are collected in Table 4.
The association of the data obtained by the three analysesallowed a deep reduction in the number of possible combi-nations, and the benefit provided by the addition of the third(HILIC) model was made clear. This method has beenproven very efficient for peptides with molar masses under400 g/mol since in most cases (nos. 1–9, 12) it provided theeffective combination sought, which was also consistentwith the sequences found in rapeseed proteins. Significantimprovement in composition prediction accuracy broughtby the addition of the HILIC model was illustrated by the
Table 3 Number of possiblecombinations of amino acidsbased on molar mass±0.2mass unit and rank on the threephysicochemical propertyordered lists for 18 peptidesin a synthetic mixture
nd cannot be determined as HCvalue is <10
Peptide MM (g/mol) Total no. of possiblecombinations
Rank in CE Rank inRPLC
Rank inHILIC
No. of combinationsin the final list
GGG 189.0 2 1 nd 1 1
LA 202.3 3 3 nd 1 1
AM 220.3 4 4 nd 1 1
AH 226.3 2 1 nd 1 1
MY 312.4 2 1 nd 1 1
LL 244.4 4 1 1 1 1
LY 294.4 3 1 1 2 1
FF 312.4 2 1 1 1 1
LW 317.4 23 3 3 3 1
LLL 357.6 20 11 3 2 1
VYV 379.4 16 9 1 1 1
KYK 437.6 42 2 nd 4 1
KKKKK 658.9 1,197 1 nd 2 1
UHWSY 702.6 1,790 323 18 102 1
TITYDL 724.8 2,956 57 19 2 1
TVTFKF 741.9 3,875 1,015 53 309 14
KNFFKE 811.9 8,568 599 36 189 27
TITYEY 788.7 10,183 1,893 nd 910 221
1946 C. Harscoat-Schiavo et al.
study of peptide no. 3. Indeed, the three combinationsmatching the RPLC and CE retention criteria correspondedto six peptide sequences found in rapeseed proteins. Con-trariwise, only one combination fulfilled the three criteria,thus corresponding to two sequences of peptides in rapeseedproteins. When only one combination matched the first twocriteria, the HILIC criterion provided confirmation of thiscombination which could correspond to two distinct sequen-ces in rapeseed proteins (nos. 4, 5, 6, 8, 9). For largerpeptides, the association of the three retention criteriaallowed a huge drop (from 88 % for no. 19 to 98 % fornos. 22 and 18) in the possible combination number. Theaddition of the third criterion allowed a massive reduction inthe number of possible combinations. Indeed, for peptidenos. 21 and 23, this reduction was as high as 66 %, from twoto three criteria. For both peptides, the actual peptide se-quence was determined by means of rapeseed protein data-bases: peptide no. 21 was identified as LEVIA and no. 23 asDVLQPT, which were respectively at rank 8 out of 12 andrank 31 out of 138 on their final lists. Prediction accuracyimprovement was even higher for peptide no. 18, with an86 % reduction from two to three criteria. However, two
among the three combinations matching all the criteriawere consistent with rapeseed protein sequences. Peptideno. 24 had already been identified by Schweizer et al. [24]as WIEFK after purification and sequencing. The HILICmodel accurately predicted its retention, with <4 %deviation between the theoretical and the experimentalretention times. WIEFK was found at rank 13 out of the106 possible combinations matching the three retentioncriteria.
All these examples have given proof of the essentialcontribution of the HILIC retention model in the identifica-tion strategy and demonstrated the absolute necessity tocombine the three models since this allowed a huge reduc-tion of the possible combination number and even some-times the actual combination identification. Moreover, thismethodology applied to a natural complex mixture analysiscould also serve to locate the presence of bioactive peptidesand help in their recovery. In fact, peptides matching thecriteria that stand in the protein sequences may be comparedwith bioactive peptide data banks, and the way to recoverspecific peptides from the mixture could be greatly enhancedby the use of the various retention models built here.
Table 4 Application of the identification strategy to a rapeseed mixed protein hydrolysate
Peptide no. Peptide mass(g/mol)
Total no. of possiblecombinations initially
No. of possiblecombinationsmatching 2 criteria
No. of possiblecombinationsmatching 3 criteria
Possible amino acid sequences aftercomparison with rapeseed protein sequences
Identification of short peptides in complex mixtures 1947
Conclusion
The HILIC separation technique was successfully applied tovarious peptides presenting a wide range of hydrophobicand hydrophilic properties. The complementary nature ofthese properties, quantified by HC and IHI coefficients,respectively, was demonstrated. Information collected fromthese orthogonal methods was thus not redundant. A modelwas established to predict the retention times of peptides inHILIC and added to the identification procedure whichconsisted of a computer-aided method of amino acid com-position determination based on the peptide masses andphysicochemical properties. The use of the selectivity char-acteristics of three complementary separation mechanisms(performed by use of RPLC, HILIC and CE) allowed sig-nificant enhancement of the identification accuracy. Theactual combination of many standard peptides in a mixturewas elucidated. This method was also successfully appliedto a complex mixture derived from mixed protein enzymatichydrolysis. The association of the three retention criteriaallowed a huge decrease in the possible combinationnumber (as high as 98 %). The use of protein databasesconfirmed first that the actual peptide retentions were satis-fyingly predicted by the various models and, second,allowed identifying the actual peptides in a significantnumber of cases.
Even though this procedure required three analyses tobe performed, up to 100 peptides were analysed in asingle run. In addition, this computer-aided method couldbecome a valuable tool, saving on the lengthy and costlysequencing steps currently performed in bioactive peptidesrecovery [4].
This approach could also be profitably combined withtandem mass spectrometry analysis, with de novo sequenc-ing and with composition-based sequencing strategy. Thelatter could be improved in the cases of insufficientfragment ion information, namely when the spectrum doesnot provide enough information to determine a uniquesequence.
Another major advantage of the proposed method isthe peptide physicochemical information provided bysuch analyses. Indeed, when complex peptide mixtures(typically protein hydrolysates) are studied, the aim isnot only to identify all the peptides in the mixture butalso, whenever possible, to find ways of recoveringpotential bioactive peptides. Since biological activitiesare sometimes related to particular properties [56], theseanalyses would help in the separation of the most activemolecules.
Acknowledgements We are grateful to Mrs M. Faure for her adviceand comments. We are also in debt to the unknown reviewers for theircontribution to the improvements of the final version of the paper.
References
1. Nii Y, Fukuta K, Yoshimoto R, Sakai K, Ogawa T (2008) BiosciBiotech Biochem 72:861–864
2. Udenigwe CC, Lin YS, Hou WC, Aluko RE (2009) J Funct Foods1:199–207
3. Aluko RE (2007) Recent Patents Biotechnol 1:260–2674. Marczak ED, Usui H, Fujita H, Yang Y, Yokoo M, Lipkowski AW,
Yoshikawa M (2003) Peptides 24:791–7985. Vanhoute M, Froidevaux R, Vanvlassenbroeck A, Lecouturier D,
Dhulster P, Guillochon D (2009) J Chromatogr B 877:1683–16886. Gauthier SF, Pouliot Y (2003) J Dairy Sci 86:E78–E877. Réhault S, Anton M, Nau F, Gautron J, Nys Y (2007) Prod Anim
20:337–3478. You SJ, Udenigwe CC, Aluko RE, Wu J (2010) Food Res Int
43:848–8559. Niehues M, Euler M, Georgi G, Mank M, Stahl B, Hensel A
(2010) Molecul Nutr Food Res 54:1851–186110. Franek F (2004) J Agr Food Chem 52:4097–410011. Chabanon G, Alves da Costa L, Farges B, Harscoat C, Chenu S,
Goergen JL, Marc A, Marc I, Chevalot I (2008) Bioresour Technol99:7143–7151
12. Amiot J, Germain L, Turgeon S, Lemay M, Ory-Salam C, AugerFA (2004) Int Dairy J 14:619–626
13. Girón-Calle J, Alaiz M, Vioque J (2010) Food Res Int Volume43:1365–1370
14. Dexter AF, Middelberg APJ (2008) Ind Eng Chem Res47:6391–6398
15. Schmelzer CH, Schops E, Ulbrich-Hofmann R, Neubert RHH,Raith K (2004) J Chromatogr A 1055:87–92
16. Gomez-Ruiz JA, Lopez-Exposito I, Pihlanto A, Ramos M, Recio I(2008) Eur Food Res Tech 227:1061–1067
18. Kafka AP, Kleffmann T, Rades T, McDowell A (2011) Int J Pharm417:70–82
19. Bruni R (2008) Comput Math Appl 55:912–92320. Spengler B (2004) J Am Soc Mass Spectrom 15:703–71421. Bertsch A, Leinenbach A, Pervukhin A, Lubeck M, Hartmer R,
Baessmann C, Elnakady YA, Müller R, Böcker S, Huber CG,Kohlbacher O (2009) Electrophoresis 30:3736–3747
22. Langsdorf M, Ghassempour A, Römpp A, Spengler B (2010) AnalBioanal Chem 398(7–8):2853–2865
31:1438–144829. Jiang Z, Smith NW, Ferguson PD, Taylor MR (2009) J Sep Sci
32:2544–255530. Jiang Z, Reilly J, Everatt B, Smith NW (2009) J Chromatogr A
1216:2439–244831. Schichtherle-Cerny H, Affolter M, Cerny C (2003) Anal Chem
75:2349–235432. Van Dorpe S, Vergote V, Pezeshki A, Burvenich C, Peremans K,
De Spiegeleer B (2010) J Sep Sci 33:728–73933. Kato M, Kato H, Eyama S, Takatsu A (2009) J Chromatogr B
877:3059–3064
1948 C. Harscoat-Schiavo et al.
34. Langrock T, Czihal P, Hoffmann R (2006) Amino Acids30:291–297
35. Nguyen HP, Schug KA (2008) J Sep Sci 31:1465–148036. Yang Y, Boysen RI, Hearn MTW (2009) J Chromatogr A
1216:5518–552437. Boersema PJ, Divecha N, Heck AJR, Mohammed S (2007) J
Proteome Res 6:937–94638. Boersema PJ, Mohammed S, Heck AJR (2008) Anal Bioanal
Chem 391:151–15939. Strege MA, Stevenson S, Lawrence SM (2000) Anal Chem
72:4629–463340. Mant CT, Hodges RS (2008) J Sep Sci 31:1573–158441. Mant CT, Hodges RS (2008) J Sep Sci 31:2754–277342. Jandera P (2008) J Sep Sci 31(9):1421–143743. Wang Y, Lu X, Xu G (2008) J Sep Sci 31:1564–157244. van Platerink CJ, Janssen HGM, Haverkamp J (2008) Anal
Bioanal Chem 391:299–30745. Liu A, Tweed J (2009) J Chromatogr B 877:1873–1881
46. Yoshida T, Okada T, Hobo T, Chiba R (2000) Chromatographia52:418–424
47. Yoshida T (2004) J Biochem Biophys Methods 60:265–28048. Quiming NS, Denola NL, Saito Y, Jinno K (2008) J Sep Sci
31:1550–156349. Jin G, Guo Z, Zhang F, Xue X, Jin Y, Liang X (2008) Talanta
76:522–52750. Wu J, Aluko RE (2007) J Pep Sci 13:63–6951. Gilar M, Jaworski A (2011) J Chromatogr A 1218:8890–889652. Messana I, Rosseti DV, Cassiano L, Misiti F, Giardina B,
Castagnola M (1997) J Chromatogr B 699:149–17153. Janini GM, Metral CJ, Issaq HJ, Muschik GM (1999) J Chromatogr
A 848:417–43354. Meek JL (1980) PNAS 77:1632–163655. Muniglia L, Nandor Kiss L, Fonteix C, Marc (2004) I Eur J
Operation Res 153:360–36956. Pripp AH, Isaksson T, Stepaniak L, Sørhaug T, Ardö Y (2005)
Trends Food Sci Technol 16:484–494
Identification of short peptides in complex mixtures 1949