Introduction to Apollo Collaborative genome annotation editing A webinar for the i5K Research Community | Dr. Carol Lee’s Lab. Monica Munoz-Torres, PhD | @monimunozto Berkeley Bioinformatics Open-Source Projects (BBOP) Lawrence Berkeley National Laboratory | University of California Berkeley | U.S. Department of Energy Dr. Carol Lee’s Lab | 23 October, 2015
Transcript
Introduction to Apollo C o l l a b o r a t i v e g e n o m e a n n o t a t i o n e d i t i n g A webinar for the i5K Research Community | Dr. Carol Lee’s Lab.
Monica Munoz-Torres, PhD | @monimunozto
Berkeley Bioinformatics Open-Source Projects (BBOP)Lawrence Berkeley National Laboratory | University of California Berkeley | U.S. Department of Energy
Dr. Carol Lee’s Lab | 23 October, 2015
OUTLINE
Web Apollo Collabora've Cura'on and Interac've Analysis of Genomes
2 OUTLINE
• Today we will discover how to extract very valuable informa'on about a genome through cura'on efforts.
APOLLO DEVELOPMENT
APOLLO DEVELOPERS 3
h*p : / /GenomeA r c h i t e c t . o r g /
Nathan Dunn
Eric Yao JBrowse, UC Berkeley
Christine Elsik’s Lab, University of Missouri
Suzi Lewis Principal Investigator
BBOP
Moni Munoz-Torres
Stephen Ficklin GenSAS,
Washington State University
Colin Diesh Deepak Unni
4
AFTER THIS TALK WE WILL...
v Be@er understand genome cura'on in the context of annota'on: assembled genome à automated annota=on à manual annota=on
v Become familiar with the environment and func'onality of the Apollo genome annota'on edi'ng tool.
v Learn to iden'fy homologs of known genes of interest in a newly sequenced genome.
v Learn about corrobora'ng and modifying automa'cally annotated gene models using available evidence in Apollo.
What to expect
A typical genome sequencing project
6
Genome Sequencing Project
Anatomy of a genome sequencing project
Experimental design, sampling.
Comparative analyses
Consensus Gene Set
Manual Annotation
Automated Annotation
Sequencing Assembly
Synthesis & dissemination.
CURATING GENOMESsteps involved
1 Genera=on of Gene Models calling ORFs, one or more rounds of gene predic'on, etc.
The gene set of an organism informs a variety of studies: • Gene number, GC%, TE composi'on, repe''ve regions. • Func'onal assignments.
• Molecular evolu'on, sequence conserva'on. • Gene families. • Metabolic pathways. • What makes an organism what it is?
What makes a bee a “bee”?
Marbach et al. 2011. Nature Methods | Shutterstock.com | Alexander Wild
Bio-‐refresher
REMEMBER... for manual annotation
To remember… Biological concepts to be@er understand manual annota'on
10 BIO-REFRESHER
• GLOSSARY from con1g to splice site
• CENTRAL DOGMA
in molecular biology • WHAT IS A GENE?
defining your goal
• TRANSCRIPTION mRNA in detail
• TRANSLATION
and other defini'ons
• GENOME CURATION steps involved
11 CURATING GENOMES
CENTRAL “DOGMA”of molecular biology
v DNA can be copied to DNA (DNA replica'on),
v DNA informa'on can be copied into mRNA (transcrip'on), and
v Proteins can be synthesized using the informa'on in mRNA as a template (transla'on).
http://www.wisegeek.com/
12 BIO-REFRESHER
WHAT IS A GENE?
v A con'nuously evolving concept paints a very complex picture of molecular ac'vity:
“A gene is a locatable region of genomic sequence, corresponding to a unit of inheritance, which is associated with regulatory regions, transcribed regions and/or other func'onal sequence regions”.
-‐ The Sequence Ontology
13 BIO-REFRESHER
WHAT IS A GENE?
v ... also long transcripts, dispersed regula1on.
“The gene is a DNA segment that contributes to phenotype and func'on. In the absence of demonstrated func'on, a gene may be characterized by sequence, transcrip'on or homology.”
-‐ The ENCODE Project
https://www.encodeproject.org/
14 BIO-REFRESHER
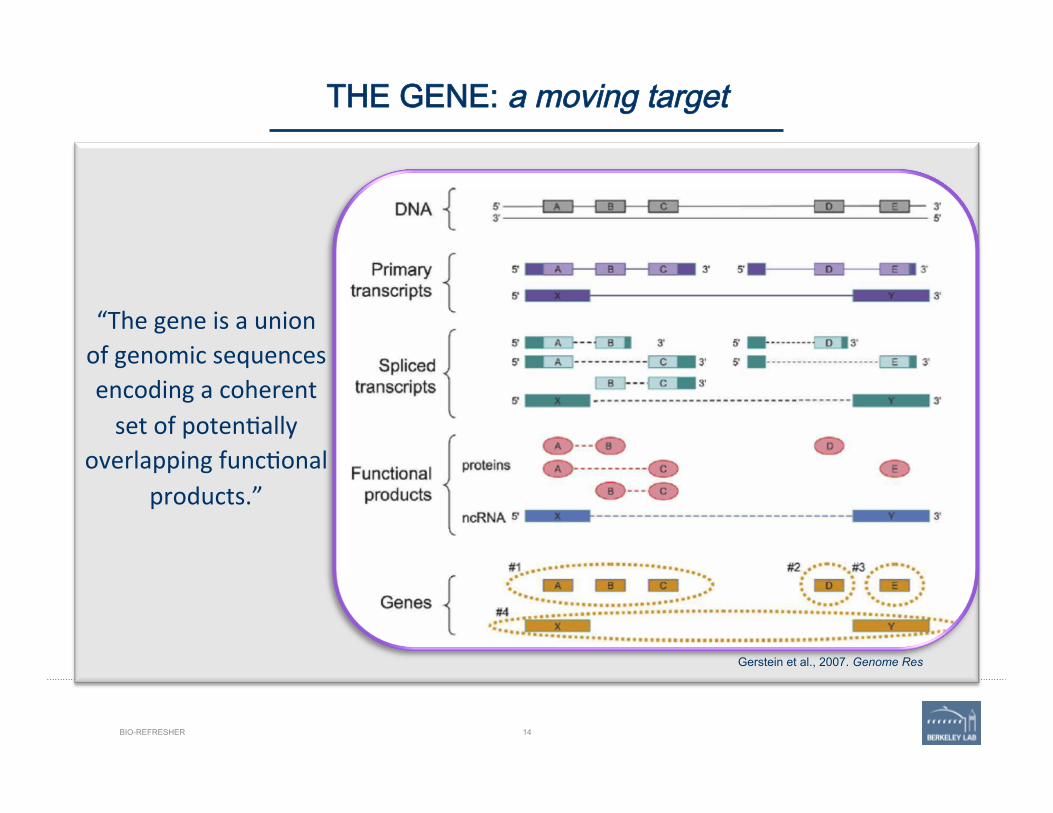
“The gene is a union of genomic sequences encoding a coherent set of poten'ally
overlapping func'onal products.”
Gerstein et al., 2007. Genome Res
THE GENE: a moving target
15 BIO-REFRESHER
TRANSLATIONreading frames
v Reading frame is a manner of dividing the sequence of nucleo'des in mRNA (or DNA) into a set of consecu've, non-‐overlapping triplets (codons).
v Three frames can be read in the 5’ à 3’ direc'on. Given that DNA has two an'-‐parallel strands, an addi'onal three frames are possible to be read on the an'-‐sense strand. Six total possible reading frames exist.
v In eukaryotes, only one reading frame per sec'on of DNA is biologically relevant at a 'me: it has the poten'al to be transcribed into RNA and translated into protein. This is called the OPEN READING FRAME (ORF) • ORF = Start signal + coding sequence (divisible by 3) + Stop signal
v The sec'ons of the mature mRNA transcribed with the coding sequence but not translated are called UnTranslated Regions (UTR); one at each end.
16
"Reading Frame" by Hornung Ákos - Wikimedia Commons
BIO-REFRESHER
TRANSLATIONreading frame
17 BIO-REFRESHER
TRANSLATIONsplice sites
v The spliceosome catalyzes the removal of introns and the liga'on of flanking exons. • introns: spaces inside the gene, not part of the coding sequence • exons: expression units (of the coding sequence)
v Splicing signals (from the point of view of an intron): • One splice signal (site) on the 5’ end: usually GT (less common: GC) • And a 3’ end splice site: usually AG • Canonical splice sites look like this: …]5’-‐GT/AG-‐3’[…
v It is possible to produce more than one protein (polypep'de) sequence from the same genic region, by alterna'vely bringing exons together= alterna=ve splicing. For example, the gene Dscam (Drosophila) has 38,000 alterna'vely spliced mRNAs = isoforms
18 BIO-REFRESHER
TRANSLATIONphase
v Introns can interrupt the reading frame of a gene by inser'ng a sequence between two consecu've codons
v Between the first and second nucleo'de of a codon
v Or between the second and third nucleo'de of a codon
"Exon and Intron classes”. Licensed under Fair use via Wikipedia
19
"Gene structure" by Daycd- Wikimedia Commons
BIO-REFRESHER
mRNAnow in your mind
• Although of brief existence, understanding mRNAs is crucial, as they will become the center of your work.
20
"Protein synthesis" by Kelvinsong - Wikimedia Commons
CURATING GENOMES
TRANSLATIONin detail
Predic'on & Annota'on
22 GENE PREDICTION & ANNOTATION
PREDICTION & ANNOTATION
v Iden'fica'on and annota'on of genome features:
• primarily focuses on protein-‐coding genes. • also iden'fies RNAs (tRNA, rRNA, long and small non-‐coding
RNAs (ncRNA)), regulatory mo'fs, repe''ve elements, etc.
In some cases algorithms and metrics used to generate consensus sets may actually reduce the accuracy of the gene’s representa'on.
CONSENSUS GENE SETS
Gene models may be organized into sets using: v combiners for automa'c integra'on of predicted sets
e.g: GLEAN, EvidenceModeler
or v tools packaged into pipelines
e.g: MAKER, PASA, Gnomon, Ensembl, etc.
GENE PREDICTION & ANNOTATION
ANNOTATIONan imperfect art
No one is perfect, least of all automated annotation. 26
New technology brings new challenges: • Assembly errors can cause fragmented
annota'ons • Limited coverage makes precise
iden'fica'on a difficult task
Image: www.BroadInstitute.org
MANUAL ANNOTATIONimproving predictions
Precise elucida=on of biological features encoded in the genome requires careful
examina=on and review.
Schiex et al. Nucleic Acids 2003 (31) 13: 3738-‐3741
Automated Predictions
Experimental Evidence
Manual Annotation – to the rescue. 27
cDNAs, HMM domain searches, RNAseq, genes from other species.
28
BIOCURATIONstructural and functional adjustments
Iden=fies elements that best represent the underlying biology and eliminates elements that reflect systemic errors of automated analyses.
Assigns func=on through compara've analysis of similar genome elements from closely related species using literature, databases, and experimental data.
MANUAL ANNOTATION
h@p://GeneOntology.org
1
2
GENOME ANNOTATIONan inherently collaborative task
GENE PREDICTION & ANNOTATION 29
Researchers oGen turn to colleagues for second opinions and insight from those with exper1se in
v Web based, integrated with JBrowse. v Supports real 'me collabora'on. v Automa'c genera'on of ready-‐made
computable data. v Supports annota'on of genes, pseudogenes,
tRNAs, snRNAs, snoRNAs, ncRNAs, miRNAs, TEs, and repeats.
v Intui've annota'on, gestures, and pull-‐down menus to create and edit transcripts and exons structures, insert comments (CV, freeform text), associate GO terms, etc.
APOLLO
h@p://GenomeArchitect.org
Con'nuous training and support for hundreds of geographically dispersed scien'sts, from diverse research communi'es, in conduc'ng manual annota'ons efforts to recover coding sequences in agreement with all available biological evidence using Apollo.
31
LESSONS LEARNED
APOLLO
• Collabora've work dis'lls invaluable knowledge
32
A LITTLE TRAINING GOES A LONG WAY!
Provided with adequate tools, wet lab scien'sts make excep'onal curators who can easily learn to maximize the genera'on of accurate,
biologically supported gene models.
APOLLO
Apollo
Sort
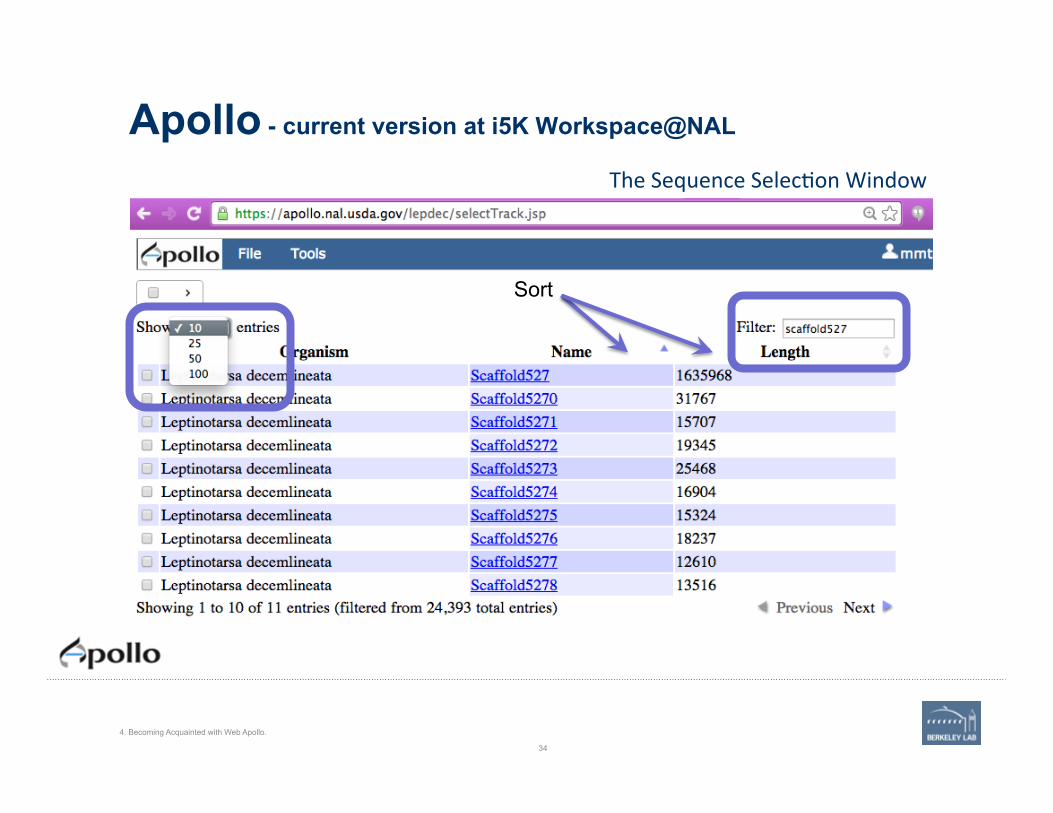
Apollo - current version at i5K Workspace@NAL
34
The Sequence Selec'on Window
4. Becoming Acquainted with Web Apollo.
34
35
APOLLOannotation editing environment
BECOMING ACQUAINTED WITH APOLLO
Color by CDS frame, toggle strands, set color scheme and highlights.
Get coordinates and “rubber band” selec'on for zooming.
Login
User-‐created annota'ons. New
annotator panel.
Evidence Tracks
Stage and cell-‐type specific transcrip'on data.
h@p://genomearchitect.org/web_apollo_user_guide
Naviga'on tools: pan and zoom Search box: go to
a scaffold or a gene model.
Grey bar of coordinates indicates loca'on. You can also select here in order to zoom to a sub-‐region.
‘View’: change color by CDS, toggle strands, set highlight.
‘File’: Upload your own evidence: GFF3, BAM, BigWig, VCF*. Add combina'on and sequence search tracks.
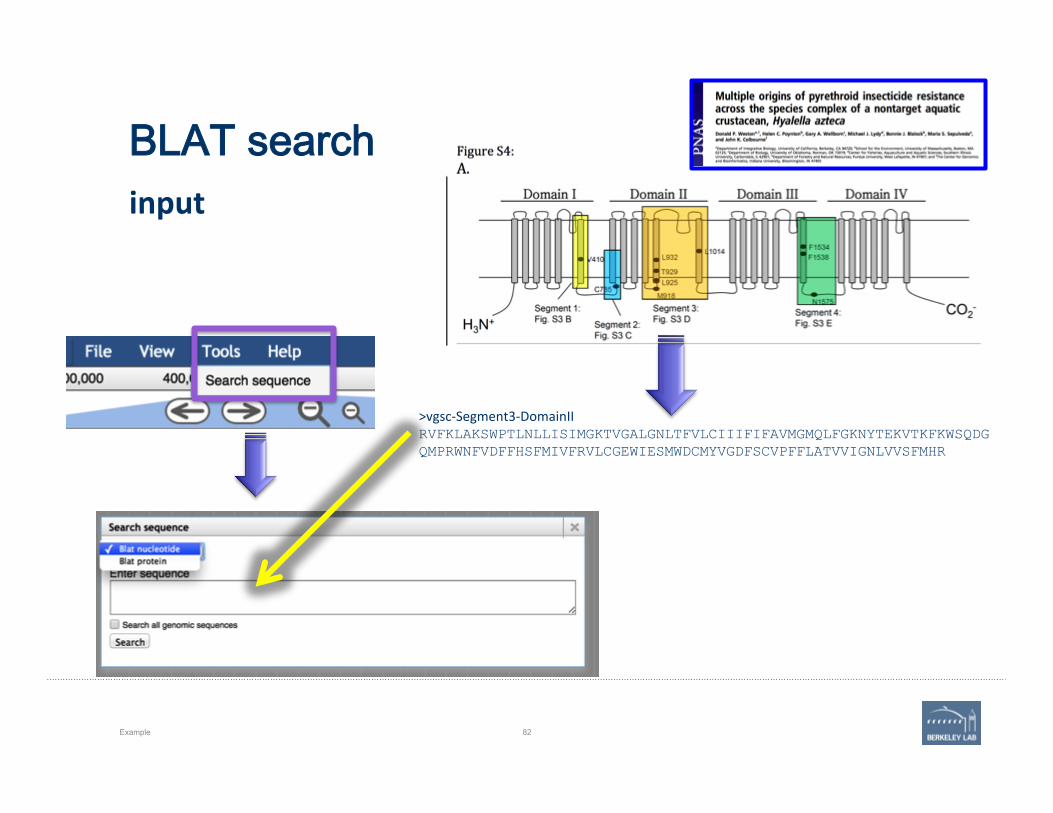
‘Tools’: Use BLAT to query the genome with a protein or DNA sequence.
Available Tracks
Evidence Tracks Area
‘User-‐created Annota'ons’ Track
Login
36
APOLLOgraphical user interface (GUI) for editing annotations
Becoming Acquainted with Web Apollo.
Cura'ng with Apollo
BECOMING ACQUAINTED WITH APOLLO 38 | 38
GENERAL PROCESS OF CURATIONmain steps to remember
1. Select or find a region of interest, e.g. scaffold. 2. Select appropriate evidence tracks to review the gene model.
3. Determine whether a feature in an exis'ng evidence track will provide a reasonable gene model to start working.
4. If necessary, adjust the gene model.
5. Check your edited gene model for integrity and accuracy by comparing it with available homologs.
6. Comment and finish.
USER NAVIGATIONremovable side dock
HIGHLIGHTED IMPROVEMENTS 39
Annotations Organism Users Groups Admin Tracks Reference Sequence
EDITS & EXPORTSannotation details, exon boundaries, data export
HIGHLIGHTED IMPROVEMENTS 40
1 2
Annotations
1
2
gene
mRNA
HIGHLIGHTED IMPROVEMENTS 41
Reference Sequences
3
FASTA
GFF3
EDITS & EXPORTSannotation details, exon boundaries, data export
3
42 | 42 BECOMING ACQUAINTED WITH APOLLO
USER NAVIGATION
Annotator panel.
• Choose appropriate evidence from list of “Tracks” on annotator panel.
• Select & drag elements from evidence track into the ‘User-‐created Annota1ons’ area.
• Hovering over annota'on in progress brings up an informa'on pop-‐up.
• Crea'ng a new annota'on
43 | 43
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
• Annota'on right-‐click menu
44 | 44
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
• ‘Zoom to base level’ op'on reveals the DNA Track.
45 | 45
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
• Color exons by CDS from the ‘View’ menu.
46 |
Zoom in/out with keyboard: shiv + arrow keys up/down
46
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
• Toggle reference DNA sequence and transla=on frames in forward strand. Toggle models in either direc'on.
Annota'on
simple cases
“Simple case”: -‐ the predicted gene model is correct or nearly correct, and
-‐ this model is supported by evidence that completely or mostly agrees with the predic'on.
-‐ evidence that extends beyond the predicted model is assumed to be non-‐coding sequence.
The following are simple modifica'ons.
49 | 49
ANNOTATING SIMPLE CASES
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
• A confirma'on box will warn you if the receiving transcript is not on the same strand as the feature where the new exon originated.
• Check ‘Start’ and ‘Stop’ signals aver each edit.
50
ADDING EXONS
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
If transcript alignment data are available & extend beyond your original annota'on, you may extend or add UTRs.
1. Right click at the exon edge and ‘Zoom to base level’.
2. Place the cursor over the edge of the exon un1l it becomes a black arrow then click and drag the edge of the exon to the new coordinate posi'on that includes the UTR.
51
ADDING UTRs
To add a new spliced UTR to an exis'ng annota'on also follow the procedure for adding an exon.
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
To modify an exon boundary and match data in the evidence tracks: select both the [offending] exon and the feature with the expected boundary, then right click on the annota'on to select ‘Set 3’ end’ or ‘Set 5’ end’ as appropriate.
In some cases all the data may disagree with the annota'on, in other cases some data support the annota'on and some of the
data support one or more alterna've transcripts. Try to annotate as many alterna've transcripts as are well supported by the data.
52
MATCHING EXON BOUNDARY TO EVIDENCE
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
Non-‐canonical splice sites flags. Double click: selec'on of feature and sub-‐features
Non-‐canonical splices are indicated by an orange circle with a white exclama'on point inside, placed over the edge of the offending exon.
Canonical splice sites:
3’-‐…exon]GA / TG[exon…-‐5’
5’-‐…exon]GT / AG[exon…-‐3’ reverse strand, not reverse-‐complemented:
forward strand
54
SPLICE SITES
Zoom to review non-‐canonical splice site warnings. Although these may not always have to be corrected (e.g GC donor), they should be flagged with a comment.
Exon/intron splice site error warning
Curated model
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
Apollo calculates the longest possible open reading frame (ORF) that includes canonical ‘Start’ and ‘Stop’ signals within the predicted exons.
If ‘Start’ appears to be incorrect, modify it by selec'ng an in-‐frame ‘Start’ codon further up or downstream, depending on evidence (proteins, RNAseq).
It may be present outside the predicted gene model, within a region supported by another evidence track.
In very rare cases, the actual ‘Start’ codon may be non-‐canonical (non-‐ATG).
55
‘Start’ AND ‘Stop’ SITES
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
1. Zoom in to clearly resolve each exon as a dis'nct rectangle.
2. Two exons from different tracks sharing the same start/end coordinates display a red bar to indicate matching edges.
3. Selec'ng the whole annota'on or one exon at a 'me, use this edge-‐matching func'on and scroll along the length of the annota'on, verifying exon boundaries against available data. Use square [ ] brackets to scroll from exon to exon. User curly { } brackets to scroll from annota'on to annota'on.
4. Check if cDNA / RNAseq reads lack one or more of the annotated exons or include addi'onal exons.
56
CHECKING EXON INTEGRITY
BECOMING ACQUAINTED WITH APOLLO SIMPLE CASES
complex cases
Evidence may support joining two or more different gene models. Warning: protein alignments may have incorrect splice sites and lack non-‐conserved regions!
1. In ‘User-‐created Annota<ons’ area shiv-‐click to select an intron from each gene model and right click to select the ‘Merge’ op'on from the menu.
2. Drag suppor'ng evidence tracks over the candidate models to corroborate overlap, or review edge matching and coverage across models.
3. Check the resul'ng transla'on by querying a protein database e.g. UniProt, NCBI nr. Add comments to record that this annota'on is the result of a merge.
58
Red lines around exons: ‘edge-‐matching’ allows annotators to confirm whether the evidence is in agreement without examining each exon at the base level.
COMPLEX CASES merge two gene predictions on the same scaffold
BECOMING ACQUAINTED WITH APOLLO COMPLEX CASES
One or more splits may be recommended when: -‐ different segments of the predicted protein align to two or more different gene families -‐ predicted protein doesn’t align to known proteins over its en're length -‐ Transcript data may support a split, but first verify whether they are alterna've transcripts.
59
COMPLEX CASES split a gene prediction
BECOMING ACQUAINTED WITH APOLLO COMPLEX CASES
DNA Track
‘User-‐created Annota=ons’ Track
60
COMPLEX CASES correcting frameshifts and single-base errors
Always remember: when annota'ng gene models using Apollo, you are looking at a ‘frozen’ version of the genome assembly and you will not be able to modify the assembly itself.
1. Apollo allows annotators to make single base modifica'ons or frameshivs that are reflected in the sequence and structure of any transcripts overlapping the modifica'on. These manipula'ons do NOT change the underlying genomic sequence.
2. If you determine that you need to make one of these changes, zoom in to the nucleo'de level and right click over a single nucleo'de on the genomic sequence to access a menu that provides op'ons for crea'ng inser'ons, dele'ons or subs'tu'ons.
3. The ‘Create Genomic Inser<on’ feature will require you to enter the necessary string of nucleo'de residues that will be inserted to the right of the cursor’s current loca'on. The ‘Create Genomic Dele<on’ op'on will require you to enter the length of the dele'on, star'ng with the nucleo'de where the cursor is posi'oned. The ‘Create Genomic Subs<tu<on’ feature asks for the string of nucleo'de residues that will replace the ones on the DNA track.
4. Once you have entered the modifica'ons, Apollo will recalculate the corrected transcript and protein sequences, which will appear when you use the right-‐click menu ‘Get Sequence’ op'on. Since the underlying genomic sequence is reflected in all annota'ons that include the modified region you should alert the curators of your organisms database using the ‘Comments’ sec'on to report the CDS edits.
5. In special cases such as selenocysteine containing proteins (read-‐throughs), right-‐click over the offending/premature ‘Stop’ signal and choose the ‘Set readthrough stop codon’ op'on from the menu.
63
COMPLEX CASES correcting frameshifts, single-base errors, and selenocysteines
BECOMING ACQUAINTED WITH APOLLO COMPLEX CASES
64 | 64
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
• Annotation right-click menu
65
Annota'ons, annota'on edits, and History: stored in a centralized database.
65
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
Follow the checklist un'l you are happy with the annota'on!
And remember to… – comment to validate your annota'on, even if you made no changes to an exis'ng model. Think of comments as your vote of confidence.
– or add a comment to inform the community of unresolved issues you think this model may have.
66 | 66
Always Remember: Apollo cura'on is a community effort so please use comments to communicate the reasons for your
annota'on. Your comments will be visible to everyone.
COMPLETING THE ANNOTATION
BECOMING ACQUAINTED WITH APOLLO
67 | 67
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
• Annotation right-click menu
68
The Annota'on Informa=on Editor
68
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
DBXRefs are database crossed references: if you have reason to believe that this gene is linked to a gene in a public database (including your own), then add it here.
69
The Annota'on Informa=on Editor
• Add PubMed IDs • Include GO terms as appropriate
from any of the three ontologies • Write comments sta'ng how you
have validated each model.
69
USER NAVIGATION
BECOMING ACQUAINTED WITH APOLLO
Checklist
• Check ‘Start’ and ‘Stop’ sites.
• Check splice sites: most splice sites display these residues …]5’-‐GT/AG-‐3’[…
• Check if you can annotate UTRs, for example using RNA-‐Seq data: – align it against relevant genes/gene family – blastp against NCBI’s RefSeq or nr
• Check for gaps in the genome.
• Addi'onal func'onality may be necessary: – merging 2 gene predic'ons on the same scaffold
– merging 2 gene predic'ons from different scaffolds
– splifng a gene predic'on – correc'ng frameshigs and other errors in the genome assembly
– annota'ng selenocysteines, correc'ng single-‐base errors, etc.
71 | 71
• Add: – Important project informa'on in the form of
comments – IDs from public databases e.g. GenBank (via
DBXRef), gene symbol(s), common name(s), synonyms, top BLAST hits, orthologs with species names, and everything else you can think of, because you are the expert.
– Comments about the kinds of changes you made to the gene model of interest, if any.
– Any appropriate func'onal assignments, e.g. via BLAST, RNA-‐Seq data, literature searches, etc.
CHECKLIST for accuracy and integrity
MANUAL ANNOTATION CHECKLIST
Cura'ng within i5K
73 i5K Workspace@NAL
THE COLLABORATIVE CURATION PROCESS AT i5K
1. A computa'onally predicted consensus gene set has been generated using mul'ple lines of evidence; e.g. LDEC_v0.5.3-‐Models
2. i5K Projects will integrate consensus computa'onal predic'ons with
manual annota'ons to produce an updated Official Gene Set (OGS):
Achtung! • If it’s not on either track, it won’t make the OGS! • If it’s there and it shouldn’t, it will s'll make the OGS!
74 i5K Workspace@NAL
THE COLLABORATIVE CURATION PROCESS AT i5K
3. In some cases algorithms and metrics used to generate consensus sets may actually reduce the accuracy of the gene’s representa'on. User your judgment and choose a different model to annotate.
4. Isoforms: drag original and alterna'vely spliced form to ‘User-‐created Annota<ons’ area.
5. If an annota'on needs to be removed from the consensus set, drag it to the ‘User-‐created Annota<ons’ area and label as ‘Delete’ on the Informa1on Editor.
6. Overlapping interests? Collaborate to reach agreement.
7. Follow guidelines for i5K Pilot Species Projects, at h@p://goo.gl/LRu1VY
Example
Example
Example 76
Cura'on example using the Hyalella azteca genome (amphipod crustacean).
What do we know about this genome?
• Currently publicly available data at NCBI: • >37,000 nucleo'de seqsà scaffolds, mitochondrial genes • 344 amino acid seqsà mitochondrion • 47 ESTs • 0 conserved domains iden'fied • 0 “gene” entries submi@ed
• Data at i5K Workspace@NAL (annota'on hosted at USDA) -‐ 10,832 scaffolds: 23,288 transcripts: 12,906 proteins
Example 77
PubMed Search: what’s new?
Example 78
PubMed Search: what’s new?
Example 79
“Ten popula'ons (3 cultures, 7 from California water bodies) differed by at least 550-‐fold in sensi=vity to pyrethroids.”
“By sequencing the primary pyrethroid target site, the voltage-‐gated sodium channel (vgsc), we show that point muta'ons and their spread in natural popula'ons were responsible for differences in pyrethroid sensi'vity.”
“The finding that a non-‐target aqua'c species has acquired resistance to pes'cides used only on terrestrial pests is troubling evidence of the impact of chronic pes=cide transport from land-‐based applica'ons into aqua'c systems.”
How many sequences are there, publicly available, for our gene of interest?
• In this case, ORF includes the high-‐scoring segment pairs (hsp), marked here in blue.
• Note that gene is transcribed from reverse strand.
Available Tracks
Example 88
Get Sequence
Example 89
http://blast.ncbi.nlm.nih.gov/Blast.cgi
Also, flanking sequences (other gene models) vs. NCBI nr
Example 90
In this case, two gene models upstream, at 5’ end.
BLAST hsps
Review alignments
Example 91
HaztTmpM006234
HaztTmpM006233
HaztTmpM006232
Hypothesis for vgsc gene model
Example 92
Editing: merge the three models
Example 93
Merge by dropping an exon or gene model onto another.
Merge by selec'ng two exons (holding down “Shiv”) and using the right click menu.
or…
Result of merging the gene models:
Example 94
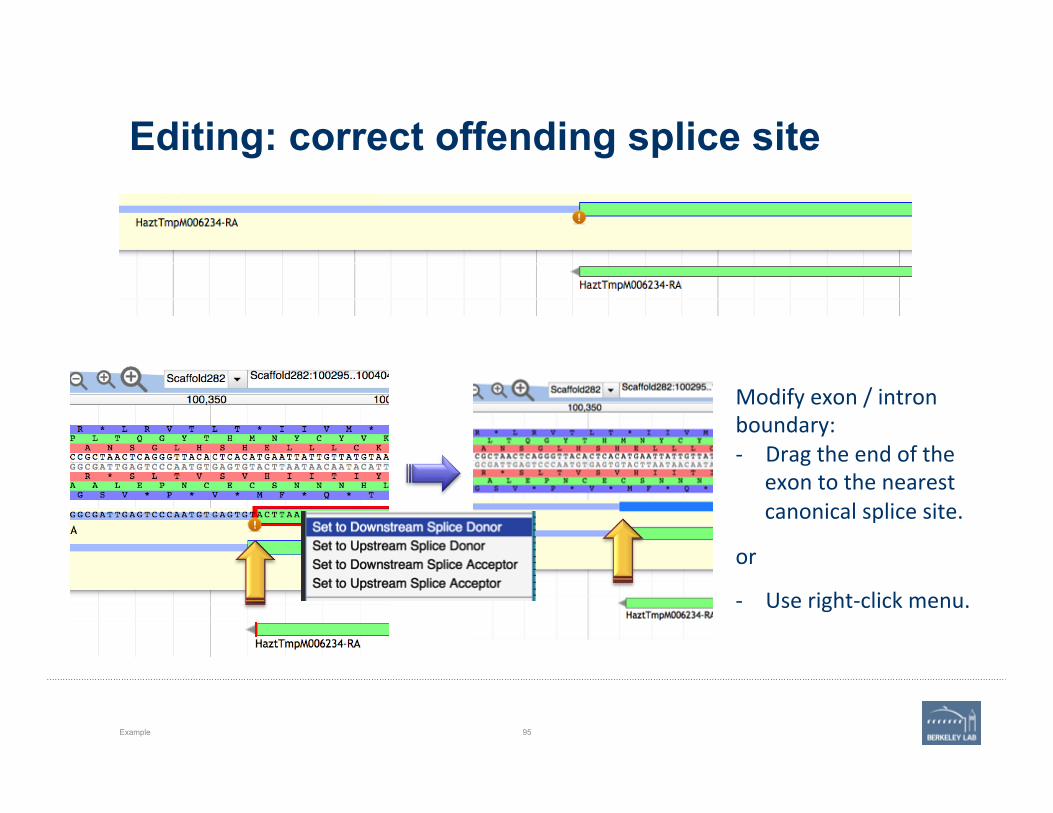
Editing: correct offending splice site
Example 95
Modify exon / intron boundary: -‐ Drag the end of the
exon to the nearest canonical splice site.
or
-‐ Use right-‐click menu.
Editing: set translation start
Example 96
Editing: delete exon not supported by evidence
Example 97
Delete first exon from HaztTmpM006233
Editing: add an exon supported by RNAseq
Example 98
• RNAseq reads show evidence in support of transcribed product, which was not predicted. • Add exon at coordinates 97946-‐98012 by dragging up one of the RNAseq reads.
Editing: adjust offending splice site using evidence
Example 99
Editing: adjust other boundaries supported by evidence
Example 100
Finished model
Example 101
Corroborate integrity and accuracy of the model: -‐ Start and Stop -‐ Exon structure and splice sites …]5’-‐GT/AG-‐3’[… -‐ Check the predicted protein product vs. NCBI nr, UniProt, etc.
Information Editor
• DBXRefs: e.g. NP_001128389.1, N. vitripennis, RefSeq
2. Contact the coordinator for each species community to receive more informa'on about how to contribute. Contact info is available on each organism’s page.
PUBLIC DEMO 105 | 105
APOLLO ON THE WEBinstructions
Public Honey bee demo available at: h@p://GenomeArchitect.org/WebApolloDemo
Demonstra'on video is available at h@ps://youtu.be/VgPtAP_fvxY
OUTLINE
Web Apollo Collabora've Cura'on and Interac've Analysis of Genomes
107 OUTLINE
• BIO-‐REFRESHER biological concepts for cura'on
• ANNOTATION automa'c predic'ons
• MANUAL ANNOTATION necessary, collabora've
• APOLLO
advancing collabora've cura'on • EXAMPLE
demos
Thank you! 108
• Berkeley Bioinforma=cs Open-‐source Projects (BBOP), Berkeley Lab: Apollo and Gene Ontology teams. Suzanna E. Lewis (PI).
• § Chris1ne G. Elsik (PI). University of Missouri.
• * Ian Holmes (PI). University of California Berkeley.
• Arthropod genomics community: i5K Steering Commi@ee (esp. Sue Brown (Kansas State)), Alexie Papanicolaou (UWS), and the Honey Bee Genome Sequencing Consor'um.
• Stephen Ficklin, GenSAS, Washington State University
• Apollo is supported by NIH grants 5R01GM080203 from NIGMS, and 5R01HG004483 from NHGRI. Both projects are also supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-‐AC02-‐05CH11231
•
• For your a*en=on, thank you!
Apollo
Nathan Dunn
Colin Diesh §
Deepak Unni §
Gene Ontology
Chris Mungall
Seth Carbon
Heiko Dietze
BBOP
Apollo: h@p://GenomeArchitect.org
GO: h@p://GeneOntology.org
i5K: h@p://arthropodgenomes.org/wiki/i5K
Thank you!
NAL at USDA Monica Poelchau Christopher Childers Gary Moore Mei-‐Ju Chen