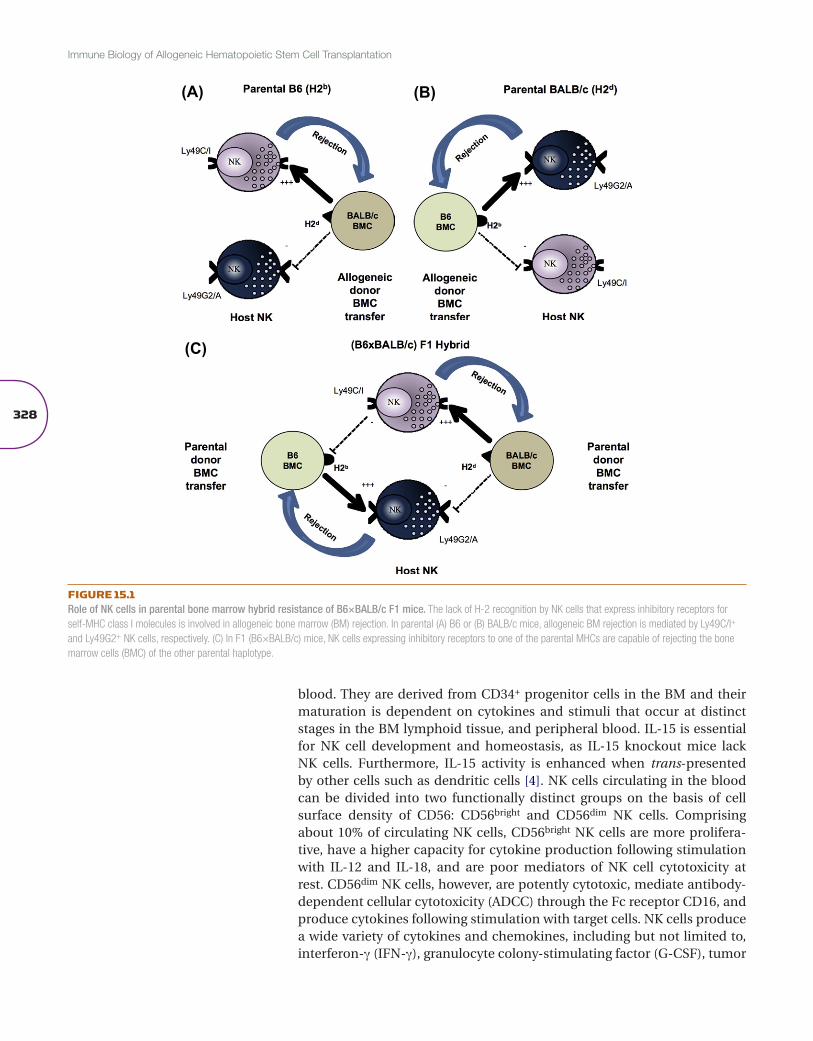
Natural killer (NK) cells were first described in mice in 1964 as an activ-ity by which lethally irradiated mice and mice without prior sensitization could resist bone marrow (BM) allografts [1]. Furthermore, this rejection ability did not follow the classical laws of transplantation in that paren-tal BM allografts were resisted by F1 hybrid recipients in a phenomenon termed “hybrid resistance” [2] (Figure 15.1). The radioresistant host effec-tor cells were capable of mediating resistance to BM but not solid tissue allografts, suggesting that a new cell type existed. The demonstration of human NK cell activity, first detected in vitro in 1975 with human periph-eral blood mononuclear cells by non-major histocompatibility complex (MHC)-restricted cytotoxicity toward transformed or virally infected target cells, heralded the era of NK cell biology. The unique receptor system uti-lized by these cells would later account for the “peculiar” immunogenet-ics of BM allograft resistance. It was these early experiments that led Karre and colleagues to formulate the “missing self hypothesis”, in which NK cell cytotoxicity is triggered by the loss of MHC class I on the tumor cells [3]. Fol-lowing this observation various families of receptors were identified on NK cells that recognize MHC class I that mediate tolerance in the host. Because of their ability to lyse tumors with aberrant MHC class I expression and pro-duce cytokines and chemokines upon activation, NK cells have immense therapeutic potential to treat cancer.
In humans, NK cells are identified by the expression of the cell adhe-sion marker CD56 and lack of the T-cell receptor, CD3, and are found in many tissues, including the spleen, BM lymph nodes, and peripheral
ch 15Natural killer cells in graft-
versus-host disease and graft-versus-leukemia
Bree FoleyDepartment of Hematology, Oncology and Transplantation, University of Minnesota, Minneapolis, Minnesota, USA
Maite AlvarezDepartment of Dermatology, University of California at Davis School of Medicine, Sacramento, California, USA
William MurphyDepartment of Dermatology, University of California at Davis School of Medicine, Sacramento, California, USA
Jeffrey S MillerDivision of Hematology, Oncology and Transplantation, University of Minnesota, Minneapolis, Minnesota, USA
328
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
blood. They are derived from CD34+ progenitor cells in the BM and their maturation is dependent on cytokines and stimuli that occur at distinct stages in the BM lymphoid tissue, and peripheral blood. IL-15 is essential for NK cell development and homeostasis, as IL-15 knockout mice lack NK cells. Furthermore, IL-15 activity is enhanced when trans-presented by other cells such as dendritic cells [4]. NK cells circulating in the blood can be divided into two functionally distinct groups on the basis of cell surface density of CD56: CD56bright and CD56dim NK cells. Comprising about 10% of circulating NK cells, CD56bright NK cells are more prolifera-tive, have a higher capacity for cytokine production following stimulation with IL-12 and IL-18, and are poor mediators of NK cell cytotoxicity at rest. CD56dim NK cells, however, are potently cytotoxic, mediate antibody-dependent cellular cytotoxicity (ADCC) through the Fc receptor CD16, and produce cytokines following stimulation with target cells. NK cells produce a wide variety of cytokines and chemokines, including but not limited to, interferon-γ (IFN-γ), granulocyte colony-stimulating factor (G-CSF), tumor
(A) (B)
(C)
FIGURE 15.1Role of NK cells in parental bone marrow hybrid resistance of B6×BALB/c F1 mice. The lack of H-2 recognition by NK cells that express inhibitory receptors for self-MHC class I molecules is involved in allogeneic bone marrow (BM) rejection. In parental (A) B6 or (B) BALB/c mice, allogeneic BM rejection is mediated by Ly49C/I+ and Ly49G2+ NK cells, respectively. (C) In F1 (B6×BALB/c) mice, NK cells expressing inhibitory receptors to one of the parental MHCs are capable of rejecting the bone marrow cells (BMC) of the other parental haplotype.
329
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
necrosis factor-α (TNF-α), transforming growth factor-β, macrophage inflammatory protein-1β (MIP-1β), and RANTES, which can modulate the adaptive immune system, recruit and stimulate other immune cells, and stimulate or inhibit hematopoiesis. NK cells enhance the immune response by eliminating virally infected or tumor cells through direct killing, medi-ated by the release of perforin and granzyme, ADCC-mediated killing through CD16, or the induction of apoptosis through Fas ligand (FasL).
NK cell receptors
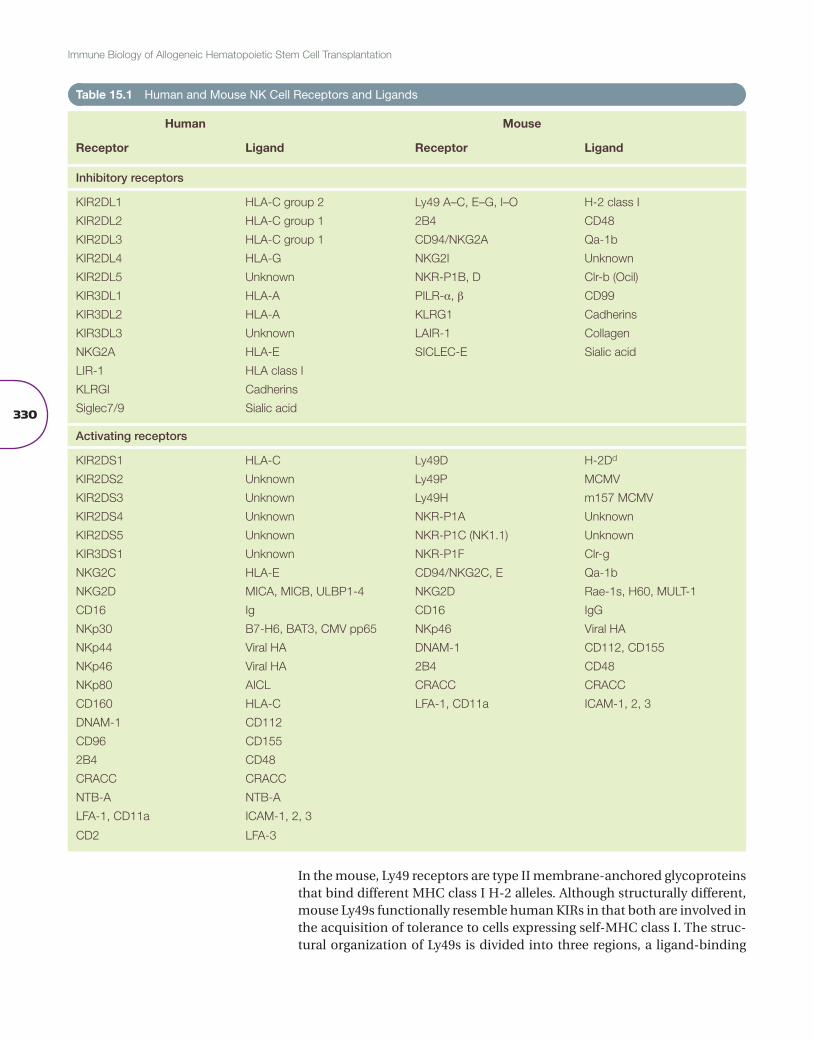
Under normal homeostatic conditions, a balance of activating and inhibitory signals tightly controls NK cell function (Table 15.1). The best-characterized family of NK cell receptors in humans is the inhibitory killer–immunoglob-ulin-like receptor (KIR) family. KIRs are type I transmembrane molecules belonging to the immunoglobulin (Ig) superfamily [5]. Genes encoding KIR are found on chromosome 19q13.4 in the leukocyte receptor complex (LRC) and are classified according to their number of immunoglobulin-like extracellular domains (two domains (2D) or three domains (3D)) and on the length of their cytoplasmic tail (long (L) or short (S)). KIRs that possess long cytoplasmic tails contain immunoreceptor tyrosine-based inhibi-tory motifs (ITIMs) and thus are inhibitory receptors. KIRs that have short cytoplasmic tails lack ITIMs, resulting in an activating function. Short-tail KIRs associate with the adaptor molecule DAP-12, which has an immuno-receptor tyrosine-based activation motif (ITAM) leading to cell activation if the KIR binds to its ligand. When the inhibitory KIRs engage their ligand, tyrosine kinase is phosphorylated, leading to the recruitment of SHP-1 or SHP-2, which in turn dephosphorylates protein substrates associated with activating receptors. KIR gene content differs between populations, but can be divided into two broad haplotypes, KIR A and KIR B haplotypes. KIR A haplotypes contain only one activating receptor, KIR2DS4, whereas KIR B haplotypes are far more varied, consisting of two or more activating recep-tors. Furthermore, KIR genes are highly polymorphic, with several alleles described for each KIR gene.
Inhibitory KIRs recognize allelic epitopes present on certain human leu-kocyte antigen (HLA) class I molecules. KIR2DL1, KIR2DL2, and KIR2DL3 recognize HLA-C alleles, with KIR2DL1 recognizing those with an Asn77 and Lys80 (HLA-C2) and KIR2DL2 and KIR2DL3 recognizing those with a Ser77 and Asn80 (HLA-C1). KIR3DL1 recognizes HLA-A and HLA-B alleles with the Bw4 motif. The ligands for KIR3DL2 remain controversial. HLA-A3 and HLA-A11 have been implicated as possible ligands, but KIR3DL2 recognition may be peptide specific. KIR2DL4 is an unusual KIR receptor as it expresses both an ITIM and an ITAM and has the capability of both activating and suppressing NK cell function. Its ligand is thought to be HLA-G.
Ligands for the activating KIRs have not been completely identified. KIR2DS1 can recognize HLA-C2 alleles, similar to its inhibitory counterpart, albeit with a much lower affinity [6]. Furthermore, KIR3DS1 has been linked to human immunodeficiency virus (HIV) progression [7] and several other activating KIRs have been linked to other infectious diseases [8]. However, current data suggest that KIR3DS1 does not directly use Bw4 as its ligand.
330
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
In the mouse, Ly49 receptors are type II membrane-anchored glycoproteins that bind different MHC class I H-2 alleles. Although structurally different, mouse Ly49s functionally resemble human KIRs in that both are involved in the acquisition of tolerance to cells expressing self-MHC class I. The struc-tural organization of Ly49s is divided into three regions, a ligand-binding
Table 15.1 Human and Mouse NK Cell Receptors and Ligands
Human Mouse
Receptor Ligand Receptor Ligand
Inhibitory receptors
KIR2DL1 HLA-C group 2 Ly49 A–C, E–G, I–O H-2 class I
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
extracellular domain, a transmembrane domain, and a cytoplasmic domain. The extensive allelic polymorphism observed in the Ly49 genes can be explained by the different regions being encoded in unique exons of the NK gene complex region in chromosome 6, which allows for multiple pos-sible rearrangements, provides high receptor diversification, and accounts for the many Ly49 receptors that have been identified [9,10]. Currently, 23 different Ly49 transcripts have been identified; among them, 13 Ly49 genes are considered inhibitory receptors based on the presence of ITIMs in their cytoplasmic domains (Ly49A, B, C, E, F, G, I, J, O, Q, S, T, and V) and 8 acti-vating because of the lack of an ITIM (Ly49D, H, L, M, P, R, U, and W) [10]. Within different mouse strains, Ly49 genes have shown high variability [9].
Several studies have suggested that Ly49 receptors can interact with mul-tiple H-2 alleles, though each may show a stronger affinity for a particular H-2 allele [11]. Ly49A, for example, exhibits a broad MHC class I cross-reactivity, but binds H-2Dd with the highest affinity. In contrast, other Ly49s, such as Ly49G2, show a more restrictive specificity for a particular MHC class I allele. It is quite interesting that Ly49D, an activating receptor, shares with the inhibitory Ly49A and Ly49G2 receptors affinity for H-2Dd. Ly49D reactivity against H-2Dd has been demonstrated through cytotoxic-ity assays in vitro and through its implication in H-2Dd BM allograft rejec-tion in vivo [12,13]. Affinity of Ly49D for H-2Dd may be lower than that of Ly49G2 and Ly49A or may require costimulation or coreceptors for its function [9]. Nevertheless, in H-2Dd strains, the coexpression of self-spe-cific inhibitory receptors was sufficient to allow self-tolerance of Ly49D+ NK cells.
The frequency and amount of Ly49 expression in a particular NK cell seem to be slightly influenced by H-2 expression in the host. However, because of the distinctive mechanism of gene regulation that results in randomized Ly49 expression, NK cells from a single host can express inhibitory recep-tors that bind different H-2 alleles, and therefore there are NK cells with inhibitory receptors able to recognize self-MHC and NK cells with inhibi-tory receptors that do not. To explain how NK activation with such a dif-ferential expression of Ly49s is regulated, a process known as NK licensing, which is discussed later, has been proposed [14].
Both humans and mice encode a family of C-type lectin-like receptors, known as the CD94/NKG2 receptor family. They are disulfide-linked heterodimers that are composed of an invariant common subunit, CD94. This is linked to a glycoprotein encoded by a gene of the NKG2 family [15]. The NKG2 family consists of four genes: NKG2A/B, NKG2C, NKG2E, and NKG2D/F (only in humans). CD94/NKG2A is an inhibitory receptor that binds to the geneti-cally invariant nonclassical MHC class I molecule—HLA-E in humans and Q1-ab in mice. As the expression of HLA-E is promoted by binding of pep-tides clipped from the leader sequence of classical HLA class I molecules, it is thought that HLA-E expression acts as a barometer of classical class I expression. The purpose of CD94/NKG2A may, therefore, be to monitor class I expression in a quantitative way, whereas KIR receptors monitor each allele individually. CD94/NKG2B appears to be a splice variant of NKG2A and also binds HLA-E. CD94/NKG2C and NKG2D are activating recep-tors. CD94/NKG2C also recognizes HLA-E, but, similar to Ly49D affinity
332
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
for H-2Dd, this recognition is weaker than for the inhibitory CD94/NKG2A receptor [16]. NKG2D is a homodimer and does not associate with CD94 like the other members of this family. NKG2D recognizes the nonclassical MHC molecules: MICA and MICB and other non-MHC molecules, ULBP1, ULBP2, and ULBP3, that are upregulated by a cell in times of stress, for example, in virus-infected cells or tumor cells. In the mouse NKG2D ligands are retinoic acid early inducible-1 (Rae-1), minor histocompatibility antigen H60, and the murine UL16-binding protein-like transcript-1 (MULT-1) [9].
Another family of immunoglobulin receptors, the leukocyte immunoglob-ulin-like receptors (LILR), are also expressed by NK cells as well as other immune cells [17], and the genes encoding these receptors are in the LRC. The LILRs deliver an inhibitory signal to NK cells. LILRB1 (LIR-1) expressed on NK cells binds to an array of classical and nonclassical HLA class I mol-ecules, including HLA-F and HLA-G. It has also been shown to bind to the cytomegalovirus (CMV)-encoded glycoprotein UL-18 [18].
The NCRs are a family of immunoglobulin-activating receptors and include the receptors NKp30 and NKp44 in humans and NKp46 in both human and mouse [19]. They have been shown to mediate NK cell cytotoxicity and lyse tumor cells. NKp30 and NKp46 are constitutively expressed on both resting and activated NK cells, whereas NKp44 is expressed only on activated NK cells. Their ligands are varied. NKp30 recognizes the tumor ligand B7-H6; the HLA-B-associated transcript 3 (BAT3), a nuclear protein released upon heat shock treatment; and the CMV protein pp65. When NKp30 engages with pp65 the NK cell is inhibited rather than activated, by dissociating CD3ζ from NKp30 and preventing activation. NKp44 and NKp46 both rec-ognize viral hemagglutinins [20,21].
Mouse and human NK cells also express other activating receptors. CD16, a member of the Fc family of receptors, binds to the Fc portion of IgG anti-bodies. Upon engagement CD16 mediates ADCC and lyses antibody-coated targets. NK cells also express a variety of activating receptors that recognize a wide range of diverse ligands [22]. DNAM-1 and CD96 have been shown to bind to CD112 and CD155, both poliovirus receptors, and mediate cell adhesion and increase NK cell killing. The glycoprotein CD160 interacts with HLA-C, triggering NK cell cytotoxicity. NKp80, a C-type lectin-like receptor, binds to AICL, which is expressed on myeloid cells and may be involved in triggering NK cell lysis of acute myeloid leukemia (AML). Finally, CD2, a cell adhesion molecule that binds to LFA-3, can act as a costimulatory molecule. A final family of receptors expressed on a wide range of cells, including NK cells, is the SLAM family of immunoglobulin-like receptors. NK cells express CD244 (2B4), which binds to CD48; this interaction generally results in NK cell activation but under certain conditions it can inhibit NK cell function [23]. Other SLAM receptors include NTB-A and CRACC, which are their own ligands, the interaction between them resulting in NK cell activation.
NK cell signaling
When an NK cell encounters a potential target, the NK cell must first adhere to the target and form an immune synapse [24]. In the absence of this adherence, NK cells are unable to lyse the cell. One example of an
333
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
adhesion marker, LFA-1, on the NK cell, can bind to ICAM-1 on the target cell and initiate signal transduction. Other NK cell receptors enhance NK cell adherence, such as CD2, CD16, and 2B4. Following adhesion to the target cell, activating receptors can now bind to their ligands. The ITAM-bearing activating receptors on NK cells use transmembrane adaptors that posses intracellular signaling domains to begin the activation cas-cade. CD16, NKp30, and NKp46 can dimerize with FcεRIγ or CD3ζ or both, whereas NKp44 uses DAP-12. The activating KIR, activating Ly49, NKG2C, and NKG2E all couple with DAP-12. Upon ligation, protein tyrosine kinases of the Src family phosphorylate ITAM-bearing subunits. This leads to recruitment of ZAP-70 and/or Syk, initiating further downstream signal-ing cascades, ultimately leading to polarization of perforin and granzyme-containing granules and release of these cytotoxic granules to lyse the target cell. The ITAM-dependent NKG2D receptor couples with DAP-10 in humans, whereas in mice it can couple to both DAP-12 and DAP-10. Upon ligation DAP-10 signaling recruits either p85 or Grb2, leading to the phos-phorylation of PI3K, when p85 binds, or SLP-76, Vav1, and PLC-y2, if Grb2 binds, ultimately leading to increased Ca2+ influx and release of cytotoxic granules [23]. Members of the SLAM family of receptors, which includes 2B4, contain a signaling motif called an immunoreceptor tyrosine-based switch motif (ITSM). Upon ligation the ITSM is recognized by three cyto-plasmic SH2-domain-containing adaptor molecules: SAP, EAT, and ERT. Mutation of SAP that results in loss of its function results in X-linked lym-phoproliferative disease. 2B4 signaling recruitment of SAP is thought to activate NK cells, whereas recruitment of EAT or ERT is thought to inhibit NK cell activation [23].
As NK cells express a wide range of activating receptors that interact with various adaptor molecules and signal through different pathways to medi-ate their effects, defects in one signaling pathway do not ultimately lead to dysfunction. Mice lacking the tyrosine kinases ZAP-70 and Syk, and there-fore having non-functional ITAMs, can still lyse tumor targets by presumably upregulating ITAM-independent signaling pathways. It is also apparent that separate pathways are involved in cytotoxicity and cytokine signal-ing, and disassociation between the abilities to degranulate and to produce cytokines may be related to differing signaling pathways. Colucci and col-leagues [25] studied the role of Vav1 in NK cell signaling and found that Vav1−/− mice develop NK cells normally, but these NK cells are capable only of target cell-induced IFN-γ production and are unable to lyse target cells. Vav1 was shown to control ERK activation and exocytosis of cytotoxic gran-ules, whereas it was clearly not involved in signaling for cytokine produc-tion. Another study investigated divergent signaling pathways by focusing on the role of CD45 in triggering cytotoxicity and cytokine production [26]. CD45−/− mice were able to lyse targets but were unable to produce IFN-γ. It was demonstrated that CD45 was required for the full activation of Syk and Vav1 and phosphorylation of JNK and p38 after ITAM activation. However, CD45 has no effect on ITAM-mediated P13K activity required for signaling of cytotoxicity. As most activating receptors can signal for both cytotoxicity and cytokine production, it is unclear if activating receptors trigger both pathways simultaneously, or if cytotoxicity and cytokine production are triggered at different signal thresholds.
334
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
Acquisition of NK cell function
NK cells can express inhibitory receptors for both self- and non-self-MHC class I molecules. Therefore, in theory, NK cells with only inhibitory receptors for non-self-MHC class I could target self cells through the “missing self” hypoth-esis. However, this was not the case, as NK cell subsets not bearing receptors for self-MHC exist. To explain how NK cells acquire tolerance to self, several groups proposed what is known as NK licensing [14], NK arming [27], or NK education [28]. These models postulate that during NK cell development and after KIR or Ly49 acquisition, contact with MHC in the environment occurs. NK cells that express inhibitory receptors for self-MHC gain functional prop-erties and become licensed. However, NK cells that lack inhibitory receptors for self-MHC remain relatively hyporesponsive. Kim and colleagues demon-strated that licensed NK cells produced higher amounts of IFN-γ upon NK1.1 stimulation in vitro compared to unlicensed NK cells [14]. Moreover, licensed NK cells also have an intrinsically greater role in the rejection of allogeneic and MHC class I-deficient BM allografts compared to unlicensed NK cells [29]. In humans, higher cytotoxic functions have also been correlated with NK cells expressing KIRs for self-HLA [28,30]. Unlicensed NK cells, although hypore-sponsive in resting stages, are able to become functional upon activation [14] and have an important role during mouse CMV infection [31]. Additionally, host unlicensed NK cells were able to reject β2m-deficient BM cells in some degree as well [29]. Interestingly, unlicensed NK cells might play an important role in the elimination of tumor cells expressing self-HLA, as unlicensed NK cells become responsive early after autologous hematopoietic stem cell trans-plantation (HSCT) and provide protection against neuroblastoma [32].
Acquisition of KIRs or Ly49s at the latest stage of NK maturation has been correlated with mature functional properties. Liao and colleagues demon-strated that mature NK cells that developed in an MHC class I-deficient envi-ronment exhibited impaired function [33]. Moreover, NK cells from mice that express a dominant-negative mutation for SHP-1 showed defects in the abil-ity to eliminate MHC class I-deficient targets [34]. Barao and colleagues [35] observed that Ly49G2 expression occurs soon after syngeneic HSCT or dur-ing activation regardless of the host MHC, suggesting all Ly49 molecules may not be the same with regard to acquisition and function. These data would suggest that Ly49G2 may be an activation marker and possibly, because of its binding capability, may play a regulatory role in NK cell function.
It has been suggested that multiple mechanisms regulate the activating potential of NK cells. Owing to the stochastic expression of Ly49s, one NK cell can display one or multiple Ly49s. The rheostat model postulates that the presence of multiple inhibitory receptors within one NK cell allows for a stronger regulation of activation and those cells exhibit higher cytotoxic function when engagement of those inhibitory receptors does not occur [36,37]. It is likely that KIR and Ly49 receptors work in concert with NKG2A, 2B4, and other inhibitory receptors in regulating NK cell subset function and it is in fact the total NK receptor expression of a subset that may generate a net positive (i.e., through binding activating receptors such as Ly49D or H as well as NKG2D) or negative (through binding inhibitory Ly49 receptors and NKG2A) effect on overall function after binding the appropriate ligands, due to the observation that NK cell subsets expressing multiple receptors exist.
335
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
NK cell memory
The capacity for immune memory has long been considered a property of T and B cells, yet our enhanced understanding of NK cell function and the receptor–ligand interactions involved has led to studies that demon-strate that NK cells may also be able to mediate immune memory. NK cells have been shown to have hapten-specific memory in mice [38]. Mice that lacked both T and B cells still mounted a strong contact-hypersensitivity response and depletion of NK cells abolished the response, suggesting that NK cells are capable of adaptive immune responses. Furthermore, hapten-specific memory was transferrable. Sun and colleagues [39] were the first to demonstrate NK-cell-mediated viral immune memory. During CMV infec-tion in mice, NK cells expressing the activating receptor Ly49H preferen-tially expand, and persist in high numbers following CMV infection, and this response is driven through interaction of Ly49H with the viral protein m157. These NK cells were able to mount a robust response to subsequent CMV infection after adoptive transfer and had higher IFN-γ mRNA tran-scripts than naïve Ly49H+ NK cells. Additionally, these memory NK cells could be isolated from mice several months after the initial infection and were still capable of mounting robust responses. Certain cytokines, IL-12 and IL-18, have also been shown to induce a population of memory NK cells [40]. Cytokine-activated NK cells were transferred into naïve hosts and upon restimulation produced significantly more IFN-γ than naïve NK cells. Characterizing NK cell memory in humans has been far more difficult; how-ever, there is some evidence to suggest that memory NK cells may exist in humans. Similar to murine CMV and Ly49H, NK cells that express the acti-vating receptor NKG2C expand following acute CMV reactivation in trans-plant recipients [41,42] and coculture with CMV-infected fibroblasts [43]. These NKG2C+ NK cells have been shown to express an inhibitory receptor for self-HLA; progressively acquire CD57, a marker of NK cell maturity; and have increased function; thus they may represent a population of long-lived memory NK cells in humans. Clonal-like expansion of NKG2C+ NK cells has been observed during the acute response to hantavirus and chikungunya virus and chronic infection with hepatitis B and C only in patients who were CMV seropositive [44,45]. As a relatively new field of NK cell biology, how NK cell memory is acquired and maintained in humans still needs to be characterized.
Role of NK cells in cancer therapeutics
Human versus mouse NK cells
The invaluable use of mouse models to study NK biology has been unques-tioned. However, certain caveats should be taken into consideration, recognizing critical differences between human and mouse NK cells, to carefully translate mouse data into human research. One of the main dif-ferences between human and mouse NK cells lies in the phenotype of NK cells. Mouse NK cells lack expression of CD56, which is the primary marker used to identify human NK cells and limits translation of mouse NK stud-ies. Furthermore, the lack of CD56 on mouse NK cells means that there are also no CD56bright and CD56dim subsets, which are present in humans. There
336
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
have been multiple attempts to find a common marker that can be used in both species. Hayakawa and colleagues [46] were able to differentiate mouse NK maturation stages through the expression of the TNF receptor family member CD27, also expressed in humans, and CD11b. Four matu-ration stages have been identified: CD11blowCD27low, CD11blowCD27high, CD11bhighCD27high, and CD11bhighCD27low. CD11blowCD27high NK cells are located in lymph nodes (LN), spleen, liver, and BM; whereas CD11bhighCD-27low are located in the spleen and liver and represent the main NK subsets in the peripheral blood and lung. In humans, a CD27high subset is found within the CD56bright NK population, while CD27low is mainly expressed on peripheral blood CD56dim NK cells. Similar to CD56bright, CD11blowCD-27high produce higher levels of cytokines than CD11bhighCD27low. However, CD11blowCD27high NK cells also display strong cytotoxic function, differ-ent from CD56bright. Nevertheless, owing to organ location and function, CD11blowCD27high and CD11bhighCD27low correspond to CD56bright and CD56dim, respectively. Additionally, there is a paucity of NK cells present in the mouse LN, whereas human CD56bright NK cells are relatively abun-dant in human LN. As CD56bright NK cells are poorly lytic and thought to predominantly mediate effector functions through production of cytokines such as IFN-γ, this suggests a significant divergence between the species with regard to possible immunoregulatory roles of NK cells.
Resting mouse NK cells, in comparison with fresh isolated human NK cells, have much lower cytotoxic functions, which necessitates the stimula-tion of mouse NK cells to reach functional capabilities. The generation of inbred mice as well as the housing conditions (specific-pathogen-free) may account for the poor lytic function of resting mouse NK cells. Additionally, mouse NK cells cultured in vitro survive for a short period of time compared with human NK cells, which can be maintained for longer with stable KIR expression [47]. These results would suggest that similar effects may occur in vivo by which mouse NK cell therapies may underestimate potential effi-cacy because of their short-lived nature.
Nevertheless, despite these differences, because of the mouse’s small size and shorter life and the accessibility to reagents and multiple transgenic and deficient models, mouse research represents a necessary platform to further advance our understanding of NK biology and provides insightful information for the clinical application of NK-based immunotherapy in human diseases.
Studies in mouse models have allowed for the analysis of multiple param-eters involved in NK maturation, activation, and function, which provide essential data to understand NK biology and have a strong potential for clinical/human translation. Mouse models have been invaluable in allow-ing for the identification of those molecules relevant for NK activation or suppression with the discovery of the Ly49 family members [14]. For example, IL-15 was first demonstrated to be required for NK maturation and activation when IL-15-deficient mice showed profound NK defects compared with IL-2-deficient mice [48]. However, it was also shown that the IL-15 requirement could be overridden after infection. Studies using IL-15- and IL-15Rα-deficient mice showed the NK population could be restored upon CMV infection [49]. Studies in mice also allowed the
337
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
identification of the various inhibitory receptors that regulate self-toler-ance and provided an explanation for “hybrid resistance,” in which parental BM allografts are resisted by their F1 hybrids [2]. The concept of NK licensing was also postulated after a study of mouse NK cells that displayed differential inhibitory receptors for MHC [14].
The promise of using NK cells in cancer immunotherapy was initially described using mouse models of allogeneic HSCT. Mouse NK studies d emonstrated the efficacy of using NK cells to promote graft-versus-tumor effects while inhibiting graft-versus-host disease (GVHD) in allogeneic HSCT [50]. As NK cells do not initiate the rejection of solid tissue allografts, it was reasonable to hypothesize that the solid organ tissues often targeted in GVHD (skin, gut, liver) would not be targets of donor NK cells. However, NK cells can be found in GVHD lesions, suggesting that they may contribute to the pathology once initiated. In nonmyeloablative conditioning, donor NK cells may suppress host hematopoiesis. Indeed, it has been suggested that donor NK cells may actually allow for less conditioning to be given to the recipient and promote donor myeloid engraftment [51]. Mouse mod-els have also demonstrated that donor-type NK cells can act as “veto” cells and inhibit host effector cells (T and NK cells) capable of mediating graft rejection [52]. Thus, the use of donor-type NK cells can promote donor engraftment not only by suppressing host hematopoietic cells but also by suppressing host resistance to the donor graft.
Another potential advantage of using NK cells is the anti-tumor capability of these cells, particularly in hematopoietic malignancies. NK cells have also been demonstrated to mediate resistance to metastatic spread in numer-ous mouse models, suggesting that solid tumors can also be potentially tar-geted. The receptors that NK cells use in recognition of tumors are focused on NKG2D and the Ly49 receptor systems. Inhibitory Ly49 molecules have been thought to inhibit NK cell anti-tumor responses. Blockade of these receptors using Fab fragments has been shown to promote anti-tumor effects without inducing myelosuppression in mouse models [53], suggesting that these approaches may be of use in the clinic. The implication of activating NK receptors, such as NKG2D or DNAM-1, in tumor surveillance has also been demonstrated in mouse models. For example, an earlier and more aggressive generation of spontaneous prostate tumors in the transgenic ade-nocarcinoma of mouse prostate model of autochthonous prostate cancer development was observed in mice defective for NKG2D [54]. Furthermore, downregulation of NKG2D expression has been detected in cancer patients and correlated with poor cytotoxic NK function. Tumor cells can promote NKG2D downregulation by multiple means, such as release of soluble NKG2D ligands, demonstrating the importance of this activating receptor in control-ling tumor growth. Accelerated tumor growth was also observed in mice defi-cient for DNAM-1 [55]. As NKG2D ligands also are expressed on proliferating normal hematopoietic cells, these may also be a cause of myelosuppression.
Finally, there is increasing evidence that NK cells play important roles in immunoregulation. There is ample evidence in mouse models that NK cells can both promote and inhibit T-cell responses, either directly or indirectly. NK cells have been demonstrated to inhibit primary immune responses through their ability to kill dendritic cells [56]. Indeed, when activated,
338
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
mouse NK cells can be detected in the lymph nodes. A pivotal study dem-onstrated that NK cells can play critical roles in viral infections by limiting T-cell responses [57]. These data suggest that while direct anti-tumor effects can occur when using activated NK cells in cancer therapy, care must be exercised, as these same cells can also impair and suppress T-cell responses that may be necessary for long-term efficacy.
NK cells in humans
The first trials in humans to harness the anti-tumor properties of NK cells focused on the use of IL-2 to activate autologous NK cells both ex vivo and in vivo. Ex vivo IL-2-stimulated autologous peripheral blood mononuclear cells (PBMCs) were infused into patients with melanoma, lymphoma, and renal cell carcinoma with additional high-dose IL-2. NK cell cytotoxicity was observed in these patients; however, these treatments were unsuccessful. Furthermore, high-dose IL-2 infusions to stimulate NK cells in vivo were also clinically disappointing and were associated with high toxicities. As a conse-quence, various studies looked at the efficacy of low-dose IL-2 infusions for a wide range of malignancies, including AML, lymphoma, and breast can-cer [58,59]. Ultimately, infusion of IL-2 or adoptive transfer of IL-2-activated autologous NK cells failed for two main reasons: competition with the recip-ient’s lymphocytes for cytokines and “space” and because autologous NK cells were inhibited by self-HLA class I. As inhibitory KIRs and their ligands were further characterized, the next approach to utilizing NK cells as immu-notherapy focused on allogeneic NK cells. These NK cells would be alloreac-tive against a target that lacked the ligand for their inhibitory receptors.
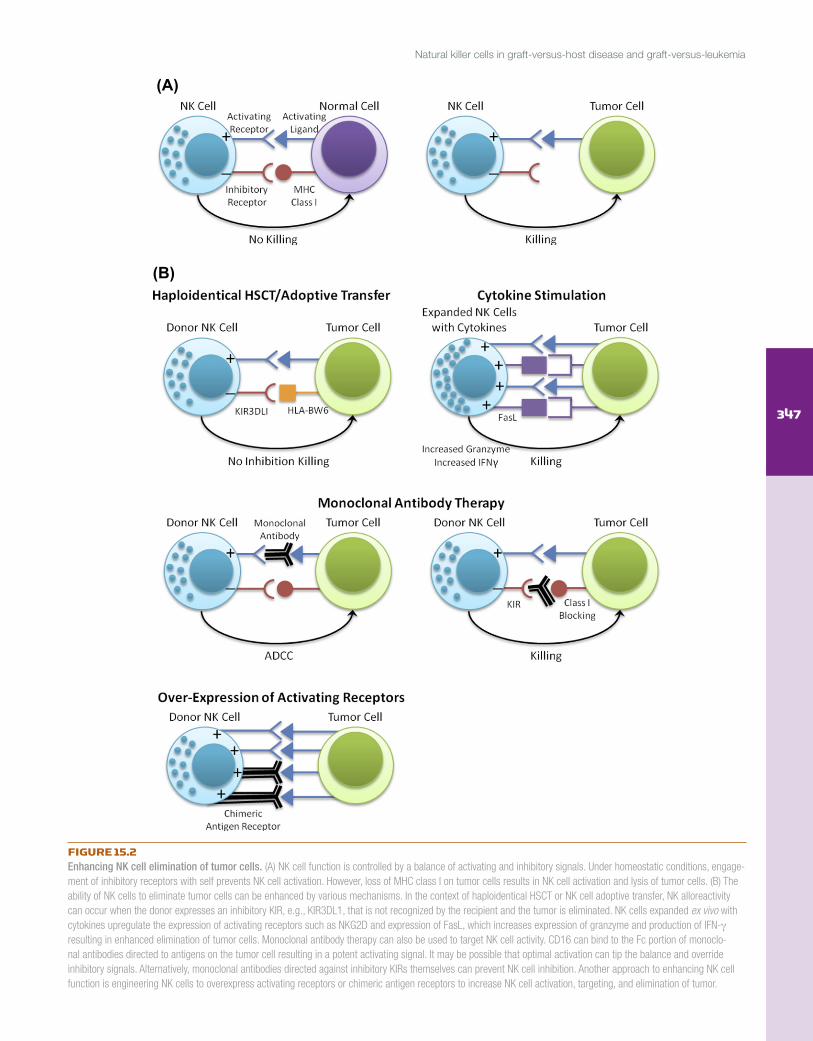
Allogeneic NK cells with the potential to mediate anti-tumor effects can be delivered in the context of an HSCT or adoptively transferred follow-ing ex vivo stimulation. The efficacy of the first approach was highlighted in a 2002 study from the Perugia University group [51], who provided evi-dence that donor alloreactive NK cells decreased graft rejection, enhanced engraftment, and mediated the graft-versus-leukemia (GVL) effect in the absence of GVHD following mismatched HSCT. The potential for NK allo-reactivity in the GVH direction was determined using what would become known as the KIR ligand incompatibility or KIR ligand mismatch model. For example the donor expresses all three ligands for the inhibitory KIR (C1, C2, and Bw4), whereas the recipient expresses only C2 and Bw4; therefore it is predicted that the donor will have C1-specific alloreactive NK cells that will not be inhibited in the recipient and can potentiate the GVL effect. This model assumes that the donor expresses the inhibitory KIRs that recognize each self-HLA ligand. Of the 92 AML and acute lymphocytic leukemia (ALL) patients who received HLA-mismatched grafts, 34 patients were KIR ligand incompatible in the GVH direction. Transplanted donor alloreactive NK cells protected against graft rejection, GVHD, and AML relapse. Without NK cell mismatch, the probability of event-free survival at 5 years was 5%, and with an NK cell mismatch the probability was 65%. This occurred only in patients with AML; there were no beneficial effects seen for patients with ALL, speculated to be due to the lack of the adhesion molecule, LFA-1, on the surface of ALL blasts [60]. Donor alloreactive NK clones could also be identified in the recipient up to 12 months after transplant. Using mouse
339
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
models it was predicted that alloreactive NK cells can prevent GVHD by eliminating recipient antigen-presenting cells (APCs), which can initiate GVHD and enhance engraftment by killing residual recipient T cells fol-lowing preparative regiments. In a follow-up study published in 2007 [61], lower rate of relapse, event-free survival, and remission were still signifi-cantly associated with NK cell alloreactivity.
On the basis of these findings several retrospective analyses were conducted with conflicting results. Giebel and colleagues [62] demonstrated a benefi-cial effect of KIR ligand mismatching studying 130 unrelated donor trans-plants, of which 20 had predicted NK cell alloreactivity. Event-free survival was markedly increased (87% vs 46%, p = 0.006). As in the Perugia study, NK cell alloreactivity was beneficial predominately for myeloid malignan-cies, yet not all studies using a similar platform agree. In a different setting of 175 unrelated mismatched transplants, there was no beneficial effect of KIR ligand incompatibility [63]. In perhaps one of the largest retrospective analyses of KIR ligand mismatching, a study that examined over 1500 unre-lated transplants found no beneficial effect and reported that KIR ligand incompatibility was associated with increased GVHD, treatment-related mortality, and shorter leukemia-free survival compared with HLA-matched transplants [64]. Improved survival was observed following HLA-matched sibling transplant in myeloid patients in which the recipient lacked a KIR ligand [65], and lower relapse following unrelated HSCT was also associated when the recipient lacked a KIR ligand.
There are several factors that may explain the disparities between the Peru-gia findings and subsequent retrospective analyses and studies. In the Perugia study, donor allografts were extensively T-cell depleted using a com-bination of CD34+ selection, soybean agglutination, and E-rosetting. It was predicted that extensive T-cell depletion was necessary for the expansion of alloreactive donor NK cells in vivo, as T cells present in the graft would compete with NK cells for cytokines and initiate GVHD before NK cells were able to eliminate residual host APCs. Two studies that examined NK cell alloreactivity in T-cell-replete HSCT found no beneficial effect of KIR ligand mismatching [66,67]. Differing pretransplant conditioning regimes and post-transplant GVHD prophylaxis may also play roles in the efficacy of NK cell alloreactivity. KIR ligand mismatching has also been studied following umbilical cord blood (UCB) HSCT. Willemze and colleagues [68] reported on the outcomes of 218 single-unit UCB transplants in AML and ALL patients. Donor–recipient pairs with KIR ligand incompatibilities showed improved leukemia-free survival and decreased relapse for patients with AML. Patients with ALL also showed a trend toward better outcomes when there was predicted KIR ligand incompatibility. In contrast, Brunstein and colleagues [69] reported a negative effect of KIR ligand incompatibilities in a cohort of 257 UCB HSCT recipients. Following myeloablative condition-ing regimens, KIR ligand incompatibilities had no effect on the occurrence of acute GVHD, relapse, or survival; however, following reduced-intensity conditioning, a higher incidence of acute GVHD and lower overall survival were seen. There were no differences in relapse.
Although these retrospective analyses used the KIR ligand incompatibility model to predict NK alloreactivity, unlike the Perugia study, the ability of the
340
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
donor to generate alloreactive NK cells in vitro could not be determined. Only donors that generated alloreactive NK cells were selected as suitable donors. Furthermore it is unknown if alloreactive donor NK cells were pres-ent in the recipient in these subsequent studies. Two other models for pre-dicting NK alloreactivity have also been studied. The KIR ligand absence model uses recipient HLA typing to predict the potential for NK alloreac-tivity. This model assumes that the respective inhibitory KIRs that would recognize the missing ligand in the recipient are present in the donor. The receptor ligand model uses donor KIR genotyping and recipient HLA typing to predict NK cell alloreactivity. This model allows for the potential for NK cells expressing a known inhibitory receptor to eliminate recipient cells that lack the receptor for that KIR. This model also allows for NK cell alloreactivity in HLA-identical transplants in which self-tolerant clones in the donor may become alloreactive in the post-transplant setting. It is becoming increasing clear that it is not merely sufficient to be able to predict NK cell alloreactiv-ity but also to be able to demonstrate it, as not all donor–recipient pairs will behave as predicted. This is most apparent with Bw4 and KIR3DL1. Rug-geri and colleagues [70] used the KIR ligand incompatibility to predict that 31 donor–recipient pairs had potential Bw4 mismatches but were able to detect Bw4-specific alloreactive NK clones in only 20 donors. Approximately 90% of individuals are KIR3DL1 positive, so a lack of KIR3DL1 may account for a small proportion of donors without Bw4-specific clones. Furthermore a common allele of KIR3DL1, KIR3DL1*004, is not expressed on the surface [71], and differing levels of surface expression of KIR3DL1 alleles have also been described that may influence generation of Bw4-specific clones; for example, KIR3DL1*01502 is a stronger allele for Bw4 than KIR3DL1*007 [72]. Also not all Bw4 alleles may interact with KIR3DL1 as predicted, and it has been demonstrated that the Bw4 alleles HLA-B*1301 and HLA-B*1302 did not bind to KIR3DL1 [73]. Furthermore HLA-A alleles that express the Bw4 epitope have been examined for their ability to bind to KIR3DL1 [73] and should also be included in analyzing potential Bw4 clones. The Bw4 epitope, which spans amino acids 77–83, can have either an isoleucine (Ile) at position 80 or a threonine. In HIV, Bw4 alleles with Ile80 have been asso-ciated with delayed progression to AIDS [7] and it could be predicted that Bw4 alleles with an Ile80 may have stronger interactions with KIR3DL1 and result in higher numbers of Bw4-specific clones.
Disparities with HLA-C mismatching predictions may also occur. Yawata and colleagues [72] examined NK cell receptor repertoire expression and function in 58 healthy individuals. They found that various combinations of HLA-C alleles and KIR genes resulted in differing levels of NK cell func-tion. There was no hierarchy observed between C2 HLA-C alleles and NK cell function, though NK cells expressing KIR2DL1*004 were poor produc-ers even when the donor expressed C2. Furthermore, Bari and colleagues [74] reported that KIR2DL1 alleles that expressed an arginine in position 245 are stronger alleles. Yawata and colleagues also demonstrated that KIR2DL3-expressing NK cells from donors who were homozygous for the C1 allele HLA-C*07 produced high levels of IFN-γ, those expressing the C1 alleles HLA-C*01, *03, *08, or *1403 produced moderate levels of IFN-γ, and those donors homozygous for HLA-C*1402 were poor producers of IFN-γ. KIR–HLA combinations that result in lower NK cell function may be due to
341
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
weak interaction between the inhibitory KIR and its cognate ligand. Indeed studies looking at NK cell education and acquisition of function suggest that inhibitory receptor interaction with its cognate ligand must reach a particular threshold to gain function and that this threshold is much higher for cytokine-producing functions than for cytotoxicity [37]. Also KIR2DL2 has been shown to be a stronger receptor for HLA-C than KIR2DL3 [75] and may also interact with C2 HLA-C alleles. In pediatric transplant recipients, KIR2DL2/3+ NK cells were poorly cytotoxic against leukemic blasts express-ing C2 owing to KIR2DL2/3 being inhibited by C2 [76].
Predicting NK cell alloreactivity is further compounded by the presence of other inhibitory and activating receptors present on donor NK cells. The inhibitory receptors NKG2A and LIR-1 may also suppress NK cell func-tion following transplantation. HLA-E, the ligand for NKG2A, is expressed on hematopoietic cells and could inhibit NK cell function even if there is a KIR mismatch especially considering that reconstituting NK cells have high NKG2A expression. Furthermore, while some tumors may downregulate class I, HLA-E usually remains intact [77]. LIR-1, which binds an array of class I alleles including HLA-G, should also be taken into consideration.
Activating KIR may be involved in the GVL effect, as KIR2DS1-expressing alloreactive NK clones have been shown to lyse targets expressing C2 and can override NKG2A-mediated inhibition [78]. In the transplant setting, Pende and colleagues [76] demonstrated KIR2DS1-mediated lysis of C2-homo-zygous leukemic blasts and confirmed that NKG2A-inhibitory signaling could be overcome. A large study of 1409 unrelated transplants examined the roles of donor and recipient KIR haplotypes and found that transplants from a KIR B haplotype donor resulted in lower relapse and improved sur-vival for patients with AML [79]. The effect was not seen for patients with ALL. As described earlier KIR A haplotypes consist predominantly of inhibi-tory KIR and only one activating KIR, KIR2DS4. KIR B haplotypes, however, have varying activating KIR gene content. To isolate which segment of the haplotype may be involved in the protection effects observed, genes in the KIR B haplotypes were divided into either centromeric or telomeric. Donors with more centromeric KIR B haplotype (Cen-B) genes were associated with the lowest level of relapse and highest overall survival. Donors were strati-fied based on KIR B haplotype gene content into three groups: best (Cen-B homozygous), better (more than two B-motifs without Cen-B/B), and neu-tral donors (no or one B-motif). It remains unclear why recipients who receive a graft from a Cen-B-homozygous donor have preferable outcomes.
Even if donor–recipient pairs are carefully selected on the basis of predicted NK alloreactivity and confirmation with in vitro lysing of recipient leukemic blasts, the transplant environment also needs to be considered. NK cells are the first lymphocyte to reconstitute following HSCT, and NK cell function varies depending on the transplant source. Reconstituting NK cells have differing receptor profiles than in the donor, which includes higher expres-sion of NKG2A and altered KIR and NCR expression and lower than normal overall function [80,81]. Therefore they can be considered developmen-tally immature NK cells, and recovery of normal receptor repertoires and function may take up to at least 1 year. Graft source also influences NK cell recovery following HSCT. Cooley and colleagues [82] compared KIR receptor
342
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
repertoires between unmanipulated BM transplants and T-cell-depleted transplants. T-cell depletion was mediated through anti-thymocyte globu-lin, elutriation, or campath. They found that KIR expression was increased in the patients who received T-cell-depleted grafts. Furthermore the pres-ence of T cells in the graft was correlated with increased GVHD and worse outcomes. However, Nguyen and colleagues [83] were able to demonstrate a beneficial effect of T cells in the graft. Looking at haploidentical HSCT they compared partial and complete T-cell depletion. Recipients who received partial T-cell-depleted grafts had lower expression of NKG2A, similar cyto-toxicity against K562 compared with donor NK cells, and higher cytotoxicity against AML blasts compared with recipients who received complete T-cell-depleted grafts. Recipients who received complete T-cell-depleted grafts had higher IFN-γ responses when stimulated with IL-12 and IL-18, which is to be expected as CD56bright NKG2A+ NK cells are more responsive to IL-12 and IL-18 stimulation [84]. These findings contradict earlier reports that T cells in the graft hinder NK cell function. This was further confirmed by a study from Foley and colleagues [85] that compared NK cell function (cytotoxicity and cytokine production) following HSCT with three different graft preps, unmanipulated (T-cell replete), CD34+ selected (T-cell depleted) without post-transplant immune suppression, and UCB. Overall NK cell function was lower following HSCT compared with donor NK cells. Surprisingly, how-ever, NK cell function was highest following T-cell-replete HSCT and this was associated with higher KIR expression. In recipients of T-cell-replete HSCT, it was demonstrated that target-induced IFN-γ production was associated with KIR and not NKG2A, as NK cells expressing only NKG2A were poor pro-ducers of IFN-γ. Furthermore IFN-γ-producing NK cells expressed a KIR that recognized self. As NKG2A-expressing NK cells were cytotoxic, it was pos-tulated that in the post-transplant setting, the interaction between NKG2A and HLA-E supports the education of NK cells to degranulate but that the signal is too weak to meet the threshold to educate for cytokine production. Therefore these studies collectively suggest that T cells may actually be ben-eficial in promoting NK cell function rather than hindering it. Activated T cells produce IL-2, which has been shown to stimulate CD56bright NK cells in the lymph nodes to produce IFN-γ and to contribute to NK cell maturity [86]. Alternatively, donor NK cells may persist in the recipient for longer than originally reported and infused mature donor NK cells may contribute to the increased function seen after T-cell-replete HSCT.
UCB transplants have become a viable alternative to BM or peripheral blood HSCT. Because of the low number of mature T cells present, the risk of acute GVHD is low and therefore HLA-mismatched units are frequently used. Despite being able to mediate cytotoxicity and induce apoptosis through Fas/FasL [87], NK cells reconstituting after UCB transplant are poor pro-ducers of IFN-γ [85]. Incomplete recovery of full NK cell effector functions may be involved in the high incidence of viral infections compared with the other types of transplant [88].
As described earlier, NK cells acquire function through a process known as NK cell education. How NK cell education influences NK cell function post-HSCT has only recently been investigated. Can an NK cell that expresses an inhibitory receptor for a nonself ligand be educated in the recipient, if the recipient sees that receptor as self, or will it remain hyporesponsive? Yu and
343
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
colleagues [89] reported that following T-cell-depleted HLA-matched HSCT, NK cells expressing a nonself KIR were hyperresponsive and capable of pro-ducing high levels of IFN-γ. These NK cells gradually became tolerized to self in the recipient by day 100 post-HSCT. Bjorklund and colleagues [90], how-ever, found that NK cells expressing KIR for nonself remained tolerant in the recipient. They studied both T-cell-replete and T-cell-depleted HLA-matched HSCT. NK cells expressing a KIR recognizing self expressed higher levels of CD107a compared with NK cells expressing a KIR that recognized nonself, similar to findings reported by Foley and colleagues [85]. Post-transplant immune suppression may partially explain the differences seen, as patients receiving T-cell-depleted HSCT in the study by Bjorklund and colleagues [90] received cyclosporin A, whereas patients in the study by Yu and colleagues [89] received no immune suppression. Cyclosporin A has been shown to alter NK cell differentiation and function [91]. Yu and colleagues also found that NK cells expressing NKG2A were hyporesponsive post-HSCT, whereas NK cells expressing NKG2A had the strongest expression of CD107a in the study by Bjorklund and colleagues. These differences could be attributed to differ-ent functional assays used in each study, in that Yu and colleagues mainly studied IFN-γ production, whereas Bjorklund and colleagues used CD107a as a marker of degranulation.
In both these studies donor and recipient were HLA matched. Haas and colleagues [92] investigated the role of NK cell education following both KIR-ligand-matched and KIR-ligand-mismatched unmanipulated HSCT. In accordance with Bjorklund and colleagues [90], NK cells adhered to the rules of education, that is, NK cells expressing a nonself KIR remained hyporesponsive in the recipient and those expressing a self KIR were func-tional. Similar to Bjorklund and colleagues [90] and Foley and colleagues [85], the subset of NK cells coexpressing a self KIR and NKG2A had the high-est expression of CD107a, consistent with the hypothesis that expression of more than one self-recognizing inhibitory receptor results in higher capac-ity for degranulation [37]. Where there was a KIR-ligand mismatch, NK cell responsiveness adhered to the HLA ligands present in the donor.
Two studies using murine models were the first to demonstrate that NK cells could be re-educated in a new host [93,94]. Educated mature NK cells, when adoptively transferred into an MHC class I-deficient mouse, become anergic to receptor stimulation. In the reverse scenario, NK cells from an MHC class I-deficient mouse gain effector functions when adoptively transferred into an MHC class I-sufficient mouse [93]. How these findings may influence NK cell functions in humans remains unclear. In the transplant setting, once donor chimerism has been established, cells of hematopoietic origin are donor derived, and the findings from Haas and colleagues [92] could imply that these cells are involved in educating reconstituting NK cells. However, adoptively transferred NK cells, which are discussed below, will encounter hematopoietic cells of recipient origin and re-education may occur.
Adoptive NK cell therapy
The second method to deliver alloreactive NK cells to the patient involves adoptive transfer of donor NK cells purified ex vivo and infused into the recipient. These NK cells are presumed to be mature and fully functional
344
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
and educated in the donor. The first trial of this approach was conducted at the University of Minnesota [95]. Forty-three patients with metastatic melanoma, metastatic renal cell carcinoma, Hodgkin’s disease, or poor prognosis AML were enrolled in the trial. PBMCs were collected from hap-loidentical related donors and CD3 depleted before being incubated over-night in IL-2. Prior to NK cell infusion, patients underwent a preparative regimen that involved three different chemotherapy preparations: high cyclophosphamide and fludarabine (Hi-Cy/Flu), low cyclophosphamide and methylprednisone, or fludarabine. Following infusion patients received IL-2 daily for 14 days. NK cell expansion was observed only for patients receiving the preparatory regimen of Hi-Cy/Flu. Successful expansion of NK cells was determined by the detection of greater than 100 NK cells/μl of blood 12–14 days after infusion. On this protocol 30% of poor progno-sis AML patients achieved a complete remission, whereas no beneficial effects were observed for the other diseases. However, this remission was not durable and patients ultimately relapsed. Since NK cells expanded only following high-dose preparative regimens, the addition of 400 cGy of total body irradiation was proposed to further deplete host immune cells and create space for donor NK cells to expand. This addition also meant that patients needed to be “rescued” with a CD34+ selected stem cell infusion. On this protocol NK cell expansion was much more successful and 75% of patients achieved measurable NK cell expansion. Furthermore leukemia clearance was observed in 66% of patients. No acute GVHD was observed, though death due to infection was common (46%). It should also be noted that, while there was induction of remission, ultimately this therapy did not result in lasting remissions. Additionally there was no influence of KIR-ligand incompatibilities.
The use of adoptive transfer of NK cells to treat various malignancies has resulted in mixed results. Shi and colleagues [96] infused haploidentical KIR-mismatched NK cells into 10 patients with relapsed multiple myeloma followed 14 days later with an autologous stem cell graft. Five patients achieved near complete remission. Bachanova and colleagues [97] treated 6 patients with non-Hodgkin’s lymphoma with infusion of haploidentical NK cells and found that NK cells expanded poorly in vivo and host T regulatory cells were significantly increased after NK cell infusion and IL-2 administra-tion. Similarly, adoptively transferred NK cells failed to expand in patients with breast and ovarian cancers and a similar increase in host T regulatory cells was also observed [98].
Expanding NK cells
In the initial report describing the clinical efficacy of adoptively transfer-ring NK cells, PBMCs were CD3 depleted and IL-2 activated overnight. After infusion IL-2 was added daily for 14 days to expand these donor NK cells in vivo. Current platforms used also involve the depletion of CD19 cells. Strategies to enhance NK cell expansion both in vivo and ex vivo have been undertaken. The addition of IL-2 is necessary to expand NK cells in vivo; however, IL-2 also activates T regulatory cells, which can suppress NK cell proliferation and NK cell cytotoxicity [99]. In an experimental tumor model using pretreatment with Ontak (denileukin diftitox) to deplete host
345
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
T regulatory cells, adoptively transferred NK cells were able to eliminate tumors in these mice and the mice survived significantly longer than mice treated with NK cells alone in the absence of pretreatment with Ontak [100]. Therefore depleting host T regulatory cells could improve NK cell expan-sion and function in humans. As IL-15 has recently been made available for clinical use, it may be preferable to IL-2. IL-15 is critical to NK cell develop-ment and homeostasis [48,84]. Trials testing the safety and toxicity of IL-15 are currently being undertaken.
The alternative is to expand NK cells ex vivo and then infuse into the patient. Several centers are working on this approach. Imai and colleagues engi-neered the class I-negative cell line K562 to express 41BBL and membrane-bound IL-15 [101]. Under GMP conditions they described on average a 1000-fold expansion of NK cells after 3 weeks in culture and retained cyto-toxic capabilities. A similar approach utilized a GMP-compatible lympho-blastoid cell line and IL-2 to expand NK cells on average 500-fold in 30 days. These expanded NK cells were shown to upregulate CD25 (IL-2R), CD48, NKG2D, and TRAIL compared with resting NK cells. They also increased secretion of many cytokines and chemokines, including IFN-γ, TNF-α, GM-CSF, and MIP-1β. Removal of IL-2 resulted in decreased expression of both NKG2D and TRAIL and multiple cytokines, including IFN-γ. These results demonstrate that ex vivo expanded NK cells may become dependent on cytokines, and may even become exhausted after prolonged expansion, so it is unclear how they may function once infused into the patient. Indeed expanded NK cells have been shown to have shorter telomeres and potential replicative senescence [102]. Recently, it has been demonstrated that K562-expressing membrane-bound IL-21 and 41BBL expanded NK cells signifi-cantly more than those expressing IL-15 [103]. Expanded NK cells did not reach senescence and had telomere lengths similar to those of resting NK cells. They also had similar levels of receptor expression compared to those NK cells expanded with IL-15, but had higher cytokine production. As these cells had less senescence this approach would be more amenable to use in the clinic. An alternative source of ex vivo-expanded NK cells is the genera-tion of NK cells from CD34+ progenitors isolated from UCB [104]. Another promising source of expanded NK cells is from embryonic stem cells [105].
Studies have also explored the use of NK cell lines for potential adoptive therapy. Both irradiated NK-92 and KHYG-1 could prove an unlimited source of cytotoxic NK cells for adoptive transfer; however, their survival in vivo is still unclear [106,107]. Irradiated NK-92 cells were used in a phase I clinical trial for renal cell cancer and melanoma with only mild infusion toxicities [108].
In addition to the expansion and adoptive transfer of unmanipulated NK cells, NK cells can also be engineered to express certain receptors to increase their ability to recognize and eliminate tumors. Chimeric antigen receptors (CARs) typically involve fusions of single-chain variable frag-ments derived from monoclonal antibodies such as CD19 and usually fused to CD3ζ transmembrane and endodomain. Addition of costimula-tory molecules such as CD28, CD137, and CD134 (OX40) increases the efficacy of CARs. NK-92 cells have been genetically modified with CARs that consist of CD20 and Her-2/neu, both fused to CD3ζ, and have shown
346
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
increased NK cell killing of CD20-positive lymphoma and breast cancer cells [109]. Pegram and colleagues [110] genetically engineered mouse NK cells with a CAR consisting of erbB2 (Her-2/neu) and CD28 fused to CD3ζ. These NK cells were adoptively transferred into Rag-1−/− mice and inhib-ited growth of erbB2+ lymphoma by directly lysing the tumors. Altvater and colleagues [111] genetically modified in vitro-stimulated NK cells from healthy donors and pediatric leukemia patients with CARs consisting of either CD19 or Gd2. They also tested 2B4 as the endodomain or a combi-nation of CD3ζ and 2B4. NK cells expressing CD19–2B4 failed to produce IFN-γ and TNF-α when stimulated but did express CD107a. They also did not upregulate expression of CD25. When combined with CD3ζ, NK cells were significantly more potent against CD19+ leukemia cells, suggesting that 2B4 can act as a costimulatory receptor to increase NK cell activity. Similarly, NK cells expressing Gd2–CD3ζ–2B4 were able to lyse resistant neuroblastoma cell lines.
Enhancing NK cell function to eliminate tumors
Several tumor-targeted monoclonal antibodies have been developed and have been used clinically to treat various malignancies (Figure 15.2). Since inhibitory KIRs bind to self-HLA class I and inhibit NK cell function, mono-clonal antibodies directed against the inhibitory KIRs have been developed. Romagne and colleagues [112] generated a human monoclonal antibody called 1-7F9 that recognizes the inhibitory KIRs KIR2DL1, KIR2DL2, and KIR2DL3. It does not cross-react with KIR3DL1. During preclinical char-acterization, it was demonstrated that 1-7F9 did not stimulate peripheral blood NK cells to degranulate or produce IFN-γ, and blocking KIR with 1-7F9 increased lysis of primary AML blasts. In transgenic mice engineered to express KIR2DL3, HLA-Cw3+ splenocytes were rejected after adding 1-7F9, and furthermore, in NOD–SCID mice, NK cells lysed autologous tumors when 1-7F9 was added. Current clinical trials are under way to investigate the efficacy of this monoclonal antibody in humans.
CD137 or 4-1BB is a costimulatory molecule of the TNF receptor family. It is expressed on activated NK cells, T cells, B cells, dendritic cells, monocytes, and neutrophils. On resting NK cells its expression is low and CD16 activa-tion can induce its expression [113]. CD137 can be activated by binding to its natural ligand or triggered with a monoclonal antibody against it. Anti-CD137 antibodies have been used in combination with other monoclonal antibodies to increase NK cell activation, which is discussed below. Artifi-cial APCs have also been engineered to express CD137L, which has been shown to increase expansion of NK cells as discussed above.
NK cells recognize antibody-coated targets through CD16 and medi-ate ADCC. Rituximab, a monoclonal antibody directed against CD20 on mature B cells, has been used to treat non-Hodgkin’s lymphoma [114]. Allelic polymorphisms within the CD16 gene have been shown to influ-ence NK-cell-mediated ADCC. One such polymorphism is at position 158, a region of the receptor that interacts with the hinge region of IgG antibod-ies, has either a phenylalanine or a valine (V) at this position, and alters NK cell binding [115]. The 158V polymorphism results in higher CD16 binding to IgG. Cartron and colleagues [116] demonstrated that the 158V
347
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
(A)
(B)
FIGURE 15.2Enhancing NK cell elimination of tumor cells. (A) NK cell function is controlled by a balance of activating and inhibitory signals. Under homeostatic conditions, engage-ment of inhibitory receptors with self prevents NK cell activation. However, loss of MHC class I on tumor cells results in NK cell activation and lysis of tumor cells. (B) The ability of NK cells to eliminate tumor cells can be enhanced by various mechanisms. In the context of haploidentical HSCT or NK cell adoptive transfer, NK alloreactivity can occur when the donor expresses an inhibitory KIR, e.g., KIR3DL1, that is not recognized by the recipient and the tumor is eliminated. NK cells expanded ex vivo with cytokines upregulate the expression of activating receptors such as NKG2D and expression of FasL, which increases expression of granzyme and production of IFN-γ resulting in enhanced elimination of tumor cells. Monoclonal antibody therapy can also be used to target NK cell activity. CD16 can bind to the Fc portion of monoclo-nal antibodies directed to antigens on the tumor cell resulting in a potent activating signal. It may be possible that optimal activation can tip the balance and override inhibitory signals. Alternatively, monoclonal antibodies directed against inhibitory KIRs themselves can prevent NK cell inhibition. Another approach to enhancing NK cell function is engineering NK cells to overexpress activating receptors or chimeric antigen receptors to increase NK cell activation, targeting, and elimination of tumor.
348
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
polymorphism was associated with higher responses to rituximab therapy in patients with follicular non-Hodgkin’s lymphoma. Other monoclonal antibodies have been developed that also mediate NK cell ADCC, includ-ing trastuzumab (Her2 on breast cancer), alemtuzumab (CD52 on chronic lymphocytic leukemia (CLL)), and cetuximab (epidermal growth factor receptor on colorectal cancer) [117], and CD16 polymorphisms have also been associated with the efficacy of using other monoclonal antibodies to mediate ADCC.
While each monoclonal antibody described can trigger NK cell function, combinations of antibodies may enhance NK cell responses. For example, engagement of inhibitory KIRs with HLA on the tumor can inhibit CD16 signaling by inhibiting tyrosine kinase activation [118]. Blockade of inhibi-tory KIRs in combination with tumor-targeted monoclonal antibodies may increase CD16-mediated ADCC. Anti-CD137 antibodies in combination with rituximab have been shown to increase degranulation and IFN-γ production [119]. Upon engagement of CD16 with rituximab-coated lymphoma cells, CD137 is upregulated on the NK cell and addition of an agonist against CD137 increased NK-cell-mediated ADCC. A similar effect was also observed using a combination of anti-CD137 and trastuzumab to eliminate breast cancer cells [119]. Other drugs such as lenalidomide, currently used in the treatment of multiple myeloma, have also been shown to enhance NK-cell-mediated ADCC in combination with rituximab [120]. However, in a later study lenalid-omide was shown to decrease CD20 expression on CLL, thereby decreasing rituximab-induced ADCC [121], so one must be careful in combining thera-pies. Lenalidomide has also been shown to be useful at increasing ADCC of targets coated with trastuzumab or cetuximab [122]. An alternative to combi-nation drug therapy is to combine NK-stimulating cytokines. IL-2, IL-12, and IL-21 have all been shown to enhance NK-cell-mediated ADCC [123]. One step further is to combine adoptive transfer of activated NK cells stimulated either alone with cytokines or with a genetically modified cell line to increase NK-cell-mediated ADCC of antibody-coated tumors.
In addition to monoclonal antibodies, bispecific antibodies have also been developed. Simply, a bispecific antibody will involve the fusion of two monoclonal antibodies, one directed at the effector cell and the other directed at the tumor cell. CD16 is commonly used to bind to the NK cell and various target-cell-specific antigens have been used, including CD19 (B-cell lymphomas) [124].
Histone deacetylase (HDAC) and proteasome inhibitors have also been shown to sensitize tumors to NK cell killing. Bortezomib, a proteasome inhibitor, has been shown to upregulate death receptors such as Fas and TRAIL-R2/DR5 [125] and induce apoptosis through Fas/FasL and TRAIL/DR5 interac-tions, both expressed by NK cells. Bortezomib has been shown to be effective in inducing apoptosis of quiescent CD34+ cells in chronic myelogenous leu-kemia patients and increasing the efficacy of ex vivo-expanded adoptively transferred autologous NK cells [126]. Depsipeptide, an HDAC inhibitor, has also been shown to sensitize tumors to NK-cell-mediated apoptosis through the upregulation of death receptors [127]. Another HDAC inhibitor, valproic acid, has been shown to induce expression of ULBP1, MICA, and MICB on AML blasts, increasing NK cell killing [128].
349
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
The role of NK cells in viral therapy
Another potential therapeutic approach to eliminating tumors is the use of oncolytic viruses. An oncolytic virus is a virus that preferentially infects, replicates in, and lyses malignant cells. In most instances, the virus is engineered to increase tumor specificity. The first report of an engineered oncolytic virus described the ability of a thymidine kinase-negative mutant herpes simplex virus-1 (HSV-1) that was attenuated for neurovirulence to treat gliomas [129]. The virus was able to lyse human glioma cell lines in vitro and inhibit the growth of human glioma in nude mice and pro-longed survival. While NK cells have anti-tumor properties, which have been discussed, NK cells also have potent antiviral properties. In fact, individuals with NK cell deficiencies are particularly susceptible to viral infections [130]. As NK cells are critical components of the host response to viral infections they may be both beneficial and a hindrance when using oncolytic viruses as a cancer therapy. For effective use of oncolytic viruses, they must be able to enter the cell, infect, and replicate before the immune response targets the virus. HSV-1- and vaccinia virus-infected cells, com-monly used as oncolytic viruses, can be rapidly lysed by NK cells, prevent-ing virus dissemination and thereby potentially preventing the efficacy of these oncolytic viruses [131]. Upon activation NK cells will also recruit other innate immune cells such as monocytes and dendritic cells and cells of the adaptive immune response, T cells and B cells, that may further impede the efficacy of the virus. To counteract the host’s antiviral immune response the addition of chemotherapy to suppress the immune system coupled with oncolytic viruses may increase the potential for this line of therapy [132]. Alternative approaches involve harnessing the potent antivi-ral properties of NK cells. This may involve viruses that upregulate ligands for NK-cell-activating receptors and downregulate ligands for the inhibi-tory receptor, e.g., HLA class I. Bergmann and colleagues [133] engineered influenza A virus to be used as a potential oncolytic virus. NKp46 expressed by all NK cells recognizes influenza A hemagglutinin on infected cells. This engineered virus was used to infect prostate cancer cells in vitro, and infected cells were eliminated by NKp46-mediated target cell lysis [134]. NKp46 activation was also shown to overcome MHC class I inhibition. An alternative approach may involve immune suppression to ensure total viral infection of the tumors, particularly in metastatic tumors, followed by adoptive transfer of activated NK cells that will be able to eradicate the infected tumor cells.
Another virus, CMV, may also play a role in eliminating malignant cells. A member of the Herpesviridae family, CMV causes asymptomatic or mild illness in healthy individuals; however, for patients immunosuppressed by HIV infection or solid organ or hematopoietic cell transplantation, CMV is a potentially life-threatening complication [135]. CMV remains latent in the host, and latent CMV reservoirs have been found in cells of the myeloid lin-eage and in endothelial cells [135]. Recently CMV reactivation after HSCT has been shown to be beneficial. Elmaagacli and colleagues [136] reported that early CMV reactivation is associated with a reduced risk of relapse in AML patients undergoing allogeneic HSCT from HLA-matched siblings or unrelated donors. All transplants were unmanipulated. The risk of leukemic
350
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
relapse was 9% at 10 years post-HSCT compared with 42% in patients who did not reactivate CMV, and CMV reactivation was not detrimental to over-all survival. It is also possible that the lower risk of relapse reported in UCB transplantation [137] is associated with CMV reactivation. CMV infection may induce expression of a ligand that activates CD8+ T cells and/or NK cells. Alternatively, the virus may act as an oncolytic virus by infecting leukemic blasts and eliminating them. In recipients of UCB HSCT, CMV reactivation has been shown to induce a more rapid reconstitution of fully functional, educated NK cells with increased survival capacity and the abil-ity to respond rapidly with cytokines [42]. In the absence of CMV reactiva-tion, NK cells remain immature and recovery of full effector functions takes at least 1 year. As we continue to understand the NK cell response to human CMV, carefully monitored CMV reactivation may actually be beneficial.
Concluding remarks
Ever since their initial identification by their ability to spontaneously reject BM allografts and lyse cells with low MHC class I expression without prior sensitization, NK cells have been considered important effector cells in eliminating aberrant cells. In the clinical setting, NK cells, both in the con-text of HSCT and adoptively transferred, have been shown to have immense therapeutic properties, including enhancing engraftment, decreasing both rejection and GVHD, and eliminating leukemic cells. The use of animal models and in vitro assays to demonstrate NK cell effectiveness at eliminat-ing cancer cells have been invaluable. Further characterization and identi-fication of NK cell receptors and their ligands on tumor cells will enable a clearer understanding of how NK cells recognize tumors and how this can be exploited in the clinical setting. As not all tumors respond to NK cells in the same manner, certain NK cell therapies may be more applicable to certain cancers. Furthermore, combinations of different therapies may be essential to enhance the efficacy of NK cell immunotherapy in vivo. Despite their discovery over 40 years ago, new and exciting areas of NK cell biology are continually emerging. The effective use of NK cells to treat cancer will only increase as we further our understanding of how NK cells gain func-tion, how the multitude of receptors expressed by NK cells control function, and how we can exploit this to eliminate tumors.
References [1] Cudkowicz G, Stimpfling JH. Hybrid resistance to parental marrow grafts: association
with the K region of H-2. Science 1964;144(3624):1339–40. [2] Cudkowicz G, Bennett M. Peculiar immunobiology of bone marrow allografts. II. Rejec-
tion of parental grafts by resistant F 1 hybrid mice. J Exp Med 1971;134(6):1513–28. [3] Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-defi-
cient lymphoma variants suggests alternative immune defence strategy. Nature 1986;319(6055):675–8.
[4] Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans- presentation promotes human NK cell development and differentiation in vivo. J Exp Med 2009;206(1):25–34.
[5] Vilches C, Parham PKIR. diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 2002;20:217–51.
[6] Stewart CA, Laugier-Anfossi F, Vely F, Saulquin X, Riedmuller J, Tisserant A, et al. Recognition of peptide–MHC class I complexes by activating killer immunoglobulin-like receptors. Proc Natl Acad Sci U S A 2005;102(37):13224–9.
351
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
[7] Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic inter-action between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet 2002;31(4):429–34.
[8] Kulkarni S, Martin MP, Carrington M. The Yin and Yang of HLA and KIR in human disease. Semin Immunol 2008;20(6):343–52.
[9] Lanier LL. NK cell recognition. Annu Rev Immunol 2005;23:225–74. [10] Dimasi N, Biassoni R. Structural and functional aspects of the Ly49 natural killer cell
receptors. Immunol Cell Biol 2005;83(1):1–8. [11] Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, et al. Variable MHC
class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2K(b). Nat Immunol 2003;4(12):1213–22.
[12] Nakamura MC, Linnemeyer PA, Niemi EC, Mason LH, Ortaldo JR, Ryan JC, et al. Mouse Ly-49D recognizes H-2Dd and activates natural killer cell cytotoxicity. J Exp Med 1999;189(3):493–500.
[13] George TC, Ortaldo JR, Lemieux S, Kumar V, Bennett M. Tolerance and alloreactivity of the Ly49D subset of murine NK cells. J Immunol 1999;163(4):1859–67.
[14] Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005;436(7051):709–13.
[15] Lazetic S, Chang C, Houchins JP, Lanier LL, Phillips JH. Human natural killer cell receptors involved in MHC class I recognition are disulfide-linked heterodimers of CD94 and NKG2 subunits. J Immunol 1996;157(11):4741–5.
[16] Vales-Gomez M, Reyburn HT, Erskine RA, Lopez-Botet M, Strominger JL. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J 1999;18(15):4250–60.
[17] Meyaard L, Adema GJ, Chang C, Woollatt E, Sutherland GR, Lanier LL, et al. LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity 1997;7(2):283–90.
[18] Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immuno-globulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 1997;7(2):273–82.
[19] Biassoni R, Bottino C, Cantoni C, Moretta A. Human natural killer receptors and their ligands. Curr Protoc Immunol 2002; Chapter 14:Unit 14.10.
[20] Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001;409(6823):1055–60.
[21] Hershkovitz O, Rosental B, Rosenberg LA, Navarro-Sanchez ME, Jivov S, Zilka A, et al. NKp44 receptor mediates interaction of the envelope glycoproteins from the West Nile and dengue viruses with NK cells. J Immunol 2009;183(4):2610–21.
[22] Bottino C, Castriconi R, Moretta L, Moretta A. Cellular ligands of activating NK recep-tors. Trends Immunol 2005;26(4):221–6.
[23] Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immu-nol 2008;9(5):495–502.
[24] Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev 2006;214:73–91.
[25] Colucci F, Rosmaraki E, Bregenholt S, Samson SI, Di Bartolo V, Turner M, et al. Func-tional dichotomy in natural killer cell signaling: Vav1-dependent and -independent mechanisms. J Exp Med 2001;193(12):1413–24.
[26] Huntington ND, Xu Y, Nutt SL, Tarlinton DM. A requirement for CD45 distinguishes Ly49D-mediated cytokine and chemokine production from killing in primary natural killer cells. J Exp Med 2005;201(9):1421–33.
[28] Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell edu-cation by inhibitory receptors for MHC class I. Immunity 2006;25(2):331–42.
[29] Sun K, Alvarez M, Ames E, Barao I, Chen M, Longo DL, et al. Mouse NK cell-mediated rejection of bone marrow allografts exhibits patterns consistent with Ly49 subset licensing. Blood 2012;119(6):1590–8.
[30] Cooley S, Xiao F, Pitt M, Gleason M, McCullar V, Bergemann TL, et al. A subpopula-tion of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood 2007;110(2):578–86.
[31] Orr MT, Murphy WJ, Lanier LL. ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol 2010;11(4):321–7.
[32] Venstrom JM, Zheng J, Noor N, Danis KE, Yeh AW, Cheung IY, et al. KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin Cancer Res 2009;15(23):7330–4.
352
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
[33] Liao NS, Bix M, Zijlstra M, Jaenisch R, Raulet D. MHC class I deficiency: susceptibility to natural killer (NK) cells and impaired NK activity. Science 1991;253(5016):199–202.
[34] Lowin-Kropf B, Kunz B, Beermann F, Held W. Impaired natural killing of MHC class I-deficient targets by NK cells expressing a catalytically inactive form of SHP-1. J Immunol 2000;165(3):1314–21.
[35] Barao I, Alvarez M, Ames E, Orr MT, Stefanski HE, Blazar BR, et al. Mouse Ly49G2+ NK cells dominate early responses during both immune reconstitution and activation independently of MHC. Blood 2011;117(26):7032–41.
[36] Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. J Immunol 2009;182(8):4572–80.
[37] Brodin P, Lakshmikanth T, Johansson S, Karre K, Hoglund P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood 2009;113(11):2434–41.
[38] O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol 2006;7(5):507–16.
[39] Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature 2009;457(7229):557–61.
[40] Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A 2009;106(6):1915–9.
[41] Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalo-virus infection. Proc Natl Acad Sci U S A 2011;108(36):14725–32.
[42] Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012;119(11):2665–74.
[43] Guma M, Budt M, Saez A, Brckalo T, Hengel H, Angulo A, et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006;107(9):3624–31.
[44] Bjorkstrom NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med 2011;208(1):13–21.
[45] Petitdemange C, Becquart P, Wauquier N, Beziat V, Debre P, Leroy EM, et al. Unconven-tional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog 2011;7(9):e1002268.
[46] Hayakawa Y, Huntington ND, Nutt SL, Smyth MJ. Functional subsets of mouse natural killer cells. Immunol Rev 2006;214:47–55.
[47] Hallett WH, Murphy WJ. Positive and negative regulation of natural killer cells: thera-peutic implications. Semin Cancer Biol 2006;16(5):367–82.
[48] Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, et al. In vivo evi-dence for a dependence on interleukin 15 for survival of natural killer cells. Blood 2002;100(10):3633–8.
[49] Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol 2009;183(5):2911–4.
[50] Asai O, Longo DL, Tian ZG, Hornung RL, Taub DD, Ruscetti FW, et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by acti-vated natural killer cells after allogeneic bone marrow transplantation. J Clin Invest 1998;101(9):1835–42.
[51] Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effective-ness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002;295(5562):2097–100.
[52] Hu B, He Y, Wu Y, Bao G, Liu H, Welniak LA, et al. Activated allogeneic NK cells as suppressors of alloreactive responses. Biol Blood Marrow Transplant 2010;16(6):772–81.
[53] Koh CY, Blazar BR, George T, Welniak LA, Capitini CM, Raziuddin A, et al. Augmenta-tion of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood 2001;97(10):3132–7.
[54] Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008;28(4):571–80.
[55] Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, et al. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med 2008;205(13):2959–64.
[56] Barber MA, Zhang T, Gagne BA, Sentman CL. NK cells negatively regulate antigen presentation and tumor-specific CTLs in a syngeneic lymphoma model. J Immunol 2007;178(10):6140–7.
353
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
[57] Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2012;481(7381):394–8.
[58] Cortes JE, Kantarjian HM, O’Brien S, Giles F, Keating MJ, Freireich EJ, et al. A pilot study of interleukin-2 for adult patients with acute myelogenous leukemia in first complete remission. Cancer 1999;85(7):1506–13.
[59] Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Trans-plant 2003;32:177–86.
[60] Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999;94(1):333–9.
[61] Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural kill-er cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood 2007;110(1):433–40.
[62] Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood 2003;102(3):814–9.
[63] Davies SM, Ruggieri L, DeFor T, Wagner JE, Weisdorf DJ, Miller JS, et al. Evaluation of KIR ligand incompatibility in mismatched unrelated donor hematopoietic transplants: killer immunoglobulin-like receptor. Blood 2002;100(10):3825–7.
[64] Farag SS, Bacigalupo A, Eapen M, Hurley C, Dupont B, Caligiuri MA, et al. The effect of KIR ligand incompatibility on the outcome of unrelated donor transplantation: a report from the center for international blood and marrow transplant research, the European blood and marrow transplant registry, and the Dutch registry. Biol Blood Marrow Trans-plant 2006;12(8):876–84.
[65] Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, et al. Improved out-come in HLA-identical sibling hematopoietic stem-cell transplantation for acute my-elogenous leukemia predicted by KIR and HLA genotypes. Blood 2005;105(12):4878–84.
[66] Lowe EJ, Turner V, Handgretinger R, Horwitz EM, Benaim E, Hale GA, et al. T-cell allore-activity dominates natural killer cell alloreactivity in minimally T-cell-depleted HLA-non-identical paediatric bone marrow transplantation. Br J Haematol 2003;123(2):323–6.
[67] Beelen DW, Ottinger HD, Ferencik S, Elmaagacli AH, Peceny R, Trenschel R, et al. Genotypic inhibitory killer immunoglobulin-like receptor ligand incompatibility enhances the long-term antileukemic effect of unmodified allogeneic hematopoietic stem cell transplantation in patients with myeloid leukemias. Blood 2005;105(6): 2594–600.
[68] Willemze R, Rodrigues CA, Labopin M, Sanz G, Michel G, Socie G, et al. KIR-ligand in-compatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia 2009;23(3):492–500.
[69] Brunstein CG, Wagner JE, Weisdorf DJ, Cooley S, Noreen H, Barker JN, et al. Negative effect of KIR alloreactivity in recipients of umbilical cord blood transplant depends on transplantation conditioning intensity. Blood 2009;113(22):5628–34.
[70] Ruggeri L, Mancusi A, Burchielli E, Perruccio K, Aversa F, Martelli MF, et al. Natural killer cell recognition of missing self and haploidentical hematopoietic transplantation. Semin Cancer Biol 2006;16(5):404–11.
[71] Pando MJ, Gardiner CM, Gleimer M, McQueen KL, Parham P. The protein made from a common allele of KIR3DL1 (3DL1*004) is poorly expressed at cell surfaces due to substitution at positions 86 in Ig domain 0 and 182 in Ig domain 1. J Immunol 2003;171(12):6640–9.
[72] Yawata M, Yawata N, Draghi M, Partheniou F, Little AM, Parham P. MHC class I-specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood 2008;112(6):2369–80.
[73] Foley BA, De Santis D, Van Beelen E, Lathbury LJ, Christiansen FT, Witt CS. The reactiv-ity of Bw4+ HLA-B and HLA-A alleles with KIR3DL1: implications for patient and donor suitability for haploidentical stem cell transplantations. Blood 2008;112(2):435–43.
[74] Bari R, Bell T, Leung WH, Vong QP, Chan WK, Das Gupta N, et al. Significant func-tional heterogeneity among KIR2DL1 alleles and a pivotal role of arginine 245. Blood 2009;114(25):5182–90.
[75] Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, Parham P. Synergistic poly-morphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J Immunol 2008;180(6):3969–79.
[76] Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti- leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009;113(13):3119–29.
354
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
[77] Lo Monaco E, Tremante E, Cerboni C, Melucci E, Sibilio L, Zingoni A, et al. Human leukocyte antigen E contributes to protect tumor cells from lysis by natural killer cells. Neoplasia 2011;13(9):822–30.
[78] Foley B, De Santis D, Lathbury L, Christiansen F, Witt C. KIR2DS1-mediated activation overrides NKG2A-mediated inhibition in HLA-C C2-negative individuals. Int Immunol 2008;20(4):555–63.
[79] Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood 2009;113(3):726–32.
[80] Nguyen S, Dhedin N, Vernant JP, Kuentz M, Al Jijakli A, Rouas-Freiss N, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: im-maturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 2005;105(10):4135–42.
[81] Vago L, Forno B, Sormani MP, Crocchiolo R, Zino E, Di Terlizzi S, et al. Temporal, quan-titative, and functional characteristics of single-KIR-positive alloreactive natural killer cell recovery account for impaired graft-versus-leukemia activity after haploidentical hematopoietic stem cell transplantation. Blood 2008;112(8):3488–99.
[82] Cooley S, McCullar V, Wangen R, Bergemann TL, Spellman S, Weisdorf DJ, et al. KIR re-constitution is altered by T cells in the graft and correlates with clinical outcomes after unrelated donor transplantation. Blood 2005;106(13):4370–6.
[83] Nguyen S, Kuentz M, Vernant JP, Dhedin N, Bories D, Debre P, et al. Involvement of mature donor T cells in the NK cell reconstitution after haploidentical hematopoietic stem-cell transplantation. Leukemia 2008;22(2):344–52.
[84] Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, et al. Dif-ferential cytokine and chemokine gene expression by human NK cells following activa-tion with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol 1999;162(8):4511–20.
[85] Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. NK cell education after allogeneic transplantation: dissociation between recovery of cytokine-producing and cytotoxic functions. Blood 2011;118(10):2784–92.
[86] Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, et al. CD-56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood 2003;101(8):3052–7.
[87] Brahmi Z, Hommel-Berrey G, Smith F, Thomson B. NK cells recover early and medi-ate cytotoxicity via perforin/granzyme and Fas/FasL pathways in umbilical cord blood recipients. Hum Immunol 2001;62(8):782–90.
[88] Barker JN, Hough RE, van Burik JA, DeFor TE, MacMillan ML, O’Brien MR, et al. Serious infections after unrelated donor transplantation in 136 children: impact of stem cell source. Biol Blood Marrow Transplant 2005;11(5):362–70.
[89] Yu J, Venstrom JM, Liu XR, Pring J, Hasan RS, O’Reilly RJ, et al. Breaking tolerance to self, circulating natural killer cells expressing inhibitory KIR for non-self HLA exhibit effector function after T cell-depleted allogeneic hematopoietic cell transplantation. Blood 2009;113(16):3875–84.
[90] Bjorklund AT, Schaffer M, Fauriat C, Ringden O, Remberger M, Hammarstedt C, et al. NK cells expressing inhibitory KIR for non-self-ligands remain tolerant in HLA-matched sibling stem cell transplantation. Blood 2010;115(13):2686–94.
[91] Wang H, Grzywacz B, Sukovich D, McCullar V, Cao Q, Lee AB, et al. The unexpected effect of cyclosporin A on CD56+CD16− and CD56+CD16+ natural killer cell subpopu-lations. Blood 2007;110(5):1530–9.
[92] Haas P, Loiseau P, Tamouza R, Cayuela JM, Moins-Teisserenc H, Busson M, et al. NK-cell education is shaped by donor HLA genotype after unrelated allogeneic hematopoi-etic stem cell transplantation. Blood 2011;117(3):1021–9.
[93] Elliott JM, Wahle JA, Yokoyama WM. MHC class I-deficient natural killer cells acquire a licensed phenotype after transfer into an MHC class I-sufficient environment. J Exp Med 2010;207(10):2073–9.
[94] Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med 2010;207(10):2065–72.
[95] Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005;105(8):3051–7.
[96] Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol 2008;143(5):641–53.
355
Natural killer cells in graft-versus-host disease and graft-versus-leukemia
[97] Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Im-munother 2010;59(11):1739–44.
[98] Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011;13(1):98–107.
[99] Barao I, Hanash AM, Hallett W, Welniak LA, Sun K, Redelman D, et al. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T cells. Proc Natl Acad Sci U S A 2006;103(14):5460–5.
[100] Salagianni M, Lekka E, Moustaki A, Iliopoulou EG, Baxevanis CN, Papamichail M, et al. NK cell adoptive transfer combined with Ontak-mediated regulatory T cell elimination induces effective adaptive antitumor immune responses. J Immunol 2011;186(6):3327–35.
[101] Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005;106(1):376–83.
[103] Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One 2012;7(1):e30264.
[104] Miller JS, McCullar V. Human natural killer cells with polyclonal lectin and immuno-globulinlike receptors develop from single hematopoietic stem cells with preferential expression of NKG2A and KIR2DL2/L3/S2. Blood 2001;98(3):705–13.
[105] Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol 2005;175(8):5095–103.
[106] Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S, et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol 2005;33(10):1160–71.
[107] Suck G, Branch DR, Keating A. Irradiated KHYG-1 retains cytotoxicity: potential for adop-tive immunotherapy with a natural killer cell line. Int J Radiat Biol 2006;82(5):355–61.
[108] Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 2008;10(6):625–32.
[109] Ngo MC, Rooney CM, Howard JM, Heslop HE. Ex vivo gene transfer for improved adop-tive immunotherapy of cancer. Hum Mol Genet 2011;20(R1):R93–9.
[110] Pegram HJ, Jackson JT, Smyth MJ, Kershaw MH, Darcy PK. Adoptive transfer of gene-modified primary NK cells can specifically inhibit tumor progression in vivo. J Immunol 2008;181(5):3449–55.
[111] Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res 2009;15(15):4857–66.
[112] Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical character-ization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009;114(13):2667–77.
[113] Lin W, Voskens CJ, Zhang X, Schindler DG, Wood A, Burch E, et al. Fc-dependent ex-pression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood 2008;112(3):699–707.
[114] Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994;83(2):435–45.
[115] Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymor-phism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoim-mune disease. J Clin Invest 1997;100(5):1059–70.
[116] Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeu-tic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002;99(3):754–8.
[117] Golay J, Introna M. Mechanism of action of therapeutic monoclonal antibodies: prom-ises and pitfalls of in vitro and in vivo assays. Arch Biochem Biophys 2012;526(2):146–53.
[118] Binstadt BA, Brumbaugh KM, Dick CJ, Scharenberg AM, Williams BL, Colonna M, et al. Sequential involvement of Lck and SHP-1 with MHC-recognizing receptors on NK cells inhibits FcR-initiated tyrosine kinase activation. Immunity 1996;5(6):629–38.
[119] Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest 2012;122(3):1066–75.
356
Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation
[120] Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res 2008;14(14):4650–7.
[121] Lapalombella R, Yu B, Triantafillou G, Liu Q, Butchar JP, Lozanski G, et al. Lenalidomide down-regulates the CD20 antigen and antagonizes direct and antibody-dependent cellular cytotoxicity of rituximab on primary chronic lymphocytic leukemia cells. Blood 2008;112(13):5180–9.
[122] Wu L, Parton A, Lu L, Adams M, Schafer P, Bartlett JB. Lenalidomide enhances antibody-dependent cellular cytotoxicity of solid tumor cells in vitro: influence of host immune and tumor markers. Cancer Immunol Immunother 2011;60(1):61–73.
[123] Roda JM, Joshi T, Butchar JP, McAlees JW, Lehman A, Tridandapani S, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res 2007;13(21):6419–28.
[124] Kipriyanov SM, Cochlovius B, Schäfer HJ, Moldenhauer G, Bähre A, Le Gall F, et al. Synergistic antitumor effect of bispecific CD19 × CD3 and CD19 × CD16 diabodies in a preclinical model of non-Hodgkin’s lymphoma. J Immunol 2002;169(1):137–44.
[125] Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, et al. Sensitiza-tion of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol 2008;180(1):163–70.
[126] Yong AS, Keyvanfar K, Hensel N, Eniafe R, Savani BN, Berg M, et al. Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood 2009;113(4):875–82.
[127] Lundqvist A, Abras SI, Schrump DS, Alvarez G, Suffredini D, Berg M, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res 2006;66(14):7317–25.
[128] Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 2008;111(3):1428–36.
[129] Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991;252(5007):854–6.
[130] Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Mi-crobes Infect 2002;4(15):1545–58.
[131] Baraz L, Khazanov E, Condiotti R, Kotler M, Nagler A. Natural killer (NK) cells prevent virus production in cell culture. Bone Marrow Transplant 1999;24(2):179–89.
[132] Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, et al. Onco-lytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med 1999;5(8):881–7.
[133] Bergmann M, Romirer I, Sachet M, Fleischhacker R, Garcia-Sastre A, Palese P, et al. A genetically engineered influenza A virus with ras-dependent oncolytic properties. Cancer Res 2001;61(22):8188–93.
[134] Ogbomo H, Michaelis M, Geiler J, van Rikxoort M, Muster T, Egorov A, et al. Tumor cells infected with oncolytic influenza A virus prime natural killer cells for lysis of resistant tumor cells. Med Microbiol Immunol 2010;199(2):93–101.
[135] Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bed-side. Clin Microbiol Rev 2009;22(1):76–98.
[136] Elmaagacli AH, Steckel NK, Koldehoff M, Hegerfeldt Y, Trenschel R, Ditschkowski M, et al. Early human cytomegalovirus replication after transplantation is associated with a decreased relapse risk: evidence for a putative virus-versus-leukemia effect in acute myeloid leukemia patients. Blood 2011;118(5):1402–12.
[137] Brunstein CG, Gutman JA, Weisdorf DJ, Woolfrey AE, Defor TE, Gooley TA, et al. Alloge-neic hematopoietic cell transplantation for hematologic malignancy: relative risks and benefits of double umbilical cord blood. Blood 2010;116(22):4693–9.