Page 1
The Pennsylvania State University
The Graduate School
Department of Chemistry
INTERMOLECULAR CONJUGATE ADDITION OF CARBON NUCLEOPHILES TO
NITROSOALKENES AND STUDIES TOWARD A TOTAL SYNTHESIS OF
ANGUSTILODINE, ALSTILOBANINE A, AND ALSTILOBANINE E
A Dissertation in
Chemistry
by
Max M. Majireck
© 2011 Max M. Majireck
Submitted in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
August 2011
Page 2
ii
The dissertation of Max M. Majireck was reviewed and approved* by the following:
Steven M. Weinreb Russell and Mildred Marker Professor of Natural Products Chemistry Dissertation Advisor Chair of Committee Kenneth S. Feldman Professor of Chemistry Raymond L. Funk Professor of Chemistry Michael A. Hickner Assistant Professor of Materials Science and Engineering Barbara J. Garrison Shapiro Professor of Chemistry Head of the Department of Chemistry
* Signatures are on file in the Graduate School.
Page 3
iii
Abstract
An efficient procedure for the alkylation of nitrosoalkenes has been developed in which
an array of potassium ester enolates were found to add in Michael fashion to various
nitrosoalkenes generated via the Denmark protocol from α-chloro-O-TBS ketoximes and α-
chloro-O-TBS aldoximes. Using this methodology, a number of different α-alkylated oxime
constructs were synthesized including systems which contain vicinal quaternary centers.
A total synthesis of the structurally unique monoterpene indole alkaloids angustilodine
(167), alstilobanine A (169), and alstilobanine E (168) has been initiated. Initial exploratory
studies identified indole-2-acetate ester enolates as suitable nucleophiles for conjugate additions
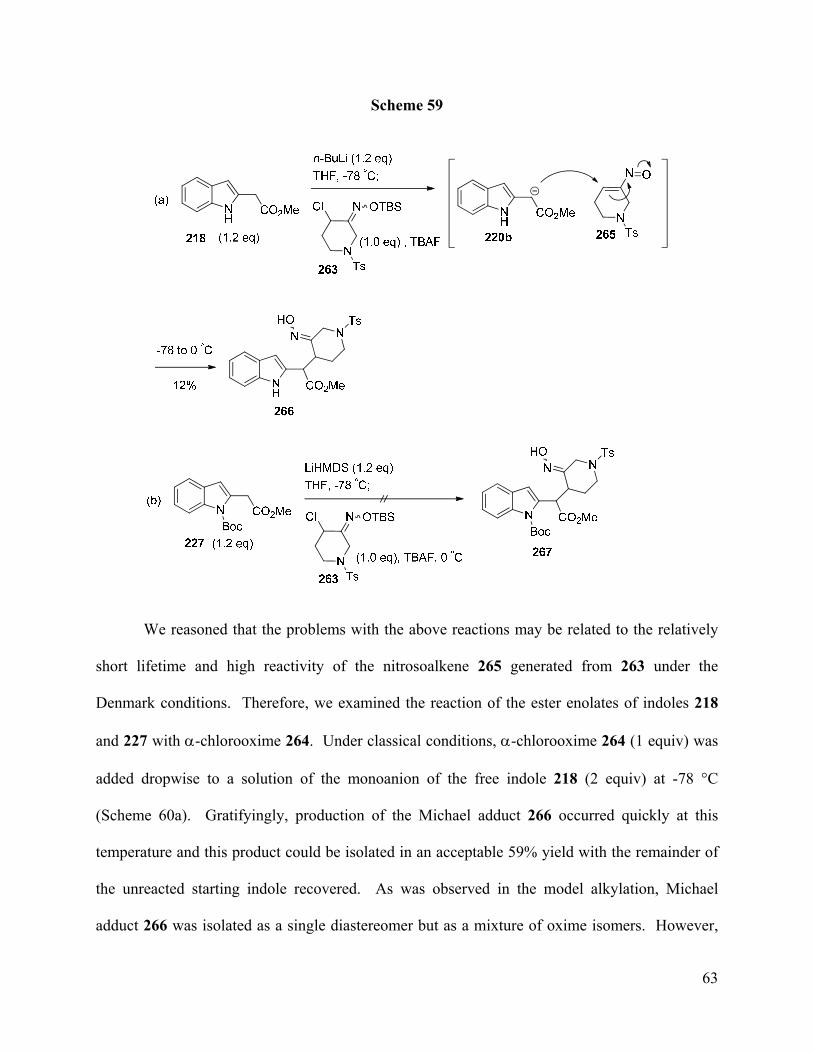
to 3-piperidone derived nitrosoalkenes. The Michael adduct 266, which contains the indole-
piperidine substructure found in alkaloids 167-169, was prepared from the conjugate addition of
the monoanion of indole-2-acetate 218 to the 3-piperidone derived nitrosoalkene 265. However,
subsequent functionalization of the indole C-3 was found to be difficult.
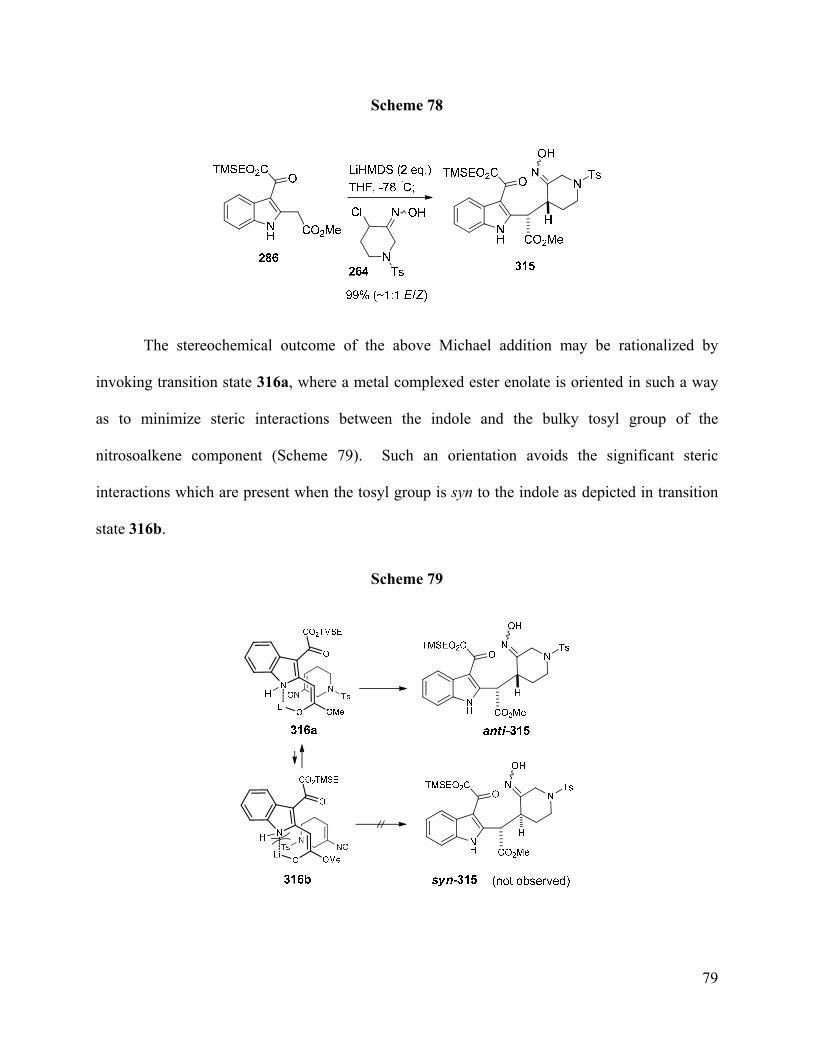
A more convergent and highly efficient reaction was developed, which employed the
conjugate addition of the dianion derived from 2,3-disubstituted indole 286, containing an
oxoacetate moiety at the indole C-3, to nitrosoalkene 265 to prepare Michael adduct 315 in
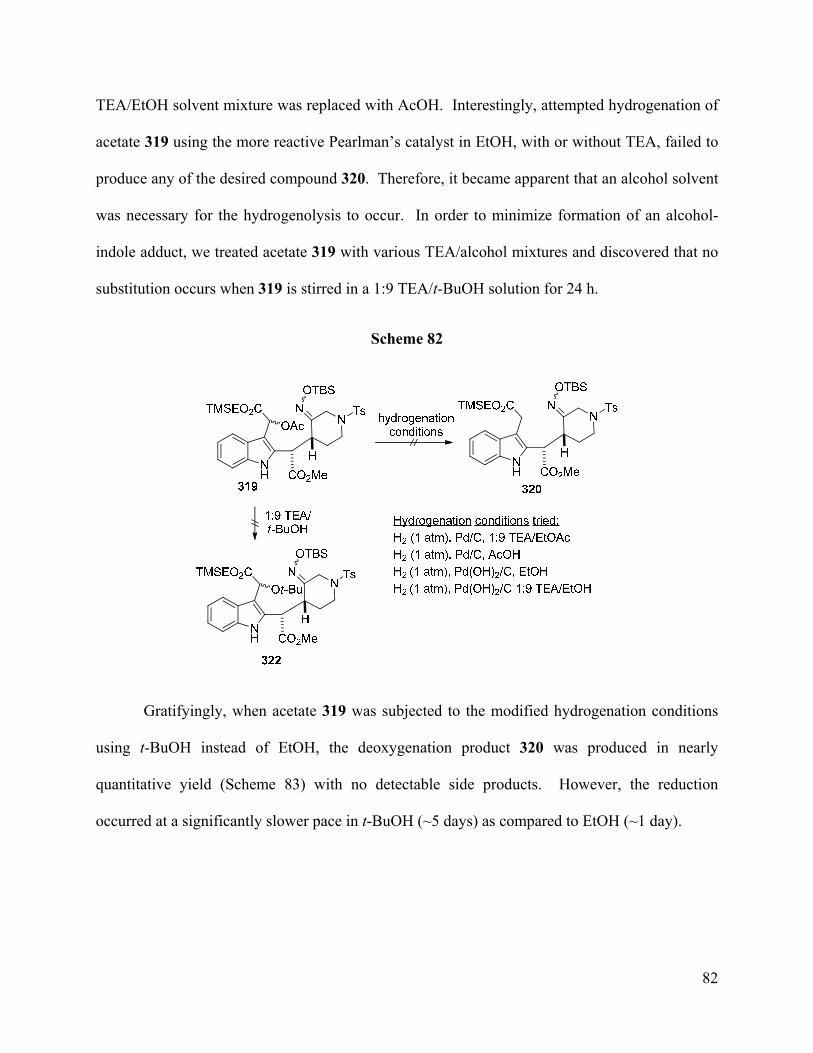
nearly quantitative yield. Additionally, an efficient stepwise deoxygenation sequence was
developed to reduce the ketone in 3-oxoacetate 315 after all attempts to directly reduce this
moiety in the related system 291 failed.
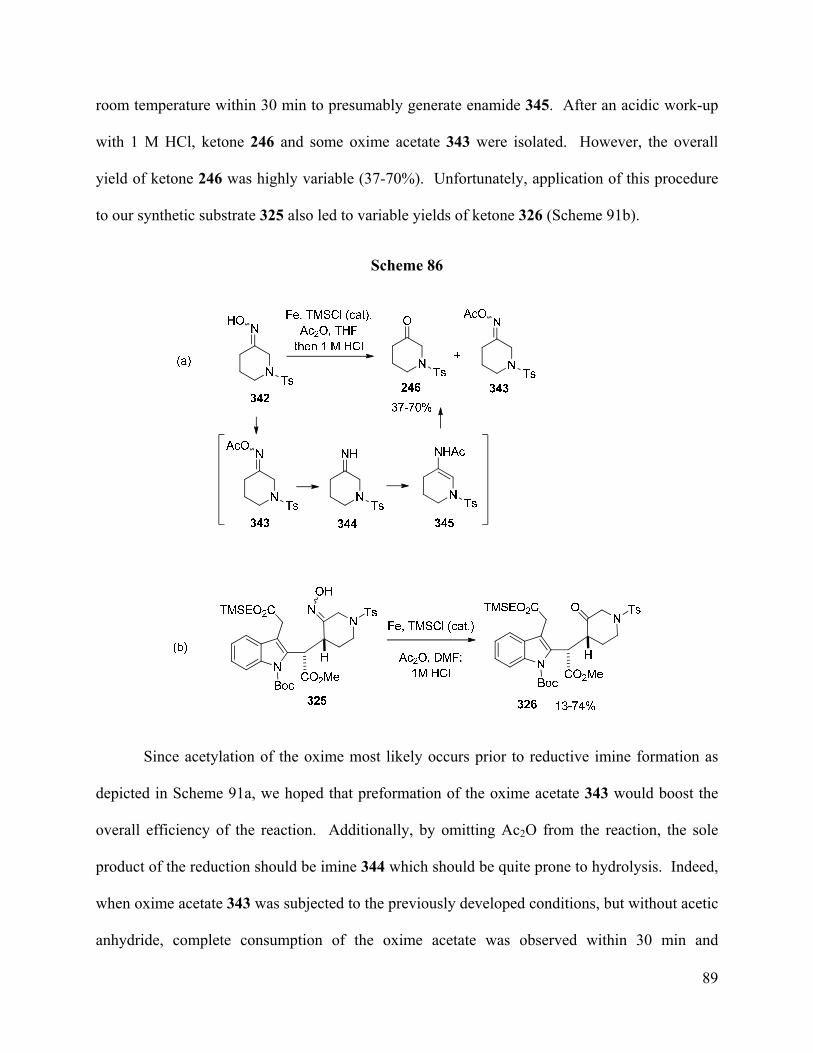
Attempted conversion of the indole containing oxime 325 to ketone 326 using standard
deoximation reactions led to low yields and/or product degradation. Therefore, a mild protocol
for the reductive deoximation of ketoxime pivalates was developed and was found to be
Page 4
iv
compatible with the indole containing ketoxime pivalates 354 and 359 as well as various other
model ketoxime pivalates. Ketone 360 was subsequently converted to the key keto-acid 361
which was subjected to Romo’s intramolecular aldol lactonization conditions to stereoselectively
form pentacyclic β-lactone 362 which contains the cis-azadecalin ring system found within the
alkaloids 167-169.
Numerous strategies have been examined for installing a C-17 hydroxymethyl group and
completing the total synthesis of alkaloids 167-169. It was discovered that intermolecular
alkylation of the enolate derived from indole ester 374 with formaldehyde produces the
hydroxymethyl compound 393 containing the undesired stereochemistry at C-17. On the other
hand, the C-17 carbon could be installed with the proper stereochemistry via a silicon-tethered
intramolecular ester enolate alkylation to provide the cyclic siloxane 408. However, numerous
attempts to oxidize siloxane 408 and its derivatives to a hydroxymethyl compound under
standard Fleming-Tamao conditions failed. Preliminary experiments using the Woerpel
modification to the Fleming-Tamao oxidation on cyclic siloxane 449 have identified this reaction
to be potentially useful for generation of the C-17 hydroxymethyl unit. Future optimization and
implementation of this procedure will provide the late stage intermediate triol 463, which will be
used to complete the total synthesis of angustilodine (167) and alstilobanine E (168). A Barton-
McCombie deoxygenation of the primary alcohol in cyclic siloxane 449 will produce siloxane
469, which should undergo a similar oxidation en route to a total synthesis of alstilobanine A
(169).
Page 5
v
TABLE OF CONTENTS
List of Figures ……….……………………………………………………………...…...…….. viii
List of Tables ……………………………………….……………………………...…..……..… ix
Acknowledgements ………….………………………………………………………………....... x
Chapter 1. Background and Introduction on Nitrosoalkenes ………………………...………….. 1
1.1 – General Information on Nitrosoalkenes …………………………………………………… 1
1.2 – Methods for Generation of Nitrosoalkenes ………………………………...……………… 1
1.2.1 – Base Promoted 1,4-Elimination from α-Heteroatom-functionalized Free Oximes …..…. 1
1.2.2 – Fluoride Promoted 1,4-Elimination from α-Halo-O-silyloximes ……………………….. 2
1.2.3 – Miscellaneous Addition and Elimination Methods ………………………...……………. 3
1.3 – Reactions of Nitrosoalkenes ………………………………………………………………. 8
1.3.1 – Tautomerization …………………………………………………………………………. 8
1.3.2 – Intramolecular Cyclizations ……………………………………………………………... 9
1.3.3 – Cycloadditions ……………………………………………………………………….… 10
1.3.3.1 – [4+2] Cycloadditions with Nitrosoalkenes as the 4π-Component ………………...…. 11
1.3.3.2 – [4+2] Cycloadditions with Nitrosoalkenes as the 2π-Component …………………… 15
1.3.3.3 – [3+2] Cycloadditions of Nitrosoalkenes ……………………………………………... 16
1.3.4 – Aromatic Substitutions ……………………………………………………………...….. 16
1.3.5 – Conjugate Additions to Nitrosoalkenes …………………………………………….….. 18
1.3.5.1 – Conventional Procedure for Conjugate Addition to Nitrosoalkenes ………………… 19
1.3.5.2 – Conjugate Addition to Nitrosoalkenes Generated via the Denmark Protocol ……..… 19
Page 6
vi
1.4 – Previous Weinreb Group Studies on Conjugate Addition of Nucleophiles to Nitrosoalkenes
……………………………………………………………………………………………..……. 21
Chapter 2 - Investigation of the Conjugate Addition of Carbon Nucleophiles to Nitrosoalkenes
Generated via the Denmark Protocol ……………………………………………………….…. 26
2.1 – Results and Discussion …………………………………………………………………... 26
Chapter 3 – Studies Toward a Total Synthesis of Angustilodine, Alstilobanine A, and
Alstilobanine E ……………………………………………………………………………….… 32
3.1 – General Background on Monoterpene Indole Alkaloids ……………………………….... 32
3.2 – Apparicine and Angustilodine-Type Alkaloids of the Aspidospermatan Subclass …….... 33
3.3 – Biosynthesis of Apparicine- and Angustilodine-Type Alkaloids ………………………... 37
3.4 – Previous Synthetic Studies on Apparicine-Type Alkaloids …………………………….... 39
3.5 – A Unified Strategy for Synthesis of Apparicine- and Angustilodine-Type Alkaloids …... 41
3.6 – First Generation Retrosynthetic Plan for the Synthesis of the Angustilodine Alkaloids .... 42
3.7 – Background on the Key Romo Aldol-Lactonization Methodology …………………….... 43
3.8 – Model Studies on the Conjugate Addition of Indole-2-acetate Enolates to Cyclic
Nitrosoalkenes ……………………………………………………………………………….…..47
3.9 – Revision to Retrosynthesis ……………………………………………………………..… 53
3.10 – Synthesis of the 3-Piperidone Nitrosoalkene Precursor ……………………………….... 54
3.11 – Conjugate Addition of Indole-2-acetate Methyl Esters to Nitrosoalkenes Derived from 3-
Piperidone …………………………………………………………………………………….... 62
3.12 – Attempted Installation of a C-3 Acetic Acid Unit on Michael Adduct 270 ………...….. 65
Page 7
vii
3.13 – Conjugate Addition of 2,3-Disubstituted Indoles to Nitrosoalkenes Revisited ……...…. 68
3.14 – Ketone Reduction of Indole-3-oxoacetates with Hydride Reagents ……………………. 74
3.15 – Stepwise Deoxygenation of Indole-3-oxoacetates ……………………………………… 78
3.16 – Development and Application of a New Reductive Deoximation Protocol ……………..83
3.17 – Synthesis of Keto Acid 361 and Subsequent Application of Romo’s Aldol Lactonization
………………………………………...………………………………………………………… 93
3.18 – Attempted Intermolecular Ester Enolate Alkylations to Install the C-17 Hydroxymethyl
Unit ………………………………………………………………………………………..…… 98
3.19 – Intramolecular Alkylation Strategy to Form a 7-Membered Ring Ether …………...…. 103
3.20 – Intermolecular Ester Enolate Alkylations Revisited ………..…………………………. 107
3.21 – A Silicon-tethered Intramolecular Ester Enolate Alkylation to Install C-17 ………..… 109
3.22 – Attempted Fleming-Tamao Oxidation of Cyclic Silyl Ethers ………………………… 115
3.23 – Reprotection of the Indole Nitrogen to Prevent a Retro Aldol Reaction …………….... 121
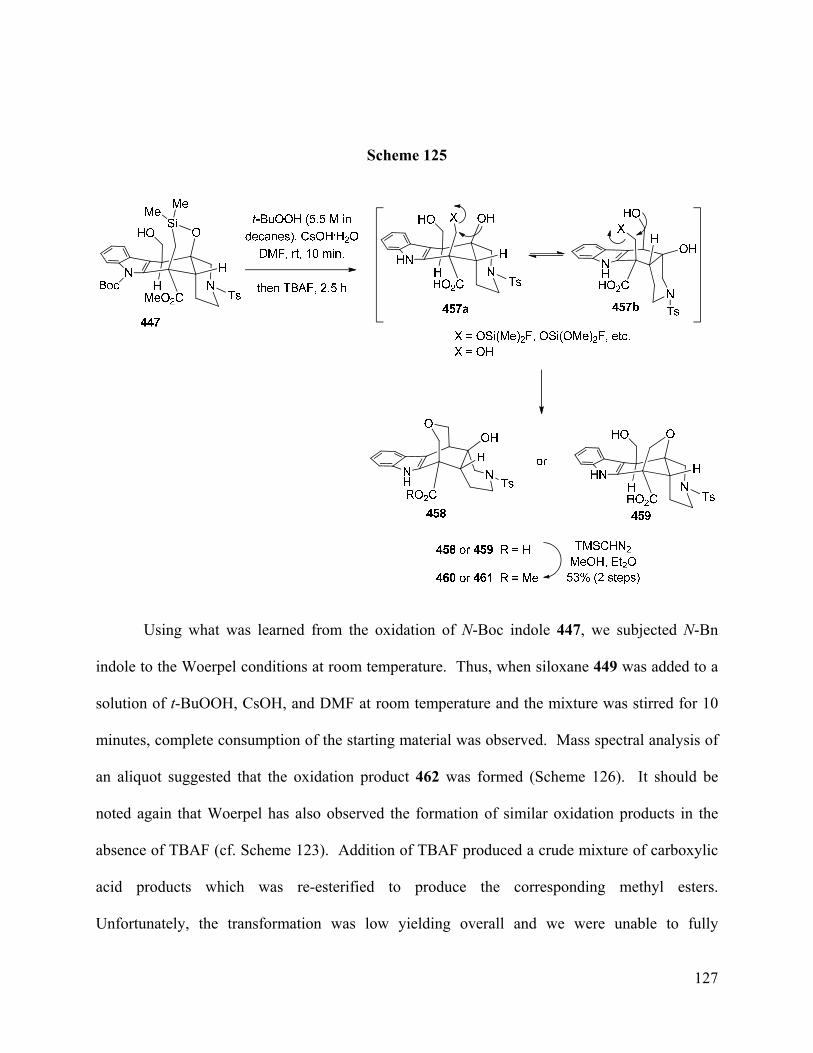
3.24 – Use of the Woerpel Modification of the Fleming-Tamao Oxidation …………………. 124
3.25 – Summary of Current Synthetic Route …………………………………………….…… 129
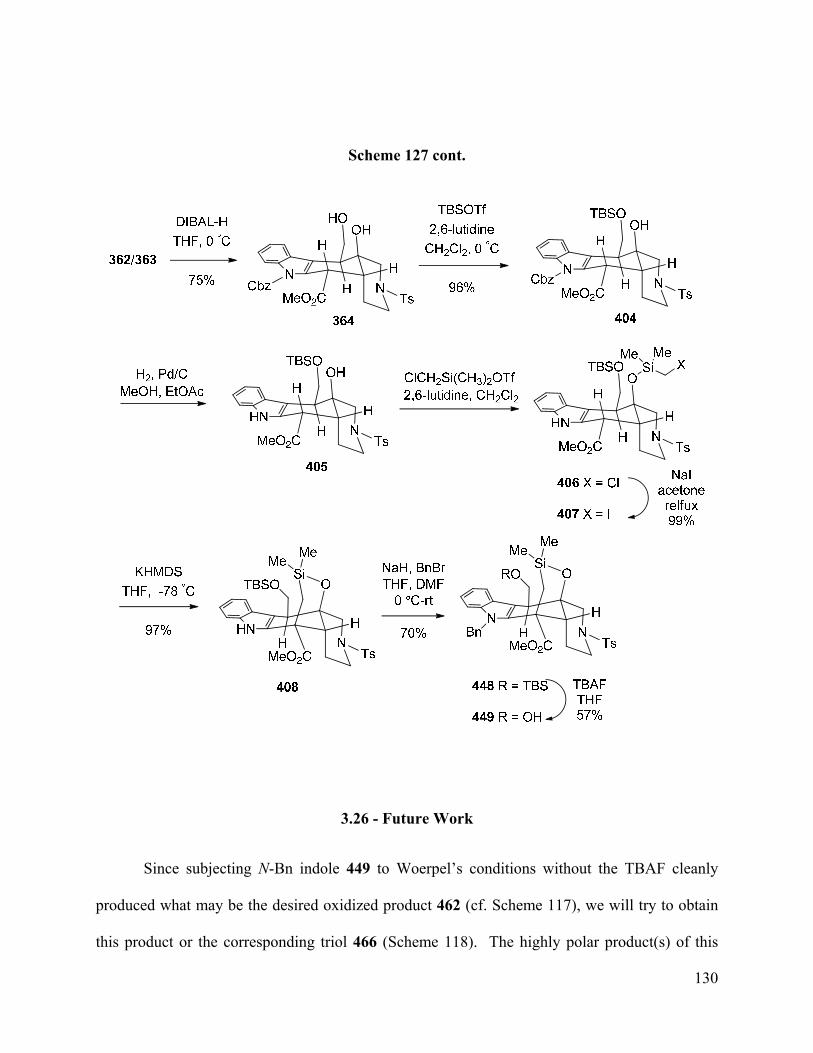
3.26 – Future Work …………………………………………………………...……………..... 130
Chapter 4 – Experimental Section ………...……………………………………….…………. 132
References and Notes ……………..…………………………………………………..………. 221
Page 8
viii
LIST OF FIGURES
Figure 1. Nitrosoalkene Structure ..………………………………………………………..…..… 1
Figure 2. Structure of 1,1-Bis-(t-butyl)-2-nitrosoethene …………………………………..…….. 9
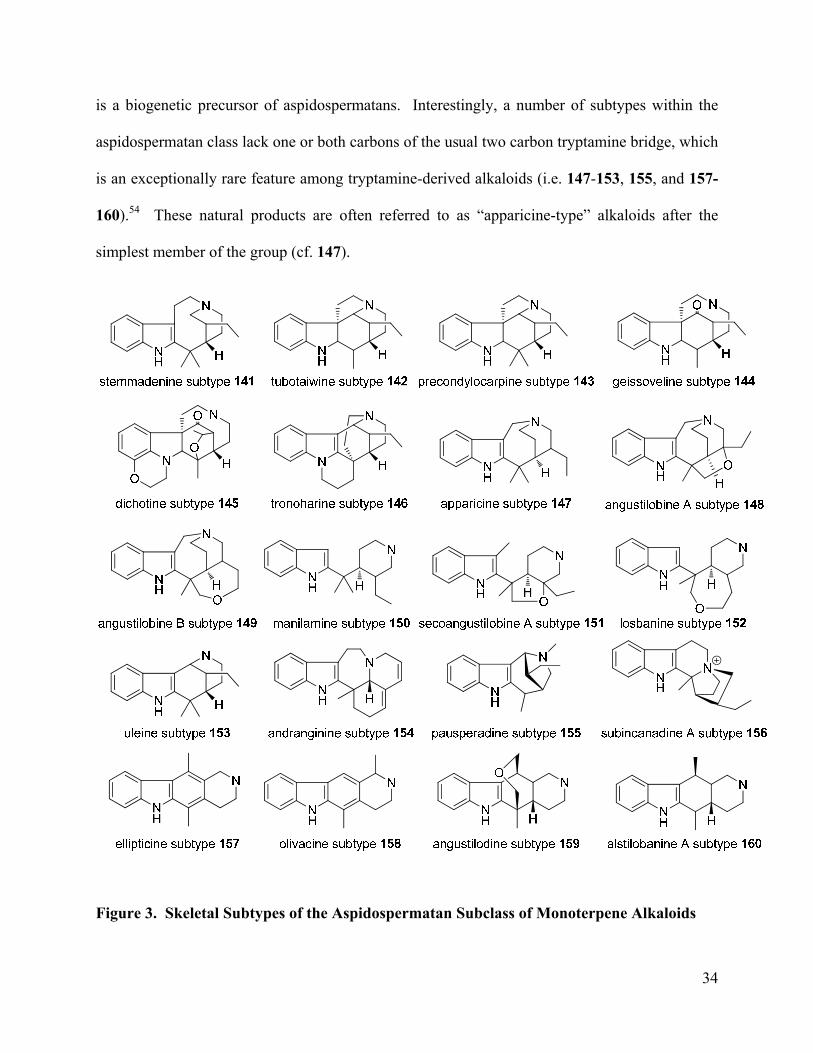
Figure 3. Skeletal Subtypes of the Aspidospermatan Subclass of Monoterpene Alkaloids ….... 34
Figure 4. Selected Apparicine-Type Alkaloids …………………………………...……………. 35
Figure 5. Angustilodine-Type Alkaloids ...………………………...……………..……….…… 36
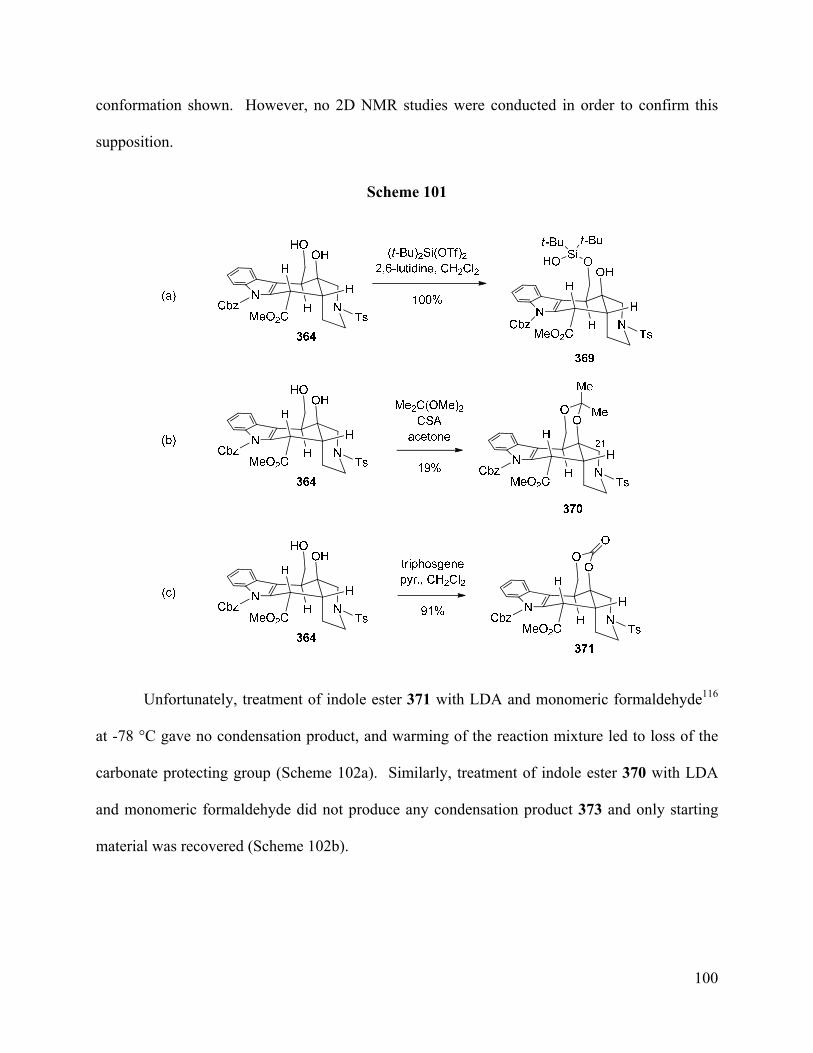
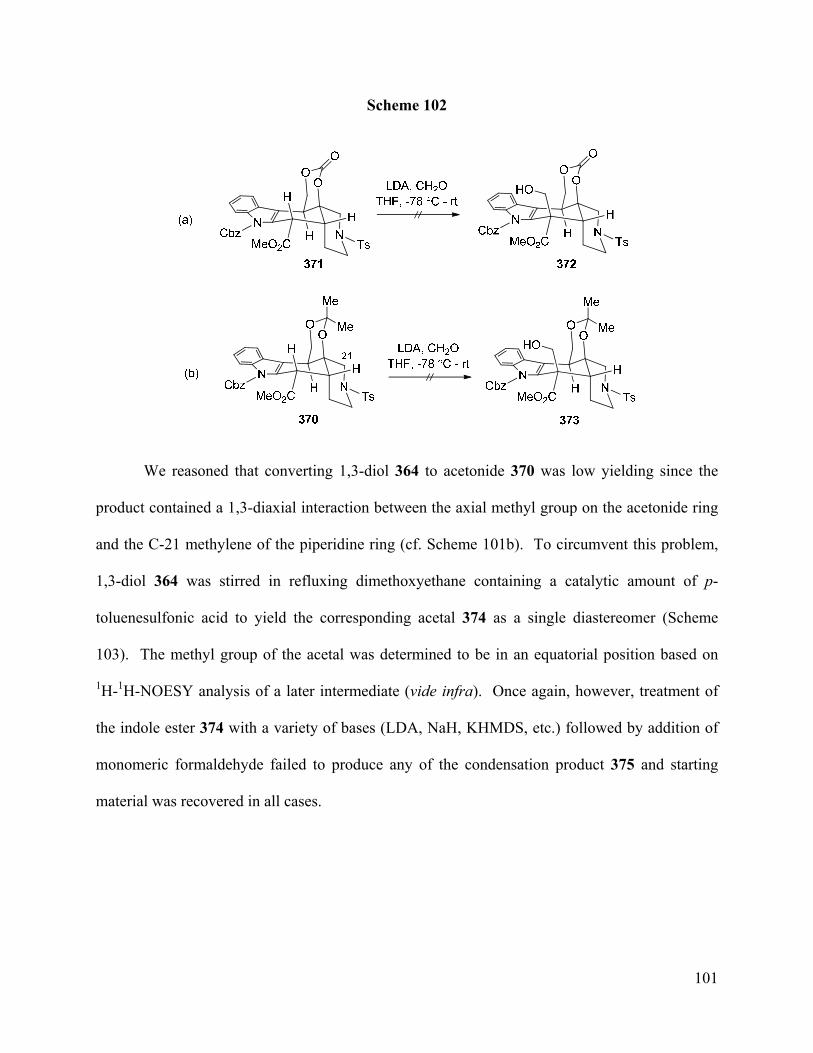
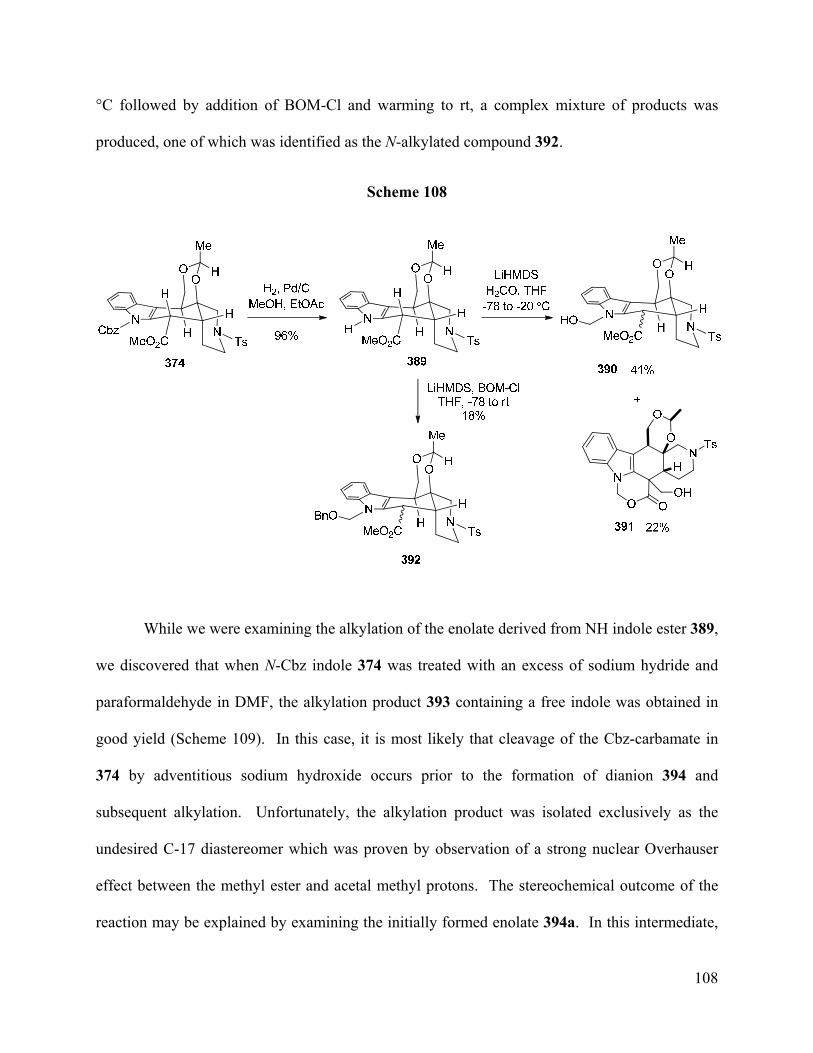
Figure 6. Conformations of Alstilobanine A (169) and Major 1,3-Diol 364 ……………..….… 98
Figure 7. ORTEP and Wireframe Structure of Compound 381 …………….………...……… 105
Figure 8. A1,3-Strain in N-Cbz Indole Enolate 385 …………………………………………… 107
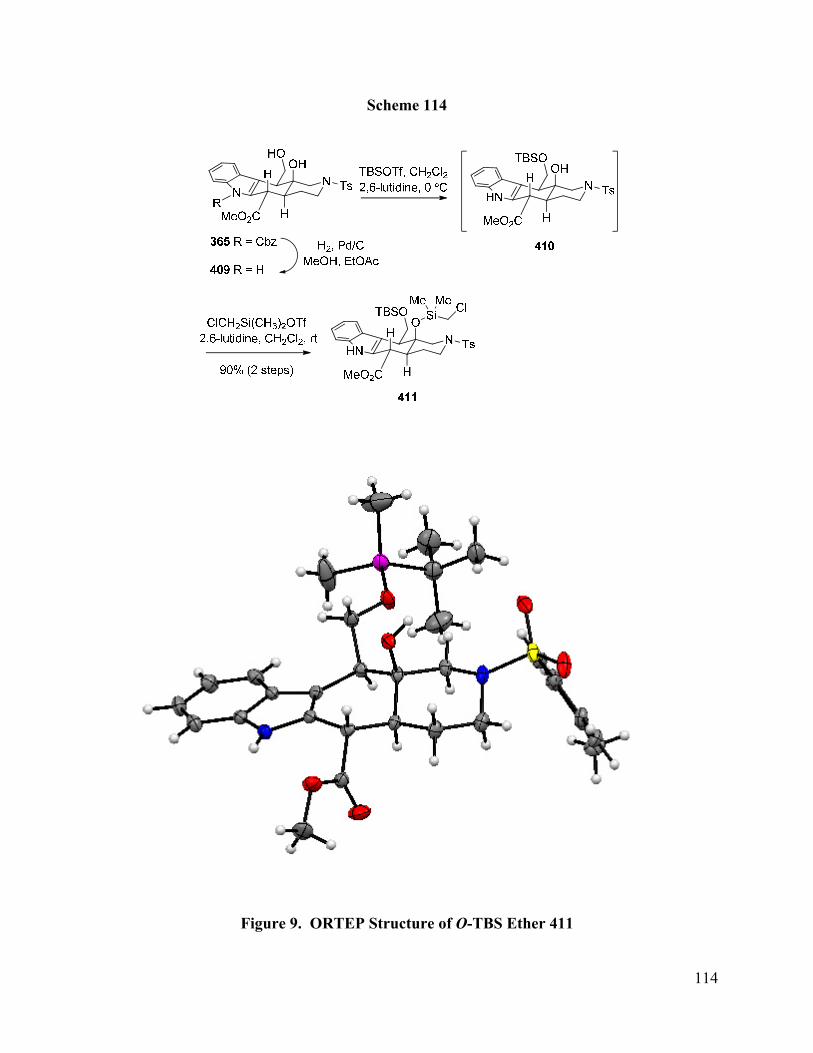
Figure 9. ORTEP Structure of O-TBS Ether 411 ...……………………………………...…… 114
Figure 10. ORTEP and Wireframe Structure of Z-357 …………………………...…..………. 186
Page 9
ix
LIST OF TABLES
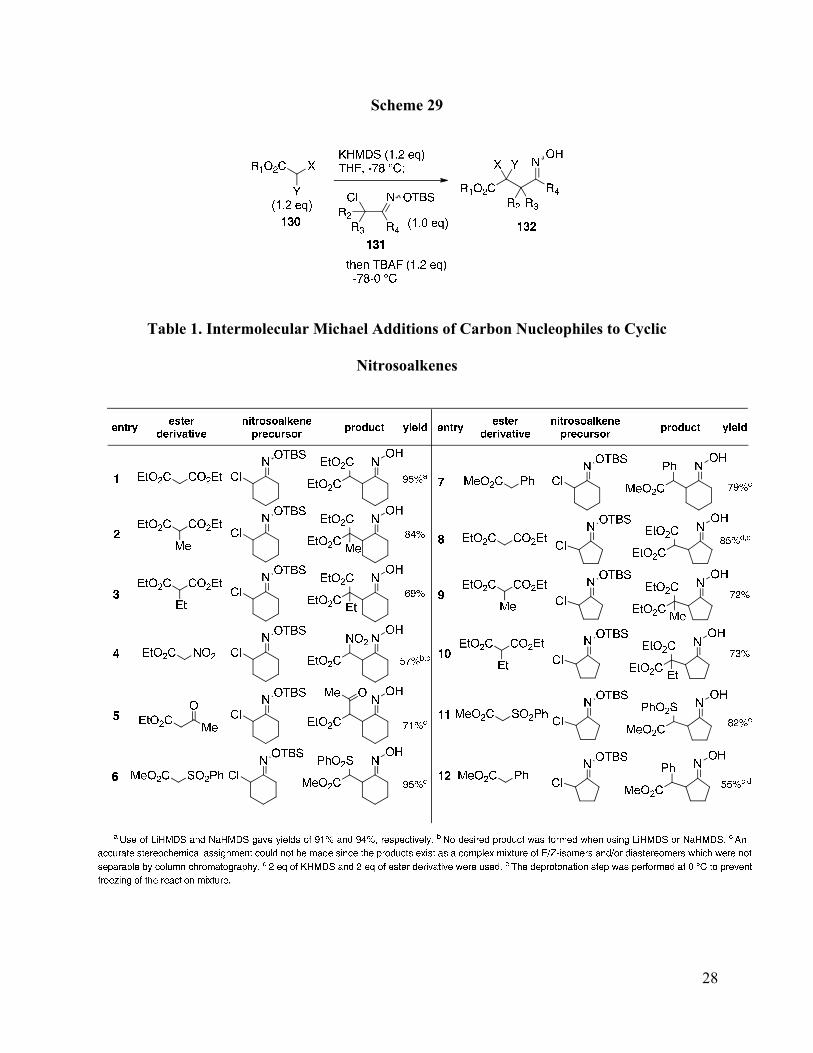
Table 1. Intermolecular Michael Additions of Carbon Nucleophiles to Cyclic Nitrosoalkenes . 28
Table 2. Intermolecular Michael Additions of Carbon Nucleophiles to Nitrosoalkenes Derived
from Aldoximes..…………………………………………………………………….…….…… 29
Table 3. Intermolecular Michael Additions of Carbon Nucleophiles to Terminal Nitrosoalkenes
..…………………………………………………………………………………………….…… 30
Table 4. Examples of β-Lactones Produced via Nucleophile-Catalyzed Aldol Lactonization ... 45
Table 5. Examples of Enantioselective Nucleophile-Catalyzed Aldol Lactonizations ….……. 46
Page 10
x
Acknowledgements
Over the course of my graduate studies, I have been very fortunate to work with a
number of great individuals. I am very grateful to my advisor, Professor Weinreb, for his overall
guidance through all aspects of my research and education. I will always appreciate the freedom
I was given to try my own ideas and also for the helpful advice during the many times I have
been stuck on a difficult research problem. I would also like to thank all of the Weinreb group
members who I have worked with over the years who have been great friends and coworkers.
A special thank you goes to my undergraduate advisor and close friend Chuck Kriley for
helping me to find my passion in synthetic chemistry and for his mentorship during my time at
Grove City College. Also, I could not be in this position today without the incredible support
and encouragement from my mom, my wife’s parents, and my siblings. I would especially like
to thank my dad, who gave me an interest in science and was my inspiration for pursuing a
career in chemistry. Most of all, I am grateful to my wife Kelly who has continually supported
me throughout graduate school despite all of the long hours, frustrations, and unforeseen
challenges that come with research. Kelly has also made a lot of personal sacrifices to allow me
to follow through with my career path and this thesis is dedicated to her.
Page 11
1
Chapter 1 – Background and Introduction on Nitrosoalkenes
1.1 – General Information on Nitrosoalkenes
In the late 19th century, nitrosoalkenes 1 were first postulated by Mathaipoulos as
transient intermediates in the reaction of α-halooximes with nucleophilic bases (Figure 1).1
Since then, the nitrosoalkene moiety has been revealed to be a highly reactive, typically unstable
functional group which usually has a lifetime on the order of seconds.2 The presence of a
nitrosoalkene may be validated in situ by spectroscopy,3 although, in general, indirect evidence
for the existence of nitrosoalkene intermediates has been gained by trapping as a Diels-Alder4 or
Michael-type adduct (vide infra).5 Additionally, some kinetic experiments have been conducted
which suggest the intermediacy of nitrosoalkenes in conjugate addition reactions.6 In very rare
cases, nitrosoalkenes which contain a bulky aryl,7 t-alkyl,8 or halo substituent9 at the β-carbon
may be isolated.
NO
R3R1
R21
Figure 1. Nitrosoalkene Structure
1.2 – Methods for Generation of Nitrosoalkenes
1.2.1 – Base Promoted 1,4-Elimination from α-Heteroatom-functionalized Oximes
Over the last 100 years, a number of different methods have been developed for the
generation of nitrosoalkenes from a variety of synthetic precursors. However, base promoted
Page 12
2
1,4-elimination of α-functionalized free oximes such as 2, where X is a suitable leaving group, is
the simplest and most frequently employed method (Scheme 1).2 In principle, the leaving group
component may be any heteroatom-based functional group which stabilizes a negative charge,
although in the vast majority of cases, a halogen (usually chlorine) is employed. Some other rare
examples of leaving groups include sulfonates,10 phenylsulfinates,11 nitrite,8 sulfoxides,12 and
oxirane oxygens.13
Scheme 1
A variety of bases may be used to effect nitrosoalkene formation. However, the type of
base employed can have a dramatic impact on the outcome of the reaction. For instance,
sparingly soluble inorganic bases, such as metal carbonates14 and hydroxides4a,15 are often used
to generate nitrosoalkenes in a slow and controlled manner in order to minimize side reactions
such as polymerization. On the other hand, soluble organic bases such as amines7,8 or
alkoxides16 may be employed for rapid generation of the nitrosoalkene. In many instances
involving nucleophilic addition to nitrosoalkenes, the nucleophilic component may also act as
the base (vide infra).5
1.2.2 – Fluoride Promoted 1,4-Elimination from α-Halo-O-silyloximes
Denmark and coworkers have developed a useful alternative to the conventional base
promoted generation of nitrosoalkenes which involves 1,4-elimination of α-chloro-silyloximes 3
Page 13
3
by fluoride (Scheme 2).13b,17 One notable advantage of this protocol is that the nitrosoalkene can
be generated at a specific point in a reaction upon addition of the fluoride source, typically
tetrabutylammonium fluoride (TBAF) or cesium fluoride. Interestingly, Denmark found that
nitrosoalkenes derived from t-butylcyclohexanone generated via this protocol are very reactive
and have approximately 3-5 times shorter half-lives than those generated from the corresponding
free α-chlorooximes and triethylamine.13b Consequently, reactions employing this method of
nitrosoalkene generation should proceed at a considerably faster rate. Denmark also discovered
that the stereochemical orientation of the chlorine and/or oxime geometry have little to no effect
on the efficiency of nitrosoalkene generation when employing either the classical or fluoride-
promoted method.13b This observation is significant since it allows for maximum flexibility in
preparation of the nitrosoalkene precursors.
Scheme 2
1.2.3 – Miscellaneous Addition and Elimination Methods
Although most nitrosoalkenes used in organic synthesis have been generated via
elimination of α-chlorooximes and α-chloro-O-silyloximes, a number of alternative methods
exist, but are rather limited in their scope. Combination of vinyl radicals with nitric oxide is a
direct, albeit usually inefficient way to form a nitrosoalkene.9,18 This method was employed by
Griffin and Haszeldine to produce the very first isolable nitrosoalkene, 1,1,2-
Page 14
4
trifluoronitrosoethylene (5) (Scheme 3) by irradiating 1,1,2-trifluoroiodoethylene (4) in the
presence of nitric oxide.
Scheme 3
One of the oldest known methods for preparation of α-chlorooximes, the most common
precursor to nitrosoalkenes, is to treat olefins with nitrosyl chloride (Scheme 4).19 The initially
formed nitroso-chloroalkane 7, which typically tautomerizes to form α-chlorooxime 8, may also
dimerize to yield appreciable amounts of diazene dioxide 9. As with the α-chlorooxime 8, the
nitrosochloride dimers 9 may be treated with nucleophilic bases to generate a common
nitrosoalkene intermediates 10 and Michael adducts 11.20
Scheme 4
Appropriately substituted silyl nitronates can be converted to nitrosoalkenes by
elimination of trialkylsiloxide (Scheme 5). For example, silyl nitronates such as 12 are
Page 15
5
converted to nitrosoalkenes such as 13 upon treatment with alkyllithium bases (Scheme 5a).21
Generally, excess alkyllithium reagent adds to the nitrosoalkene intermediate in a Michael
fashion. More recently, it was discovered that treatment of β-nitroesters such as 15 with N,O-
bis(trimethylsilyl)acetamide (BSA) provides silyl nitronate intermediates 16 which undergo
spontaneous elimination to the corresponding nitrosoalkenes such as 17 (Scheme 5b).22 This
type of nitrosoalkene generation protocol has been used in hetero-Diels-Alder cycloadditions
with enol ethers to yield cycloadducts such as 18 (vide infra).
Scheme 5
Interestingly, the addition of the sulfoxonium ylide 19 to aryl nitrile oxides such as 20
can produce nitrosoalkene intermediates 21 following the expulsion of dimethyl sulfoxide
(Scheme 6). Typically, in this type of reaction the intermediate nitrosoalkene 21 will react with
excess ylide 19 to ultimately produce adducts such as 22 and 23.23
Page 16
6
Scheme 6
In certain cases, ring fragmentations can lead to nitrosoalkene intermediates.
Abramovitch24 and others25 have observed both the thermal and photochemical decomposition of
2-azidopyridine N-oxides such as 24 to form oxazines 26 and nitrones 27 (Scheme 7). It is most
likely that these products are generated from a dienylnitroso intermediate like 25.
Scheme 7
Another, ring opening reaction to generate nitrosoalkenes is by reaction of α-bromo-
oxazinones with hydroxide (Scheme 8).26 For example, reaction of 28 with sodium hydroxide
leads to the nitrosoalkene tautomer 30 in high yield. In the few substrates examined,
nitrosoalkene formation appears to occur with good efficiency. However, no examples of using
intermediates like 29 in either Michael additions or cycloadditions have been reported.
Scheme 8
Page 17
7
Retro-Michael reactions27 and retro-Diels-Alder reactions,28 especially those that undergo
ring cleavage to produce more stable systems (i.e. aromatic rings) have been postulated to
proceed via nitrosoalkene intermediates (Scheme 9). However, it appears that the reversibility of
these types of reactions affects the overall ability to generate the nitrosoalkene intermediate
stoichiometrically. For example, Gilchrist and coworkers showed that thermal fragmentation of
indolinooxazine 31 generates methyl 2-nitrosoacrylate (33) which can be trapped with indole or
n-pentanol to produce adducts 34 or 35, respectively.
Scheme 9
Very recently, Hsieh and Dong have developed a novel Pd-catalyzed reduction of 2,2-
bis(aryl)-1-nitroalkenes to nitrosoalkenes en route to the synthesis of 3-arylindoles (Scheme 10).
Mechanistically, this reaction most likely proceeds via the palladium bound nitrosoalkene 38, a
product of decarboxylation from the initially formed palladacycle 37. Further cyclization and a
proton shift leads to the N-hydroxyindole 40, which is reduced further to indole 41 by the Pd-
catalyst and a second equivalent of CO.
Page 18
8
Scheme 10
NO2 NOO
[Pd]O Ln
Pd(OAc)2,1,10-phenanthrolineCO, DMF, 110 C
NO[PdLn]
-CO2
NO
[LnPd]
NOH
[LnPd]
CO
-CO2, -[PdLn]
HN
97%
36 37 38 39
40 41
1.3 – Reactions of Nitrosoalkenes
1.3.1 - Tautomerization
Probably the most common degradation pathway for nitrosoalkenes which bear an allylic
hydrogen is tautomerization.29 For example, Kisan and Prtizkow found that treatment of either
α-chlorooxime 42 or α-sulfatooxime 43 with triethylamine (TEA) produces a blue-colored
solution (λmax = 740 nm) corresponding to formation of nitrosoalkene 44.29a In the absence of
nucleophiles, this solution fades to colorless within 15-30 min, which most likely is due to a
conversion of nitrosoalkene 44 to tautomers 45 and 46.
Page 19
9
Scheme 11
Nitrosoalkene systems without allylic hydrogens tend to be more stable since
tautomerization is not possible. For example, nitrosoalkene 47, which lacks allylic hydrogens
exists as an isolable blue solid with a melting point of 38 °C (Figure 2).8
Figure 2. Structure of 1,1-Bis-(t-butyl)-2-nitrosoethene
1.3.2 – Intramolecular Cyclizations
Another unimolecular mode of decomposition that may be observed for nitrosoalkenes is
an intramolecular 4π-electrocyclization to form oxazetes 48 (Scheme 12). Typically, these
highly strained intermediates undergo fragmentation to produce the corresponding nitriles 50 and
carbonyl compounds 49. For example, treatment of α-chlorooximes 51 and 53 with NaHCO3
yields benzonitrile and benzophenone, respectively (Scheme 12b,c).3
Page 20
10
Scheme 12
Another noteworthy unimolecular reaction involving intramolecular cyclizations of
dienylnitroso compounds to form oxazines and nitrones was previously discussed (cf. Scheme 7).
1.3.3 – Cycloadditions
Cycloadditions are one of the most synthetically useful reactions involving nitrosoalkenes
and therefore much attention has been directed to this area. In the majority of cases,
nitrosoalkenes tend to participate as the 4π component in inverse electron demand hetero-Diels-
Alder cycloadditions (Scheme 13, path A). However, several instances where the N-O π-bond
acts as the dienophile have also been documented (path B). Although the C-C double bond of
nitrosoalkenes could theoretically participate as a dienophile, there are no known examples of
products arising from path C. However, such products, if formed, may undergo a facile [3,3]
sigmatropic rearrangement to form the products of a path A type cycloaddition. Products of
[3+2] cycloadditions (path D) have only been isolated as side products in Diels-Alder reactions.
Page 21
11
However, recent developments have uncovered favorable pathways for synthesizing nitrones via
formal [3+2] cycloadditions (vide infra).
Scheme 13
1.3.3.1 - [4+2] Cycloadditions with Nitrosoalkenes as the 4π-Component
Over the years, both inter- and intramolecular hetero-Diels-Alder cycloadditions in which
a nitrosoalkene is the 4π-component have developed into a versatile set of reactions for the
construction of highly functionalized 1,2-oxazines. A number of different electron rich olefins
can be utilized with a variety of electrophilic nitrosoalkenes in inverse electron demand [4+2]-
cycloadditions.15,30 Suitable dienophiles for this reaction include, among others, enol ethers,
enamines, allyl silanes, allenes, and even fullerenes.31 In a representative example, Reissig and
coworkers demonstrated the highly electrophilic 1-nitroso-1-trifluoromethylethylene (56)
undergoes hetero-Diels-Alder cycloadditions with allyl trimethylsilane, 3,4-dihydro-2H-pyran,
Page 22
12
and methoxyallene to give the corresponding cycloadducts in modest to good overall yield
(Scheme 14).32
Scheme 14
CF3
NO
NO
Me3Si
NOO
NOMeO
CF3
NBr
HONa2CO3MeOtBu SiMe3
O
MeO
31%
70%
92%
CF3
CF3
CF3
55 56
57
58
59
In some instances, the [4+2]-cycloadducts obtained in these reactions may actually be
formed via stepwise pathways. For example, Gilchrist has shown that furans and enamines add
efficiently to α-nitrosostyrene to give the products of a formal [4+2]-cycloaddition (Scheme 15).
Since a number of other dienophiles failed in this reaction (i.e. cyclopentene, anthracene, 1,3-
diphenylisobenzofuran, and dimethyl acetylenedicarboxylate), Gilchrist postulated that
formation of a normal electron demand Diels-Alder adduct 61 occurs initially and subsequent
[3,3] sigmatropic rearrangement produces 62.33 In seminal work, Gilchrist also demonstrated
that enamines react particularly well with nitrosoalkenes to give formal Diels-Alder adducts.
However, such reactions most likely proceed via an initial Michael addition to produce an
oximate-iminium ion species 63 which undergoes further cyclization to produce the formal
[4+2]-cycloadduct 64. Recent theoretical calculations on these types of reactions by Domingo et
al. further reinforce this hypothesis.34
Page 23
13
Scheme 15
Despite the potential synthetic utility of 1,2-oxazines in complex molecule synthesis,35
surprisingly few examples of hetero-Diels-Alder reactions with nitrosoalkenes to construct
natural products or other complex systems are found in the literature. However, Gallos et al.
have recently synthesized the cytotoxic alkaloid (±)-crispine (69) utilizing a nitrosoalkene
hetero-Diels-Alder cycloaddition as the key step. Thus, a mixture of α-bromooxime 66 and
Na2CO3 was stirred in neat ethyl vinyl ether to provide cycloadduct 67, which contains all atoms
necessary for completion of the natural product. Next, a reductive cyclization of the C=N bond
in 67 provided the tricyclic product 68 which was converted to the natural product in two
additional reductive transformations.
Page 24
14
Scheme 16
Denmark has explored the utility of intramolecular [4+2]-cycloadditions of
nitrosoalkenes.21 It was found that α-chloro-O-silyloximes tethered to a methylenol ether such
as 70 are capable of undergoing efficient cycloadditions to produce tricyclic oxazine compounds
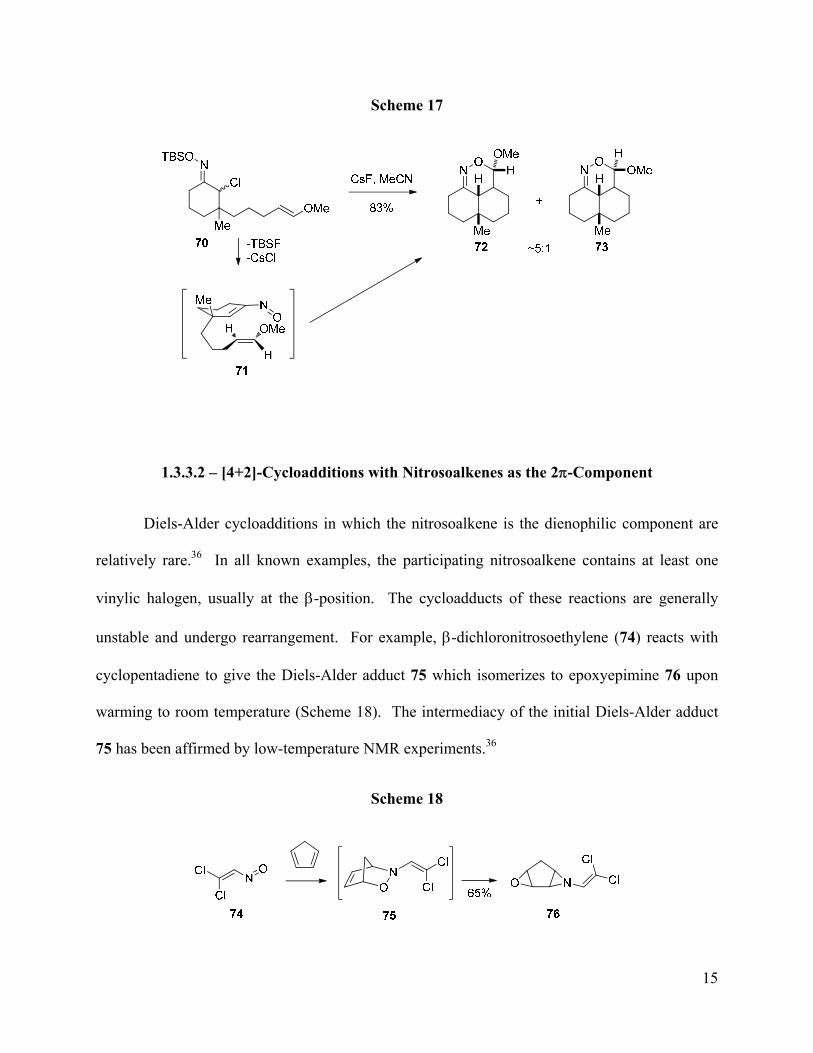
such as 72 and 73 with good levels of diastereoselectivity (Scheme 17). Use of the sparingly
soluble cesium fluoride was critical for successful reactions to occur since rapid generation of the
nitrosoalkene with TBAF led to inferior results. Interestingly, use of Z-enol ethers led to
considerably lower yields, which may be due to a preference for the methoxy group in transition
state 71 to be endo to the nitrosoalkene.
Page 25
15
Scheme 17
1.3.3.2 – [4+2]-Cycloadditions with Nitrosoalkenes as the 2π-Component
Diels-Alder cycloadditions in which the nitrosoalkene is the dienophilic component are
relatively rare.36 In all known examples, the participating nitrosoalkene contains at least one
vinylic halogen, usually at the β-position. The cycloadducts of these reactions are generally
unstable and undergo rearrangement. For example, β-dichloronitrosoethylene (74) reacts with
cyclopentadiene to give the Diels-Alder adduct 75 which isomerizes to epoxyepimine 76 upon
warming to room temperature (Scheme 18). The intermediacy of the initial Diels-Alder adduct
75 has been affirmed by low-temperature NMR experiments.36
Scheme 18
Page 26
16
1.3.3.3 – [3+2]-Cycloadditions of Nitrosoalkenes
Until recently, the only instances of nitrosoalkenes participating in [3+2]-cycloadditions
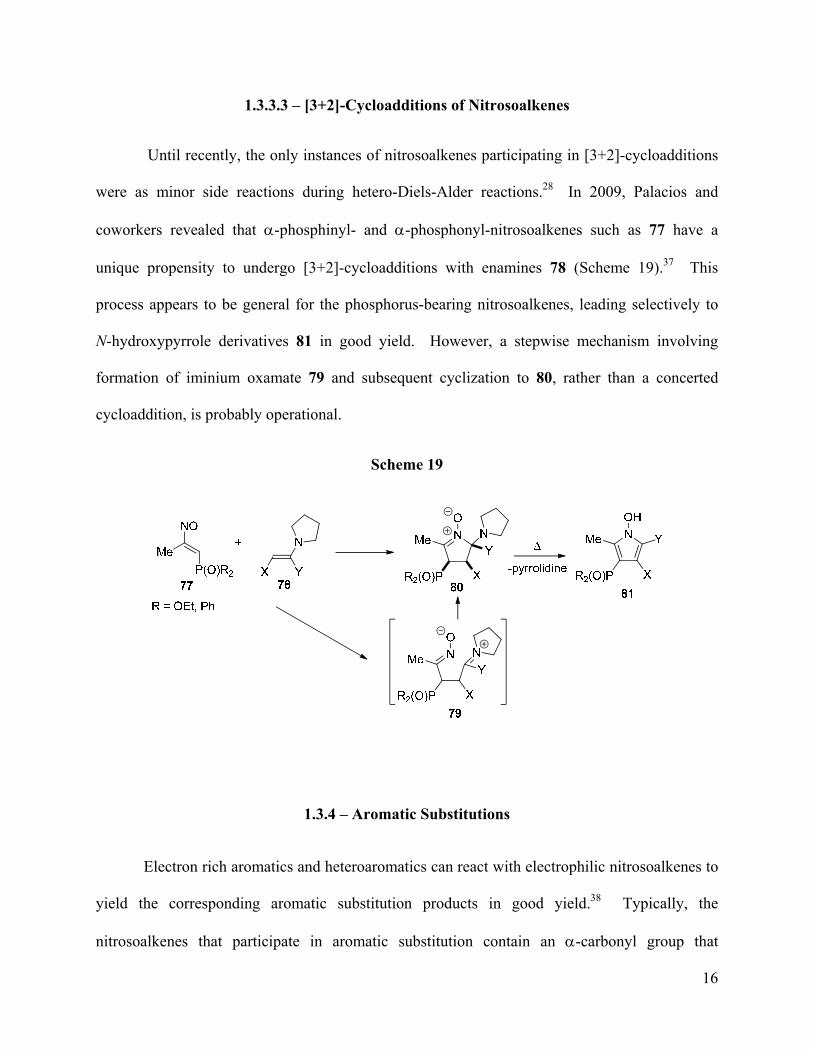
were as minor side reactions during hetero-Diels-Alder reactions.28 In 2009, Palacios and
coworkers revealed that α-phosphinyl- and α-phosphonyl-nitrosoalkenes such as 77 have a
unique propensity to undergo [3+2]-cycloadditions with enamines 78 (Scheme 19).37 This
process appears to be general for the phosphorus-bearing nitrosoalkenes, leading selectively to
N-hydroxypyrrole derivatives 81 in good yield. However, a stepwise mechanism involving
formation of iminium oxamate 79 and subsequent cyclization to 80, rather than a concerted
cycloaddition, is probably operational.
Scheme 19
1.3.4 – Aromatic Substitutions
Electron rich aromatics and heteroaromatics can react with electrophilic nitrosoalkenes to
yield the corresponding aromatic substitution products in good yield.38 Typically, the
nitrosoalkenes that participate in aromatic substitution contain an α-carbonyl group that
Page 27
17
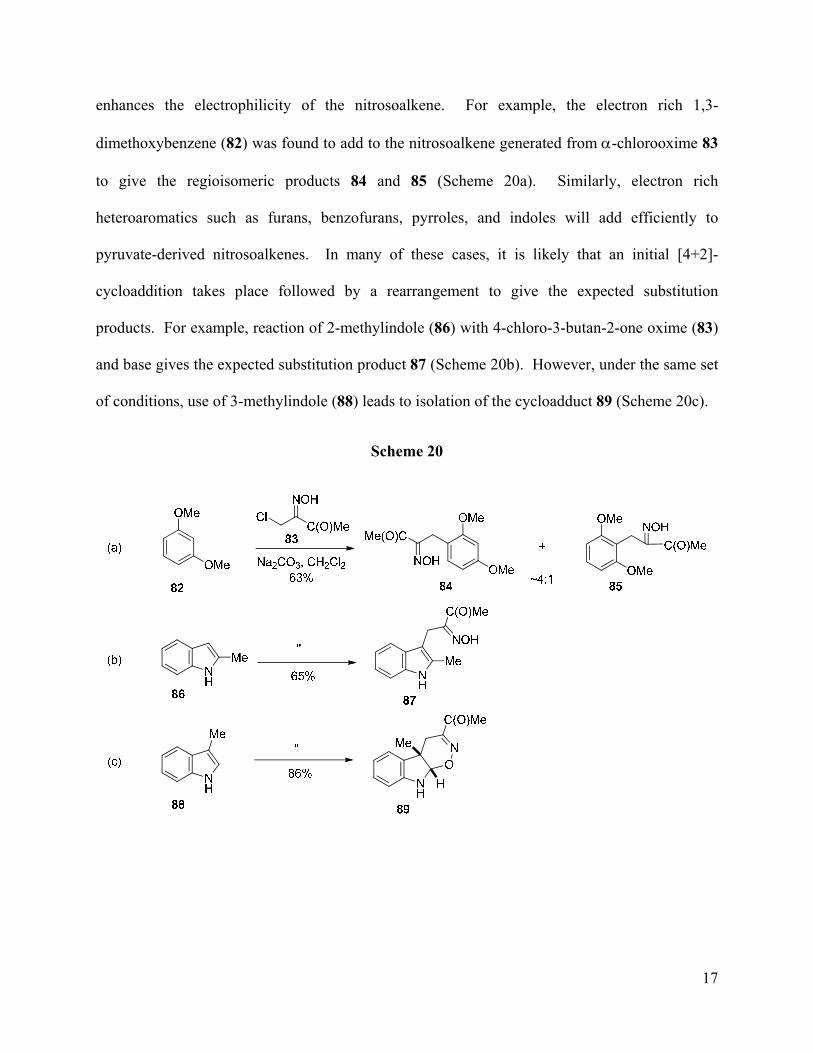
enhances the electrophilicity of the nitrosoalkene. For example, the electron rich 1,3-
dimethoxybenzene (82) was found to add to the nitrosoalkene generated from α-chlorooxime 83
to give the regioisomeric products 84 and 85 (Scheme 20a). Similarly, electron rich
heteroaromatics such as furans, benzofurans, pyrroles, and indoles will add efficiently to
pyruvate-derived nitrosoalkenes. In many of these cases, it is likely that an initial [4+2]-
cycloaddition takes place followed by a rearrangement to give the expected substitution
products. For example, reaction of 2-methylindole (86) with 4-chloro-3-butan-2-one oxime (83)
and base gives the expected substitution product 87 (Scheme 20b). However, under the same set
of conditions, use of 3-methylindole (88) leads to isolation of the cycloadduct 89 (Scheme 20c).
Scheme 20
Page 28
18
1.3.5 – Conjugate Additions to Nitrosoalkenes
Conjugate addition of nucleophiles to nitrosoalkene intermediates provides a useful but
underutilized means for the synthesis of α-functionalized carbonyls (Scheme 21). In this
manner, the nitrosoalkene 1 can act as an enolonium ion surrogate (92),39 allowing for the
umpolung construction of α-functionalized carbonyls 91 as compared to standard enolate
chemistry. Although several direct SN2 displacements of α-haloketones,40,41 and to a lesser
extent α-haloaldehydes,42 have been documented, this method is not general since many
nucleophiles preferentially add to the electrophilic C-O π-bond. Furthermore, treatment of α-
haloketones with malonates and other nucleophilic bases can induce undesirable Favorskii
rearrangements.43
Scheme 21
A wide array of hetero- and carbon-based nucleophiles have been shown to add in a
Michael fashion to nitrosoalkenes. Heteroatom nucleophiles found to participate in this reaction
include alcohols, acetates, amines, azides, nitrites, and thiols.2,5 Carbon nucleophiles include
ester and ketone enolates, β-dicarbonyls, malononitrile, acetylides, sulfoxonium ylides (cf.
scheme 6) and alkyl or aryl Grignard reagents.2,5 As mentioned above, carbon nucleophiles such
as enamines and enol ethers, which give formal [4+2]-cycloadducts, most likely react through a
stepwise conjugate addition/cyclization pathway (cf. Scheme 15).
Page 29
19
1.3.5.1 – Conventional Procedure for Conjugate Addition to Nitrosoalkenes
In the vast majority of cases, two or more equivalents of the nucleophile is added to a free
α-halooxime since the first equivalent effects a 1,4-elimination to generate the nitrosoalkene.
For example, in seminal studies on nucleophilic additions to α-chlorocycloalkanone oximes,
Ohno et al. showed that subjection of oxime 93 to a large excess of either morpholine, sodium
azide, sodium nitrite, phenylacetylenemagnesium bromide, or diethyl malonate sodium salt
produced the corresponding Michael adducts 94 in good yields (Scheme 22).5
Scheme 22
1.3.5.2 – Conjugate Addition to Nitrosoalkenes Generated via the Denmark Protocol
Despite the propensity of nitrosoalkenes to participate in Michael additions with a large
variety of nucleophiles, nitrosoalkene methodology has found surprisingly little use in organic
synthesis. This may be due in part to the fact that the classical procedure for nitrosoalkene
generation, which generally requires two or more equivalents of nucleophile, is inefficient,
Page 30
20
particularly if the nucleophile is valuable. However, this problem can be overcome by
generating nitrosoalkenes via the Denmark protocol in which only 1 equivalent of nucleophile is
required. For example, Barrett and coworkers demonstrated that a single equivalent of the
valuable anomeric alcohol 95 adds to one equivalent of the nitrosoalkene generated from α-
chloro-O-TBS-oxime 96 to yield glycoside 97 (Scheme 23a).44 Surprisingly, this type of
strategy has found only sporadic use in related Michael additions (vide infra).45 While the
majority of these examples involve addition of oxygen, nitrogen, or sulfur nucleophiles, only a
few instances involving the addition of a carbon nucleophile have been documented. In one such
case, Hassner and coworkers showed that the sodium salt of allyl malonate 98 adds to 1-
nitrosobutene, generated in situ from α-bromo-O-TMS-oxime 99 and TBAF, to give adduct 100
in good yield (Scheme 23b).45e However, since 3 equivalents of the malonate nucleophile were
employed in this case, little efficiency was demonstrated in this particular alkylation.
Scheme 23
Page 31
21
1.4 – Previous Weinreb Group Studies on Conjugate Addition of Nucleophiles to
Nitrosoalkenes
In recent years, the Weinreb group has begun to systematically investigate Michael
additions to nitrosoalkenes in order to explore their potential as enolonium ion equivalents. The
preliminary investigations were aimed at developing the first intramolecular conjugate additions
of tethered nucleophiles to nitrosoalkenes to form fused and bridged ring systems.46 To access
the requisite substrates for this process, two reactions developed in the Weinreb group were
relied upon, namely, the ring closing metathesis of vinyl chlorides47 and the regioselective
oxidation of vinyl chlorides to α-chloroketones.48 Thus, diene 101 was converted to cyclic vinyl
chloride 102 using Grubb’s 2nd generation catalyst (Scheme 24). Next, treatment of 102 with
sodium hypochlorite and acetic acid in acetone produced α-chloroketone 103 and subsequent
oximation with O-TBS-hydroxylamine provided α-chloro-O-TBS-oxime 104 as an
inconsequential mixture of diastereomers and geometric isomers. Since nitrosoalkenes have very
short lifetimes, deprotonation of the tethered malonate with NaHMDS was performed first,
followed by addition of TBAF to unveil the nitrosoalkene functionality. After warming the
reaction mixture to 0 °C, the [2.2.2]-bicyclic product 105 was produced in 74% yield.
Page 32
22
Scheme 24
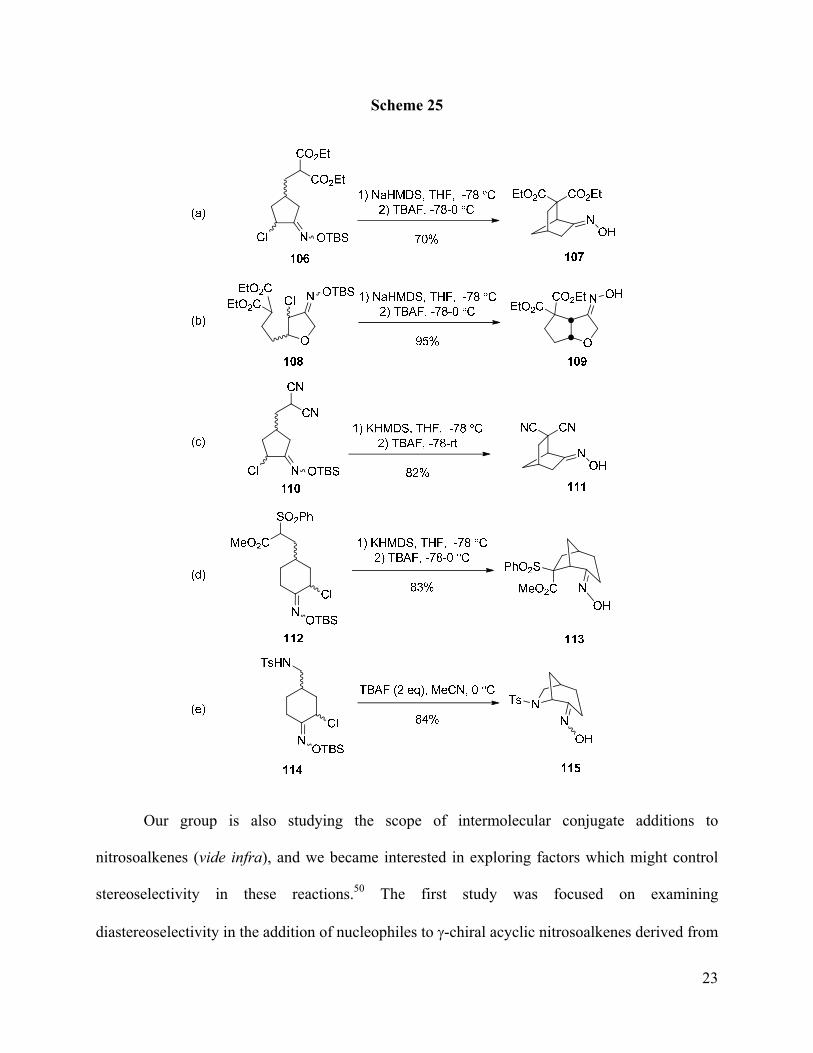
Examples of other ring systems produced by this methodology are shown in Scheme 25.
Through this method, various bridged and ring systems can be constructed in good yields.
Interestingly, other types of soft carbanions have been shown to participate in these additions
(Scheme 25, entries c,d). In general, use of NaHMDS or KHMDS for formation of the
carbanions led to superior results than with LiHMDS. Additionally, a sulfonamide nucleophile
was used for construction of an azabicyclic ring system (Scheme 25, entry e).49
Page 33
23
Scheme 25
Our group is also studying the scope of intermolecular conjugate additions to
nitrosoalkenes (vide infra), and we became interested in exploring factors which might control
stereoselectivity in these reactions.50 The first study was focused on examining
diastereoselectivity in the addition of nucleophiles to γ-chiral acyclic nitrosoalkenes derived from
Page 34
24
aldoximes (Scheme 26). It was found that conjugate addition of malonate nucleophiles derived
from 117 to the nitrosoalkene 118 derived from 116 led to exclusive formation of anti adducts
119. Assuming that the nitrosoalkene reacts via the presumably lower energy E-configuration,
formation of the anti products would arise from Burgi-Dunitz attack of the nucleophile on the
preferred Felkin-Ahn-type conformation 118b where the phenyl group is perpendicular to the
alkene and the methyl group is “inside”. To firmly establish the relative stereochemistry of
119a, a thermal intramolecular 1,3-dipolar cycloaddition was effected to provide the cis-fused
isoxazolidine 120. Conversion of the free amine to the tosylate then provided the crystalline
compound 121, whose stereochemistry was elucidated using X-ray diffraction.
Scheme 26
Several additional systems related to 188 were examined and shown also to undergo
diastereoselective conjugate additions with various nucleophiles. For example, addition of the
potassium salt of diethyl α-allylmalonate (117a) to the β-alkoxy nitrosoalkene derived from 122
led exclusively to anti product 123, again via a similar Felkin-Ahn-type process (Scheme 27a).
Page 35
25
This result rules out a polar Felkin-Ahn mechanism in which the alkoxy substituent would be
perpendicular to the alkene instead of the phenyl group (cf. Scheme 26). To examine whether a
chelation effect could be observed in the methoxy series, the corresponding lithium salt of α-
allyl diethyl malonate was used in this reaction. However, the adduct 123 was again produced
exclusively with the same anti-stereochemistry but in a somewhat lower yield. Similarly, it was
discovered that N-methyltoluensulfonamide potassium salt adds to the nitrosoalkene derived
from 124 to also yield the corresponding anti product 125 (Scheme 27b).
Scheme 27
Page 36
26
Chapter 2 – Results and Discussion
2.1 – Investigation of the Conjugate Addition of Carbon Nucleophiles to Nitrosoalkenes
Generated via the Denmark Protocol
As part of our ongoing studies on utilizing nitrosoalkenes as enolonium ion equivalents in
organic synthesis (see Section 1.4), we became interested in systematically exploring the
alkylation of nitrosoalkenes produced via the Denmark protocol. Since only a few examples of
this strategy have been documented in the literature,44,45 and since this strategy would be well
suited for use with valuable nucleophiles (such as in a total synthesis project), we were prompted
to explore a range of carbon nucleophiles and nitrosoalkenes that could participate in this
reaction.51
Since conjugate additions to nitrosocycloalkenes are relatively rare, we decided to
conduct our initial optimization experiments using nitrosocyclohexene derived from the α-
chloro-O-TBS-oxime 127 (Scheme 28). We first tested the enolate of methyl phenylacetate
(126) as the nucleophilic component in these reactions. Thus, deprotonation of methyl
phenylacetate (126) (1.2 equiv) with KHMDS at -78 °C followed by sequential addition of α-
chloro-O-TBS-oxime 127 (1.0 equiv) and TBAF (1.2 equiv) and warming to 0 °C provided the
Michael adduct 128 in 79% yield. It should be noted that allowing the reaction mixture to warm
to room temperature prior to aqueous workup leads to formation of oxazanone 129 and various
decomposition byproducts.
Page 37
27
Scheme 28
We next sought to examine these general reaction conditions (Scheme 29) using various
ester-containing nucleophiles and nitrosoalkene precursors. This work was performed in
collaboration with Puhui Li and Jason Witek of our group. Using nitrosocyclohexene, several
additional nucleophiles were found to participate effectively in the Michael addition (Table 1).
Use of malonates in the Michael reaction gave adducts in reasonable to excellent yields (entries
1-3). Additionally, a β-ketoester, α-sulfonyl ester, and an α-nitroester were all identified as
compatible nucleophiles (entries 4-6). Nitrosocyclopentene also performed well in these
reactions, albeit in slightly lower yields compared to the cyclohexanone-derived nitrosoalkenes
(entries 7-12). In certain cases, the type of base employed affects the outcome of the reaction.
For example, the potassium salt of ethyl nitroacetate was found to react with nitrosocyclohexene
to yield the corresponding Michael adduct in 57% yield (entry 4). However, when KHMDS was
replaced with either LiHMDS or NaHMDS, none of the desired Michael adduct was produced.
In contrast, use of LiHMDS or NaHMDS as the base with diethyl malonate and
nitrosocyclohexene provided the Michael adduct in nearly the same yields as with KHMDS
(91% and 94%, respectively, entry 1).
Page 38
28
Scheme 29
Table 1. Intermolecular Michael Additions of Carbon Nucleophiles to Cyclic
Nitrosoalkenes
Page 39
29
We also examined the use of nitrosoalkenes derived from aldoximes since conjugate
additions to such species are not well studied45 and also because the direct α-alkylation of
aldehydes by conventional enolate chemistry is inherently difficult.52 It was found that α-chloro
hydrocinnamaldehyde-derived nitrosoalkenes react efficiently with malonates and methyl
phenylacetate anions (Table 2, entries 1-5). Similarly, addition of malonate and methyl
phenylacetate enolates to the exocyclic nitrosoalkene derived from cyclohexane carboxaldehyde
proceeded in good yields (entries 6-9). To our delight, we discovered that use of α-alkyl
malonates in this reaction produces vicinal quaternary centers in good yields (entries 7-8).
Table 2. Intermolecular Michael Additions of Carbon Nucleophiles to Nitrosoalkenes
Derived from Aldoximes
Page 40
30
Terminal nitrosoalkenes, such as those derived from chloromethyl ketones, are typically
employed as heterodienes in Diels-Alder cycloadditions.2 Since only a few reactions involving
conjugate addition to terminal nitrosoalkenes have been reported, we wished to extend our
methodology to these systems. In this instance, both malonate and methyl phenyl acetate
enolates added efficiently to produce the corresponding Michael adducts in good overall yield
(Table 3, entries 1-3).
Table 3. Intermolecular Michael Additions of Carbon Nucleophiles to Terminal
Nitrosoalkenes
When phenylsulfonyl acetonitrile (133) and α-chloro-O-TBS-oxime 127 were subjected
to the above standard conditions only the unstable adduct 135 was isolated, which results from
an intramolecular cyclization of the intermediate oxime anion onto the cyano group of 134.53
Although the reaction appears to occur cleanly on analysis by thin layer chromatography, the
moderate yield is likely due to the chromatographic instability of 135.
Page 41
31
Scheme 29
While the above methodology worked well for soft ester containing nucleophiles, it was
somewhat surprising that simple ester or ketone enolates such as those prepared from t-butyl
acetate or acetophenone did not participate in the Michael addition despite ample precedent that
such nucleophiles add efficiently to nitrosoalkenes generated under classical conditions from α-
chlorooximes.2 We had also attempted to use nitrosoalkene systems derived from α-chloro-O-
TBS-oxime 136 as a means to form α-quaternary centers. However, in all cases examined no
Michael addition could be effected and only the nitrosoalkene tautomer 137 was isolated. This is
probably due to the fact that nitrosoalkene 44 undergoes relatively fast tautomerization (cf.
Scheme 11),29a and also since nitrosoalkenes generated via the Denmark protocol tend to have
shorter lifetimes relative to those generated from α-chlorooximes.13b
Scheme 30
Page 42
32
Chapter 3. Studies Toward a Total Synthesis of Angustilodine, Alstilobanine A, and
Alstilobanine E
3.1 – General Background on Monoterpene Indole Alkaloids
Monoterpene indole alkaloids, which are usually comprised of a 3-(2-aminoethyl)indole
(tryptamine) appended to a single C9- or C10-terpene unit, constitute one of the largest classes of
natural products known.54 Considering the vast range of structural diversity found within this
class of alkaloids, it is rather remarkable that all members arise biogenetically from strictosidine
(140), which itself is a condensation product of tryptamine (138) and secologanin (139) (Scheme
31).55
Scheme 31
Scheme 32 depicts the biogenetic interrelationship of the ten main skeletal types of
monoterpene indole alkaloids from strictosidine as the biosynthetic origin. It is worth noting that
the tryptamine fragment of these alkaloids generally shows little or no modification. Most of the
observed modifications typically include oxygenation of the indole nucleus whereas the
tryptamine bridge is less often affected (vide infra). The C9- or C10-monoterpene moiety adds
significant structural diversity to this class of indole alkaloids since the terpene skeleton can
undergo a myriad of oxidations, rearrangements, and cyclizations to produce a wide array of
Page 43
33
different skeletal types. Thus, the large structural diversity displayed by monoterpene indole
alkaloids can be primarily attributed to variation in the monoterpene unit.
Scheme 32
3.2 – Apparicine and Angustilodine-Type Alkaloids of the Aspidospermatan Subclass
The aspidospermatan subclass of monoterpene indole alkaloids currently includes about
20 unique skeletal subtypes that have all been isolated from the Apocynaceae family of plants
(Figure 3).56,57 Most of the structural subtypes found within this class are derived from
stemmadenine, which itself is grouped under the aspidospermatan subclass despite the fact that it
Page 44
34
is a biogenetic precursor of aspidospermatans. Interestingly, a number of subtypes within the
aspidospermatan class lack one or both carbons of the usual two carbon tryptamine bridge, which
is an exceptionally rare feature among tryptamine-derived alkaloids (i.e. 147-153, 155, and 157-
160).54 These natural products are often referred to as “apparicine-type” alkaloids after the
simplest member of the group (cf. 147).
Figure 3. Skeletal Subtypes of the Aspidospermatan Subclass of Monoterpene Alkaloids
Page 45
35
Several interesting members of the apparicine subclass are shown in Figure 4. The
characteristic 1-azabicyclo[4.2.2]decane ring system of apparicine (161) is conserved among
some members of this group (i.e. 162-164). Additionally, higher degrees of oxidation are found
in congeners such as (E)-vallesamine (162), in addition to alstonamine (163) and angustilobine A
(164) which contain an additional ring. Some seco- and seco-nor derivatives such as nor-6,7-
secoangustilobine A (165) and 6,7-seco-19,20-α-epoxyangustilobine B (166) have also been
isolated. Over 20 additional alkaloids related to this subtype are found in a number of different
flowering plants of the Apocynaceae family. Many of these alkaloids are biologically active and
exhibit a broad pharmacological profile.
Figure 4. Selected Apparicine-Type Alkaloids
In 2004, Kam and Choo isolated a new alkaloid, angustilodine (167), which contains a
unique skeletal type, from the leaves of the Malayan plant Alstonia angustiloba (Figure 5).58
The structure of angustilodine was determined by spectroscopic analysis to include an indole
appended to a cis-azadecalin ring system interfused with a 7-membered ring bridging ether.
Page 46
36
More recently, Morita and coworkers discovered the N-demethyl derivative alstilobanine E (168)
as well as alstilobanine A (169), which lacks the bridging oxepane ring found in 167 and 168.59
The alstilobanines were found to possess modest vasorelaxant activity. For clarity, compounds
167-169 will be referred to “angustilodine-type” alkaloids.
Figure 5. Angustilodine-Type Alkaloids
Interestingly, the Morita group determined through 1H-1H NOESY correlations in
CD3OD solution that the piperidine ring in alstilobanine E (168) exists in a boat conformation.
On the other hand, Kam and Choo draw the piperidine ring in angustilodine (167) in the chair
form. However, it is unclear if this chair conformation is only assumed, since no 1H-1H NOESY
correlations within the piperidine ring were reported and there is no discussion of this point. It is
quite possible that the piperidine ring in angustilodine (167) actually exists in a similar boat
Page 47
37
conformation as alstilobanine E (168). The piperidine ring of alstilobanine A (169), on the other
hand, was determined by 1H-1H NOESY experiments to exist in a chair form.
3.3 – Biosynthesis of Apparicine- and Angustilodine-Type Alkaloids
Both apparicine and angustilodine-type alkaloids probably originate biogenetically from
stemmadenine (170) (Scheme 33).60 As illustrated in the transformation of stemmadenine (170)
to (E)-vallesamine (162), an initial oxidation of 170 to the corresponding N-oxide 171 is
followed by Potier-Polonovski fragmentation60c to the azafulvene iminium intermediate 172.
Excision of the iminium carbon would produce amino azafulvene 173 which undergoes an
intramolecular Mannich addition to afford (E)-vallesamine (162). Additional decarboxylations,
dehydrations, and/or oxidative ring closures can be invoked in the biosynthesis of the related
apparicine alkaloids (cf. Figure 4). For example, a concerted decarboxylative-dehydration of
(E)-vallesamine (162) produces apparicine (161). It should also be noted that partial synthesis of
both apparicine (from pericine) and vallesamine (from stemmadenine) have been achieved,
further supporting the feasibility of this biosynthetic hypothesis.61 Additionally, it was
demonstrated by feeding experiments that radio-labeled stemmadenine (170) and (E)-
vallesamine (162) are both converted into apparicine (161).60a,b
Page 48
38
Scheme 33
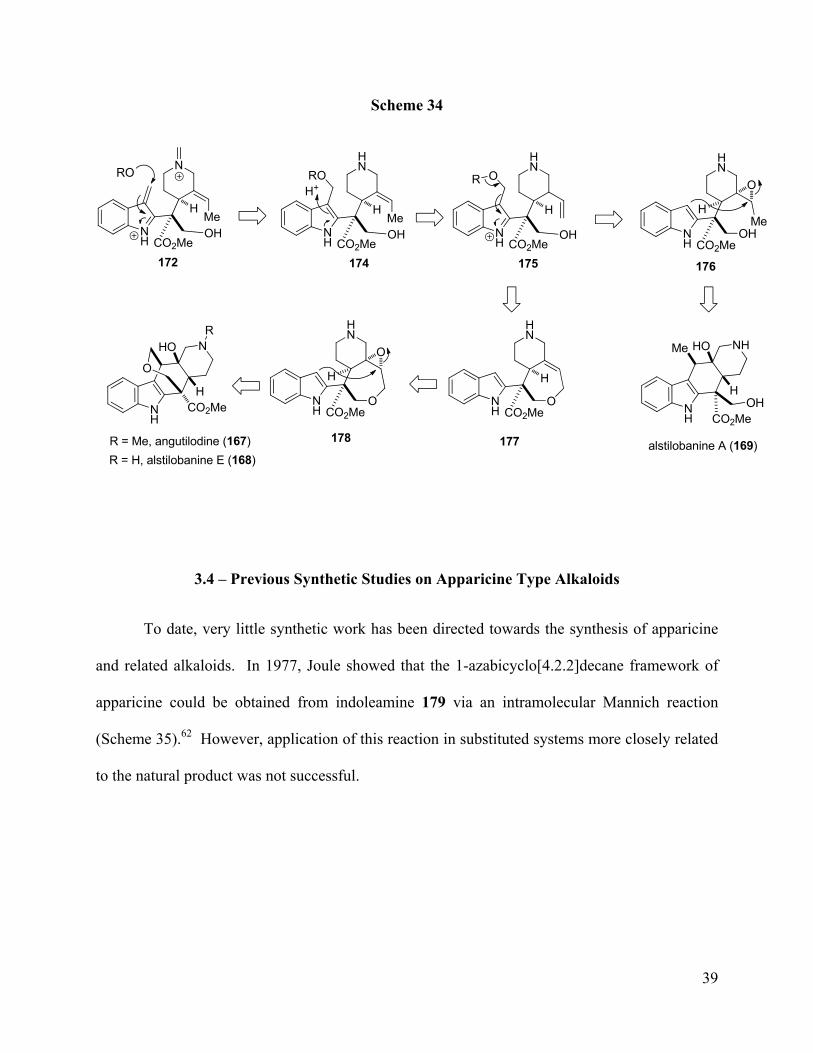
Similarly, the biogenesis of angustilodine-type alkaloids is believed to also proceed from
the fragmentation of stemmadenine (Scheme 34). In this case, the Potier-Polonovski
fragmentation product 172 (cf. Scheme 33) may undergo nucleophilic addition to the azafulvene
to generate indole 174. To produce the alstilobanine E skeleton, an isomerization/epoxidation of
the olefinic moiety in 175 and rearomatization of the indole first produces intermediate 176.
Intramolecular attack of the nucleophilic indole C-3 carbon onto the epoxide of 174 would
generate alstilobanine A (169). The angustilodine skeleton may be generated from 175
following cyclization of the primary alcohol onto the alkene to give the 7-membered ring ether
176. A similar intramolecular epoxide addition of indole 178 would lead to angustilodine (167)
and alstilobanine A (168).
Page 49
39
Scheme 34
172
NH CO2Me
H
OH
N
Me
RO
NH CO2Me
H
OH
HN
RO
Me
H+
NH CO2Me
H
OH
HN
O
NH CO2Me
H
HN
ONH CO2Me
H
HN
O
O
NH
NR
HO
H
O
CO2Me
R
174 175
177178R = Me, angutilodine (167)R = H, alstilobanine E (168)
176
NH CO2Me
H
HN
OHMe
O
NH
NHHO
H
Me
CO2MeOH
alstilobanine A (169)
3.4 – Previous Synthetic Studies on Apparicine Type Alkaloids
To date, very little synthetic work has been directed towards the synthesis of apparicine
and related alkaloids. In 1977, Joule showed that the 1-azabicyclo[4.2.2]decane framework of
apparicine could be obtained from indoleamine 179 via an intramolecular Mannich reaction
(Scheme 35).62 However, application of this reaction in substituted systems more closely related
to the natural product was not successful.
Page 50
40
Scheme 35
NH
HN
O
ONH
N
O
1) H2CO, AcOH
2) HCl, 35%
180179
It was not until 2009 that the Bennesar group reported the first synthesis of apparicine,
which is the only member of this class of alkaloids to have been prepared to date.63 The
successful route featured a ring-closing metathesis of diene 181 to provide the tricyclic
compound 182 (Scheme 36). Next, alkene isomerization to produce 183, followed by Boc-
removal and alkylation of the resulting free amine with tosylate 184 provided the indole 185.
Finally, formation of the azabicyclic core via an intramolecular Heck reaction gave apparicine
(161) but in only 15% yield.
Scheme 36
Page 51
41
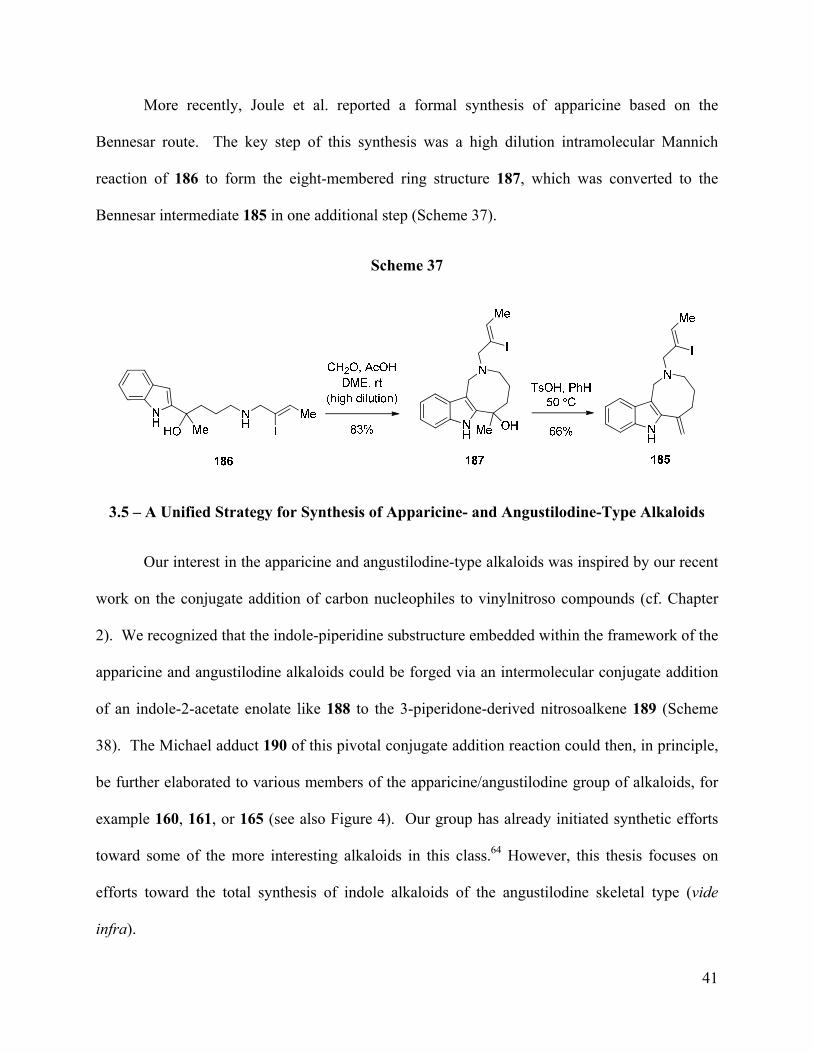
More recently, Joule et al. reported a formal synthesis of apparicine based on the
Bennesar route. The key step of this synthesis was a high dilution intramolecular Mannich
reaction of 186 to form the eight-membered ring structure 187, which was converted to the
Bennesar intermediate 185 in one additional step (Scheme 37).
Scheme 37
3.5 – A Unified Strategy for Synthesis of Apparicine- and Angustilodine-Type Alkaloids
Our interest in the apparicine and angustilodine-type alkaloids was inspired by our recent
work on the conjugate addition of carbon nucleophiles to vinylnitroso compounds (cf. Chapter
2). We recognized that the indole-piperidine substructure embedded within the framework of the
apparicine and angustilodine alkaloids could be forged via an intermolecular conjugate addition
of an indole-2-acetate enolate like 188 to the 3-piperidone-derived nitrosoalkene 189 (Scheme
38). The Michael adduct 190 of this pivotal conjugate addition reaction could then, in principle,
be further elaborated to various members of the apparicine/angustilodine group of alkaloids, for
example 160, 161, or 165 (see also Figure 4). Our group has already initiated synthetic efforts
toward some of the more interesting alkaloids in this class.64 However, this thesis focuses on
efforts toward the total synthesis of indole alkaloids of the angustilodine skeletal type (vide
infra).
Page 52
42
Scheme 38
3.6 – First Generation Retrosynthetic Plan for the Synthesis of the Angustilodine Alkaloids
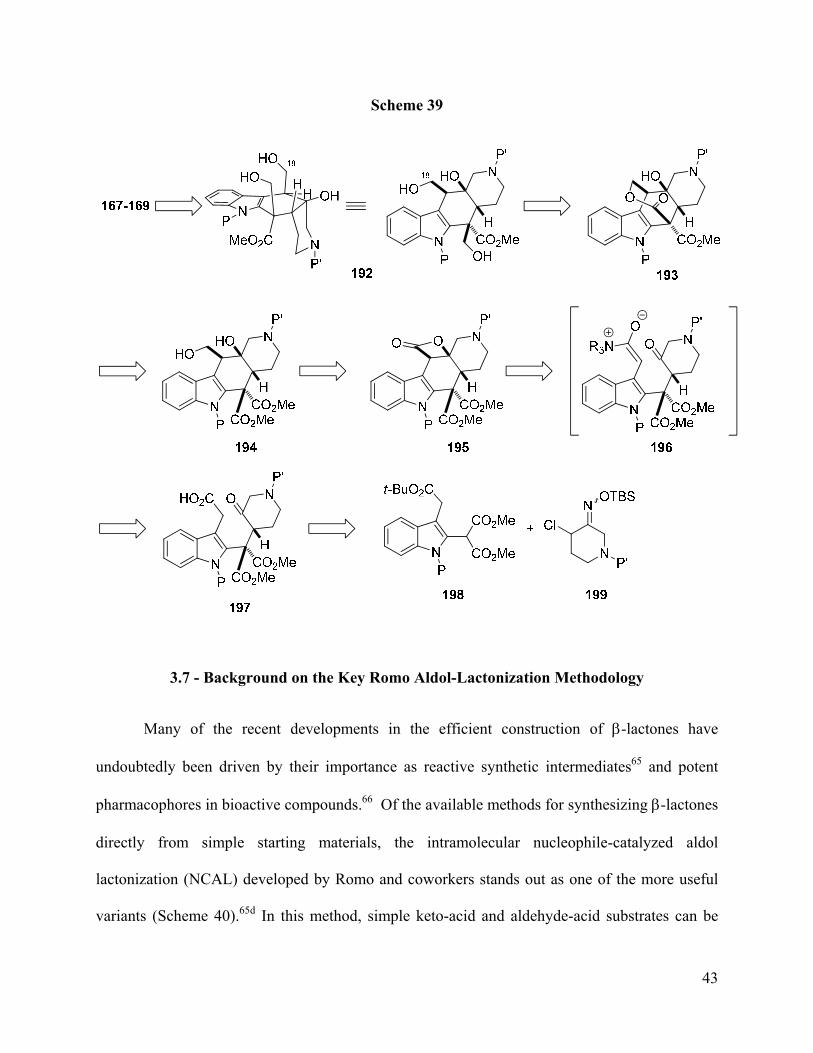
At the onset of our study, we planned to access all three angustilodine alkaloids 167-169
through the common late stage intermediate 192 (Scheme 39). Thus, regioselective
cyclodehydration of 192 via the natural product-like conformation shown would form the
bridging seven membered ring ether found in angustilodine (167) and alstilobanine E (168).
Alternatively, a selective Barton-McCombie deoxygenation of the C-18 alcohol in 192 would
provide the alstilobanine A (169) structure. In order to access triol 192, a selective lactonization
of the diester 194 would provide lactone 193, which in turn would be chemoselectively reduced
to afford 192. The 1,3-diol 194 would be produced directly from β-lactone 195. In a key
reaction, the keto-acid 197 would be converted to the cis-azadecalin β-lactone 195 via
ammonium-enolate 196 using Romo’s aldol lactonization methodology (vide infra). Finally,
construction of the indole-piperidone substructure in keto-acid 197 would be accomplished via
conjugate addition of an indole-2-malonate 198 to a nitrosoalkene generated from α-chlorooxime
199 using our methodology.
Page 53
43
Scheme 39
3.7 - Background on the Key Romo Aldol-Lactonization Methodology
Many of the recent developments in the efficient construction of β-lactones have
undoubtedly been driven by their importance as reactive synthetic intermediates65 and potent
pharmacophores in bioactive compounds.66 Of the available methods for synthesizing β-lactones
directly from simple starting materials, the intramolecular nucleophile-catalyzed aldol
lactonization (NCAL) developed by Romo and coworkers stands out as one of the more useful
variants (Scheme 40).65d In this method, simple keto-acid and aldehyde-acid substrates can be
Page 54
44
converted to β-lactones in one-pot in the presence of a carboxylic acid activator and a
nucleophilic amine catalyst. Mechanistically, the process begins with activation of the
carboxylic acid 200 and subsequent transacylation by the nucleophilic catalyst to produce an acyl
ammonium species 202. Enolization to ammonium enolate 203 and subsequent attack of this
enolate on the carbonyl group produces diastereomeric products 204 and 205, the former of
which cannot cyclize to a β-lactone due to ring strain. However, a retro aldol of 204 regenerates
ammonium enolate 203. Lactonization of intermediate 205, however, leads to the β-lactone 207
and regenerates the nucleophilic amine catalyst.
Scheme 40
Initially, this process was only applicable to substrates containing electrophilic
aldehydes.67 However, the Romo group discovered that use of a modified Mukaiyama salt for
carboxylic acid activation enabled participation of less electrophilic aliphatic ketones (Table 4).68
For example, using the combination of 2-bromo-N-propylpyridinium triflate and 4-
pyrrolidinopyridine (PPY), bicyclic and tricyclic fused β-lactones can be constructed with high
diastereoselectivity (Table 4, entries 1-4). It should be noted that a cis-decalin containing the
same relative stereochemistry as found in the angustilodine cis-azadecalin substructure can be
Page 55
45
produced, with no detectable amounts of the corresponding trans-decalin, from a racemic keto-
acid (entry 4). Additionally, a complex amide containing substrate, which was utilized in the
total synthesis of (-)-salinosporamide A, could be employed to a produce highly substituted β-
lactone (entry 5).68g
Table 4. Examples of β-Lactones Produced via Nucleophile-Catalyzed Aldol Lactonization
Since the Romo NCAL process proceeds via an ammonium enolate intermediate (cf.
Scheme 40), it is possible to induce asymmetry in this type of reaction. Very recently, the Romo
group was able to exploit this feature of the NCAL reaction by employing a chiral cyclic
Page 56
46
isothiourea derivative as the nucleophilic catalyst.69 Thus, using TsCl as the activating agent,
(S)-HBTM as the nucleophilic catalyst, and 1 equivalent of LiCl to accelerate the rate of the
reaction, various keto-acids were converted to their corresponding β-lactones with good
enantioselectivity (Table 5). Omission of LiCl in this reaction improves the enantioselectivity
slightly but the overall yields of the β-lactone products are significantly diminished.
Table 5. Examples of Enantioselective Nucleophile-Catalyzed Aldol Lactonizations
3.8 – Model Studies on the Conjugate Addition of Indole-2-acetate Enolates to Cyclic
Nitrosoalkenes
At the onset of our work we sought to identify a suitable indole containing nucleophile to
participate in the proposed nitrosoalkene Michael addition (cf. Scheme 39). Since we expected
Page 57
47
that preparation of the requisite 4-chloro-3-piperidone oxime 199 might be challenging, we opted
to use the α-chlorooxime derived from cyclohexanone as a model system. Ideally, it was hoped
that a fully substituted indole such as 210, containing the required C-3 acetic acid functionality
would prove to be compatible with the conjugate addition methodology (Scheme 41). Thus,
synthesis of 210 was accomplished via treatment of the known indole 20870 with t-butyl
hypochlorite to form a 3-chloroindolenine intermediate71 followed by addition of thallium
malonate 209.72 Unfortunately, deprotonation of indole malonate 210 using either K2CO3 or
LiHMDS (1 equiv) followed by addition of α-chlorooxime 211 (1 equiv) failed to produce the
desired Michael adduct 212 and only the starting indole was recovered.
Scheme 41
We reasoned that the failure to alkylate the fully functionalized indole 210 may be due, in
part, to the competitive deprotonation of the indole NH. To avoid this issue, we attempted a
number of different methods to protect the indole with alkyl or sulfonyl groups. However, this
protection proved to be less straightforward than anticipated. For example, attempted
methylation of the indole nitrogen with dimethyl sulfate led to exclusive methylation at the
malonate active methine (Scheme 42). Additionally, deprotonation of indole 210 with a variety
of bases followed by the addition of benzenesulfonyl chloride or TsCl gave no reaction.
Page 58
48
Scheme 42
A second potential issue with using an indole nucleophile such as 210 is the presence of
acidic α-protons on the C-3 acetate. To see whether removal of the C-3 acetate would facilitate
the Michael reaction, dimethyl indole-2-malonate (214)73 was prepared and employed as the
nucleophile in the model conjugate addition reaction (Scheme 43). Unfortunately, formation of
the monoanion of 214 (2 equiv), followed by addition of α-chlorooxime 211 (1 equiv), led
exclusively to the C-3 alkylated indole 215. Once again, we met with the same difficulty
protecting the indole nitrogen of 214 via alkylation or sulfonylation (cf. Scheme 42). In this
system, it was found that the indole nitrogen could be protected as a Boc-carbamate 216, but this
process is accompanied with concomitant acylation of the malonate group to yield 217 as the
major product.74
Scheme 43
Page 59
49
At this point, it became clear that use of an indole-2-malonate nucleophile in the
conjugate addition reaction was problematic. We reasoned that indole-2-acetic acid methyl ester
(218) might act as a better partner in these Michael additions since the derived ester enolate
would be more nucleophilic than a malonate enolate. However, in order to effect a conjugate
addition using this substrate, it was anticipated that formation of the dianion would be necessary
since deprotonation at the presumably more acidic NH of the indole would occur first. It was
also unclear at this point whether, under classical conditions, a single equivalent of the dianion of
218 could serve as the base to deprotonate the α-chlorooxime and the nucleophile to form a
Michael adduct with the resulting nitrosoalkene. To examine this possibility, the indole 218 (1
equiv) was treated with n-BuLi (2 equiv) at -78 °C to produce the corresponding dianion
(Scheme 44). Next, 1 equivalent of α-chlorooxime 211 was added dropwise and the resulting
mixture was stirred at -78 °C for 1 h. Gratifyingly, the desired Michael adduct 219 was isolated
from the reaction, albeit in only 20% unoptimized yield.
Scheme 44
Page 60
50
While attempting to optimize the reaction utilizing the dianion of 218, we serendipitously
discovered due to an error in calculating stoichiometry that reacting 2 equivalents of the
monoanion (instead of the intended 1 equivalent of the dianion) with 1 equivalent of α-
chlorooxime 211 led to clean production of the Michael adduct in 78% yield and with almost
complete recovery of the excess starting indole ester 218. This fortunate observation suggested
that the indole monoanion probably exists as an equilibrating mixture of the initially formed and
presumably more stable anion 220a and the more nucleophilic enolate anion 220b (Scheme 45).
While either anion could deprotonate α-chlorooxime 211 to form the transient nitrosoalkene 221,
only 220b adds to nitrosoalkene 221 in a Michael fashion to produce C-alkylation product 219
exclusively. It should also be noted that Michael adduct 219 was determined by 1H and 13C
NMR analysis to be a single diastereomer but an E/Z-mixture of oxime isomers. The relative
stereochemistry of the two stereocenters in 219 could not be determined. However, this
stereochemistry is inconsequential to the total synthesis (vide infra).
Scheme 45
Page 61
51
Because the monoanion of 218 could be employed, we found that it was also possible to
use the Denmark procedure to generate nitrosoalkene 221. Thus, addition of α-chloro-O-TBS-
oxime 127 (1 equiv) to a -78 °C solution of the monoanion derived from 218 (1 equiv) followed
by the addition of TBAF and warming to 0 °C led to formation of Michael adduct 219 in 42%
unoptimized yield (Scheme 46).
Scheme 46
After successfully synthesizing the model indole-cyclohexanone oxime system, our next
goal was to identify a set of mild conditions by which the oxime functionality of 219 could be
converted to the corresponding ketone. While this operation could, in principle, be performed at
various stages in the synthesis, our main concern was that the majority of known deoximation
protocols, which are either hydrolytic or oxidative, might be incompatible with the sensitive
indole and amino groups (vide infra).105 Therefore, we opted to utilize a mild, reductive
procedure involving low-valent titanium (Scheme 47).75 Thus, when oxime 219 was treated with
TiCl3 all of the starting oxime was converted to ketone 222 as observed by TLC analysis.
Page 62
52
However, subjecting the crude product to flash column chromatography on silica gel provided
the ketone 222 in 63% yield and a cyclocondensation product 223 in 13% yield. Furthermore,
when the purified ketone 222 was dissolved in CDCl3, complete conversion to the tetracyclic
pyrrole 223 was observed (presumably due to a trace of HCl).
Scheme 47
In light of this result, we decided to determine if it was possible to use an N-protected
indole in the nitrosoalkene conjugate addition. As noted before, attempts at N-alkylation and N-
sulfonylation of earlier indole systems proved to be fruitless (cf. Schemes 42 and 43). Similar to
these previous results, we found that it was also very difficult to protect the indole nitrogen with
alkyl and sulfonyl groups. For example, attempted N-methylation of indole 218 with NaH (1
equiv.) and MeI only produced a mixture of C-alkylated products 224 and 225 along with N,C-
dimethylated product 226 (Scheme 48). Similar reactivity was observed using other
electrophiles (i.e. BOM-Cl, PMB-Cl, etc). This problem is most likely due to equilibration of the
initially formed indole nitrogen anion with the more reactive C-2 enolate (cf. Scheme 45).
Scheme 48
Page 63
53
On the other hand, we were pleased to find that Boc-protection in this system proceeds
cleanly to give Boc-indole 227 in high yield (Scheme 48). Alkylation of the monoanion derived
from indole 227 (1.2 equiv) with 1.0 equivalent of the nitrosoalkene derived from α-chloro-O-
TBS-oxime 127 via the Denmark protocol led to clean production of adduct 228. Furthermore,
reductive cleavage of the oxime in 228 with TiCl3 proceeded without incident to give ketone 229
in an unoptimized 48% yield.
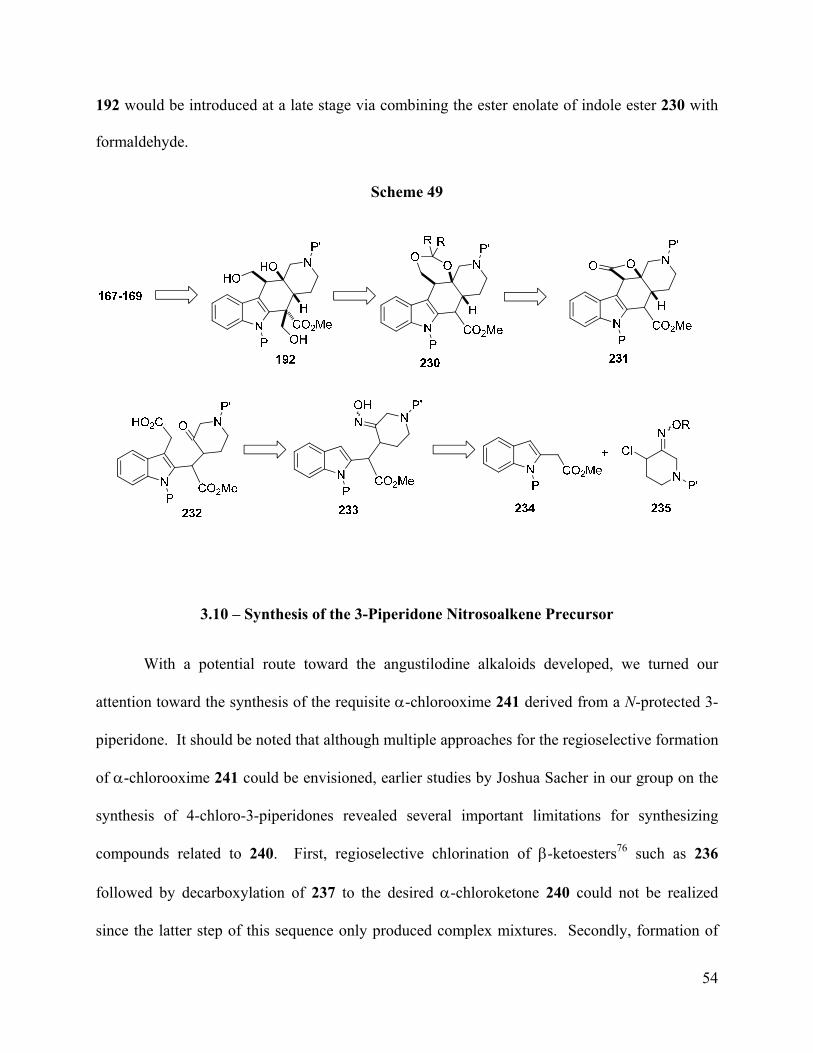
3.9 – Revision to Retrosynthesis
After identifying indole-2-acetate enolates as viable nucleophiles for the key conjugate
addition reaction, our retrosynthetic plan required some revision. Thus, conjugate addition of the
monoanion derived from indole ester 234 with the nitrosoalkene derived from 235 would
produce adduct 233 (Scheme 49). At this point, the acetic acid unit would be introduced via
alkylation at the nucleophilic indole C-3 of indole 233. Finally, the C-17 hydroxymethyl unit of
Page 64
54
192 would be introduced at a late stage via combining the ester enolate of indole ester 230 with
formaldehyde.
Scheme 49
3.10 – Synthesis of the 3-Piperidone Nitrosoalkene Precursor
With a potential route toward the angustilodine alkaloids developed, we turned our
attention toward the synthesis of the requisite α-chlorooxime 241 derived from a N-protected 3-
piperidone. It should be noted that although multiple approaches for the regioselective formation
of α-chlorooxime 241 could be envisioned, earlier studies by Joshua Sacher in our group on the
synthesis of 4-chloro-3-piperidones revealed several important limitations for synthesizing
compounds related to 240. First, regioselective chlorination of β-ketoesters76 such as 236
followed by decarboxylation of 237 to the desired α-chloroketone 240 could not be realized
since the latter step of this sequence only produced complex mixtures. Secondly, formation of
Page 65
55
α-chloroketones utilizing our previously disclosed strategy47,48 was not possible since oxidation
of vinyl chlorides such as 239 with sodium hypochlorite under various conditions failed to
provide 240. Finally, it was observed that 3-piperidones containing a basic nitrogen (i.e. P =
alkyl) tended to be unstable and thus could not be converted to an α-chlorooxime 240. A survey
of the literature provided additional examples of the instability of enolizable α-aminoketones
containing a basic nitrogen.77
Scheme 50
Given the above information, it seemed prudent to install the chlorine atom at the latest
possible stage so as to avoid subjecting any labile chlorinated compounds to multiple synthetic
steps. Initially, we wished to utilize an enol ether derived from a 3-piperidone in an
electrophilic chlorination to provide the α-chloroketone regioselectively. Engler and Wanner
have shown that methyl enol ethers 244 can be formed with modest to good regioselectivity from
3-piperidones 242 in a two step procedure (Scheme 51a).78 Thus, conversion of 3-piperidones
242 to ketals 243 and subsequent elimination of methanol with AlCl3 and TEA provided methyl
enol ethers 244 as the major regioisomers. It was reported that the nature of the N-sulfonyl
group affects the regioselectivity of the elimination step and use of the N-benzylsulfonyl group
gave the best results.78 Thus, we synthesized methyl enol ether 244c by this method, but as a 3:1
Page 66
56
mixture of regioisomers. Unfortunately, treatment of 244c with NCS under standard conditions79
did not provide the desired α-chloroketone 245 and only a complex mixture was obtained.
Scheme 51
Next, we attempted to synthesize the TMS-enol ether 247 from N-tosyl-3-piperidone80
(246) by trapping the corresponding kinetic enolate80,81 with TMS-Cl. However, only a complex
mixture of products was isolated which contained a significant amount of toluenesulfinic acid
(251). This decomposition pathway is likely the result of equilibration of the initially formed
kinetic enolate 249a with the thermodynamic enolate 249b, which eliminates toluenesulfinic acid
to produce imine 250.80 Additionally, direct addition of N-chlorosuccinimide to the enolate
solution did not yield any of the desired α-chloroketone 248.
Page 67
57
Scheme 52
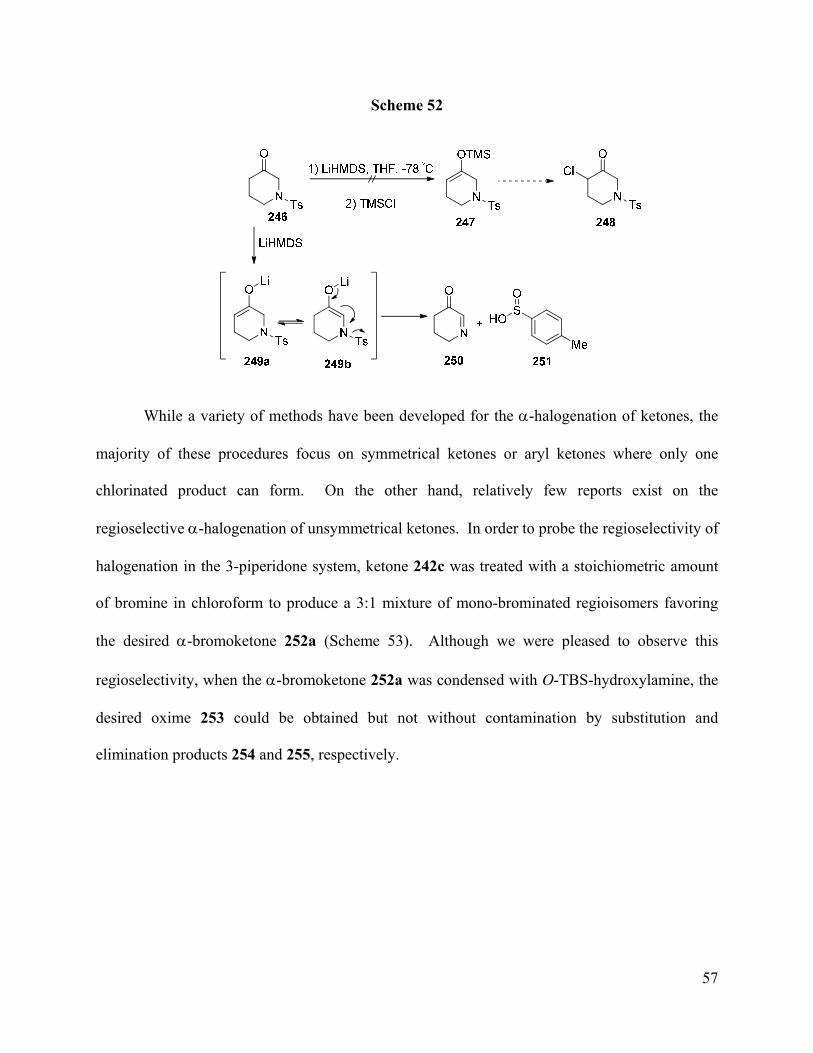
While a variety of methods have been developed for the α-halogenation of ketones, the
majority of these procedures focus on symmetrical ketones or aryl ketones where only one
chlorinated product can form. On the other hand, relatively few reports exist on the
regioselective α-halogenation of unsymmetrical ketones. In order to probe the regioselectivity of
halogenation in the 3-piperidone system, ketone 242c was treated with a stoichiometric amount
of bromine in chloroform to produce a 3:1 mixture of mono-brominated regioisomers favoring
the desired α-bromoketone 252a (Scheme 53). Although we were pleased to observe this
regioselectivity, when the α-bromoketone 252a was condensed with O-TBS-hydroxylamine, the
desired oxime 253 could be obtained but not without contamination by substitution and
elimination products 254 and 255, respectively.
Page 68
58
Scheme 53
Encouraged by the α-bromination of ketone 242c, we sought to develop an analogous
protocol for α-chlorination of N-sulfonyl-protected 3-piperidones. However, a process which
avoided the use of chlorine gas was highly preferable. Early studies by Masilamani and Rogic
have showed that sulfuryl chloride is an effective reagent for the mono-chlorination of
symmetrical cyclohexanones and displays reactivity similar to elemental chlorine.82 Fortunately,
treatment of N-tosyl-3-piperidone (246) with sulfuryl chloride produced a mixture of α-
chloroketones 256a and 256b with good regioselectivity (~5-7:1 by analysis of the crude
product) and the desired α-chloroketone regioisomer 256a could be separated from 256b by
column chromatography (Scheme 54). It should also be noted that when using fresh bottles of
sulfuryl chloride, only 1 equivalent of the chlorinating reagent was required. Since sulfuryl
chloride gradually decomposes to sulfur dioxide and chlorine upon standing, additional
equivalents of this reagent must be used from older bottles. However, we have found that as
many as 8 equivalents could be used if older reagents (~1 year stored at room temperature) were
employed with no apparent effect on the outcome of the reaction.
Page 69
59
Scheme 54
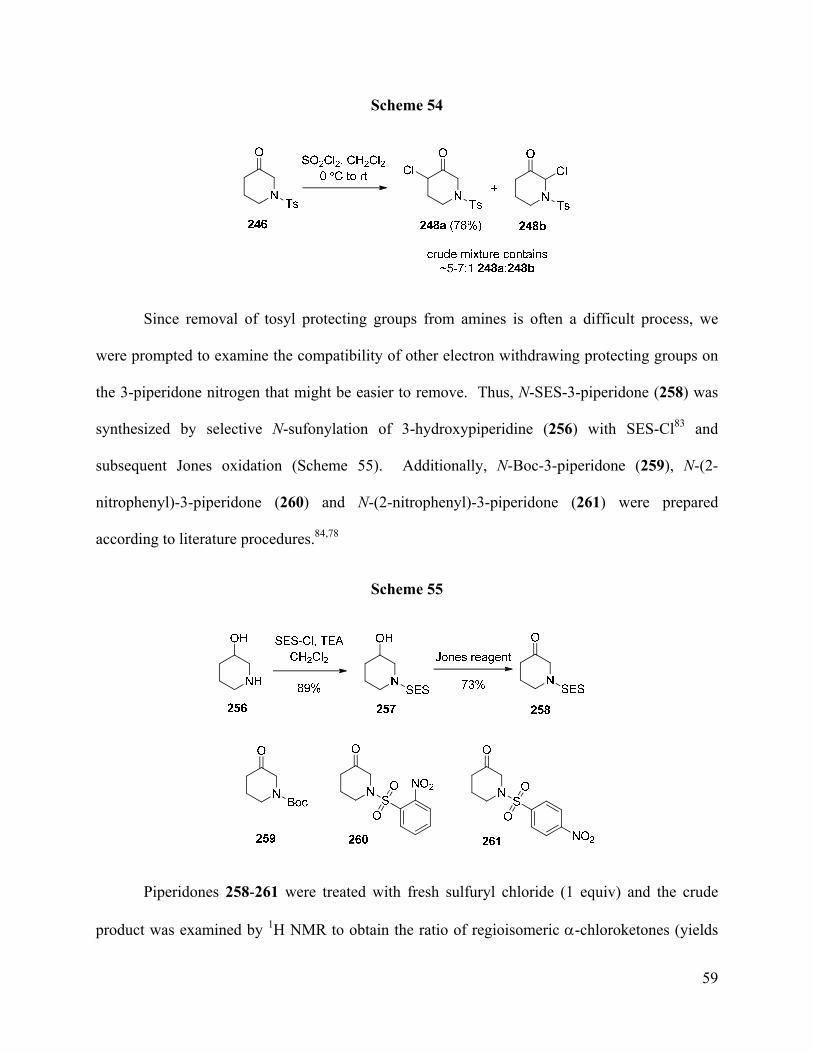
Since removal of tosyl protecting groups from amines is often a difficult process, we
were prompted to examine the compatibility of other electron withdrawing protecting groups on
the 3-piperidone nitrogen that might be easier to remove. Thus, N-SES-3-piperidone (258) was
synthesized by selective N-sufonylation of 3-hydroxypiperidine (256) with SES-Cl83 and
subsequent Jones oxidation (Scheme 55). Additionally, N-Boc-3-piperidone (259), N-(2-
nitrophenyl)-3-piperidone (260) and N-(2-nitrophenyl)-3-piperidone (261) were prepared
according to literature procedures.84,78
Scheme 55
Piperidones 258-261 were treated with fresh sulfuryl chloride (1 equiv) and the crude
product was examined by 1H NMR to obtain the ratio of regioisomeric α-chloroketones (yields
Page 70
60
were not determined). Interestingly, chlorination of N-Boc-3-piperidone (259) led to exclusive
formation of the undesired regioisomer B (Scheme 56). Treatment of all three N-sulfonyl-3-
piperidones 258, 260, and 261 with sulfuryl chloride produced the desired 4-chloro-3-piperidone
A as the major regioisomer but in somewhat a lower A/B ratios than with N-tosyl-3-piperidone
(cf. Scheme 54).
Scheme 56
At a later time, an alternative protocol for the regioselective chlorination of ketone 246
was developed by Pradeep Chauhan in our group. By this method, α-chloroketone 248a could
be produced exclusively from 3-piperidone 246 by treatment with NCS and Amberlyst-15 in
EtOAc (Scheme 57).85 In this case, direct isolation of the product by filtration of the reaction
mixture through a pad of Celite yields a mixture of primarily enol tautomer 262 and some of the
desired ketone 248a as observed by 1H NMR of the crude product. However, subjecting this
mixture to subsequent oximation reactions led to poor results. In order to obtain the keto-form
248a exclusively, silica gel was added to the reaction mixture after complete consumption of the
ketone 246 and the resulting mixture was stirred for 2 h prior to workup.
Page 71
61
Scheme 57
Our next objective was to find suitable conditions for oximation of ketone 248a.
Formation of O-TBS-oxime 263 from 3-piperidone 248a was accomplished in high yield by
treatment of 248a with O-TBS-hydroxylamine and PPTS (Scheme 58). Conversion of α-
chloroketone 248a to the corresponding α-chlorooxime 264 using the standard basic conditions
was not possible because of the base lability of the α-chlorooxime. However, oximation of 248a
under acidic conditions provided the desired α-chorooxime 264 in good yield. Unfortunately,
purification of 264 via silica gel chromatography led to some degradation of the product.
Although, the crude oxime 264 could be used in subsequent conjugate addition reactions (vide
infra), some residual acetic acid in the crude material affected the overall yield. Furthermore,
washing the crude product from this oximation reaction with aqueous sodium bicarbonate led to
product degradation presumably via formation of the corresponding nitrosoalkene. Because of
these problems, we developed a neutral protocol in which α-chloroketone 248a (1.0 equiv) and
hydroxylamine hydrochloride (1.1 equiv) are dissolved in DMSO and stirred at room
temperature to yield α-chlorooxime 264 in high purity which can be used without further
purification.
Page 72
62
Scheme 58
3.11 – Conjugate Addition of Indole-2-acetate Methyl Esters to Nitrosoalkenes Derived
from 3-Piperidone
With oxime derivatives 263 and 264 in hand, our efforts were now directed at finding a
suitable indole ester nucleophile in the key conjugate addition. Thus, the monoanion of indole
218 (1.2 equiv) was generated at -78 °C and then α-chloro-O-TBS-oxime 263 (1.0 equiv) was
added followed by TBAF to generate the nitrosoalkene 265 (Scheme 59). After warming the
reaction mixture to 0 °C and stirring for 2 h, the desired Michael addition of ester enolate 220b
to nitrosoalkene 265 occurred to provide adduct 266, albeit in only 12% yield. Surprisingly,
addition of the enolate derived from N-Boc indole 227, which fared well in the model alkylation
(cf. Scheme 48), to the nitrosoalkene 265 derived from 263 via the Denmark protocol did not
produce any of the Michael adduct 267.
Page 73
63
Scheme 59
We reasoned that the problems with the above reactions may be related to the relatively
short lifetime and high reactivity of the nitrosoalkene 265 generated from 263 under the
Denmark conditions. Therefore, we examined the reaction of the ester enolates of indoles 218
and 227 with α-chlorooxime 264. Under classical conditions, α-chlorooxime 264 (1 equiv) was
added dropwise to a solution of the monoanion of the free indole 218 (2 equiv) at -78 °C
(Scheme 60a). Gratifyingly, production of the Michael adduct 266 occurred quickly at this
temperature and this product could be isolated in an acceptable 59% yield with the remainder of
the unreacted starting indole recovered. As was observed in the model alkylation, Michael
adduct 266 was isolated as a single diastereomer but as a mixture of oxime isomers. However,
Page 74
64
the relative chemistry of the two stereocenters in 266 was not determined. Next, α-chlorooxime
264 (1 equiv) was added to a solution of the monoanion of N-Boc indole 227 (2 equiv).
However, very little of the desired Michael adduct 267 was produced at -78 °C or upon warming
the reaction mixture to 0 °C (Scheme 60b).
Scheme 60
Before moving forward with our revised plan (cf. Scheme 49), we wanted to see if a
deoximation of the 3-piperidone oxime was possible in this system. Once again, however,
unmasking of the carbonyl functionality of 266 via titanium (III)-mediated deoximation led to a
mixture of the desired ketone 268 and cyclocondensation product 269 (Scheme 61). Selective
protection of the indole as a Boc-carbamate was not possible at this stage since O-acylation of
the oxime occurred preferentially. For example, treatment of 266 with 2 equivalents of Boc2O
and TEA produced a 1:1 mixture of O-Boc oxime 270 and bis-Boc compound 271. Use of 1
Page 75
65
equivalent of Boc2O in this reaction led to exclusive formation of the O-Boc oxime 270 in
modest yield.
Scheme 61
3.12 – Attempted Installation of a C-3 Acetic Acid Unit on Michael Adduct 270
To move forward in the synthesis, installation of an acetic acid substituent onto C-3 of
the indole was required. Nucleophilic addition of the electron rich C-3 of indole in 270 to an α-
haloacetate derivative appeared to be the most straightforward approach (Scheme 62).
Following this step and subsequent indole nitrogen protection, we hoped to simultaneously
hydrolyze the ester and oxime functionalities to give the key keto-acid 273. Unfortunately,
treatment of indole 270 with a variety of bases (LDA, NaH, K2CO3, etc.) followed by addition of
t-butyl bromoacetate produced no desired C-3 alkylation product 272. Usually, only
Page 76
66
decomposition of the starting indole 270 was observed. It is possible that an enolate
equilibration of the indoyl anion with the ester enolate complicates the reaction (cf. Scheme 45).
As an alternative, we turned to Qin’s indole cyclopropanation/ring opening strategy to
functionalize C-3 of the indole 270. However, indole 270 was found to be unreactive toward
both Cu86 and Rh87-carbenoids generated from t-butyl diazoacetate. Instead, rapid dimerization
of the metal-carbenoids led to production of E- and Z-di-t-butyl fumarate.
Scheme 62
At the same time that we were examining the above strategy, we explored another C-3
alkylation process utilizing allylic electrophiles (Scheme 63). In this case, it was hoped that after
C-3 allylation and indole protection, oxidative cleavage of both the alkene and the oxime in 274
using KMnO4 under acidic conditions would provide keto-acid 273 in a single operation.
However, as before, no alkylation product 274 was observed when 270 was treated with a variety
of bases followed by addition of allyl bromide. Additionally, application of Tamaru’s method
for indole C-3 allylation using a π-allyl palladium species failed to provide 274.88
Page 77
67
Scheme 63
Since installation of a 2-carbon unit at C-3 was proving to be difficult, we turned our
attention to 1-carbon homologations. Toward this end, we found that subjection of indole 270 to
a Vilsmaeir-Haack formylation provided 275 in good yield (Scheme 64). We had hoped that
Wittig homologation of 275 using ylide 27689 would afford the methylenol ether 277 and a
subsequent one-pot deoximation/oxidation90 would produce the key keto-acid 273. However, the
aldehyde moiety of 275 was unreactive toward phosphonium ylide 276. This failure to react may
be due to the fact that aldehyde 275 is actually a vinylogous amide which may be deprotonated
by ylide 276.
Scheme 64
Page 78
68
Alternatively, we found that treatment of indole 270 with dimethylamine hydrochloride
and formalin under acidic conditions led to a clean conversion to the gramine derivative 278
which was used crude in the following step due to its instability. Quaternization of the gramine
278 with MeI in the presence of KCN produced the desired nitrile product 279 in low yield along
with the corresponding deacylated oxime 280. Because it would be necessary to reprotect the
oxime before subsequent operations, this route was abandoned.
Scheme 65
3.13 – Conjugate Addition of 2,3-Disubstituted Indoles to Nitrosoalkenes Revisited
Since we were unable to develop an effective route for installation of a C-3 acetic acid
unit into indole 270, we decided to explore the potential of a different convergent approach
utilizing a 2,3-disubstituted indole nucleophile in the key nitrosoalkene conjugate addition step.
Thus, placement of an oxoacetate moiety at C-3 of the indole nucleophile would be
accomplished by reacting indole 218 with oxalyl chloride and treating the resulting adduct with
an appropriate alcohol to form an oxoacetate ester 283 (Scheme 66).91 Next, a conjugate
Page 79
69
addition of the ester enolate of indole derivative 283 to the nitrosoalkene derived from α-
chlorooxime 282 would provide adduct 281. Following reductive removal of the oxoacetate
ketone oxygen, hydrolysis of the ester to a carboxylic acid, and conversion of an oxime to a
ketone to provide keto ester 232, the synthesis would proceed as previously planned (cf. Scheme
49). It is also worth noting that indoyl C-3 ketones similar to 281 have previously been reduced
to the corresponding methylene compounds in one step.94
Scheme 66
First, for maximum flexibility in the synthesis, we attempted to prepare several indoles of
type 283 with differentially protected carboxylic acids. Thus, indole 218 was treated with oxalyl
chloride to cleanly provide adduct 284, which was not isolated but reacted in situ with either t-
BuOH, BnOH, or 2-trimethylsilylethanol. Whereas both BnOH and 2-trimethylsilylethanol
(TMSE-OH) added efficiently to acid chloride 284 to provide the corresponding esters 285 and
286 respectively in good yields, the sterically hindered t-BuOH gave a complex mixture of
products. Using acid chloride 284 with t-BuOK and heating did not improve the outcome of the
esterification.
Page 80
70
Scheme 67
Additionally, indole malonate 214 was converted to the benzyl oxoacetate 288 in an
analogous manner (Scheme 68). Unfortunately, addition of the model α-chlorooxime 211 (1
equiv) to either the monoanion (2 equiv) or dianion (1 equiv) of indole malonate 288 did not
produce any adduct 289 and most of the starting indole was recovered in both cases.
Scheme 68
Due to the high cost of 2-trimethylsilylethanol, our initial experiments using the indole-3-
oxoacetate system were carried out using the benzyl ester 285. Additionally, we decided to use
the N-Ns-piperidone-oxime 290 (Ns = 2-nitrophenylsulfonyl) since these types of sulfonamides
are, in general, easier to cleave than other aryl sulfonamides.92 At this point, we were unsure if
Page 81
71
the presence of the C-3 oxoacetate moiety would change the equilibrium behavior of the
corresponding monoanion as observed in earlier systems (cf. Scheme 45). Previously, it was
noted that using 2 equivalents of an indole ester monoanion 218 gave a higher yielding and
cleaner reaction in the alkylation of nitrosocyclohexene than with 1 equivalent of the
corresponding dianion (cf. Scheme 44). In this case, we were pleased to find that reacting the
dianion of 285 (1 equiv) with α-chlorooxime 290 (1 equiv) provided adduct 291 in 81% yield
(Scheme 69). Using 2 equivalents of the monoanion derived from indole 285 in the conjugate
addition reaction provided adduct 291 in approximately the same yield. Once again, these results
indicate a monoanion equilibration similar to that observed in the earlier system (cf. Scheme 45).
In the present case, we believe the dianion 293a first deprotonates the oxime 290 providing
nitrosoalkene 294 and an equilibrium mixture of the anion 293b and the presumably more
nucleophilic anion 293c. The latter anion 293c then reacts with the nitrosoalkene 294 to produce
Michael adduct 291 as a single diastereomer and a 1:1 mixture of oxime geometric isomers.
Based on the results of a closely related conjugate addition reaction, the relative stereochemistry
of 291 has been assigned as shown (vide infra). Silyl oxime 292 was subsequently prepared in
nearly quantitative yield by treatment of oxime 291 with TBSCl and imidazole.
Page 82
72
Scheme 69
During the study of the above conjugate addition (cf. Scheme 69), we also examined the
use of N-protected indole ester monoanions in this key reaction. However, use of the N-
protected indole substrates in the key conjugate addition reaction had a deleterious effect on the
outcome of the reaction. For example, when the model α-chlorooxime 211 was added to 2
equivalents of the monoanion of Boc-protected indole ester 295, two products were formed
(Scheme 70). The desired adduct 296 and the minor adduct 297, resulting from double
alkylation, was isolated as a ~4:1 mixture.
Page 83
73
Scheme 70
We also examined the effect of an electron donating alkyl protecting group on the indole
nitrogen. Not surprisingly, it was difficult to directly alkylate the indole nitrogen (Scheme 71).
For example, treatment of indole ester 286 with sodium hydride in DMF followed by addition of
PMB-Cl only led to α-alkylation of the ester (entry a). Similarly, deprotonation of indole ester
285 with a weaker base and addition of PMB-Cl provided a similar α-ester alkylation product
299 (entry b).
Scheme 71
Page 84
74
Since direct alkylation of the indole nitrogen was problematic, N-methyl protected indole
301, which we previously attempted to prepare (cf. Scheme 48), was synthesized from N-
methylindole (300) in 4 steps following a literature procedure (Scheme 72).93 The indole 301
was treated with oxalyl chloride followed by BnOH to provide the C-3 oxoacetate 302.
However, addition of the monoanion of N-methyl protected indole 302 (2 equiv) to the
nitrosoalkene derived from α-chlorooxime 290 (1 equiv) provided the desired impure adduct 303
in only 14% impure yield along with double alkylation product 304 in 8% yield.
Scheme 72
3.14 – Ketone Reduction of Indole-3-oxoacetates with Hydride Reagents
After identifying an efficient conjugate addition reaction using 2,3-disubstituted indole
285 and α-chlorooxime 290 (cf. Scheme 69), we turned our attention to removing the ketone
oxygen of the 3-oxoacetate functionality in indole-3-oxoacetate 292. In order to do this, we
envisioned that an initial reduction of the ketone in 292 with a metal hydride would provide
Page 85
75
alkoxide 305 (Scheme 73). We believed that this intermediate would spontaneously eliminate to
yield azafulvene 306, which would be reduced by a second equivalent of hydride to provide 307.
Scheme 73
A number of indole-2- and 3-oxoacetates have been deoxygenated using triethylsilane
and TFA and we first examined these conditions in our system.94 However, when oxoacetate
292 was subjected to TFA and triethylsilane, the desired product 307 was not observed, but
rather tetracycle 309 was isolated in moderate yield and as a single diastereomer of unknown
configuration (Scheme 74). This product is likely the result of an acidic cleavage of the oxime
moiety and a concomitant reduction of both the ketone and indole functionalities to give indoline
ketone intermediate 308. Cyclocondensation of the ketone onto the indoline nitrogen leads to
tetracycle 309. Running the reaction at lower temperatures had little effect on the outcome of
this reaction.
Page 86
76
Scheme 74
We had hoped that placement of a protecting group on the indole nitrogen would serve
the dual purpose of preventing both the reduction at the indole and the cyclocondensation of the
newly generated ketone. Thus, NH-indole 292 was converted to the methyl carbamate 310.
However, treatment of 310 with TFA and triethylsilane did not generate any of the desired
reduced product and only the free oxime 311 was isolated (Scheme 75).
Scheme 75
Page 87
77
We also examined several alternative hydride reduction methods to effect the
deoxygenation of oxoacetate 292. For example, it was discovered that treatment of 292 with
NaBH4 (1 equiv) provided alcohol 312, which was quite stable and could be chromatographed.
In order to effect complete reduction to the methylene compound 307, an excess of NaBH4 was
added to oxoacetate 292. However this led to a complex mixture of products, one of which was
identified as the aldehyde 313. Addition of Lewis95 or protic91b acids to the reaction medium in
order to facilitate formation of an azafulvene did not improve the outcome. Several attempts
were made to mesylate the alcohol moiety of 312 in the presence of NaBH4 in order to expedite
generation of an azafulvene intermediate, but this method also failed. Finally, some attempted
reductions of the alcohol 312 using P2I4,96 Ph3P/I2,97 or a Barton-McCombie sequence98 failed to
provide any of the desired product 307.
Scheme 76
It was discovered that deoxygenation of the oxoacetate 292 could be achieved, albeit in
low yield and with concomitant indole reduction, by treating 292 with NaBH3CN/ZnI2 in
Page 88
78
refluxing 1,2-dichloroethane,99 followed by refluxing the crude material in MeOH to break up
the resulting amine borane complex (Scheme 77). Reoxidation of indoline 314 to indole 307 was
attempted using DDQ but was unsuccessful.100
Scheme 77
3.15 – Stepwise Deoxygenation of the Indole-3-oxoacetates
Since deoxygenation of the oxoacetate functionality in 292 using hydride reagents proved
to be unsuccessful, and since the presence of the benzyl ester of the oxoacetate and
nitrophenylsulfonamide functionalities in 292 precluded the use of catalytic hydrogenation, we
turned to the modified substrate 315 with which we could explore the latter process. Thus, using
the methodology previously developed, addition of the dianion of indole 286 to the nitrosoalkene
generated from α-chlorooxime 264 provided Michael adduct 315 as a single diastereomer and a
1:1 mixture of oxime isomers in excellent yield (Scheme 78). The relative stereochemistry of
315 was subsequently established by X-ray analysis on a derivative (vide infra).
Page 89
79
Scheme 78
The stereochemical outcome of the above Michael addition may be rationalized by
invoking transition state 316a, where a metal complexed ester enolate is oriented in such a way
as to minimize steric interactions between the indole and the bulky tosyl group of the
nitrosoalkene component (Scheme 79). Such an orientation avoids the significant steric
interactions which are present when the tosyl group is syn to the indole as depicted in transition
state 316b.
Scheme 79
Page 90
80
Prior to the deoxygenation sequence, oxime 315 was converted to OTBS ether 317 in
good yield (Scheme 80). Next, treatment of oxoacetate 317 with NaBH4 (1 equiv) cleanly
provided the alcohol 318 in nearly quantitative yield. However no reduction occurred when
either ketone 315 or alcohol 317 was subjected to standard hydrogenation conditions (H2 (1 atm),
Pd/C, EtOH).101,93 On the other hand, in a reductive hydrogenation of a similar system, Hlasta
and coworkers had found that converting the alcohol to an acetate and then subjecting this
compound to hydrogenation with Pd/C in a 1:9 TEA/EtOH solution was necessary for reduction
to occur (presumably via an azafulvene). Therefore, alcohol 318 was acetylated in high yield to
provide the acetate 319. Gratifyingly, treatment of the activated compound 319 under Hlasta’s
hydrogenation conditions93 provided the desired reduction product 320, albeit with some minor
impurities which co-eluted with 320 during purification by column chromatography. We
suspected from 1H NMR analysis of the products that the impurities may be a side product
resulting from the addition of EtOH to the azafulvene intermediate.
Scheme 80
Page 91
81
To investigate the nature of these side products, indoles E-319 and Z-319 were
individually treated with a 1:9 TEA/EtOH mixture in the absence of H2 and Pd/C (Scheme 81)
leading to adducts E-321 and Z-321 which result from the attack of EtOH on to the azafulvene
intermediate. Indeed, EtOH adducts E- and Z-321 were confirmed to be the side products of the
above hydrogenation by comparison of 1H NMR spectra. It was also discovered that in all of the
TLC mobile phases examined, the ethanol adducts E-321 and Z-321 had identical Rf values as
the reduction products E-320 and Z-320, respectively. Thus, we were unable to separate the side
products 321 from the desired deoxygenation products 320.
Scheme 81
Since the contamination of deoxygenation products 320 with ethanol adducts 321
compromised the efficiency of later steps in the synthesis, we needed to develop an alternative
hydrogenation protocol. To determine if an alcohol solvent was necessary for the hydrogenation
of acetate 319, the EtOH was replaced with EtOAc, but no reaction occurred during several
hours (Scheme 82). However, if EtOH was then added to the hydrogenation reaction medium,
the product 320 began to form. Additionally, the acetate 319 was not reduced when the
Page 92
82
TEA/EtOH solvent mixture was replaced with AcOH. Interestingly, attempted hydrogenation of
acetate 319 using the more reactive Pearlman’s catalyst in EtOH, with or without TEA, failed to
produce any of the desired compound 320. Therefore, it became apparent that an alcohol solvent
was necessary for the hydrogenolysis to occur. In order to minimize formation of an alcohol-
indole adduct, we treated acetate 319 with various TEA/alcohol mixtures and discovered that no
substitution occurs when 319 is stirred in a 1:9 TEA/t-BuOH solution for 24 h.
Scheme 82
Gratifyingly, when acetate 319 was subjected to the modified hydrogenation conditions
using t-BuOH instead of EtOH, the deoxygenation product 320 was produced in nearly
quantitative yield (Scheme 83) with no detectable side products. However, the reduction
occurred at a significantly slower pace in t-BuOH (~5 days) as compared to EtOH (~1 day).
Page 93
83
Scheme 83
Before moving forward in the synthesis, we decided to investigate toluenesulfonamide
320 as a model substrate for probing the eventual removal of the N-Ts protecting group. Since
removal of this protecting group would be one of the final steps in our synthesis, we needed to
find a deprotection method that would be compatible with the indole and ester functionalities that
are present in the natural product (cf. Scheme 49). We were pleased to find that treatment of
indole 320 with sodium naphthalenide produced the free amine 323 in 55% unoptimized yield
(Scheme 84).
Scheme 84
3.16 - Development and Application of a New Reductive Deoximation Protocol
Based on our previous observations (cf. Schemes 47 and 61), it was evident that
protection of the indole nitrogen of 320 was required before converting the oxime to a ketone in
Page 94
84
order to avoid cyclocondensation between the ketone and nitrogen of the indole. Furthermore,
since we were unable to install N-sulfonyl or a N-alkyl protecting groups in a related system (cf.
Schemes 48), we opted to protect the indole as a Boc-carbamate. Thus, indole 320 was
converted to N-Boc indole 324 in good yield (Scheme 85). Next, treatment of O-TBS oxime 324
with TBAF for 1 minute at 0 °C led to loss of the TBS group and produced free oxime 325 in
93% yield. Stirring the reaction mixture for longer time periods led to degradation of the
product, presumably due to side reactions of basic TBAF with the Boc-carbamate102 and/or
TMSE groups. For example, when the same desilylation reaction was run for 15 min, oxime 325
was isolated in only 18% yield.
Scheme 84
Previously using model oximes, we had identified TiCl3 as a suitable reagent for
deoximations in the presence of the sensitive indole functionality (cf. Schemes 47, 48 and 61).
Unfortunately, when the more complex oxime 325 was subjected to the same conditions the
desired ketone 326 was isolated in only 26% yield (Scheme 86). Several other deoximation
protocols were tested but were largely ineffective. For example, oxidative cleavage of the oxime
Page 95
85
with Dess Martin periodinane103 produced the ketone 326 in low yield along with multiple side
products. Similarly, use of KMnO4104
to cleave the oxime led to a complex mixture of products
with only a small amount ketone observed by thin layer chromatography. Finally, an acidic
hydrolysis of the oxime with acetic acid was attempted.105a However, only slow formation of the
ketone product 326 was observed at room temperature and heating of the reaction mixture
generated a complex mixture of products.
Scheme 86
Based on the above difficulty in converting oxime 325 to ketone 326, we were prompted
to develop new deoximation methodology that might be applicable to sensitive alkaloid systems
such as 325. The majority of existing deoximation protocols,105 which are generally run under
hydrolytic or oxidative conditions, are often incompatible with indole and/or amine functionality
found in many alkaloids. Therefore, we sought to develop a mild, reductive deoximation
protocol that would be compatible with such systems.
Page 96
86
In this regard, we were inspired by Burke’s methodology for reductive conversion of
oximes to enamides with iron powder and acetic anhydride in hot toluene (Scheme 87).106 For
example, when propiophenone oxime (327) was heated in a mixture of Fe, Ac2O, AcOH, and
PhMe at 75 °C, the enamide 328 was isolated in high purity as a mixture of geometric isomers
(no yield was reported). Interestingly, in the absence of added AcOH, the reductive amidation
occurs at a slower rate.
Scheme 87
This method was later improved by Zhang, who found that the Fe(0)-mediated reductive
amidation could be effected at room temperature in DMF when a catalytic amount of TMSCl
was added to the reaction mixture (Scheme 88a).107 For example, α-oxygenated aryl ketoximes
329 were converted under these mild conditions to the corresponding enamides 330 in good
yields. More recently, our group expanded the scope of this reaction by employing acid
chlorides and chloroformates of various types to produce enamides and enecarbamates (Scheme
88b).108 For example, in the presence of benzoyl chloride, α-tetralone oxime 331 was converted
to enamide 332 in good yield. Alternatively, when ethyl chloroformate was used as the acylating
agent enecarbamate 333 was generated in good yield. Additionally, non-conjugated enamides
and enecarbamates can be prepared by this methodology. However, these compounds were
found to be quite unstable toward hydrolysis and chromatography and were generally used
without purification in subsequent reactions (vide infra).
Page 97
87
Scheme 88
Mechanistically, it is most likely that these types of reactions involve two single electron
reductions (Scheme 89).109 Thus, following formation of the O-acyl oxime 336, a single electron
reduction produces the iminyl radical 337 Subsequent single electron reduction of iminyl radical
337 generates the iminyl anion 338 which is then protonated to yield the imine 339. Capture of
the acylating agent 335 by the enamine tautomer 340 leads to the N-acyl enamine product 341.
Scheme 89
Page 98
88
Deoximation reactions which are promoted by Fe(0) have been previously reported. 110,111
However, these methods are generally run under harsh conditions (i.e. Fe and conc. HCl in
refluxing MeOH). It occurred to us that a modification of the above Burke/Zhang oxime
reduction procedures (Cf. Schemes 87-89), in which the acylating reagent is omitted, may
produce an imine intermediate such as 339 which would be easily hydrolyzed to the
corresponding carbonyl compound upon aqueous work-up.
To explore the feasibility of this idea, we treated the model 3-piperidone oxime 342 with
Fe powder and a catalytic amount of TMSCl in MeOH, THF, or PhMe (Scheme 90). However,
no ketone 246 was produced at room temperature or at reflux for several hours and only E/Z
isomerization of the oxime was observed.
Scheme 90
In the earlier study, we had observed that the enamides and enecarbamates derived from
aliphatic ketoximes were very susceptible to hydrolysis.108 Since we needed to obtain the
corresponding ketone 246 from oxime 342, we turned our attention to forming the enamide in
situ and then purposely hydrolyzing this intermediate to the corresponding ketone (Scheme 91a).
Thus, when Ac2O and a catalytic amount of TMSCl was added to a solution of the model oxime
342 and Fe powder (10 equiv) in THF, complete consumption of the starting material occurred at
Page 99
89
room temperature within 30 min to presumably generate enamide 345. After an acidic work-up
with 1 M HCl, ketone 246 and some oxime acetate 343 were isolated. However, the overall
yield of ketone 246 was highly variable (37-70%). Unfortunately, application of this procedure
to our synthetic substrate 325 also led to variable yields of ketone 326 (Scheme 91b).
Scheme 86
Since acetylation of the oxime most likely occurs prior to reductive imine formation as
depicted in Scheme 91a, we hoped that preformation of the oxime acetate 343 would boost the
overall efficiency of the reaction. Additionally, by omitting Ac2O from the reaction, the sole
product of the reduction should be imine 344 which should be quite prone to hydrolysis. Indeed,
when oxime acetate 343 was subjected to the previously developed conditions, but without acetic
anhydride, complete consumption of the oxime acetate was observed within 30 min and
Page 100
90
production of ketone 246 was observed by TLC analysis (via in situ hydrolysis of the
intermediate imine 344). Aqueous work-up led to isolation of the ketone 246 in moderate yields
(~60-70%) as well as some of the free oxime 342. Surprisingly, however, this reaction was
irreproducible and in several runs very little of the oxime acetate 343 was converted to ketone
246. After some investigation, it was discovered that the successful reactions had been run in
flasks which had previously been soaked in concentrated nitric acid and then thoroughly rinsed
with water to remove the iron oxide residues of previous runs. Thus, we realized that a trace
amount of acid was catalyzing the conversion of 343 to 246. Indeed, addition of a catalytic
amount of various acids (nitric acid, trifluoroacetic acid, and p-toluenesulfonic acid) increased
the amount of ketone produced. However use of a small amount of glacial acetic acid gave the
best results. When these new conditions were applied to the complex indole oxime acetate 346,
available in high yield from acetylation of oxime 325, the ketone was produced in a reasonable
57 % yield along with some deacetylated oxime 325.
Scheme 92
Page 101
91
Since we observed some deacetylation of the ketoxime acetate 346 in this reaction to
form the unreactive free oxime 325, we decided to explore the use of other less labile O-acyl-
groups in this reaction that might circumvent this problem. This phase of the study was
conducted by Jason Witek of our group. Initially, 4-phenylcyclohexanone oxime was O-acylated
with several different groups that we hoped would expedite imine formation and/or inhibit
deacylation to the unreactive free oxime (Scheme 93). Once again, the oxime acetate 347 was
converted to ketone 351 in reasonable yield but with formation of some free oxime by-product.
Use of benzoate 348 and ethyl carbonate 349 in the deoximation procedure led to lower yields of
ketone 351. On the other hand, use of the pivalate 350 provided ketone 351 in 88% isolated
yield and with no detectable amount of the free oxime.
Scheme 93
Based on the above optimization studies, a general procedure for deoximation was
developed (Scheme 94).112 Thus, a catalytic amount of glacial AcOH (1 drop) and TMSCl (1
drop) are sequentially added to a mixture of oxime pivalate 352 (1 eq) and Fe powder (10 eq) at
room temperature. The mixture is stirred for 30 min, diluted with water, stirred for an additional
15 min and then worked up to provide the ketone 353. These general conditions were applied to
several types of ketoxime pivalates shown in Table 5. It should be noted that aldoxime pivalates
Page 102
92
were found to be poor substrates under the general deoximation conditions since formation of
nitriles was favored over production of the corresponding aldehydes.
Scheme 94
Table 5. Conversion of Ketoxime Pivalates to the Corresponding Ketones
Gratifyingly, subjecting the indole oxime pivalate 354 to the general deoximation
conditions provided ketone 326 in 84% yield (Scheme 89).
Page 103
93
Scheme 89
3.17 – Synthesis of Keto Acid 361 and Subsequent Application of Romo’s Aldol
Lactonization
With an efficient route to ketone 326 now developed, all that remained to access the key
keto acid substrate for the Romo aldol lactonization was to selectively cleave the 2-
trimethylsilylethyl ester to the corresponding carboxylic acid. However, attempted removal of
the 2-trimethylsilylethyl ester in 326 using several fluoride-based methods only led to
decomposition of the starting material, perhaps due to the presence of the Boc-carbamate which
is sensitive to fluoride.102 Alternatively, it was found that treatment of 326 with TFA led to the
desired cleavage of the 2-trimethylsilylethyl ester.113 However, concomitant loss of the Boc
protecting group was unavoidable and only cyclodehydration product 356 was isolated from the
reaction (Scheme 96). Therefore, a more acid stable protecting group for the indole nitrogen was
required.
Page 104
94
Scheme 96
At this stage of the synthesis, the oxime isomers of NH indole 320 were separated and
individually subjected to Cbz-protection conditions to provide N-Cbz indoles E- and Z-357 in
identical 89% yields (Scheme 97). This separation was required because in these acylation
reactions, some unreacted NH indole remains and the chromatographic polarity of NH indole Z-
320 is approximately the same as N-Cbz indole E-357, which makes purification difficult. Slow
evaporation of an EtOAc solution of Z-357 provided crystals that were suitable for X-ray
analysis141 (see Experimental Section for ORTEP of Z-357 (pg. 186)) and the relative
stereochemistry of the compound was determined to be as shown (cf. Scheme 79).
Page 105
95
Scheme 97
Moving forward, the recombined mixture of O-TBS-oximes 357 was treated with TBAF
buffered with AcOH to prevent side reactions resulting from the basic fluoride to give the free
oximes 358 (cf. Scheme 85). Subsequent conversion of oximes 358 to the oxime pivalates 359
and reductive deoximation using our Fe-mediated protocol provided ketone 360 in good yield.
Finally, 2-trimethylsilylethyl ester 360 was smoothly converted to the pivotal keto acid 361 in
92% yield by treatment with TFA.112
Page 106
96
Scheme 98
With keto acid 361 in hand, we were now ready to attempt the key Romo aldol
lactonization. Thus, slow addition of the keto acid 361 to a mixture 2-bromo-N-
propylpyridinium triflate, Hunig’s base, and PPY in dichloromethane led to smooth conversion
to the pentacyclic β-lactones which were isolated as a chromatographically inseparable 8:1
mixture of the desired cis-azadecaline 362 and the trans-azadecalin 363 (Scheme 92). This
diastereomeric β-lactone mixture was treated with DIBAL-H to provide a 7.5:1 mixture of 1,3-
diols 364 and 365 which were now separable by column chromatography.
Page 107
97
Scheme 99
Extensive 2D-NMR analysis (HMBC, HMQC, COSY, and NOESY) was conducted in
order to determine the structure and conformation of the cis-azadecalin 1,3-diol 364. It should
be noted that 1,3-diol 364 is similar in structure to alstilobanine A (169), which in solution has
the piperidine ring in a chair conformation (169a) and where the C-19 tertiary hydroxyl group is
axial relative to that ring (Figure 6). However, azadecalin 364 is in the opposite conformation in
which the C-20 alcohol is equatorial to the piperidine ring. The difference in ring conformations
between 364 and 169 may be due to the presence of the C-16 hydroxy methyl group in
alstilobanine A. This substituent may cause the piperidine ring of alstilobanine A to exist as the
cis-azadecalin conformer 169a, rather than a conformation like 169b in which the C-20 hydroxyl
group has an energetically penalizing 1,3-diaxial interaction with the C-16 hydroxymethyl group.
Conversely, the piperidine ring in 1,3-diol 364a may exist in the opposite conformer relative to
alstilobanine A to avoid torsional strain between the C-16-C/C-22 and C-14/C-15 bonds that
Page 108
98
would exist in a conformation like 364b. The structure of the trans-azadecalin system was later
established by X-ray analysis (vide infra).
Figure 6. Conformations of Alstilobanine A (169) and Major 1,3-Diol 364
3.18 – Attempted Intermolecular Ester Enolate Alkylations to Install the C-17
Hydroxymethyl Unit
Our plan at this stage of the synthesis was to add formaldehyde (or an equivalent) to the
upper face of an enolate derived from 366 which could exist in conformations 367a or 367b
(Scheme 100). It was our anticipation that the electrophile might add from the top, least
congested face of either enolate, although such attack would be more likely from the natural
product-like conformation 367b, where the C20-O bond is in the equatorial position.
Page 109
99
Conversely, addition of electrophiles to the top face of conformation 367a may be hindered by a
developing 1,3-diaxial interaction between the C20-O bond and the incoming electrophile.
Scheme 100
Initial attempts were made to directly install the C-17 hydroxymethyl group into 1,3-diol
364. Unfortunately, only starting material was recovered upon treatment of 364 with potassium
carbonate and formalin. Treatment of 364 with NaH (3 equiv) and paraformaldehyde in DMF
only led to decomposition. Therefore, we opted to protect the 1,3-diol of 364 at this stage.
Treatment of 364 with the di-t-butylsilyl bis(triflate)114 and 2,6-lutidine only led to silylation of
the primary alcohol to yield silanol 369 (Scheme 101a). However, subjecting 364 to a mixture of
2,2-dimethoxypropane and acetone under acidic conditions provided acetonide 370 in low yield
(Scheme 101b). Unfortunately, we were unable to optimize this reaction. Alternatively, reacting
1,3-diol 364 with triphosgene115 produced the cyclic carbonate 371 in high yield (Scheme 101c).
The protected 1,3-diols 370 and 371 are assumed to maintain the unnatural cis-azadecalin
Page 110
100
conformation shown. However, no 2D NMR studies were conducted in order to confirm this
supposition.
Scheme 101
Unfortunately, treatment of indole ester 371 with LDA and monomeric formaldehyde116
at -78 °C gave no condensation product, and warming of the reaction mixture led to loss of the
carbonate protecting group (Scheme 102a). Similarly, treatment of indole ester 370 with LDA
and monomeric formaldehyde did not produce any condensation product 373 and only starting
material was recovered (Scheme 102b).
Page 111
101
Scheme 102
We reasoned that converting 1,3-diol 364 to acetonide 370 was low yielding since the
product contained a 1,3-diaxial interaction between the axial methyl group on the acetonide ring
and the C-21 methylene of the piperidine ring (cf. Scheme 101b). To circumvent this problem,
1,3-diol 364 was stirred in refluxing dimethoxyethane containing a catalytic amount of p-
toluenesulfonic acid to yield the corresponding acetal 374 as a single diastereomer (Scheme
103). The methyl group of the acetal was determined to be in an equatorial position based on
1H-1H-NOESY analysis of a later intermediate (vide infra). Once again, however, treatment of
the indole ester 374 with a variety of bases (LDA, NaH, KHMDS, etc.) followed by addition of
monomeric formaldehyde failed to produce any of the condensation product 375 and starting
material was recovered in all cases.
Page 112
102
Scheme 103
Since we thought that it might be possible that the highly reactive monomeric
formaldehyde was polymerizing at a faster rate than it was reacting with the ester enolate derived
from 374, the alkylation was attempted using various halomethyl ethers that could later be
converted to a hydroxymethyl group (i.e. BOM-Cl, PMB-Cl, MOM-Cl) (Scheme 104).
Unfortunately, all alkylation attempts failed with these electrophiles and starting material was
recovered in the majority of cases.
Scheme 104
Page 113
103
3.19 – Intramolecular Alkylation Strategy to Form a 7-Membered Ring Ether
Since we were having difficulty installing the C-17 hydroxymethyl group via
intermolecular additions, we were prompted to see whether we could construct the 7-membered
bridging ether through an intramolecular alkylation strategy (Scheme 105). For example,
intramolecular attack of the enolate derived from indole ester 378 onto the tethered halomethyl
ether would directly form the bridging ether 377.117 In turn, the halomethyl ether of 378 could
be generated directly from the methylthiomethyl (MTM) ether 379 by a number of known
methods.118
Scheme 105
To explore conversion of the primary alcohol in diol 364 to a methylthiomethyl ether, a
Pummerer alkylation using DMSO/Ac2O was first tested.119 However, these attempts
predominately led to oxidation of the primary alcohol to give the corresponding aldehyde. On the
other hand, the method of Kyler, which utilizes benzoyl peroxide and dimethyl sulfide, produced
MTM-ether 380 in reasonable yield (Scheme 106).120 Subsequent TBS-protection of the tertiary
Page 114
104
alcohol provided the crystalline compound 381, which was unambiguously shown to exist in the
unnatural azadecalin conformation by X-ray crystallography (Figure 7).141
Scheme 106
Page 115
105
Figure 7. ORTEP and Wireframe Structure of Compound 381
Page 116
106
Initial attempts to convert the methyl thiomethyl ether 381 to an iodomethyl ether using
MeI118d failed, and in all cases loss of the MTM group to yield primary alcohol 384 was observed
(Scheme 107). However, treatment of MTM-ether 381 with sulfuryl chloride119 cleanly
produced the chloromethyl ether 382. This compound proved to be quite sensitive and could not
be chromatographed or even dissolved in CDCl3 without conversion to primary alcohol 384
(presumably due to traces of HCl and H2O). However, concentration of the reaction mixture in
vacuo gave essentially pure chloromethyl ether 382 as determined by 1H NMR analysis of the
crude material in C6D6. Unfortunately, all attempts to effect an intramolecular alkylation of 382
failed and primary alcohol 384 was usually the only product obtained from the reaction.
Scheme 107
N
OTBS
HTs
HNCbz
H
MeO2C
O
N
OTBS
HTs
HNCbz
H
MeO2C
OSO2Cl2
CH2Cl2, 0 C
conditions
N
OTBS
HTs
HNCbz
H
MeO2C
OH
N
NO
CO2Me
OTBS
H
Ts
Cbz
381 382
384383
ClMeS
conditions tried:LiHMDS, THF, -78 C to rtNaH, THF, 0 to 50 CLDA, THF, -78 C to rtLDA, HMPA, THF, -78 C to rt
Page 117
107
3.20 – Intermolecular Ester Enolate Alkylations Revisited
When planning our synthesis, we had anticipated that formation of the requisite C-17
hydroxymethyl system at this stage would be challenging. However, we were surprised to find
that absolutely no C-17 alkylation products were observed in any of the above reactions. In light
of these failures, we reasoned that the main problem may lie in formation of the ester enolate
itself. Inspection of molecular models suggested that the ester enolate of 385 might be unstable
due to A1,3 strain121 with the adjacent Cbz-carbamate (Figure 8). Although we had considered
this potential problem earlier, it seemed reasonable that the enolate could perhaps assume a
conformation in which A1,3 strain would be minimized.
Figure 8. A1,3-Strain in N-Cbz Indole Enolate 385
In order to test this hypothesis, the Cbz-carbamate moiety of 374 was removed via
hydrogenolysis to yield NH indole 389 in 96% yield (Scheme 108). Indeed, treatment of the free
indole 389 with LiHMDS (3.5 eq) followed by addition of monomeric formaldehyde produced
several alkylation products. Two of these products were identified as hemiaminal 390 and cyclic
aminal 391 which also contained a hydroxymethyl group of unknown configuration. It should be
noted that Overman and coworkers have successfully α-hydroxymethylated a similar indole ester
by this procedure.73 When indole ester 389 was treated with 1.0 equivalent of LiHMDS at -78
Page 118
108
°C followed by addition of BOM-Cl and warming to rt, a complex mixture of products was
produced, one of which was identified as the N-alkylated compound 392.
Scheme 108
While we were examining the alkylation of the enolate derived from NH indole ester 389,
we discovered that when N-Cbz indole 374 was treated with an excess of sodium hydride and
paraformaldehyde in DMF, the alkylation product 393 containing a free indole was obtained in
good yield (Scheme 109). In this case, it is most likely that cleavage of the Cbz-carbamate in
374 by adventitious sodium hydroxide occurs prior to the formation of dianion 394 and
subsequent alkylation. Unfortunately, the alkylation product was isolated exclusively as the
undesired C-17 diastereomer which was proven by observation of a strong nuclear Overhauser
effect between the methyl ester and acetal methyl protons. The stereochemical outcome of the
reaction may be explained by examining the initially formed enolate 394a. In this intermediate,
Page 119
109
attack by the enolate on the electrophile from the desired top face is hindered by a 1,3-diaxial
interaction between the incoming electrophile and the C20-O bond. Furthermore,
conformational inversion of the azadecalin system to an alstilobane A-type conformation 394b
may be prevented by A1,3-strain between the enolate and the C14/C15 bond of the adjacent
piperidine ring.
Scheme 109
3.21 – A Silicon-Tethered Intramolecular Ester Enolate Alkylation to Install C-17
Given the above result, it was unlikely that we could install the C-17 hydroxymethyl
substituent with the requisite configuration via an intermolecular alkylation. However, since all
derivatives of the 1,3-diol 364 presumably exist in the unnatural cis-azadecalin conformation (cf.
Figure 6), we reasoned that the C-17 hydroxymethyl group could be installed using an
intramolecular alkylation of a halomethylsilane like 397 (Scheme 110).122 The cyclic siloxane
Page 120
110
396 produced by this process could then undergo a Fleming-Tamao oxidation123 to generate the
C-17 hydroxymethyl moiety with the proper stereochemistry.
Scheme 110
To investigate this strategy, the tertiary alcohol moiety of 380 was smoothly converted to
the chloromethyl(dimethyl)silyl protected derivative 398 by treatment with an excess of
chloromethyl(dimethyl)silyl triflate and 2,6-lutidine in refluxing CH2Cl2 (Scheme 111).124 Next,
a Finkelstein reaction converted chloride 398 to the iodide 399. However, Cbz-removal via
catalytic hydrogenation was not possible on any of the synthetic intermediates which contained
the catalyst poisoning sulfide. Use of Bennesar’s procedure125 involving tributyltin radical
mediated Cbz-cleavage appeared to provide some of the desired deprotected product derived
from 380. However, this material contained several impurities and could not be used in
subsequent reactions. Similarly, base catalyzed methanolysis126 of the N-Cbz indole 380 failed
to provide the desired NH indole. With N-Cbz indole 399 in hand, we attempted to effect the
desired intramolecular ester enolate alkylation, but unsurprisingly, based on the results discussed
above, no reaction occurred and only the starting indole was recovered.
Page 121
111
Scheme 111
One of our strategies for synthesizing alstilobanine A (169), which lacks the 7-membered
bridging ether, was to deoxygenate the C-18 primary alcohol using a Barton-McCombie
sequence. Toward this end, xanthate ester 401 was prepared in acceptable yield using standard
conditions (Scheme 112). However subjecting 401 to radical deoxygenation in refluxing toluene
only led to formation of the oxetane 403 in low yield, presumably via an SN2-pathway.
Alternatively, we attempted to employ Myers protocol for the selective deoxygenation of
primary alcohols in the presence of secondary and tertiary alcohols, but unfortunately this
reaction failed in our system.127
Page 122
112
Scheme 112
Fortunately, selective protection of the primary alcohol in 364 could be achieved with
TBSOTf and 2,6-lutidine to provide OTBS ether 404 (Scheme 113). Next, removal of the Cbz-
protecting group via hydrogenolysis occurred cleanly to give NH indole 405 and subsequent
silylation of the tertiary alcohol with chloromethyl(dimethyl)silyl triflate produced
chloromethylsilane 406. Refluxing 406 in a solution of NaI and acetone provided iodide 407 in
nearly quantitative yield. To our delight, treatment of the indole ester 407 with KHMDS (3
equiv) effected cyclization at C-16 to produce the silyl ether 408 in excellent yield.
Page 123
113
Scheme 113
At the same time that we were developing the above route for the synthesis of siloxane
408, we were also testing an alternative one-pot strategy for eventual introduction of the two silyl
groups into intermediate 364 to make 406. For this study, we used the minor trans-azadecalin
1,3-diol 365 as a model system. Thus, the N-Cbz-protecting group in 365 was removed via
hydrogenolysis and the crude NH indole 409 was treated with TBSOTf and 2,6-lutidine (Scheme
114). After complete consumption of the starting indole 409 as monitored by TLC,
chloromethyl(dimethyl)silyl triflate and additional 2,6-lutidine was added to the reaction mixture
to ultimately provide the differentially bis-O-silylated product 411 in 90% yield over 2 steps. In
one of the runs of this reaction, not all of the monosilylated intermediate 410 was converted to
disilylated product 414. Compound 410 was isolated and found to be crystalline and analysis of
this derivative by X-ray diffraction141 confirmed the structure of a trans-azadecalin ring system
in 365 (Figure 9).
Page 124
114
Scheme 114
Figure 9. ORTEP Structure of O-TBS Ether 411
Page 125
115
3.22 – Attempted Fleming-Tamao Oxidation of Cyclic Silyl Ethers
After successfully forming the challenging C-16/C-17 bond using a silicon tethered
intramolecular alkylation, the next step was to convert the siloxane into the corresponding
hydroxymethyl compound. While numerous methods exist for this transformation, we decided
to focus on oxidations run in basic media since we reasoned that the peroxide anion would be
less likely to oxidize the nucleophilic indole than under neutral or acidic conditions. For easier
monitoring of the reaction, the TBS group was first removed from 408 by treatment with TBAF
to provide primary alcohol 412 (Scheme 115). Unfortunately, when silane 412 was subjected to
the standard Tamao conditions, no reaction occurred at room temperature or upon refluxing the
reaction mixture (entries A and B). However, when KF was replaced with TBAF, the 1,3-diol
414 was isolated as the sole product (entry C). Thus, it is likely that the oxidation occurs to
initially generate the triol 413, but under the basic conditions a retro-aldol reaction takes place to
ultimately produce 1,3-diol ester 414. In order to examine whether more neutral conditions
would provide the triol, 412 was treated with KHF2 and H2O2 in hot DMF, but only a complex
mixture was obtained (entry D). Interestingly, subjecting the OTBS protected substrate 408 to
the same conditions as entry C led to no reaction (entry E). In a parallel reaction, the same
substrate was treated under similar conditions but without TBAF (entry F). Initially, no reaction
occurred at room temperature, but when a large excess of hydrogen peroxide was added, and the
reaction mixture was refluxed for 2 days, two new products were formed in low yield which
were tentatively assigned as hydroxymethyl compounds 415 and 416 resulting from over-
oxidation and/or elimination.
Page 126
116
Scheme 115
N
O
HTs
HN
MeO2C
SiMe
Me
RO
N
OH
HTs
HN
MeO2C
HOH
TBAFTHF, 0 C90%
408 R = TBS
412 R = H
Fleming-Tamaooxidation
413
414
Oxidation Conditions
KHCO3, KF, 30% H2O2MeOH/THF, rt
KHCO3, KF, 30% H2O2MeOH/THF, reflux
KHCO3, TBAF, 30% H2O2MeOH/THF, rt
KHF2, 30% H2O2DMF, 80 C
KHCO3, TBAF, 30% H2O2MeOH/THF, rt
KHCO3, 30% H2O2MeOH/THF, reflux
Result
no reaction
no reaction
414 (58%)
decomposition
no reaction
415 and 416
Entry
A
B
C
D
E
F
H
H
Indole
412
412
412
412
408
408
NH
NTsOH
H
TBSO
CO2MeOH
NH
NTs
H
TBSO
CO2MeOH
415
416
N
HOH
MeO2C
N
Ts
H
HHO
HO
One of the drawbacks of the Fleming-Tamao oxidation is that sterically hindered
siloxanes, such as in our system, are often difficult to oxidize.123c In order to enhance the
reactivity of our system, some attempts were made to open the cyclic silyl ether 412 to the more
reactive silanol 418 or silyl fluoride 417 (Scheme 116). Unfortunately, treatment of 412 with
excess TBAF at room temperature produced no reaction after several days and heating the
reaction mixture led to complete decomposition (entries A and B).128 Treatment of the silane
Page 127
117
412 with silica gel failed to produce the desired silanol 418 (entry C).129 Additionally, the
siloxane was found to be stable under basic conditions for several days (entry D). Finally, acidic
hydrolysis of the Si-O bond in 412 was attempted, but 412 was stable in AcOH (entry E) and
treatment with TFA only produced the trifluoroacetate 419 (entry F).
Scheme 116
As seen above, the cyclic silyl ether 412 was very stable under a variety of conditions.
Since siloxanes derived from tertiary alcohols tend to be less reactive toward hydrolysis130 and
oxidation than the corresponding siloxanes derived from secondary and primary alcohols,123c we
decided to move the silicon tether to the primary alcohol. In this case, the silyl group should be
more prone to both hydrolysis and oxidation compared to the sterically hindered siloxane 412.
Thus, the primary alcohol of 414 was silylated with a chloromethyl(dimethyl)silyl group to yield
Page 128
118
chloromethylsiloxane 420 (Scheme 117). This compound, which was very acid sensitive, was
immediately treated with TMSOTf and 2,6-lutidine to give the desired disilyl compound 421 as
well as some of the bis-OTMS product 422. When compound 421, was treated with KHMDS, a
mixture of several products was obtained which included the intramolecular alkylation products
423 and 424, as well as the ring-opened product 425 in an overall low yield. Unfortunately, we
were unable to optimize this reaction to produce any of the cycloalkylation products 423-425 in
higher yield.
Scheme 117
To improve the acid stability of the silicon tether, we decided at this point to use a
chloromethyl(diphenyl)silyl group instead. Thus, the 1,3-diol 414 was selectively silylated with
Page 129
119
chloromethyl(diphenyl)silyl chloride to give 426, which was considerably more stable than 420
(Scheme 118). After trimethylsilyl protection of the tertiary alcohol 426, indole ester 427 was
treated with KHMDS to effect an intramolecular alkylation. Surprisingly, only a trace amount of
what is presumably the cycloalkylation product 428 was observed via mass spectrometry but
very little material could be isolated (<10% of the mass balance).
Scheme 118
Since we had already developed an efficient route to form the cyclic dimethylsilyl ether
408 (cf. Scheme 113), we decided to prepare the analogous diphenylsilyl ether since, in general,
diphenylsiloxanes are more prone to undergo a Fleming-Tamao oxidation than the corresponding
dialkylsiloxanes.131 Moreover, a protodesilylation of one or both of the phenyl groups could, in
principle, be effected to produce even more reactive silyl intermediates for the oxidation.123c
In order to install the chloromethyl(diphenyl)silyl group onto the tertiary alcohol of 405,
it was necessary to synthesize diphenylsilyl triflate 430, which to the best of our knowledge was
Page 130
120
previously unknown. Thus, treatment of chloromethyl triphenylsilane (429)132 with TfOH in
refluxing 1,2-dichloroethane produced the desired triflate in an unoptimized 50% yield (Scheme
119).133
Scheme 119
Silylation of tertiary alcohol 405 with chloromethyl(diphenyl)silyltriflate and subsequent
conversion of the chloride to the corresponding iodide provided iodomethylsilane 432 in good
yield (Scheme 120). However, we were surprised to find that treatment of 432 with KHMDS led
to only a very small amount of the desired product 433 and, once again, very little material was
isolable (<15% of the mass balance).
Scheme 120
Page 131
121
3.23 – Reprotection of the Indole Nitrogen to Prevent a Retro Aldol Reaction
As discussed above, we had discovered an oxidation/retro-aldol sequence involving
cyclic silyl ether 412 (cf. Scheme 115). However, based on our previous observations, we
reasoned that it might be possible to prevent the retro-aldol process from occurring if the indole
nitrogen is acylated during the Fleming-Tamao oxidation since formation of an enolate
intermediate would be disfavored due to A1,3 strain (cf. Figure 7). Toward this end, the Cbz-
protecting group was reintroduced onto the indole nitrogen of 408, albeit in low yield (Scheme
121a). However, Boc-protection of the indole 408 was achieved in high yield to provide N-Boc
indole 446 (Scheme 121b). In addition, since we were concerned that the Cbz- and Boc-
protecting groups may too labile under the required basic conditions of the Fleming-Tamao
oxidation, a N-benzyl protected analogue 448 was also prepared from indole 408 (Scheme 121c).
Additionally, the primary OTBS groups of 444, 446, and 448, were removed with TBAF to yield
the corresponding primary alcohols 445, 447, and 449, respectively.
Page 132
122
Scheme 121
When removing the O-TBS-protecting group from the N-Boc indole 446, we observed
formation of a significant amount of the NH-indole as a side product. Therefore, due to the
sensitivity of the Boc-carbamate to fluoride, N-Boc indole 447 was subjected to buffered
Fleming-Tamao oxidation conditions (Scheme 122), but unfortunately no reaction occurred at
room temperature or at reflux (entries A and B). We also examined the use of stronger oxidants
with the N-Boc indole 447. However, when 447 was treated with m-CPBA and KF in DMF
Page 133
123
only starting material was recovered (entry C). On the other hand, treatment of 447 with
peracetic acid (formed in situ from 30% H2O2 and Ac2O) led to a small amount of the over-
oxidized product assumed to be diol 451 (entry D). N-Cbz indole 444 was heated in a mixture of
H2O2, KF, and DMF but no reaction occurred (entry E). Finally, N-Bn indole 449 was subjected
to the same conditions that promoted an oxidation/retro-aldol of the corresponding NH indole
412 (cf. Scheme 115), but no reaction occurred at room temperature or on heating the reaction
mixture.
Scheme 122
Page 134
124
3.24 – Use of the Woerpel Modification of the Fleming-Tamao Oxidation
Woerpel has developed a useful modification for the oxidation of sterically hindered
siloxanes.134 For example, when the oxasilacyclopentane 452 was treated under standard
conditions (aqueous H2O2, KHCO3, and KF) only starting material was recovered (Scheme 123).
Additionally, none of the other usual modifications to the Fleming-Tamao oxidation were
successful with this substrate.132,122a However, it was found that when oxasilacyclopentane 452
was added to a mixture of t-BuOOH (14 equiv) and CsOH.H2O (12 equiv) in DMF at room
temperature, an oxidation occurred to produce dioxasilacyclohexane 453, which could be
isolated from the reaction mixture. However, to complete the transformation, TBAF (5 equiv)
was added to 453 and the resulting reaction mixture was stirred for 8 h at 75 °C to provide 1,3-
diol 454. It should be noted that in the absence of TBAF or when less than 5 equivalents of the
fluoride source is used, not all of the dioxasilacyclohexane 453 was converted to the 1,3-diol
454. Notably, this protocol has been used in the successful oxidations of generally unreactive
substrates such as alkoxysilanes derived from tertiary alcohols and even silanes bearing four
alkyl substituents.
Scheme 123
Although this method has been demonstrated to be effective in simple systems, we had
initially avoided using these highly basic conditions with our substrates which contained base
Page 135
125
sensitive ester and carbamate functionalities. However, with N-Bn protected indole 449 in hand,
we were prompted to examine Woerpel’s conditions even though we expected that the methyl
ester would likely be converted to the acid. When siloxane 449 was subjected to Woerpel’s
standard conditions, all of the starting material was consumed and a crude mixture containing
primarily the retro-aldol product 455 was obtained. For full characterization, this material was
converted to the methyl ester 456 using TMSCHN2 in MeOH (Scheme 124).135 This result is
somewhat surprising since we expected that the presence of the N-benzyl group should slow the
retro-aldol pathway due to the A1,3 strain present in intermediate enolate 457.
Scheme 124
It was evident that modification of the Woerpel conditions would be necessary in order
to avoid the undesired retro-aldol pathway following the initial Fleming-Tamao oxidation. As
we were studying the above N-Bn indole system, we decided to also examine N-Boc indole 447,
which we had in hand, under Woerpel’s conditions. We anticipated that hydrolysis of the Boc-
protecting group and methyl ester would occur, but we were curious to see if the retro-aldol
Page 136
126
reaction would take place if we ran the oxidation at room temperature. In the initial test reaction,
5 mg of the N-Boc indole 447 was subjected to the Woerpel conditions at room temperature and
the reaction was stirred for 2.5 h. Analysis of the crude product mixture by mass spectrometry
suggested that only a small amount of the desired triol 457 (X = OH) was present. However, we
were surprised to find that the primary molecular ion peak in the spectrum corresponded to a
product resulting from dehydration of the triol 457. For characterization purposes, the crude
mixture was treated with TMSCHN2 and MeOH to provide 2 mg of the corresponding methyl
ester of the dehydration product. Based on the 1H NMR spectrum of this product, it appears that
the major product is a cyclic ether, which could be the result of an intramolecular etherification
via an intermediate like 457a or 457b. However, due to lack of material, it has not been
determined whether the ether is the desired 7-membered ring 458 or the undesired 5-membered
ring 459. Unfortunately, running this reaction on a larger scale using 30 mg of the starting
siloxane 447 was less efficient, and once again only a very small amount of the apparent
dehydration product was obtained.
Page 137
127
Scheme 125
Using what was learned from the oxidation of N-Boc indole 447, we subjected N-Bn
indole to the Woerpel conditions at room temperature. Thus, when siloxane 449 was added to a
solution of t-BuOOH, CsOH, and DMF at room temperature and the mixture was stirred for 10
minutes, complete consumption of the starting material was observed. Mass spectral analysis of
an aliquot suggested that the oxidation product 462 was formed (Scheme 126). It should be
noted again that Woerpel has also observed the formation of similar oxidation products in the
absence of TBAF (cf. Scheme 123). Addition of TBAF produced a crude mixture of carboxylic
acid products which was re-esterified to produce the corresponding methyl esters.
Unfortunately, the transformation was low yielding overall and we were unable to fully
Page 138
128
characterize the products beyond obtaining mass spectra. In this experiment, two components
were obtained by preparative thin layer chromatography which both had the same molecular
weight corresponding to the isomeric cyclodehydration products 464 and 465. However, the
structure of these products could not be confirmed since not enough material was produced for
1H NMR analysis. Additionally, a peak with the molecular weight for the acid triol 463 was
observed in the mass spectrum of the crude product mixture from the oxidation reaction.
Unfortunately, none of the corresponding methyl ester triol 466 was isolated from the
esterification reaction which is probably due to its high polarity.
Scheme 126
Page 139
129
3.25 – Summary of Current Synthetic Route
Scheme 127
Page 140
130
Scheme 127 cont.
3.26 - Future Work
Since subjecting N-Bn indole 449 to Woerpel’s conditions without the TBAF cleanly
produced what may be the desired oxidized product 462 (cf. Scheme 117), we will try to obtain
this product or the corresponding triol 466 (Scheme 118). The highly polar product(s) of this
Page 141
131
oxidation will be purified by preparative HPLC (MeCN/H2O gradient + 1% TFA) which will
likely furnish only triol 463 due to the presumed acid lability of the dimethyldioxysilane group in
462. Following esterification of the carboxylic acid, a cyclodehydration136 of the triol 466,
which should exist in the alstilobanine A conformation (cf. Figure 6), will be performed to
furnish a 7-membered cyclic ether. Removal of the Ts- and Bn-protecting groups from this
compound with Na/napthalenide will provide alstilobanine E (168). It should be noted that
removal of the Ts-protecting group on an earlier intermediate 320 was accomplished using
Na/napthalenide (cf. Scheme 84). A selective N-methylation of 168 will produce angustilodine
(167). To access alstilobanine A (169), a Barton-McCombie deoxygenation of primary alcohol
449 will be performed. Finally, an oxidation/esterification sequence and deprotection will
provide alstilobanine A (169).
Scheme 118
Page 142
132
Chapter 4 – Experimental Section
General Methods. All reactions were carried out under an argon atmosphere in flame-
dried glassware using standard Schlenk techniques unless otherwise noted. Anhydrous
tetrahydrofuran, diethyl ether, dichloromethane, and toluene were obtained from a solvent
dispensing system equipped with alumina drying columns. Additional solvents and reagents
were used as obtained from commercial sources without further purification. Flash column
chromatography was performed using EM Science silica gel 60 (230-400 mesh). Preparative
thin layer chromatography was performed using 0.5 or 1.0 mm EM Science silica gel 60 PF254
plates. 1H and 13C NMR spectra were obtained on a Bruker CDPX-300, DPX-300, AMX-360, or
DRX-400 MHz spectrometer. Infrared spectra were obtained on a Perkin-Elmer 1600 FTIR.
Nominal mass spectra were obtained on an Applied Biosystems 150EX. High resolution mass
spectra were obtained on a Waters LCT Premier time-of-flight (TOF) mass spectrometer. X-Ray
data was collected on a Bruker SMART APEX CCD area detector system.
General Procedure for Conversion of α-Chloroketones to α-Chloro-O-TBS-oximes.
To a stirred solution of the α-chloroketone (1.0 eq) in CH2Cl2 (ca. 0.5 M) was added PPTS (0.05
equiv.), 4Å powdered molecular sieves (~15 mg per mmol of α-chloro ketone), followed by O-
TBS-hydroxylamine (1.1-2.0 equiv.). The resulting mixture was stirred until complete
Page 143
133
consumption of the starting α-chloroketone was observed (typically 1-2 days) and then filtered
through a Celite pad washing with CH2Cl2. The filtrate was concentrated in vacuo to give a
residue which was purified by flash column chromatography on silica gel using a mixture of
ethyl acetate and hexanes to give the α-chloro-O-TBS-oxime.
2-Chlorocyclohexanone O-(tert-Butyldimethylsilyl) Oxime (127). α-Chloro-O-TBS-
oxime 127 was obtained using the above general experimental procedure with the following
quantities: α-chlorocyclohexanone (1.0 mL, 8.19 mmol), CH2Cl2 (15 mL), PPTS (105 mg, 0.41
mmol), 4Å powdered molecular sieves (~100 mg), and O-TBS-hydroxylamine (2.54 g, 16.4
mmol). 2.18 g, 100%, ~2:1 mixture of oxime isomers. 1H NMR (400 MHz, CDCl3) δ 5.69 (s,
0.33H), 4.77 (s, 0.67H), 3.27 (d, J = 14.6 Hz, 0.67H), 2.56 (td, J = 13.9, 4.7, 0.33H), 2.35 (d, J =
14.2 Hz, 0.33H) 2.22-1.81 (m, 4.67 H), 1.66-1.63 (m, 1H), 1.43-1.32 (m, 1H), 1.09-0.95 (m, 9H),
0.18 (s, 6H).
2-Chlorocyclopentanone O-(tert-Butyldimethylsilyl) Oxime. The title compound was
obtained using the above general experimental procedure with the following quantities: α-
chlorocyclopentanone (100 mg, 0.77 mmol), CH2Cl2 (1.5 mL), PPTS (10 mg, 0.04 mmol), 4Å
Page 144
134
powdered molecular sieves (~12 mg), and O-TBS-hydroxylamine (147 mg, 0.95 mmol). 172 mg,
81%, ~2:1 mixture of oxime isomers. 1H NMR (300 MHz, CDCl3) δ 4.82 (dd, J = 2.6, 0.8 Hz,
0.33H), 4.59 (dd, J = 4.6, 2.8 Hz, 0.67H), 2.51-2.45 (m, 1H), 2.26-2.15 (m, 1H), 1.93-1.88 (m,
3H), 1.70-1.67 (m, 1H), 0.79-0.76 (m, 9H), 0.00--0.02 (m, 6H); LRMS-ES+ m/z (relative
intensity) 248 (MH+, 30).
2-Chloro-3-phenylpropanal O-(tert-Butyldimethylsilyl) Oxime. The title compound
was obtained using the above general experimental procedure with the following quantities: 2-
chloro-3-phenylpropanal137 (1.0 g, 5.93 mmol), CH2Cl2 (10 mL), PPTS (76 mg, 0.30 mmol), 4Å
powdered molecular sieves (~70 mg), and O-TBS-hydroxylamine (1.0 g, 6.52 mmol). 1.24 g,
70%, ~2:1 mixture of oxime isomers. 1H NMR (300 MHz, CDCl3) δ 7.20-7.08 (m, 6H), 5.30
(m, 0.33H) 4.59 (q, J = 7.2 Hz, 0.67H), 3.12-3.02 (m, 2H), 0.77-0.75 (m, 9H), 0.05-0.00 (m, 6H).
(E)-1-Chlorocyclohexanecarboxaldehyde O-(tert-Butyldimethylsilyl) Oxime. The
title compound was obtained using the above general experimental procedure with the following
quantities: 1-chlorocyclohexanecarbaldehyde138 (120 mg, 0.82 mmol), CH2Cl2 (1.5 mL), PPTS
(10 mg, 0.04 mmol), 4Å powdered molecular sieves (~10 mg), and O-TBS-hydroxylamine (140
Page 145
135
mg, 0.90 mmol). 136 mg, 60%. 1H NMR (300 MHz, CDCl3) δ 7.36 (s, 1H), 1.85-1.40 (m, 10H),
0.77 (s, 9H), -0.01 (s, 6H); LRMS-ES+ m/z (relative intensity) 276 (MH+, 15).
1-Chloro-4-phenylbutan-2-one O-(tert-Butyldimethylsilyl) Oxime. The title
compound was obtained using the above general experimental procedure with the following
quantities: 1-chloro-4-phenylbutan-2-one139 (86 mL, 0.47 mmol), CH2Cl2 (1 mL), PPTS (6 mg,
0.02 mmol), 4Å powdered molecular sieves (~6 mg), and O-TBS-hydroxylamine (81 mg, 0.52
mmol). 81 mg, 55%, ~2:1 mixture of oxime isomers. 1H NMR (300 MHz, CDCl3) δ 7.17-7.02
(m, 5H), 4.12 (s, 1.3H), 3.83 (s, 0.7H), 2.77-2.54 (m, 4H), 0.81 (s, 3H), 0.77 (s, 6H), 0.04 (s,
2H), -0.01 (s, 4H); LRMS-ES+ m/z (relative intensity) 312 (MH+, 25).
General Procedure for Intermolecular Michael Additions of Carbon Nucleophiles to
Nitrosoalkenes. To a -78 °C solution of ester derivative 130 (0.46 mmol) in THF (1 mL) was
added KHMDS (917 μL, 0.5 M in PhMe, 0.46 mmol). The resulting solution was then stirred
for 45 min at that temperature. The O-TBS-oxime 131 dissolved in THF (0.38 mmol in 0.3 mL
of THF) was added slowly over 1 min, followed by the dropwise addition of TBAF (458 μL, 1.0
Page 146
136
M in THF, 0.46 mmol) over 3 min. The resulting solution was immediately transferred to a 0 °C
ice bath and stirred for an additional 2 h. The reaction mixture was diluted with conc. aqueous
NH4Cl and EtOAc. The organic layer was separated and the aqueous layer was extracted with
EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give
a residue, which was purified by flash column chromatography on silica gel eluting with a
mixture of ethyl acetate and hexanes. Isolated yields of conjugate addition products 132 are
shown in Scheme 132 and Tables 1-3.
(E) and (Z)-Methyl 2-(2-(Hydroxyimino)cyclohexyl)-2-phenylacetate (128). 1H NMR
(300 MHz, CDCl3, ~3:1 mixture of oxime isomers) δ 8.09 (br s, 1H), 7.40-7.26 (m, 5H), 4.09 (d,
J = 10.6 Hz, 0.25H), 3.78 (d, J = 11.3 Hz, 0.75H), 3.68 (s, 1H), 3.59 (s, 2H), 3.30 (dt, J = 12.4,
3.0 Hz, 0.75H), 3.16-3.09 (m, 0.25H), 3.00 (td, J = 11.1, 4.0 Hz, 0.75H), 2.62-2.57 (m, 0.25H),
1.99-1.39 (m, 7H); 13C NMR (75 MHz, CDCl3) δ 174.4, 173.8, 161.9, 160.1, 137.6, 137.3,
129.2, 129.1, 129.0, 128.9, 128.7, 128.0, 127.8, 52.5, 52.2, 45.8, 45.1, 31.5, 30.9, 26.7, 26.5,
25.4, 25.0, 23.4, 23.2; LRMS-ES+ m/z (relative intensity) 262 (MH+, 80).
Page 147
137
(E)-Ethyl 2-(2-(Hydroxyimino)cyclohexyl)-2-(ethylester)acetate (Table 1, entry 1).
1H NMR (300 MHz, CDCl3) δ 8.09 (br s, 1H), 4.22-4.11 (m, 4H), 3.69 (d, J = 10.2 Hz, 1H),
3.20-3.16 (m, 1H), 3.01 (td, J = 10.6, 4.0 Hz, 1H), 1.93-1.78 (m, 4H), 1.60-1.42 (m, 3H), 1.28-
1.20 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 169.1, 168.7, 160.4, 61.83, 61.82, 53.8, 42.6, 31.2,
26.3, 25.2, 24.7, 14.5, 14.1; LRMS-ES+ m/z (relative intensity) 272 (MH+, 100).
(E)-Ethyl 2-(2-(Hydroxyimino)cyclohexyl)-2-(ethylester)-2-methylacetate (Table 1,
entry 2). 1H NMR (300 MHz, CDCl3) δ 7.54 (br s, 1H), 4.13-4.00 (m, 4H), 3.30-3.25 (m, 1H),
3.09-3.01 (m, 1H), 1.85-1.72 (m 3H), 1.59-1.26 (m, 7H), 1.18-1.08 (m, 6H); 13C NMR (75
MHz, CDCl3) δ 171.9, 171.8, 158.9, 61.8. 61.7, 56.1, 47.5, 30.2, 26.4, 26.3, 25.1, 17.0, 14.4,
14.3; LRMS-ES+ m/z (relative intensity) 286 (MH+, 80).
(E)-Ethyl 2-(2-(Hydroxyimino)cyclohexyl)-2-(ethylester)-2-ethylacetate (Table 1,
entry 3). 1H NMR (300 MHz, CDCl3) δ 7.71 (br s, 1H), 4.12-3.99 (m, 4H), 3.20 (dt, J = 13.5,
Page 148
138
4.0 Hz, 1H), 2.74 (dd, J = 11.4, 3.9 Hz, 1H), 2.16-2.07 (m, 1H), 1.95-1.30 (m, 8H), 1.19-1.13 (m,
6H), 0.84 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.8, 171.7, 159.0, 61.3, 61.1,
59.5, 48.6, 31.4, 28.1, 26.70, 26.69, 25.4, 14.44, 14.35, 10.3; LRMS-ES+ m/z (relative intensity)
300 (MH+, 50).
(E) and (Z)-Ethyl 2-(2-(Hydroxyimino)cyclohexyl)-2-nitroacetate (Table 1, entry 4).
1H NMR (300 MHz, CDCl3, ~1:1 mixture of oxime isomers) δ 7.00 (br s, 1H), 5.38 (d, J = 9.9
Hz, 0.5H), 5.34 (d, J = 9.8, 0.5H), 4.30-4.15 (m, 2H), 3.31-3.13 (m, 2H), 1.86-1.71 (m, 4H),
1.48-1.17 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 164.7, 163.7, 159.0, 158.4, 90.0, 88.0, 63.4,
63.3, 43.3, 43.1, 29.6, 29.2, 25.94, 25.88, 25.4, 25.1, 24.7, 14.2; LRMS-ES+ m/z (relative
intensity) 245 (MH+, 75).
Ethyl 2-(2-(Hydroxyimino)cyclohexyl)-3-oxobutanoate (Table 1, entry 5). 1H NMR
(300 MHz, CDCl3, complex mixture of diastereomers and/or oxime isomers) δ 4.58-4.25 (m,
2H), 3.00-2.71 (m, 3H), 2.45-2.03 (m, 4H), 1.82-1.31 (m, 9H); LRMS-ES+ m/z (relative
intensity) 242 (MH+, 75).
Page 149
139
(E)-Methyl 2-(Phenylsulfonyl)-2-(2-(hydroxyimino)cyclohexyl)acetate (Table 1,
entry 6). 1H NMR (300 MHz, CDCl3, ~2:1 mixture of oxime isomers) δ 8.00 (br s, 1H), 7.95-
7.85 (m, 2H), 7.69-7.64 (m, 1H), 7.57-7.51 (m, 2H), 4.62 (d, J = 6.5 Hz, 0.33H), 4.29 (d, J =
11.0 Hz, 0.67H), 3.60 (s, 1H), 3.27 (s, 2H), 3.23-3.11 (m, 2H), 2.85-2.63 (m, 1H), 2.20-2.15 (m,
1H), 1.89-1.49 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 167.0, 165.9, 160.3, 159.2, 138.8, 138.4,
134.7, 134.6, 129.53, 129.51, 129.41, 129.38, 72.7, 70.1, 53.1, 53.0, 42.5, 41.1, 32.2, 30.5, 26.4,
26.1, 25.1, 25.0, 24.4, 23.8; LRMS-ES+ m/z (relative intensity) 326 (MH+, 50).
(E)-Ethyl 2-(2-(Hydroxyimino)cyclopentyl)-2-(ethylester)acetate (Table 1, entry 8).
1H NMR (300 MHz, CDCl3) δ 8.15 (br s, 1H), 4.18-4.01 (m, 4H), 3.53 (d, J = 8.0 Hz, 1H), 3.15-
3.09 (m, 1H), 2.55-2.50 (m, 1H), 2.34-2.28 (m, 1H), 1.95-1.79 (m, 2H), 1.64-1.55 (m, 2H), 1.20-
1.14 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 168.8, 168.5, 166.2, 62.0, 61.8, 53.8, 42.7, 29.5,
27.5, 22.7, 14.44, 14.37; LRMS-ES+ m/z (relative intensity) 258 (MH+, 90).
Page 150
140
(E)-Ethyl 2-(2-(Hydroxyimino)cyclopentyl)-2-(ethylester)-2-methylacetate (Table 1,
entry 9). 1H NMR (300 MHz, CDCl3) δ 7.95 (br s, 1H), 4.13-4.00 (m, 4H), 3.29-3.22 (m, 1H),
2.65-2.55 (m, 1H), 2.23-2.14 (m, 1H), 1.93-1.85 (m, 1H), 1.79-1.73 (m, 1H), 1.54-1.42 (m, 2H),
1.34 (s, 3H), 1.15-1.05 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 171.7, 171.5, 165.4, 61.9, 61.8,
56.2, 47.5, 28.9, 28.4, 22.6, 17.6, 14.4, 14.3; LRMS-ES+ m/z (relative intensity) 272 (MH+, 90).
(E)-Ethyl 2-(2-(Hydroxyimino)cyclopentyl)-2-(ethylester)-2-ethylacetate (Table 1,
entry 10). 1H NMR (300 MHz, CDCl3) δ 8.04 (br s, 1H), 4.13-3.99 (m, 4H), 3.10-3.05 (m, 1H),
2.62 (dd, J = 18.5, 8.1 Hz, 1H), 2.20-2.14 (m, 1H), 2.00-1.88 (m, 3H), 1.74-1.64 (m, 2H), 1.50-
1.38 (m, 1H), 1.17-1.12 (m, 6H), 0.86 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) � 171.23,
171.15, 61.5, 61.4, 60.7, 47.2, 29.1, 28.5, 28.3, 22.7, 14.4, 10.2; LRMS-ES+ m/z (relative
intensity) 286 (MH+, 70).
(E) and (Z)-Methyl 2-(2-(Hydroxyimino)cyclopentyl)-2-(phenylsulfonyl) acetate
(Table 1, entry 11). 1H NMR (300 MHz, CDCl3, ~1:1 mixture of oxime isomers) δ 7.88-7.45
Page 151
141
(m, 6H), 4.51 (d, J = 4.1 Hz, 0.5H), 3.97 (d, J = 10.3 Hz, 0.5H), 3.44 (s, 1.5H), 3.33 (s, 1.5H),
3.29-3.23 (m, 1H), 2.58-2.16 (m, 3H), 1.91-1.51 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 166.6,
166.5, 165.8, 165.2, 139.1, 138.2, 134.7, 134.6, 129.51, 129.46, 129.2, 73.1, 70.0, 53.2, 53.0,
41.9, 40.9, 30.7, 28.0, 27.2, 26.7, 23.2, 22.9.
(E) and (Z)-Methyl 2-(2-(Hydroxyimino)cyclopentyl)-2-phenylacetate (Table 1,
entry 12). 1H NMR (300 MHz, CDCl3, ~2:1 mixture of oxime isomers) δ 8.05 (br s, 1H), 7.24-
7.14 (m, 5H), 3.88 (d, J = 7.4 Hz, 0.33H), 3.57 (s, 2H), 3.56 (s, 1H), 3.47 (d, J = 10.4 Hz,
0.67H), 3.45-3.27 (m, 0.67 H), 3.05-2.95 (m, 0.33H), 2.56-2.47 (m, 1H), 2.40-2.30 (m, 1H),
1.81-1.60 (m, 2H), 1.50-1.44 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 174.2, 173.4, 167.2, 167.1,
137.84, 137.81, 129.1, 129.0, 128.7, 127.9, 127.7, 54.3, 52.8, 52.6, 52.4. 46.8, 46.3, 30.6, 29.6,
27.9, 27.6, 22.6, 22.4; LRMS-ES+ m/z (relative intensity) 248 (MH+, 50).
(E)-Diethyl 2-(1-(Hydroxyimino)-3-phenylpropan-2-yl)malonate (Table 2, entry 1).
1H NMR (300 MHz, CDCl3) δ 7.91 (br s, 1H), 7.54-7.51 (m, 1H), 7.34-7.18 (m, 5H), 4.26-4.19
(m, 4H), 3.59 (d, J = 7.1 Hz, 1H), 3.40-3.35 (m, 1H), 2.93-2.89 (m, 2H), 1.32-1.26 (m, 6H); 13C
Page 152
142
NMR (90 MHz, CDCl3) δ 168.1, 167.8, 151.4, 137.9, 129.3, 129.2, 128.62, 128.56, 126.74,
126.70, 61.8, 61.7, 53.7, 41.2, 36.7, 14.1.
(E)-Diethyl 2-(1-(Hydroxyimino)-3-phenylpropan-2-yl)-2-methylmalonate (Table 2,
entry 2). 1H NMR (300 MHz, CDCl3) δ 7.76 (br s, 1H), 7.24-7.04 (m, 6H), 4.15-3.99 (m, 4H),
3.14 (ddd, J = 10.9, 7.9, 3.0 Hz, 1H), 2.85-2.62 (m, 2H), 1.40 (s, 3H), 1.19-1.01 (m, 6H); 13C
NMR (75 MHz, CDCl3) δ 171.3, 171.2, 150.9, 139.5, 129.6, 129.0, 128.8, 126.8, 126.7, 62.14,
62.10, 57.2, 47.1, 35.4, 18.6, 14.5, 14.4; LRMS-ES+ m/z (relative intensity) 322 (MH+, 60).
(E)-Diethyl 2-(1-(Hydroxyimino)-3-phenylpropan-2-yl)-2-ethylmalonate (Table 2,
entry 3). 1H NMR (300 MHz, CDCl3) δ 7.67 (br s, 1H), 7.29-7.09 (m, 6H), 4.25-4.16 (m, 4H),
3.14-3.03 (m, 2H), 2.55-2.46 (m, 1H), 2.02-1.84 (m, 2H), 1.32-1.17 (m, 6H), 0.83 (t, J = 7.4 Hz,
3H); 13C NMR (75 MHz, CDCl3) δ 170.9, 170.8, 151.0, 139.7, 129.7, 129.0, 128.8, 126.7, 61.8,
61.1, 45.6, 36.1, 27.9, 14.6, 9.0; LRMS-ES+ m/z (relative intensity) 336 (MH+, 40).
Page 153
143
(E) and (Z)-Methyl 3-Benzyl-4-(hydroxyimino)-2-phenylbutanoate (Table 2, entry
4). 1H NMR (300 MHz, CDCl3, ~2:1 mixture of oxime isomers) δ 7.91 (br s, 0.66H), 7.65 (br s
0.33H), 7.25-6.84 (m, 11H), 3.60-3.55 (m, 1H), 3.49 (s, 1H), 3.43 (s, 2H), 3.25-3.16 (m, 1H),
2.71-2.49 (m, 2H), 2.36-2.24 (m, 1H); LRMS-ES+ m/z (relative intensity) 298 (MH+, 75).
(E)-Diethyl 2-Allyl-2-(1-(hydroxyimino)-3-phenylpropan-2-yl)malonate (Table 2,
entry 5). 1H NMR (360 MHz, CDCl3) δ 7.81 (s, 1H), 7.38 (d, J = 8.4, 1H), 7.30-7.17 (m, 5H),
5.86-5.77 (m, 1H), 5.17-5.12 (m, 2H), 4.31-4.24 (m, 4H), 3.23-3.12 (m, 2H), 2.81-2.63 (m, 3H),
1.36-1.22 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 170.4, 170.3, 151.0, 139.6, 132.7, 129.7,
128.8, 126.8, 119.6, 62.0, 60.8, 46.0, 39.0, 35.9, 14.6.
(E)-Diethyl 2-(1-((Hydroxyimino)methyl)cyclohexyl)malonate (Table 2, entry 6). 1H
NMR (300 MHz, CDCl3) δ 8.01 (br s, 1H), 7.63 (s, 1H), 4.15-4.07 (m, 4H), 3.51 (s, 1H), 1.99-
Page 154
144
1.82 (m, 2H), 1.58-1.40 (m, 8H), 1.19 (t, J = 7.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 167.7,
155.8, 61.7, 60.6, 42.1, 33.2, 26.0, 22.3, 14.5; LRMS-ES+ m/z (relative intensity) 286 (MH+,
60).
(E)-Diethyl 2-(1-((Hydroxyimino)methyl)cyclohexyl)-2-methylmalonate (Table 2,
entry 7). 1H NMR (300 MHz, CDCl3) δ 7.92 (s, 1H), 7.43 (s, 1H), 4.07 (q, J = 7.1 Hz, 4H), 1.99
(d, J = 12.4 Hz, 2H), 1.53-1.44 (m, 6H), 1.34-1.23 (m, 5H), 1.13 (t, J = 7.1 Hz, 6H); 13C NMR
(75 MHz, CDCl3) δ 171.3, 158.5, 61.8, 61.6, 45.4, 30.1, 26.1, 22.9, 18.7, 14.4; LRMS-ES+ m/z
(relative intensity) 300 (MH+, 60).
(E) and (Z)-Diethyl 2-(1-((Hydroxyimino)methyl)cyclohexyl)-2-ethylmalonate (Table
2, entry 8). 1H NMR (300 MHz, CDCl3, ~3:1 mixture of oxime isomers) δ 7.78 (s, 0.75H), 7.72
(s, 0.25H), 7.37 (s, 0.75H), 7.17 (s, 0.25H), 4.18-4.06 (m, 4H), 1.96-1.80 (m, 4H), 1.57-1.38 (m,
8H), 1.21-1.17 (m, 6H), 0.82-0.76 (m, 3H); LRMS-ES+ m/z (relative intensity) 314 (MH+, 60).
Page 155
145
(E)-Methyl 2-(1-((Hydroxyimino)methyl)cyclohexyl)-2-phenylacetate (Table 2, entry
9). 1H NMR (300 MHz, CDCl3) δ 7.99 (s, 1H), 7.42 (s, 1H), 7.24-7.13 (m, 5H), 3.59 (s, 1H),
3.55 (s, 3H), 1.80-1.20 (m, 10H); 13C NMR (75 MHz, CDCl3) δ 170.9, 153.5, 132.7, 128.6,
126.4, 126.1, 59.7, 50.3, 41.5, 32.2, 30.7, 24.2, 20.8, 20.3; LRMS-ES+ m/z (relative intensity)
276 (MH+, 40).
(E)-5,5-Bis(ethyl ester)-1-phenylhexan-3-one Oxime (Table 3, entry 1). 1H NMR
(300 MHz, CDCl3) δ 8.81 (br s, 1H), 7.19-7.05 (m, 5H), 4.17-3.94 (m, 4H), 2.73-2.62 (m, 4H),
2.46 (dd, J = 8.8, 5.6 Hz, 2H), 1.35 (s, 3H), 1.16-1.08 (m, 6H); 13C NMR (75 MHz, CDCl3) δ
172.3, 157.1, 141.6, 128.8, 128.7, 126.5, 62.1, 61.9, 52.7, 39.6, 31.8, 31.2, 25.1, 20.5, 14.4;
LRMS-ES+ m/z (relative intensity) 336 (MH+, 75).
(E)-5,5-Bis(ethyl ester)-1-phenylheptan-3-one Oxime (Table 3, entry 2). 1H NMR
(300 MHz, CDCl3) δ 8.01 (br s, 1H), 7.21-7.07 (m, 5H), 4.10-4.01 (m, 4H), 2.78-2.70 (m, 4H),
2.47 (dd, J = 8.7, 5.5 Hz, 2H), 1.95 (q, J = 7.6 Hz, 2H), 1.16-1.10 (m, 6H), 0.70 (t, J = 7.6 Hz,
Page 156
146
3H); 13C NMR (75 MHz, CDCl3) δ 171.6, 157.3, 141.6, 128.9, 128.7, 126.5, 61.8, 57.0, 36.1,
31.9, 31.4, 31.1, 25.8, 14.6, 14.4, 9.1; LRMS-ES+ m/z (relative intensity) 350 (MH+, 80).
(E)-Methyl 4-(Hydroxyimino)-2,6-diphenylhexanoate (Table 3, entry 3). 1H NMR
(300 MHz, CDCl3) δ 8.20 (br s, 1H), 7.22-7.09 (m, 10H), 3.88 (dd, J = 10.2, 4.8 Hz, 1H), 3.55
(s, 3H), 2.95 (dd, J = 16.6, 10.2, 1H), 2.72 (dd, J= 10.6, 6.8, 2H), 2.52 (dd, J = 9.2, 5.9, 2H), 2.36
(dd, J = 16.6, 4.8 Hz, 1H);��13C NMR (75 MHz, CDCl3) δ 174.2, 159.1, 141.6, 139.0, 129.2,
128.9, 128.7, 128.2, 128.0, 126.6, 52.7, 48.3, 38.6, 31.8, 31.1; LRMS-ES+ m/z (relative
intensity) 312 (MH+, 50).
4-(Phenylsulfonyl)-5,6,7,8-tetrahydro-4aH-benzo[c][1,2]oxazin-3-amine (135). 1H
NMR (300 MHz, CDCl3) δ 7.90-7.83 (m, 2H), 7.55-7.43 (m, 3H), 4.90 (br s, 2H), 2.41-2.06 (m,
4H), 1.67-1.59 (m, 5H); LRMS-ES+ m/z (relative intensity) 293 (MH+, 60).
Page 157
147
Dimethyl 2-(3-(2-(tert-Butoxy)-2-oxoethyl)-1H-indol-2-yl)malonate (210). To a
stirred solution of indole 20870 (100 mg, 0.43 mmol), TEA (72 μL, 0.51 mmol), and THF (15
mL) at -78 °C was added tert-butyl hypochlorite (60 μL, 0.51 mmol). The resulting solution was
stirred for 30 min at -78 °C and then dimethyl malonate thallium salt72 was added (209 mg, 0.62
mmol). The resulting solution was stirred for 7 h at rt and then filtered through a pad of Celite.
The filtrate was concentrated in vacuo to give a residue which was purified by flash
chromatography on silica gel (15% EtOAc/hexanes) to afford indole-2-malonate 210 (99 mg,
63%). 1H NMR (300 MHz, CDCl3) δ 9.17 (s, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.0 Hz,
1H), 7.25-7.15 (m, 2H), 5.22 (s, 1H), 3.80 (s, 6H), 3.72 (s, 2H), 1.44 (s, 9H); 13C NMR (75 MHz,
CDCl3) δ 170.9, 168.0, 136.2, 127.9, 126.3, 123.1, 120.2, 119.4, 111.7, 108.8, 81.3, 53.6, 49.5,
32.0, 28.4; LRMS-ES+ m/z (relative intensity) 379 (M+NH4+, 30).
Dimethyl 2-(3-(2-(tert-Butoxy)-2-oxoethyl)-1H-indol-2-yl)-2-methylmalonate (213).
To a stirred solution of indole 210 (24.5 mg, 0.07 mmol), potassium carbonate (14 mg, 0.10
mmol), and acetone (1 mL) was added dimethyl sulfate (9 μL, 0.09 mmol). The resulting
mixture was stirred at rt for 20 h and then diluted with water and EtOAc. The organic layer was
dried over MgSO4 and concentrated in vacuo to give a residue, which was purified by
Page 158
148
preparative thin layer chromatography (25% EtOAc/hexanes, eluted three times for complete
separation) to give methyl malonate 213 (14 mg, 55%) and recovered starting indole 210 (11 mg,
45%). 1H NMR (300 MHz, CDCl3) δ 9.77 (s, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.39 (d, J = 8.0 Hz,
1H), 7.21 (t, J = 7.0, 1H), 7.13 (t, J = 7.0 Hz, 1H), 3.83 (s, 6H), 3.64 (s, 2H), 1.99 (s, 3H), 1.45
(s, 9H); 13C NMR (75 MHz, CDCl3) δ 171.7, 171.0, 134.8, 131.5, 129.0, 122.7, 120.1, 119.3,
111.6, 106.7, 81.1, 54.0, 53.7, 32.0, 28.5, 24.5; LRMS-ES+ m/z (relative intensity) 398 (M+Na+,
75).
Dimethyl 2-(3-(2-(Hydroxyimino)cyclohexyl)-1H-indol-2-yl)malonate (215). Method
A. To a -78 °C solution of indole malonate 21473 (100.0 mg, 0.404 mmol) in THF (1 mL) was
added LiHMDS (1.0 M in THF, 404 μL, 0.404 mmol) and the resulting solution was stirred for
30 min. A solution of oxime 211 (30 mg, 0.202 mmol) in THF (0.5 mL) was added dropwise
over 3 min and the reaction mixture was stirred for 1.5 h at -78 °C and then 1 h at 0 °C. The
reaction mixture was diluted with NH4Cl(aq), the organic layer was separated, and the aqueous
layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and
concentrated in vacuo to give a residue which was purified by preparative thin layer
chromatography (30% EtOAc/hexanes) to afford Michael adduct 215 (less polar oxime isomer -
29.4 mg, 40%; more polar oxime isomer - 33.2 mg, 46%). Less polar oxime isomer: 1H NMR
(300 MHz, CDCl3) δ 11.34 (s, 1H), 7.71 (s, 1H), 7.57-7.46 (m, 1H), 7.23-6.93 (m, 3H), 5.59 (s,
Page 159
149
1H), 3.81-3.72 (m, 6H), 3.47-3.42 (m, 1H), 2.61 (dd, J = 12.7, 3.0 Hz, 1H), 2.05-1.95 (m, 1H),
1.80-1.19 (m, 6H). More polar oxime isomer: 1H NMR (300 MHz, CDCl3) δ 9.03 (s, 1H), 7.54
(d, J = 7.9 Hz, 1H), 7.37 (d, J = 7.4 Hz, 1H), 7.18 (d, J = 7.1 Hz, 1H), 7.07 (d, J = 7.0 Hz, 1H),
5.02 (s, 1H), 3.77 (s, 3H), 3.74 (s, 3H), 3.66 (dd, J = 12.5, 7.7 Hz, 1H), 3.48 (d, J = 14.5 Hz, 1H),
2.12-1.58 (m, 8H). Mixture of oxime isomers: LRMS-ES+ m/z (relative intensity) 359 (MH+,
100).
Method B. To a stirred solution of indole malonate 214 (50 mg, 0.20 mmol) in THF (1
mL) at -78 °C was added n-BuLi (2.0 M in hexanes, 100 μL, 0.20 mmol). The resulting solution
was stirred for 45 min and a solution of oxime 127 (46 mg, 0.18 mmol, in 0.5 mL THF) was
added followed by dropwise addition of TBAF (1.0 M in THF, 210 μL, 0.21 mmol). After
stirring for 25 min at -78 °C, the reaction mixture was transferred to a 0 °C ice bath and stirred
for an additional 2 h. The reaction mixture was quenched with NH4Cl(aq), the organic layer was
separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were
dried over MgSO4 and concentrated in vacuo to afford a residue which was purified by flash
chromatography on silica gel (25% EtOAc/hexanes) to give Michael adduct 215 (19 mg, 26%) as
a mixture of oxime isomers.
Dimethyl 2-(1-(tert-Butoxycarbonyl)-1H-indol-2-yl)malonate (216) and 1-tert-Butyl
1,1-Dimethyl (1-(tert-Butoxycarbonyl)-1H-indol-2-yl)methanetricarboxylate (217). To a
stirred solution of indole malonate 21473 (100 mg, 0.40 mmol), Boc2O (135 mg, 0.61 mmol), and
Page 160
150
CH2Cl2 (1.5 mL) at 0 °C was added TEA (96 μL, 0.69 mmol) followed by DMAP (5 mg, 0.04
mmol). The resulting solution was stirred at rt for 90 min and then diluted with NaHCO3(aq).
The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The
combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue
which was purified by flash chromatography on silica gel (15% EtOAc/hexanes) to yield N-Boc
indole 216 (18 mg, 13%) and bis-Boc indole 217 (133 mg, 74%). N-Boc indole 216: 1H NMR
(300 MHz, CDCl3, ~5:1 mixture of Boc-rotamers) δ 8.09-8.06 (m, 1H), 7.56-7.53 (m, 1H), 7.36-
7.25 (m, 2H), 6.57 (t, J = 0.9 Hz, 1H), 5.53 (d, J = 0.9 Hz, 1H), 3.85 (s, 6H), 1.70 (s, 9H). Bis-
Boc indole 217: 1H NMR (300 MHz, CDCl3) δ 8.19-8.16 (m, 1H), 8.08-8.05 (m, 1H), 7.40-7.26
(m, 2H), 6.82 (s, 1H), 3.77 (s, 6H), 1.68 (s, 9H), 1.64 (s, 9H).
Methyl 2-(2-(Hydroxyimino)cyclohexyl)-2-(1H-indol-2-yl)acetate (219). Method A.
To a stirred solution of indole ester 218140 (40 mg, 0.21 mmol) in THF (0.75 mL) at -78 °C was
added n-BuLi (2.15 M in hexanes, 90 μL, 0.20 mmol). The resulting solution was stirred for 45
min and a solution of α-chlorooxime 211 (16.4 mg, 0.11 mmol, in 0.25 mL THF) was added
dropwise over 20 min. After stirring for an additional 45 min at -78 °C, the reaction mixture was
quenched with NH4Cl(aq), the organic layer was separated, and the aqueous layer was extracted
with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to
produce a residue which was purified by flash chromatography on silica gel (25%
Page 161
151
EtOAc/hexanes) to give Michael adduct 219 (26 mg, 78% based on starting oxime 211) as ~9:1
mixture of oxime isomers and recovered starting indole (23 mg, 58% based on starting indole
218). 1H NMR (300 MHz, CDCl3) Major oxime isomer: δ 8.56 (s, 1H), 7.57 (d, J = 7.8 Hz,
1H), 7.35 (d, J = 8.0 Hz, 1H), 7.17 (t, J = 8.1 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 6.42 (s, 1H) 4.10
(d, J = 10.7 Hz, 1H), 3.70 (s, 3H), 3.15-3.10 (m, 1H), 3.01 (td, J = 10.4, 4.3 Hz, 1H), 2.10-2.04
(m, 1H), 1.80-1.41 (m, 5H), 1.28-1.22 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 173.9, 161.6,
136.7, 133.6, 128.2, 122.5, 120.7, 120.2, 111.3, 103.3, 52.9, 47.1, 45.2, 31.7, 26.4, 24.8, 24.4;
LRMS-ES+ m/z (relative intensity) 301 (MH+, 50).
Method B. To a stirred solution of indole ester 218140 (50 mg, 0.26 mmol) in THF (0.75
mL) at -78 °C was added n-BuLi (2.15 M in hexanes, 123 μL, 0.26 mmol). The resulting
solution was stirred for 45 min and a solution of α-chlorooxime 127 (69 mg, 0.26 mmol, in 0.25
mL THF) was added followed by dropwise addition of TBAF (1.0 M in THF, 260 μL, 0.26
mmol). The reaction mixture was stirred for 1 h at -78 °C and then quenched with NH4Cl(aq).
The organic layer was separated, the aqueous layer was extracted with EtOAc, and the combined
organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was
purified by flash chromatography on silica gel (25% EtOAc/hexanes) to yield Michael adduct
219 (44 mg, 55%) as a mixture of E and Z-oxime isomers and recovered starting indole (21 mg,
42%).
Page 162
152
Methyl 2-(1H-Indol-2-yl)-2-(2-oxocyclohexyl)acetate (222) and Methyl 2,3,4,10-
Tetrahydro-1H-indolo[1,2-a]indole-11-carboxylate (223). To a stirred solution of oxime 219
(40.0 mg, 0.13 mmol), ammonium acetate (126 mg, 1.63 mmol), water (4 mL), and THF (4 mL)
was added TiCl3 solution (Aldrich, 10 wt% TiCl3, 20-30 wt% HCl, 422 μL, 0.33 mmol). The
resulting solution was stirred for 18 h and then extracted with ether. The combined organic
layers were washed with NaHCO3(aq), dried over MgSO4, and then concentrated in vacuo to give
a residue which was purified by flash chromatography on silica gel (15-25% EtOAc/hexanes) to
afford ketone 222 (24 mg, 63%) and tetracyclic pyrrole 223 (4.5 mg, 13%). Ketone 222: 1H
NMR (300 MHz, CDCl3) δ 8.43 (s, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.37-7.34 (m, 2H), 7.22-7.10
(m, 2H), 6.41-6.40 (m, 1H), 4.02 (d, J = 9.6 Hz, 1H), 3.75 (s, 3H), 3.23-3.15 (m, 1H), 3.00-2.95
(m, 1H), 2.86 (tt, J = 8.1, 1.6 Hz, 1H), 2.50-2.41 (m, 2H), 1.96-1.60 (m, 3H); 13C NMR (75
MHz, THF-D8) δ 209.5, 172.6, 137.2, 134.2, 129.0, 121.2, 119.9, 119.4, 111.0, 101.3, 53.1, 51.5,
46.0, 42.1, 32.0, 28.5, 25.6; LRMS-ES+ m/z (relative intensity) 286 (MH+, 40). Pyrrole 223: 1H
NMR (300 MHz, CDCl3) δ 7.49-7.46 (m, 1H), 7.40-7.30 (m, 2H), 7.15 (td, J = 7.4, 1.3 Hz, 1H),
4.04 (s, 2H), 3.87 (s, 3H), 2.97 (t, J = 6.2 Hz, 2H), 2.85 (tt, J = 6.12, 1.7 Hz, 2H), 1.96-1.90 (m,
2H), 1.87-1.81 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.4, 141.7, 141.2, 135.2, 127.9, 126.4,
123.84, 123.76, 123.5, 111.5, 106.9, 51.1, 31.4, 23.8, 23.5, 23.3, 23.1; LRMS-ES+ m/z (relative
intensity) 268 (MH+, 100).
Page 163
153
tert-Butyl 2-(2-Methoxy-2-oxoethyl)-1H-indole-1-carboxylate (227). To a stirred
solution of indole ester 218140 (100 mg, 0.53 mmol), Boc2O (177 mg, 0.79 mmol), and CH2Cl2 (2
mL) was added TEA (125 μL, 0.90 mmol) followed by DMAP (6.5 mg, 0.05 mmol). The
resulting solution was stirred for 70 min at rt and then diluted with NaHCO3(aq). The organic
layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic
layers were dried over MgSO4 and concentrated in vacuo to yield a residue which was purified
by flash chromatography on silica gel (10% EtOAc/hexanes) to give N-Boc indole 227 (126 mg,
82%). 1H NMR (300 MHz, CDCl3) δ 8.15-8.12 (m, 1H), 7.54-7.51 (m, 1H), 7.35-7.21 (m, 2H),
6.51 (s, 1H), 4.08 (s, 2H), 3.75 (s, 3H), 1.70 (s, 9H).
tert-Butyl 2-(1-(2-(Hydroxyimino)cyclohexyl)-2-methoxy-2-oxoethyl)-1H-indole-1-
carboxylate (228). To a stirred solution of indole ester 227 (63.0 mg, 0.22 mmol) in THF (1
mL) at -78 °C was added LHMDS (1.0 M in THF, 220 μL, 0.22 mmol). The resulting solution
was stirred for 35 min and a solution of α-chlorooxime 127 (48 mg, 0.18 mmol, in 0.5 mL THF)
was added followed by dropwise addition of TBAF (1.0 M in THF, 220 μL, 0.22 mmol). The
reaction mixture was transferred to a 0 °C ice bath and stirred for an additional 1 h and 45 min
and then quenched with NH4Cl(aq). The organic layer was separated and the aqueous layer was
Page 164
154
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to yield a residue, which was purified by flash chromatography (15-25% EtOAc/hexanes)
to give Michael adduct 228 (57.7 mg, 79%) as a ~2:1 mixture of oxime isomers. 1H NMR (300
MHz, CDCl3) δ 8.12-8.06 (m, 1H), 7.54-7.49 (m, 1H), 7.33-7.19 (m, 2H), 6.68 (s, 0.66H), 6.63
(s, 0.33H), 5.29 (d, J = 10.9 Hz, 0.66 H), 5.24 (d, J = 8.2 Hz, 0.33H), 3.71 (s, 1H), 3.66 (s, 2H),
3.25-3.19 (m, 0.66H), 3.11-3.04 (m, 1H), 2.95-2.90 (m, 0.33H), 2.19-1.99 (m, 1H), 1.90-1.63 (m,
12H), 1.57-1.38 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 173.5, 173.0, 161.6, 160.5, 150.9,
150.8, 137.9, 137.1, 136.9, 129.3, 129.3, 124.4, 124.3, 123.3, 123.1, 120.84, 120.77, 116.2,
116.0, 110.3, 109.3, 85.1, 84.8, 60.9, 52.7, 52.5, 46.8, 44.8, 44.5, 31.0, 30.6, 28.7, 28.6, 26.6,
26.4, 25.4, 24.7, 24.2; LRMS-ES+ m/z (relative intensity) 401 (MH+, 100).
tert-Butyl 2-(2-Methoxy-2-oxo-1-(2-oxocyclohexyl)ethyl)-1H-indole-1-carboxylate
(229). To a stirred solution of oxime 228 (25.0 mg, 0.062 mmol), ammonium acetate (59 mg,
0.77 mmol), water (2.5 mL), and THF (2.5 mL) was added TiCl3 solution (Aldrich, 10 wt%
TiCl3, 20-30 wt% HCl, 200 μL, 0.15 mmol). The resulting solution was stirred for 17 h and then
extracted with ether. The organic layers were washed with NaHCO3(aq), dried over MgSO4, and
concentrated in vacuo to give a residue, which was purified by flash chromatography on silica
gel (10-15% EtOAc/hexanes) to afford ketone 229 (11.5 mg, 48%) as a ~4:1 mixture of
diastereomers or Boc-rotamers. 1H NMR (300 MHz, CDCl3) Only the major peaks are reported:
δ 8.06 (d, J = 8.3 Hz, 1H), 7.52-7.50 (m, 1H), 7.31-7.22 (m, 2H), 6.56 (s, 1H), 5.13 (d, J = 9.6
Page 165
155
Hz, 1H), 3.67 (s, 3H), 3.42-3.32 (m, 1H), 2.55-2.50 (m, 2H), 2.16-2.12 (m, 1H), 1.80-1.65 (m,
14H); LRMS-ES+ m/z (relative intensity) 386 (MH+, 50).
4-Chloro-1-tosylpiperidin-3-one (248a). Method A. To a stirred solution of readily
prepared 1-tosylpiperidin-3-one (246)80 (2.00 g, 7.90 mmol) in CH2Cl2 (100 mL) at 0 °C was
added sulfuryl chloride (2.0 mL, 23.7 mmol) and the solution was stirred for 17 h gradually
warming to rt. An aqueous solution of NaHCO3 was added to the reaction and the mixture was
stirred vigorously for 30 min. The organic layer was separated and the aqueous layer was
extracted with CH2Cl2. The combined organic layers were washed with water and brine, dried
over MgSO4, and concentrated in vacuo to give a solid which was purified by flash
chromatography on silica gel (1:1:1 hexanes/CH2Cl2/EtOAc) to yield α-chloroketone 248a (1.77
g, 78%). 1H NMR (300 MHz, CDCl3) δ 7.69 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 4.35
(t, J = 5.0 Hz, 1H), 3.84 (s, 2H), 3.53-3.50 (m, 1H), 3.42-3.38 (m, 1H), 2.53-2.48 (m, 4H), 2.27-
2.22 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 195.1, 145.0, 132.9, 130.5, 128.1, 58.6, 53.5, 42.3,
34.0, 22.0; LRMS-ES+ m/z (relative intensity) 288 (MH+, 100); HRMS-ES+ (C12H18ClN2O3S)
calcd 305.0729 (M+NH4+), found 305.0727.
Method B. To a stirred solution of piperidone 246 (4.00 g, 31.6 mmol) in EtOAc (250
mL) was added Amberlyst-15 (19.5 g) followed by NCS (3.88 g, 56.9 mmol) and the resulting
mixture was stirred at rt for 20 h. Silica gel (~3 g) was added and the resulting mixture was
stirred for an additional 2 h, filtered, and concentrated in vacuo to give a residue, which was
Page 166
156
purified by flash column chromatography on silica gel (2:2:1-1:1:1 hexanes/CH2Cl2/EtOAc) to
afford the α-chloroketone 248a (2.71 g, 59%).
4-Chloro-1-nosylpiperidin-3-one (Scheme 56, entry b). To a stirred solution of 1-
nosylpiperidin-3-one (260)78 (609 mg, 2.14 mmol) in CH2Cl2 (10 mL) at 0 °C was added sulfuryl
chloride (650 μL, 7.50 mmol) and the solution was stirred for 18 h gradually warming to rt. An
aqueous solution of NaHCO3 was added and the mixture was stirred vigorously for 5 min. The
organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined
organic layers were washed with water and brine, dried over MgSO4, and concentrated in vacuo
to give a solid which was purified by flash chromatography on silica gel (1:1:1
hexanes/CH2Cl2/EtOAc) to afford the title compound (437 mg, 64%) as a mixture of keto and
enol tautomers. 1H NMR (300 MHz, CDCl3, + 1 drop of trifluoroacetic acid to generate the
ketone exclusively) δ 8.14-8.04 (m, 1H), 7.86-7.71 (m, 3H), 4.57-4.51 (m, 1H), 4.33-4.02 (m,
2H), 3.85-3.74 (m, 2H), 2.65-2.58 (m, 1H), 2.38-2.31 (m, 1H); LRMS-ES+ m/z (relative
intensity) 319 (MH+, 50).
Page 167
157
4-Chloro-1-tosylpiperidin-3-one O-(tert-Butyldimethylsilyl) Oxime (263). To a stirred
solution of α-chloroketone 248a (110 mg, 0.38 mmol) in CH2Cl2 (3 mL) was added PPTS (5 mg,
0.02 mmol), 4Å powdered molecular sieves (25 mg), and then O-TBS-hydroxylamine (59 mg,
0.38 mmol). The resulting mixture was stirred for 14 h at rt and then filtered through a Celite
plug washing with CH2Cl2. The filtrate was concentrated in vacuo to give a residue which was
purified by flash column chromatography on silica gel (5% EtOAc/hexanes) to yield the α-
chloro-O-TBS-oxime 263 (67 mg, 42%) as a ~8:1 mixture of oxime isomers. 1H NMR (300
MHz, CDCl3) Only the major oxime isomer peaks are reported: δ 7.71 (d, J = 8.1 Hz, 2H), 7.35
(d, J = 7.8 Hz, 2H), 5.22 (d, J = 14.4 Hz, 1H), 4.79 (s, 1H), 3.82 (d, J = 12.6 Hz, 1H), 3.18-3.06
(m, 2H), 2.46 (s, 3H), 2.20-2.01 (m, 2H), 0.95 (s, 9H), 0.20 (s, 6H); LRMS-ES+ m/z (relative
intensity) 418 (MH+, 90).
4-Chloro-1-tosylpiperidin-3-one Oxime (290). To a stirred solution of N-Ns-4-
chloropiperdone oxime (129 mg, 0.41 mmol) in DMSO (1 mL) was added H2NOH.HCl (34 mg,
0.49 mmol) and the resulting solution was stirred for 18 h at rt. The reaction mixture was diluted
with water (5 mL) and extracted repeatedly with CHCl3. The combined organic layers were
washed with water and brine and dried over MgSO4. Removal of the solvent in vacuo provided
Page 168
158
α-chlorooxime 290 (119 mg, 88%) as a ~2:1 mixture of oxime isomers. This material was used
in subsequent reactions without further purification. 1H NMR (300 MHz, CDCl3) δ 8.29 (s,
0.33H), 8.21 (s, 0.67H), 8.11-8.05 (m, 1H), 7.80-7.67 (m, 3H), 5.61 (s, 0.33H), 5.19 (d, J = 15.3
Hz, 0.67H), 4.85 (t, J = 3.1 Hz, 0.67H), 4.30 (d, J = 15.0 Hz, 0.33H), 3.99-3.90 (m, 1.33 H), 3.70
(d, J = 15.4 Hz, 0.67H), 3.59-3.54 (m, 1H), 2.34-2.17 (m, 2H).
4-Chloro-1-tosylpiperidin-3-one Oxime (264). To a stirred solution of α-chloroketone
248a (5.40 g, 18.8 mmol) in DMSO (50 mL) was added H2NOH.HCl (1.46 g, 20.6 mmol) and
the resulting solution was stirred at rt for 18 h. The reaction mixture was diluted with water (300
mL) and extracted repeatedly with CH2Cl2. The combined organic layers were then washed with
water, brine, and then dried over MgSO4. Removal of the solvent in vacuo provided pure α-
chlorooxime 264 (5.49 g, 97%) as a white solid which was used in the following reaction without
further purification. 1H NMR (300 MHz, CDCl3, ~2:1 E/Z mixture of oxime isomers) δ 7.81-
7.70 (m, 2H), 7.40-7.38 (d, J = 8.2 Hz, 2H), 5.53 (m, 0.33H), 5.15 (d, J = 14.2 Hz, 0.67H), 4.77
(t, J = 3.0 Hz, 0.67H), 4.30-4.27 (m, 0.33 H), 3.85-3.80 (m, 1H), 3.45 (d, J = 13.6 Hz, 0.33H),
3.23 (d, J = 14.5 Hz, 0.67H), 3.11 (td, J = 11.9 Hz, 3.0 Hz, 0.67H), 2.95 (m, 0.33H), 2.48 (m,
3H), 2.28-2.08 (m, 2H).
Page 169
159
Methyl 2-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-(1H-indol-2-yl)acetate (266).
To a stirred solution of indole ester 218140(691 mg, 3.65 mmol) in THF (11 mL) at -78 °C was
added LiHMDS (1.0 M in THF, 3.65 mL, 3.65 mmol). The resulting solution was stirred for 30
min and a solution of α-chlorooxime 264 (553 mg, 1.83 mmol, in 7 mL of THF) was added
dropwise over 8 min. After stirring the mixture for an additional 90 min at -78 °C the reaction
was quenched with NH4Cl(aq). The organic layer was separated, and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to give a residue which was purified by flash chromatography on silica gel (15-50%
EtOAc/hexanes) to afford Michael adduct 266 (491 mg, 59%) as a ~3:1 mixture of oxime
isomers and recovered starting indole (454 mg, 66%). 1H NMR (400 MHz, CDCl3, ~3:1 mixture
of oxime isomers) δ 8.83 (s, 1H), 8.60 (br s, 1H), 7.69-7.09 (m, 8H), 6.36 (s, 0.75 H), 6.29 (s,
0.25 H), 4.89 (d, J = 14.8 Hz, 0.75 H), 4.56 (d, J = 15.0 Hz, 0.25H), 3.98 (d, J = 10.3 Hz, 1 H),
3.69 (s, 0.75 H), 3.67 (s, 2.25 H), 3.60-3.46 (m, 1H) , 3.20 (d, J = 14.4 Hz, 1H), 3.02 (td, J =
10.7, 4.7 Hz, 1H), 2.76-2.72 (m, 1H), 2.45-2.41 (m, 4H), 1.63-1.59 (m, 1H); 13C NMR (75 MHz,
CDCl3) δ 173.4, 153.4, 144.5, 136.7, 133.2, 132.5, 130.4, 130.3, 128.4, 128.1, 122.6, 120.7,
120.5, 111.5, 46.6, 46.5, 45.3, 42.3, 42.1, 28.1, 22.0; LRMS-ES+ m/z (relative intensity) 456
(MH+, 95); HRMS-ES+ (C23H26N3O5S) calcd 456.1593 (MH+), found 456.1571.
Page 170
160
tert-Butyl 2-(1-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-1H-
indole-1-carboxylate (267). To a -78 °C solution of N-Boc indole 227 (32.8 mg, 0.113 mmol)
in THF (1 mL) was added LiHMDS (113 μL, 0.11 mmol, 1.0 M in THF) and the resulting
solution was stirred for 30 min. A solution of α-chlorooxime 264 (17.2 mg, 0.057 mmol) in
THF (1 mL) was added dropwise over 8 min. The reaction mixture was transferred to an ice-
bath, stirred for 1.5 h, and then quenched with NH4Cl(aq). The organic layer was separated and
the aqueous layer was extracted with EtOAc. The combined organic layers were dried over
MgSO4 and concentrated in vacuo to give a residue which was purified by flash chromatography
on silica gel (15-50% EtOAc/hexanes) to afford Michael adduct 267 (4.3 mg, 7%) as a ~2:1
mixture of oxime isomers. 1H NMR (300 MHz, CDCl3) δ 8.07-8.00 (m, 1H), 7.74-7.68 (m, 3H),
7.47-7.22 (m, 5H), 6.56 (s, 0.66 H), 6.53 (s, 0.33H), 5.22 (d, J = 10.5 Hz, 0.66H), 5.15 (d, J = 7.1
Hz, 0.33H), 4.99 (d, J = 14.4 Hz, 0.66H), 4.58-4.52 (m, 0.33H), 3.71-3.52 (m, 5H), 3.12-2.89 (m,
2H), 2.48-2.42 (m, 4H), 1.73-1.58 (m, 10H); LRMS-ES+ m/z (relative intensity) 556 (MH+, 75).
Methyl 2-(3-(((tert-Butoxycarbonyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(1H-indol-2-
yl)acetate (270). To a stirred solution of indole 266 (21.0 mg, 0.046 mmol), Boc2O (10 mg,
Page 171
161
0.046 mmol), and CH2Cl2 (1 mL) was added TEA (20 μL, 0.14 mmol) followed by DMAP (0.6
mg, 5 μmol). The resulting solution was stirred for 1 h at rt and then diluted with NaHCO3(aq).
The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The
combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue
which was purified by flash chromatography on silica gel (25-50% EtOAc/hexanes) to yield O-
Boc oxime 270 (14.2 mg, 55%,) as a single O-Boc oxime isomer assumed to be the less hindered
(E)-compound. 1H NMR (400 MHz, CDCl3) δ 9.5 (s, 1H), 7.63 (d, J = 8.2 Hz, 2H), 7.53 (d, J =
7.8 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.17 (t, J = 7.1 Hz, 1H), 7.08 (t, J
= 7.2 Hz, 1H), 6.28 (s, 1H), 4.87 (d, J = 15.1 Hz, 1H), 4.52 (d, J = 5.2 Hz, 1H), 3.37 (s, 3H),
3.66-3.63 (m, 1H), 3.24 (d, J = 15.2 Hz, 1H), 3.04 (dt, J = 12.2 , 5.1 Hz, 1H), 2.83 (td, J = 12.2,
3.6 Hz, 1H), 2.37 (s, 3H), 2.14-2.10 (m, 1H), 1.64 (s, 9H), 1.48-1.40 (m, 1H); LRMS-ES+ m/z
(relative intensity) 556 (MH+, 100).
Methyl 2-(3-(((tert-Butoxycarbonyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-formyl-
1H-indol-2-yl)acetate (275). To a stirred solution of indole 270 (73 mg, 0.13 mmol) in DMF
(0.5 mL) at 0 °C was added POCl3 (31 μL, 0.33 mmol). The resulting solution was stirred at rt
for 17 h, diluted with EtOAc, and washed with NaHCO3(aq) followed by brine. The organic layer
was dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash
chromatography on silica gel (15-40% EtOAc/hexanes) to afford 3-formylindole 275 (54 mg,
Page 172
162
71%). 1H NMR (300 MHz, CDCl3) δ 10.9 (s, 1H), 10.2 (s, 1H), 8.09-8.06 (m, 1H), 7.64 (d, J =
8.3 Hz, 2H), 7.48-7.45 (m, 1H), 7.34-7.26 (m, 4H), 5.04 (d, J = 3.4 Hz, 1H), 4.91 (d, J = 15.3
Hz, 1H), 3.77-3.67 (m, 4H), 3.19-3.14 (m, 2H), 2.78 (td, J = 12.1, 3.5 Hz, 1H), 2.38 (s, 3H),
2.35-2.26 (m, 1H), 1.95-1.85 (m, 1H), 1.66 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 185.0, 170.9,
158.8, 152.9, 144.8, 141.1, 135.7, 133.0, 130.3, 128.0, 127.0, 124.1, 122.9, 119.3, 114.9, 112.5,
85.8, 64.8, 60.9, 53.2, 45.5, 43.8, 43.3, 28.2, 21.9; LRMS-ES+ m/z (relative intensity) 584
(MH+, 75).
Benzyl 2-(2-(2-Methoxy-2-oxoethyl)-1H-indol-3-yl)-2-oxoacetate (285). To a stirred
solution of indole ester 218140 (150 mg, 0.79 mmol) in Et2O (10 mL) at 0 °C was added oxalyl
chloride (350 μL, 4.0 mmol). After stirring the mixture for 30 min, benzyl alcohol (1.6 mL, 15.9
mmol) was added followed by the slow and careful addition of TEA (3.3 mL, 23.8 mmol) over 5
min. The resulting thick slurry was stirred vigorously at rt for 2 h and then quenched with water.
The organic layer was separated and the aqueous layer extracted with Et2O. The combined
organic layers were washed with water and brine, dried over MgSO4, and concentrated in vacuo
to give a viscous oil. This material was purified by flash column chromatography on silica gel
(20-40% EtOAc/hexanes) to give indole-3-oxoacetate 285 (196 mg, 71%). 1H NMR (400 MHz,
CDCl3) δ 10.43 (s, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.49-7.47 (m, 2H), 7.40-7.38 (m, 4H), 7.24 (t, J
= 7.8 Hz, 1H), 7.14 (t, J = 7.6 Hz, 1H), 5.46 (s, 2H), 4.26 (s, 2H), 3.79 (s, 3H); 13C NMR (75
MHz, CDCl3) δ 181.9, 171.1, 166.0, 142.5, 135.3, 134.9, 129.4, 129.2, 129.1, 126.0, 124.0,
Page 173
163
123.4, 120.2, 112.3, 110.4, 68.2, 60.9, 53.0, 32.9; LRMS-ES+ m/z (relative intensity) 352 (MH+,
80).
2-(Trimethylsilyl)ethyl-2-(2-(2-methoxy-2-oxoethyl)-1H-indol-3-yl)-2-oxoacetate
(286). To a stirred solution of indole 218140 (1.00 g, 5.3 mmol) in Et2O (200 mL) at 0 °C was
added oxalyl chloride (1.88 mL, 21.1 mmol). After stirring the mixture for 30 min, 2-
trimethylsilyl ethanol (12.4 mL, 84.6 mmol) was added followed by the slow and careful
addition of TEA (17.7 mL, 127 mmol) over 5 min. The resulting thick slurry was stirred
vigorously at rt for 2 h and then quenched with water. The organic layer was separated and the
aqueous layer extracted with Et2O. The combined organic layers were washed with water and
brine, dried over MgSO4, and concentrated in vacuo to give a viscous red oil. This material was
purified by flash chromatography on silica gel (20-40% EtOAc/hexanes) to give indole-3-
oxoacetate 286 (1.26 g, 66%). 1H NMR (300 MHz, CDCl3) δ 10.50 (s, 1H), 7.83-7.78 (m, 1H),
7.45-7.40 (m, 1H), 7.30-7.23 (m, 2H), 4.56-4.50 (m, 2H), 4.32 (s, 2H), 3.82 (s, 3H), 1.22-1.16
(m, 2H), 0.10 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 182.5; 171.3, 166.5, 142.3, 135.4, 126.1,
124.0, 123.4, 120.3, 112.3, 110.5, 65.2, 53.1, 32.9, 17.8, -1.1; LRMS-ES+ m/z (relative intensity)
362 (MH+, 80); HRMS-ES+ (C18H24NO5Si) calcd 362.1424 (MH+), found 362.1416.
Page 174
164
Dimethyl 2-(3-(2-(Benzyloxy)-2-oxoacetyl)-1H-indol-2-yl)malonate (288). To a
stirred solution of indole malonate 21473 (77.7 mg, 0.314 mmol) in Et2O (5 mL) at 0 °C was
added oxalyl chloride (112 μL, 1.26 mmol). After stirring the mixture for 30 min at 0 °C and 1 h
at rt, benzyl alcohol (520 μL, 5.03 mmol) was added followed by the slow and careful addition
of TEA (1.05 mL, 7.54 mmol) over 5 min. The resulting thick slurry was stirred vigorously at rt
for 2 h and then quenched with water. The organic layer was separated and the aqueous layer
was extracted with Et2O. The combined organic layers were washed with water and brine, dried
over MgSO4, and concentrated in vacuo to give a viscous oil. This material was purified by flash
column chromatography on silica gel (25-40% EtOAc/hexanes) to give indole-3-oxoacetate 288
(65.5 mg, 51%). 1H NMR (300 MHz, CDCl3) δ 10.25 (s, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.50-
7.39 (m, 6H), 7.29-7.25 (m, 1H), 7.14 (t, J = 7.3 Hz, 1H), 6.11 (s, 1H), 5.48 (s, 2H), 3.81 (s, 6H);
LRMS-ES+ m/z (relative intensity) 410 (MH+, 90).
Benzyl 2-(2-(1-(3-(Hydroxyimino)-1-nosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-1H-
indol-3-yl)-2-oxoacetate (291). To a -78 °C solution of indole 285 (110 mg, 0.31 mmol) in THF
(2 mL) was added LiHMDS (630 μL, 0.63 mmol, 1.0 M in THF) and the resulting solution was
stirred for 50 min. A solution of oxime 290 (104 mg, 0.31 mmol) in THF (1 mL) was then added
Page 175
165
dropwise over 1 min. The reaction mixture was stirred for 2 h at -78 °C and then quenched with
NH4Cl(aq). The organic layer was separated and the aqueous layer was extracted with EtOAc.
The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a
residue, which was purified by flash chromatography on silica gel (50% EtOAc/hexanes) to yield
Michael adduct 291 (165 mg, 81%) as a ~1:1 mixture of oxime isomers and recovered starting
indole 285 (21 mg, 19%). 1H NMR (300 MHz, CDCl3) δ 9.95 (s, 0.5H), 0.89 (s, 0.5H), 8.04 (s,
0.5H), 7.96 (dd, J = 7.5, 1.7 Hz, 0.5H), 7.92-7.88 (m, 1H), 7.65-7.60 (m, 1H), 7.52-7.39 (m,
8.5H), 7.25-7.20 (m, 1.5H), 7.15-7.10 (m, 1H), 5.50 (d, J = 6.3 Hz, 0.5H), 5.45 (s, 2H), 5.17 (d, J
= 8.5 Hz, 0.5H), 4.97 (d, J = 16.1 Hz, 0.5H), 4.67 (d, J = 15.8 Hz, 0.5H), 4.11-4.07 (m, 0.5H),
4.02 (d, J = 15.9 Hz, 0.5H), 3.76-3.68 (m, 4H), 3.45-3.42 (m, 0.5H), 3.32-3.26 (m, 1H), 3.13-
3.09 (m, 0.5H), 2.06-2.02 (0.5H), 1.84-1.60 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 182.8,
182.4, 172.8, 171.9, 165.8, 165.6, 153.03, 153.00, 148.5, 148.1, 143.13, 143.08, 135.6, 135.5,
134.84, 134.79, 134.4, 134.2, 132.2, 132.1, 131.9, 131.8, 131.3, 131.2, 129.6, 129.5, 129.4,
129.3, 129.20, 129.18, 125.8, 125.5, 124.6, 125.5, 124.4, 124.3, 123.5, 123.2, 120.3, 120.0,
112.6, 112.4, 111.4, 111.2, 68.4, 68.3, 64.9, 53.4, 53.3, 44.8, 44.5, 44.0, 43.5, 43.3, 42.7, 42.2,
41.7, 31.0, 28.0, 27.8, 19.6; LRMS-ES+ m/z (relative intensity) 649 (MH+, 100).
Benzyl 2-(2-(1-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-nosylpiperidin-4-yl)-2-
methoxy-2-oxoethyl)-1H-indol-3-yl)-2-oxoacetate (292). To a stirred solution of oxime 291
Page 176
166
(165 mg, 0.25 mmol), imidazole (70 mg, 1.0 mmol), and CH2Cl2 (5 mL) was added TBS-Cl (103
mg, 0.66 mmol). The resulting mixture was stirred at rt for 4 h and then diluted with 1 M HCl.
The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The
combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue.
This material was purified by flash chromatography on silica gel (25-50% EtOAc/hexanes) to
give O-TBS-oxime 292 (186 mg, 96%) as a ~1:1 mixture of oxime isomers. 1H NMR (300 MHz,
CDCl3) δ 9.87 (s, 0.5H), 9.84 (s, 0.5H), 7.96-7.89 (m, 1H), 7.67-7.64 (m, 1H), 7.54-7.36 (m,
8.5H), 7.32-7.15 (2.5H), 5.50 (d, J = 7.5 Hz, 0.5H), 5.46 (s, 2H), 5.13-5.07 (m, 1H), 4.92 (d, J =
15.8 Hz, 0.5H), 3.77-3.65 (m, 5H), 3.52-3.48 (m, 0.5H), 3.29-3.12 (m, 1.5H), 2.04-1.98 (m,
0.5H), 1.88-1.70 (m, 1.5H), 0.99 (s, 9H), 0.31 (s, 1.5H), 0.26 (s, 1.5H), 0.23 (s, 1.5H), 0.21 (s,
1.5H); 13C NMR (75 MHz, CDCl3) δ 182.8, 182.3, 172.5, 170.9, 165.8, 165.7, 157.5, 156.8,
148.6, 148.2, 143.9, 143.5, 135.5, 135.4, 134.88, 134.86, 134.4, 134.3, 132.1, 131.9, 131.8,
131.7, 131.2, 131.1, 129.6, 129.5, 129.4, 129.3, 129.21, 129.19, 125.8, 125.6, 124.6, 124.5,
124.4, 124.2, 123.5, 123.2, 120.5, 120.0, 112.4, 112.3, 111.19, 111.17, 68.3, 68.2, 53.2, 53.1,
45.0, 44.9, 44.2, 43.5, 43.4, 43.0, 42.7, 28.3, 27.6, 26.32, 26.25, 18.3, 18.2, -4.2, -4.6, -4.7;
LRMS-ES+ m/z (relative intensity) 763 (MH+, 100).
tert-Butyl 3-(2-(Benzyloxy)-2-oxoacetyl)-2-(2-methoxy-2-oxoethyl)-1H-indole-1-
carboxylate (295). To a stirred solution of indole 285 (26.0 mg, 0.074 mmol), Boc2O (18 mg,
0.081 mmol), and CH2Cl2 (1 mL) was added TEA (21 μL, 0.15 mmol) followed by DMAP (1
Page 177
167
mg, 0.007 mmol). The resulting solution was stirred at rt for 13 h and then diluted with
NaHCO3(aq). The organic layer was separated and the aqueous layer was extracted with CH2Cl2.
The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a
residue which was purified by flash chromatography on silica gel (10-30% EtO/hexanes) to
afford N-Boc indole 295 (13.6 mg, 41%). 1H NMR (300 MHz, CDCl3) δ 8.11 (d, J = 8.5 Hz,
1H), 7.55 (d, J = 7.9 Hz, 1H), 7.46-7.29 (m, 6H), 7.19 (t, J = 7.2 Hz, 1H), 5.44 (s, 2H), 4.55 (s,
2H), 3.71 (s, 3H), 1.69 (s, 9H); LRMS-ES+ m/z (relative intensity) 452 (MH+, 50).
tert-Butyl 3-(2-(Benzyloxy)-2-oxoacetyl)-2-(1-(2-(hydroxyimino)cyclohexyl)-2-
methoxy-2-oxoethyl)-1H-indole-1-carboxylate (296). To a -78 °C solution of indole 295 (13.6
mg, 0.030 mmol) in THF (0.5 mL) was added LiHMDS (30 μL, 0.030 mmol, 1.0 M in THF) and
the resulting solution was stirred for 30 min. A solution of oxime 211 (2.2 mg, 0.015 mmol) in
THF (0.5 mL) was then added dropwise over 1 min. The reaction mixture was stirred for 2 h at -
78 °C and then quenched with NH4Cl(aq). The organic layer was separated and the aqueous layer
was extracted with EtOAc. The combined organic layers were dried over MgSO4 and
concentrated in vacuo to give a residue. This material was purified by flash chromatography on
silica gel (15-25% EtOAc/hexanes) to give Michael adduct 296 (8.0 mg, ~95%) as a ~3:1
mixture of oxime isomers contaminated with a small amount of the bis-alkylation product 297.
1H NMR (300 MHz, CDCl3) δ 8.06-7.95 (m, 1H), 7.64 (br s, 0.5H), 7.48-7.27 (m, 7H), 7.20-7.18
(m, 1H), 6.81 (br s, 0.5H), 5.97-5.95 (d, J = 7.8 Hz, 0.5H), 5.54-5.43 (m, 1.5H), 3.61-3.56 (m,
Page 178
168
3H), 3.45-3.37 (m, 1H), 1.93-1.63 (m, 14H), 1.43-1.13 (m, 6H); LRMS-ES+ m/z (relative
intensity) 563 (MH+, 75) with bis-alkylation product: 674 (MH+, 40).
Methyl 3-(4-Methoxyphenyl)-2-(3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)acetyl)-1H-
indol-2-yl)propanoate (298). To a stirred solution of indole 286 (54.4 mg, 0.151 mmol) and
DMF (1 mL) at 0 °C was added NaH (60% dispersion in mineral oil, 6.6 mg, 0.17 mmol) and the
resulting solution was stirred at rt for 30 min. p-Methoxybenzyl chloride (23 μL, 0.17 mmol)
and TBAI (5.6 mg, 0.015 mmol) were added and the reaction was stirred for 5 h at rt before
diluting with water and EtOAc. The organic layer was separated and the aqueous layer was
extracted with EtOAc. The combined organic layers were washed with water and brine, then
dried over MgSO4 and concentrated in vacuo to give a residue. This material was purified by
preparative thin layer chromatography (50% EtOAc/hexanes) to give alkylation product 298
(29.8 mg, 41%) and recovered starting indole 286 (18.9 mg, 35%). 1H NMR (400 MHz, CDCl3)
δ 10.08 (s, 1H), 7.79-7.77 (m, 1H), 7.43-7.41 (m, 1H), 7.31-7.28 (m, 2H), 7.09 (d, J = 8.6 Hz,
2H), 6.81 (d, J = 8.6 Hz, 2H), 5.05 (dd, J = 9.4, 4.6 Hz, 1H), 4.61-4.50 (m, 2H), 3.80 (s, 3H),
3.64 (s, 3H), 3.35 (dd, J = 13.4, 4.5 Hz, 1H), 3.16 (dd, J = 13.3, 9.5 Hz, 1H), 1.28-1.18 (m, 2H),
0.11 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 182.5, 174.6, 166.5, 159.1, 146.4, 135.2, 130.5,
129.5, 126.1, 124.1, 123.4, 120.4, 114.3, 112.3, 109.5, 65.2, 55.6, 52.9, 46.2, 40.9, 17.8, -1.1;
LRMS-ES+ m/z (relative intensity) 336 (MH+, 100).
Page 179
169
Methyl 2-(3-(2-(Benzyloxy)-2-oxoacetyl)-1H-indol-2-yl)-3-(4-methoxyphenyl)
propanoate (298). To a stirred solution of indole 285 (41 mg, 0.12 mmol), K2CO3 (24 mg, 0.18
mmol) and MeCN (3 mL) was added p-methoxybenzyl chloride (24 μL, 0.18 mmol) and the
resulting solution was stirred at reflux for 6 h. After cooling to rt, the reaction mixture was
diluted with water and EtOAc. The organic layer was separated, and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to give a residue. This material was purified by preparative thin layer chromatography
(50% EtOAc/hexanes) to afford alkylation product 299 (26 mg, 47%). 1H NMR (400 MHz,
CDCl3) δ 10.08 (s, 1H), 7.54-7.49 (m, 3H), 7.41-7.39 (m, 4H), 7.26 (t, J = 7.4 Hz, 1H), 7.14 (t, J
= 7.3 Hz, 1H), 7.07 (d, J = 8.5 Hz, 2H), 6.81 (d, J = 8.6 Hz, 2H), 5.50 (ABq, J = 21.2, 12.0 Hz,
2H), 3.80 (s, 3H), 3.63 (s, 3H), 3.34 (dd, J = 13.4, 4.7 Hz, 1H), 3.15 (dd, J = 13.4, 9.4 Hz, 1H).
Benzyl 2-(2-(2-Methoxy-2-oxoethyl)-1-methyl-1H-indol-3-yl)-2-oxoacetate (302). To
a stirred solution of indole ester 30193 (604 mg, 2.97 mmol) in Et2O (100 mL) at 0 °C was added
oxalyl chloride (1.06 mL, 11.9 mmol). After stirring the mixture for 30 min, benzyl alcohol
(4.92 mL, 47.6 mmol) was added followed by the slow and careful addition of TEA (9.94 mL,
71.3 mmol) over 5 min. The resulting thick slurry was stirred vigorously at rt for 2 h and then
Page 180
170
quenched with water. The organic layer was separated and the aqueous layer was extracted with
Et2O. The combined organic layers were washed with water and brine, dried over MgSO4, and
concentrated in vacuo to give a viscous oil which was purified by flash chromatography on silica
gel (25% to 50% EtOAc/hexanes) to afford indole-3-oxoacetate 302 (911 mg, 84%). 1H NMR
(400 MHz, CDCl3) δ 7.59 (d, J = 8.0 Hz, 1H), 7.47-7.45 (m, 2H), 7.38-7.35 (m, 3H), 7.24 (d, J =
3.8 Hz, 2H), 7.15-7.10 (m, 1H), 5.42 (s, 2H), 4.16 (s, 2H), 3.66 (s, 3H), 3.54 (s, 3H); 13C NMR
(75 MHz, CDCl3) δ 181.6, 169.1, 166.1, 143.3, 137.4, 135.1, 129.4, 129.2, 129.1, 125.7, 123.5,
120.3, 110.7, 110.4, 68.1, 60.8, 52.9, 32.2, 30.4; LRMS-ES+ m/z (relative intensity) 366 (MH+,
100).
Methyl 6-(2-(Benzyloxy)-2-oxoethyl)-2-nosyl-2,3,4,5,5a,6-hexahydro-1H-
pyrido[4',3':4,5]pyrrolo[1,2-a]indole-5-carboxylate (309). To a solution of indole 292 (43.0
mg, 0.056 mmol) in TFA (0.5 mL) was added triethylsilane (18 μL, 0.113 mmol). The resulting
solution was stirred for 4 h at rt and then concentrated in vacuo. The resulting residue was
dissolved in EtOAc and washed with NaHCO3(aq). The organic layer was separated and the
aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4
and concentrated in vacuo to give a residue which was purified by preparative thin layer
Page 181
171
chromatography (60% EtOAc/hexanes) to yield tetracycle 209 (10.4 mg, 31%). 1H NMR (300
MHz, CDCl3) δ 7.88-7.85 (m, 1H), 7.68-7.52 (m, 4H), 7.35-7.33 (m, 6H), 7.22-7.10 (m, 2H),
5.13 (s, 2H), 4.78 (q, J = 7.4 Hz, 1H), 4.12-4.0 (m, 1H), 3.89-3.60 (m, 5H), 3.45-3.39 (m, 1H),
3.29-3.21 (m, 1H), 3.08 (dd, J = 13.6, 8.0 Hz, 1H), 2.19-1.97 (m, 2H); 13C NMR (75 MHz,
CDCl3) δ 171.9, 171.5, 148.5, 137.2, 136.3, 134.2, 132.5, 132.3, 132.1, 131.2, 129.0, 128.8,
128.7, 124.6, 122.4, 120.3, 119.5, 110.2, 102.3, 67.0, 53.7, 53.0, 46.3, 45.5, 43.5, 42.9, 30.6,
25.8; LRMS-ES+ m/z (relative intensity) 604 (MH+, 100).
(Z)-Methyl 3-(2-(Benzyloxy)-2-oxoacetyl)-2-(1-(3-(((tert-butyldimethylsilyl)-
oxy)imino)-1-nosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-1H-indole-1-carboxylate (310).
To a stirred solution of indole 292 (22.0 mg, 0.029 mmol) and THF (0.5 mL) at -78 °C was
added LiHMDS (1.0 M in THF, 29 mL, 0.029 mmol) and the resulting solution was stirred for
30 min. Methyl chloroformate (12 μL, 0.15 mmol) was added and the reaction mixture was
stirred for 18 h gradually warming to rt before quenching with water. The organic layer was
separated and the aqueous layer was extracted with EtOAc. The combined organic layers were
dried over MgSO4 and concentrated in vacuo to give a residue which was purified by preparative
thin layer chromatography (30-40% EtOAc/hexanes) to affored methyl carbamate 310 (9.0 mg,
38%). 1H NMR (300 MHz, CDCl3) δ 8.11 (d, J = 8.5 Hz, 1H), 7.89-7.87 (m, 1H), 7.58-7.52 (m,
3H), 7.45-7.42 (m, 5H), 7.34 (t, J = 8.4 Hz, 1H), 7.22 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 8.0 Hz,
Page 182
172
1H), 5.72 (d, J = 9.7 Hz, 1H), 5.43 (ABq, J = 16.8, 11.9 Hz, 2H), 4.08 (s, 3H), 3.90-3.87 (m,
1H), 3.70-3.68 (m, 1H), 3.63-3.57 (m, 4H), 3.03 (d, J = 14.9 Hz, 1H), 2.58 (d, J = 14.6 Hz, 1H),
2.13-2.07 (m, 1H), 0.98-0.86 (m, 1H) 0.71 (s, 9H), -0.08 (s, 3H), -0.26 (s, 3H); LRMS-ES+ m/z
(relative intensity) 821 (MH+, 100).
Methyl 3-(2-(Benzyloxy)-2-oxoacetyl)-2-(1-(3-(hydroxyimino)-1-nosyl-piperidin-4-
yl)-2-methoxy-2-oxoethyl)-1H-indole-1-carboxylate (311). To a stirred solution of indole 310
(9.0 mg, 0.011 mmol) in TFA (0.5 mL) was added triethylsilane (3.5 μL, 0.022 mmol). The
resulting solution was stirred for 4 h at rt and then concentrated in vacuo. The resulting residue
was dissolved in EtOAc and washed with NaHCO3(aq). The organic layer was separated and the
aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4
and concentrated in vacuo to give a residue which was purified by preparative thin layer
chromatography (50% EtOAc/hexanes) to yield free oxime 311 (6.7 mg, 86%) as a ~1:1 mixture
of diastereomers. 1H NMR (300 MHz, CDCl3) δ 8.10-8.07 (m, 1H), 7.91-7.89 (m, 1H), 7.61-
7.54 (m, 3H), 7.47-7.37 (m, 6H), 7.29-7.14 (m, 2H), 5.66 (d, J = 9.4 Hz, 1H), 5.44 (s, 2H), 4.82
(d, J = 15.7 Hz, 1H), 4.06 (s, 3H), 3.97-3.85 (m, 1H), 3.64-3.55 (m, 5H), 3.08 (d, J = 15.7 Hz,
1H), 2.49-2.43 (m, 1H), 2.24-2.15 (m, 1H), 1.85-1.70 (m, 1H); LRMS-ES+ m/z (relative
intensity) 707 (MH+, 50).
Page 183
173
Methyl 2-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-(3-(2-oxo-2-(2-(trimethylsilyl)-
ethoxy)acetyl)-1H-indol-2-yl)acetate (315). To a -78 °C solution of indole 286 (5.80 g, 16.1
mmol) in THF (100 mL) was added LiHMDS (1.0 M in THF, 32.1 mL, 32.1 mmol) and the
resulting solution was stirred for 30 min. A solution of oxime 264 (4.86 g, 16.1 mmol) in THF
(30 mL) was then added dropwise over 3 min. The reaction mixture was stirred for 2 h at -78 °C
and then quenched with NH4Cl(aq). The organic layer was separated and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to afford a residue which was purified by flash chromatography on silica gel (50%
EtOAc/hexanes) to give Michael adduct 315 (10.00 g, 99%). 1H NMR (300 MHz, CDCl3)
δ 10.31 (s, 0.5H), 10.23 (s, 0.5H), 8.90 (s, 0.5H), 8.66 (s, 0.5H), 7.79-7.58 (m, 3H), 7.39-7.17
(m, 5H), 5.59 (d, J = 5.9 Hz, 0.5H), 5.13 (d, J = 8.6 Hz, 0.5H), 4.93 (d, J = 14.5 Hz, 0.5H), 4.60-
4.50 (m, 2H), 3.69-4.47 (m, 4H) 3.37 (p, J = 5.7 Hz, 0.5H), 3.20 (d, J = 14.6 Hz, 0.5 H); 3.10 (q,
J = 7.3 Hz, 0.5H), 2.97-2.85 (m, 1H), 2.44-2.28 (m, 3H), 2.08-2.03 (m, 0.5H), 1.78-1.76 (m,
0.5H), 1.41-1.31 (m, 0.5H), 0.11-0.07 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 183.6, 183.0,
172.6, 171.9, 166.5, 166.5, 152.8, 152.7, 144.61, 144.57, 143.5, 143.4, 135.83, 135.80, 130.4,
130.3, 128.06, 127.96, 125.8, 125.6, 124.4, 124.2, 123.4, 123.2, 120.0, 119.6, 112.9, 112.7,
111.2, 111.1, 65.33, 65.29, 53.3, 53.2, 45.4, 44.9, 44.2, 43.4, 42.9, 42.7, 42.3, 41.9, 27.9, 27.8,
22.0, 21.9, 17.8, 14.6, 14.2, -1.1; LRMS-ES+ m/z (relative intensity) 666 (M+K+, 100); HRMS-
ES+ (C30H41N4O8SSi) calcd 645.2414 (M+NH4+), found 645.2445.
Page 184
174
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(2-oxo-
2-(2-(trimethylsilyl)ethoxy)acetyl)-1H-indol-2-yl)acetate (317). To a solution of oxime 315
(10.00 g, 15.9 mmol), imidazole (4.43 g, 64.4 mmol), and CH2Cl2 (600 mL) was added TBS-Cl
(6.50 g, 41.8 mmol). The resulting mixture was stirred for 17 h at rt and then diluted with 1 M
HCl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The
combined organic layers were dried over MgSO4 and concentrated in vacuo to yield a residue
which was purified by flash chromatography on silica gel (15-30% EtOAc/hexanes) to give O-
TBS-oxime 317 (9.88 g, 84%). 1H NMR (300 MHz, CDCl3) δ 9.91 (s, 0.5H), 9.79 (s, 0.5H),
7.77-7.61 (m, 3H), 7.41-7.21 (m, 5H) 5.56 (d, J = 5.7 Hz, 0.5H), 5.06-4.97 (m, 1H), 4.84 (d, J =
15.0, 0.5H), 4.54-4.45 (m, 2H), 3.69-3.60 (m, 4H), 3.41 (dt, J = 7.1, 12.6 Hz, 0.5H), 3.30 (d, J =
15.0 Hz, 0.5H), 3.18 (d, J = 14.9 Hz, 0.5H), 3.00-2.92 (m, 1H), 2.85-2.70 (m, 0.5H), 2.42 (s,
1.5H) 2.32 (s, 1.5H), 2.06-1.98 (m, 0.5H), 1.85-1.70 (m, 0.5H), 1.65-1.55 (m, 0.5H), 1.40-1.35
(m, 1H), 1.20-1.13 (m, 2H), 0.99 (m, 9H), 0.31-0.20 (m, 6H), 0.10-0.07 (m, 9H); 13C NMR (75
MHz, CDCl3) δ 183.4, 182.8, 172.6, 171.0, 166.3, 166.1, 157.9, 156.8, 144.40, 144.37, 143.6,
143.4, 135.5, 135.4, 133.9, 133.6, 130.20, 130.15, 128.0, 127.9, 126.0, 125.7, 124.4, 124.2,
123.5, 123.2, 120.6, 120.0, 112.4, 112.2, 111.3, 65.1, 53.2, 53.0, 45.2, 44.1, 43.5, 43.3, 42.9,
28.1, 27.7, 26.34, 26.28, 21.93, 21.85, 18.3, 18.2, 17.79, 17.76, -1.1, -4.2, -4.6, -4.8; LRMS-ES+
m/z (relative intensity) 780 (M+K+, 100); HRMS-ES+ (C36H52N3O8SSi2) calcd 742.3014 (MH+),
found 742.3005.
Page 185
175
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(1-
hydroxy-2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetate (318). To a stirred
solution of indole-3-oxoacetate 317 (6.07 g, 8.18 mmol) in MeOH (70 mL) and THF (70 mL) at
0 °C was added NaBH4 (378 mg, 9.79 mmol). The resulting solution was stirred for 1 h at 0 °C
and then diluted with NH4Cl(aq). The organic layer was separated and the aqueous layer was
extracted with CH2Cl2. The combined organic layers washed with water, brine, and then dried
over MgSO4. Removal of the solvent in vacuo provided alcohol 318 (6.05 g, 99%) which was
used in the following reaction without further purification. 1H NMR (300 MHz, CDCl3) δ 8.99
(s, 0.5H), 8.81-8.78 (m, 0.5H), 7.66-7.60 (m, 3H), 7.33-7.07 (m, 5H), 5.39-5.28 (m, 1H), 5.15-
5.0 (m, 0.5H), 4.95-4.85 (m, 0.5H), 5.54-4.50 (m, 0.5H), 4.35-4.11 (m, 2.5H), 3.68-3.25 (m, 6H),
3.20-2.90 (m, 1.5H), 3.70-3.60 (m, 0.5H), 2.45-2.36 (m, 3H), 1.95-1.80 (m, 0.5H), 1.65-1.20 (m,
2H), 1.01-0.87 (m, 11H), 0.27-0.17 (m, 6H), 0.04--0.05 (m, 9H); 13C NMR (75 MHz, CDCl3) δ
174.6, 174.5, 174.3, 173.2, 173.1, 171.8, 157.6, 157.5, 156.9, 156.7, 144.3, 136.24, 136.17,
135.7, 134.0, 133.9, 131.4, 130.2, 128.1, 128.0, 126.4, 126.2, 126.1, 123.2, 122.8, 120.8, 120.4,
119.7, 119.6, 119.3, 112.62, 112.56, 112.2, 112.0, 111.6, 66.8, 66.5, 66.2, 65.2, 64.94, 64.89,
53.9, 53.0, 52.9, 52.8, 45.4, 45.3, 44.8, 44.2, 44.2, 43.4, 43.3, 43.0, 42.8, 42.6, 42.5, 42.3, 42.1,
28.4, 27.8, 26.4, 26.3, 22.0, 21.9, 18.4, 18.3, 17.8, 17.6, -1.2, -4.5, -4.7, -4.9; LRMS-ES+ m/z
(relative intensity) 782 (M+K+, 100); HRMS-ES+ (C36H54N3O8SSi2) calcd 744.3170 (MH+),
found 744.3148.
Page 186
176
Methyl 2-(3-(1-Acetoxy-2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)-2-(3-
(((tert-butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)acetate (319). The alcohol 318
(6.05 g, 8.13 mmol) was dissolved in Ac2O (40 mL) and pyridine (40 mL) and the mixture was
stirred at rt for 26 h. The solvent was removed in vacuo and the resulting crude product was
purified by flash chromatography on silica gel (15-25% EtOAc/hexanes) to give acetate 319
(5.84 g, 91%). For characterization purposes, E-319 and Z-319 were separated for NMR
analysis. E-319 (more polar oxime isomer, ~3:1 mixture of acetoxy diastereomers): 1H NMR
(400 MHz, CDCl3) δ 9.07 (s, 0.75H), 9.03 (s, 0.25H), 7.75-7.71 (m, 1H), 7.64 (d, J = 8.2 Hz,
2H), 7.32-7.13 (m, 5H), 6.21 (s, 0.25H), 6.18 (s, 0.75H), 4.97-4.88 (m, 1H), 4.52 (d, J = 5.9 Hz,
0.25H), 4.48 (d, J = 5.5 Hz, 0.75H), 4.28-4.20 (m, 1H), 4.09-4.02 (m, 1H), 3.67-3.55 (m, 4H),
3.41-3.31 (m, 2H), 3.00-2.93 (m, 1H), 2.39-2.36 (m, 3H), 2.14 (s, 3H), 1.95-1.70 (m, 3H), 1.60-
1.40 (m, 2H), 0.98 (s, 9H), 0.29-0.18 (m, 6H), 0.00--0.02 (m, 9H); 13C NMR (90 MHz, CDCl3)
δ 171.0, 170.8, 170.6, 170.5, 169.1, 157.3, 144.0, 135.3, 135.2, 133.7, 133.5, 132.2, 131.7, 129.8,
127.6, 126.2, 126.1, 122.7, 120.3, 119.3, 111.2, 108.0, 107.8, 67.7, 64.2, 53.4, 52.5, 44.8, 43.2,
42.9, 42.7, 42.4, 42.1, 28.0, 26.0, 25.9, 21.5, 20.9, 20.7, 17.9, 17.3, -1.56, -1.59, -4.8, -5.1; Z-319
(less polar oxime isomer, ~2:1 mixture of acetoxy diastereomers): 1H NMR (300 MHz, CDCl3)
δ 8.73-8.71 (m, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.35-7.14 (m, 5H), 6.24-
6.22 (m, 1H), 5.02 (t, J = 7.1 Hz, 1H), 4.34-4.07 (m, 3H), 3.66-3.53 (m, 4H), 3.15-3.07 (m, 2H),
2.72 (d, J = 11.3, 3.2 Hz, 1H), 2.45 (s, 3H), 2.20-2.18 (m, 3H), 1.58-1.39 (m, 2H), 1.00-0.89 (m,
12H), 0.26 (s, 3H), 0.20 (s, 3H), 0.02--0.04 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 173.1, 173.0,
170.9, 170.8, 169.3, 169.2, 156.7, 144.4, 136.1, 136.0, 133.8, 133.6, 132.4, 132.2, 130.2, 128.1,
126.7, 123.5, 123.4, 121.1, 120.2, 111.5, 108.54, 108.50, 68.4, 68.0, 64.7, 64.5, 53.9, 53.0, 52.9,
45.4, 44.4, 44.1, 42.8, 42.6, 42.5, 28.5, 28.0, 26.4, 22.0, 21.2, 21.1, 18.4, 17.7, 17.6, -1.2, -4.75, -
Page 187
177
4.89, -4.92; mixture of E- and Z-319: LRMS-ES+ m/z (relative intensity) 824 (M+K+, 25);
HRMS-ES+ (C38H56N3O9SSi2) calcd 786.3276 (MH+), found 786.3286.
(Z)-Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(1-
ethoxy-2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetate (Z-321). Indole Z-319
(23.3 mg, 0.030 mmol) was dissolved in 10:1 EtOH/TEA (1.1 mL) and the solution was stirred
for 22 h at rt. The solution was and concentrated in vacuo to give a residue which was purified
by flash chromatography on silica gel (30% EtOAc/hexanes) to give ethanol adduct Z-321 (17.2
mg, 75%) as a ~1:1 mixture of ethoxy-diastereomers. 1H NMR (300 MHz, CDCl3) δ 8.37 (s,
0.5H), 8.33 (s, 0.5H), 7.83-7.80 (m, 1H), 7.68 (d, J = 7.8 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.28-
7.25 (m, 1H), 7.21-7.11 (m, 2H), 5.12-5.00 (m, 1H), 4.43-4.08 (m, 3H), 3.65-3.58 (m, 4H), 3.52-
5.48 (m, 1H), 3.15-3.10 (m, 2H), 2.75-2.65 (m, 1H), 2.45 (s, 3H), 1.62-1.40 (m, 3H), 1.30-1.21
(m, 2H), 1.00-0.93 (m, 12H), 0.25-0.19 (m, 6H), 0.04-0.01 (m, 9H); LRMS-ES+ m/z (relative
intensity) 810 (M+K+, 50).
(E)-Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(1-
ethoxy-2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetate (E-321). Using the
above procedure with the E-oxime isomer E-319 (22.0 mg, 0.028 mmol) afforded ethanol adduct
E-321 (14.5 mg, 67%) as a ~1:1 mixture of ethoxydiastereomers. 1H NMR (300 MHz, CDCl3)
δ 9.03 (s, 0.5H), 8.99 (s, 0.5H), 7.73-7.70 (m, 1H), 7.66-7.63 (m, 2H), 7.31-7.25 (m, 3H), 7.21 (t,
Page 188
178
J = 8.0 Hz, 1H), 7.13 (t, J = 7.2 Hz, 1H), 5.21 (s, 0.5H), 5.15 (s, 0.5H), 5.01 (t, J = 15.8 Hz, 1H),
4.82 (d, J = 5.6 Hz, 0.5H), 4.75 (d, J = 5.6 Hz, 0.5H), 4.23-4.16 (m, 1H), 4.09-3.99 (m, 1H),
3.67-3.55 (m, 4.5H), 3.49-3.44 (m, 1.5H), 3.37-3.32 (m, 1.5H), 3.24 (d, J = 15.9 Hz, 0.5H), 2.95-
2.88 (m, 1H), 2.37 (s, 1.5H), 2.36 (s, 1.5H), 1.97-1.86 (m, 1H), 1.41-1.28 (m, 1H), 1.23-1.16 (m,
3H), 1.00 (s, 4.5H), 0.99 (s, 4.5H), 0.96-0.87 (m, 2H), 0.31 (s, 1.5H), 0.28 (s, 1.5H), 0.20 (s, 3H),
-0.01 (s, 9H); LRMS-ES+ m/z (relative intensity) 810 (M+K+, 60).
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-(3-(2-oxo-
2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetate (320). To a solution of acetate 319
(413 mg, 0.525 mmol) and t-BuOH (12.5 mL) under an argon atmosphere was added Pd/C (10
wt% Pd on carbon, 84 mg, 0.079 mmol). The resulting mixture evacuated and backfilled with H2
from a balloon and then TEA (1.4 mL) was added. The mixture was warmed to 30 °C and
stirred for 4 days. An additional portion of TEA (350 μL) was added and the reaction mixture
was stirred for another 24 h. The reaction mixture was filtered through a pad of Celite eluting
with EtOAc and the filtrate was concentrated in vacuo to afford a residue, which was purified by
flash chromatography on silica gel (10-25% EtOAc/hexanes) to give indole E-320 (190 mg) and
indole Z-320 (184 mg, combined 98% yield). 1H NMR (300 MHz, CDCl3, ~1:1 mixture of
oxime isomers) δ 8.81 (s, 0.5H), 8.43 (s, 0.5H), 7.69-7.61 (m, 3H), 7.36-7.14 (m, 5H), 5.10 (d, J
= 14.3 Hz, 0.5H), 4.88 (d, J = 14.9 Hz, 0.5H), 4.39 (d, J = 6.0 Hz, 0.5H), 4.22-4.10 (m, 2.5H),
Page 189
179
3.71-3.59 (m, 6.5H), 3.41(d, J = 15.1 Hz, 0.5H), 3.31 (p, J = 5.9 Hz, 0.5H), 3.12-2.96 (m, 1.5H)
2.48 (s, 1.5H), 2.40 (s, 1.5H), 1.92-1.88 (m, 0.5H), 1.63-1.59 (m, 1.5H), 1.04-0.99 (m, 11H),
0.28-0.20 (m, 6H), 0.08-0.05 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 173.3, 172.0, 171.9, 171.8,
157.5, 156.8, 144.3, 136.1, 135.8, 134.0, 133.7, 130.4, 130.20, 130.16, 128.7, 128.4, 128.2,
128.04, 128.80, 126.0, 123.0, 122.6, 120.5, 120.0, 119.5, 119.2, 111.4, 108.6, 107.9, 65.6, 63.5,
52.9, 52.8, 45.6, 45.0, 44.2, 43.4, 43.0, 42.9, 42.6, 42.3, 34.7, 32.4, 32.2, 30.9, 30.8, 30.1, 29.8,
28.1, 27.9, 26.4, 26.4, 23.2, 22.0, 21.9, 21.7, 18.5, 18.3, 17.84, 17.79, 14.6, -1.1, -4.5, -4.69, -
4.74, -4.88; LRMS-ES+ m/z (relative intensity) 766 (M+K+, 75); E-320: HRMS-ES+
(C36H57N4O7SSi2) calcd 745.3487 (M+NH4+), found 745.3478. Z-320: HRMS-ES+
(C36H54N3O7SSi2) calcd 728.3221 (MH+), found 728.3224.
Methyl 2-(3-(((tert-Butyldimethylsilyl)oxy)imino)piperidin-4-yl)-2-(3-(2-oxo-2-(2-
(trimethylsilyl)ethoxy)ethyl)-1H-indol-2-yl)acetate (323). A stock solution of sodium-
naphthalenide was prepared by adding Na (39 mg, 1.7 mmol) to a stirred solution of naphthalene
(222 mg, 1.72 mmol) and 1,2-dimethoxyethane (2 mL). The resulting solution was stirred for 30
min and then 1 mL of the stock sodium-naphthalenide solution was added dropwise over 10 min
to a -78 °C solution of indole 320 (25.0 mg, 0.034 mmol) in 1,2-dimethoxyethane (2 mL). The
reaction mixture was quenched with NH4Cl(aq) and then concentrated in vacuo. The resulting
residue was taken up in EtOAc and washed with water. The organic layer was dried over
Page 190
180
MgSO4 and concentrated in vacuo to give a residue, which was purified by flash chromatography
on silica gel (85% EtOAc/hexanes + 1% TEA) to afford amine 323 (10.8 mg, 55%). 1H NMR
(360 MHz, CDCl3, ~2:1 mixture of oxime isomers) δ 8.08 (s, 0.33H), 8.37 (s, 0.66H), 7.65-7.60
(m, 1H), 7.35-7.10 (m, 3H), 4.58-4.35 (m, 2H), 4.27-4.08 (m, 3H), 3.75-3.65 (m, 5H), 3.53-3.48
(m, 0.66H), 3.30-3.12 (m, 1.33H), 3.10-2.96 (m, 2H), 2.78-2.68 (m, 1H), 1.93-1.88 (m, 1H),
1.60-1.48 (m, 2H), 1.05-0.98 (m, 9H), 0.30-0.15 (m, 6H), 0.04--0.01 (m, 9H); LRMS-ES+ m/z
(relative intensity) 574(MH+, 100).
tert-Butyl 2-(1-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-
methoxy-2-oxoethyl)-3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate
(324). To a stirred solution of indole 320 (1.00 g, 1.37 mmol), Boc2O (1.80 g, 8.24 mmol), and
CH2Cl2 (60 mL) at 0 °C was added TEA (2.90 mL, 20.6 mmol) followed by DMAP (17 mg, 0.14
mmol). The resulting solution was stirred at rt for 16 h and then diluted with NaHCO3(aq). The
organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined
organic layers were dried over MgSO4 and concentrated in vacuo to yield a residue which was
purified by flash chromatography on silica gel (10-15% EtOAc/hexanes) to give N-Boc indole
324 (1.00 g, 88%) as a ~1:1 mixture of oxime isomers. Z-324: 1H NMR (300 MHz, CDCl3,
mixture of BOC-rotamers) δ 8.01-7.94 (m, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.57-7.43 (m, 1H),
7.35-7.25 (m, 4H), 5.60-5.22 (m, 2H), 4.15-4.06 (m, 2H), 3.67-3.56 (m, 6H), 3.45-3.38 (m, 1H),
Page 191
181
2.90 (d, J = 13.9 Hz, 1H), 2.64 (t, J = 8.7 Hz, 1H), 2.47 (s, 3H), 1.67-1.65 (m, 9H), 1.54-1.15 (m,
4H), 0.98-0.90 (m, 9H), 0.21-0.16 (m, 6H), 0.03 (s, 9H); LRMS-ES+ m/z (relative intensity) 828
(MH+, 80). E-324: 1H NMR (300 MHz, CDCl3) δ 7.94 (d, J = 7.5 Hz, 1H), 7.70 (d, J = 8.2 Hz,
2H), 7.48 (d, J = 7.5 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.32-7.20 (m, 2H), 4.18-4.12 (m, 2H),
3.75-3.46 (m, 8H), 3.09-3.01 (m, 1H), 2.55-2.50 (m, 1H), 2.47 (s, 3H), 2.03-1.92 (m, 1H), 1.67
(s, 9H), 1.29-1.25 (m, 1H), 1.01-0.91 (m, 3H), 0.76 (s, 9H), 0.03 (s, 9H), -0.08 (s, 3H), -0.18 (s,
3H); 13C NMR (90 MHz, CDCl3) δ 171.3, 170.6, 150.5, 143.7, 135.4, 133.0, 129.8, 129.4, 127.9,
124.5, 122.7, 118.9, 116.0, 115.0, 84.4, 63.2, 52.0, 42.8, 42.0, 30.7, 28.2, 27.4, 26.3, 25.8, 21.6,
17.9, 17.4, -1.5, -5.5, -5.6; LRMS-ES+ m/z (relative intensity) 828 (MH+, 75); HRMS-ES+
(C41H62N3O9Si2S) calcd 828.3745 (MH+), found 828.3746.
tert-Butyl 2-(1-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-3-
(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (325). To a stirred solution
of O-TBS-oxime 324 (500 mg, 0.604 mmol) and THF (9 mL) at 0 °C was added TBAF (1.0 M
in THF, 604 μL, 0.604 mmol). The resulting solution was stirred for 1 min and then
immediately diluted with NH4Cl(aq). The organic layer was separated and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to give a residue which was purified by flash column chromatography on silica gel (30%
EtOAc/hexanes) to afford free oxime 325 (401 mg, 93%) as a ~1:1 mixture of oxime isomers. Z-
Page 192
182
325: 1H NMR (400 MHz, CDCl3, mixture of BOC-rotamers) δ 8.03-7.95 (m, 1H), 7.69 (d, J =
8.0 Hz, 1H), 7.60-7.40 (m, 2H), 7.36-7.25 (m, 4H), 5.69-5.15 (m, 2H), 4.15-4.07 (m, 2H), 3.68-
3.58 (m, 5H), 3.37-3.35 (m, 1H), 2.95-2.90 (m, 1H), 2.65-2.63 (m, 1H), 2.47 (s, 3H), 1.67-1.54
(m, 10H), 1.43-1.21 (m, 2H), 1.01-0.87 (m, 3H), 0.04 (s, 9H); LRMS-ES+ m/z (relative
intensity) 714 (MH+, 50). E-325: 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.1 Hz, 1H), 7.69
(d, J = 8.1 Hz, 2H), 7.47 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.1 Hz, 2H), 7.30-7.21 (m, 2H), 4.17-
4.12 (m, 2H), 3.80-3.74 (m, 1H), 3.67-3.55 (m, 5H), 3.50-3.30 (m, 2H), 3.13-3.07 (m, 1H), 2.48-
2.45 (m, 4H), 2.35-2.30 (m, 1H), 2.12-2.06 (m, 1H), 1.73-1.57 (m, 10H), 1.05-0.95 (m, 3H), 0.03
(s, 9H); LRMS-ES+ m/z (relative intensity) 714 (MH+, 100); HRMS-ES+ (C35H48N3O9SiS) calcd
714.2881 (MH+), found 714.2867.
tert-Butyl 2-(2-Methoxy-2-oxo-1-(3-((pivaloyloxy)imino)-1-tosylpiperidin-4-yl)ethyl)-
3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (354). Oxime 325 (149
mg, 0.209 mmol) was dissolved in pivalic anhydride (1 mL) and pyridine (2 mL) and the
solution was stirred at rt for 36 h. The volatiles were removed in vacuo and the resulting crude
product was purified by flash chromatography on silica gel (15-25% EtOAc/hexanes) to give
oxime pivalate 354 (120 mg, 72%) as a complex mixture of oxime isomers and Boc-rotamers.
1H NMR (360 MHz, CDCl3) δ 8.08 (d, J = 7.8 Hz, 0.33H), 7.96-7.91 (m, 0.67H), 7.69-7.65 (m,
2H), 7.59 (d, J = 7.6 Hz, 0.66H), 7.47 (d, J = 7.0 Hz, 0.33H), 7.40-7.25 (m, 4H), 5.72-5.67 (m,
Page 193
183
0.5H), 5.47-5.45 (m, 0.5H), 5.10-5.03 (m, 1H), 4.22-4.02 (m, 2.5H), 3.80-3.57 (m, 6H), 3.40-
3.30 (m, 0.5H), 3.15-3.04 (m, 1H), 2.80-2.64 (m, 1H), 2.49 (s, 3H), 1.71-1.66 (m, 10H), 1.55-
1.53 (m, 1H), 1.38-1.35 (m, 9H), 1.01-0.97 (m, 3H), 0.05 (s, 9H).
General Procedure for the Conversion of Ketoxime Pivalates to Ketones. To a
solution of oxime pivalate 352 (0.10 mmol) in THF (1 mL) was added iron powder (55.8 mg, 1.0
mmol) followed by glacial AcOH (1 drop, cat.) and TMSCl (1 drop, cat.). After stirring at rt for
30 min, the reaction mixture was diluted with H2O (1 mL) and stirred for an additional 15 min.
The liquid phase was separated from the remaining Fe powder using a pipette and transferred to
a separatory funnel. The Fe powder was then washed with EtOAc (3 x 2 mL) and the washings
were added to the separatory funnel. The organic layer was separated and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to give a residue which was purified by flash column chromatography on silica gel eluting
with a mixture of ethyl acetate and hexanes. Known ketones were characterized by comparison
to their reported 1H NMR spectra (cf. Table 5).
Page 194
184
tert-Butyl 2-(2-Methoxy-2-oxo-1-(3-oxo-1-tosylpiperidin-4-yl)ethyl)-3-(2-oxo-2-(2-
(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (326). Following the above general
experimental procedure at a 0.03 M concentration, ketoxime pivalate 354 (30 mg, 0.038 mmol)
was converted to ketone 326 (22 mg, 84%). 1H NMR (300 MHz, CDCl3, mixture of Boc-
rotamers). δ 7.97-7.90 (m, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.69-7.64 (m, 1H), 7.38 (d, J = 8.4 Hz,
2H), 7.34-7.28 (m, 2H), 4.34-4.05 (m, 3H), 3.66-3.59 (m, 7H), 3.41-3.36 (m, 1H), 2.86 (td, J =
11.3, 3.9 Hz, 1H), 2.49 (s, 3H), 1.68-1.46 (m, 11H), 1.04-0.91 (m, 3H), 0.07-0.03 (m, 9H);
LRMS-ES+ m/z (relative intensity) 716 (M+NH4+, 100); HRMS-ES+ (C35H50N3O9SiS) calcd
716.3037 (M+NH4+), found 716.3013.
2-(5-(Methoxycarbonyl)-2-tosyl-2,3,4,6-tetrahydro-1H-pyrido[4',3':4,5]pyrrolo[1,2-
a]indol-6-yl)acetic Acid (356). To a stirred solution of TMSE-ester 326 (21.5 mg, 0.0308
mmol) in CH2Cl2 (2.5 mL) was added TFA (100 μL). The resulting solution was stirred for 1.5 h
at rt, an additional portion of TFA (500 μL) was added, and the solution was stirred for an
additional 3 h. The solvent was removed in vacuo to give a residue which was purified by flash
column chromatography on silica gel (40% EtOAc/hexanes + 1% AcOH) to afford tetracyclic
Page 195
185
pyrrole 356 (9.3 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.1 Hz, 2H) 7.54-7.52
(m, 1H), 7.39-7.31 (m, 3H), 7.20-7.17 (m, 2H), 4.60-4.55 (m, 2H), 3.84 (s, 3H), 3.78-3.75 (m,
1H). 3.66-3.59 (m, 1H), 3.48-3.38 (m, 2H), 2.96-2.95 (m, 2H), 2.74 (br s, 1H), 2.44-2.36 (m,
4H); LRMS-ES+ m/z (relative intensity) 481 (MH+, 100).
Benzyl 2-(1-(3-(((tert-Butyldimethylsilyl)oxy)imino)-1-tosylpiperidin-4-yl)-2-
methoxy-2-oxoethyl)-3-(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate
(357). To a –78 °C stirred solution of indole Z-320 (830 mg, 1.14 mmol) and THF (20 mL) was
added KHMDS (0.5 M in THF, 2.5 mL, 1.25 mmol). The resulting solution was stirred for 45
min and Cbz-Cl (212 μL, 1.48 mmol) was added. The solution was stirred for 12 h at –78 °C
and then quenched with NH4Cl(aq). The organic layer was separated and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to give a residue which was purified by flash column chromatography on silica gel (10-
15% EtOAc/hexanes) to yield N-Cbz-indole Z-357 (870 mg, 89%). An identical procedure was
followed using indole E-320 (820 mg, 1.13 mmol) to prepare N-Cbz-indole E-357 (860 mg,
89%). Z-357: 1H NMR (300 MHz, CDCl3, mixture of Cbz-rotamers) δ 8.14-8.02 (m, 1H), 7.68-
7.13 (m, 12H), 5.72-5.51 (m, 1.6H), 5.33-5.16 (m, 2.4H), 4.18-4.10 (m, 2H), 3.67-3.50 (m, 6H),
3.16-3.11 (m, 1H), 2.72-2.67 (m, 1H), 2.51-2.43 (m, 3H), 2.25-2.15 (m, 1H), 1.15-1.10, 1.04-
0.93 (m, 12H), 0.27-0.19 (m, 6H), 0.03 (s, 9H), (m, 1H). E-357: 1H NMR (300 MHz, CDCl3)
Page 196
186
δ 8.01-7.98 (m, 1H), 7.68 (d, J = 8.3 Hz, 2H), 7.49-7.46 (m, 3H), 7.42-7.33 (m, 5H), 7.26-7.16
(m, 2H), 5.41 (s, 2H), 4.17-4.07 (m, 2.5H), 3.80-3.71 (m, 1H), 3.65-3.60 (m, 3H), 3.54-3.37 (m,
4.5H), 3.03-2.95 (m, 1H), 2.50-2.38 (m, 4H), 2.01-1.90 (m, 1H), 0.99-0.87 (m, 3H), 0.77 (s, 9H),
0.02 (s, 9H), -0.03 (s, 3H), -0.17 (s, 3H). Mixture of E- and Z-357: LRMS-ES+ m/z (relative
intensity) 862 (MH+, 90); HRMS-ES+ (C44H60N3O9SSi2) calcd 862.3589 (MH+), found
862.3585. Slow evaporation of a solution of Z-357 in EtOAc provided crystals which were
analyzed by X-ray diffraction (Figure 9).141
Figure 10. ORTEP and Wireframe Structures of Z-357
Page 197
187
Benzyl 2-(1-(3-(Hydroxyimino)-1-tosylpiperidin-4-yl)-2-methoxy-2-oxoethyl)-3-(2-
oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (358). To a stirred solution of
O-TBS-oxime 357 (685 mg, 0.794 mmol) and THF (37 mL) was added AcOH (370 μL)
followed by TBAF (1.0 M in THF, 1.19 mL, 1.19 mmol). The resulting solution was stirred
overnight at rt and then diluted with NH4Cl(aq). The organic layer was separated and the aqueous
layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and
concentrated in vacuo to give a residue which was purified by flash column chromatography on
silica gel (30% EtOAc/hexanes) to afford free oxime 358 (590 mg, 99%) as a complex mixture
of oxime isomers and Cbz-rotamers. 1H NMR (300 MHz, CDCl3) δ 8.56-7.29 (m, 13H), 5.74-
4.91 (m, 2.7H), 4.23-4.13 (m, 2.4H), 3.70-3.50 (m, 5.7H), 3.25-3.10 (m, 1.4H), 2.52-2.45 (3.6H),
2.28-2.25 (m, 0.5H) , 1.95-1.85 (m, 0.5H), 1.60-1.40 (m, 2H), 1.33-1.17 (m, 1H),1.00-0.90 (m,
3H), 0.10-0.01 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 173.4, 172.2, 172.0, 170.9, 153.3, 151.8,
144.4, 144.3, 136.2, 135.0, 133.4, 132.7, 131.7, 130.3, 130.2, 129.9, 129.8, 129.4, 129.2, 129.03,
128.99, 128.8, 128.3, 128.2, 127.4, 126.0, 125.7, 123.9, 123.1, 120.4, 119.6, 119.5, 117.6, 116.3,
111.4, 108.6, 70.2, 69.3, 69.1, 64.2, 63.9, 63.7, 53.0, 52.6, 46.0, 45.4, 44.3, 44.1, 42.8, 42.4, 42.3,
40.9, 31.4, 30.9, 30.8, 30.4, 28.9, 27.8, 26.4, 26.1, 22.03, 22.00, 18.4, 17.8, 17.7, 1.5, -1.1, -2.5, -
3.1; LRMS-ES+ m/z (relative intensity) 748 (MH+, 75); HRMS-ES+ (C38H46N3O8SSi2) calcd
748.2724 (MH+), found 748.2690.
Page 198
188
Benzyl 2-(2-Methoxy-2-oxo-1-(3-((pivaloyloxy)imino)-1-tosylpiperidin-4-yl)ethyl)-3-
(2-oxo-2-(2-(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (359). The oxime 358
(550 mg, 0.735 mmol) was dissolved in Piv2O (6 mL) and pyridine (12 mL) and the mixture was
warmed to 60 °C. After stirring the mixture for 12 h at that temperature, the solvent was
removed in vacuo using PhMe to azeotropically remove the excess Piv2O. The resulting crude
product was purified by flash chromatography on silica gel (15-30% EtOAc/hexanes) to give
oxime-pivalate 359 (527 mg, 94%) as a complex mixture of oxime isomers and Cbz-rotamers.
1H NMR (300 MHz, CDCl3) δ 9.01 (s, 0.3H), 8.13-7.95 (m, 0.5H), 7.67-7.11 (m, 12.2H), 5.69-
4.89 (m, 2H), 4.19-4.14 (m, 2H), 3.77-3.50 (m, 6H), 3.35-3.10 (m, 2H), 2.83-2.70 (m, 1H), 2.52-
2.43 (m, 3H), 2.30-2.20 (m, 0.5H), 1.85-1.50 (m, 1.5H), 1.38-1.26 (m, 11H), 1.03-0.97 (m, 2H),
0.06-0.00 (m, 9H). 13C NMR (75 MHz, CDCl3) δ 174.6, 174.1, 171.6, 171.3, 170.8, 161.2,
160.8, 152.1, 151.8, 144.7, 144.6, 135.9, 135.8, 135.3, 135.1, 133.2, 132.5, 132.2, 130.4, 130.2,
129.7, 129.5, 129.4, 129.2, 129.113, 129.08, 128.8, 128.4, 128.2, 127.7, 125.7, 125.5, 123.9,
123.5, 119.8, 119.3, 118.2, 116.5, 116.5, 116.2, 69.7, 69.1, 64.0, 63.9, 52.8, 52.7, 45.8, 44.6,
43.5, 42.2, 40.6, 39.2, 38.9, 31.3, 31.2, 29.0, 27.7, 27.4, 22.0, 17.8, 17.6, 1.5, -1.1; LRMS-ES+
m/z (relative intensity) 849 (M+NH4+, 25); HRMS-ES+ (C43H57N4O10SSi) calcd 849.3565
(M+NH4+), found 849.3587.
Page 199
189
Benzyl 2-(2-Methoxy-2-oxo-1-(3-oxo-1-tosylpiperidin-4-yl)ethyl)-3-(2-oxo-2-(2-
(trimethylsilyl)ethoxy)ethyl)-1H-indole-1-carboxylate (360). To a solution of oxime pivalate
359 (81.0 mg, 0.097 mmol) in THF (2 mL) at 0 °C was added iron powder (54 mg, 0.97 mmol)
followed by AcOH (1 drop, cat.) and TMSCl (1 drop, cat.). After stirring for 30 min, the
reaction mixture was diluted with H2O (2 mL) and stirred for an additional 15 min. The liquid
phase was separated from the remaining Fe powder using a pipette and transferred to a
separatory funnel. The Fe powder was then washed with EtOAc and the washings were added to
the separatory funnel. The organic layer was separated and the aqueous layer was extracted with
EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give
a residue which was purified by flash column chromatography on silica gel (25%
EtOAc/hexanes) to afford ketone 360 (64.4 mg, 90%). 1H NMR (300 MHz, CDCl3) δ 8.09 (d, J
= 7.6 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.58-7.20 (m, 10H), 5.40 (ABq, J = 106.0, 11.8 Hz, 2H),
4.93 (d, J = 6.6 Hz, 1H), 4.18-4.11 (m, 2H), 4.02 (d, J = 13.8 Hz, 1H), 3.71-3.64 (m, 3H), 3.50
(s, 3H), 3.34 (q, J = 9.9 Hz, 1H), 3.21 (d, J = 14.1 Hz, 1H), 2.51-2.43 (m, 4H), 1.55-1.44 (m,
1H), 1.30-1.22 (m, 1H), 1.00-0.94 (m, 2H), 0.04 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 201.9,
171.6, 170.6, 151.9, 144.7, 135.8, 135.0, 132.5, 132.1, 130.3, 129.7, 129.4, 129.2, 128.8, 128.43,
128.35, 127.7, 125.8, 124.0, 119.7, 117.8, 116.3, 69.2, 64.2, 56.4, 52.8, 49.1, 45.6, 40.3, 31.1,
27.9, 27.7, 27.5, 22.0, 17.7, -1.1; LRMS-ES+ m/z (relative intensity) 750 (M+NH4+, 75); HRMS-
ES+ (C38H48N3O9SSi) calcd 750.2881 (M+NH4+), found 750.2878.
Page 200
190
2-(1-((Benzyloxy)carbonyl)-2-(2-methoxy-2-oxo-1-(3-oxo-1-tosylpiperidin-4-
yl)ethyl)-1H-indol-3-yl)acetic Acid (361). To a stirred solution of TMSE-ester 360 (332 mg,
0.453 mmol) in CH2Cl2 (12 mL) was added TFA (3 mL). The resulting solution was stirred for
16 h at rt. The solvent was removed in vacuo to give a residue which was purified by flash
column chromatography on silica gel (40% EtOAc/hexanes + 1% AcOH) to yield carboxylic
acid 361 (274 mg, 96%). 1H NMR (300 MHz, CDCl3) δ 8.10 (d, J = 7.9 Hz, 1H), 7.80 (d, J =
8.3 Hz, 2H), 7.66-7.17 (m, 10H), 5.41 (ABq, J = 101.7, 11.9 Hz, 2H), 4.91 (d, J = 6.8 Hz, 1H),
4.03 (d, J = 13.8 Hz, 1H), 3.70 (br s, 2H), 3.60-3.49 (m, 4), 3.38 (p, J = 7.2 Hz, 1H), 3.22 (d, J =
13.7 Hz, 1H), 2.50-2.45 (m, 4H), 1.45-1.27 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 202.1, 177.7,
171.6, 151.9, 144.7, 135.7, 134.9, 132.6, 132.4, 130.3, 129.6, 129.5, 129.2, 128.3, 125.9, 124.1,
119.5, 117.0, 116.3, 69.3, 56.3, 52.9, 49.1, 45.5, 40.5, 30.4, 27.9, 22.0, 21.2; LRMS-ES+ m/z
(relative intensity) 633 (MH+, 100); HRMS-ES+ (C33H36N3O9S) calcd 650.2172 (M+NH4+),
found 650.2142.
β-Lactones 362 and 363. To a solution of 2-bromo-N-propylpyridinium triflate68b (196
mg, 0.503 mmol), PPY (76 mg, 0.503 mmol), and CH2Cl2 (9 mL) was added DIPEA (118 μL,
Page 201
191
0.670 mmol). To this mixture, a solution of keto-acid 361 (212 mg, 0.335 mmol) in CH2Cl2 (4
mL) was added via syringe-pump over 1 h. The resulting solution was stirred for 20 h and then
concentrated in vacuo to give a residue, which was purified by flash column chromatography on
silica gel (40% EtOAc/hexanes) to afford β-lactones 362 and 363 (190 mg, 92%) as an ~8:1
mixture of inseparable diastereomers. FTIR (film) 1835 cm-1; 1H NMR (400 MHz, CDCl3, ~8:1
mixture of diastereomers (only the major diastereomer 362 peaks are reported)) δ 8.05 (d, J = 5.5
Hz, 1H), 7.68 (d, J = 6.1 Hz, 2H), 7.62 (d, J = 5.9 Hz, 1H), 7.49-7.30 (m, 9H), 5.48 (d, J = 9.0
Hz, 1H), 5.34 (d, J = 9.1 Hz, 1H), 4.78 (s, 1H), 4.43 (d, J = 4.4 Hz, 1H), 3.89 (d, J = 8.9 Hz, 1H),
3.52-3.49 (m, 4H), 2.98 (d, J = 8.9 Hz, 1H), 2.57 (q, J = 5.5 Hz, 1H), 2.51-2.43 (m, 4H) 13C
NMR (75 MHz, CDCl3, ~8:1 mixture of diastereomers (only the major diastereomer 362 peaks
are reported) δ 171.0, 165.8, 152.2, 144.9, 137.0, 134.9, 132.7, 130.4, 130.3, 129.5, 129.22,
129.16, 128.2, 127.2, 126.1, 124.2, 119.1, 115.9, 110.9, 75.9, 69.7, 55.0, 53.5, 52.5, 51.3, 45.2,
43.6, 38.8, 25.6, 25.5, 22.0; LRMS-ES+ m/z (relative intensity) 615 (MH+, 100). HRMS-ES+
(C33H34N3O8S) calcd 632.2067 (M+NH4+), found 632.2053.
1,3-Diols 364 and 365. To a 0 °C solution of β-lactones 362 and 363 (62.8 mg, 0.102
mmol) was added DIBAL-H (1.0 M in PhMe, 511 μL, 0.511 mmol). The resulting solution was
stirred for 1.5 h, then poured into a stirring solution of NH4Cl(aq) and the mixture was stirred for
an additional 2 h. The organic layer was separated and the aqueous layer was extracted with
Page 202
192
EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give
a residue which was purified by preparative thin layer chromatography on silica gel (50%
EtOAc/hexanes) to afford major diol 364 (41.8 mg, 66%) and minor diol 365 (5.6 mg, 9%).
Major 1,3-diol 364: 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 7.8 Hz, 1H), 7.67-7.64 (m, 3H),
7.49-7.47 (m, 2H), 7.43-7.28 (m, 7H), 5.47 (d, J = 12.0 Hz, 1H), 5.29 (d, J = 12.1 Hz, 1H), 4.80-
4.75 (m, 1H), 4.68 (d, J = 4.0 Hz, 1H), 4.50-4.45 (m, 1H), 3.77 (br s, 1H), 3.65 (s, 1H), 3.50 (s,
3H), 2.47 (s, 3H), 2.30-2.22 (m, 2H), 2.15-2.10 (m, 1H), 1.99 (br s, 1H), 1.67 (s, 1H), 1.60-1.54
(m, 1H), 1.30-1.26 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 172.9, 152.0, 144.3, 137.1, 135.1,
133.3, 130.3, 130.2, 129.2, 129.1, 128.5, 128.1, 128.0, 125.2, 123.8, 119.4, 116.4, 114.0, 73.1,
69.3, 60.7, 54.3, 52.1, 46.1, 44.6, 43.6, 36.7, 24.9, 22.0; LRMS-ES+ m/z (relative intensity) 619
(MH+, 100); HRMS-ES+ (C33H35N2O8S) calcd 619.2114 (MH+), found 619.2150. Minor 1,3-
diol 365: 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 7.9 Hz, 1H), 7.67-7.55 (m, 3H), 7.48-7.28
(m, 9H), 5.43 (q, J = 8.4 Hz, 2H), 4.65-4.53 (m, 2H), 4.33-4.30 (m, 1H), 4.10 (br s, 1H), 3.54 (s,
2H), 3.51 (s, 3H), 2.46-2.43 (m, 4H), 2.42-2.38 (m, 2H), 2.36-2.26 (m, 1H), 1.81-1.76 (m, 1H),
1.70-1.65 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 173.6, 151.9, 144.3, 137.0, 135.1, 133.3,
130.3, 129.30, 129.25, 129.2, 128.3, 128.2, 128.0, 125.4, 123.9, 119.5, 116.6, 72.5, 71.1, 69.3,
61.3, 57.5, 54.2, 52.7, 45.4, 25.7, 22.0; LRMS-ES+ m/z (relative intensity) 619 (MH+, 100);
HRMS-ES+ (C33H35N2O8S) calcd 619.2114 (MH+), found 619.2138.
Page 203
193
Bis-tert-Butylsilanol 369. To a solution of 1,3-diol 364 (16 mg, 0.026 mmol) and 2,6-
lutidine (9 μL, 0.078 mmol) in CH2Cl2 (1 mL) was added di-t-butylsilyl bis(triflate) (11 μL,
0.034 mmol). The resulting mixture was stirred for 1 h at rt and then washed with 1 M HCl. The
organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined
organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was
purified by flash chromatography on silica gel (25% EtOAc/hexanes) to yield silanol 369 (21
mg, 100%). 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 7.4 Hz, 1H), 7.70 (d, J = 8.2 Hz, 2H),
7.45-7.31 (m, 10H), 5.42 (s, 1H), 5.39 (ABq, J = 86.1, 12.2 Hz, 2H), 4.96 (dd, J = 11.5, 3.0 Hz,
1H), 4.70 (dd, J = 5.7, 2.2 Hz, 1H), 4.57 (d, J = 10.6 Hz, 1H), 4.18-4.07 (m, 2H), 3.78 (d, J =
11.3 Hz, 1H), 3.66 (br s, 1H), 3.50 (s, 3H), 2.52 (s, 3H), 2.26 (d, J = 10.1 Hz, 1H), 2.12-2.05 (m,
2H), 1.65-1.54 (m, 2H), 1.32-1.22 (m, 1H), 1.00 (s, 9H), 0.44 (s, 9H); LRMS-ES+ m/z (relative
intensity) 777 (MH+, 100).
Cyclic Acetonide 370. To a stirred solution of 1,3-diol 364 (10.0 mg, 0.016 mmol), 4 Å
molecular sieves (10 mg), and acetone (2 mL) was added Amberlyst-15 (10 mg). The resulting
Page 204
194
mixture was stirred for 15 h and additional Amberlyst-15 (50 mg) was added. After stirring for
an additional 18 h, the reaction mixture was filtered through a pad of Celite and concentrated in
vacuo to give a residue. This material was purified by preparative thin layer chromatography on
silica gel (25% EtOAc/hexanes) to give acetonide 370 (1.6 mg, 15%) and recovered 1,3-diol 364
(8.0 mg, 80%). 1H NMR (300 MHz, CDCl3) δ 8.12 (d, J = 7.2 Hz, 1H), 7.70 (d, J = 8.2 Hz, 2H),
7.50-7.38 (m, 8H), 7.32-7.27 (m, 2H), 5.40 (ABq, J = 60.3, 12.3 Hz, 2H), 4.61-4.51 (m, 3H),
4.44-4.39 (m, 1H), 3.79 (d, J = 11.5 Hz, 1H), 3.51-3.48 (m, 4H), 2.49 (s, 3H), 2.25-2.22 (m, 2H),
2.10-2.02 (m, 1H), 1.54 (s, 3H), 1.28-1.17 (m, 2H), 1.15 (s, 3H); LRMS-ES+ m/z (relative
intensity) 659 (MH+, 60).
Cyclic Carbonate 371. To a stirred solution of 1,3-diol 364 (11.5 mg, 0.0186 mmol) and
pyridine (22 μL, 0.26 mmol) in CH2Cl2 (1 mL) at 0 °C was added triphosgene (11 μL, 0.017
mmol). The resulting mixture was stirred for 18 h gradually warming to rt and then quenched
with 1 M HCl. The organic layer was separated and the aqueous layer was extracted with
EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give
a residue which was purified by flash chromatography on silica gel (30% EtOAc/hexanes) to
afford carbonate 371 (10.9 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 7.3 Hz, 1H),
7.68 (d, J = 8.3 Hz, 2H), 7.53-7.32 (m, 10H), 5.40 (ABq, J = 39.0, 12.1 Hz, 2H), 5.09 (d, J =
12.0 Hz, 1H), 4.91 (dd, J = 12.0, 2.9 Hz, 1H), 4.63 (dd, J = 6.0, 2.0 Hz, 1H), 4.28 (dd, J = 11.9,
Page 205
195
1.4 Hz, 1H), 4.01 (br s, 1H), 3.88-3.84 (m, 1H), 3.50 (s, 3H), 2.49 (s, 3H), 2.46-2.40 (m, 1H),
2.32 (td, J = 12.2, 3.1 Hz, 1H), 1.71-1.59 (m, 2H), 1.42-1.37 (m, 1H); LRMS-ES+ m/z (relative
intensity) 662 (M+NH4+, 100).
6-Benzyl 5-Methyl 11a-Hydroxy-11-(hydroxymethyl)-2-tosyl-3,4,4a,5,11,11a-
hexahydro-1H-pyrido[4,3-b]carbazole-5,6(2H)-dicarboxylate (374). To a solution of 1,3-diol
364 (12.0 mg, 0.019 mmol) in MeCH(OMe)2 (1 mL) was added a catalytic amount of p-
TsOH.H2O (<1 mg) and the resulting mixture was placed in a preheated oil bath at 80 °C. After
stirring for 30 min, the solution was concentrated in vacuo to give a residue which was purified
by column chromatography on silica gel (30% EtOAc/hexanes) to yield the acetal 374 (11.6 mg,
93%). 1H NMR (300 MHz, CDCl3) δ 8.12 (br d, J = 6.1 Hz, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.52-
7.48 (m, 3H), 7.46-7.38 (5H), 7.31-7.27 (m, 2H), 5.50 (d, J = 12.1 Hz, 1H), 5.34 (d, J = 10.3 Hz,
1H), 5.07 (q, J = 5.0 Hz, 1H), 4.78-4.66 (m, 3H), 4.36 (dd, J = 12.3, 3.0 Hz, 1H), 3.84-3.80 (m,
1H), 3.52 (s, 3H), 3.40 (br s, 1H), 2.49 (s, 3H), 2.27 (td, J = 12.2, 2.9 Hz, 1H), 2.19-2.06 (m,
2H), 1.70-1.55 (m, 1H), 1.31-1.23 (m, 1H), 1.20 (d, J = 4.9 Hz, 3H); 13C NMR (75 MHz,
CDCl3) δ 172.9, 152.3, 144.5, 135.4, 133.5, 131.5, 130.4, 129.3, 129.13, 129.06, 128.3, 127.9,
124.8, 123.4, 119.5, 116.5, 116.3, 93.3, 72.5, 69.1, 65.3, 53.9, 52.1, 50.4, 46.2, 44.5, 43.3, 29.7,
23.7, 22.0, 21.6; LRMS-ES+ m/z (relative intensity) 645 (MH+, 50).
Page 206
196
Methyl Thiomethyl Ether 380. Benzyol peroxide (278 mg, 1.11 mmol) was added
portionwise over approximately 20 min to a 0 °C solution of 1,3-diol 364 (86 mg, 0.14 mmol)
and dimethyl sulfide (163 μL, 2.22 mmol) in MeCN (6 mL). The resulting solution was stirred
for 13 h while maintaining a temperature of 0 to 5 °C. The reaction mixture was diluted with 1:1
Na2SO3(aq) and NaHCO3(aq), the organic layer was separated, and the aqueous layer was extracted
with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to
give a residue which was purified by flash column chromatography on silica gel (gradient 25-
40% EtOAc/hexanes) to afford sulfide 380 (64 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 8.15-
8.09 (m, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 6.7 Hz, 2H), 7.51-7.48 (m, 2H), 7.43-7.37 (m,
5H), 7.30-7.26 (m, 2H), 5.49 (d, J = 11.7 Hz, 1H), 5.33-5.28 (m, 1H), 4.85 (d, J = 10.6 Hz, 1H),
4.70-4.69 (m, 1H), 4.58 (q, J = 9.5 Hz, 2H), 4.47 (s, 1H), 4.24-4.14 (m, 2H), 3.82-3.80 (m, 2H),
3.51 (s, 3H), 2.48 (s, 3H), 2.27-2.21 (m, 1H), 2.16-2.13 (m, 1H), 1.74 (s, 3H); LRMS-ES+ m/z
(relative intensity) 679 (MH+, 75); HRMS-ES+ (C35H39N2O8S2) calcd 679.2148 (MH+), found
679.2134.
Page 207
197
TBS Ether 381. A sealed tube was charged with a solution of alcohol 380 (50.9 mg,
0.0750 mmol) and 2,6-lutidine (438 μL, 3.75 mmol) in CH2Cl2 (5 mL). TBSOTf (357 μL, 1.87
mmol) was added to the solution and the tube was sealed and heated at 40 °C with stirring for 24
h. After cooling to the mixture to rt, additional 2,6-lutidine (438 μL, 3.75 mmol) and TBSOTf
(357 μL, 1.87 mmol) were added and the mixture heated for 24 h at 40 °C. After cooling to rt,
the reaction mixture was washed with 1 M HCl, the organic layer was separated, and the aqueous
layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and
concentrated in vacuo to give a residue which was purified by flash chromatography on silica gel
(5-20% EtOAc/hexanes) to afford TBS ether 381 (43.8 mg, 74%). 1H NMR (400 MHz, CDCl3)
δ 8.15 (d, J = 6.7 Hz, 1H), 7.90 (d, J = 6.9 Hz, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.45-7.43 (m, 2H),
7.41-7.34 (m, 5H), 7.32-7.37 (m, 2H), 5.51 (d, J = 12.1 Hz, 1H), 5.20 (d, J = 11.3 Hz, 1H), 4.84
(s, 2H), 4.54 (d, J = 3.5 Hz, 1H), 4.49 (d, J = 10.3 Hz, 1H), 4.10 (dd, J = 10.5, 4.2 Hz, 1H), 4.02
(dd, J = 10.5, 6.0 Hz, 1H), 3.77-3.72 (m, 2H), 3.52 (s, 3H), 2.48 (s, 3H), 2.33 (s, 3H), 2.19-2.11
(m, 2H), 2.09-2.03 (m, 1H), 1.53 (qd, J = 13.3, 4.5 Hz, 1H), 1.23-1.20 (m, 1H), 0.78 (s, 9H),
0.14 (s, 3H), 0.10 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.9, 151.9, 144.3, 137.2, 135.2,
133.0, 130.1, 129.1, 129.04, 129.03, 128.8, 128.3, 124.8, 123.3, 120.9, 118.4, 117.9, 115.7, 75.8,
75.2, 69.0, 67.8, 55.3, 52.1, 46.5, 45.9, 43.7, 37.4, 26.2, 24.8, 22.0, 19.0, 14.7, -1.1, -1.4; LRMS-
ES+ m/z (relative intensity) 810 (MH+, 20). Slow evaporation of a solution of TBS ether 381 in
1:1 MeOH/CHCl3 provide crystals which were analyzed by X-ray diffraction.141
Page 208
198
Chloromethyl Ether 382. To a stirred solution of sulfide 381 (13.9 mg, 0.0175 mmol)
in CH2Cl2 (1.5 mL) at 0 °C was added sulfuryl chloride (0.1 M in CH2Cl2, 19 μL, 0.019 mmol)
and the solution was stirred for 30 min until TLC analysis indicated complete consumption of the
starting material. The solution was then concentrated in vacuo to give essentially pure
chloromethyl ether 382 which was used immediately in subsequent reactions. 1H NMR (400
MHz, C6D6) δ 7.89 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.25-7.10 (m, 8H), 6.91 (d, J =
8.2 Hz, 2H), 5.73 (d, J = 5.7 Hz, 1H), 5.48 (d, J = 5.7 Hz, 1H), 5.32 (d, J = 12.3 Hz, 1H), 4.96 (d,
J = 12.1 Hz, 1H), 4.88-4.85 (m, 1H), 4.71 (d, J = 10.6 Hz, 2H), 4.55 (dd, J = 11.0, 3.6 Hz, 1H),
4.40 (dd, J = 11.0, 7.2 Hz, 1H), 4.03 (p, J = 3.2 Hz, 1H), 3.71-3.64 (m, 1H), 2.28-2.24 (m, 2H),
2.00- 1.97 (m, 4H), 1.91-1.85 (m, 2H), 0.85 (s, 9H), 0.24 (s, 3H), 0.12 (s, 3H).
Cyclic Acetal Indole 389. To a solution of Cbz-carbamate 374 (11.6 mg, 0.0180 mmol)
in MeOH (2 mL) and EtOAc (2 mL) under an argon atmosphere was added Pd/C (10 wt%, 4.1
mg, 0.0039 mmol). The resulting mixture was evacuated and backfilled three times with H2
from a balloon and the resulting suspension was stirred for 2 h. The reaction mixture was then
filtered through a pad of Celite with EtOAc and the filtrate was concentrated in vacuo to give a
Page 209
199
residue. This material was purified by flash column chromatography on silica gel (30%
EtOAc/hexanes) to give NH indole 389 (9.1 mg, 99%). 1H NMR (300 MHz, CDCl3) δ 9.13 (s,
1H), 7.72 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 7.9 Hz, 1H), 7.41-7.38 (m, 3H), 7.20-7.12 (m, 2H),
5.07 (q, J = 5.0 Hz, 1H), 4.82 (d, J = 12.2 Hz, 1H), 4.68 (dd, J = 11.3, 1.6 Hz, 1H), 4.59 (dd, J =
4.5, 2.2 Hz, 1H), 4.34 (dd, J = 12.2, 3.2 Hz, 1H), 3.82-3.77 (m, 4H), 3.39 (br s, 1H), 2.49 (s, 3H),
2.36 (td, J = 12.0, 3.2 Hz, 1H), 2.25-2.18 (m, 2H), 1.60-1.55 (m, 2H), 1.20 (d, J = 4.9 Hz, 3H);
LRMS-ES+ m/z (relative intensity) 511 (MH+, 100).
Hydroxymethyl Compound 393. To a stirred suspension of paraformaldehyde (4.3 mg,
0.140 mmol) and ester 374 (6.0 mg, 0.009 mmol) in DMF (0.5 mL) was added NaH (60%
dispersion on mineral oil, 1.9 mg, 0.005 mmol) and the resulting mixture was stirred for 2 h at rt.
The reaction mixture was diluted with brine and EtOAc. The organic layer was separated and
washed with water and then brine. The organic layer was dried over MgSO4 and concentrated in
vacuo to give a residue which was purified by flash column chromatography on silica gel (75%
EtOAc/hexanes) to give β-hydroxy ester 394 (4.7 mg, 93%). 1H NMR (400 MHz, CDCl3) δ 8.91
(s, 1H), 7.70 (d, J = 6.1 Hz, 2H), 7.57 (d, J = 6.0 Hz, 1H), 7.40-7.36 (m, 3H), 7.20 (t, J = 5.5 Hz,
1H), 7.12 (t, J = 5.8 Hz, 1H), 5.03 (q, J = 3.7 Hz, 1H), 4.83 (d, J = 9.0 Hz, 1H), 4.67 (d, J = 8.1
Hz, 1H), 4.37 (dd, J = 9.1, 2.1 Hz, 1H), 3.86-3.78 (m, 2H), 3.68 (s, 3H), 3.39-3.37 (m, 1H), 2.49
(s, 3H), 2.39-2.26 (m, 2H), 2.21-2.16 (m, 2H), 1.54-1.41 (m, 2H), 1.10 (d, J = 3.7 Hz, 3H); 13C
Page 210
200
NMR (75 MHz, CDCl3) δ 175.9, 144.5, 137.3, 133.3, 132.8, 130.3, 128.0, 126.0, 122.3, 119.7,
119.4, 111.8, 108.2, 93.1, 73.6, 68.7, 65.8, 52.8, 50.4, 49.5, 47.7, 46.2, 30.0, 22.7, 22.0, 21.3;
LRMS-ES+ m/z (relative intensity) 541 (MH+, 30); HRMS-ES+ (C28H36N3O7S) calcd 558.2274
(M+NH4+), found 558.2265.
Chloromethyl(dimethyl)silane 398. To a solution of crude alcohol 380 (4.8 mg, 0.0071
mmol) and 2,6-lutidine (38 mg, 0.35 mmol) in CH2Cl2 (0.5 mL) was added
chloromethyl(dimethyl)chlorosilane (35 μL, 0.18 mmol). The resulting mixture was refluxed for
1 h, cooled to rt, and then washed with 1 M HCl. The organic layer was separated and the
aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4
and concentrated in vacuo to give a residue which was purified by flash chromatography on
silica gel (5-25% EtOAc/hexanes) to afford chloromethylsilane 398 (5.0 mg, 90%). 1H NMR
(400 MHz, CDCl3) δ 8.12 (d, J = 7.4 Hz, 1H), 7.84-7.82 (m, 1H), 7.69 (d, J = 8.2 Hz, 2H), 7.48-
7.46 (m, 2H), 7.42-7.33 (m, 5H), 7.31-7.25 (m, 2H), 5.50 (d, J = 12.1 Hz, 1H), 5.26 (d, J = 12.0
Hz, 1H), 4.83 (ABq, J = 12.7, 11.6 Hz, 2H), 4.53 (d, J = 10.2 Hz, 1H), 4.48 (dd, J = 5.5, 2.3 Hz,
1H), 4.16 (dd, J = 3.9, 10.4, 1H), 3.89 (dd, J = 10.4, 7.0 Hz, 1H), 3.81-3.78 (m, 1H), 3.73 (br d, J
= 10.6 Hz, 1H), 3.51 (s, 3H), 2.74 (s, 2H), 2.48 (s, 3H), 2.31 (s, 3H), 2.23-2.13 (m, 2H), 2.08-
2.04 (m, 1H), 1.54 (qd, J = 13.3, 4.3 Hz, 1H), 1.27-1.23 (m, 1H), 0.30 (s, 3H), 0.28 (s, 3H); 13C
NMR (100 MHz, CDCl3) δ 172.7, 144.4, 135.3, 132.9, 130.1, 129.12, 129.09, 129.0, 128.8,
Page 211
201
128.2, 124.9, 123.3, 120.7, 117.2, 115.9, 75.8, 69.1, 67.4, 55.4, 54.2, 52.1, 46.4, 46.2, 43.6, 36.7,
32.5, 31.2, 29.8, 29.7, 24.7, 22.0, 14.8, -0.01, -0.04; LRMS-ES+ m/z (relative intensity) 802
(M+NH4+, 20).
Iodomethyl(dimethyl)silane 399. A sealed tube was charged with chloride 388 (9.5 mg,
0.0121 mmol), acetone (1 mL), and then NaI (45 mg, 0.30 mmol). The vessel was sealed and the
resulting mixture was heated at reflux and stirred for 14 h. After cooling to rt, the reaction
mixture was diluted with ether and the organic phase was washed with water followed by
Na2S2O3(aq). The aqueous layers were extracted with ether and the combined organic layers were
dried over MgSO4 and concentrated in vacuo to give a residue. This material was purified by
flash chromatography on silica gel (30% EtOAc/hexanes) to give iodide 399 (7.8 mg, 74%). 1H
NMR (300 MHz, CDCl3) δ 8.13 (br d, J = 7.2, 1H), 7.87-7.84 (m, 1H), 7.70 (d, J = 8.1 Hz, 2H),
7.48-7.37 (m, 7H), 7.32-7.23 (m, 2H), 5.51 (d, J = 12.1 Hz, 1H), 5.26 (br d, J = 10.8 Hz, 1H), 4.8
(s, 2H), 4.56-4.53 (m, 2H), 4.19-4.11 (m, 1H), 3.92 (dd, J = 10.3, 6.9 Hz, 1H), 3.83-3.72 (m,
2H), 3.52 (s, 3H), 2.50 (s, 3H), 2.32 (s, 3H), 2.25-2.00 (m, 3H), 1.95 (s, 2H), 1.58 (qd, J = 13.2,
4.8 Hz, 1H), 1.29-1.23 (m, 1H), 0.36 (s, 3H), 0.35 (s, 3H); LRMS-ES+ m/z (relative intensity)
894 (M+NH4+, 30).
Page 212
202
TBS Ether 404. To a solution of 1,3-diol 364 (39.7 mg, 0.0642 mmol) anf 2,6-lutidine
(75 μL, 0.64 mmol) in CH2Cl2 (3 mL) at 0 °C was added TBSOTf (61 μL, 0.32 mmol). The
resulting mixture was stirred for 1 h and then washed with 1 M HCl. The organic layer was
separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were
dried over MgSO4 and concentrated in vacuo to give a residue which was purified by flash
chromatography on silica gel (30% EtOAc/hexanes) to afford O-TBS-ether 404 (45 mg, 96%).
1H NMR (300 MHz, CDCl3) δ 8.14 (d, J = 9.2 Hz, 1H), 7.70-7.67 (m, 2H), 7.59-7.56 (m, 1H),
7.48-7.34 (m, 7H), 7.30-7.25 (m, 2H), 5.50 (d, J = 7.8 Hz, 1H), 5.29 (d, J = 12.2 Hz, 1H), 4.73-
4.69 (m, 2H), 4.51 (dd, J = 11.2, 1.7 Hz, 1H), 4.15 (d, J = 10.2 Hz, 1H), 3.77 (br d, J = 11.6 Hz,
1H), 3.66-3.64 (m, 1H), 3.50 (s, 3H), 2.47 (s, 3H), 2.22-2.17 (m, 2H), 2.10-2.03 (m, 1H), 1.63-
1.53 (m, 2H), 0.60 (s, 9H), 0.04-0.03 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 173.1, 152.0, 144.3,
135.4, 130.2, 129.1, 129.0, 128.9, 128.6, 128.0, 124.7, 123.1, 119.9, 116.1, 115.6, 72.8, 69.0,
61.5, 54.7, 52.0, 46.2, 45.1, 43.5, 36.1, 30.1, 26.1, 25.7, 24.7, 22.0, 18.0, 1.4, -5.2, -5.7; LRMS-
ES+ m/z (relative intensity) 733 (MH+, 100); HRMS-ES+ (C39H49N2O8SSi) calcd 733.2979
(MH+), found 733.2961.
Page 213
203
Chloromethyl(dimethyl)silane 406. To a solution of N-Cbz-indole 404 (45.0 mg, 0.061
mmol) in MeOH (1.5 mL) and EtOAc (1.5 mL) under an argon atmosphere was added Pd/C (10
wt%, 6.5 mg, 0.006 mmol). The resulting mixture was evacuated and backfilled three times with
H2 from a balloon and the resulting suspension was stirred for 2.5 h. The reaction mixture was
then filtered through a pad of Celite with EtOAc and the filtrate was concentrated in vacuo to
give NH indole 405, which was used in the next step without further purification. 1H NMR (400
MHz, CDCl3) δ 9.14 (s, 1H), 7.69 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 7.8 Hz, 1H), 7.40-7.33 (m,
3H), 7.18 (t, J = 6.2 Hz, 1H), 7.13 (t, J = 8.1 Hz, 1H), 5.70 (s, 1H), 4.64-4.52 (m, 3H), 4.19 (dd,
J = 11.4, 1.7 Hz, 1H), 3.80 (s, 3H), 3.77-3.68 (m, 1H), 3.69-3.67 (m, 1H), 2.48 (s, 3H), 2.31-2.18
(m, 3H), 1.54-1.41 (m, 2H), 0.67 (s, 9H), 0.09 (s, 3H), 0.07 (s, 3H); LRMS-ES+ m/z (relative
intensity) 599 (MH+, 75); HRMS-ES+ (C31H43N2O6SSi) calcd 599.2611 (MH+), found 599.2585.
To a solution of crude alcohol 405 and 2,6-lutidine (270 μL, 1.23 mmol) in CH2Cl2 (2.5
mL) was added chloromethyl(dimethyl)silyl triflate (289 μL, 2.47 mmol). The resulting mixture
was stirred for 1.5 h at rt and then washed with 1 M HCl. The organic layer was separated and
the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over
MgSO4 and concentrated in vacuo to afford a residue, which was purified by flash
chromatography on silica gel (20% EtOAc/hexanes) to give chlormethyl(dimethyl)silane 496
(32.9 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 7.67-7.62 (m, 3H), 7.40-7.33 (m,
3H), 7.20-7.12 (m, 2H), 4.73 (dd, J = 10.1, 1.5 Hz, 1H), 4.45-4.39 (m, 2H), 3.89 (t, J = 10.1 Hz,
Page 214
204
1H), 3.82 (s, 3H), 3.78-3.70 (m, 2H), 2.77 (ABq, J = 13.8, 5.0 Hz, 2H), 2.48 (s, 3H), 2.26-2.17
(m, 3H), 1.51-1.42 (m, 1H), 1.37-1.26 (m, 1H). 1.04 (s, 9H), 0.32-0.28 (m, 12H); 13C NMR (75
MHz, CDCl3) δ 173.4, 144.1, 136.8, 133.4, 130.1, 128.7, 128.1, 127.0, 121.9, 119.81, 119.78,
111.8, 108.2, 76.2, 64.0, 60.8, 56.8, 52.9, 47.7, 46.4, 39.8, 31.5, 26.8, 24.2, 22.0, 21.5, 19.2, 14.6,
0.2, -0.1, -4.6, -5.0; LRMS-ES+ m/z (relative intensity) 705 (MH+, 50).
N
O
HTs
HHN
H
MeO2C
TBSO SiMe
IMe
Iodomethyl(dimethyl)silane 407. A sealed tube was charged with chloride 406 (77.4
mg, 0.110 mmol), acetone (5 mL) and then NaI (411 mg, 2.74 mmol). The vessel was sealed and
the resulting mixture was heated at reflux and stirred for 15 h. After cooling to rt, the reaction
mixture was diluted with ether and the organic phase was washed with water followed by
Na2S2O3(aq). The aqueous layers were extracted with ether and the combined organic layers were
dried over MgSO4 and concentrated in vacuo to give a residue. This material was purified by
flash chromatography on silica gel (20% EtOAc/hexanes) to give iodide 407 (84.4 mg, 97%). 1H
NMR (300 MHz, CDCl3) δ 9.17 (s, 1H), 7.69-7.64 (m, 3H), 7.40-7.47 (m, 3H), 7.21-7.11 (m,
2H), 4.73 (d, J = 9.9 Hz, 1H), 4.48-4.42 (m, 2H), 3.92 (t, J = 9.8 Hz, 1H), 3.82 (s, 3H), 3.78-3.70
(m, 2H), 2.48 (s, 3H), 2.28-2.16 (m, 3H), 2.03 (q, J = 8.7 Hz, 2H), 1.50-1.28 (m, 3H), 1.05 (s,
9H), 0.38-0.28 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 173.4, 144.1, 136.8, 133.4, 130.1, 128.7,
128.1, 127.0, 121.8, 119.8, 111.7, 108.2, 76.2, 64.0, 56.8, 52.8, 47.8, 46.4, 39.9, 39.8, 26.8, 24.2,
22.0, 19.2, 1.3, 0.7, -4.5, -4.9, -12.4; LRMS-ES+ m/z (relative intensity) 797 (MH+, 40).
Page 215
205
Cyclic Silyl Ether 408. To a solution of indole ester 407 (36.9 mg, 0.0463 mmol) at -78
°C was added KHMDS (0.1 M solution in THF, 1.39 mL, 0.139 mmol). The resulting mixture
was stirred for 1 h at -78 °C, then transferred to a ice bath and stirred for an additional 15 min.
The reaction mixture was diluted with NH4Cl(aq), the organic layer was separated and the
aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4
and concentrated in vacuo to give a residue, which was purified by flash chromatography on
silica gel (25% EtOAc/hexanes) to afford pentacycle 408 (31.0 mg, 97%). 1H NMR (300 MHz,
CDCl3) δ 9.00 (s, 1H), 8.06 (d, J = 7.08 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.39-7.34 (m, 3H),
7.21 (t, J = 7.7 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 4.50 (d, J = 10.7 Hz, 1H), 5.60 (dd, J = 10.4,
4.8 Hz, 1H), 4.17 (dd, J = 10.5, 5.1 Hz, 1H), 3.80-3.72 (m, 5H), 2.47 (s, 3H), 2.34-2.26 (m, 1H),
2.08 (d, J = 11.1 Hz, 1H), 1.91-1.86 (m, 1H), 1.53-1.41 (m, 3H), 1.06 (s, 9H), 0.97-0.85 (m, 2H),
0.29 (s, 3H), 0.27 (s, 3H), 0.10 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 176.1, 143.8, 137.8, 133.6,
132.1, 130.0, 128.4, 127.1, 122.4, 121.8, 119.6, 111.5, 110.4, 75.5, 63.8, 55.6, 52.8, 49.9, 46.9,
46.7, 40.5, 30.1, 28.0, 26.7, 25.5, 22.0, 19.1, 2.23, 2.16, -4.8, -4.9; LRMS-ES+ m/z (relative
intensity) 669 (MH+, 30); HRMS-ES+ (C34H49N2O6SSi2) calcd 669.2850 (MH+), found
669.2857.
Page 216
206
Minor Free Indole O-TBS ether 410. 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.67
(d, J = 8.1 Hz, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.35-7.32 (m, 3H), 7.16 (t, J = 7.1 Hz, 1H), 7.11 (t,
J = 7.3 Hz, 1H), 5.67 (s, 1H), 4.56-4.42 (m, 2H), 3.68 (s, 3H), 3.53-3.45 (m, 3H), 2.58 (d, J =
10.4 Hz, 4H), 2.45 (s, 3H), 1.70 (br s, 1H), 1.62-1.59 (m, 1H), 0.69 (s, 9H), 0.06 (s, 3H), -0.17 (s,
3H). Slow evaporation of a solution of TBS ether 410 in 1:1 MeOH/CHCl3 provide crystals
which were analyzed by X-ray diffraction (cf. Figure 9).141
Primary Alcohol 412. To a stirred solution of O-TBS ether 408 (15.0 mg, 0.022 mmol)
and THF (1 mL) at 0 °C was added TBAF (1.0 M in THF, 67 μL, 0.067 mmol). The resulting
solution was stirred for 1 h and then diluted with NH4Cl(aq). The organic layer was separated and
the aqueous layer was extracted with EtOAc. The combined organic layers were dried over
MgSO4 and concentrated in vacuo to give a residue which was purified by flash column
chromatography on silica gel (25% EtOAc/hexanes) to afford alcohol 412 (11.2 mg, 90%). 1H
NMR (360 MHz, CDCl3) δ 8.96 (s, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.41-
7.37 (m, 3H), 7.24 (t, J = 7.1 Hz, 1H), 7.16 (t, J = 7.2 Hz, 1H), 4.51-4.32 (m, 3H), 3.94-3.91 (m,
1H), 3.82 (s, 3H), 3.78 (t, J = 5.0 Hz, 1H), 2.55-2.49 (m, 4H), 2.33 (t, J = 6.3 Hz, 1H), 2.22 (d, J
Page 217
207
= 11.2 Hz, 1H), 2.02-1.97 (m, 1H), 1.60-1.50 (m, 2H), 1.37-1.27 (m, 2H), 0.12 (s, 3H), -0.53 (s,
3H); LRMS-ES+ m/z (relative intensity) 555 (MH+, 50).
Trifluoroacetate 419. To a solution of O-TBS-ether 408 (6.5 mg, 0.0097 mmol) in
CH2Cl2 (0.75 mL) was added TFA (0.25 mL) and the resulting mixture was stirred for 20 h at rt.
Concentration of the crude mixture in vacuo gave a residue which was purified by preparative
thin layer chromatography (4:2:1 hexanes/CH2Cl2/EtOAc) to give trifluoroacetate 412 (6.0 mg,
95%). 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 10.1 Hz,
2H), 7.42 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 8.3 Hz, 2H), 7.25 (t, J = 7.7 Hz, 1H), 7.16 (t, J = 7.2
Hz, 1H), 5.02-4.99 (m, 2H), 4.17-4.07 (m, 2H), 3.85-3.80 (m, 4H), 2.47 (s, 3H), 2.37 (td, J =
11.4, 3.3 Hz, 1H), 2.12 (d, J = 11.7 Hz, 1H), 2.00 (dd, J = 12.0, 4.1 Hz, 1H), 1.56-1.29 (m, 4H),
0.14 (s, 3H), -0.48 (s, 3H); LRMS-ES+ m/z (relative intensity) 651 (MH+, 100).
Retro-aldol Product 414. To a stirred solution of cyclic silyl ether 412 (1.0 mg, 0.002
mmol) and KHCO3 (10 mg, 0.010 mmol) in 1:1 MeOH/THF (250 μL), was added TBAF (1.0 M
Page 218
208
in THF, 40 μL, 0.040 mmol) followed by hydrogen peroxide (30 wt% in water, 150 μL). The
reaction mixture was stirred for 16 h and then diluted with Na2SO3(aq) and EtOAc. The organic
layer was separated and the aqueous layer was extracted with EtOAc. The combined organic
layers were dried over MgSO4 and concentrated in vacuo to give a residue which was purified by
preparative thin layer chromatography (50% EtOAc/hexanes) to afford retro-aldol product 414
(0.5 mg, 58%). 1H NMR (300 MHz, CDCl3) δ 9.27 (s, 1H), 7.70 (d, J = 8.3 Hz, 2H), 7.63 (d, J =
7.6 Hz, 1H), 7.44 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.26-7.16 (m, 2H), 5.13 (s, 1H),
4.79 (d, J = 12.6 Hz, 1H), 4.56 (dd, J = 4.7, 2.2 Hz, 1H), 4.16 (dd, J = 11.2, 1.8 Hz, 1H), 3.82 (s,
3H), 3.78-3.75 (m, 1H), 3.71-3.68 (m, 1H), 2.47 (s, 3H), 2.32-2.22 (m, 3H), 1.85-1.81 (m, 1H),
1.61-1.47 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 172.4, 144.2, 137.0, 133.5, 131.1, 130.2,
129.0, 128.0, 127.4, 126.4, 122.3, 120.3, 119.0, 112.1, 105.2, 73.5, 60.9, 54.8, 52.8, 46.2, 45.8,
39.9, 37.3, 24.2, 22.0; LRMS-ES+ m/z (relative intensity) 484 (MH+, 100).
Chloromethyl(dimethyl)silane 420. To a solution of N-Cbz-indole 364 (102 mg, 0.165
mmol) in MeOH (2 mL) and EtOAc (2 mL) under an argon atmosphere was added Pd/C (10
wt%, 17.5 mg, 0.0165 mmol). The resulting mixture was evacuated and backfilled three times
with H2 from a balloon and the resulting suspension was stirred for 2 h. The reaction mixture
was filtered through a pad of Celite with EtOAc and the filtrate was concentrated in vacuo to
afford NH-indole 414, which was used in the next step without further purification. 1H NMR
Page 219
209
(300 MHz, CDCl3) δ 9.27 (s, 1H), 7.70 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 7.6 Hz, 1H), 7.44 (d, J =
7.9 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.26-7.16 (m, 2H), 5.13 (s, 1H), 4.79 (d, J = 12.6 Hz, 1H),
4.56 (dd, J = 4.7, 2.2 Hz, 1H), 4.16 (dd, J = 11.2, 1.8 Hz, 1H), 3.82 (s, 3H), 3.78-3.75 (m, 1H),
3.71-3.68 (m, 1H), 2.47 (s, 3H), 2.32-2.22 (m, 3H), 1.85-1.81 (m, 1H), 1.61-1.47 (m, 3H); 13C
NMR (75 MHz, CDCl3) δ 172.4, 144.2, 137.0, 133.5, 131.1, 130.2, 129.0, 128.0, 127.4, 126.4,
122.3, 120.3, 119.0, 112.1, 105.2, 73.5, 60.9, 54.8, 52.8, 46.2, 45.8, 39.9, 37.3, 24.2, 22.0;
LRMS-ES+ m/z (relative intensity) 484 (MH+, 100).
To a solution of the above crude 1,3-diol 414 and 2,6-lutidine (19 μL, 0.17 mmol) in
CH2Cl2 (2.5 mL) was added chloromethyl(dimethyl)chlorosilane (22 μL, 0.17 mmol) at 0 °C.
The resulting mixture was stirred for 2 h at 0 °C and then 1 h at rt. The reaction mixture was
diluted with NH4Cl(aq), the organic layer was removed, and the aqueous layer was extracted with
EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give
a residue which was purified by flash chromatography on silica gel (25-75% EtOAc/hexanes) to
afford chloromethyl dimethylsilyl ether 420 (95 mg, 98%). 1H NMR (300 MHz, CDCl3) δ 8.13
(s, 1H), 7.74-7.60 (m, 3H), 7.38-7.33 (m, 3H), 7.25-7.13 (m, 2H), 4.59-4.50 (m, 3H), 3.79-3.71
(m, 4H), 3.51 (d, J = 3.7 Hz, 2H), 2.73-2.58 (m, 3H), 2.49-2.42 (m, 4H), 1.97-1.91 (m, 1H),
1.69-1.57 (m, 2H), 0.15 (s, 3H), 0.10 (s, 3H); LRMS-ES+ m/z (relative intensity) 591 (MH+, 25).
Page 220
210
N
OTMS
HTs
HHN
H
MeO2C
OSiMe Me
Cl
N
OTMS
HTs
HHN
H
MeO2C
TMSO
Chloromethyl(dimethyl)silane 421. To a solution of tertiary alcohol 420 (95.0 mg,
0.161 mmol) and 2,6-lutidine (113 μL, 0.966 mmol) in CH2Cl2 (2 mL) was added TMSOTf (88
μL, 0.17 mmol) at 0 °C. The resulting mixture was stirred for 2 h at 0 °C and then 1 h at rt. The
reaction mixture was filtered through a silica gel plug eluting with 30% EtOAc/hexanes. The
filtrate was concentrated in vacuo to give a residue which was dissolved in CH2Cl2 and washed
with NH4Cl(aq). The organic layer was dried over MgSO4 and concentrated in vacuo to give a
residue. This material was purified by flash chromatography on silica gel (30% EtOAc/hexanes)
to yield chloromethyl dimethylsilyl ether 421 (37.5 mg, 35%) which was contaminated with a
small amount of bis-TMS product 422. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 7.68 (d, J =
8.5 Hz, 2H), 7.63 (d, J = 7.7 Hz, 1H), 7.38 (d, J = 7.9 Hz, 2H), 7.23-7.11 (m, 2H), 4.77 (dd, J =
10.3, 1.7 Hz, 1H), 4.49 (dd, J = 10.1, 4.0 Hz, 1H), 4.38-4.34 (m, 1H), 3.99 (t, J = 9.9 Hz, 1H),
3.81 (s, 3H), 3.79-3.70 (m, 2H), 3.03 (ABq, J = 16.7, 13.8 Hz, 2H), 2.48 (s, 3H), 2.26-2.14 (m,
3H), 1.50-1.27 (m, 2H), 0.44 (s, 6H), 0.16 (s, 9H); LRMS-ES+ m/z (relative intensity) 663 (MH+,
20).
Bis-TMS product 422. 1H NMR (300 MHz, CDCl3) δ 9.12 (s, 1H), 7.68 (d, J = 8.3 Hz,
2H), 7.62 (d, J = 7.6 Hz, 1H), 7.39-7.35 (m, 3H), 7.20-7.09 (m, 2H), 4.84 (dd, J = 10.1, 1.7 Hz,
1H), 4.39-4.24 (m, 2H), 3.87 (t, J = 9.8 Hz, 1H), 3.81 (s, 3H), 3.74-3.70 (m, 2H), 2.48 (s, 3H),
2.28-2.12 (m, 3H), 1.50-1.34 (m, 2H), 0.28 (s, 9H), 0.16 (s, 9H); LRMS-ES+ m/z (relative
intensity) 629 (MH+, 90).
Page 221
211
Chloromethyl(diphenyl)silane 426. To a solution of 1,3-diol 414 (16.0 mg, 0.033
mmol) and imidazole (6.8 mg, 0.099 mmol) inCH2Cl2 (1 mL) was added
chloromethyl(diphenyl)chlorosilane142 (14 μL, 0.050 mmol). The resulting mixture was stirred
for 1 h, diluted with CH2Cl2, and then washed with ice-cold 0.1 M HCl. The organic layer was
dried over MgSO4 and concentrated in vacuo to give a residue, which was purified by preparative
thin layer chromatography (50% EtOAc/hexanes) to afford chloromethyl diphenylsilylether 426
(16.3 mg, 69%). 1H NMR (300 MHz, CDCl3) δ 9.21 (s, 1H), 7.67 (d, J = 6.5 Hz, 2H), 7.53-7.34
(m, 13H), 7.18-7.08 (m, 2H), 6.99-6.93 (m, 1H), 4.80 (s, 1H), 4.70 (dd, J = 11.1, 3.0 Hz, 1H),
4.60-4.52 (m, 2H), 4.30 (dd, J = 11.3, 1.7 Hz, 1H), 3.81 (s, 3H), 3.76-3.74 (m, 2H), 3.12 (ABq, J
= 59.2, 14.3 Hz, 2H), 2.47 (s, 3H), 2.34-2.21 (m, 3H), 1.53-1.43 (m, 2H); LRMS-ES+ m/z
(relative intensity) 715 (MH+, 50).
Chloromethyl(diphenyl)silane 427. To a solution of tertiary alcohol 426 (12.5 mg,
0.0175 mmol) and 2,6-lutidine (20 μL, 0.175 mmol) in CH2Cl2 (1 mL) was added TMSOTf (16
μL, 0.087 mmol). The resulting mixture was stirred for 45 min at rt, diluted with CH2Cl2 and
Page 222
212
then washed with ice-cold 0.1 M HCl. The organic layer was dried over MgSO4 and
concentrated in vacuo to give a residue which was purified by preparative thin layer
chromatography (30% EtOAc/hexanes) to yield chloromethyl silane 427 (10.9 mg, 79%) and the
bis-TMS side product 422 (3.7 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 7.92-7.84
(m, 4H), 7.70 (d, J = 8.3 Hz, 2H), 7.59-7.48 (m, 6H), 7.37 (d, J = 7.9 Hz, 2H), 7.29 (d, J = 8.8
Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 6.80 (t, J = 8.0 Hz, 1H), 6.61 (d, J = 8.0 Hz, 1H), 4.87 (dd, J =
10.3, 1.9 Hz, 1H), 4.42 (dd, J = 10.1, 3.8 Hz, 1H), 4.35 (dd, J = 4.2, 2.0 Hz, 1H), 4.20-4.12 (m,
1H), 3.83-3.77 (m, 5H), 3.62 (ABq, J = 75.9 Hz, 14.0 Hz, 2H), 2.53 (s, 3H), 2.29-2.18 (3H),
1.50-1.29 (m, 2H), 0.15 (s, 9H); LRMS-ES+ m/z (relative intensity) 787 (MH+, 25).
Chloromethyl(diphenylsilyl) Trifluoromethanesulfonate (430). Triflic acid (159 μL,
1.76 mmol) was added to a stirred solution of chloromethyl(triphenyl)silane (429)132 (670 mg,
1.76 mmol) in 1,2-dichloroethane (4 mL). The resulting solution was refluxed for 30 min and
the 1,2-dichloroethane was removed by distillation at atmospheric pressure. The triflate 430 was
isolated by vacuum distillation (410 mg, 50%, bp 102-105 °C / 0.25 mmHg). FTIR (film) 1260,
1174, 1037 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.75-7.36 (m, 10H), 3.57 (s, 3H); 13C NMR (75
MHz, CDCl3) δ 135.6, 133.0, 129.1, 126.7, 118.7 (JC,F = 316 Hz), 25.5; 19F NMR (282 MHz,
CDCl3) δ -76.6.
Page 223
213
Chloromethyl(diphenyl)silane 431. A sealed tube was charged with tertiary alcohol
405 (27.0 mg, 0.045 mmol) and 2,6-lutidine (105 μL, 0.90 mmol) in CH2Cl2 (2 mL).
Chloromethyl(diphenyl)silyl triflate (171 mg, 0.45 mmol) was added, the vessel was sealed, and
the resulting mixture was heated at 40 °C for 16 h. After cooling to rt, the reaction mixture was
washed with 1 M HCl. The organic layer was separated and the aqueous layer was extracted
with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo to
afford a residue which was purified by flash chromatography on silica gel (40% EtOAc/hexanes)
to give chloromethylsilane 431 (23.7 mg, 63%). 1H NMR (300 MHz, CDCl3) δ 9.18 (s, 1H),
7.79 (d, J = 7.6 Hz, 1H), 7.64-7.30 (m, 15H), 7.28-7.23 (m, 2H), 4.68 (dd, J = 10.1, 1.3 Hz, 1H),
4.54 (dd, J = 10.6, 4.4 Hz, 1H), 4.40 (dd, J = 4.3, 1.9 Hz, 1H), 4.27 (dd, J = 10.5, 8.9 Hz, 1H),
3.84-3.79 (m, 1H), 3.70 (s, 3H), 3.64-3.58 (m, 1H), 3.31 (s, 2H), 2.53 (s, 3H), 2.14 (d, J = 10.2
Hz, 1H), 2.01 (dt, J = 12.6, 4.0 Hz, 1H), 1.88 (td, J = 11.4, 1.9 Hz, 1H), 1.44-1.16 (m, 2H), 1.05
(s, 9H), 0.37 (s, 3H), 0.32 (s, 3H); LRMS-ES+ m/z (relative intensity) 829 (MH+, 15).
Iodomethyl(diphenyl)silane 432. A sealed tube was charged with chloride 431 (23.7
mg, 0.0286 mmol), acetone (3 mL) and then NaI (107 mg, 0.714 mmol). The vessel was sealed
Page 224
214
and the resulting mixture was heated at 40 °C and stirred for 17 h. The reaction mixture was
cooled to rt, additional NaI (200 mg, 1.33 mmol) was added, and the resulting mixture was
stirred for 5 h at 40 °C. After cooling to rt, the reaction mixture was diluted with ether and the
organic phase was washed with water followed by Na2S2O3(aq). The aqueous layer was extracted
with ether and the combined organic layers were dried over MgSO4 and concentrated in vacuo to
give a residue. This material was purified by flash column chromatography on silica gel (30%
EtOAc/hexanes) to give iodide 432 (25.4 mg, 97%). 1H NMR (300 MHz, CDCl3) δ 9.17 (s, 1H),
7.79 (d, J = 7.6 Hz, 1H), 7.63-7.30 (m, 15H), 7.22-7.14 (m, 2H), 4.68 (d, J = 10.1 Hz, 1H), 4.54
(dd, J = 10.6, 4.5 Hz, 1H), 4.39-4.27 (m, 2H), 3.91-3.79 (m, 2H), 3.68 (s, 3H), 3.60-3.56 (m,
1H), 2.53 (s, 3H), 1.95-1.83 (m, 2H), 1.47-1.15 (m, 3H), 1.06 (s, 9H), 0.39 (s, 3H), 0.37 (s, 3H);
LRMS-ES+ m/z (relative intensity) 921 (MH+, 15).
N-Cbz Indole 444. To a stirred solution of indole 408 (10 mg, 0.015 mmol) in THF (300
μL) at –78 °C was added KHMDS (0.1 M in THF, 300 μL, 0.030 mmol). The resulting solution
was stirred for 45 min and then Cbz-Cl (11 μL, 0.075 mmol) was added. The solution was
stirred for 5 h at –78 °C and then 12 h at rt before diluting with NH4Cl(aq) and EtOAc. The
organic layer was separated and the aqueous layer was extracted with EtOAc. The combined
organic layers were dried over MgSO4 and concentrated in vacuo to give a residue which was
purified by preparative thin layer chromatography (30% EtOAc/hexanes) to afford N-Cbz-indole
Page 225
215
444 (3.4 mg, 28%) and recovered starting indole 408 (6.4 mg, 64%). 1H NMR (300 MHz,
CDCl3) δ 8.20 (d, J = 7.4 Hz, 1H), 8.11 (d. J = 7.7 Hz, 1H), 7.69 (d, J = 8.2 Hz, 2H). 7.52-7.25
(m, 9H), 5.45 (ABq, J = 52.0, 12.1 Hz, 2H), 4.34 (d, J = 11.2 Hz, 1H), 4.21 (dd, J = 9.3, 3.9 Hz,
1H), 4.04 (dd, J = 10.9, 4.4 Hz, 1H), 3.73-3.70 (m, 2H), 3.31 (s, 3H), 2.48 (s, 3H), 2.16-2.01 (m,
2H), 1.81 (s, 2H), 1.72-1.48 (m, 2H), 1.19-1.13 (m, 1H), 1.06 (s, 9H), 0.28 (s, 3H), 0.25 (s, 3H),
0.14 (s, 3H), -0.43 (s, 3H); HRMS-ES+ (C42H55N2O8Si2S) calcd 803.3218 (MH+), found
803.3223.
Primary Alcohol N-Boc-Indole 447. To a stirred solution of indole 408 (15.8 mg, 0.024
mmol) and triethylamine (66 μL, 0.47 mmol) in CH2Cl2 (2 mL) was added Boc2O (53 mg, 0.24
mmol) and then DMAP (~0.5 mg, cat.). The resulting mixture was warmed to 35 °C, stirred for
35 h, and then diluted with NaHCO3(aq). The organic layer was separated and the aqueous layer
was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and
concentrated in vacuo to give a residue, which was used directly in the following step. For
characterization purposes a sample was purified by flash chromatography on silica gel (20%
EtOAc/hexanes). 1H NMR (300 MHz, CDCl3) δ 8.17 (d, J = 7.0 Hz, 1H), 8.05 (d, J = 8.2 Hz,
1H), 7.69 (d, J = 8.3 Hz, 2H), 7.37-7.23 (m, 4H) 4.34 (d, J = 9.7 Hz, 1H), 4.21 (dd, J = 10.8, 3.9
Hz, 1H), 4.05 (dd, J = 10.8, 4.3 Hz, 1H), 3.73-3.68 (m, 2H), 3.56 (s, 3H), 2.47 (s, 3H), 2.20-2.10
(m, 1H), 2.02 (d, J = 11.3 Hz, 1H), 1.85 (ABq, J = 16.6, 15.2 Hz, 2H), 1.68 (s, 9H), 1.61-1.51
Page 226
216
(m, 1H), 1.20-1.14 (m, 2H), 1.05 (s, 9H), 0.28 (s, 3H), 0.24 (s, 3H), 0.13 (s, 3H), -0.43 (s, 3H);
13C NMR (75 MHz, CDCl3) δ 175.3, 151.0, 143.9, 136.7, 133.6, 133.4, 130.0, 128.8, 128.3,
124.7, 122.7, 121.8, 118.1, 116.1, 84.7, 74.5, 63.6, 55.2, 52,4, 52.0, 48.8, 46.7, 40.8, 30.1, 28.6,
27.8, 26.6, 26.1, 23.5, 22.0, 19.0, 2.3, 2.0, -4.8, -5.2; LRMS-ES+ m/z (relative intensity) 769
(MH+, 80).
To a stirred solution of the above crude O-TBS-oxime 446 and THF (2 mL) was added
TBAF (1.0 M in THF, 115 μL, 0.115 mmol). The resulting solution was stirred for 10 h at rt and
additional TBAF was added (230 μL, 0.230 mmol). The solution was stirred for 5 h longer and
then diluted with NH4Cl(aq). The organic layer was separated and the aqueous layer was
extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in
vacuo to afford a residue which was purified by flash column chromatography on silica gel (25-
40% EtOAc/hexanes) to give primary alcohol 447 (15.0 mg, 97%). 1H NMR (300 MHz, CDCl3)
δ 8.09-8.02 (m, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.40-7.31 (m, 4H), 4.46-4.38 (m, 1H), 4.29-4.21
(m, 2H), 3.94-3.89 (m, 1H), 3.70 (t, J = 3.4 Hz, 1H), 3.60 (s, 3H), 2.58 (dd, J = 7.7, 6.3 Hz, 1H),
2.49 (s, 3H), 2.45-2.38 (m, 1H), 2.18 (d, J = 12.1 Hz, 1H), 1.91-1.80 (m, 3H), 1.70 (s, 9H), 1.31-
1.25 (m, 2H), 0.15 (s, 3H), -0.04 (s, 3H); LRMS-ES+ m/z (relative intensity) 655 (MH+, 30).
Page 227
217
N-Benzyl Indole 448. To a stirred solution of indole 408 (32.6 mg, 0.0487 mmol) in
DMF (0.8 mL) and THF (0.5 mL) at 0 °C was added NaH (60% dispersion in mineral oil, 5.6
mg, 0.15 mmol). The resulting mixture was stirred for 30 min and benzyl bromide (53 μL, 0.44
mmol) was added. The reaction mixture was stirred for 20 min at 0 °C and then 20 min at rt. The
reaction mixture was diluted with water and Et2O, the organic layer was separated, and the
aqueous layer was extracted with Et2O. The combined organic layers were washed with water,
brine, dried over MgSO4, and concentrated in vacuo to give a residue which was purified by
flash column chromatography on silica gel (25% EtOAc/hexanes) to afford N-benzyl indole 448
(25.8 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 8.18-8.14 (m, 1H), 7.71 (d, J = 8.3 Hz, 2H),
7.35 (d, J = 8.0 Hz, 2H), 7.27-7.23 (m, 3H), 7.12-7.10 (m, 2H), 6.99-6.97 (m, 3H), 5.12 (ABq, J
= 56.0, 17.4 Hz, 2H), 4.52 (dd, J = 11.2, 1.7 Hz, 1H), 4.28 (dd, J = 10.6, 4.8 Hz, 1H), 4.18 (dd, J
= 10.6, 4.8 Hz, 1H), 3.83 (t, J = 4.7 Hz, 1H), 3.77-3.74 (m, 1H), 3.40 (s, 3H), 2.47 (s, 3H), 2.24-
2.19 (m, 1H), 2.03 (d, J = 11.2 Hz, 1H), 1.77-1.65 (m, 3H), 1.20-1.15 (m, 1H), 1.08-1.06 (s,
10H), 0.29 (s, 3H), 0.27 (s, 3H), 0.07 (s, 3H), -0.54 (s, 3H); LRMS-ES+ m/z (relative intensity)
759 (MH+, 30).
Page 228
218
Primary Alcohol 449. To a stirred solution of O-TBS-oxime 408 (25.8 mg, 0.034
mmol) and THF (2 mL) was added TBAF (1.0 M in THF, 340 μL, 0.340 mmol). The resulting
solution was stirred for 15 h and then diluted with NH4Cl(aq). The organic layer was separated
and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over
MgSO4 and concentrated in vacuo to give a residue which was purified by flash column
chromatography on silica gel (25-40% EtOAc/hexanes) to afford primary alcohol 449 (12.6 mg,
57%). 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 8.3 Hz, 2H), 7.37 (d,
J = 7.6 Hz, 2H), 7.29-7.25 (m, 3H), 7.16-7.10 (m, 2H), 7.05-6.95 (m, 3H), 5.15 (ABq, J = 40.2,
17.8 Hz, 2H), 4.56-4.37 (m, 3H), 3.95-3.91 (m, 1H), 3.81-3.79 (m, 1H), 3.40 (s, 3H), 2.47-2.36
(m, 5H), 2.15 (d, J = 11.9 Hz, 1H), 1.81-1.65 (m, 3H), 1.23 (d, J = 14.4 Hz, 1H), 1.00-0.90 (m,
1H), 0.09 (s, 3H), -0.53 (s, 3H); LRMS-ES+ m/z (relative intensity) 645 (MH+, 50).
Retro-aldol Product 456. To a stirred solution of t-butyl hydroperoxide (70% in water,
35 μL, 0.25 mmol) in DMF (200 μL) at 0 °C was added cesium hydroxide monohydrate (35 mg,
0.21 mmol). The resulting mixture was stirred at rt for 10 min and then cyclic silyl ether 449 (3
mg, 0.005 mmol) in DMF (200 μL) was added dropwise. After stirring the mixture for 10 min,
Page 229
219
TBAF (1.0 M in THF, 88 μL, 0.088 mmol) was added and the resulting mixture was heated at 75
°C and stirred for 6.5 h. After cooling the mixture to rt, solid Na2S2O3 was added and the solvent
was removed in vacuo. The resulting residue was diluted with EtOAc and water. The organic
layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic
layers were washed with water then brine, dried over MgSO4, and concentrated in vacuo to give
a residue which was used in subsequent reactions without further purification.
The above crude residue was dissolved in MeOH (500 μL) and cooled to 0 °C. TMS-
diazomethane (100 μL) was added and the resulting mixture was stirred for 45 min at 0 °C and
45 min at rt. The volatiles were removed in vacuo to give a residue which was purified by
preparative thin layer chromatography (50% EtOAc/hexanes) to give the retro-aldol product 456
(1 mg, 37%). 1H NMR (300 MHz, CDCl3) δ 7.72 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 8.2 Hz, 2H),
7.35 (d, J = 8.0 Hz, 2H), 7.31-7.16 (m, 6H), 6.88 (d, J = 7.6 Hz, 2H), 5.30 (ABq, J = 116.6, 17.4
Hz, 2H), 5.05-4.96 (m, 1H), 4.72 (d, J = 13.6 Hz, 1H), 4.39 (t, J = 10.9 Hz, 1H), 3.95-3.86 (m,
1H), 3.75-3.74 (m, 1H), 3.56 (s, 3H), 3.37-3.34 (m, 1H), 2.46-2.34 (m, 6H), 2.11-2.00 (m, 1H),
1.02-0.88 (m, 3H); LRMS-ES+ m/z (relative intensity) 575 (MH+, 50).
Cyclic Ether Product 459 or 461. To a stirred solution of t-butyl hydroperoxide (5.5 M
in decane, 19 μL, 0.11 mmol) in DMF (300 μL) at 0 °C was added cesium hydroxide
monohydrate (15 mg, 0.092 mmol). The resulting mixture was stirred at room rt for 10 min and
Page 230
220
then cyclic silyl ether 447 (5 mg, 0.008 mmol) in DMF (400 μL) was added dropwise. After
stirring the mixture for 10 min, TBAF (1.0 M in THF, 38 μL, 0.038 mmol) was added and the
resulting mixture was stirred at rt for 2.5 h. Solid Na2S2O3 was added and the solvent was
removed in vacuo. The resulting residue was diluted with EtOAc and water. The organic layer
was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers
were washed with water then brine, dried over MgSO4, and concentrated in vacuo to give a
residue which was used in the subsequent reaction without further purification.
The above crude residue was dissolved in MeOH (500 μL) and Et2O (200 μL). TMS-
diazomethane (100 μL) was added and the resulting mixture was stirred for 1.5 h at rt. The
volatiles were removed in vacuo to give a residue which was purified by preparative thin layer
chromatography (50% EtOAc/hexanes) to afford a dehydration product assumed to be cyclic
ether 459 or 461 (2 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 9.05 (s, 1H), 7.72 (d, J = 8.2 Hz,
2H), 7.62 (d, J = 7.7 Hz, 1H), 7.40-7.36 (m, 3H), 7.23-7.13 (m, 2H), 4.75 (d, J = 10.8 Hz, 1H),
4.41 (tt, J = 8.9, 2.7 Hz, 1H), 4.25-4.20 (m, 4H), 4.02-3.98 (m, 1H). 3.89 (s, 3H), 3.75 (dd, J =
7.0, 3.0 Hz, 1H), 2.50-2.46 (m, 5H), 2.39 (dd, J = 9.4, 3.0 Hz, 1H), 2.32 (dd, J = 12.7, 3.0 Hz,
1H), 1.72-1.55 (m, 2H). Due to the small amount of material, the aliphatic region (1.6-1.2 ppm)
was complex due to overlap of water and grease peaks. LRMS-ES+ m/z (relative intensity) 497
(MH+, 25).
Page 231
221
References and Notes
1 Mathaipoulos, G. Chem. Ber. 1898, 31, 2396.
2 (a) Gilchrist, T. L. Chem. Soc. Rev. 1983, 11, 53. (b) Lyapkalo, I. M.; Ioffe, S. L. Russ. Chem.
Rev. 1998, 67, 467.
3 Francotte, E.; Merenyi, R.; Vladenbulcke-Coyette, B.; Viehe, H.-G. Helv. Chim. Acta 1981, 64,
1208.
4 (a) Ciattoni, P.; Rivolta, L. Chim. Ind. (Milan) 1967, 1186. (b) Hobold, W.; Prietz, U.;
Pritzkow, W. J. Prakt. Chem. 1969, 311, 260.
5 Ohno, M.; Naruse, N. Bull. Chem. Soc. Jpn. 1966, 39, 1129.
6 Kaiser, E. T.; Smith, J. H.; Heidema, J. H. J. Am. Chem. Soc. 1972, 94, 9276.
7 Hobold, W.; Prietz, U.; Pritzkow, W. J. Prakt. Chem. 1969, 311, 260.
8 Weiser, K.; Berndt, A. Angew. Chem. Int. Ed. 1975, 14, 70.
9 Griffin, C. E.; Haszeldine, R. N. J. Chem. Soc. 1960, 1398.
10 Kisan, W.; Pritskow, W. J. Prakt. Chem., 1978, 320, 59.
11 Trost, B. M.; Barret, D. Tetrahedron 1996, 52, 6903.
12 Corkins, H. G.; Storace, L.; Osgood, E. R. Teterahedron Lett. 1980, 21, 2025.
13 (a) Corey, E. J.; Melvin, L. S.; Haslanger, M. F. Tetrahedron Lett. 1975, 16, 3117. (b)
Denmark, S. E.; Dappen, M. S. J. Org. Chem. 1984, 49, 798.
14 (a) Dornow, A.; Jordan, H. D.; Chem. Ber. 1961, 94, 76. (b) Faragher, R.; Gilchrist. T. L. J.
Chem. Soc., Perkin Trans. 1, 1979, 249.
15 Lemos, A.; Lourenco, J. P. Arkivoc 2010, 170.
16 Ohno, M.; Naruse, N.; Torimitsu, S.; Okamoto, M. Bull. Chem. Soc. Jpn. 1966, 39, 1119.
Page 232
222
17 (a) Denmark, S. E.; Dappen, M. S.; Sternberg, J. A. J. Org. Chem. 1984, 49, 4741. (b)
Denmark, S. E.; Dappen, M. S.; Sear, N. L.; Jacobs, R. T. J. Am. Chem. Soc. 1990, 112, 3466.
18 (a) Sherwood, A. G.; Gunning, H. E. J. Am. Chem. Soc. 1963, 85, 3506. (b) Surzur, J. -M.;
Dupuy, C.; Betrand, M. P.; Nouguier, R. J. Org. Chem., 1972, 37, 2782.
19 (a) Tilden, W. A.; Shenstone, W. A. J. Chem. Soc. 1877, 554. (b) Beckham, L. J.; Fessler, W.
A.; Kise, M. A. Chem. Rev. 1951, 48, 319. (c) Kadzyauskas, P. P.; Zefirov, N. S. Russ. Chem.
Rev. 1968, 37, 543.
20 (a) Wallach, O. Liebigs Ann. Chem. 1899, 306, 278. (b) Prtizkow, W.; Schaefer, H.; Pabst, P.;
Ebenroth, A.; Beger, J. J. Prakt. Chem., 1965, 29, 123.
21 Colvin, E. W.; Robertson, A. D.; Seebach, D.; Beck, A. K. J. Chem. Soc., Chem. Commun.
1981, 952.
22 (a) Ioffe, S. L.; Lyapkalo, I. M.; Tishkom, A. A.; Danilenko, V. M.; Strelenko, Y. A.;
Tartakovsky, V. A. Tetrahedron 1997, 53, 13085. (b) Ustinov, A. V.; Dilman, A. D.; Ioffe, S.
L.; Belyakov, P. A.; Strelenko, Y. U. Russ. Chem. Bull., Int. Ed. 2002, 51, 1457.
23 (a) Bravo, P.; Gaudiano, G.; Umani-Ronchi, A. Gazz. Chim. Ital. 1967, 97, 1664. (b) Bravo,
P.; Gaudiano, G.; Ponti, P. P.; Ticozzi, C. Tetrahedron 1972, 28, 3845. (c) Hayashi, Y.;
Watanabe, T.; Oda, R. Tetrahedron Lett. 1970, 11, 605. (d) Faragher, R.; Gilchrist, T. L. J.
Chem. Soc., Perkin Trans. 1 1977, 1196.
24 (a) Abramovitch, A. B.; Cue, B. W. J. Am. Chem. Soc. 1976, 98, 1478. (b) Abramovitch, A.
B.; Shinkai, I.; Cue, B. W.; Ragan, A.; Atwood, J. L. J. Heterocycl. Chem. 1976, 13, 415.
25 Dirlam, J. P.; Cue, B. W.; Gombatz, K. J. J. Org. Chem. 1978, 43, 76.
26 Silveira, A.; Satra, S. K. J. Org. Chem., 1979, 44, 873.
27 Ranganathan, S.; Singh, B. B.; Panda, C. S. Tetrahedron 1977, 33, 2415.
Page 233
223
28 Davies, D. E.; Gilchrist, T. L. J. Chem. Soc., Perkin Trans. 1 1983, 1479.
29 (a) Kisan, W.; Pritzkow, W. J. Prakt. Chem. 1978, 320, 59. (b) Gilchrist, T. L., Harris, C. J.;
King, F. D.; Peek, M. E., Rees, C. W. J. Chem. Soc., Perkin Trans. 1 1976, 2161.
30 Ranganathan, S.; Singh, B. B.; Panda, C. S. Tetrahedron, 1977, 33, 2415.
31 Yang, H.-T.; Ruan, X.-J.; Miao, C.-B.; Xi, H.-T.; Jiang, Y.; Meng, Q.; Sun, X.-Q. Tetrahedron
Lett. 2009, 50, 7337.
32 (a) Zimmer, R.; Reissig, H.-U.; J. Org. Chem. 1992, 57, 339. (b) Zimmer, R.; Reissig, H.-U.
Liebigs Ann. Chem. 1991, 553.
33 Faragher, R.; Gilchrist, T. L. J. Chem. Soc., Chem. Commun. 1976, 581.
34 Domingo, L. R.; Picher, M. T.; Arroyo, P. Eur. J. Org. Chem. 2006, 2570.
35 See for example: Zimmer, R.; Buchholz, M.; Collas, M.; Angermann, J.; Homann, K.;
Reissig, H.-U. Eur. J. Org. Chem. 2010, 4111.
36 Francotte, E.; Merenyi, Viehe, H.-G. Angew. Chem. Int. Ed. 1978, 17, 936.
37 De los Sandos, J. M.; Ignacio, R.; Aparicio, D.; Palacios, F.; Ezpeleta, J. M. J. Org. Chem.
2009, 74, 3444.
38 Gilchrist, T. L.; Roberts, T. G. J. Chem. Soc., Perkin Trans. 1 1983, 1283.
39 (a) Fuchs, P. L. J. Org. Chem. 1976, 41, 2935. (b) Wender, P. A.; Erhardt, J. M.; Letendre, L.
J. J. Am. Chem. Soc. 1981, 103, 2114.
40 (a) Zeigler, F. E.; Chliwner, I.; Fowler, K. W.; Kanfer, S. J.; Kuo, S. J.; Sinha, N. D. J. Am.
Chem. Soc. 1980, 102, 790. (b) Tsuboi, S.; Shinhama, K.; Takeda, A. J. Heterocycl. Chem. 1988,
25, 523. (c) Herman, T.; Carlson, R. Tetrahedron Lett. 1989, 30, 3657. (d) Caussanel, F.;
Deslongchamps, P.; Dory, Y. L. Org. Lett. 2003, 5, 4799. (e) Ishizaki, M.; Satoh, H.; Hoshino,
Page 234
224
O.; Nishitani, K. Hara, H. Heterocycles 2004, 63, 827. (f) Fortin, D.; Gaudette, F.; Marsault, E.;
Deslongchamps, P. Tetrahedron 2001, 57, 4167.
41 For cross-coupling reactions of α-chloroketones with alkylzinc halides see: Malosh, C. F.;
Ready, J. M. J. Am. Chem. Soc. 2004, 126, 10240.
42 For examples see: (a) Takeda, A.; Sadao, T.; Oota, Y. J. Org. Chem. 1973, 38, 4148. (b)
Kirrmann, A.; Chancel, P.; Vignalow, M.; Federlin, P. Bull. Soc. Chim. Fr. 1950, 707.
43 Sakai, T.; Tabata, H.; Takeda, A. J. Org. Chem. 1983, 48, 4618.
44 Trewertha, G.; Burrows, J. N.; Barrett, A. G. M. Tetrahedron Lett. 2005, 46, 3553.
45 (a) Hassner, A.; Murthy, K. S. K. Tetrahedron Lett. 1987, 28, 683. (b) Padwa, A.; Chiacchio,
U.; Dean, D. C.; Schoffsatll, A. M.; Hassner, A.; Murthy, K. S. K. Tetrahedron Lett. 1988, 29,
4169. (c) Hassner, A.; Maurya, R.; Mesko, E. Tetrahedron Lett. 1988, 29, 5313. (d) Hassner, A.;
Murthy, K. S. K.; Padwa, A.; Bullock, W. H.; Stull, P. D. J. Org. Chem. 1988, 53, 5063. (e)
Hassner, A.; Maurya, R. Tetrahedron Lett. 1989, 30, 5803. (f) Hassner, A.; Murthy, K. S. K.;
Padwa, A.; Chiacchio, U.; Dean, D. C.; Schoffstall, A. M. J. Org. Chem. 1989, 54, 5277. (g)
Hassner, A.; Maurya, R.; Friedman, O.; Gottlieb, H. E.; Padwa, A.; Austin, D. J. Org. Chem.
1993, 58, 4539.
46 Korboukh, I.; Kumar, P.; Weinreb, S. M. J. Am. Chem. Soc. 2007, 129, 10342.
47 (a) Chao, W.; Weinreb, S. M. Org. Lett. 2003, 5, 2505. (b) Chao, W.; Meketa, M. L.; Weinreb,
S. M. Synthesis 2004, 2058.
48 VanBrunt, M. P.; Ambenge, R. O.; Weinreb, S. M. J. Org. Chem. 2003, 68, 3323.
49 Kumar, P.; Li, P.; Korboukh, I.; Wang, T. L.; Yennawar, H., Weinreb, S. M. J. Org. Chem.
2011, 76, 2094.
50 Witek, J. A.; Weinreb, S. M. Org. Lett. 2011, 13, 1258.
Page 235
225
51 Li, P.; Majireck, M. M.; Witek, J. A., Weinreb S. M. Tetrahedron Lett. 2010, 51, 2032.
52 (a) Modern Carbonyl Chemistry J. Otera, Ed.; Wiley-WCH, Weinheim, 2000. (b) Caine, D. In
Comprehensive Organic Synthesis, Trost, B. M., Ed.; Pergamon Press, New York, 1991, Vol. 2.
(c) Evans, D. A. In Asymmetric Synthesis, Morrison, J. D., Ed.; Academic Press, New York,
1983, Vol. 3. (d) Job, A.; Janeck, C. F.; Bettray, W.; Peters, R.; Enders, D. Tetrahedron 2002,
58, 2253.
53 For examples of similar compounds see: (a) Salah Eldin, A. M. Heteroatom Chem. 2003, 14,
612. (b) Abdelrazek, F. M. Synth. Commun. 2005, 35, 2251. (c) Abdelrazek, F. M.; Ghozlan, S.
A.; Michael, F. A. J. Heterocycl. Chem. 2007, 44, 63. (d) Hammouda, M.; Abou Zeid, Z. M.;
Metwally, M. A. Chem. Heterocycl. Compd. 2008, 44, 985.
54 (a) Cordell, G. A. Introduction to Alkaloids: A Biogenetic Approach; Wiley-Interscience: New
York, 1981. (b) Pelletier, S. W. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S.
W., Ed.; Wiley: New York, 1983; Vol. 1.
55 (a) Bisset, N. G. In Indoles and Biogenetically Related Alkaloids; Phillipson, J. D., Zenk, M.
H., Eds.; Academic Press: London, 1980. (b) Herbert, R. B. In The Chemistry of Heterocyclic
Compounds. Indoles Part 4, The Monoterpenoid Indole Alkaloids; Saxton, J. E., Ed.; Wiley:
New York, 1983. (c) Atta-ur-Rahman; Basha, A. Biosynthesis of Indole Alkaloids; Clarendon
Press: Oxford, 1983. (d) Kutchan, T. M.; Phytochemistry 1993, 32, 493.
56 (a) Ishikura, M., Yamada, K.; Abe, T. Nat. Prod. Rep. 2010, 27, 1630. (b) Ishikura, M.,
Yamada, K. Nat. Prod. Rep. 2009, 26, 803. (c) Higuchi, K.; Kawasaki, T. Nat. Prod. Rep. 2007,
24, 843. (d) Kawasaki, T.; Higuchi, K. Nat. Prod. Rep. 2005, 22, 761. (e) Somei, M.; Yamada,
F. Nat. Prod. Rep. 2005, 22, 73. (f) Somei, M.; Yamada, F. Nat. Prod. Rep. 2004, 21, 278. (g)
Somei, M.; Yamada, F. Nat. Prod. Rep. 2003, 20, 216. (h) Hibino, S.; Choshi, T. Nat. Prod.
Page 236
226
Rep. 2002, 19, 148. (i) Hibino, S.; Choshi, T. Nat. Prod. Rep. 2001, 18, 66. (j) Lounasmaa, M.;
Tolvanen, A. Nat. Prod. Rep. 2000, 17, 175. (k) Leonard, J. Nat. Prod. Rep. 1999, 16, 319. (l)
Toyota, M.; Ihara, M. Nat. Prod. Rep. 1998, 15, 327. (m) Ihara, M.; Fukumoto, K. Nat. Prod.
Rep. 1997, 14, 413. (n) Saxton, J. E. Nat. Prod. Rep. 1997, 14, 559. (o) Ihara, M.; Fukumoto, K.
Nat. Prod. Rep. 1996, 13, 241. (p) Saxton, J. E. Nat. Prod. Rep. 1996, 13, 327. (q) Ihara, M.;
Fukumoto, K. Nat. Prod. Rep. 1995, 12, 277. (r) Saxton, J. E. Nat. Prod. Rep. 1995, 12, 385. (s)
Saxton, J. E. Nat. Prod. Rep. 1994, 11, 493. (t) Saxton, J. E. Nat. Prod. Rep. 1993, 10, 349. (u)
Saxton, J. E. Nat. Prod. Rep. 1992, 9, 393. (v) Saxton, J. E. Nat. Prod. Rep. 1991, 8, 251. (w)
Saxton, J. E. Nat. Prod. Rep. 1990, 7, 191. (x) Saxton, J. E. Nat. Prod. Rep. 1989, 6, 433. (y)
Saxton, J. E. Nat. Prod. Rep. 1989, 6, 1. (z) Saxton, J. E. Nat. Prod.Rep. 1987, 4, 591. (aa)
Saxton, J. E. Nat. Prod. Rep. 1986, 3, 353. (bb) Saxton, J. E. Nat. Prod. Rep. 1985, 2, 49. (cc)
Saxton, J. E. Nat. Prod. Rep. 1984, 1, 21.
57 Hon, L. K. Ph.D. Thesis, University of Malaya, Kuala Lumpur, 2008.
58 Kam, T.-S.; Choo, Y.-M. Helv. Chim. Acta 2004, 87, 366.
59 Koyama, K.; Hirasawa, Y.; Zaima, K.; Hoe, T. C.; Chan, K.-L., Morita, H. Biorg. Med. Chem.
2008, 16, 6483.
60 (a) Kutney, J. P.; Nelson, V. R.; Wigfield, D. C. J. Am. Chem. Soc. 1969, 91, 4278. (b)
Kutney, J. P.; Nelson, V. R.; Wigfield, D. C. J. Am. Chem. Soc. 1969, 91, 4279. (c) Ahond, A.;
Cave, A.; Kan-Fan, C.; Langlois, Y.; Potier, P. J. Chem. Soc., Chem. Commun. 1970, 517. (d)
Scott, A. I.; Yeh, C. L.; Greenslade, D. J. Chem. Soc., Chem. Commun. 1978, 947. (e) Potier, P.
In Indole and Biogenetically Related Alkaloids; Phillipson, J. D.; Zenk, M. H., Eds.; Academic
Press: London, 1980; Chapter 8.
Page 237
227
61 (a) Scott, A. I.; Yey, C.-L.; Greenslade, D. J. Chem. Soc., Chem. Commun. 1978, 947. (b) Lim,
K.-H.; Low, Y.-Y.; Kam, T.-S. Tetrahedron Lett. 2006, 47, 5037.
62 (a) Scopes, D. I. C.; Allen, M. S.; Hignett, G. J.; Wilson, N. D. V.; Harris, M.; Joule, J. A. J.
Chem. Soc., Perkin Trans. 1 1977, 2376. (b) Joule, J. A.; Allen, M. S.; Bishop, D. I.; Harris, M.;
Hignett, G. J.; Scopes, D. I. C.; Wilson, N. D. V. Indole and Biogenetically Related Alkaloids;
Academic Press: London, 1980; pp 229-247.
63 (a) Bennesar, M.-L.; Zulaica, E.; Sole, D.; Alonso, S. Chem. Commun. 2009, 3372. (b)
Bennesar, M.-L.; Zulaica, E.; Sole, D.; Roca, T.; Garcia-Diaz, D.; Alonso, S. J. Org. Chem.
2009, 74, 8359. (c) Bennesar, M.-L.; Zulaica, E.; Sole, D.; Alonso, S. Tetrahedron 2007, 63,
861.
64 Chauhan, P. S. Unpublished work.
65 (a) Yang, H. W.; Romo, D. Tetrahedron 1999, 55, 6403. (b) Orr, R. K.; Calter, M. A.
Tetrahedron 2003, 59, 3545. (c) Paull, D. H.; Weatherwax, A.; Letcka, T. Tetrahedron 2009,
65, 6771. (d) Purohit, V. C.; Matla, A. S.; Romo, D. Heterocycles 2008, 76, 949.
66 Lowe, C.; Vederas, J. C. Org. Prep. Proc. Int. 1995, 27, 305 and references cited.
67 (a) Wynberg, H.; Staring, E. G. J. J. Am. Chem. Soc. 1982, 104, 166. (b) Wynberg, H. Top.
Stereochem. 1986, 16, 87.
68 (a) Oh, S. H.; Cortez, G. S.; Romo, D. J. Org. Chem. 2005, 70, 2835. (b) Henry-Riyad, H.;
Lee, C.; Purohit, V. C.; Romo, D. Org. Lett. 2006, 8, 4363. (c) Ma, G.; Nguyen, H.; Romo, D.
Org. Lett. 2007, 9, 2143. (d) Purohit, V. C.; Matla, A. S.; Romo, D. J. Am. Chem. Soc. 2008,
130, 10478. (e) Nguyen, H.; Ma, G.; Romo, D. Chem. Commun. 2010, 46, 4803. (f) Morris, K.
A.; Arendt, K. M.; Oh, S. H.; Romo, D. Org. Lett. 2010, 12, 3764. (g) Nguyen, H.; Ma, G.;
Gladysheva, T.; Fremgen, T.; Romo, D. J. Org. Chem. 2011, 76, 2.
Page 238
228
69 Leverett, C. A.; Purohit, V. C.; Romo, D. Angew. Chem. Int. Ed. 2010, 49, 9479.
70 Richter, J. M.; Whitefield, B. W.; Maimone, T. J.; Lin, D. W.; Castroviejo, M. P.; Baran, P. S.
J. Am. Chem. Soc. 2007, 129, 12857.
71 (a) Kuehne, M. E.; Hafter, R. J. Org. Chem. 1978, 43, 3702. (b) Parsons, R. L.; Berk, J. D.;
Kuehne, M. E. J. Org. Chem. 1993, 58, 7482. (c) Schkeryantz, J. M.; Woo, J. C. G.;
Siliphaivanh, P.; Depew, K. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1999, 121, 11964.
72 (a) Taylor, E. C.; Hawks, G. H., III; McKillop, A. J. Am. Chem. Soc. 1968, 90, 2421. (b)
Taylor, E. C.; McKillop, A. Acc. Chem. Res. 1970, 3, 338.
73 (a) Martin, C. L.; Overman, L. E.; Rohde, J. M. J. Am. Chem. Soc. 2010, 132, 4894. (b)
Martin, C. L.; Overman, L. E.; Rohde, J. M. J. Am. Chem. Soc. 2008, 130, 7568.
74 Grehn, L.; Gunnarsson, K.; Ragnarsson, U. Acta Chem. Scand. B 1986, 40, 745.
75 Wildsmith, E.; Timms, G. H. Tetrahedron Lett. 1971, 12, 195.
76 De Kimpe, N.; Brunet, P. Synthesis 1990, 595.
77 (a) D’hooghe, M.; Baele, J.; Contreras, J.; Boelens, M.; De Kimpe, N. Tetrahedron Lett. 2008,
49, 6039. (b) Aszodi, J.; Rowlands, D. A.; Mauvais, P.; Collette, P.; Bonnefoy, A.; Lampilas, M.
Bioorg. Med. Chem. Lett. 2004, 14, 2489. (c) Van den Branden, S.; Compernolle, F.; Hoornaert,
G. J. J. Chem. Soc., Perkin Trans. 1 1992, 1035. (d) Easton, N. R.; Bartron, L. R.; Meinhofer, F.
L.; Fish, V. B. J. Am. Chem. Soc. 1953, 75, 2086.
78 Engler, T. A.; Wanner, J. J. Org. Chem. 2000, 65, 2444.
79 Lee, J.; Oh, J.; Jin, S.; Choi, J.-R.; Atwood, J. L.; Cha, J. K. J. Org. Chem. 1994, 59, 6955.
80 Brosius, A. D.; Overman, L. E.; Schwink, L. J. Am. Chem. Soc. 1999, 121, 700.
81 (a) Garst, M. E.; Bonfiglio, J. N.; Grudoski, D. A.; Marks, J. J. Org. Chem. 1980, 45, 2307.
(b) Garst, M. E.; Bonfiglio, J. N.; Grudoski, D. A.; Marks, J. Tetrahedron Lett. 1978, 19, 2671.
Page 239
229
82 Masilamani, D.; Rogic, M. M. J. Org. Chem. 1981, 46, 4486.
83 (a) Weinreb, S. M.; Chase, C. E.; Wipf, P.; Venkatraman, S. Org. Synth. 1998, 75, 161. (b)
Weinreb, S. M.; Demko, D. M.; Lessen, T. A. Tetrahedron Lett. 1986, 27, 2099.
84 Kehler, J.; Stensbol, T. B.; Krogsgaard-Larsen, P. Biorg. Med. Chem. Lett. 1999, 9, 811.
85 Meshram, H. M.; Reddy, P. N.; Sadashiv, K.; Yadav, J. S. Tetrahedron Lett. 2005, 46, 623.
86 Johansen, M. B.; Kerr, M. A. Org. Lett. 2010, 12, 4956.
87 (a) Zhang, M.; Huang, X.; Shen, L.; Qin, Y. J. Am. Chem. Soc. 2009, 131, 6013. (b) Shen, L.;
Zhang, M.; Wu, Y.; Qin, Y. Angew. Chem. Int. Ed. 2008, 47, 3618. (c) He, B.; Song, H.; Du, Y.;
Qin, Y. J. Org. Chem. 2008, 74, 298. (d) Yang, J.; Wu, H.; Shen, L.; Qin, Y. J. Am. Chem. Soc.
2007, 129, 13794. (e) Song, H.; Yang, J.; Chen, W.; Qin, Y. Org. Lett. 2006, 8, 6011. (f) Yang,
J.; Song, H.; Xiao, X.; Wang, J.; Qin, Y. Org. Lett. 2006, 8, 2187.
88 Kimura, M.; Futamata, M.; Mukai, R.; Tamaru, Y. J. Am. Chem. Soc. 2005, 127, 4592.
89 Kempf, D. J.; Condon, S. L. J. Org. Chem. 1990, 55, 1390.
90 (a) Morris, D. J.; Hayes, A. M.; Wills, M. J. Org. Chem. 2006, 71, 7035. (b) Kunetsky, R. A.;
Dilman, A. D.; Tsvaygboym, K. P.; Ioffe, S. L.; Strelenko, Y. A.; Tartakovsky, V. A. Synthesis
2003, 1339.
91 For examples, see: (a) McKew, J. C.; Foley, M. A.; Thakker, P.; Behnke, M. L.; Lovering, F.
E.; Sum, F.-W.; Tam, S.; Wu, K.; Shen, M. W. H.; Zhang, W.; Gonzalez, M.; Liu, S.;
Mahadevan, A.; Sard, H.; Khor, S. P.; Clark, J. D. J. Med. Chem. 2006, 49, 135. (b) Raucher, S.;
Klein, P. J. Org. Chem. 1986, 51, 123.
92 (a) Kan, T.; Fukuyama, T. Chem. Commun. 2005, 353. (b) Fukuyama, T.; Jow, C.-K.;
Cheung, M. Tetrahedron Lett. 1995, 36, 6373.
Page 240
230
93 Hlasta, D. J.; Luttinger, D.; Perrone, M. H.; Silbernagel, M. J.; Ward, S. J.; Haubrich, D. R. J.
Med. Chem. 1987, 30, 1555.
94 For examples see: (a) Han, Q.; Dominguez, C.; Stouten, P. F. W.; Park, J. M.; Duffy, D. E.;
Galemmo, R. A., Jr.; Rossi, K. A.; Alexander, R. S.; Smallwood, A. M.; Wong, P. C.; Wright,
M. M.; Leuttgen, J. M.; Knabb, R. M.; Wexler, R. R. J. Med. Chem. 2000, 43, 4398. (b) Batt, D.
G.; Qiao, J. X.; Modi, D. P.; Houghton, G. C.; Pierson, D. A.; Rossi, K. A.; Luettgen, J. M.;
Knabb, R. M.; Jadhav, P. K.; Wexler, R. R. Biorg. Med. Chem. Lett. 2004, 12, 5269.
95 Khripach, V. A.; Zhabinskii, V. N.; Kuchto, A. I.; Fando, G. P.; Zhiburtovich, Y. Y.; Lyakhov,
A. S.; Govorova, A. A.; Groen, M. B.; van der Louw, J.; de Groot, A. Steroids 2004, 69, 511.
96 Collot, V.; Schmitt, M.; Marwah, P.; Bourguignon, J.-J. Heterocycles 1999, 51, 2823.
97 Hynd, G.; Ray, N. C.; Finch, H.; Montana, J. G.; Cramp, M. C.; Harrison, T. K.; Arienzo, R.;
Blaney, P.; Griffon, Y.; Middlemiss, D. US Patent 20080306109, 2008.
98 (a) Barton, D. H. R.; Blundell, P.; Dorchak, J.; Jang, D. O.; Jaszberenyi, J. C. Tetrahedron
1991, 43, 8969. (b) Barton, D. H. R.; Motherwell, W. B.; Stange, A. Synthesis 1981, 743.
99 (a) Tran, Y. S.; Kwon, O. Org. Lett. 2005, 7, 4289. (b) Lau, C. K.; Dufresne, C.; Belanger, P.
C.; Pietre, S.; Scheigetz, J. J. Org. Chem. 1986, 51, 3038.
100 Tomioka, Y.; Nagahiro, C.; Nomura, Y.; Maruoka, H. J. Heterocycl. Chem. 2003, 40, 121.
101 Cao, L.-L.; Wang, D.-S.; Jiang, G.-F.; Zhou, Y.-G. Tetrahedron Lett. 2011, 52, 0000.
102 Routier, S.; Sauge, L.; Ayerbe, N.; Coudert, G.; Merour, J.-Y. Tetrahedron Lett. 2002, 43,
589.
103 Chaudhari, S. S.; Akamanchi, K. G. Synthesis 1999, 760.
104 Wali, A.; Ganeshpure, P. A.; Satish, S. Bull. Chem. Soc. Jpn. 1993, 66, 1847.
Page 241
231
105 For general reviews on deoximations: (a) Corsaro, A.; Chiacchio, U.; Pistara, V. Synthesis
2001, 1903. (b) Corsaro, A.; Chiacchio, U.; Pistara, V. Curr. Org. Chem. 2009, 13, 482. (c)
Sahu, S.; Sahu, S.; Patel, S.; Dash, S.; Mishra, B. K. Ind. J. Chem. 2008, 47B, 259.
106Burk, M. J.; Casey, G.; Johnson, N. B. J. Org. Chem. 1998, 63, 6084.
107 Zhu, G.; Casalnuovo, A. L.; Zhang, X. J. Org. Chem. 1998, 63, 8100.
108 Sun, C.; Weinreb, S. M. Synthesis 2006, 3585.
109 (a) Boar, R. B.; McGhie, J. F.; Robinson, M.; Barton, D. H. R.; Horwell, D. C.; Stick, R. V. J.
Chem. Soc., Perkin Trans. 1 1975, 1237. (b) Boivin, J.; Schiano, Anne-Marie, Zard, S. Z.
Tetrahedron Lett. 1992, 33, 7849.
110 Pradhan, P. K.; Dey, S.; Jaisankar, P.; Giri, V. S. Synth. Commun. 2005, 35, 913.
111 For cleavage of N-O bonds in oxazolines with Fe(0), see: (a) Jiang, D.; Peng, J.; Chen, Y.
Org. Lett. 2008, 10, 1695. (b) Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181.
112 Majireck, M. M.; Witek, J. A.; Weinreb, S. M. Tetrahedron Lett. 2010, 51, 3555.
113 Van den Broek, L. A. G. M.; Breuer, M. L.; Liskamp, R. M. J.; Ottenheijm, H. C. J. J. Org.
Chem. 1987, 52, 1511.
114 (a) Trost, B. M.; Caldwell, C. G. Tetrahedron Lett. 1981, 22, 4999. (b) Corey, E. J.; Hopkins,
P. B. Tetrahedron Lett. 1982, 23, 4871.
115 (a) Burk, R. M.; Roof, M. B. Tetrahedron Lett. 1993, 34, 395. (b) Kang, S.-K.; Jeon, J.-H.;
Nam, K.-S.; Park, C.-H.; Lee, H.-W. Synth. Commun. 1994, 24, 305.
116 Schlosser, M.; Jenny, T.; Guggisberg, Y. Synlett 1990, 704.
117 For similar intramolecular alkylations see: (a) Danishefsky, S. J.; Vaughn, K.; Gadwood, R.;
Tsuzuki, K. J. Am. Chem. Soc. 1981, 103, 4136. (b) Danishefsky, S. J.; Vaughn, K.; Gadwood,
R.; Tsuzuki, K. J. Am. Chem. Soc. 1980, 102, 4262. (c) Iwata, C.; Suzuki, K.; Aoki, S.-I.;
Page 242
232
Okamura, K.; Yamashita, M.; Takahashi, I.; Tanaka, T. Chem. Pharm. Bull. 1986, 34, 4939. (d)
Kieczykowski, G. R.; Schlessinger, R. H. J. Am. Chem. Soc. 1978, 100, 1938. (e) Della, E. W.;
Tsanaktsidis, J. Aust. J. Chem. 1985, 38, 1705. (f) Kubyshkin, V. S.; Mykhailiuk, P. K.; Ulrich,
A. S.; Komarov, I. Synthesis 2009, 3327. (g) Liu, H.-J.; Llinas-Brunet, M. Can. J. Chem. 1988,
66, 528.
118 (a) Okada, Y.; Wang, J.; Yamamoto, T.; Mu, Y.; Yokoi, T. J. Chem. Soc., Perkin Trans. 1
1996, 2139. (b) Benneche, T.; Undheim, K. Acta Chem. Scand. 1983, 37, 93. (c) Benneche, T.;
Strande, P.; Wiggen, U. Acta Chem. Scand. 1988, 43, 74. (d) Watanabe, H.; Mori, N.; Itoh, D.;
Kitahara, T.; Mori, K. Angew. Chem. Int. Ed. 2007, 46, 1512.
119 Fleming, F. F.; Zhang, Z.; Wang, Q.; Steward, O. W. J. Org. Chem. 2003, 68, 7646.
120 Medina, J. C.; Salomon, M.; Kyler, K. S. Tetrahedron Lett. 1988, 29, 3773.
121 Malhotra, S. K.; Johnson, F. J. Am. Chem. Soc. 1965, 87, 5493.
122 Iwamoto, M.; Miyano, M.; Utsugi, M.; Kawada, H.; Nakada, M. Tetrahedron Lett. 2004, 45,
8647.
123 For reviews on the Fleming-Tamao oxidation see: (a) Tamao, K. Proc. Jpn. Acad., Ser. B
2008, 84, 123. (b) Mullins, R. J.; Jolley, S. L.; Knapp, A. R. In Name Reactions for Functional
Group Transformations; Li, J. J., Corey, E. J., Eds.; Wiley: Hoboken, 2007. (c) Fleming, I.
Chemtracts: Org. Chem. 1996, 9, 1. (c) Jones, G. R.; Landais, Y. Tetrahedron 1996, 52, 7599.
(d) Tamao, K. In Adv. Silicon Chem. 1996, 3, 1. (d) Schinzer, D. In Organic Synthesis
Highlights II; Waldmann, H.; Ed. New York, 1995. (e) Colvin, E. W. In Comprehensive
Organic Synthesis; Trost, B. M.; Fleming, I.; Eds. Pergamon Press, Oxford, 1991, Vol. 7.
124 For preparation of chloromethyldimethylsilyl triflate see: Bassindale, A. R.; Borbaruah, M.;
Glynn, S. J.; Parker, D. J.; Taylor, P. G. J. Chem. Soc., Perkin Trans. 2 1999, 2099.
Page 243
233
125 Bennesar, M.-L.; Roca, T.; Padulles, A. Org. Lett. 2003, 5, 569.
126 Shieh, W.-C.; Xue, S.; McKenna, J.; Prasad, K.; Repic, O.; Blacklock, T. Tetrahedron Lett.
2006, 47, 5645.
127 Myers, A. G.; Movassaghi, M.; Zheng, B. J. Am. Chem. Soc. 1997, 119, 8572.
128 Devineau, A.; Grosbois, M.; Carletti, I.; Vacher, B. J. Org. Chem. 2009, 74, 757.
129 Nagano, H.; Hara, S. Tetrahedron Lett. 2004, 45, 4329.
130 Davies, J. S.; Higginbotham, C. L.; Tremeer, E. J.; Brown, C.; Treadgold, R. C. J. Chem.
Soc., Perkin Trans. 1 1992, 3043.
131 For examples, see: (a) Terauchi, M.; Abe, H.; Tovey, S. C.; Dedos, S. G.; Taylor, C. W.; Paul,
M.; Trusselle, M.; Potter, B. V. L.; Matsuda, A.; Shuto, S. J. Med. Chem. 2006, 49, 1900. (b)
Tenenbaum, J. M.; Woerpel, K. A. Org. Lett. 2003, 5, 4325.
132 Kobayashi, T.; Pannell, K. H. Organometallics 1991, 10, 1960.
133 Uhlig, W. J. Organomet. Chem. 1993, 452, 29.
134 (a) Smitrovich, J. H.; Woerpel, K. A. J. Org. Chem. 1996, 61, 6044. (b) Bodnar, P. M.;
Palmer, W. S.; Shaw, J. T.; Smitrovich, J. H.; Sonnenberg, J. D.; Presley, A. L.; Woerpel, K. A.
J. Am. Chem. Soc. 1995, 117, 10575.
135 Kuhnel, E.; Laffan, D. D. P.; Lloyd-Jones, G. C.; del Campo, T. M.; Shepperson, I. R.;
Slaughter, J. L. Angew. Chem. Int. Ed. 2007, 46, 7075.
136 (a) Bull, J. R.; Steer, L. M. Tetrahedron 1991, 47, 7377. (b) Chen, Z.; Venkatesan, A. M.;
Dos Santos, O.; Delos Santos, E.; Dehnhardt, C. M.; Ayral-Kaloustian, S.; Ashcroft, J.;
McDonald, L. A.; Mansour, T. S. J. Org. Chem. 2010, 75, 1643. (c) For a recently developed
cyclopropenium-mediated cyclodehydration of diols, see: Kelly, B. D.; Lambert, T. H. Org. Lett.
2011, 13, 740.
Page 244
234
137 Racemic 2-chloro-3-phenylpropanal was synthesized from hydrocinnamaldehyde, NCS, and
D,L-proline following a modified literature procedure: Halland, N.; Braunton, A.; Bachmann,
S.; Marigo, M.; Jorgenson, K. A. J. Am. Chem. Soc. 2004, 126, 4790.
138 Masschelein, K. G. R.; Stevens, C. V. Tetrahedron Lett. 2008, 49, 4336.
139 Ram, R. N.; Manoj, T. P. J. Org. Chem. 2008, 73, 5633.
140 Mahboobi, S.; Bernauer, K. Helv. Chim. Acta 1988, 71, 2034.
141 We thank Hemant Yennawar of the Penn State X-Ray Small Molecule Crystallographic
Facility for structure determinations of compounds Z-357, 381, and 410.
142 Allen, J. M.; Aprahamian, E. A. S.; Shechter, H. J. Org. Chem. 2002, 67, 3561.
Page 245
VITA
Max M. Majireck
Max M. Majireck was born in 1983 and raised in the South Hills of Pittsburgh, PA. After
graduating from South Park High School in 2001, he attended Grove City College where he
earned his B.S. in biochemistry in 2005. His undergraduate research with Charles E. Kriley
focused on the synthesis and characterization of novel transition metal-phosphine coordination
complexes. While enrolled at Grove City College, Max also conducted summer research
internships at Purdue University with Ian P. Rothwell studying organometallic synthesis and at
Consol Energy developing technology for reducing mercury emissions from coal-fired power
plants. In the Fall of 2005, he joined the laboratory of Steven M. Weinreb where he worked on
the total synthesis of complex alkaloids and developing new methodologies for organic
synthesis. Upon completion of his graduate studies, Max will begin a postdoctoral appointment
with Stuart L. Schreiber at Harvard University and the Broad Institute of MIT and Harvard.