International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Secretariat, Chemin des Mines 9, P.O. Box 195, 1211 Geneva 20, Switzerland Telephone: +41 (22) 338 32 06 - [email protected], http://www.ich.org ICH Procedures Endorsed by the ICH Steering Committee on June 10 2015
Transcript
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
ICH Secretariat, Chemin des Mines 9, P.O. Box 195, 1211 Geneva 20, Switzerland
i ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015
ICH PROCEDURES Document History
Circular (SC Report)
Date
ICH7/64F Steering Committee Meeting (Final Report) Yokohama, November 2004:
The Steering Committee endorsed the ICH Procedures, with some corrections to be given.
November 2004
ICH7/98F Steering Committee Meeting (Final Report) Brussels, May 2005.
The Steering Committee re-endorsed the revised consolidated ICH Procedures document, following amendments on page 2 (definition of the Interested Parties in the Glossary and page 27 (Annex 8)).
The ICH Procedures document is to be reviewed annually.
May 2005
ICH7/130F Steering Committee Teleconference (Final Report), September 2005.
The Steering Committee adopted the edited “Revised ICH Procedures” at the 21 September 2005 SC teleconference. The new version includes the following addition:
A section on Roles and Responsibilities of Technical Coordinators
21 September 05
ICH7/196F Steering Committee Meeting (Revised Report), Chicago October 2006.
The Steering Committee endorsed the Revised 2006 ICH Procedures document. The new version includes the following additions:
Updates to general text; An expanded Glossary Annex on MedDRA PtC Working Group; Annex on Implementation Working Group; Notes on Format and Style of the ICH Guidelines.
October 2006
ICH15/29F Steering Committee Meeting in Yokohama, October-November 2007.
The Steering Committee endorsed the Revised 2007 ICH Procedures document. The new version includes the following additions:
Expansion of Annex on MedDRA PtC WG; Section on the Role of ICH Observers; New Template for Step 4 Sign-off Sheet, where Step 4 is
reached without Step 2/Step 3; Rule for Co-Rapporteurs/Co-Group Leaders should be
from different regions/different parties;
November 2007
ii
Responsibility of the EWG/IWG Rapporteur to develop presentation for ICH library upon reaching Step2/Step4;
Q3C Maintenance Procedure: clarification on rotation of Regulator and Topic Leader.
ICH17/13 Steering Committee Meeting in Brussels, November 2008.
The Steering Committee endorsed the Revised 2008 ICH Procedures document. The new version includes the following additions:
An enhanced section on SC responsibilities; Integration of GCG Principles and Procedures into
Annex on GCG; Rule for Informal WGs and face-to-face meetings; An enhanced section on the development of
addendum; Definition for Invited Technical Expert.
November 2008
ICH19/24 Steering Committee Meeting in St. Louis, October 2009.
The Steering Committee endorsed the Revised 2009 ICH Procedures document. The new version includes the following additions:
Procedure for nomination of alternates to EWGs by ICH Observers and Interested Parties;
Clarification of WG responsibilities: need for EWGs to reach consensus on work plans, reports and other documents prior to provision to the SC;
Concept of “Feasability for Testing with information on consultation;
Addition of note to Annex 15 (Notes on the Format and Style of ICH Guidelines on the use of the term “sex” versus “gender”;
Clarification on the funding policy for speakers invited to participate to GCG-endorsed events (Ref: Annex 12 of ICH Procedures);
Use of Department of Health to designate Chinese Taipei.
October 2009
ICH21/20 The new version includes the following additions:
Addition to glossary of definition of DRA & DoH Addition to glossary of definition of SENTRI;; Addition of Annex 17 on General principles to
consider for the organisation of ICH meetings; Addition of Annex 18 which provides a work plan
template for EWGs/IWGs. Reference to this template is made in Annexes 6 and 16.
November 2010
ICH 23/15 The new version includes the following additions:
Addition to glossary of definition of EAC; Revision of description of Maintenance Procedure
(Category 4) to include: (1) Q4B maintenance procedure; and (2) process for the correction of out-
November 2011
iii
of-date information (e.g., out-of-date references, links etc…);
Addition of Annex 5 on Q4B maintenance procedure; Addition to Annex 6 (Principles for Increasing the
Efficiency of EWG/IWG Working Practices) of procedure related to ICH expert presentations on ICH topics;
Updating of Annex 11 (GCG Principles & Procedures) to reflect: (1) the November 2010 SC decision to open-up ICH technical working groups to participation from technical experts nominated by RHIs/DRAs/DoH; and (2) the June 2011 SC decision to invite the EAC as a RHI;
Addition of Annex 13 which includes the Considerations and Criteria (approved by the SC in November 2010) and the Rules and Procedures (approved by the SC in March 2011) relating to the participation of RHIs/DRAs/DoH in ICH technical working groups;
Updating of Annex 15 (Notes on the Format and Style of ICH Guidelines) to reflect the November 2010 SC decision regarding Glossaries within ICH Guidelines;
Changing references to “EC” to “EU” (e.g., sign-off templates etc…).
ICH 25/14 Steering Committee Meeting in San Diego, in November 2012.
The Steering Committee endorsed the 2012 ICH Procedures document. The new version includes the following changes agreed by the SC in Fukuoka in June 2012:
Restructuring of the Step process (Step 2a/2b), Nomination and role of the EWG/IWG Regulatory
Chair, Role of the EWG/IWG Rapporteur, Adoption of a Concept Paper without the support of
all six ICH parties.
The new version includes also the following addition:
Addition of Annex 14 (Rules and Procedures and modus operandi) which provides guidance for RHI Representatives attending ICH Meetings.
November 2012
ICH 26/26 Steering Committee Meeting in La Hulpe, Brussels, in June 2013.
The Steering Committee endorsed the 2013 ICH Procedures document. The new version includes the following additions:
Addition to Annexes 1 and 2 of a paragraph on invitation of liaison(s) to participate in EWG/IWG activities,
Addition of Annex 16 (Notes on the Format and Style of ICH Technical Document) which provides guidance
June 2013
iv
to ICH Topic Rapporteurs when drafting technical documents
ICH 27/18 Steering Committee Meeting in Osaka, in November 2013.
M2 proposal regarding replacement of Feasibility Testing with ‘Step 2 for Testing’;
Addition/clarification of definitions for PtC documents, Considerations documents, Q&As;
Revision of the GCG Procedures to reflect SC decision to recast the GCG
November 2013
ICH 29/30 Steering Committee Meeting in Lisbon, in November 2014
Clarification of Addendum Format;
Revision of the Q3C Maintenance Procedure;
Use of ICH design (including its logo) by ICH Parties;
Removal of Annex 20 (work plan)
Changes related to the promotion of Health Canada and Swissmedic as SC members (see main changes summarized in the box below);
Addition of Annex 20 which explain the Impact of Swissmedic and Health Canada Promotion to SC Member
November 2014
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 1
Annex 6: Principles for increasing the efficiency of EWG/IWG Working Practices ............ 27
Annex 7: ICH Business Plan .................................................................................................. 33
Annex 8: Role and Responsibilities of the Steering Committee, the Coordinators, the Technical Coordinators and the Observer .............................................................. 34
Annex 9: Standard Operating Procedures for Organisation of Interim Meetings .................. 40
Annex 11: Global Cooperation (GC) Principles and Procedures ........................................... 43
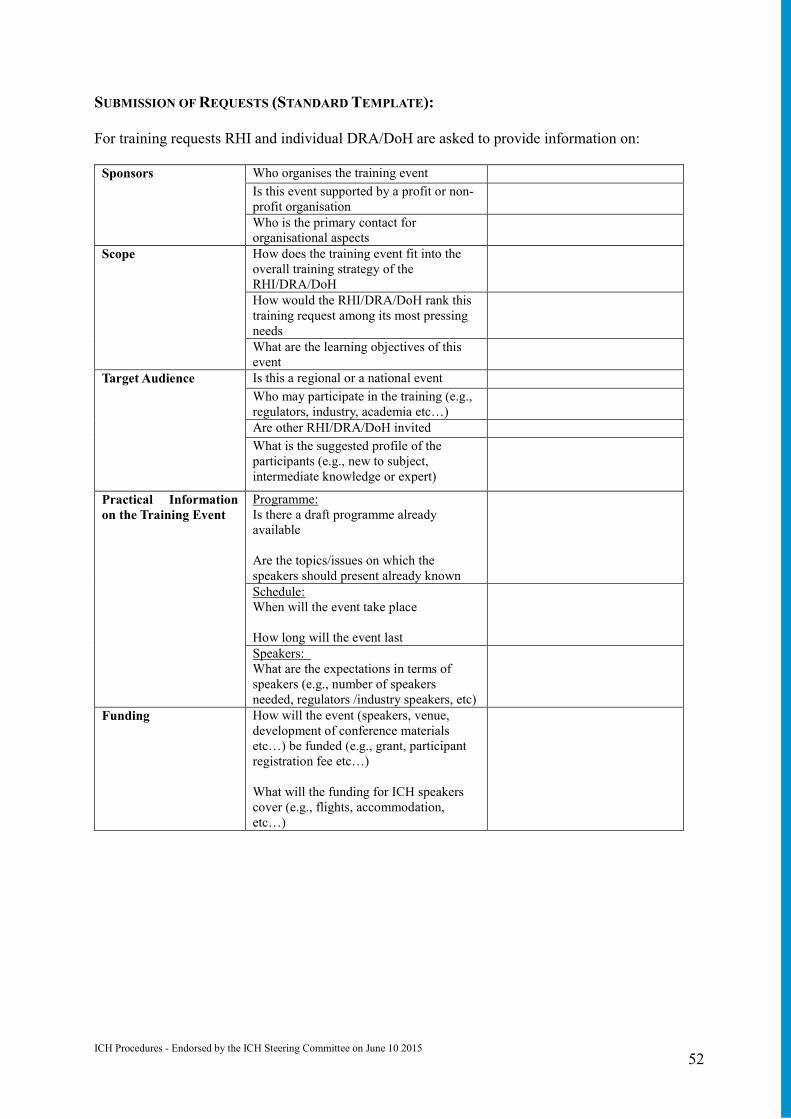
Annex 12: Global Cooperation (GC) Procedure on Training Activities: Selection, Prioritization, Coordination of RHI & DRA/DoH Requests ................................ 48
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 2
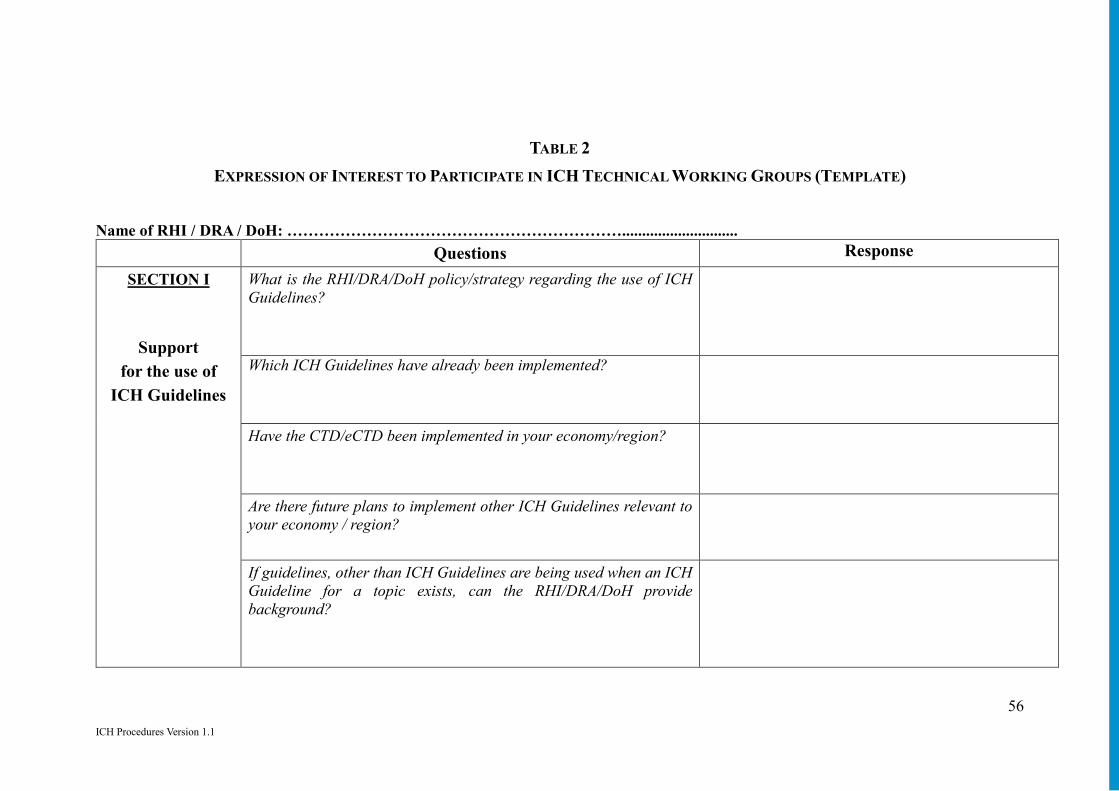
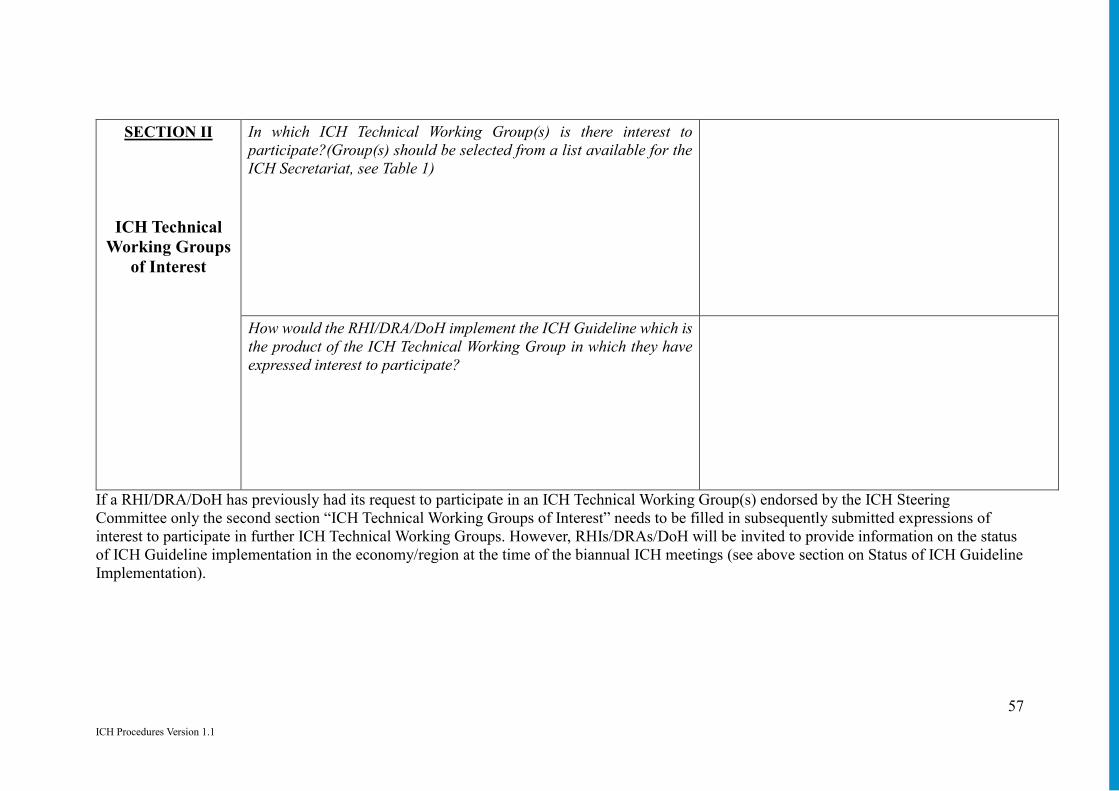
Annex 13: Participation of RHIs and DRAs/DoH in ICH Technical Working Groups to Promote Use of ICH Guidelines ........................................................................... 53
Annex 14: Guidance Document for RHI Representatives attending ICH Meetings. ............. 59
Annex 15: MedDRA Points to Consider (PtC) Working Group ............................................ 61
Annex 16: Notes on the Format and Style of Technical Documents
Annex 17: Notes on the Format and Style of ICH Guidelines ............................................... 63
Annex 18: Templates for Step 2 and Step 4 Sign-off Sheets, including template for Sign-off Sheet where Step 4 is reached without Step 2/Step .............................................. 71
Annex 19: General principles to consider for the organisation of ICH meetings .................. 89
Annex 20: Impact of Swissmedic and Health Canada Promotion to SC member
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 3
Glossary
APEC: Asia-Pacific Economic Cooperation, and one of the Regional Harmonisation Initiatives (RHI).
ASEAN: Association of Southeast Asian Nations, and one of the Regional Harmonisation Initiatives (RHIs).
Brainstorming Group: Is a group that discusses the need for harmonisation within specific scientific domains. The outcome of Brainstorming sessions is recommendations for topics for ICH harmonisation, for Steering Committee consideration. ICH Parties nominate representatives to the group and, unless otherwise specified by the Steering Committee, the ICH membership is limited to two representatives per Party per working group and one representative per ICH Observer, and also if applicable, one per Interested Party. The Steering Committee officially designates a Group Leader among the representatives designated by the ICH Parties. In some cases, both a Group Leader and a Co-Group Leader may be appointed. Whenever possible, Co-Group Leaders should be from different regions and should not both be from a Regulatory or Industry Party.
Business Plan: After consideration of a Concept Paper, the Steering Committee may request the development of a Business Plan, outlining the costs and benefits of harmonising the topic proposed by the Concept Paper. The Business Plan is complimentary to the Concept Paper and focuses in particular on regulatory feasibility. The template for the Business Plan was adopted at the Washington meeting in June 2004 (see Annex 7).
Concept Paper: Describes the perceived problem and the issues to be resolved, and is the trigger of all ICH activities. A Concept Paper for a new harmonisation activity can be submitted by any ICH Party or Observer to the Steering Committee (see Section 2).
Considerations Document: Provides specific scientific considerations on a topic and does not require a sign-off approval, but do require discussion and endorsement by the Steering Committee. Considerations documents were developed by the following discussion groups: Gene Therapy Discussion Group (GTDG), and ICH & Women Discussion Group.
CTD: Common Technical Document
Discussion Group: Is a group established to discuss specific scientific considerations or views i.e., Gene Therapy Discussion Group (GTDG), and ICH & Women Discussion Group. ICH Parties nominate representatives to the group and, unless otherwise specified by the Steering Committee, the official membership is limited to two representatives per Party per working group and one representative per ICH Observer, and also if applicable, one per Interested Party.
DRA: Drug Regulatory Authority.
DoH: Department of Health.
eCTD: Electronic Common Technical Document.
EAC: East African Community, and one of the Regional Harmonisation Initiatives (RHIs).
EFPIA: European Federation of Pharmaceutical Industries and Associations, and one of the ICH Parties.
EFTA: European Free Trade Association, one of the ICH Observers until 2014. EFTA was then replaced by Swissmedic (Swiss Agency for Therapeutic Products). The other members of EFTA are part of the European Economic Area (EEA) and therefore follow the EU rules. EMA: European Medicines Agency.
ESTRI: Electronic Standards for the Transfer of Regulatory Information.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 4
EU: European Union (the EU is represented by the European Commission, and EMA and its expert committees), and one of the ICH Parties.
EWG: Expert Working Group is charged with developing a harmonised guideline that meets the objectives in the Concept Paper and Business Plan. ICH Parties nominate representatives and, unless otherwise specified by the Steering Committee, the official membership is limited to two representatives per Party per working group and one representative per ICH Observer, and also if applicable, one per Interested Party (see Annex 1).
FDA: US Food and Drug Administration, and one of the ICH Parties.
GCC: Gulf Cooperation Council, and one of the Regional Harmonisation Initiatives (RHIs).
GC: Global Cooperation (see Annexes 11 and 12).
GCG: Global Cooperation Group (see Annex 11).
GTDG: Gene Therapy Discussion Group.
Health Canada: is represented by the Health Products and Food Branch (HPFB), and one of the ICH Parties.
ICH Coordinators: Are nominated by ICH Parties and are fundamental to the smooth running of the ICH. An ICH Coordinator acts as the main contact point with the ICH Secretariat. Due to the structural differences within the EU and MHLW, ICH Technical Coordinators are also designated from the EMA and PMDA respectively. They support their respective ICH Coordinators and facilitate every action of the Steering Committee members in the region, mainly by applying their scientific knowledge. Their roles include acting as a contact point between the experts within EMA or PMDA and the ICH Coordinator at the main regulatory body and as a contact point with the ICH Secretariat (see Annex 8).
ICH Observer: World Health Organisation (WHO).
ICH Parties: Are the founding Regulatory Parties (EU, FDA, MHLW), and the founding Industry Parties (EFPIA; PhRMA, JPMA). Since June 2014, Health Canada and Swissmedic have been promoted as Parties to the SC.
ICH Secretariat: Operates in Geneva, Switzerland, is responsible for the day-to-day management of ICH. It provides administrative support to the ICH Steering Committee by coordinating the activities of ICH. The Secretariat is primarily concerned with preparations for, and documentation of, meetings of the Steering Committee as well as coordination of preparations for Working Group (EWG, IWG, informal WG) and Discussion Group meetings. The ICH Secretariat also provides administrative support for the Global Cooperation-related activities and MedDRA (see Annex 8).
ICH Steering Committee: Is the body that governs the ICH, determines the policies and procedures for ICH, selects topics for harmonisation and monitors the progress of harmonisation initiatives. Each of ICH Parties has up to two seats on the ICH Steering Committee. The ICH Observer (WHO) nominates a non-voting participant to attend the ICH Steering Committee Meetings. IFPMA also participates as a non-voting member (see Annex 8).
IFPMA: International Federation of Pharmaceutical Manufacturers and Associations; provides the ICH Secretariat.
IGPA: International Generic Pharmaceutical Alliance, and one of the ICH Interested Parties.
Informal Working Group: Is formed prior to any official ICH harmonisation activity with the objectives of developing/finalising a Concept Paper, as well as developing a Business Plan. ICH Parties nominate representatives and, unless otherwise specified by the Steering Committee, the official membership is limited to two representatives per Party per working group and one
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 5
representative per ICH Observer, and also if applicable, one per Interested Party (see Annex 3).
Interested Parties: Those organisations that are expected to implement or to be regulated by the outcome of ICH efforts (World Self-Medication Industry – WSMI, International Generic Pharmaceutical Alliance – IGPA, joint representative of bio industries of the ICH regions, and other Interested Parties as determined by the Steering Committee over time).
Invited Technical Expert: From time to time the ICH Steering Committee may identify an organisation or regulatory authority with expertise relevant to an ICH harmonisation activity. Upon invitation from the Steering Committee, the organisation or regulatory authority may nominate an expert to provide technical input. Invited Technical Experts are invited to participate in all activities of the Working Group to which they are nominated e.g., face-to-face meetings, work by e-mail, tele/webconferences etc…
IWG: Implementation Working Group is tasked to develop Q&As to facilitate implementation of existing guidelines. the ICH Parties nominate representatives and, unless otherwise specified by the Steering Committee, the official membership is limited to two representatives per party per working group and one representative per ICH Observer, and also if applicable, one per Interested Party (see Annex 2).
JMO: Japanese Maintenance Organisation (is a partner of the MSSO and is responsible for maintaining and distributing MedDRA in Japan).
JPMA: Japan Pharmaceutical Manufacturers Association, and one of the ICH Parties.
MedDRA: Medical Dictionary for Regulatory Activities Terminology; developed under the auspices of ICH and maintained by MSSO.
MedDRA Management Board: Is appointed by the ICH Steering Committee to oversee the activities of the "Maintenance and Support Services Organisation" (MSSO) for MedDRA, and ensure that the MSSO is meeting the various needs of MedDRA users. The Management Board is composed of one representative from ICH Parties and one representative from MHRA. WHO is present as a non-voting member. The IFPMA acts as a non-voting Observer on the Management Board.
MedDRA Points to Consider Document: Provides a best practice approach for the use of MedDRA.
MedDRA Points to Consider Working Group: Works to develop best practice initiatives related to the use of MedDRA, and to maintain the released PtC documents in parallel with MedDRA version updates (see Annex 15).
MHLW: Ministry of Health, Labour and Welfare, Japan, and one of the ICH Parties.
MSSO: Maintenance and Support Services Organisation (serves as the repository, maintainer, and distributor of MedDRA).
Options Paper: May be requested by the Steering Committee to outline the options in terms of the benefits, costs and risks of outsourcing specific tasks to organisations external to the ICH process i.e. consideration of the use of vocabulary services for the maintenance of complex terminology, or the use of SDOs (Standards Development Organisations) for the development of technical standards (M2 Message Standards).
PANDRH: Pan American Network on Drug Regulatory Harmonisation, and one of the Regional Harmonisation Initiatives (RHIs).
PhRMA: Pharmaceutical Research and Manufacturers of America, and one of the ICH Parties.
PMDA: Pharmaceuticals and Medical Devices Agency (Japan).
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 6
Point to Consider (PtC) document: Provides further clarification to both industry and regulators on how to use/implement ICH Guidelines and Standards by supplementing an already existing ICH guidance and its Questions & Answers document.
POC: Proof of Concept provides evidence that demonstrates ultimate feasibility i.e., development by the M5, M2 and E2B(R3) EWGs of a POC Plan to test the viability of using M2’s message specifications to exchange information. Determining Proof of Concept, and alignment of a Guideline with the POC results is necessary for regional implementation to occur.
Questions and Answers document: Provides additional guidance and advice when necessary in the form of Questions and Answers "Q&As" to help the interpretation of certain ICH harmonised Guidelines, in order to ensure a smooth and consistent implementation in the three ICH regions and beyond and to allow that certain practical issues be overcome.
Rapporteur: The Steering Committee (SC) officially designates a Rapporteur when a new ICH Topic is formally adopted. The Rapporteur will be one of the representatives of the ICH Parties. In some cases, both a Rapporteur and a Co-Rapporteur may be appointed. A Co-Rapporteur is nominated to assist the Rapporteur in his different tasks (see Annex 6). The Rapporteur is usually a representative from the Party which proposed the topic originally, and he takes the lead for the day-to-day work of the EWG/IWG. If the Rapporteur is a representative from one of the ICH Industry Parties, the rapporteurship will have to be transferred to an ICH Regulatory Party (usually from the same region) after Step 2a consensus is reached. Throughout the development of the draft guideline, it is the responsibility of the Rapporteur (/Co-Rapporteur) to keep an up-to-date action plan and timetable, with clear deliverables and deadlines. The Rapporteur (/Co-Rapporteur) shall regularly present reports to the Steering Committee (SC), focusing in particular on the scientific aspects of the document under development.
Recommendations Document: Due to the information technology (IT) nature of the M2 EWG’s work on Electronic Standards for the Transfer of Regulatory Information (ESTRI), some of their activities result in Recommendations. These Recommendations do not undergo the formal ICH step process, so as to allow for flexible change as both science, and technologies evolve. They are agreed in the EWG, signed by all parties of the EWG, and are approved and signed off by the ICH Steering Committee. Current M2 Recommendations are posted on the ESTRI website (http://estri.ich.org).
Regulatory Chair: The Regulatory Parties of the Steering Committee (SC) officially designate a Regulatory Chair when a new ICH Topic is formally adopted. The Regulatory Chair ensures timely execution of the ICH process and adherence to the Concept Paper and Business Plan, including scope and timelines. The Regulatory Chair shall work in close collaboration with the Rapporteur. If the Rapporteurship is taken by an ICH Regulatory Party, the Regulatory Chair may also be appointed as the Rapporteur as an option of the Regulatory Party which is taking the role of the Regulatory Chair and upon agreement of the SC. The Regulatory Chair shall regularly present reports to the SC, in collaboration with the Rapporteur, focusing in particular on the timelines, adherence to scope and conflicting views if they arise (see Annex 6).
RHI: Regional Harmonisation Initiative, founded on the principle of harmonising drug regulation across a defined group of non-ICH countries (see Annex 11).
Topic leader/Deputy Topic Leader: ICH Parties shall nominate representatives to Expert Working Groups (EWG, see Annex 1) / Implementation Working Groups (IWG, see Annex 2). Unless otherwise specified by the Steering Committee (SC), the official membership of EWG/IWG shall be limited to two representatives per Party per working group (one expert shall be designated as Topic Leader and the other as Deputy Topic Leader), and one representative
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 7
per ICH Observer (WHO), and also if applicable one per Interested Party (World Self-Medication Industry - WSMI, International Generic Pharmaceutical Alliance - IGPA, and other interested parties as determined by the Steering Committee). It is the responsibility of the Topic leader/Deputy Topic Leader to officially represent a consolidated view of their party during any ICH interactions (e-mails, tele/webconferences and face-to-face meetings).
SADC: Southern African Development Community, and one of the Regional Harmonisation Initiatives (RHIs).
SENTRI: Standards Everyone Needs for the Transfer of Regulatory Information; SENTRI activities are undertaken by the M2 EWG which is responsible for monitoring and recommending technology to see if it is useful to ICH’s scope.
Swissmedic: Swiss regulatory agency, and one of the ICH Parties.
WHO: World Health Organisation, and the ICH Observer.
WSMI: World Self-Medication Industry, and one of the ICH Interested Parties.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 8
1. INTRODUCTION
The original ICH process involved a step-wise progression of guidelines. The original process has evolved to include maintenance activities, as an essential part of the ICH procedure. The importance of having a sound maintenance mechanism in place has been shown during the progress of several ICH topics (e.g., Q3C guideline1 on “Residual Solvents”, M2 recommendations, Q4B Annexes2).
The Importance of monitoring the Implementation of ICH Guidelines:
In addition to the maintenance activity, it is also important to have proper procedures in place for the revision (modification) of existing guidelines and to assist their implementation (through Q&A documents).
The benefits of harmonising technical requirements can only be reached if the various ICH Guidelines are implemented and interpreted in a consistent way across regions.
The following factors may hinder or challenge a smooth and consistent implementation of ICH Guideline:
- New technical information and/or scientific progress requiring some updating and maintenance to existing ICH Guideline;
- New guideline(s) being generated individually at a regional/national level; - Divergent local/regional interpretation of certain statements and/or requirements from ICH
harmonised Guidelines.
It is essential to prevent any future disharmony in harmonised technical areas, through careful monitoring of the use of ICH Guideline at regional/national level and through early collaboration and exchange of information on newly emerging issues. It should be noted that the Steering Committee is not constrained by these procedures to undertake a specified activity when necessary to respond to a specific need, e.g., brainstorming plenaries, etc.
It is the responsibility of all ICH Parties3 to report back any difficulties in the implementation of existing harmonised guidelines or any problems resulting from inaccurate or divergent interpretation at a local, national or regional level. The Steering Committee (SC) may also wish to consider any issues brought forward by the ICH Observer and Interested Parties (World Self-Medication Industry – WSMI, International Generic Pharmaceutical Alliance - IGPA, and other Interested Parties as determined by the Steering Committee). However, one of the ICH Parties should bring the matter to the SC table. If the topic is adopted, the Regulatory Chair will be chosen among the ICH Regulatory Parties and the Rapporteur among all ICH Parties.
When an issue is identified within a region, it should be communicated to the ICH Secretariat, which shall then inform the Coordinators of all ICH Parties and the Observer, asking them to consider whether the issue should be brought to the attention of the SC.
ICH Coordinators are expected to liaise with their relevant experts when necessary, in order to carry out a preliminary assessment of the issue(s) and elaborate proposed solutions.
For issues that cannot be resolved at a local level, the SC should discuss whether the concerned ICH Guideline requires either a simple maintenance or a more fundamental implementation activity or revision.
Depending on the urgency of the matter, the SC may be consulted via e-mail or tele/webconference, in order to consider whether any further SC discussions on the issue require the presence of specific experts.
1 See Annex 4 2 See Annex 5 3 The ICH Parties are EU, MHLW, FDA, Swissmedic, Canada, EFPIA, JPMA, and PhRMA.,
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 9
The implementation and maintenance of ICH Guideline should be an essential agenda item of any meeting or tele/webconference of the SC.
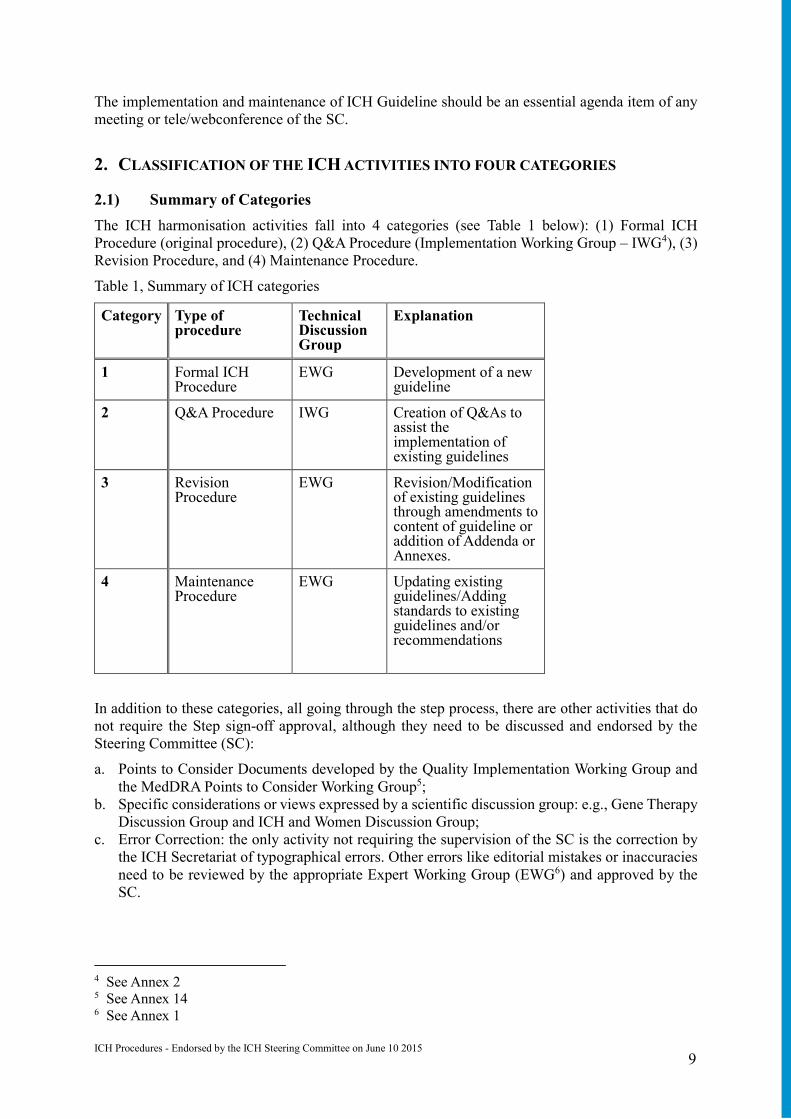
2. CLASSIFICATION OF THE ICH ACTIVITIES INTO FOUR CATEGORIES
2.1) Summary of Categories
The ICH harmonisation activities fall into 4 categories (see Table 1 below): (1) Formal ICH Procedure (original procedure), (2) Q&A Procedure (Implementation Working Group – IWG4), (3) Revision Procedure, and (4) Maintenance Procedure.
Table 1, Summary of ICH categories
Category Type of procedure
Technical Discussion Group
Explanation
1 Formal ICH Procedure
EWG Development of a new guideline
2 Q&A Procedure IWG Creation of Q&As to assist the implementation of existing guidelines
3 Revision Procedure
EWG Revision/Modification of existing guidelines through amendments to content of guideline or addition of Addenda or Annexes.
4 Maintenance Procedure
EWG Updating existing guidelines/Adding standards to existing guidelines and/or recommendations
In addition to these categories, all going through the step process, there are other activities that do not require the Step sign-off approval, although they need to be discussed and endorsed by the Steering Committee (SC):
a. Points to Consider Documents developed by the Quality Implementation Working Group and the MedDRA Points to Consider Working Group5;
b. Specific considerations or views expressed by a scientific discussion group: e.g., Gene Therapy Discussion Group and ICH and Women Discussion Group;
c. Error Correction: the only activity not requiring the supervision of the SC is the correction by the ICH Secretariat of typographical errors. Other errors like editorial mistakes or inaccuracies need to be reviewed by the appropriate Expert Working Group (EWG6) and approved by the SC.
4 See Annex 2 5 See Annex 14 6 See Annex 1
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 10
2.2) ICH harmonisation activities before Step 17
2.2.1) Preparation of a Concept Paper
The Concept Paper is the trigger of all ICH harmonisation activities. This provides a short summary of the proposal (maximum 2 pages) and provides the information indicated below:
Type of Harmonisation Action Proposed (e.g., a new harmonised guideline /recommendation, or a revision of an existing guideline – indicating the category of procedure).
Statement of the Perceived Problem: Brief description with an indication of the magnitude of the problems currently caused by a lack of harmonisation, or - in the case of new scientific developments - anticipated if harmonisation action is not taken.
Issues to be Resolved: A summary of the main technical and scientific issues, which require harmonisation.
Background to the Proposal: Further relevant information, e.g., the origin of the proposal, references to publications, and discussions in other fora.
Type of Expert Working Group: Recommendation on whether the EWG8 (if needed) should be an extended EWG - for topics with implications beyond new drug research.
If necessary, further documentation and reports may be annexed to the Concept Paper.
2.2.2) Endorsement by the Steering Committee
A Concept Paper can be submitted by any ICH Party or Observer to the Steering Committee.
If the Steering Committee agrees to the topic that is being proposed for harmonisation, it may decide to establish an informal Working Group9 to develop a Concept Paper.
The ICH Party responsible for proposing the topic originally will lead the informal Working Group in developing the Concept Paper. When considering the Concept Paper, the Steering Committee should take the following points into account:
a. Objectives and Expected Outcome of the harmonisation action;
b. Categories of ICH process;
c. Composition of the EWG or IWG appointed to discuss the technical issues10;
d. Set a Timetable and Action Plan for the EWG/IWG.
The Steering Committee should discuss intensively the necessity to develop a new ICH Guideline or to revise an existing ICH Guideline. When a Q&A document could cover the proposed concerns, then the Q&A Procedure should be preferred.
As guidance for this discussion, the following points are of importance:
a. Any changes to the content of an existing ICH Guideline are considered as “Revision of the Guideline (Category 3)”.
b. Changes to the content of a Guideline means “addendum of a new paragraph” and/or “partial replacement of the sentence“, but does not mean “adding an identified list of standards (i.e., level of residual solvents)” and/or “correction of a typographical error.”
c. If the Steering Committee agrees to the topic that is being proposed for harmonisation, it may decide to establish an informal Working Group11 to develop a Concept Paper
When the Steering Committee supports the adoption of the Concept Paper, the proposed action can be initiated (Start of Step 1). An EWG or IWG12 will be established to discuss the proposed items.
7 See Annex 6 8 See Annex 1 9 See Annex 3 10 See Annexes 1 and 2
12 See Annexes 1 and 2
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 11
The Concept Paper may need to be revised to reflect the SC discussion and conclusions.
2.2.3) ICH Business Plan
The Business Plan, whose template was adopted at the Washington meeting in June 2004, should be submitted to the Steering Committee upon request13.
Concept papers will be developed and proposed by one or several ICH Parties or the ICH Observer for initial consideration by the Steering Committee. At this initial stage a Business Plan will not be required although one may be provided at the discretion of the proposing party. If the Steering Committee agrees that a topic may warrant further consideration and a Business Plan has not been provided and agreed, an informal Working Group will be formed and will work through e-mail, tele/webconference and, exceptionally, face-to-face meetings. The first tasks of the informal Working Group will be to finalise a Concept Paper and develop and agree a Business Plan. The revised Concept Paper and Business Plan will be sent prior to and presented to the next SC webconference or face-to-face meeting.
2.2.4) Decisions by the Steering Committee
In principle, the agreement of the ICH Parties is necessary for initiating any ICH harmonisation activities. However, as an exceptional case when ICH Party consensus cannot be achieved, the three ICH Founding Regulatory Parties can jointly decide to adopt a Concept Paper considering that Regulators have the ultimate responsibility to ensure the protection of public health and have the responsibility for issuing regulatory guidelines.
3. ICH PROCESS FOR EACH CATEGORY14 The following chapter describes the detailed procedure within each category after adoption of a topic by the ICH Steering Committee. The five-step process, which proved successful for the first phase of ICH activities, is maintained.
3.1) Formal ICH Procedure (Category 1) by EWG15
This procedure corresponds to the original ICH process and was used for more than a decade, and it now includes some additional explanation on each activity. The Steering Committee may decide to follow an accelerated procedure for new topics, when necessary. To this end, the Steering Committee adopted the Streamlined Procedure 16 (final version dated October 21, 2002) in Washington in September 2002.
Step 1: Consensus building
The process of consensus building begins when the Steering Committee adopts a Concept Paper as a new topic. The EWG is established by the nomination of the Working Group members by the ICH Parties, the ICH Observer and, where applicable, by RHIs and DRAs/DoH or other Interested Parties. Health Canada and Swissmedic are not obliged to send technical experts to all EWGs/IWGs.17 At the same time, the Regulatory Steering Committee designates the Regulatory Chair, and the Steering Committee designates the Rapporteur (and Co-Rapporteur).
As requested in the Concept Paper, an EWG shall be established. The Rapporteur prepares an initial draft of the technical document18, based on the objectives set out in the Concept Paper, and in
13 See Annex 7 14 See Annex 18 for templates of Sign-off Sheets 15 See Annex 1 16 See Annex 10 17 See Annex 20 18 See Annex 17 for notes on the style and format of ICH Guidelines
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 12
consultation with experts designated to the EWG. The initial draft and successive revisions are circulated for comments within the EWG, giving fixed deadlines for receipt of those comments.
To the extent possible, the consultation will be carried out by correspondence, using fax and e-mail. Face-to-face meetings of the EWG will normally only take place at the time and venue of the biannual SC meetings. Additional, formal meetings of the ICH EWG need to be agreed, in advance, by the Steering Committee.
Interim reports are made by the EWG Regulatory Chair and Rapporteur at each meeting of the Steering Committee.
When consensus is reached within the EWG, the technical experts of the EWG will sign the Step 1 Experts Sign-off sheet.
The consensus text approved by all the ICH Parties’ members in the EWG is signed-off by the experts as Step 1 Technical Document.
If consensus is reached within the agreed timetable the Step 1 Experts Technical Document with EWG signatures is submitted to the Steering Committee to request adoption under Step 2a of the ICH process.
In exceptional circumstances where the EWG cannot come to full consensus on all aspects of the technical document, the Regulatory Chair with support of the Rapporteur will provide a report indicating the extent of agreement reached and highlighting the points on which there are differences among the parties. Experts from all ICH Parties represented on the EWG will have the opportunity to explain their position to the Steering Committee.
The Regulatory Chair with support of the Rapporteur will recommend a potential resolution to the Steering Committee (such as preparing a technical document that includes the different alternatives which are supported by the Experts or minority opinions).
The Steering Committee may then:
Allow an extension of the timetable, on the basis that the EWG can give assurances that consensus could be reached within a short, specified period;
Request the EWG to develop a technical document that includes the different alternatives which are supported by the experts or minority opinions;
Decide to suspend or abandon the harmonisation project.
When consensus is reached on a technical document that includes the different alternatives which are supported by the experts or minority opinions is finalised, the EWG will sign the Step 1 Experts Sign-off sheet.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress including the ICH step process19.
Step 2a: Confirmation of consensus on the Technical Document
Step 2a is reached when the Steering Committee agrees, based on the report of the EWG, that there is sufficient scientific consensus on the technical issues for the Technical Document or recommendation to proceed to the next stage of regulatory consultation.
The consensus text approved by the Steering Committee is signed-off by the Steering Committee as Step 2a Final Technical Document.
Irrespective of whether or not Health Canada and Swissmedic will have appointed technical experts in the Working Group, the SC Members of Health Canada and Swissmedic will be invited to sign-
19 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 13
off the Step 2a as ICH Parties20.
In the unlikely situation where consensus cannot be reached but the three ICH SC Founding Regulatory Parties (EU, FDA and MHLW) are in agreement, they will proceed with the Step sign-off21.
The technical document will be made public on the ICH website.
Step 2b: Adoption of the draft Guideline
On the basis of the technical document, the ICH Regulatory Parties will take the actions they deem necessary to develop the “draft Guideline”.
The consensus text approved by the ICH Regulatory Parties is signed-off by the ICH Regulatory Parties as Step 2b Draft Guideline.
Irrespective of whether or not Health Canada and Swissmedic will have appointed technical experts in the Working Group, the SC Members of Health Canada and Swissmedic will be invited to sign-off the Step 2b as ICH Parties22.
In the unlikely situation where consensus cannot be reached but the three ICH SC Founding Regulatory Parties (EU, FDA and MHLW) are in agreement, they will proceed with the Step sign-off23.
Step 3: Regulatory consultation and discussion
a) Regional regulatory consultation
At this stage, the Step 2b draft Guideline leaves the ICH process and becomes the subject of normal wide-ranging regulatory consultation in the regions. In the EU it is published as a draft CHMP Guideline, in Japan it is translated and issued by MHLW for internal and external consultation and in the USA it is published as draft guidance in the Federal Register. Swissmedic is referring input to the EU consultation. In Canada, Health Canada also solicits comments on draft ICH guidances.
The difference from normal, national/regional procedures for consultation on draft guidelines are that the Regulatory Parties exchange information on the comments they have received in order to arrive at a single, harmonised Guideline. Also, there is an opportunity for Industry Associations and Regulatory Authorities in other regions to comment on the draft consultation documents, which are distributed by the ICH Secretariat via the ICH website.
b) Discussion of regional consultation comments
After obtaining all regulatory consultation results, the EWG who organised the discussion for consensus building will be resumed. If the Rapporteur was designated from an industry party until Step 2b, then a new Rapporteur will be appointed from the regulatory party, preferably from the same region as the previous Rapporteur. The same procedure described in Step 1 is used to address the consultation results. The draft document to be generated as a result of the Step 3 phase is called Step 3 Experts Draft Guideline.
c) Finalisation of Step3 Experts Draft Guideline
If, after due consideration of the consultation results by the EWG, consensus is reached amongst the experts from the ICH Regulatory Parties on a revised version of the Step 2b Final Draft Guideline, the Step 3 Experts Draft Guideline is signed by the EWG experts of the ICH Regulatory Parties. The Step 3 Document with regulatory EWG signatures is submitted to the Steering
20 See Annex 20 21 See Annex 20 22 See Annex 20 23 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 14
Committee to request adoption as Step 4 of the ICH process.
This Step 3 Document with regulatory EWG signatures is named Step 3 Draft Guideline, and this sign-off is to be called Step 3 Experts Sign-off.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress including the ICH step process24.
Where complete consensus has not been achieved within the agreed time frame, the Regulatory Chair in support of the Rapporteur will make a report to the Steering Committee indicating the extent of agreement reached and highlighting the points on which differences between the parties remain. Experts from all ICH Parties represented on the EWG will have the opportunity to explain their position to the Steering Committee. The Steering Committee may then:
Allow an extension of the time frame, if the EWG can give assurances that consensus could be reached within a short, specified period;
Decide to abandon the current draft and resume the discussion from Step 1; Decide to suspend or abandon the harmonisation project.
Step 4: Adoption of an ICH Harmonised Guideline
Step 4 is reached when the Steering Committee agrees, on the basis of the report from the Regulatory Chair and the Regulatory Rapporteur of the EWG, that there is sufficient consensus on the draft ICH Guideline.
This endorsement is based on the signatures from the ICH Regulatory Parties affirming that the Guideline is recommended for adoption by the regulatory bodies of the ICH regions.
In the event that one or more ICH Parties representing Industry have strong objections to the adoption of the guideline, on the grounds that the revised draft departs substantially from the original consensus, or introduces new issues, the ICH Regulatory Parties may agree that a revised document should be submitted for further consultation. In this case, the EWG discussion may be resumed.
The Step 4 Final Document is signed-off by the SC signatories for the ICH Regulatory Parties as an ICH Harmonised Guideline at Step 4 of the ICH process.
Irrespective of whether or not Health Canada and Swissmedic will have appointed technical experts in the Working Group, the SC Members of Health Canada and Swissmedic will be invited to sign-off the Step 4 as ICH Parties25.
In the unlikely situation where consensus cannot be reached but the three ICH SC Founding Regulatory Parties (EU, FDA and MHLW) are in agreement, they will proceed with the Step sign-off26.
Step 5: Implementation
Having reached Step 4, the harmonised Guideline moves immediately to the final step of the process that is the regulatory implementation. This step is carried out according to the same national/regional procedures that apply to other regional regulatory guidelines and requirements, in the EU, Japan, the USA, Canada, Switzerland and beyond.
Information on the regulatory action taken and implementation dates are reported back to the Steering Committee and published by the ICH Secretariat on the ICH website.
Streamlined Procedure
24 See Annex 20 25 See Annex 20 26 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 15
The Steering Committee may decide to follow an accelerated procedure for new topics, when necessary. To this end, the Steering Committee adopted the Streamlined Procedure27 (final version dated October 21, 2002) in Washington in September 2002.
Decisions by the Steering Committee and consensus by EWG
In principle, the agreement of all ICH Parties is necessary for initiating a formal ICH procedure. In such circumstances, the rules to take decisions as outlined above apply.
However, in the exceptional cases where the ICH Regulatory Parties have adopted a Concept Paper without the support of all ICH Industry Parties, ICH Industry Parties which have not supported the adoption of the concept paper may decide whether they nominate experts to the EWG.
Step 2 for Testing (Optional)
Step 2 for Testing is an optional ICH step during which ICH is testing the proposed implementation guide/standard/specification against the ICH requirements (e.g. business, technical, system and functional requirements) to confirm technical adequacy. ICH will publish a consensus document on its website for review of the proposed implementation guide/standard/specification by the public. The document will be published in English only and not required to be translated into Japanese. The primary focus, however, is ICH testing but comments will be considered from external parties. Step 2 for Testing may be repeated if considered necessary. The duration of Step 2 for Testing is flexible and may be set based upon the timescale allowed by the project of concern (e.g. SDO ballot timelines or target for ICH Step 2). Step 2 for Testing is particular relevant to an EWG that develops an ICH Implementation Guide as part of an SDO project where the ICH step process is aligned with SDO processes. Step 2 for Testing is conducted to assess technical feasibility of proposed SDO solutions prior to ICH Step 2 because there is a greater ability to influence the degree of modification of technical solutions at this stage of development rather than at later stages. Step 2 for Testing is distinguished from general feasibility testing in the sense that feasibility testing can be conducted at any time during the development of a technical standard in an informal way.
3.2) Q&A Procedure by Implementation Working Group (Category 2)28
Implementation activities should be undertaken when additional guidance and advice are considered necessary to help the interpretation of certain ICH harmonised Guidelines in specific circumstances, in order to ensure a smooth and consistent implementation in the ICH regions and beyond and to allow that certain practical issues be overcome.
It is recommended that particularly in the case of major implementation activities, the Steering Committee (SC) consider whether a Business Plan is required.
In addition to problems identified during the Step 5 phase (regional implementation) some time after the finalisation of the Guidelines at the ICH level, there are also certain instances where additional work and discussions are already considered necessary right after the Step 4 Sign-off (e.g., the CTD and the e-CTD).
(1) Additional implementation guidance/advice in the form of Q&As
Additional implementation guidance/advice are usually developed in the form of Questions and Answers (“Q&As”). The development and adoption of these Q&As follow an established process described under section 3(ICH Process for each category).
(2) Procedure to review and clear Q&A documents
The Q&A process is intended to be a mechanism by which questions received from stakeholders 27 See Annex 10 28 See Annex 2
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 16
are collected, analysed, reformulated and ultimately used as model questions for which standard answers are developed and posted on the ICH website.
The incoming questions will not be answered individually. They will rather serve to highlight areas that need additional clarification and will be used to develop a model question that will be answered in the Q&A document.
Any question sent to the mailbox of the ICH website, or raised by any of the ICH Parties, and/or by the ICH Observer, will be brought to the attention of the appropriate IWG.
The regional questions and issues should first be handled by the Regulatory Party of the concerned region then shared and evaluated within the IWG, finally the proposed answer is presented to the SC for approval/endorsement before publication on the ICH website.
The IWG Rapporteur in collaboration with the Regulatory Chair will send the questions to the members of his IWG. Based on this information, the IWG will prepare model questions and their responses for presentation at the SC meeting. Based on the level of guidance given by the answers, the IWG will assess whether the Q&A document should be a Step 2b Document and published for comments or should be a Step 4 Document and published as final. The document should be Step 2b if, by the answers provided, it sets forth substantial new interpretations of the guideline(s). The document should be a Step 4 if, by the answers provided, it sets forth existing practices or minor changes in the interpretation or policy of the guideline(s).
Each IWG presents its draft Q&A document to the SC meeting (including regional regulatory legal review process) and makes recommendations to the SC on the status of the document (Step 2b or 4).29
The SC concurs with the Q&A document and its (Step) status.
The document will then follow the normal path of a Step 2b / Step 4 Document:
- Step 2b: upon agreement of technical content through sign-off by the experts of the ICH Parties as Step 1 and by the SC members of the ICH Parties as Step2a, the Regulatory SC members will sign the Q&A document (as Step 2b). The document will then be published for comments in the ICH regions and beyond.
- Step 4: the Regulatory experts will sign the Q&A document as Step 3 Final Document and then the Regulatory SC members will sign it as Step 4.
Each region should develop internal procedures to deal with the case of absence of the experts at the time of sign-off, which could include the possibility for the Steering Committee to sign-off the Step 2a / Step 3 on behalf of the IWG experts.
The Experts/Final Q&A document will be posted on the ICH website five working days after the SC meeting.
This procedure is intended to provide results quickly and efficiently using the minimum amount of resources consistent with the achievement of a scientifically valid result.
3.3) Revision Procedure (Category 3)
If an adopted guideline needs to be revised, then the formal ICH step procedure should take place rather than the Q&A process. Any revision or modification to an existing ICH Guideline should fall into the “Revision Procedure” category*.
There are two approaches for revision of an existing ICH Guideline under this category. The first approach involves amendments being made directly to the content of the existing guideline e.g., in cases where the scientific/technical content is no longer up-to-date or valid. The second approach is where the existing text in the original guideline is not modified, but instead an Addendum or
29 It is recommended that the IWG be represented while the Steering Committee is discussing the Q&A document to address the technical questions that may arise.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 17
Annex to that guideline is developed. The latter approach is used where no amendments to the content of the existing guideline are necessary but there is a need to provide further complementary guidance.
The format of the Addendum is to be recommended by the Expert Working Group for SC consideration and endorsement. The clarifying text can either be added at the end of the current ICH Guideline, or after the relevant paragraph(s) within the original guideline when the purpose of the Addendum is to clarify specific section(s) of the ICH Guideline. The later should be used to avoid many cross references and for an easier read of the guideline. The clarifying content added after specific sections of the guideline should be formatted in a specific way to facilitate its distinction from the original text by the reader.
The “Revision Procedure” is almost identical to the formal ICH procedure (Category 1), i.e., 5 ICH steps. The only difference compared to Category 1 is the final outcome: in Category 3, the final outcome will be a revised version of a current existing guideline, whereas in Category 1 the final outcome is a new guideline.
In cases where an Addendum or Annex has been developed, upon reaching Step 4 the Addendum or Annex is added to the existing guideline resulting in a revised guideline.
The revision of a guideline is designated by the letter R1 after the usual denomination of the guideline. When a guideline is revised more than once, the document will be named R2, R3, R4, (etc.) at each new revision.30
In some cases, during the process of creation of a Q&A document, the revision of the original guideline may be considered necessary. In this case, the IWG Regulatory Chair and the Rapporteur, when presenting the Concept Paper to the Steering Committee, may also request the setting-up of an EWG to discuss the document modifications. To increase efficiency, the same members as those forming the IWG may develop both the Q&A document and revise the ICH Guideline.
In the case of Q4B, topic-specific Annexes are developed to provide information on how pharmacopoeial texts can be used at a national/regional level. Each Annex is issued as a stand-alone companion document to the Q4B guideline, with each Annex assigned a number in sequential order e.g., Annex 1, Annex 2, Annex 3 etc…
3.4) Maintenance Procedure (Category 4)
This procedure specifically applies to Q3C residual solvents 31 , Q4B Annexes 32 and M2 Recommendations.
M2 Recommendations constitute an exceptional case, because no Step 2b Document is required. However, the Steering Committee may request further clarification. In such cases, a Step 2b document may be necessary.
Updates to the Q3C Guideline (Parent Guideline or Addenda) and the Q4B Annexes are considered as revisions and are designated by the letter R.
Each new version of the M2 Recommendations is designated by a different version number.
The Maintenance Procedure also extends to any ICH Guideline which contains out-of-date information (e.g., out-of-date references, links etc…) which can be updated by the ICH Secretariat without the establishment of an EWG. Such updates require SC approval and are also considered revisions and assigned the letter R.
30 A history box on the cover page details all the changes made to the guideline. 31 See Annex 4 32 See Annex 5
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 18
4. ADDITIONAL ACTIVITIES DURING THE COURSE OF ICH HARMONISATION
ACTIVITIES33
During the course of the ICH harmonisation activities outlined above, the Steering Committee may authorize the EWGs/IWGs to carry out other tasks intended to provide additional information complementary to a topic that is undergoing one of the above categories of harmonisation.
Such tasks may include the development of an Options Paper or Points to Consider Document. In the case of groups (i.e., M2/M5/E2B(R3) EWGs) involved in working towards enabling the transfer of regulatory information by electronic means, it may be necessary to conduct a Proof of Concept (POC). In the case of M2/M5/E2B(R3), the POC concerns testing the viability of using M2’s message specifications to exchange information.
5. OTHER ACTIVITIES
Some ICH activities cannot fit in the above-mentioned four Procedures. As of May 2005, 2 activities are considered under this category, i.e., Error Correction and Considerations.
1) Error Correction
The ICH Secretariat may correct obvious typographical errors. In this case, no approval from the Steering Committee is required.
In some cases where more substantial corrections are needed (e.g., editorial mistakes, errors/inaccuracies), a technical expert discussion may be necessary. This case would therefore undergo the Revision Procedure (Category 3).
Editorial mistakes (i.e., changes in the wording, the grammar in order to keep with consistency and clarity) and errors/inaccuracies (i.e., wrong meaning needing correction) are therefore corrected by the EWG and require approval by the Steering Committee.
2) Considerations
Under this section enter specific considerations or views expressed by a scientific discussion group: e.g., Gene Therapy and the ICH and Women. Although these topics have not been adopted as formal ICH topics, ICH supports them and the outcome of their discussions. These are posted as “Considerations” on the ICH website.
6. GUIDELINE WITHDRAWAL
Under exceptional circumstances an ICH Guideline may be withdrawn (e.g., Q1F). Such action requires substantial justification and endorsement by the Steering Committee.
33 See Glossary for an expanded definition of the terms discussed in this section.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 19
Annex 1: Expert Working Group (EWG)
ICH Parties nominate representatives on EWGs (Expert Working Groups). Note that Health Canada and Swissmedic are not required to appoint technical experts in all EWGs. Unless otherwise specified by the Steering Committee (SC), the Membership of EWG/IWG shall be limited to two representatives per ICH Party per Working Group (one expert shall be designated as Topic Leader and the other as Deputy Topic Leader), and one representative per ICH Observer, and also if applicable one per Interested Party (World Self-Medication Industry - WSMI, International Generic Pharmaceutical Alliance - IGPA, and other Interested Parties as determined by the SC). The ICH Observer and Interested Parties may nominate an alternate member to the Working Group (WG) whom shall be added on the email list. If the representative for the Observer or Interested Parties is unable to participate to a tele/webconference or a face-to-face meeting, the alternate may replace the representative at the tele/webconferences or face-to-face meetings.
ICH does not cover the cost of travel or accommodation for EWG participants. Participation is at the expense of the Party or Observer concerned.
The Topic Leaders/Deputy Topic Leaders will participate in the EWG discussions and be the point of contact for any consultation carried out between meetings by correspondence, fax, e-mail etc. It is the responsibility of the Topic leader/Deputy Topic Leader to officially represent a consolidated view from their Party, during any ICH interactions (e-mails, tele/webconferences and face-to-face meetings).
The ICH Regulatory Parties officially designate a Regulatory Chair from the ICH Regulatory Parties. Also the SC officially designates a Rapporteur among the Topic Leaders designated by the ICH Parties, when a new ICH Topic is formally adopted. In some cases, both a Rapporteur and a Co-Rapporteur may be appointed. Whenever possible, Co-Rapporteurs should be from different regions and should not both be from a Regulatory or Industry Party.
In order for a tele/webconference or face-to-face meeting to be considered official, all ICH Parties shall be represented, at least by one delegate. However, the absence of one of the Observer or Interested Parties from an EWG meeting will not prevent the meeting from taking place and decision to be made.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress including the ICH step process34.
Where appropriate, additional experts may contribute to the discussion but the official voice of each delegation rests with Topic Leader and his Deputy. ICH Regulatory Parties may nominate additional personnel (e.g. "MHLW Officials" for MHLW/PMDA) to the EWG mailing list for information only.
In the case that external expertise may be helpful, and subject to SC approval, the EWG/IWG may consider inviting one or two liaisons from an entity outside of ICH to participate in EWG/IWG to facilitate communication between ICH and the entity. The level of participation would be decided by the SC (e.g., tele/webconference, emails, and face-to-face meetings).
For logistical purposes, it is essential that in preparation for any official face-to-face meeting, each ICH Party communicate the names of its representatives to the ICH Secretariat, and that the host Association is informed of each Party delegation well in advance of the meetings. The ICH Secretariat shall keep records of experts’ nominations.
N.B.: Any face-to-face meetings of EWGs will be subject to decision by the SC. EWGs shall not systematically meet in conjunction with every SC if not justified.
34 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 20
With a view to keeping down organisational and logistical costs of the ICH Process, EWGs should meet face-to-face only when necessary and justified and when sufficient discussion materials are available.
Interim face-to-face meetings (i.e., EWG meetings outside the regular ICH SC weeks) should be exceptional, and only when there is an absolute necessity in order for the topic to meet its assigned objectives in time (see Annex 9).
ICH EWGs are encouraged to make better use of e-mail communication to progress draft Guidelines between face-to-face meetings and tele/webconferences.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 21
Annex 2: Implementation Working Group (IWG)
Similarly to EWGs, ICH Parties nominate representatives on IWGs (Implementation Working Groups). Note that Health Canada and Swimedic are not required to appoint technical experts in all IWGs. Unless otherwise specified by the Steering Committee (SC) the official membership of an IWG shall be limited to two representatives per party per working group (one expert shall be designated as Topic Leader and the other as Deputy Topic Leader), and one representative per ICH Observer, and also if applicable one per Interested Party (World Self-Medication Industry - WSMI, International Generic Pharmaceutical Alliance - IGPA, and other Interested Parties as determined by the SC). The ICH Observer and Interested Parties may nominate an alternate member to the Working Group (WG) whom shall be added on the email list. If the representative for the Observer or Interested Parties is unable to participate to a tele/webconference or a face-to-face meeting, the alternate may replace the representative at the tele/webconferences or face-to-face meetings.
ICH does not cover the cost of travel or accommodation for IWG participants. Participation is at the expense of the party or observer concerned.
The Topic Leaders/Deputy Topic Leaders will participate in the IWG discussions and be the point of contact for any consultation carried out between meetings by correspondence, fax, e-mail etc. It is the responsibility of the Topic leader/Deputy Topic Leader to officially represent a consolidated view from their party, during any ICH interactions (e-mails, tele/webconferences and face-to-face meetings).
The ICH Regulatory Parties officially designate a Regulatory Chair from the ICH Regulatory Parties. Also the Steering Committee officially designates a Rapporteur among the Topic Leaders designated by the ICH Parties, when a new ICH Topic is formally adopted. In some cases, both a Rapporteur and a Co-Rapporteur may be appointed. Whenever possible, Co-Rapporteurs should be from different regions and should not both be from a Regulatory or Industry Party.
In order for a tele/webconference or face-to-face meeting to be considered official, all ICH Parties shall be represented, at least by one delegate. However, the absence of one of the Observer or Interested Parties from an IWG meeting will not prevent the meeting from taking place.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress including the ICH step process35.
Where appropriate, additional experts may contribute to the discussion but the official voice of each delegation rests with the two representatives. ICH Regulatory Parties may nominate additional personnel (e.g. "MHLW Officials" for MHLW/PMDA) to the EWG mailing list for information only.
In the case that external expertise may be helpful, and subject to SC approval, the EWG/IWG may consider inviting one or two liaisons from an entity outside of ICH to participate in EWG/IWG to facilitate communication between ICH and the entity. The level of participation would be decided by the SC (e.g., tele/webconference, emails, face-to-face meetings).
For logistical purposes, it is essential that in preparation for any official face-to-face meeting, each party communicate the names of its representatives to the ICH Secretariat, and that the host Association is informed of each party delegation well in advance of the meetings. The ICH Secretariat shall keep records of experts’ nominations.
N.B.: Any face-to-face meetings of IWGs will be subject to decision by the SC.
35 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 22
IWGs shall not systematically meet in conjunction with every SC if not justified.
With a view to keeping down organisational and logistical costs of the ICH Process, IWGs should meet face-to-face only when necessary and justified and when sufficient discussion materials are available.
Interim face-to-face meetings (i.e., IWG meetings outside the regular ICH SC weeks) should be exceptional, and only when there is an absolute necessity in order for the topic to meet its assigned objectives in time (see Annex 9).
ICH IWGs are also encouraged to make better use of e-mail communication to progress draft Guidelines between face-to-face meetings and tele/webconferences.
In the case of CTD, a close link between CTD-Quality/ Safety/ Efficacy and electronic-CTD is mandatory. In such a case, an Implementation Coordination Group (ICG) may be established to oversee the work of each IWG.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 23
Annex 3: Informal Working Group
An informal Working Group is frequently formed prior to an official ICH harmonisation activity with the objectives of developing/finalising a Concept Paper, as well as developing a Business Plan. As a principle, informal Working Groups should work by e-mail and tele/webconference and should not need to meet face-to-face. ICH Parties nominate representatives on informal Working Groups. Unless otherwise specified by the Steering Committee (SC), the official membership of an informal Working Group shall be limited to two representatives per ICH Party per working group (one expert shall be designated as Topic Leader and the other as Deputy Topic Leader), and one representative per ICH Observer, and also if applicable one per Interested Party.
The Topic Leaders/Deputy Topic Leaders will participate in the informal Working Group discussions and be the point of contact for any consultation carried out between meetings by correspondence, fax, e-mail etc. It is the responsibility of the Topic Leader/Deputy Topic Leader to officially represent a consolidated view from their Party, during any ICH interactions (e-mails and tele/webconferences).
An expert from the Party responsible for proposing the topic originally shall be nominated Group Leader and will lead the efforts of the informal Working Group. In some cases, both a Group Leader and a Group Co-Leader may be appointed, particularly in cases where the topic proposed for harmonisation originated from more than one ICH Party. Whenever possible, Co-Leaders should be from different regions and should not both be from a Regulatory or Industry Party.
Additional representatives may contribute to the discussion but the official voice of each delegation rests with the two representatives. No upper limit of representatives is set per party but names should be submitted to the ICH Secretariat. The ICH Secretariat shall keep records of experts’ nominations.
The entire membership of an informal Working Group shall be copied on e-mails and invited to participate in tele/webconferences.
For a tele/webconference to be considered official, all ICH Parties need to be represented, at least by one delegate. However, the absence of one of the Observer or Interested Parties from an informal Working Group meeting will not prevent the meeting from taking place. If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress including the ICH step process36.
In general the working practices as outlined in Annex 6 (Principles for Increasing the Efficiency of EWG/IWG Working Practices) may be applied, where appropriate, to an informal Working Group.
36 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 24
Annex 4: Q3C Maintenance Procedure
There are data/proposals related to Q3C that are submitted directly to the ICH Secretariat, with supporting information either through an ICH regional Coordinator or an RHI and DRA/DoH representative. A classic example is a proposal of a Permitted Daily Exposure (PDE) for a new solvent or a revised PDE for an already classified solvent.
This information should be based on significant toxicity data from studies such as genotoxicity studies, repeat-dose studies, reproductive toxicity studies, carcinogenicity studies and/or other relevant toxicology studies. Single-dose toxicity data alone are not sufficient. The toxicity data should be of sufficient quality to calculate a PDE.
The ICH Secretariat will initially share the proposal with the Regulatory Topic Leader who will be one of the regulatory members of the ICH available for two-year terms e.g., FDA (2017-2018, 2027-2028 etc…), MHLW (2023-2024, 2033-2034 etc…), EU (2015-2016, 2025-2026 etc), Health Canada (2019-2020, 2029-2030 etc…) and Swissmedic (2021-2022; 2031-2032 etc…).
If the proposal for maintenance is supported by the regulatory Topic Leader, the ICH Secretariat will subsequently notify the ICH Steering Committee (SC), Coordinators, and the ICH Observer that the ad hoc Q3C Expert Working Group (EWG) should be re-established to consider a proposal/Concept Paper for Q3C maintenance.
The Q3C EWG will count up to two members (one chemist and one toxicologist) nominated by EU; MHLW; FDA; Health Canada; Swissmedic; EFPIA; JPMA; and PHRMA, ,one member nominated by the Interested Parties (WSMI, IGPA and other Interested Parties as determined by the ICH Steering Committee) one per pharmacopoeia and one per RHIs/DRAs/DoH, if requested. As appropriate, the ICH Observer (WHO) may be invited to join the working group.
The revision of an established PDE will be considered only on presentation of previously unrecognised toxicity data sufficient to result in a significant change, or because of convincing evidence that the existing data used to calculate a PDE are invalid. Minor changes in a PDE will not be considered. The Regulatory Topic Leader, with the consensus of the EWG members, will assign data reviews and request subsequent recommendations to the EWG.
The Regulatory Topic Leader will ordinarily rely on correspondence or teleconferencing to avoid unnecessary travel. Based on the discussion, with requests for further information to the proposing group and/or individual as appropriate, the Regulatory Topic Leader will prepare an assessment report based on EWG’s approval with a recommendation to accept, with or without modifications, or reject the proposed PDE. Ideally, this activity would occur at the rate of 2 residual solvents per calendar year. For particular residual solvents, it is anticipated that a period of six months from receipt of the toxicological information by the Regulatory Topic Leader to the recommendation of a Step2a Technical Document to the SC will be necessary.
After endorsement by the SC, either at the next formal meeting or earlier if feasible, the recommendation of the Q3C EWG will be published in each region for public comment (Step 3 of the ICH process). In addition, the proposal will be provided to each pharmacopoeia for their publication.
After closure of the public comment period, the Regulatory Topic Leader may convene a meeting of the Q3C EWG or will rely on correspondence or teleconferencing to consider the comments and finalise the proposal for the new/revised PDE. The final recommendation for the new/revised PDE and implementation is then forwarded to the SC for approval. Implementation will follow regional practices. With approval of the ICH SC, the change will be provided to the pharmacopoeias at
In Brussels in February 2002, the SC has harmonised this procedure.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 25
regional/national level for publication.
When an existing PDE is revised or a PDE for a new residual solvent is recommended by the EWG, approval by the ICH Steering Committee is required. Once approval occurs, the information should be disseminated as quickly as possible to all ICH participants and other members of the chemical and pharmaceutical communities. It is recommended that the following actions should be taken by the SC to ensure rapid transmission of the new information:
Publish relevant information on the ICH website; Request publication of revisions by the pharmacopoeias of the ICH regions in their Forums
or websites; Request that each member publish the new solvent PDE information on its respective
websites.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 26
Annex 5: Q4B Annex Maintenance Procedure
The ICH Q4B Guideline Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions reached Step 5 in November 2007. Subsequently, the individual topic-specific Annexes reached Step 5 in accordance with the dates listed on the ICH website. Because the inputs to the Q4B process were from the Pharmacopoeial Discussion Group (PDG) harmonisation process, it is recognised that the pharmacopoeial texts could be updated as technology and requirements change, or for other reasons. Because changes to the pharmacopoeial texts could have an impact on the interchangeability assessment contained in the Annexes, it is necessary to have a maintenance procedure for updating the Annexes when needed. The Pharmacopoeias (e.g., JP/Ph.Eur./USP) publish updates to the status of chapters in the PDG harmonisation work programme. Because of the potential impact of these chapters, the status of the work programme is regularly monitored by interested stakeholders, including industry. If PDG or any of the pharmacopoeias make revisions to any chapter that is the subject of an ICH Q4B Annex an assessment of the change(s) should be conducted by interested stakeholders, to determine whether a revision to the Annex may be necessary. As a result of this assessment, a recommendation from any stakeholder, including regulators, industry, or PDG, to revise the Annex will be communicated to ICH (for example through the ICH website), so that all ICH Parties constituent are alerted. Following consideration by the ICH Parties and with the ICH Steering Committee (SC) approval, an ad-hoc group including at least one representative from ICH Parties and Observer also invited, will formally review the revision proposal and, if necessary, make a recommendation to revise the Annex. The evaluation and revision work will be completed electronically through use of email and web-based technology. Any Annex revision would follow the appropriate ICH process.
Approved by the SC in Fukuoka on November 10, 2010.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 27
Annex 6: Principles for increasing the efficiency of EWG/IWG Working Practices
This annex is included to help increase the efficiency of the ICH processes. It is acknowledged that this annex duplicated text in other parts of this procedures document, however, such duplication is intentional as this Annex should be given to new Regulatory Chairs, Rapporteurs, Co-Rapporteurs, Topic Leaders and Deputy Topic Leaders as a short guide.
I. Role of the ICH Steering Committee in the selection of ICH topics and supervision of projects
It is essential that every ICH project be supervised by the Steering Committee (SC), in order to make sure that it is properly managed.
The SC decides on the adoption of every ICH project (whether a new topic, or maintenance of an existing guideline, or a specific implementation work). The SC also endorses the creation of EWG/IWG (Expert Working Group/ Implementation Working Group) when these are deemed necessary.
Any ICH Party or ICH Observer (WHO) is welcomed to submit a proposal for a new ICH activity to the SC. For this purpose it shall develop a Concept Paper and share it with the other ICH Parties and the ICH Observer well in advance of an ICH SC meeting. The Concept Paper should discuss on the following aspects:
Type of Harmonisation Action Proposed (e.g., a new harmonised guideline / recommendation, or a revision of an existing guideline – indicating the category of procedure)
Statement of the Perceived Problem: Brief description with an indication of the magnitude of the problems currently caused by a lack of harmonisation, or - in the case of new scientific developments - anticipated if harmonisation action is not taken.
Issues to be Resolved: A summary of the main technical and scientific issues, which require harmonisation.
Background to the Proposal: Further relevant information, e.g., the origin of the proposal, references to publications, discussions in other fora.
Type of Expert Working Group: Recommendation on whether the EWG37 (if needed) should be an extended EWG - for topics with implications beyond new drug research.
If ICH Parties agree to the topic that is being proposed for harmonisation, the SC may endorse the establishment of an informal Working Group to develop a Concept Paper. The party responsible for proposing the topic originally will normally lead the informal Working Group in developing the Consensus Concept Paper. The SC may also request the development of a Business Plan.
The Business Plan should include among others the issue expected to be tackled, required resources, time frame, likely health, social and financial benefits and how the results will be evaluated. The Business Plan shall particularly focus on the regulatory feasibility, and would complement the Concept Paper, which focuses on the issue and scope. It is essential that the Business Plan set out a detailed action plan and a timetable with clear deliverables and deadlines, and include concrete milestones (scientific, technical and regulatory). The time frame for the delivery of this document will have to be endorsed by the SC.
Before formally adopting any new ICH project or topic, the final Concept Paper, and (where requested) the Business Plan must be endorsed by the SC.
Each ICH Party should be in a position to present a consolidated opinion on any new ICH
Approval by the SC in Yokohama on 18 November 2004. 37 See Annex 1
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 28
project during the SC discussion. For this purpose, SC members from each individual Party shall make sure that the proposal has been reviewed internally by appropriate scientific, technical, regulatory (and for the authorities, legal) committee(s), well in advance of an ICH SC meeting.
In principle, the agreement of the ICH Parties is necessary for initiating any ICH harmonisation activities. However, as an exceptional case when ICH Party consensus cannot be achieved, the three ICH Founding Regulatory Parties can jointly decide to adopt a Concept Paper considering that Regulators have the ultimate responsibility to ensure the protection of public health and have the responsibility for issuing regulatory guidelines.
The SC shall control the progress of work on any specific ICH project, and in particular adherence to the timelines and milestones. For this purpose, each ICH Party should be in a position to have the documents, which comprise the milestones, properly reviewed by their relevant internal committee(s) well in advance of a SC meeting. (N.B. In other word, Working Groups must give feedback sufficiently in advance of a SC meeting to allow each ICH party to consult their relevant committee(s) and come well prepared and advised for the SC discussion.)
The SC shall systematically decide to suspend a topic or even completely stop it if the regulatory environment or the technical issues are such that a useful guideline cannot be finalised, or cannot be completed in a reasonable period of time.
II. Expert Working Groups (EWGs) and Implementation Working Groups (IWGs)
1. Membership:
ICH Parties nominate representatives on EWGs (Expert Working Groups) / IWGs (Implementation Working Groups). Note that Health Canada and Swimedic are not required to appoint technical experts in all IWGs. Unless otherwise specified by the SC, the Membership of EWG/IWG shall be limited to two representatives per ICH Party per Working Group (one expert shall be designated as Topic Leader and the other as Deputy Topic Leader), and one representative for the ICH Observer, and also if applicable one for Interested Parties (World Self-Medication Industry - WSMI, International Generic Pharmaceutical Alliance - IGPA, and other interested parties as determined by the SC). The ICH Observer and Interested Parties may nominate an alternate member to the Working Group (WG) whom shall be added on the email list. If the representative for the Observer or Interested Parties is unable to participate to a tele/webconference or a face-to-face meeting, the alternate may replace the representative at the tele/webconferences or face-to-face meetings. ICH does not cover the cost of travel or accommodation for EWG/IWG participants.
Participation is at the expense of the party concerned.
Where appropriate, additional experts may contribute to the discussions but the official voice of each delegation rests with the Topic Leader and his Deputy. For logistical purposes, it is essential that in preparation for any official face-to-face meeting, each party communicate the names of its representatives to the ICH Secretariat, and that the host Association is informed of each Party delegation well in advance of the meetings. The ICH Secretariat shall keep records of experts’ nominations.
2. EWGs/IWGs Working Rules
2.1 Designation of the EWG/IWG Regulatory Chair
The regulatory members of the SC officially designate a Regulatory Chair when a new ICH Topic is formally adopted. The Regulatory Chair will be one of the representatives of the ICH Regulatory Parties.
2.2 Role and Responsibilities of the Regulatory Chair
The role of the Regulatory Chair is to ensure timely execution of the ICH process and adherence to
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 29
the Concept Paper and Business Plan, including scope and timelines. The Regulatory Chair shall work in close collaboration with the Rapporteur.
Responsibilities of the Regulatory Chair include:
1. Ensuring timeframes are met and work is within the scope of the EWG/IWG mandate. 2. Collaborating with the Rapporteur in developing a work plan that is consistent with the
scope and time frame of the SC-approved Concept Paper and Business Plan. 3. Regularly reporting to SC on progress of the EWG/IWG regarding timeliness, adherence to
scope and conflicting views if they arise and ensuring that all expert perspectives are reflected in the documents presented to the SC.
4. If conflict arises, working with the Rapporteur to achieve consensus within the EWG/IWG by reconciling divergent views; if the Regulatory Chair and the Rapporteur fail to achieve consensus, the Regulatory Chair will elevate the issue to the SC for resolution as early as possible.
5. Addressing the behaviour of any expert within the EWG/IWG that is disruptive or is not constructive, in consultation with the Regulatory Chair’s Coordinator.
6. Deciding when it is necessary to document significant differences of position or conflicting views among members of the EWG/IWG and will work on this task with the assistance of the Rapporteur.
The SC reserves the right to replace a Regulatory Chair, when it considers that this is necessary for the topic to progress according to the plan.
2.3 Designation of the EWG/IWG Rapporteur
When a new ICH Topic is formally adopted, the SC appoints the Topic Rapporteur among the Topic Leaders designated by the ICH Parties. (N.B. In exceptional cases, a Co-Rapporteur may also be appointed to assist the Rapporteur.) Whenever possible, Co-Rapporteurs should be from different regions and should not both be from a Regulatory or Industry Party. If the Rapporteur is a representative from one of the Industry Parties, the rapporteurship will then have to be transferred to a Regulatory Party (usually from the same region) after Step 2b is reached. When the rapporteurship is to be taken by a Regulatory Party, the Regulatory Chair may also be appointed as the Rapporteur as an option of the Regulatory Parties which is taking the role of the Regulatory Chair and upon agreement of the SC.
2.4. Role and Responsibilities of the EWG/IWG Rapporteur
The role of the Rapporteur is to serve as the scientific co-chair, to facilitate and manage scientific and technical activities of the EWG/IWG, reconciling scientific differences of opinion, in order to produce an ICH document with the scientific and technical content that is in accordance with SC decisions/expectations. The Rapporteur shall work in close collaboration with the Regulatory Chair.
Responsibilities of the Rapporteur include:
1. Development of a detailed work plan, in collaboration with the Regulatory Chair that will achieve the technical objectives outlined in the SC-approved Concept Paper and Business Plan and contains clear technical deliverables and associated deadlines; the updated work plan, approved by the whole EWG, shall be provided ahead of Coordinator/SC webconference for SC consideration.
2. Responsible for day-to-day management including setting deadlines, assigning work to the members of the EWG/IWG and assuring all ICH Parties’ views are incorporated into documents and presentations as appropriate.
3. The Rapporteur shall seek to reconcile scientific and technical differences among
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 30
EWG/IWG members.
4. The Rapporteur shall make sure that the views of the different parties are reflected in an appropriate and fair manner in any outcomes of the EWG/IWG work.
5. The Rapporteur shall regularly present reports to the SC, on the technical and scientific aspects of the document under development.
6. Upon reaching Step 2a or Step 4, the Rapporteur shall ensure the development of a presentation for review by EWG/IWG members, and provision to the ICH Secretariat to be included in a library of presentations and training materials made available on the ICH public website.
The SC reserves the right to replace a Rapporteur, when it considers that this is necessary for the topic to progress according to the plan.
2.5 Responsibilities of the EWG/IWG Members (Topic Leaders and Deputy Topic Leaders)
1. The Topic Leaders/Deputy Topic Leaders will participate in the EWG/IWG discussions and be the point of contact for any consultations carried out between meetings by correspondence, fax, e-mail etc.
2. The entire membership EWG/IWG shall be invited to take part in the regular tele/webconferences or face-to-face meetings.
3. It is the responsibility of the Topic Leader/Deputy Topic Leader to officially represent a consolidated view from the party they represent, during any ICH interactions (e-mails, tele/webconferences and face-to-face meetings). Each party/delegation is therefore expected to have had appropriate preparatory discussion prior to the ICH interaction.
4. The EWG/IWG members shall provide their technical contribution in a constructive manner to the EWG/IWG discussions. They are to play an active role in the consensus building process and should refrain from adopting disruptive behaviour.
5. For the sake of fairness, each Party/Observer should have an equal opportunity to provide their views during tele/webconferences, videoconferences and face-to-face meetings, irrespective of the size of their delegation.
6. At the end of the Step 3 phase (regional consultation), the comments received by each of the ICH Regulatory Parties shall be consolidated by respective regulatory EWG/IWG members and provided to the Rapporteur for distribution to the members of the EWG/IWG.
2.6 Interactions between EWG/IWG Members
EWGs/IWGs are encouraged to make use of modern communication technologies for their interactions, in addition to the usual face-to-face meetings (i.e., e-mails, web-conferencing systems, regular teleconferences or videoconferences, etc.).
In order for a tele/webconference or face-to-face meeting to be considered official, ICH Parties need to be represented, at least by one delegate. If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress including the ICH step process38.
Any face-to-face meetings of EWGs/IWGs will be conditional on a decision by the ICH SC).
N.B.: With a view to keeping down organisational and logistical costs of the ICH Process,
EWGs/IWGs should meet face-to-face only when necessary and justified and when sufficient discussion materials are available.
38 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 31
Interim face-to-face meetings (i.e., IWG/EWG meetings outside the regular ICH SC weeks) should be exceptional, and only when there is an absolute necessity in order for the topic to meet its assigned objectives in time (see Annex 9).
3. Consensus Building and Decision Making Process
N.B. The ICH stepwise process is currently described on the ICH website. The ICH Guidelines are developed through a five-step procedure, which is based on a consensus-building exercise. Steps 1, 2a/b, 3 and 4 constitute the decision-making steps.
Step1/Step 2a/Step2b consensus: At Step 1, designated experts of the ICH Parties are asked to confirm their agreement with the technical document by formal sign-off, which is then officially endorsed through sign-off by the ICH SC members from the ICH Parties at Step 2a and by the Regulatory SC members at Step2b.
Experts from the ICH Observer (WHO), and where appropriate from Interested Parties, who have participated in an EWG/IWG, may also wish to add their sign-off on a voluntary basis, with a view to confirming their support to the guideline. However, the absence of signature from the Observer or Interested Parties will never lead to a suspension of the guideline.
As per SC decision in November 201439, experts of Health Canada and Swissmedic remain invited to sign-off the Step 1 Technical Document as ICH Observers for Working Groups established prior to June 2014.
For Working Group(s) established after June 2014, Irrespective of whether or not Health Canada and Swissmedic will have appointed technical experts in the Working Group, the SC Members of Health Canada and Swissmedic will be invited to sign-off the Step 2a / 2b / 4 as ICH Parties.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress.
In the unlikely situation where consensus cannot be reached but the three ICH SC Founding Regulatory Parties (EU, FDA and MHLW) are in agreement, they will proceed with the Step sign-off.
Step3/Step 4 consensus: At the end of the Step 3 phase (regional consultation phase), the regional comments received by each of the Regulatory ICH Parties are reviewed by the all members of the EWG/IWG. When the EWG/IWG reach consensus on the final text of the guideline, the ICH Regulatory Parties confirm their agreement by formal Step 3 Sign-off, which is then officially endorsed through Step 4 sign-off by the SC members from the ICH Regulatory Parties.
As per SC decision in November 201440, for Working Groups established prior to June 2014 experts of Health Canada and Swissmedic remain invited to sign-off the Step 3 Experts Draft Guideline as ICH Observers.
For Working Group(s) established after June 2014, and selected by the SC Members of Health Canada and Swissmedic, their expert(s) will be invited to sign-off the Step 3 Experts Draft Guideline as ICH Parties.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working Group meeting, the work of the group can still progress. It is nonetheless the expectation that all permanent Members will attend all face-to-face meetings. Health Canada and Swissmedic commit to support the progress made by the group.
In the unlikely situation where consensus cannot be reached but the three ICH SC Founding
39 See Annex 20
40 See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 32
Regulatory Parties (EU, FDA and MHLW) are in agreement, they will proceed with the Step sign-off.
N.B. In the case of a Q&A document developed in the context of an implementation activity, the IWG will assess, based on the level of guidance given by the Q&As, whether the document should follow a complete ICH stepwise procedure and be published for comments as a Step 2b Q&A document, or whether it can be progressed directly to Step 4 of the process and published as a final Q&A document.
4. Additional Points for Consideration by Rapporteurs and EWG/IWG Members
Presentations on ICH Topics
Presentations made formally by Regulatory Chairs, Rapporteurs or EWG/IWG experts officially on behalf of ICH (e.g., GC webconferences etc…) should be prepared using the ICH slide template (available from the ICH Secretariat) and the presentation should be reviewed and agreed by all EWG/IWG members. Where a Regulatory Chair, Rapporteur or EWG/IWG expert has been invited to present on an ICH topic, but not in relation to any event formally supported by ICH, the ICH slide template should not be used. In such cases, it should be clear that the Regulatory Chair, Rapporteur or EWG/IWG expert is providing their own (or their party/organisation/company) views based on their experience participating in the ICH process; however consideration should still be given as a courtesy to inviting fellow EWG/IWG members to review the presentation.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 33
Annex 7: ICH Business Plan
Introduction
A Business Plan may be requested by the ICH Steering Committee.
Concept papers will be developed and proposed by one or several ICH Parties (including WHO – ICH Observer) for initial consideration by the ICH Steering Committee (SC). At this initial stage a Business Plan will not be required although one may be provided at the discretion of the proposing Party(ies). If the SC agrees that a topic may warrant further consideration and a Business Plan has not been provided and agreed, an informal EWG / IWG will be formed and the informal Working Group will work through e-mail, tele/webconference and, exceptionally, face-to-face meetings. The first tasks of the informal EWG / IWG will be to finalise a Concept Paper and develop and agree a Business Plan. The revised Concept Paper and Business Plan will be sent prior to and presented to the next SC tele/webconference or face-to-face meeting.
Agreed template for ICH Business Plan 1. The issue and its costs
What problem/issue is the proposal expected to tackle?
What are the costs (social/health and financial) to our stakeholders associated with the current situation or associated with “non action”?
2. Planning
What are the main deliverables?
What resources (financial and human) would be required?
What is the time frame of the project?
What will be the key milestones?
3. The impacts of the project
What are the likely benefits (social, health and financial) to our key stakeholders of the fulfilment of the objective?
What are the regulatory implications of the proposed work – is the topic feasible (implementable) from a regulatory standpoint?
4. Post-hoc evaluation
How and when will the results of the work be evaluated?
Approval by the SC in Washington DC in June 2004 and revised in Yokohama in November 2004. Revision consists in renaming the “Topic Justification and Project Plans” to Business Plan.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 34
Annex 8: Role and Responsibilities of the Steering Committee, the Coordinators, the Technical Coordinators and the ICH Observer
ICH Steering Committee – Role and Responsibilities
BACKGROUND & DEFINITION:
The ICH Steering Committee was established in April 1990, when the ICH was initiated. The ICH Steering Committee (SC) is defined as the body that governs the ICH. ICH Parties have up to two seats on the SC. Since the outset of ICH, the World Health Organization (WHO) has participated as an Observer to the SC. IFPMA also participates as a non-voting member.
STEERING COMMITTEE MEMBERSHIP41
Voting Regulatory ICH Parties
Japan
o MHLW/PMDA
Europe
o EU/EMA
United States of America
o CDER/CBER FDA
Canada
o Health Products and Food Branch
Switzerland
o Swissmedic
Voting Industry ICH Parties
Japan
o JPMA
Europe
o EFPIA
United States of America
o PhRMA
ICH Observer
o WHO
Non-Voting Member
o IFPMA
41 In June 2014, in Minneapolis, Health Canada and Swissmedic were promoted Steering Committee members.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 35
TASKS:
1. Governance of ICH
The SC is responsible for the governance of ICH. This includes deciding on the adoption of every ICH project, whether a new topic, maintenance of an existing guideline, or a specific implementation work.
The SC is responsible for determining all policies and procedures of ICH. This includes the endorsement of the ICH Procedures document which is reviewed annually and recommended to the SC by ICH Coordinators for endorsement at the autumn meeting each year.
The SC meets usually twice a year with the location rotating between the ICH regions. SC meetings are chaired by the Regulatory ICH Party of the region where the meeting takes place. In order for a face-to-face meeting to be considered official, all ICH Parties need to be represented, at least by one SC member. In the event that both SC members from one ICH Party are unable to participate in a face-to-face meeting, an alternate may be nominated to replace the official SC members at the face-to-face meeting.
Six to eight weeks prior to ICH SC meetings a tele/webconference is organised to prepare for the meeting. The SC should reach agreement on which Working Groups will meet at the face-to-face meeting, and raise any issues to be tabled at the meeting. Tele/webconferences are chaired by the regulatory ICH Party of the region where the next meeting will take place. In order for a tele/webconference to be considered official, all ICH Parties need to be represented, at least by one SC member. In the event that both SC members from one ICH Party are unable to participate in a tele/webconference, an alternate may be nominated to replace the official SC members at the tele/webconference. Similar principles are applied if an ad hoc SC tele/webconference needs to be organised.
The SC is invited by the ICH Secretariat to comment on the draft reports of all SC meetings and tele/webconferences, and is responsible for adopting them as final.
2. Selection of Topics for Harmonisation
Any ICH Party or the Observer is welcomed to submit a proposal for a new ICH activity (i.e., new topic, maintenance of an existing guideline, or a specific implementation work) to the SC in the form of a Concept Paper. The SC is responsible for reaching agreement on the need for harmonisation of the proposed topic, and may endorse the establishment of an informal Working Group to develop a Consensus Concept Paper, which would then be for SC approval. The SC may also request the development of a Business Plan.
As an exceptional case when ICH Party consensus cannot be achieved, the three Founding ICH Regulatory Parties can jointly decide to adopt a Concept Paper considering that Regulators have the ultimate responsibility to ensure the protection of public health to issue regulatory guidelines.
3. Establishment of ICH Working Groups
The SC is responsible for endorsing the establishment of all ICH Working Groups (e.g., informal Working Groups, EWGs, IWGs etc…) and confirming the Leadership/Regulatory Chairmanship/Rapporteurship and the invitation of Interested Parties to participate.
4. Monitoring Working Group Activities and Providing Guidance
Once an ICH Working Group has been established, the SC is responsible for endorsing the work plan of a Working Group and monitoring the progress of its harmonisation activities.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 36
The SC is responsible for providing guidance to Working Groups, both on an ongoing basis and as requested by Working Groups. Guidance requested by a Working Group between ICH SC meetings may necessitate the organisation of an ad hoc SC tele/webconference.
5. Signing-Off of ICH Documents42
The SC is responsible for confirming its agreement with all ICH documents through written sign-off procedure.
Step 2a is reached when the SC agrees, based on the report of the EWG, that the technical document provides all the necessary scientific and technical information to proceed to the next stage of preparation of the draft guideline or recommendation This agreement is confirmed by at least one of the SC members for ICH Parties signing their assent. As an exceptional case, when ICH Party consensus cannot be achieved, the three ICH Founding Regulatory Parties can jointly decide to sign-off the technical document.
Step 2b is reached when the SC agrees that there is sufficient consensus on the draft guideline or recommendation to proceed to the next stage of regulatory consultation. This agreement is confirmed by at least one of the SC members for each of the Regulatory ICH Parties signing their assent. As an exceptional case, when ICH Party consensus cannot be achieved, the three ICH Founding Regulatory Parties can jointly decide to sign-off the technical document.
Step 4 is reached when the SC agrees, on the basis of the reports from the Regulatory Chair and Rapporteur of the Working Group, that there is sufficient consensus on the scientific and technical issues. This agreement is confirmed by at least one of the SC members for each of the Regulatory ICHParties signing their assent. As an exceptional case, when ICH Party consensus cannot be achieved, the three ICH Founding Regulatory Parties can jointly decide to sign-off the technical document.
6. ICH Communication/branding
The ICH branding is used by the ICH Secretariat for communication with the public through documentation reviewed by the Steering Committee (e.g., website, press releases, Concept Papers …). Each Steering Committee Party owns the ICH branding and can make use of it for their communication on ICH in its region but it should be clear that the documentation is developed by its own Party; consideration should still be given as a courtesy to inform other ICH Parties of these publications under ICH branding (e.g., inform other Parties under agenda item Communication about ICH during bi-annual meetings).
ICH Coordinators – Role and Responsibilities
BACKGROUND & DEFINITION:
ICH Coordinators are designated by ICH Parties and play a fundamental role in smooth running of ICH. Their roles include to act as the main contact point with the ICH Secretariat and to ensure that ICH documents are distributed to the appropriate persons within the area of their responsibility. Each party has established a Contact Network of experts within their own organisation or region in order to ensure that, in the discussions, they reflect the views and policies of the Party they represent. ICH Coordinators manage and coordinate through the Contact Network to liaise and
42 Irrespective of whether or not Health Canada and Swissmedic will have appointed technical experts in the Working Group, the SC Members of Health Canada and Swissmedic will be invited to sign-off the Step 2a / 2b / 4 as ICH Parties. See Annex 20
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 37
carry out necessary issues. The way in which this network operates differs according to the administrative structure of the party concerned.
TASKS:
1. Liaison among experts, Steering Committee and the Secretariat
Establish a Contact Network of representatives and experts within their own organisation
Be the main contact point between the expert and the ICH Secretariat
Be the initial contact point for the Regulatory Chair/Rapporteur of its party when they have an issue to be raised to the SC.
Convey comments and requests from experts to the ICH Secretariat and other as appropriate
Ensure proper distribution of ICH information, documents and actions to the appropriate individuals from their party (SC Members, Topic Leaders, Experts, and any other representatives) within the area of their responsibility.
2. Tele/webconferences
2.1 Before a tele/webconference
Notify the Secretariat of issues and topics to be discussed
Consult with relevant experts on various topics and issues for discussion in order to be prepared to convey information as represented during the tele/webconference
2.2 During a tele/webconference
Give oral report on status and/or party’s position on every issue and topic under discussion
Take notes on actions for the responsible topics (ex. If Co-Rapporteurs are designated from two parties, Coordinators from both parties will take responsibility for actions)
2.3 After a tele/webconference
Review and comment on draft report of the tele/webconference circulated by ICH Secretariat respecting the designated deadline
Ensure proper follow up on actions by their respective Party/organisation within assigned deadlines
3. Face-to-face Meetings
3.1 Before a face-to-face meeting
Notify the Secretariat about items/issues/topics for inclusion in the SC Agenda, at least one month prior to the meeting whenever possible
Distribute meeting announcements to representatives of their respective Party
Verify, discuss and distribute the meeting schedules to all representatives concerned. Comment on the draft schedule as appropriate
Nominate representatives for their Party (Topic Leader, Deputy Topic Leader, experts, etc.) for each topic under discussion
Check the preliminary draft agendas (SC meeting, Coordinators meeting, ICG or Regulators meeting as appropriate…)
3.2 During a face-to-face meeting
Ensure that appropriate information is conveyed to the expert of their region.
Help the ICH Secretariat in the preparation of the draft provisional minutes
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 38
of the SC meeting (i.e., by providing notes, suggestions, comments and specific wording, in a continuous way during the meeting)
Confirm the list of actions endorsed by the Steering Committee on each topic and subject
One of the Coordinators from the host region to elaborate the Press Release and other Coordinators to participate in its clearance
3.3 After a face-to-face meeting
Ensure appropriate follow-up on every subject according to the list of actions endorsed at SC level
Review the provisional report of the SC meeting distributed by the ICH Secretariat after the meeting, and coordinate comments from their Party (collect and consolidate comments from their respective representatives as appropriate) respecting the designated deadline
ICH Technical Coordinators – Role and Responsibilities
BACKGROUND & DEFINITION:
ICH Technical Coordinators are designated from EMA for EU and PMDA for MHLW. They support the ICH Coordinator and facilitate every action of the Steering Committee members in the region, mainly by applying their scientific knowledge. Their roles include acting as a contact point between the experts within EMA or PMDA and the ICH Coordinator at the main regulatory body, and as a contact point with the ICH Secretariat.
TASKS:
Participate in the Steering Committee meeting and Coordinators meeting for supporting the role & responsibilities of ICH coordinator in their region
Propose appropriate experts to the main regulatory body for each topic in liaison with the relevant members
Contact person between the experts within EMA or PMDA and ICH Coordinator at the main regulatory body
Liaise with experts during the Steering Committee and provide the important messages to the SC member of his Party
Ensure that an appropriate colleague read the draft guidelines to check for compliance with regional regulatory documents
Check the guidelines and the comments during discussion in ICH and before publication
ICH Observer – Role and Responsibilities
Since the formation of the ICH initiative in 1990, the World Health Organization (WHO) has held the official standing of Observer to ICH. WHO is a specialised health agency of the United Nations governed by 194 Member States and bound to a constitution.
General role of ICH Observer:
- The ICH Observer actively contributes to the work of ICH by participation in all ICH bodies.
o The ICH Observer is a (non-voting) member of the ICH Steering Committee. o It can designate experts to participate in all technical discussions.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 39
- The ICH Observer can submit proposals for new ICH topics to the Steering Committee.
- If proposed by one of the ICH Parties, the ICH Observer may be asked by the Steering Committee to take over additional tasks.
- WHO represents the views of members states and as such serves as a conduit for information dissemination to and comments from non-ICH member states regarding ICH and ICH Guideline.
- WHO will consider ICH Guidelines following its internal procedures.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 40
Annex 9: Standard Operating Procedures for Organisation of Interim Meetings
Interim face-to-face meetings (i.e., IWG/EWG meetings outside the regular ICH SC weeks) should be exceptional, and only when there is an absolute necessity in order for the topic to meet its assigned objectives in time.
Approval to organise an interim meeting: Any interim face-to-face meetings of EWGs/IWGs will be subject to decision by the ICH Steering Committee (SC). These meetings should remain the exception. The Working Group interested in organising an interim meeting must present at the time of the SC face-to-face meeting or tele/webconference, their reasons, including expected achievement, clear Business Plan and a possible location for the meeting as well as plausible time period of a meeting if possible.
Approval by the ICH Regulatory Parties: If an interim meeting is proposed and adopted during a SC meeting without prior discussion, ICH Regulatory Parties may request additional time for the final approval due to budgetary reasons. When this occurs, the Regulatory Coordinators of the requesting Parties must inform the Secretariat of their final approval strictly respecting the deadline date. The ICH Secretariat may start the nomination process prior to the final approvals from ICH Regulatory Parties in order to inform the experts in due time on a possible meeting.
Scheduling and arrangements: Possible dates of the meeting are arranged by the Rapporteurs, in collaboration with the Regulatory Chairs, by contacting the Topic Leaders of each Party, including Observer and other participants, if there are any. ICH Parties must be represented, at least by one delegate, in order for a face-to-face meeting to be considered as an official ICH interim meeting. If a nominated Health Canada or Swissmedic expert exceptionally is not participating in an interim Working Group meeting, the work of the group can still progress including the ICH step process.
Location of the meeting must be arranged together with the ICH Coordinators of the concerned region and the ICH Secretariat (e.g., if the meeting is agreed to take place in London, the EU and EFPIA ICH Coordinators may look for a meeting room at EMA).
The costs of the venue of the interim meeting are covered either by the industry association of the host region or by the ICH Regulatory Party. The funding should be agreed upon in advance and the financing party should be directly involved in any funding decisions associated with the meeting. (N.B.: The costs relating to the interim meeting may include audio-visual equipment and catering costs for lunches and coffee breaks, but will not include a set-up of simultaneous translation.) The costs of travel and accommodation of individual experts will be borne by the party which nominated those experts (following normal ICH funding rules).
Follow-up after the meeting: After the meeting, the Regulatory Chair and the Rapporteur are requested to provide a report (summarising the progress made, the achievements and conclusions reached, the list of actions with clear deadlines and responsible individuals). Draft reports shall be circulated to the EWG/IWG members for discussion and adoption. Approved reports shall be sent by the Rapporteur to the ICH Secretariat for circulation to the SC and Coordinators.
Approval by the Steering Committee in Yokohama on 18 November 2004.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 41
Annex 10: Streamlined Procedure
PURPOSE: To develop a guideline to a short deadline in response to an emerging health care problem.
DESCRIPTION: When it is critical for an ICH country/region to develop a guideline that other ICH partners share an interest in, then the task could be undertaken under the auspices of ICH. Under such circumstances the Steering Committee (SC) would grant the use of the streamlined procedure in order to make the process as short and efficient as possible.
In addition to time constraints, the following conditions are required to make a document eligible for the streamlined procedure:
I. The presence of an emerging health issue, such as: A health problem that affects many persons A significant change in state of art of science.
II. A draft or final document should already exist in one of the ICH regions (including the Observer) that would provide a strong foundation for the development of the ICH guideline.
As there should be consensus from ICH Parties that the draft document would be the starting point in the development of the ICH Guideline, no Concept Paper would be necessary and the country(ies)/region(s) originating the document would lead the EWG responsible in developing the guideline. However, this does not obviate the need for a Business Plan.
If the starting document originates from an ICH Regulatory Party and if the ICH Industry Parties expresses little interest in the potential guideline and in participating in its development, then the ICH regulators could continue developing the document autonomously, without industry aid.
The SC will consider proposed streamlined procedure guidelines on a case-by-case basis.
The Streamlined Step Process: Upon approval of a streamlined guideline the SC: Confirms the objectives and expected outcome of the harmonisation action Confirms the composition of the EWG, which can include outside experts if additional expertise
is needed and Sets a timetable and Action Plan with extremely tight deadlines.
Then, the ICH Parties designates a Topic Leader, as in the normal process and the region originating the documents would nominate a Rapporteur, and one of the ICH Regulatory Parties would nominate a Regulatory Chair.
Except for the absence of a Concept Paper in the streamlined procedure, the step process is the same as the normal ICH process. The form of communication to be used for the sign-off will be fax and/or email.
Reminder of the step process: In principle, the agreement of the ICH Parties is necessary for initiating any ICH harmonisation
activities. However, as an exceptional case when ICH Party consensus cannot be achieved, the three ICH Founding Regulatory Parties can jointly decide to adopt a Concept Paper considering that Regulators have the ultimate responsibility to ensure the protection of public health and have the responsibility for issuing regulatory guidelines.
Step 1: Consensus building between the experts. The Rapporteur circulates the existing document to its group for comments and discussion. As the document has been agreed to in principle, the comments are unlikely to be major. The experts reach
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 42
consensus on the document and sign-off on it. Steps 2a and 2b: The SC and the ICH Regulatory Parties of the SC, respectively, signs-off (by
fax/email if possible). Step 3: Regulatory consultation: Publication for comments in each of the ICH regions (comment
period may be shortened to accommodate regulatory needs and timetables) After addressing all regulatory consultation results, the EWG regulatory experts reach consensus on the Step 3 Experts Draft Guideline and sign-off on it.
Step 4: Adoption of a harmonised Guideline: The SC reaches consensus and the regulatory authorities sign-off (by fax if possible).
Safeguard Clause: In case of unexpected delays in the procedure that would jeopardize reaching consensus and finalising the ICH Guideline on time, the country/ region from which the document originated may withdraw the document from the ICH process in order to meet its own deadlines at any time during the process in consultation with the other parties.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 43
Annex 11: Global Cooperation (GC) Principles and Procedures
Background
The Global Cooperation Group (GCG) was formed as a subcommittee of the ICH Steering Committee (SC) in March 1999 in response to a growing interest in ICH beyond the three ICH regions. The original purpose of the GCG was to make information available on ICH, ICH activities, and ICH Guidelines to any country or company that requested such information. In this regard, the GCG created a set of principles to guide its activities in responding to requests for information.
In November 2003, new Terms of Reference and rules were endorsed with the aim of establishing partnerships with Regional Harmonisation Initiatives (RHIs) and promoting a better understanding of ICH Guidelines. Based on Criteria of Invitation (see below) the GCG invited qualifying RHI to attend the GCG meetings.
From November 2005, RHIs were invited to listen to43 technical topics44 both at the level of the Expert Working (EWG) Groups, Implementation Working Groups (IWG), Discussion Groups and the SC.
In October 2007, the ICH SC discussed the need for a change to the GCG principles and procedures in order to fully realise the GCG objectives and thereby contribute to achieving a number of important goals related to improving public health.
The SC identified these goals to be:
o to reduce country and regional differences in technical requirements that impact on the availability and cost of new medicines;
o to promote international movement of pharmaceuticals that are safe, effective and of high quality;
o to promote the conduct of clinical trials and data collection that meet international standards.
The SC endorsed an expansion of the GCG and agreed to invite representatives from individual Drug Regulatory Authorities (DRAs) and Department of Health (DoH) to participate. Based on a number of Considerations for Invitation (see below) the SC invited a number of individual DRA/DoH to participate in the GCG. Representatives of individual DRA/DoH will enjoy the same access as RHI at the biannual ICH meetings and would similarly be invited to technical topics both at the level of EWGs, IWGs, Discussion Groups and the SC. The participation of individual DRA/DoH would be distinct and complementary to the participation of official RHI representatives/observers.
In November 2010, the ICH SC finalised a Considerations and Criteria document (See Annex 13, Section 2) and in March 2011 endorsed Rules and Procedures (see Annex 13, Section 1) related to the opening up of ICH technical Working Groups to participation from technical experts from RHIs and DRAs/DoH. Technical experts nominated by RHIs/DRAs/DoH will be invited to participate in ICH Technical Working Groups in the same manner as experts from WHO (ICH Observer). In recognition of their contribution to the development of an ICH Guideline, RHI/DRA/DoH experts will be invited to sign-off Step 1 Technical Document and Step 3 ICH Draft Guidelines.
In November 2012, the ICH SC endorsed a guiding document developed by the RHIs entitled “Rules and Procedures and modus operandi” providing guidance to RHI Representatives attending ICH meetings (see Annex 14).
In San Diego, in November 2012, the SC decided to create a modified GCG format that would
43 The ICH SC decided in May 2005 that RHI representatives would be invited to listen to technical
meetings for the purpose of gaining an enhanced understanding of the ICH process. 44 The Secretariat is responsible for providing the list of topics open to RHI.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 44
allow more time for in-depth and focused discussions among regulators while also incorporating an abbreviated industry session that retains the most valuable, non-redundant elements of the GCG and the Regulators Forum. In June 2013 in Brussels/la Hulpe, the SC adopted the new format for the GCG which was incorporated into a standing agenda item of the SC agenda and entitled Global Cooperation (GC) session. The GC session maintains current GCG arrangements regarding RHIs/DRAs/DoH participation and the need for SC endorsement of proposed activities. About half a day (4h) is now allocated to the GC session to discuss on the implementation of ICH Guidelines and the training.
RHIs and DRAs/DoH Membership To date, the Global Cooperation (GC) session gathers SC members from the all ICH Parties (EU/EMA, MHLW/PMDA, FDA, Swissmedic, Health Canada, EFPIA, JPMA, PhRMA), the ICH Observer (WHO) and IFPMA; in addition to representatives from six Regional Harmonisation Initiatives (RHIs) and representatives from eight individual Regulatory Authorities and Department of Health (DRAs/DoH).
The two Co-chairs of the GC session are nominated among the SC members with one Co-chair from Industry and one from a ICH Regulatory Authority. The GC Co-chairs should also come from different regions. In June 2006, the ICH SC recommended that the GC Co-chairs rotate every two years (following the fall meeting) and that a staggered rotation of the Co-Chairs occur to ensure continuity.
o Invited Harmonisation Initiatives The SC invited RHI to nominate representatives to participate in the ICH meetings (e.g., ICH SC meeting including its Global Cooperation session, EWGs/IWGs meetings, MedDRA Management Board Special session). The SC considered the following criteria as a basis for the invitations:
Considerations for Invitation to ICH 1. The initiative should be founded on the principle of harmonising drug regulation across a
defined group of countries.
2. The initiative should be science-based, with clear scientific harmonisation objectives.
3. The initiative should be currently active with meetings/activities regularly scheduled.
4. The initiative will have available, or establish, a mechanism to disseminate information on its activities with the ICH to its members.
To date, representatives (or observers) from the following RHI now attend the ICH meetings (e.g., ICH SC meeting including its Global Cooperation session, EWGs/IWGs meetings, MedDRA Management Board Special session): Asia-Pacific Economic Cooperation (APEC LSIF 45 ), Association of Southeast Asian Nations (ASEAN ACCSQ PPWG46) as observer, East African Community (EAC), Gulf Cooperation Council (GCC), Pan American Network on Drug Regulatory Harmonization (PANDRH) and Southern African Development Community (SADC).
RHI may nominate one or two representative(s)/observer(s)47 to attend the ICH meetings.
o Invited Drug Regulatory Authorities and Department of Health The ICH SC invited a number of individual Drug Regulatory Authorities and Department of Health
45 APEC LSIF: APEC Life Sciences Innovation Forum 46 ASEAN ACCSQ PPWG: ASEAN Consultative Committee on Standards and Quality, Pharmaceutical Product Working Group 47 RHI can send a second representative/observer on the condition that this individual is officially
nominated and that his/her expenses are covered by the region.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 45
(DRAs/DoH) from countries beyond ICH to nominate representatives to participate in ICH-related activities: Considerations for Invitation
1. Use or intended use of ICH Guidelines.
2. Whether a country or region is a source of active pharmaceutical ingredients, medicinal products or clinical data for the ICH regions.
To date, the following DRA/DoH have been invited to attend the ICH meetings (e.g., ICH SC meeting including its Global Cooperation session, EWGs/IWGs meetings, MedDRA Management Board Special session): Australia, Brazil, China, Chinese Taipei, India, Russia, Republic of Korea and Singapore. DRA/DoH may nominate one or two representatives to attend the ICH meetings.
o Other Invitees On a case-by-case basis the ICH SC will invite other organisations or initiatives not meeting the criteria defined above to present at the GC session on a specific topic of interest and relevance to GC members.
GC activities
Purpose The GCG was created to facilitate the dissemination of information beyond ICH regions / parties concerning the operation, organisation and products of ICH. With the adoption of a new framework for cooperation with other regions48, the GC is engaged in a more active and interactive manner with non-ICH regions / parties. This new engagement promotes a better understanding of the underlying principles and intent of ICH Guidelines as well as the challenges faced by countries and regions beyond ICH in their utilisation.
Mission Statement The scope of activities undertaken by the GC members is embodied in the mission statement adopted by all parties in May 2005:
To promote a mutual understanding of regional harmonisation initiatives in order to facilitate the harmonisation process related to ICH Guidelines regionally and globally, and to facilitate the capacity of drug regulatory authorities and industry to utilise them.
Principles of Transparency and Openness Key principles underlying the decisions and work of the GC members are transparency and openness.
The outcome of GC activities, such as the Terms of Reference, reports, presentations, etc., is available to the public on the ICH website49. Furthermore, questions can be addressed to GC members through the same website. A ‘GC Members’ section of the ICH website has also been established as a repository for non-public documents, including draft reports and a calendar of training and harmonisation activities.
Openness is achieved by identifying with the RHI and DRA/DoH topics and process issues associated with harmonisation for discussion, collaboration and potential development into a joint program of activities and opening the SC and technical meetings to the invited RHI and DRA/DoH representatives.
48 Terms of Reference, November 2003 49 www.ich.org
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 46
Strategy on Training and Capacity-building In 2006, the GCG implemented a strategy for addressing the training and capacity50 needs of RHI and for ensuring the most effective use of resources, opportunities and the realisation of desired outcomes.
The GCG defined principles and recommendations in responding to training requests. Whenever possible, training initiatives directed through the GC should:
Optimally, be regionally-based, with flexibility to consider nationally-based training, if deemed appropriate
Be coordinated amongst ICH Parties and RHI and leverage existing regional training activities and events
Leverage the experience and resources of non-profit training organisations
Be planned and reviewed on a periodic basis
Take full advantage of appropriate training modalities/technologies.
With the decision of the ICH SC in October 2007 to invite representatives of individual DRA/DoH to participate in the GCG it was agreed to extend the training strategy to also apply to requests from individual DRA/DoH. However, in line with the original principles of the training strategy it was agreed that regionally-based training events should be given a higher priority than country specific events.
Please see Annex 12 for full GC Procedure on Training Activities: Selection, Prioritization, Coordination of RHI & DRA/DoH Requests.
ICH Meetings
Global Cooperation Session The GC sessions of the SC take place at the same time and location as the ICH technical and SC meetings, usually twice a year. GC tele/webconferences are held between face-to-face meetings to maintain progress on action items and to prepare for upcoming meetings.
Representatives/observers of the RHI and DRA/DoH hold their respective pre-meeting prior to the GC session of the SC meeting to exchange information and views (on topics of common interest, implementation of ICH Guidelines and Training) and to prepare for the GC session.
Preparation for ICH EWGs and SC meetings Representatives/observers of the RHI and DRA/DoH are asked to register for sessions of technical topics they wish to listen to based on the list of open topics51 provided by the ICH Secretariat.
The SC recommended that a pre-meeting be arranged between the Regulatory Chair and Rapporteur and the RHI and DRA/DoH representatives/observers, when necessary, with the objective of briefing the RHI and DRA/DoH on both the work of the EWG and the relevant ICH procedures and logistics. New RHI and DRA/DoH representatives/observers should be provided excerpts of the ICH procedures document and, whenever possible, should be met by representatives of the ICH Secretariat or one of the GC Co-chairs prior to the start of ICH meetings to clarify administrative and procedural matters.
In the event that RHI or DRA/DoH representatives/observers are unable to attend a particular ICH meeting, alternates should be versed on ICH procedures, on ICH and RHI/DRA/DoH matters.
50 Strategy document on Training and Capacity-building, endorsed by the ICH SC and GCG in June
2006. 51 The list of topics open to RHI and DRA/DoH is endorsed by the ICH Coordinators before being
circulated to RHI and DRA/DoH.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 47
Funding
The ICH Secretariat budget covers GC-related activities. Most GC expenses covered by the ICH Secretariat budget are related to the travel-related costs of the RHI representatives/observers. The ICH Secretariat budget covers all transportation and accommodation expenses associated with one representative/observer per RHI participating in ICH meetings. The expenses of a second RHI representative/observer are covered by the region. DRA/DoH representatives cover their own travel and accommodation expenses. The SC can offer technical assistance on an ad hoc basis to training activities, but no funding is available to organise training sessions.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 48
Annex 12: Global Cooperation (GC) Procedure on Training Activities: Selection, Prioritisation, Coordination of RHI & DRA/DoH Requests
BACKGROUND:
This procedure on training activities aims at improving the selection, prioritisation and coordination of requests for training activities and capacity-building events from Regional Harmonisation Initiatives (RHIs) and individual Drug Regulatory Authorities and Department of Health (DRAs/DoH) considered during the GC session of the ICH meetings52. This procedure also aims at achieving greater transparency regarding events in which ICH experts are asked to participate.
The GC sessions were established as an evolution of the GCG that was created to facilitate the dissemination of information beyond ICH regions / parties concerning the operation, organisation and products of ICH. With the adoption of a new framework for cooperation with other regions53, the GCG was engaged in a more active and interactive manner with non-ICH regions / parties. This new engagement promotes a better understanding of the underlying principles and intent of ICH Guidelines as well as the challenges faced by both RHI and individual DRA/DoH in their utilisation.
The scope of activities undertaken by the GCG was embodied in the Mission Statement adopted by all ICH Parties in May 2005:
To promote a mutual understanding of regional harmonisation initiatives in order to facilitate the harmonisation process related to ICH Guidelines regionally and globally, and to facilitate the capacity of drug regulatory authorities and industry to utilse them.
In 2006, the GCG implemented a strategy for addressing the training and capacity54 needs of RHI and for ensuring the most effective use of resources, opportunities and the realisation of desired outcomes.
The GCG defined principles and recommendations in responding to training requests. Whenever possible, training initiatives directed through the GCG should:
Optimally, be regionally-based, with flexibility to consider nationally-based training, if deemed appropriate
Be coordinated amongst ICH Parties and RHI and leverage existing regional training activities and events
Leverage the experience and resources of non-profit training organisations
Be planned and reviewed on a periodic basis
Take full advantage of appropriate training modalities/technologies.
With the decision of the SC in October 2007 to invite representatives of individual DRA/DoH to participate in the GCG it was agreed to extend the training strategy to also apply to requests from individual DRA/DoH. However, in line with the original principles of the training strategy it was
52 This procedure does not preclude the consideration by GCG of requests for training by DRA/DoH
that are not participating in the GCG. 53 Revised GCG Terms of Reference, November 2003 54 Strategy document on Training and Capacity-building, endorsed by the ICH SC and GCG in June
2006
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 49
agreed that regionally-based training events should be given a higher priority than country specific events.
In June 2013, the GCG adopted a new format. The GC sessions of the ICH SC were established to allow interactive discussions on implementation of ICH Guidelines in the ICH regions and beyond, and training as main focus.
PROCEDURE FOR ORGANISATION OF TRAINING:
1. TRAINING PLAN
RHI and individual DRA/DoH are encouraged to submit to the ICH Secretariat, one-month prior to the ICH meetings, an updated two-year training plan which would outline the overall training strategy relating to ICH Guidelines for the RHI/DRA/DoH and list any training events for which the ICH SC would be requested to consider providing speakers.
The training plan would be circulated to the GC members by the ICH Secretariat for discussion at the GC session of the ICH SC meeting. The two-year training plan will act as a supporting document to assist the ICH SC to prioritise training requests in the context of the overall training strategy of RHI/DRA/DoH.
2. SUBMISSION OF INDIVIDUAL TRAINING REQUESTS (STANDARD TEMPLATE)
All training requests should be submitted to the ICH Secretariat using a standard template (see below). For training requests RHI and individual DRA/DoH are asked to provide information on:
1. Sponsors: Who organises the training event Is this event supported by a profit or non-profit organisation Who is the primary contact for organisational aspects
2. Scope: How does the training event fit into the overall training strategy of the RHI/DRA/DoH? How would the RHI/DRA/DoH rank this training request among its most pressing needs What are the learning objectives of this event
3. Target audience: Is this a regional or a national event Who may participate in the training (e.g., regulators, industry, academia etc…) Are other RHI/DRA/DoH invited What is the suggested profile of the participants (e.g., new to subject, intermediate knowledge or expert)
4. Practical information on the training event: Programme: Is there a draft programme already available? Are the topics/issues on which the speakers should present already known? Schedule: When will the event take place? How long will the event last Speakers: What are the expectations in terms of speakers (e.g., number of speakers needed, regulators/industry speakers, etc)?
5. Funding: How will the event (speakers, venue, development of conference materials etc…) be funded (e.g., grant, participant registration fee etc…)?
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 50
What will the funding for ICH speakers cover (e.g., flights, accommodation, etc…, please note that ICH SC endorsement does not necessary mean that the ICH party pays its own travel expenses)?
Where necessary, the ICH Secretariat will provide assistance to RHI/DRA/DoH to complete the template. The ICH Secretariat will work with RHI/DRA/DoH to ensure that all requests contain the appropriate information before circulation for ICH SC consideration.
3. REVIEW CRITERIA FOR THE ICH SC
The ICH SC reviews requests based on the following criteria:
1. Sponsors: Is the event leveraging the input of a training organisation? Do the funding arrangements preclude participation of regulators?
2. Scope: Is the event within the scope of the GC? Is the training event integrated into the regional/national training strategy of the RHI/DRA/DoH? What are the expected benefits for the RHI/DRA/DoH? Does the event conflict with WHO or other training initiatives?
3. Target audience: Is the training nationally or regionally based?55 Does the event promote interaction, support and a common understanding of the ICH Guidelines amongst the countries and stakeholders within a given region?
4. Appropriateness: Is the event appropriate to cover the objectives set by the RHI/DRA/DoH?
4. PRIORITISATION BY THE ICH SC
At the GC session of the SC meeting, the ICH SC will consider the two-year training plans and requests for training. Prioritisation would be based on assessment against the review criteria outlined above.
5. ORGANISATION OF TRAINING
In general the host RHI/DRA/DoH is responsible for the organisation of training events. It should be noted that the role of the ICH Secretariat extends only to acting as a point of contact between the host and the ICH parties/experts involved in the training. As a principle it should also be noted that following all GC-endorsed training events training materials/presentations from the event will be made available for re-use on the ICH Public website (see also Section 7).
Development of agenda and program: Once a training request is approved by the ICH SC, the ICH SC will nominate an ICH lead party or lead parties. The host RHI(s)/DRA/DoH is responsible for developing the draft programme with input sought from the ICH lead party/parties. Work should progress via e-mail exchange and, if necessary, tele/webconferences. The ICH Secretariat will periodically monitor the progress of GC-actioned training requests.
Selection of ICH experts:
55 As a principle regionally-based training events should be given a higher priority than country
specific events.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 51
Once the type of expert(s) required has been decided the ICH Secretariat will work with the ICH Coordinators to identify the expert(s) to participate in the training event. The role of the ICH Coordinators is to identify speakers within their Party and region. The ICH Secretariat will also keep a record of the ICH experts who participated in RHI/DRA/DoH training events. As a general rule, the Rapporteur of the topic of the training event should be consulted during the development of the event.
Logistics/Conference Management: The host RHI(s)/DRA/DoH is responsible for the meeting logistics e.g., organisation of venue – organisation of equipment for meeting rooms etc…; identification of hotel(s) - making block booking if appropriate; sending invitations to speakers; collection of abstracts/CVs; printing of program, background documents etc…; promotion of event - development/releasing of announcements; organisation of translation - translation of documents, interpreters; organisation of registration of participants - name tags etc.. Coordinating with the ICH Secretariat, the host should ensure that the ICH experts nominated as speakers are provided with all the necessary information on the event logistics.
Information on ICH: Training that has been endorsed through this training procedure should normally include the ICH logo in all promotional material (this does not exclude the additional use of other logos). Those speaking on ICH topics should consider to use the ICH logo on their slides as appropriate. To frame the ICH training event an ICH expert participating in the event should be asked to present on ICH in general. The slides for this presentation will be provided by the ICH Secretariat and consist of a general set of slides with background information on ICH and the GC-related activities.
6. ASSESSMENT AND FEEDBACK
It is important to monitor and evaluate the training outcomes in order to assess whether training objectives were achieved and to identify lessons learnt for future events. Therefore the host RHI(s)/DRA/DoH is requested to submit a short report on the event to the ICH SC. ICH speakers may provide feedback as appropriate and will also be advised of any important feedback from the host RHI(s)/DRA/DoH that would be of help for future presentations.
7. LEVERAGING RESOURCES FOR TRAINING
In order to better leverage resources the GC members should take into consideration the following points for GC training events:
- Could training materials and programmes developed for previous training events be reused.
- In cases where stand-alone training is requested (i.e., not part of a bigger event) could the training be conducted remotely by webinar.
- Could some training events be opportunities for a “train the trainer” approach i.e., other RHI/DRA/DoH could send participants who would be trained and could then return to their regions to give training.
The presentations/training materials will be for reuse and for posting on the working area of the GC, and where appropriate on the ICH public website. Any errors identified in the presentations/training materials should be corrected before posting on the working area of the GC/ICH public website. Following GC-endorsed training events, speakers/trainers involved in the events will be asked to sign a standard form provided by the Secretariat granting ICH and the host RHI/DRA/DoH permission to publish their training materials/presentations on their respective websites.
ICH Procedures - Endorsed by the ICH Steering Committee on June 10 2015 52
SUBMISSION OF REQUESTS (STANDARD TEMPLATE): For training requests RHI and individual DRA/DoH are asked to provide information on:
Sponsors Who organises the training event Is this event supported by a profit or non-profit organisation
Who is the primary contact for organisational aspects
Scope How does the training event fit into the overall training strategy of the RHI/DRA/DoH
How would the RHI/DRA/DoH rank this training request among its most pressing needs
What are the learning objectives of this event
Target Audience
Is this a regional or a national event Who may participate in the training (e.g., regulators, industry, academia etc…)
Are other RHI/DRA/DoH invited What is the suggested profile of the participants (e.g., new to subject, intermediate knowledge or expert)
Practical Information on the Training Event
Programme: Is there a draft programme already available Are the topics/issues on which the speakers should present already known
Schedule: When will the event take place How long will the event last
Speakers: What are the expectations in terms of speakers (e.g., number of speakers needed, regulators /industry speakers, etc)
Funding How will the event (speakers, venue, development of conference materials etc…) be funded (e.g., grant, participant registration fee etc…) What will the funding for ICH speakers cover (e.g., flights, accommodation, etc…)
53
ICH Procedures Version 1.1
Annex 13: Participation of RHIs and DRAs/DoH in ICH Technical Working Groups to Promote Use of ICH Guidelines
1. RULES AND PROCEDURES
Expression of Interest to Participate in ICH Technical Working Groups
Regional Harmonisation Initiatives (RHIs) and Drug Regulatory Authorities / Department of Health (DRAs/DoH) interested to participate in ICH Technical Working Groups should inform the ICH Secretariat in which ICH Technical Working Groups there is interest to participate (for list see Table 156) and in addition should provide information to inform the ICH Steering Committee on their respective initiative/authority’s use of ICH Guidelines. It is understood that RHI/DRA/DoH are in various stages with regards to the use / implementation of ICH Guidelines. Using the “Expression of Interest to Participate in ICH Technical Working Groups” template (see Table 2), please try to provide as much of the requested information as possible.
ICH Steering Committee Consideration of RHI/DRA/DoH Expressions of Interest
The ICH Steering Committee (SC) will consider individual expressions of interest based on the considerations and criteria set forth in the document entitled “Participation of RHIs and DRAs/DoH in ICH Technical Working Groups to Promote Use of ICH Guidelines”, which was approved by the SC in November 2010 (see Section 2 of this Annex). The ICH Secretariat will inform individual RHIs/DRAs/DoH of Steering Committee considerations in relation to their expressed interest.
Participation of Experts in ICH Technical Working Groups
RHI/DRA/DoH representatives will nominate a main Focal Point to act as a coordinator between the ICH Secretariat and their experts. Technical experts nominated by RHIs/DRAs/DoH will be invited to participate in ICH Technical Working Groups in the same manner as experts from the ICH Observer (WHO).
Contact information (name, title, position, e-mail address, work address, phone number etc…) for RHI/DRA/DoH experts should be provided to the ICH Secretariat by the RHI/DRA/DoH Focal Point. This information will be provided to the Regulatory Chair and the Rapporteur of the relevant ICH Technical Working Group.
For the purposes of continuity, the intention should be that the same nominated RHI/DRA/DoH expert participates for the duration of the ICH Technical Working Group’s work to develop an ICH Guideline. However, if a RHI/DRA/DoH needs to replace its expert with another expert this information should be conveyed to the ICH Secretariat by the Focal Point. The departing expert should brief the new expert on the status of the ICH Guideline development to facilitate the smooth integration of the new expert into the ICH Technical Working Group.
One expert per RHI/DRA/DoH may participate in ICH Technical Working Group discussions (e.g., face-to-face meetings, tele/webconferences etc…); however an alternate expert may also be nominated. This alternate expert would be copied on ICH Technical Working Group e-mails for informational purposes so that if the official expert is unable to participate to a tele/webconference or a face-to-face
56 Table 1 is only provided as an example. The list is maintained by the Secretariat as appropriate.
54
ICH Procedures Version 1.1
meeting, the alternate may replace the official expert at the tele/webconference or face-to-face meeting.
RHI/DRA/DoH experts will be copied on all ICH Technical Working Group e-mails and should attend tele/webconferences and face-to-face meetings. It should be noted that in line with ICH Procedures, for a tele/webconference or face-to-face meeting to be considered official, ICH Parties need to be represented, at least by one delegate. The absence of experts from RHIs/DRAs/DoH, the ICH Observer or any Interested Parties will not prevent the tele/webconference/meeting from taking place.
The RHI/DRA/DoH Focal Point will confirm to RHI/DRA/DoH experts (once information is available from the ICH Secretariat), whether or not their Technical Working Group will meet face-to-face at the next ICH meeting. If meeting, the RHI/DRA/DoH Focal Point will provide all meeting information to their experts. To facilitate the organisation of the meeting by the host ICH Party, the RHI/DRA/DoH Focal Point will provide to the ICH Secretariat a list of their delegation and ensure that experts return their meeting registration form at the latest by the deadline indicated.
The RHI/DRA/DoH Focal Point will provide to new nominated RHI/DRA/DoH experts a written summary on general ICH Procedures in preparation of their first face-to-face meeting.
New RHI/DRA/DoH experts, in addition to other new experts from the ICH Parties, ICH Observer, and Interested Parties, will be invited to participate in a briefing session on ICH Procedures at the time of their first face-to-face meeting. It is important that all experts are informed on ICH Procedures.
In recognition of their contribution to the development of a guideline, RHI/DRA/DoH experts will be invited to sign-off Step 1 technical document and Step 3 ICH Draft Guideline. Such sign-off will be on a voluntary basis, with a view to confirming their support to the guideline. However, the absence of signature from RHI/DRA/DoH experts will not lead to a suspension of the guideline.
Confirmation of Status of ICH Guideline Implementation
Standing items on the agenda of the biannual ICH Steering Committee meetings include an “Update on ICH Guideline Implementation” and “General Implementation Issues”. These agenda items provide the ICH Parties and the ICH Observer the opportunity to inform the SC on the status of ICH Guideline implementation in their respective countries/regions and share any issues being experienced with the implementation of an ICH Guideline in their economy/region.
RHIs/DRAs/DoH actively participating in ICH Technical Working Groups should submit to the SC a written report of the status of ICH Guideline Implementation in their countries/regions as an update to the information provided when they originally expressed interest to participate in ICH Technical Working Groups. This information should be provided in writing to the ICH Secretariat by the focal point one month in advance of ICH meetings. Any issues with ICH Guideline implementation may also be raised.
TABLE 1 (EXAMPLE)
LIST OF TECHNICAL WORKING GROUPS FOR EXPERT NOMINATIONS FROM
RHI/DRA/DOH
55
ICH Procedures Version 1.1
ICH Quality Groups
Q3D
Q11 EWG
ICH Safety Groups
S10 EWG
M7 EWG
M3(R2) IWG
ICH Efficacy Groups
E2C(R2) EWG
E14 IWG
Other Groups
MedDRA PtC WG
56
ICH Procedures Version 1.1
TABLE 2
EXPRESSION OF INTEREST TO PARTICIPATE IN ICH TECHNICAL WORKING GROUPS (TEMPLATE)
Name of RHI / DRA / DoH: ……………………………………………………….............................
Questions Response
SECTION I
Support
for the use of
ICH Guidelines
What is the RHI/DRA/DoH policy/strategy regarding the use of ICH Guidelines?
Which ICH Guidelines have already been implemented?
Have the CTD/eCTD been implemented in your economy/region?
Are there future plans to implement other ICH Guidelines relevant to your economy / region?
If guidelines, other than ICH Guidelines are being used when an ICH Guideline for a topic exists, can the RHI/DRA/DoH provide background?
57
ICH Procedures Version 1.1
SECTION II
ICH Technical Working Groups
of Interest
In which ICH Technical Working Group(s) is there interest to participate?(Group(s) should be selected from a list available for the ICH Secretariat, see Table 1)
How would the RHI/DRA/DoH implement the ICH Guideline which is the product of the ICH Technical Working Group in which they have expressed interest to participate?
If a RHI/DRA/DoH has previously had its request to participate in an ICH Technical Working Group(s) endorsed by the ICH Steering Committee only the second section “ICH Technical Working Groups of Interest” needs to be filled in subsequently submitted expressions of interest to participate in further ICH Technical Working Groups. However, RHIs/DRAs/DoH will be invited to provide information on the status of ICH Guideline implementation in the economy/region at the time of the biannual ICH meetings (see above section on Status of ICH Guideline Implementation).
58
2. GENERAL CONSIDERATIONS AND CRITERIA FOR PARTICIPATION OF RHIS AND
DRAS/DOH IN ICH TECHNICAL WORKING GROUPS
Background
This document sets out considerations and criteria by which experts from Drug Regulatory Authorities (DRAs) / Department of Health (DoH) and Regional Harmonisation Initiatives (RHIs) may participate in ICH technical working groups (such as Expert Working Groups, Implementation Working Groups and other groups).
General Considerations
1. Qualifying DRAs/DoH and RHIs are entitled to send one expert each to a technical working group, subject to one time consideration of overall size of the technical working group by the ICH Steering Committee (SC) prior to the first meeting of the group. Participation in ongoing technical working groups will be considered on a case by case basis primarily depending on the stage of discussion and the size of the technical working group.
2. Experts from DRAs/DoH or RHIs that participate in ICH-related activities will be permitted to participate in technical working groups in the same manner as experts from the ICH Observer (WHO) provided the DRA or RHI meets the criteria established for such participation. Note: Representatives from DRAs/DoH and RHIs would continue to play a distinct role from experts from qualifying DRAs/DoH and RHIs in technical working groups.
3. Experts from DRAs/DoH and RHIs would be designated as such, and would exclusively represent their specific economy or region and would be expected to report back to their respective economy/region. A process document specifies the procedures for nomination and approval of participation (see Table 2).
Criteria for the DRAs/DoH and RHIs to nominate experts to participate in technical working groups:
1. Demonstrated support for the use of ICH Guidelines, 2. Experts to possess appropriate knowledge, 3. Commitment that nominated experts participate in all activities of the working
group (including meetings, tele/webconferences and email communications) and present views based on consultations within the RHI or economy (in the case of a DRA/DoH),
4. Self-finance travel and accommodation costs of experts, 5. Timely registration for meetings.
59
Annex 14: Guidance Document for RHI Representatives attending ICH Meetings: Rules and Procedures and modus operandi
Background Information The RHI participation in the Global Cooperation session of the Steering Committee (SC) is based on the following principles:
1. Harmonisation of technical requirements for the registration and control of
pharmaceutical products is imperative in view of the increase in globalisation in
development and manufacturing of pharmaceutical products.
2. The work undertaken by the ICH is of most value to allow medicines regulatory
authorities to be better and faster in assessing pharmaceutical products.
The ICH-GC mission statement is to “promote a mutual understanding of regional harmonisation initiatives in order to facilitate harmonisation processes related to ICH Guidelines regionally and globally and to strengthen the capacity of drug regulatory authorities and industry to implement them”. The driving force behind the harmonisation initiatives is the need to speed up access to pharmaceutical products. In 1999, the creation of the ICH Global Cooperation Group (GCG) was aimed at promoting a better understanding of ICH Guidelines in order to encourage their acceptance and adoption in the non-ICH countries and regions. The ICH managed to develop highly technical Guidelines that could not have been achieved by individual regional blocks within a short period. The implementation of ICH Guidelines is mandatory in ICH countries and encouraged in countries and regions beyond ICH. The Regional Harmonisation Initiatives’ (RHI) participation in ICH activities is a remarkable opportunity for the member countries to establish partnerships with ICH and improve their regulatory frameworks, practices, acceptance and adoption of ICH Guidelines in non-ICH countries.
RHI Guidance 1. The participation of Regional Harmonisation Initiatives’ (RHI) in ICH activities is
aimed at:
Gaining common understanding on the ICH process and products;
Gaining the knowledge and the know how to implement technical
guidelines properly; and
Sharing the RHIs activities.
2. RHI representatives should review and understand the ICH process for developing
guidelines.
3. RHI representatives should discuss the implementation of the ICH Guidelines
within their region.
4. RHI representative are encouraged to determine the feasibility of implementing the
ICH Guideline(s) in the particular region in view of the available resources.
5. RHI representative are encouraged to develop training plans for their region(s) and
requests for training to the ICH SC should be in line with training needs for the
RHI.
6. The RHI representatives are encouraged to coordinate with the ICH Secretariat for
hosting GC-endorsed trainings in their region(s).
60
7. RHI representatives are encouraged to share its ICH Guideline implementation
experiences and issues with the GC members.
8. RHI representatives are encouraged to share regulatory harmonisation experiences
with the GC members.
61
Annex 15: MedDRA Points to Consider (PtC) Working Group
History: The ICH Points-to-Consider (PtC) Working Group (WG) was established in 1999 with the scope of developing a PtC document on Good MedDRA Selection Practices. In late 2003, the ICH Steering Committee (SC) agreed to extend the remit of the PtC WG from their role of advising on coding standards to a broader role of advising on standards for data output. The PtC WG uses the ICH platform to develop best practice initiatives related to the use of MedDRA. The group also works to maintain the released PtC documents in parallel with MedDRA version updates.
PtC activities – Concept Paper: When proposing a new PtC document, the PtC WG will be asked to develop a draft Concept Paper with detailed information on the scope, need, benefits, deliverables, cost, time frame, membership, for consideration by the MedDRA Management Board (MB), followed by SC approval.
Endorsement of PtC documents: The PtC documents are not subject to regional implementation, but rather provide a best practice approach. Generally the PtC WG releases a new version of the PtC documents for every version of MedDRA. PtC documents with major changes (i.e., significant new documents, new concepts in existing documents) will be signed off by the ICH Parties of the PtC WG and by the MedDRA MB. PtC documents with minor changes (e.g., simple revisions) will be signed-off by Rapporteur/Co-Rapporteur. Once signed-off, the PtC documents are available for public consultation. Any comments are forwarded to the PtC WG and will be taken into consideration for the release of the next version of MedDRA.
62
Membership:
The PtC WG is similar to other ICH Working Groups, in that the ICH Parties nominates up to two experts, one in the role of Topic Leader and the other as a Deputy Topic Leader. The PtC WG usually also includes an expert from both MSSO and JMO, as well as one representative from WHO (ICH Observer).
Working Procedures:
The PtC WG has an on-going mandate from the SC to work by tele/webconference/e-mail. The group is asked to report at MB tele/webconference when there is a need for a face-to-face meeting. Justification will need to be provided for all face-to-face meetings, for MB consideration, followed by SC approval.
The PtC WG usually meets during the SC week. However, some flexibility is allowed for the scheduling of PtC WG, as the necessity for holding meetings depends on the feedback received from Users and the time of release of MedDRA (March and September). The usual maintenance of both PtC documents on term selection and on data retrieval & presentation does not require frequent face-to-face meetings. A large part of the work is done by correspondence, and major and only complex changes to MedDRA are discussed during face-to-face meetings. The Rapporteur is asked to report on progress and issues to the MedDRA Management Board on a regular basis. Unresolved issues will be brought to the attention of the SC.
Designation of the Rapporteur / Co-Rapporteur:
The nomination of Rapporteur/Co-Rapporteur proceeds via consultation for candidate(s) among the ICH Coordinators of ICH Parties. The PtC WG should also be consulted and invited to discuss their leadership. The MedDRA MB will be asked to approve the nomination and the SC will be informed of the nomination. The Rapporteur/Co-Rapporteur should not be of the same region/affiliation (industry vs. regulatory authority) at any one time.
The MedDRA MB will reassess the term of Rapporteurship, as needed.
63
Annex 16: Notes on the Format and Style of ICH Technical Documents
The following guidance, which has been agreed by the ICH Steering Committee is intended to be provided to, and followed by, ICH Topic Rapporteurs when drafting technical documents. The objective is to develop a document that will include all the necessary scientific and technical information which will be the basis for development or the revision of an ICH draft Guideline. The structure of the document will contain the same main elements as the guideline, but the content and subsections will differ in order to meet the objectives of a technical document. It is thus recommended that, to the extent possible, all technical documents should contain the following sections:
Table of Contents
1. INTRODUCTION
1.1 Objective(s)
1.2 Background
1.3 Scope of the Technical Document
1.4 General Principles
2. Technical Document
2.1 Scientific and technical discussion
2.2 Recommendations for the draft Guideline
3 GLOSSARY
NOTES ON THE FORMAT
1. INTRODUCTION
1.1 Objective(s)
Brief introductory comments giving the purpose for which the technical document has been drafted. (For new topics this should be based on the “Problem Statement/Objectives” in the concept paper).
1.2 Background
This section should include information which helps to put the technical document and the future guideline into context, including, for example, whether it is to be read in conjunction with any other guideline (rule or regulation). It may be useful to highlight existing differences in requirements which are relevant to the objectives of the guidelines, but should not include subjective observations or statements which appear critical of existing requirements or practices.
1.3 Scope of the Technical Document
This should be included in order to define the application of the technical document and future guideline, but it may not be appropriate for some Topics, in which case it would be
64
optional. The scope should state the range of tests and/or products to which the guideline applies, and state whether there are any exclusions. For example, the scope of the first Stability guideline was limited to tests on new chemical entities and their formulations; existing formulations and variations were excluded.
1.4 General Principles
This section should be included if there are general considerations that apply to the scientific area/issue that is addressed by the technical document. For example, the Impurities guideline includes a section on the “Classification of Impurities” which would be appropriate to a “General Principles” section.
The level of detail of these general principles with respect to scientific and technical considerations may be more than in a draft guideline itself.
2. Technical Document
2.1. Scientific and technical discussion
This section should describe the relevant scientific and technical information that needs to be taken into account when developing a guideline(s).
2.2. Recommendations for the draft guideline
This section should contain operative considerations, giving specific recommendations and guidance on technical matters, for example the way in which tests should be carried out and/or the way data should be presented. The format and structure of this section (headings, sub-headings etc.) should be similar to the anticipated structure of the draft guideline. It will vary according to the nature of the subject, but there should be a clear separation between:
Specific guidance and recommendations
Commentaries on the guidance
Technical documents should always contain a rationale supporting a specific guidance or recommendation. If different options or approaches were considered in the expert group, they should be included in the text along with a discussion on their respective pros and cons. For example, if different tests are available for the same purpose or different ways to present data, the strengths and weaknesses of the various approaches should be discussed.
It is preferable to include supplementary information within the technical document and differentiate it by the use of a combination of formatting and headings (“Note:”, “Example:” etc.). The extensive use of footnotes or end notes is not recommended.
2.3. Minority opinions
The document should either indicate that there are no minority opinions or include these opinions under this section.
3. GLOSSARY
Consideration should always be given to the need for a glossary defining the way in which certain terms are used for the purpose of the technical document. Where possible, the glossary should be defined at the outset and it is important to cross-refer to Glossaries in all other ICH Guidelines in order to ensure consistent definitions. A compilation of Glossary definitions from existing ICH Q, S, E and M Guidelines is available from the ICH Secretariat. As far as possible, already existing definitions should be used.
65
References
References to published papers and the scientific literature may be included in the section on scientific and technical discussions. These references should however be limited to scientific reviews or authoritative publications considered as pivotal for the development of the technical document. The only references which should normally appear in the text are to existing regulatory guidelines related to the ICH harmonisation initiative.
66
Annex 17: Notes on the Format and Style of ICH Guidelines
The following guidance, which has been agreed by the ICH Steering Committee is intended to be provided to, and followed by, ICH Topic Regulatory Chairs and Rapporteurs when drawing up or revising ICH drafts. The objective is to adopt a more uniform structure for future ICH Guidelines. The format that is proposed is based on a review of the main elements in the existing guidelines. It is recommended that, as far as possible all guidelines should consist of the following sections:
Table of Contents
1. INTRODUCTION
1.1 Objective(s) of the Guideline
1.2 Background
1.3 Scope of the Guideline
1.4 General Principles
2. GUIDELINES
3 GLOSSARY
NOTES ON THE FORMAT
1. INTRODUCTION
1.1 Objective(s) of the Guideline
Brief introductory comments giving the purpose for which the guideline has been drawn up. (For new Topics this should be based on the “Problem Statement/Objectives” in the concept paper).
1.2 Background
This section should include information which helps to put the guideline into context, including, for example, whether it is to be read in conjunction with any other guideline (rule or regulation). It may be useful to highlight existing differences in requirements which are relevant to the objectives of the guidelines, but should not include subjective observations or statements which appear critical of existing requirements or practices.
1.3 Scope of the Guideline
This should normally be included in order to define the application of the guideline but it may not be appropriate for some Topics, and is therefore optional. The scope should state the range of tests and/or products to which the guideline applies, and state whether there are any exclusions. For example, the scope of the first Stability Guideline was limited to tests on new chemical entities and their formulations; existing formulations and variations were excluded.
1.4 General Principles
This section should be included if there are general considerations which apply to the whole technical area which is addressed by the guideline. For example, the Impurities guideline
67
includes a section on the “Classification of Impurities” which would be appropriate to a “General Principles” section.
2. GUIDELINES
This is the “operative” section of the text, giving specific recommendations and guidance on technical matters, for example the way in which tests should be carried out and/or the way data should be presented. The format and structure of the guideline (headings, sub-headings etc.) will vary according to the nature of the subject, but there should be a clear separation between:
Specific guidance and recommendations
Commentaries on the guidance
Notes and examples, which do not form part of the specific guidelines, should be clearly differentiated from the “operative” text. The extensive use of footnotes or end notes is not, however, recommended. It is preferable to include the supplementary information within the guideline but differentiate it by the use of a combination of formatting and headings (“Note:”, “Example:” etc.)
3. GLOSSARY
Consideration should always be given to the need for a glossary defining the way in which certain terms are used for the purpose of the guideline. Where possible, the glossary should be defined at the outset and it is important to cross-refer to Glossaries in all other ICH Guidelines in order to avoid conflicting definitions. A compilation of Glossary definitions from existing ICH Q, S, E and M Guidelines is available from the ICH Secretariat. If appropriate, an already existing definition should be used. In cases where more than one definition already exists, a unified definition should be created if deemed appropriate. The unified definition should be considered for use in subsequent guidelines if appropriate.
POINTS TO AVOID
References
References to published papers and the scientific literature are not usually appropriate for a document which is destined to end up as a regulatory guideline. The only references which should normally appear in the text are to existing regulatory guidelines related to the ICH harmonisation initiative.
Summary and Conclusions
It is not necessary to summarise the effect and implications of the guideline in the ICH text, as this is not appropriate for a document which is destined to become a regulatory guideline. The “Concept Paper” - suitably revised once the guideline has been developed - will provide the vehicle for a commentary on and résumé of the subject.
Description of the ICH process
Reference should not be made in the texts themselves to discussions in the Expert Working Group or the “Steps” in the ICH process. This will be given elsewhere. The texts are ultimately destined for publication as official regulatory guidelines, etc. where such information would not be appropriate.
Use of the term sex versus gender
68
Starting from October 29, 2009, the term sex should be used in all ICH documents and future revisions to denote the biogenetic differences that distinguish males and females.
STYLE
Guidelines should be in clear plain English free from jargon. Guidelines should normally be written in the “third person”, predominantly in the passive
tense. The imperative tense is not appropriate (e.g., “Retain all samples for 5 years”). The first person must never be used e.g., “In our opinion...” “We agreed....”
The final texts will become regulatory guidelines with a similar role to legal documents. Clarity is more important than literary style. The repetitive use of a phrase or word is acceptable in order to convey the correct meaning and changes should not be made merely for the sake of variety.
The following gives guidance on the use of the terms “must”, “should” and “may” in the English language drafts. Please note that translation of these terms into Japanese is particularly difficult as many similar terms exist. It is therefore important that the meaning is unambiguous in the original text.
must: (e.g., “Data must be provided on....”) is used where the item is mandatory. It would not normally be used in a “guideline” where, by definition, there should be flexibility. When a mandatory term is appropriate, “must” should be used in preference to “shall” (“Data shall be provided on....”)
should: (e.g., “Data should be provided on...”) is used where it is a strong recommendation, but not an absolute requirement. It leaves open the possibility that the requirement could be waived if there is adequate justification. The verb “ought” has a similar meaning but is rarely correct in a formal document (i.e., “Data ought to be provided on...” is not appropriate.)
may: (e.g., “Data may be provided on ...”) is used where there is a clear option regarding the provision of information. Such statements would often be qualified by an “if” clause. (“Data may be needed to demonstrate ... if .......”). The verb “might” has a similar meaning, but implies a slightly weaker option (i.e., “Data might be needed on ...”).
69
EXAMPLES FROM EXISTING GUIDELINES
1. INTRODUCTION
1.1 Objective(s) of the Guideline
Example taken from Clinical Trials in Special Populations: Geriatrics It is important to ensure that clinical testing programs are carried out according to harmonised guidelines based on agreed ethical and scientific principles so that the international development of valuable innovative drugs is achieved with maximum efficiency. Harmonisation in relation to medicines for geriatric populations is an important issue because the total population of the elderly will increase significantly in the coming years in Europe, Japan and the USA. The use of drugs in this population requires special consideration due to the frequent occurrence of underlying diseases, concomitant drug therapy and the consequent risk of drug interaction.
1.2 Background
Example taken from: Pharmacokinetics: Guidance for Repeated Dose Tissue Distribution Studies
A comprehensive knowledge of the absorption, distribution, metabolism, and elimination of a compound is important for the interpretation of pharmacology and toxicology studies. Tissue distribution studies are essential in providing information on distribution and accumulation of the compound and/or metabolites especially in relation to potential sites of action. This information may be useful for designing toxicology and pharmacology studies and for interpreting the results of these experiments. In the EU, US, and Japan, there has been a general agreement on the need to conduct single dose tissue distribution studies as part of the preclinical package. These studies often provide sufficient information about tissue distribution. There has been no consistent requirement for repeated dose tissue distribution studies. However, there may be circumstances when assessments after repeated dosing may yield important information. This paper provides guidance on circumstances when repeat dose tissue distribution studies should be considered and on the conduct of such studies.
1.3 Scope of the Guideline
Example taken from Stability Testing of New Drugs and Products
The guideline primarily addresses the information required in Registration Applications for new molecular entities and associated drug products. This guideline does not currently seek to cover the information required for abbreviated or abridged applications, variations, clinical trial applications, etc. The choice of test conditions defined in this guideline is based on an analysis of the effects of climatic conditions in the three areas of the EU, Japan and the USA.
1.4 General Principles
Example taken from Impurities in New Drug Substances
Impurities may be classified into the following categories: Organic Impurities (Process and Drug Related)
Inorganic Impurities
Residual Solvents
Organic impurities may arise during the manufacturing process and/or storage of the new drug substance. They may be identified or unidentified, volatile or non-volatile, and include: Starting Materials
70
By-Products
Intermediates
Degradation Products
Reagents, Ligands and Catalysts
Inorganic impurities may derive from the manufacturing process. They are normally known and identified and include: Reagents, Ligands and Catalysts
Heavy Metals
Inorganic Salts
Other Materials (e.g., Filter Aids, Charcoal, etc.) Solvents are organic or inorganic liquids used during the manufacturing process. Since these are generally of known toxicity, the selection of appropriate controls is easily accomplished.
II. GUIDELINES
Example taken from Carcinogenicity: Dose Selection for Carcinogenicity Studies of Therapeutic Agents edited to include the Note with the text.
Pharmacodynamic endpoints in high dose selection The utility and safety of many therapeutics depend on their pharmacodynamic receptor selectivity. Pharmacodynamic endpoints for high dose selection will be highly compound-specific and are considered for individual study designs based on scientific merits (Note 10). The high dose selected should not produce disturbances of physiology or homeostasis but should produce a pharmacodynamic response in dosed animals which would preclude further dose escalation and compromise the validity of the study.
Note
When using any new endpoint, either pharmacokinetic, pharmacodynamic or toxicity based for high dose selection it is necessary to carefully consider, prior to carcinogenicity study initiation, if the endpoint can insure the acceptability of the carcinogenicity study.
III. GLOSSARY
Example taken from Reproductive Toxicology: Detection of Toxicity to Reproduction for Medicinal Products
Developmental toxicity: Any adverse effect induced prior to attainment of adult life. It includes effects induced or manifested in the embryonic or fetal period and those induced or manifested post-natally. Embryotoxicity, fetotoxicity, embryo-fetal toxicity: Any adverse effect on the conceptus resulting from prenatal exposure, including structural or functional abnormalities or postnatal manifestations of such effects. Terms like "embryotoxicity", "fetotoxicity" relate to the time point/-period of induction of adverse effects, irrespective of the time of detection.
Annex 18: Templates for Steps 1, 2a, 2b, 3 and Step 4 Sign-off Sheets, including template for Sign-off Sheet where Step 4 is reached without Step 2/Step 3
Topic Reference: STEP 1 – EXPERTS
For Working Groups established PRIOR June 2014
GUIDELINE TITLE
Consensus on a technical document to be submitted to the ICH Steering Committee under Step 1 of the ICH Process
Step 1 technical document signed-off by the DESIGNATED EXPERTS FROM THE ICH EXPERT WORKING GROUP
The official ICH procedure specifies that a Step 1 technical document can be submitted to the SC for endorsement when the designated experts of the six ICH parties reach consensus
57 Due to its structure and the representation of both CBER and CDER in ICH, FDA may nominate two Topic Leaders, one per Center (depending on the scope of the topic).
Topic Reference: STEP 1 – EXPERTS
For Working Groups established PRIOR June 2014
GUIDELINE TITLE
All additional experts who participate in the Working Group are invited to sign-off the Step 1 technical document in recognition of their contribution to the discussion.
The signatories of Interested Parties who participated in the working group will be included.
The signatories of RHIs/DRAs/DoH technical experts will be included if nominated to participate in the Working Group.
Topic Reference: STEP 1 – EXPERTS
For Working Groups established AFTER June 2014
GUIDELINE TITLE
Consensus on a technical document to be submitted to the ICH Steering Committee under Step 1 of the ICH Process
Step 1 technical document signed-off by the DESIGNATED EXPERTS FROM THE ICH EXPERT WORKING GROUP
The official ICH procedure specifies that a Step 1 technical document can be submitted to the SC for endorsement when the designated experts of the ICH parties reach consensus and sign
All additional experts who participate in the Working Group are invited to sign-off the Step 1 technical document in recognition of their contribution to the discussion.
FDA ................................................................................ .................................................................. ................................
Health Canada ..................................................... ........................................... .....................
58 At Step 2b, the Steering Committee transmits the document to the regional regulatory agencies for wider, formal consultation in accordance with their normal internal or external consultation procedure.
Topic Reference: STEP 3 – REGULATORY EXPERTS
For Working Groups established PRIOR June 2014
GUIDELINE TITLE
Conclusion of Step 3 of the ICH Process59 Step 3 experts draft Guideline signed-off by the
DESIGNATED EXPERTS FROM THE ICH EXPERT WORKING GROUP
The official ICH procedure specifies that a Step 3 experts draft Guideline can be submitted to the SC for adoption as an ICH Harmonised Guideline when the designated
experts of the ICH Regulatory Parties reach consensus and sign the Step 3.
59 The comments received by the ICH Regulatory Parties on the regional consultation on the Step 2b Guideline have been considered for the preparation of a Step 3 experts draft Guideline which, once signed-off by the experts designated by the regulatory parties, will be submitted to the Steering Committee for adoption as a harmonised guideline (Step 4 of the Process). 60 Due to its structure and the representation of both CBER and CDER in ICH, FDA may nominate two Topic Leaders, one per Center (depending on the scope of the topic).
Topic Reference: STEP 3 – REGULATORY EXPERTS
For Working Groups established PRIOR June 2014
GUIDELINE TITLE
All additional experts who participate in the Working Group are invited to sign the Step 3 Experts Draft Guideline in recognition of their contribution to the discussion.
The signatories of RHIs/DRAs/DoH technical experts will be included if nominated to participate in the Working Group.
Topic Reference: STEP 3 – REGULATORY EXPERTS
For Working Groups established AFTER June 2014
GUIDELINE TITLE
Conclusion of Step 3 of the ICH Process61 Step 3 experts draft Guideline signed-off by the
DESIGNATED EXPERTS FROM THE ICH EXPERT WORKING GROUP
The official ICH procedure specifies that a Step 3 experts draft Guideline can be submitted to the SC for adoption as an ICH Harmonised Guideline when the designated
experts of the ICH Regulatory Parties reach consensus and sign the Step 3.
Health Canada ................................................... .......................................... .....................
61 The comments received by the ICH Regulatory Parties on the regional consultation on the Step 2b Guideline have been considered for the preparation of a Step 3 experts draft Guideline which, once signed-off by the experts designated by the regulatory parties, will be submitted to the Steering Committee for adoption as a harmonised guideline (Step 4 of the Process). 62 Due to its structure and the representation of both CBER and CDER in ICH, FDA may nominate two Topic Leaders, one per Center (depending on the scope of the topic).
All additional experts who participate in the Working Group are invited to sign the Step 3 Experts Draft Guideline in recognition of their contribution to the discussion.
63 Step 4 is reached following regional and national consultation (Step 3), when the Step 3 experts draft Guideline has been signed-off by the experts designated by the regulatory parties (Step 3 Regulatory Experts) and the harmonised Guideline is recommended for adoption to the regulatory bodies.
Topic Reference: STEP 3 – REGULATORY EXPERTS
For Working Groups established PRIOR June 2014
TITLE
Step 3 of the ICH Process64 without public consultation
Step 3 Experts Document signed-off by the
DESIGNATED EXPERTS FROM THE ICH EXPERT WORKING GROUP
The official ICH procedure specifies that a Step 3 Document can be submitted to the SC
for adoption as an ICH Harmonised Guideline when the Designated Experts of the ICH
regulatory parties reach consensus and sign the Step 3.
64 Once signed-off by the experts designated by the regulatory parties, this document will be
submitted to the Steering Committee for adoption as a harmonised guideline (Step 4) without public consultation.
65 Due to its structure and the representation of both CBER and CDER in ICH, FDA may nominate two Topic Leaders, one per Center (depending on the scope of the topic).
Topic Reference: STEP 3 – REGULATORY EXPERTS
For Working Groups established PRIOR June 2014
TITLE
Step 3 of the ICH Process without public consultation
All additional experts who participate in the Working Group are invited to sign the Step
3
Experts Draft Guideline in recognition of their contribution to the discussion.
67 Step 4 is reached - in this case without regional and national consultation - when the Step 3 Experts Draft Guideline has been signed-off by the experts designated by the regulatory parties (Step 3 Regulatory Experts) and the harmonised guideline is recommended for adoption to the regulatory bodies.
Annex 19: General principles to consider for the organisation of ICH meetings
The following bullets outline general principles agreed by the ICH Steering Committee (SC) at its
meeting in Tallinn, Estonia in June 2010 aimed at strengthening the procedures for the organisation
of ICH meetings. It is acknowledged that there may be exceptions to these principles:
Effort should be made to adhere to a 6-month cycle for ICH meetings (allowing an appropriate
regulatory consultation period);
Coordinators tele/webconference should be organised at least 3 months before an ICH meeting
and the SC tele/webconference six to eight weeks before;
EWGs should be requested to update their work plans (see Annex 18 for work plan template)
prior to Coordinator/SC tele/webconferences to enable Coordinators/SC to make informed
decisions ;
Twelve should be considered as the cap for the number of parallel WGs which can be approved
to meet, not including meetings such as SC, MedDRA Management Board, Regulators Forum
etc....;
The SC should try to confirm at ICH SC meetings whether or not a WG should meet face-to-
face at the next ICH meeting - the agreed cap on the number of WGs which can meet (see above)
should be respected;
The SC tele/webconference (six to eight weeks before) should be the cut-off point for the
approval of WGs to help the host with preparations for meetings and allow experts to finalise
travel plans;
Before any decisions are taken in relation to new topics (e.g., approval of new Concept Papers),
substantive SC discussion should take place, preferentially at a face-to-face SC meeting;
Documents for approval (e.g., Concept Papers) should be circulated at least two weeks before
meetings to facilitate consultation with experts/committees and preferably at least four weeks
before meetings if the topic of concern is tabled for SC discussion for the first time.
Annex 20: Impact of Swissmedic and Health Canada Promotion to SC Member
Once agreement will be reached on the ICH reforms, a major review of the ICH Procedures will be
conducted. During the transitional period, flexibility will be applied.
The SC regulators agreed to make a few changes to the ICH Procedures, effective as of the Lisbon
meeting held in November 2014, to take into consideration the promotion of Swissmedic and Health
Canada as SC Members in June 2014. The following describes the procedures that will apply:
(1) For Working Groups established prior to June 2014:
The current rules for Experts sign-off will apply. The technical experts of Health Canada
and Swissmedic will remain invited to sign-off the Step 1 Technical Document and Step 3
Experts Draft Guideline as ICH Observers;
The SC Members of Health Canada and Swissmedic will now be invited to sign-off the Step
2a Final Technical Document / 2b Draft Guideline / 4 Harmonised Guideline as ICH
Parties.
(2) For Working Groups established from June 2014:
The SC Members of Health Canada and Swissmedic will be invited to confirm which
Working Group(s) (established since their promotion as SC Members) they will contribute
actively by nominating technical expert(s) to the Working Group;
For Working Group(s) selected by the SC Members of Health Canada and Swissmedic, their
technical expert(s) will be invited to sign-off the Step 1 and Step 3 as ICH Parties;
Irrespective of whether or not Health Canada and Swissmedic will have appointed technical
experts in the Working Group, the SC Members of Health Canada and Swissmedic will be
invited to sign-off the Step 2a / 2b / 4 as ICH Parties.
Consensus building approach:
At the level of the Working Groups, if Health Canada and Swissmedic nominate technical experts,
it is expected that they will participate actively in the efforts of the Working Group with their views
taken into consideration as is the case for the six Founding SC Parties.
If a nominated Health Canada or Swissmedic expert exceptionally is not participating in a Working
Group meeting, the work of the group can still progress. It is nonetheless the expectation that all
permanent Members will attend all face-to-face meetings. Health Canada and Swissmedic commit
to support the progress made by the group.
As per the current procedures, if at any time during the step process, there are issues encountered
by the Working Group to reach consensus, the Regulatory Chair will elevate it at the SC level for
consideration and direction. If following further expert discussion based on SC direction, consensus
can still not be reached at the expert level, the Regulatory Chair will again elevate it at the SC level.
In the unlikely situation where consensus cannot be reached but the three ICH SC Founding
Regulatory Parties (EU, FDA and MHLW) are in agreement, they will proceed with the Step sign-
off.
Information on groups in which Health Canada and Swissmedic are not participating:
Health Canada and Swissmedic are not obliged to send experts to all Working Groups. For those
Working Groups to which they are not sending experts, appropriate communication / information
sharing68 on the progress of the work of these groups will be set up by the ICH Secretariat.
Such a mechanism will assist Health Canada and Swissmedic to take informed decisions at the SC
level and provide a basis for the implementation of the guidelines.
In addition, such a mechanism could serve the same purpose for future members of the Assembly,
who will only send experts to a few Working Groups.
68 E.g. inclusion in e-mail distribution lists, access to working documents, telephone conferences or informational webinars during the development process