Biochem. J. (1987) 247, 547-554 (Printed in Great Britain) Klebsiella pneumoniae nitrogenase Inhibition of hydrogen evolution by ethylene and the reduction of ethylene to ethane Gillian A. ASHBY, Michael J. DILWORTH* and Roger N. F. THORNELEYt A.F.R.C. Unit of Nitrogen Fixation, University of Sussex, Brighton BNI 9RQ, U.K. Ethylene (C2H4) inhibited H2 evolution by the Mo-containing nitrogenase of Klebsiella pneumoniae. The extent of inhibition depended on the electron flux determined by the ratio of Fe protein (Kp2) to MoFe protein (Kpl) with KC2H4 = 409 kPa ([Kp2]/[Kpl] = 22: 1) and K'2H4 = 88 kPa ([Kpl]/[Kp2] = 21: 1) at 23 °C at pH 7.4. At [Kp2]/[Kpl] = 1: 1, inhibition was minimal with C2H4 (101-kPa). Extrapolation of data obtained when C 2H4 was varied from 60 to 290 kPa indicates that at infinite pressure of C2H4 total inhibition of H2 evolution should occur. C24 inhibited concomitant S2042- oxidation to the same extent that it inhibited H2 evolution. Although other inhibitors of total electron flux such as CN- and CH3NC uncouple MgATP hydrolysis from electron transfer, C2H4 did not affect the ATP/2e ratio. Inhibition of H2 evolution by C2H4 was not relieved by CO. C2H4 was reduced to C2H6 at [Kp2]/[Kpl] ratios > 5: 1 in a reaction that accounted for no more than 1 % of the total electron flux. These data are discussed in terms of the chemistry of alkyne and alkene reduction on transition-metal centres. INTRODUCTION The nitrogenase of Klebsiella pneumoniae comprises two metalloproteins, the MoFe protein (Kp 1, Mr 218000) and the Fe protein (Kp2, Mr 67000). In the presence of MgATP and a reductant (flavodoxin in vivo, Na2S204 in vitro), nitrogenase catalyses the reduction of N2 to 2NH3. Under optimal conditions, the reduction of N2 is accompanied by the stoichiometric reduction of 2H+ to H2 and the hydrolysis of 16 MgATP to 16 MgADP + 16 Pi (for recent reviews on the structure and mechanism of nitrogenase see Burgess, 1985; Lowe et al., 1985; Orme-Johnson, 1985; Stephens, 1985; Thorneley & Lowe, 1985). Although a comprehensive mechanism for nitro- genase-catalysed H2 evolution and N2 reduction has been developed (Lowe & Thorneley, 1984a, b; Thorneley & Lowe, 1984a, b), it does not consider the mechanism of reduction of other substrates (for a review of these and their reduction products see Burgess, 1985). The most important of these is acetylene (C2H2), which is reduced to ethylene (C2H4) (Dilworth, 1966; Schollhorn & Burris, 1967), and forms the basis for a widely used assay of nitrogenase activity both in vivo and in vitro (Koch & Evans, 1966; Hardy et al., 1968). An understanding of the way C2H2 is reduced is therefore useful in defining the chemistry occurring at the active site of nitrogenase. The relationships between H2 evolution, C2H2 reduc- tion and N2 reduction under various conditions of electron flux through the enzyme are not well understood (Hageman & Burris, 1980; Wherland et al., 1981). Before a detailed analysis can be made of pre-steady- state and steady-state kinetic data for C2H2 reduction to C2H4 and for concomitant H2 evolution, it is necessary to determine the affinity of the MoFe protein not only for the substrate C2H2 but also for the potential product inhibitor C24. A transient form of the MoFe protein with bound C2H4 has been detected at low concentrations by e.p.r. spectroscopy in assays run at 10 °C with low electron flux (Kp2/Kpl molar ratio of 1:3) (Lowe et al., 1978). The binding constants for C2H2 (15 /iM and 13 mM) and C2H4 (1.3mM) determined under these conditions at 10 °C are clearly inappropriate for computer simulations using the Lowe-Thorneley model, which uses rate constants determined at 23 'C. At 10 'C and low electron flux, C2H4 stimulated H2 evolution by approx. 7500 rather than inhibiting it (Lowe et al., 1978), an observation that cannot be accommodated in the Lowe- Thorneley model in its present form. There is therefore evidence of C24 interacting chemically with the enzyme, but few data on the nature of that interaction. The conventional Mo-containing nitrogenases reduce C2H2 to C2H4, but further reduction to ethane (C2H6) or methane (CH4) has not been detected (Hardy et al., 1968). However, the recently discovered vanadium- containing nitrogenase from Azotobacter chroococcum (Robson et al., 1986) does reduce a significant proportion of C2H2 to C2H6 (Dilworth et al., 1987). A reduction product from nitrogenase that inhibits total electron flux and MgATP hydrolysis is potentially useful for preparing high concentrations of MoFe protein in a state that, during substrate reduction, is only present at a low concentration as a transient species. The spectroscopic characterization of such a complex could provide information about the site(s) of substrate and product binding and the level of reduction of the MoFe protein in the catalytic cycle when product is released. In the present paper we describe the conditions under which C2H4 can behave as such an inhibitor. We also show that C2H4 is slowly reduced to C2H6 at high electron flux. MATERIALS AND METHODS Nitrogenase The nitrogenase component proteins from Klebsiella pneumoniae (oxytoca) N.C.I.B. 12204 were purified and Vol. 247 547 Abbreviations used: Kpl and Kp2, MoFe protein and Fe protein components respectively of Klebsiella pneumoniae nitrogenase. * Permanent address: School of Environmental and Life Sciences, Murdoch University, Murdoch, Western Australia 6150, Australia. t To whom correspondence should be addressed.
Transcript
Biochem. J. (1987) 247, 547-554 (Printed in Great Britain)
Klebsiella pneumoniae nitrogenaseInhibition of hydrogen evolution by ethylene and the reduction of ethylene to ethane
Gillian A. ASHBY, Michael J. DILWORTH* and Roger N. F. THORNELEYtA.F.R.C. Unit of Nitrogen Fixation, University of Sussex, Brighton BNI 9RQ, U.K.
Ethylene (C2H4) inhibited H2 evolution by the Mo-containing nitrogenase of Klebsiella pneumoniae. Theextent of inhibition depended on the electron flux determined by the ratio of Fe protein (Kp2) to MoFeprotein (Kpl) with KC2H4 = 409 kPa ([Kp2]/[Kpl] = 22: 1) and K'2H4 = 88 kPa ([Kpl]/[Kp2] = 21: 1) at23 °C at pH 7.4. At [Kp2]/[Kpl] = 1: 1, inhibition was minimal with C2H4 (101-kPa). Extrapolation of dataobtained when C2H4 was varied from 60 to 290 kPa indicates that at infinite pressure of C2H4 total inhibitionof H2 evolution should occur. C24 inhibited concomitant S2042- oxidation to the same extent that itinhibited H2 evolution. Although other inhibitors of total electron flux such as CN- and CH3NC uncoupleMgATP hydrolysis from electron transfer, C2H4 did not affect the ATP/2e ratio. Inhibition of H2 evolutionby C2H4 was not relieved by CO. C2H4 was reduced to C2H6 at [Kp2]/[Kpl] ratios > 5: 1 in a reaction thataccounted for no more than 1 % of the total electron flux. These data are discussed in terms of the chemistryof alkyne and alkene reduction on transition-metal centres.
INTRODUCTIONThe nitrogenase of Klebsiella pneumoniae comprises
two metalloproteins, the MoFe protein (Kp 1, Mr218000) and the Fe protein (Kp2, Mr 67000). In thepresence of MgATP and a reductant (flavodoxin in vivo,Na2S204 in vitro), nitrogenase catalyses the reduction ofN2 to 2NH3. Under optimal conditions, the reduction ofN2 is accompanied by the stoichiometric reductionof 2H+ to H2 and the hydrolysis of 16 MgATP to16 MgADP + 16 Pi (for recent reviews on the structureand mechanism of nitrogenase see Burgess, 1985; Loweet al., 1985; Orme-Johnson, 1985; Stephens, 1985;Thorneley & Lowe, 1985).Although a comprehensive mechanism for nitro-
genase-catalysed H2 evolution and N2 reduction has beendeveloped (Lowe & Thorneley, 1984a, b; Thorneley &Lowe, 1984a, b), it does not consider the mechanism ofreduction of other substrates (for a review of these andtheir reduction products see Burgess, 1985). The mostimportant of these is acetylene (C2H2), which is reducedto ethylene (C2H4) (Dilworth, 1966; Schollhorn & Burris,1967), and forms the basis for a widely used assay ofnitrogenase activity both in vivo and in vitro (Koch &Evans, 1966; Hardy et al., 1968). An understanding of theway C2H2 is reduced is therefore useful in defining thechemistry occurring at the active site of nitrogenase.The relationships between H2 evolution, C2H2 reduc-
tion and N2 reduction under various conditions ofelectron flux through the enzyme are not well understood(Hageman & Burris, 1980; Wherland et al., 1981).Before a detailed analysis can be made of pre-steady-state and steady-state kinetic data for C2H2 reduction toC2H4 and for concomitant H2 evolution, it is necessary todetermine the affinity of the MoFe protein not only forthe substrate C2H2 but also for the potential productinhibitor C24.A transient form of the MoFe protein with bound
C2H4 has been detected at low concentrations by e.p.r.
spectroscopy in assays run at 10 °C with low electron flux(Kp2/Kpl molar ratio of 1:3) (Lowe et al., 1978). Thebinding constants for C2H2 (15 /iM and 13 mM) andC2H4 (1.3mM) determined under these conditions at10 °C are clearly inappropriate for computer simulationsusing the Lowe-Thorneley model, which uses rateconstants determined at 23 'C. At 10 'C and low electronflux, C2H4 stimulated H2 evolution by approx. 7500rather than inhibiting it (Lowe et al., 1978), anobservation that cannot be accommodated in the Lowe-Thorneley model in its present form. There is thereforeevidence ofC24 interacting chemically with the enzyme,but few data on the nature of that interaction.The conventional Mo-containing nitrogenases reduce
C2H2 to C2H4, but further reduction to ethane (C2H6) ormethane (CH4) has not been detected (Hardy et al.,1968). However, the recently discovered vanadium-containing nitrogenase from Azotobacter chroococcum(Robson et al., 1986) does reduce a significant proportionof C2H2 to C2H6 (Dilworth et al., 1987).A reduction product from nitrogenase that inhibits
total electron flux and MgATP hydrolysis is potentiallyuseful for preparing high concentrations ofMoFe proteinin a state that, during substrate reduction, is only presentat a low concentration as a transient species. Thespectroscopic characterization of such a complex couldprovide information about the site(s) of substrate andproduct binding and the level of reduction of the MoFeprotein in the catalytic cycle when product is released. Inthe present paper we describe the conditions under whichC2H4 can behave as such an inhibitor. We also show thatC2H4 is slowly reduced to C2H6 at high electron flux.
MATERIALS AND METHODSNitrogenaseThe nitrogenase component proteins from Klebsiella
pneumoniae (oxytoca) N.C.I.B. 12204 were purified and
Vol. 247
547
Abbreviations used: Kpl and Kp2, MoFe protein and Fe protein components respectively of Klebsiella pneumoniae nitrogenase.* Permanent address: School of Environmental and Life Sciences, Murdoch University, Murdoch, Western Australia 6150, Australia.t To whom correspondence should be addressed.
G. A. Ashby, M. J. Dilworth and R. N. F. Thorneley
(b)-
I
(aI''1)
211-
Fig. 1. Gas chromatograms ofC24 before and after purification
C2H4 was purified as described in the Materials andmethods section. Samples of cylinder and purified C24were diluted 72-fold into Ar and 0.5 ml samples werechromatographed. (a) Cylinder C2H4. Peak 1 (C2H6),attenuation x 80; peak 2 (C2H4), attenuation x 32 x 103.(b) Purified C2H4' Peak I (C2H6), attenuation x 80(< 0.005 % C2H6); peak 2 (C2H4), attenuation x 32 x 103.
assayed as previously described (Thorneley & Lowe,1983). Kpl and Kp2 proteins had specific activities at30 °C of 1800 and 1450 nmol of C2H4 produced/min permg of protein respectively.
GasesCylinder argon, hydrogen and carbon monoxide (Air
Products, Walton-on-Thames, Surrey, U.K.), methane(British Oxygen Co., London S.W.19, U.K.) and etliane(Messer Griesheim, Dusseldorf, W. Germany) were usedwithout further purification. Cylinder ethylene (AirProducts) was purified as described below. Acetylene wasprepared by the action of water on CaC2 (BDHChemicals, Poole, Dorset, U.K.). Gases were added byback-filling evacuated serum vials fitted with Subasealrubber closures (Ar and C2H4) or by injection with gas-tight syringes (C2H2, CH, H2 and CO).Ethylene purification
Ethylene was purified by selective absorption intoacidic Hg(CIO4)2 solution, from which it was sub-
sequently released by addition of LiCl (Young et al.,1952). A 250 ml portion of 0.5 M-Hg(C104)2 in 2 M-HCl04 was placed in a 2-litre round-bottomed flask withsockets for two stopcocks and a 250 ml separatingfunnel, and cooled to 0 °C in an ice bath. The flask wasevacuated and refilled twice with argon, and then withcylinder ethylene containing 0.060% (v/v) ethane, andsealed. The absorption of ethylene was monitoredmanometrically as the solution was stirred, with absorp-tion complete in 2 h. The flask was then evacuated andrefilled twice with argon, and then fully evacuated andsealed. The ice bath was removed and a 4-fold molarexcess of LiCl was added slowly from the separatingfunnel as a 4 M solution that had been sparged withargon. Gas evolution was rapid; the flask was leftovernight to complete gas production. The resultantethylene contained less than 0.0005 % (v/v) ethane; noother C2, C3 or C4 hydrocarbon was detectable in it. Fig.1 shows gas chromatograms of ethylene before and afterpurification. Details of the chromatography and detec-tion procedures are given below.
Assay procedureAssays were performed in a shaking water bath at
23 °C and unless otherwise stated the assay mixturecontained in a final volume of I ml 21.5 ,umol of MgCl2,45 ,umol of Hepes, pH 7.4, 16 ,tmol of phosphocreatine,30 ,smol of Na2S204, 8 ,tmol of ATP and 66 gcg ofcreatine kinase under various mixtures of Ar, C2H2,C2H49 H2 and CO in the gas phase (6.3 ml). Assays werestarted by syringe addition of pre-mixed Kpl and Kp2(0.05 ml) to give the concentrations quoted in the Figurelegends and text. Assays were stopped with 0.1 ml of30% (w/v) trichloroacetic acid, except for those in whichATP hydrolysis was to be measured, when 0.3 ml of0.5 M-EDTA, pH 7.4, was used.
Assays under hyberbaric C2H4 (202 and 303 kPa)required a modified procedure. The assays were preparedas described above under C2H4 (101 kPa) and equili-brated at 23 'C. Within 5 s after the injection of theprotein mix, the requisite volumes of C2H4 to give totalpressures of 202 (6.3 ml) and 303 kPa (12.6 ml) wereinjected. No leakages of gas occurred through thepunctured rubber closure during the course of the assayterminated as described above. The recovery of aninternal standard (0.1 ml of CH4) added to the vialbefore the injection of hyperbaric C24 was used toverify that correction of the gaseous products recoveredby a factor equal to the pressure in atmospheres wasvalid.Water (0.5 ml) was injected into each vial containing a
quenched assay, and 0.5 ml samples of gas were assayedfor H2 on a Pye Series 204 gas chromatograph fitted witha 1 m x 6 mm-diam. column of molecular sieve 5A(80-100 mesh). The column was operated at 100 'C withAr (20 ml/min) as carrier gas and H2 was detected witha katharometer.CH4, C2H6, C2H4 and C2H2 were measured by flame
ionization detection following chromatography on aI m x 6 mm-diam. column of chromatographic aluminaprepared as described by Smith & Dowdell (1973) andoperated with a 45 ml/min flow of N2 at 110 'C (Smith& Restall, 1971). C2H6 and C214 were completelyresolved on this column: 60 pmol of C216 is easilymeasured.
Linear time courses for product formation were
1987
II
548
.1
Klebsiella pneumoniae nitrogenase and ethylene
obtained within the range 5-40 min under all experi-mental conditions, except in those experiments whereNa2S204 oxidation was monitored (see below).
ATP hydrolysis assayATP hydrolysis during the nitrogenase assay was
determined by measuring creatine released from phospho-creatine. Ion-exchange resin (AG l-X8; Bio-Rad Labora-tories, Richmond, CA, U.S.A.) was washed with 4 M-HCI and then exhaustively with water. The liquid contentof EDTA-quenched assay vials after gas analysiswere allowed to flow through individual columns(2 cm x 6 mm diam.) of AG1-X8 resin, followed by twowashes of vials and columns with 0.5 ml portions ofwater. S2042- and its oxidation products, ATP, phos-phocreatine, Hepes, EDTA and protein were adsorbedto the resin, leaving creatine quantitatively recoverable inthe eluate. The resulting solutions were collected inweighed 30 ml Universal bottles and re-weighed todetermine the sample volumes. Samples were then takenfor creatine determination by the method of Ennor(1957).
Dithionite oxidationAssays in which S2042- oxidation was to be determined
were stopped with 0.1 ml of 36% (w/v) formaldehyde,and residual S2042- was measured by iodometric titrationof the formaldehyde adduct (Li et al., 1982). The initialNa2S204 concentration in such assays was 7.5 mm ratherthan the normal 30 mm.
ReagentsAll salts were purchased from BDH Chemicals, and
biochemicals from Sigma Chemical Co., Poole, Dorset,U.K.
RESULTS AND DISCUSSIONInhibition of H2 evolution by C2H4
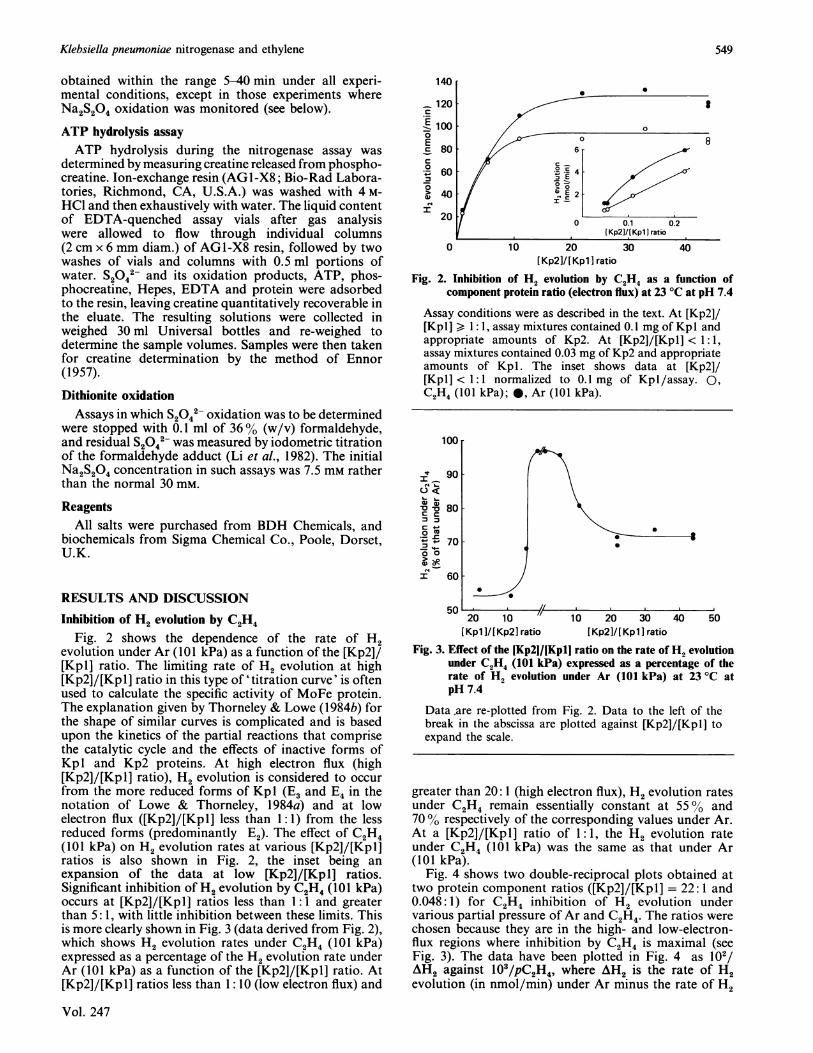
Fig. 2 shows the dependence of the rate of H2evolution under Ar (101 kPa) as a function of the [Kp2]/[Kpl] ratio. The limiting rate of H2 evolution at high[Kp2]/[Kpl] ratio in this type of 'titration curve' is oftenused to calculate the specific activity of MoFe protein.The explanation given by Thorneley & Lowe (1984b) forthe shape of similar curves is complicated and is basedupon the kinetics of the partial reactions that comprisethe catalytic cycle and the effects of inactive forms ofKpl and Kp2 proteins. At high electron flux (high[Kp2]/[Kpl] ratio), H2 evolution is considered to occurfrom the more reduced forms of Kp 1 (E3 and E4 in thenotation of Lowe & Thorneley, 1 984a) and at lowelectron flux ([Kp2]/[Kpl] less than 1:1) from the lessreduced forms (predominantly E2). The effect of C24(101 kPa) on H2 evolution rates at various [Kp2]/[Kpl]ratios is also shown in Fig. 2, the inset being anexpansion of the data at low [Kp2]/[Kpl] ratios.Significant inhibition of H2 evolution by C2H4(01 kPa)occurs at [Kp2]/[Kpl] ratios less than 1: 1 and greaterthan 5: 1, with little inhibition between these limits. Thisis more clearly shown in Fig. 3 (data derived from Fig. 2),which shows H2 evolution rates under C2H4 (101 kPa)expressed as a percentage of the H2 evolution rate underAr (101 kPa) as a function of the [Kp2]/[Kpl] ratio. At[Kp2]/[Kpl] ratios less than 1:10 (low electron flux) and
140
z120
Ec 80
E 100 6
060E
20
20 ~~ ~~~~00.1 0.2/Kp2I/[ Kp1 ] ratio
0 10 20 30 40[Kp2]/[Kpl ] ratio
Fig. 2. Inhibition of H2 evolution by C2H4 as a function ofcomponent protein ratio (electron flux) at 23 °C at pH 7.4
Assay conditions were as described in the text. At [Kp2]/[Kpl] > 1: 1, assay mixtures contained 0.1 mg of Kpl andappropriate amounts of Kp2. At [Kp2]/[Kpl] < 1:1,assay mixtures contained 0.03 mg of Kp2 and appropriateamounts of Kpl. The inset shows data at [Kp2]/[Kpl] < 1:1 normalized to 0.1 mg of Kpl/assay. 0,C2H4 (101 kPa); 0, Ar (101 kPa).
1001
cJ
-
.) .L D-
'a0C c
c +-CX
43 4--0 0
Ia4I:
90
801-
70 F
60 [0
20 10[ Kp1 ]/ [ Kp2] ratio
10 20 30 40 50[Kp2]/[ Kpl ] ratio
Fig. 3. Effect of the IKp2l/[Kpl] ratio on the rate of H2 evolutionunder C2H4 (101 kPa) expressed as a percentage of therate of H2 evolution under Ar (101 kPa) at 23 °C atpH 7.4
Data are re-plotted from Fig. 2. Data to the left of thebreak in the abscissa are plotted against [Kp2]/[Kpl] toexpand the scale.
greater than 20: 1 (high electron flux), H2 evolution ratesunder C2H4 remain essentially constant at 55 % and700 respectively of the corresponding values under Ar.At a [Kp2]/[Kpl] ratio of 1:1, the H2 evolution rateunder C2H4 (101 kPa) was the same as that under Ar(101 kPa).
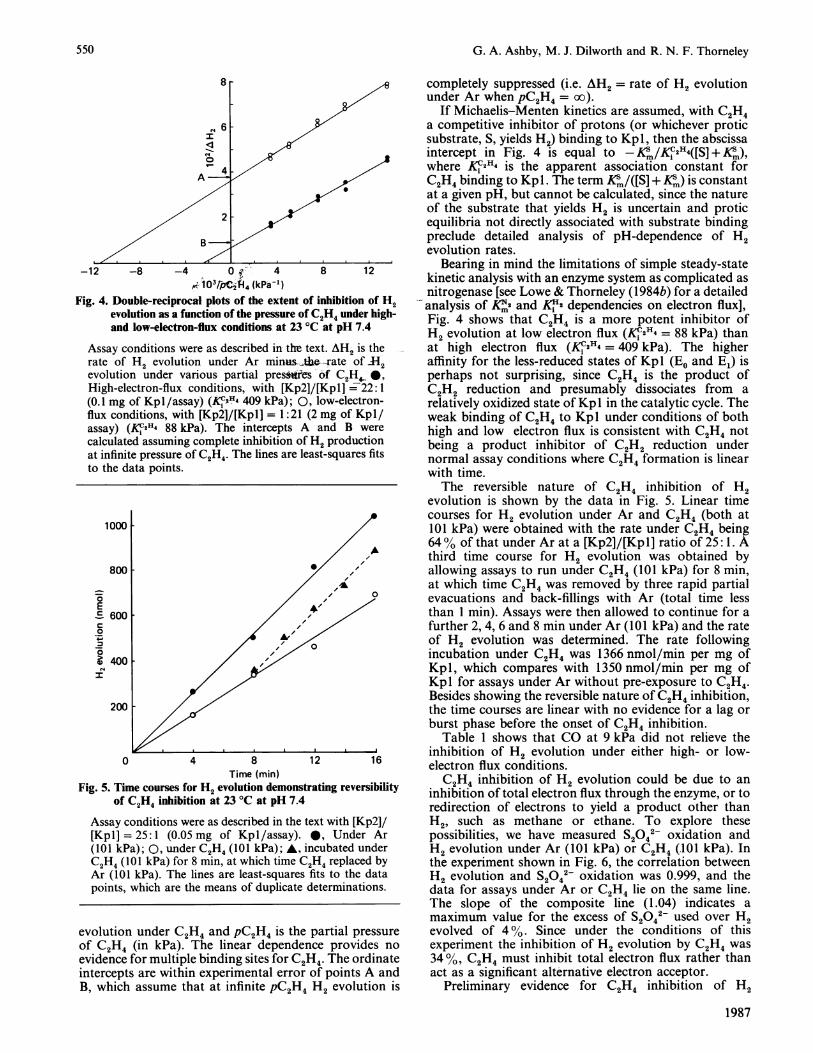
Fig. 4 shows two double-reciprocal plots obtained attwo protein component ratios ([Kp2]/[Kpl] = 22:1 and0.048: 1) for C2H4 inhibition of H2 evolution undervarious partial pressure of Ar and C2H4 The ratios werechosen because they are in the high- and low-electron-flux regions where inhibition by C2H4 is maximal (seeFig. 3). The data have been plotted in Fig. 4 as 102/AH2 against 103/pC2H4, where AH2 is the rate of H2evolution (in nmol/min) under Ar minus the rate of H2
Vol. 247
549
G. A. Ashby, M. J. Dilworth and R. N. F. Thorneley
-12 -8 -4 0 4 8 120;1O3/Pii4 (kPaf)
Fig. 4. Double-reciprocal plots of the extent of inhibition of H2evolution as a function of the pressure of C2H4 under high-and low-electron-flux conditions at 23 °C at pH 7.4
Assay conditions were as described in the text. AH2 is therate of H2 evolution under Ar minus-e--rate of 112
evolution under various partial presWes of C2H4High-electron-flux conditions, with [Kp2]/[Kpl] = 22:1(0.1 mg of Kpl/assay) (KliH4 409 kPa); 0, low-electron-flux conditions, with [Kp2]/[Kpl] = 1: 21 (2 mg of Kpl/assay) (KC2H4 88 kPa). The intercepts A and B were
calculated assuming complete inhibition of H2 productionat infinite pressure of C2HA. The lines are least-squares fitsto the data points.
1000
800
Es 600c
0
.0O 400I
200
0
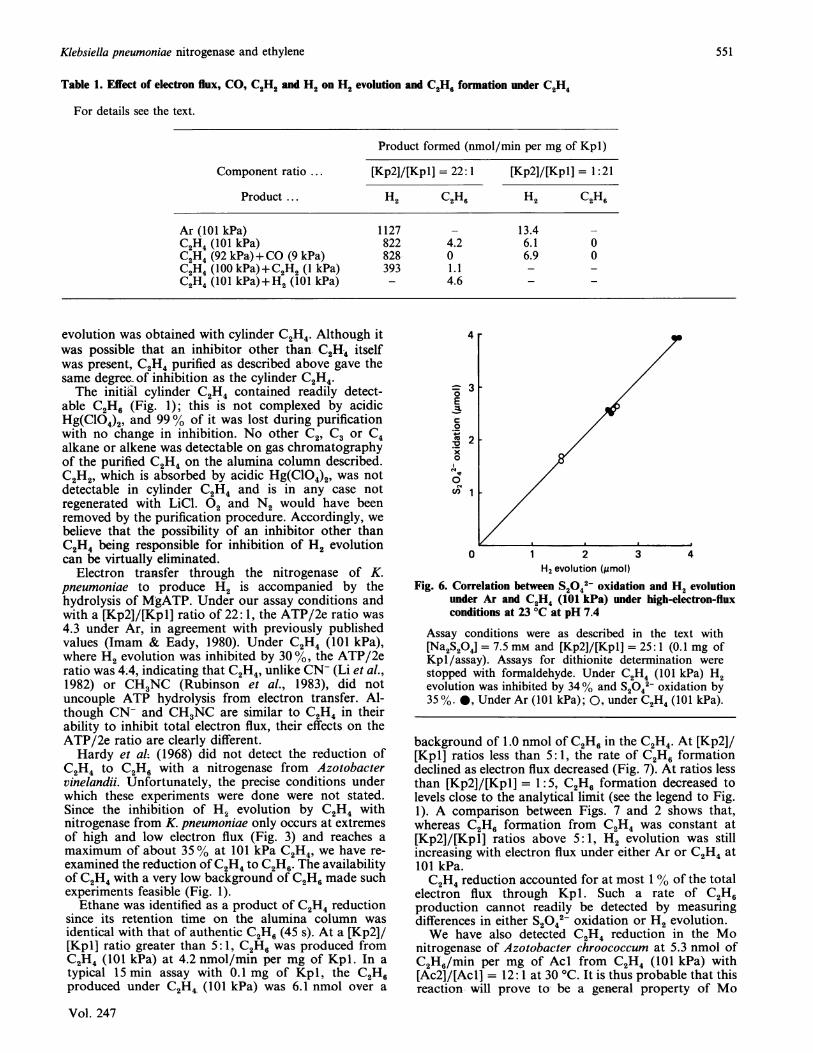
0 4 8 12 16Time (min)
Fig. 5. Time courses for H2 evolution demonstrating reversibilityof C24 inhibition at 23 °C at pH 7.4
Assay conditions were as described in the text with [Kp2]/[Kpl] = 25:1 (0.05 mg of Kpl/assay). *, Under Ar(101 kPa); 0, under C2H4 (101 kPa); A, incubated underC2H4 (101 kPa) for 8 min, at which time C2H4 replaced byAr (101 kPa). The lines are least-squares fits to the datapoints, which are the means of duplicate determinations.
evolution under C2H4 and pC2H4 is the partial pressureof C2H4 (in kPa). The linear dependence provides no
evidence for multiple binding sites for C2H4. The ordinateintercepts are within experimental error of points A andB, which assume that at infinitepC2H4 H2 evolution is
completely suppressed (i.e. AH2 = rate of H2 evolutionunder Ar when pC2H4 = oo).
If Michaelis-Menten kinetics are assumed, with C2H4a competitive inhibitor of protons (or whichever proticsubstrate, S, yields H2) binding to Kpl, then the abscissaintercept in Fig. 4 is equal to - Km/K,2H4([S] + Km),where Kf2H4 is the apparent association constant forC2H4 binding to Kpl . The term KSm/([S] + Klm) is constantat a given pH, but cannot be calculated, since the natureof the substrate that yields H2 is uncertain and proticequilibria not directly associated with substrate bindingpreclude detailed analysis of pH-dependence of H2evolution rates.
Bearing in mind the limitations of simple steady-statekinetic analysis with an enzyme system as complicated asnitrogenase [see Lowe & Thorneley (1984b) for a detailedanalysis of Km2 and K,'2 dependencies on electron flux],Fig. 4 shows that C2H4 is a more potent inhibitor ofH2 evolution at low electron flux (K,2H4 = 88 kPa) thanat high electron flux (K,2H4 = 409 kPa). The higheraffinity for the less-reduced states of Kpl (Eo and E1) isperhaps not surprising, since C24 is the product ofC22 reduction and presumably dissociates from arelatively oxidized state ofKp 1 in the catalytic cycle. Theweak binding of C2H4 to Kpl under conditions of bothhigh and low electron flux is consistent with C2H4 notbeing a product inhibitor of C2H2 reduction undernormal assay conditions where C24 formation is linearwith time.The reversible nature of C24 inhibition of H2
evolution is shown by the data in Fig. 5. Linear timecourses for H2 evolution under Ar and C2H4 (both at101 kPa) were obtained with the rate under C2H4 being64% of that under Ar at a [Kp2]/[Kpl] ratio of 25: 1. Athird time course for H2 evolution was obtained byallowing assays to run under C2H4 (101 kPa) for 8 min,at which time C2H4 was removed by three rapid partialevacuations and back-fillings with Ar (total time lessthan 1 min). Assays were then allowed to continue for afurther 2, 4, 6 and 8 min under Ar (101 kPa) and the rateof H2 evolution was determined. The rate followingincubation under C24 was 1366 nmol/min per mg ofKpl, which compares with 1350 nmol/min per mg ofKpl for assays under Ar without pre-exposure to C24.Besides showing the reversible nature of C2H4 inhibition,the time courses are linear with no evidence for a lag orburst phase before the onset of C24 inhibition.
Table 1 shows that CO at 9 kPa did not relieve theinhibition of H2 evolution under either high- or low-electron flux conditions.
24 inhibition of H2 evolution could be due to aninhibition of total electron flux through the enzyme, or toredirection of electrons to yield a product other thanH2, such as methane or ethane. To explore thesepossibilities, we have measured S2042- oxidation andH2 evolution under Ar (101 kPa) or C2H4 (.101 kPa). Inthe experiment shown in Fig. 6, the correlation betweenH2 evolution and S2042- oxidation was 0.999, and thedata for assays under Ar or C24 lie on the same line.The slope of the composite line (1.04) indicates amaximum value for the excess of S2042- used over H2evolved of 4 %. Since under the conditions of thisexperiment the inhibition of H2 evolution by C2H4 was34%/, C2H4 must inhibit total electron flux rather thanact as a significant alternative electron acceptor.
Preliminary evidence for C2H4 inhibition of H2
1987
550
Klebsiella pneumoniae nitrogenase and ethylene
Table 1. Effect of electron flux, CO, C2H2 and H2 on H2 evolution and C2H6 formation under C24
For details see the text.
Product formed (nmol/min per mg of Kpl)
Component ratio ... [Kp2]/[Kpl] = 22:1 [Kp2]/[Kpl] = 1:21
evolution was obtained with cylinder C24 Although itwas possible that an inhibitor other than C2H4 itselfwas present, C2H4 purified as described above gave thesame degree of inhibition as the cylinder C2H4.The initial cylinder C2H4 contained readily detect-
able C2H6 (Fig. 1); this is not complexed by acidicHg(CI04)2, and 99 % of it was lost during purificationwith no change in inhibition. No other C2, C3 or C4alkane or alkene was detectable on gas chromatographyof the purified C2H4 on the alumina column described.C2H2, which is absorbed by acidic Hg(C104)2, was notdetectable in cylinder C24 and is in any case notregenerated with LiCl. 02 and N2 would have beenremoved by the purification procedure. Accordingly, webelieve that the possibility of an inhibitor other thanC2H4 being responsible for inhibition of H2 evolutioncan be virtually eliminated.
Electron transfer through the nitrogenase of K.pneumoniae to produce H2 is accompanied by thehydrolysis of MgATP. Under our assay conditions andwith a [Kp2]/[Kpl] ratio of 22:1, the ATP/2e ratio was4.3 under Ar, in agreement with previously publishedvalues (Imam & Eady, 1980). Under C2H4 (101 kPa),where H2 evolution was inhibited by 30 %, the ATP/2eratio was 4.4, indicating that C2H4, unlike CN- (Li et al.,1982) or CH3NC (Rubinson et al., 1983), did notuncouple ATP hydrolysis from electron transfer. Al-though CN- and CH3NC are similar to C2H4 in theirability to inhibit total electron flux, their effects on theATP/2e ratio are clearly different.Hardy et aL (1968) did not detect the reduction of
C24 to C2H6 with a nitrogenase from Azotobactervinelandii. Unfortunately, the precise conditions underwhich these experiments were done were not stated.Since the inhibition of H2 evolution by C2H4 withnitrogenase from K. pneumoniae only occurs at extremesof high and low electron flux (Fig. 3) and reaches amaximum of about 35% at 101 kPa C24 we have re-examined the reduction ofC24 to C2H6. The availabilityof C2H4 with a very low background of C2H6 made suchexperiments feasible (Fig. 1).
Ethane was identified as a product of C2H4 reductionsince its retention time on the alumina column wasidentical with that of authentic C2H6 (45 s). At a [Kp2]/[Kpl] ratio greater than 5:1, C2H6 was produced fromC2H4 (101 kPa) at 4.2 nmol/min per mg of Kpl. In atypical 15 min assay with 0.1 mg of Kpl, the C2H6produced under C2H4. (101 kPa) was 6.1 nmol over a
Vol. 247
4
_3/E
:1
c
.20
0cnl
0 1 2 3 4H2 evolution (,umol)
Fig. 6. Correlation between S2042- oxidation and H2 evolutionunder Ar and C2H4 (101 kPa) under high-electron-fluxconditions at 23 °C at pH 7.4
Assay conditions were as described in the text with[Na2S204] = 7.5 mM and [Kp2]/[Kpl] = 25:1 (0.1 mg ofKpl/assay). Assays for dithionite determination werestopped with formaldehyde. Under C2H4 (101 kPa) H2evolution was inhibited by 34 % and S2042- oxidation by35%. *, Under Ar (101 kPa); 0, under C2H4 (101 kPa).
background of 1.0 nmol of C2H6 in the C2H4. At [Kp2]/[Kpl] ratios less than 5:1, the rate of C2H6 formationdeclined as electron flux decreased (Fig. 7). At ratios lessthan [Kp2]/[Kpl] = 1:5, C2H6 formation decreased tolevels close to the analytical limit (see the legend to Fig.1). A comparison between Figs. 7 and 2 shows that,whereas C26 formation from C2H4 was constant at[Kp2]/[Kpl] ratios above 5:1, H2 evolution was stillincreasing with electron flux under either Ar or C2H4 at101 kPa.C2H4reduction accounted for at most l% of the total
electron flux through Kpl. Such a rate of C2H6production cannot readily be detected by measuringdifferences in either S2042- oxidation or H2 evolution.We have also detected C2H4 reduction in the Mo
nitrogenase of Azotobacter chroococcum at 5.3 nmol ofC2H6/min per mg of Acl from C2H4 (101 kPa) with[Ac2]/[Acl] = 12:1 at 30 'C. It is thus probable that thisreaction will prove to be a general property of Mo
551
G. A. Ashby, M. J. Dilworth and R. N. F. Thorneley
0.5
0~~~~~~~~~~~~~*' 0.4
ES 0.3c0
E 0.20
0.1
0 10 20 30 40[Kp2]/[Kpl ] ratio
Fig. 7. Effect of component protein ratio on the rate of C2H6formation from C24 (101 kPa) at 23 °C at pH 7.4
Assay conditions were as described in the text. At [Kp2]/[Kpl] > 1:1 assay mixtures contained 0.1 mg of Kpl andthe appropriate amounts of Kp2. At [Kp2]/[Kpl] < 1:1,assay mixtures contained 0.03 mg of Kp2 and the requisiteamounts of Kpl (data normalized to 0.1 mg of Kpl/assay).
nitrogenases. Although evidence for the reduction ofC2H4 to C2H6 was sought when the C22-reduction assaywas first developed (Dilworth, 1966; Schollhorn & Burris,1967; Hardy et al., 1968), probable reasons why it wasnot detected are (a) that it was masked by C2H6contamination of their C2H4, (b) that their pC2H4 wasinsufficient, and (c) their nitrogenases were operating attoo low electron flux. In addition, C2H6 formation fromC2H4 is strongly inhibited by C2H2 (Table 1), so thatC2H6 is not formed in CAH -reduction assays with Monitrogenases. Finally, the column packings commonlyused to separate C2H2 and C2H4 rapidly (Porapak N andPorapak T) do not resolve C2H4 and C2H6.A double-reciprocal plot of the rates ofC2H6 formation
as a function of the partial pressure of C2H4 in the rangebetween 62 and 295 kPa with a [Kp2]/[Kpl] = 22:1yielded an apparent KHm24 of 130 kPa. This value ismarkedly different from the Ki214 of 409 kPa calculatedfrom the H2 evolution data under similar conditions (seeFig. 4 and above). Thus, with a C24 pressure of approx.400 kPa, total electron flux and H2 evolution would beinhibited by only ca. 50 % whereas the rate of C26would be almost maximal.
Table 1 shows that H2 (101 kPa) did not inhibit norsignificantly enhance C2H6 formation from C24(101 kPa). In this respect C2H4 reduction to C2H6resembles the reduction of other substrates by nitro-genase, with the exception of N2, which is thought tobind by displacement of H2 and for which H2 is acompetitive inhibitor (Lowe & Thorneley, 1984a).CO is not a substrate for wild-type nitrogenase, but
inhibits the reduction of all substrates except H+; theonly other exception is the partial inhibition of H2formation by CO observed with the nitrogenase fromnifV mutants of K. pneumoniae (McLean et al., 1983).CO also relieves the inhibition of total electron fluxinduced by CN- and CH3NC (Li et al., 1982; Rubinsonet al., 1983). CO did not relieve the inhibition of H2evolution induced by C24 under either high- or low-electron-flux conditions (Table 1). However, CO (9 kPa)
completely inhibited C26 formation from C2H4(Table 1).The differential response to CO suggests that CO and
C2H4 can both be bound at the same time. Lowe et al.(1978) obtained evidence for two C2H2-binding sites onKpl from the dependence of amplitudes of transiente.p.r. signals on the partial pressure of C2H2. Burgess(1985) has reviewed other evidence for two classes of siteon the MoFe protein that bind reducible substrates and/or inhibitors such as CH3NC, HCN and CN-. We do notwish to speculate on the precise nature of the bindingsites except to note that two co-ordination positions onthe same metal atom could accommodate our observa-tions.Lowe et al. (1978) found no evidence for C24 binding
to Kpl in its resting state, i.e. no change in the e.p.rsignal with features at g = 4.3, 3.7 and 2.01. However,they did detect a new e.p.r. signal (gav. = 2.042) in thepresence of C2H4 under turnover conditions whenprotons were reduced to H2. This implies that CAH bindsto a transient reduced state of Kpl. The coupling ofelectron transfer to protonation to yield metal hydride(s)at the active site is a key postulate in the Lowe-Thorneleymodel for nitrogenase action (Lowe & Thorneley, 1984a,and references cited therein). The transient presence ofmetal hydrides may be linked to the inhibition of H2evolution by C2H4 and the formation of C2H6 byreaction of the type shown in Scheme 1. This Scheme isnot intended to be a comprehensive mechanism anddoes not include two binding sites for C2H2,C2H4 or CO binding to give a mixed CO plus C2H4species that does not evolve H2 or produce C2H6. It isbased on known chemistry in that reversible formationof metal-alkyl complexes from hydride-alkene com-plexes of transition-metal ions by a fl-eliminationmechanism is well understood (Alt & Eicher, 1982).Further reduction of the co-ordinated alkyl can yield freealkane (Nakamura & Otsuka, 1972).
There is some indirect evidence for C2H4 binding to amonohydride form of Kpl (E1H in Scheme 2 of Lowe &Thorneley, 1984a). Pre-steady-state rapid-quench experi-ments on C2H2 reduction show that three electrons aretransferred to Kpl before C2H4 is released (D. J. Lowe &R. N. F. Thorneley, unpublished work). This is consis-tent with C2H2 reduction via a metal-alkyl intermediatethat undergoes fl-elimination to yield a hydride-ethylenecomplex (E1H-C2H4) before dissociation of the product,C2H4. Evidence for this type of mechanism occurringwith molybdenum-alkyne complexes has recently beenobtained in these laboratories (R. L. Richards & N.Kashef, unpublished work) and has previously beendiscussed by Schrauzer et al. (1982). Restricted rotationabout the carbon-carbon bond of the metal-alkylintermediate due to steric constraints imposed by eitherthe protein or other ligands at the metal site couldaccount for the high proportion of cis-dideuteroethyleneproduced when C2H2 is reduced by nitrogenase in 2H20(Dilworth, 1966; Hardy et al., 1968; Kelly, 1969).However, reduction of propyne in 2H20 yields cis- andtrans-dideuteropropene in the ratio approx. 2: 1, indicat-ing cis-addition to bound alkene is not obligatory(McKenna et al., 1979).The failure to detect C2H6 as a product of C2H2
reduction does not exclude a metal-alkyl structure for acommon intermediate in C2H2 reduction to C2H4 and inC2H4 reduction to C2H6' Scheme 1 shows how this can
1987
552
Klebsiella pneumoniae nitrogenase and ethylene 553
H + C2H2 HICH
1 1C2H4 2e7/2H+
HI/CH2
---------I&SlowM CH TMC2CH *->M+C2H6mN""I I M-CH2-CH,H/e C
Scheme 1. Mechanism for C2 formation from C2H4 involving ametal-alkyl intermediate, and inhibition by C2H2,which is reduced only to C2H4
occur and also how C H2 (1 kPa) could strongly inhibitC26 formation from C2H4 (100 kPa) (see Table 1). Inthe presence of C22, the concentration of the metal-alkyl intermediate (M-CH2CH3) that slowly yieldsC2H6 on reduction is decreased by suppression ofits formation from the hydride-ethylene complex(MH-C2H4). This happens because C2H2 effectivelydisplaces C2H4 to yield the hydride-acetylene complex(MH-C2H2). Since the rate of reduction of the hyd-ride-acetylene complex (MH-C2H2) is slow [limitedby the dissociation of Kp20X (MgADP)2 from Kpl,k = 6.4 s-1, for each electron (Thorneley & Lowe, 1983)1,the intermediate MH-C2H2, and others not shown inScheme 1 (possibly metal hydride-carbynes and/or-carbenes), accumulate at the expense of M-CH2CH3and MH-C2H4.
Thorneley & Lowe (1983) compared the steady-staterates of H+, C2H2 and N2 reduction with the inde-pendently measured rate of protein dissociation (k=6.4s-') that occurs after each electron transfer fromKp2(MgATP)2 to Kp1 and concluded that proteindissociation limits the rate of product formation. Thelow rate of C2H6 formation and inhibition of totalelectron flux shows that this is not the case when C24 isthe substrate. Thus reduction of co-ordinated ethyl toyield C2H6 must be slow compared with the rate ofprotein dissociation (k = 6.4 s-1).The one-electron-reduced form ofKp 1 (E1H), which is
shown in Scheme 1 to bind C2H2 and C2H4, does notevolve H2 in the steady state or on quenching with acid(Lowe & Thorneley, 1984a). Formation of E1H-C2H4prevents formation of E2H2 (the first form ofKp 1 able toevolve H2 in Scheme 2 of Lowe & Thorneley, 1984a),thereby inhibiting H2 evolution and total electron flux.This is also consistent with the failure of H2 (101 kPa) toinhibit C2HA formation, since reversible binding of H2 isonly considered to be possible, when catalysed by N2, onthe more reduced forms E3H3 and E4H4 (Lowe &Thorneley, 1984a, b).The ability of C2H4 to inhibit total electron flux,
MgATP hydrolysis and H2 evolution while being reducedto C2H6 at a low rate makes it potentially a valuable toolfor investigating the active site of nitrogenase. H2evolution reactions normally prevent the generation of
no more than low concentrations of reduced forms ofKpl. C2H4 at only moderate pressures should, bypreventing H2 evolution, allow such reduced forms witheither C2H4 alone or C2H4 plus CO bound to reachconcentrations adequate for spectroscopic analysis. In-deed, under conditions of low electron flux, C24 willcompletely suppress H2 evolution while reduction toC2H6 occurs at an undetectable rate, with the result thatall the Kpl should be converted into a state with C2H4bound.
We thank Dr. R. L. Richards, Dr. D. J. Lowe and Dr. R. R.Eady for helpful discussions and Professor J. R. Postgate andProfessor G. J. Leigh for their comments on the manuscript.Azotobacter chroococcum nitrogenase component proteins werea gift from Dr. R. R. Eady and Mrs. M. Eldridge.
REFERENCESAlt, H. G. & Eicher, M. E. (1982) Angew. Chem. Int. Ed. Engl.
21, 205-206Burgess, B. K. (1985) in Metal Ions in Biology Series vol. 7:Molybdenum Enzymes (Spiro, T. G., ed.), pp. 161-219,Wiley-Interscience, New York
Dilworth, M. J. (1966) Biochim. Biophys. Acta 127, 283-294Dilworth, M. J., Eady, R. R., Robson, R. L. & Miller, R. W.
(1987) Nature (London) 327, 167-168Ennor, A. H. (1957) Methods Enzymol. 3, 850-856Hageman, R. V. & Burris, R. H. (1980) Biochim. Biophys.Acta 591, 63-75
Hardy, R. W. F., Holsten, R. D., Jackson, E. K. & Burns,R. C. (1968) Plant Physiol. 43, 1185-1207
Imam, S. & Eady, R. R. (1980) FEBS Lett. 110, 35-38Kelly, M. (1969) Biochim. Biophys. Acta 191, 527-540Koch, B. & Evans, H. J. (1966) Plant Physiol. 41, 1748-1750Li, J.-G., Burgess, B. K. & Corbin, J. L. (1982) Biochemistry
21, 4343-4402Lowe, D. J. & Thorneley, R. N. F. (1984a) Biochem. J. 224,
877-886Lowe, D. J. & Thorneley, R. N. F. (1984b) Biochem. J. 224,
895-901Lowe, D. J., Eady, R. R. & Thorneley, R. N. F. (1978)
Biochem. J. 173, 277-290Lowe, D. J., Thorneley, R. N. F. & Smith, B. E. (1985) in
Metallo Proteins (Harrison, P., ed.), vol. 1, pp. 207-249,Macmillan, London
McKenna, C. E., McKenna, M.-C. & Huang, C. W. (1979)Proc. Natl. Acad. Sci. U.S.A. 76, 4773-4777
McLean, P. A., Smith, B. E. & Dixon, R. A. (1983) Biochem.J. 211, 589-597
Nakamura, A. & Otsuka, S. (1972) J. Am. Chem. Soc. 94,1886-1894
Orme-Johnson, W. H. (1985) Annu. Rev. Biophys. Chem. 14,419-459
Robson, R. L., Eady, R. R., Richardson, T. H., Miller, R. W.,Hawkins, M. & Postgate, J. R. (1986) Nature (London) 322,388-390
Rubinson, J. F., Corbin, J. L. & Burgess, B. K. (1983)Biochemistry 22, 6260-6268
Sch6llhorn, R. & Burris, R. H. (1967) Proc. Natl. Acad. Sci.U.S.A. 58, 213-216
Schrauzer, G. N., Hughes, L. A. & Strampach, N. (1982) inProceedings Int. Conf. Chemistry and Uses of Mo 4th(Berry, H. F. & Mitchell, P. C. H., eds.), pp. 145-149, ClimaxMolydenum Co., Ann Arbor
Smith, K. A. & Dowdell, R. J. (1973) J. Chromatogr. Sci. 11,655-658
Vol. 247
554 G. A. Ashby, M. J. Dilworth and R. N. F. Thorneley
Smith, K. A. & Restall, S. W. (1971) J. Soil Sci. 22, 430-433
Stephens, P. J. (1985) in Metal Ions in Biology Series vol. 7:Molybdenum Enzymes (Spiro, T. G., ed.), pp. 117-159,Wiley-Interscience, New York
Thorneley, R. N. F. & Lowe, D. J. (1983) Biochem. J. 215,393-403
Thorneley, R. N. F. & Lowe, D. J. (1984a) Biochem. J. 224,886-894
Thorneley, R. N. F. & Lowe, D. J. (1984b) Biochem. J. 224,903-909
Thorneley, R. N. F. & Lowe, D. J. (1985) in Metal Ions inBiology Series vol. 7: Molybdenum Enzymes (Spiro, T. G.,ed.) pp. 221-284, Wiley-Interscience, New York
Wherland, S., Burgess, B. K., Stiefel, E. L. & Newton, W. E.(1981) Biochemistry 20, 5132-5140
Young, R. E., Pratt, H. K. & Biale, J. B. (1952) Anal. Chem.24, 551-555