Page 1
08/03/2011
1
Inherited Eye Disease
Michel MichaelidesMoorfields Eye Hospital &
UCL Institute of Ophthalmology
Single gene disorders affecting the retina
Inherited retinal dystrophies• clinically heterogeneous• variable visual loss • bilateral symmetrical retinal
abnormalities• AD, AR, XL & mitochondrial
inheritance • considerable heterogeneity even
within these subtypes
Inherited retinal dystrophies• phenotypically variable
•onset•rate of progression•severity• fundus appearance•fundus autofluorescence•electrophysiology•psychophysics
Page 2
08/03/2011
2
Inherited retinal diseases
• stationary versus progressive• predominant rod vs. cone• macular vs. periphery• structural developmental disorders
Disorders• Retinitis Pigmentosa *
– progressive rod photoreceptor demise• Cone dystrophy / Cone-Rod dystrophy• Achromatopsia *
– Stationary cone dysfunction• Congenital night blindness *
– Stationary rod dysfunction• Macular Dystrophies
– Best Disease
a-wave
a-wave
b-wave
b-wave
PERG
P50
N95
EOG
ERG
PERG
Electrophysiology and retinal structure
Standard full-field ERGs
No significant contribution from the macula
Dark-adapted Light-adapted
Page 3
08/03/2011
3
Pattern Electroretinography
contrast response
reflects macularfunction
no significant contribution fromthe peripheral retina
P50
N95
Inheritance• Autosomal dominant• Autosomal recessive• X-linked recessive• Mitochondrial (16,569bp)
• X-linked dominant (incontinentia pigmenti, Aicardi)• Digenic (bi- or triallelic)
– RDS +/- & ROM1 +/- -> RP (Kajiwara, Dryja Science 1994) – Bardet-Biedl Syndrome (Katsanis Science 2001)
• Rules– If male to male not X-linked– If male to anyone not mitochondrial
Number of single gene disorders affecting retinal function
http://www.retnet.org
• 46 year old male• Night blindness since teens• Mainstream education• Driving until 20s• Progressive loss of field• Family history of vision loss (mother,
maternal uncle, maternal grand-father)
Page 5
08/03/2011
5
Autosomal Dominant RP
• Rhodopsin gene (RP4) chromosome 3q• Mutation
– c.1040c->t– p.Pro347Leu
Rod opsin – 25000 /µm membrane, 108 / outer segment
• Night-blindness• Fit and well, no medication history
• Acuities 6/6 6/6 (emmetrope)• Mild restriction of visual field
14 year old boy
Page 6
08/03/2011
6
IV:8
III:6III:5
IV:9 IV:10
II:3 II:4
III:2 III:4III:3
IV:6 IV:7
III:1
IV:1 IV:5IV:4IV:3
V:2
IV:2
V:1
II:5 II:6
I:1 I:2
II:1 II:2
14 YRS
glaucoma
Page 7
08/03/2011
7
IV:10
III:8III:7
IV:11 IV:12
II:4 II:5
III:4 III:6III:5
IV:8 IV:9
III:3
IV:3 IV:7IV:6IV:5
V:3
IV:4
V:2
II:6 II:7
I:1 I:2
II:2 II:3II:1
III:2III:1
IV:2IV:1
V:1
14 YRS
IVS 6 + 3 A>G
Exon 6
Splicing factor PRPF31
RP11• 19q ADRP Vithana et al. (2001) PRPF31• one of four splicing genes causative of
ADRP –– PRPF8 – RP13, 17p– PRPF3 – RP18, 1q– Pim-1 kinase Activating protein – RP9, 7p
• non-penetrance of carriers is due to allele from non-affected parent.
ADRP genes• rod opsin - RP4 - 3q• NRL - RP27 14q #• PRPF8 - RP13 - 17p• PRPF31 - RP11 - 19q *• PRPF3 - RP18 - 1q• PAP1 - RP9 - 7p *• IMPDH1 - RP10 - 7q • NR2E3 – 15q #• RDH12 – 14q #• TOPORS – RP31 – 9q• RDS - RP7 - 6p • ORP1 - RP1 8q *
Red – rod specific expressionBlue – splicing factorDark blue – other ubiquitously expressed geneGreen – expressed in both rods and cones, rods more susceptible to mutation# - other alleles cause recessive disease
http://www.retnet.org
Page 8
08/03/2011
8
Non-syndromic ARRP genes
– USH2A– NR2E3– PDE6A– PDE6B– LRAT– MERTK – central macular hyperfluorescent plaque– CNGA1– CNGB1– CERKL– RLBP1 – white dots when early, gyrate-like appearance later– RGR– RDH12– SAG– TULP1– AIPL1– CRB1 – perivascular sparing, retinal thickening and RPE pigment– GUCY2D - severe, photophobia common, preserved retinal appearance– RPE65 - absent retinal autofluorescence– RPGRIP1– ABCA4 - starts with macular dystrophy– CYP4V2 - intraretinal crystals– NRL – similar to NR2E3 disease– SPATA7– CEP290– LCA5– EYS– IDH3B– PCDH21
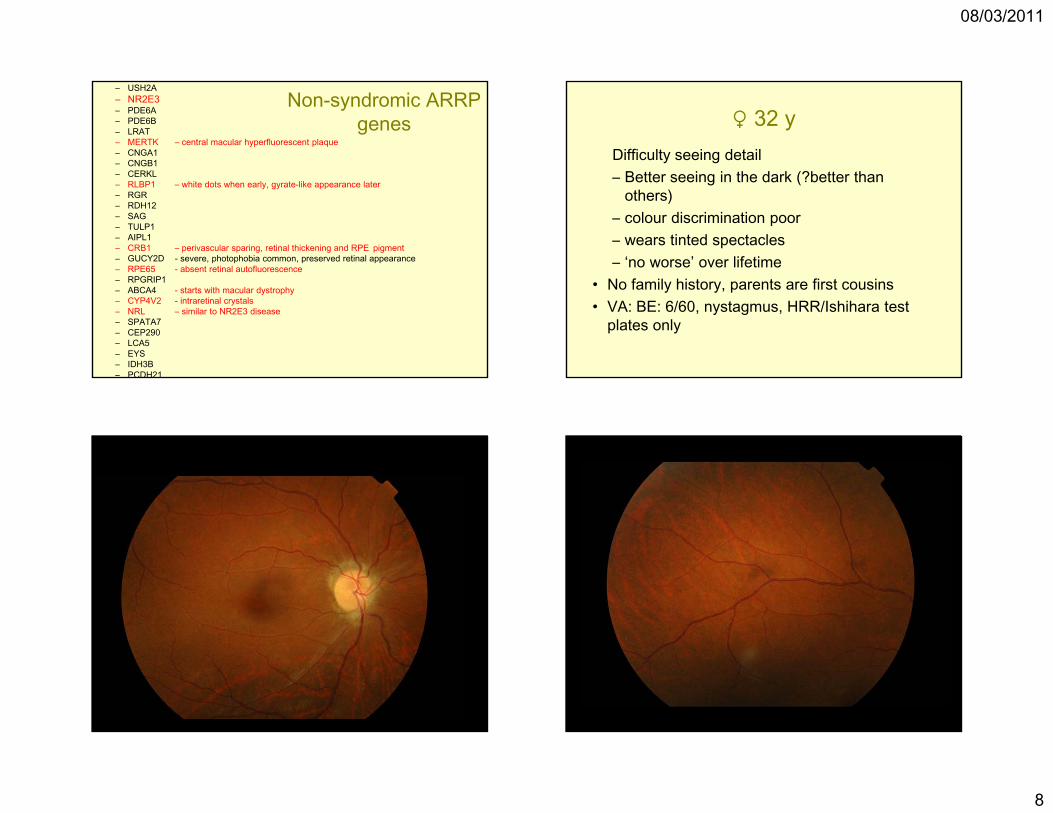
♀ 32 yDifficulty seeing detail– Better seeing in the dark (?better than
others)– colour discrimination poor– wears tinted spectacles– ‘no worse’ over lifetime
• No family history, parents are first cousins• VA: BE: 6/60, nystagmus, HRR/Ishihara test
plates only
Page 10
08/03/2011
10
RE
LE
N
55586
dark -adapted light -adapted Molecular Genetics
CNGA3 (2q11) : cone -subunit of the cGMP-gated (CNG) cation channel
CNGB3 (8q21-q22) : cone -subunit of the CNG cation channel
GNAT2 (1p13) : cone -subunit of transducin
Chromosome 14 (isodisomy) CNGB3
PDE6C (10q24) : cone -subunit of cGMP-phosphodiesterase (PDE)
Inherited macular dystrophies• progressive central visual loss• clinically heterogeneous• variable severity• bilateral symmetrical macular
abnormalities• AD, AR, XL & mitochondrial
inheritance • dysfunction not always limited to
macula
Page 11
08/03/2011
11
Stargardt disease• AR retinal dystrophy• macular atrophy• white flecks at level of RPE• abnormal autofluorescent material
in RPE• lipofuscin accumulation in RPE• abnormal pattern ERG• normal / cone / cone-rod
ffERG
Stargardt disease
Age 14
VA 6/60 6/36
Page 12
08/03/2011
12
10 year brother
VA 6/18 6/18
AF imaging in STGD disease
ABCA4 gene• Locus 1p21• 50 exons• highly polymorphic• expressed in rod and cone
photoreceptors• encodes ABC transporter protein
involved in removing all-trans retinal from OS discs
Allikmets et al Nat Genet 1997;17:8269-81
ABCA4 mutations and retinal disease
nullnull
null
normal
RP/CORD
STGD
STGD null
ENVIRONMENT
AMDmissense
Page 13
08/03/2011
13
A2E formation in RPE
Sparrow, Janet R. (2003) Proc. Natl. Acad. Sci. USA 100, 4353-4354
A2E is major fluorophore of lipofuscin
Stargardt disease
Associated with accumulation of A2E in RPE
High levels of A2E :-damages cell membranesaffects lysosomal functionresults in release of pro-apoptotic proteins from mitochondria
Sparrow, Janet R. (2003) Proc. Natl. Acad. Sci. USA 100, 4353-4354
Therapy in mouse modelSlow visual cycle:limit light exposureisotretinoin (13-cis retinoic acid)
Page 14
08/03/2011
14
Travis et al. (2007) Annu. Rev. Pharmacol. Toxicol. 47, 469-512
Fenretinide
Stargardt disease• primary abnormality is in photoreceptors
• defective ATP dependent transport mechanism
• leads to accumulation of A2E and lipofuscin in RPE
• secondary photoreceptor atrophy
Therapeutic agents may be targeted at the ATP dependent transport mechanism, slowing visual cycle or A2E formation
Subject OS• age 5• nyctalopia from birth• Light staring from 2/52• Squint at 6/12• Poor vision noted from 10/12• Decreased VF from 1 yr• No nystagmus
Page 15
08/03/2011
15
Lancelot: 2001
Acland G. M et al., 2001. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28, 92-95.
RPE65… is a candidate for gene
therapy
Gene therapy in patients with RPE65 mutations 2008
Maguire et al – NEJM May 2008Bainbridge et al - NEJM May 2008Cideciyan et al – PNAS Sept 2008
RPE65 trials – comparisons
• Subjective improvement in dark vision, 1 – 2 weeks, in two studies
• Improved visual function in 7/9 patients.• ERG, retinal structure (AF OCT)
unchanged• One significant complication – macular
hole (Maguire et al)
Visually-guided mobility: Subject #3; 6 months following surgery
Page 16
08/03/2011
16
Gene therapy future• RPE65
– Longer follow up– Younger patients
• RPE disease– MERTK, BEST1, REP1, LRAT, CYP4V2,
RLBP1• Photoreceptor disease• Dominant disease (knock-down replacement
strategy)• Assessing efficacy in a timely fashion is a
significant challenge
Inherited Retinal Dystrophies• wide heterogeneity
• therapy may be possible in progressive disorders
• novel therapies will be directed at patients with known genotype
• knowledge of disease mechanisms will dictate therapeutic approaches
• disorders must be well characterised
• natural history must be known