Inhibition of Mammalian Target of Rapamycin AugmentsLipopolysaccharide-Induced Lung Injury and Apoptosis
Jill A. Fielhaber,* Scott F. Carroll,† Anders B. Dydensborg,‡ Mitra Shourian,†
Alexandra Triantafillopoulos,* Sharon Harel,* Sabah N. Hussain,*,x Maxime Bouchard,‡
Salman T. Qureshi,†,1 and Arnold S. Kristof*,x,1
Acute lung injury during bacterial infection is associated with neutrophilic inflammation, epithelial cell apoptosis, and disruption of
the alveolar-capillary barrier. TLR4 is required for lung injury in animals exposed to bacterial LPS and initiates proinflammatory
responses in part via the transcription factor NF-kB. Ligation of TLR4 also initiates a proapoptotic response by activating IFN-b
and STAT1-dependent genes. We recently demonstrated that mammalian target of rapamycin (mTOR), a key controller of cell
growth and survival, can physically interact with STAT1 and suppress the induction of STAT1-dependent apoptosis genes. We
therefore hypothesized that the mTOR inhibitor rapamycin would increase LPS-induced apoptosis and lung injury in vivo.
Rapamycin increased lung injury and cellular apoptosis in C57BL/6J mice exposed to intratracheal LPS for 24 h. Rapamycin
also augmented STAT1 activation, and the induction of STAT1-dependent genes that mediate cellular apoptosis (i.e., Fas, caspase-
3). LPS-induced lung injury was attenuated in STAT1 knockout mice. In addition, LPS and IFN-b–induced apoptosis was absent
in cultured cells lacking STAT1, and, unlike in wild-type cells, a permissive effect of rapamycin was not observed. In contrast to its
effect on STAT1, rapamycin inhibited NF-kB activation in vivo and reduced selected markers of inflammation (i.e., neutrophils in
the bronchoalveolar lavage fluid, TNF-a). Therefore, although it inhibits NF-kB and neutrophilic inflammation, rapamycin
augments LPS-induced lung injury and apoptosis in a mechanism that involves STAT1 and the induction of STAT1-dependent
apoptosis genes. The Journal of Immunology, 2012, 188: 000–000.
Acute lung injury in patients with viral or bacterialpneumonia accounts for significant morbidity and mor-tality in hospitalized patients. The pathogenesis of acute
lung injury is complex and involves the recruitment of neutrophils,the elaboration of inflammatory cytokines, and apoptosis of epi-thelial cells. Together, these lead to disruption of the alveolarepithelial barrier, pulmonary edema, and abnormalities in gasexchange (1).In animal models of Gram-negative sepsis or pneumonia, the
initiation of lung injury requires ligation of the TLR4 by bacterialLPS and the induction of proinflammatory and proapoptotictranscriptional programs (2). Known signal transduction pathwaysdownstream of TLR4 include myeloid differentiation primaryresponse gene 88 (MyD88)-dependent and independent signalingintermediates (3). Via MyD88, TLR4 activates the transcriptionfactor NF-kB and the transcription of proinflammatory and sur-
vival genes. However, TLR4 also triggers the MyD88-independentactivation of IFN regulatory factor 3 (IRF-3), a transcription factor
that rapidly induces the production of IFN-b (IFN-b). IFN-b
then induces apoptosis and antimicrobial genes in a feed-forward
autocrine-paracrine signaling pathway that requires the tran-
scription factor STAT1. STAT1 is required for the synthesis of
several key apoptosis regulators that mediate lung injury, includ-
ing CD95/Fas and inducible NO synthase (1, 4, 5). Genetically
engineered mice with defects in IFN-b (6) or STAT1 (7) exhibit
deficient antibacterial innate immune responses.Consistent with an important role for apoptosis in acute lung
injury, signaling pathways that regulate cell survival and prolif-
eration have been implicated in the genesis of acute lung injury
(e.g., PI3K, NF-kB, p53) (8–10). One such signaling intermediate
is mammalian target of rapamycin (mTOR), a highly conserved
and ubiquitously expressed controller of cell growth, proliferation,
and survival (11). mTOR senses growth or metabolic signals (e.g.,
ATP, oxygen, amino acids, glucose, reactive oxidant species) and
exerts anabolic effects by stimulating protein synthesis and ribo-
somal biogenesis, enhancing cell proliferation, and promoting cell
survival (12). Rapamycin (i.e., sirolimus [Rapamune]) is a specific
inhibitor of mTOR and is a commonly used pharmacologic tool
for the study of mTOR biology. In addition, rapamycin is ap-
proved by the U.S. Food and Drug Administration for immuno-
suppression in transplant patients, cancer chemotherapy, and local
prevention of coronary artery stent thrombosis (13). In this study,
we used rapamycin to dissect the relative contribution of mTOR
during the evolution of LPS-induced lung injury.In regard to protein synthesis and cell growth, known mTOR
effectors include p70 S6 kinase (S6K), 4E-BP1, and Akt; however,
mTOR can also modify gene transcription by directly regulating
transcription factors (11). For example, by stimulating NF-kB,
mTOR potentiates cell survival responses (14). We recently iden-
*Critical Care Division, Department of Medicine, McGill University Health Centre–Royal Victoria Hospital, Montreal, Quebec H3A 1A1, Canada; †Centre for the Studyof Host Resistance, McGill University, McGill University Health Centre–MontrealGeneral Hospital, Montreal, Quebec H3G 1A4, Canada; ‡Biochemistry Department,Goodman Cancer Centre, McGill University, Montreal, Quebec H3A 1A3, Canada;and xMeakins-Christie Laboratories, Respiratory Division, Department of Medicine,McGill University, Montreal, Quebec H2X 2P2, Canada
1S.T.Q. and A.S.K. contributed equally to this work.
Received for publication November 3, 2010. Accepted for publication February 21,2012.
This work was supported by Canadian Institutes of Health Research Operating GrantsMOP-81259 (to S.T.Q.) and MOP-69007 (to A.S.K.).
Address correspondence and reprint requests to Dr. Arnold S. Kristof, McGill Uni-versity Health Centre, Royal Victoria Hospital, 687 Pine Avenue West, L3.05, Mon-treal, QC, Canada H3A 1A1. E-mail address: [email protected]
Abbreviations used in this article: BAL, bronchoalveolar lavage; EB, Evans Blue; IRF,IFN regulatory factor; mTOR, mammalian target of rapamycin; S6K, p70 S6 kinase.
Copyright� 2012 by The American Association of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1003655
Published March 26, 2012, doi:10.4049/jimmunol.1003655 by guest on M
tified a physical and functional interaction between mTOR andSTAT1 (15). In intact cells, inhibition of mTOR kinase activitylead to increased STAT1 nuclear content and transcription of IFN-sensitive apoptosis genes (16). Based on these studies, we hy-pothesized that mTOR blockade with rapamycin would augmentLPS-induced injury and apoptosis in the lung, as well as theSTAT1 transcriptional response in vivo. In this study, we show thatrapamycin increases acute lung injury and apoptosis in mice ex-posed to LPS. Increased injury and cell death were associated withelevated levels of activated STAT1 and STAT1-dependent apo-ptosis genes, but reduced NF-kB activation and markers of in-flammation (e.g., neutrophils and TNF-a in the bronchoalveolarlavage fluid). In vivo and in cultured cells, STAT1 was required forLPS-induced lung injury and apoptosis, and the proapoptotic ef-fect of rapamycin was absent in STAT1-deficient cells. Our find-ings indicate that mTOR is an endogenous suppressor of STAT1,proapoptotic responses, and lung injury, independent of NF-kB–mediated proinflammatory responses.
Materials and MethodsMaterials
STAT1-deficient (U3A) cells and their wild-type control (2fTGH) wereobtained from Dr. G. Stark (Cleveland Clinic) and propagated as describedpreviously (17). Escherichia coli O55:B55 LPS was purchased fromSigma-Aldrich. IFN-b was obtained from PBL IFNSource. Rapamycinwas purchased from Biomol. Hexadecyltrimethylammonium bromide ando-dianisidine dihydrochloride were purchased from Sigma-Aldrich. Absagainst phospho-p70 S6 kinase (pS6K Thr 389), p70 S6 kinase (S6K),phospho-S6 (Ser 235/236), phospho-p65 (Ser 536), IkBa, S6, and cleavedcaspase-3 were purchased from Cell Signaling Technology. The Ab againstphospho-STAT1 (pSTAT1 Ser 727) and caspase-3 were purchased fromMillipore and Becton Dickenson, respectively. Anti–b-tubulin, b-actin,and anti STAT1 Abs were purchased from Sigma-Aldrich and Santa Cruz,respectively. Goat anti-rabbit and anti-mouse IgG HRP conjugates wereobtained from Jackson ImmunoResearch. Cytokine ELISA kit for TNF-awas obtained from R&D Systems.
Animal preparation
Animal protocols were approved by the animal care committee at McGillUniversity (Montreal, Canada). Six-week-old male C57BL/6J, BALB/cSTAT1 +/+, or BALB/c STAT12/2 mice were injected i.p. with rapamy-cin (1.5 mg/kg) or control 6 h before tracheotomy and intratracheal in-stillation of saline or LPS (50 ml of 1 mg/ml solution). There were noperiprocedural deaths, and animals were killed by CO2 asphyxiation 24 hlater for assays of lung injury, apoptosis, and gene expression, as indicatedbelow. BALB/c and C57BL/6J wild-type mice were obtained from Harlanand Charles River; BALB/c STAT12/2 mice were originally derived byDr. J. Durbin (18) and obtained from Dr. A. Koromilas (McGill Univer-sity). Genotyping of STAT1 alleles was performed on mouse ear punchDNA isolated by alkaline lysis. Three primer sequences (59-TAATGTTT-CATAGTTGGATATCAT-39), (59-GAGATAATTCACAAAATCAGAGAG-39), (59-CTGATCCAGGCAGCGTTG-39) were used in a single PCR re-action to identify the wild-type allele (product size 142 bp) or the knockoutallele (product size 342 bp).
Measures of lung injury
For lung wet-to-dry ratio, the lungs were removed en bloc, gently blotted toremove excess blood, weighed, and dried at 60˚C for 72 h. The wet-to-drymass ratio was calculated for each excised lung. For lung histopathology,whole lungs were inflated at a constant pressure of 25 cm H2O via thetrachea with 10% neutral buffered formalin, removed en bloc, and storedin 10% formalin for 24 h before the preparation of paraffin-embeddedtissue blocks. Microscope slides were prepared by the McGill Life Sci-ences Centre Histopathology Core using 1-mm transverse sections. Rep-resentative H&E-stained sections were prepared for histopathologicanalysis. For measures of alveolar-capillary leak, the Evans Blue (EB)dye technique was used as described previously (19). Tetrasodium saltof EB dye was diluted to a concentration of 5 mg/ml in Dulbecco’s PBSand conjugated to 4% of BSA, before filtering through a 0.22-mmmembrane. Twenty-two hours after LPS instillation, the mice wereinjected with 20 mg/kg EB solution via the tail vein. Two hours later, themice were sacrificed by CO2 asphyxiation, and the pulmonary circulation
was flushed with 10 ml cold Dulbecco’s PBS. Lungs were harvested,snap-frozen in liquid nitrogen, and kept at 280˚C. Samples were weigh-ed, and EB was extracted from the lungs as described (19). Two volumesof formamide were added before incubation at 60˚C for 18 h, centrifu-gation for 30 min at 12,000 3 g, and measurement of absorbance at 620nm and 740 nm by spectrophotometry. Dilution standards for EB andblank samples were prepared in 50% formamide, and a lung-specificcorrection factor was applied to the homogenate values (A620[EB] = A620 2[1.1649 3 A740] + 0.004) as described (19). For each sample, the homog-enate concentrations are expressed as micrograms of EB per gram of lungtissue.
Measures of apoptosis
For TdT-mediated dUTP nick end labeling (TUNEL) assay, histopathologyslides were rehydrated in PBS for 10 min before boiling in citrate buffer(100 mM citrate, pH 6.0, 0.05% Tween) for 5 min. TUNEL staining wasperformed using the In Situ Cell Death Detection Kit (Roche Diagnostics)according to the manufacturer’s protocol. Coverslips were applied usingmounting solution (Dako Cytomation) containing the nuclear stain DAPI,and fluorescence was detected using an Olympus 370 inverted fluores-cence microscope. The number of TUNEL (FITC)-positive cells per high-power field (magnification 3400) was determined after acquisition ofimages using charge-coupled device camera and Metamorph software. Fordetection of cleaved caspase-3, immunohistochemical detection was per-formed on a Discovery XT automatic immunostainer (Ventana MedicalSystems). Following deparaffinization and Ag retrieval, slides were incu-bated for 2 h at room temperature with 1:100 anti-cleaved caspase-3 Ab(Biocare Medical) in PBS, washed and incubated with biotin-conjugatedanti-rabbit Ab (Jackson ImmunoResearch). Streptavidin-HRP, 3,3-dia-minobenzidine detection kit, and avidin-biotin blocking kit were usedaccording to the manufacturer’s instructions (Ventana Medical Systems).The sections were counterstained with hematoxylin before application ofa bluing reagent.
Assessment of bronchoalveolar lavage and lung neutrophilcontent
Bronchoalveolar lavage (BAL) fluid was obtained by cannulating the tra-chea and then lavaging the lungs four times with 0.5 ml ice-cold PBS. BALfluid samples were pooled for each mouse and centrifuged onto glass slidesat 1000 rpm for 10 min using Shandon Cytospin 2. The slides were fixed andstained with Diff Quick (Dade Behring) before counting the number ofneutrophils per high-power field (400 3 magnification) by light micros-copy.
For lung neutrophil content, myeloperoxidase activity was measured inwhole lung lysates as described previously (20). Lung samples were ho-mogenized in homogenization buffer (0.5% hexadecyltrimethylammoniumbromide, 5 mM EDTA, 50 mM potassium phosphate [pH 6.0], 1 mMphenylmethanesulphonyl fluoride, 1 mg/ml aprotinin, 1 mg/ml leupeptin)before sonication (three 20-s pulses), and centrifugation at 30003 g for 30min. After measurement of protein concentration (Bradford assay), 10 mlof each sample was added to 250 ml assay buffer (0.005% hydrogen per-oxide, 0.5 mM o-dianisidine dihydrochloride, 100 mM potassium phos-phate [pH 6.0]), before measurement of absorbance at 460 nm every 20 sfor 10 min by spectrophotometry. Data for myeloperoxidase activity arepresented as fold change in relative absorbance units per second per mi-crogram of protein.
Detection of lung mRNA
Whole lungs of lavaged micewere removed en bloc and then stored at220˚Cin RNA Later (Invitrogen) prior to RNA extraction. Lungs were mechan-ically disrupted in 1 ml Trizol reagent (Invitrogen) per 50–100 mg of tissueusing a Brickman homogenizer. RNA was purified by phenol-chloroformextraction per the manufacturer’s protocol. cDNA was generated by re-verse transcription from 2 mg RNA (Superscript II; Invitrogen). Real-timePCR was performed on 1 ml cDNA using Power SYBR Green UniversalPCR master mix (ABI), per the manufacturer’s instructions. The followingsequences were used for forward and reverse real-time PCR primers: Fas(forward) 59-AGGACTGCAAAATGAATGGG-39, (reverse) 59-GGGTGC-AGTTTGTTTCCACT-39; caspase-3 (forward) 59-CATTTATGGGACAA-ATGGGC-39, (reverse) 59-CCGTCCTTTGAATTTCTCCA-39; 45S rRNA(forward) 59-GACACGCTGTCCTTTCCCTA-39, (reverse) 59-GTCTGAC-ACGCAGCAAAGTC-39; STAT1 (forward) 59-CTTGTGTTGAATCCCG-AACC-39, (reverse) 59-AGCTCGAACCACTGTGACATC-39. PCR reac-tions were carried out for 45 cycles (ABI 7500 Real Time PCR System).Results are expressed as fold induction in mRNA levels (6 SEM) as cal-culated by the DDCt method (21).
Whole lungs of lavaged mice were removed en bloc, snap frozen in liquidnitrogen, and stored at 280˚C prior to protein extraction. Lungs weremechanically disrupted using a Brickman mechanical homogenizer inhomogenization buffer (20 mM Tris [pH 8.0], 0.5% Nonidet P-40, 1 mMphenylmethanesulphonyl fluoride, 50 mM NaF, 1 mg/ml aprotinin, 1 mg/mlleupeptin, 100 mM sodium orthovanadate). Homogenates were snap frozenon dry ice, thawed, and cleared by centrifugation at 16,000 3 g for 30 minat 4˚C. For detection of proteins in cultured cell lines, 2fTGH or U3A cellswere washed once with cold PBS and incubated for 15 min on ice inhomogenization buffer. After freezing and thawing, cells were homoge-nized on ice and cleared (1000 3 g for 5 min). Supernatants were furtherseparated (16,000 3 g for 30 min) to generate particulate-free lysates. Formouse lung homogenates and cultured cell lysates, supernatants wereassayed for protein content by Bradford assay. Equal amounts of proteinwere separated by SDS-PAGE and transferred to nitrocellulose membranebefore immunoblotting with primary Abs as indicated. Membranes wereincubated with anti-rabbit or anti-mouse IgG HRP conjugated Abs anddeveloped using Super-Signal West Pico chemiluminescence detection kit(Pierce). Images of Western blot films were acquired using an Alpha Im-ager (Innotech), and analyzed with Alpha Ease FC software (version4.1.0). Integrated band density for the indicated protein was obtained usingthe spot density and auto-background functions, and was normalized tothat of b-tubulin or b-actin for each sample. Levels of TNF-a in the BALfluid were determined using a commercially available ELISA kit (R&DSystems), according to the manufacturer’s instructions.
Detection of cell number in vitro
U3A or 2fTGH cells were washed twice with PBS before incubating with0.2% crystal violet in 25% methanol for 15 min. Excess stain was removedby washing four times with PBS. Stained cells were solubilized by mixingwith 1% SDS solution for 15 min. The OD of each sample (100 ml) wasdetermined at 570 nm and 620 nm using a SpectraMax M2 microplatereader (Molecular Devices).
Statistical analysis
Statistical analysis was performed using JMP 8.0.1 software (SAS Institute)Student t test, or ANOVA with Tukey–Kramer multiple comparisons testswas applied to grouped data for each experiment. A p value , 0.05 wasconsidered statistically significant.
ResultsRapamycin enhances LPS-induced lung injury
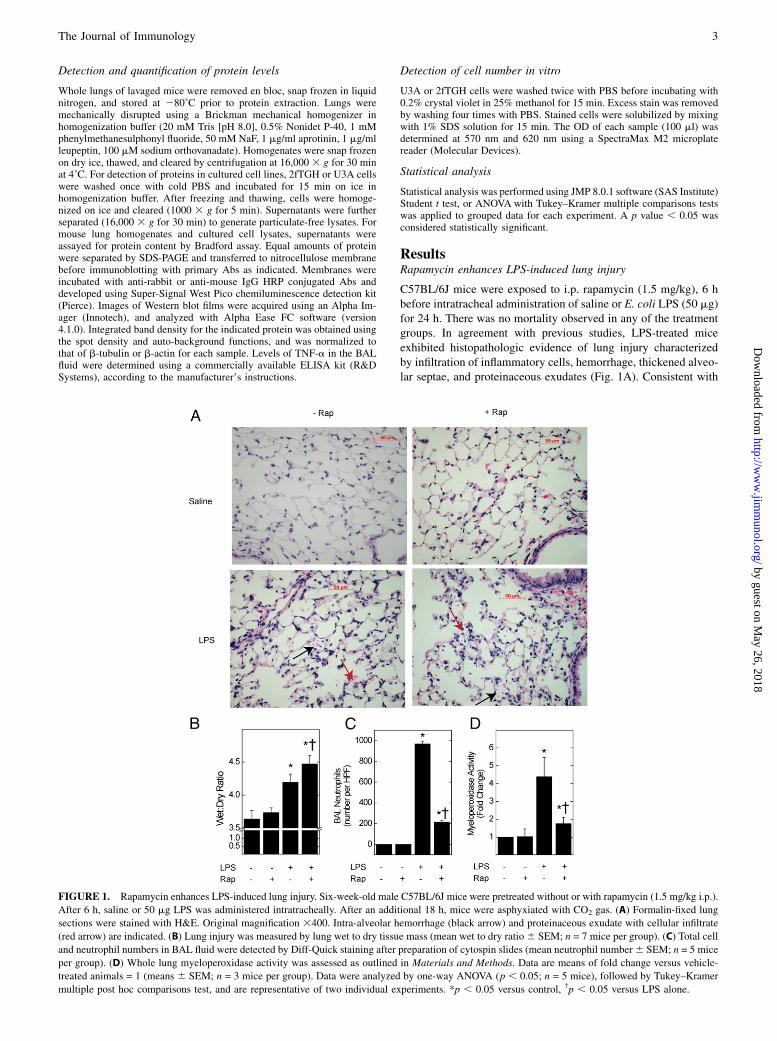
C57BL/6J mice were exposed to i.p. rapamycin (1.5 mg/kg), 6 hbefore intratracheal administration of saline or E. coli LPS (50 mg)for 24 h. There was no mortality observed in any of the treatmentgroups. In agreement with previous studies, LPS-treated miceexhibited histopathologic evidence of lung injury characterizedby infiltration of inflammatory cells, hemorrhage, thickened alveo-lar septae, and proteinaceous exudates (Fig. 1A). Consistent with
FIGURE 1. Rapamycin enhances LPS-induced lung injury. Six-week-old male C57BL/6J mice were pretreated without or with rapamycin (1.5 mg/kg i.p.).
After 6 h, saline or 50 mg LPS was administered intratracheally. After an additional 18 h, mice were asphyxiated with CO2 gas. (A) Formalin-fixed lung
sections were stained with H&E. Original magnification 3400. Intra-alveolar hemorrhage (black arrow) and proteinaceous exudate with cellular infiltrate
(red arrow) are indicated. (B) Lung injury was measured by lung wet to dry tissue mass (mean wet to dry ratio6 SEM; n = 7 mice per group). (C) Total cell
and neutrophil numbers in BAL fluid were detected by Diff-Quick staining after preparation of cytospin slides (mean neutrophil number6 SEM; n = 5 mice
per group). (D) Whole lung myeloperoxidase activity was assessed as outlined in Materials and Methods. Data are means of fold change versus vehicle-
treated animals = 1 (means 6 SEM; n = 3 mice per group). Data were analyzed by one-way ANOVA (p , 0.05; n = 5 mice), followed by Tukey–Kramer
multiple post hoc comparisons test, and are representative of two individual experiments. *p , 0.05 versus control, †p , 0.05 versus LPS alone.
intratracheal administration, the injury was not observed homo-genously throughout the lungs. In contrast to a previous report(22), there was no inhibitory effect of rapamycin in LPS-inducedlung injury by histopathology.To quantify lung injury, we next determined the effect of
rapamycin on LPS-induced pulmonary edema by whole-lung wet-to-dry ratio. Consistent with previous studies, wet-to-dry ratio was3.7 in control animals and increased by 17% in LPS-exposedanimals (Fig. 1B). Administration of rapamycin led to a statisti-cally significant increase in lung wet-to-dry ratio in animals ex-posed to LPS (4.5 versus 4.2; p , 0.05). In contrast to lung injury,the number of neutrophils in the BAL fluid and lung myeloper-oxidase activity was reduced by rapamycin, suggesting the in-volvement of a mechanism other than neutrophilic recruitment orinflammation (Fig. 1C, 1D).
Rapamycin enhances LPS-induced cellular apoptosis
Previous studies demonstrated a requirement for apoptosis andthe death receptor Fas/CD95 in LPS-induced lung injury (23,24). Moreover, we recently reported the suppression of STAT1-dependent genes (e.g., Fas, caspase-1, inducible NO synthase) bymTOR (16), and reasoned that inactivation of mTOR would en-hance apoptosis in vivo. In the current study, administrationof intratracheal LPS significantly increased levels of cleavedcaspase-3 (8.4 versus 4.2 positive cells per high-power field; p ,0.05; Fig. 2A). Rapamycin further enhanced cleaved caspase-3levels in animals exposed to LPS (13.6 versus 8.4 positive cellsper high-power field; p , 0.05). By TUNEL staining, LPS in-creased cellular apoptosis compared with that in control animals(2.5 versus 0.3 TUNEL-positive cells per high-power field; p ,0.05; Fig. 2B). Rapamycin further increased apoptosis in LPS-treated mice (4.4 versus 2.5 TUNEL-positive cells per high-power field; p , 0.05). These results indicate that the augment-ing effect of rapamycin on LPS-induced lung injury correlateswith its effect on apoptosis in the lung.
STAT1 is required for the enhancing effect of rapamycin oncellular apoptosis
We previously demonstrated an enhancing effect of rapamycinon the induction of STAT1-dependent proapoptotic genes (16).To demonstrate that STAT1 is required for the effect of rapa-mycin on LPS and IFN-b–induced cellular apoptosis, we used aSTAT1-deficient human cell line (U3A) and its wild-type control(2fTGH). Inhibition of mTOR activity with rapamycin, or incu-bation with LPS and IFN-b, significantly increased cleavedcaspase-3 levels in wild-type (2fTGH) cells (Fig. 3A, lanes 4 and5). The addition of LPS and IFN-b further increased cleavedcaspase-3 levels in rapamycin-treated cells compared with cellsexposed to rapamycin alone (Fig. 3A, lane 6 versus 4). In contrast,rapamycin failed to significantly induce or enhance cleavage ofcaspase-3 in STAT1-deficient (U3A) cells (Fig. 3A, lanes 10 and12). Corresponding densitometry data demonstrate statisticallysignificant augmentation of LPS/IFN-b–induced apoptosis in con-trol, but not STAT1-deficient, cells (Fig. 3A, bottom panel).We next used crystal violet staining to quantify changes in cell
number in cultures exposed to rapamycin, LPS/IFN-b, or both. Inwild-type cells exposed to rapamycin for 40 h, there was a markeddecrease in cell number, likely because of its known inhibitoryeffect on cell proliferation (Fig. 3B) (25). The addition of LPS/IFN-b to rapamycin led to further decreased cell number, whichcorrelated with increased cleaved caspase-3 levels (Fig. 3A, lane6). LPS and IFN-b did not decrease cell number in STAT1-deficient cells, perhaps because of a failure to induce apoptosis(Fig. 3A, lane 11). In contrast to control cells, LPS and IFN-b did
not further decrease cell number when added to rapamycin-treatedSTAT1-deficient cells (Fig. 3B). These results indicate that theincremental effect of LPS and IFN-b on rapamycin-induced ap-optosis, and reduction in cell number, requires STAT1.To demonstrate that STAT1 is required for LPS-induced lung
injury in vivo, we used BALB/c STAT1 knockout (STAT12/2) orwild-type (STAT1+/+) mice exposed to intratracheal LPS. More-over, we measured intravascular EB dye transit from the vascu-lature into the alveolar space (i.e., alveolar capillary leak) asanother index of acute lung injury. In wild-type mice, intratrachealLPS led to a significant increase in EB dye leak (Fig. 3C); theLPS-induced leak was absent in STAT12/2 mice.
Rapamycin enhances the expression of genes involved inapoptosis and lung injury
Through TLR4, LPS leads to the activation of STAT1-dependentapoptosis genes (26, 27). We therefore determined the effect ofrapamycin on the expression of Fas and caspase-3 mRNA. Con-sistent with previous studies, expression of Fas was increased, andthat of caspase-3 reduced, 24 h after exposure of mice to LPS (Fig.4) (28, 29). In mice exposed to LPS, rapamycin significantly in-creased Fas mRNA levels compared with mice exposed to LPSalone. Similarly, rapamycin prevented the reduction in caspase-3mRNA levels observed in LPS-treated mice.
FIGURE 2. Rapamycin enhances LPS-induced apoptosis in the lung.
Lungs were collected, fixed in formalin, and sectioned before detection of
(A) cleaved caspase-3 (cleaved Casp-3) by immunohistochemistry or (B)
TUNEL by immunofluorescence, and detection of positively stained cells
per high-power field (original magnification 3400; n = 5 high-power fields
per section from each of three mice). A representative image of cleaved
caspase-3–stained cells (brown) in LPS-treated mice is shown in the top
panel. *p , 0.05 versus control, †p , 0.05 versus LPS alone.
The proapoptotic effects of TLR4 receptor ligation are mediatedprimarily by the induction of IFN-b and autocrine-paracrine ac-tivation of STAT1 (26, 30–32). We reasoned that the increase inSTAT1-dependent genes by rapamycin in vivo (Fig. 4) correlateswith additional activation of STAT1. By Western blot analysis ofwhole lung homogenates, the levels of phosphorylated (activated)and total STAT1 were significantly increased in mice exposed toLPS for 24 h (Fig. 5A, lane 2 versus 1). Phospho-STAT1 and totalSTAT1 levels were further increased by rapamycin (lane 4 versus2). As was the case for caspase-3 (Fig. 4), STAT1 mRNA levelswere increased in mice exposed to LPS and rapamycin, but notLPS alone (Fig. 5B). Thus, the additional enhancement of lunginjury, apoptosis, and induction of apoptosis genes caused byrapamycin correlates with elevated levels of activated STAT1 inmice exposed to LPS.
Rapamycin attenuates the activation of NF-kB by LPS
In contrast to STAT1, previous work demonstrated inhibition ofNF-kB by rapamycin, NF-kB–dependent inflammatory mediators,and neutrophil recruitment (14, 22). We therefore confirmed thatrapamycin blocks NF-kB activation in mice exposed to LPS and
rapamycin, despite the observed increase in STAT1 activation andapoptosis. As expected, LPS increased phosphorylation of theNF-kB p65 subunit, and reduced IkBa levels; rapamycin blockedLPS-induced phosphorylation of p65 and reversed the degradationof IkBa (Fig. 6A, 6B). In agreement with previous studies, LPSincreased BAL TNF-a protein levels (33). Consistent with its ef-fect on neutrophil recruitment and NF-kB signaling, rapamycininhibited TNF-a induction by LPS when measured in the BALfluid (Fig. 6C). These results indicate that rapamycin promotesa proapoptotic, STAT1-dependent transcriptional program whilesuppressing inflammatory modulators such as NF-kB, neutrophils,and TNF-a.
Rapamycin attenuates mTORC1 effectors and ribosomalbiogenesis
mTOR nucleates two protein complexes: mTOR complex 1(mTORC1) and mTOR complex 2 (mTORC2) (12). Rapamycinpotently and specifically inhibits mTORC1, which controls cellgrowth and ribosomal biogenesis by activating p70 S6 kinase. Therapamycin-insensitive mTORC2 controls cell proliferation andcytokinesis in part by activating Akt and protein kinase C-a (34,35). We next determined whether the effect of rapamycin on lunginjury and apoptosis accompanied blockade of mTORC1. In con-
FIGURE 3. STAT1 is required for rapamycin enhancement of LPS and IFN-b–induced apoptosis in vitro and LPS-induced lung vascular leak in vivo.
2fTGH or U3A cells were incubated with LPS or IFN-b, or both, in the absence or presence of rapamycin for (A) 24 h or (B) 40 h. In (A), cleaved caspase-3
levels (CCasp3) in cell lysates were assessed by Western blot. Data were quantified by band densitometry (means of cleaved caspase-3 integrated pixel
density normalized to b-actin 6 SEM; n = 3 experiments). Means were significantly different by two-way ANOVA (p , 0.0001). *p , 0.05 versus LPS or
control, †p , 0.05 versus LPS/IFN-b by Tukey–Kramer multiple post hoc comparisons test. In (B), to assess cell number, cells were stained with crystal
violet and the OD of solubilized dye was measured. Changes in OD at 570 nm after correction for the OD at 620 nm are shown (mean 6 SEM; n = 4
independent experiments each performed in triplicate). Means were significantly different by two-way ANOVA (p , 0.01). By Tukey–Kramer multiple
post hoc comparisons test: *p, 0.05 versus control; †p, 0.05 versus rapamycin alone or control; ns, not significant versus rapamycin alone. (C) Wild-type
(STAT1+/+) or knockout (STAT12/2) mice were exposed to saline or 50 mg LPS administered intratracheally. After 22 h, EB dye was injected systemically,
before asphyxiation with CO2 gas 2 h later. EB dye leak was measured as described inMaterials and Methods. Data are expressed as micrograms of EB dye
per gram of lung (mean 6 SE; n = 8–10 mice per group). *p , 0.05 versus control by Student t test.
trol or LPS-treated mice, rapamycin abolished the phosphoryla-tion of p70 S6 kinase (Thr 389) or S6 (Ser 235/236), which aremTORC1-sensitive phosphorylation sites (Fig. 7A). Rapamycindid not significantly affect phosphorylation of Akt at Serine 473,which is an mTORC2-sensitive site (Fig. 7A). Consistent withblockade of mTORC1 activity, rapamycin significantly decreasedlevels of 45S pre-rRNA, indicating inhibition of the p70 S6kinase-dependent rate-limiting step in ribosomal biogenesis (Fig.7B). Thus, enhancement of apoptosis and lung injury by rapa-mycin coincides with its blockade of mTORC1 effectors oftranslation initiation and ribosomal biogenesis.
DiscussionIn this study, we demonstrate that rapamycin can enhance lunginjury and cellular apoptosis in mice exposed to the TLR4 ligandLPS. The additional cell death produced by rapamycin correlatedwith increased lung injury and the amplification of apoptosis genes,but not markers of inflammation. The proapoptotic transcriptionfactor STAT1 was increased in rapamycin-treated animals, whereas
the activation of the proinflammatory and antiapoptotic tran-scription factor NF-kB was attenuated. In cultured cells and mice,STAT1 was required for apoptosis and lung injury, respectively.Rapamycin did not cause or permit apoptosis in STAT1-deficientcells. These results indicate that inactivation of mTOR can sen-sitize the proapoptotic response to bacterial products and thatenhanced lung injury is one manifestation. Consistent with thisobservation, complications of rapamycin therapy include con-ditions characterized by dysregulated lung injury and repairsuch as interstitial pneumonitis, bronchiolitis obliterans organizingpneumonia, and pulmonary fibrosis (36). Moreover, other con-ditions that reduce mTOR activity, such as nutritional deprivation,might also predispose to tissue injury during microbial infection.Using rapamycin to dissect the role of mTOR in acute lung
injury, our studies reveal an important role for STAT1 in the in vivoand in vitro proapoptotic effects of LPS. Previous studies showedthat physical interactions between TLR4 and its signaling adaptorproteins form the molecular basis for these distinct responses (3).Ligation of TLR4 by LPS recruits MyD88, which in turn activatesNF-kB and proinflammatory genes. In addition to MyD88, re-cruitment of the adaptor protein TIR-domain–containing adapter-inducing IFN-b activates IRF-3 and the synthesis of IFN-b. Thesubsequent activation of STAT1 is required for the induction ofMyD88-independent genes induced by LPS (e.g., IP10, RANTES,IRF-1) (37). Our data indicate that inactivation of mTOR enhancesMyD88-independent signaling (i.e., STAT1, apoptosis) while at-tenuating MyD88-dependent processes (i.e., NF-kB, neutrophilrecruitment, TNF-a release).Inhibition of mTOR promoted STAT1 activation under con-
ditions of attenuated S6K activity (Fig. 7), suggesting a directeffect of mTOR on transcriptional control mechanisms. The effectof mTOR on IFN signaling and innate immune function waspreviously thought to occur primarily via its effectors p70 S6kinase or 4E-BP1 and the control of protein synthesis (38, 39).However, recent studies indicated a direct molecular link betweenmTOR and transcription factors. Inactivation of mTOR enhancedSTAT1 nuclear levels in human lung epithelial cells and amplifiedthe induction of STAT1-dependent apoptosis genes by IFN-g (16).mTOR also regulated NF-kB, although its effect depended on thecell type or model. In vitro studies using cultured monocytes ex-posed to rapamycin revealed increased NF-kB activity and TNF-alevels; in contrast, NF-kB and TNF-a were suppressed by rapa-mycin in the lungs of mice exposed to LPS (22, 40). In a separatestudy, rapamycin increased mortality in mice exposed to i.p. LPS,and this was associated with enhanced levels of caspase-1, an NF-kB-dependent protein, but decreased TNF-a levels (41). In thisstudy, we investigated STAT1 and NF-kB simultaneously, and
FIGURE 4. Inhibition of mTOR augments the expression of STAT1-
dependent proapoptotic genes. Mice were treated as indicated, total lung
RNAwas extracted, and mRNA levels of Fas or caspase-3 were quantified
by real-time PCR. Changes in mRNA levels are expressed as fold change
relative to control mRNA levels = 1 (DDCt method; mean fold-induction6SEM; n = 5 mice per experimental condition). *p , 0.05 versus control,†p , 0.05 versus LPS alone.
FIGURE 5. Rapamycin enhances STAT1 signaling. (A) STAT1 phosphorylation in lung homogenates was assessed by Western blot. Data were quantified
by band densitometry and are expressed as fold change in phospho-STAT1 S727 or STAT1 versus control = 1 (means 6 SEM; n = 6 mice per group).
Phospho-STAT1 S727 or STAT1 integrated pixel density was first normalized to that for b-tubulin in each sample. (B) STAT1 mRNA levels were assessed
by real-time PCR. Changes in mRNA levels are expressed as fold change relative to control mRNA levels = 1 (DDCt method; mean fold induction6 SEM;
n = 5 mice per condition).*p , 0.05 versus control, †p , 0.05 versus LPS alone.
confirmed inhibition of NF-kB by rapamycin in vivo. However, incontrast to NF-kB, rapamycin augmented STAT1 activity and thetranscription of STAT1-dependent apoptosis genes.
The augmenting effect of rapamycin on lung injury and apo-ptosis correlated with activation of STAT1, and inhibition of NF-
kB. Of note, a recent report also described an inhibitory effect ofrapamycin on NF-kB, TNF-a, and neutrophil recruitment in LPS-
treated mice (22); however, despite the expected antisurvival ef-fect of NF-kB blockade, and in contrast to the current study, lunginjury was reported to be blocked by rapamycin. Possible ex-
planations for this discrepancy include differences in the timing ofrapamycin administration and high dose (two 5-mg/kg doses 6 h
prior to and concurrent with LPS administration in the previousstudy versus one 1.5-mg/kg dose 6 h prior to LPS in the current
study). Moreover, the former report focused on neutrophilic in-flammation and did not address MyD88-independent TLR4 sig-naling responses or cellular apoptosis. Finally, our in vivo and
in vitro data demonstrate that STAT1 is required for LPS-inducedlung injury and apoptosis (Fig. 3). The data also support an in-
hibitory role for mTOR in the induction of STAT1- and LPS-induced tissue injury. Further genetic dissection of the effects
rapamycin in vivo will be required to reconcile the disparatefindings in regard to lung injury.Although both neutrophils (neutrophil hypothesis) and epithe-
lial cell death (epithelial cell hypothesis) appear to play a role,there is a debate on the relative importance of each (42). In the
current study, the inhibitory effect of rapamycin on NF-kB, TNF-a, and bronchoalveolar fluid neutrophil levels did not correlatewith its enhancement of apoptosis, STAT1, or lung injury. More-
over, the inhibitory effect of rapamycin on NF-kB and TNF-amight be consistent with enhanced lung injury given the known
prosurvival (i.e., antiapoptotic) effects of NF-kB and TNF-a (43).In addition to effects on cell survival, the TNF-a lectin-bindingdomain reduced acute lung injury in part by promoting alveolar
liquid clearance (33). Our results suggest that mTOR selectivelysuppresses MyD88-independent signaling and cellular apoptosis.
This paradigm is supported by in vitro studies demonstratinga physical and functional association between mTOR and STAT1
(16). Other inhibitors of mTOR activity include reduced essentialamino acids, glucose, oxygen, or growth factors. The mTOR-STAT1 signaling axis may therefore represent an important in-
FIGURE 6. Rapamycin blocks LPS-induced acti-
vation of NF-kB. (A) Phosphorylation of p65 and
(B) degradation of IkBa were quantified in lung ho-
mogenates by Western blot and band densitometry.
Data were quantified by band densitometry and are
expressed as fold change in phospho-p65 S536, total
p65 or IkBa (means 6 SEM; n = 6 mice per group).
Phospho-p65 S536, total p65, or IkBa integrated pixel
density were first normalized to that for b-tubulin in
each sample. (C) TNF-a levels in BAL fluid were
quantified by ELISA (means of protein levels 6 SEM;
n = 7 mice per group). *p , 0.05 versus control, †p ,0.05 versus LPS alone.
FIGURE 7. Rapamycin attenuates mTORC1 effectors and ribosomal bio-
genesis. (A) Activation of mTORC1 targets p70 S6K and S6, or the mTORC2
target Akt, was assessed by Western blot. (B) 45S pre-rRNA levels were
assessed by real-time PCR. Changes in mRNA levels are expressed as fold
change relative to controlmRNA levels =1 (mean fold induction6SEM;n=5
mice per group). *p, 0.05 versus control, †p, 0.05 versus LPS alone.
terface between metabolism, innate immunity, and lung injuryin vivo.
AcknowledgmentsWe thank Dr. S. Levine and Dr. J. Moss (National Institutes of Health) for
advice and critical review of the manuscript, Dr. A. Koromilas for pro-
viding STAT1 knockout mice, I. Angers for conducting Evans Blue dye
leak experiments, and Dr. S. Vidal (McGill University) for providing
real-time PCR primer sequences.
DisclosuresThe authors have no financial conflicts of interest.
References1. Martin, T. R., N. Hagimoto, M. Nakamura, and G. Matute-Bello. 2005. Apo-
ptosis and epithelial injury in the lungs. Proc. Am. Thorac. Soc. 2: 214–220.2. Chaudhuri, N., M. K. Whyte, and I. Sabroe. 2007. Reducing the toll of in-
flammatory lung disease. Chest 131: 1550–1556.3. Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol.
4: 499–511.4. Kim, H. S., and M. S. Lee. 2007. STAT1 as a key modulator of cell death. Cell.
Signal. 19: 454–465.5. Kristof, A. S., P. Goldberg, V. Laubach, and S. N. Hussain. 1998. Role of in-
ducible nitric oxide synthase in endotoxin-induced acute lung injury. Am. J.Respir. Crit. Care Med. 158: 1883–1889.
6. Mancuso, G., A. Midiri, C. Biondo, C. Beninati, S. Zummo, R. Galbo,F. Tomasello, M. Gambuzza, G. Macrı, A. Ruggeri, et al. 2007. Type I IFNsignaling is crucial for host resistance against different species of pathogenicbacteria. J. Immunol. 178: 3126–3133.
7. Varinou, L., K. Ramsauer, M. Karaghiosoff, T. Kolbe, K. Pfeffer, M. Muller, andT. Decker. 2003. Phosphorylation of the Stat1 transactivation domain is requiredfor full-fledged IFN-gamma-dependent innate immunity. Immunity 19: 793–802.
8. Liu, G., Y. J. Park, Y. Tsuruta, E. Lorne, and E. Abraham. 2009. p53 Attenuateslipopolysaccharide-induced NF-kappaB activation and acute lung injury. J.Immunol. 182: 5063–5071.
9. Imai, Y., K. Kuba, G. G. Neely, R. Yaghubian-Malhami, T. Perkmann, G. vanLoo, M. Ermolaeva, R. Veldhuizen, Y. H. Leung, H. Wang, et al. 2008. Identi-fication of oxidative stress and Toll-like receptor 4 signaling as a key pathway ofacute lung injury. Cell 133: 235–249.
10. Yum, H. K., J. Arcaroli, J. Kupfner, R. Shenkar, J. M. Penninger, T. Sasaki,K. Y. Yang, J. S. Park, and E. Abraham. 2001. Involvement of phosphoinositide3-kinases in neutrophil activation and the development of acute lung injury. J.Immunol. 167: 6601–6608.
11. Laplante, M., and D. M. Sabatini. 2009. mTOR signaling at a glance. J. Cell Sci.122: 3589–3594.
12. Wullschleger, S., R. Loewith, and M. N. Hall. 2006. TOR signaling in growthand metabolism. Cell 124: 471–484.
13. Huang, S., M. A. Bjornsti, and P. J. Houghton. 2003. Rapamycins: mechanism ofaction and cellular resistance. Cancer Biol. Ther. 2: 222–232.
14. Ghosh, S., V. Tergaonkar, C. V. Rothlin, R. G. Correa, V. Bottero, P. Bist,I. M. Verma, and T. Hunter. 2006. Essential role of tuberous sclerosis genesTSC1 and TSC2 in NF-kappaB activation and cell survival. Cancer Cell 10:215–226.
15. Kristof, A. S., J. Marks-Konczalik, E. Billings, and J. Moss. 2003. Stimulation ofSTAT1-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J. Biol. Chem. 278:33637–33644.
16. Fielhaber, J. A., Y. S. Han, J. Tan, S. Xing, C. M. Biggs, K. B. Joung, andA. S. Kristof. 2009. Inactivation of mammalian target of rapamycin increasesSTAT1 nuclear content and transcriptional activity in a4- and protein phos-phatase 2A-dependent fashion. J. Biol. Chem. 284: 24341–24353.
17. McKendry, R., J. John, D. Flavell, M. Muller, I. M. Kerr, and G. R. Stark. 1991.High-frequency mutagenesis of human cells and characterization of a mutantunresponsive to both alpha and gamma interferons. Proc. Natl. Acad. Sci. USA88: 11455–11459.
18. Durbin, J. E., R. Hackenmiller, M. C. Simon, and D. E. Levy. 1996. Targeteddisruption of the mouse Stat1 gene results in compromised innate immunity toviral disease. Cell 84: 443–450.
19. Moitra, J., S. Sammani, and J. G. Garcia. 2007. Re-evaluation of Evans Blue dyeas a marker of albumin clearance in murine models of acute lung injury. Transl.Res. 150: 253–265.
20. Laudes, I. J., R. F. Guo, N. C. Riedemann, C. Speyer, R. Craig, J. V. Sarma, andP. A. Ward. 2004. Disturbed homeostasis of lung intercellular adhesionmolecule-1 and vascular cell adhesion molecule-1 during sepsis. Am. J. Pathol.164: 1435–1445.
21. Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expressiondata using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.Methods 25: 402–408.
22. Lorne, E., X. Zhao, J. W. Zmijewski, G. Liu, Y. J. Park, Y. Tsuruta, andE. Abraham. 2009. Participation of mTOR Complex 1 in TLR2 and TLR4 In-duced Neutrophil Activation and Acute Lung Injury. Am. J. Respir. Cell Mol.Biol. 41: 237–245.
23. Matute-Bello, G., C. W. Frevert, W. C. Liles, M. Nakamura, J. T. Ruzinski,K. Ballman, V. A. Wong, C. Vathanaprida, and T. R. Martin. 2001. Fas/Fas li-gand system mediates epithelial injury, but not pulmonary host defenses, inresponse to inhaled bacteria. Infect. Immun. 69: 5768–5776.
24. Matute-Bello, G., R. K. Winn, M. Jonas, E. Y. Chi, T. R. Martin, and W. C. Liles.2001. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implicationsfor acute pulmonary inflammation. Am. J. Pathol. 158: 153–161.
25. Kristof, A. S., G. Pacheco-Rodriguez, B. Schremmer, and J. Moss. 2005.LY303511 Acts via PI3-kinase Independent Pathways to Inhibit Cell Prolifera-tion via mTOR- and non-mTOR-dependent Mechanisms. J. Pharmacol. Exp.Ther. 314: 1134–1143.
26. Ruckdeschel, K., G. Pfaffinger, R. Haase, A. Sing, H. Weighardt, G. Hacker,B. Holzmann, and J. Heesemann. 2004. Signaling of apoptosis through TLRscritically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J. Immunol. 173:3320–3328.
27. Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P. F. Muhlradt, S. Sato, K. Hoshino,and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independentpathway and results in activation of IFN-regulatory factor 3 and the expres-sion of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167: 5887–5894.
28. Bilban, M., F. H. Bach, S. L. Otterbein, E. Ifedigbo, J. C. d’Avila, H. Esterbauer,B. Y. Chin, A. Usheva, S. C. Robson, O. Wagner, and L. E. Otterbein. 2006.Carbon monoxide orchestrates a protective response through PPARgamma.Immunity 24: 601–610.
29. Murray, R. Z., F. G. Wylie, T. Khromykh, D. A. Hume, and J. L. Stow. 2005.Syntaxin 6 and Vti1b form a novel SNARE complex, which is up-regulated inactivated macrophages to facilitate exocytosis of tumor necrosis Factor-alpha. J.Biol. Chem. 280: 10478–10483.
30. Jung, D. Y., H. Lee, B. Y. Jung, J. Ock, M. S. Lee, W. H. Lee, and K. Suk. 2005.TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia:a critical role of IFN-beta as a decision maker. J. Immunol. 174: 6467–6476.
31. De Trez, C., B. Pajak, M. Brait, N. Glaichenhaus, J. Urbain, M. Moser,G. Lauvau, and E. Muraille. 2005. TLR4 and Toll-IL-1 receptor domain-containing adapter-inducing IFN-beta, but not MyD88, regulate Escherichiacoli-induced dendritic cell maturation and apoptosis in vivo. J. Immunol. 175:839–846.
32. Kaiser, W. J., and M. K. Offermann. 2005. Apoptosis induced by the toll-likereceptor adaptor TRIF is dependent on its receptor interacting protein homotypicinteraction motif. J. Immunol. 174: 4942–4952.
33. Lucas, R., A. D. Verin, S. M. Black, and J. D. Catravas. 2009. Regulators ofendothelial and epithelial barrier integrity and function in acute lung injury.Biochem. Pharmacol. 77: 1763–1772.
34. Sarbassov, D. D., S. M. Ali, D. H. Kim, D. A. Guertin, R. R. Latek,H. Erdjument-Bromage, P. Tempst, and D. M. Sabatini. 2004. Rictor, a novelbinding partner of mTOR, defines a rapamycin-insensitive and raptor-indepen-dent pathway that regulates the cytoskeleton. Curr. Biol. 14: 1296–1302.
35. Sarbassov, D. D., D. A. Guertin, S. M. Ali, and D. M. Sabatini. 2005. Phos-phorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science307: 1098–1101.
36. Vahid, B., and P. E. Marik. 2008. Pulmonary complications of novel antineo-plastic agents for solid tumors. Chest 133: 528–538.
37. Bjorkbacka, H., K. A. Fitzgerald, F. Huet, X. Li, J. A. Gregory, M. A. Lee,C. M. Ordija, N. E. Dowley, D. T. Golenbock, and M. W. Freeman. 2004. Theinduction of macrophage gene expression by LPS predominantly utilizesMyd88-independent signaling cascades. Physiol. Genomics 19: 319–330.
38. Colina, R., M. Costa-Mattioli, R. J. Dowling, M. Jaramillo, L. H. Tai,C. J. Breitbach, Y. Martineau, O. Larsson, L. Rong, Y. V. Svitkin, et al. 2008.Translational control of the innate immune response through IRF-7. Nature 452:323–328.
39. Platanias, L. C. 2005. Mechanisms of type-I- and type-II-interferon-mediatedsignalling. Nat. Rev. Immunol. 5: 375–386.
40. Weichhart, T., G. Costantino,M. Poglitsch,M. Rosner,M. Zeyda, K.M. Stuhlmeier,T. Kolbe, T. M. Stulnig, W. H. Horl, M. Hengstschlager, et al. 2008. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29:565–577.
41. Schmitz, F., A. Heit, S. Dreher, K. Eisenacher, J. Mages, T. Haas, A. Krug,K. P. Janssen, C. J. Kirschning, and H. Wagner. 2008. Mammalian target ofrapamycin (mTOR) orchestrates the defense program of innate immune cells.Eur. J. Immunol. 38: 2981–2992.
42. Perl, M., J. Lomas-Neira, C. S. Chung, and A. Ayala. 2008. Epithelial cell ap-optosis and neutrophil recruitment in acute lung injury-a unifying hypothesis?What we have learned from small interfering RNAs. Mol. Med. 14: 465–475.
43. Karin, M., and F. R. Greten. 2005. NF-kappaB: linking inflammation and im-munity to cancer development and progression. Nat. Rev. Immunol. 5: 749–759.











![Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation … · 2017. 8. 25. · AKT/mammalian target of rapamycin (PI3K/AKT/ mTOR) [1]. This pathway controls numerous cellular](https://static.documents.pub/doc/80x56/60af5eaa6ab71f4bc15363aa/targeting-of-pi3kaktmtor-pathway-to-inhibit-t-cell-activation-2017-8-25-aktmammalian.jpg)













