Iridium-Catalyzed Reactions Involving Transfer Hydrogenation, Addition, N-Heterocyclization, and Alkylation Using Alcohols and Diols as Key SubstratesIridium-Catalyzed Reactions Using Alcohols and Diols as Key SubstratesYasushi Obora,* Yasutaka Ishii*Department of Chemistry and Materials Engineering, Faculty of Chemistry, Materials and Bioengineering and ORDIST, Kansai University, Suita, Osaka 564-8680, JapanFax +81(6)63394026; E-mail: [email protected]; E-mail: [email protected] 24 August 2010
Abstract: This account gives an overview of iridium-catalyzedreactions developed by our group using mainly alcohols and diolsas substrates. In the presented reactions, the iridium catalyst servesas a hydrogen acceptor from the alcohols giving iridium hydride,which is a key transient species. Herein, we report hydrogenation,alkylation, esterification, N-heterocyclization, and coupling reac-tions using alcohols and diols as reagents.
1 Introduction2 Hydrogenation of a,b-Unsaturated Carbonyl Compounds
and Alkenes3 Synthesis of Vinyl and Allyl Ethers4 Synthesis of g,d-Unsaturated Carbonyl Compounds5 a-Alkylation Using Alcohols and Diols as Alkylating
Agents5.1 a-Alkylation of Methyl Ketones5.2 a-Alkylation of Arylacetonitriles5.3 a-Alkylation of Active Methylene Compounds5.4 a-Alkylation of Acetates6 b-Alkylation (Guerbet Reaction)7 Oxidative Esterification8 N-Heterocyclization8.1 Synthesis of Quinolines from Amino Alcohols and Ketones8.2 Synthesis of Benzo[h]quinolines and Benzoindole Deriva-
tives from Naphthylamines and Diols9 Coupling Reaction of Alcohols with Alkynes9.1 Synthesis of Homoallylic Alcohols9.2 Synthesis of a,b-Unsaturated Ketones (Hydroacylation)10 Concluding Remarks
Key words: iridium, alcohols, transfer hydrogenations, alkylations,coupling
1 Introduction
Since the discovery of Crabtree’s cationic iridium com-plex [Ir(cod)(py)(PCy3)2]
+ (cod = cycloocta-1,5-diene,py = pyridine, Cy = cyclohexyl), which was found to be avery active catalyst for the hydrogenation of tetrasubsti-tuted amidoalkenes,1 various organoiridium complexeshave been widely employed in catalytic hydrogenations ofalkenes.2–8 The effectiveness of iridium catalysts has beensuccessfully extended to transfer hydrogenation using al-cohols, and their catalytic performance has attracted much
attention. The reaction involves hydrogen transfer fromthe alcohol to the iridium center to form an iridium hy-dride species as the key intermediate. Subsequently, thehydrogen atom of the resulting iridium hydride transfersto another hydrogen acceptor. Thus, the creation of vari-ous novel catalytic systems can be realized using alcoholsas substrates. To date, various review papers regarding iri-dium- and ruthenium-catalyzed transfer hydrogenationreactions have been published.9–18 In this account, wesummarize our recent research on iridium-catalyzed trans-formations using alcohols and diols involving transfer hy-drogenation which include hydrogenation, alkylation,esterification, N-heterocyclization, and coupling reac-tions.
2 Hydrogenation of a,b-Unsaturated Carbonyl Compounds and Alkenes
The reduction of the carbonyl group of a,b-unsaturatedcarbonyl compounds to give allylic alcohols using alco-hols as a hydrogen source has been well studied.19–27 Incontrast, limited examples have been reported on thechemoselective reduction of the alkenic double bond ofconjugated enones through transfer hydrogenation fromalcohols.28–30 We found that the [Ir(cod)Cl]2/dppp/cesiumcarbonate (Cs2CO3) [dppp = 1,3-bis(diphenylphosphi-no)propane] system serves as an efficient catalyst for thereduction of a,b-unsaturated carbonyl compounds usingpropan-2-ol as a hydrogen source.31 Thus, the treatment of4-phenylbut-3-en-2-one (1) (0.5 mmol) with propan-2-ol(2) (5 mmol) under the influence of a catalytic amount of[Ir(cod)Cl]2 (2 mol%), dppp (2 mol%), and Cs2CO3 (2mol%) in toluene (0.5 mL) at 80 °C for 4 hours gave 4-phenylbutan-2-one (3) with 100% selectivity and 93%conversion (Scheme 1).
Scheme 1
Table 1 summarizes the results for the hydrogenation ofvarious a,b-unsaturated carbonyl compounds using the[IrCl(cod)]2/dppp/Cs2CO3 system. A variety of a,b-unsat-
Ph
O
Ph
OOH [Ir(cod)Cl]2/dppp (cat.)+
1 2 3
Cs2CO3
toluene, 80 °C, 4 h
93% conversion 100% selectivity
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 31
urated ketones were selectively reduced at the carbon–carbon double bond giving the corresponding saturatedketones in good to excellent yields (entries 1–6). The[IrCl(cod)]2/dppp/Cs2CO3 system was also found to pro-mote the hydrogenation of simple alkenes. For example,styrene was hydrogenated to give ethylbenzene in excel-lent yield (entry 7).
Spogliarich and co-workers reported a detailed study ofthe hydrogenation reaction of a,b-unsaturated carbonylcompounds, and the chemoselectivity of the hydrogena-tion was found to be dependent on the steric properties ofthe phosphine used, as evidenced by solution NMR spec-troscopy.32,33 The authors reported that an [IrH5(PR3)2]species was formed in the presence of bulkier phosphinesunder a hydrogen atmosphere and this complex was foundto function as the catalyst for the selective hydrogenationof the carbon–carbon double bond. On the other hand, inthe presence of less-bulky phosphines, such as dieth-yl(phenyl)phosphine, mer-[IrH3(PR3)3] was formed andcatalyzed the reduction of the carbonyl group exclusive-ly.33 Furthermore, Tani et al. reported that simple alkynesand alkenes were reduced by a hydrido(methoxo)iridiumcomplex prepared from [Ir(bpbp)Cl]2 [bpbp = 2,2¢-bis(diphenylphosphino)-1,1¢-biphenyl] with methanol.34
These results suggest that an iridium hydride complex isformed during the course of our reaction.
Thus, the reaction may be initiated by the coordination ofan iridium hydride complex, generated in situ from the[IrCl(cod)]2/dppp complex and propan-2-ol in the pres-ence of Cs2CO3, to the a,b-unsaturated carbonyl com-pound, followed by hydrogen transfer to the conjugateddouble bond to give the saturated carbonyl compound.
3 Synthesis of Vinyl and Allyl Ethers
Vinyl ethers are important raw materials in the productionof glutaraldehyde,35,36 as well as vinyl polymericmaterials37 containing oxygen, and are expected to de-grade easily in nature. Vinyl ethers are prepared practical-ly by the reaction of acetylene with alcohols under severeconditions at high pressures (2–5 MPa) and temperatures(180–200 °C) in the presence of potassium hydroxide(KOH) as a catalyst (Reppe process).38 We found that the[IrCl(cod)]2 catalyst promotes efficiently a new type ofexchange reaction between vinyl acetate (4) and alcohols5, including phenols, leading to the corresponding vinylethers 6 (Scheme 2).39 Usually, the acid-catalyzed ex-change reaction between alcohols and vinyl acetate resultsin alkyl acetates, along with the formation of acetalde-hyde, which is derived readily by isomerization from vi-nyl alcohol.
Yasushi Obora was born in1969 in Shizuoka, Japan,and received his B.Sc.(1991) and Ph.D. degrees(1995) from Gifu Universi-ty. After working as a post-doctoral fellow (1995–1997) at Northwestern Uni-versity, USA, with Profes-sor T. J. Marks, he moved to
the National Institute ofMaterials and ChemicalResearch, AIST, Japan(1997–1999). In 1999, hejoined the research group ofProfessor Yasushi Tsuji atthe Catalysis Research Cen-ter, Hokkaido University, asResearch Associate. In2006, he was appointed to
Associate Professor atKansai University. He re-ceived the Shionogi Awardin Synthetic Organic Chem-istry, Japan, in 2007. Hiscurrent research interests in-clude the development ofnew homogeneous catalysisand organometallic chemis-try.
Yasutaka Ishii was born inOsaka, Japan, in 1941. Hereceived his B.A. (1964)and M.S. degrees (1967)from Kansai University. In1967, he was appointed As-sistant Professor at KansaiUniversity. In 1971, he re-ceived his Ph.D. degreeworking under the supervi-sion of Professor MasayaOgawa. He was a postdoc-toral fellow working withProfessor Louis S. Hegedusat Colorado State Universi-ty, USA, between 1980 and1981. In 1990, he was ap-
pointed to full Professor atKansai University, Japan.Since 2009, he has beenEmeritus Professor atKansai University. He re-ceived the Japan PetroleumInstitute Award for Distin-guished Papers in 1987, theDivisional Award (OrganicSynthesis) of the ChemicalSociety of Japan in 1999,the Award of Synthetic Or-ganic Chemistry, Japan, in1999, the Award of the Ja-pan Petroleum Institute in2002, the Green and Sus-tainable Chemistry Award:
Minister of Education,Sports, Culture, Science andTechnology Prize in 2003,and the Chemical Society ofJapan Award for TechnicalDevelopment in 2004. Hiscurrent research interests in-clude the development ofpractical oxidation reactionsusing molecular oxygen andhydrogen peroxide, homo-geneous catalysis, petro-chemistry, and organo-metallic chemistry directedtoward organic synthesis.
The following is a typical iridium-catalyzed reaction: vi-nyl acetate (2 mmol) was allowed to react with octan-1-ol(1 mmol) in the presence of [IrCl(cod)]2 (0.01 mmol,1 mol%) and sodium carbonate (Na2CO3) (0.6 mmol) intoluene (1 mL) at 100 °C for 2 hours, giving n-octyl vinylether in almost quantitative yield. It is noteworthy that theiridium-catalyzed reaction stopped at the stage of vinylether formation, while in the palladium-catalyzed version,
the vinyl ethers formed would tend to react further with anadditional alcohol to give acetals rather than vinylethers.40
Under the optimized reaction conditions, a wide variety ofvinyl ether derivatives could be synthesized using this iri-dium-catalyzed method (Figure 1). This catalytic vinyla-tion system was found to be applicable to the generalsynthesis of vinyl ethers from secondary and tertiary alco-hols.
Figure 1
To obtain further mechanistic insight, phenol-d was al-lowed to react with vinyl acetate under these conditions.As a result, no deuterium incorporation was observed inthe resulting phenyl vinyl ether. This suggests that the re-action pathway may proceed via intermediate 8, which re-sults from the reaction of [IrCl(cod)]2, vinyl acetate (4),and alcohol 5 under the influence of Na2CO3 (Scheme 3).Release of alkyl vinyl ether 6 from intermediate 8 givesrise to acetoxyiridium complex 9, which then reacts withalcohol 5, leading to alkoxyiridium complex 10. Subse-quent coordination of vinyl acetate (4) to complex 10 fol-lowed by insertion regenerates 8.
Scheme 3
Table 1 Transfer Hydrogenation of Various a,b-Unsaturated Car-bonyl Compounds and Alkenes Catalyzed by [Ir(cod)Cl]2
a
Entry Substrate Product Conversion (%)
Selectivity (%)
1 99 98
2b 96 94
3c,d 79 96
4 99 95
5d 90 98
6c,d 78 96
7e 99 96
8d 68 92
9d 64 84
a The substrate (0.5 mmol) was allowed to react with propan-2-ol (5.0 mmol) in the presence of a catalytic amount of [IrCl(cod)]2 (0.01 mmol, 2 mol%), dppp (0.01 mmol, 2 mol%), and Cs2CO3 (0.01 mmol, 2 mol%) in toluene (0.5 mL) at 80 °C for 4 h.b Using [IrCl(cod)]2 (1 mol%), dppp (1 mol%), and Cs2CO3 (1 mol%).c Using Cs2CO3 (1 mol%).d After 15 h.e Using [Ir(cod)Cl]2 (4 mol%), dppp (4 mol%), and Cs2CO3 (4 mol%).
O O
O O
O O
O O
O O
O O
Scheme 2
ROHO
O
RO OH
O
+[IrCl(cod)]2 (cat.)
toluene, 100 °C
+
4 5 6
Na2CO3
7
n-C8H17
O
99%O
91%
S
92%
O
88%O
87%
O O4
O
O
75%
O
86%
O
94%
O
95%
O
98%
O4
78%
(GC yield)
LnIr OR
ROHAcOH
OR
OLnIrO
LnIr OAc
ROHAcO
AcO RO8
10 9
[IrCl(cod)]2/Na2CO3
+
4 5
64
5
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 33
This method could be extended to the synthesis of alkylallyl ethers from alcohols and allyl acetate. Thus, iridiumcationic complex [Ir(cod)2]
+BF4– catalyzed the allylation
of alcohols 11 with allyl acetate (12) to afford allyl ethers13 (Scheme 4).41 Allyl ethers are important compoundsand are frequently used as monomers for the production ofpolymeric materials and as starting materials for Claisenrearrangements.42,43 The above protocol allows the syn-thesis of allyl ethers from allylic acetates and simple alco-hols.
Scheme 4
For instance, the reaction of allyl acetate with octan-1-olin the presence of a catalytic amount of [Ir(cod)2]
+BF4– af-
forded allyl n-octyl ether in quantitative yield. Allyl car-boxylates were also prepared in good yields by theexchange reaction between carboxylic acids and allylacetate.
On the basis of these results, a variety of alcohol deriva-tives were allowed to react with allyl acetate under similarconditions to give the corresponding products as shown inFigure 2.
Figure 2
The reaction would proceed via the formation of a p-allyl-iridium complex, followed by nucleophilic attack of thealcohol (Scheme 5).
Scheme 5
4 Synthesis of g,d-Unsaturated Carbonyl Com-pounds
We previously reported the rearrangement of allyl homo-allyl ethers to g,d-unsaturated aldehydes induced by the[IrCl(cod)]2 complex.44 Alternatively, allyl vinyl ethers
can be directly prepared by the vinylation of allylic alco-hols with vinyl acetate, providing the start of a very attrac-tive route to g,d-unsaturated carbonyl compounds becauseallylic alcohols are easily prepared. The one-pot synthesisof g,d-unsaturated carbonyl compounds from allylic alco-hols and vinyl or isopropenyl acetate was achieved usingthe [IrCl(cod)]2 catalyst. The reaction proceeds via in-situ-generated allyl vinyl ethers, e.g. intermediate 16(Scheme 6), followed by Claisen rearrangement of the re-sulting ether.45
Thus, the reaction of trans-2-methyl-3-phenylprop-2-en-1-ol (14) (0.5 mmol) with isopropenyl acetate (15) (6mmol) in the presence of [IrCl(cod)]2 (2 mol%) andCs2CO3 (5 mol%) at 100 °C for 3 hours, followed by thereaction at 140 °C for 15 hours, afforded 5-methyl-4-phe-nylhex-5-en-2-one (17) in 83% yield (Scheme 6).
Scheme 6
Transfer isopropenylation of alcohol 14 with substrate 15is induced by the [IrCl(cod)]2 complex to afford ether 16,followed by Claisen rearrangement to give g,d-unsaturat-ed carbonyl compound 17.44
5 a-Alkylation Using Alcohols and Diols as Alkylating Agents
5.1 a-Alkylation of Methyl Ketones
The a-alkylation of enolates derived from ketones withalkyl halides is an important and frequently used methodfor forming new carbon–carbon bonds in organic synthe-sis.46 However, the main disadvantage of this methodolo-gy is the formation of undesirable waste salts. If the a-alkylation of enolates derived from ketones with alkyl ha-lides could be replaced by the direct reaction of ketoneswith alcohols, this method would provide a very usefulwaste-free and environmentally benign route to a-alkyla-tion.
We found that the [IrCl(cod)]2 complex combined withtriphenylphosphine (Ph3P) and KOH efficiently catalyzedthe selective a-alkylation of ketones with alcohols.47 Be-cause alcohols are employed as the alkyl source, thismethodology provides an efficient, clean route to a-alkyl-ated ketones from methyl ketones without the formationof any waste products other than water. For instance, thereaction of octan-2-one (18) (2 mmol) with butan-1-ol(19) (4 mmol) in the presence of [IrCl(cod)]2 (0.02 mmol),Ph3P (0.04 mmol), and KOH (0.2 mmol) at 100 °C for 4
hours without any solvent afforded dodecan-6-one (20) in80% yield (Scheme 7). It is noteworthy that the alkylationproceeded with complete regioselectivity at the less-hin-dered side of octan-2-one, namely at the methyl group; theregioselectivity of the conventional alkylation of enolateswith halides is very difficult to control, producing usuallythe a- and a¢-alkylated products. The reaction is promotedby only a catalytic quantity of a base, such as KOH, in theabsence of both a hydrogen acceptor and a solvent.
Scheme 7
It is thought that the above reaction is a novel route to var-ious aliphatic ketones, which are obtained in mainly highyields by selecting the ketones and alcohols employed(Figure 3).
Figure 3
Our strategy was successfully extended to the reaction be-tween ketones and a,w-diols which provides a very conve-nient synthetic tool for preparing w-hydroxy ketones anddiketones.48 The selectivity to give w-hydroxy ketones bymonoalkylation or diketones by double alkylation wasfound to be controlled by varying the starting ratio of me-thyl ketone to a,w-diol.
The following is a typical example of the synthesis ofdiketones: the reaction of acetophenone (21) (10 mmol)with hexane-1,6-diol (22) (2 mmol) in the presence of[IrCl(cod)]2 (0.1 mmol, 5 mol%), Ph3P (0.4 mmol, 20mol%), and KOH (0.4 mmol) without any solvent at 100°C for 15 hours gave the double-alkylation product 1,10-diphenyldecane-1,10-dione (23) in 86% isolated yield
(Scheme 8), along with a small amount of the aldol con-densate of 21, 1,3-diphenylbut-2-en-1-one (10%).
On the basis of the above result, several methyl ketoneswere reacted with various a,w-diols under these condi-tions, giving the corresponding diketones in high yields(Figure 4).
Figure 4
As mentioned above, by varying the starting ratio of me-thyl ketone to a,w-diol, w-hydroxy ketones could also beobtained with high selectivity. Thus, the reaction of meth-yl ketones with excess a,w-diols provided w-hydroxyketones using the same protocol as above. As a typical ex-ample, acetophenone (21) (2 mmol) was allowed to reactwith 4 equivalents of hexane-1,6-diol (22) (8 mmol) underthe influence of [IrCl(cod)]2 (0.05 mmol), Ph3P (0.2mmol), and KOH (0.2 mmol) in 1,4-dioxane (1.0 mL) at100 °C for 15 hours to give selectively w-hydroxy ketone24 in high yield (87%) (Scheme 9). The formation ofdiketone 23 under these conditions was suppressed to 5%yield. In spite of the use of excess a,w-diol 22, whichserves as a hydrogen donor to 21, the formation of the hy-drogenation product 1-phenylethanol was in less than 3%yield.
Scheme 9
Because the reaction using excess a,w-diols results in vis-cous liquids that are difficult to stir magnetically, thereaction to prepare some w-hydroxy ketones was carriedout using a solvent, such as 1,4-dioxane.
80%
[IrCl(cod)]2 (cat.)
100 °C, 4 hKOH
O
O
+ n-BuOHPh3P
18 19
20
88% 86%
80%
86%
81%
71%
47%
96%
88%
Ph
O
n-BuPh
O
n-Bu
On-Bu
O
n-C3H7
O
n-Bu
n-C6H11
O
Ph
Ph
O
Ph
n-C6H11
O
Ph Ph
O
81%
n-C6H13
O
84%n-C6H13
O
Ph
(isolated yield)
Scheme 8
Ph
O
+ HO OH6
[IrCl(cod)]2 (cat.)
100 °C, 15 h21 22
Ph
O
23
Ph6
O
86%
Ph3P
KOH
10 mmol 2 mmol
84%
Ph Ph
O
7
O
Ph Ph
O
8
O
Ph Ph
O
9
O
Ph Ph
O
10
O
n-Pr n-Pr
O
6
O
n-Bu n-Bu
O
6
O
n-C6H13 n-C6H13
O
6
O
88% 91%
85% 69%
71% 75%(isolated yield)
Ph
O
+ HO OH6
[IrCl(cod)]2 (cat.)
100 °C, 15 h21 22
Ph3P
KOH
2 mmol 8 mmol
Ph OH
O
24
6
87%1,4-dioxane
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 35
Under these conditions, the reaction of methyl ketoneswith various aliphatic a,w-diols afforded the correspond-ing w-hydroxy ketones in good to excellent yields(Figure 5). Because this type of compound is difficultgenerally to prepare by a one-step reaction, this strategygives a simple direct approach to w-hydroxy ketones.
Figure 5
The above method was successfully applied to the doublealkylation of acetone with a,w-diols, resulting in a novelsynthetic tool for producing a,w-dimethyl diketones.49
Diketones such as hexadecane-2,15-dione (HDDO) areone of the attractive precursors of macrocyclic musks.50
Thus, the reaction of acetone (25) (10 mmol) with decane-1,10-diol (26) (1 mmol) in the presence of [IrCl(cod)]2
(0.05 mmol) combined with Ph3P (0.15 mmol) and KOH(0.4 mmol) in toluene (0.5 mL) at 100 °C for 2 hours re-sulted in HDDO (27) in 90% yield (Scheme 10). Underthese reaction conditions, no monoalkylated product, i.e.13-hydroxytridecan-2-one, was detected.
Scheme 10
Figure 6 shows several a,w-dimethyl diketones preparedfrom acetone and various a,w-diols. It is interesting tonote that this use of the strategy confirms a direct route toa,w-dimethyl diketones by a one-step reaction usingreadily available starting materials, like acetone and diols.
The above a-alkylation reaction is thought to proceedthrough the following sequential reactions involving three
key steps, as similarly proposed in reviews:9–18 (i) hydro-gen transfer from alcohol 28 to an iridium complex, giv-ing aldehyde 29 and an iridium hydride intermediate, (ii)base-catalyzed aldol condensation between the resultingaldehyde 29 and ketone 30 to form a,b-unsaturated ketone31, and (iii) selective hydrogenation of 31 by the iridiumhydride complex generated during the course of the reac-tion, leading to a-alkylated ketone 32 (Scheme 11).
Scheme 11
To obtain information on the pathway of the a-alkylationreaction, the alkylation of acetophenone (21) (1 mmol)with butan-1-ol-d9 (C4D9OH) (33) (4 mmol) was exam-ined under the conditions shown in Scheme 12.
Scheme 12
As a result, the 1H NMR spectrum showed that a 63:37mixture of deuterium-incorporated 1-phenylhexan-1-one-d7 34 and 35 was formed. The formation of these deuter-ated compounds is rationally explained by the followingreaction pathway involving the generation of an iridiumdihydride as a key intermediate (Scheme 13).
The iridium complex (LnIr) is subjected to oxidative addi-tion by C4D9OH (33), followed by b-hydride eliminationto give butanal-d8 (36) and LnIrHD. Under the basic con-ditions, the deuterium atoms at the a-position of interme-diate 36 may undergo rapid proton–deuterium exchangewith C4D9OH (33), via enolate formation, to give eventu-ally butanal-d6 (37). Aldol condensation with the ketone,i.e. acetophenone (21), forms 1-phenylhex-2-en-1-one-d6
(38), followed by hydrogenation with LnIrHD to lead todeuterated products 34 and 35. Deuterium exchange at thea-position of the resulting compound 35 with formed wa-ter and/or butanol seems to contribute to the preferentialformation of 34 rather than 35.
5.2 a-Alkylation of Arylacetonitriles
Grigg and co-workers reported the a-monoalkylation ofarylacetonitriles with alcohols using [Cp*IrCl2]2 (Cp* =pentamethylcyclopentadienyl) in the presence of KOH.51
We successfully extended this strategy to the doublealkylation of arylacetonitriles with a,w-diols.48
For instance, phenylacetonitrile (39) (10 mmol) was al-lowed to react with pentane-1,5-diol (40) (2 mmol) in thepresence of [Cp*IrCl2]2 (0.05 mmol) and Cs2CO3 (0.4mmol) at 160 °C for 15 hours to afford the double-alkyla-tion product 2,8-diphenylnonanedinitrile (41) in 93%yield (Scheme 14). The reaction is highly chemoselectiveand no monoalkylation product was detected at all underthese conditions.
Scheme 14
By reacting various arylacetonitriles and a,w-diols in thepresence of the [Cp*IrCl2]2 catalyst combined withCs2CO3, the corresponding diaryl-containing dinitrileswere obtained in moderate to excellent yields (Figure 7).
5.3 a-Alkylation of Active Methylene Com-pounds
The iridium catalytic system is applicable to the a-alkyla-tion of active methylene compounds, such as alkyl cy-anoacetates.52 As a typical example, n-butyl cyanoacetate(42) (1 mmol) was allowed to react with butan-1-ol (19)(2 mmol) in the presence of [IrCl(cod)]2 (0.05 mmol, 5mol%) and Ph3P (0.2 mmol, 20 mol%) in p-xylene (1 mL)at 130 °C for 15 hours, giving n-butyl 2-cyanohexanoate(43) in 96% yield (Scheme 15). The reaction affordedonly the saturated alkyl cyanoacetate, although in the ru-thenium-catalyzed reaction of alkyl cyanoacetates withaldehydes by Murahashi and co-workers, (E)-a,b-unsatur-ated nitriles were formed.53,54 In contrast to the above-mentioned a-alkylation of methyl ketones with alcohols,
the reaction is efficiently promoted without any base. Co-ordination of the nitrile to the iridium catalyst may assistthe facile activation of the a-carbon–hydrogen bond ofsubstrate 42.
Scheme 15
Under the optimized reaction conditions, the a-alkylationof n-butyl cyanoacetate (42) with various alcohols wassuccessfully achieved, affording the corresponding satu-rated a-alkylated cyano compounds in moderate to excel-lent yields (Figure 8).
In addition, products obtained from the a-alkylation reac-tion of various active methylene compounds with butan-1-ol are shown in Figure 9.
The above iridium-catalyzed a-alkylation of active meth-ylene compounds with alcohols was extended to the reac-tion with a,w-diols.49 For example, the reaction of n-butylcyanoacetate (42) (1 mmol) with dodecane-1,12-diol (44)(3 mmol) in the presence of [IrCl(cod)]2 (0.05 mmol, 5mol%) combined with Ph3P (0.2 mmol) in 1,4-dioxane at120 °C for 20 hours produced n-butyl 2-cyano-14-hydroxy-tetradecanoate (45) in 82% yield (Scheme 16).
This method provides a very attractive selective route to2-cyano-w-hydroxy-substituted carboxylates (Figure 10)using various a,w-diols, giving the precursors of syntheti-cally useful w-hydroxy-substituted carboxylic acids.
Among those w-hydroxy-substituted carboxylic acids, 15-hydroxypentadecanoic acid (CPDA) is an important pre-cursor of the most widely produced macrocyclic syntheticmusk lactone cyclopentadecanolide (CPDL).55 Several
Ph CN + HO OH5
4039
[Cp*IrCl2]2 (cat.)
160 °C, 15 hno solvent
Ph Ph
CN
415
CN
Cs2CO3
89%
Figure 7
48%
Ph Ph
CN
3
CN
84%
Ph Ph
CN
4
CN
70%
Ph Ph
CN
6
CN
67%
Ph Ph
CN
7
CN
85%
CN
5
CN
89%
CN
5
CN
80%
CN
5
CN
MeO OMe
Cl Cl
(isolated yield)
NCOn-Bu
O+
[IrCl(cod)]2 (cat.)Ph3P
p-xylene130 °C, 15 h CN
On-Bu
O
42 19 4396%
n-BuOH
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 37
methods for the synthesis of CPDL from CPDA via in-tramolecular cyclization have been reported.56–58
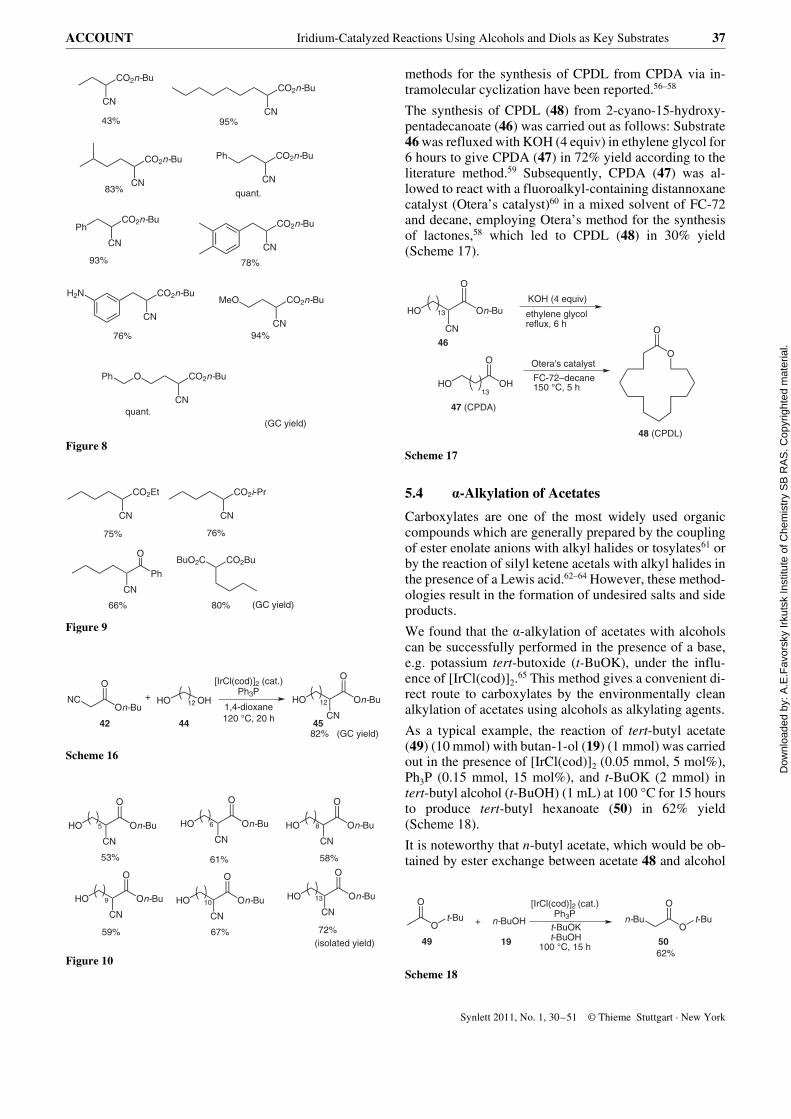
The synthesis of CPDL (48) from 2-cyano-15-hydroxy-pentadecanoate (46) was carried out as follows: Substrate46 was refluxed with KOH (4 equiv) in ethylene glycol for6 hours to give CPDA (47) in 72% yield according to theliterature method.59 Subsequently, CPDA (47) was al-lowed to react with a fluoroalkyl-containing distannoxanecatalyst (Otera’s catalyst)60 in a mixed solvent of FC-72and decane, employing Otera’s method for the synthesisof lactones,58 which led to CPDL (48) in 30% yield(Scheme 17).
Scheme 17
5.4 a-Alkylation of Acetates
Carboxylates are one of the most widely used organiccompounds which are generally prepared by the couplingof ester enolate anions with alkyl halides or tosylates61 orby the reaction of silyl ketene acetals with alkyl halides inthe presence of a Lewis acid.62–64 However, these method-ologies result in the formation of undesired salts and sideproducts.
We found that the a-alkylation of acetates with alcoholscan be successfully performed in the presence of a base,e.g. potassium tert-butoxide (t-BuOK), under the influ-ence of [IrCl(cod)]2.
65 This method gives a convenient di-rect route to carboxylates by the environmentally cleanalkylation of acetates using alcohols as alkylating agents.
As a typical example, the reaction of tert-butyl acetate(49) (10 mmol) with butan-1-ol (19) (1 mmol) was carriedout in the presence of [IrCl(cod)]2 (0.05 mmol, 5 mol%),Ph3P (0.15 mmol, 15 mol%), and t-BuOK (2 mmol) intert-butyl alcohol (t-BuOH) (1 mL) at 100 °C for 15 hoursto produce tert-butyl hexanoate (50) in 62% yield(Scheme 18).
It is noteworthy that n-butyl acetate, which would be ob-tained by ester exchange between acetate 48 and alcohol
19, was not detected in the above reaction at all underthese conditions. Here, the use of acetates that are reluc-tant to undergo the ester exchange (e.g., tert-butyl acetate)is important to achieve the desired alkylation reaction. Inaddition, the alkylation was markedly influenced by thebase employed, and the use of 2 equivalents of t-BuOKbased on alcohol 19 gave the best result.
Under these conditions, the a-alkylation of tert-butyl ace-tate with various primary alcohols afforded the corre-sponding tert-butyl carboxylates in good to excellentyields (Figure 11).
Figure 11
To elucidate the role of t-BuOH as the solvent, the reac-tion time-course of tert-butyl acetate (49) with alcohol 19without the use of a solvent was compared with that in t-
BuOH. The reaction without the solvent reached an equi-librium of tert-butyl hexanoate (50) and n-butyl acetate(51) at an early stage (within 1 h). This observation sug-gests that the alkylation of tert-butyl acetate (49) withbutan-1-ol (19) and the ester-exchange reaction betweenacetate 49 and alcohol 19, producing n-butyl acetate (51),are competitively promoted. In contrast, in the reactionusing t-BuOH as a solvent, the ester exchange was rapidlyinduced in 0.5 hours to form n-butyl acetate (51), whichthen gradually decreased with the increasing formation ofproduct 50. This observation indicates that the ester ex-change proceeds faster than the alkylation, and the result-ing acetate 51 undergoes exchange with t-BuOH, whichexists in excess as the solvent, to regenerate substrate 49,which then reacts with alcohol 19 to form tert-butyl hex-anoate (50).
Indeed, the reaction of tert-butyl acetate (49) with n-butylacetate (51) was carried out under these conditions andgave 50 in 70% yield (Scheme 19), which is a comparableresult with that of the reaction of 49 with butan-1-ol (19)(74% yield, cf. Scheme 18). This finding shows that 51undergoes the exchange reaction in t-BuOH to leave bu-tan-1-ol (19), which subsequently serves as the alkylatingagent of 49 leading to the desired product 50.
The reaction mechanism may be explained by the follow-ing pathway (Scheme 20): First, ester exchange betweentert-butyl acetate (49) and alcohol 19 using t-BuOK oc-curs to generate n-butyl acetate (51), competing with theiridium-catalyzed dehydrogenation of 19 generatingbutanal and an iridium hydride complex. The resulting bu-tanal of the latter reaction undergoes base-catalyzed aldolcondensation with 49 to form an a,b-unsaturated ester,which then reacts with the iridium hydride to give product50 and the iridium catalyst. Meanwhile, n-butyl acetate
O
O
t-Bu
89%
O
O
t-BuR
R = n-C6H13
R = n-Bu
R = n-C8H17
R = H
R = Me
R = OMe
R = CF3
R = Cl
R
82%
62%
75%
78%
82%
64%
69%
67%
67%
R = CyCH2
R = Ph(CH2)3
(isolated yield)
Scheme 19
Ot-Bu +
Ot-Bu
100 °C, 15 h49 51 50
n-Bu
O [IrCl(cod)]2 (cat.)Ph3P
O
70% (GC yield)
On-Bu
O
t-BuOK t-BuOH
Scheme 20
n-BuOH
O
O
n-Bu
Ot-Bu
O
Ot-Bu
O
n-Bun-Pr
4919
50
51
hydrogen-transfer
cycleLnIr LnIrH2
base
H2O
esterexchange
aldolcondensation
t-BuOH
O
O
t-Bu
n-Pr O
H
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 39
(51), generated in situ by ester exchange, is converted into50 by base-catalyzed exchange in t-BuOH. Hence, in thereaction without t-BuOH, the alkylation stopped at thestage of the formation of 51 because of the difficulty of theregeneration of butan-1-ol (19) from 51.
This reaction was applied to the synthesis of a fragrantcompound, ethylene brassylate (1,4-dioxacycloheptade-cane-5,17-dione, Musk T) (55), which is currently manu-factured on a large scale as a synthetic perfume with amusk odor.55,66–68 The reaction of tert-butyl acetate (49)(20 mmol) with nonane-1,9-diol (52) (1 mmol) under theabove conditions gave di-tert-butyl tridecane-1,13-dioate(53). Subsequent hydrolysis of 53 to give dicarboxylicacid 54, followed by the reaction of 54 according to thereported method,68 led to product 55 in 17% yield(Scheme 21).
Scheme 21
6 b-Alkylation (Guerbet Reaction)
The Guerbet reaction is recognized as a useful synthetictool for obtaining b-alkylated alcohols through the self-condensation/dimerization of primary alcohols, as exem-plified by the conversion of butan-1-ol into 2-ethylhexan-1-ol.69 We found that the [Cp*IrCl2]2 and [IrCl(cod)]2
complexes were efficient catalysts of the Guerbet reactionof primary alcohols to afford b-alkylated alcohols.70
Several examples of iridium- and ruthenium-catalyzedGuerbet reactions of alcohols have also been reported.71,72
As an example of our method, the reaction of butan-1-ol(19) (2 mmol) in the presence of [Cp*IrCl2]2 (0.02 mmol,1 mol%), KOH (0.8 mmol, 40 mol%), and octa-1,7-diene(0.2 mmol, 10 mol%) in p-xylene (0.5 mL) at 120 °C for4 hours produced 2-ethylhexan-1-ol (56) in 93% yield(Scheme 22). In this reaction, the addition of a base and asmall amount of a hydrogen acceptor (e.g., octa-1,7-di-ene) was needed. Among the iridium complexes used,[Cp*IrCl2]2 was found to give the best result, but other iri-dium complexes, such as [IrCl(cod)]2 and [Ir(OH)(cod)]2,
also showed high catalytic activities. Needless to say, thisreaction did not proceed without an iridium catalyst and abase.
A variety of primary alcohols have been shown to under-go the iridium-catalyzed Guerbet reaction under theseconditions to give the corresponding condensed dimer al-cohols in moderate to excellent yields (Figure 12). Thismethod is an alternative direct route to b-alkylated prima-ry alcohols, which are usually prepared by the aldol con-densation of aldehydes, followed by hydrogenation.
Figure 12
As a successful extension of this methodology, ethanolwas efficiently converted into butan-1-ol and higher alco-hols.73 Butan-1-ol is an important chemical feedstock andis widely utilized as a solvent and for the production ofpolymeric materials.74 Our method provides an alternativeapproach to butan-1-ol, which is conventionally manufac-tured by the oxo process using propene with hydrogen andcarbon monoxide, followed by the hydrogenation of bu-tyraldehyde.74–77
The most-successful result for the formation of butan-1-olusing the iridium-catalyzed Guerbet reaction is shown inScheme 23. Thus, ethanol (57) (2 mL, 34 mmol) was al-lowed to react with [Ir(acac)(cod)] (acac = acetylaceto-nate) (0.01 mol%), dppp (0.01 mol%), and sodiumethoxide (5 mol%) in a pressured tube and stirred at roomtemperature for 2 hours for the preactivation of the cata-lyst; then, the mixture was heated at 120 °C for 15 hours.A turnover number (TON, based on Ir used) for the forma-tion of butan-1-ol (19) of 1220 was attained with 41%conversion of ethanol. Under these reaction conditions,product 19 was formed with 51% selectivity as the majorproduct, along with 2-ethylbutan-1-ol (58), hexan-1-ol(59), 2-ethylhexan-1-ol (56), and octan-1-ol (60) as minorproducts (Scheme 23).
The above iridium-catalyzed Guerbet reaction, exempli-fied by the conversion of ethanol (57) into butan-1-ol (19),can be rationally explained by the following sequentialpathway (Scheme 24): First the iridium catalyst serves asa hydrogen acceptor from substrate 57 to give acetalde-hyde and an iridium hydride species. Then, the formed al-dehyde reacts via base-catalyzed aldol condensation togive crotonaldehyde and water. Finally, the unsaturatedaldehyde undergoes selective hydrogenation by the iridi-um hydride complex to give the desired product.
Scheme 24
7 Oxidative Esterification
Esters are produced conventionally by the Fischer esteri-fication of acids with alcohols78 or by the Tishchenko re-action of aldehydes.79–81As an alternative method, thehomogeneous catalytic transformation of primary alco-hols into esters has been investigated using [Pd(OAc)2],
82
[Ru(CO)12],83 and [RuH2(PPh3)4]
84–86 catalysts. Suzukiet al. and Williams and co-workers also reported thereaction of primary alcohols to give esters using (penta-methylcyclopentadienyl)iridium aminoalkoxide and[RuH2(CO)(PPh3)3]/xantphos catalysts in the presence ofa hydrogen acceptor, such as butan-2-one and crotono-nitrile, respectively.87,88 Here, we show our findings onthe [IrCl(coe)2]2-catalyzed (coe = cyclooctene) oxidativedimerization of alcohols to produce esters using air as anoxidant.89
As a typical example of our method, the reaction of octan-1-ol (60) (2 mmol) in the presence of [IrCl(coe)2]2 (0.06mmol, 3 mol%) at 95 °C for 15 hours in open air producedn-octyl octanoate (61) in 82% yield (Scheme 25). The
[IrCl(cod)]2 complex showed similar catalytic activity to-ward this reaction and gave product 61 in 76% yield. It isnoteworthy that this reaction proceeded without any base,solvent, and hydrogen acceptor, such as acetone. Thus,this method provides an environmentally clean route toesters from primary alcohols.
Scheme 25
On the basis of these results, the oxidative dimerization ofvarious primary alcohols and the lactonization of diolswere carried out in the presence of [IrCl(coe)2]2 under airto give the corresponding esters in moderate to excellentyields (Table 2).
A plausible reaction pathway is shown in Scheme 26. Thereaction is initiated by the formation of an iridium hydrideand an aldehyde via hydrogen transfer from the alcohol tothe iridium catalyst. Then, the formation of a hemiacetalderived from the alcohol and aldehyde, followed by dehy-drogenation by the iridium catalyst affords the desired es-ter and the iridium dihydride complex. The iridiumdihydride complex reacts with oxygen in the air to liberatewater. Previously, we reported that [Cp2ZrH2] efficientlycatalyzes the Tishchenko reaction of aldehydes to give es-ters through an alkoxyzirconium species, but not a hemi-acetal.90 However, we propose that the above reactionpasses through the formation of a hemiacetal as a transientintermediate because the [IrCl(coe)2]2 complex did notcatalyze the Tishchenko reaction of aldehydes alone in theabsence of alcohols.
The above oxidative esterification should be carried out at95 °C under open air conditions. Therefore, lower alco-hols, such as ethanol, were difficult to convert into ethylacetate using this method. Because ethyl acetate is a veryimportant feedstock, the development of an efficientmethod to access ethyl acetate from ethanol is highly de-sired. We found that the oxidative dimerization of ethanol(57) to give ethyl acetate (62) proceeded smoothly at
Scheme 23
OH
[Ir(acac)(cod)] (cat.)
r.t. to 120 °COH +
OH
+ OH + OH+ OH
19 58
59 56 60
57
dppp1,7-octadiene
NaOEt
41% conversion2 mL
TON 1220selectivity 51%
TON 464selectivity 15%
TON 464selectivity 15%
TON 261selectivity 7%
TON 87selectivity 2%
(34 mmol)
OH
LnIr
[LnIrH2]
H
O
OH19
257
H
O2
2
2
base
aldol condensation
H2O
R OH
[IrCl(coe)2]2 (cat.)
95 °C, 15 h
open air
R O R
O
6061without base and solvent(R = C7H15)
82% (GC yield)
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 41
room temperature in the presence of [Cp*IrCl2]2 com-bined with 2-(methylamino)ethanol using acetone as ahydrogen acceptor (Scheme 27).91
The experimental procedure was as follows: a mixture ofethanol (57) (2 mmol) and acetone (1 mL, 13.6 mmol) was
stirred in the presence of [Cp*IrCl2]2 (0.04 mmol, 2mol%), 2-(methylamino)ethanol (0.12 mmol, 6 mol%),and Cs2CO3 (0.2 mmol, 10 mol%) at room temperature for24 hours, giving ethyl acetate (62) in 85% yield with 96%selectivity.
The addition of a catalytic amount of 2-(methylami-no)ethanol to [Cp*IrCl2]2 is indispensable to achieve thereaction. To elucidate the effect of 2-(methylamino)etha-nol as an additive, the reaction of ethanol (57) catalyzedby [Cp*IrCl2]2 with several analogues was examined(Table 3). Like 2-(methylamino)ethanol, 2-(ethylami-no)ethanol efficiently promoted the reaction (cf. entries 1and 2). However, the yield of ethyl acetate (62) decreasedwith the increasing length of the alkyl group of the alkyl-amino moiety (entries 3 and 4), and the reaction involvingthe addition of 2-anilinoethanol or 2-aminoethanol was
Table 2 Oxidative Dimerization of Primary Alcohols To Give Esters and Lactonization of a Diol To Give a Lactone Catalyzed by [IrCl(coe)2]2
a
Entry Substrate Product Yield (%)
1 90
2 94
3b 51
4 56
5 80
6 76
7 61
8c 88
a The alcohol (2 mmol) was allowed to react in the presence of [IrCl(coe)2]2 (3 mol%) in open air at 95 °C for 15 h.b After 20 h.c The reaction was performed in the presence of Na2CO3 (5 mol%), Cy3P (10 mol%), and toluene (1 mL).
sluggish (entries 5 and 6, respectively). No reaction tookplace with ethane-1,2-diamine (entry 7).
Suzuki et al. reported that [Cp*Ir(OCH2CPh2NH)] is anefficient catalyst for the direct conversion of alcohols intoesters.87 The above reaction employing 2-amino-2,2-diphenylethanol, prepared independently, and potassiumcarbonate as additives was found to give a comparable re-sult to that using our system (Table 3, entry 8). This resultsuggests that a similar iridium aminoalkoxide complexwould be formed, even in our catalytic system.
In addition, we tried the reaction on a multigram scale us-ing alcohol 57 (2 g) and a lower amount of [Cp*IrCl2]2
(0.5 mol%), 2-(methylamino)ethanol (1 mol%), andCs2CO3 (3 mol%) in acetone (5.5 mL) at room tempera-ture for 48 hours; product 62 was obtained in high yield(1.53 g, 80% yield).
This method can be applied to the methyl esterification ofethanol using methanol. Under these conditions, ethanol(57) (2 mmol) was allowed to react in the presence ofmethanol (63) (10 mmol) to afford methyl acetate (64) in82% yield along with the formation of ethyl acetate (62)in 6% yield (Scheme 28).
8 N-Heterocyclization
8.1 Synthesis of Quinolines from Amino Alco-hols and Ketones
Nitrogen-containing aromatic compounds, such as quino-lines, pyrroles, and indoles, are very important substancesin synthetic organic chemistry for the production of phar-maceuticals, herbicides, and dyes.92 For the synthesis ofquinolines, the coupling of 2-aminobenzaldehyde with aketone, namely the Friedländer reaction, is one of themost widely used methods because of its high selectivityto give the desired products.93,94 On the other hand,Friedländer-type reactions using 2-aminobenzyl alcoholare far more beneficial because the substrate is cheaperand more-stable than 2-aminobenzaldehyde. Therefore,several modified Friedländer reactions using 2-aminoben-zyl alcohol have been reported.95–97
Here, we report that our iridium-catalyzed transfer hydro-genation system can be applied to quinoline synthesis bythe reaction of 2-aminobenzyl alcohol with ketones in thepresence of small amounts of [IrCl(cod)]2 and a base un-der solvent-free conditions (e.g., Scheme 29).98
Scheme 29
Thus, treatment of 2-aminobenzyl alcohol (65) (2 mmol)with acetophenone (21) (4 mmol) in the presence of cata-lytic amounts of [IrCl(cod)]2 (0.02 mmol, 1 mol%), Ph3P(0.08 mmol, 4 mol%), and KOH (0.4 mmol, 20 mol%) at100 °C for 3 hours without a solvent gave 2-phenylquino-line (66) in 90% yield. In this reaction, 1 equivalent of ke-tone 21 was found to act as a hydrogen acceptor and wasconverted into 1-phenylethanol.
The iridium-catalyzed reaction of amino alcohol 65 withvarious ketones under the solvent-free conditions afford-ed the corresponding quinoline derivatives (Table 4).
In contrast to the reaction of 2-aminobenzyl alcohol (65)with ketones, the coupling reaction of benzyl alcohol (67)with cycloheptanone (68) failed to proceed selectivelyand led to a complex mixture (Scheme 30, cf. Table 4, en-try 3). These results suggest that the quinoline may besynthesized via the formation of a ketimine, such as 69,obtained from 2-aminobenzyl alcohol and the ketone.Subsequent hydrogen transfer from the ketimine to an iri-
Table 3 Effect of Additives on the Iridium-Catalyzed Oxidative Dimerization of Ethanol (57) To Give Ethyl Acetate (62)a
Entry Additive GC Yield (%) Selectivity (%)b
1 MeNH(CH2)2OH 85 96
2 EtNH(CH2)2OH 83 97
3 n-PrNH(CH2)2OH 73 88
4 n-BuNH(CH2)2OH 48 87
5 PhNHC2H4OH 6 24
6 H2NC2H4OH 16 50
7 H2NC2H4NH2 no reaction –
8c H2NCPh2CH2OH 82 93
a Ethanol (2 mmol) was stirred in the presence of [Cp*IrCl2]2 (2 mol%), the additive (6 mol%), and Cs2CO3 (10 mol%) in acetone (1 mL) at r.t. for 24 h.b Selectivity based on [(yield of 62)/(conversion of 57)] × 100.c Using K2CO3 as the base.
Scheme 28
OH
57OMe
O
+
6482% (GC yield) 6% (GC yield)
+ MeOH
2 mmol 10 mmol62
acetone (1 mL)r.t., 24 h, under Ar
[Cp*IrCl2]2/MeNH(CH2)2OH (cat.)Cs2CO3
63O
O
N Ph
OH
NH2Ph
O
100 °C, 3 hKOH
+[IrCl(cod)]2/Ph3P (cat.)
65 21 6690% (GC yield)
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 43
dium complex gives the corresponding aldehyde and irid-ium hydride, and the aldehyde is subjected tointramolecular aldol condensation to afford the quinolinederivative, e.g. 71 (Scheme 31).
This catalytic system could be applied to pyrrole synthe-sis. Thus, the reaction of 2-(methylamino)ethanol (72) (2mmol) with propiophenone (73) (4 mmol) in the presenceof [IrCl(cod)]2 (0.03 mmol), 1,1¢-bis(diphenylphosphi-no)ferrocene (dppf) (0.06 mmol), and KOH (0.8 mmol) at120 °C for 3 hours under solvent-free conditions afforded1,3-dimethyl-2-phenyl-1H-pyrrole (74) in 70% yield(Scheme 32).
8.2 Synthesis of Benzo[h]quinolines and Benzo-indole Derivatives from Naphthylamines and Diols
As an alternative methodology for the synthesis of quino-line and indole derivatives, the ruthenium-catalyzed N-cyclization of anilines with diols has been reported.99,100
We found that benzo[h]quinolines and benzoindole deriv-atives could be synthesized in high yields by the iridium-catalyzed direct cyclization of naphthylamines with 1,3-and 1,2-diols, respectively.101
As an example, using the preparation of benzo[h]quino-line, a mixture of 1-naphthylamine (75) (5 mmol) and pro-pane-1,3-diol (76) (2 mmol) was allowed to react in thepresence of iridium(III) chloride trihydrate (IrCl3·3H2O)(0.10 mmol), BINAP [BINAP = rac-2,2¢-bis(diphe-nylphosphino)-1,1¢-biphenyl] (0.15 mmol), and Na2CO3
(0.16 mmol) under air at refluxing temperature (169 °C)in mesitylene for 15 hours, giving benzo[h]quinoline (77)in 96% isolated yield (Scheme 33). The reaction proceed-ed more rapidly under an oxygen atmosphere (1 atm)compared with that under air, whereas the reaction undernitrogen decreased the yield to 55%. These results indi-cate that the oxidation step is an important one in the re-action.
On the basis of these results, the reaction of 1-naphthyl-amine derivatives with propane-1,3-diol (76) was exam-ined under the standard conditions (Table 5).
Benzoindole derivatives were also synthesized from 1-naphthylamines with various 1,2-diols using the samestrategy (Table 6). It is noteworthy that the reactions withnaphthalene-1,5-diamine gave the monocyclization prod-ucts, i.e. benzo[h]quinolin-7-amine (Table 5, entry 4) and2,3-dimethyl-1H-benzo[g]indol-6-amine (Table 6, entry8) were formed exclusively; the double cyclization prod-ucts were not obtained.
The iridium-catalyzed quinoline synthesis from ketonesand 2-aminobenzyl alcohols (Section 8.1)98 has beenshown to proceed via the formation of imines, followed byhydrogenation by an iridium hydride generated during thereaction course to give cyclic amines (see Scheme 31).For the ruthenium(III) chloride catalyzed quinoline syn-thesis from aniline and a 1,3-diol reported by Tsuji etal.,100 the reaction path is proposed to proceed via the for-mation of 3-anilinopropan-1-ol (78) (Figure 13). No in-tramolecular cyclization of amino alcohol 78 takes placedirectly, but N,N¢-diphenylpropane-1,3-diamine (79) maybe formed from 78 and aniline. Then, the ruthenium-
Table 4 Iridium-Catalyzed Coupling Reaction of Amino Alcohol 65 with Several Ketones under Solvent-Free Conditions
catalyzed intermolecular cyclization of 79 with anilineforms quinoline.
To obtain more information regarding the reaction mech-anism of our iridium-catalyzed benzo[h]quinoline synthe-sis, we prepared 3-(1-naphthylamino)propan-1-ol (80),and both the cyclization of 80 and the reaction of 80 with1-naphthylamine (75) were performed under the influenceof IrCl3·3H2O and BINAP (Schemes 34 and 35). In con-
trast to the above-mentioned ruthenium-catalyzed reac-tion of amino alcohol 78 in which the intramolecularcyclization of 78 to give quinoline does not occur, the iri-dium-catalyzed reaction of 80 produced benzo[h]quino-line (77) in 43% yield (Scheme 34). In addition, thereaction of amino alcohol 80 with 1-naphthylamine (75)led to product 77 in 72% yield, and substrate 75 was re-covered in 19% (Scheme 35).
The product distribution of benzo[h]quinoline (77) de-rived from the intramolecular cyclization of amino alco-hol 80 was found to be different from that derived fromthe reaction of 80 and 1-naphthylamine (75). These resultssuggest that the reaction mechanism for the formation ofbenzo[h]quinoline (77) in Scheme 34 is different fromthat of the reaction shown in Scheme 35.
Indeed, when the reaction of substrate 80 with amine 75was performed under a lower reaction temperature (150°C), the yield of 77 was only 3%, but N,N¢-di-1-naphthyl-propane-1,3-diamine (81) was obtained in 58% yield.This result suggests that diamine 81 is also a probable in-termediate in our iridium-catalyzed reaction. Thus, di-amine 81 was independently prepared and the reaction ofsubstrate 81 was carried out under the same conditions toproduce an approximately equimolar amount of product77 in 76% yield and amine 75 in 83% yield (Scheme 36).
Scheme 36
In addition, to investigate the reactivity and selectivity ofthe diamine intermediates, N-1-naphthyl-N¢-phenylpro-pane-1,3-diamine (82) was prepared and allowed to reactunder the above iridium-catalyzed conditions(Scheme 37). As a result, selective N-heterocyclizationtook place to produce benzo[h]quinoline (77) in 69%yield together with aniline (83) in 65% yield; the yield ofquinoline (84) was only 7%.
Based on these results, two reaction pathways are pro-posed for the formation of benzo[h]quinoline (77) in theabove iridium-catalyzed reaction (Schemes 38 and 39). Inboth mechanisms, the reaction is initiated by the iridium-
Table 5 Synthesis of Benzo[h]quinolines by the Reaction of Vari-ous Naphthylamines with Propane-1,3-diol (76) Using an IrCl3/BINAP Catalyst
catalyzed dehydrogenation of diol 76 to give an aldehyde,which readily reacts with 1-naphthylamine (75) to give animine intermediate followed by hydrogenation by the in-situ-generated iridium hydride leading to amino alcohol80. Then, one possible reaction pathway is that the result-ing compound 80 reacts further with 75, after the forma-tion of aldehyde 85, to give imine 86 (Scheme 38). Theformation of the desired product 77 from intermediate 86may be explained by a similar reaction pathway to thatproposed by Tsuji et al.100 However, another possible re-action pathway involves the direct intramolecular cycliza-
tion of 80 to give product 77 via the iridium-catalyzedreaction of aldehyde 85 (Scheme 39).
9 Coupling Reaction of Alcohols with Alkynes
9.1 Synthesis of Homoallylic Alcohols
The transition-metal-catalyzed addition of alcohols toalkynes, which is generally referred to as a hydroalkoxyl-ation reaction, is an important methodology that leads to awide variety of oxygen-containing compounds.102 We
Table 6 Synthesis of Benzoindole Derivatives by the Reaction of Various Naphthylamines with 1,2-Diols Using an IrCl3/BINAP Catalysta
a Using a procedure similar to that described for the synthesis of benzo[h]quinoline (see above).b 75 (10 mmol) was used.c Triphenylphosphine (0.2 mmol, 10 mol%) was used instead of BINAP.
previously reported the addition of water and alcohols toterminal alkynes using an iridium complex combined witha phosphite and a Lewis acid to give ketones and ketals.103
However, Krische and co-workers reported ruthenium-and iridium-catalyzed transformations of alcohols with
dienes, allyl acetates, allenes, and alkynes to afford ho-moallylic alcohols.15,104–109
Here, we show our findings on the iridium-catalyzed cou-pling of alkynes, such as 1-arylprop-1-ynes, with primaryalcohols leading to secondary homoallylic alcohols asproducts through the formation of a p-allyl(hydrido)iridi-um complex as a possible key intermediate.110
For instance, butan-1-ol (19) (1 mmol) was allowed toreact with 1-phenylprop-1-yne (87) (2 mmol) in the pres-ence of [Ir(OH)(cod)]2 (0.05 mmol, 5 mol%) combinedwith tri-n-octylphosphine (0.30 mmol) in toluene (1 mL)at 100 °C for 15 hours, giving 3-phenylhept-1-en-4-ol(88) in 95% isolated yield (Scheme 40). The reaction ishighly stereoselective and afforded the anti-isomer exclu-sively.
Under optimized conditions, the reactions of various pri-mary alcohols with 1-arylprop-1-ynes were carried out toafford the corresponding homoallylic alcohols in good toexcellent yields (Figure 14). The methyl substituent onthe alkyne plays a crucial role in achieving this reaction.
Scheme 37
82
NH
NH
IrCl3⋅3H2O/BINAP (cat.)
Na2CO3mesitylenereflux, 15 hunder O2
N
7769% (GC yield)
+
NH2
8365% (GC yield)
+
NH2
N
+
847% (GC yield)
7513% (GC yield)
Scheme 38
HN N
80
HN OH
LnIr
HN H
O
75 H2O
75 + 76LnIr
LnIr
NH
HN IrLn
+
HN
75LnIr
aromatization
cyclization
77
85
86
N
Scheme 39
LnIr
HN H
O
HN
OH
HN
– H2O
77
– LnIr
aromatization
– H2
80 LnIr HN H
OLnIr
85
N
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 47
Thus, no coupling product was obtained from the reactionof oct-4-yne or 1-phenylbut-1-yne.
To obtain further information on the reaction pathway, wecarried out the reaction of secondary alcohols, such asbutan-2-ol (89), with 87 under these reaction conditions.As a result, allylbenzene (90) was formed in quantitativeyield with concomitant formation of butan-2-one (91)(Scheme 41). This result indicates that the alcohol be-haves as a hydrogen source, and transfer hydrogenation tothe iridium complex occurs to form an iridium hydridespecies, followed by the formation of a p-allyliridiumcomplex as a key intermediate.
Scheme 41
For further evidence of the formation of a p-allyliridiumspecies in this reaction, Esteruelas et al. reported the stoi-chiometric transformation of alk-2-ynes (including aprop-1-yne derivative) using an osmium dihydride com-plex, leading to p-allyl(hydrido)osmium complexes.111
Thus, our above study demonstrates the catalytic transfor-mation of 1-arylprop-1-ynes with primary alcoholsthrough the possible formation of a p-allyl(hydrido)iridi-um complex as an intermediate.
On the basis of these experimental results, the reactionmechanism of Scheme 42 can be postulated. The iridiumcatalyst initially serves as the hydrogen acceptor from al-
cohol 92 giving aldehyde 93 and an iridium hydride.Then, alkyne 94 inserts into the iridium–hydrogen bond ofthe iridium hydride to form alkenyl(hydrido)iridium spe-cies 95. Intermediate 95 is subjected to hydrogenation bythe iridium hydride, followed by abstraction of hydrogenfrom the methyl group of the alkyne, resulting in the for-mation of p-allyl(hydrido)iridium species 96.111 Finally,complex 96 reacts with aldehyde 93 to give homoallylicalcohol 98 as the product via the formation of six-mem-bered transition state 97.
Scheme 42
9.2 Synthesis of a,b-Unsaturated Ketones (Hydroacylation)
The above-mentioned iridium-catalyzed coupling reac-tion of 1-arylprop-1-ynes with primary alcohols is also ap-plicable to the synthesis of a,b-unsaturated ketones,realizing overall the intermolecular hydroacylation ofalkynes with aldehydes.112 To date, the hydroacylation ofalkynes has been mainly achieved with intramolecularreactions,113–118 and intermolecular alkyne hydroacylationhas been mostly limited to reactions with substrates bear-ing directing groups.119–124 The intermolecular hydroacyl-ation of alkynes with simple aldehydes without directinggroups has been far less explored.125 Ruthenium-cata-lyzed hydroacylation reactions of alkynes with alcohols oraldehydes were more-recently reported by Krische andco-workers.126
Using our method, the reaction of benzyl alcohol (99) (0.5mmol) and but-2-yne (100) (1 mmol) in the presence of[IrCl(cod)]2 (0.025 mmol, 5 mol%) combined with tri-n-octylphosphine (0.1 mmol, 20 mol%) in toluene (1 mL) at120 °C for 15 hours gave 2-methyl-1-phenylbut-2-en-1-one (101) in 92% isolated yield (Scheme 43).
Under the optimized conditions, the reaction of variousprimary alcohols with alk-2-ynes was examined and af-forded the corresponding hydroacylation products in goodto excellent yields (Figure 15).
Figure 15
In this reaction, the iridium catalyst serves as a hydrogenacceptor from alcohols to give aldehydes, which are pro-posed to react with the alk-2-yne to afford a,b-unsaturatedketones. Therefore, the reaction of aromatic aldehydeswith alk-2-ynes under these reaction conditions was ex-amined. For instance, the reaction of benzaldehyde (0.5mmol) (102) with but-2-yne (2 mmol) (100) in the pres-ence of [IrCl(cod)]2 (0.025 mmol, 5 mol%) and tri-n-octylphosphine (0.1 mmol, 20 mol%) in toluene (1 mL) at120 °C for 15 hours gave a,b-unsaturated ketone 101 in84% isolated yield (Scheme 44).
Scheme 44
Similarly, various aromatic aldehydes reacted with but-2-yne to produce the corresponding a,b-unsaturated ketonesin good yields (Figure 16).
To elucidate the reaction pathway for the formation of thehydroacylation product as an a,b-unsaturated ketone, wemonitored the reaction time-course of alcohol 99 with alk-2-yne 100, and homoallylic alcohol 103 and b,g-unsatur-ated ketone 104 were observed during the early stages ofthe reaction. Ketone 104 was then converted into a,b-unsaturated ketone 101 as the final product (Scheme 45).
Scheme 45
To obtain further experimental evidence regarding thereaction mechanism, the reaction of homoallylic alcohol103 with alk-2-yne 100 was carried out and gave 2-methyl-1-phenylbut-2-en-1-one (101) in 74% yield(Scheme 46).
Scheme 46
Furthermore, when 4-methylpent-2-yne (105) bearing abulky isopropyl group was used, the desired hydroacyl-ation product 106 was not formed at all and b,g-unsaturat-ed ketone 107 was obtained exclusively in 81% yield(Scheme 47).
The above-mentioned results strongly suggest that the re-sulting hydroacylation products are obtained through theformation of homoallylic alcohols and b,g-unsaturated ke-tones as intermediates. Thus, we suppose that this reactionproceeds by dehydrogenation of homoallylic alcohol 108,derived from the reaction involving the p-allyl(hydri-
76% (54:46) 85% (60:40)
63% (79:21) 71% (81:19) 77% (78:22)
O
O O
O O
O S
ClMeO
89% (57:43)
O
(E:Z) ratio
(isolated yield)
[IrCl(cod)]2 (cat.)P(n-Oct)3
toluene 120 °C, 15 h
Me Me+ Ph
O
84%E/Z = 9:91
102 100 101
HPh
O
[IrCl(cod)]2P(n-Oct)3
toluene120 °C
Ph
O
Ph
OH
Ph
O
103 104 101
99
100
+
Figure 16
80%, E/Z = 57:43 86%, E/Z = 14:86
72%, E/Z = 84:16
70%, E/Z = 70:30 86%, E/Z = 55:45
O
O O
O O
O S
ClMeO
81%, E/Z = 43:57
O
(isolated yield)
Ph
O
Ph
OH
103 101
+
74%E/Z = 60:40
100
[IrCl(cod)]2 (cat.)P(n-oct)3
toluene 120 °C, 15 h
Me Me
Scheme 47
Mei-Pr+
[IrCl(cod)]2 (cat.) P(n-Oct)3
toluene 120 °C, 15 h
Ph
O
i-Pr
Ph
O
i-Pr
+
Ph OH99 105
106 1070% 81%
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 49
do)iridium intermediate mentioned in Section 9.1, leadingto b,g-unsaturated ketone 109, which undergoes isomer-ization to produce 110 as the final product (Scheme 48).
Scheme 48
10 Concluding Remarks
In this account, we have reported various organic transfor-mations using alcohols and diols which include hydroge-nation, alkylation, N-heterocyclization, and couplingreactions with alkynes. In the presented reactions, the for-mation of an iridium hydride generated through transferhydrogenation from alcohols is a key intermediate. Thesereactions provide efficient and greener processes com-pared with the conventional methods. Therefore, we be-lieve that the further application and broadening of thescope of this protocol will contribute to catalytic organicchemistry and industrial process chemistry.
Acknowledgment
We acknowledge that these studies were carried out in collaborationwith the co-workers at Kansai University listed in the references.These studies were supported in part by a Grant-in-Aid for Scienti-fic Research from MEXT (Ministry of Education, Culture, Sports,Science and Technology), the Research for the Future Program ofthe Japan Society for the Promotion of Science, and the High-TechResearch Center Project for Private Universities and the StrategicProject to Support the Formation of Research Bases at Private Uni-versities (matching fund subsidy from MEXT).
References
(1) Crabtree, R. H. Acc. Chem. Res. 1979, 12, 331.(2) Oro, L. A.; Cabeza, J. A.; Cativiela, C.; Díaz de Villegas,
M. D.; Meléndez, E. J. Chem. Soc., Chem. Commun. 1983, 1383.
(3) Cabeza, J. A.; Cativiela, C.; Díaz de Villegas, M. D.; Oro, L. A. J. Chem. Soc., Perkin Trans. 1 1988, 1.
(5) Roseblade, S. J.; Pfaltz, A. C. R. Chim. 2007, 10, 178.(6) Stoke, G.; Kahne, D. E. J. Am. Chem. Soc. 1983, 105, 1072.(7) Kainz, S.; Brinkmann, A.; Leitner, W.; Pfaltz, A. J. Am.
Chem. Soc. 1999, 121, 6421.(8) Vázquez-Serreno, L. D.; Owens, B. T.; Buriak, J. M. Chem.
Commun. (Cambridge) 2002, 2518.(9) Ishii, Y.; Obora, Y.; Sakaguchi, S. In Iridium Complexes in
Organic Synthesis; Oro, L. A.; Claver, C., Eds.; Wiley: New York, 2009, 251–275.
(10) Dobereiner, G. E.; Crabtree, R. H. Chem. Rev. 2010, 110, 681.
(11) Guillena, G.; Ramón, D. J.; Yus, M. Chem. Rev. 2010, 110, 1611.
(12) Guillena, G.; Ramón, D. J.; Yus, M. Angew. Chem. Int. Ed. 2007, 46, 2358.
(13) Hamid, M. H. S. A.; Slatford, P. A.; Williams, J. M. J. Adv. Synth. Catal. 2007, 349, 1555.
(14) Nixon, T. D.; Whittlesey, M. K.; Williams, J. M. J. Dalton Trans. 2009, 753.
(15) Bower, J. F.; Kim, I. S.; Patman, R. L.; Krische, M. J. Angew. Chem. Int. Ed. 2009, 48, 34.
(16) Ishii, Y.; Sakaguchi, S. Bull. Chem. Soc. Jpn. 2004, 77, 909.(17) Takeuchi, R.; Kezuka, S. Synthesis 2006, 3349.(18) Fujita, K.; Yamaguchi, R. Synlett 2005, 560.(19) Bianchini, C.; Gonsalvi, L.; Peruzzini, M. In Iridium
Complexes in Organic Synthesis; Oro, L. A.; Claver, C., Eds.; Wiley: New York, 2009, 55–106.
(20) Mizugaki, T.; Kanayama, Y.; Ebitani, K.; Kaneda, K. J. Org. Chem. 1988, 63, 2378.
(21) Bianchini, C.; Peruzzini, M.; Farnetti, E.; Kašper, J.; Graziani, M. J. Organomet. Chem. 1995, 488, 91.
(22) Bhaduri, S.; Sharma, K. J. Chem. Soc., Chem. Commun. 1988, 173.
J. Org. Chem. 1991, 56, 261; and references cited therein.(62) Reetz, M. T. Angew. Chem. Int. Ed. Engl. 1982, 21, 96.(63) Nishimoto, Y.; Yasuda, M.; Baba, A. Org. Lett. 2007, 9,
Arpe, H.-J., Eds.; Wiley-VCH: Weinheim, 2003.(75) Frohning, C. D.; Kohlpaintner, C. W.; Bohnen, H.-W. In
Applied Homogeneous Catalysis with Organometallic Compounds, Vol. 1; Cornils, B.; Herrman, W. A., Eds.; Wiley-VCH: Weinheim, 2002, 31.
(76) New Synthesis with Carbon Monoxide; Bahrmann, H.; Cornils, B.; Frohning, C. D.; Mullen, A.; Falbe, J., Eds.; Springer: Berlin, 1980.
(77) Maitlis, P. M.; Haynes, A. In Metal-Catalysis in Industrial Organic Processes; Chiusoli, G. P.; Maitlis, P. M., Eds.; Royal Society of Chemistry: Cambridge, 2006, 114.
(78) For example, see: Haslam, E. Tetrahedron 1980, 36, 2409.(79) Seki, T.; Nakajo, T.; Onaka, M. Chem. Lett. 2006, 35, 824.(80) Ooi, T.; Ohmatsu, K.; Sasaki, K.; Miura, T.; Maruoka, K.
Tetrahedron Lett. 2003, 44, 3191.
(81) Gnanadesikan, V.; Horiuchi, Y.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2004, 126, 7782.
(82) Tamaru, Y.; Yamada, Y.; Inoue, K.; Yamamoto, Y.; Yoshida, Z. J. Org. Chem. 1983, 48, 1286.
(83) Blum, Y.; Reshef, D.; Shvo, Y. Tetrahedron Lett. 1981, 22, 1541.
(84) Murahashi, S.-I.; Naota, T.; Ito, K.; Maeda, Y.; Taki, H. J. Org. Chem. 1987, 52, 4319.
(85) Murahashi, S.-I.; Ito, K.; Naota, T.; Maeda, Y. Tetrahedron Lett. 1981, 22, 5327.
(86) Cho, C. S.; Kim, B. T.; Kim, T.-J.; Shim, S. C. Tetrahedron Lett. 2002, 43, 7987.
(91) Yamamoto, N.; Obora, Y.; Ishii, Y. Chem. Lett. 2009, 38, 1106.
(92) Sundberg, R. J. In Kirk-Othmer Encyclopedia of Chemical Technology, Vol. 14; Kroschwitz, J. I.; Howe-Grand, M., Eds.; Wiley: New York, 1995.
(93) Hsiao, Y.; Rivera, N. R.; Yasuda, N.; Hughes, D. L.; Reider, P. J. Org. Lett. 2001, 3, 1101.
(94) Yadav, J. S.; Reddy, B. V. S.; Premalatha, K. Synlett 2004, 963.
(95) Cho, C. S.; Kim, B. T.; Kim, T.-J.; Shim, S. C. Chem. Commun. (Cambridge) 2001, 2576.
(96) Cho, C. S.; Kim, B. T.; Choi, H.-J.; Kim, T.-J.; Shim, S. C. Tetrahedron 2003, 59, 7997.
(97) Motokura, K.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Tetrahedron Lett. 2004, 45, 6029.
(98) Taguchi, K.; Sakaguchi, S.; Ishii, Y. Tetrahedron Lett. 2005, 46, 4539.
(99) Tsuji, Y.; Huh, K.-T.; Watanabe, Y. Tetrahedron Lett. 1986, 27, 377.
(100) Tsuji, Y.; Huh, K.-T.; Watanabe, Y. J. Org. Chem. 1987, 52, 1673.
(101) Aramoto, H.; Obora, Y.; Ishii, Y. J. Org. Chem. 2009, 74, 628.
(102) For a review, see: Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079.
(103) Hirabayashi, T.; Okimoto, Y.; Saito, A.; Morita, M.; Sakaguchi, S.; Ishii, Y. Tetrahedron 2006, 62, 2231.
(104) Patman, R. L.; Williams, V. M.; Bower, J. F.; Krische, M. J. Angew. Chem. Int. Ed. 2008, 47, 5220.
(105) Kim, I. S.; Ngai, M.-Y.; Krische, M. J. J. Am. Chem. Soc. 2008, 130, 6340.
(106) Shibahara, F.; Bower, J. F.; Krische, M. J. J. Am. Chem. Soc. 2008, 130, 14120.
(107) Kim, I. S.; Ngai, M.-Y.; Krische, M. J. J. Am. Chem. Soc. 2008, 131, 2514.
(108) Kim, I. S.; Ngai, M.-Y.; Krische, M. J. J. Am. Chem. Soc. 2008, 130, 14891.
(109) Patman, R. L.; Chaulagain, M. R.; Williams, V. M.; Krische, M. J. J. Am. Chem. Soc. 2009, 131, 2066.
(110) Obora, Y.; Hatanaka, S.; Ishii, Y. Org. Lett. 2009, 11, 3510.(111) Esteruelas, M. A.; Hernandez, Y. A.; López, A. M.; Oliván,
M.; Oñate, E. Organometallics 2007, 26, 2193.(112) Hatanaka, S.; Obora, Y.; Ishii, Y. Chem. Eur. J. 2010, 16,
1883.(113) Fu, G. C.; Tanaka, K. J. Am. Chem. Soc. 2001, 123, 11492.(114) Fu, G. C.; Tanaka, K. Angew. Chem. Int. Ed. 2002, 41, 1607.(115) Tanaka, K.; Fu, G. C. J. Am. Chem. Soc. 2002, 124, 10296.
Dow
nloa
ded
by: A
.E.F
avor
sky
Irku
tsk
Inst
itute
of C
hem
istr
y S
B R
AS
. Cop
yrig
hted
mat
eria
l.
ACCOUNT Iridium-Catalyzed Reactions Using Alcohols and Diols as Key Substrates 51