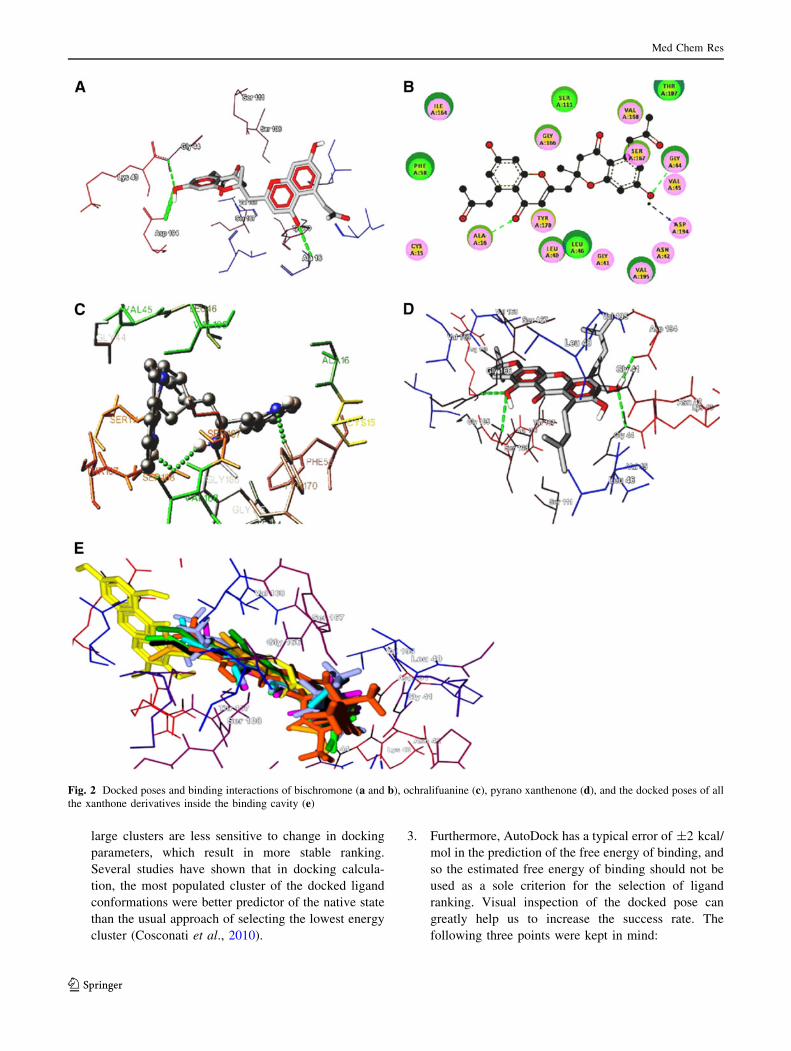
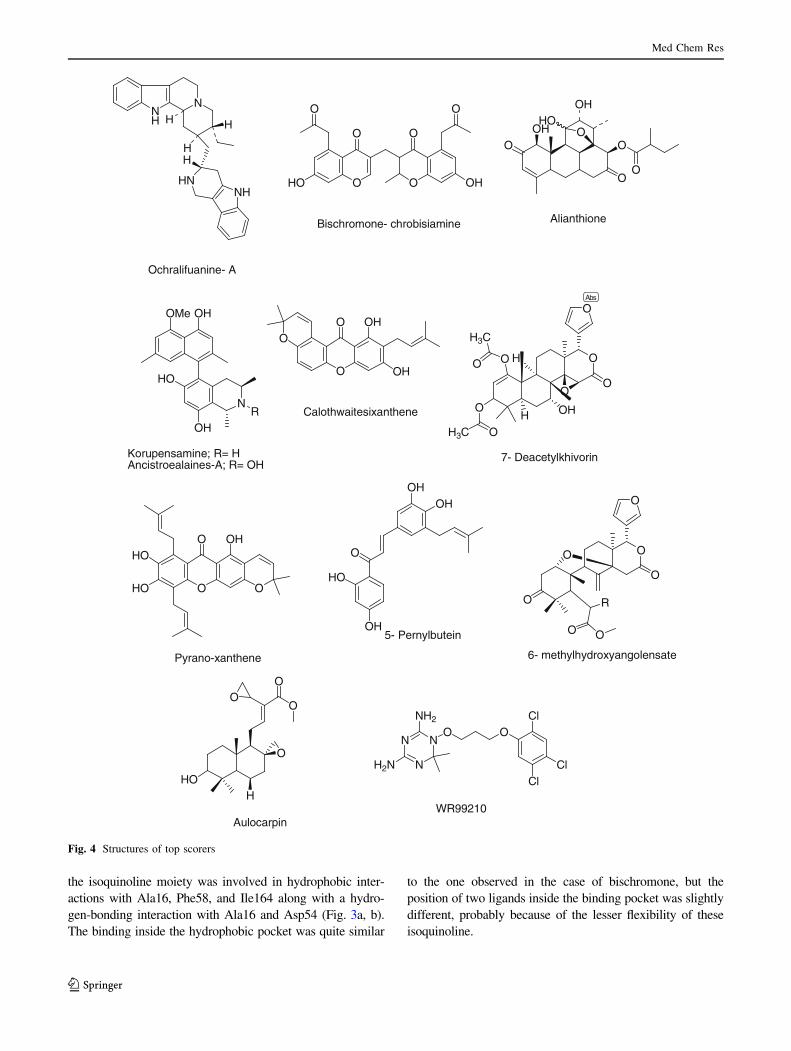
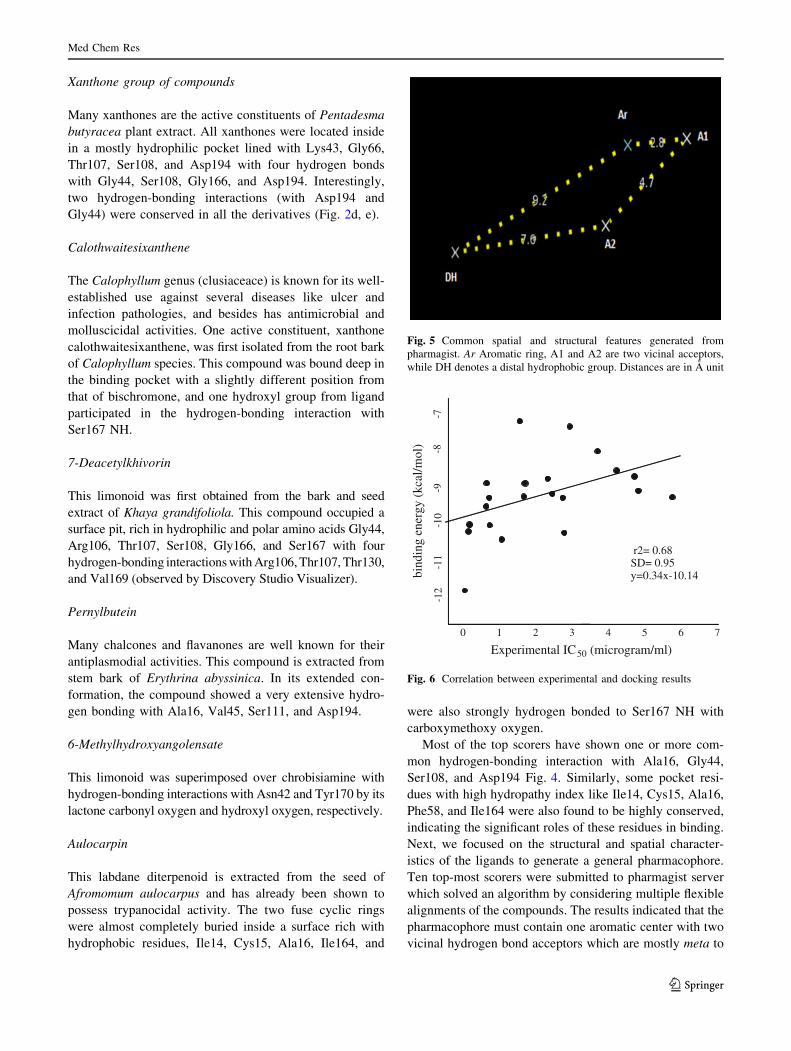
ORIGINAL RESEARCH In silico docking studies of bioactive natural plant products as putative DHFR antagonists Manoj Kumar • Anuradha Dagar • V. K. Gupta • Anuj Sharma Received: 28 December 2012 / Accepted: 28 May 2013 Ó Springer Science+Business Media New York 2013 Abstract In a bid to come up with effective natural plant product-based antagonist in antimalarial chemotherapy, we have built an in-house library of 185 compounds. The binding site of Plasmodium wild-type DHFR (1J3I) was explored computationally using AutoDock. The top-screened com- pounds revealed some novel scaffolds, relative to the general folate template, with micromolar to nanomolar inhibition constants. Further structural optimization subjected to the actual synthesis of these inhibitors can improve their efficacy as better candidates in the drug design pipeline. Keywords Malaria Á AutoDock Á Molecular docking Á DHFR Á Natural product Introduction Reported as one of the leading cause of death, malaria is a major health problem around the world. World Malaria report 2011 indicates that about 3.3 billion people, almost half of the world’s population, are at the risk of malaria with 216 million cases and 6,55,000 malarial deaths (in 2010) (World Malaria Report 2011, WHO, Fact Sheet). It is also predicted that global warming and demographic changes will lead to an increase in the distribution of clinical malarial cases in the Western world, including Europe and North America (Bathurst and Hatchel, 2006). Despite a significant number of antimalarials developed in the latter half of the twentieth century, there is a crying need for novel scaffolds, because of the genesis and spread of drug-resistant strains. Drugs, which have been worst affec- ted by resistant strains include chloroquine, dihydrofolate reductase (DHFR) inhibitors, cycloguanil and pyrimeth- amine (Dondorp et al., 2010). Thanks to the advancements achieved in the field of proteomics and genomics, the biol- ogy of the parasite is now well understood, and this has greatly aided in the rationale-based drug design (Malcom et al., 2002; Laurence and Michael, 2002). The dihydrofolate domain of the bifunctional enzyme dihydrofolate reductase-thymidylate synthase of Plasmo- dium falciparum (PfDHFR-TS) is an immensely important target in antimalarial chemotherapy. This enzyme maintains the intracellular level of tetrahydrofolate cofactor which is important for cell proliferation and cell growth. Inhibition of DHFR blocks the NADPH-dependent reduction of dihydrofolate to tetrahydrofolate and thus prevents DNA synthesis, resulting in cell death (Schnell et al., 2004). The receptor had been designed through homology mod- eling several times, before the crystal structure of the mole- cule complexed with the third generation folate inhibitor WR99210 was resolved in 2003. A series of antifolates such as cycloguanil (a dihydrotriazine), methotrexate (a diamin- opteridine), pyrimethamine, and trimethoprim (diaminopyr- imidines) were docked within the binding site of wild-type DHFR with significant binding constant and free energy. (Warhust, 1998; Lemke et al., 1999; Rastelli et al., 2000; Defino et al., 2002; Yuvaniyama et al., 2003). However, considering resistance against time-tested molecules, we strongly believe that many more scaffolds need to be tested and hypothesized against DHFR. To actualize this aim, we found no better source than mother nature itself as it produces molecules with unmatched Electronic supplementary material The online version of this article (doi:10.1007/s00044-013-0654-9) contains supplementary material, which is available to authorized users. M. Kumar Á A. Dagar Á V. K. Gupta Á A. Sharma (&) MedChemLab, Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee 247667, Uttarakhand, India e-mail: [email protected]; [email protected]123 Med Chem Res DOI 10.1007/s00044-013-0654-9 MEDICINAL CHEMISTR Y RESEARCH
Transcript
ORIGINAL RESEARCH
In silico docking studies of bioactive natural plant productsas putative DHFR antagonists
Manoj Kumar • Anuradha Dagar • V. K. Gupta •
Anuj Sharma
Received: 28 December 2012 / Accepted: 28 May 2013
� Springer Science+Business Media New York 2013
Abstract In a bid to come up with effective natural plant
product-based antagonist in antimalarial chemotherapy, we
have built an in-house library of 185 compounds. The binding
site of Plasmodium wild-type DHFR (1J3I) was explored
computationally using AutoDock. The top-screened com-
pounds revealed some novel scaffolds, relative to the general
folate template, with micromolar to nanomolar inhibition
constants. Further structural optimization subjected to the
actual synthesis of these inhibitors can improve their efficacy
Reported as one of the leading cause of death, malaria is a
major health problem around the world. World Malaria
report 2011 indicates that about 3.3 billion people, almost
half of the world’s population, are at the risk of malaria
with 216 million cases and 6,55,000 malarial deaths (in
2010) (World Malaria Report 2011, WHO, Fact Sheet). It
is also predicted that global warming and demographic
changes will lead to an increase in the distribution of
clinical malarial cases in the Western world, including
Europe and North America (Bathurst and Hatchel, 2006).
Despite a significant number of antimalarials developed
in the latter half of the twentieth century, there is a crying
need for novel scaffolds, because of the genesis and spread of
drug-resistant strains. Drugs, which have been worst affec-
ted by resistant strains include chloroquine, dihydrofolate
reductase (DHFR) inhibitors, cycloguanil and pyrimeth-
amine (Dondorp et al., 2010). Thanks to the advancements
achieved in the field of proteomics and genomics, the biol-
ogy of the parasite is now well understood, and this has
greatly aided in the rationale-based drug design (Malcom
et al., 2002; Laurence and Michael, 2002).
The dihydrofolate domain of the bifunctional enzyme
dihydrofolate reductase-thymidylate synthase of Plasmo-
dium falciparum (PfDHFR-TS) is an immensely important
target in antimalarial chemotherapy. This enzyme maintains
the intracellular level of tetrahydrofolate cofactor which is
important for cell proliferation and cell growth. Inhibition
of DHFR blocks the NADPH-dependent reduction of
dihydrofolate to tetrahydrofolate and thus prevents DNA
synthesis, resulting in cell death (Schnell et al., 2004).
The receptor had been designed through homology mod-
eling several times, before the crystal structure of the mole-
cule complexed with the third generation folate inhibitor
WR99210 was resolved in 2003. A series of antifolates such
as cycloguanil (a dihydrotriazine), methotrexate (a diamin-
opteridine), pyrimethamine, and trimethoprim (diaminopyr-
imidines) were docked within the binding site of wild-type
DHFR with significant binding constant and free energy.
(Warhust, 1998; Lemke et al., 1999; Rastelli et al., 2000;
Defino et al., 2002; Yuvaniyama et al., 2003).
However, considering resistance against time-tested
molecules, we strongly believe that many more scaffolds
need to be tested and hypothesized against DHFR. To
actualize this aim, we found no better source than mother
nature itself as it produces molecules with unmatched
Electronic supplementary material The online version of thisarticle (doi:10.1007/s00044-013-0654-9) contains supplementarymaterial, which is available to authorized users.
M. Kumar � A. Dagar � V. K. Gupta � A. Sharma (&)
MedChemLab, Department of Chemistry, Indian Institute of
Technology Roorkee, Roorkee 247667, Uttarakhand, India