Increasing Sequence Correlation Limits the Efficiency of Recombination in a Multisite Evolution Model S. Gheorghiu-Svirschevski, I. M. Rouzine, and J. M. Coffin Department of Microbiology and Molecular Biology, Tufts University The accumulation of preexisting beneficial alleles in a haploid population, under selection and infrequent recombination, and in the absence of new mutation events is studied numerically by means of detailed Monte Carlo simulations. On the one hand, we confirm our previous work, in that the accumulation rate follows modified single-site kinetics, with a timescale set by an effective selection coefficient s eff as shown in a previous work, and we confirm the qualitative features of the de- pendence of s eff on the population size and the recombination rate reported therein. In particular, we confirm the existence of a threshold population size below which evolution stops before the emergence of best-fit individuals. On the other hand, our simulations reveal that the population dynamics is essentially shaped by the steady accumulation of pairwise sequence correlation, causing sequence congruence in excess of what one would expect from a uniformly random distribution of alleles. By sequence congruence, we understand here the opposite of genetic distance, that is, the fraction of monomorphic sites of specified allele type in a pair of genomes (individual sequences). The effective selection coefficient changes more rapidly with the recombination rate and has a higher threshold in this parameter than found in the previous work, which neglected correlation effects. We examine this phenomenon by monitoring the time dependence of sequence correlation based on a set of sequence congruence measures and verify that it is not associated with the development of linkage disequilibrium. We also discuss applications to HIV evolution in infected individuals and potential implications for drug therapy. Introduction Genomic recombination is a pervasive biological mechanism. It occurs in living taxa of all levels of complex- ity, from viruses and prokaryotes to highest order mammals and gymnosperms. Yet its benefits for the survival and propagation of a species are not always clear. Whether re- combination is advantageous or not for a certain population seems to depend on a complex set of conditions involving, with equal weight, the nature of intragenome interactions between various beneficial alleles, the structure and size of the population itself, and the type of environment shap- ing the evolution of the population. It is frequently argued, for instance, that the exchange of genetic material via recombination (sexual reproduction) expands the range of individual variability that provides the substrate for natural selection (Burt 2000). However, according to few-site models for very large populations in a stationary environment, such an increase in variability arises only under negative linkage disequilibrium (Otto and Lenormand 2002) or negative epistasis (Barton 1995a; Otto and Lenormand 2002). Still because the effects of linkage and epistasis can change with the character of the environ- ment, it was found that recombination may yet increase var- iability under rapidly changing environmental conditions, regardless of epistasis (Barton 1995a; Burger 1999). Perhaps the most quoted advantage of recombination in finite populations is that the exchange of alleles between members of a population may promote the accumulation and fixation of beneficial mutations, counteracting the neg- ative effect of chromosomal linkage on the response to se- lection (Fisher 1930; Muller 1932). Beneficial mutations survive selection and random drift only if they arise in a sufficiently large number of high-fitness individuals. If the population is not large enough and/or the selection pres- sure is not high enough, the population will experience an excessive buildup of deleterious mutations known as Muller’s ratchet (Felsenstein 1974), as well as a slow down in the fixation of beneficial mutations that have to compete for representation in the population (clonal interference ef- fect). Recombination has the potential to counteract these effects by bringing together, in the same genome, distinct beneficial mutations that otherwise would likely remain seg- regated. This old idea (Fisher 1930; Muller 1932) has re- ceived quantitative support, both analytically (Crow 1965; Hill and Robertson 1966; Felsenstein 1974; Barton 1995b) and numerically (Hey 1998), typically in models with two or few linked sites. Then again, recombination can just as well dissociate beneficial mutations in highly fit individuals, producing less-fit progeny with diminished chances of survival. In the context of an early deterministic model with multiplicative selection and no linkage disequi- librium, such events were shown to erase entirely the pos- itive effects of recombination on the fixation of beneficial mutations (Maynard-Smith 1968). The reasons for such divergent conclusions can be traced eventually to various disparities in the assumptions of the different models employed. The enormous complex- ity of the corresponding in vivo processes calls necessarily for a number of drastic simplifications, starting with the type and size of genome (haploid vs. diploid, 2-allele vs. multiallele, 2 or 3 site vs. multisite or even infinite length), the size of the population (finite vs. infinite, constant vs. variable) and its initial configuration, low or high rate of mutation (when present), type and strength of selection, mechanism and rate of recombination, stochastic or deter- ministic kinetics, etc. Given the wide range of options and the enduring complexity of the problem, even under strong simplification, it is not surprising that different models lead to different conclusions. The overall impression is, how- ever, that models that take into account the effects of finite population size tend to predict an advantage of recombina- tion in the progressive evolution of organisms (Crow 1965; Hill and Robertson 1966; Felsenstein 1974; Barton 1995b). Key words: recombination, selection, model, numerical, simulation, correlation. E-mail: [email protected]. Mol. Biol. Evol. 24(2):574–586. 2007 doi:10.1093/molbev/msl189 Advance Access publication November 30, 2006 Ó The Author 2006. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: [email protected]Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840 by guest on 14 February 2018
Transcript
Increasing Sequence Correlation Limits the Efficiency of Recombination ina Multisite Evolution Model
S. Gheorghiu-Svirschevski, I. M. Rouzine, and J. M. CoffinDepartment of Microbiology and Molecular Biology, Tufts University
The accumulation of preexisting beneficial alleles in a haploid population, under selection and infrequent recombination,and in the absence of newmutation events is studied numerically by means of detailedMonte Carlo simulations. On the onehand, we confirm our previous work, in that the accumulation rate follows modified single-site kinetics, with a timescale setby an effective selection coefficient seff as shown in a previous work, and we confirm the qualitative features of the de-pendence of seff on the population size and the recombination rate reported therein. In particular, we confirm the existenceof a threshold population size below which evolution stops before the emergence of best-fit individuals. On the other hand,our simulations reveal that the population dynamics is essentially shaped by the steady accumulation of pairwise sequencecorrelation, causing sequence congruence in excess of what one would expect from a uniformly random distribution ofalleles. By sequence congruence, we understand here the opposite of genetic distance, that is, the fraction of monomorphicsites of specified allele type in a pair of genomes (individual sequences). The effective selection coefficient changes morerapidly with the recombination rate and has a higher threshold in this parameter than found in the previous work, whichneglected correlation effects. We examine this phenomenon by monitoring the time dependence of sequence correlationbased on a set of sequence congruence measures and verify that it is not associated with the development of linkagedisequilibrium. We also discuss applications to HIV evolution in infected individuals and potential implications for drugtherapy.
Introduction
Genomic recombination is a pervasive biologicalmechanism. It occurs in living taxa of all levels of complex-ity, from viruses and prokaryotes to highest order mammalsand gymnosperms. Yet its benefits for the survival andpropagation of a species are not always clear. Whether re-combination is advantageous or not for a certain populationseems to depend on a complex set of conditions involving,with equal weight, the nature of intragenome interactionsbetween various beneficial alleles, the structure and sizeof the population itself, and the type of environment shap-ing the evolution of the population.
It is frequently argued, for instance, that the exchangeof genetic material via recombination (sexual reproduction)expands the range of individual variability that provides thesubstrate for natural selection (Burt 2000). However,according to few-site models for very large populationsin a stationary environment, such an increase in variabilityarises only under negative linkage disequilibrium (Otto andLenormand 2002) or negative epistasis (Barton 1995a; Ottoand Lenormand 2002). Still because the effects of linkageand epistasis can change with the character of the environ-ment, it was found that recombination may yet increase var-iability under rapidly changing environmental conditions,regardless of epistasis (Barton 1995a; Burger 1999).
Perhaps the most quoted advantage of recombinationin finite populations is that the exchange of alleles betweenmembers of a population may promote the accumulationand fixation of beneficial mutations, counteracting the neg-ative effect of chromosomal linkage on the response to se-lection (Fisher 1930; Muller 1932). Beneficial mutationssurvive selection and random drift only if they arise in asufficiently large number of high-fitness individuals. If the
population is not large enough and/or the selection pres-sure is not high enough, the population will experience anexcessive buildup of deleterious mutations known asMuller’s ratchet (Felsenstein 1974), as well as a slow downin the fixation of beneficial mutations that have to competefor representation in the population (clonal interference ef-fect). Recombination has the potential to counteract theseeffects by bringing together, in the same genome, distinctbeneficial mutations that otherwisewould likely remain seg-regated. This old idea (Fisher 1930; Muller 1932) has re-ceived quantitative support, both analytically (Crow1965; Hill and Robertson 1966; Felsenstein 1974; Barton1995b) and numerically (Hey 1998), typically in modelswith two or few linked sites. Then again, recombinationcan just as well dissociate beneficial mutations in highlyfit individuals, producing less-fit progeny with diminishedchances of survival. In the context of an early deterministicmodel with multiplicative selection and no linkage disequi-librium, such events were shown to erase entirely the pos-itive effects of recombination on the fixation of beneficialmutations (Maynard-Smith 1968).
The reasons for such divergent conclusions can betraced eventually to various disparities in the assumptionsof the different models employed. The enormous complex-ity of the corresponding in vivo processes calls necessarilyfor a number of drastic simplifications, starting with thetype and size of genome (haploid vs. diploid, 2-allele vs.multiallele, 2 or 3 site vs. multisite or even infinite length),the size of the population (finite vs. infinite, constant vs.variable) and its initial configuration, low or high rate ofmutation (when present), type and strength of selection,mechanism and rate of recombination, stochastic or deter-ministic kinetics, etc. Given the wide range of options andthe enduring complexity of the problem, even under strongsimplification, it is not surprising that different models leadto different conclusions. The overall impression is, how-ever, that models that take into account the effects of finitepopulation size tend to predict an advantage of recombina-tion in the progressive evolution of organisms (Crow 1965;Hill and Robertson 1966; Felsenstein 1974; Barton 1995b).
Mol. Biol. Evol. 24(2):574–586. 2007doi:10.1093/molbev/msl189Advance Access publication November 30, 2006
� The Author 2006. Published by Oxford University Press on behalf ofthe Society for Molecular Biology and Evolution. All rights reserved.For permissions, please e-mail: [email protected]
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
One always hopes that approaching the problem with a gen-erous enough set of assumptions will eventually allow a uni-fied understanding of now seemingly antagonistic results.
With this position in mind, we (Rouzine et al. 2003;Rouzine and Coffin 2005) have recently formulated a novelanalytical approach to a 2 allele, multisite model of evolu-tion in haploid populations with a simple fitness landscape.Variants of this type of model have proved useful in quan-titative studies of the evolution of RNA viruses and bacteria(Tsimring et al. 1996; Aranson et al. 1997; Kessler et al.1997; Ridgway et al. 1998). The key observation of our ap-proach is that, in a great variety of situations, the distributionof genomes (individual sequences) according to fitness(or the number of less-fit alleles) can be approximated asa quasi-deterministic ‘‘solitary wave’’ with stochastic edges,propagating in fitness space at a slowly changing speed. Themethod allows the derivation of closed-form expressions forbasic kinetic quantities, valid over a broad range of modelparameters. Hence, it can provide estimates of the evolutionrate for a large variety of theoretical and experimentalsituations. The framework was first applied to a study ofMuller’s ratchet and Fisher–Muller–Hill–Robertson effectunder weak selection and mutation (forward, backward, andcompensatory), in the absence of recombination (Rouzineet al. 2003). For a wide range of population sizes, it wasfound that the evolution rate has a logarithmic dependenceon the selection coefficient and the population size N.In particular, the model recovered successfully the resultof the single-site model in the infinite population limit,the rate of accumulation of beneficial alleles limited byFisher–Muller–Hill–Robertson effect at intermediate N,a steady state at a critical value of N, and Muller’s ratchetin small populations.
The same approach was employed recently in an anal-ysis of evolution under recombination and weak selection,in the absence of mutations (Rouzine and Coffin 2005), us-ing the approximation that positions of deleterious allelesdo not correlate within and between genomes of given fit-ness. The prediction of a quasi-deterministic solitary wavewas found to hold for this model as well. The magnitude ofthe corresponding accumulation rate followed single-sitetype kinetics, with the effects of recombination, selection,and random drift combined into an effective selection co-efficient seff. The effective coefficient seff was smaller thanthe single-site selection coefficient s, but increased with therecombination rate, confirming that recombination counter-acts only partially the Hill–Robertson effect introduced bylinkage. Notably, seff, and therefore the entire dynamics,was shown to display a well-defined transition with increas-ing population size N or recombination rate r. Below a cer-tain critical size Nc’ðN=rÞ1=2; the population evolvedeventually into a clone and the accumulation of beneficialalleles stopped, corresponding to a null value for seff. Forpopulation sizes larger than Nc, beneficial alleles accumu-lated at a steady rate, until such alleles became fixed at allsites. In this case, the effective selection coefficient ac-quired a logarithmic dependence on N and approachedthe single-site selection coefficient s at large enough N.
The current paper presents detailed numerical tests ofthe model and conclusions of Rouzine and Coffin (2005) bymeans of Monte Carlo simulation. We perform a careful
numerical analysis of the accumulation kinetics, with spe-cific emphasis on the emergence of intergenome correlationfor homologous sites throughout the population. Intergen-ome correlation is assessed by means of pairwise sequencecongruence, defined as the fraction of monomorphic sites ofspecified allele type in a pair of genomes. Thus, sequencecongruence, as a measure of genetic similarity, is the ‘‘op-posite’’ of the genetic distance. We examine the interplaybetween the buildup of sequence congruence and the accu-mulation of better-fit alleles by monitoring the time depen-dence of a complementary set of sequence congruencefunctions and of linkage disequilibrium. Particular attentionis also given to the suitability of the single-site form of theaccumulation rate expression and to the dependence ofthe effective selection coefficient on the population sizeand the recombination rate. As before, we apply our studyto the evolution of HIV populations and the probable roleof recombination in the emergence of drug resistance.
Model and MethodsModel
Wemodel the viral population as a collection ofN hap-loid genomes, represented by sets of L sites (bases or basepairs) each and subject to discrete time Wright–Fisher evo-lution with recombination, random drift, and selection forbetter-fit genotypes. For simplicity, each site is assumed todisplay only one of 2 alleles: better fit or less fit. We furtherdisregard any epistasis, as well as all mutation events. Del-eterious mutation events are neglected under the assump-tion that the accumulation rate of better-fit sites, in theparameter range of interest, is much larger than the mutationrate. Beneficial mutation events are also neglected becauseall beneficial alleles are assumed to preexist. This simplifi-cation allows us to consider only significant sites thatpresent at least one copy of a better-fit allele in the initialpopulation. The appearance of new significant sites at latertimes can be ignored, and the effective genome length L isfixed accordingly for the duration of the evolution. Thepopulation size N is also assumed to be constant in time.
The initial population is subjected to a discretized evo-lution with a time step of one generation. Each generation,a (small) number of randomly sampled pairs of genomesundergoes recombination, and the entire population is sub-sequently replaced by sampled progeny. In the samplingprocess, individual genomes are substituted by their directdescendants in random numbers according to a Poisson dis-tribution, under the constraint of a constant population size.The average number of progeny for any given genome isproportional to its fitness, which is determined on the as-sumption that each expressed nonresistant allele diminishesthe chances of survival by a factor e�s, slightly less than unit(s � 1). In other words, log fitness is proportional to thenumber k of less-fit sites, by the selection coefficient s(log fitness 5 �sk, no epistasis).
The model recombination process assumes thatrecombinant genome pairs are selected randomly and se-quentially with a probability 1/N0, such that each genomehas an equal chance of undergoing recombination, at a rater 5 N/N0. Each generation, the randomly sampled genomepairs undergo a preset number of sequence crossovers to
Sequence Correlation Limits Effect of Recombination 575
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
produce pairs of descendent strands. Both descendents ofa recombinant pair are retained in the evolving population(even though in a real population only one descendant is pro-duced, the difference hardly matters, as only unusuallyhighly fit recombinants influence evolution). We note thatbecause of this latter assumption, our parameter r shouldnot be thought of as a probability, because it may well hap-pen that r. 1. In such a case, there is a nonzero probabilitythat some descendents of genomes already involved in a re-combinant event become themselves involved in recombi-nation within the same generation. In the case of HIV, therate r is determined by the frequency of coinfection of atarget cell by 2 (or more) viral particles. More precisely,r represents the average number of coinfection events expe-rienced by a target cell during one generation. Assuming thateach such coinfection event results in the recombinationof the 2 viral strands, the average number of recombinantencounters per generation in the entire population amountsto ;rN/2.
Model Parameters for HIV Populations In Vivo
The model and the model parameters are tailored to fitour particular interest in HIV evolution. The details dependon the timescale considered. For long observation periods,102 to 103 generations, the number of polymorphic sites ison the order L ; 100. For short-term evolution, the effec-tive genome length L (number of drug-sensitive polymor-phic sites) may be much smaller. The effective size of anHIV population, N, is set by the number of infected cells onthe productive pathway. According to existing estimatesbased on models that include selection (Chun et al.1997; Rouzine and Coffin 1999; Frost et al. 2000), in anaverage patient, this number is N; 106 or larger. However,the time required for our Monte Carlo simulations increasesat least quadratically with the population size, and simula-tions become rather inefficient at N. 105. We compromiseon somewhat smaller population sizes, 105 or fewer. A sim-ilar predicament constrains our choice of allele composi-tion. We consider cases when only a small fraction f0 ofthe alleles presented by significant sites at the onset of evo-lution are better fit. This initial condition corresponds to 2experiments: 1) reversion of deleterious alleles in untreatedpatients soon after transmission due to the transmission bot-tleneck and 2) amplification of drug-resistant alleles afterstart of drug therapy. But a very low initial fraction ofbetter-fit alleles requires higher N to avoid their loss,Nf0s � 1, and leads again to inefficiently long simulationtimes. We prefer to balance against lengthy simulations byconsidering initial compositions with f0 ; 10�2 randomlydistributed drug-resistant alleles. In the absence of initialallele loss, simulation results are weakly sensitive to f0.
Variation of the selection coefficient s among differentsites is ignored, replaced by a representative value deter-mined by the timescale of the experiment under consider-ation (t ; 1/s). Indeed, given a timescale, beneficial allelesat sites with much larger s will be fixed rapidly and similaralleles at sites with much smaller s will not have enoughtime to accumulate. Short-term evolution, spanning 10–100generations, suggests s ; 0.1, whereas longer timescales,stretching over hundreds and thousands of generations,
imply s ; 10�2 (Rouzine and Coffin 1999). Muchsmaller values of s may be dismissed because the corre-sponding timescales exceed the average duration of HIVinfections. Larger values s ; 1, corresponding to shortdurations of the order of several generations, are also ir-relevant because such sites can be regarded as conservedon longer timescales. Our simulations use typical values ofs 5 0.1–0.01.
In each round of infection, 2 RNA templates carried bya virus particle are reverse transcribed into a new provirus.Recombination occurs when the reverse transcriptaseswitches between the 2 available templates and incorporatesfragments of geneticmaterial from both parental strands intothe newly transcribedDNAgenomes. It is assumed that a rel-atively large number, ;10, of such switches (crossovers)occurs before the reverse transcription process is completed(Dang et al. 2004; Levy et al. 2004), with the result that theprogeny provirus carries a well-mixed blend of sequencesfrom the parental RNA strands. Two different virus ge-nomes can recombine only if they coinfect the same cell.If the density of newly infected cells is low in the tissue,as in persistent HIV infection, the rate r of recombinationfor any one genome becomes proportional to the numberN of infected cells, r 5 N/N0. The factor 1/N0 representsthe probability that any randomly selected pair of genomessucceeds in coinfection; alternatively, N0 is the extrapolatedpopulation size for which the recombination rate becomes 1.It depends on the total number of cells permissive for virusreplication and their distribution in tissue. Whereas an esti-mate, r ; 1, has been obtained in some untreated patientsbased on the sampling of double HIV DNA positive cells(Jung et al. 2002), conclusive experimental measurementsof the rate r in productively infected (RNApositive) cells forgivenN have yet to be performed. Unfortunately, this leavesa good deal of uncertainty concerning the relevant values forbothN and r in real patients. For instance, the above estimateof rmay be too high if most coinfecting virions originate inneighboring cells and therefore are genetically uniform.Similarly, the estimate for N, which is based on a 2-sitemodel, may have to be adjusted both to account for a higherfrequency of observed crossovers and for the multisite na-ture of evolution. In the following, we use values on the or-der N0 ; 103 to 105, which may be lower than the typicalvalue in an untreated patient but offer a reasonable compro-mise in terms of simulation time. We note that N0 can bevaried in vitro by changing the culture conditions, specifi-cally, the number, density, and type of permissive cells. Themodel is equally applicable for other partially sexual pop-ulations, such as yeast, in which the recombination rater is regulated independently of N by external factors.
Because in the initial population the position of bene-ficial alleles within genomes is random and any crossoverevents during recombination at later times also occur in ran-dom positions, it could be expected that the better-fit allelesremain randomly distributed at any time during the evo-lution (Rouzine and Coffin 2005). In such an approxima-tion, the viral population kinetics can be monitored by the
576 Gheorghiu-Svirschevski et al.
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
time-dependent frequency of genomes with k less-fit alleles,f(k, t). It has been found previously (Rouzine et al. 2003;Rouzine and Coffin 2005) that, under the assumptions dis-cussed above, the main part of the distribution f(k, t) followsa quasi-deterministic kinetics. By contrast, the edges of thedistribution (beyond which all fitness classes are empty), es-pecially the leading edge toward small k, advance at a ratedetermined by the stochastic formation of best-fit sequencesthrough recombination. Away from the edges f(k, t) is ex-pected to follow kinetics of the form
df ðk; tÞdt
5 ðe�sðk��kÞ � 1Þf ðk; tÞ1Rðk; tÞ; ð1Þ
where the first term on the right hand side describes the se-lection process, whereas the generic term R(k, t) subsumesthe effects of recombination. The quantitye�s�k5
Pk e
�skf ðkÞ accounts for the normalization of the se-lection contribution to constant population size. If the pro-file of f(k, t) is always narrow enough that sjk � �kj,1;e�sðk��kÞ’1� sðk � �kÞ; and the normalization factor be-comes es
�k’esÆkæ; where Ækæ5P
k kf ðk; tÞ is the averagenumber of less-fit alleles, and the kinetic equation (1) ac-quires the simpler form
df ðk; tÞdt
5 � sðk � ÆkæÞf ðk; tÞ1Rðk; tÞ: ð2Þ
In this limit, we retrieve immediately the well-knownFisher’s law for the evolution of Ækæ:
dÆkædt
5 � sVar½k�; ð3Þ
with Var½k�5Æðk � ÆkæÞ2æ being the variance of the numberk of deleterious alleles (Haigh 1978). The recombinationterm R(k, t) does not appear in equation (3) because the re-combination process does not change the number of allelesof one type or another, as given by
RdkkRðk; tÞ50; and R
does not contribute directly to changes in Ækæ: Its contribu-tion is concealed in the explicit form of the variance Var[k].
At infinite N or small N0, the expression of Var[k] canbe further inferred without direct knowledge of the recom-bination term R, recalling that in this case the initial random(binomial) distribution of mutations ðÆkæðt50Þ5L� 1Þ re-mains random (binomial) throughout the evolution. Indeed,in either case, the average number of recombination eventsper generation, rN/2 5 N2/2N0, becomes infinitely large,and such strong recombination maintains a perfectly ran-dom (binomial) redistribution of alleles throughout thepopulation. As in the single-site case, the correspondingvariance is given by
Var½k�5 Ækæð1� Ækæ=LÞ: ð4Þ
Substitution of equation (4) into equation (3) yields a stan-dard deterministic evolution equation for the single-siteallelic frequency Ækæ=L:
At finite N and large N0, the binomial distribution ofalleles (eq. 4), and therefore the deterministic expression forthe evolution rate, equation (3), does not apply directly for 3reasons as follows.
(i) Because better-fit alleles at different sites are selectedconcurrently and because new sequences are producedat a limited rate, the number k of less-fit sites correlatesbetween different genomes. Recombination producesbest-fit individual genomes that spread gradually overthe population, then the process is repeated. As a result,at a given Ækæ; the distribution of k among genomes ismore narrow than given by equation (4), and the evolu-tion rate, correspondingly, is smaller than that given byequation (3). In order to determine the correct varianceVar[k], one has to specify the expression for the recom-bination term R(k, t) in equation (2). In the approximationthat the positions of individual sites are completely ran-dom within a genome and, unlike their total number, donot correlate between different genomes, R(k, t) can beexpressed in terms of f(k, t), as a quadratic functional(Rouzine and Coffin 2005). Matching the rate of gener-ation of new best-fit genomes (i.e., those with smallest k)to the evolution rate of the deterministic part of fitnessdistribution, we (Rouzine and Coffin 2005) obtained
Var½k�5 ðseff=sÞÆkæð1� Ækæ=LÞ; ð5aÞ
where seff is given by
seff 5 slnðNrÞ
ln½ðNsÞ2Ækæð1� Ækæ=LÞ=ðNrÞ�;
1=N � r � sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiÆkæð1� Ækæ=LÞ
Thus, the binomial dependence of Var[k] on Ækæ; equation(4), is mostly preserved, but Var[k] is decreased bya factor seff/s that becomes one at N5N orr;s
at N;N0sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiÆkæð1� Ækæ=LÞ
p; if r and N are proportional.
Accordingly, the deterministic evolution equation (3)formally holds, but with a smaller effective selection co-efficient seff that absorbs the effects of finite populationsize, linkage (Hill–Robertson effect), and recombination.
(ii) The approximation (Rouzine and Coffin 2005) that thepositions of beneficial alleles, for a given number persequence, do not correlate between genomes may beinaccurate. Even if the initial distribution of alleles iscompletely random, correlations between genomesmay develop later because pairs of recombining genomesshare sequence segments inherited from common ances-tors. Suppose, for example, that 2 genomes are producedby recombination between 2 pairs of parental genomes. Ifone parent sequence is common for both recombinants,approximately a quarter of their sequence will be iden-tical. The frequency of common ancestral sequences islimited by the combined effect of selection and randomsampling. At this time, we are not able to provide goodtheoretical estimates for the magnitude of this effect.However, the direct numerical simulations presentedbelow will show that the effective selection coefficient
Sequence Correlation Limits Effect of Recombination 577
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
approximation provides very good estimates for the timedependence of Ækæ and the variance Var[k], but equation(5b) for seff has to be modified substantially, by a factorthat is a nontrivial functional of the model parameters N,L, r, s.
(iii) equation (5a) for the variance Var[k] has to be adjusted,in addition, for the fixation and loss of better-fit allelesbecause monomorphic sites do not contribute to fluctua-tions of k. If klost(t) and kfix(t) denote, respectively, thenumber of lost and fixed better-fit sites at time t, thesemodifications bring equation (5a) for Var[k] to the form
Var½k�5 ðseff=sÞðÆkæ
� kfixðtÞÞ 1� Ækæ� kfixðtÞL� klostðtÞ � kfixðtÞ
� �: ð6Þ
Numerical Implementation
TheMonte Carlo simulations were performed with ournewly developed microPopulation software package(Gheorghiu-Svirschevski S, Rouzine I, unpublished data).The corresponding algorithm of the Wright–Fisher kineticsis rather simple. The model population is formally repre-sented by an array of N strings with L binary variables each.The initial array is constructed site by site according to abinomial distribution with an average of f0 � L beneficial al-leles per genome. Each Monte Carlo generation (iteration)carries the population array through one session of recom-bination events and one session of selection. During therecombination step, parent genome pairs, selected sequen-tially and randomlywith a probability 1/N0, undergo a presetnumber of M 5 10 crossovers each, at randomly selectedpositions along the genome length. No recombination hot-spots are used in our simulations. (A random number ofcrossover events would be a better assumption. However,because the results are not sensitive to the value chosenfor M, we can safely assume that they are not sensitive tofluctuations of M either). The resulting population is thensubmitted to a selection algorithm based on the well-known‘‘broken stick’’ algorithm. The unit interval 0; 1½ � is dividedinto N segments, each segment corresponding to one ge-nome in the population. The length of each segment isset proportional to the fitness of the matching genome. Thenthe N genomes surviving into the next generation are se-lected by N random drawings of a random variable uni-formly distributed on 0; 1½ �; according to the particularsegment on which each drawing places the random variable.
Calculated Quantities
Our numerical calculations have targeted a 3-fold task.First, we performed a detailed numerical check on the ac-
tual validity of the inferred kinetics described by equation(6). The distribution f(k, t) of genomes with given number kof less-fit alleles was monitored throughout the run, alongwith the corresponding average Ækæ; its time derivativedÆkæ=dt; and the variance Var[k]. For the same purpose,we alsomonitored the time dependence of the number of lostand fixed better-fit alleles. Second, we paid particular atten-tion to similarity in pairs of sequences. To give quantitativeexpression to this phenomenon, we defined 3 complemen-tary measures of the degree of sequence congruence (theopposite of the genetic distance) in the following way. Forany given pair of genomes i and j, we denote nlf and nbfthe number of monomorphic less-fit and better-fit sites,respectively, and denote npoly the number of polymorphicsites. Observe that nlf1nbf1npoly5L: For each type ofallele, we define a pair congruence measure Clf(bf)(i, j) asthe ratio of monomorphic sites for that type of allele to thetotal number of sites expressing the type in at least one ofthe genomes. That is,
Clfði; jÞ5nlf
nlf 1 npoly5
nlfL� nbf
;Cbfði; jÞ5nbf
nbf 1 npoly
5nbf
L� nlf
:
ð7Þ
Then each degree of sequence congruence is averaged overall possible pairs in the population,
Clf 5 ÆClfði; jÞæ; Cbf 5 ÆCbfði; jÞæ: ð8Þ
We also define a total sequence congruence Ctot by takinginto account all monomorphic sites, irrespective of the typeof allele expressed. In this case, Ctotði; jÞ5 nlf1nbf
L and
Ctot 5 ÆCtotði; jÞæ; ð9Þ
where the average is again taken over all possible pairs ofgenomes. Note that all measures defined in equations(8–9) are positive and normalized to 1, 0 � Clf ;Cbf ;Ctot � 1: Value 1–Ctot is the average pairwise geneticdistance.
In our third numerical task, we checked the time de-pendence of linkage disequilibrium throughout the modeldynamics. Linkage disequilibrium was monitored via the2-site Lewontin measure D’(a, b) (Lewontin 1964) aver-aged at every given time over all pairs of sites separatedby a prescribed mutual distance d,D#5ÆD#ða; a1dÞæ:Herea and b5 a1 d, 1 � a; b � L; label 2 arbitrary sites of theviral genome separated by d intermediate positions. TheLewontin disequilibrium is calculated as follows. Iff11(a, b) denotes the probability that both site a and siteb contain less-fit alleles, f1(a) denotes the probability ofa less-fit allele at site a alone, and Dða; bÞ5f11ða;bÞ�f1ðaÞf1ðbÞ; then
FIG. 1.—Monte Carlo simulations of the evolution of genomes: (a) Time dependence of the average distribution of genomes with k less-fit mutations,f(k, t), for the listed parameters. The recombination rate per genome is r5 N/N0 5 0.5, where the characteristic population size is N0 5 104. The averagewas computed over 10 runs, with identical initial conditions, Ækæ5L� 1;modeling the initial state of an HIV population in untreated low-viremia patientsor at the beginning of antiretroviral therapy. Alternating colors label successive times in generations. (b) Time dependence of the average number of less-fitalleles Ækæ; the normalized reversion rate s�1dÆkæ=dt; the variance of k, Var[k], and the time dependence of the average numbers of lost and fixed less-fitalleles for the parameter set shown in (a). Color coding is shown. (c) Same as in (b), but for a smaller population size. Parameters are shown. Therecombination rate per genome is r 5 N/N0 5 0.3. (d) Variation with the population size N of the average mutation number Ækfinalæ of final clonesand of the width stdðÆkfinalæÞ of the corresponding distribution. L, s, and N0 are shown.
!
578 Gheorghiu-Svirschevski et al.
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
Sequence Correlation Limits Effect of Recombination 579
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
D#ða; bÞ5Dða;bÞDmax
; if Dða; bÞ.0Dða;bÞjDmin j
; if Dða; bÞ,0
(; ð10Þ
where Dmax 5 minff1(a)(1 � f1(b)), (1�f1(a))f1(b)g is themaximum value ofD(a, b) over all possible distributions ofalleles in genomes at fixed f1(a) and f1(b), and Dmin 5maxf�f1(a) f1(b),�(1� f1(a))(1� f1(b))g is the minimumof its absolute value.With the normalization shown in equa-tion (10), the total disequilibrium is bounded, as given byjD#ðdÞj � 1:
ResultsKinetics of Better-Fit Allele Accumulation
The typical observed evolution of the distributionf(k, t) for a sufficiently large population size N and randominitial distribution of alleles is shown in figure 1a (averageover 10 runs with an identical initial population). Parametervalues are defined in the figure. Figure 1b shows the cor-responding time dependence (averaged over 10 runs) of Ækæ;s�1jdÆkæ=dtj and Var[k], as well as the time-dependent num-bers of lost and fixed better-fit alleles, klost and kfix. The 3
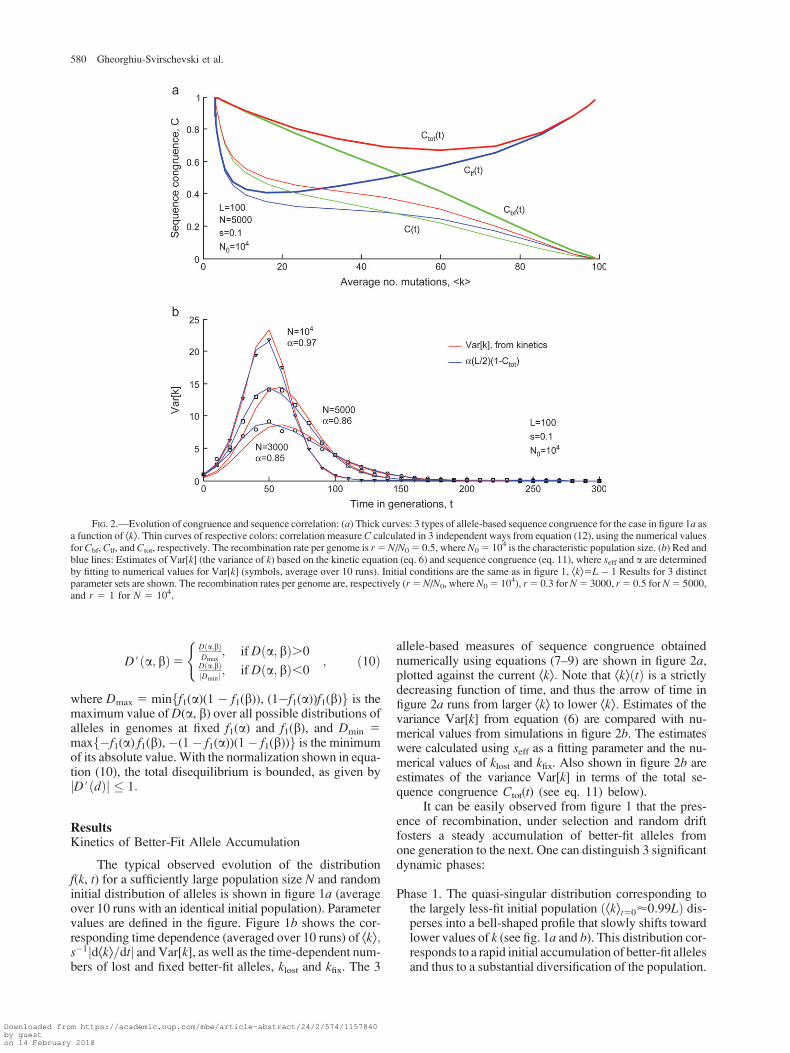
allele-based measures of sequence congruence obtainednumerically using equations (7–9) are shown in figure 2a,plotted against the current Ækæ: Note that ÆkæðtÞ is a strictlydecreasing function of time, and thus the arrow of time infigure 2a runs from larger Ækæ to lower Ækæ: Estimates of thevariance Var[k] from equation (6) are compared with nu-merical values from simulations in figure 2b. The estimateswere calculated using seff as a fitting parameter and the nu-merical values of klost and kfix. Also shown in figure 2b areestimates of the variance Var[k] in terms of the total se-quence congruence Ctot(t) (see eq. 11) below).
It can be easily observed from figure 1 that the pres-ence of recombination, under selection and random driftfosters a steady accumulation of better-fit alleles fromone generation to the next. One can distinguish 3 significantdynamic phases:
Phase 1. The quasi-singular distribution corresponding tothe largely less-fit initial population ðÆkæt50’0:99LÞ dis-perses into a bell-shaped profile that slowly shifts towardlower values of k (see fig. 1a and b). This distribution cor-responds to a rapid initial accumulation of better-fit allelesand thus to a substantial diversification of the population.
FIG. 2.—Evolution of congruence and sequence correlation: (a) Thick curves: 3 types of allele-based sequence congruence for the case in figure 1a asa function of Ækæ: Thin curves of respective colors: correlation measure C calculated in 3 independent ways from equation (12), using the numerical valuesfor Cbf, Clf, andCtot, respectively. The recombination rate per genome is r5N/N05 0.5, whereN05 104 is the characteristic population size. (b) Red andblue lines: Estimates of Var[k] (the variance of k) based on the kinetic equation (eq. 6) and sequence congruence (eq. 11), where seff and a are determinedby fitting to numerical values for Var[k] (symbols, average over 10 runs). Initial conditions are the same as in figure 1, Ækæ5L� 1 Results for 3 distinctparameter sets are shown. The recombination rates per genome are, respectively (r5N/N0, whereN05 104), r5 0.3 for N5 3000, r5 0.5 for N5 5000,and r 5 1 for N 5 104.
580 Gheorghiu-Svirschevski et al.
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
By the end of this transient period, the distribution f(k)develops a relatively large number of individual k-groups,most comprising a sizeable number of genomes.
Phase 2. The bell-shaped distribution continues to shift to-ward lower numbers of k, this time at a considerably ac-celerated but slowly varying rate (fig. 1a and b). Notably,the distribution profile varies only slightly during thisphase (fig. 1a, 40 � t � 100); that is, as pointed outin our earlier work (Rouzine et al. 2003; Rouzine andCoffin 2005), the propagation of f(k, t) can be approxi-mated as a solitary wave.
Phase 3. The remaining less-fit alleles continue to be elim-inated until (almost) completely purged. In other words,the better-fit alleles undergo gradual fixation, whereasthe bell-shaped profile of f(k) shrinks until it eventuallyreduces to a single stationary group at kfinal’0:
At lower population sizes, this general pattern is al-tered by the loss of better-fit alleles during Phase 2 (fig.1c). In this case, the ongoing accumulation of better-fit al-leles is limited by the diminished number of polymorphicsites. Beyond the solitary wave regime, the loss of better-fitalleles declines rapidly until it eventually halts, whereastheir fixation also begins at random available sites. As inthe case of larger N, the latter process continues until com-plete elimination of nonfixed less-fit alleles, with the resultthat the entire population is reduced finally to a clone, withkfinal.0 randomly positioned less-fit sites (fig. 1a). Thenumber kfinal of less-fit sites surviving in any particular finalclone samples a bell-shaped distribution centered on an av-erage position Ækfinalæ (results not shown). Figure 1d showsthat, for fixed recombination and selection rates, Ækfinalæ de-pends on the population size N. It decreases toward k 5 0with increasing N, whereas the width of its correspondingdistribution shrinks, such that in sufficiently large popula-tions the final clone displays invariably the best-fit genomethat can evolve (kfinal 5 0).
As was checked in all simulations that we have run, thekinetics of allele accumulation proves to be in excellentagreement with the general prescription of Fisher’s law,equation (3), that the speed of evolution is proportionalto the width of the distribution f(k, t) (see 2 examples infig. 1b and c). Figure 2b shows that we also find very goodagreement with the modified one-site kinetics of equation(6), based on the effective selection coefficient approach.The dependence of seff on N and other parameters is furtherdiscussed in the next section.
Sequence Congruence
The emergence of congruent sequence segmentsthroughout the population was followed by means of the3 measures defined in equations (7–9). A typical outputis shown in figure 2a, plotted against the current averagenumber of less-fit sites Ækæ:
The overall sequence congruence Ctot and the congru-ence of less-fit sites Clf both display a nonmonotonous de-pendence, with a rather shallowminimum betweenmaximalunit values at the beginning and at the end of the evolution(fig. 2a). Unit congruence corresponds to (almost) uniformfinal and initial populations. During most of the evolu-tion, the congruence of less-fit sites Clf decreases due to the
constant accumulation of better-fit alleles. The final increasein Clf is related to the shrinking of the distribution f(k, t)(fig. 1a) and the emergence of the final clone. The increaseof total congruence during the second half of evolutionwitnesses the increasing congruence of better-fit sites.
The congruence of better-fit sites Cbf increases monot-onously with Ækæ throughout the evolution, from an initialnull value for Ækæ’99 (f0 5 0.01, L 5 100) at t 5 0 tothe unit value marking the establishment of the final clonalpopulation. Interestingly, in the parameter range we tested,for example, N0 5 104, the time dependence of Cbf(t) is ap-proximately linear throughout the evolution. Although wedo not have a theoretical understanding of this feature, itis worth pointing out that the approximately linear depen-dence of Cbf can be exploited for early anticipation ofÆkfinalæ and therefore of the ratio of better-fit/less-fit sitesin the final clone.
Connection between the Genetic Distance and theVariance of the Fitness Distribution
The total congruence Ctot correlates closely with thevariance of the distribution f(k, t). Figure 2b shows thata proportionality of the form
Var½k�5 aL
2ð1� CtotÞ ð11Þ
holds with excellent accuracy over the entire range of pop-ulation sizes, with a a constant coefficient close to unit, a50.85–0.97. We note that Lð1� CtotÞ represents the averageHamming distance within the population. Relation (11) im-plies, then, that the rate of evolution, equation (3), is pro-portional to the average Hamming distance, as
dÆkædt
5 � saL
2ð1� CtotÞ: ð12Þ
The origin of equation (11) for a 5 1 can be understoodunder the assumption that the distribution of less-fit alleleson polymorphic sites within any pair of genomes is random,as it is the case, for example, when nonfixed alleles are ran-domly distributed in the population. To this end considerthe following expression for the variance Var[k]:
Var½k�5 Æðk � ÆkæÞ2æ5 1
2N2
XN
i; j5 1
ðki � kjÞ2; ð13Þ
where i and j label individual genomes. For each pair (i, j),let npoly denote the number of polymorphic sites betweenthe respective genomes and let nlf be the correspondingnumber of monomorphic mutant sites, so thatki1kj5npoly12nlf : Then each term ki � kj
� �2can be rewrit-
ten as 4 Dki � npoly=2� �2
; where Dki5ki � nlf denotes thenumber of polymorphic mutant sites in genome i. But be-cause mutations in pairs of polymorphic sites are distributedwith equal chance (1/2) between the 2 sites, npoly=2 is justthe average number of polymorphic mutations that can becontributed by genome i for fixed npoly, and one hasðki � kjÞ254ðDki � DkiÞ2: Further, if Dki does not correlatebetween genomes, for example, less-fit alleles for a fixednumber of polymorphic sites are distributed randomly
Sequence Correlation Limits Effect of Recombination 581
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
between the 2 genomes, from the binomial distribution wehave, on average, ÆðDki � DkiÞ2æ5Dki=25npoly=4: By def-inition npoly5L 1� Ctotði; jÞð Þ; and from equation (13), itfollows that Var½k�5 L
21� Ctotð Þ: Correlations between
the numbers Dki of polymorphic mutations, associated withcorrelations between fitness values of different genomes[Rouzine and Coffin 2005], lead to ðDki � DkiÞ2,Dki=2;which accounts for a,1 in equation (11). We note that
in the limit of large N, when L21� Ctotð Þ’Ækæ 1� Ækæ
L
� �;
coefficient a approaches the ratio p 5 seff/s, as definedin Rouzine and Coffin (2005), a’p; whereas at lower N,equation (11) still holds, but a 6¼ p (see fig. 2b).
Linkage Disequilibrium
The initial population has a random allele makeup,with null pairwise linkage disequilibrium as defined byequation (10). Representative tests of pairwise linkage dis-equilibrium at later times are shown in figure 3 for 2 typical
FIG. 3.—Evolution of linkage disequilibrium: (a) Sample calculation of linkage disequilibrium for pairs of sites d sites apart as a function of theaverage number of less-fit mutations, Ækæ: Parameters are shown. The recombination rate per genome is r5N/N05 0.3, whereN05 104. (b) Same as in (a)for a different set of parameters. The recombination rate is r 5 N/N0 5 0.3.
582 Gheorghiu-Svirschevski et al.
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
sets of model parameters and various linkage distancesbetween sites of a pair, d. The quantity D’(d), calculatedaccording to equation (10), exhibits only small, statisticallyinsignificant fluctuations from a null value, irrespectiveof the distance d. In particular, this result shows that thepositions of alleles of a specific type (beneficial or delete-rious) do not correlate along genomes, although alleles(either deleterious or beneficial) do correlate betweenhomologous sites on different genomes (across the popula-tion), as described in the previous paragraph. We note inthis regard that the experimental observation of linkage dis-equilibrium can be attributed to random mutation eventsthat generate new beneficial alleles (Rouzine and Coffin1999), as well as to epistasis. In the present model, epistasisis absent, and all the alleles are assumed to preexist.
Dependence of the Evolution Rate and the SequenceCongruence on N and Other Parameters
As documented in figure 2b, the analytic approachbased on the effective selection coefficient seff, equation(6), provides a good fit to the simulated results for the kinet-ics of Var[k] and Ækæ: As can be seen in figure 4, for a fixedcharacteristic population size N0 and raw selection coeffi-cient s, the average over all runs of the ratio seff/s shows a log-arithmic increase with the population size N and reaches anupper plateau at the deterministic value of 1 for large enoughN. In this limit, recombination is so effective that sites that
are statistically independent at the onset evolve indepen-dently henceforth. At lower population sizes, seff/s alsoshows a tendency toward a plateau. Closer examination re-veals that the onset of this lower plateau correlates with thegradual disappearance of simulation runs that end in thepopulation with maximum fitness, Ækæ 5 0 (i.e., for a smallenough population size, the final clones start to include withcertainty some less-fit alleles). To emphasize this feature, wecalculated, in addition, a fractional ratio s#eff=s; defined asthe contribution to the average seff/s arising exclusively fromthe fraction of runs arriving at the best-fit population (fig. 4).Whereas at sufficiently large N the fractional ratio s#eff=s isidentical to seff/s, at the lower population sizes, where runsthat end in the best-fit population gradually disappear, thefraction displays an abrupt drop and reaches a null valueat some critical population size Nc. This behavior may beregarded as a phase transition from all-best-fit final clonesto partially better-fit final clones.
We observe also that the increase of seff/s with popu-lation size N is closely matched by a similar decrease insequence congruence, as given by seff=s;1� Cmin
lf (fig. 4).Furthermore, the ratio sa/2 of the evolution rate to the av-erage Hamming distance L(1 � Ctot) in equation (12) de-pends only weakly on the population size (result not shown).
Finally, we note that our results do not greatly dependon the number of crossovers, M, per genome per recombi-nation event. Although the majority of our simulations useda valueM5 10, we verified that the dependence of seff on N
FIG. 4.—Effective selection coefficient versus population size: (a) Dependence of the normalized effective selection coefficient, seff=s; on populationsize (averages over 10–20 runs) for listed parameters. Blue curve: seff=s averaged over all runs. Dotted cyan curve: analogous parameter s#eff=s; includingonly runs that end in best-fit genomes ðÆkæt/N50Þ: Red curve: complement of the minimum (in time) less-fit allele congruence, 1� Cmin
lf : The recom-bination rate per genome varies with N as r 5 N/N0. (b) Same as in (a), for smaller genome size (left) or weaker selection (right).
Sequence Correlation Limits Effect of Recombination 583
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
does not change much in the limitM/N: For instance, fora rather high recombination rate, N0 5 103 and long ge-nomes, L 5 1000, an increase in the number of crossoversproduced a shift in the critical population size fromNc’300; for M 5 10, to Nc’200; for M/N:
DiscussionEvolution Rate versus Accumulation of SequenceCorrelation
A previous approach (Rouzine and Coffin 2005) to thepresent selection/recombination model considered onlycorrelations between the number k of less-fit alleles in dif-ferent genomes but neglected allele correlations at homol-ogous sites. Under this approximation, it was found that therate of evolution was described by the effective selectioncoefficient approach, equations (3 and 5a), with coefficients#eff given by equation (5b). The ratio s#eff=s was found tohave a threshold in the population size at Nc;N
1=20 ; equa-
tion (5b), below which evolution stopped shortly after itsonset ðs#eff=s50Þ:
The present Monte Carlo results confirm the existenceof a threshold inN for s#eff=s (fig. 4). Also, for fixedN0 and s,the fractional ratio s#eff=s shows an increase with populationsize. There are, however, a number of differences betweenthe previous approximation and the present simulations.First, the transition between s#eff50 and s#eff5s occurs ina more narrow interval of lnN. Second, the value of the crit-ical population size Nc is higher than;N
1=20 : Third, the cur-
rent simulations indicate that the accumulation of pairwisecongruence, in excess of what one would expect from a uni-formly random distribution of alleles, plays an essential rolein the evolution of a viral population (fig. 2a). At the sametime, pairwise linkage disequilibrium between sites withinthe same genome does not emerge (fig. 3).
A qualitative understanding of correlation buildup canbe gained on noticing that 2 genomes must correlate if theyshare segments originating in an identical ancestral clone.To quantify this effect, let us introduce the ‘‘identity by de-scent’’ C of a pair of sequences, as the fraction of sites thathave a common ancestor, that is, are identical by descent.Two homologous sites in a pair of sequences descend fromeither 2 different genomes in the initial population or thesame genome. In the second case, a site belongs to one ofC�L congruent sites. Notice that, in the absence of mutation(as is the case in our model), sites identical by descent nec-essarily carry identical alleles, but the opposite is not true.
Each of the observable congruences defined in equa-tions (7–9) can be related in a simple fashion to the averageidentity by descent with respect to the initial population, C,under the approximation that the positions of alleles in non-congruent segments of genomes do not correlate. Indeed, ifwe denote f5Ækæ=L; then one has, respectively,
1� Clf 5 ð1� CÞ 1� f2
1� ð1� f Þ2�
; ð14aÞ
1� Cbf 5 ð1� CÞ 1� ð1� f Þ2
1� f2
� ; ð14bÞ
and
1� Ctot 5 ð1� CÞð1� f2 � ð1� f Þ2Þ: ð14cÞ
In our model, we expect C to increase monotonously intime, from a vanishing value, Cðt50Þ’0; for the uncorre-lated initial population at f’1; to some finite valueCðt/NÞ � 1; for the final clone at f 5 0. With this con-jecture, it can be shown from equation (12) that the se-quence congruences will have most of the qualitativefeatures (monotony, minima, asymptotic trend at f 5 0and f 5 1) displayed in figure 2a.
One can use equation (14) to compare values of theidentity by descent C obtained in 3 independent ways. Infigure 2a, we show the 3 values of C calculated in thisway and numerical values for Clf, Cbf, and Ctot for a param-eter value set near the transition point in N, where seff/s ;0.5. We observe that 1) the 3 curves for C are close, whichconfirms the assumption that sites not identical by descentdo not correlate and 2) the average value of C in the intervalof f is not small, demonstrating the connection between se-quence correlation and the kinetic properties of population.We note that, at small f, Clf can be used as a crude approx-imation for C, equation (14a).
The effective selection coefficient can be expressed interms of the identity by descent C. It is sufficient to note thatsites identical by descent (and therefore having identical al-leles) do not contribute, on average, to the variance Var[k]of the number of deleterious mutations becauseVar½k�51=2N2
Pi;jðki � kjÞ2: Hence, for a population size
N sufficiently large to avoid any loss of better-fit alleles andneglecting correlation in k between genomes (Rouzine andCoffin 2005), one can write
Var½k�’ð1� CÞÆkæ 1� ÆkæL
� �: ð15Þ
Note that equations (15 and 14c) produce equation (12) fora 5 1. This interpretation is essentially based on the as-sumption that partial correlation between 2 genomes at in-dividual sites does not occur: sites either have a commonancestor or do not correlate at all. In this regime, equation(15) and equation (5a) allow one to identify
seffs’ 1� C: ð14Þ
Furthermore, as the population size increases, so does thenumber of clones represented in the population, whereas thefrequency of genomes belonging to any given clonedecreases. This relationship means that the accumulationof identity by descent decreases, and therefore, C decreasesmonotonously with N. Thus, equation (14) implies, in turn,that seff increases monotonously with N, in agreement withour numerical observations.
Relevance for HIV Populations
Our choice of numerical values for the model param-eters (L 5 10–100, N , 105, N0 5 103 to 105, s 5 10�2 to10�1, initial frequency of better-fit alleles ;10�2) was tai-lored for a quantitative understanding of long-term HIV
584 Gheorghiu-Svirschevski et al.
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
evolution, particularly for the reversion of deleterious al-leles in untreated low-viremia patients. Our results are alsorelevant for improvement of the existing antiviral therapytargeted at delay of emergence of drug-resistant variants.In this case, the initial state corresponds to the small viruspopulation surviving drug depletion and harboring a verylow number of better-fit, drug-resistant alleles (frequency; 10�2). The subsequent evolution at constant populationsize models the initial period of accumulation of drug-resistant alleles at a low viral load, before the onset of un-controlled rebound.
Current drug cocktails that achieve . 99.99% virusdepletion target a small number of sites in HIV genome.The level of resistance (fitness increase) sufficient for virusreplication under drug is not evident from the level of de-pletion and has to be estimated from a population dynamicsmodel. A recent work of one of us (Rouzine et al. 2006)estimates that the decrease of the wild-type virus fitnessdue to drug by a factor of 10–20 may be sufficient toachieve such a deep level of depletion. Because L ; 2–3mutations enable virus to replicate effectively under drug,the selection coefficient under therapy is large for these crit-ical mutations, s 5 ln(10–20)/L 5 0.7–1.5. Thus, the pres-ent simulations, using large L and small s, do not apply toevolution of the critical resistance mutations under the cur-rent therapy regiments. As shown in Rouzine and Coffin(2005) using a separate argument, in this case, resistant mu-tations become fixed in the surviving population withina few generations, unless the effective population size isextremely low, Nc , 30–45.
On the other hand, both the analytic results (Rouzineand Coffin 2005) and the present simulations suggest thatsuch drastic depletion levels are not necessary for success-ful control of resistance development. According to ourmodel, if the number of drug target sites is increased to,for example, L5 10 (which somewhat exceeds the numberof currently known drug targets), we predict that the re-bound of resistant strains will require much larger popula-tion sizes. The selection coefficient per critical resistant site,in this case, is smaller, s 5 ln(10–20)/L 5 0.2–0.3, and thepresent theory applies approximately. Our previous esti-mate of the critical population size is Nc’N
1=20 (Rouzine
and Coffin 2005). The present numerical results, figure4, show that the actual critical population size, below whichcompletely resistant strains do not materialize with cer-tainty, is considerably larger, as given by the empiricalrelationship Nc ; (15–30) N
1=20 for N0 5 103 to 105, L
5 10–100, and s 5 0.1. Using the only available estimateof recombination rate r 5 N/N0 ; 1 (Jung et al. 2002) andthe estimate of population size in an average patient N ;106 (Rouzine and Coffin 1999), we obtain N0 ; 106 and Nc
; 104. The value of r may be overestimated because mostcoinfecting virions may originate from nearby cells and begenetically uniform. The estimate of N also has to be up-dated to account for a higher number of crossovers thanthought previously and for the multisite nature of evolution(the quoted estimate was based on a 2-site model). How-ever, even assuming that the value of N0 is 2 orders of mag-nitude smaller, that is, N0 5 104, we still obtain Nc ; 1000(fig. 4), much larger than the critical depletion level for cur-rently used drug cocktails, Nc ; 30.
In summary, our current results strongly reinforce theconclusion set forth in the previous study (Rouzine andCoffin 2005) that successful long-term antiretroviral ther-apy could be achieved with significantly reduced dosage,provided the number of target sites is sufficiently large.
Acknowledgments
This work was supported by grants K25 AI 01811 toI.R. and R01 CA 089441 to J.M.C. S.G. was supported bytraining grant T32 AI007389. J.M.C. was an AmericanCancer Society Research Professor with support from theGeorge Kirby Foundation.
Literature Cited
Aranson I, Tsimring L, Vinokur V. 1997. Evolution on a ruggedlandscape: pinning and aging. Phys Rev Lett. 79:3298.
Barton NH. 1995a. A general model for the evolution of recom-bination. Genet Res. 65:123–144.
Barton NH. 1995b. Linkage and the limits to natural selection.Genetics. 140:821–841.
Burger R. 1999. Evolution of genetic variability and the advantageof sex and recombination in changing environments. Genetics.153:1055–1069.
Burt A. 2000. Perspective: sex, recombination, and the efficacy ofselection—was Weismann right? Evol Int J Org Evol. 54:337–351.
Chun TW, Carruth L, Finzi D, et al. [15 co-authors]. 1997. Quan-tification of latent tissue reservoirs and total body viral load inHIV-1 infection. Nature. 387:183–188.
Crow JF, Kimura M. 1965. Evolution in sexual and asexual pop-ulations. Am Nat. 9:439–450.
Dang Q, Chen J, Unutmaz D, Coffin JM, Pathak VK, Powell D,KewalRamani VN, Maldarelli F, Hu WS. 2004. NonrandomHIV-1 infection and double infection via direct and cell-mediated pathways. Proc Natl Acad Sci USA. 101:632–637.
Felsenstein J. 1974. The evolutionary advantage of recombination.Genetics. 78:737–756.
Fisher RA. 1930. The genetical theory of natural selection.Oxford: Oxford University Press.
Frost SD, Nijhuis M, Schuurman R, Boucher CA, Brown AJ.2000. Evolution of lamivudine resistance in human immuno-deficiency virus type 1-infected individuals: the relative rolesof drift and selection. J Virol. 74:6262–6268.
Haigh J. 1978. The accumulation of deleterious genes in apopulation—Muller’s ratchet. Theor Popul Biol. 14:251–267.
Hey J. 1998. Selfish genes, pleiotropy and the origin of recombi-nation. Genetics. 149:2089–2097.
Hill WG, Robertson A. 1966. The effect of linkage on the limits toartificial selection. Genet Res. 8:269–294.
Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U,Meese E, Wain-Hobson S, Meyerhans A. 2002. Multiplyinfected spleen cells in HIV patients. Nature. 418:144.
Kessler DA, Ridgway D, Levine H, Tsimring L. 1997. Evolutionon a smooth landscape. J Stat Phys. 87:519.
Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamicsof HIV-1 recombination in its natural target cells. Proc NatlAcad Sci USA. 101:4204–4209.
Lewontin RC. 1964. The interaction of selection and linkage. I.General considerations; heterotic models. Genetics. 49:49–67.
Maynard-Smith J. 1968. Evolution in sexual and asexual popula-tions. Am Nat. 102:469–473.
Muller HJ. 1932. Some genetic aspects of sex. Am Nat. 66:118–138.
Sequence Correlation Limits Effect of Recombination 585
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018
Otto SP, Lenormand T. 2002. Resolving the paradox of sex andrecombination. Nat Rev Genet. 3:252–261.
Ridgway D, Levine H, Kessler DA. 1998. Evolution on a smoothlandscape: the role of bias. J Stat Phys. 90:191.
Rouzine I, Wakeley J, Coffin J. 2003. The solitary wave of asexualevolution. Proc Natl Acad Sci USA. 100:587–592.
Rouzine IM, Coffin JM. 1999. Linkage disequilibrium test impliesa large effective population number for HIV in vivo. Proc NatlAcad Sci USA. 96:10758–10763.
Rouzine IM, Coffin JM. 2005. Evolution of human immunodefi-ciency virus under selection and weak recombination. Genet-ics. 170:7–18.
Rouzine IM, Sergeev RA, Glushtsov AI. 2006. Two types ofcytotoxic lymphocyte regulation explain kinetics of immuneresponse to human immunodeficiency virus. Proc Natl AcadSci USA. 103:666–671.
Tsimring LS, Levine H, Kessler DA. 1996. RNA virus evolutionvia a fitness-space model. Phys Rev Lett. 76:4440.
Pekka Pamilo, Associate Editor
Accepted November 21, 2006
586 Gheorghiu-Svirschevski et al.
Downloaded from https://academic.oup.com/mbe/article-abstract/24/2/574/1157840by gueston 14 February 2018