ORIGINAL RESEARCH Identification and Analyses of miRNA Genes in Allotetraploid Gossypium hirsutum Fiber Cells Based on the Sequenced Diploid G. raimondii Genome Qin Li a , Xiang Jin a , Yu-Xian Zhu a,b, * a The State Key Laboratory of Protein and Plant Gene Research, College of Life Sciences, Peking University, Beijing 100871, China b National Center for Plant Gene Research (Beijing), Beijing 100101, China Received 13 February 2012; revised 25 April 2012; accepted 25 April 2012 Available online 18 May 2012 ABSTRACT The plant genome possesses a large number of microRNAs (miRNAs) mainly 21e24 nucleotides in length. They play a vital role in regulation of target gene expression at various stages throughout the whole plant life cycle. Here we sequenced and analyzed w10 million non-coding RNAs (ncRNAs) derived from fiber tissue of the allotetraploid cotton (Gossypium hirsutum) 7 days post-anthesis using ncRNA-seq technology. In terms of distinct reads, 24 nt ncRNA is by far the dominant species, followed by 21 nt and 23 nt ncRNAs. Using ab initio prediction, we identified and characterized a total of 562 candidate miRNA gene loci on the recently assembled D 5 genome of the diploid cotton G. raimondii. Of all the 562 predicted miRNAs, 22 were previously discovered in cotton species and 187 had sequence conservation and homology to homologous miRNAs of other plant species. Nucleotide bias analysis showed that the 9th and 1st positions were significantly conserved among different types of miRNA genes. Among the 463 putative miRNA target genes, most significant up/down-regulation occurred in 10e20 days post-anthesis, indicating that miRNAs played an important role during the elongation and secondary cell wall synthesis stages of cotton fiber development. The discovery of new miRNA genes will help understand the mechanisms of miRNA generation and regulation in cotton. KEYWORDS: Cotton; Genome; Micro RNA; Deep sequencing; Microarray 1. INTRODUCTION Cotton belongs to the genus Gossypium, which contains 5 tetraploid (2n ¼ 4x) and more than 45 diploid (2n ¼ 2x) species (Bowers et al., 2003). Upland cotton (Gossypium hirsutum, AADD, 2n ¼ 4x ¼ 52), constituting over 90% of the world’s cotton lint production, is thought to have undergone an allopolyploidization event about 1e2 million years ago (MYA), involving both A and D genome species (Wendel and Albert, 1992). The progenitor of G. raimondii (D 5 D 5 , 2n ¼ 2x ¼ 26) is considered the contributor of the D sub- genome while ancestors of G. arboreum (AA, 2n ¼ 2x ¼ 26) may contributed the A sub-genome to G. hirsutum (Paterson et al., 2004; Chen et al., 2007). Cotton fibers are single-cell trichomes differentiated from ovule epidermis. Indexed by the number of days post-anthesis (DPA), fiber development process consists of four distinctive but overlapping stages: initiation (0e3 DPA), elongation (3e20 DPA), secondary cell wall deposition (15e45 DPA) and maturation (40e60 DPA) (Kim and Triplett, 2001; Ji et al., 2003). The unicellular structure of cotton fiber cell renders it an ideal model for studying cell elongation, differentiation and also for deciphering the regulatory machinery involved in cellulose biosynthesis (Qin and Zhu, 2011). Based on microarray hybridization of 12,233 Uni-ESTs obtained from * Corresponding author. Tel: þ86 10 6275 1193. E-mail address: [email protected](Y.-X. Zhu). Available online at www.sciencedirect.com Journal of Genetics and Genomics 39 (2012) 351e360 JGG 1673-8527/$ - see front matter Copyright Ó 2012, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, and Genetics Society of China. Published by Elsevier Limited and Science Press. All rights reserved. doi:10.1016/j.jgg.2012.04.008
Transcript
Available online at www.sciencedirect.com
Journal of Genetics and Genomics 39 (2012) 351e360
JGG
ORIGINAL RESEARCH
Identification and Analyses of miRNA Genes in AllotetraploidGossypium hirsutum Fiber Cells Based on the Sequenced Diploid
G. raimondii Genome
Qin Li a, Xiang Jin a, Yu-Xian Zhu a,b,*
a The State Key Laboratory of Protein and Plant Gene Research, College of Life Sciences, Peking University, Beijing 100871, ChinabNational Center for Plant Gene Research (Beijing), Beijing 100101, China
Received 13 February 2012; revised 25 April 2012; accepted 25 April 2012
Available online 18 May 2012
ABSTRACT
The plant genome possesses a large number of microRNAs (miRNAs) mainly 21e24 nucleotides in length. They play a vital role inregulation of target gene expression at various stages throughout the whole plant life cycle. Here we sequenced and analyzed w10 millionnon-coding RNAs (ncRNAs) derived from fiber tissue of the allotetraploid cotton (Gossypium hirsutum) 7 days post-anthesis usingncRNA-seq technology. In terms of distinct reads, 24 nt ncRNA is by far the dominant species, followed by 21 nt and 23 nt ncRNAs.Using ab initio prediction, we identified and characterized a total of 562 candidate miRNA gene loci on the recently assembled D5
genome of the diploid cotton G. raimondii. Of all the 562 predicted miRNAs, 22 were previously discovered in cotton species and 187 hadsequence conservation and homology to homologous miRNAs of other plant species. Nucleotide bias analysis showed that the 9th and 1stpositions were significantly conserved among different types of miRNA genes. Among the 463 putative miRNA target genes, mostsignificant up/down-regulation occurred in 10e20 days post-anthesis, indicating that miRNAs played an important role during theelongation and secondary cell wall synthesis stages of cotton fiber development. The discovery of new miRNA genes will help understandthe mechanisms of miRNA generation and regulation in cotton.
KEYWORDS: Cotton; Genome; Micro RNA; Deep sequencing; Microarray
1. INTRODUCTION
Cotton belongs to the genus Gossypium, which contains 5tetraploid (2n ¼ 4x) and more than 45 diploid (2n ¼ 2x)species (Bowers et al., 2003). Upland cotton (Gossypiumhirsutum, AADD, 2n ¼ 4x ¼ 52), constituting over 90% of theworld’s cotton lint production, is thought to have undergonean allopolyploidization event about 1e2 million years ago(MYA), involving both A and D genome species (Wendel andAlbert, 1992). The progenitor of G. raimondii (D5D5,2n ¼ 2x ¼ 26) is considered the contributor of the D sub-
1673-8527/$ - see front matter Copyright � 2012, Institute of Genetics and Develop
Published by Elsevier Limited and Science Press. All rights reserved.
doi:10.1016/j.jgg.2012.04.008
genome while ancestors of G. arboreum (AA, 2n ¼ 2x ¼26) may contributed the A sub-genome to G. hirsutum(Paterson et al., 2004; Chen et al., 2007).
Cotton fibers are single-cell trichomes differentiated fromovule epidermis. Indexed by the number of days post-anthesis(DPA), fiber development process consists of four distinctivebut overlapping stages: initiation (0e3 DPA), elongation(3e20 DPA), secondary cell wall deposition (15e45 DPA) andmaturation (40e60 DPA) (Kim and Triplett, 2001; Ji et al.,2003). The unicellular structure of cotton fiber cell renders itan ideal model for studying cell elongation, differentiationand also for deciphering the regulatory machinery involvedin cellulose biosynthesis (Qin and Zhu, 2011). Based onmicroarray hybridization of 12,233 Uni-ESTs obtained from
mental Biology, Chinese Academy of Sciences, and Genetics Society of China.
352 Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
102,000 expressed sequence tags (ESTs) of cotton cDNAlibrary, we previously identified 778 cDNAs that were pref-erentially expressed during the fast fiber-elongation period,with ethylene biosynthesis being the most significantly up-regulated pathway (Shi et al., 2006). Further studies showedthat biosynthesis of very-long-chain fatty acids (VLCFAs),production of reactive oxygen species and pectin polymersmay also be required for fiber cell elongation (Qin et al., 2007;Mei et al., 2009; Pang et al., 2010).
MicroRNAs (miRNAs) are mainly 21e24 nucleotide-longnon-coding RNAs (Bartel, 2004; Baulcombe, 2004; Chapmanand Carrington, 2007). In general, miRNA gene loci ingenome are transcribed by RNA polymerase II into stem-loopprecursors (pre-miRNAs), which are excised by DICER-LIKEprotein in plants (Ambros et al., 2003; Kurihara andWatanabe, 2004). Mature miRNAs were then loaded intoAgonaute complexes for cleavage to produce 21e24nucleotide-long short sequences involved in target mRNAdegradation (Llave et al., 2002; Ambros et al., 2003; Li et al.,2010). Allen et al. (2005) revealed the miRNA-directedphasing of trans-acting siRNA biogenesis and gene regulationin plants. Chuck et al. (2007) reported that a tandem miRNAcaused the heterochronic maize mutant Corngrass1. The 22-nucleotide miR4376 was found to mediate the cleavage of anautoinhibited Ca2þ-ATPase, ACA10, which played a criticalrole in tomato reproductive growth (Wang et al., 2011). Also,miRNA was found to act as translational repressors of APE-TALA2duringArabidopsisflower development (Aukerman andSakai, 2003; Chen, 2004). Qiu et al. (2007) and Zhang et al.(2007) first identified miRNAs and their targets in cotton.Kwak et al. (2009) studied miRNAs expression during cottonfiber development in wild type (WT) and fuzz/lintless ( flmutantin the WT background) mutant using Solexa deep sequencing.
The fundamental feature of plant miRNAs is the preciseexcision of miRNA/miRNA* duplex from the stem of a stem-loop precursor which is predicted from genomic DNA orknown ESTs (Meyers et al., 2008). Due to the deficiencyof complete cotton genome sequences and limited ESTcoverage of non-coding transcriptome, only 34 stem-loopprecursors of G. hirsutum were recorded in miRBase(Griffiths-Jones et al., 2008; www.mirbase.org) comparedwith 291 of Arabidopsis thaliana, 362 of Glycine max, 635 ofMedicago truncatula and 234 of Populus trichocarpa (www.mirbase.org). Kwak et al. (2009) identified 22 conservedcandidate miRNA families including 111 members. UsingncRNA-seq and EST database, Pang et al. (2009) reported 25cotton miRNA precursors, of which 4 were identified for thefirst time. Wang et al. (2011) revealed 7 cotton fiberinitiation-related and 36 novel miRNAs. In this study, usingthe D5 sub-genome sequence as the template, we revealed theexistence of 562 cotton miRNA gene loci with authenticstem-loop structures on genome and expression evidence inribo-genome. We also analyzed the cotton transcriptome overdifferent fiber development stages to find the expressionpatterns of several sets of potential target genes that may beregulated by miRNAs.
2. MATERIALS AND METHODS
2.1. Plant growth and sample preparation
Cotton plants (G. hirsutum cv. Xuzhou 142) were grown ina fully automated walk-in growth rooms. Fiber tissue washarvested 7 days post-anthesis and frozen in liquid nitrogenuntil being used for RNA extraction. Total RNA from cottontissue was extracted using a modified protocol (Ji et al., 2003)excluding polyvinylpyrrolidone in the extraction buffer, whichwould cause RNA degradation.
2.2. Non-coding RNA deep sequencing
Small RNA molecules from 18 to 30 nucleotides in lengthwere amplified and isolated from 15% polyacrylamide gel.The purified sample was used directly for cluster generationand sequencing analysis using the Illumina Genome Analyzer(Illumina, Inc., USA) according to the manufacturer’sinstructions. Raw sequencing data was deposited to NCBIGEO (GSE27697).
2.3. miRNA identification
We first performed genome-wide scanning to detect inver-ted repeat sequences as candidates in the D5 sub-genome. Wethen used the RNAfold program of the Vienna RNA Packageto construct stem-loop structures of the putative pre-miRNAs.The maximal energy for stem-loop structure was set to�18 kCal/mol and at least 19 nucleotides must form pairs withwhat was found in the genome. Less than 3 asymmetric bulgeswere allowed in the secondary structure for miRNA identifi-cation. Candidate pre-miRNAs were mapped with ncRNA-seqdata using mireap (http://sourceforge.net/projects/mireap/) toestimate mature miRNA and miRNA* sequences. At last,candidate miRNAs were aligned with miRNAs from otherplant species in miRBase (Griffiths-Jones et al., 2008; www.mirbase.org) for sequence conservation and homology withmaximum mismatch of 3 nucleotides.
2.4. siRNA identification
Small interfering RNAs (siRNAs) are 22e24 nucleotideslong double-strand ncRNAs. Each strand of siRNA is 2 ntlonger than the other on the 30 end. We aligned raw sequencingreads with each other in pairs to find ncRNAs that satisfy thesecriteria.
2.5. Identification of other non-coding RNAs
For transfer RNAs (tRNAs), ribosomal RNAs (rRNAs),small nuclear RNAs (snRNAs) and small nucleolar RNAs(snoRNAs), we searched against Rfam (http://rfam.sanger.ac.uk) database and NCBI GenBank database using BLASTNsoftware (E -value < 0.01).
353Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
2.6. Genome mapping of non-coding RNAs
We used CASHX 2.3 software to map ncRNA-seq reads inthe genome of G. raimondii. We used perfect match parametersthat allowed no single mismatch in the sequence in question.Multi position mappings in the genome were treated asdifferent matches. Assembled G. raimondii genome sequenceswere obtained from the cotton genome browser (http://cotton.cbi.pku.edu.cn/).
2.7. miRNA targets prediction
Fig. 1. Length distribution of non-coding RNAs obtained by ncRNA-seq using
small RNA samples extracted from 7 DPA G. hirsutum fiber cells.
All ncRNAs with greater than 26 nucleotides in length (26e30 nt) were
combined because they constitute only a very small percentage.
We used psRNATarget (http://plantgrn.noble.org/psRNATarget/) to predict miRNA targets in upland cotton. MaturemiRNA sequences detected in ncRNA-seq data and supportedby genomic structures were used as queries to map cotton ESTdata with NCBI GenBank accession numbers DR452281eDR463972. The maximum energy allowed to be unpaired withthe target site was set to �25 kCal/mol with at least 20 nucle-otides perfectly mapped in the genomic sequence.
2.8. Analyses of predicted miRNA targets in cDNAmicroarray
We used cDNA microarray data with NCBI GEO accessionnumber GSE2901 to estimate the expression profiles ofpotential miRNA target genes. We used MeV 4.8 software(Saeed et al., 2006) with Hierarchical Clustering method togenerate gene clusters according to their expression profilechanges during different stages of cotton fiber development.
3. RESULTS
3.1. Length distribution of ncRNAs obtained fromG. hirsutum fiber cells
Table 1
Annotation and genome-wide mapping of non-coding RNAs
Type of
molecules
Raw reads Distinct
reads
Total length
(Mb)
% mapped
in the genome
miRNA 568,777 647 12.0 100
siRNA 477,336 88,300 10.6 49.1
To characterize ncRNAs in cotton fiber cells, we sequenceda library that was derived from total RNAs extracted from7 DPA G. hirsutum cv. Xuzhou 142 fibers. We produced11,582,792 raw sequence reads using Illumina GenomeAnalyzer (Table S1). After removal of adaptors sequences,contaminants, polyA sequences and reads shorter than 18nucleotides, we obtained 10,428,441 (90% of raw reads) cleanreads with 18e30 nucleotides in length (Tables S1 and S2).
In cotton fiber cells, the most abundant ncRNAs are 24nucleotides long,which take up48.4%of the total count, followedby 21 nt (14.7%), 23 nt (13.8%) and 22 nt (9.2%) species (Fig. 1and Table S2). In A. thaliana, 24 nt ncRNAs mainly consist ofsmall interfering RNAs (siRNAs) that were associated withrepeats and transposons in the genome (Lu, 2005). The highproportion of 24 nt ncRNA in fiber cells may suggest that siRNAsare also accumulated during rapid cotton fiber elongation.
rRNA 452,024 33,127 9.9 46.8
tRNA 402,235 11,533 0.7 78.4
3.2. Most ncRNAs can bemapped onto theD5 sub-genome
snRNA 1964 1107 0.04 62.2
snoRNA 1548 651 0.03 61.8
The annotated G. hirsutum ncRNAs sequences, including
miRNAs, siRNAs, rRNAs, tRNAs, snRNAs and snoRNAs,
snoRNAs, were aligned with the recently assembled 775.2-Mb G. raimondii genome sequences. For accuracy, weallowed not even a single mismatch in the sequence. Asa result, 78.4% of the 402,235 annotated tRNAs were map-ped in the genome, followed by snRNAs (62.2%) andsnoRNAs (61.8%) (Table 1), indicating high sequenceconservation for these three ncRNA families between G.hirsutum and G. raimondii. 49.1% of the siRNAs and 46.8%of the rRNAs showed perfect matches in the D5 sub-genome(Table 1). Because all miRNAs were predicted with referenceto the G. raimondii genome to locate the stem-loop structure,they were 100% mapped in the D5 sub-genome (Table 1). Wealso mapped 25 cotton miRNA precursors reported by Panget al. (2009) with the D5 sub-genome sequences and 21matches were found in the genome with identity �99%(Table S3).
3.3. Identification of 562 candidate miRNA loci on D5
sub-genome and 187 homologous miRNA genes
The fundamental feature of plant miRNAs is the preciseexcision of miRNA/miRNA* duplex from the stem of
354 Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
a stem-loop precursor which is predicted primarily fromgenomic DNA sequences (Meyers et al., 2008). We scannedthe assembled 775.2-Mb D5 sub-genome for inverted repeatsequences as candidates of potential miRNA loci. Thesecandidates were then folded in lowest energy formation toconstruct stem-loop structures of putative pre-miRNAs. Wethen mapped all ncRNA-seq reads (w10 million) onto thestems of the putative stem-loop structures to estimate maturemiRNA and/or miRNA* sequences (Fig. 2 and Table S4). Atotal of 562 miRNA loci, that each of them formed classicalstem-loop structure and was supported by ncRNA-seq attranscription level, were identified from the D5 sub-genome.Among the 562 miRNA loci, 46 were identified withmiRNA* sequences (Table S4), 187 had sequence conserva-tion to homologous miRNAs of other plant species (Table S5)and 8 of them formed into miRNA/miRNA* duplexes withexact 2 nucleotide overhang (Fig. 2).
Fig. 2. Different stem-loop structures of eight pre-miRNAs with both mature miR
Red, mature miRNA sequence; green, miRNA* sequence. The number above o
ncRNA-seq.
3.4. Different miRNAs may have different nucleotidebiases along the sequence
As shown in Fig. 3, high G or C, especially G at the 9thnucleotide position is observed in all four kinds of miRNAsexcept for 21 nt long miRNAs. In 22 and 23 nt miRNAs,greater than 80% of the nucleotides at the 9th position is G orC and in 24 nt miRNAs (Fig. 3). Nucleotide bias at the 9thpositions, which constitute the “tail” of the “seed” sequence,may suggest that nucleotides at this position is important forassociation of miRNA sequences with the Agonaute complex.Usually, the sequence motif including the 2nd to the 9thnucleotides is called the “seed” sequence, which is thecore-binding region of miRNA to the Agonaute complex(Ambros et al., 2003). Also, analyses showed that the 1stnucleotide in 24 nt miRNAs tends to be G/C, whereas in allother miRNAs, this position is more likely occupied by U
NA and miRNA* strands detected in ncRNA-seq data set.
r below each strand represents number of reads obtained for the strand in
Fig. 3. Analyses of nucleotide bias at each position along 21 (A), 22 (B), 23 (C) and 24 nt (D) miRNAs and ncRNAs. , A; , U; , G; , C.
355Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
(Fig. 3). Nucleotide position 12 in 21-nt miRNAs and position13 in all other three miRNAs are highly conserved with G. Thelast two nucleotide positions in 21 and 23 nt miRNA speciesare often occupied by G, whereas in 22 and 24 nt miRNA
species, these positions are conserved with U or C (in case ofthe last position in 24 nt miRNA). We suggest that thesenucleotide positions might also play important roles duringmiRNA recognition and binding to Agonaute complex.
356 Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
3.5. Prediction and expression profiling of 463 miRNAtarget genes
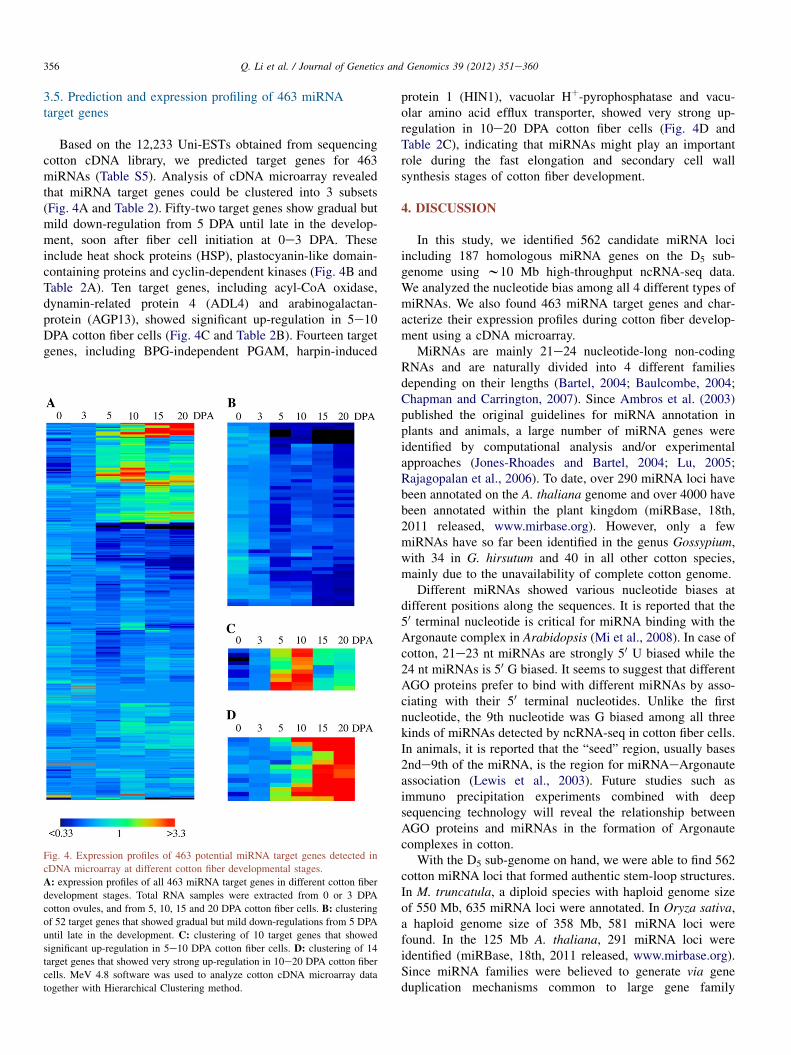
Based on the 12,233 Uni-ESTs obtained from sequencingcotton cDNA library, we predicted target genes for 463miRNAs (Table S5). Analysis of cDNA microarray revealedthat miRNA target genes could be clustered into 3 subsets(Fig. 4A and Table 2). Fifty-two target genes show gradual butmild down-regulation from 5 DPA until late in the develop-ment, soon after fiber cell initiation at 0e3 DPA. Theseinclude heat shock proteins (HSP), plastocyanin-like domain-containing proteins and cyclin-dependent kinases (Fig. 4B andTable 2A). Ten target genes, including acyl-CoA oxidase,dynamin-related protein 4 (ADL4) and arabinogalactan-protein (AGP13), showed significant up-regulation in 5e10DPA cotton fiber cells (Fig. 4C and Table 2B). Fourteen targetgenes, including BPG-independent PGAM, harpin-induced
Fig. 4. Expression profiles of 463 potential miRNA target genes detected in
cDNA microarray at different cotton fiber developmental stages.
A: expression profiles of all 463 miRNA target genes in different cotton fiber
development stages. Total RNA samples were extracted from 0 or 3 DPA
cotton ovules, and from 5, 10, 15 and 20 DPA cotton fiber cells. B: clustering
of 52 target genes that showed gradual but mild down-regulations from 5 DPA
until late in the development. C: clustering of 10 target genes that showed
significant up-regulation in 5e10 DPA cotton fiber cells. D: clustering of 14
target genes that showed very strong up-regulation in 10e20 DPA cotton fiber
cells. MeV 4.8 software was used to analyze cotton cDNA microarray data
together with Hierarchical Clustering method.
protein 1 (HIN1), vacuolar Hþ-pyrophosphatase and vacu-olar amino acid efflux transporter, showed very strong up-regulation in 10e20 DPA cotton fiber cells (Fig. 4D andTable 2C), indicating that miRNAs might play an importantrole during the fast elongation and secondary cell wallsynthesis stages of cotton fiber development.
4. DISCUSSION
In this study, we identified 562 candidate miRNA lociincluding 187 homologous miRNA genes on the D5 sub-genome using w10 Mb high-throughput ncRNA-seq data.We analyzed the nucleotide bias among all 4 different types ofmiRNAs. We also found 463 miRNA target genes and char-acterize their expression profiles during cotton fiber develop-ment using a cDNA microarray.
MiRNAs are mainly 21e24 nucleotide-long non-codingRNAs and are naturally divided into 4 different familiesdepending on their lengths (Bartel, 2004; Baulcombe, 2004;Chapman and Carrington, 2007). Since Ambros et al. (2003)published the original guidelines for miRNA annotation inplants and animals, a large number of miRNA genes wereidentified by computational analysis and/or experimentalapproaches (Jones-Rhoades and Bartel, 2004; Lu, 2005;Rajagopalan et al., 2006). To date, over 290 miRNA loci havebeen annotated on the A. thaliana genome and over 4000 havebeen annotated within the plant kingdom (miRBase, 18th,2011 released, www.mirbase.org). However, only a fewmiRNAs have so far been identified in the genus Gossypium,with 34 in G. hirsutum and 40 in all other cotton species,mainly due to the unavailability of complete cotton genome.
Different miRNAs showed various nucleotide biases atdifferent positions along the sequences. It is reported that the50 terminal nucleotide is critical for miRNA binding with theArgonaute complex in Arabidopsis (Mi et al., 2008). In case ofcotton, 21e23 nt miRNAs are strongly 50 U biased while the24 nt miRNAs is 50 G biased. It seems to suggest that differentAGO proteins prefer to bind with different miRNAs by asso-ciating with their 50 terminal nucleotides. Unlike the firstnucleotide, the 9th nucleotide was G biased among all threekinds of miRNAs detected by ncRNA-seq in cotton fiber cells.In animals, it is reported that the “seed” region, usually bases2nde9th of the miRNA, is the region for miRNAeArgonauteassociation (Lewis et al., 2003). Future studies such asimmuno precipitation experiments combined with deepsequencing technology will reveal the relationship betweenAGO proteins and miRNAs in the formation of Argonautecomplexes in cotton.
With the D5 sub-genome on hand, we were able to find 562cotton miRNA loci that formed authentic stem-loop structures.In M. truncatula, a diploid species with haploid genome sizeof 550 Mb, 635 miRNA loci were annotated. In Oryza sativa,a haploid genome size of 358 Mb, 581 miRNA loci werefound. In the 125 Mb A. thaliana, 291 miRNA loci wereidentified (miRBase, 18th, 2011 released, www.mirbase.org).Since miRNA families were believed to generate via geneduplication mechanisms common to large gene family
CM023A03 1.40E-03 Nodulin MtN21 family protein [Arabidopsis thaliana]
P-value < 0.005.
358 Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
formation (Elemento et al., 2002), the number of miRNA lociobtained from the 880 Mb D5 sub-genome is comparablewith other plant species. Because similar whole genomeduplication (WGD) events were suggested to happen indifferent Gossypium species before the polyploidization of Aand D sub-genomes (Senchina et al., 2003; Blanc and Wolfe,2004; Fawcett et al., 2009), we predict that there will be moremiRNA loci on the larger A sub-genome (w1.7 Gb of haploidG. arboretum, Hendrix and Stewart, 2005) and the allotetra-ploid G. hirsutum genome might contain more than 1000miRNA loci.
The expression pattern of miRNAs varies greatly amongtissues and development stages in cotton (Kwak et al., 2009;
Pang et al., 2009; Wang et al., 2011). So using mixture ofcotton fibers in different development stages, or even differenttissues would be helpful for better profiling of cotton miRNAs.
A recent study predicted 223 targets of cotton miRNAsfrom the expressed sequence tags derived mainly from cottonfibers and ovules in the early stages of fiber development (�3DPA to 7 DPA) (Pang et al., 2009). They pointed out thatmany miRNAs accumulating at lower levels in fibers (7 DPA)and fiber-bearing ovules (3 DPA) than immature ovules (�3DPA) may active the expression of target genes, such asNAM-like, ARF3/ARF4-like, Class III HA-zip protein-likeand TAS3-like, which were required for fiber cell elongation.Accordingly, we reported 24 miRNA target genes that were
359Q. Li et al. / Journal of Genetics and Genomics 39 (2012) 351e360
strongly up-regulated either in 5e10 DPA or 10e20 DPAcotton ovules (Table 2). Some of the potential miRNA targetgenes were previously known to be required for fiber devel-opment. For example, the harpin-induced protein 1 (HIN1)was involved in multiple signaling pathways including sali-cylic acid, jasmonic acid and ethylene in cotton (Miao et al.,2010). The vacuolar Hþ-pyrophosphatase (AVP1) gene wasable to increase fiber yield in field conditions (Pasapula et al.,2011). The dynamin-related protein, which is a GTP bindingprotein required for membrane trafficking (Kang et al., 2003),was associated with cotton fiber development in a chromo-somal substitution line (CS-B22sh) (Wu et al., 2008). Theseresults collectively suggest that miRNAs may serve as nega-tive regulators of target genes mainly in the elongation andsecondary cell wall synthesis stages and are important for thequality of fiber cells.
SUPPLEMENTARY DATA
Table S1. Deep sequencing and primary analyses of G.hirsutum ncRNAs.
Table S2. Length distribution of ncRNAs detected by deepsequencing.
Table S3. Sequence alignment of 21 cotton miRNAprecursors with D5 sub-genome.
Table S4. 562 miRNA precursors predicted on D sub-genome sequences using ncRNA-seq.
Table S5. Expression analyses, ortholog classificationand target prediction of strand-specific and strand-unspecificmiRNAs.
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.jgg.2012.04.008.
REFERENCES
Allen, E., Xie, Z., Gustafson, A.M., Carrington, J.C., 2005. microRNA-
directed phasing during trans-acting siRNA biogenesis in plants. Cell 121,