Journal of Taibah University for Science 10 (2016) 685–699
Available online at www.sciencedirect.com
ScienceDirect
Kinetics and deactivation of a dual-site heterogeneous oxide catalystduring the transesterification of crude jatropha oil with methanol
M.A. Olutoye a,∗, B.H. Hameed b
a Department of Chemical Engineering, Federal University of Technology, P.M.B. 65, Minna, Nigeriab School of Chemical Engineering, Engineering Campus, Universiti Sains Malaysia, 14300 Nibong Tebal, Penang, Malaysia
Received 13 July 2015; received in revised form 14 October 2015; accepted 28 October 2015Available online 27 November 2015
bstract
In this work, a dual-site heterogeneous catalyst was used to study the kinetics of the transesterification of crude jatropha oil withethanol in batch experiments. Experimental data were obtained between 150 and 182 ◦C at a constant molar ratio of alcohol to
il (11:1) and at a set catalyst concentration (3.32 wt% based on weight of oil). Kinetic modelling was performed with an assumedseudo-first order to describe the rate and catalyst deactivation, which suitably fit the experimental data. The model parametersere determined for both the mass transfer controlled and reaction regimes. The triglyceride (TG) conversion, methyl ester (ME)
ormation, and system thermodynamics were also evaluated. Based on the calculated values, within an acceptable range, thequilibrium constant, Ke = 1.42; activation energy, Ea = 161 kJ/mol; and free energy, �G = −1286 J/mol indicate that the developedodel adequately described the methanolysis of the oil, which may be useful in reactor design and process simulation. The values
hat were calculated from the kinetic equations agree well with the experimental values, and the results help to understand andredict the behaviour of dual-site catalysts in practical applications for the production of biodiesel.
2015 The Authors. Production and hosting by Elsevier B.V. on behalf of Taibah University. This is an open access article underhe CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
In recent years, the synthesis of fatty acid methylsters (FAME) has become important, as research hasntensified on the utilization of vegetable oils and ani-
al fat derivatives for liquid fuel (biodiesel) production
C BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
eactivation
[1–5]. FAME can be obtained from renewable feedstock,such as vegetable oils or animal fats, by the process oftransesterification with alcohols in the presence of anacid or an alkaline catalyst. Biodiesel is a clean-burningfuel that is nontoxic and biodegradable and is consid-ered the fuel of the future [6]. Biodiesel is referred toas neat fuel (B100) in its pure form or as the blendedform (B20 or B80) when mixed with petroleum diesel.Biodiesel can be utilized in compression ignition enginesunder a variety of operating conditions. B100 contains
behalf of Taibah University. This is an open access article under the
no petroleum fuels and emits virtually no sulphur, aro-matics, particulates, or carcinogenic compounds; thusit is a safer alternative to petroleum diesel. Biodieselcan be used in all conventional diesel engines, and its
686 M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699
nd R3 a
Scheme 1. 3-Step reversible reactions of triglyceride, where R1, R2, a
performance and engine durability closely match thoseof petroleum diesel, requiring no modifications in fuelhandling and delivery systems [7,8]. Three consecutiveand reversible reactions are believed to occur during thetransesterification of vegetable oils and fats, convertingto intermediate products that are formed during the reac-tion: diglycerides (DGs), monoglycerides (MGs), andthe final methyl ester product. The reaction scheme isshown in Scheme 1.
The various parameters that influence the synthesisof FAME have been investigated by many authors dueto the increasing industrial application of methyl esters[7]. These parameters include the molar ratio of alcoholto vegetable oil, catalyst type and loading (wt% of oil),reaction temperature, agitation intensity and presence ofimpurities, such as free fatty acids and moisture content.There are a number of kinetic studies on the transesterifi-cation of esters with alcohol catalyzed by homogeneousbase catalysts, but very little information is available onthe kinetics of transesterification reactions by solid-basecatalysts [9]. In particular, few studies have investigatedthe transesterification reactions of vegetable oils andFFAs with the application of a dual-site composite
catalyst. Hattori et al. [10] proposed a mechanism for thetransesterification of ethyl acetate with different alco-hols using a variety of solid-base catalysts, particularlyalkaline-earth-metal oxides. However, this mechanistic
re long chains of carbon and hydrogen atoms (fatty acid chains).
study provided neither values of activation energies norrate constants. Recently, a kinetic model was proposedto describe the transesterification of ethyl acetate withmethanol catalyzed by a heterogeneous magnesiumoxide catalyst [11,12]. This model was based on a three-step ‘Eley–Rideal’ type of mechanism applied in liquidphase, where methanol adsorption is assumed to berate determining. The activation energy was 20 kJ/mol,with a methanol adsorption equilibrium coefficient of3.13 × 10−3 m3/mol. The objective of this study was toevaluate the use of a dual-site heterogeneously catalyzedtransesterification reaction in batch reactors under pre-determined conditions using the kinetic model based onthe three-step ‘Eley–Rideal’ type mechanism assumingmethanol adsorption as a rate-determining step. Thekinetics and deactivation studies of a dual-site catalystin the transesterification of CJO (non-edible stock) areimportant for determining reactor design parameters andadvancing the stage of efficient biodiesel production.
In addition, a kinetic model for the reaction oftriglyceride over solid catalyst was developed, and thedeactivation characteristic during the transesterificationand esterification reactions with methanol was aimed to
be observed. The influence of various reaction param-eters on the catalyst activity and kinetic rate wereevaluated. Because the kinetics of transesterification andesterification reactions are increased using basic and
aibah U
attct
2
2
pMmoogAZ(wTstyrat
2
cttcTmmt2guTt
2
dMTE
M.A. Olutoye, B.H. Hameed / Journal of T
cidic catalysts, respectively, it is interesting to inves-igate the use of a dual-site catalyst for reactions of bothhe transesterification of vegetable oils and the esterifi-ation of FFAs because of the wide industrial use of thisype of catalyst and its significance for reactor design.
. Experimental
.1. Materials
Crude Jatropha (Jatropha curcas Linnaeus) oil sup-lied by Telegamadu Sdn. Bdh., Butterworth, Penang,alaysia, was used for this study. The acid value,oisture content and saponification values of the
il were 14.47%, 3.28% and 193 mg of KOH/g ofil, respectively. Methanol (HPLC, analytical reagentrade 99.9%) from Merck (Malaysia) was used.l(NO3)3·9H2O (99%), Mg(NO3)2·6H2O (≥99%),n(NO3)2·6H2O (≥98%) and analytical-grade NH4OH≥85%) that were used to synthesize the catalystsere purchased from Sigma–Aldrich Pty Ltd., Malaysia.he other chemicals that were used in the experiment,uch as acetonitrile (≥99%), and acetone (≥99.9%) forhe Ultra-Fast Liquid Chromatography (UFLC) anal-sis, were purchased from Merck (Malaysia). Theseeagents were used without further purification for cat-lyst synthesis and to measure the kinetics of therans-esterification of crude jatropha oil.
.2. Catalyst preparation
The dual-site catalyst (MgZnAlO) was prepared byo-precipitation using a 25% ammonia water solution ashe precipitating medium. The mixture was stirred con-inuously for 6 h under constant heat at 80 ◦C until aompletely homogenized milky solution was obtained.he basic strength was determined by the indicatorethod, which gives a pH value range of 8–9 and wasaintained throughout the catalyst preparation. The mix-
ure was filtered, dried, and calcined at 461 ◦C for 4 h5 min. The catalyst powder was stored in a well-closedlass desiccator to keep it moisture-free before it wassed for the kinetic and trans-esterification experiments.he detailed procedure for the preparation and charac-
erization has been previously reported [13].
.2.1. CharacterizationThe characterization of the catalyst by N2 adsorption-
esorption isotherm at 77 K was performed using aicromeritics ASAP 2020 gas adsorption analyser.
he surface area was calculated using the Brunauer–mmett–Teller (BET) equation, and the mean pore
niversity for Science 10 (2016) 685–699 687
diameter was obtained by applying the Barret–Joyner–Halenda (BJH) method to the desorption branch. Ther-mogravimetric measurement (TG/DTG) was carried outwith a thermo balance SETARAM (accuracy ± 0.04 �g)thermal analyser from 298 K to 1073 K at a heating rateof 293 K min−1 under an N2 atmosphere. The structureof the catalyst was checked by an X-ray diffractometer(Philips PW 1710) with Cu K� (Ni-filtered) radia-tion. The particle microstructures were studied on aPhilips XL30S model Scanning Electron Microscopy(SEM). The element composition was analyzed usingan energy-dispersive X-ray detector (EDX) that wasmounted on the microscope. Fourier transform infrared(FT-IR) spectra were recorded for the active surfacefunctional groups. The spectra were recorded in therange of 4000–400 cm−1.
2.3. Determination of the surface acidity andbasicity of the catalysts
The surface basicity and acidity are important solidcatalyst properties that influence its activity. The surfaceproperties were evaluated by titration after a displace-ment reaction, in which the adhered free OH− andH+ on the solid catalyst were treated with acidic andbasic species to protonate and displace from the sur-face through neutralization. Briefly, to a catalyst sample(1 g) was applied 25 mL of 0.1 M NaOH and HCl intwo separate batch experiments. The mixture in 100-mL glass beakers was mixed thoroughly for 24 h. Thecontents at the end of reaction were allowed to settle andwere filtered, after which the filtrate was titrated with a0.1 M solution of NaOH and HCl using a phenolthaleinindicator. The measurements and the evaluation of theresults were determined from the acidic or basic indica-tor molecules on the solid catalyst surface to count acidor base sites, establish the dual-site component of eitherLewis or Brönsted characteristics, and determine the sitestrength in relation to known properties of the substance.The surface acidity was determined from the expressionin Eqs. (1) and (2)
Acid concentration, CA
(�mole
g
)
= nNaOH(initial) − nNaOH(final)
mass of catalyst(1)
and (�mole
)
Base concentration, CB
g
= nHCl(initial) − nHCl(final)
mass of catalyst(2)
aibah U
688 M.A. Olutoye, B.H. Hameed / Journal of T
2.4. Equipment and reaction conditions
The transesterification reaction and kinetic experi-ments were carried out in an automated high-pressure,high-temperature batch reactor (PARR Instruments,4843) with a maximum capacity of 300 mL, PID, athermocouple that regulates the temperature insidethe reactor, a heating chamber, and water cooling andpressure devices. A long shaft rod with an impellerblade mounted at the tip provided efficient stirring of themixture.
The molar ratio of methanol to crude jatropha oilthat was used in all of the kinetic experiments was11:1, which represents the optimum conditions [13].Other reaction conditions included a catalyst loading of3.32 wt% oil. The temperature and time of reaction werevaried, at 150, 160, 170, and 182 ◦C over 0–360 min,respectively. All of the experiments were run at themethanol vapour pressure that built up in the autoclavereactor (no external pressure was supplied). An impellerspeed of 300 rpm (max) was applied throughout the reac-tion to produce uniform dispersion and to avoid masstransfer limitations.
2.5. Experimental procedure for the kinetic study
The reactor was charged with a mixture of 38 mLof methanol, 82 mL of CJO and 2.424 g of solid cata-lyst for each experimental batch. The experiment wastimed immediately the stirrer was turned on, and it con-tinued throughout the batch process over different timeintervals of 0, 5, 10, 15, 30, 60, 180, and 360 min. Atthe completion of each batch process, the samples wereimmediately quenched in a cold water bath to further stopthe reaction between participating species in the reac-tor and thereafter discharged. The product (a mixture offatty acid methyl esters and glycerol) was centrifugedand then separated before being sent for HPLC analysis.In all of the kinetic experiments, 1 h was used for theequipment to reach the target set temperature. Thus, theexperiments were initialized for all of the batch runs withTG, methanol and the catalyst charged into the reactorand run for 1 h until the target temperature was reached.At this point, the reaction was stopped and quenched withcooling water and the content analyzed for FAME. TheTG conversions that were obtained at other experimentalpoints were determined based on the initialization, and
the reaction was allowed to run at the target temperatureover a specified period. The experiments were plannedto enable the determination of reaction rate constantsand to study the effect of the molar concentration
niversity for Science 10 (2016) 685–699
of alcohol to oil ratio, time and temperature onconversion.
2.6. Separation and analysis of fatty acids, FAME,MG, DG and TG in crude jatropha oil
CJO was analyzed by the high-performance Ultra-Fast Liquid Chromatograph (UFLC model LC20ACseries, Schimadzu, Japan). The same procedures wereused to analyze all of the product samples. The LC sys-tem was equipped with a split/splitless injection systemwith a column dimension 250 mm × 4.6 mm, HypersilGold. The method that was used for the quantitationof products was based on the isocratic (fixed gradient)pump mode with mobile phases A (Acetone) and B (Ace-tonitrile) at 36.5% and 63.5%, respectively. The pumpwas set to a flow rate of 1 mL/min, and the injectorvolume was 60 �L. The total analysis time was 60 minwith column C18 at an oven temperature of 30 ◦C. TheUFLC method that was used in the present study wassuccessfully validated in-house with repeated (n = 10; dfapproximately 1:200) analyses of the CJO sample thatwas used in transesterification. For each sample deter-mination, 20 �L was mixed with 3820 �L of acetone togive a dilution factor (df) of approximately 1:200. Thecalibration curve (figure not shown) was obtained for theCJO using different concentrations based on the weightof the oil and prepared in the range of 0.01–1.5 g/10 mLof acetone and the component peak area. Because ofthe difference in the partition coefficient between thesolid and liquid phases, four eluates containing a mixtureof fatty acid and fatty acid methyl ester, partial glyce-rides (mono- and di-glycerides) and triglycerides wereseparated in this order using the solvent system ace-tone/acetonitrile ratio. The TG concentration was thencalculated based on the regression equation as expressedin Eq. (3)
Concentration, CTG
( g
L
)= 5 × 10−7A + 2.1727 (3)
where A is the peak area of the TG component as deter-mined by UFLC and the coefficient of regression (R2)given as 0.9625. The obtained fractions were character-ized as FA + FAME, MG, DG, and TG, but the sampleswere further characterized for FA to determine the actualester content. Based on the change in the percentage
composition of TG before and after the reaction, theconversion values were obtained. The contents of theother minor components in CJO were neglected in theconversion calculation.
M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699 689
e conve
3
3d
tpttcS
i
T
o
A
wg
iatciaaedr(akr
−
kfi
face reaction is the rate-limiting step (RLS). Excess ofmethanol (≥11:1 methanol:TG) was used to compensatefor the slow reaction rate, and it would be expected that
Scheme 2. Triglycerid
. Results and discussion
.1. Reaction kinetic model and analysis of kineticata
Batch kinetic experiments were carried out to studyhe kinetics of transesterification at various reaction tem-eratures and times, while other parameters, includinghe initial reactant molar ratio and catalyst concentra-ion, were kept constant. The simplified reaction for theonversion of triglycerides to products is presented incheme 2.
The reactants and products in Scheme 2 are presentedn coded form and written as
G + MeOH = ME + GLY (4)
r
+ 3Bkf
�kr
3P + D (5)
here A, triglyceride; B, alcohol; P, FAME and D,lycerol.
Although more than one species is involvedn the rate-determining step of this reaction,s shown in Scheme 1, a pseudo-first order reac-ion is proposed. Initially, CP = 0 and CD = 0, and theoncentration-dependent rate function, −rA = fn(Cj),s defined, where j represents the reacting species And B. Thus, a general rate can be written for Eq. (5)s −rA = kACα
ACβB. However because B is present in
xcess, its concentration remains virtually constanturing the course of the reaction. Thus, the rate ofeaction depends on the concentration of A in the systemthe limiting reactant). In the absence of both externalnd internal resistance to mass transfer, the intrinsicinetics can be determined using the pseudo-first-ordereaction, that is
dCA
dt= kmam(CA,b − CA,s) (6)
m, am, CA,b = CA, and CA,s are the mass transfer coef-cient, external surface area, concentration in the bulk
rsion to methyl ester.
liquid and concentration on the catalyst surface, respec-tively. If the transport process is much smaller thanthe surface reaction process, it is rate determining, andCA,s = 0. In addition, when k1 = kmam, we have
−dCA
dt= k1(CA)
and by integration are given
ln
[CAo
CA
]= k1t
or
−ln(1 − XA) = k1t (7)
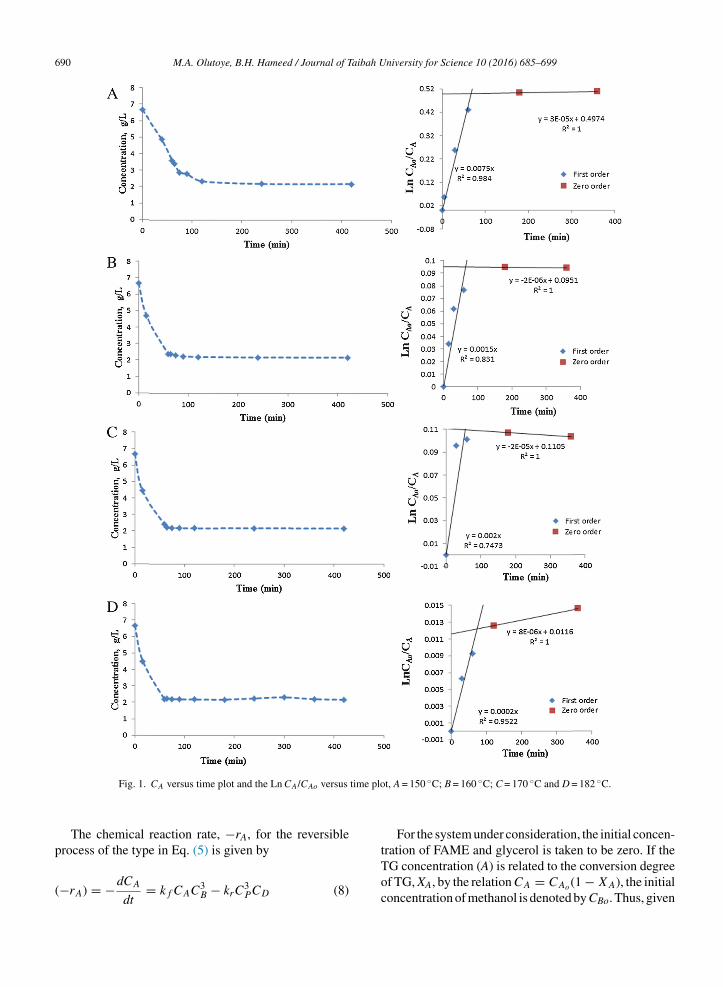
where XA is the fractional conversion of A. The plot of−ln(1 − XA) against t was used to determine k1, fromwhich km was also obtained. To obtain a first approx-imation of the kinetic model, it was assumed that thetrans-esterification reaction behaved like a first-orderirreversible reaction. Therefore, if this model is valid,a plot of −ln(1 − XA) versus t will be a straight line, andthe value of the slope will be k1, according to Eq. (7). Thereaction orders with respect to the triglyceride and thealcohol obey zero and pseudo-first orders with well-fitteddata, indicating a shift from lower order to higher orderas the reactant concentration decreases in the system,as illustrated in Fig. 1A–D. As shown, the experimen-tal data followed a linear trend with a correlation factorR2 of 0.984 and with a slope value of 0.0075 (min−1)with respect to Fig. 1A. The irreversibility of this reac-tion is favoured with excess of methanol, which induceslow concentrations of triglycerides in the equilibriumcomposition. This determination suggests that the sur-
under this condition that the reaction order with respectto methanol approaches zero. The above observation issupported by the works of Chantrasa et al. [14] and Casaset al. [15].
690 M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699
time plo
Fig. 1. CA versus time plot and the Ln CA/CAo versus
The chemical reaction rate, −rA, for the reversible
process of the type in Eq. (5) is given by
(−rA) = −dCA
dt= kf CAC3
B − krC3PCD (8)
t, A = 150 ◦C; B = 160 ◦C; C = 170 ◦C and D = 182 ◦C.
For the system under consideration, the initial concen-
tration of FAME and glycerol is taken to be zero. If theTG concentration (A) is related to the conversion degreeof TG, XA, by the relation CA = CAo (1 − XA), the initialconcentration of methanol is denoted by CBo. Thus, given
M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699 691
Table 1Values of experimental and kinetic model parameters.
Activation energy Ea (103 J/mol) 26.103, 161.42Equilibrium constant of TG adsorption KA (cm3/mol) 0.121Equilibrium constant of the overall reaction Ke 1.42Diffusivity TG-MeOH system De (m2/s) 6.83 × 10−10
Thiele modulus ϕs 0.373Effectiveness factor η 0.93Gibb’s free energy �G (J/mol) −1285.61A θAd
Cf
C
C
a
wTtci
3
elfTdstt
k
dsorption coefficient
B = CBo − 3 CAoXA, CP = 3 CAoXA and CD = CAoXA, itollows that:
Ao
dXA
dt= kf CAo(1 − XA)(CBo − 3CAoXA)
− 3kr(CAoXA)2 − rA
= CAo(1 − XA)(CBo − 3CAoXA)
− 3kr(CAoXA)2 (9)
At equilibrium, rA = 0; therefore, Eq. (9) becomes
Ao(1 − XAe)(CBo − 3CAoXAe) − 3kr(CAoXAe)2 = 0
(10)
nd after re-arrangement becomes
3CAoX2Ae
(1 − XAe)[CBo − 3CAoXAe]= Ke (11)
here XAe represent equilibrium fractional conversion.able 1 shows the kinetic and thermodynamic parame-
ers that were obtained for the process under the reactiononditions for all of the experiments that were performedn this work.
.2. The thermodynamics of process
For each step of the reversible reaction, a dynamicquilibrium can be established in accordance with theaw of mass action such that at equilibrium, the rate of theorward reaction equals the rate of the reverse reaction.he thermodynamics process based on the experimentalata was used to quantify the heat effects in the reactingystems and to calculate the equilibrium conversion –
he maximum conversion that could be obtained duringhe reaction. Thus, re-writing Eq. (8), we have
f CAC3B = krC
3PCD (12)
s 0.211
where kf and kr are the reaction rate constants for theforward and reverse reactions, respectively, and CB, CP
and CD are the actual concentrations of methanol, FAMEand glycerol, respectively:
At equilibrium, −rA = 0,(C3
PCD
CAC3B
)= Ke = kf
kr
(13)
where Ke is the dimensionless equilibrium constant interms of concentration, and Eqs. (12) and (13) combineto give
(−rA) = kf
(CAC3
B − C3PCD
Ke
)= 0 (14)
For the reversible reaction as stated in the stoichio-metric equation of the reaction being considered, theequilibrium constant Ke at temperature T was evaluatedfrom the change in the Gibb’s free energy expressed asa function of forward and backward rate in terms of
�G◦i 〈T 〉 = −RT (ln K〈T 〉) (15)
K = exp
(−�G
◦i
RT
)(16)
�G◦i = 3G
◦P + G
◦D − 3G
◦B − G
◦A (17)
�G◦i is the standard Gibb’s free energy of formation of
a given species G◦i . The relationship between the change
in Gibb’s free energy and the enthalpy, H, and entropy,S, of this system is thus defined by
�G = �H − T�S (18)
The summary of all of the calculations for the kineticand thermodynamic parameters is presented in Table 1.
The value of equilibrium constant for the TG methanol-ysis was Ke = 1.42. Thus, species A and B are dominantin the reaction mixture, and Ke >1 indicates that FAMEproduction is favoured.
692 M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699
e FA m
Scheme 3. Alcohol exchange scheme with th
3.3. Catalyst deactivation model and mechanism
Eley–Rideal and Langmuir–Hinshelwood kineticswere fitted to the batch reaction data for the heteroge-neous catalyst. The kinetics are assumed to involve theadsorption of one reactant, in this case the alcohol, whichis then attacked by the other reactant from the liquidphase. The reaction that occurs through the Eley–Ridealmechanism is between a protonated fatty acid and themethanol coming from the liquid phase. Scheme 3 showsthe transesterification reaction of triglycerides over asolid base catalyst. The scheme shows an initiation stepof an exchange between fatty acid and a protonatedmethanol, which is followed by the Eley–Rideal surfacereaction step that involves the protonated fatty acid andmethanol to produce protonated methyl ester. Water isalso formed in this step due to the large moisture contentthat is present in the feedstock.
Partial glyceride (MG and DG) complexes are formedthat have both alcohol and ester functionality; thus, forthe back reaction, it was assumed that the partial gly-cerides are adsorbed on the catalyst surface and thattheir alcohol functionality reacts with that of the esterproduct. The surface coverage of the adsorbed speciesis represented by θAds, and the relationship between theadsorbed and bulk species is derived using the Langmuirisotherm. The Langmuir isotherm relates the adsorptionand desorption of the species from the surface, as givenin Eq. (19), which applies to all species being adsorbed.This result is based on Langmuir–Hinshelwood kinetics,which requires the adsorption of both reactants onto adja-cent sites on the catalyst surface. The reaction takes placeon the catalyst surface, and both products then desorbs:
θAds = KAdsCA
1 + KAdsCA
(19)
where KAds is the adsorption equilibrium constant, andCA is the triglyceride concentration. The value of theadsorption coefficient was calculated from experimentaldata for the transesterification of CJO with methanol, aspresented in Table 1. This coefficient was assumed to beconstant for all species.
The transesterification of high-molecular-weightTG with methanol in a batch reactor using a dual-sitesolid catalyst is a highly complex chemical reaction andyields various conversions to products. The results of
oiety of TG for basic-site catalytic reaction.
these conversions are presented in Fig. 2, where it wasobserved generally that from the start of the reaction,the conversion to FAME increases as the DG, MGand TG values decrease in the reaction system. Thereare various assumptions underlying the models forreactions taking place in closed reactors, and the modeldevelopment for the process will include both steady-and unsteady-state predictions. The kinetic equationwas developed by investigating the general behaviourof the system in terms of one variable influencinganother based on theoretical knowledge of the systemwith relevant physical information in the form ofconservation laws and rate equations. Assumptionsfor the development of the kinetic equation based onthe Langmuir approach are (a) all of the surfaces ofthe catalyst have the same activity for adsorption, thatis, the surface is energetically uniform; (b) there isno interaction between adsorbed molecules; (c) alladsorption events occur by the same mechanism, andeach adsorbed complex has the same structure; (d)the extent of adsorption is less than one completemonomolecular layer on the surface; and (e) the TGmolecule is in contact with the catalyst and undergoesa three-stage process of adsorption (rAds = rA): surfacereaction (rS), desorption (rD = −rAds = rP) and that nearequilibrium, in which all of the rates rA, rS, rD are equal.
If TG is adsorbed on the catalyst surface for the con-version of triglycerides to products (neglecting the otherreactions in the series), then
TG(liquid) + Xk1�k′
1
TG · X(ads) (20)
TG represents species A, and the mechanisms fromwhich these equations were obtained are summarized infive consecutive steps. Using Langmuir–Hinshelwoodkinetics with the surface reaction being the RLS, theobserved reaction would be
1. A + Xk1−→A · X
rA = k1CAC̄v − k′1C̄A (21)
2. B + Xk1−→B · X
rA = k1CBC̄v − k′1C̄B (22)
M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699 693
F of CJOC .
wtas
3
4
at
tf
ig. 2. FAME and partial glyceride yield during the transesterification = % DG content at T = 170 ◦C and D = % TG converted at T = 170 ◦C
The net rate of adsorption, rA, gives
rA = k1CA(C̄m − C̄) − k′1C̄A
rA = k1
[CAC̄v − 1
Kads
C̄A
] (23)
here C̄v is the concentration of vacant sites. If a simul-aneous reaction is considered and if species A and Bre adjacent to each other, the adsorption process andurface reaction of the type given below occur:
. A · X + Bk2−→P · X
rS = k2C̄ACB − k′2C̄P (24)
If the reaction takes place between adsorbed A andadsorbed B, then
. A · X + B · Xk3−→P · X + X (25)
From the reactions above, it is assumed that only thedsorbed A immediately adjacent to adsorbed B will react
o yield the products.
If C̄m, molar concentration of total sites and θB, frac-ion of total surface occupied by B (methanol), then θv,raction of the total surface that is vacant.
, A = % FAME content at T = 170 ◦C; B = % MG content at T = 170 ◦C;
The net rate expression for the reaction gives
rS = k3C̄A
C̄B
C̄m
− k′3C̄P
C̄v
C̄m
rS = k3
C̄m
[C̄AC̄B − 1
KS
C̄PC̄v
] (26)
If the resistance is negligible at the surface reactionwith respect to others, the process will occur at equilib-rium, rs = 0
KS =(
CPCv
CACB
)e
(27)
If the resistance is high such that it is rate controlling,then rate = rS in Eq. (26) above:
Desorption.For the process being considered, let the desorption
process take the form
5. P · Xk′
4−→P + X
rP = k′4C̄P − k4CPC̄v
rP = −k4
[CPC̄v − 1
K4C̄P
] (28)
aibah University for Science 10 (2016) 685–699
694 M.A. Olutoye, B.H. Hameed / Journal of T
Eq. (28) shows the desorption process is the reverseof the adsorption process. X represents the vacant sites,P denotes the methyl ester, and A·X, B·X and P·X arethe adsorbed species. The concentrations of the adsorbedspecies can be obtained considering that the reaction rateconstant for the formation of the intermediates is largecompared to the reaction rate. Thus,rA
kads
≈ rB
kB
≈ 0, (29)
which represents a pseudo-equilibrium; therefore,
C̄Ae = KACAC̄v (30)
C̄Be = KBCBC̄v (31)
With K4 = Kd, at equilibrium, rP = 0; then, the equa-tion of desorption gives the expression for C̄P :
C̄P = K4CPC̄V (32)
It must be noted that
C̄m = C̄V + C̄A + C̄B + C̄P (33)
Therefore, combining Eqs. (26) and (30)–(32) andassuming that the surface reaction controls, then
C̄V = C̄m
[1 + KACA + KBCB + KdCP ](34)
Substituting for C̄v, the surface reaction rate will begiven by
rate = rS = C̄2v
C̄m
(KAKBCACB − KdCP
KS
)(35)
In this study, the Cv was based on the summation ofthe surface acidity and basicity as determined by Eqs.(1) and (2).
Although A and B are in the liquid phase, diffu-sion into the interior surface of the catalyst is requiredaccording to the postulated mechanism above throughthe processes of adsorption, surface reaction and desorp-tion in and out of the catalyst to the bulk of the liquid.The overall rate of the reaction could be controlled by oneor some of these processes, depending on their relativemagnitude. To determine the controlling mechanism, thekinetics and equilibrium constants in Table 1 were deter-mined from the experimental data. The temperature inthis case was not used to establish the controlling mech-anism because of the dependence and variation of theequilibrium constant with temperature.
3.4. External mass transfer effects on the kinetics
The rate-determining step in this heterogeneouskinetic study involved species being transported from
Fig. 3. Schematic process of solid–liquid transport showing catalystpores (enlarged).
a bulk solution to the catalyst particle or occurred in theform of transport within the particle to the active sitesor in the reaction itself, as depicted by the diagram inFig. 3.
If component A enters the pore of the catalyst inthe system through constant agitation and resulted inproduction or disappearance by chemical reaction, theamount of A in moles in the pore is given by the productof the cross-sectional area of contact and the number ofmoles if the catalyst surface is assumed to be spherical.Thus, NA in moles, NA(4πr2), is the amount of materialon a spherical surface radius r. If the disappearance of Atakes place at the �r surface by chemical reaction, thenthe amount of materials that disappeared at this surfaceis given by −rA(4πr2)�r, where rA is the rate. In addi-tion, if the effective diffusivity for species A in the porousmedium is defined by
NAr = −De
dCA
dr(36)
CA, concentration of A contained within the pores andDe, effective diffusivity as a function of concentration,temperature and catalyst pore structure. Neglecting anyvolume change with reaction (liquid phase), the conver-sion of CJO, XA, is given by XA = 1 − CA/CAo, where CA
and CAo are the CJO molar concentration at a particulartime and initially, respectively.
Because A was used as the limiting reactant, its con-centration may not be uniform within the catalyst particledue to intra-particle diffusional resistance, and the alco-hol concentration is almost uniform. To account for this
resistance, differential equations for intra-particle diffu-sion with reaction on the surface of the catalyst weresolved. The different steps in which these were achievedare presented in Eqs. (20)–(35). The occurrence of active
aibah U
sTarctb
w
ϕ
ϕ
pg
rio
η
a
r
fco
tCtcoe0nItiaitei
M.A. Olutoye, B.H. Hameed / Journal of T
ites on the catalyst surface is supported by the work ofrionfetti et al. [16], who reported the characterizationnd mechanism of regeneration. Assuming a first-ordereaction occurring in a spherical particle of radius rs, theoncentration of A at any radial distance r from the cen-re of the catalyst to its concentration on the surface wille expressed by Eq. (37)
CA
CAs
= rs
r
sinh(3ϕs(r/rs))
sinh3ϕs
(37)
here
s = rs
3
√k1ρp
De
(38)
s, Thiele-type modulus for spherical pellet; k1 is theseudo-first-order rate constant and ρp, particle density,/cm3. If
rp
rs= η (39)
p, reaction rate for the pellet; rs, diffusion rate of reactantnto the pellet, and η, effectiveness factor, then rp = ηrs
r rp = ηk1CAs
= 1
ϕs
(1
tanh 3ϕs
− 1
3ϕs
)(40)
From the above, rp can be obtained from rp = ηk1CAs
s
p = 1
ϕs
(1
tanh 3ϕs
− 1
3ϕs
)k1CAs (41)
Depending on the relative magnitudes of mass trans-er coefficient km as obtained from Eq. (6) and ηk1, theontrolling resistance was evaluated. These values andther important determinations are presented in Table 1.
The influence of intra-particle mass transfer resis-ances on the dual-site-catalyzed transesterification ofJO was evaluated by first calculating the value of
he Thiele modulus for the catalyst (assumed spheri-al pellet) ϕs = 0.373, which explains the significancef diffusion and chemical reaction. Clearly, ϕs < 1 andffectiveness factor η → 1 with a calculated value of.93 indicate that the intra-particle mass transport haso effect on the rate or chemical reaction controls.nternal diffusion is negligible. Although limited massransfer could be assumed for the reaction within thenvestigated temperature range, the limitations that wereccounted for in the effectiveness factor were included
n the developed kinetic model. If the initial rate isaken as the maximum rate for each experiment, theffective diffusivity of TG in methanol liquid phases 6.83 × 10−10 m2/s obtained from the Wilke–Chang
niversity for Science 10 (2016) 685–699 695
equation. In addition, transesterification reactions arebimolecular and reversible, and the calculated param-eters in which a pseudo first-order, irreversible, andisothermal reaction were justifiable because methanolwas always in considerable excess (>11:1 molar ratiowith CJO), which tends to benefit the forward reaction,and the equilibrium conversion in all of the experimentswas >67%.
3.5. The mechanism of the dual-site catalystreaction
3.5.1. Acidic-site-catalyzed processThe long FA moieties in TG can be esterified by
alcohols in the presence of a suitable acidic catalyst, asillustrated in Scheme 4. Fatty acids can occur in nature inthe free (unesterified) state and are often found as esterslinked to glycerol. The dual-site nature of the catalystemployed for this study is demonstrated from the plot ofrate versus time, as shown in Fig. 1, where the profilegave two rate constants with different R2. The higher R2
was the rate-determining step. Briefly, it is believed thatthe initial step is the protonation of the TG (the R1-, R2-,or R3-chain) to give an oxonium ion that thereafter under-goes an exchange reaction with an alcohol to give theintermediate dihydric complex; this in turn can cause theloss of a proton to produce an ester. It is proposed that theacid site of this dual catalyst will exchange hydrogen ionswith the other components that are involved in the reac-tion and that are adsorbed on the catalyst surface. Thisexchange will enhance the absorption equilibrium andsubsequently give rise to successive protonic exchangeequilibrium reactions. Because methanol is in excess, itis preferably adsorbed to the TG, and most of the activesites of the catalyst are occupied by the acid-protonatedmethanol.
The process is characterized by reversible steps, andthe equilibrium point of the reaction is displaced tothe right in the presence excess alcohol. However, inthe presence of water (a stronger electron donor thanaliphatic alcohols), the forward reaction in the esterifica-tion of FA is not favoured and may hinder the formationof an intermediate. Ester exchange or transesterifica-tion occurs under similar conditions. In the reactionunder consideration, the initial protonation of the esteris followed by adding the exchanging alcohol to give apartially distorted intermediate complex that dissociateseasily via transition states to give the ester product. A side
reaction, the dissociation of the intermediate by hydrol-ysis, due to the presence of water could take place andwas excluded in the reaction system. The water contentof the TG that was used for this study is quite high; thus,
696 M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699
R C
O
OH
H+
-H+
R C
+OH
OH + R1 O H R C OH
OH
+OR1H
R C OH
OH
+OR1H
H+
-H+
R C OH
OH
OR1
H+
-H+
R C+OH
:O
OR1
H
R C+OH
:O
OR1
H
R C
+OH
OR1
+ H2O
H+
-H+
H
H
1 2
2 3 4
R C
O
OR1
6
FA mo
45
Scheme 4. Alcohol exchange scheme with the
hydrolysis cannot be completely eliminated, as reportedby Suwannakarn et al. [17]. The kinetic study gave goodconversion to FAME of >97% with the water content.Fig. 4 shows the plot of FAME yield and other convertedproducts. The addition of water in the system may be use-ful to increase the polarity. The overall reaction schemeshowed how the acidic end of the dual-site catalyst couldhave influenced both the esterification and transesterifi-cation steps to produce methyl esters from crude jatropha
oil. The 3-stepwise reversible processes occurred overthe dual-site catalyst with excess methanol via a three-phase medium. In addition, as reported by Berrios et al.[18], the evaporation of water through the oil when TG
0
10
20
30
40
50
60
70
80
90
100
0 50 100 150 200 250 300 350 400 450
FAM
E y
ield
, %
Time (min)
T150T160T170T182
Fig. 4. Plot of % FAME yield versus time.
iety of TG for the acid-site catalytic reaction.
are broken down will lead to the formation of partialglycerides and FFAs in the reaction medium.
3.5.2. Basic-site-catalyzed processAs shown in Scheme 3, the reaction mechanism is pro-
posed for the base-catalyzed transesterification of oils atthe basic site. The alcohol that was used for the reactionproduced an anionic −OR “intermediate that can dis-sociate back to the original ester or form a new esterproduct if excess alcohol is used. The excess −OR”anion will shift the equilibrium point of the reactionin the forward direction. In a similar reaction, an unes-terified fatty acid will be converted to the carboxylateion RCOO− in basic medium, and this is not subject tonucleophilic attack by alcohols because of its negativecharge. Transesterification will then proceed by the pro-posed mechanism with the basic catalyst (basic site) butnot with esterification. As in the case of acid-catalyzedreactions, the presence of water will cause a dissocia-tion of the intermediate component irreversibly to thefree acid. Thus, a base-catalyzed transesterification ofesters requires excess alcohol in the absence of waterfrom the reaction medium to obtain a high yield of prod-ucts. In this study, the FAME, DG, MG and TG yields asanalyzed using the HPLC are presented in Table 2. The
results showed high conversion for FAME; however, anearly complete conversion of the TG as the temper-ature increased was observed, indicating a higher rateorder and the predomination of the chemical reaction.
M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699 697
Table 2Products and yields of transesterification reactions using the dual-site catalyst.
Temperature (◦C) Time (min) Monoglyceride (%) Diglyceride (%) Triglyceride (%) FAME (%)
The evaluation of the effect of temperature is verymportant in kinetic studies because temperature is use-ul for calculating the activation energy of the reaction.n this regard, the effect of temperature on the rate ofeaction was studied by conducting the reaction at 150,60, 170, and 182 ◦C, having previously determined theptimum conditions for CJO methanolysis. Represen-ative results are shown in Fig. 4. The ester conversionncreased with increasing reaction temperature. How-ver, the equilibrium conversion was nearly equal in theange of temperatures that were considered in this work.
similar observation was reported by Delgado et al. [19]ith a conversion value of greater than 70%. Generally,
n most transesterification reactions, the equilibrium con-tant is a weak function of the temperature because ofhe small value of the heat of reaction. This fact is cor-oborated by the work of Sanz et al. [20]. Arrhenius plotsere constructed (figure not shown) and used to estab-
ish the activation energy and the pre-exponential factor
s presented in Table 1. The high values of activationnergy support the fact that there was no external andnternal mass transfer resistance and that the reactionas kinetically controlled for transesterification. Two
0.60 0.02 97.14
distinctive slopes were calculated from the Arrheniusplot, corresponding to activation energies of Ea of thefirst-order and zero-order kinetics. The change in acti-vation energy in relation to the order as the temperaturevaries suggests a change in the reaction mechanism or therate-limiting step. This change could be due to the dualsite being involved in both esterification and transesteri-fication. Based on the activation energy results, thesereactions did not appear to be pore diffusion limitedunder the employed reaction conditions. Stamenkovicand co-workers [21] observed a similar trend as theresults presented in this study. In addition, these authorsfound that the rate constants increased when the reactiontemperature increased.
3.7. Catalyst reusability and deactivation studies
Heterogeneous catalytic reactions are usuallyaffected by deactivation, although they have the poten-tial to be recovered, regenerated and reused. In thepresent work, the catalyst that was used was employed
under similar conditions of reaction temperature,concentration, and catalyst loading to generate a plotof conversion against time. Deactivation studies werecarried out for the catalyst that was employed in this
698 M.A. Olutoye, B.H. Hameed / Journal of Taibah University for Science 10 (2016) 685–699
talyst a
for the transesterification of palm oil, Appl. Catal. A: Gen. 370
Fig. 5. Conversion of TG as a function of time using the dual-site caand e,f = 182 ◦C (regeneration and reuse runs).
study. Fig. 5 shows the results of triglyceride conversionfor five successive 6-h reaction cycles. The catalystactivity reactions were carried out in the reactor asdescribed earlier in this article except that the conditionswere fixed at optimum values. Batch runs were preparedfor the catalyst, and the reaction was quenched to beconsistent with the kinetic study experiments. The reac-tion mixture samples were centrifuged prior to analysisto separate the catalyst from the reactant mixture and tostop the reaction. For the catalyst deactivation studies,the reaction were started as previously described [13].The observed slight decrease in the conversion forthe reusability runs could be due to the depositionof carbonaceous residues from reactants, products orintermediates (coking); the chemisorption of impuritiesof the feed stream (poisoning); and losses that occurredduring the handling process. The regeneration andreusability test on the catalyst for five cycles showedthat there was no appreciable decline in the conversionand confirms the catalyst activity with minimal surfacedeactivation.
4. Conclusions
The kinetics of the transesterification of CJO with
methanol using a dual-site (Alumina-Mg-Zn) cata-lyst was studied. The reaction follows both zero- andfirst-order rate models in the formation of ester. Fur-ther observations from both the concentration-time and
t various temperatures, a = 150 ◦C; b = 160 ◦C; c = 170 ◦C; d = 182 ◦C
rate-time plots revealed a shift in the order of the reactionfrom low (zero) to high (first) order, and the reaction wasfound to occur between an adsorbed methanol moleculeand a TG molecule in the bulk liquid phase. As a result, itwas concluded that the dual sites catalyst offers a viableand suitable alternative to the existing heterogeneous cat-alysts for the transesterification of CJO with methanolunder the stated conditions with equilibrium conversion,Ke, greater than 67%; an equilibrium constant of TGadsorption, KA, of 0.121 (cm3/mol); and a diffusivityof the TG-MeOH system, De, of 6.83 × 10−10 (m2/s).The catalyst is simple in preparation, separates easilyfrom product mixture and is thus recommended for theindustrial production of biodiesel.
Acknowledgments
The authors acknowledge the research grant providedby the Ministry of Higher Education, Malaysia, under theFundamental Research Grant Scheme (FRGS) with theAccount No: 203/PJKIMIA/6071206.
References
[1] B.O. Aderemi, B.H. Hameed, Alum as a heterogeneous catalyst
[3] A.A. Apostolakou, I.K. Kookos, C. Marazioti, K.C. Angelopoulo,Techno-economic analysis of a biodiesel production process fromvegetable oils, Fuel Process. Technol. 90 (2009) 1023–1103.
[4] R. Sarin, M. Sharma, A.A. Khan, Terminalia belerica Roxb. seedoil: a potential biodiesel resource, Bioresour. Technol. 101 (2010)1380–1384.
[5] S. Pinzi, L.M. Gandía, G. Arzamendi, J.J. Ruiz, M.P. Dorado,Influence of vegetable oils fatty acid composition on reactiontemperature and glycerides conversion to biodiesel during trans-esterification, Bioresour. Technol. 102 (2011) 1044–1050.
[6] X. Yu, Z. Wen, H. Li, S. Tu, J. Yan, Transesterification of Pistaciachinensis oil for biodiesel catalyzed by CaO-CeO2 mixed oxides,Fuel 90 (2011) 1868–1874.
[7] M.A. Olutoye, B.H. Hameed, KyMg1−xZn1+xO3 as a heteroge-neous catalyst in the transesterification of palm oil to fatty acidmethyl esters, Appl. Catal. A: Gen. 371 (2009) 191–198.
[8] Z. Wan, B.H. Hameed, Transesterification of palm oil to methylester on activated carbon supported calcium oxide catalyst, Biore-sour. Technol. 102 (2011) 2659–2664.
[9] T.F. Dossin, M. Reyniers, R.J. Berger, G.B. Marin, Simulationof heterogeneously MgO-catalyzed transesterification for fine-chemical and biodiesel industrial production, Appl. Catal. B:Environ. 67 (2006) 136–148.
10] H. Hattori, M. Shima, H. Kabashima, Alcoholysis of ester andepoxide catalyzed by solid bases, Stud. Surf. Sci. Catal. 130(2000) 3507–3512.
11] T.F. Dossin, M. Reyniers, G.B. Marin, Kinetics of hetero-geneously MgO-catalyzed transesterification, Appl. Catal. B:Environ. 62 (2006) 35–45.
12] P. Patil, V.G. Gude, S. Pinappu, S. Deng, Transesterificationkinetics of Camelina sativa oil on metal oxide catalysts underconventional and microwave heating conditions, Chem. Eng. J.168 (2011) 1296–1300.
[
niversity for Science 10 (2016) 685–699 699
13] M.A. Olutoye, B.H. Hameed, Synthesis of fatty acid methylester from crude jatropha (Jatropha curcas Linnaeus) oil usingaluminium oxide modified Mg-Zn heterogeneous catalyst, Biore-sour. Technol. 102 (2011) 6392–6398.
14] A. Chantrasa, N. Phlernjai, J.G. Goodwin, Kinetics of hydrotal-cite catalyzed transesterification of tricaprylin and methanol forbiodiesel synthesis, Chem. Eng. J. 168 (2011) 333–340.
15] A. Casas, et al., Kinetics of chemical interesterification ofsunflower oil with methyl acetate for biodiesel and triacetinproduction, Chem. Eng. J. (2011), http://dx.doi.org/10.1016/j.cej.2011.05.037.
16] C. Trionfetti, S. Crapanzan, I.V. Babich, K. Seshan, L. Lefferts,Lithium ions incorporation in MgO for oxidative dehydro-genation/cracking of propane: active site characterization andmechanism of regeneration, Catal. Today 145 (2009) 19–26.
17] K. Suwannakarn, E. Lotero, K. Ngaosuwan, J.G. Goodwin,Simultaneous free fatty acid esterification and triglyceride trans-esterification using a solid acid catalyst with in situ removal ofwater and unreacted methanol, Ind. Eng. Chem. Res. 48 (2009)2810–2818.
18] M. Berrios, M.A. Martin, A.F. Chica, A. Martin, Study of esteri-fication and transesterification in biodiesel production from usedfrying oils in a closed system, Chem. Eng. J. 160 (2010) 473–479.
19] P. Delgado, M.T. Sanz, S. Beltran, Kinetic study for esterifica-tion of lactic acid with ethanol and hydrolysis of ethyl lactateusing ion-exchange resin catalyst, Bioresour. Technol. 126 (2007)111–118.
20] M.T. Sanz, S.M. Beltran, J.L. Cabezas, J. Coca, Autocatalyzedand ion-exchange-resin-catalyzed esterification kinetics of lactic
acid with methanol, Ind. Eng. Chem. Res. 41 (2002) 512–517.
21] O.S. Stamenkovic, Z.B. Todorovic, M.L. Lazic, V.B. Veljkovic,D.U. Skala, Kinetics of sunflower oil methanolysis at low tem-peratures, Bioresour. Technol. 99 (2008) 1131–1140.