Research Collection Doctoral Thesis Ueber die Synthese einiger Pyrrolidin- und Piperidin-Derivate Author(s): Suter, Theo Publication Date: 1944 Permanent Link: https://doi.org/10.3929/ethz-a-000096237 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript
Research Collection
Doctoral Thesis
Ueber die Synthese einiger Pyrrolidin- und Piperidin-Derivate
Es sei mir gestattet, an dieser Stelle meinem sehr verehrten
leider zu früh verstorbenen Lehrer
Herrn Prof. Dr. Robert Eder
meinen besten Dank auszusprechen für seine Anregungen und sein
stetes Interesse an der vorliegenden Arbeit.
4
Inhaltsverzeichnis
Einleitung 7
Theoretischer Teil 10
I. Dolantin und ähnliche Derivate des 4-Phenyl-piperidins ... 10
II. Methoden zur Herstellung stickstoffhaltiger Heterocyclendurch katalytische Hydrierung von Cyanderivaten ... 16
1. Einleitung ... 16
2. Mechanismus der Nitril-Hydrierung 17
3. Herstellung cyclischer Produkte durch katalytische Hy¬
drierung von Cyanderivaten .......20
a) Hydrierung von Ketonitrilen. . . .
21
a-Ketonitrile 22
ß-Ketonitrile ....
22
y-Ketonitrile 25
5-Ketonitrile 27
b) Hydrierung von Dinitrilen.......
28
III. Eigene Versuche zur Synthese von Pyrrolidin- und Piperi-din-Derivaten 29
A. Kondensationen 32
Herstellung von Phenyl-cyan-essigsäureäthylester ... 32
Herstellung von a-Phenyl-a-cyan-bernsteinsäurediäthyl-
äthylester 33
Herstellung von a-Phenyl-a-cyan-bernsteinsäurediäthyl-ester 34
Herstellung von a-Phenyl-a-cyan-y-iacetyl-buttersäure-äthylester 34
Kondensationen von Phenylcyanessigester mit Aethylen-chlorhydrin ....
35
Weitere Kondensationsversuche mit Phenylcyanessig-ester 3(J
Kondensation von Phenylcyanessigester mit Chloraceto-
nitril .37
B. Katalytische Hydrierungen 38
Herstellung von 2-Methyl-4-phenyl-pyrrolidin-4-carbonsäureäthylester 38
Herstellung von 2-Oxo-4-phenyl-pyrrolidin-4-carbonsäure-
äthyleiter 40
5
Herstellung von 2-Methyl-5-phenyl-piperidin-5-earbon-säureäthylesier 41
Hydrierung von Phenyl-cyan-ß-oxyäthyl-essigsäureäthyl-ester (?) 42
Tabelle der Hydrierungsversuche ...... 47
Experimenteller Teil 49
A. Kondensationen 49
Herstellung von Phenyl-cyan-essigsäureäthylester .... 49
Herstellung von a-Phenyl-a-cyan-ß-acetyl-propionsäureätnyl-ester.... 50
Herstellung von a-Phenyl-a-cyan-bernsteinsäurediäthylester .51
Herstellung von 3-Keto-butanol-(l) ...... 52
Herstellung von l-Chlor-butanon-(3)t
52
Herstellung von a-Phenyl-a-cyan-y-acetyl-butteraäureäthyl-ester....
. .... .52
Kondensationen von Phenylcyanessigester mit Aethylenchlor-hydrin ... 53
Kondensation von Phenylcyanessigester mit Chloracetonitril.
59
11 Katalytische Hydrierungen 60
Herstellung von 2-Methyl-4-phenyl-pyrrolidin-4-carbonsäure-äthylester (Form A) (50
Herstellung von 2-Methyl-4-c\clohexyl-pyrrolidin-4-earbon-säureäthylester ... 61
Herstellung von 2-Methyl-4-phenyl-pyrrolidin-4-carbonsäure-äthylester (Form B) 62
Herstellung von 2-Methyl-2-phenyl-3-amino-propionsäure-äthylester
........... 63
Herstellung von 2-Oxo-4-phenyl-pyrrolidin-4-carbonsäure-äthylester ...... 64
Herstellung von 2-Methyl-5-phenyl-piperidin-5-carbonsäure-äthylester (Form A) 64
Herstellung von 2-Methyl-5-phenyl-piperidin-5-carbonsäure-äthylester (Form B) 65
Hydrierung von Phenyl-cyan-ß-oxyäthyl essigsäureäthylester 66
C. Unterscheidende Farbreaktionen von Pyrrolidin- und Piperi-din-Derivaten 67
Zusammenfassung . . ..
68
6
Einleitung
Morphin, als wichtigstes Opium-Alkaloid, wurde schon 1806
von Sertürner isoliert. Die Aufklärung seiner Konstitution gelang
allerdings erst viel später: 1923 stellten Qullütid und Robinson1)die heute allgemein anerkannte Strukturformel des Morphins auf:
die 1927 auch von Schöpfe) bestätigt wurde. Eine Totalsynthesedes Morphins erscheint bei der komplizierten Struktur dieses Al¬
kaloids als sehr schwer durchführbar, und sie ist bis heute auch
noch nicht gelungen.Durch Umwandlungen am natürlichen Morphin wurde eine ganze
Reihe neuer Derivate hergestellt, die recht verschiedene pharma¬
kologische Wirkungen aufweisen. Aus den Ergebnissen der pharma¬
kologischen Prüfung dieser Umwandlungsprodukte wurde dann
versucht, Schlüsse über Zusammenhänge zwischen chemischer Kon¬
stitution und pharmakologischer Wirkung zu ziehen. Trotz des heute
vorliegenden grossen experimentellen Materials ist es aber noch
nicht gelungen, mit Sicherheit alle Momente abzuklären.
Neben Umwandlungsprodukten des natürlichen Morphins wurden
von verschiedenen Forschern neue Substanzen synthetisiert, die nur
einen Teil des Morphin-Moleküls darstellen oder einen solchen
Teil enthalten. Es erscheint denkbar, dass auf diesem Wege neue
Körper gefunden werden können, in denen die guten und erwünsch¬
ten Eigenschaften des Morphins erhalten geblieben, die uner¬
wünschten und schädlichen dagegen verschwunden sind. Bis heute
sind allerdings noch keine Produkte gewonnen worden, die das"
Morphin in seinen erwünschten Wirkungen vollwertig, und dabei
ohne unerwünschte Nebenwirkungen, ersetzen könnten. Dass durch
i) Soc. 123, 980 (1923)2) A 452, 211 (1927).
7
Synthese von, im Vergleich zu einem natürlichen Vorbild, einfâcher aufgebauten Körpern auch schöne Erfolge erreicht werden
können, beweist u. a. das Gebiet der Lokalanaesthetica, wo es nachder Konstitutionsaufklärung des Cocains gelang, eine ganze An¬zahl neuer Lokalanaesthetica zu schaffen, die im Vergleich zum
Cocain einfacher konstituiert sind, ihm gegenüber Vorzüge zeigen,und sich auch in der Praxis weitgehend durchgesetzt haben1).
Betrachtet man das vielfältige Tatsachenmaterial, das zur Ab¬
klärung der Zusammenhänge zwischen chemischer Konstitution und
pharmakologischer Wirkung bei verschiedenartigen chemischenStoffen gesammelt wurde, so kann man feststellen, dass sich solche
Zusammenhänge meist nur in begrenzten Gebieten finden lassen.Der synthetisch tätige Chemiker wird bei der Aufstellung einer
neuen Arbeitshypothese vorteilhaft solche Zusammenhänge berück¬
sichtigen. Er muss sich dabei jedoch vor zu starker Beeinflussunghüten, denn eine kleine Aenderung in einem Molekül, z. B. die
Verschiebung nur einer Methylgruppe, kann für dessen Wirksam¬keit ausschlaggebend sein 2).
Betrachtet 'man die Strukturformel des Morphins, so stellt man
fest, dass sich das Stickstoffatom im Morphin sowohl in ß- alsauch in y-Abstand zum Phenylkern befindet.
N—CH3 N—CH3 N—CH3
OH
,3-Abstand y Abstand
Es konnte daher von Interesse sein, Körper herzustellen, welchedieser Tatsache Rechnung tragen, d. h. Substanzen, die einen
Phenylkern enthalten und ein cyclisch gebundenes Stickstoffatomin ß- und y-Abstand dazu. Dies ist der Grundgedanke zur Darstel¬lung der nachstehend besprochenen Phenyl-pyrrolidin-Derivate,
die als einfachste Körper dieser Bedingung entsprechen.
1) \gl. 7.. B. Eisleb, „Vom Cocain zum Pantooain", Medizin und Chemie
(I.G.Fai'beniindustiw A.G.) 2, 364 (J934).2) vgl. a. Läuger und Martin, „Uebei Iigalen, Ein Beitrag zui Konstituhons-
spezifität", Schvv.med Wschrft. 73, 399 (19438
Dass die Herstellung solcher Substanzen von Interesse sein kann,bewies uns das Erscheinen des l-Methyl-4-phenyl-piperidin-4-car-bonsäureäthylesters von Eisleb1), der als Chlorhydrat unter demNamen „Dolantin" zwecks arzneilicher Verwendung in den Handel
gelangt ist.
COOC2H5
^n-ch,Dolantm
Das Dolantin weist neben spasmolytischen Wirkungen analge¬tische Eigenschaften auf und ist in letzterer Hinsicht dem Morphinnahe verwandt. Als 4-Phenyl-piperidinderivat entspricht es, wie
nachstehend noch näher ausgeführt wird, auch strukturmässigeinem Teilstück des Morphinmoleküls. Zum Unterschied von den
Phenyl-pyrrolidin-Derivaten, deren Synthese wir beabsichtigten undbei denen das Stickstoffatom sich wie beim Morphin in |3- undY-Abstand zum Phenylkern, aber in einem Fünferring, befindet,ist es beim Dolantin nur in y-Abstand und wie beim Morphin ineinem Sechserring vorhanden.
In Analogie zum Dolantin, dessen C-Atom 4 des heterocyclischenRinges neben dem Phenylkern noch mit einer veresterten Carbon¬
sâuregruppe verbunden ist, sollten unsere Pyrrolidinderivate an dem
mit dem Phenylkern verbundenen C-Atom auch eine veresterte
Carbonsäuregruppe tragen. Beim Dolantin zeigte sich nämlich,dass beim Fehlen dieser Carbonsäuregruppe die analgetischenEigenschaften ganz verschwanden, wahrend die spasmolytischeneine Schwächung erfuhren. Es ist daher denkbar, dass die Verhält¬
nisse bei den Pyrrolidinderivaten ähnlich sein werden.
h DIU' 079 281 dei I G.Farliemndustnc \.G. (1939.
9
Theoretischer Teil
/. Dolantin und ähnliche Derivate
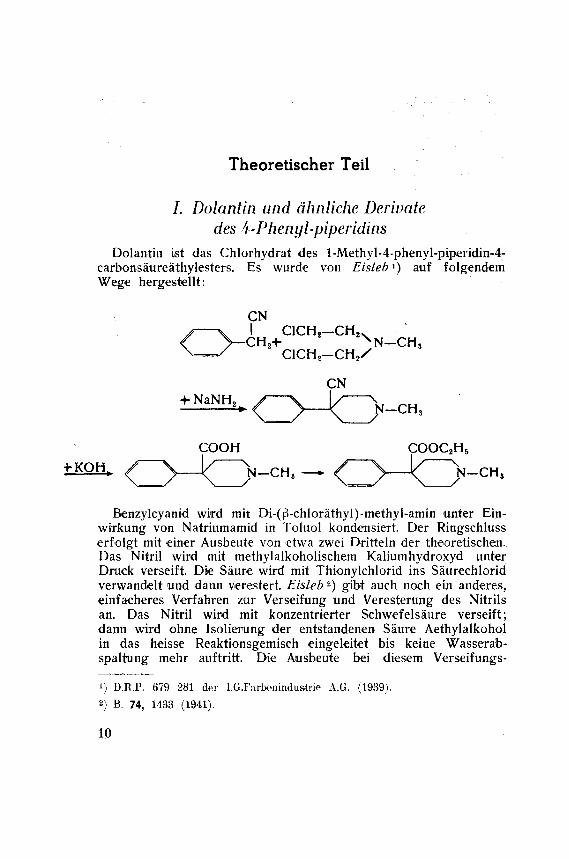
des b-Phenyl-piperidinsDolantin ist das Chlorhydrat des l-Methyl-4-phenyl-piperidin-4-
carbonsäureäthylesters. Es wurde von Eislebx) auf folgendemWege hergestellt:
CN
y y I ClCHs—CH2v< V-CH.+ NN-CH,n—/ C1CH2-CH2/
CN
+ NaNH2 y \ \/ \
COOH COOC2H5
Benzylcyanid wird mit Di-(ß-chloräthyl)-methyl-amin unter Ein¬
wirkung von Natriumamid in Toluol kondensiert. Der Ringschlusserfolgt mit einer Ausbeute von etwa zwei Dritteln der theoretischen.
Das Nitril wird mit methylalkoholischem Kaliumhydroxyd unter
Druck verseift. Die Säure wird mit Thionylchlorid ins Säurechlorid
verwandelt und dann verestert. Eisleb2) gibt auch noch ein anderes,einfacheres Verfahren zur Verseifung und Veresterung des Nitrils
an. Das Nitril wird mit konzentrierter Schwefelsäure verseift;dann wird ohne Isolierung der entstandenen Säure Aethylalkoholin das heisse Reaktionsgemisch eingeleitet bis keine Wasserab-
spaltung mehr auftritt. Die Ausbeute bei diesem Verseifungs-
i) D.R.P. 679 281 der I.G.Farbonindustric A.G. (1939).
2) B. 74, 1433 (1941).
10
und Veresterungsverfahren soll 95°/o der Theorie betragen. Das
Chlorhydrat wurde durch Zusatz von alkoholischer Salzsäure zu
einer ätherischen Lösung der Base ausgefällt.Das zur Dolantin-Synthese benötigte Di-(ß-chloräthyl)-methyl-
amin lässt sich durch Chlorierung von Di-(ß-oxyäthyl)-methyl-amin mit Thionylchlorid darstellen. Zu einem orientierenden Ver¬
such stellten wir Di-(ß-oxyäthyl)-methyl-amin nach Knorr1) her.
Die Chlorierung mit Thionylchlorid nahmen wir nach Prelog und
Stepan2) vor. Beim Arbeiten mit dem Di-(ßichloräthyl)-methyl-amin zeigte sich, dass diese Substanz ein Hautgift von yperit-ähnlicher Wirkung ist, also mit gewissen Vorsichtsmassmahmen
gehandhabt werden muss. Die Wirkung ist zwar wesentlich schwä¬
cher als beim Yperit, aber längere Einwirkung kann doch zu einer
Sensibilisierung führen, wie dies bei mir der Fall war. Da an
einer Hand starke Ekzeme auftraten, musste ich das Arbeiten mit
dieser Substanz für einige Zeit ganz einstellen.
Zur Bezeichnung Dolantin muss erwähnt werden, dass Eisleb •>)früher das Chlorhydrat des Piperidinoäthanolesters der N-ß-Meth-oxyäthyl-p-amino-benzoesäure
CHjOCHj—CH,—NH—/ \- COOCH2-CH2-N > HCl
als Dolantin bezeichnet hat. Diese Substanz wurde im Laufe der
Arbeiten, die zur Herstellung des Pantocains führten, untersucht.
Sie gelangte nicht in den Handel, da sich das Pantocain wirksamer
erwies. Dadurch war der Name für das heute als Dolantin be¬
zeichnete Produkt frei. Es muss noch bemerkt werden, dass für
Dolantin in England der Name Pethidin und in den U.S.A. der
Name Demerol gebraucht wird.
Eisleb und Schaumann4) berichten, dass sie das Dolantin bei
Versuchen zur Herstellung eines neuen Spasmolyticums, ausgehendvom Atropin, fanden. Die basische Gruppe sollte aus dem alkoho¬
lischen Teil, wie sie beim Atropin vorliegt, in den Säureteil des
Moleküls verlegt werden.
!) B. 22, 2088 (1889); B. 31, 1070 (1898).
2) Coll.Trav.chim.Tschécoslovaqme 7, 93 (1935); C. 1935 II 2817.
Die Substanzen A und B erwiesen sich auch als spasmolytischwirksam. Das Dolantin, zu dem der Schritt ja dann klein war,
zeigte dann überraschenderweise neben spasmolytischen Eigen¬schaften von der Art des Atropins und Papaverins, auch noch eine
zentralanalgetische Wirkung von morphinahnlichem Charakter.
Diese letztere Wirkung wird verständlich, wenn man die Formel des
Dolantins in gleicher Weise wie die Morphinformel aufzeichnet:
OH N0
MoiphiJi
OH
C-OC2H5
o
Dolantin
Man sieht deutlich, dass der Qrundkörper des Dolantins, das
l-Methyl-4-phenyl-piperiidin, auch ein Bestandteil des Morphinmole¬küls ist.
Eisleb1) stellte eine ganze Reihe von Derivaten des 4-Phenyl-piperidins her; sie erwiesen sich jedoch bei der pharmakologischenPrüfung durch Schaumann2) dem Dolantin wirkungsmässig unter-
1) Medn/jn und Chemie (LG Farbenindustne A.G.) 4, 213 (1942).*i \rehiv JexpPathoJ und Phnimakol 196, 109 (1940).
12
legen. Durch systematische Aenderung der einzelnen Gruppen im
Dolantin-Molekül wurde deren Einfluss auf die Wirkung bestimmt.
Der Grundkörper des Dolantins, das l-Methyl-4-phenyl-piperi-din, das aus der Carbonsäure durch Erhitzen über den Schmelz¬
punkt erhalten wird, weist verminderte spasmolytische Wirksamkeit
und keine analgetische Wirkung auf.
Aendert man den Alkohol mit dem die l-Methyl-4-phenyl-pipe-ridin-4-carbonsäure verestert ist, so nimmt die spasmolytische Wirk¬samkeit vom Methyl- über den Aethyl-, n-Butyl-, Glycerin-, Phenyl-zum Benzylester zu, während die analgetischen Eigenschaften beim
Aethylester ihr Maximum haben.
Das l-Methyl-4-phenyl-4-cyan-piperidin zeigt nur etwa einen Fünf -
tel der Dolantin-Wirkung. Das durch partielle Verseifung des Ni-
trils hergestellte Säureamid hat stark abgeschwächte spasmolytischeund keine analgetische Wirkung. Das Diäthyl-amid hat eine 2-
bis 3mal bessere spasmolytische, aber auch keine analgetischeWirkung.
Eisleb ^) stellte auch l-Methyl-4-phenyl-piperidyl-(4)-ketone her,und zwar durch Umsetzung des l-Methyl-4-phenyl-4-cyan-piperidinsmit magnesiumorganischen Verbindungen und Zerlegung der ent¬
standenen Ketimidzwischenprodukte mit verdünnter Säure.
CN
O-fO-cH. ^O-O"^»-C=NMgHlg C=0
NR R
Diese Ketone weisen auch eine gute Wirkung, wie die Ester, auf.
Das Maximum der Analgesie liegt beim n-Propyl-keton, dessen
analgetische Wirksamkeit der des Dolantins gleichkommt. Das Maxi¬
mum an spasmolytischer Wirkung, die 4- bis 10-fache des
Dolantins, weist das Phenyl-keton auf, dafür hat es fast keine anal¬
getische Wirksamkeit mehr.
Ersetzt man die Ketogruppe durch eine primäre, sekundäre
oder tertiäre Alkoholgruppe, so zeigen diese Carbinole ganz allge¬mein eine schlechtere Wirksamkeit als die Ketone und die Ester.
Bei den Estern wurde die Substitution am Stickstoffatom abge-
i] DRP 713 746 dei IG.L'arbwiindustrie A.G. (1941)
13
wandelt: das N-Aethyl-dérivât ist fast gleich stark wirksam wie das
N-Methyl-derivat (Dolantin). Mit zunehmender Grösse des Sub-
stituenten am N-Atom nimmt im allgemeinen auch die analgetischeund spasmolytische Wirksamkeit ab.
Die am Piperidin-Stickstoff nicht substituierte Nor-Verbin-
dung wurde aus l-Benzyl-4-phenyl-piperidin-4-carbonsäureäthylesterdurch Abspaltung der Benzylgruppe mit Palladium und Wasserstoff
erhalten; sie weist geringere spasmolytische und wesentlich ge¬
ringere analgetische Wirksamkeit auf. Am Stickstoffatom nicht
substituierte Derivate wurden von Eisleb1) auch auf folgende Weise
hergestellt: p-Toluolsulfon-di-(ß-chlorathyl)-amid wird mit Benzyl-cyanid kondensiert und das entstandene l-p-Toluolsulfon-4-phenyl-piperidin-4-carbonsäurenitril verseift und verestert. Die am Stick¬
stoffatom nicht substituierten Ester, Ketone und Carbinole sind
weniger wirksam als die entsprechenden N-Methyl-Derivate.Es zeigte sich, dass das Dolantin den anderen untersuchten 4-
Phenyl-piperidin-derivaten an Vielseitigkeit der Wirkung deutlich
überlegen war. Das Dolantin hat eine komplexe, sowohl neurotropewie myotrope spasmolytische Wirkung, die sich deutlich in seinem
Antagonismus gegen Histamin ausdrückt. Beim Histaminkrampfdes isolierten Meerschweinchendarmes wirkt Dolantin 4mal stärker
als Atropin und lOmal stärker als Papaverin.Die analgetische Wirksamkeit von Dolantin ist bei Mausen 7- bis
lOmal kleiner als die von Morphin. Wie beim Morphin tragenMause nach Dolantin-Injektion ihren Schwanz s-förmig gekrümmtüber dem Rücken.
Die Toxizität des Dolantins ist ungefähr 3i/2mal grösser als
die des Morphins. Die D.L. 50-) beträgt bei subcutaner Injektionan Mäusen beim Dolantin 150 mg/kg, beim Morphin 530 mg/kg.
Die Vergiftungssymptome sind wenig charakteristisch; die Tiere
gehen unter Krämpfen an Atemlähmung ein, ohne dass Anzeichen
einer narkotischen Wirkung zu beobachten sind. Im Vordergrunddes Vergiftungsbildes steht eine zentrale Erregung, die sich bei
Mäusen in der charakteristischen Schwanzhaltung äussert. Diese
zentrale Enthemmung zeigt sich auch bei Katzen, die nach sub¬
cutaner Dolantininjektion halluzinieren und ausserordentlich schreck¬
haft und bösartig werden.
In der Medizin hat sich Dolantin bei Krampfzuständen der glat¬ten Muskulatur und in der Gynäkologie eingeführt. Anfänglichwurden keine Beobachtungen über eine rauschgiftartige Wirkung
!) DRP 695 216 dei I G.Faibcnintlustiic A.ü. (1940)2) d.i che Dosis, bei dei 5()Oo dei Versuchstier' eingehen
14
des Dolantins gemacht, was Schaumann *) zu folgender Theorie
veranlasste: Morphin ist ein Rauschgift, da man es auch als Phenyl-äthylamin-derivat, mit ß-Abstand zwischen Phenylkern und N-
Atom, anfassen kann. Als einfachstes solches Rauschgift führt
Schaumann das Mezcalin, Trimethoxy-phenyl-äthylamin, an. Beim
Dolantin fehlt die beim Morphin vorhandene Methylenbrücke zwi¬
schen Phenyl- und Piperidinring, Phenylkern und N-Atom stehen
nur in y-Abstand; dies sollte der Grund für das Fehlen rausch¬
giftartiger Eigenschaften beim Dolantin sein. Es zeigte sich dann
aber doch, dass auch Dolantin Süchtigkeit erzeugen kann, eine
Möglichkeit, auf die z. B. Vogt'2) hingewiesen hatte. Fälle von
Dolantin-Sucht werden z. B. von Schwarte3), Kucher 4) und Lung¬witz 6) beschrieben.
Um das Dolantin-Patent von Eisleb zu umgehen, stellten Jensen
und Lundquist6) l-Methyl-4-phenyl-4-oxypiperidin her.
/CH2—CH2—COOC2H6 / V
CH3-Nf —CH3-N C=0
NCH2-CH2-COOCsH5N '
«!dÄcH,-(OH
Dieses Carbinol zeigt ein Fünftel der spasmolytischen Wirksam¬
keit des Dolantins, eine analgetische Wirkung fehlt. Der Plan von
Jensen und Lundquist war, die Oxygruppe durch Halogen zu er¬
setzen, um dann durch Umsetzung mit NaCN zu dem aus der
Dolantin-Synthese bekannten Nitril zu gelangen. Die Halogenderi¬vate waren jedoch nicht herstellbar; es wurde bei der Behandlungmit PBr3 oder SOCl2 Wasser abgespalten unter Bildung von 1-
Methyl-4-phenyl-tetrahydropyridin. Aus diesem Grunde wurde das
Carbinol mit verschiedenen Carbonsäuren verestert. Der dem Do¬
lantin isomere Propionsäureester ist analgetisch 5- bis lOmal wirk¬
samer als Dolantin, während seine spasmolytische Wirksamkeit nur
ein Viertel beträgt. Die D.L.50 beträgt 50 mg/kg, die Toxizität
ist also 3mal grösser als beim Dolantin und lOV^mal grösser als
beim Morphin.
M Archiv f. exp. Pathol, und Phannakol. 196. 109 (1940).
//. Methoden zur Herstellungstickstoffhaltiger Heterocyclen durch kcüalytische
Hydrierung von Cyanderiuaten
1) Einleitung
Die Einführung von Aminogruppen in organische Verbindungenkann auf mannigfaltige Weise geschehen. Verschiedene Methoden
führen über Zwischenprodukte, wie Nitro- und Nitrosokörper,Oxime, Hydrazone oder Nitrile, deren Reduktion dann das ge¬wünschte Amin liefert. Diese Reduktionen lassen sich sowohl mit
chemischen Mitteln, mit nascierendem Wasserstoff, als auch mit
molekularem Wasserstoff unter Benützung eines Katalysators durch¬
führen. Das Verfahren der katalytischen Hydrierung findet immer
grössere Verbreitung, da es den grossen Vorteil besitzt, ausser dem
Katalysator und eventuell benötigten Lösungsmitteln keinerlei
fremde Substanzen in das Reaktionsgemisch einzuführen. Bei der
Aufarbeitung können daher die Hydrierungsprodukte mit geringemArbeitsaufwand rein erhalten werden. Beide Arbeitsweisen führen
allerdings in den meisten Fallen zu einem Gemisch von primären,sekundären und tertiären Aminen, wobei je nach dem Ausgangs¬material und den Arbeitsbedingungen die eine oder andere Kompo¬nente bevorzugt wird.
Die Verhältnisse werden noch komplizierter, wenn in einem
Molekül ausser der bei der Reduktion entstehenden Aminogruppenoch Atomgruppen vorhanden sind, die mit dieser Aminogruppereagieren können. Diese sekundäre Reaktion kann unter Umständen
zu einem intramolekularen Ringschluss führen; aber auch die Kon¬
densation mehrerer Moleküle zu höheren Polymerisationsprodukten,wohl meist unerwünschter Natur, ist möglich. Ob bei Hydrierungensolche sekundäre Reaktionen auftreten oder nicht, hängt weitgehendvon den Arbeitsbedingungen ab. So erhielten Ruggli und Preis¬
werk l) bei der Hydrierung von Nitro-p-phenylen-diacrylsäure mit
Raney-Nickel bei Zimmertemperatur Amino-p-phenylen-diacrylsäurein einer Ausbeute von 88% der Theorie, bei 75" jedoch Hydro-carbostyril-7-propionsaure in einer Ausbeute von 70°o der Theorie.
'i Heh. 22, 478 (1939)
16
rrcH-cooHHOOC-CH=CH NH.
X\/CH=CH-COOH *>/<$
*SHOOC-CH=CH/N/sN02 A^
HOOC—CH2-CH
XVN
o
Dies diene als Beispiel einer intramolekularen Ringbildung, wie sie
verhältnismässig häufig anzutreffen ist.
2) Mechanismus der Nitril-Hydrierung
Bei der katalytischen Hydrierung von Nitrilen, die uns im fol¬
genden beschäftigen soll, werden meistens Gemische von primären,sekundären und tertiären Aminen erhalten. Wenn die Möglichkeiteiner Ringbildung besteht, so entsteht bei der Hydrierung von
Cyanverbindungen immer diejenige Form, welche für eine Ring¬bildung am geeignetsten ist. Diese Erscheinung wurde von Rapeund Bernstein *) als Regel erkannt. Der Mechanismus der kata¬
lytischen Nitril-Hydrierung wurde von verschiedenen Forschern zu
deuten versucht. Grundsätzlich muss man dabei zwei verschiedene
Arbeitsmethoden, die zu ungleichen Resultaten und Erklärungenführen, unterscheiden:
a) Hydrierung in wasserhaltigen Medien,
b) Hydrierung in wasserfreien, indifferenten Medien.
Petal und Gerum2) erhielten bei der Hydrierung von Benzo»-
nitril in wässerig-alkoholischer Lösung bei Zimmertemperatur und
i) Helv. )3, 457 (1930)2) B 42, 1553 (1909)
17
in Gegenwart von kolloidalem Palladium primäre und sekundäre
Base, Benzaldehyd, Ammoniak und unverändertes Nitril. Zur Er¬
klärung stellten sie folgende Hypothese auf. Zuerst wird das Ni¬
tril bis zum Imin hydriert, welches durch Wasser hydrolysiertwird zu Benzaldehyd und Ammoniak. Drei Moleküle Aldehyd rea¬
gieren dann mit zwei Molekülen Ammoniak unter Bildung von Hy-drobenzamid, welches dann bei weiterer Hydrierung in Benzyl-und Dibenzylamin gespalten wird.
Nach obigem Reaktionsschema ist die Bildung des sekundären
Amins auf den intermediär gebildeten Aldehyd zurückzuführen.
Rupe und Hodel1) hydrierten Benzonitril bei Raumtemperaturmit Nickel-Katalysator (auf Tonpulver als Trägersubstanz) in wäs¬
serigem Alkohol '- Essigester als Lösungsmittel und erhielten da¬
bei auch die sekundäre Base. Auch diese Verfasser nehmen an,
dass das Nitril zuerst zum Imin hydriert wird; dieses soll dann teils
zum primären Imin weiterhydriert, teils zu Aldehyd und Ammoniak
hydrolysiert werden. Primäres Amin und Aldehyd sollen sich zu
einer Schiffschen Base kondensieren, deren Hydrierung dann das
sekundäre Amin ergibt.
R__CN f- H2 -> R-CH = NH ^ H. -> R—CH8NH,
j H20
NH, R—CHO | R—CH.NH2-> R—CH = N—CH2—R
1 ~ H2
R—CH2-NH—CH,—R
Rape und Hodel konnten experimentell zeigen, dass bei Blok-
kierung des Aldehyds durch Phenylhydrazin die Bildung von se¬
kundären Amin fast ganz aufhört. Nach Angaben von Mignonac ")ist das Phenylhydrazon des Benzaldehydes schwer hydrierbar, wäh-
i) Helv 6, 865 (1923).
•*; Ann. chim (11) 2, 225 (1934)
18
rend Phenylhydrazin selbst bei der katalytischen Hydrierungleicht zu Anilin und Ammoniak gespalten wird.
Der Beweis der Entstehung einer Schiffschen Base, die dann zum
sekundären Amin weiterhydriert wird, konnte durch Rupe und Be¬
cherer l) erbracht werden. Bei der Hydrierung des ß-Naphtoni-triles erhielten sie fast kein sekundäres Amin, dagegen gelang es
ihnen die Schiffsche Base rein zu isolieren, da diese sich infolgeihrer Schwerlöslichkeit der weiteren Hydrierung entzieht.
Auch Skita2), der sich in verschiedenen Publikationen mit der
katalytischen Hydrierung beschäftigt hat, fasst seine Versuchsergeb¬nisse der katalytischen Hydrierung von Nitrilen in folgendemSchema, das mit der Theorie von Rupe übereinstimmt, zusammen:
iC=NOH
Oxime
- C^N
Nitrile
H,
H.,
>C=NH
ïCH—NH2 prim. Amin
H9 ;c=o
(;CH)2NH sek. Amin
H2 5C=0
y
(;CH)3N tert. Amin
Um bei der Hydrierung von Nitrilen primäre Amine zu erhalten,empfiehlt Skita in Gegenwart von Ammoniaküberschuss zu arbeiten,oder den entstehenden Aldehyd durch Phenylhydrazin, nach Rupe,zu blockieren. Als beste Methode empfiehlt er die katalytische Hy¬drierung in Gegenwart eines acylierenden Mittels vorzunehmen, um
so das primäre Amin als Acylverbindung vor einer weiteren Re¬
aktion zu schützen.
Bei den vorstehend beschriebenen Reaktionsmechanismen spieltder durch Hydrolyse des Imins gebildete Aldehyd die entschei¬
dende Rolle für die Bildung sekundärer und tertiärer Amine. Da
aber auch bei der Hydrierung in wasserfreien Medien sekundäre
i) Helv. 6, 880 (1923).-') Forsch.-Ber. Zellwoäk"
('. 1942 II 2781.
und Kunstseide-Ring G.m.b.H. No. 1, 42 (19421;
19
Amine entstehen, muss es noch andere Reaktionsmöglichkeitengeben.
Mignonac1) hydrierte Benzonitril in absolutem Alkohol. Wurde
die Hydrierung nach Aufnahme von 1 Mol Wasserstoff unter¬
brochen, so fand man Benzylamin und Benzalbenzylamin, also die
Schiffsche Base, welche durch Reaktion von Imin und primäremAmin unter Ammoniakabspaltung entstanden sein muss.
R -CN 1 H, > R CH=NH j H, -> R~~CH2NHS
R—CH,NH,
NH3 - R-CH-=N~CH2--R H2- > R- CHä—NH-CH2-R
Diese Erklärung des Reaktionsmechanismus wurde auch von Braun,
Blessing und Zobel2) gegeben. Dass diese Deutung recht allge¬meine Gültigkeit hat, konnte in einer neuen Arbeit von Zam3) ge¬
zeigt werden, der bei der Hydrierung aliphatischer Nitrile auch
Schiffsche Basen isolieren konnte.
Da bei der Bildung von sekundären und tertiären Aminen immer
intermediär eine Ammoniakabspaltung stattfinden muss, wurde von
verschiedenen Forschern, u. a. Mignonac4), empfohlen, bei der
Hydrierung Ammoniak zuzusetzen. Dadurch Hess sich in verschie¬
denen Fällen die Ausbeute an primärem Amin steigern.
3) Herstellung cyclischer Produkte durch
katalytische Hydrierung von C y and er i v ate n
Nachdem wir nun vorstehend den Reaktionsmechanismus der
katalytischen Nitrilhydrierung in kurzen Zügen besprochen haben,wollen wir unsere Aufmerksamkeit solchen Hydrierungen zuwenden,die direkt zu cyclischen Aminen führen.
Paden und Adkins 5) geben folgende Zusammenstellung von Sub¬
stanztypen, bei deren Hydrierung cyclische Amine erhalten werden
können :
i) Ann.chim. (11) 2, 225 (1934).
2) B. 56, 1988 (1923)
) Diss. E.T.H. Zürich 1944.
*) Bril.Pat. 282 083; D.B.P. 541229.
•>) Am. Soc. 58, 2487 (1936)
20
R-(CH,)n-R R-(CH2) „-COOR'
R~-(CH2)n-C=0 R-(CH2)n~CHOH-R'
R'
n = 2, 3 oder 4;
R = _cn, —CONH2, —CONHR', -C—NH2, —C—NHR',! !
i I
—C—NO, —C—N02 —C=NOH, —C=N—N=.
Die Bedingungen und die Leichtigkeit solcher Ringschlussreak¬tionen durch Hydrierung sind natürlich von Fall zu Fall sehr
verschieden und hängen auch stark von der Wahl des Katalysatorsab. Bei den meisten dieser intramolekularen Ringschlüsse handelt
es sich um Reaktionen zwischen Aminogruppen unter sich, oder
mit Oxygruppen oder Carbonsäureestern, wobei diese reagierendenGruppen teilweise auch erst während der Hydrierung entstehen
können.
In der vorliegenden Arbeit (siehe Experimenteller Teil) wird eine
präparativ brauchbare Methode zur Herstellung von Pyrrolidin-und Piperidin-Derivaten, durch Hydrierung entsprechender Keto-
nitrile, angegeben.
a) Hydrierung von Ketonitrilen
Bei der Hydrierung von Ketonitrilen wurde in vielen Fallen die
Entstehung von cyclischen Hydrierungsprodukten beobachtet. Wi¬
ley und Adkins l) geben an, dass bei Ketonitrilen die Cyangruppevor der Ketogruppe hydriert wird. Als Hydrierungsprodukte erhiel¬
ten sie Aminoketone, Aminoalkohole, cyclische Amine und als
Nebenprodukte manchmal stickstoffreie cyclische Aether, mit der
gleichen Anzahl C-Atome wie das Ketonitril. Sie schreiben diesen
Aethern die Form von substituierten Furanen zu, ohne jedoch diese
Annahme mit Sicherheit beweisen zu können. Zur Erklärung der
Bildung dieser Furanderivate muss eine Wanderung des Sauerstoff-
i) Am Soc 60, 914 (1938)
21
atoms angenommen werden (siehe nachstehend). Diese stickstoff¬
freien Aether wurden bei der Hydrierung von verschiedenen ß-Keto-nitrilen in Mengen von 20—25°,o erhalten, neben den entsprechen¬den Aminoalkoholen. Ihre Bildung ist stark von der Reduktions¬
temperatur abhängig und damit von der Hydrierungsgeschwindig¬keit. Sie entstehen am leichtesten bei 40—150°, während bei tie¬
ferer Temperatur hauptsächlich Ketoamine, und bei höherer
Aminoalkohole erhalten werden. Es ist noch zu erwähnen, dass die
amerikanische Forschergruppe von Adkins die Hydrierungen nor¬
malerweise mit Raney-Nickell) '-) als Katalysator und unter Druck
ausführt.
Die Hydrierung von Ketonitrilen führt, je nach dem Abstand
zwischen Cyan- und Ketogruppe, zu ganz verschiedenen Produkten.
Im folgenden wollen wir nun einige Beispiele für die einzelnen
Typen besprechen.
a-Ketonitrile:
Das einfachste a-Ketonitril, Acetylcyanid, wurde von LJrlass3) mit
Pd-BaSCvKatalysator in benzolischer Lösung hydriert. Als Re¬
duktionsprodukte erhielt er Dimethyläthylenglykol und Methylamin.Es trat also eine Spaltung des Acetylcyanids zwischen der Cyan-und Ketogruppe ein, mit anschliessender Hydrierung der Spalt¬stücke. Bei der Hydrierung von Benzoylcyanid erhielt Urlass ana¬
log als Reduktionsprodukte Hydrobenzoin, Isohydrobenzoin und
Methylamin. Diese beiden a-Ketonitrile wurden also bei der Hy¬drierung zuerst aufgespalten und die Spaltstücke dann jedes für
sich hydriert.
ß-Ketonitrile:
Urlass machte Hydrierungsversuche mit Benzoylacetonitril oder
Phenacylcyanid CeH5—CO—CH2—CN. Bei der Hydrierung mit Pd-
CaC03-Katalysator wurde nur die sekundäre Base, Di-(3-phenyl-3-oxy-propyl)-amin (CeH5—CHOH—CH2CH2)2NH, und Ammoniak
gefunden. Um die Beteiligung der Keto- und Cyangruppe an der
Bildung des Hydrierungsproduktes festzustellen, wurden auch Hy¬drierungsversuche mit dem Hydrazid und dem Phenylhydrazon an¬
gestellt. Das Ergebnis war merkwürdig, denn es fand keine Wasser¬
stoffaufnahme, auch nicht in der Nitrilgruppe statt. Urlass kam
M U.S.Pat 1628 190 (1927]-) Covert und Adkins, Ain. Sor 54, 4116 (1932).
J) Diss Umv Leipzig 1931
22
zum Schluss, dass das Phenacylcyanid eine Umlagerung zum 5-
Phenyl-isoxazol erleidet, dessen Hydrierung dann nach folgendemSchema erfolgt:
C6H,-CO-CH2CN
C6H6-C CH
I+ 2H,
C6H5—CH
O\ /
NH
CH2ICH,
+H2
CeH;,—CHOH—CH2CH2 NH2
Nach der Annahme von Claisen ') werden Isoxazole mit besetz¬
ter a- aber unbesetzter y-Stellung mit Alkali zu den isomeren Cyan-ketonen aufgespalten. Urlass führte deshalb einen Hydrierungsver¬such in alkalischer Lösung durch, der unter nur geringer Wasser¬
stoffaufnahme verlief und zu einer Molekülspaltung in Benzaldehydund niedere aliphatische Amine, wahrscheinlich Aethylamin, das
wegen zu geringer Menge nicht identifiziert werden konnte, führte.
Da Urlass bei den Hydrierungsversuchen immer nur die sekun¬
däre Base erhielt, führte er auch einige Versuche in Essigsäure¬anhydridlösung durch. Er erhielt dabei das Acetylderivat des
Phenylmilchsäurenitrils C6H5—CHOCOCH3-CH2CN. Als Neben¬
produkt fand er Di-N-acetyl-phenyl-propyl-1-amin CeH5CH2CH2-
CH2N(COCH3)2. Die Hydrierung des Phenacylcyanids verläuft
also wahrscheinlich immer zuerst über das Phenylmilchsäurenitril.
Rupe, Metzger und Vogler2) hydrierten Cyan-aoetyl-harnstoffund erhielten als Reduktionsprodukt Uracil nach folgendemSchema ;
/NHOC CO
HSC NH,\
CN
•H,
N^OC CO
H.C^
H,Q
-NH3
/NHOC CO
NH,
CH=NH
H,C
-H20
\NH2
CHO
,NJ
OC CO
HC NH
r) B. 24, 3916 (1891); B 36, 3664 (19031.
•*) Helv 8, 848 (1925)
23
Rape und Heckendornx) und Rape und Pieper-) hydrierten Ben-
zyliden-cyan-essigester in nicht wasserfreier Lösung und erhielten
als Hauptprodukt den Halbaldehyd des Benzylmalonsäureestersund daneben noch Benzyl-ß-amino-propionsäure nach folgendemSchema:
C6H5—CH=C—COOC2H5+ 2H.
CCH.-CH2—CH-COOC2H,
CN
C6H5-CH2—CH—COOCH,+ H20
CH=NH
-f H2 -j- H2Q
CHO
+ NHj
C6H5-CH2—CH—COOH
CH2NH2
Bei der Hydrierung von o-Nitrobenzyliden-cyan-essigester durch
Rupe und Heckendorn >) wurde nur die Nitrogruppe reduziert. Dasentstandene Amidocyanderivat kondensierte sich aber sofort zum
2-Aminochinolin-3-carbonsäureester, der tautomer reagieren kann:
CN +3H^ ( |NO,
-2H.O N^NH, NH
Wiley und Adkins3) erhielten bei der Hydrierung einiger ß-Ketonitrile die entsprechenden Aminoalkohole und daneben nochstickstoffreie cyclische Aether. Beim 2-Aethyl-4-methyl-3-keto-va-leronitril erhielten sie 30»/o Aminoalkohol und 20°<o cyclischenAether, beim 2-n-Propyl-4-methyl-3-keto-valeronitril erhielten sie
54«/o Aminoalkohol und 25»/o cyclischen Aether, beim 2-n-Propyl-3-keto-önanthonitril 50o/o Aminoalkohol und 24°'o cyclischen Aether.Für diese cyclischen Aether nimmt Adkins die Form von Furan-derivaten an, deren Bildung sich, unter Annahme einer Wanderung
Die katalytische Hydrierung von y-Ketonitrilen führt in den
meisten Fällen zur Bildung von Pyrrolidinderivaten, die z. T. mit
M Helv- 12, 637 (19291
25
sehr guter Ausbeute entstehen. Vollmann und Schlofferl) hydrier¬ten Lävulinsäurenitril mit einem Kobaltkatalysator unter erhöhtemDruck und erhöhter Temperatur. Sie erhielten in guter Ausbeute a-
Methylpyrrolidin, daneben noch a-Methylpyrrol.Rupe und Pieper2) erhielten bei der Hydrierung von Phenyl-
cyan-brenztraubensäure-äthylester, ein ß,y-Diketonitril, sozusagenquantitativ 4-Phenyl-2,3-diketo-pyrrolidin.
C,H5-CH-CN2 H
C8Hä-CH-CHs-NHg_Q H QH
CO-COOQH,*
CO—COOC2H5>
C(,H-, -CH—CH2> NH
CO—CO
Winans und Adkins ") erhielten bei der Hydrierung von ß-Phenyl-ß-cyan-propionsäureäthylester mit Nickelkatalysator unter Druckbei 50—90° mit 89<>/o Ausbeute 4-Phenyl-pyrrolidon. Die analogausgeführte Hydrierung von ß-Cyan-propionsäureäthylester ergabmit einer Ausbeute von 38<>'o Pyrrolidon.Rupe und Gisiger^) hydrierten a-Phenyl-ß-benzoyl-propionsäure-
nitril. Bei der Hydrierung in der Kälte erhielten sie die Pyrrolin-base und in der Wärme die Pyrrolidinbase, 2,4-Diphenyl-pyrrolidin.
C6H5—CO—CH2—CH—C6H5
CN
C6H£,—C=CH—CH—C6H5momentan I i
NH CH,- H30
CeH,—CH—CH,-CH—C6Hr,+ H2
> NH CH2
Bei der Hydrierung von Cyan-benzoesäureestern durch Rupe und
Bernstein5) lieferten die m- und p-Verbindung überwiegend sekun¬däre Base, während bei der o-Verbindung Phtalimidin entstand.
Rape und Stern2) hydrierten a,ß-Diphenyl-ß-phenacyl-propion-säurenitril. Das Nitril liegt in zwei isomeren Formen vor, von
denen jedoch nur die eine hydriert zu werden scheint, die andere
Form wird bei der Hydrierung umgelagert und dann auch hydriert.Als Reduktionsprodukt wird 2,4,5-Triphenylpiperidin erhalten.
CH—CH.—CO
<0^h-cn
Zusammenfassung :
Bei a-Ketonitrilen tritt bei der Hydrierung meist eine Spaltungdes Moleküls zwischen Keto- und Cyangruppe ein, mit nachfol-
i> Holv. 9, 980 (1926).
-') Helv. 10, 859 (1927)
27
gender Hydrierung der Bruchstücke. Bei ß-Ketonitrilen werden sehr
verschiedenartige Hydrierungsprodukte erhalten, durch normale Re¬duktion entstehen Aminoketone und Aminoalkohole, daneben werdenaber auch stickstoffreie Produkte gebildet. Bei der Hydrierung von
"f-Ketonitrilen werden meist Pyrrolidinderivate erhalten und bei der
Hydrierung von &-Ketonitrilen in vielen Fällen Piperidinderivate.Bei grösserem Abstand zwischen Keto- und Cyangruppe wird dieTendenz zu Ringschlüssen wieder geringer. Dies entspricht jaauch der bei anderen Ringschlussreaktionen gemachten Erfahrung,dass sich Fünf- und Sechsringe am leichtesten bilden.
b) Hydrierung von Dinitrilen
Auch bei der Hydrierung von Dinitrilen wurde die Entstehungcyclischer Amine beobachtet. Ruggli und Mitarbeiterl) erhielten beider Hydrierung von o-Xylylen-dicyanid mit einem Nickelkatalysa¬tor unter Druck mit 50« o Ausbeute Benzo-hexa-methylen-imm oder
Homo-tetrahydro-isochinolin; die Hydrierung der m- und p-Ver-bindung führte zu den entsprechenden Diaminen.
CH,-CN
+3H2 /v(CH.-CH=NH
CH.-CN
CH.-CH
\N
CH.-CH/
V^CHr-CHj-NH,
+ H20
-NH3
yxXHjj—CH,N
CH,-CH,
NH
') Helv 18, 1388 (1936)
28
///. Eigene Versuche zur Synthese von Pyrrolidin-und Piperidin-Derivaten
Auf Grund der in der Einleitung kurz dargelegten Ueber-
legungen, setzten wir uns die Synthese von Verbindungen mit dem
Skelett des 4-Phenyl-pyrrolidins zum Ziel.
In Analogie zum Dolantin sollte der 4-Phenyl-pyrrolidin-4-car-bonsäureäthylester (VI) hergestellt werden.
Die im vorstehenden Abschnitt I erwähnte sehr elegante Syn¬these des Dolantins durch Eisleb lässt sich nicht zur Herstellungvon Pyrrolidinderivaten verwenden, da die in diesem Falle benötig¬ten a-Halogenalkyl-ß-halogenalkyl-amine,
z. B. C1CH,-CH»
C1CH2>N CH
nicht leicht zugänglich sind.
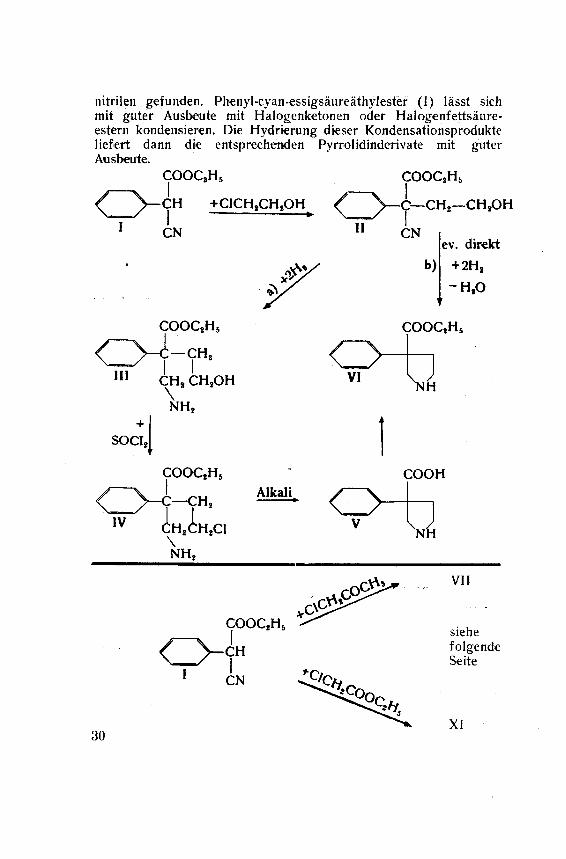
Ein anderer Weg, der denkbar erschien, wird durch folgendeFormeln illustriert (siehe Seite 30, Formeln I VI):
Ausgehend vom Phenyl-cyan-essigsaureäthylester (I) soll durch
Kondensation mit Aethylenchlorhydrin Phenyl-cyan-ß-oxyäthyl-essigsaureäthylester (II) hergestellt werden. Die katalytische Hy¬
drierung von II würde dann nach a) zum Aminoalkohol III führen,oder nach b) unter direkter Bildung des Pyrrolidinringes zum 4-
Phenyl-pyrrolidin-4-carbonsäureäthylester (VI). Für den Fall der
Hydrierung nach a) war vorgesehen den Weg über das Chlorid IV
und die Pyrrolidincarbonsäure V einzuschlagen.Diese Synthese erwies sich jedoch als nicht durchführbar, da
die Reindarstellung der Substanz II nur in sehr geringer Mengemöglich war. Bei der Hydrierung von II wurden dann auch nicht
die Substanzen III oder VI erhalten, sondern Polymerisationspro¬dukte. Nach Durchführung einer Reihe von Versuchen, über die
nachstehend berichtet wird, wurde dieser Weg verlassen.
Es wurde nun eine brauchbare Methode zur Herstellung von
Pyrrolidinderivaten in der katalytischen Hydrierung von y-Keto-
^
29
nitrilen gefunden. Phenyl-cyan-essigsäureäthylester (I) lässt sich
mit guter Ausbeute mit Halogenketonen oder Halogenfettsäure¬estern kondensieren. Die Hydrierung dieser Kondensationsprodukteliefert dann die entsprechenden Pyrrolidinderivate mit guterAusbeute.
COOC8H5 COOQH5
<^)>~CH +C1CH,CH8QHL /~~\_c—CH*—CH,OH
CN
III
+
SOCIg
IV
COOC2Hs
—CH,I ICH, CH8OH\NHj
COOC,H6
C—CH,Ah
CH,v
NH,
Cl
Alkali
II
VI
O
CN
b)
ev. direkt
+ 2H,
-H.O
COOQH5
>IH
COOH
\l
oCOOC2H6
CH
ICN
30
VII
siehe
folgendeSeite
XI
VII
COOC8H5
I—CH8
CN CO—CH3
-H.O \ /
VIII
COOC8H5
H CH,
COOC8H5
CH.ACOOC2H5
+ 2H,
C8H5OHIX
CN COOC2H5V-^\0
Phenyl-cyan-essigester (I) lässt sich leicht mit Chloraceton zum u-
Phenyl-a-cyan-ß-acetyl-propionsäureäthylester (VII) kondensieren,dessen katalytische Hydrierung führt dann zum 2-Methyl-4-phenyl-pyrrolidin-4-carbonsäureäthylester (VIII). Ebenso leicht lässt sich
Phenyl-cyan-essigester (I) mit Chloressigsäureäthylester zum a-
Phenyl-a-cyan-bernsteinsäurediäthylester (IX) kondensieren, dessen
Hydrierung den 2-Oxo-4-phenyl-pyrrolidin-4-earbonsâureàthylester(X) liefert.
Um die Brauchbarkeit dieser Methode zur Herstellung analogerPiperidinderivate aus b-Ketonitrilen zu prüfen, kondensierten wir
Phenyl-cyan-essigester (I) mit ß-Chloräthyl-methylketon zum a-
Phenyl-a-cyan-y-acetyl-buttersäureäthylester (XI), dessen Hydrie¬rung 2-Methyl-5-phenyl-piperidin-5-carbonsaureäthylester (XII) er¬
gab.
COOC8H6
I
CH
ICN
COOC,H5
CH2—CH\
XCN HO7
+ C1CH2-CH! -CO -CH3
COOC2H5
C-CH.+ 3H,:rFnö* CH,
XI XIINH
Die durch Hydrierung von Ketonitrilen erhaltenen Pyrrolidin-und Piperidinderivate enthalten am C-Atom 2 immer eine Seiten¬
kette !), die von der Wahl des Ketonitriles abhängig ist.
M vgl .inch die Zusammonstellurm iibi'i llviluciurm \(m Ketumtnlen, 21
31
Die vorstehend skizzierte Ringschlussmethode durch Hydrierunglâsst sich also gut zur Herstellung von 2-substituierten Pyrrolidin-und Piperidinderivaten, sowie zur Herstellung von a-Pyrrolidonenund a-Piperidonen benutzen.
A. Kondensationen
Herstellung von Phenyl-cyan-essigsäure-äthylester
Als Ausgangsmaterial zu unseren Synthesen diente der Phenyl-cyan-essigsäureäthylester, der auch schon zur Synthese des „Nir-vanols"1) benutzt wurde. Dieser Ester wurde zuerst von HesslerJ)hergestellt. Eine ähnliche Methode wurde von Bodroux ^) beschrie¬ben. Wir versuchten die Herstellung des Esters nach diesen An¬
gaben, erhielten aber nur relativ schlechte Ausbeuten zwischen 40
und 45°o; daneben entstanden in grösserem Masse nicht destillier¬
bare harzige Rückstande. Bei einem unserer Kondensationsversuche
von Benzylcyanid mit Kohlensaurediäthylester in Toluol mit Na-
triumalkoholat wurde gar kein Phenylcyanessigester erhalten, son¬
dern nur ein undestillierbarer Rückstand, der direkt kristallisierte.
Durch Umkristallisieren aus Alkohol wurden weisse Kristallnadeln
vom F: 265° erhalten. Es handelte sich um a, a'-Diphenyl-a, a'-di-
cyan-aceton, das durch Kondensation von zwei Molekülen Phenyl¬cyanessigester entstanden ist. Das von uns erhaltene Produkt hatte
die gleichen Eigenschaften wie das von Chamberlain, Chap, Doyleund Spaulding4) beschriebene. Da sich bei diesen Versuchen garkeine neuen Beobachtungen ergaben, haben wir auf ihre Beschrei¬
bung im experimentellen Teil verzichtet.
Die besten Ausbeuten an Phenylcyanessigester wurden bei der
Kondensation von Benzylcyanid mit Kohlensäurediäthylester in ab¬
solutem Aether mit Natriumamid als Kondensationsmittel erhalten.
Cretcher und Nelson 3) geben eine Arbeitsvorschrift für diese Kon¬
densation, nach der sie Phenylcyanessigester mit einer Ausbeute
von 70,3»/o der Theorie erhalten haben. Bei einem ersten Versuch
nach dieser Vorschrift erhielten wir eine Ausbeute von 63,7°/o d. Th.
Wir stellten fest, dass bei dieser Kondensation jede Spur von
!) z. B DR.P. 309 508 (1918).2) Am Chem.J 32, 119 (1904)
>) C j 151, 1358, (1910)4) Am.Soc 57, 352 (1935).~->\ Am Soc 50, 2760 (1928)
32
Feuchtigkeit sorgfältig vermieden werden muss. Die Reaktion,die ziemlich stark exotherm verläuft, muss so langsam durchge¬führt werden, dass keine zu starke Selbsterwärmung eintritt. Bei
unsorgfältigem Arbeiten wird in erheblichem Masse Diphenyl-dicyan-aceton gebildet. Es gelang uns, nach der im experimentellenTeil angegebenen Vorschrift, die Ausbeute an Phenyl-cyan-essigesterauf 79,5°/o d. Th. zu erhöhen.
Das zur Kondensation verwendete Natriumamid lässt sich gefahr¬los in Portionen im Porzellanmörser pulverisieren; die Zerkleine¬
rung wird erleichtert, wenn man vorher Reibschale und Pistill
leicht anwärmt.
Herstellung von a-Phenyl-a-cyan-^-acetyl-proplonsäureäthylester
Dieser Ester (VII) kann durch Kondensation der Natrium¬
verbindung des Phenylcyanessigesters (I) mit Chloraceton her¬
gestellt werden. Die Natriumverbindung entsteht beim Kochen
von Phenylcyanessigester mit metallischem Natrium in absolutem
Aether. Sie lässt sich nach Chamberlain und Mitarb.1) mit guterAusbeute mit Halogenalkylen umsetzen. Wir arbeiteten entsprechendbei der Umsetzung der Natriumverbindung mit Chloraceton und
erhielten den a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester mit
einer Ausbeute von 69°o d. Th. Störend bei dieser Arbeitsweise ist
die Langsamkeit der Umsetzung. Die Kondensation nimmt 3 Tagein Anspruch: 1 Tag zur Bildung der Natriumverbindung und wei¬
tere 2 Tage zur Umsetzung mit der Halogenverbindung. Im Laufe
unserer Arbeiten wurde uns dann eine Publikation von Wldeqvlst2),über die Herstellung von Phenyl-cyan-substituierten Carbonsäuren,bekannt. Wideqvist kondensiert Phenylcyanessigester in alkoholi¬
scher Lösung mit Halogenfettsäureestern unter Einwirkung von Na-
triumalkoholat. Bei unseren Versuchen zeigte sich dann, dass unser
Ester (VII) sich entsprechend diesem Verfahren mit einem Zeit¬
aufwand von nur 6 Stunden für die Kondensation und einer Aus¬
beute von 77i/2°/o d. Th. herstellen lässt. Das erhaltene Produkt
weist nach beiden Verfahren den gleichen Reinheitsgrad auf. Die
bessere Ausbeute und die kürzere Dauer der Kondensation in alko¬
holischer Lösung lässt sich durch die Löslichkeit der Reaktions¬
teilnehmer in Alkohol und durch die höhere Temperatur des Re¬
aktionsgemisches erklären. Im Falle der alkoholischen Lösung wird
Phenylcyanessigester mit einer Lösung von Natriumalkoholat um-
gesetzt, die Natriumverbindung ist in Alkohol beträchtlich löslich,daher findet auch die Kondensation mit der Halogenverbindung in
Lösung statt; erst das entstehende Natriumchlorid scheidet sich als
unlösliche Verbindung ab. Bei der Kondensation in Aether handelt
es sich dagegen immer um eine Reaktion zwischen einer gelöstenund einer suspendierten Komponente.Der im Hochvakuum frisch destillierte a-Phenyl-«- cyan-ß-acetyl-
propionsäureäthylester ist eine hellgelbe, sehr viskose Flüssigkeit,die nach einigem Stehen zu kristallisieren beginnt und einen
Schmelzpunkt von 51—52" aufweist. Durch Umkristallisieren aus
Petroläther Hess sich der Schmelzpunkt auf 571/2° steigern, wobei
jedoch bei der Mikroanalyse keine Unterschiede zwischen dem
nur destillierten und dem noch umkristallisierten Produkt festge¬stellt wurden. Die angegebenen Werte für Refraktion und Dichte
wurden im flüssigen Ester, unmittelbar nach der Destillation be¬stimmt.
Herstellung von a-Phenyl- a-cyan-bernsteinsäurrdiäthylester
Der Ester (IX) wurde von WideqvisO) hergestellt, aber nicht
näher untersucht. Wir arbeiteten bei der Herstellung dieses Esters
nach den Angaben dieses Autors, erhielten aber eine etwas bessere
Ausbeute.
Herstellung von a-Phenyl- a-cyan-^-acetyl-buttersäureäthylester
Dieser Ester (XI) wurde durch Kondensation der Natriumver¬
bindung von Phenylcyanessigester mit ß-Chloräthyl-methyl-ketonin absolutem Aether hergestellt. Das benötigte ß-Chloräthyl-methyl-keton wurde durch Chlorierung von 3-Keto-butanol-(l) mit kon¬
zentrierter Salzsäure nach Décombe2) hergestellt. Das Ketobutanolstellten wir durch Kondensation von Aceton mit Formaldehydnach einem Patent der Firma Bayer & Co.6) her. Das Ketobutanolist eine ziemlich leicht polymerisierende Flüssigkeit; in der Literatur
ist es nirgends genauer charakterisiert worden. Bei der Kondensa¬tion von Aceton mit Formaldehyd werden auch keine einheitlichen
Kondensationsprodukte erhalten, Morgan, Megson und Pepper4)geben eine ganze Tabelle solcher Kondensationsprodukte an. White
i) Svensk kern. Tidskr. 54, 88 (1942
2j C. r. 202, 1685 (1936).3) D.R.P. 223 207 (1910).'-) Chem. & Ind (London) 57, 885 (1938).
34
und Howard1) geben für 3-Ketobutanol einen Kp12: 70—71° an,
während im alten Patent ein Kp30: 109—110° genannt wird; nach
Ansicht der erwähnten Autoren soll es sich bei dem höher sieden¬
den Produkt, das auch von uns erhalten wurde, um nicht ganz
reines Ketobutanol handeln. Wir haben das bei der Kondensation
von Aceton und Formaldehyd erhaltene Produkt nicht näher unter¬
sucht, sondern unmittelbar nach der Destillation zum ß-Chloräthyl-methyl-keton chloriert. Die Umsetzung mit der Natriumverbindungdes Phenylcyanessigesters erfolgte in absolutem Aether, entspre¬chend dem weiter oben beschriebenen Beispiel.Der a-Phenyl-a- cyan-y-acetyl-buttersäureäthylester ist eine farb¬
lose, nicht erstarrende Flüssigkeit. Bei der Bestimmung der Mole¬
kularrefraktion zeigte sich, dass dieses Ketonitril in der Enolform
vorliegen muss. Für das Vorhandensein der Enoldoppelbindungsprach auch die intensive Gelbfärbung mit Tetranitromethan. Bei
der katalytischen Hydrierung wurde die zur Hydrierung einer
Doppelbindung benötigte Menge Wasserstoff in sehr kurzer Zeit
aufgenommen (siehe Fig. 1, S. 44), bei der Weiterhydrierung der
Cyangruppe erfolgte dann die Wasserstoffaufnahme bedeutend
langsamer. Wurde die Hydrierung nach Aufnahme von 1 Mol
Wasserstoff unterbrochen, so ergab das Hydrierungsprodukt keine
Gelbfärbung mehr mit Tetranitromethan. Alles spricht daher für
die Enolform und nicht die Ketoform.
Bei der mikroanalytischen CH-Bestimmung konnten bei diesem
Ester, aus nicht abgeklärten Gründen, keine übereinstimmenden
Resultate erhalten werden. Die Substanz scheint rein zu sein, da
sich auch bei mehrfacher Destillation ihre physikalischen Konstan¬
ten nicht änderten.
Kondensationen von Phenylcyanessigester mit Aethylenchlorhydrin
Das Ziel dieser Kondensationsversuche war die Herstellung von
führte im allgemeinen zu wenig einheitlichen Produkten. Betrach¬
tet man die Versuchsergebnisse, so findet man aber doch zwei Sub¬
stanzen, von verschiedenem Siedepunkt, die auf Grund ihrer Mole¬
kularrefraktion als die gewünschte Verbindung angesprochen wer¬
den könnten, wobei die Möglichkeit bestände, dass es sich um
Isomere handelt. Es wurde eine Reihe von Mikroanalysen ausge¬
führt, die aber wenig übereinstimmende Werte ergaben und auch
von den berechneten Werten teils erheblich abwichen. Als kristalli-
i) So<" 62, 25 (1943), (' 1943 II 713
35
siertes Derivat wurde ein 3,5-Dinitrobenzoat hergestellt, das, ana¬
lysenrein umkristallisiert, bei 2 verschiedenen Versuchen keineübereinstimmenden Analysenwerte gab, obwohl beide Präparateden gleichen Schmelzpunkt hatten und keine Depression ergaben.Der Versuch die Oxygruppe mit Thionylchlorid durch Chlor zu er¬
setzen war erfolglos. Das Ausgangsmaterial wurde quantitativwieder zurückerhalten. Diese Chlorierungsversuche hatten aber das
überraschende Ergebnis, dass nach einem solchen Versuch die
Analyse des regenerierten Produktes auf Phenyl-cyan-ß-oxyäthyl-essigsäureäthylester stimmte; bei diesem Präparat wurde auch ein
sehr gut stimmender MD-Wert gefunden.Da es aber auf keine Weise gelang, den gewünschten Ester II
mit Sicherheit zu identifizieren und genau zu charakterisieren, wurdedie Herstellung dieses Esters aufgegeben. Es ist wahrscheinlich,dass bei der Kondensation der Ester in 2 stereoisomeren Formen
gebildet wird, die sich eventuell schlecht trennen lassen. Aber alsweitere Erschwerung ist auch anzunehmen, dass sich Polymerisa¬tionsreaktionen abspielen. Der bei einigen Versuchen beträchtlicheHarzanfall spricht dafür.
Weitere Kondensationsversuche mit Phenylcyanessigester
Da die Hydrierung eines y-Ketonitriles zu einem in 2-Stellungsubstituierten Pyrrolidinderivat führt, wollten wir ein analogesCyanderivat mit einer Aldehydgruppe an Stelle der Ketogruppe her¬
stellen. Es ist denkbar, dass die Aldehydgruppe mit der bei der
Hydrierung gebildeten Aminogruppen unter Wasserabspaltung und
Ringschluss zu einem in 2-Stellung unsubstituierten Pyrrolidinderi¬vat reagiert:
CN CHO
-C CH,
CH2 CHO
\NH2
Zur Einführung der Aldehydgruppe wollten wir Phenylcyanessig¬ester mit Chloressigsäure zum a-Phenyl-a-cyan-bernsteinsäuremono-äthylester kondensieren. Die Säuregruppe sollte ins Säurechlorid
H,0
C— CH,
ICH, CH,
"^NH
36
übergeführt und dieses nach Rosenmund zum Aldehyd reduziert
werden. Dieser Plan scheiterte daran, dass sich Phenylcyanessig-ester und Chloressigsäure nicht kondensieren Hessen.
Zur Einführung der Aldehydgruppe wurde nun die Kondensa¬
tion von Phenylcyanessigester mit Chloracetal versucht. Die Versei¬
fung des Acetals hätte dann die gewünschte Aldehydgruppe er¬
geben. Auch diese Kondensation Hess sich nicht durchführen.
Es wurde noch versucht die Kondensation mit Chloracetamid
auszuführen, aber auch in diesem Falle war das Ergebnis negativ.Da diese erfolglosen Kondensationsversuche in gleicher Weise
wie die schon beschriebenen Kondensationen mit Halogenketonenund Halogenfettsäureestern ausgeführt wurden, haben wir darauf
verzichtet sie im experimentellen Teil zu beschreiben.
Kondensation von Phenylcyanessigester mit Chloracetonitrü
Eine weitere Möglichkeit zur Herstellung von 4-Phenyl-pyrrolidin-4-carbonsäureäthylester (VI) wäre wahrscheinlich die Hydrierung des
entsprechenden Dinitriles1), des a-Phenyl-a,ß-dicyan-propionsaure-äthylesters,
COOC2H5 COOC2H.
-CH2+ 4H2
-NH,
CN CN VINH
zu dessen Herstellung Phenylcyanessigester mit Chloracetonitrükondensiert wurde. Der Ester konnte aber nicht rein erhalten wer¬
den. Das bei der Destillation erhaltene Produkt erwies sich als ein
Gemisch von a-Phenyl-a,ß-dicyan-propionsäureäthylester und Phe-
nylbernsteinsäuredinitril. Da das Destillationsprodukt rasch er¬
starrte, wurde es durch Umkristallisieren aus Petroläther weiter
gereinigt. Das so erhaltene kristalline Produkt vom F: 69,5° er¬
wies sich dann als reines Phenylbernsteinsäuredinitril, für das in
der Literatur ein Schmelzpunkt von 68—69° angegeben wird. Die
Herstellung von a-Phenyla,ß-dicyan-propionsäureäthylester er¬
scheint daher ziemlich aussichtslos, da dieses Produkt anschei¬
Bei der katalytischen Hydrierung des y-Ketonitriles VII wird mit
sehr guter Ausbeute das Pyrrolidinderivat VIII erhalten.
COOC*H6
VII I I VIIICN CO—CH,
Bei unseren Hydrierungsversuchen bekamen wir den 2-Methyl-4-phenyl-pyrrolidin-4-carbonsäureäthylester in zwei stereoisomeren
Formen, die verschiedene Siedepunkte, Refraktion und Dichte
haben. Merkwürdigerweise hatten die Chlorhydrate beider Basen
den gleichen Schmelzpunkt (F: 137°), und eine Mischprobe zeigteauch keine Depression. Die aus den Chlorhydraten in Freiheit ge¬setzten Basen erwiesen sich jedoch wieder identisch mit den ur¬
sprünglichen. Wir haben willkürlich die Base mit tieferem Siede¬
punkt, kleinerer Refraktion und Dichte als Form A, die Base mit
höherem Siedepunkt, grösserer Refraktion und Dichte als Form B
bezeichnet. Die Siedepunktsdifferenz beträgt ca. 8°/0,7 mm, bei der
Refraktion ist die Differenz ca. 0,01, bei der Dichte ca. 0,02. Beide
Basen sind optisch inaktiv. Es handelt sich wahrscheinlich um eine
cis/trans-Isomerie. Das Pyrrolidinderivat VIII enthält zwei asym¬metrische C-Atome, die Atome 2 und 4, die mögliche Isomerenzahl
wäre daher 4. Es ist aber wahrscheinlich, dass für das Auftreten
der von uns gefundenen zwei Isomeren die Asymmetrieverhält¬nisse am C-Atom 2 ausschlaggebend sind. Bei dem zur Hydrierunggelangenden Ketonitril wäre das C-Atom, das nachher im Pyrroli-dinring Nummer 4 hat, schon asymmetrisch; beim Ketonitril wurde
jedoch niemals das Auftreten von Isomeren bemerkt. Für die Bil¬
dung der Isomeren bei der katalytischen Hydrierung spricht auch,dass man aus dem gleichen Ketonitril, je nach den Hydrierungsbe¬dingungen, willkürlich die Base der Form A oder der Form B er¬
halten kann. Dies bedingt jedoch das C-Atom 2 als Asymmetrie¬zentrum.
Die ersten Hydrierungsversuche führten wir mit Platinoxyd(Kahlbaum) und Feinsprit als Lösungsmittel durch. Die Hydrie¬rungen verliefen sehr langsam und unvollständig. Die Wasserstoff-
Hi
38
aufnähme kam nach Aufnahme von 20—23,5% der für 3 Mol be¬
rechneten Menge zum Stillstand. Die Hydrierungsdauer bis zum
Stillstand war sehr lange und betrug bis zu 15 Tagen.Wir fanden dann in einer Arbeit von Schroder *) die Angabe, dass
bei der Hydrierung von Estern des Mandelsäurenitrils mit Palla¬
dium-Katalysatoren in neutralen Lösungsmitteln vorwiegend oder
ausschliesslich Arylacetonitrile erhalten wurden, also die Cyan-gruppe nicht hydriert wird. Wurde aber in Gegenwart einer star¬
ken Säure hydriert, so wurde in jedem Falle Phenyläthylamin und
kein Benzylcyanid als Hydrierungsprodukt gefunden. Die Cyan-gruppe Hess sich also mit Palladium-Katalysatoren nur in Gegen¬wart von Säuren hydrieren.Da in unserem Falle die Hydrierung mit Platinoxyd in neu¬
tralem Lösungsmittel auch nicht vollständig erfolgte, setzten wir
die zur Bildung des Chlorhydrates der Pyrrolidinbase nötige Mengekonzentrierte Salzsäure dem Hydrierungsgemisch zu. Nach die¬
sem Säurezusatz erfolgte die Aufnahme der berechneten 3 Mol
Wasserstoff. Die Hydrierungsgeschwindigkeit war immer noch re¬
lativ klein; bei einer Katalvsatormenge von 2ia°'o durchschnittlich
5 Tage. Wie aus Tabelle I (S. 47) ersichtlich ist, steigt die Hydrie¬rungsgeschwindigkeit mit wachsender Katalysatormenge. Es zeigtesich dann auch, dass beim Ueberschreiten von 2% Katalysator die
Hydrierung nach Aufnahme von 3 Mol Wasserstoff nicht zum Still¬
stand kommt. Bei 10°/o Katalysator wurde der Stillstand erst nach
Aufnahme von 6 Mol (Versuch 21, Tab. 1) erreicht. Der Phenyl-kern wird noch zum Cyclohexylderivat hydriert.
Bei der Hydrierung mit Platinoxyd in Alkohol r Salz¬
säure erhält man direkt das Chlorhydrat der Pyrrolidinbase, das
durch Umkristallisieren aus Alkohol Aether gereinigt werden kann
(Chlorhydrat der Form A).Bei der Hydrierung mit Raney-Nickel in Alkohol ohne
Säurezusatz erhält man die freie Base der Form B. Das Chlor¬
hydrat wurde durch Einleiten von trockenem Salzsäuregas in eine
Lösung der Base in absolutem Aether gefällt. Die Umkristallisation
erfolgte aus Alkohol/Aether. Auch bei dieser Hydrierungsart ist die
Wasserstoffaufnahmegeschwindigkeit abhängig von der Katalysator¬
menge. Die Hydrierung hört aber nach Aufnahme von 3 Mol
Wasserstoff in jedem Falle auf und führt nicht zu einer Kernhydrie¬
rung. Die Ausbeute ist bei diesem Hydrierungsverfahren besser als
bei der direkten Hydrierung zum Chlorhydrat mit Platinoxyd in
saurer alkoholischer Lösung.
1 ) Diss Hansische l'niv Hamburg 1940
39
Entsprechend den Hydrierungsversuchen mit Platinoxyd in alko¬
holischer Lösung, die einen Sàurezusatz zur Hydrierung erforderten,machten wir einen entsprechenden Versuch mit Raney-Nickelund Sàurezusatz. Es wurde aber kein Pyrrolidinderivat erhal¬
ten. Der bei dieser Hydrierung bemerkte Acetaldehydgeruch, das Er¬
gebnis der Molekularrefraktion und der Mikroanalyse, lassen uns eine
Aufspaltung des Moleküls bei der Hydrierung annehmen. a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester würde so zu 2-Methyl-2-phenyl-3-amino-propionsaureäthylester und Acetaldehyd hydriertwerden.
COOC,H5 COOC,Hs
<o4-ch. ^ oC-CH, + CH.CHO
VII I I CH2CN CO—CH3 |
NHS
Der zu unseren Hydrierungsversuchen benötigte Raney-Nickelwurde zu jedem Versuch frisch bereitet, durch Zersetzen der Nickel-
Aluminium-Legierung !) mit 4°oiger Natronlauge, Auswaschen mit
Wasser und Aethanol.
Herstellung von 2-Oxo-4-phenyl-pyrrolidin-4-carbonsäureäthylester
Bei der Hydrierung des y-Ketonitriles IX mit Raney-Nickel in
Feinsprit entsteht mit quantitativer Ausbeute das Pyrrolidonderi-vat X.
COUCH, COOC8H5
Wo
I*
-CtH5OH \ /
XCN COOC,H6
Die Hydrierung geht relativ rasch im Vergleich zur Herstellungdes Pyrrolidinderivates ; in 24 Stunden werden 2 Mol Wasserstoff
aufgenommen, und die Hydrierung bleibt stehen.
Ein entsprechender Hydrierungsversuch mit Platinoxyd in Alko¬hol 4 Salzsäure lieferte den identischen Pyrrolidonester X, wie er
bei der Hydrierung mit Raney-Nickel erhalten wurde. Es ist also
r) Von Hrn. Gilman, Chattanooga USA freundlichst zui Verfügung gestellt.
40
keine isomere Form entstanden wie beim Pyrrolidinderivat. Dieser
Befund scheint uns auch zu bestätigen, dass beim Pyrrolidinderivatdas C-Atom 2 als Asymmetriezentrum angesprochen werden muss.
Beim Pyrrolidinderivat ist das C-Atom 2 wegen der C=0 Doppel¬bindung nicht mehr asymmetrisch; in diesem Falle musste auch
erwartet werden, dass keine Isomeren entstehen.
Der Hydrierungsversuch mit Platinoxyd wurde im experimen¬tellen Teil nicht beschrieben, da er zu keinen neuen BeobachtungenAnlass gab. Entsprechend der Hydrierung zu Pyrrol idinderivatenbleibt die Wasserstoffaufnahme nicht bei 2 Mol stehen, so dass man
Wir hydrierten das in der Enolform vorliegende b-Ketonitril XI
und erhielten so das Piperidinderivat XII.
COOC8H5 C00CSH5
XI XII
Auch diese Base XII wurde in zwei stereoisomeren Formen erhalten,die eine Siedepunktsdifferenz von ca. 6°/0,8 mm, eine Differenz der
Refraktion von ca. 0,008 und der Dichte von ca. 0,015 aufweisen.
In Uebereinstimmung zum Pyrrolidinderivat bezeichnen wir (auchwillkürlich) die tiefer siedende Base, die auch den kleineren Bre¬
chungsindex und die kleinere Dichte hat, als Form A, und die Base
mit dem höheren Siedepunkt, grösserer Refraktion und Dichte als
Form B. Beide Basen sind optisch inaktiv. Das für die Isomerie-
verhältnisse beim Pyrrolidinderivat gesagte gilt auch für das Pi¬
peridinderivat, das sich ja nur durch eine CH2-Gruppe mehr im
heterocyclischen Ring vom Pyrrolidinderivat unterscheidet.
Es muss erwähnt werden, dass bei der Hydrierung mit Platin¬
oxyd (2,8o/o) in Alkohol - Salzsäure die Hydrierung erst zum Still¬
stand kam, als 17°'o zu viel Wasserstoff (17°o der für 3 Mol be¬
rechneten Menge) aufgenommen waren. Es ist daher möglich, dass
die bei diesem Versuch gefundene 2. Fraktion mit höherem Siede¬
punkt, deren Molekularrefraktionswert schlecht ist, bereits zum
Teil im Phenylkern hydriert wurde. Entsprechend dem Befund beim
41
Pyrrolidinderivat ist anzunehmen, dass bei genügend grosser Kata¬
lysatormenge die Hydrierung bis zum Cyclohexylderivat gehenkann.
Hydrierung von Phenyl-cyan-\S-oxyäthyl-essigsäureäthylester (?)
Wie bei der Beschreibung der Versuche zur Herstellung dieses
Oxynitriles dargelegt wurde, gelang es uns nicht, mit Sicherheitzu beweisen, dass wir diese Verbindung überhaupt erhalten haben.
Auf Grund unserer Versuche, bei denen wir ja zwei verschiedeneProdukte mit verschiedenem Siedepunkt und verschiedener Refrak¬tion und Dichte erhielten, schien es uns doch interessant einige Hy¬drierungsversuche zu unternehmen. Es gelangten dazu solche Frak¬
tionen zur Verwendung, bei denen wir auf Grund der gefundenenMolekularrefraktionen annehmen konnten, dass sie vielleicht dem
Phenyl-cyan-ß-oxyäthyl-essigsäureäthylester entsprechen würden. Im
experimentellen Teil sind zwei typische Hydrierungsversuche mit
Raney-Nickel beschrieben.
Die Ergebnisse dieser Versuche lassen uns vermuten, dass bei der
Hydrierung zwei Moleküle kondensiert werden, unter Abspaltunggewisser Gruppen.Im ersten Versuch mit dem tiefer siedenden Nitril mit der klei¬
neren Refraktion finden wir für das Hydrierungsprodukt eine mög¬liche Bruttoformel Cä2H.,oON2 aus dem Analysenergebnis:
C,2H30ON2 ber. C 78,06«A> H 8,94<Vo N 8,27o/0gef. C 77,94o/o H 8,58o/o N 7,930/0
Es ist uns aber nicht möglich, eine Deutung dieses Reaktionsver¬laufes zu geben.
Im Falle der Hydrierung des höher siedenden Nitriles mit dem
grösseren Brechungsindex finden wir, dass ein Hydrierungsproduktmit der Bruttoformel CsjH^OjN dem Ergebnis der Analyse sehr
gut entsprechen würde. Für die Bildung dieses Hydrierungspro¬duktes könnten wir uns, allerdings als unbewiesene Hypothese,folgendes Reaktionsschema denken:
2 C13H150,N f 4 H2 = C31H„0+N C2H,OH +H20 + NH,
42
CH-CH,OH
C—CN
cooc,h5
COOC2H,f
NC—C-
HOH2C—CH,
+4H,
-C2H5OH
-NHS
-H20
v(vj £{-]o ÇOOC2H5V
o-\
/
O-CHa-CH,
C24H2704N
Für dieses Hydrierungsprodukt, das einen Lactonring enthalten
müsste, errechnet sich eine MD ber.: 108,68. Aus Refraktion und
Dichte unseres Hydrierungsproduktes erhalten wir:
1. ng : 1,5410 d{6 : 1,1228 MD gel: 109,15
2. n1o : 1,5470 d]6 : 1,1456 MD gef.: 108,92
Diese Werte würden also die angedeutete Möglichkeit nicht aus-
schliessen. Wir betonen aber nochmals, dass diese Erklärung des
Reaktionsverlaufes eine unbewiesene Hypothese ist.
43
ccm H2
500 r
60 120 Minuten
Fig. 1. Hydrierung von a-Phenyl-a-cyan-y-acetyl-buäersäureäthyl-ester
44
rJ Mol H
Fig. 2. Hydrierung von a-Phenyl-a-cyan-y-acetyl-buäersäureäfhy/-ester (Enol), und von et- Phenyl-a-cyan-ß-acety/-propion-säureäthylester (Keton)
% von 3 Mol H,
12 24 Stunden
Fig. 3. Hydrierung von u-PK<enyl-v.-cyan-$-acetyl-propionsäwreäthyl-ester (ohne Säurezusatz mit Platinoxyd)
16 = 1,0 o/o Pr02 (17,0»)20 = 2,2 o/o
„ (11,50)17 = 2,42<tf> „ (12,5°)19 = 2,9 o,0
„ (12,0°)45
90^0 von 3 Mol H
Fig. 4. Hydrierung von a-Phenyl-a-cyan:$-acetyl-propionsäureäthyl-ester (mit Säurezusatz mit Platinoxyd)
Ver¬ such Nr.CCOXt^CTi-^OCMTHCOCCXCit-i-(CMi—li-ti-HCMCOCMCMCMCMCMCMCO
Die Hydrierungskurven auf Seite 44 (Fig. 1) zeigen den Be¬
ginn der Hydrierung von a-Phenyl-a-cyan-y-acetyl-buttersäureäthyl-ester (Hydrierung 31 und 32), der in der Enolform vorliegt. DerKnick in den Kurven nach Aufnahme der zur Absättigung der Enol-
Doppelbindung erforderlichen Menge Wasserstoff (1 Mol) ist deut¬lich sichtbar.
Die Kurven auf Seite 45 (Fig. 2) zeigen den Hydrierungsver¬lauf des enolisierten a-Phenyl-a-cyan-y-acetyl-buttersäureäthylesters(Enol) und des nicht enolisierten a-Phenyl-a-cyan-ß-acetyl-propion-säureäthylesters (Keton). Es handelt sich um zwei Hydrierungenmit Platinoxyd (Hydrierung 22 und 31). Da die Kurve für das
nicht enolisierte Ketonitril ganz gleichmässig verläuft, können dar¬
aus keine Folgerungen über den Hydrierungsmechanismus gezogenwerden. Es kann nicht ausgesagt werden, ob zuerst die Cyangruppereduziert wird, die gebildete Aminogruppe dann mit der Ketogruppeunter Wasserabspaltung den Ringschluss unter Eintritt einer
Doppelbindung in den Ring vollzieht, wobei dann anschliessend
diese Doppelbindung hydriert wird, oder ob nach der Cyangruppedie Ketogruppe zum Alkohol hydriert wird und dann erst die Cycli-sierung unter Wasserabspaltung erfolgt.
Fig. 3 (S. 45) und Fig. 4 (S. 46) geben eine graphische Dar¬
stellung des Zusammenhanges zwischen Katalysatormenge und Hy¬drierungsgeschwindigkeit bei Platinoxyd ohne und mit Säurezusatz.
Auf den Tabellen 1 und 2 (S. 47) wurden nicht alle ausgeführ¬ten Hydrierungsversuche angegeben, da sich die Ergebnisse bei
einer Anzahl der Versuche vollständig decken. Die Anordnung der
Versuche auf den Tabellen erfolgte nach prozentual steigender Ka¬
talysatormenge, um den Einfluss der Katalysatormenge auf die Hy¬drierungsgeschwindigkeit zu zeigen. Der Einfluss der Hydrierungs¬temperatur, die nicht bei allen Versuchen gleich war, auf die Hydrie¬rungsgeschwindigkeit muss beim Vergleich der Angaben berück¬
sichtigt werden.
48
Experimenteller Teil1 )
A. Kondensationen
Herstellung von Phenyl-cyan-esslgsäureäthylester
In einen Dreihalskolben von U/2 Liter Inhalt mit Rührwerk,Rückflusskühler und Tropftrichter werden 420 ccm (= 300 g)absoluter Aether und 42 g fein pulverisiertes Natriumamid ge¬
geben. Es ist sehr wichtig, bei dieser Kondensation unter Feuchtig-keitsausschluss zu arbeiten. Unter gutem Rühren werden dann 117g(1 Mol) frisch destilliertes Benzylcyanid (Kp12: 106°) innert einer
Stunde zugetropft. Dabei färbt sich das Gemisch unter Selbster¬
wärmung dunkelbraun. Nach Beendigung des Zutropfens wird
während 30 Minuten zum Sieden erhitzt. Dann wird die Heizungabgestellt und das Reaktionsgemisch während 45 Minuten ab¬
kühlen gelassen. Während 31/2 Stunden werden nun 150 g (114 Mol)Kohlensäurediäthylester (Kp: 124°) unter ständigem Rühren zuge¬
tropft. Der Kohlensäureester muss so langsam zugefügt werden,um eine übermässige Selbsterwärmung des Reaktionsgemischeszu vermeiden. Nachdem aller Kohlensäureester eingetropft ist, wird
noch während 45 Minuten auf dem Wasserbad gekocht. Nach Ab¬
lauf dieser Zeit wird der Kolben sofort mit Eis abgekühlt. Unter
ständigem Rühren gibt man ca. 500 ccm 2n-Salzsäure zu, zur Zer¬
setzung des bei der Kondensation gebildeten Natriumalkoholates.
Nach fünf Minuten wird der ganze Kolbeninhalt in einen Scheide¬
trichter gespült, geschüttelt und sorgfältig gegen Lackmus neutrali¬
siert. Der abgetrennte neutrale wässerige Anteil wird nochmals mit
frischem Aether ausgeschüttelt. Die vereinigten ätherischen Lösun¬
gen werden über Na2SO! getrocknet. Nach Entfernung des Aethers
wird im Vakuum destilliert. Als erste Fraktion werden ca. 30 g
Kohlensäurediäthylester zurückgewonnen; das entspricht dem
v\ Mol Ueberschuss, das zur Kondensation verwendet werden muss.
1) Samtliche Schmelzpunkte sind korrigiert; die Bestimmung eifolgto mit dem
MikroschmeJzpunktapparat nach Fuchs der Firma C. Reichert, Wien.
Die Mikroanalysen wuiden ausgeführt in der mikroanalytischen Abteilung
des 01 ganisch-chemischen Laboratoriums der E.T.H. Zürich, Leitung
H. Gubser und W. Manser, und in der mikroanalytischen Abteilung des
organisch-technischen Laboratorums der E.T.H. Zürich, von Frl. Dr.
E. Pfanner.49
Man erhält 150 g Phenyl-cyan-essigsäureäthyiester vom Kpu: 158°,was einer Ausbeute von 79,5°,o der Theorie entspricht. Der Esterist eine wasserklare, leicht viskose Flüssigkeit von angenehmemester- und zugleich nitrilartigem Geruch.
no 1,5069 di5: 1,0965 MD ber.: 50,87
MD gef.: 51,34Im Destillationsrückstand wurden Kristalle von Diphenyl-dicyan-aceton gefunden, die nach einmaligem Umkristallisieren aus Alkoholeinen Schmelzpunkt von 265° hatten.
Herstellung von a-Phenyl-a,-cyan-$-acetylproplonsäureäthylester
In einen Dreihalskolben mit Rührwerk, Rückflusskühler und
Tropftrichter werden 2,3 g (! f0 Mol) Natriumschnitzel in 150 ccm
absoluten Aether gegeben. Dann lässt man 19 g (Vio Mol) Phenyl-cyan-essigsäureäthylester eintropfen und kocht während 24 Stundenauf dem Wasserbad. Während dieser Zeit verschwindet das Na¬trium unter Bildung der ätherunlöslichen weissen Natriumverbin¬
dung des Phenylcyanessigesters. Jetzt lässt man 10,5 g (1,25 gUeberschuss) frisch destilliertes Chloraceton (Kp: 118°) eintropfenund erhitzt während weiteren zwei Tagen unter stetem Rühren.Das Gemisch reagiert zum Schluss neutral. Nach dem Erkaltenwird mit stark verdünnter Salzsäure schwach angesäuert, ausge¬schüttelt und die ätherische Lösung neutralgewaschen. Nach Trock¬
nung über Na2S04 wird der Aether abdestilliert. Bei der anschlies¬senden Vakuumdestillation wird nach einem geringen Vorlauf eine
Hauptfraktion vom Kp„, : 158° aufgefangen. Man erhält 17 g a-
Phenyl-a-cyan-ß-acetyl-propionsäureäthylester, was einer Ausbeutevon 69»'o d. Th. entspricht. Dieser Ester ist eine hellgelbe, sehrviskose Flüssigkeit.
ni6: 1,5101 d"
: 1,1302 MD ber.: 64,74
MD gef.: 64,88
3,238 mg Substanz gaben 8,149 mg C02 und 1,810 mg H30CuH1503N (245,27) ber. C 68,55<>o H 6,16o«
gef. C 68,68o/o H 6,26<>/oNach einigem Stehen erstarrt der Ester zu einer strahlig kristal¬
linen Masse, F: 51—52°. Durch Umkristallisieren aus Petroläthererhält man sehr schöne, weisse, seidenglänzende, lange, feine Na¬deln vom F: 57,5°. Zur Analyse wurde 6 Stunden bei 35°/0,l mm
5,938 mg Substanz gaben 0,309 ccm N bei 17°/710 mm
CuH1503N (245,27) ber. C 68,550,0 H 6,16<>/o N 5,72o„
gef. C 68,70.o,o H 6,08o/0 N 5,72o„
Die vorstehend beschriebene Kondensation kann auch auf etwas
andere Weise, rascher und mit besserer Ausbeute, durchgeführtwerden. In der gleichen Apparatur werden 2,3 g Natrium in
100 ccm absolutem Alkohol gelöst. Dann werden 19 g Phenyl-cyanessigester zugegeben und eine Stunde lang zum Sieden erhitzt.
In das siedende Reaktionsgemisch lässt man dann 10,5 g Chlor-
aceton eintropfen, wobei sofort Natriumchlorid-Ausscheidung be¬
ginnt. Nach 4 Stunden lässt man den Alkohol abdestülieren, was
in einer Stunde geschehen ist und lässt dann erkalten. Das neutral
reagierende Gemisch wird in Aether aufgenommen und mit Wasser
ausgeschüttelt. Nach Trocknung über Na2S04 wird der Aether ab¬
destilliert. Bei der folgenden Vakuumdestillation erhält man 19 g
a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester vom Kp02: 153°,
entsprechend einer Ausbeute von 771/2°o der Theorie.
Herstellung von a-Phenyl-a-cyan-bernstelnsäuredläthylester
In einem Dreihalskolben mit Rührwerk, Rückflusskühler und
Tropftrichter werden 2,3 g (' \0 Mol) Natrium in 100 ccm absolu¬
tem Alkohol gelöst. Dann werden 19 g ^/îo Mol) Phenylcyanessig-ester zugetropft, wobei sich die Lösung rötlich färbt. Nach einer
Stunde werden 12,5 g (\',0 Mol) frisch destillierter Chloressig-saureäthylester (Kp: 144°) zugetropft. Das Reaktionsgemisch wird
wahrend 2 Stunden unter stetem Rühren auf dem Wasserbad er¬
wärmt, es trübt sich sehr bald durch Natriumchlorid-Ausscheidung.Während einer weiteren Stunde wird der Alkohol abdestilliert. Nach
dem Erkalten wird in Aether aufgenommen. Die ätherische Lösungwird mit verdünnter Salzsäure ausgeschüttelt, dann neutralge¬waschen und über Na^S04 getrocknet. Nach dem Abdestülieren des
Aethers bleiben 28 g hellgelbes, dickliches Oel. Bei der Vakuum¬
destillation erhält man 23,25 g a-Phenyl-a-cyan-bernsteinsäuredi-äthylester vom Kp0. : 153°, entsprechend einer Ausbeute von 84,5°o
der Theorie. Der Ester ist eine farblose, sehr viskose Flüssigkeit.
150 g Aceton und 50 g 40°oige Formaldehydlösung wurden mit
3 g Kaliumcarbonat (in wenig Wasser gelöst) versetzt und tinterRühren auf dem Wasserbad auf 30—35° erwärmt. Diese Tempera¬tur hielt sich einige Minuten lang, um dann in einigen Stunden
wieder auf Zimmertemperatur zu sinken. Das Reaktionsgemischwurde mit verdünnter Salzsäure neutralisiert, vom wässerigen An¬
teil abgehoben und das überschüssige Aceton im Wasserstrahlva¬
kuum abgesaugt. Im Kolben blieb ein farbloses, viskoses Oel zu¬
rück, das bei der Vakuumdestillation nach einem geringen Vorlauf
14 g Ketobutanol vom KpM: 108—110° ergab. Das entspricht einer
Ausbeute von 23,8»/o d. Th. bezogen auf die eingesetzte MengeFormaldehyd. Das 3-Keto-butanol-(l) ist ein färb- und geruchlosesOel, das nicht näher untersucht wurde.
Herstellung von l-Chlor-butanon-(3)
In 35 ccm (dreifacher Ueberschuss) konzentrierte Salzsäure wur¬
den unter Eiskühlung 14 g Ketobutanol eingetropft. Das Reaktions¬
gemisch wurde 30 Minuten unter Kühlung gerührt, während wei¬
teren 30 Minuten unter Weglassung der Kühlung. Dann wurde das
Gemisch auf Eis gegossen und ausgeäthert. Nach dem Trocknen
über Na2S04 wurde der Aether abdestilliert und das Produkt der
Vakuumdestillation unterworfen. Als Hauptfraktion wurden 8,5 g
l-Chlor-butanon-(3) oder ß-Chloräthyl-methyl-keton vom Kp25:65—68° erhalten, entsprechend einer Ausbeute von 50°o d. Th. Das
Chloräthyl-methylketon ist eine farblose, lacrimogene Flüssigkeit.n*2 : 1,4600 d» : 1,1330 MD ber.: 25,50
MD gef.: 25,75
Herstellung von a-Phenyl-a-cyan-^-acetyl-buttersäureäthylester
In einen Dreihalskolben mit Rührwerk, Rückflusskühler und
Tropftrichter wurden 1,7 g Natriumschnitzel in absoluten Aether
gegeben. Dann wurden 14,4 g Phenylcyanessigester eingetropft und
zur Bildung der Natriumverbindung 24 Stunden auf dem Wasser¬bad erwärmt. Nun wurden 8,36 g ß-Chloräthyl-methylketon zuge¬tropft und weitere 24 Stunden zum Sieden erhitzt. Das Reaktions¬
gemisch färbte sich dabei dunkel unter Bildung von etwas äther¬unlöslichem Harz. Nach dem Erkalten wurde die ätherische Lösungmit verdünnter Salzsäure schwach angesäuert, mit Wasser neutral-
52
gewaschen, von den unlöslichen harzigen Anteilen (0,5 g) abfil¬
triert und über Na2S04 getrocknet. Nach der Entfernung des
Aethers wurde im Vakuum destilliert. Dabei wurden folgende Frak¬
tionen erhalten:
1. Kp12 : 50— 60° 0,90 g n20 . •1,4455
2. Kp12 : 130—158» 4,60 g„20 .
"d 1,5050
3. KPi,o : 140—165° 2,84 g n20 . 1,5097
4. KPW : 164—165° 8,92 g n21 . 1,5155 d? 1,1071
17,26 g
Nach den Siedepunkten und den Brechungsindices zu schliessen,
besteht Fraktion 1 aus Chloräthylmethylketon, Fraktion 2 aus Phe¬
nylcyanessigester, Fraktion 3 aus einem Gemisch von 2 -\- 4 und
Fraktion 4 ist der gesuchte a-Phenyl-a-cyan-y-acetyl-buttersäure-äthylester. Für Fraktion 4 wird eine Molekularrefraktion Mß :
70,64 gefunden. Die berechnete Molekularrefraktion beträgt für die
Ketoform MD : 69,27, und für die Enolform MD: 70,40. Aus der
guten Uebereinstimmung der MQ-Werte für die Enolform muss
geschlossen werden, dass dieses Ketonitril in der Enolform vor¬
liegt. Die Ausbeute beträgt, nach Abzug der nicht umgesetzten
Zur Herstellung von Phenyl-cyan-ß-oxyäthyl-essigsäureäthylesterwurde Phenylcyanessigester unter verschiedenen Bedingungen mit
Aethylenchlorhydrin kondensiert.
53
1. In einem Bromierungskolben wurden 2,17 g Natrium in 10 ccm
absolutem Alkohol gelöst. Dann wurden 17,85 g Phenylcyanessig¬ester und 7,6 g Aethylenchlorhydrin zugefügt. Das Gemisch wurde
unter häufigem Umschütteln während 4 Stunden auf dem Wasser¬bad erwärmt. Nach dem Erkalten wurde in Aether aufgenommen,mit verdünnter Salzsäure ausgeschüttelt und neutralgewaschen.Nach dem Trocknen über Na2S04 wurde der Aether abdestilliert;es blieben 18,3 g rohes Reaktionsprodukt zurück. Im Wasserstrahl¬vakuum konnte nichts destilliert werden; bei der Destillation im
Hochvakuum wurden folgende Fraktionen erhalten:
1,1108
1. KPl 2: 154--160° 2,25 g
2. KPW : 166--168° 5,82 g n22 .
"d'1,5002 df
' 4
3. KPo,3: 145--152» 0,20 g „18 . 1,5232
4. KP„,5 : 152--154» 0,74 g ng: 1,5269
5. KP0,5 : 155--165» 0,88 g n«: 1,5295
9,89 g
Es blieb ein beträchtlicher, nicht destillierbarer, verharzter Rück¬stand. Für die Fraktion 2 findet man MD : 61,60, MD ber.: 61,63(für Phenyl-cyan-ß-oxyäthyl-essigsäureäthylester).
II. In einem Bromierungskolben wurden 6,1 g Natrium in
150 ccm absolutem Alkohol gelöst. Der Natriumalkoholatlösungwurden zuerst 50 g Phenylcyanessigester und dann 21,3 gAethylenchlorhydrin zugefügt. Die Lösung beginnt sich durch
Natriumchloridausscheidung bald zu trüben. Es wurde 4 Stundenam Rückfluss gekocht, dann der Alkohol abdestilliert. Nach demErkalten wurde in Aether aufgenommen, mit verdünnter Salzsäure
ausgeschüttelt und neutralgewaschen. Nach dem Trocknen über
Na2S04 wurde der Aether abgedampft und das Produkt destilliert:
Von Fraktion 8 wurde eine weitere Probe mehrfach destilliert, ohne
dass sich Refraktion und Dichte änderten. Eine Probe vom
Kp01: 149° wurde analysiert:
4,222 mg Substanz gaben 10,60 mg CO, und 2,37 mg H20CuH15OsN ber. C 66,9~3o'o H 6,48°o
gef. C 68,51 o'o H 6,28"„
Zwecks Ueberführung in einen 3,5-Dinitrobenzoesäureester wurde
eine Probe der Fraktion 8 mit einer Lösung von Dinitrobenzoyl-chlorid in absolutem Toluol versetzt und nach Zusatz einiger Trop¬fen Pyridin 3 Stunden stehen gelassen. Nach Verdünnung mit abso¬
lutem Aether wurde zuerst mit verdünnter Salzsäure, dann mit ver¬
dünnter Lauge und zuletzt mit Wasser gewaschen. Der Ester wurde
aus der Grenzschicht abgesaugt, gewaschen und getrocknet. Beim
Umkristallisieren aus Ligroin/Toluol wurde eine schwerlösliche
Komponente vom F: 203—205°, wahrscheinlich 3,5-Dinitrobenzoe-säure, und eine leichter lösliche vom F: 123° gefunden. Dieser
Teil änderte seinen Schmelzpunkt nach mehrfachem Umkristalli¬
sieren nicht weiter. Zur Analyse wurde 12 Stunden bei 100°/0,1 mm
III. In einem Bromierungskolben wurden 18,4 g Phenylcyanessig-ester und 7,85 g Aethylenchlorhydrin in 100 ccm absolutem To¬
luol gelöst. Dazu wurden 3,8 g fein pulverisiertes Natriumamid
gegeben. Das Gemisch färbte sich gelb unter leichter Selbsterwär¬
mung. Es wurde dann 16 Stunden auf 120° erwärmt, wobei lebhafte
Ammoniakentwicklung erfolgte. Nach dieser Zeit wurde das Toluol
abdestilliert und das Produkt in Aether aufgenommen. Nach dem
Ausschütteln mit Salzsäure wurde neutralgewaschen, über Na2S04
getrocknet und der Aether entfernt. Es blieben 19 g hell rot¬
braunes Produkt. Bei der Destillation wurden folgende Fraktionen
erhalten:
55
1. Kp13 : 116-157« 3,22 g ng : 1,51402. Kp05: 135—150» 0,68 g 1,50803. Kp0'6 : 150—156" 3,66 g 1,5077
4. Kp10: 161 —170° 4,48 g 1,5370
12,07gCa. 5 g verharzter Rückstand.
MD ber.: 61,63; MD gef. 3: 62,30 4: 62,68
Von Fraktion 3 wurde eine Probe nach einjähriger Lagerung im
Dunkeln zur Analyse destilliert. Das Analysenpräparat hatte fol¬
gende Konstanten: Kpo^: 151°
n* : 1,5012'
df : 1,1037 MD ber.: 61,63
MD gef.: 62,29
19,19 mg Substanz gaben 47,67 mg C02 und 10,99 mg H20C13H1503N (233,26) ber. C 66,93o/0 H 6,48<>/o
gef. C 67,79o/o H 6,41 o/„
Mit einer Probe der Fraktion 3 wurde, wie oben beschrieben, der
3,5-Dinitrobenzoesäureester hergestellt. Es wurde ein kristallisiertesProdukt vom F: 123° erhalten, das mit dem Dinitrobenzoat aus Ver¬such II keine Depression gab. Zur Analyse wurde 12 Stunden bei
3,008 mg Substanz gaben 0,318 ccm N bei 15°/723 mm
C20H17O8N3 (427,36) ber. C 56,21 o/0 H 4,01 «/o N 9,83o/0gef. C 55,530/0 H 3,24o/0 N ll,92o/0
IV. In einem Dreihalskolben (500 ccm) mit Rührwerk, Rück¬flusskühler und verschliessbarer Einfüllöffnung wurden 38,7 gPhenylcyanessigester und 16,5 g Aethylenchlorhydrin in 150 ccm
absolutem Toluol gelöst. Dazu wurden unter Kühlung in zwei Por¬tionen 8,5 g fein pulverisiertes Natriumamid gegeben. Nach 30
Minuten wurde langsam auf 120° geheizt und diese Temperaturbis zum Aufhören der Ammoniakentwicklung, nach ca. H/2 Stunden,beibehalten. Dann wurde abgekühlt, die Toluollösung direkt mitverdünnter Salzsäure ausgeschüttelt und hierauf neutralgewaschen.Nach dem Trocknen über Na2S04 wurde das Toluol abdestilliert.Bei der Destillation wurden folgende Fraktionen erhalten:
d^ : 1,1169
1,1628
56
1. Kp16 115--162« 7,12 gnl6,5
J1D 1,5133
2. KPo,7 117--152" 5,48 gn16.5
11D1,5061 d'4'8 1,1026
3. KPo,7 152--158" 1,39 gn16-5
11D1,5061 d14.8 1,1144
4. KPo,8 158--159» 4,00 g "d 1,5095 d!4'54
1,1311
5. KPo,8 160--166» 8,79 gn17
11D1,5345 d]5,5
41,1992
26,78 g
Ca. 10 g verharzter Rückstand.
MD ber.: 61,63; M» gef. 2:62,90 3:
Fraktion 5, die bei diesem Versuch
wurde nochmals destilliert:
62,24 4:61,63 5:60,53
die Hauptmenge darstellt,
1.
2.
3.
4.
KPo,4:KP0,5:KP0,5 :
KPo,5:
135—140»
142—152»
152—153»
153—157»
0,81 g
1,21 g
4,49 g
1,70 g
8,21 g
11,5D
11,5D
12
D
13
D
1,5202 d£'1,5275 d{4'2
d'4,54
1,5375
1,5405 d'5'14
1,0917
1,1562
1,1969
1,2043
M0 ber.: 61,63; MD gef. 2:62,91 3:60,77 4:60,70Von Fraktion 3 wurde eine Probe zur Analyse destilliert. Das
Analysenpräparat hatte folgende Konstanten: Kp„„: 148°
4,022 mg Substanz gaben 0,255 ccm N bei 15°/718 mmC13H1503N (233,26) ber. C 66,93»A> H 6,48o/o N 6,01 •>/«
gef. C 69,60»o H 5,02<>/o N 7,10»-,,
V. Es wurden zwei Versuche zur Kondensation von Aethylen-chlorhydrin mit der Natriumverbindung des Phenylcyanessigestersin absolutem Aether gemacht. Bei beiden Versuchen wurde nur
unverändertes Ausgangsmaterial und verharzte Produkte gefunden.Die Versuchsausführung entsprach der weiter oben beschriebenen
Kondensation mit Halogenketonen.
VI. Es wurde eine Reihe von Kondensationsversuchen mit A1C13als Kondensationsmittel in verschiedenen Lösungsmitteln durchge¬führt. Da alle Versuche negativ verlaufen sind, soll nicht näher
darauf eingegangen werden.
57
VII. In einem Dreihalskolben mit Rührwerk, Rückflusskühler und
Tropftrichter wurden 2,3 g Natrium in 100 ccm absolutem Alkohol
gelöst. Es wurden 20 g Phenylcyanessigester zugegeben und %Stunde gekocht. Dann wurden 15 g Aethylenchlorhydrin langsam in
die siedende Lösung eingetropft. Die gelbe Lösung trübt sich bald
durch Natriumchloridausscheidung. Nach 41/2 Stunden wurde eine
Stunde lang der Alkohol abdestilliert, dann das Reaktionsgemischabgekühlt und in Aether aufgenommen. Die ätherische Lösungwurde mit verdünnter Salzsäure schwach angesäuert, dann neutral¬
gewaschen und über Na2SOt getrocknet. Nach Entfernung des
Aethers wurde im Vakuum destilliert:
1 Kpr, 70—132" 5,02 g
2. KPo>7 115—150" 6,49 g nj6, : 1,5087 d7 1,0955
3- Kp0>4 140—145» 7,41 g nj* : 1,5057 d16 1,1121
4- KPo.4 144—145" 1,44 g nj6 : 1,5025 d164
1,1189
5. Kp07 173—178" 2,97 g n* : 1,5445 d'64
1,2002
23,33 g
Bei dieser Kondensation
MD ber.: 61,63; MQ gef
war die Verharzung minimal.
2:63,48 3:62,29 4:61,58 5:61
Fraktion 3 wurde nochmals destilliert:
1. Kp04: 140—145" 2,55 g n£5 : 1,5060 d «•* 1,1067
2- KPo',4 144-145" 4,47 g n1D5'5 : 1,5029 d]5'54
1,1127
7,02 g
MD ber.: 61,63; MQ gef. 1:62,62 2:61,96
Aus Fraktion 5 wurde ein Analysenpräparat von folgenden Kon¬
Versuche zur Chlorierung der Oxygruppedes Phenyl-cyan-fi-oxyathyl-essigsäureäthytesters
Es wurde bei einigen der vorerwähnten Fraktionen versucht, die
Oxygruppe mit Thionylchlorid in Toluol oder Chloroform zu chlo¬
rieren. Versuche mit und ohne Erwärmen hatten stets das gleicheErgebnis: bei der Destillation wurde quantitativ das Ausgangs¬material zurückerhalten. Interessant ist, dass ein aus Fraktion 4 von
Versuch VII erhaltenes Produkt, nach der erfolglosen Behandlungmit Thionylchlorid, erstmals eine stimmende Analyse auf Phenyl-cyan-ß-oxyäthyl-essigsäureäthylester lieferte: Kp04: 146°
C10H8N2 (156,18) ber. C 76,90o/0 H 5,17<>/ogef. C 77,04o/o H 5,10o/o
Es handelt sich also um Phenylbernsteinsäuredinitril.
B. Katalytische Hydrierungen
Da die katalytischen Hydrierungen im allgemeinen immer gleichausgeführt und aufgearbeitet wurden, wird für jeden Substanztypnur ein Beispiel ausführlich angegeben. Die übrigen Resultate
wurden in Tabellenform (siehe S. 47) zusammengefasst.
Herstellung von 2-Methyl-4-phenyl-pyrrolidin-4-carbonsäureäthylester (Form A)
Direkte Herstellung des Chlorhydrates
5 g a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester wurden in
250 ccm Feinsprit + 3 ccm konzentrierte Salzsäure gelöst. Nach
60
Zugabe von 0,1046 g Platinoxyd als Katalysator wurde in einer
Wasserstoffatmosphäre geschüttelt. Nach 4Va Tagen kam die Hy¬
drierung zum Stillstand nach Aufnahme von 1635 ccm Wasserstoff,was einer Menge von 3 Mol entspricht (berechnet 1625 ccm
15°/732 mm). Nach dem Abfiltrieren des Katalysators wurde der
Alkohol im Vakuum abdestilliert. Es blieb ein schwach gelblichesOel zurück, das nach eintägigem Stehen auszukristallisieren begann.Beim Anspritzen mit Aether wurden 4,17 g Chlorhydrat als weisses
Kristallpulver erhalten. Das entspricht einer Ausbeute von 75,8»(>der Theorie. Das Chlorhydrat hat nach dem Umkristallisieren aus
Alkohol/Aether einen Schmelzpunkt von 137°; es kristallisiert in
strahligen Gebilden. Das Chlorhydrat ist in Wasser sehr leicht und
mit neutraler Reaktion löslich. Auf die Zunge gebracht, schmeckt
es bitter und erzeugt eine kurz dauernde schwache Anaesthesie.
Herstellung von 2-Methyl-4-cyclohexyl-pyrrolidln-4-carbonsäureäthylester
100 mg reinster a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester(F: 57,5°) wurden in 50 ccm Feinsprit + 2 Tropfen konzentrierte
61
Salzsaure gelost und nach Zugabe von 10 mg Platinoxyd hydriert.Die Hydrierung kam nach Aufnahme von 60,2 ccm Wasserstoffin 5 Tagen zum Stillstand, das entspricht einer Menge von 6 Mol
(berechnet 60,0 ccm bei 11,5°/ 736 mm). Nach dem Abfiltrieren des
Katalysators und Entfernung des Alkohols im Vakuum wurde sofortdas kristallisierte Chlorhydrat des 2-Methyl-4-cyclohexyl-pyrroli-din-4-carbonsäureäthylesters erhalten, das nach dem Umkristalli¬sieren aus Alkohol/Aether den F: 157—158° hatte.Zur Analyse wurde 6 Stunden bei 105°/0,1 mm getrocknet.
19,01 mg Substanz gaben 42,45 mg C02 und 16,06 mg H20C„HmO,NC1 (275,82) ber. C 60,96° o H 9,50°<o
gef. C 60,94oo H 9,45o/„Die diesem Chlorhydrat entsprechende freie Base wurde wegender vorhandenen geringen Substanzmenge nicht hergestellt.
Herstellung von 2-Meiliyl-4-phenyl-pyrrolidin-4-carbonsäureäthylester (Form B)
Herstellung der freien Base
10 g a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester wurden in250 ccm Feinsprit gelöst. Nach Zugabe von Raney-Nickel (bereitetaus 20 g Ni-Al-Legierung) als Katalysator wurde in einer Wasser¬
stoffatmosphäre geschüttelt. Da die Wasserstoffaufnahmegeschwin¬digkeit nach Aufnahme von -
,der berechneten Menge sehr klein
wurde, erwärmten wir den Hydrierungskolben während 6 Stundenauf 40—50" mittels einer Hydrierheizplatte, worauf die Wasser¬stoffaufnähme wieder in schnellerem Tempo erfolgte. In 3i/;> Tagenkam die Hydrierung nach Aufnahme von 3255 ccm Wasserstoff zum
Stillstand, was einer Menge von 3 Mol entspricht (berechnet 3250 ccmbei 17,5° 724 mm). Nach dem Abfiltrieren des Katalysators wurdeder Alkohol im Vakuum abdestilliert. Es blieb ein schwach gelb¬liches Oel zurück, das bei der Destillation im Hochvakuum 9,15 gPyrrolidincarbonsäureester vom Kp0?: 127,5° ergab, was einer Aus¬
beute von 96o/o der Theorie entspricht. Die Base ist farblos und
dünnflüssig, von ester- und etwas pyrrolidinartigem Geruch. In
Wasser ist die Base wenig löslich, mit stark alkalischer Reaktion.
n£ : 1,5212 dj1 : 1,0714 MQ ber.: 66,30
MD gef.: 66,33
19,52 mg Substanz gaben 51,74 mg C02 und 14,03 mg H30CUH,A,N (233,30) ber. C 72,07°/o H 8,21 >
gef. C 72,33°o H 8,04o/„
62
Herstellung des Chlorhydrates
Das Chlorhydrat wird durch Einleiten von trockenem HCl-Oas
in eine Lösung der Base in absolutem Aether gefällt. Es wird als
weisse Kristallmasse in einer Ausbeute von ca. 90°/o d. Th. ge¬
wonnen. Durch Umkristallisation aus Alkohol/Aether wird es in
weissen strahligen Kristallen vom F: 137° erhalten. Eine Misch¬
probe mit dem gleich hoch schmelzenden Chlorhydrat der Form A
gibt keine Depression. Das Chlorhydrat der Form B zeigt auch
sonst die gleichen Eigenschaften wie das der Form A, ergibt aber
bei der Rückführung in die freie Base wieder die Base B.
CuH20O2NCl (269,76) ber. C 62,33»o H 7,47«,,gef. C 62,25»0 H 7,40» „
Herstellung von 2-Methyl-2-phenyl-3~amino-
propionsdureäthylester
5 g a-Phenyl-a-cyan-ß-acetyl-propionsäureäthylester wurden in
250 cem Feinsprit + 3 cem konzentrierte Salzsäure gelöst und nach
Zugabe von Raney-Nickel (bereitet aus 5 g Ni-Al-Legierung) hy¬driert. In 8 Tagen wurden 1550 cem Wasserstoff, entsprechend3 Mol (berechnet 1556 cem bei 17,5°/727 mm), aufgenommen. Beim
Oeffnen des Kolbens fiel ein starker Geruch nach Acetaldehyd auf.
Nach Entfernung des Katalysators und des Lösungsmittels konnte
kein kristallisiertes Produkt direkt isoliert werden. Da der Rück¬
stand durch Nickelsalze ziemlich stark gefärbt war, wurde mit ver¬
dünnter Sodalösung versetzt und mit Aether ausgeschüttelt. Die
ätherische Lösung wurde mit wenig Wasser gewaschen und über
Na2SOi getrocknet. Nach dem Abdestillieren des Aethers wurden
bei der Hochvakuumdestillation zwei Fraktionen erhalten:
C12H1702N (207,26) ber. C 69,53«o H 8,27o/0gef. C 69,54o/o H 8,21 o/o
Es muss also 2-Methyl-2-phenyl-3-amino-propionsäureäthylester(C12H1702N) entstanden sein.
Herstellung von 2-Oxo-4-phenyl-pyrrolidin-4-carbonsäureäthylester
5 g a-Phenyl-a-cyan -bernsteinsäurediäthylester wurden in
150 ccm Feinsprit gelöst und nach Zugabe von Raney-Nickel (berei¬tet aus 10 g Ni-Al-Legierung) hydriert. Die Wasserstoffaufnähmekam nach Aufnahme von 995 ccm in 24 Stunden zum Stillstand,was der Menge von 2 Mol (berechnet 1000 ccm bei 18°/726 mm)entspricht. Nach dem Abfiltrieren des Katalysators wurde der Alko¬
hol im Vakuum abdestilliert. Es blieb ein farbloses Oel zurück,das rasch auskristallisierte. Es wurden 4,2 g 2-Oxo-4-phenyl-pyrro-lidin-4-carbonsäureäthylester (4-Phenyl-pyrrolidon-4-carbonsäure-äthylester) erhalten, entsprechend der theoretischen Menge. AusAethanol kristallisiert dieser Pyrrolidonester in weissen Blättchen
vom F: 122°. Er hat einen angenehm blumigen Geruch, der viel¬
leicht am meisten an den von Spiraea erinnert. Der Pyrrolidonesterist in Wasser sehr wenig und mit schwach saurer Reaktion löslich.
19,61 mg Substanz gaben 47,99 mg C02 und 11,25 mg H20C^H^OsN (233,26) ber. C 66,93<>/o H 6,48o0
gef. C 66,780« H 6,42o0
Herstellung von 2-Methyl-5-phenyl-piperhdin-5-carbonsäureäthylester (Form A)
2,5 g a-Phenyl-a-cyan-y-acetyl-buttersäureäthylester wurden in125 ccm Feinsprit -f 1,5 ccm konzentrierte Salzsäure gelöst. Nach
Zugabe von 70,8 mg Platinoxyd als Katalysator wurde in einer
Wasserstoffatmosphäre geschüttelt. Die einem Mol Wasserstoff
entsprechende Menge wurde in 20 Minuten aufgenommen, was
die Anwesenheit einer leicht hydrierbaren Doppelbindung beweist.
Durch die Wasserstoffaufnahme wird also auch bestätigt, dassdieses b-Ketonitril in der Enolform vorliegt. Die weitere Hydrie¬rung erfolgte dann viel langsamer, um nach Aufnahme von 896 ccm
Wasserstoff in 3 Tagen aufzuhören. Die für 3 Mol berechnete
Menge beträgt 767ccm bei 20° 721 mm; es wurden also 17o0 zu
viel Wasserstoff aufgenommen. Nach dem Abfiltrieren des Kataly¬sators wurde der Alkohol im Vakuum abdestilliert. Es blieben 2,7 ghelles, viskoses Oel, das zur Förderung der Kristallisation mit abso¬
lutem Aether angespritzt wurde. Da das Hydrochlorid auch nach
64
mehrtägigem Stehenlassen nicht kristallisierte, wurde mit verdünn¬
ter Natriumbicarbonatlösung versetzt und ausgeäthert. Nach Trock¬
nung der ätherischen Lösung über Na^Ot wurde der Aether ab¬
destilliert und das Produkt im Vakuum fraktioniert:
C1SH1702N (219,27) ber. C 71,20o,o H 7,82»„ N 6,39«ogef. C 73,24o/o H 6,79<>o N 3,64«„
gef. C 73,08o/o H 6,70o0
Es scheint bei der Hydrierung eine Kondensation von 2 Molekülen
Oxynitril eingetreten zu sein, unter Abspaltung von Alkohol, Am¬
moniak und Wasser. Das Hydrierungsprodukt hätte dann die
Formel :
C24H2,04N (393,46) ber. C 73,26o« H 6,92o„ N 3,56o0
gef. C 73,24o« H 6,79o0 N 3,64o0
C. Unterscheidende Farbreaktionen
von Pyrrolidin-und Piperidin-Derivaten
Da Morphin mit Formalin-Schwefelsàure (Marquis-Rtagtn?,) eine
rotviolette Farbe gibt, prüften wir das Verhalten von Dolantin
und von unseren Pyrrolidin- und Piperidinderivaten bei dieser Farb¬
reaktion.
Einige mg der zu untersuchenden Substanz wurden in 3—4 ccm
konzentrierter Schwefelsäure gelöst und nach Zugabe von 4 Trop¬fen Formaldehydlösung (40°oig) leicht erwärmt. Die Farbe wurde
bei Tageslicht und eine allfällige Fluorescenz unter der Quarz¬
lampe bestimmt.
Morphin gibt eine rotviolette Farbe, Fluorescenz tritt keine auf.
Dolantin gibt eine lachs- bis granatrote Färbung, unter der
Quarzlampe fluoresciert die Lösung intensiv blau bis violett.
2-Methyl-4-phenyl-pyrrolidin-4-carbonsäureäthylester gibt eine
Bordeaux-rote Färbung, unter der Quarzlampe fluoresciert diese Lö¬
sung intensiv ziegelrot.
67
1-Oxo-4-phenyl-pyrrolidin-4-carbonsäureäthylester gibt eine kar¬minrote Färbung; unter der Quarzlampe bemerkt man eine inten¬siv ockergelbe Fluorescenz.
2-Methyl-5-phenyl-piperidin-5-carbonsäureäthylester gibt eine rot¬braune Farbe; die Fluorescenz dieser Lösung unter der Quarzlampeist intensiv braunrot.
Eine alkoholische Lösung der erwähnten Substanzen ist bei
Tageslicht farblos und wasserklar. Bei Betrachtung unter der
Quarzlampe zeigen die Lösungen der erwähnten Substanzen alleeine schwache bläuliche Fluorescenz.
Zusammenfassung
I. Es wird eine Uebersicht über Dolantin und Dolantin-ähnlicheDerivate des 4-Phenyl-piperidins gegeben.
II. Es wird eine Zusammenstellung der Literatur über die Deu¬tung des Mechanismus der katalytischen Hydrierung von Nitrilengegeben. Anhand der Literatur wird die Bildung cyclischer Derivatebei der katalytischen Hydrierung von Cyan-Derivaten, insbesonderevon Ketonitrilen, besprochen.
III. In eigenen Versuchen wurden einige neue Ketonitrile durchKondensation von Phenyl-cyan-essigsäureäthylester mit Halogen-ketonen und Halogenfettsäureestern hergestellt. Es wurden Konden¬sationsversuche mit weiteren halogenierten Substanzen und Phenyl-cyan-essigsäureäthylester durchgeführt. Es wurden einige neue
Pyrrolidin- und Piperidin-Derivate durch katalytische Hydrierungvon Ketonitrilen gewonnen. Der Zusammenhang zwischen Kataly¬satormenge und Hydrierungsgeschwindigkeit wurde untersucht.
IV. Es werden unterscheidende Farbreaktionen für die gewon¬nenen Pyrrolidin- und Piperidin-Derivate im Vergleich zu Dolantinund Morphin angegeben.
68
LEBENSLAUF
Ich wurde am 16. Juni 1916 als Sohn von Ernst und Luise
Suter-von Zabern in Zürich geboren und besitze das Bürgerrecht
von Rüfenach (Aargau).
Meine Schulzeit verbrachte ich in Zürich, Gent (Belgien), Lau¬
sanne, Düsseldorf, Trogen und wieder in Zürich, wo ich im Herbst
1934 am kantonalen Gymnasium die Maturitätsprüfung bestand.
Anschliessend begann ich meine Studien an der E.T.H. mit zwei
Semestern an der Abteilung für Maschineningenieurwesen. Dann
trat ich an die Abteilung für Chemie über, wo ich mir im Herbst
1940 das Diplom als Ingenieur-Chemiker erwarb. Nach einer Unter¬
brechung durch Militärdienst begann ich im April 1941 die vor¬
liegende Arbeit am Pharmazeutischen Institut der E.T.H. unter