96
Laboratory investigation of mitochondrial disease Johannes A. Mayr („Hans“) Department of Paediatrics Paracelsus Medical University, Salzburg [email protected] www.mito-center.org
| Date post: | 09-Dec-2018 |
| Category: |
Documents |
| Upload: | phamkhuong |
| View: | 215 times |
| Download: | 0 times |

Laboratory investigation of
mitochondrial disease
Johannes A. Mayr („Hans“)
Department of PaediatricsParacelsus Medical University, Salzburg
www.mito-center.org


Mitochondria in disease
• Inherited metabolic disorders
(1/5000)
• Neurodegenerative disorders
(Parkinson, Alzheimer, etc.)
• Tumours
• Aging
• …

Reactions in mitochondria leading to
ATP from pyruvate
Mitochondrial Energy Metabolismdefinition
>100 enzymes

Ou
ter M
em
bran
e
Cytosol
Lactate
NADHNAD+
Mitochondrial Matrix
Inner Membrane
Metabolite-
Protein-ADP/ATP-
Redox-Transport
NADH
NADH
NADH
Krebs-cycle
FADH2 FAD
ATP ADP+Pi
Oxidative phosphorylation
V
H+
H+
IIIH+
I
NADH NAD+
IIcyt.c
H+
IV
H2O O2
Q
PDHC NADHAcetyl-CoA
PyruvateGlucoseGlycolysis
PyruvateATP
Fatty acids,Ketone bodies

Glycolysis, anaerobic2 ATP/glucose
Oxidation in mitochondriaca. 30 ATP/glucose
Energy Metabolismenergetic yield: anaerobic versus aerobic

Ou
ter M
em
bran
e
Cytosol
Lactate
NADHNAD+
Mitochondrial Matrix
Inner Membrane
Metabolite-
Protein-ADP/ATP-
Redox-Transport
NADH
NADH
NADH
Krebs-cycle
FADH2 FAD
ATP ADP+Pi
Oxidative Phosphorylation
V
H+
H+
IIIH+
I
NADH NAD+
IIcyt.c
H+
IV
H2O O2
Q
PDHC NADHAcetyl-CoA
PyruvateGlucoseGlycolysis
PyruvateATP
Fatty acids,Ketone bodiesPyruvate oxidation
Krebs-cycle
Respiratory chain ATP-synthesis
Transmembrane-transport
Lipide-pattern(e.g. cardiolipin)
Cofactors(e.g. coenzyme Q)
DEFECTSMotilityfission, fusion
Toxic substances(e.g. H2S in ETHE1)
Anaplerosis

EnzymesPyruvate oxidationKrebs cycleRespiratory chainATP synthesis
Biogenesis of mitochondriaMitochondrial replication, transcription, translationImport and assembly of proteinsTransport (import+export) of metabolitesFission and fusionLipid metabolismRegulation of the amount of mitochondria
Mitochondrial Energy Metabolismpossible different kinds of defects
CofactorsThiamine, biotin, FeS-Clusters, lipoic acid, etc.
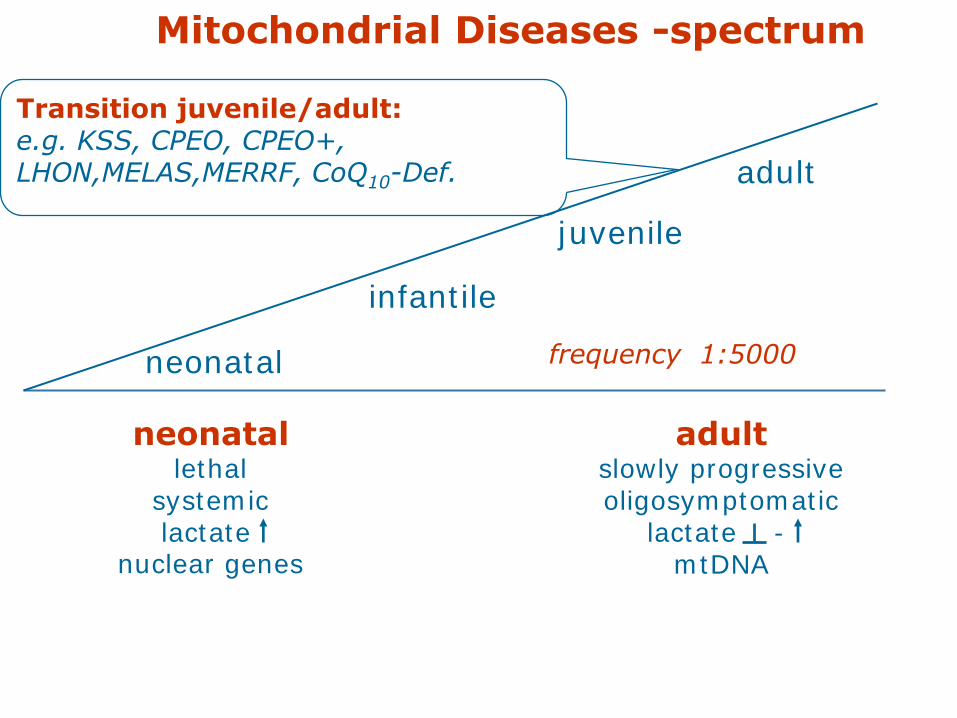
neonatallethal
systemiclactate
nuclear genes
adultslowly progressiveoligosymptomatic
lactate -mtDNA
Mitochondrial Diseases -spectrum
juvenile
adult
infantile
neonatal frequency 1:5000
Transition juvenile/adult:e.g. KSS, CPEO, CPEO+,LHON,MELAS,MERRF, CoQ10-Def.
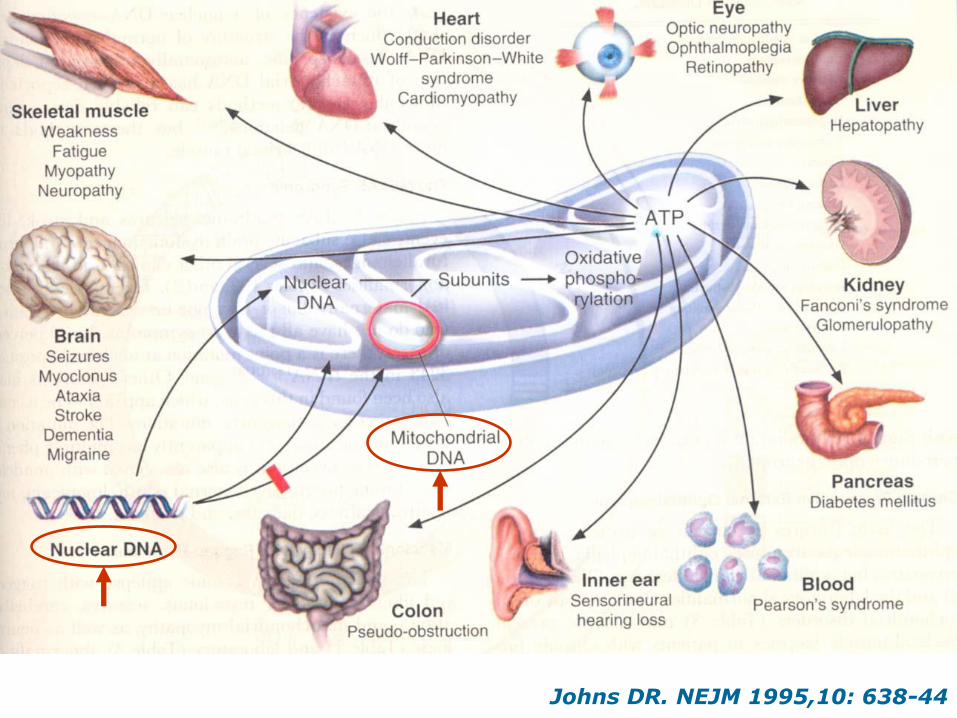
Johns DR. NEJM 1995,10: 638-44

Genetics of mitochondrial disorders
Nuclear genome (>98% of proteins)
Inheritance: Mendelian (autosomal
recessive, autosomal dominant), X-linked,
sporadic, somatic
Mitochondrial genome (13 proteins)
Inheritance: Maternal, sporadic, somatic

mitochondrium
I II III IV V
nucleus
endopl. reticulum
~1000 mitochondrial
proteins
nukl. DNA
mtDNAreplication
mtDNA
transcription
translationassembly
13 mitochondrial proteins22 tRNAs2 rRNAs

oute
r m
em
bra
ne
inner membrane
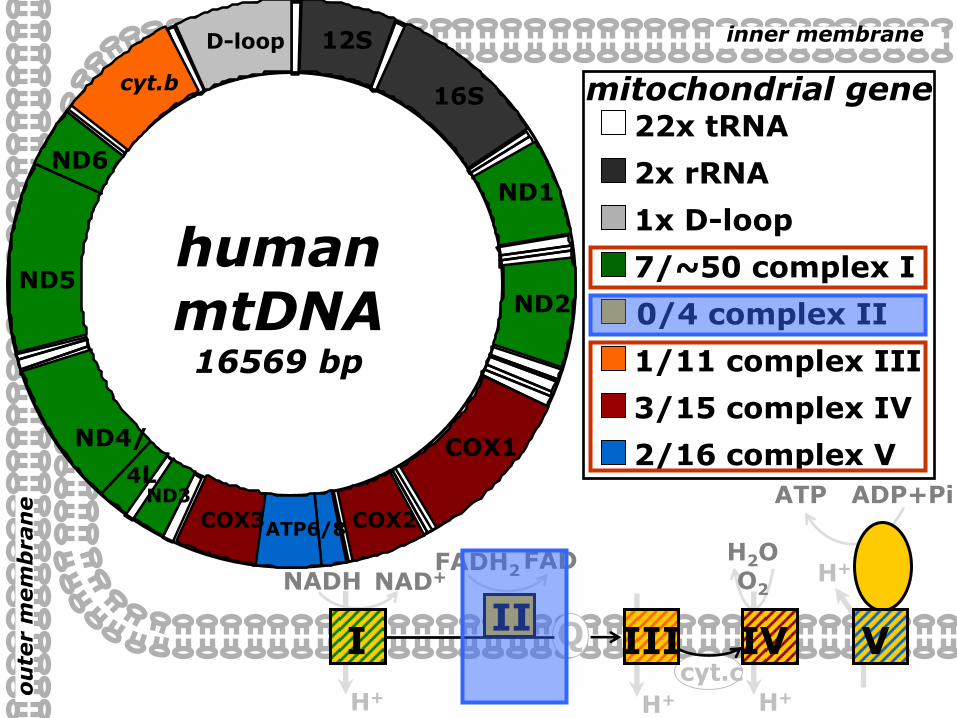
humanmtDNA16569 bp
22x tRNA
mitochondrial genescyt b
12S
16S
2x rRNA
D-loop
1x D-loop
H+H+
NADH NAD+
0/4 complex II
II
FADH2 FAD
cyt.c
Q
H+
H2O O2H+
ATP ADP+Pi
ATP6/8
2/16 complex V
V
COX1
COX2COX3
3/15 complex IV
IV
ND1
ND2
ND3
ND4/
4L
ND5
ND6
7/~50 complex I
I
cyt.b
1/11 complex III
III

Special features of mtDNAmutations
• Inherited from the oozyte
• Random segregation of mutations in
the germ line
• Variable copy number per cell
• Cell cycle independent replication
• Quantitative mutations (depletion)
• Heteroplasmy (wildtype and mutations)
• Threshold phenomenon
• Higher mutation rate

Middle Europe: H, V South Europe: J, K
Nord Europe: T, U, X Middle East: J, N
Africa: L, L1, L2, L3, L3* Asia: A, B, C, D, E, F, G (M is made up of C, D, E, and G) Native American: A, B, C, D and a small proportion of X
Haplogrupsof mitochondrial DNA

Mutations of the mtDNAheteroplasmy - gradual mutation load
100% normal 10% normal

0102030405060708090
100
0 20 40 60 80 100Mutation load [%]
Fu
ncti
on
[%
]Mutations of the mtDNA
heteroplasmy - threshold
mtDNA depletion

Problems we face in the diagnosisof mitochondrial diseases
Family doctorPediatrician
Neuropediatrician
Laboratory 1, screening routine
Laboratory 2, Biochemistry
Metabolic specialist (second opinion)
(Rebiopsy)
Internet knowledge
PATIENT
Clinical geneticist
RadiologistFirst information
Genetic counselling
„definite“„probable“„possible“DIAGNOSIS
Final information
Laboratory 3, Genetics
Histochemistry, EM
Biopsy

Guidelines issued by the Working Group on Pediatric metabolic disorders (APS)
„Diagnostic and treatment approaches to mitochondriopathies
in children and adolescents“
*According to the prescription of the Guideline-Manual of AWMF and ÄZQ
http://www.uni-duesseldorf.de/WWW/AWMF/ll/index.html
Results of three conferences of experts (n=17)adopted by the APS and Society for Neuropediatrics
Current status of the guideline development:Development stage 2*

Suspicion of mitochondriopathy
Anamnesis
Bodily fluids - basal invest.
Physical examination including
neurological status
MITO?
MITO?
CNS?, PNS?
Muscle?
MRI (MRS), cerebrospinal fluid-lactate
Abandonment of
mito. diagnostics
CNS, PNS,
muscles Syndrome?
Encephalo-(myo)-neuropathies,
no defined syndrome
Syndrome-specific
Genetics diagnostics?
Genetically defined
syndrome?
Mutation
investigation
Blood?
Genetics in respect
of muscle biopsy
Genetically
defined
syndrome?
Specific diagnostics
Blood, urine?
1
Apparatus-based organ examinations,
stress tests, expanded laboratory
diagnostics
Muscle biopsy (+skin)
Biochemistry, histology
Unambiguous
diagnosis?
Specific genetics
appropriate?
Diagnosis
1
2
1
Resumé
MITO?
Re-enter further on
Apparatus-based organ examinations,
expanded laboratory diagnostics
Biopsy of the affected tissue
(+skin, muscles)
Non-neurological,
other organs
1
MITO?
MITO?Abandonment of
mito. diagnostics
Abandonment of
mito. diagnostics
Abandonment of
mito. diagnostics
Therapy
Abandonment of
mito. diagnostics
CNS-, PNS-, Muscle-involvement
MITO?Abandonment of
mito. diagnostics
Genetics, blood
Specific diagnostics, blood, urine
positive?
Specific genetics –
appropriate? Genetics, tissue
1
Biopsy of theaffected tissue? Note: We refer to the problems
associated with a biopsy
2
Syndrome with central nervous system,
peripheral nervous system, muscular involvement
2

Suspicion of mitochondriopathy
Anamnesis
Bodily fluids - basal invest.
Physical examination including
neurological status
MITO?
MITO?
CNS?, PNS?
Muscle?
MRI (MRS), cerebrospinal fluid-lactate
Abandonment of
mito. diagnostics
CNS, PNS,
muscles Syndrome?
Encephalo-(myo)-neuropathies,
no defined syndrome
Syndrome-specific
Genetics diagnostics?
Genetically defined
syndrome?
Mutation
investigation
Blood?
Genetics in respect
of muscle biopsy
Genetically
defined
syndrome?
Specific diagnostics
Blood, urine?
1
Apparatus-based organ examinations,
stress tests, expanded laboratory
diagnostics
Muscle biopsy (+skin)
Biochemistry, histology
Unambiguous
diagnosis?
Specific genetics
appropriate?
Diagnosis
1
2
1
Resumé
MITO?
Re-enter further on
Apparatus-based organ examinations,
expanded laboratory diagnostics
Biopsy of the affected tissue
(+skin, muscles)
Non-neurological,
other organs
1
MITO?
MITO?Abandonment of
mito. diagnostics
Abandonment of
mito. diagnostics
Abandonment of
mito. diagnostics
Therapy
Abandonment of
mito. diagnostics
CNS-, PNS-, Muscle-involvement
MITO?Abandonment of
mito. diagnostics
Genetics, blood
Specific diagnostics, blood, urine
positive?
Specific genetics –
appropriate? Genetics, tissue
1
Biopsy of theaffected tissue? Note: We refer to the problems
associated with a biopsy
2
Syndrome with central nervous system,
peripheral nervous system, muscular involvement
2
www.aps-med.de
www.mito-center.org
Guidelines for diagnostics and therapy

Diagnostic cascade
Suspicious symptoms
SyndromesOrgan
involvement
Metabolites
Ergometry, loading tests
Investigation for organ involvement
Biopsy
Biochemistry Morphology
Molecular geneticsmtDNA, nuclear Genes

Diagnostic cascade
Suspicious symptoms
SyndromesOrgan
involvement
Metabolites
Ergometry, loading tests
Investigation for organ involvement
Biopsy
Biochemistry Morphology
Molecular geneticsmtDNA, nuclear Genes

SyndromesSuspicious symptoms
Organ involvement
Cardiomyopathies
(dilatative,
hypertrophic, Barth)
Myopathies
Hepatopathies
(mtDNA depletion)
Nephropathies
(Fanconi syndrom)
Peripheral
neuropathies
Endocrinopathies
Clinics of mitochondrial diseases
Leigh
Depletion
Pearson
Alpers
Barth
MELAS
MERRF
NARP
LHON
KSS
CPEO
MNGIE
Muscular hypotonia
Exercise intolerance
Convulsions
Ataxia and other
cerebellar
symptoms
Brainstem
involvement
Hearing loss
Short stature
Ptosis ...

Metabolites
Suspicious symptoms
SyndromesOrgan
involvement
Metabolites
Loading tests, ergometry Ergometry, loading tests
Investigation for organ involvement
Biopsy
Biochemistry Morphology
Molecular geneticsmtDNA, nuclear Genes
Plasma/Serum:lactate, pyruvate, alaninecreatinethymidine, deoxyuridine3-hydroxybutyrate/acetoacetateCSF:lactate, alanineUrine:methylmalonic acid3-methylglutaconic acidethylmalonic acidKrebs cycle metabolites

Ou
ter M
em
bran
e
Cytosol
Lactate
NADHNAD+
Mitochondrial Matrix
Inner Membrane
Metabolite-
Protein-ADP/ATP-
Redox-Transport
NADH
NADH
NADH
Krebs-cycle
FADH2 FAD
ATP ADP+Pi
Oxidative Phosphorylation
V
H+
H+
IIIH+
I
NADH NAD+
IIcyt.c
H+
IV
H2O O2
Q
PDHC NADHAcetyl-CoA
PyruvateGlucoseGlycolysis
PyruvateATP
Fatty acids,Ketone bodies

Lactate in physiologyCori cycle
Gerty and Carl CoriNobel Prize in Physiology or Medicine 1947
anaerobic ATP in muscle

Lactate in physiologyCori + alanine cycle
Cave: Plasma lactate and alanine are in correlation with liver function

Boumezbeur et al., J Neurosci. 2010;30:13983-91
Lactate in physiologyfuel in brain metabolism
10% under basal plasma lactate conditions60% at supraphysiological plasma lactate concentrations
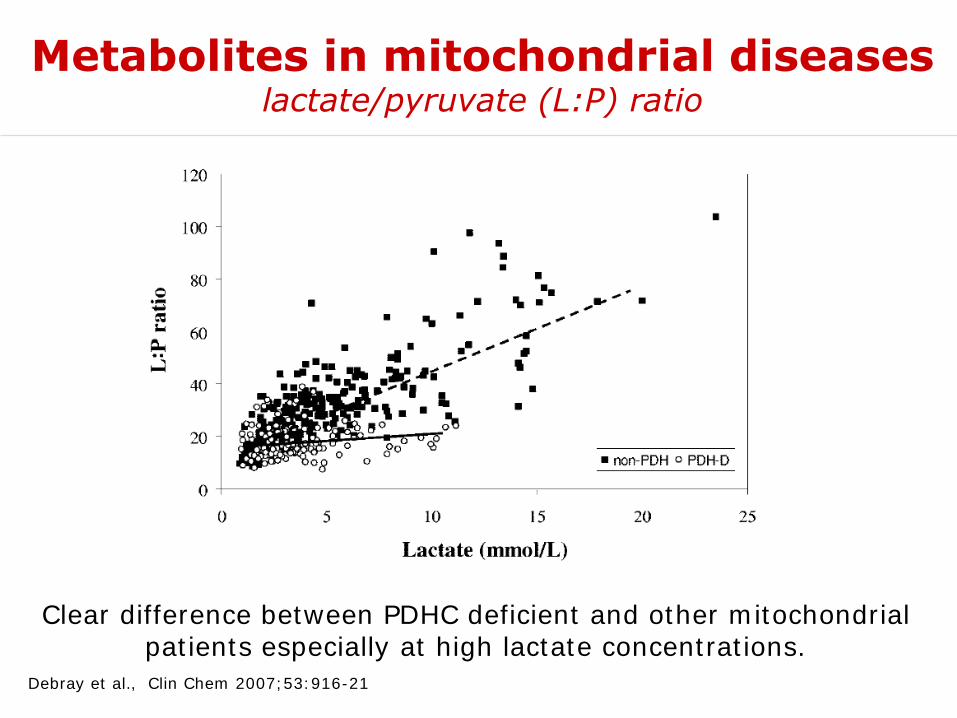
Debray et al., Clin Chem 2007;53:916-21
Metabolites in mitochondrial diseaseslactate/pyruvate (L:P) ratio
Clear difference between PDHC deficient and other mitochondrial patients especially at high lactate concentrations.

Ou
ter M
em
bran
e
Cytosol
Lactate
NADHNAD+
Mitochondrial Matrix
Inner Membrane
Metabolite-
Protein-ADP/ATP-
Redox-Transport
NADH
NADH
NADH
Krebs-cycle
FADH2 FAD
ATP ADP+Pi
Oxidative phosphorylation
V
H+
H+
IIIH+
I
NADH NAD+
IIcyt.c
H+
IV
H2O O2
Q
PDHC NADHAcetyl-CoA
PyruvateGlucoseGlycolysis
PyruvateATP
Fatty acids,Ketone bodies

Shaham et al., Proc Natl Acad Sci U S A 2010;107:1571-5
Metabolites in mitochondrial diseases
H+
IIIH+
I
NADH NAD+
cyt.cH+
IV
H2O O2
Q
rotenone antimycin A

Shaham et al., Proc Natl Acad Sci U S A 2010;107:1571-5
Metabolites in mitochondrial diseaseselevation of creatine
creatine elevation also found by: Boenzi et al., J Inherit Metab Dis 2011;34(Suppl. 3):S160

Metabolites in mitochondrial diseaseselevation of creatine
Shaham et al., Proc Natl Acad Sci U S A 2010;107:1571-5

Metabolites in mitochondrial diseasesthymidine, deoxyuridine in MNGIE
MNGIE ... Mitochondrial NeuroGastroIntestinal Encephalomyopathy (TYMP)
Martí et al., Clin Chem 2004;50:120-4
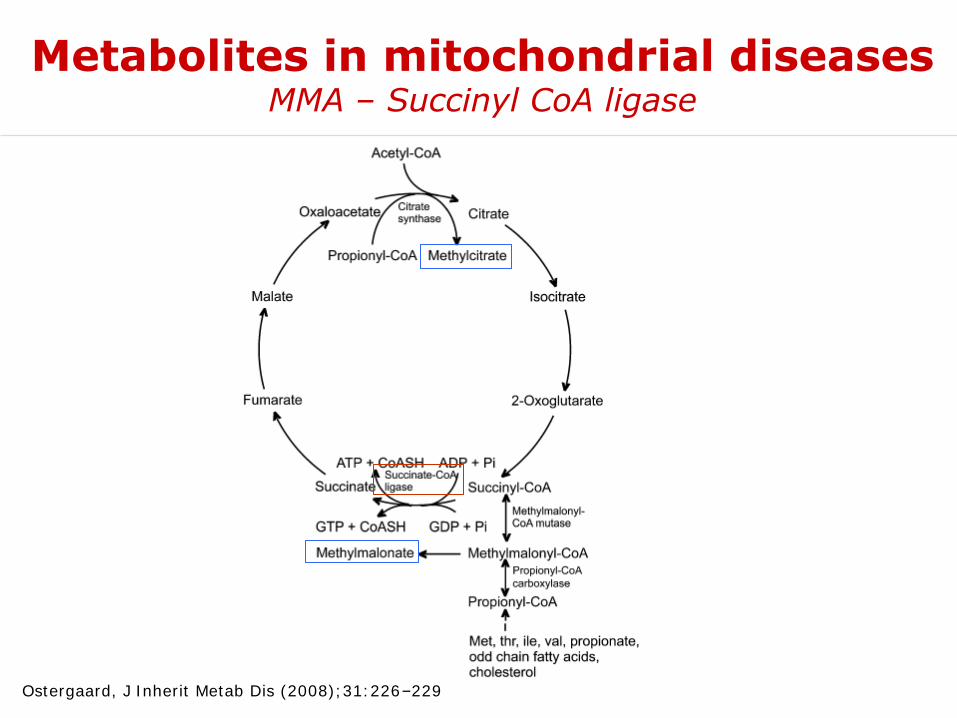
Metabolites in mitochondrial diseasesMMA – Succinyl CoA ligase
Ostergaard, J Inherit Metab Dis (2008);31:226–229

Metabolites in mitochondrial diseases3-methylglutaconic acid
Type IV 3-MGA: TMEM70, ATP5EPOLG, SUCLA2, mtDNA deletions
Wortmann, J Inherit Metab Dis (2010)
Loading tests, ergometry
Suspicious symptoms
SyndromesOrgan
involvement
Metabolites
Ergometry, loading tests
Investigation for organ involvement
Biopsy
Biochemistry Morphology
Molecular geneticsmtDNA, nuclear Genes
Ergometry (spiroergometry)Exercise physiology (near infrared
spectroscopy, 31PMRS)
Fasting testPre, postprandial lactateGlucose-, alanine- etc. pyruvate-loading

Organ involvement
Suspicious symptoms
SyndromesOrgan
involvement
Metabolites
Ergometry, loading tests
Investigation for organ involvement
Biopsy
Biochemistry Morphology
Molecular geneticsmtDNA, nuclear Genes
CNS: MRI/MRS, PET?Heart: ECHO/ECG Eye: ERGMuscle Nerve: EMG/NLG etcKidney, endocrine system etc.

Lactate
6 a old boy with myopathy1H-magnetic resonance spectroscopy

A3243G mutation (MELAS)
4 years later: „stroke-like episode“

Feedback questions 1
1. Mitochondrial diseases have been described by the statement “... any disease, any organ, any age …”. Please comment this statement.
2. Which unique features of the mitochondrial genome do you remind?
3. Which metabolites are useful for the diagnosis of mitochondrial disease?
Please prepare answers together with your neighbour (10 min).

Biopsy
Suspicious symptoms
SyndromesOrgan
involvement
Metabolites
Ergometry, loading tests
Investigation for organ involvement
Biopsy
Biochemistry Morphology
Molecular geneticsmtDNA, nuclear Genes
Muscle: Open – Needle ?Fresh – frozen ?
Skin - fibroblastsOther tissues (Liver, heart muscle...)

Tissuefor the analysis of mitochondrial energy metabolism
1. Clinically affected tissue
2. If not possible, other tissue (muscle, fibroblasts)
Amount: Depends on investigations planned (biochemistry 20-100 mg, histology 10-50 mg, electron microscopy 10-50 mg, myoblast cultivation 10 mg, back-up 50 mg), 200 mg muscle

Muscle biopsyNeedle-biopsy under vacuum
Ø = 5 mm
Typical yield: 70-200 mg muscle

approx. 40 mg muscle
~2 rice grains
Needle biopsy:•Higher acceptance for a biopsy
•No anaesthesia in severely affected children•Miniaturized biochemical procedure necessary

Methodsanalysis of mitochondrial energy metabolism
Histological & histochemical staining, electron microscopy
Functional investigations (unfrozen cells)
Enzyme investigations
Blue Native electrophoresis
Immunological methods (western blot, immunohistochemistry, dip sticks)
Membrane potential
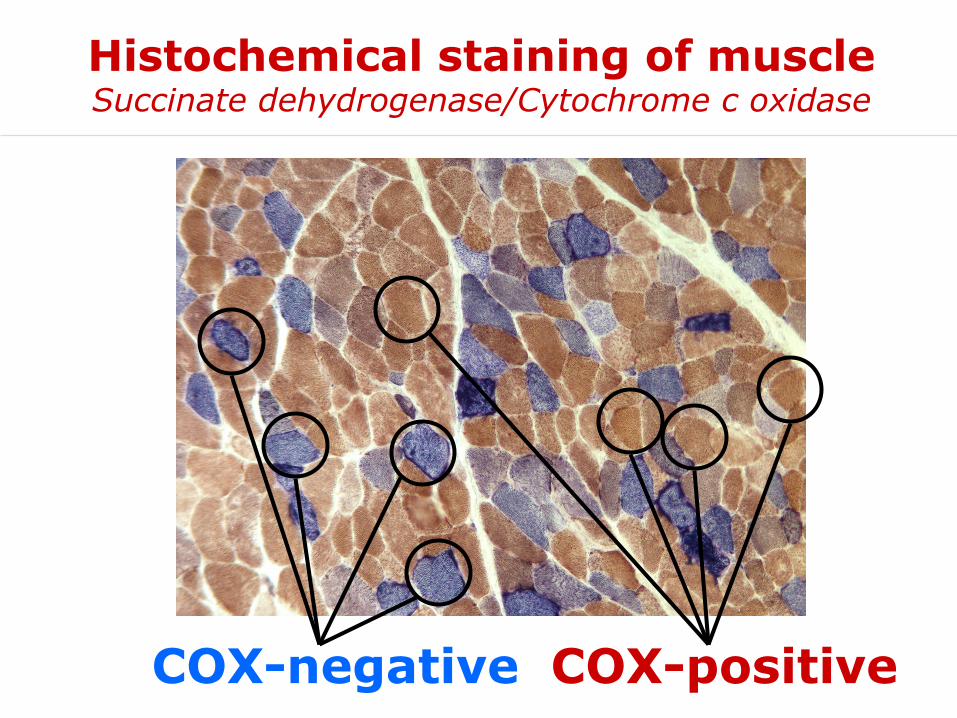
Histochemical staining of muscleSuccinate dehydrogenase/Cytochrome c oxidase
COX-negative COX-positive

Methods:
Measurement of
O2 (polarography, fluorimetry)
ATP (endpoint, luciferase-mediated)
CO2 (14C-labelled substrates)
Functional investigationsunfrozen biopsy tissue

ou
ter m
em
bran
e
Cytosol
mitochondrial matrix
inner membrane
NADH
NADH
NADH
Krebs-cycle
FADH2 FAD
ATP
oxidative phosphorylation
V
H+
H+
IIIH+
I
NADH NAD+
IIcyt.c
H+
IV
H2O O2
Q
PDHC NADHacetyl-CoA
pyruvatepyruvate
malate
14CO2
ADP+PiADP+Pi
[1-14C]-pyruvate
malate
ADP, Pi

Functional investigationsunfrozen biopsy tissue
Advantages:
• Measurement of transport processes
• Total activity of all enzymes of the mitochondrial energy metabolism
• Investigation of the Krebs cycle
• First results within few hours

Functional analysis – patientreport substrate oxidation
Activity in relation toprotein
Activity in relation tomarker enzyme
Clinical synopsis of patient 1:Neonatal onset hypertrophic cardiomyopathy, muscular hypotonia. Elevation of lactate up to 10 mmol/L.Excretion of 3-methylglutaconic acid in urine.Muscle biopsy at the age of 2 months.
II. Substrate oxidation analysis form 600g supernatant of fresh muscle homogenate[nmol/h/mg protein] normal range [nmol/h/mUnit CS] normal range
[1-14C]pyruvate+malate 127 6 263 - 900 P+M/CS 0,56 6 1,54 - 3,55
[1-14C]pyruvate+carnitine 298 302 - 856 P+C/CS 1,32 6 1,65 - 3,66
[1-14C]pyruvate+malate-ADP 48 32 - 102 P+M-ADP/CS 0,21 0,21 - 0,41
[1-14C]pyruvate+malate+CCCP 565 304 - 889 P+M+C/CS 2,50 1,31 - 3,11
[1-14C]pyruvate+malate+atractyloside 110 5 19 - 90 P+M+A/CS 0,49 0,16 - 0,55
[U-14C]malate+pyruvate+malonate 123 6 282 - 874 M+P+Mal/CS 0,54 6 1,56 - 3,87
[U-14C]malate+acetylcarn.+malonate 176 6 273 - 678 M+AC+Mal/CS 0,78 6 1,16 - 2,82
[U-14C]malate+acetylcarn.+arsenite 159 156 - 378 M+AC+As/CS 0,70 0,57 - 1,52
[1,4-14C]succinate+acetylcarnitine 47 6 167 - 488 G+AC/CS 0,21 6 1,36 - 2,53

Functional analysis – ATP synthesissubstrate oxidation of intact muscle-mitochondria
Patients with defective ATP-synthesis
pyruvate + malate + CCCP
pyruvate + malate + ADP
Respiratory chain(w/o ATP-synthesis)
Oxidative phosphorylation(incl. ATP-synthesis)
CCCP/ADP-activation
rati
o0
1
2
3
4
5
6
7
8
9
P1

Synthesis of ATPvia oxidative phosphorylation
Fo
H+
PiC
H+ Pi
ANT
ADP ATP
F1
Pi ADP+ ATP
ATP synthesisapparatus
1. F1Fo ATP synthase
2. Adenine-nucleotide-translocator, ANT
3. Mitochondrial phosphate-carrier, PiC

340 nm
ATP ADP + P
ATPase
pyruvate PEP
pyruvate kinase
lactate dehydrogenase
lactateNAD
NADH
Enzyme investigationse.g. oligomycin-sensitive ATPase (complex V)

Tris pH 8.0 40 mM
MgCl2 5 mM
KCl 10 mM
lactate dehydrogenase 4 U/ml
pyruvate kinase 4 U/ml
NADH 0.2 mM
PEP 2 mM
ATP 0.5 mM
BSA 0.1 %
antimycin A 1 µM
FCCP 3 µM
Conc.Substance
Oligomycin-sensitive ATPaseassay mixture

• 10 s sonification to open the mitochondrial membranes
• Follow NADH decrease
at 340 nm 3-5 min.
• Inhibition with
oligomycin.
Oligomycin-sensitive ATPaseassay mixture

Oligomycin-sensitive ATPasedecrease at 340 nm
start measurement after sonification
inhibition with oligomycin

• 1st derivative:
Total activity
Oliogomycin inhibited
*specific enzyme activity:oligomycin-sensitive ATPase
*
Oligomycin-sensitive ATPasedecrease at 340 nm

Enzyme activitiesreport
I. Enzyme activities form 600g supernatant of fresh muscle homogenate[mUnit/mg protein] normal range [mUnit/mUnit CS] normal range
Citrate synthase (CS) 226 150 - 325
Complex I (CI) 18 6 28 - 76 CI/CS 0,08 6 0,17 - 0,31
Complex I+III (C13) 60 64 - 218 C13/CS 0,27 0,24 - 0,81
Complex II (CII) 78 39 - 102 CII/CS 0,35 0,23 - 0,41
Complex II+III (C23) 119 93 - 180 C23/CS 0,52 0,55 - 0,67
Complex III (CIII) 564 426 - 762 CIII/CS 2,49 2,12 - 3,09
Cytochrome c oxidase (COX) 583 452 - 889 COX/CS 2,58 2,59 - 3,12
Complex V (CV) 5 6 70 - 397 CV/CS 0,02 6 0,47 - 1,47
Pyruvate dehydrogenase (PDHC) 10,5 6,1 - 19,8 PDHC/CS 0,046 0,034 - 0,079
1. Decrease of complex V (oligomycin-sensitive ATPase)2. Less severe: defect of complex I
Activity in relation toprotein
Activity in relation tomarker enzyme

Blue Native PAGE muscle tissue
Complex I -
F1Fo-ATP synthase -
Complex III -
Cytochrome c Oxidase -
C P1
1. Decrease of complex V (oligomycin-sensitive ATPase)2. Less severe: defect of complex I

Genetic cause of the diseasehomozygosity mapping
control
patient
INTRON 2 EXON 3
Cízková et al., Nat Genet 2008;40:1288-90
TMEM70, mitochondrial transmembrane protein

Activitie
s a
t 25°C
-37°C
[U
/g N
CP
])
1
10
100
1000Lab 1 (37°C)
Lab 2 (30°C)
Lab 3 (37°C)
Lab 4 (30°C)
Lab 5 (25°C)
Lab 6 (30°C)
Lab 7 (37°C)
Lab 8 (25°C)
Lab 9 (30°C)
Lab 10 (37°C)
Lab 11 (25°C)
Lab 12 (30°C)
C I
C I+
III
C II+
III
CO
X
CS
AT
Pase
NC
P
C III
C II
PD
HC
6
2
Cave: comparison of enzyme activitiesinternational multicenter trial of 12 diagnostic laboratories
Gellerich et al., Mitochondrion 2004;4:427-39

Feedback questions 2
1. Which tissue should be analysed to identify a mitochondrial disease?
2. Which biochemical methods are used for the identification of mitochondrial disorders?
3. What are advantages of the investigation of intact mitochondria?
Please prepare answers together with your neighbour (10 min).

• Girl, normal birth after uneventful pregnancy.• Neonatal hearing test revealed congenital deafness• 8 weeks, lack of head control, truncal hypotonia.• Plasma lactate 1.3 to 8.9 mmol/L (normal 0.5-2.2 mmol/L).
• Aminoaciduria, glucosuria, renal loss of phosphate and urate indicated proximal tubulopathy.
• MRI at the age of 10 weeks was normal.• 3 months, poor sucking and recurrent vomiting necessitated continuous nasogastric tube feeding
• She died under palliative care at the age of 4 month.
Case Aclinical synopsis

Case Ainvestigation of a muscle biopsy
• Histological: ragged red fibers and fat accumulation, severe decrease of cytochrome c oxidase staining
-

% o
f n
orm
al
16%22%
101%
31%23%
11%
34%
0%
100%
com
plex
I/C
S
com
plex
I+III/CS
com
plex
II/CS
com
plex
II+
III/CS
com
plex
III/C
S
cyto
chro
me c ox
idas
e/CS
oligom
ycin-s
ens. A
TPas
e/CS
Case Aenzymes in relation to citrate synthase
ou
ter
mem
bran
einner membrane
humanmtDNA16569 bp
22x tRNA
mitochondrial genecyt b
12S
16S
2x rRNA
D-loop
1x D-loop
H+H+
NADH NAD+
0/4 complex II
II
FADH2FAD
cyt.c
Q
H+
H2O O2
H+
ATP ADP+Pi
ATP6/8
2/16 complex V
V
COX1
COX2COX3
3/15 complex IV
IV
ND1
ND2
ND3
ND4/
4L
ND5
ND6
7/~50 complex I
I
cyt.b
1/11 complex III
III

control
mitochondrial DNA nuclear DNA
patientmtDNA-content:
4% of control
Case Aquantitation of mtDNA content

Case Amutation in RRM2B
Acham-Roschitz et al., Mol Genet Metab 2009;98:300-4
Yen et al. Clin Cancer Res 2003; 9:4304-8
NDP dNDP
Ribonucleotide reductase(cytosolic enzyme)
ribonucleotides
deoxyribonucleotides

MNGIE
Copeland WC. Annu Rev Med 2008,59:309-24
Encephalomyopathykidney
Encephalo-myopathy,mild MMA
Myopathy Hepato-cerebral
symptoms
SUCLG1
Mitochondrial DNA depletion

• A girl was born after normal pregnancy.• Free walking at the age of 17 months.• At 7 years referred to gastroenterologist due to loss of appetite, weakness and slight growth retardation.
• Discrete muscular atrophy.• Slightly elevated transaminases, lactate dehydrogenase and creatine kinase.
• Bicycle ergometry showed an endurance performance of 70% of age-matched controls, increase in lactate from 4.0 to 8.1 mmol/l
• Myopathic pattern in electromyographic investigation.
Case Bclinical synopsis

Case Bmuscle biopsy investigations
Gömöri staining:ragged-red fibres
Cytochrome c oxidase (brown)
succinate dehydrogenase
(blue)
Electron microscopy:large mitochondria with tubular cristae

Case Bmuscle biopsy investigations
Gömöri staining:ragged-red fibres
Cytochrome c oxidase (brown)
succinatedehydrogenase
(blue)
Electron microscopy:large mitochondria with tubular cristae

ou
ter
mem
bran
einner membrane
humanmtDNA16569 bp
22x tRNA
mitochondrial genecyt b
12S
16S
2x rRNA
D-loop
1x D-loop
H+H+
NADH NAD+
0/4 complex II
II
FADH2FAD
cyt.c
Q
H+
H2O O2
H+
ATP ADP+Pi
ATP6/8
2/16 complex V
V
COX1
COX2COX3
3/15 complex IV
IV
ND1
ND2
ND3
ND4/
4L
ND5
ND6
7/~50 complex I
I
cyt.b
1/11 complex III
III

Case B - Single fibre PCRmutation in a mitochondrial tRNA gene
Mayr et al., Neuromuscul Disorders 2006;16:874-77
DHPLC
mitochondrialtRNAGlu
sequenceanalysis
single muscle fibre
dissection+
mutation quantification

Patient C – Case reportfamiliy of Turkish origin, non consanguineous
Findings
Sex
Birth weight [g]
Hypertrophic CMP
Muscular hypotonia
Hyperlactatemia [mmol/l]
Poor feeding
Death at the age of
Organic and amino acids,
carnitine, acylcarnitine,
Enzymes: OXPHOS, PDHC
patient C2
female
2870
+
+
4-17
+
9 months
normal
normal
patient C
female
2600
+
+
4-15
+
6 months
normal
normal
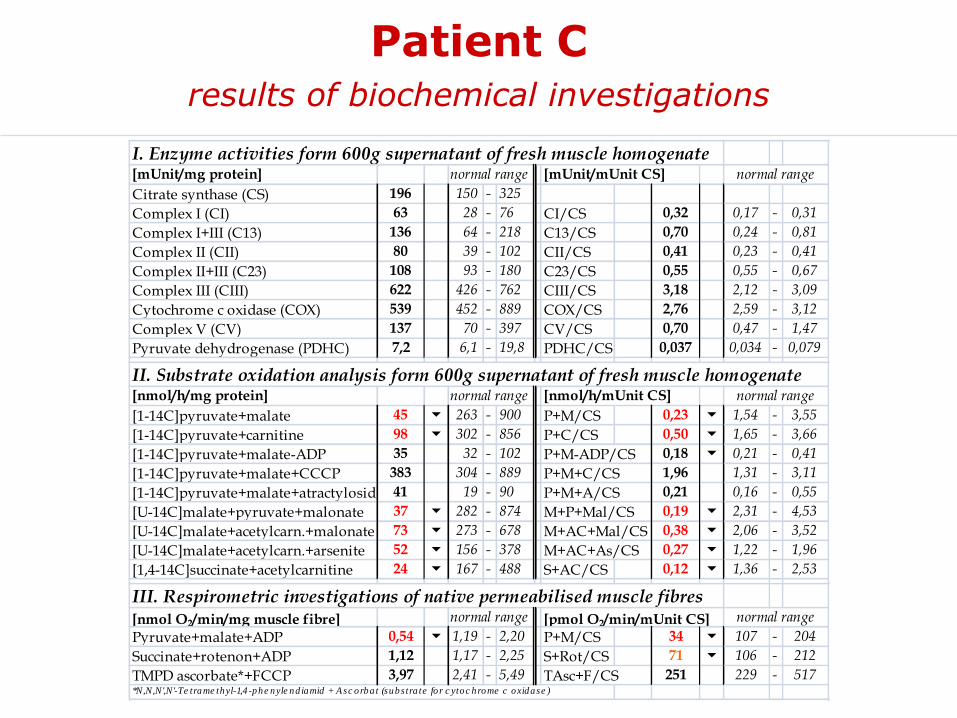
Patient Cresults of biochemical investigations
I. Enzyme activities form 600g supernatant of fresh muscle homogenate[mUnit/mg protein] normal range [mUnit/mUnit CS] normal range
Citrate synthase (CS) 196 150 - 325
Complex I (CI) 63 28 - 76 CI/CS 0,32 0,17 - 0,31
Complex I+III (C13) 136 64 - 218 C13/CS 0,70 0,24 - 0,81
Complex II (CII) 80 39 - 102 CII/CS 0,41 0,23 - 0,41
Complex II+III (C23) 108 93 - 180 C23/CS 0,55 0,55 - 0,67
Complex III (CIII) 622 426 - 762 CIII/CS 3,18 2,12 - 3,09
Cytochrome c oxidase (COX) 539 452 - 889 COX/CS 2,76 2,59 - 3,12
Complex V (CV) 137 70 - 397 CV/CS 0,70 0,47 - 1,47
Pyruvate dehydrogenase (PDHC) 7,2 6,1 - 19,8 PDHC/CS 0,037 0,034 - 0,079
II. Substrate oxidation analysis form 600g supernatant of fresh muscle homogenate[nmol/h/mg protein] normal range [nmol/h/mUnit CS] normal range
[1-14C]pyruvate+malate 45 6 263 - 900 P+M/CS 0,23 6 1,54 - 3,55
[1-14C]pyruvate+carnitine 98 6 302 - 856 P+C/CS 0,50 6 1,65 - 3,66
[1-14C]pyruvate+malate-ADP 35 32 - 102 P+M-ADP/CS 0,18 6 0,21 - 0,41
[1-14C]pyruvate+malate+CCCP 383 304 - 889 P+M+C/CS 1,96 1,31 - 3,11
[1-14C]pyruvate+malate+atractyloside 41 19 - 90 P+M+A/CS 0,21 0,16 - 0,55
[U-14C]malate+pyruvate+malonate 37 6 282 - 874 M+P+Mal/CS 0,19 6 2,31 - 4,53
[U-14C]malate+acetylcarn.+malonate 73 6 273 - 678 M+AC+Mal/CS 0,38 6 2,06 - 3,52
[U-14C]malate+acetylcarn.+arsenite 52 6 156 - 378 M+AC+As/CS 0,27 6 1,22 - 1,96
[1,4-14C]succinate+acetylcarnitine 24 6 167 - 488 S+AC/CS 0,12 6 1,36 - 2,53
III. Respirometric investigations of native permeabilised muscle fibres[nmol O2/min/mg muscle fibre] normal range [pmol O2/min/mUnit CS] normal range
Pyruvate+malate+ADP 0,54 6 1,19 - 2,20 P+M/CS 34 6 107 - 204
Succinate+rotenon+ADP 1,12 1,17 - 2,25 S+Rot/CS 71 6 106 - 212
TMPD ascorbate*+FCCP 3,97 2,41 - 5,49 TAsc+F/CS 251 229 - 517*N,N,N',N'-Te trame thyl-1,4 -phe nyle nd iamid + A sc orba t (subs tra te fo r c ytoc hrome c oxidase )

Functional analysis muscle vs. fibroblasts
ATP synthesis in patient C
Patient C: Tissue specific defect of thesynthesis of ATP
CC
CP
-vs.
AD
P-s
tim
ula
tio
n
Muscle Fibroblasts
Patient C
0
1
2
3
4
5
6
7
8
9
PC PC-2

Synthesis of ATPvia oxidative phosphorylation
Fo
H+
PiC
H+ Pi
ANT
ADP ATP
F1
Pi ADP+ ATP
ATP synthesisapparatus
1. F1Fo ATP synthase
2. Adenine-nucleotide-translocator, ANT
3. Mitochondrial phosphate-carrier, PiC

Mitochondrial Phosphate CarrierSLC25A3 gene on chromosome 12
3AIsoform A
3BIsoform B
“mutually exclusivealternative splicing”
Heart, skeletal muscle
Fibroblasts and other tissues

Patient 4: Mutation in the Exon 3Amitochondrial phosphate carrier, SLC25A3
A highly conserved glycine is replaced by glutamic acid.
Mayr et al., Am J Hum Genet 2007;80:478-84
Patient C Exon-3A E
Human PiC Exon-3A YSCDYGSGRFFILCGLGGIISCGTTHTALVPLDLVKCRMQ 94
Human PiC Exon-3B YSCEFGSAKYYALCGFGGVLSCGLTHTAVVPLDLVKCRMQ 93
Xenopus trop. FSCEYGSGTFYAYCGFGGILSCGLTHTAVVPLDLVKCRLQ 92
Drosphila mela. DSCEFGSTKYFALCGIGGILSCGTTHTFVVPLDLVKCRLQ 91
S.pombe KTLQLYTPQYYGLCTLGGLLACGTTHSAITPLDLIKCRKQ 52
S.cerevisiae-PIC2 RKIQLYTKEFYATCTLGGIIACGPTHSSITPLDLVKCRLQ 47
Arabidopsis thal. KGIEMYSPAFYAACTFGGILSCGLTHMTVTPLDLVKCNMQ 107
S.cerevisiae-MIR1 -AIPQYSVSDYMKFALAGAIGCGSTHSSMVPIDVVKTRIQ 46
homology : : :.* :.** ** :.*:*::* . *
16
543
2
Growth on YP glycerol (3%)1: wild type2: mp (mir1pic2)3: mp hPICA-E (mutant)4: mp hPICA-G (wild type)5: mp yMIR1-E (mutant)6: mp yMIR1-G (wild type)

• A boy, born after uneventful pregnancy at term.• At 2 years he became dizzy and vertiginous and developed a gait disturbance but recovered.
• Several similar episodes during the next years with spontaneous remission.
• 10 ½ years admitted to hospital with loss of speech• Seizure with clonic jerks of his arms.• Cerebellar and bulbar affection with dysarthria, intention tremor, confusion and episodic ataxia.
• Ophthalmoplegia and nystagmus were noted and there was spasticity right accented.
• MRI which was normal before showed now changes in the white matter of the cerebellum.
Case Dclinical synopsis

• MR-Spectroscopy of the cerebellum revealed a soft decrease of N-acetylaspartate und an increase of choline, lactate was normal.
• Respiratory insufficiency needing artificial ventilation for 6 days.
• He recovered and is mentally normal, attending high school, present age 17 years.
• Lactate was elevated up to 4.4 mmol/L in plasma and 3.3 mmol/L in the cerebrospinal fluid during the crisis otherwise normal.
• Histologically normal muscle biopsy.
Case Dclinical synopsis (continued)

Case Dmuscle biopsy investigations
I. Enzyme activities form 600g supernatant of fresh muscle homogenate[mUnit/mg protein] normal range [mUnit/mUnit CS] normal range
Citrate synthase (CS) 244 150 - 338
Complex I (CI) 40 28 - 76 CI/CS 0,16 0,14 - 0,35
Complex I+III (C13) 103 49 - 218 C13/CS 0,42 0,24 - 0,81
Complex II (CII) 86 39 - 102 CII/CS 0,35 0,23 - 0,41
Complex II+III (C23) 164 65 - 180 C23/CS 0,67 0,30 - 0,67
Complex III (CIII) 342 351 - 939 CIII/CS 1,40 1,45 - 3,76
Cytochrome c oxidase (COX) 540 306 - 889 COX/CS 2,21 1,45 - 3,47
Complex V (CV) 170 86 - 257 CV/CS 0,70 0,42 - 1,26
Pyruvate dehydrogenase (PDHC) 8,2 5,3 - 19,8 PDHC/CS 0,034 0,026 - 0,079
II. Substrate oxidation analysis form 600g supernatant of fresh muscle homogenate[nmol/h/mg protein] normal range [nmol/h/mUnit CS] normal range
[1-14C]pyruvate+malate 183 6 263 - 900 P+M/CS 0,75 6 1,54 - 3,55
[1-14C]pyruvate+carnitine 195 6 302 - 856 P+C/CS 0,80 6 1,65 - 3,66
[1-14C]pyruvate+malate-ADP 54 32 - 102 P+M-ADP/CS 0,22 0,21 - 0,41
[U-14C]malate+pyruvate+malonate 235 6 282 - 874 M+P+Mal/CS 0,96 6 1,56 - 3,87
[U-14C]malate+acetylcarn.+malonate 414 273 - 678 M+AC+Mal/CS 1,70 1,16 - 2,82
[U-14C]malate+acetylcarn.+arsenite 225 156 - 378 M+AC+As/CS 0,92 0,57 - 1,52
[U-14C]glutamate+acetylcarnitine 337 167 - 488 G+AC/CS 1,38 0,90 - 2,06
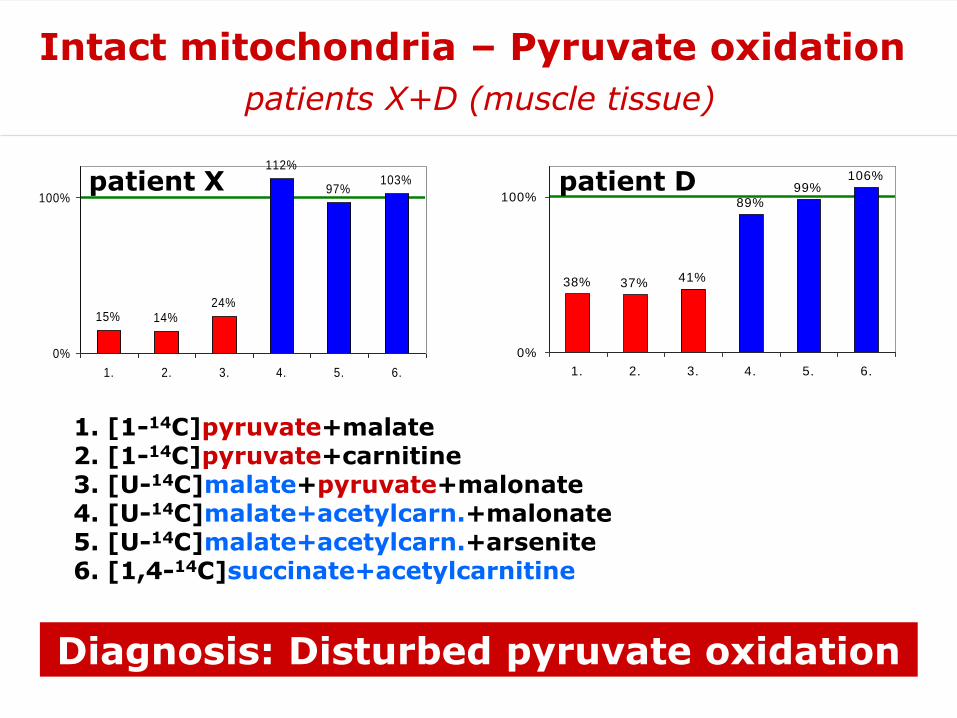
1. [1-14C]pyruvate+malate2. [1-14C]pyruvate+carnitine3. [U-14C]malate+pyruvate+malonate4. [U-14C]malate+acetylcarn.+malonate5. [U-14C]malate+acetylcarn.+arsenite6. [1,4-14C]succinate+acetylcarnitine
Diagnosis: Disturbed pyruvate oxidation
15% 14%24%
112%
97%103%
0%
100%
1. 2. 3. 4. 5. 6.
patient X
38% 37% 41%
89%99%
106%
0%
100%
1. 2. 3. 4. 5. 6.
patient D
Intact mitochondria – Pyruvate oxidation
patients X+D (muscle tissue)

15% 14%24%
112%
97%103%
0%
100%
1. 2. 3. 4. 5. 6.
patient X
38% 37% 41%
89%99%
106%
0%
100%
1. 2. 3. 4. 5. 6.
patient D
142%
162%
119%
172%
82%
107% 108%
143%
16%
0%
100%
CS C I C I+III C II C II+III C III COX ATPase PDHC
PDHC: 16% residual activityE1 Gen: CGA->GGA, Arg263Gly
105%
83%
101%
115%
143%
70%
92% 96%
84%
0%
100%
CS C I C I+III C II C II+III C III COX ATPase PDHC
PDHC-activity is normal (84%)E1 ,E1β,E2,E3BP,PDP1: normal
Intact mitochondria – Pyruvate oxidation
patients X+D (muscle tissue)

C1 C1 P3 P3
E2 -E3BP -
E1α -E1β -
su OSCP -
- 132
- 78
- 45
- 32
- 18
Protein [µg]
C2 PD P2 C3
5 510 10 10 10 10 10 kDa
Western blot analysis
subunits of pyruvate dehydrogenase
Normal pattern of PDHC subunits in patient D.

E2
E3BP
Smolle et al., J Biol Chem 2006;281:19772-80
30x E1α subunit30x E1β60x E2 12x E3BP12x E3PD-Kinase (PDK)PD-Phosphatase (PDP)PDP regulatory protein
total size: ~10 MDalton!
PDHC - structure

[TPP]
CO2
CH3-C-COOH
=O
[CH3-C=TPP]-
OH
[CH3-C-S-LipSH]
=O
CH3-C-S-CoA
=O
CoA-SH
[Lip(SH)2]
[LipS2]
FAD
FADH2
NAD+
NADH + H+
pyruvate acetyl-CoA
E1E2
E3
Pyruvate dehydrogenase
reaction mechanism
PDHC reaction depends on cofactors

0
1
2
3
PD
HC
ac
t. -
TP
P
[mU
nit
s/g
pro
tein
]
0.00
0.05
0.10
0.15
0.20
rati
o:
PD
HC
ac
tivit
y -
/+T
PP
Patients ControlsPatients Controls
Pyruvate dehydrogenase
thiamine pyrophosphate dependency

TPP TMP
Thiamine
Control
Patient D
Standard
TPP
TMP Thiamine
TPPThiamine
Pyruvate dehydrogenase
thiamine pyrophosphate dependency
Muscle 600g sup. [nmol/g prot.]
TPP TMP Thiamine
P2 8.8 0.1 1.5P3 9.5 0.1 3.0Patient D 9.3 0.2 1.9Controls (n=9)Mean ± SD 58.7 ± 12.6 1.1 ± 0.4 0.9 ± 1.4
range 41.6 - 81.6 0.4 - 1.7 0.2 - 4.4
Blood [nmol/L] TPP TMP Thiamine
P3 68.0 2.2 21.4P4 50.4 1.2 5.3Patient D 96.9 18.1 67.5Controls (n=10)Mean ± SD 190.9 ± 41.5 6.2 ± 1.3 10.7 ± 6.1
range 132.2 - 271.2 4.1 - 8.8 5.0 - 26.4
Deficiency of TPP in muscle and blood.

Thiamine(TMP)
Thiamine
TPK1
SLC19A2Pla
sm
a m
em
bra
ne
SLC19A3
TPP
TPP
TransketolaseTPP
TPP
BCKDHTPP
PDHCTPP
α-KGDHTPP
SLC25A19
ATP AMP
Mitochondrion

Affected Individuals P3+P4 P1+P2 Patient D
Missense mutation P H S
H. sapiens 33 LWNKALLRACADGGANRLYDI .. LVSTSNTYDGS 224
M. musculus 33 LWKKALLRACADGGANHLYDL .. LVSTSNTYDGS 224
D. rerio 33 LWSKAQIRACADGGANHLYRL .. LVSTSNTYEDH 224
D. melanogaster 116 LWKNAAVRCAVDGGSNHWRDF .. MVSTSNTYATE 316
A. thaliana 40 LWEHAKLRLCADGGANRIYDE .. LISTSNLVKEE 239
S. cerevisiae 56 IWKLHDLKVCADGAANRLYDY .. RVSSSNRFVGD 293
Clostr. tetani 20 ELKDSDIIIAADKGAEALYKC .. GLGVSNEIKEN 195
Staph. aureus 18 AKSNEGKWGGVDRGALILLKH .. TLTISNEIESL 193
Consensus . .* .: : **
Mutation analysisthiamine pyrophosphokinase, TPK1 gene
Mayr et al., Am J Hum Genet 2011;89:806-12

GAPDH
TPKIsoform aIsoform b
Prot. [µg] 5 10 5 10 10 510 510 5
P2 C1 P3 C2 Patient D
Western blot analysisthiamine pyrophosphokinase
Mayr et al., Am J Hum Genet 2011;89:806-12

• “... any disease, any organ, any age ...”
• enzymes – biogenesis – cofactors
• mitochondrial DNA – nuclear DNA
• systematic clinical and laboratory analyses
• functional investigations: enzymes, proteins, intact mitochondrial membranes
Summarydefects in the mitochondrial energy metabolism

















