Page 1
1
INTERNATIONAL JOURNAL OF
PHARMACEUTICAL INNOVATIONS
Official Research Publication of
Nova college of Pharmaceutical Education and Research
Affiliated to Jawaharlal Nehru Technological University Kakinada,
Approved by Pharmacy Council of India and AICTE New Delhi.
Jupudi, Ibrahimpatnam- 521456, Krishna district, Andhra Pradesh.
Email id: [email protected]
Volume 1, 2014
Page 2
2
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Editorial Board
Editor in-Chief
Dr. P. Selvam M.Pharm, Ph.D., FNABS, (NCPER)
Associate editor
M. Tejaswi M. Pharm.(NCPER)
Editorial Board Members
Dr. G. Narasimhan M.Pharm, Ph.D (Harvard University, USA)
Dr. Andrea Brancle ,Ph.D (Cadiff University, UK)
Dr. C. N. Ramchand., Ph.D., ( Laila Pharmaceutical., Pvt Ltd. India)
Dr. D. Selvakumar , M.Pharm., Ph.D (Wayne StateUniversity, USA).
Dr. M. Chandramohan ,M.D ,Ph.D (Madurai)
Dr. N. Murugesh B Sc, MBBS., M.Sc., Ph.D (Madurai)
Dr. P. Vasanthraj, M.Pharm, Ph.D (AIMST, Malaysia)
Dr. R. Govindharajan, M.Pharm., Ph.D (Head Dabur R&D centre, Dubai)
Dr. K. Prabhu M.Pharm, Ph.D (Nova College of Pharmacy, Jangareddygudem)
Dr. Kasthur Reddy, M.Sc, PhD (Mako Research Labs, Hyderabad)
Dr. P. Venkatesh, M.Pharm ,Ph.D ( Senior Principal Scientist, Orchid R&D centre, Chennai)
Dr. Alagasamy, M.Pharm, Ph.D (Hyderabad)
Dr. D.Sriram, M.Pharm, Ph.D, ( BITS, Hyderabad)
Dr. Subashini M.Pharm, Ph.D, (MSU University, Kuala lumbur, Malaysia)
Page 3
3
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
INSTRUCTIONS TO AUTHORS
The International Journal of Pharmaceutical Innovations is a e-Journal, which publishes innovative
research papers, reviews, mini-reviews, short communications and notes dealing with Pharmaceutical
Sciences (Pharmaceutical Technology, Pharmaceutics, Biopharmaceutics, Pharmacokinetics,
Pharmaceutical/Medicinal Chemistry, Computational Chemistry and Molecular Drug Design,
Pharmacognosy and Phytochemistry, Pharmacology, Pharmaceutical Analysis, Pharmacy Practice,
Clinical and Hospital Pharmacy, Cell Biology, Genomics and Proteomics, Pharmacogenomics,
Bioinformatics and Biotechnology of Pharmaceutical Interest). All manuscripts are subject to rapid peer
review. Those of high quality (not previously published and not under consideration for publication
in another journal) will be published without delay.
Manuscript Format
The preferred format of all manuscript is MS Word. Illustrations (figures) and images must be inserted in
the last of after all the text material.
Preparation of Manuscript
Mode of Presenting Paper is English. Each manuscript should be typed single-spaced on A4 (8.5" × 11")
paper size with 1 inch margins. It should be arranged in the following order: Title, Abstract, Keywords,
Introduction, Materials and Methods, Results, Discussion and References.
Title Page
Title page should contain title of the paper in bold face, title case (font size 14), names of the authors in
normal face, upper case (font size 12) followed by the address in normal face lower case. The author to
whom all correspondence be addressed should be denoted by an asterisk mark. The title should be as
short as possible and precisely indicate the nature of the work in the communication. Names of the
authors should appear as initials followed by surnames. At the bottom left corner of the title page,
please mention “*Address For correspondence” and provide a functional e-mail address. Address of the
corresponding author to whom all correspondence may be sent should be given only if it is different from
the address already given under authors' names.
Abstract
Should start on a new page after the title page and should be typed in single-space to distinguish it from
the Introduction. Abstracts should briefly reflect all aspects of the study, as most databases list mainly
abstracts. The manuscript should have an abstract 150- 250 words.
Keywords
Provide four to six appropriate key words after abstract
Page 4
4
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Introduction
A short introduction of the research problem followed by a brief review of literature and objective
of the research.
Materials and Methods Describe the materials used in the experiment, year of experimentation, site etc. Describe the methods
implied for collection of data in short.
Results and Discussion
This segment should focus on the fulfillment of stated objectives as given in the introduction. It should
contain the findings presented in the form of tables, figures and photographs.
References
Should be numbered consecutively in the order in which they are first mentioned in the text (not in
alphabetic order). Identify references in text, tables and legends by Arabic numerals in superscript.
References cited only in tables or figure legends should be numbered in accordance with the sequence
established by the first identification in the text of the particular table or figure.
Journal Articles
Shashi A, Jain SK and Pandey M: In-vitro evaluation of antilthiatic activity of seeds of Dolichos
biflorus and roots of Asparagus racemosus . International Journal of Plant Sciences 2008; 1:67-71.
A Book
Kalia AN: A Text Book of Industrial Pharmacognosy. CBS Publishers & Distributors, First Edition
2005.
A Chapter in a Book
Nadkarni KM: Indian Materia Medica. Popular Prakashan, Mumbai, Edition 3, Vol. I, 2000:
242-246.
Illustrations
All the tables and figures should be after the text at suitable place. Only MS word table format should be
used for preparing tables. Tables should show lines separating rows and columns. Tables should be
numbered consecutively in Arabic numerals and bear a brief title in capital letters normal face. Tables
should not be very large that they run more than one A4 sized page. Table format should be as follow:
Phytochemical Analysis of successive extract of............
Chemical Constituent Aqueous Extract Ethanolic Extract
Page 5
5
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Abbrevations, Units Etc The journal strictly follows the rules defined in the IUPAC Manual of symbols and terminology for
physicochemical quantities and units. Short Communication The journal publishes exciting findings, preliminary data or studies that did not yield enough
information to make a full paper as short communications. These have the same format requirements as
full papers but are only up to 10 pages in length in total. Short Communications should have
subtitles such as Introduction, Materials and Methods, Results and Discussion- all these have to be
merged into the running text. Short Communications preferably should have only 3-4 illustrations.
Review Artcile: Should be about 15-30 pages long, contain up-to-date information, comprehensively cover relevant
literature and preferably be written by scientists who have in-depth knowledge on the topic. All format
requirements are same as those applicable to full papers. Submission of Manuscript All manuscripts (must be in English and in MS Word format) and should be submitted via our online
system or through e-mail at [email protected] as an attachment for quick evaluation. Copyright and Permission Submission is a representation that the manuscript has not been published previously and is not under
consideration for publication elsewhere. Authors would be required to sign a form (to be supplied by the
Editor) transferring copyright before the manuscript can be published. Ethical matter Authors publishing results from in vivo experiments involving animals or humans should state whether
due permission for conduction of these experiments was obtained, from the relevant ethics committees,
in the Materials and Methods section. In addition, authors wishing to publish research work involving
human studies should also send a notary verified letter of approval from the Ethics Committee or the
Institutional Review Board. Authors are requested to send their research articles strictly according to the given format
mentioned in the guidelines to the authors.
Email id: [email protected]
Page 6
6
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Contents
Invited article:
DESIGN, MOLECULAR MODELLING STUDIES ON CHLOROQUINE AND ITS
ANALOGUES AS INHIBITORS OF HCV NS5B RNA POLYMERASE
M. ChandraMohan, P. Selvam , J. Pranitha, N. Saravanan Prabhu, M. Suresh Kumar Pg no: 5-20
Research article:
DISSOLUTION ENHANCEMENT OF PIOGLITAZONE HYDROCHLORIDE BY SOLID
DISPERSION TECHNIQUE
T. Divya , S. Pranav ----- Pg no : 21-41
AN ATTEMPT TO ISOLATE INDUSTRIALLY IMPORTANT MICROORGANISM FROM
DIVERSE SAMPLES COLLECTED FROM SEMELING, KEDAH, MALAYSIA
P. Vasanth Raj, S.A. Dhanaraj, Saw Pei Jie, Sim Poh Pei, Vanushya a/p Alagasan, Tien Ja She,
Loh Mon Ying, Tan Te Wei Pg no : 42-55
Short communications :
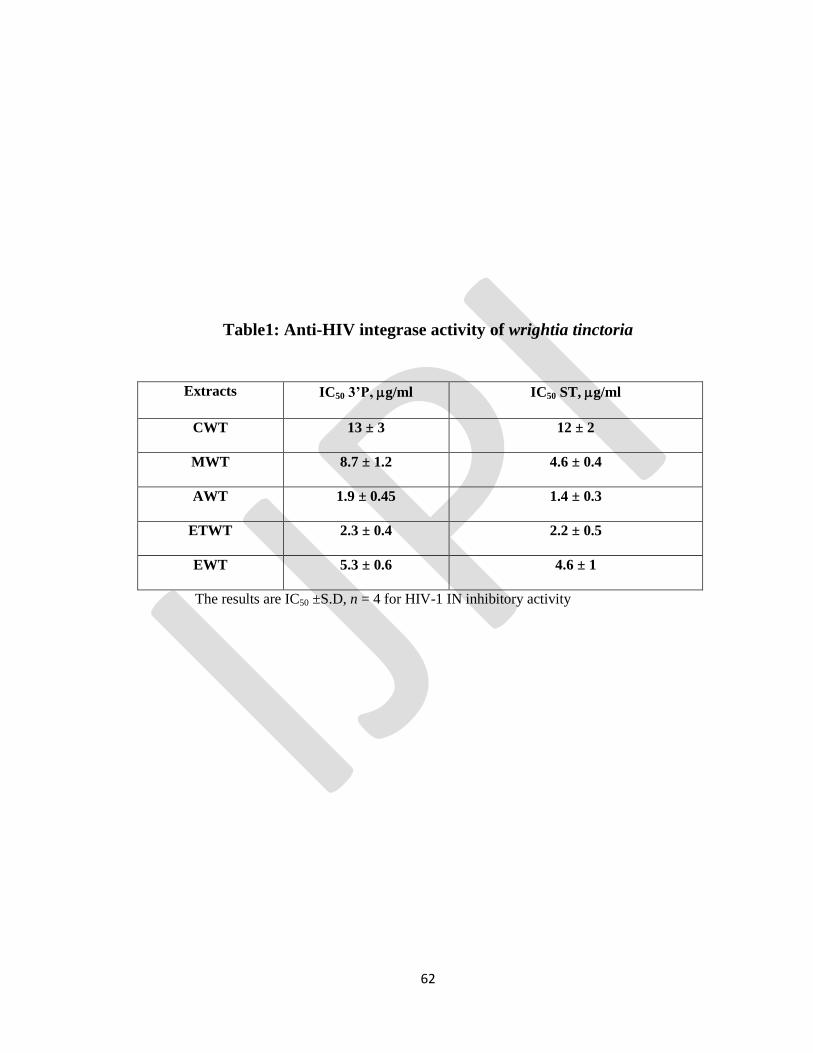
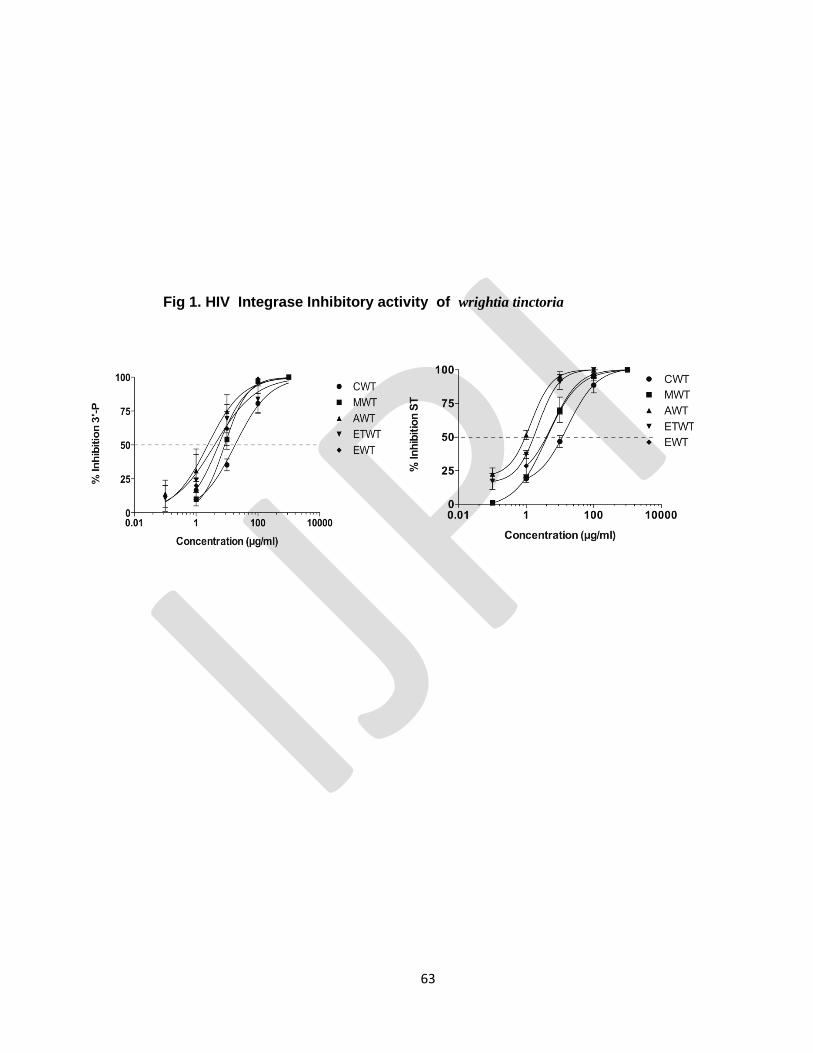
STUDIES OF HIV INTEGRASE INHIBITORY ACTIVITY OF WRIGHTIA TINCTORIA
Periyasamy Selvam, Kasthuraiah Maddali , Christophe marchand
, Yves Pommier Pg no : 55-63
DRUG DISCOVERY AND DEVELOPMENT-CURRENT STATUS AND FUTURE FOCUS
N. Somashekara., P. Jagadeesh., C.N. Ramchand Pg no : 64-67
Page 7
7
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Invited Article
DESIGN, MOLECULAR MODELLING STUDIES ON CHLOROQUINE AND ITS
ANALOGUES AS INHIBITORS OF HCV NS5B RNA POLYMERASE
M. ChandraMohan 1, P. Selvam
2, J. Pranitha
3, N. Saravanan Prabhu
3, M. Suresh Kumar
3
1 Bharat Ratna Kamarajar liver Hospital and Research Centre, Madurai-625020,
2Nova College of Pharmaceutical Education and Research, Jupudi, Krishna DT, AP,
3 Centre for Bioinformatics, School of Life Sciences, Pondicherry University, Puducherry-605014.
Page 8
8
ABSTRACT
Hepatitis C virus (HCV) a member of the Flavivirus genus is a negative strand RNA virus that is
estimated to have infected and rendering 170 million people as carrier of HCV globally. Ribavirin and
interferon combination therapy is now available to treat HCV viral infection. Due to high rate of viral
drug resistance, we need new targeted therapy to combat HCV. Structure-based drug design methods
utilize knowledge of three dimensional structure of an enzyme to develop some novel inhibitors of
HCV. NS5B protein encoded RNA dependent RNA polymerase activity has a critically important role in
HCV RNA replication and so considered as an attractive therapeutic target for designing newer classes of
antiviral compounds. Present work is to investigate the molecular modeling studies of quinoline
derivatives such as Chloroquine, Hydroxychloroquine and Primaquine with HCV RNA polymerase.
Modelling studies were done after the three quinoline derivatives 3D structures were converted from 2D;
using the Schrodinger Maestro the compounds were docked to the active site residues of HCV RNA
Polymerase which involves interaction of Lipophilic compounds. They formed hydrophobic interaction
with quinoline rings of Chloroquine and Primaquine and with the alkyl groups of hydroxy chloroquine.
Results of the molecular modelling studies indicate that the quinoline derivatives strongly interact with
the residues in the primer grip site of the polymerase.
Page 9
9
INTRODUCTION
The World Health Organization has estimated that there are 170 million HCV carriers in the world with a
substantial risk of developing liver cirrhosis and/or liver cancer. There is currently neither an effective
vaccine nor anti- HCV-specific therapeutics for HCV infection. Development of anti-HCV agents is thus
a matter of much importance. HCV is a positive single stranded RNA virus whose genome encodes one
polyprotein, which is processed to structural and non-structural proteins by host and viral proteases. A
non-structural protein at the polyprotein C terminus, NS5B, includes the RNA-dependent RNA
polymerase responsible for HCV replication. This RNA-dependent RNA polymerase is validated target
for the development of newer anti-HCV therapeutic agent.
Chloroquine is a versatile bioactive agent and reported to possess antiviral activity against human
immunodeficiency virus (Romanelli et al., 2004), Hepatitis A virus (Superti et al., 1987; Bishop, 1998),
and severe acute respiratory syndrome virus (Keyaerts et al, 2004) infections. The documented antiviral
activities of chloroquine are by the following mechanisms (1) Viral particles entry into target cells by
endocytosis is blocked by chloroquine (Carrillo et al.,1985 ; Zeichhardt, et al., 1985),
(2) As
lysosomotrophic agent chloroquine accumulates in lysosome of host cell and increases the lysosomal pH
and renders lysosomal enzymes inactive and prevent the viral decoating (Carrillo et al., 1984), (3)
Intercalating action; chloroquine intercalates the HBV-DNA polymerase and inhibits the enzyme activity
of the virus and blocks the multiplication of HBV (Hirschman, S. Z.1974; Garfinkel, E. 1978), (4)
Chloroquine inhibits Duck hepatitis B virus super coiled DNA (DHBV-sc DNA) (Civitico et al., 1990)
and Duck hepatitis B surface antigen (DHBsAg) (Offensperger, et al., 1991). Also Chloroquine inhibits
the replication of HIV-1 and 2 at clinically achievable concentration and the anti HIV activity is by (5)
inhibition of synthesis of surface antigen GP120 of HIV (Savarino et al., 2001) (6) by inhibition of HIV
Integrase (Fesen et al., 1993) (7) Chloroquine also inhibits the replication of Ribonucleic Acid (RNA) of
Page 10
10
Sindbis virus (Cassell et al., 1984). Inhibition of normal proteolytic processing of Flavivirus Pr.M Protein
(Randolph et al., 1990) is taken as the eighth mode of antiviral action of Chloroquine. The eight modes of
antiviral activity of Chloroquine made us to look for antiviral action for Primaquine an analogue of
Chloroquine and found that Primaquine has antiviral actions against HIV by blocking HIV Integrase, and
New Castle Virus by blocking the synthesis of its RNA and release of its progeny( Burdick et al, 1974).
We have previously reported antiviral activities of chloroquine and hydroxy chloroquine against
Hepatitis C virus in Huh-5-2 cells (Bartenschlager 2002; Chandramohan et al., 2006) and Primaquine
against Severe Acute Respiratory Syndrome (SARS-CoV) in Vero cells (Chandramohan et al., 2009) In
view of many modes of antiviral activities of chloroquine and primaquine against many human
pathogenic viruses; the present work is to evaluate the binding studies of quinoline derivatives against
HCV RNA polymerase activity by molecular modelling studies.
METHODOLOGY
Molecular docking studies were performed using Schrödinger Maestro v8.0, and Glide v4.5.19.
Chloroquine, Primaquine and Hydroxy chloroquine structures were obtained as 2D and then converted
into 3D structures and optimized using the Maestro. The three compounds were preprocessed with
LigPrep at pH 7. Generated 3D structures of ligand were used to dock to the active site residues of HCV
RNA polymerase.The 3D structure of protein chosen for molecular docking study is 1Z4U. The protein
was prepared and optimized using the Protein Preparation Wizard tool supplied with Schrödinger Glide
(Friesner et al., 2004; Halgren et al., 2004). Using this tool, hydrogen atoms were added to the RNA
polymerase, the protonation states for histidine residues were optimized, crystallographic waters not
deemed to be important for ligand binding were deleted, and the entire protein was minimized. The
OPLS2005 force field was used for all calculations. Previous to this process the missing residues of the
protein were modeled using the PrimeX module. Next, a grid was prepared for docking the three ligands
Page 11
11
into HCV RNA Polymerase using the Receptor Grid Generation tool in Glide. The binding site was
defined as the residues located within 10 Å from Tyr448 of the macromolecule. To compensate for the
rigid representation of the receptor, the Van der Waals radii of the atoms with partial charges lower than
0.25 were scaled by the factor 0.8 to generate the receptor grid. No constraints were defined for the
docking runs. For this single-ligand automated docking, the Simple Precision (SP) mode in Glide was
selected. Default settings were used for docking. The final ligand poses were selected based on the Glide
empirical docking scores (GlideScore). The highest-scoring docking pose returned for each of the ligand
was considered for interaction studies. Docked result was analyzed using Glide Pose Viewer and with
Pymol21
(DeLano 2002). All conformations of the docked compounds were compared with the preceding
reports ( Pfefferkorn et al., 2005a,b).
RESULT AND DISCUSSION
Interaction study of Lipophilic compounds with the Active residues of HCV RNA polymerase
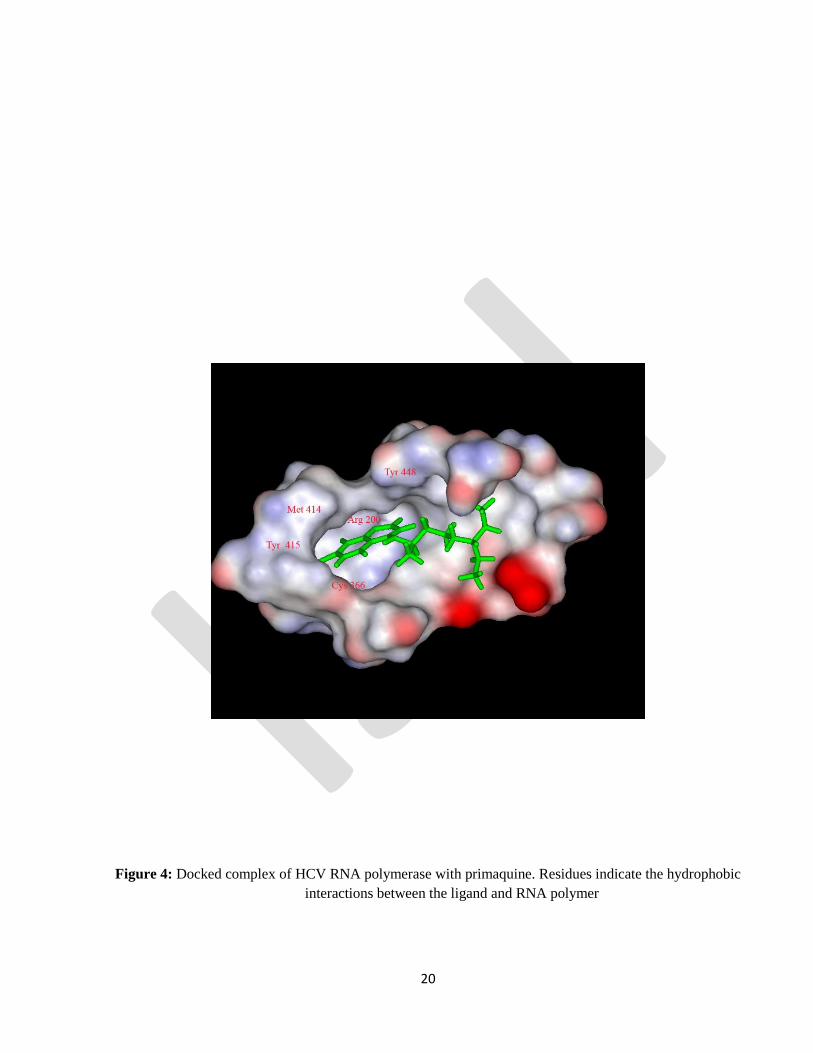
Primaquine: Preceding reports suggest that amino acids in the binding pocket are hydrophobic in nature
which forms hydrophobic interaction with the lipophilic compounds. Due to the aromatic side chain and
non polar side chain groups of amino acids of binding pocket such as Tyr 448, Tyr 415, and Met 414 and
Leu 384 form hydrophobic interaction with the quinoline ring of Primaquine. The highest-scoring pose
for the Primaquine protein complex obtained from this SP Glide docking study had a docking score of -
6.66 kcal/mol. Other residues such as Cys 366 and Arg 200 also involve in hydrophobic interaction (
Pfefferkorn et al., 2005a,b) . The ligand has established 2 hydrogen bond interactions with the protein
residues. The H25 of the ligand has established a hydrogen bond with the hydroxyl oxygen in the residue
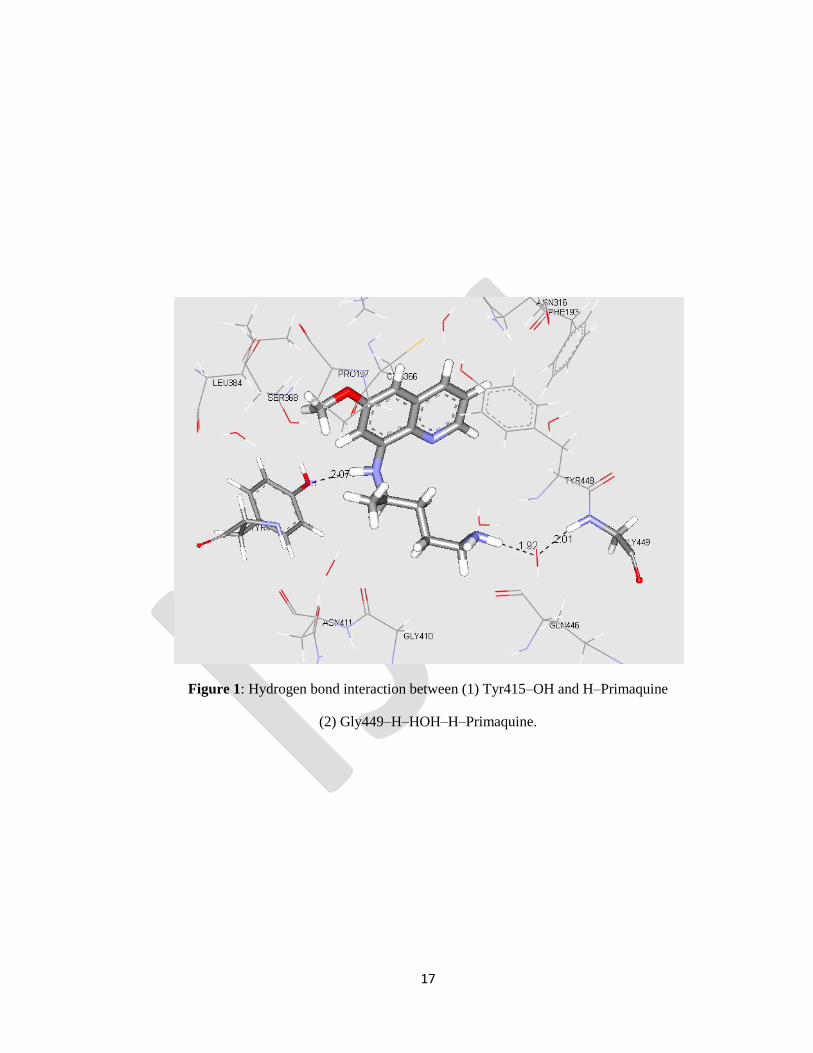
Tyr415.9 (Fig.1 and 4).
Page 12
12
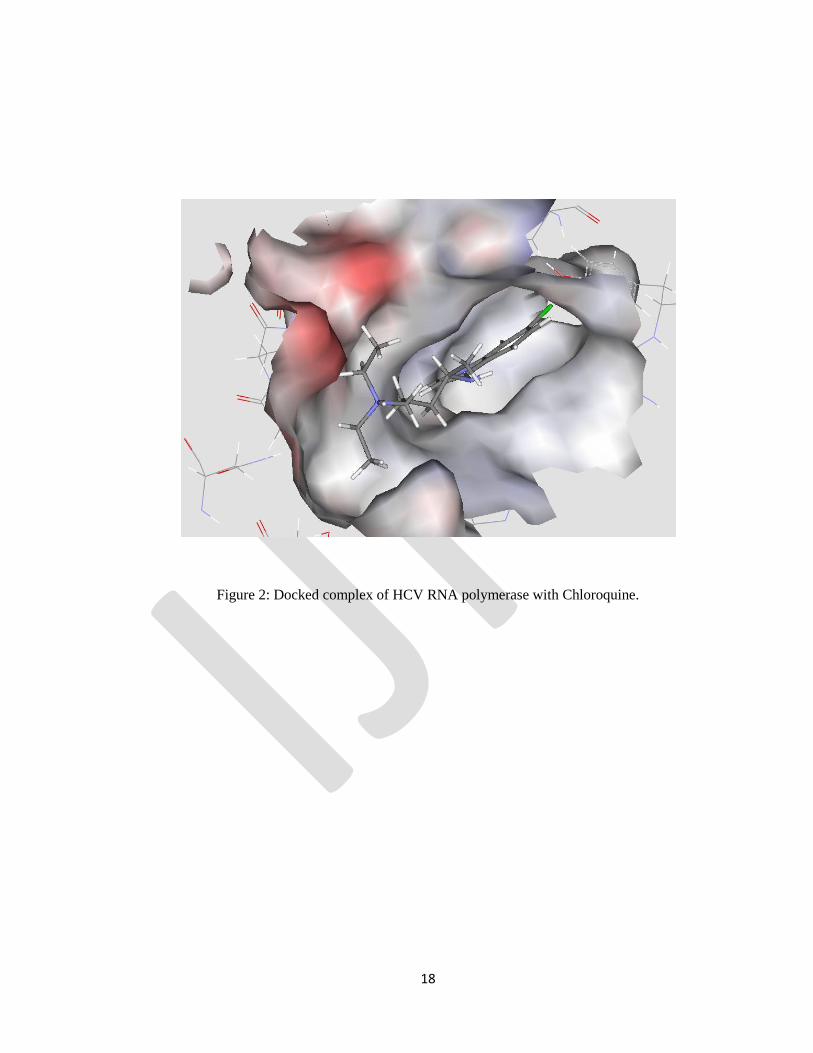
Chloroquine: The highest scoring pose for the Chloroquine-protein complex obtained from the SP Glide
docking result had a score of -5.35kcal/mol. Amino acids such as Arg200, Cys366, Tyr448, Ile447,
Pro197, Leu384 and Tyr415 form hydrophobic interaction with quinoline ring of Chloroquine (Fig.2).
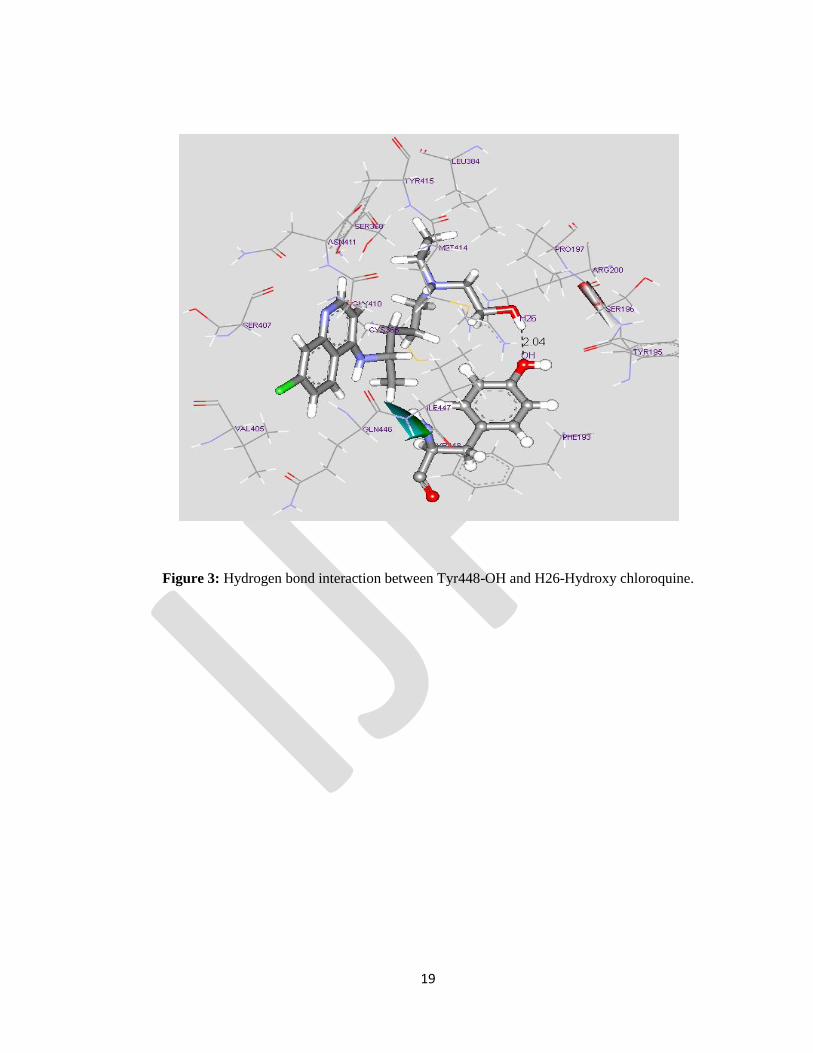
Hydroxy chloroquine: Hydroxyl oxygen of the Tyrosine 448 residue establishes a hydrogen bond with
the hydrogen moiety of Hydroxy chloroquine (Tyr448-OH…H26–Hydroxychloroquine) spanning of a
distance of 2.04Ao
(Fig.3). Other amino acids in the binding pocket show hydrophobic interaction with
the alkyl group of the hydroxy choloroquine ring. The interaction between the lipophilic compounds with
the active site residues of HCV RNA polymerase was preserved among the Lipophilic compounds (
Pfefferkorn et al., 2005a,b)
CONCLUSION
With the results on hand we may suggest or infer that the biochemical configuration of both the ligand
and the virus, along with their lipophilicity and hydrophobicity may determine the docking site and the
level of bonding. This may dictate the efficiency of the drug action and the susceptibility of the virus.
Possibly this docking and binding of pockets in the HCV RNA polymerase active site residues was the
mode of action in our work published in 2006. Now the new mode of action on HCV – NS5B polymerase
targeted action is being established.
Having noted “Eight” modes of antiviral activity for chloroquine against many human pathogenic
viruses, this molecular modelling and showing the binding of quinoline compound may add up the
modes of action to “Nine” and name chloroquine as a versatile, unique and promising antiviral agent. The
antiviral activity of lipophilic amine and acidotropic amine (both happen to be Chloroquine) had been
documented in 1987 and 1990 respectively. Chloroquine documented for anti-HCV activity with
inhibition of viral entry (Usman et al., 2011). Ferroquine (FQ), an antimalarial ferrocenic analog of
Page 13
13
chloroquine, is a novel inhibitor of HCV (Vausselin et al., 2013). FQ potently inhibited HCV infection of
hepatoma cell lines by affecting an early step of the viral life cycle. Recently our group also
demonstrated chloroquine inhibits the replication of Human Noro virus (HNV) at 24 + 2.2 ug/ml and
cytotoxicity was found to be 98 + 5.5 ug/ml (unpublished data).
REFERENCE:
Bishop, N. E. Examination of potential inhibitors of hepatitis A virus uncoating. Intervirology 1998, 41,
261-71.
Burdick, J. R.; Durand, D. P. Primaquine diphosphate: inhibition of Newcastle disease virus replication.
Antimicrob Agents Chemother 1974, 6, 460-4.
Bartenschlager, R. Hepatitis C virus replicons: potential role for drug development. Nature reviews 2002,
1, 911-6.
Cassell, S.; Edwards, J.; Brown, D. T. Effects of lysosomotropic weak bases on infection of BHK-21
cells by Sindbis virus. J Virol 1984, 52, 857-64.
Carrillo, E. C.; Giachetti, C.; Campos, R. H. Effect of lysosomotropic agents on the foot-and-mouth
disease virus replication. Virology 1984, 135, 542-5.
Carrillo, E. C.; Giachetti, C.; Campos, R. Early steps in FMDV replication: further analysis on the effects
of chloroquine. Virology 1985, 147, 118-25.
Chandramohan, M.; Vivekananthan , S. C.; Sivakumar, D.; Selvam, P.; Neyts , J.; Katrien, G.; De Clercq,
E. Preliminary report of anti-hepatitis C virus activity of chloroquine and hydroxychloroquine in huh-5-2
cell line. Indian Journal of Pharmaceutical Sciences 2006, 68, 538-540.
Page 14
14
Chandramohan, M.; Selvam, P.; Vivekananthan, S. C.; Sivakumar, D.; Keyaerts, E.; Van Ranst, M.;
Vijgen, L.; Neyts, J. In vitro inhibition of Severe Acute Respiratory Syndrome Coronavirus by
Primaquine. International Journal of Chemical Sciences. 2009, 2(7), 1195-1198.
Civitico, G.; Wang, Y. Y.; Luscombe, C.; Bishop, N.; Tachedjian, G.; Gust, I.; Locarnini, S. Antiviral
strategies in chronic hepatitis B virus infection: II. Inhibition of duck hepatitis B virus in vitro using
conventional antiviral agents and supercoiled-DNA active compounds. J Med Virol 1990, 31, 90-7.
DeLano, W. L. The PyMOL Molecular Graphics System. 2002.
Friesner, R. A.; Banks, J. L.; Murphy, R. B.; Halgren, T. A.; Klicic, J. J.; Mainz, D. T.; Repasky, M. P.;
Knoll, E. H.; Shelley, M.; Perry, J. K.; Shaw, D. E.; Francis, P.; Shenkin, P. S. Glide: a new approach for
rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of
Medicinal Chemistry 2004, 47, 1739-49.
Fesen, M. R.; Kohn, K. W.; Leteurtre, F.; Pommier, Y. Inhibitors of human immunodeficiency virus
integrase. Proceedings of the National Academy of Sciences of the United States of America 1993, 90,
2399-403.
Halgren, T. A.; Murphy, R. B.; Friesner, R. A.; Beard, H. S.; Frye, L. L.; Pollard, W. T.; Banks, J. L.
Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database
screening. Journal of Medicinal Chemistry 2004, 47, 1750-9.
Hirschman, S. Z.; Garfinkel, E. Inhibition of hepatitis B DNA polymerase by intercalating agents. Nature
1978, 271, 681-3.
Keyaerts, E.; Vijgen, L.; Maes, P.; Neyts, J.; Van Ranst, M. In vitro inhibition of severe acute respiratory
syndrome coronavirus by chloroquine. Biochemical and Biophysical Research communications 2004,
323, 264-8.
Page 15
15
Offensperger, W. B.; Offensperger, S.; Walter, E.; Blum, H. E.; Gerok, W. Inhibition of duck hepatitis B
virus infection by lysosomotropic agents. Virology 1991, 183, 415-8.
Pfefferkorn, J. A.; Greene, M. L.; Nugent, R. A.; Gross, R. J.; Mitchell, M. A.; Finzel, B. C.; Harris, M.
S.; Wells, P. A.; Shelly, J. A.; Anstadt, R. A.; Kilkuskie, R. E.; Kopta, L. A.; Schwende, F. J. Inhibitors
of HCV NS5B polymerase. Part 1: Evaluation of the southern region of (2Z)-2-(benzoylamino)-3-(5-
phenyl-2-furyl)acrylic acid. Bioorganic & medicinal chemistry letters 2005, 15, 2481-6.
Pfefferkorn, J. A.; Nugent, R.; Gross, R. J.; Greene, M.; Mitchell, M. A.; Reding, M. T.; Funk, L. A.;
Anderson, R.; Wells, P. A.; Shelly, J. A.; Anstadt, R.; Finzel, B. C.; Harris, M. S.; Kilkuskie, R. E.;
Kopta, L. A.; Schwende, F. J. Inhibitors of HCV NS5B polymerase. Part 2: Evaluation of the northern
region of (2Z)-2-benzoylamino-3-(4-phenoxy-phenyl)-acrylic acid. Bioorganic & Medicinal Chemistry
letters 2005, 15, 2812-8.
Randolph, V. B.; Winkler, G.; Stollar, V. Acidotropic amines inhibit proteolytic processing of flavivirus
prM protein. Virology 1990, 174, 450-8.
Romanelli, F.; Smith, K. M.; Hoven, A. D. Chloroquine and hydroxychloroquine as inhibitors of human
immunodeficiency virus (HIV-1) activity. Current Pharmaceutical Design 2004, 10, 2643-8.
Savarino, A.; Gennero, L.; Chen, H. C.; Serrano, D.; Malavasi, F.; Boelaert, J. R.; Sperber, K. Anti-HIV
effects of chloroquine: mechanisms of inhibition and spectrum of activity. AIDS (London, England)
2001, 15, 2221-9.
Superti, F.; Seganti, L.; Orsi, N.; Divizia, M.; Gabrieli, R.; Pana, A. The effect of lipophilic amines on
the growth of hepatitis A virus in Frp/3 cells. Archives of Virology 1987, 96, 289-96.
Usman A Ashfaq, Tariq Javed, Sidra Rehman, Zafar Nawaz and Sheikh Riazuddin Lysosomotropic
agents as HCV entry inhibitors. Virology Journal 2011, 8:163: 2-6
Page 16
16
Vausselin T, Calland N, Belouzard S, Descamps V, Douam F, Helle F, François C, Lavillette D, Duverlie
G, Wahid A, Fénéant L, Cocquerel L, Guérardel Y, Wychowski C, Biot C, Dubuisson J. The antimalarial
ferroquine is an inhibitor of hepatitis C virus. Hepatology. 2013 Jul;58(1):86-97
Zeichhardt, H.; Wetz, K.; Willingmann, P.; Habermehl, K. O. Entry of poliovirus type 1 and Mouse
Elberfeld (ME) virus into HEp-2 cells: receptor-mediated endocytosis and endosomal or lysosomal
uncoating. The Journal of General Virology 1985, 66 ( Pt 3), 483-92.
Page 17
17
Figure 1: Hydrogen bond interaction between (1) Tyr415–OH and H–Primaquine
(2) Gly449–H–HOH–H–Primaquine.
Page 18
18
Figure 2: Docked complex of HCV RNA polymerase with Chloroquine.
Page 19
19
Figure 3: Hydrogen bond interaction between Tyr448-OH and H26-Hydroxy chloroquine.
Page 20
20
Figure 4: Docked complex of HCV RNA polymerase with primaquine. Residues indicate the hydrophobic
interactions between the ligand and RNA polymer
Page 21
21
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
DISSOLUTION ENHANCEMENT OF PIOGLITAZONE HYDROCHLORIDE BY
SOLID DISPERSION TECHNIQUE
T. DIVYA 1, S. PRANAV 1*
1Maliba Pharmacy College, Bardoli-Mahuva road,Dist. Surat (Gujarat), India - 394 350
Address for correspondence:
Maliba Pharmacy College, Bardoli-Mahuva road,
Dist. Surat (Gujarat), India - 394 350
[email protected]
Page 22
22
ABSTRACT
The present study focuses on the formulation of immediate release tablets containing solid dispersion of
Pioglitazone hydrochloride (PGH), a poorly soluble drug used in the treatment of type II diabetes. The solid
dispersions (SDs) were prepared by melting method using hydrophilic carriers namely polyethylene glycol
(PEG) 6000 and Poloxamer 188 individually in different ratios. The prepared SDs were evaluated for
solubility, drug content, Fourier transform infra red spectroscopy (FTIR), differential scanning calorimetry
(DSC), X-ray diffraction (XRD), and scanning electron microscopy (SEM). The solid dispersions were
formulated into immediate release tablets by wet granulation method. The tablets were evaluated for weight
variation, thickness, hardness, friability, assay, in vitro disintegration time and in vitro drug release. The
hypoglycaemic response of the optimized formulation was also studied in Albino wistar rats. The solubility of
PGZ was enhanced Results of SDs showed enhanced solubility of in 0.1 N HCl. FTIR, DSC, XRD and SEM
studies confirmed the formation of solid dispersion and conversion of drug from crystalline to amorphous. He
optimized batch of SDs showed better in vitro dissolution profile compared to the marketed tablets. In vivo
studies exhibited better hypoglycaemic activity than marketed tablets. The formulations were proved to be
stable after three months accelerated stability studies.
Keyword(s): Pioglitazone hydrochloride, solid dispersion, solubility, dissolution, PEG 6000 and Poloxamer
188
Page 23
23
INTRODUCTION
Oral administration is the most popular route for systemic effects due to its ease of ingestion, avoidance of
pain, versatility and most importantly patient compliance. Oral bioavailability of a drug depends on its solubility
and/or dissolution rate. Majority of the new chemical entities (NCE) and several existing drug molecules possess poor
solubility, thereby limiting their potential uses and increasing the difficulty of formulating bioavailable drug
delivery systems.1 There are various techniques employed to improve the solubility of poorly soluble
drugs. e.g. particle size reduction: micronization, nanosuspension; modification of the crystal habit:
polymorphs, pseudopolymorphs; drug dispersion in carriers: eutectic mixtures, solid dispersions, solid
solutions; complexation: use of complexing agents; solubilization by surfactants: microemulsions, self
microemulsifying drug delivery systems.2
Solid dispersion (SD) is one of the widely used approaches to improve the solubility of poorly water soluble
drugs. A solid dispersion can be defined as a dispersion of one or more drugs in an inert hydrophilic carrier or
matrix in the solid state. Once the solid dispersion is exposed to aqueous media, the hydrophilic carrier
dissolves spontaneously, releasing the drug as very fine, colloidal particles. This leads to greatly enhanced
surface area, as a result of which, the dissolution rate and bioavailability of poorly water-soluble drugs are
expected to be high.
SDs can be prepared by number of methods like melting method, solvent evaporation, spray drying, fusion
method, etc.3 Among all the methods, melting method seems quite advantageous compared to other methods.
This method avoids the exposure to hazardous organic solvents. The carrier having low melting point can be
selected so that heat exposure to the drug can also avoided. The method is simple compared to others and has
industrial applicability.4
PGH is a thiazolidinedione class of antidiabetic agent that decreases insulin resistance in the periphery and in
the liver resulting in increased insulin-dependent glucose disposal and decreased hepatic glucose output. PGH
is a potent and highly selective agonist for peroxisome proliferator-activated receptor (PPAR)-gamma.
Activation of PPAR-gamma receptors regulates the transcription of insulin-responsive genes involved in the
control of glucose production, transport, and utilization. In this way, PGH enhances tissue sensitivity to
insulin. In general, rapid gastrointestinal (GI) absorption is required for oral hypoglycaemic drugs, in order to
prevent a sudden increase in blood glucose level after food intake in patient with diabetes mellitus. However,
PGH exhibits poor aqueous solubility and its absorption is dissolution rate limited. This eventually limits it
oral bioavaibility and therapeutic efficacy.5
Page 24
24
The aim of the present investigation was to enhance the solubility of a poorly soluble drug, PGH, using solid
dispersion technique and thereby improve its dissolution characteristics. The solid dispersions were prepared
using hydrophilic carriers: PEG 6000, and Poloxamer 188. The solid dispersions were characterized by FTIR,
DSC and XRD techniques. The solid dispersions were compressed into immediate release tablets for oral
delivery. These tablets were evaluated for the quality control parameters and the dissolution rate. The
therapeutic efficacy of the prepared formulations was checked by in vivo in Albino wistar rats by measuring
blood glucose levels.
Materials and Methods
PGH was received as a gift sample from Torrent Research Centre, Ahmedabad, India. PEG 6000 and
Poloxamer 188 were purchased from Balaji drugs, Surat, India. Lactose monohydrate, starch, magnesium
stearate, talc and PVP K 30 were obtained from S.D Fine Chemicals Ltd., Mumbai, India. All other chemicals
and reagents used were of analytical grade.
Preformulation studies
The overall objective of preformulation testing is to generate information useful in developing the formulation
which is stable and acceptable. Physicochemical properties of the bulk drug like physical appearance,
solubility, bulk density, tapped density, compressibility index and drug-excipient compatibility were
studied.
Estimation of Pioglitazone hydrochloride
10 mg of PGH was dissolved in 10 ml of methanol and the volume was adjusted up to 100 ml with 0.1N
hydrochloric acid (100µg/ml)(Stock solution). The above solution was diluted with 0.1 N HCl to obtain the
series of dilutions containing 10, 15, 20, 25, 30, 35 and 40µg/ml of PGH. The absorbances of the above
solutions were measured on a Shimadzu UV-Visible Spectrophotometer at 269.5 nm. The calibration curve
was plotted and it was used for the estimation of PGH.
Preparation of solid dispersions
SDs of PGZ was prepared by melting method using PEG 6000 and Poloxamer 188 as hydrophilic carriers.
Three different weight ratios of drug: carrier of 1:1, 1:2, 1:3 and 1:4 were used (Table 1). A weighed amount
of carrier was taken in a porcelain dish and heated up to its melting point (60˚C for PEG 6000 and 55˚C for
Poloxamer 188) on a water bath. The drug was dispersed into molten carrier with continuous stirring. The
Page 25
25
molten mass was removed from water bath and cooled on the ice bath till solidification. Then the solid mass
was pulverized and sifted through 40 # sieve.
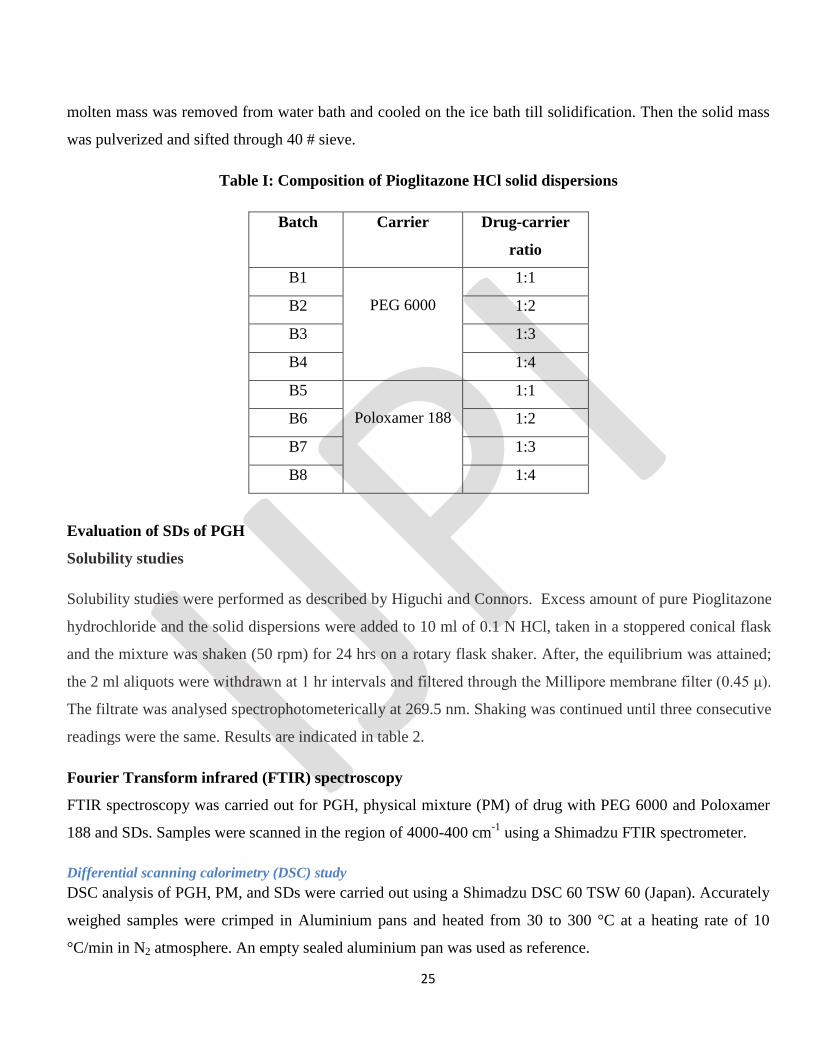
Table I: Composition of Pioglitazone HCl solid dispersions
Batch Carrier Drug-carrier
ratio
B1
PEG 6000
1:1
B2 1:2
B3 1:3
B4 1:4
B5
Poloxamer 188
1:1
B6 1:2
B7 1:3
B8 1:4
Evaluation of SDs of PGH
Solubility studies
Solubility studies were performed as described by Higuchi and Connors. Excess amount of pure Pioglitazone
hydrochloride and the solid dispersions were added to 10 ml of 0.1 N HCl, taken in a stoppered conical flask
and the mixture was shaken (50 rpm) for 24 hrs on a rotary flask shaker. After, the equilibrium was attained;
the 2 ml aliquots were withdrawn at 1 hr intervals and filtered through the Millipore membrane filter (0.45 μ).
The filtrate was analysed spectrophotometerically at 269.5 nm. Shaking was continued until three consecutive
readings were the same. Results are indicated in table 2.
Fourier Transform infrared (FTIR) spectroscopy
FTIR spectroscopy was carried out for PGH, physical mixture (PM) of drug with PEG 6000 and Poloxamer
188 and SDs. Samples were scanned in the region of 4000-400 cm-1
using a Shimadzu FTIR spectrometer.
Differential scanning calorimetry (DSC) study
DSC analysis of PGH, PM, and SDs were carried out using a Shimadzu DSC 60 TSW 60 (Japan). Accurately
weighed samples were crimped in Aluminium pans and heated from 30 to 300 °C at a heating rate of 10
°C/min in N2 atmosphere. An empty sealed aluminium pan was used as reference.
Page 26
26
X-ray diffraction (XRD) study
Crystallinity of PGH and SDs was determined using Analytical XRD (Bruker: D2 phaser). XRD patterns of
PGH and SDs were recorded at room temperature on Bruker’s D8 advance diffractometer (Bruker, Germany)
at 40 kV, 40 mA. Analysis was performed in a continuous mode with a step size of 0.02° and step time of 1
sec over an angular range of 10-80° 2θ. Obtained diffractograms were analyzed with DIFFRACplus EVA
(version 9.0) diffraction software.
Scanning electron microscopy
The surface morphology of PGZ and SDs were examined using a scanning electron microscope (JEOL Model
JSM - 6390LV, JEOL Ltd, Tokyo, Japan). The powders were fixed on a brass stub using double-sided
adhesive tape. The scanning electron microscope was operated at an acceleration voltage of 30 kV.
Percentage yield
Determination of % practical yield is useful to determine the efficiency of a preparation technique. The
practical yield is calculated by using following equation:
% Practical yield = (Weight of prepared solid dispersions × 100)/Theoretical weight
Estimation of drug content in solid dispersion
From each batch of SD, quantity equivalent to 15 mg of the Pioglitazone was taken into a 100 ml conical
flask containing 0.1 N HCl. The solution was diluted with 0.1 N HCl and filtered. The absorbance of this
solution was measured at 269.5 nm using UV spectrophotometer.
FORMULATION DEVELOPMENT
Preparation of immediate release tablets containing PGZ SDs
Wet granulation method was selected to formulate immediate release tablet containing SDs of PGZ. The
composition of different batches is shown in table II.
SDs, Lactose monohydrate and Starch were passed through 40 # mesh sieve prior to mixing. The ingredients
were mixed in a polythene bag for 5 minutes and granulated with binder solution of PVP K 30 in Isopropyl
alcohol. Dough mass was passed through 20 # sieve and granules were dried at 40ᵒC in Hot air oven. The
dried granules were passed through 30# mesh sieve. Talc and magnesium stearate were passed through 60#
sieve and then mixed with dried granules for 2 minutes. The lubricated blend was compressed into tablets
Page 27
27
using a rotary tablet press (Rimek, Minipress I) equipped with 8 mm standard concave punches plain on both
sides.
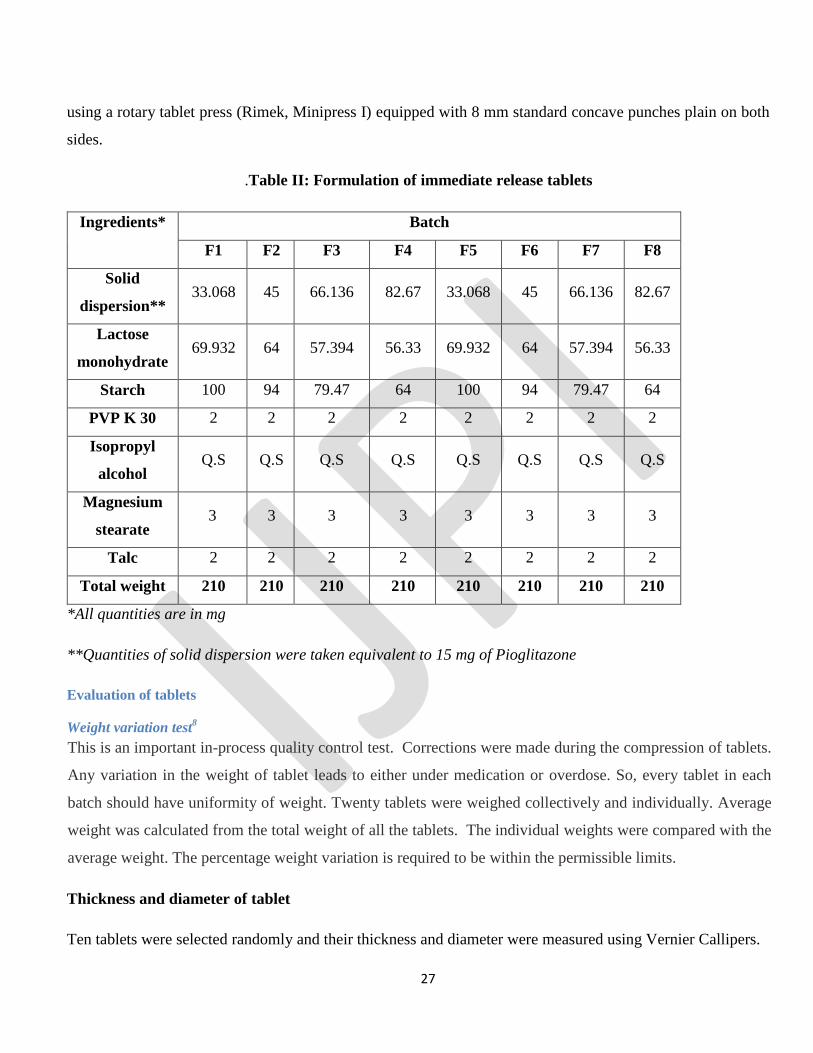
.Table II: Formulation of immediate release tablets
Ingredients* Batch
F1 F2 F3 F4 F5 F6 F7 F8
Solid
dispersion** 33.068 45 66.136 82.67 33.068 45 66.136 82.67
Lactose
monohydrate 69.932 64 57.394 56.33 69.932 64 57.394 56.33
Starch 100 94 79.47 64 100 94 79.47 64
PVP K 30 2 2 2 2 2 2 2 2
Isopropyl
alcohol Q.S Q.S Q.S Q.S Q.S Q.S Q.S Q.S
Magnesium
stearate 3 3 3 3 3 3 3 3
Talc 2 2 2 2 2 2 2 2
Total weight 210 210 210 210 210 210 210 210
*All quantities are in mg
**Quantities of solid dispersion were taken equivalent to 15 mg of Pioglitazone
Evaluation of tablets
Weight variation test8
This is an important in-process quality control test. Corrections were made during the compression of tablets.
Any variation in the weight of tablet leads to either under medication or overdose. So, every tablet in each
batch should have uniformity of weight. Twenty tablets were weighed collectively and individually. Average
weight was calculated from the total weight of all the tablets. The individual weights were compared with the
average weight. The percentage weight variation is required to be within the permissible limits.
Thickness and diameter of tablet
Ten tablets were selected randomly and their thickness and diameter were measured using Vernier Callipers.
Page 28
28
Hardness
Hardness (diametric crushing strength) is the force required to break a tablet across the diameter. The
hardness of a tablet is an indication of its strength. The tablet should be stable to mechanical stress during
handling and transportation. The hardness was tested using Monsanto hardness tester. The average of the five
determinations was determined and reported.
Friability8
Roche friabilator was used to measure the friability of the tablets. It was rotated at a rate of 25 rpm. Tablets
equivalent to weight 6.5 gms were weighed collectively and placed in the friabilator. In the friabilator, the
tablets were exposed to rolling, resulting from free fall of tablets within the chamber of the friabilator. After,
100 rotations (i.e. in 4 minutes), the tablets were taken out from the friabilator, dedusted and the intact tablets
were again weighed collectively.
In-vitro disintegration8
Disintegration time of the tablet was observed with the help of disintegration test apparatus (ED 2L
Electrolab) consisting of a basket rack assembly with 1000 ml beaker, a thermostatic arrangement for heating
the beaker between 35 ᵒC to 39 ᵒC, and a device for raising and lowering the basket in the immersion fluid at
a constant rate between 29 and 32 cycles per min. Six tablets were randomly selected and in vitro
disintegration was determined. using disintegration apparatus.
Assay
The prepared tablets were tested for their drug content. Ten tablets were finely powdered; quantities of the
powder equivalent to 30 mg of PGH were accurately weighed and transferred to a 100 ml of volumetric flask.
Volume was made up to 100 ml mark with 0.1 N HCl. Solution was then filtered to remove insoluble
excipients. The absorbance of the resulting solution was measured at 269.5 nm using a Shimadzu UV 1800
against 0.1 N HCl as blank. The linearity equation obtained from calibration curve was used for estimation of
PGH in the tablet formulation.
In vitro drug release8
In vitro dissolution studies of in-house tablets and marketed tablets (Piolem -15, Alembic Pharmaceuticals,
Vadodara) were carried out using IP dissolution apparatus 1, paddle (Electrolab Dissolution Tester TDT-08P)
at 75 rpm in 900 ml of 0.1 N HCl. Aliquots of 5 ml were withdrawn at specified time intervals of 5, 10, 15,
30, 45 and 60 min and replaced with fresh media. The samples were filtered, diluted with dissolution media
and analyzed spectrophotometerically at 269.5 nm. The dissolution studies were carried out in triplicate.
Page 29
29
Similarity factor (f2 analysis):
Similarity factor between the reference and the test product was determined.
Stability study9
Accelerated stability studies of batches were carried out at 400C/75% RH for 3 months using stability
chamber Oswald JRIC 11B. Physical stability was analyzed by change in appearance and chemical stability
was analyzed by the change in the assay and % cumulative drug release after 3 month.
In vivo study
Animals
Male wistar rats of (250 - 300 g) were under standard conditions, maintained on a 12 h light/dark cycle and
had free access to food and water up to the time of experimentation. The animals were acclimatized to the
laboratory environment 1 h before the experiments. Animals were randomly distributed into groups of 6
animals each. All experiments were conducted during the light period (08.00-16.00 h). All the protocols were
approved by the Institutional Animal Ethical Committee (IAEC; Protocol No.: MPC/IAEC/10/2013.
Method 10
Fasted rats were divided into two groups with six rats each and were treated orally in the following
manner.
Group I (Reference): Treated with suspension of marketed PGZ tablets
Group II (Test): Treated with suspension of prepared PGZ tablets containing SDs
The rats were orally loaded with 1.25 g/kg of 25% (w/v) glucose solution. After glucose administration, the
rats were treated with PGZ equivalent to 6 mg/kg body weight from suspension of developed and marketed
immediate release tablets of PGZ. The suspension was administered orally to each group of rat using
stomach intubation. Blood samples were collected through tail vein immediately prior to commencement of
treatment and at 15, 30, 45, 60 and 75 minutes after glucose challenge. The blood glucose level was
determined using the glucose-measuring instrument Accu-Chek Sensor Comfort (Roche Diagnostics GmbH,
Germany).
Page 30
30
RESULTS AND DISCUSSION
Preformulation studies
The drug appeared as white to off white crystalline powder, it is poorly water soluble (0.042 mg/ml), bulk
density and the tapped density of the pure drug (PGH) was found to be 0.491 g/ml and 0.748 g/ml
respectively. The compressibility index and the Hausner's ratio of the pure drug were found to be 34.36% and
1.52 respectively. These values suggested that poor flow characteristics ruling out the possibility of
employing direct compression for manufacturing of tablets. Wet granulation was employed as a method for
tablet manufacturing. No interaction between drug and excipients suggested the suitability of the selected
excipients.
Preparation of Solid Dispersions: The solid dispersions were prepared using PEG 6000 and Poloxamer 188
as the hydrophilic carriers by the melting method. All the prepared batches of solid dispersions were found to
be non-hygroscopic, free flowing powders. All the prepared solid dispersions exhibited uniformity of drug
content. The drug content of the solid dispersions was found in the range: 95% to102%. the % yield of all
formulations was found in the range of 93-99% (Table III).
Evaluations of SDs
Solubility studies
The aqueous solubility of Pioglitazone hydrochloride was found to be 0.042 mg/ml suggesting a strong need
to enhance the solubility of PGH. Our results are in agreement with the findings of other authors. Therefore,
the solid dispersion technique was employed for improving the solubility of PGH. Improvement of several
hundreds of folds in solubility of PGH was observed with the SDs (Table III). This could be attributed to the
formation of soluble complexes between the water soluble polymeric carriers and poorly water soluble
drugs. The values of solubilities suggested that as the amount of the carrier increased, the solubility was
found of PGH was found to increase.
Page 31
31
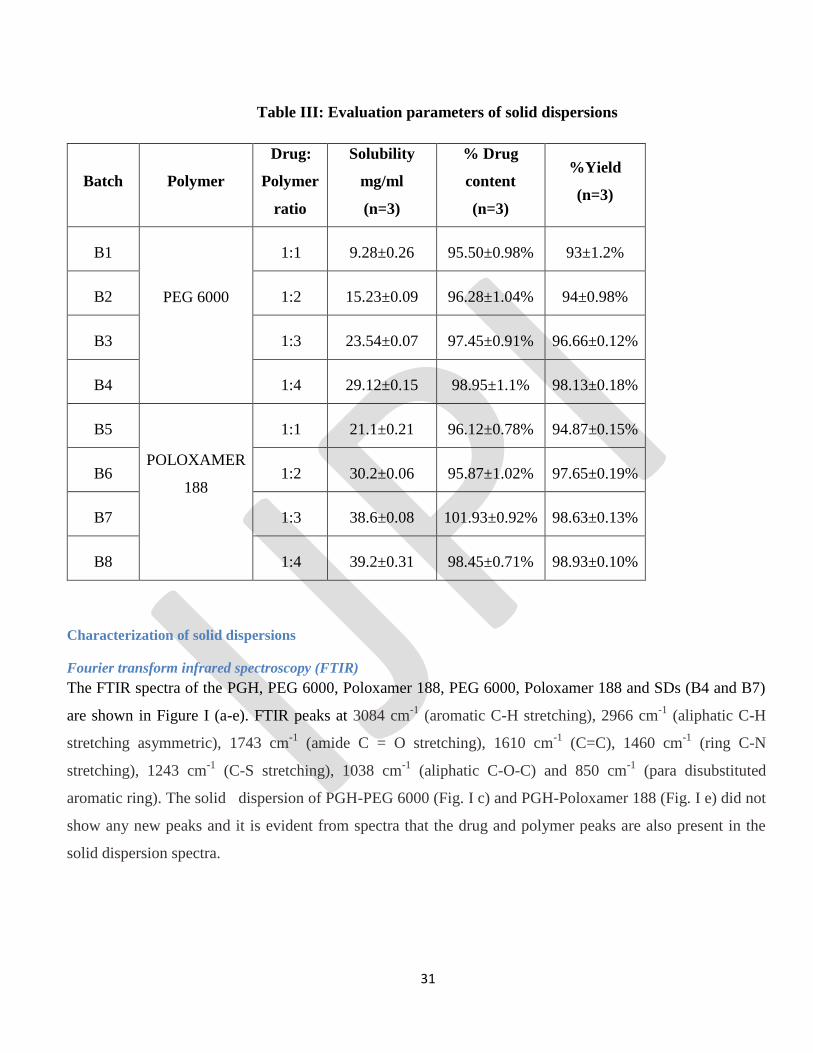
Table III: Evaluation parameters of solid dispersions
Batch Polymer
Drug:
Polymer
ratio
Solubility
mg/ml
(n=3)
% Drug
content
(n=3)
%Yield
(n=3)
B1
PEG 6000
1:1 9.28±0.26 95.50±0.98% 93±1.2%
B2 1:2 15.23±0.09 96.28±1.04% 94±0.98%
B3 1:3 23.54±0.07 97.45±0.91% 96.66±0.12%
B4 1:4 29.12±0.15 98.95±1.1% 98.13±0.18%
B5
POLOXAMER
188
1:1 21.1±0.21 96.12±0.78% 94.87±0.15%
B6 1:2 30.2±0.06 95.87±1.02% 97.65±0.19%
B7 1:3 38.6±0.08 101.93±0.92% 98.63±0.13%
B8 1:4 39.2±0.31 98.45±0.71% 98.93±0.10%
Characterization of solid dispersions
Fourier transform infrared spectroscopy (FTIR)
The FTIR spectra of the PGH, PEG 6000, Poloxamer 188, PEG 6000, Poloxamer 188 and SDs (B4 and B7)
are shown in Figure I (a-e). FTIR peaks at 3084 cm-1
(aromatic C-H stretching), 2966 cm-1
(aliphatic C-H
stretching asymmetric), 1743 cm-1
(amide C = O stretching), 1610 cm-1
(C=C), 1460 cm-1
(ring C-N
stretching), 1243 cm-1
(C-S stretching), 1038 cm-1
(aliphatic C-O-C) and 850 cm-1
(para disubstituted
aromatic ring). The solid dispersion of PGH-PEG 6000 (Fig. I c) and PGH-Poloxamer 188 (Fig. I e) did not
show any new peaks and it is evident from spectra that the drug and polymer peaks are also present in the
solid dispersion spectra.
Page 32
32
(a)
(b)
(c)
(d)
(e)
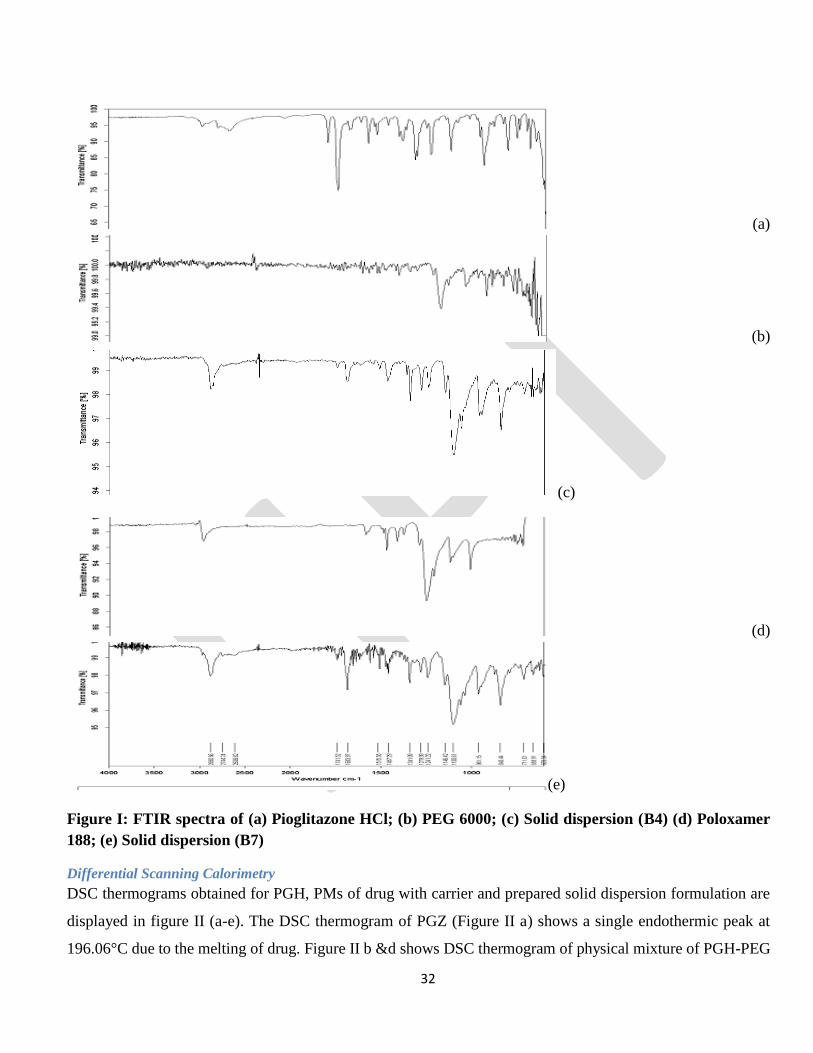
Figure I: FTIR spectra of (a) Pioglitazone HCl; (b) PEG 6000; (c) Solid dispersion (B4) (d) Poloxamer
188; (e) Solid dispersion (B7)
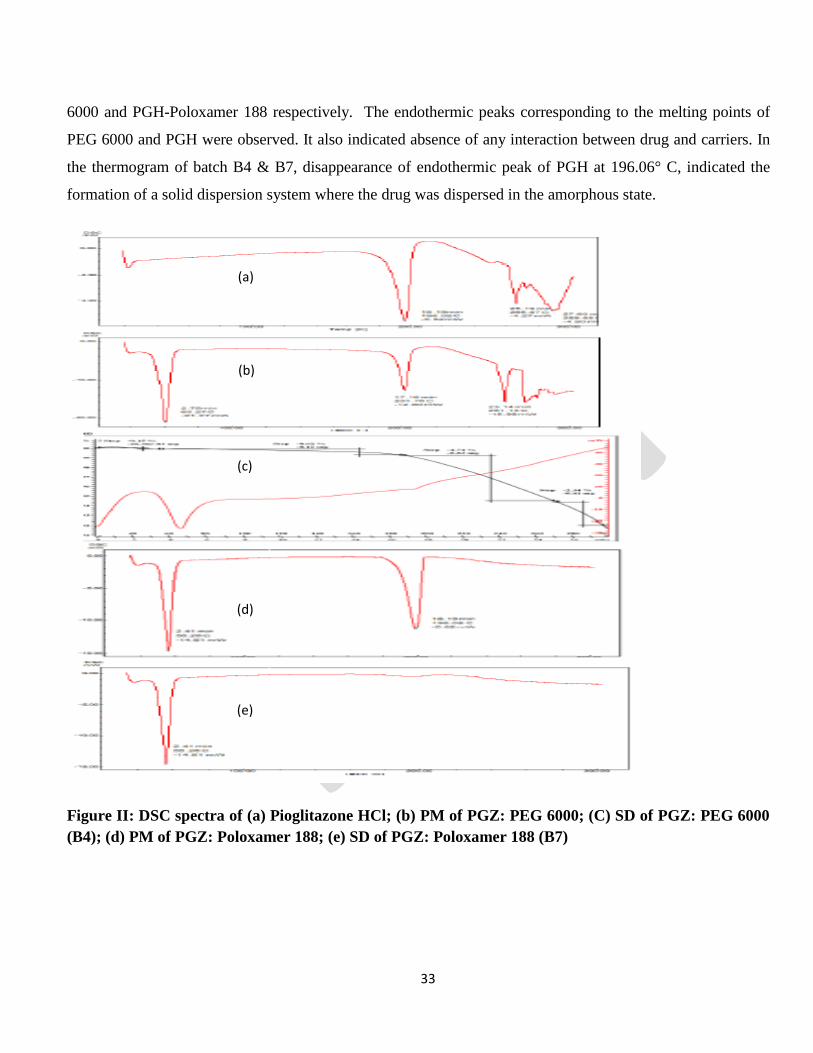
Differential Scanning Calorimetry
DSC thermograms obtained for PGH, PMs of drug with carrier and prepared solid dispersion formulation are
displayed in figure II (a-e). The DSC thermogram of PGZ (Figure II a) shows a single endothermic peak at
196.06°C due to the melting of drug. Figure II b &d shows DSC thermogram of physical mixture of PGH-PEG
Page 33
33
6000 and PGH-Poloxamer 188 respectively. The endothermic peaks corresponding to the melting points of
PEG 6000 and PGH were observed. It also indicated absence of any interaction between drug and carriers. In
the thermogram of batch B4 & B7, disappearance of endothermic peak of PGH at 196.06° C, indicated the
formation of a solid dispersion system where the drug was dispersed in the amorphous state.
Figure II: DSC spectra of (a) Pioglitazone HCl; (b) PM of PGZ: PEG 6000; (C) SD of PGZ: PEG 6000
(B4); (d) PM of PGZ: Poloxamer 188; (e) SD of PGZ: Poloxamer 188 (B7)
(a)
(b)
(c)
(d)
(e)
Page 34
34
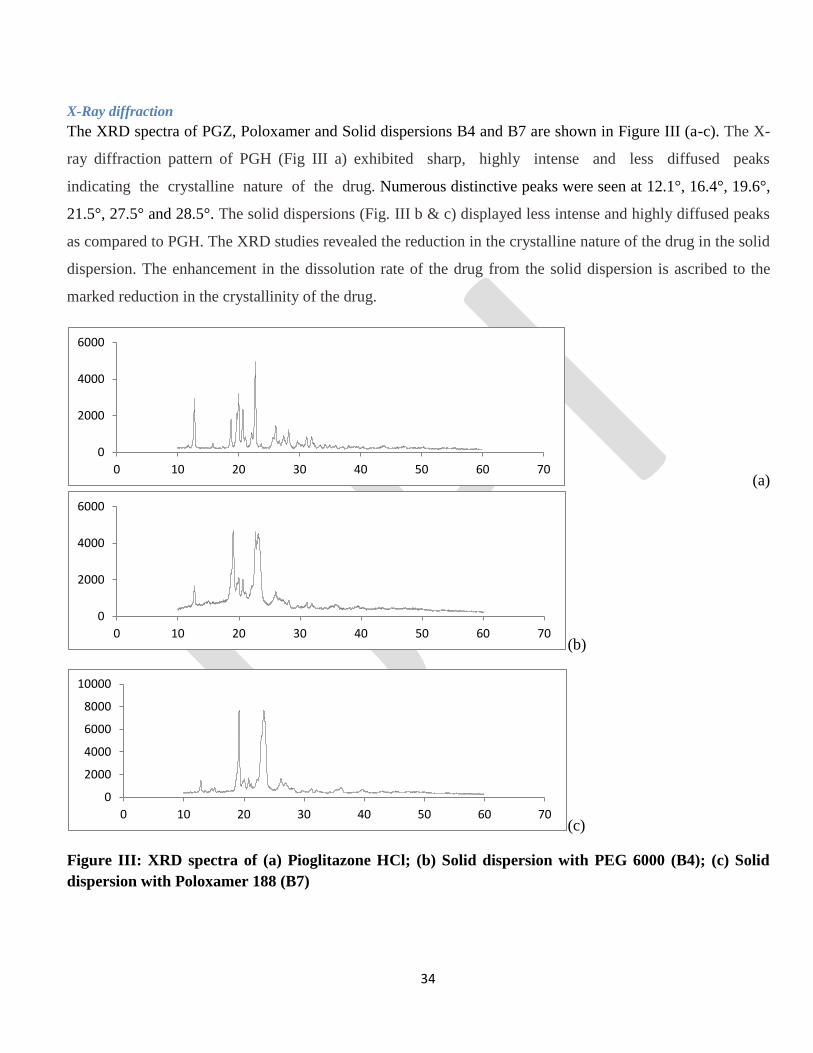
X-Ray diffraction
The XRD spectra of PGZ, Poloxamer and Solid dispersions B4 and B7 are shown in Figure III (a-c). The X-
ray diffraction pattern of PGH (Fig III a) exhibited sharp, highly intense and less diffused peaks
indicating the crystalline nature of the drug. Numerous distinctive peaks were seen at 12.1°, 16.4°, 19.6°,
21.5°, 27.5° and 28.5°. The solid dispersions (Fig. III b & c) displayed less intense and highly diffused peaks
as compared to PGH. The XRD studies revealed the reduction in the crystalline nature of the drug in the solid
dispersion. The enhancement in the dissolution rate of the drug from the solid dispersion is ascribed to the
marked reduction in the crystallinity of the drug.
(a)
(b)
(c)
Figure III: XRD spectra of (a) Pioglitazone HCl; (b) Solid dispersion with PEG 6000 (B4); (c) Solid
dispersion with Poloxamer 188 (B7)
0
2000
4000
6000
0 10 20 30 40 50 60 70
0
2000
4000
6000
0 10 20 30 40 50 60 70
0
2000
4000
6000
8000
10000
0 10 20 30 40 50 60 70
Page 35
35
Evaluations of immediate release tablets
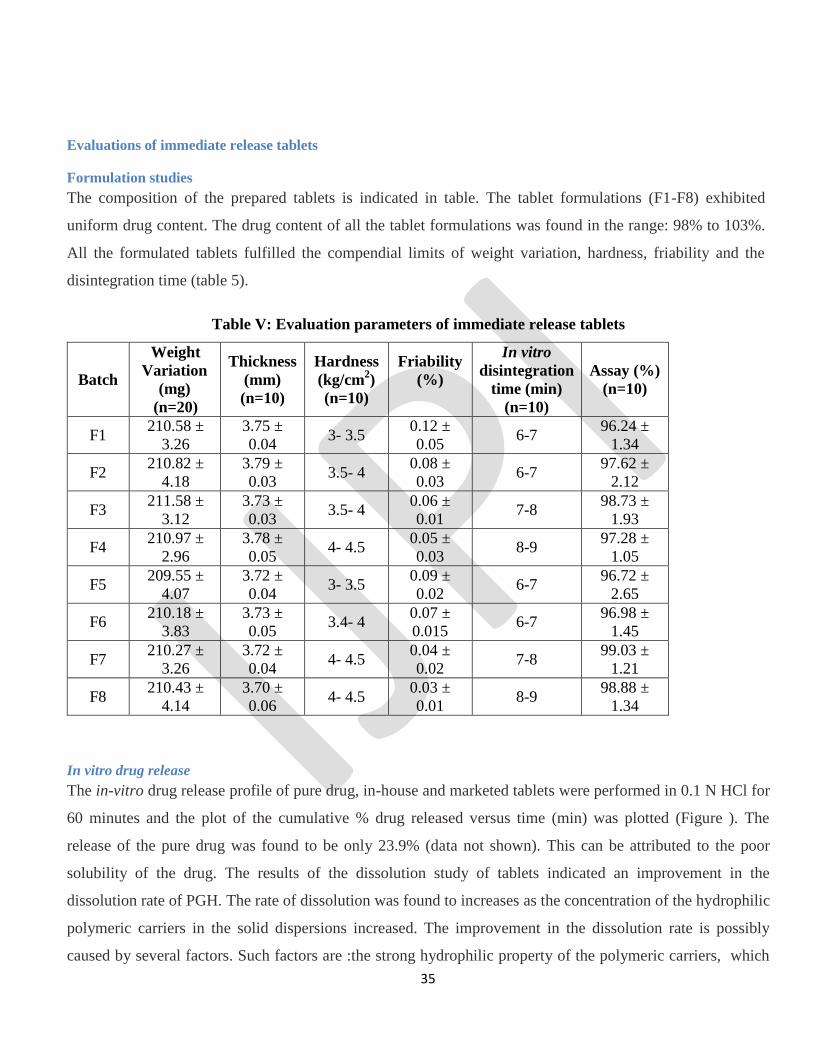
Formulation studies
The composition of the prepared tablets is indicated in table. The tablet formulations (F1-F8) exhibited
uniform drug content. The drug content of all the tablet formulations was found in the range: 98% to 103%.
All the formulated tablets fulfilled the compendial limits of weight variation, hardness, friability and the
disintegration time (table 5).
Table V: Evaluation parameters of immediate release tablets
Batch
Weight
Variation
(mg)
(n=20)
Thickness
(mm)
(n=10)
Hardness
(kg/cm2)
(n=10)
Friability
(%)
In vitro
disintegration
time (min)
(n=10)
Assay (%)
(n=10)
F1 210.58 ±
3.26
3.75 ±
0.04 3- 3.5
0.12 ±
0.05 6-7
96.24 ±
1.34
F2 210.82 ±
4.18
3.79 ±
0.03 3.5- 4
0.08 ±
0.03 6-7
97.62 ±
2.12
F3 211.58 ±
3.12
3.73 ±
0.03 3.5- 4
0.06 ±
0.01 7-8
98.73 ±
1.93
F4 210.97 ±
2.96
3.78 ±
0.05 4- 4.5
0.05 ±
0.03 8-9
97.28 ±
1.05
F5 209.55 ±
4.07
3.72 ±
0.04 3- 3.5
0.09 ±
0.02 6-7
96.72 ±
2.65
F6 210.18 ±
3.83
3.73 ±
0.05 3.4- 4
0.07 ±
0.015 6-7
96.98 ±
1.45
F7 210.27 ±
3.26
3.72 ±
0.04 4- 4.5
0.04 ±
0.02 7-8
99.03 ±
1.21
F8 210.43 ±
4.14
3.70 ±
0.06 4- 4.5
0.03 ±
0.01 8-9
98.88 ±
1.34
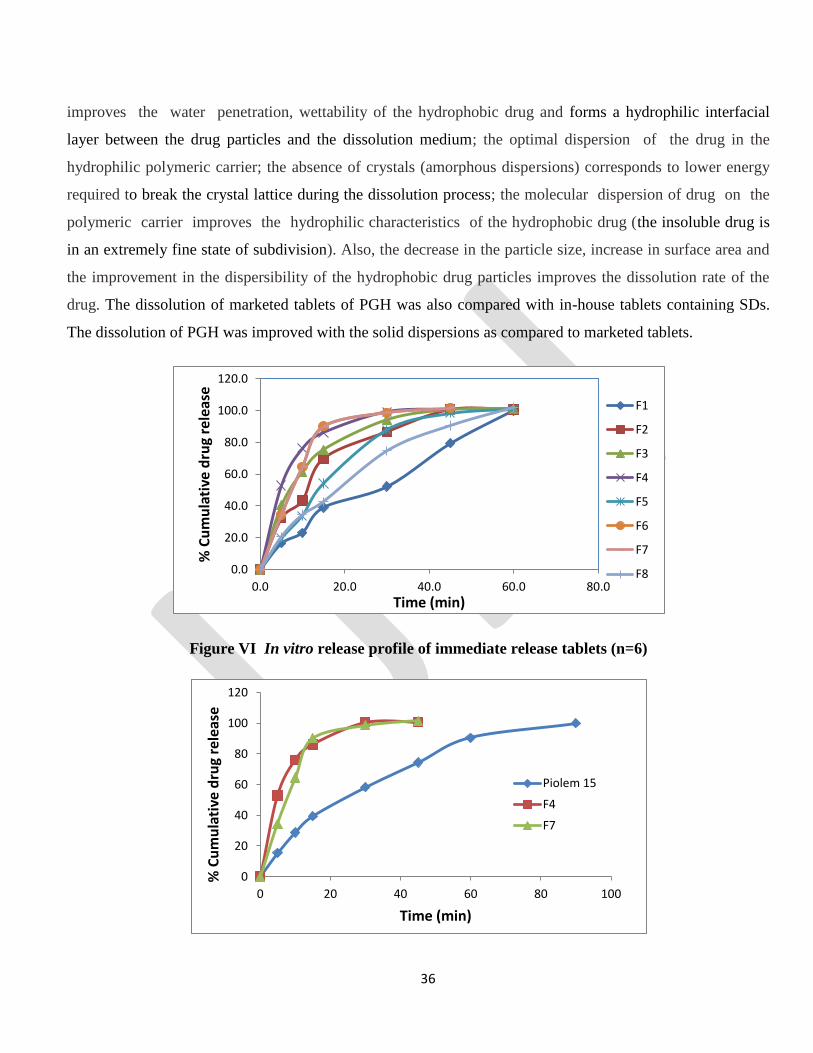
In vitro drug release
The in-vitro drug release profile of pure drug, in-house and marketed tablets were performed in 0.1 N HCl for
60 minutes and the plot of the cumulative % drug released versus time (min) was plotted (Figure ). The
release of the pure drug was found to be only 23.9% (data not shown). This can be attributed to the poor
solubility of the drug. The results of the dissolution study of tablets indicated an improvement in the
dissolution rate of PGH. The rate of dissolution was found to increases as the concentration of the hydrophilic
polymeric carriers in the solid dispersions increased. The improvement in the dissolution rate is possibly
caused by several factors. Such factors are :the strong hydrophilic property of the polymeric carriers, which
Page 36
36
improves the water penetration, wettability of the hydrophobic drug and forms a hydrophilic interfacial
layer between the drug particles and the dissolution medium; the optimal dispersion of the drug in the
hydrophilic polymeric carrier; the absence of crystals (amorphous dispersions) corresponds to lower energy
required to break the crystal lattice during the dissolution process; the molecular dispersion of drug on the
polymeric carrier improves the hydrophilic characteristics of the hydrophobic drug (the insoluble drug is
in an extremely fine state of subdivision). Also, the decrease in the particle size, increase in surface area and
the improvement in the dispersibility of the hydrophobic drug particles improves the dissolution rate of the
drug. The dissolution of marketed tablets of PGH was also compared with in-house tablets containing SDs.
The dissolution of PGH was improved with the solid dispersions as compared to marketed tablets.
Figure VI In vitro release profile of immediate release tablets (n=6)
0.0
20.0
40.0
60.0
80.0
100.0
120.0
0.0 20.0 40.0 60.0 80.0
% C
um
ula
tive
dru
g re
leas
e
Time (min)
F1
F2
F3
F4
F5
F6
F7
F8
0
20
40
60
80
100
120
0 20 40 60 80 100
% C
um
ula
tive
dru
g re
leas
e
Time (min)
Piolem 15
F4
F7
Page 37
37
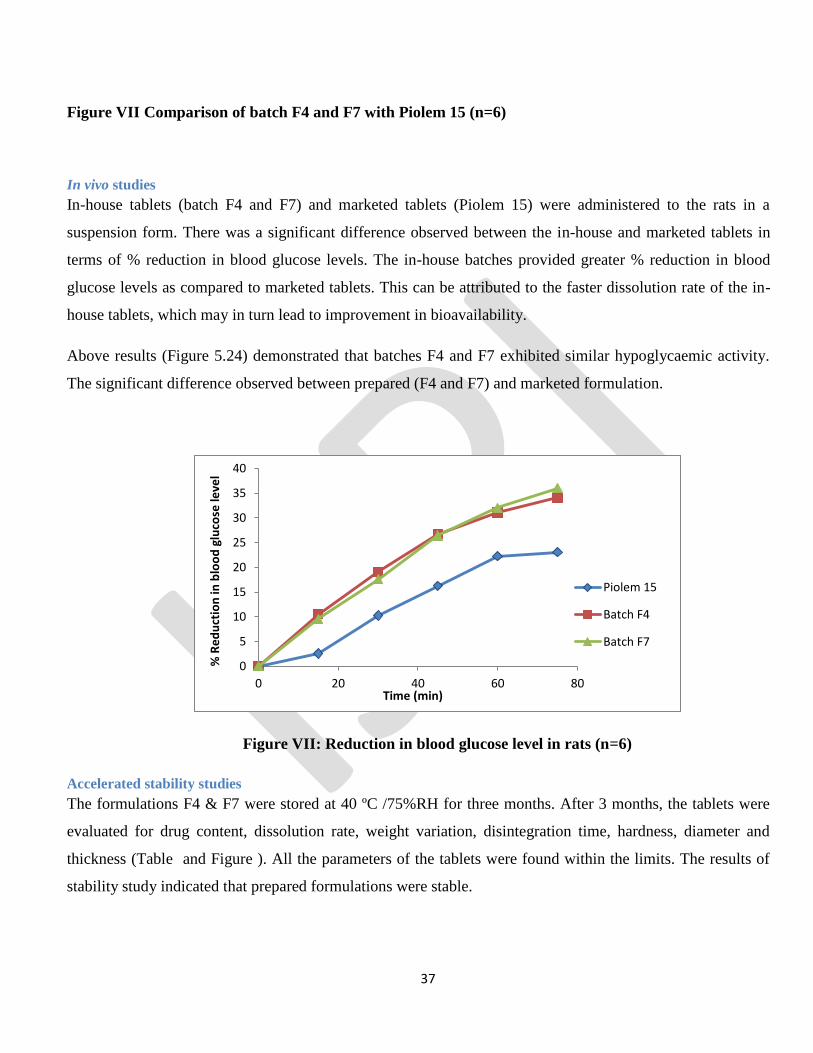
Figure VII Comparison of batch F4 and F7 with Piolem 15 (n=6)
In vivo studies
In-house tablets (batch F4 and F7) and marketed tablets (Piolem 15) were administered to the rats in a
suspension form. There was a significant difference observed between the in-house and marketed tablets in
terms of % reduction in blood glucose levels. The in-house batches provided greater % reduction in blood
glucose levels as compared to marketed tablets. This can be attributed to the faster dissolution rate of the in-
house tablets, which may in turn lead to improvement in bioavailability.
Above results (Figure 5.24) demonstrated that batches F4 and F7 exhibited similar hypoglycaemic activity.
The significant difference observed between prepared (F4 and F7) and marketed formulation.
Figure VII: Reduction in blood glucose level in rats (n=6)
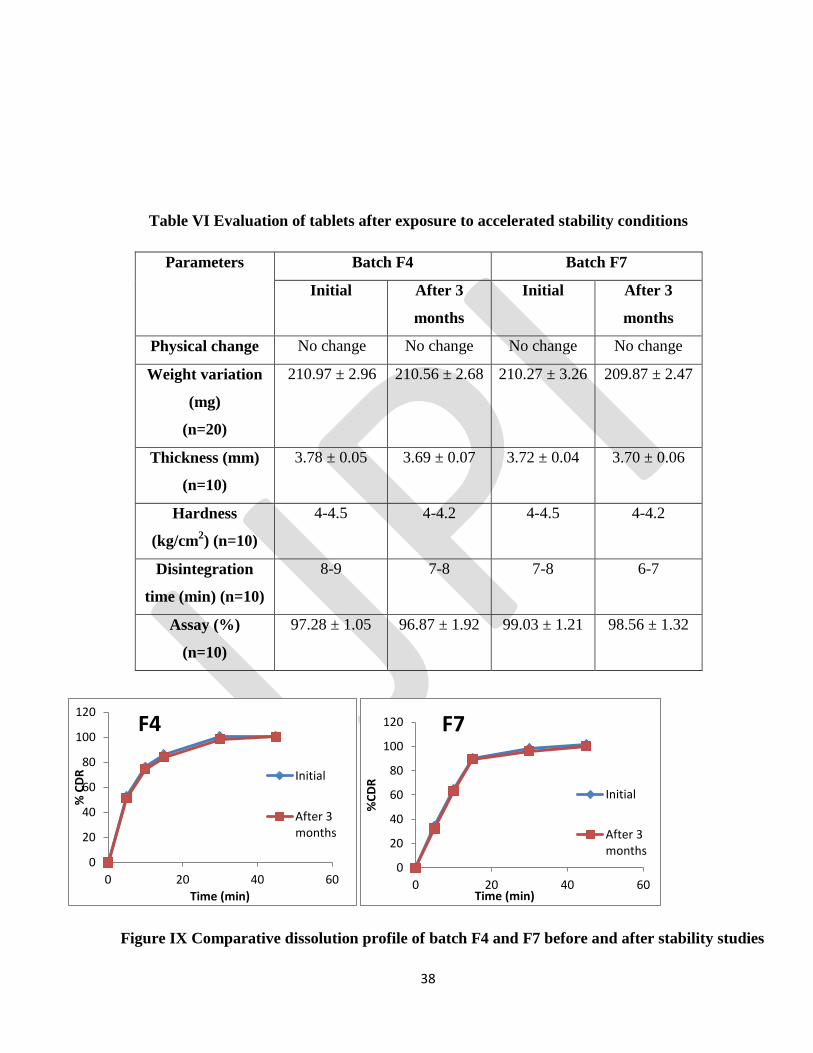
Accelerated stability studies
The formulations F4 & F7 were stored at 40 ºC /75%RH for three months. After 3 months, the tablets were
evaluated for drug content, dissolution rate, weight variation, disintegration time, hardness, diameter and
thickness (Table and Figure ). All the parameters of the tablets were found within the limits. The results of
stability study indicated that prepared formulations were stable.
0
5
10
15
20
25
30
35
40
0 20 40 60 80
% R
ed
uct
ion
in b
loo
d g
luco
se le
vel
Time (min)
Piolem 15
Batch F4
Batch F7
Page 38
38
Table VI Evaluation of tablets after exposure to accelerated stability conditions
Parameters
Batch F4 Batch F7
Initial After 3
months
Initial After 3
months
Physical change No change No change No change No change
Weight variation
(mg)
(n=20)
210.97 ± 2.96 210.56 ± 2.68 210.27 ± 3.26 209.87 ± 2.47
Thickness (mm)
(n=10)
3.78 ± 0.05 3.69 ± 0.07 3.72 ± 0.04 3.70 ± 0.06
Hardness
(kg/cm2) (n=10)
4-4.5 4-4.2 4-4.5 4-4.2
Disintegration
time (min) (n=10)
8-9 7-8 7-8 6-7
Assay (%)
(n=10)
97.28 ± 1.05 96.87 ± 1.92 99.03 ± 1.21 98.56 ± 1.32
Figure IX Comparative dissolution profile of batch F4 and F7 before and after stability studies
0
20
40
60
80
100
120
0 20 40 60
% C
DR
Time (min)
F4
Initial
After 3months
0
20
40
60
80
100
120
0 20 40 60
%C
DR
Time (min)
F7
Initial
After 3months
Page 39
39
There were no significant changes observed in the physical parameters, assay and dissolution of the tablets
after 3 months storage at 400C/75% RH.
Conclusion:
The solid dispersions of PGH were successfully formulated by melting method using hydrophilic carriers like
PEG 6000 and Poloxamer 188 in various ratios. The characterization studies of SDs indicated the conversion
of PGH from crystalline to amorphous form. The prepared solid dispersions were further formulated into
immediate release tablets by wet granulation method. The in vitro drug release profiles of developed tablets
were compared to marketed tablets of PGH. The tablets containing SDs of PGH showed higher dissolution
rate than pure drug and marketed tablets (Piolem 15). The dissolution rate enhancement of the in-house
tablets based on solid dispersions was further substantiated by the in vivo studies in Albino wistar rats.
Pharmacodynamic evaluation suggested greater reduction in % blood glucose by the in-house tablets as
compared to marketed tablets. The in-house tablet formulations were found to be stable after accelerated
stability studies. Thus, the dissolution rate improvement of PGH by solid dispersions may be useful in
enhancing the oral bioavailability of the drug, which may in turn lead to reduction in dose; dose related side
effects; cost of the therapy and improved patient compliance.
REFERENCES:
1. Brahmankar DM, Jaiswal SB. Biopharmaceutics and Pharmacokinetics. Delhi, India: Vallabh
Prakashan, 2nd
ed. 2009; pp 27-28.
2. Gribbon P, Andreas S. High-throughput drug discovery: what can we expect from HTS? Drug
Discovery Today. 2005; 10(1):17-22.
3. Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;
60:1281-1302.
4. Patel T, Patel LD. Enhancement of dissolution of Fenofibrate by Solid dispersion Technique. Int. J.
Res. Pharm. Sci.2010: 1(2); 127-132.
5. Lobenberg R, Amidon GL, Modern bioavailability, bioequivalence and biopharmaceutics
classification system. New scientific approaches to international regulatory standards. Eur J Pharm
Biopharm. 2000; 50(3):20.
6. Badry ME, Fetih G. Improvement of solubility and dissolution rate of indomethacin by solid
dispersions in gelucire 50/13 and peg 4000. Saudi Pharmaceutical Journal. 2009:17(3); 219-230.
Page 40
40
7. Higuchi T, Connors KA. Phase solubility techniques. Adv Anal Chem Instrum 1965; 4: 117-212.
8. Govt of India, Ministry of Health and family welfare. Indian Pharmacocpoeia, 2010. The Indian
Pharmacopoeia Commission, Ghaziabad. 2010: pp 187-193, 1916-1917.
9. Q2AR1: Validation of analytical procedures: Text and methodology, (2005). ICH harmonised
tripartite guideline: ICH Harmonised Tripartite Convention, 17.
10. Cretti A, Brunato B, Zenti MG, Tosi F, Muggeo M, Bonora E, Bonadonna RC. A novel tool to assess
ß-cell function during the oral glucose tolerance test (OGTT). Diabetes. 2000; 49(Suppl. 1): A89.
11. Savjani KT, Gajjar AK, Savjani JK. Drug Solubility: Importance and Enhancement Techniques. ISRN
Pharm. 2012; 195727.
12. Yu LX, Amidon GL, Polli JE. Biopharmaceutics classification system: the scientific basis for
biowaiver extension. Pharm Research. 2002; 19:921-925.
13. Fahr A, Liu X. Drug delivery strategies for poorly water-soluble drugs. Drug Delivery. 2007;
4(4):403-416.
14. Yalkowsky S. Techniques of Solubilization of Drugs. Drugs and the pharmaceutical sciences
(Volume 12) 1981. Yalkowsky S. (Ed). Marcel Dekker, New York vii.
15. Paolo Brunetti, “Pioglitazone–current profile”, The British Journal of Diabetes and Vascular disease,
2002; 2(1):S16- S22.
16. Assessment report. Pioglitazone Actavis. European Medicines Agency. EMA/210151/2012:1-27.
17. Bando Hiroto, Omachi Yoshihiro, “Coated Preparation”, US20100166853, July 2010: 1-18.
18. Chiou W, Riegelman S. Pharmaceutical Applications of Solid Dispersion Systems. Journal of
Pharmceutical Sciences. 1971; 60(9): 12-81.
19. Dixit AK, Singh RP, Singh S. Solid Dispersion - A Strategy for Improving the Solubility of Poorly
Soluble Drugs. International Journal of Research in Pharmaceutical and Biomedical Sciences. 2012:
3(2); 960-967.
20. Kumar SD, Bihari GV, Suresh P. Solubility improvement using solid dispersion; strategy, mechanism
and characterization: responsiveness and prospect way outs. International Research Journal of
Pharmacy. 2011;2:55-60.
21. Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of
poor water soluble drugs. Drug discovery today. 2007;12(23):1068-1075.
22. Chaturvedi A, Verma A. Solubility enhancement of poorly water soluble drugs by solid dispersion. Int
Pharm Sci Res. 2012;3:26-34.
Page 41
41
23. Chauhan B, Shimpi S, Paradkar A. Preparation and evaluation of glibenclamide-polyglycolized
glycerides solid dispersions with silicon dioxide by spray drying technique. European Journal of
Pharmaceutical Sciences. 2005;26: 219–230.
24. Frizon F, Eloy JO, Donaduzzi CM. Dissolution rate enhancement of loratidine in polyvinylpyrrolidine
K 30 solid dispersions by solvent methods. Journal of Powder technology. 2012:235:532-539.
25. The United Pharmacopoeia 32 and national formulary 27. The United States Pharmacopoeial
Convention, Rockville 2009.
Page 42
42
Volume 1, 2014 NTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
An Attempt to isolate industrially important microorganism from diverse samples
collected from Semeling, Kedah, Malaysia
P. Vasanth Raj, S.A. Dhanaraj, Saw Pei Jie, Sim Poh Pei, Vanushya a/p Alagasan,
Tien Ja She, Loh Mon Ying, Tan Te Wei
Faculty of Pharmacy, AIMST University, Semeling, Bedong, Malaysia 08100
Corresponding Author:
Dr. P. Vasanth Raj
Faculty of Pharmacy
AIMST University
Semeling, Bedong, Malaysia 08100
Phone: +604- 429 8000, Fax: +604- 429 8009
Email: [email protected]
Page 43
43
Abstract:
An attempt was made to isolate industrially important microorganisms from diverse samples collected
from Semeling, Kedah, Malaysia. Samples were aseptically collected from cafeteria soil, lake water,
rotten wood soil, mangrove swamp, plastic bag and sewage water. Primary screening experiments such as
antibiotic screening, cellulose utilization, oil utilization and plastic degrading properties were checked.
All the nine isolates were investigated for biochemical test like indole test, methyl red test, Macconkey
test, Celluose utilization test. Morphological testing such as gram staining, motility test, Mannitol semi
solid agar method were investigated. An attempt for optimization of cellulose medium was also made.
Total of nine isolates were obtained from primary screening. Eight samples were able to utilize cellulose,
one sample produced zone of inhibition (antibiotic screening). One sample was able to utilize plastic and
three samples showed positive result in oil medium. This Project has covered the major concerns about
the nine different isolates, their morphology, biochemical characteristics, capability of producing specific
enzymes, identification tests and uses. Further studies on cellulose optimization, batch level and scale up
have to be explored. On long term studies, this project will contribute in the field of environmental and
industrial biotechnology.
1. Introduction
Microorganism was known to have undiscovered benefits to the industrial sectors. However, the lack of
initiative to discover deeper on the earthly microbes has become a hindrance for the industries to
improve. Thus in this research project, we are motivated to learn more on their usage. There are various
applications of microbes in industry. We selected few important areas of research such as.
1.1 Plastic biodegradation:
Lack of degradability and the closing of landfill sites as well as growing water and land pollution
problems have led to concern about plastics. With excessive use of plastics and increasing pressure being
placed on capacities available for plastic waste disposal, the need for biodegradable plastics and
biodegradation of plastic wastes has assumed increasing importance in the last few years. (Aamer Ali
Shah et al 2008) In view of this, the biodegradation of plastics has been studied extensively for the past
three decades. Some types of plastic have shown to be biodegradable and their degradation mechanisms
have progressively become clearer. (Masayuki Shimao 2001) It is possible to say that if any
microorganism that able to grow in plastic medium has the ability to degrade the plastic.
1.2 Oil biodegradation:
Crude oil continues to be used as the principle source of energy and play an important role in the global
environment pollutant consideration. On the other hand, oil remains as a major source of energy in the
next several decades, because reliable alternative energy consumption has not yet been substituted.
(Trindade et al, 2005 Al-Saleh and Obuekwe.2005). The biodegradation of crude oil by microorganism is
one of the primary ways for eliminating crude oil from contaminated sites and appears to be the most
environmentally friendly method of removal oil pollutant. (Korda et al 1997, Kapley 1999,Del Arco and
Page 44
44
Dabe Franca, 2001: Barathi and Vasudevan, 2001 ). It shows that any microorganism that able to grow
in oil-medium able to utilize oil for growth.
1.3 Cellulose utilisation:
The cellulose constitutes the major form of stocking glucose obtained through photosynthesis and in the
same time the major component of solar energy conversion to the biomass. Because of its highly ordered
structure, the cellulose is very hard to be degraded and that is why it is unusable and stocked in nature as
waste. The capacity to degrade the natural cellulose implies the synthesis of the entire cellulolytic system.
Cellulose has been used by man for centuries, however, its enormous potential as renewable source of
energy was recognized only after cellulose degrading enzymes “cellulases” has been identified (Bhat &
Bhat, 1997) These components act synergistically in the conversion of cellulose to glucose (Eveleigh
1987). It is possible to say that any microorganism that able to grow in cellulose medium has the ability
to degrade cellulose.
1.4 Antibiotic Screening:
The activity of antibiotic may be demonstrated under suitable condition by inhibitory effect of
microorganisms. A reduction of antimicrobial activity will also reveal the subtle changes not
demonstrable by chemical method. Accordingly, microbial and biology assays remain generally the
standard for resolving doubt with respect to possible loss of activity.
2. Methodology
2.1 Collection of Samples
The samples were aseptically collected from cafeteria soil, AIMST lake water, cafeteria water, rotten
wood soil, mangrove swamp, plastic bag, and sewage water.
2.2 Test for Plastic Degrading Microorganism
Plastics are man-made long chain polymeric molecules (Scott, 1999). More than half a century ago
synthetic polymers started to substitute natural materials in almost every area and nowadays plastic have
become an indispensable part of our life. The plastic we use today are made from inorganic and organic
raw materials, such as carbon, silicon, hydrogen, nitrogen, oxygen and chloride. The basic materials used
for making plastics are extracted from oil, coal and natural gas. (Syemour, 1989). In 1980’s, scientist
started to look if plastic could be designed to become susceptible to microbial attack, making them
degradable in a microbial active environment. Biodegradable plastic opened the way for new
considerations of waste management strategies since these materials are designed to degrade under
environmental conditions or in municipal and industrial biological waste treatment facilities (Augusta et
al, 1992; Witt et al., 1997).
Page 45
45
2.3 Test for Oil Degrading Bacteria
Biosurfactant or surface-active compounds are a heterogeneous group of surface active molecules
produced by microorganisms, which either adhere to cell surface or are excreted extracellular in the
growth medium (Fietcher 1992; Zajic and Stiffens, 1994; Makker and Cameotra, 1998). These molecules
reduce surface tension and Critical Micelle Dilution (CMD) in both aqueous solutions and hydrocarbon
mixtures. These properties create microemulsion in which micelle formations occur where hydrocarbons
can solubilize in water or water in hydrocarbons (Banat, 1995). Several types of biosurfactant have been
isolated and characterized, including glycolipids, phospholipids, lipopeptides, natural lipids, fatty acids,
lipopolysacharides and other fully characterized. The majority of known biosurfactants are synthesized
by microorganisms grown on water immiscible hydrocarbons, but some have been produced on such
water-soluble substrates as glucose, glycerol and ethanol (ABU-Ruwaida et al., 1991). Chemically-
synthesized surfactants have been used in the oil industry to aid cleanup of oil spills, as well as to
enhance oil recovery from oil reservoirs. Biosurfactant have special advantage over their commercially
manufactured counterparts because of their lower toxicity, biodegradable nature and effectiveness at
extreme temperature, pH, salinity and ease of synthesis.
2.4 Test for Cellulose Degrading Bacteria
A key role in the decomposition and transformation of organic matter is attributed to bacteria, especially
of relatively complex and recalcitrant plant residues, such as polysaccharides as well as aromatic
compounds in different terrestrial or semi-terrestrial habitats. According to the complexities of habitats
and physiological capabilities of organisms, a range of approaches is available in order to assess the
composition, diversity and activities of soil microbial communities, including 16S rDNA analysis, assays
of substrate utilization profiles and assays of enzyme activities. As there is still a general need to cultivate
micro-organisms isolated from soil in order to study their role in soil biochemical processes, a
combination of both culture-dependent and culture-independent methods is suggested. Most studies of
aerobic cellulolytic soil bacteria involve techniques based on dilution plating, which is a quick,
inexpensive and reliable technique, though necessarily selective since an unknown part of the indigenous
microbial population is considered to be non-culturable. Moreover, a range of different cellulose
substrates is used to detect cellulolytic activity in vitro, such as soluble, substituted cellulose derivatives,
colloidal or amorphous cellulose, insoluble micro-crystalline cellulose and cellulose acetate films.
Several reports have shown incomplete cellulose systems of soil bacteria such as Bacillus species with
the main activity being that of endo-cellulase (carboxymethyl-cellulase).
2.5 Antimicrobial Test
There are two general methods employed, the cylinder plate assay and the turbidimetric assay. The first
depends upon the diffusion upon the antibiotic from a vertical cylinder through a solidified agar layer in a
petri dish or plate to extent such that growth of the added microorganism is prevented entirely in a
circular area or “zone” around the cylinder containing a solution of antibiotic. The turbidimetric method
depends upon the inhibition of growth of a microbial culture in a uniform solution of an antibiotic in a
Page 46
46
fluid medium that is favourable to its rapid growth in the absence of the antibiotic. (Brian D. Gilbert, vol
30)
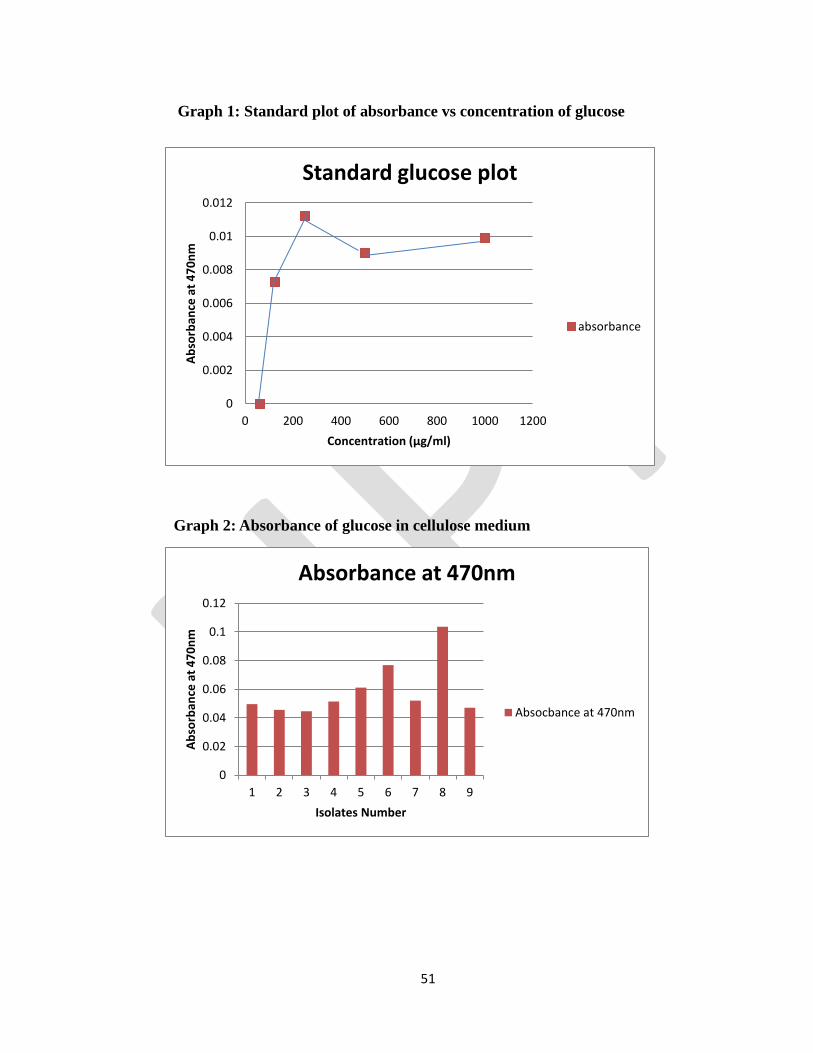
2.6 Optimization of Cellulose
Cellulose is the most abundant carbohydrate found in plants that provides structural integrity to their cell
walls. It is composed of a linear polymer of D-glucose linked by β-1,4 bonds which can be degraded by
the cellulase enzyme complex. Bacterial and fungal plant pathogens utilize cellulases to breach the plant
cell wall for nutrient acquisition and colonization. Spectrophotometric methods previously used to detect
cellulase activity were based on the reducing sugar method described by Miller (1959). The main
objective of these studies was to determine the presence of cellulase-producing enzyme in bacteria, and
the amount of cellulose utilized. This can be done by determining the glucose present in cellulose broth
with the help of UV Spectrometer.
3. Results and Discussion:
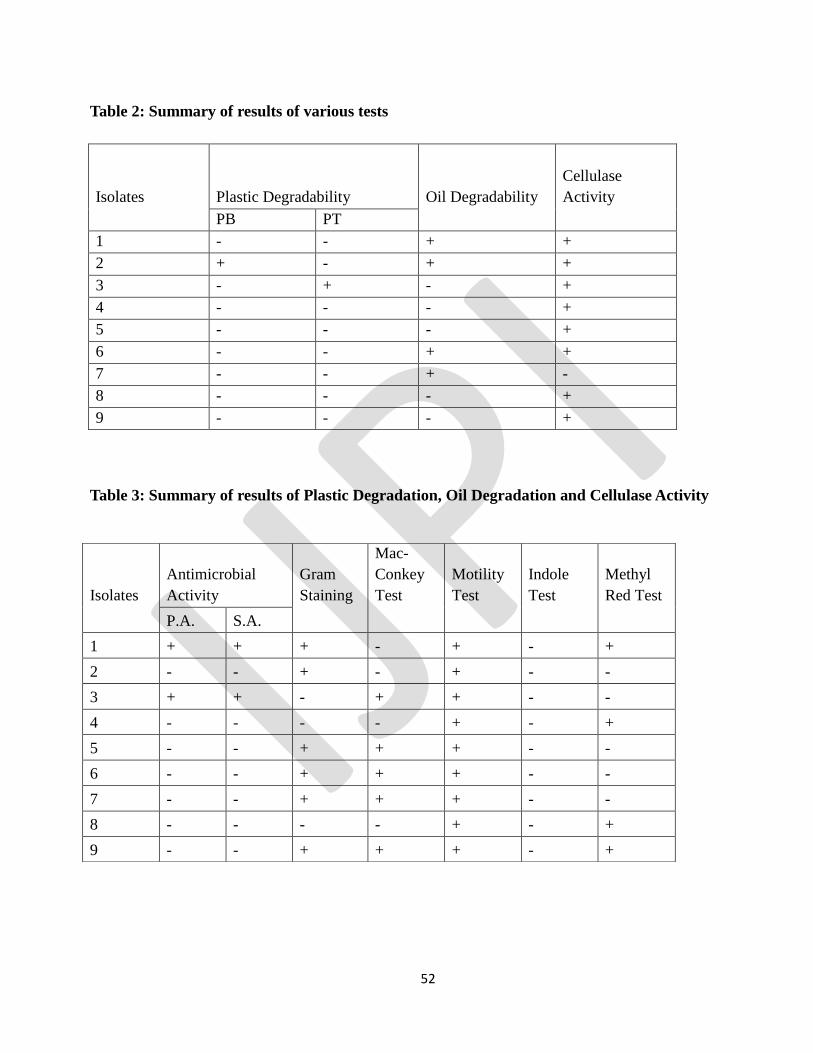
3.1 Collection of Samples
The samples were aseptically collected from cafeteria soil, AIMST lake water, cafeteria water, rotten
wood soil, mangrove swamp, plastic bag, and sewage water.
Table 1: Different types of isolates
ISOLATE
NUMBER
NAME Sample code
1 Cafeteria soil antimicrobial CSA 1
2 AIMST lake water near guardhouse ALWG2
3 Cafeteria water CW 3
4 Rotten wood soil RWS 4
5 Mangrove soil MS 5
6 Big plastic BP 6
7 Sewage water SW 7
8 Small plastic soil SPS(A) 8
9 Small plastic soil SPS(O) 9
3.2 Test for Plastic Degrading Microorganism
As the results show after a few days of incubation, growth was observed in the medium that even consists
of only basal salt nutrient medium, therefore it is possible to say that the microorganism able to utilize the
plastic bag for growth. Day 5 result shows that isolates number 2 able to grow in petri dish with only
plastic bag. While the petri dish number 5,6, and 7 shows grow of fungi. Therefore it is possible to say
that isolate number 2 had the ability to grow in plastic bag medium, it should be further studied to
confirm the properties of the microorganism. While for the medium that consists of toothbrush fiber as
the main source for bacteria growth shows no contamination. Day 5 results of the isolate number 3 also
showed growth.
Page 47
47
It shows that growth of fungus in the coffee dusk for petri dish that consists of plastic bag that not treated
with UV light. Yet the actual result of bacterial degrading plastic unable to identify because the holes and
pores unable to identify under microscope, it should be studied under scanning electro-microscope
(SEM).
Figure 1: Plastic Degrading Activity
3.3 Test for Oil Degrading Bacteria
The samples from the various sources carried several oil degrading microorganisms. Utilization of crude
oil as a substrate by isolated microorganism is shown by increasing the number of cells. Observations of
both turbidity and viable cell count during the experimental period indicate that isolated microorganism
can utilize crude oil as carbon substrate. The growth dynamics of the organisms was determined by the
optical densities, and total viable count.This marked variation is obvious due to the difference in the
samples sites, environmental condition and nutrient availability and also difference in time of exposure
with the pollutants. The results were shown in Figures and reflect the ability of isolated microorganism to
degrade and utilize crude oil as a source of carbon and energy. This technique was used in several studies
to show the ability of bacteria for utilizing crude oil. (Bola, 2006; Emtiazi and Shakarami, 2004).
In the similar investigation by Rahman (2002) in England, the total viable count method used to confirm
the ability of different kind of bacteria for mineralizing crude oil. The comparison of the statistics
obtained in this study and other similar study shows that isolated microorganism can use crude oil and
degrading that better than many kind of bacterial that isolated previously.
The isolates 1, 2, 6 and 7 found to produce biosurfactants. As mentioned before, the work forms part of
an ongoing program. The figure obtained so far suggest that the inoculums would be potentially useful
for treating wastewaters containing oils of both plant and animal origins. Isolation of the responsible gene
and subsequent PCR amplification may prove useful to artificially produce environmentally friendly
surfactants. In future studies involving biosurfactants and bioremediation technologies, these four isolates
may be helpful. Further understanding of the metabolic process of this organism on the crude oil will
Page 48
48
increase possibilities of developing models and strategies for removing crude oil pollutants from oil-
impacted environments.
Figure 2: Oil Degrading Activity
3.4 Test for Cellulose Degrading Bacteria
The samples from the various sources carried several cellulose degrading microorganisms. Utilization of
cellulose as a substrate by isolated microorganism is shown by increasing the number of cells.
Observations of both turbidity and viable cell count during the experimental period indicate that isolated
microorganism can utilize cellulose (isolates 1, 2, 3, 4, 5, 6, 8, 9). The growth dynamics of the organisms
was determined by the optical densities and total viable count.This marked variation is obvious due to the
difference in the samples sites, environmental condition and nutrient availability and also difference in
time of exposure with the pollutants. The results were shown in isolates 1, 2, 3, 4, 5, 6, 8, 9 and reflect
the ability of isolated microorganism to degrade and utilize cellulose. The cellulolytic activities of
bacteria are mostly screened using agar-plate techniques with a variety of substrates, such as
carboxymethyl-cellulose (CMC). This technique was used in several studies to show the ability of
bacteria for utilizing cellulose. (Bola, 2006; Emtiazi and Shakarami, 2004). The comparison of the
statistics obtained in this study and other similar study shows that isolated microorganism can use
cellulose and degrading that better than many kind of bacterial that isolated previously.
As mentioned before, the work forms part of an ongoing program. The figure obtained so far suggest that
the inoculums would be potentially useful. Isolation of the responsible gene and subsequent PCR
amplification may prove useful. More surveys of the degradation potentials of cellulolytic soil bacteria
are required to further validate this function. In future studies involving cellulose, these eight isolates
may be helpful.
Page 49
49
Figure 3: Cellulose Degrading Activity
3.5 Antimicrobial Test
We have used the agar cup method to evaluate the antibacterial activity. It is one of the non-automated in
vitro bacterial susceptibility tests. This classic method yields a zone of inhibition in mm result for the
amount of antimicrobial agents that is needed to inhibit growth of specific microorganisms. It is carried
out in petri plates. Agar cup plate method is simple and less sensitive. They are tremendously advantages
in cases of analyses a large number of sample. The basic principle involves diffusion of the vitamins
though the agar medium and can be adapted from the crude to more sophisticated and quantitative assay.
In cup plate method a number of cups a definite diameter are bored (with the use of glass cylinders or
specially design apparatus) in the agar plate made with the proper assay medium and seeded with the
vitamin requiring organisms. Aliquots of standards and isolates are pipette into the cups as quickly as
possible and the plates are incubated overnight at 37 °C. The vitamins in the cups diffuse out into the
surrounding agar producing a zone of growth proportional to the concentration. At the end of the
incubation period the growth zones are measured either with a ruler or needle-pointed vernier calipers.
Isolate 1 showed positive result in this test, means it produces zone of inhibition.
Page 50
50
Figure 4: Antimicrobial Activity
3.6 Optimization of Cellulose
Based on graph (1), the highest absorbency of glucose is shown at the concentration of 250µg/ml, and the
lowest absorbency is at 62.5µg/ml. The aim of finding out standard glucose plot was for comparison
purposes. Due to some problem related to our instrumentation, the obtained graph was not shown in a
perfect straight line. Another concern was that the glucose concentration in cellulose broth was too high,
thus we were unable to compare with standard glucose plot. However, it was proven that cellulose was
utilized, and degraded into glucose.
The utilization of cellulose broth to detect the presence of glucose was successfully shown. The amount
of glucose produced was detected using UV Spectrometer, and was identified through the standard
glucose graph.
Page 51
51
Graph 1: Standard plot of absorbance vs concentration of glucose
Graph 2: Absorbance of glucose in cellulose medium
0
0.02
0.04
0.06
0.08
0.1
0.12
1 2 3 4 5 6 7 8 9
Ab
sorb
ance
at
47
0n
m
Isolates Number
Absorbance at 470nm
Absocbance at 470nm
0
0.002
0.004
0.006
0.008
0.01
0.012
0 200 400 600 800 1000 1200
Ab
sorb
ance
at
47
0n
m
Concentration (µg/ml)
Standard glucose plot
absorbance
Page 52
52
Table 2: Summary of results of various tests
Isolates Plastic Degradability Oil Degradability
Cellulase
Activity
PB PT
1 - - + +
2 + - + +
3 - + - +
4 - - - +
5 - - - +
6 - - + +
7 - - + -
8 - - - +
9 - - - +
Table 3: Summary of results of Plastic Degradation, Oil Degradation and Cellulase Activity
Isolates
Antimicrobial
Activity
Gram
Staining
Mac-
Conkey
Test
Motility
Test
Indole
Test
Methyl
Red Test
P.A. S.A.
1 + + + - + - +
2 - - + - + - -
3 + + - + + - -
4 - - - - + - +
5 - - + + + - -
6 - - + + + - -
7 - - + + + - -
8 - - - - + - +
9 - - + + + - +
Page 53
53
4. Conclusion
This Project has covered the major concerns about the nine different isolates, their morphology,
biochemical characteristics, capability of producing specific enzymes, identification tests and uses. There
are a large number of tests which are used to determine their morphology, namely gram staining and
hanging drop method. These methods have brought great results as the morphology of all the isolates
were identified clearly. Moving on to the biochemical tests, we adopted methods like Mac-conkey test,
methyl red and Indole test which has given us a clearer picture on how the isolates react with different
chemicals. Followed by that was some identification test such as motility test, and antimicrobial test.
Next, utilizing different mediums like cellulose medium, plastic medium, and basal oil medium enabled
us to find out more details on their capability of degrading cellulose, plastics, and oil. This will also lead
us to find out their uses and benefits to our environment. We basically worked on Primary and Secondary
screening of samples. Due to time constrain, we leave the secondary screening experiments to be
explored further. With our output, increased understanding and contribution this project will contribute
in the field of environmental and industrial biotechnology for the society.
5. Acknowledgement:
We thank AIMST University, Malaysia for financial support and infrastructure provided to carry out this
study.
6. References
Aamer Ali Shah, Fariha Hasan, Abdul Hameed, Safia Ahmed (2008) Biotechnology Advances.
Biological degradation of plastics: A comprehensive review, pp 246-265.
Aamer Ali Shah. Biological degradation of plastic: A comprehensive review. Islamabad, Pakistan; 2008.
ABU-Ruwaida A S, Banat M, Haditirto, Salem S, Kadri A (1991). Isolation of biosurfactant producing
bacteria product characterization and evaluation. Acta Biotech, 11(4): 315-24.
Achouak, W., Christen, R., Barakat, M., Martel, M.-H., Heulin, T.: Burkholderia caribensis sp. Nov., an
expolysaccharide-producing bacterium isolated from vertisol microaggregates in Martinique. Int. J. Syst.
Bacteriol. 49, 787-794 (1999).
Alfred E. Brown (2005). Benson’s Microbiological Applications- Laboratory Manual in General
Microbiology. Physiological Characteristics: Oxidation and Fermentation Tests, pp 249-256.
Athnasopoulos, N., 1991. Biodegradation of textile wastewaters. In: Martin, A.M. (Ed.), Biological
Degradation of Wastes. Elsevier Applied Science, London, pp. 389-411.
Augusta J, Muller RJ, Widdecke H. Biologisch abbaubare Kunststoffe; Testverfahren und
Beurteilungskriterien. Chern Ing Tech 1992; 64:410-5
Page 54
54
Babu S, Vaidya P A N, Bal A S et al (1996). Kinetic of biosurfactant production by Pseu-domonas
aeruginosa strain BS2 from industrial wastes. Biotech letters, 18(13): 263-68.
Bacillus subtilis C9. J Fermentation and Bioengin, 84(1): 41-46.
Barathi,S. Vasudevan N (2001). Utilization of petroleum hydrocarbons by Pseudomonas fluorescens
isolated from petroleum contaminated soil, 26:413-416
Barer, M.R., Harwood, C.R.: Bacterial viability and culturability, pp. 93-137. In: Advances in Microbial
Physiology, Vol. 41 (R.K. Poole, ed.), London, Academic Press, Inc. 1999.
Béguin, P., Aubert, J.-P.: The biological degradation of cellulose. FEMS Microbial. Rev. 13, 25-58
(1994).
Bergquist, P.L., Gibbs, M.D., Morris, D.D., Te’o, V.S., Saul, D.J., Morgan, H.W.: Molecular diversity of
thermophilic cellulolytic and hemicellulolytic bacteria – minireview. FEMS Microbiol. Ecol. 28, 99-110
(1999).
Brock, T.D., Madigan, M.T., 1991. Biology of Microorganisms, 6th
edn. Prentice-Hall, New Jersey.
Coughlan, M.P., Ljungdahl, L.G.: Comparative biochemistry of fungal and bacterial cellulolytic enzyme
systems, pp. 11-30. In: Biochemistry and Genetics of Cellulose Degradation, FEMS Symposium 43 (J.-P.
Aubert, P. Beguin, J. Millet, eds.), London, Academic Press, Inc. 1988.
Del Arco, J.P.De Franca, F.P (2001). Influence of oil contamination levels on hydrocarbon
biodegradation in sandy sediment. Environ. Pollut. 1 (10): 515-519
Handbook of Physiological Methods: Culture Methods and Growth Measurements, edited by Janet
R.Stein (pg 427 – 428)
IMViC test. Retrieved 3rd
March 2013, from
http://www.eplantscience.com/botanical_biotechnology_biology_chemistry/biotechnology_methods/micr
obiology/imvic_test.php
James G. Cappuccino, Natelie Sherman (2008). Microbiology-A Laboratory Manual, 8th
edition. IMVic
Test, pp 165-174.
John P. Harley, Lansing M. Prescott (1996). Laboratory Exercises in Microbiology, 3rd
Edition. Proteins,
Amino Acids, and Enzymes II: The IMVic Tests, pp 90-94.
Kapley, A, Heman J Chhatre, J.P Shanker R Chakrabrati K (1999) Osmotolerane and hydrocarbon
degradation by a genetically engineered Microbial consortium. Bioresour. Technol, 61:241-245
John P. Harley, Lensing M. Prescott (1996) Laboratory Exercise in Microbiology (pg 17-18, 149- 151)
Robert A. Pollack Laboratory Exercise in Microbiology.
Page 55
55
M. Charitha Devi and M. Sunil Kumar (2012) Journal of Microbiology and Biotechnology Research,
Production, Optimization and Partial purification of Cellulase by Aspergillus niger fermented with paper
and timber sawmill industrial wastes. Pp120-128
Masayuki Shimao (2001) Biodegradation of plastics.
Materials and method retrieved from Brazilian Journal of Microbiology, Braz. J.
Microbiol. vol.37 no.2 São Paulo Apr./June 2006
Nam Sun Wang. Cellulose Degradation. Retrieved from
http://www.eng.umd.edu/~nsw/ench485/lab4.htm
Pharmaceutical Microbiology Kakarani
Rajashree Patil and U. S. Bagde (2012). African Journal of Biotechnology Vol. 11(31).Isolation of
polyvinyl chloride degrading bacterial strains from environmental samples using enrichment culture
technique, pp. 7947-7956
Retrieved from http://www.austincc.edu/microbugz/macconkey_agar.php
Scott G. Polymers in modern life. Polymers and Environment. Cambridge, UK: The Royal Society of
Chemitstry; 1999.
Scott G. Polymers in modern life. Polymers and Environment. Cambridge, UK: The Royal Society of
Chemitstry; 1999.
Seymour RB. (1989)Polymer science before & after 1899: notable development during the lifetime of
Maurtis Dekker. J Macromol Sci Chem 26: 1023-32.
Seymour RB. (1989)Polymer science before & after 1899: notable development during the lifetime of
Maurtis Dekker. J Macromol Sci Chem; 26: 1023-32.
S. Brandt Rose and Ruth E. Miller (1939) Studies with the Agar Cup-Plate Method: I. A Standardized
Agar Cup-Plate Technique,Journal of Bacteriology, 38(5):525.
Tian Shuangqi, Wang Zhenyu, Fan Ziluan, Zuo Lili and Wang Jichang (2011). African Journal of
Biotechnology Vol. 10(37). Determination methods of cellulase activity, pp. 7122-7125.
Brian. D. Gilbert, Ph.D. Scientist U.S Pharmacopeial Forum: Volume No 30(3), (pg 1002)
W.M. Jurick II, I. Vico, B.D. Whitaker, V.L. Gaskins and W.J. Janisiewicz (2012). Plant Pathology
Journal II (1)c. Application of the 2-Cyanoacetamide Method for Spectrophotometric Assay of Cellulase
Enzyme Activity, pp 38-41.
Witt U, Muller RJ, Deckwer WD. Biodegradation behavior and material properties of aliphatic/aromatic
polyesters of commercial importance. J Environ Poly Degrade 1997; 15:81-9
Witt U, Muller RJ, Deckwer WD. Biodegradation behavior and material properties of aliphatic/aromatic
polyesters of commercial importance. J Environ Poly Degrade 1997; 15:81-9
Page 56
56
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Short Communication
Studies on HIV Integrase inhibitory activity of Wrightia tinctoria (Roxb) R.Br
P. Selvam1, Kasthuraiah Maddali
2, Christophe marchand
2, Yves Pommier
2
1Nova College of Pharmaceutical Education and Research, Jupudi-515 721, Krishna DT.A.P
2Laboratory of Molecular Pharmacology, National Cancer Institute, Bethesda, Maryland 20892, USA
* For correspondance
Email: [email protected]
Page 57
57
Wrightia tinctoria (WT) leaf extracts have been studied against inhibition of HIV-1 integrase activity. Chloroform,
ether, ethanol, methanol and aqueous extracts of Wrightia tinctoria were investigated for both 3’processing (3’P)
and strand transfer (ST) process of HIV-1 integrase enzymatic activity. All extracts exhibited significant inhibitory
activity against HIV-1 integrase enzyme (3’P 1.9-12 µg/ml and ST: 2.2-12 µg/ml). The aqueous extract (AWT)
displayed potent inhibitory activity against both step of HIV Integrase enzymatic activity (3’P IC50:1.9±0.451
µg/ml and ST IC50: 1.4 ± 0.3 µg/ml).
Keywords: Wrightia tinctoria, HIV Integrase activity, Aqueous extract
INTRODUCTION
Acquired immunodeficiency syndrome (AIDS) is a life threatening and debilitating disease state caused by
retrovirus HIV. Three different classes of chemotherapeutic agents are generally combined to block the replication
of human immunodeficiency virus type 1 (HIV-1) responsible for AIDS and to prevent the occurrence of
resistance: reverse transcriptase inhibitors (RTI), protease inhibitors (PRI), and fusion inhibitors. This widespread
triple combination therapy is referred to as HAART [highly active antiretroviral therapy] ( Richman DD. 2001).
HAART effectively inhibits HIV replication to such an extent that the virus becomes undetectable in the blood.
However, it fails to eradicate viruses that are integrated in the host genome or that persist in cellular and
anatomical “reservoirs”. In addition, prolonged drug exposure led to HIV drug resistance, thus reducing patients’
therapeutically available options (Cohen J. 2002). Based on above fact and the toxicity profile of available of
antiretroviral agents have fueled the discovery of drugs against additional molecular targets. Among them, HIV
integrase (IN), which has no cellular counterpart and crucial enzyme HIV replication, has been intensely studied
over the past 15 years (Fesen et al., 1993; Pommier, et al., 2005; Dayam et al., 2008).
Integration occurs via a sequence of reactions, which start with the IN-mediated cleavage of terminal dinucleotide
from the 3'- end of the viral cDNA (termed “3'-processing”, 3'-P) shortly after reverse transcriptions in the
cytoplasm. Following transfer of the resulting processed viral cDNA into the nucleus, IN catalyzes the insertion of
both ends into target cellular host DNA. That second reaction is referred as “strand transfer” (ST). In the past 15
years, a range of natural and synthetic compounds have been identified as inhibitors of recombinant IN enzyme in
Page 58
58
biochemical assays. Interestingly, polyhydroxylated aromatics and diketo compounds were among the first
inhibitors identified. (Fesen et al., 1993 & 1994). HIV integrase has recently been fully validated as a therapeutic
target with the first FDA approved IN inhibitor Raltegravir (Evering T. H, Markowitz M.2007)
Wrightia tinctoria (Roxb) R.Br is an important medicinal plant used in the Indian system of medicine for the
treatment of variety of diseases (Bigoniya et al., 2008) and it reported to possess analgesic (Reddy et al., 2002)
antifertility ( Keshri et al., 2008), cytotoxicity (Kawamoto et al., 2003), hemostasis (Rajesh et al., 2003), anti-ulcer
activity (Bigoniya et al., 2006) and Hepatoprotective (Selvam et al., 2011). Indole derivatives such as isatin,
indirubin, indigotin and tryphanthrin are the principle active constituents of wrightia tinctoria (Muruganandhan
et al., 2000) which may responsible for board spectrum biological activity. Review of literature revealed that
antiviral activity of Wrightia tinctoria against the replication of HIV is relatively less explored. The present study
is designed to find the inhibitory activity of different extracts of Wrightia tinctoria (WT) against the HIV-1
integrase enzymatic activity.
Extraction: Leaf parts of Wrightia tinctoria (Apocynaceae) were collected in and around Tenkasi, Tamilnadu,
India and were dried in shade, subjected to hot continuous percolation using ether, chloroform, methanol, ethanol
and water. The ether (EWT), chloroform (CWT), methanol (MWT) and ethanol (ETWT) and aqueous extracts
(AWT) of Wrightia tinctoria were concentrated by distillation and dried under vaccum.
Anti-HIV Integrase assays.
Integrase Catalytic Assay (Marchand et al., 2001): Integrase reactions were carried out by mixing 20 nM DNA
with 400 nM IN in a buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MgCl2, 14 mM 2-mercaptoethanol, and the
drug of interest or 10% DMSO. Reactions were incubated at 37°C for 1 h and quenched by addition of an equal
volume of gel loading dye (formamide containing 1% SDS, 0.25% bromophenol blue, and xylene cyanol).
Reaction products were separated in 20% polyacrylamide denaturing sequencing gels. Dried gels were visualized
using the Typhoon 8600 (GE Healthcare, Piscataway, NJ). Densitometry analyses were performed using
ImageQuant software from GE Healthcare
Page 59
59
RESULTS AND DISCUSSION
The ether (EWT), chloroform (CWT), methanol (MWT) and ethanol (ETWT) and aqueous(AWT) leaf extracts
of Wrightia tinctoria have been evaluated for HIV Integrase enzyme inhibitory activity (Table 1; Fig 1.). All the
extracts were investigated for both 3’ processing (3’P) and strands transfer reaction (ST) of HIV integrase
enzymatic activity (Fig. 1). All extracts exhibited inhibitory activity against HIV-1 integrase enzyme (3’P 1.9-12
µg/ml and ST: 2.2-12 µg/ml). The aqueous extract (AWT) displayed significant activity against both step of HIV
In enzymatic activity (3’P IC50:1.9 ± 0.451 µg/ml and ST IC50:1.4 ± 0.3 µg/ml). Indirubin-3'-monoxime (Heredia et
al.,2005) a derivative of a Chinese antileukemia medicine isatis tinctoria inhibits HIV-1 replication and another
isatin containing medicinal plant Isatis indigotica also reported to posses anti HIV activity (Liu et al., 2007).
Anti-HIV and Cytotoxicity of Wrightia tomentosa also demonstrated by our preliminary studies (Selvam et al.,
2012). To our best knowledge, this is the first time to report the anti-HIV-1 Integrase activities of these Indian
plants locally known as wrightia tinctoria. The isolation of active compounds possessing anti-HIV-1 activities
from wrightia tinctoria are now in progress.
REFERENCE
Bigoniya P., Rana A.C. and Agrawal G.P. 2006: Evaluation of the antiulcer activity of hydroalcoholic extract
of Wrightia tinctoria bark in experimentally induced acute gastric ulcers on rat; Nig. J Nat. Pro. Med., 10, 36-
40.
Bigoniya P, Shukla A, Agrawal G.P , Rana A C.2008. Pharmacological Screening of Wrightia tinctoria Bark
Hydro-Alcoholic Extract. Asian J. Exp. Sci.,22: 235-244
Cohen J. 2002. Therapies. Confronting the limits of success. Science 296: 2320-24.
Dayam R, Gundla R, Al-Mawsawi LQ, Neamati, N. 2008. HIV-1 integrase inhibitors: 2005-2006 update.
Med. Res. Reveiw. 28:118–154.
Page 60
60
Evering T. H, Markowitz M.2007. Raltegravir (MK-0518): an integrase inhibitor for the treatment of HIV-1.
Drugs Today 43: 865–877.
Fesen MR, Kohn KW, Leteurtre F, Pommier Y. 1993. Inhibitors of human immunodeficiency virus integrase.
Proc. Natl. Acad. Sci. U.S.A. 90: 2399-03.
Fesen MR, Pommier Y, Leteurtre F, Hiroguchi S, Yung J, Kohn KW. 1994. Inhibition of HIV-1 integrase
by flavones, caffeic acid phenethyl ester (CAPE) and related compounds. Biochem. Pharmacol. 48:595–608.
Heredia A., Davis C., Bamba D., Le N., Gwarzo, MY, Sadowska M., Gallo RC, and Redfield RR. Indirubin-
3-monoxime, a derivative of a Chinese antileukemia medicine, inhibits P- TEFb funcation and HIV-l
replication. AIDS 2005, 19:2087-2095
Keshri G, Kumar S, Kulshreshtha DK, Rajendran SM, Singh MM. 2008. Postcoital interceptive activity of
Wrightia tinctoria in Sprague-Dawley rats: a preliminary study. Contraception 78: 266-70
Kawamoto S, Koyano T, Kowithayakorn T, Fujimoto H, Okuyama E, Hayashi M, Komiyama K, Ishibashi M.
2003. Wrightiamines A and B, two new cytotoxic pregnane alkaloids from Wrightia javanica. Chem Pharm
Bull . 6:737-9.
Marchand C, Neamati N, Pommier Y. 2001. In vitro human immunodeficiency virus type 1 integrase assays.
Methods Enzymol. 340:624–633.
Muruganandhan AV, Battacharya SK, Ghosal S. 2000. Indole and flavanoid constituents of Wrightia tinctoria.
Indian J Chem 39:125-131.
Mumthaz, P. Selvam.P., Exploration of Hepatoprotective activity on fruit extracts of Wrightia tinctoria against
CCl4 induced toxicity. International Research Journal of Pharmacology and Pharmacotherapeutics.2,178-
181,2012.
Page 61
61
Liu JF, Jiang ZY, Wang RR, Chen JJ, Zhang XM, Ma YB. 2007. Isatisine A, a novel alkaloid with an
unprecedented skeleton from leaves of Isatis indigotica. Org Lett.9:4127-30.
Periyasamy Selvam, Christophe Pannecouque, E. De. Clercq . Studies on Anti-HIV and Cytotoxicity of
Wrightia tomentosa. International Journal of Pharmacy and Analytical Research. 1;8-11:2012
Pommier, Y, Johnson, A. A, Marchand, C. 2005. Integrase inhibitors to treat HIV/AIDS. Nat. Review. Drug
Discovery 4:236-248.
Rajesh R, Shivaprasad HV, Gowda CD, Nataraju A, Dhananjaya BL, Vishwanath BS. 2007. Comparative
study on plant latex proteases and their involment in hemostasis: a special emphasis on clot inducing and
dissolving properties.planta Med 73:1061-7.
Reddy YS, Venkatesh S, Ravichandran T, Murugan V, Suresh B. 2002; Antinociceptive activity of Wrightia
tinctoria bark. Fitoterapia 73:421-3.
Richman DD. 2001. HIV chemotherapy. Nature 410: 995–1001.
Page 62
62
Table1: Anti-HIV integrase activity of wrightia tinctoria
Extracts IC50 3’P, g/ml IC50 ST, g/ml
CWT 13 ± 3 12 ± 2
MWT 8.7 ± 1.2 4.6 ± 0.4
AWT 1.9 ± 0.45 1.4 ± 0.3
ETWT 2.3 ± 0.4 2.2 ± 0.5
EWT 5.3 ± 0.6 4.6 ± 1
The results are IC50 ±S.D, n = 4 for HIV-1 IN inhibitory activity
Page 63
63
Fig 1. HIV Integrase Inhibitory activity of wrightia tinctoria
Page 64
64
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Drug Discovery and Development-Current status and Future Focus
N. Somashekara., P. Jagadeesh., C.N. Ramchand
Laila Pharmaceuticals Pvt.Ltd., Vijayawada, A.P
Page 65
65
Short Note
Drug is “a substance intended for use in the diagnosis, cure, mitigation, treatment, or prevention of
disease or as a component of a medication” (The Merriam-Webster online dictionary). Drugs generally
affect the biological processes and must be present in the body at an adequate concentration for it to act at
the specific site, where it can exert its effect. Drug must be easily absorbed, metabolized, and eliminated
from the body without causing any injury. The process of developing a new drug consists of two main
stages called discovery and development. The objective of drug discovery is to find synthetic compounds
or natural products that will cure or ameliorate diseases (Mart et al., (2006). The discovery stage is key
step which includes target selection, lead identification, while the development stage includes Preclinical
studies, clinical trials, manufacturing and marketing of the developed drug and managing the life cycle of
the drug. Target may be a biomolecule such as gene or protein, which can potentially be affected by a
drug molecule. Once the target is identified, a promising lead molecule can be developed which in turn
can be used therapeutically. The lead is optimized by further analyzing and screening through in vitro , ex
vivo and in vivo techniques to get the drug molecule, which will further go for preclinical development to
access pharmaco kinetics, pharmaco dynamics and toxicity. Once the preclinical studies are over, the
drug will go through phase-I, phase-II and Phase III development program. After starting with thousands
of compounds, only a few drug candidates continue into the development stage for clinical trials and
manufacturing (Lifenget al., 2008)
The process of discovering drug by serendipitous approach was improved to a more rational approach.
Rational drug design is a method for developing new pharmaceuticals that typically involves the
elucidation of fundamental physiological mechanisms. Rational drug design is aimed at the targeted
discovery and optimization of new drugs based on detailed structural and chemical information of the
protein. Unlike the earlier approaches to drug discovery, by trial-and-error testing of chemical substances
Page 66
66
on animals, and matching the apparent effects to treatments, rational drug design begins with a
knowledge of specific chemical responses in the body, and tailoring combinations of these to fit a
treatment profile. It thus combines the quest for a scientific understanding of natural phenomena with the
design of useful technology and hence integrates epistemic and practical aims of research and
development. The rational method usually involves a theoretical understanding of which protein is
targeted by the drug, how the drug acts on it, and which mechanisms lead to the desired therapeutic
effects (Matthias Adam, 2005). Another different approach to drug development was to use the body’s
own biological molecules as disease treatments. Antibodies, cytokines, and other immune system
substances can be produced in the laboratory for use in disease treatment.
There are different technologies which enabled successful drug discovery process. The technologies such
as High throughput screening, Virtual screening, combinatorial chemistry. Although there was little
success, there is a steep fall the in identifying and developing new chemical entities (NCE’s). The recent
trend on development of NCE’s is declining year by year ending with only few NCE’s and the future
graph of NCE’s may be worse.
The future focus in the drug discovery and development are mainly concentrated on Biologics,
Biosimilars, Analogs , Cell therapy, Antibodies. Also reverse pharmacology approach to drug discovery
is taking leading path. The resources required for new drug discovery are enormous. The time to bring a
drug to market averaging 14 -15 years and at the same time the average cost to bring a drug to market is
more than $1billion (Dimasi., 2001a). Pharmaceuticals companies are trying hard to reduce the time to
market the drugs. Result of once such effort is contract research, which will enable pharmaceutical
companies to outsource the part of drug discovery and development to contract research organizations.
Contract research has benefitted pharmaceutical companies to have accesses to skills sets of contract
research organization and also achieve quicker time to market.
Page 67
67
References:
1. Marta Artal-Sanz, Liesbeth de Jong and NektariosTavernarakis ,(2006), Caenorhabditiselegans: A
versatile platform for drug discovery, Biotechnol. J. 2006, 1, 1405–1418
2. Lifeng Kang, Bong GeunChung,Robert Langer and Ali Khademhosseini, (2008) Microfluidics for
drug discovery and development: From target selection to product lifecycle management, Drug
Discovery Today _ Volume 13, Numbers 1/2 January 2008
3. Matthias Adam, Studies in History and Philosophy of Biological and Biomedical Sciences 36,
(2005)
4. Dimasi, J.A. (2001a). Risks in new drug development is approval success rates for investigational
drugs. Clin.Pharmacol.Ther. 69, 297-307 .
Page 68
68
Volume 1, 2014 INTERNATIONAL JOURNAL OF PHARMACEUTICAL INNOVATIONS
Contents
Invited article:
DESIGN, MOLECULAR MODELLING STUDIES ON CHLOROQUINE AND ITS
ANALOGUES AS INHIBITORS OF HCV NS5B RNA POLYMERASE
M. ChandraMohan, P. Selvam , J. Pranitha, N. Saravanan Prabhu, M. Suresh Kumar Pg no: 5-20
Research article:
DISSOLUTION ENHANCEMENT OF PIOGLITAZONE HYDROCHLORIDE BY SOLID
DISPERSION TECHNIQUE
T. Divya , S. Pranav ----- Pg no : 21-41
AN ATTEMPT TO ISOLATE INDUSTRIALLY IMPORTANT MICROORGANISM FROM
DIVERSE SAMPLES COLLECTED FROM SEMELING, KEDAH, MALAYSIA
P. Vasanth Raj, S.A. Dhanaraj, Saw Pei Jie, Sim Poh Pei, Vanushya a/p Alagasan, Tien Ja She,
Loh Mon Ying, Tan Te Wei Pg no : 42-55
Short communications :
STUDIES OF HIV INTEGRASE INHIBITORY ACTIVITY OF WRIGHTIA TINCTORIA
Periyasamy Selvam, Kasthuraiah Maddali , Christophe marchand
, Yves Pommier Pg no : 55-63
DRUG DISCOVERY AND DEVELOPMENT-CURRENT STATUS AND FUTURE FOCUS
N. Somashekara., P. Jagadeesh., C.N. Ramchand Pg no : 64-67