Perspectives in Diabetes Lipotoxicity in the Pathogenesis of Obesity-Dependent NIDDM Genetic and Clinical Implications Roger H. Unger We review evidence that increased tissue levels of fatty acyl CoA cause the p-cell abnormalities of nondiabetic obesity and ultimately result in obesity-dependent diabe- tes. Nondiabetic obesity in Zucker rats is characterized by hypersecretion of insulin at normal fasting and sub- fasting glucose concentrations. This is a result of P-cell hyperplasia and increased low K m glucose usage and oxidation. These abnormalities, the hyperinsulinemia, the hyperplasia of P-cells, i.e., its in vitro equivalent, enhanced bromodeoxyuridine incorporation, and the in- creased low 7ir m glucose usage can be induced by culturing normal islets with 2 mmol/l free fatty acids (FFAs). Once obese Zucker diabetic fatty rats become diabetic, glucose- stimulated insulin secretion (GSIS) is absent and P-cell GLUT2 reduced. Islet triglyceride (TG) content is in- creased 10-fold, probably reflecting increased FFA deliv- ery (plasma FFA levels >1.5 mmol/l) beginning about 2 weeks before the onset of diabetes. These p-cell abnor- malities, GSIS loss, GLUT2 loss, and TG accumulation, are prevented by reducing plasma FFAs by caloric restric- tion and by nicotinamide injection. The loss of GSIS and the accumulation of TGs, but not the GLUT2 loss, can be induced in vitro in normal islets cultured in a 2 mmol/l FFA-containing medium, but prediabetic islets seem far more vulnerable to FFA-induced functional impairment and TG accumulation. It is proposed that in uncompli- cated obesity, increased lipid availability (FFA levels <1.5 mmol/l) induces both hyperinsulinemia and insulin resistance in parallel fashion, thereby maintaining nor- moglycemia. A further increase in substrate overload impairs p-cell compensation for insulin resistance and hyperglycemia appears. Diabetes 44:863-870, 1995 From the Department of Internal Medicine, Gilford Laboratories, Center for Diabetes Research at the University of Texas Southwestern Medical Center at Dallas, and the Department of Veterans Affairs Medical Center at Dallas, Texas. Address correspondence and reprint requests to Dr. Roger H. Unger, University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Blvd., Dallas, TX 75235-8854. Received for publication 21 December 1994 and accepted in revised form 5 April 1995. BrdU, bromodeoxyuridine; FACoA, fatty acyl CoA; FFA, free fatty acid; GSIS, glucose-stimulated insulin secretion; NIDDM, non-insulin-dependent diabetes mel- litus; TG, triglyceride; ZDF, Zucker diabetic fatty. T he U.S. faces a health problem of alarming propor- tions as a consequence of a progressive increase in the incidence of obesity. As of 1990, 30% of Amer- icans were obese, up from 25% in 1980 (1). Given the well-established linkages between obesity and age and non-insulin-dependent diabetes mellitus (NIDDM) (2), it fol- lows that the prevalence of overt NIDDM in the U.S. must be increasing in proportion to the population's expanding girth and advancing age. Recently estimated to be approaching 7% (3), the true prevalence of NIDDM may be even higher. The morbidity and spiraling costs generated by this disease make a more complete understanding of its pathogenesis a goal of the highest priority. Fundamental to such understanding is the issue of the pathogenic role of insulin resistance in obesity and NIDDM (4,5). Epidemiological evidence that obesity exerts a dose effect on the incidence of NIDDM raises the possibility that the insulin resistance of obesity is sufficient in and of itself to cause the disease. In Japan, where obesity is rare, the prevalence of NIDDM has been estimated to be 1% (6), yet at least 30% of massively obese Sumo wrestlers develop NIDDM in later life (H. Hirose, personal communication). Similarly, among Americans weighing >46% of ideal body weight, the prevalence of fasting hyperglycemia and abnor- mal oral glucose tolerance approaches 50% (7). On the other hand, 50% of insulin-resistant individuals never develop overt NIDDM. This suggests that insulin resistance by itself is not sufficient to cause NIDDM and that a second defect is required (4,5,8-10). Because the onset of overt NIDDM is invariably accompanied by a loss of glucose-stimulated insu- lin secretion (GSIS) (11-16) and, at least in rodents, by a reduction in p-cell GLUT2 (9,10,13-16), glucose incompe- tence of p-cells has been viewed as the second defect (14). If it is, the familial clustering of NIDDM (17) would imply that P-cell glucose incompetence, like insulin resistance, is inher- ited. If we accept this premise, we must next determine the relationship of the glucose incompetence to the insulin resistance. Are they the result of two independent but coinciding primary defects, one intrinsic to p-cells, the other extrinsically expressed in insulin's target tissues; or is one defect primary and the other secondary to it; or are both defects secondary to a single primary abnormality? Herein DIABETES, VOL. 44, AUGUST 1995 863 Downloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 March 2022
Transcript
Perspectives in DiabetesLipotoxicity in the Pathogenesis of Obesity-DependentNIDDMGenetic and Clinical ImplicationsRoger H. Unger
We review evidence that increased tissue levels of fattyacyl CoA cause the p-cell abnormalities of nondiabeticobesity and ultimately result in obesity-dependent diabe-tes. Nondiabetic obesity in Zucker rats is characterizedby hypersecretion of insulin at normal fasting and sub-fasting glucose concentrations. This is a result of P-cellhyperplasia and increased low Km glucose usage andoxidation. These abnormalities, the hyperinsulinemia,the hyperplasia of P-cells, i.e., its in vitro equivalent,enhanced bromodeoxyuridine incorporation, and the in-creased low 7irm glucose usage can be induced by culturingnormal islets with 2 mmol/l free fatty acids (FFAs). Onceobese Zucker diabetic fatty rats become diabetic, glucose-stimulated insulin secretion (GSIS) is absent and P-cellGLUT2 reduced. Islet triglyceride (TG) content is in-creased 10-fold, probably reflecting increased FFA deliv-ery (plasma FFA levels >1.5 mmol/l) beginning about 2weeks before the onset of diabetes. These p-cell abnor-malities, GSIS loss, GLUT2 loss, and TG accumulation,are prevented by reducing plasma FFAs by caloric restric-tion and by nicotinamide injection. The loss of GSIS andthe accumulation of TGs, but not the GLUT2 loss, can beinduced in vitro in normal islets cultured in a 2 mmol/lFFA-containing medium, but prediabetic islets seem farmore vulnerable to FFA-induced functional impairmentand TG accumulation. It is proposed that in uncompli-cated obesity, increased lipid availability (FFA levels<1.5 mmol/l) induces both hyperinsulinemia and insulinresistance in parallel fashion, thereby maintaining nor-moglycemia. A further increase in substrate overloadimpairs p-cell compensation for insulin resistance andhyperglycemia appears. Diabetes 44:863-870, 1995
From the Department of Internal Medicine, Gilford Laboratories, Center forDiabetes Research at the University of Texas Southwestern Medical Center atDallas, and the Department of Veterans Affairs Medical Center at Dallas, Texas.
Address correspondence and reprint requests to Dr. Roger H. Unger, Universityof Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Blvd., Dallas,TX 75235-8854.
Received for publication 21 December 1994 and accepted in revised form 5 April1995.
The U.S. faces a health problem of alarming propor-tions as a consequence of a progressive increase inthe incidence of obesity. As of 1990, 30% of Amer-icans were obese, up from 25% in 1980 (1). Given
the well-established linkages between obesity and age andnon-insulin-dependent diabetes mellitus (NIDDM) (2), it fol-lows that the prevalence of overt NIDDM in the U.S. must beincreasing in proportion to the population's expanding girthand advancing age. Recently estimated to be approaching 7%(3), the true prevalence of NIDDM may be even higher. Themorbidity and spiraling costs generated by this disease makea more complete understanding of its pathogenesis a goal ofthe highest priority.
Fundamental to such understanding is the issue of thepathogenic role of insulin resistance in obesity and NIDDM(4,5). Epidemiological evidence that obesity exerts a doseeffect on the incidence of NIDDM raises the possibility thatthe insulin resistance of obesity is sufficient in and of itself tocause the disease. In Japan, where obesity is rare, theprevalence of NIDDM has been estimated to be 1% (6), yet atleast 30% of massively obese Sumo wrestlers developNIDDM in later life (H. Hirose, personal communication).Similarly, among Americans weighing >46% of ideal bodyweight, the prevalence of fasting hyperglycemia and abnor-mal oral glucose tolerance approaches 50% (7). On the otherhand, 50% of insulin-resistant individuals never develop overtNIDDM. This suggests that insulin resistance by itself is notsufficient to cause NIDDM and that a second defect isrequired (4,5,8-10). Because the onset of overt NIDDM isinvariably accompanied by a loss of glucose-stimulated insu-lin secretion (GSIS) (11-16) and, at least in rodents, by areduction in p-cell GLUT2 (9,10,13-16), glucose incompe-tence of p-cells has been viewed as the second defect (14). Ifit is, the familial clustering of NIDDM (17) would imply thatP-cell glucose incompetence, like insulin resistance, is inher-ited.
If we accept this premise, we must next determine therelationship of the glucose incompetence to the insulinresistance. Are they the result of two independent butcoinciding primary defects, one intrinsic to p-cells, the otherextrinsically expressed in insulin's target tissues; or is onedefect primary and the other secondary to it; or are bothdefects secondary to a single primary abnormality? Herein
DIABETES, VOL. 44, AUGUST 1995 863
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
LIPOTOXICITY OF OBESITY-DEPENDENT NIDDM
we review evidence that in obesity-dependent NIDDM theinsulin resistance of target tissues and the glucose incompe-tence of P-cells are both caused by a single abnormalityextrinsic to (B-cells. We examine the idea that the extrinsicabnormality is an increase in the delivery of free fatty acids(FFAs) to tissues. This includes the target tissues of insulinand the islets of Langerhans.
ADIPOCYTE-B-CELL RELATIONSHIPS IN NORMOGLYCEMICOBESITYPhysiological versus pathophysiological hyperlipaci-demia. Normally FFAs provide an alternative fuel to glucoseto spare glucose for cerebral requirements and during pro-longed fasting to conserve body proteins that would other-wise be used as a source of gluconeogenic substrate. Thisglucose-sparing action of FFAs thus prolongs survival duringstarvation (18). (FFAs, which can stimulate insulin secretionin the fed state [19-21], do not do so in glucopenic condi-tions such as fasting. During prolonged fasting the fJ-cellsnormally become refractory to nonglucose secretagogues, aspart of the defense against hypoglycemia.) Conversely, in thefed state the powerful antilipolytic action of insulin sup-presses FFA levels, thereby permitting ingested glucose to bemetabolized without FFA-induced hindrance.
In obesity plasma FFAs are not fully suppressed by feeding(22,23), either because of insensitivity of adipocytes to theantilipolytic action of insulin (23) or because of the increasein adipocyte mass (24) or both. An elevation of fatty acyl CoAin muscle could cause resistance to the glucoregulatoryaction of insulin (25-27), as reviewed by McGarry (28,29).Despite insulin resistance, glucose tolerance remains normalbecause (3-cells somehow sense the level of insulin secretionrequired for full compensation. One possible explanation forthe perfect match between insulin secretion and insulinresistance is that both are caused by the same abnormality.High tissue levels of FFAs qualify as such an abnormality.First, plasma levels of FFAs are elevated in obesity (22-24,30-32). Second, chronic exposure of cultured islets toFFAs causes basal hypersecretion of insulin (33). Third, FFAlevels are high in obesity because they are not fully sup-pressed by the hyperinsulinemia they induce (23,34), i.e.,there is attenuation of the feedback between (3-cells andadipocytes that normally would prevent the coexistence ofhigh insulin and high FFA levels. This hypothesis is depictedin Fig. 1A(S-cells in normoglycemic obesity. Zucker fatty rats pro-vide an ideal model in which to study the mechanism ofP-cell hypersecretion in obesity. Like obese humans, theserats exhibit high basal insulin secretion, exaggerated insulinresponses (9), and relative hyperlipacidemia (35). Whenperfused with glucose in concentrations at or below thenormal fasting range, the isolated pancreases of nondiabeticobese rats secrete 10 times as much insulin as those of leancontrols (13,35). In normal islets insulin secretion is turnedoff when glucose is perfused at concentrations <4.2 mmol/1;this is not the case in islets of obese rats, which secrete asmuch insulin at 1.4 mmol/1 glucose as normal islets secrete at5.6 mmol/1 glucose (35) (Fig. 2).
To determine if this hypersecretion is the result of (3-cellhyperplasia, we compared the f$-cell volume fraction andislet DNA content of obese and lean groups. The volumefraction of (3-cells in pancreases of the obese rats was 3.9times normal and their DNA content per islet was 3.5 times
AObesity
Pre-NIDDM
BObesityNIDDM
t Tissue
FACoA
Skeletal MuscleInsulin
ResistanceObesity
Skeletal MuscleInsulin
Resistance
p-Cells
Hyperinsulinemia
tTissue
FACoA P-Cells
Hyperinsulinemia
FIG. 1. The lipotoxic hypothesis. A: In obese rats without diabetes thereis a relative increase in plasma FFA levels (>0.5 to <1.5 mmol/1) andpresumably in the tissue levels of fatty acyl CoA (FACoA). Thishyperlipacidemia may be a consequence of the expanded adipocyte massor of insensitivity of adipocytes to the antilipolytic action of insulin orboth. The presumed increase in tissue FFA levels throughout the bodyinterferes with normal glucose metabolism at multiple levels. In targettissues of insulin, such as muscle, this interference is referred to asinsulin resistance. In islets, the increased FFA levels stimulate insulinsecretion and induce p-cell proliferation and expansion of low Kmglucose metabolism, which increases basal insulin secretion andpotentiates insulin responses to all stimuli. Since FFA-induced changesin tissues are proportional to the levels of FFAs, the insulin resistanceand the insulin hypersecretion are perfectly matched and glucosetolerance remains normal. B: In obese rats that became diabetic, FFAsrose to still higher levels (>1.5 mmol/1) and "marbleization" of tissuesoccurs as FACoA is increasingly esterified to TGs. In muscle thisintensifies insulin resistance, while the islets, which have alreadyresponded fully to more moderate FFA overload, are incapable of furtherincreases in insulin secretion to match insulin resistance. The FFAoverload interferes with glucose metabolism and impairs the capacity ofP-cells to respond to postprandial hyperglycemia. Hyperinsulinemia atthis point no longer matches the increases in insulin resistance andNIDDM begins.
normal (35). However, this degree of hyperplasia could notfully explain the increase in basal insulin secretion, whichwas 10 times normal, nor could it account for the functionaldata in Fig. 2, indicating that in obese rats insulin secretionis not fully suppressed during glucopenia. Given the well-accepted relationships between glucose metabolism andinsulin secretion (36-38), one might expect an increase inlow Km glucose metabolism in islets that hypersecrete insu-lin during glucopenia. In fact, we observed an eightfoldgreater rate of glucose oxidation and usage in the islets fromobese rats (35) (Table 1). When glucose concentration waslowered from 5.6 to 1.4 mmol/1, glucose utilization declinedby 65% in normal islets, compared with only 11% in islets
1.4mM 2.8mM 4.2mM 5.6mM
5
£
300
200
100
20 40
TIME (min)60 80
FIG. 2. Insulin secretion by isolated pancreases (mean ± SE) of obeseZucker (O, n = 6) or lean Wistar rats (•, n = 5) during perfusion withglucose in concentrations in the fasting (5.6 and 4.2 mmol/1) orsubfasting (2.8 and 1.4 mmol/1) ranges. Note the leftward shift of theglucose-insulin dose-effect curve (35). IRI, immunoreactive insulin.
864 DIABETES, VOL. 44, AUGUST 1995
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
1TABLE 1Glucose usage
Wistarn
Zuckern
P
(5-[3H]glucose-*3H
1.4 mmol/1
19.5 ± 4.66
155.4 ± 26.46
<0.001
2O) and glucose oxidation
Glucose usage (pmol • h"
2.8 mmol/1
16.9 ± .94
193.5 ± 46.24
<0.05
R.H. UNGER
([U-14C]glucose->14CO2) at subfasting and fasting glucose concentrations
"^ islet"1)
5.6 mmol/1
55.4 ± 14.24
215.9 ± 8.43
<0.001
Glucose(pmol • h
2.8 mmol/1
7.7 ± 2.03
105.9 ± 5.03
<0.001
oxidation" ^ islet"1)
5.6 mmol/1
10.7 ± 3.33
135.4 ± 1.63
<0.001
Data are means ± SE. From Milburn et al. (35).
from obese rats. This combination of enhanced low Km
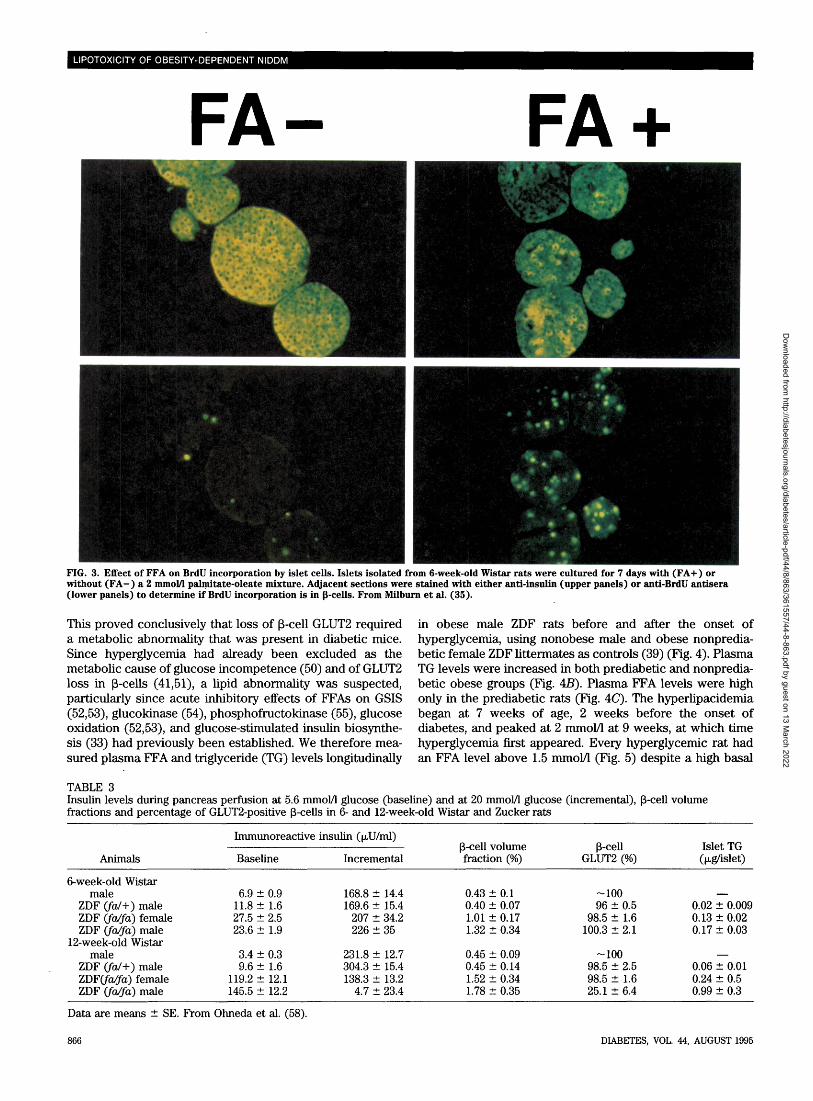
glucose metabolism and p-cell hyperplasia appears to ex-plain the hypersecretion of insulin at fasting and glucopenicconcentrations of glucose by islets of obese Zucker rats.Can increased FFA levels induce the P-cell phenotypeof normoglycemic obesity? If insulin hypersecretion result-ing from P-cell hyperplasia and increased low Km glucosemetabolism in islets is caused by high FFA levels, theseabnormalities should be inducible in normal rat islets byexposing them to comparable concentrations of FFAs. Zhouand Grill (33) previously reported a fourfold increase ininsulin released at 3 mmol/1 glucose from normal rat isletscultured for 48 h in FFAs. We repeated their study andobserved a doubling of insulin secretion in normal isletsperifused with 3 mmol/1 glucose after 7 days in culture in thepresence of 2 mmol/1 FFA and 2% bovine serum albumin (39).We also noted an FFA concentration-dependent increase inglucose usage at glucose concentrations of 2.8 and 5.6mmol/1 in islets cultured in 1 and 2 mmol/1 FFAs (35) (Table2). Finally, in normal islets cultured in 2 mmol/1 FFAs, wefound an increase in bromodeoxyuridine (BrdU) incorpora-tions from 2.3 ± 0.3 per islet in controls to 7.4 ±1.1 per islet,evidence that FFA stimulates DNA replication (35) (Fig. 3).Thus, long-chain FFAs can induce in normal islets the samefunctional, metabolic, and morphometric abnormalities thatoccur in P-cells of obese rats.
ADIPOCYTE-B-CELL RELATIONSHIPS IN OBESITY-DEPENDENT DIABETESP-cells in obesity-dependent diabetes. Before the onsetof diabetes, which begins in Zucker diabetic fatty (ZDF) ratsbetween 7 and 10 weeks of age (40), the p-cells of obeseprediabetic male ZDF rats are functionally and morphologi-cally indistinguishable from those of nonprediabetic obesefemale littermates. Basal insulin secretion is high (Table 3,6-week-old rats), GLUT2 is present in -100% of p-cells, andthe P-cell mass is enlarged. However, at the onset of NIDDM<60% of p-cells are GLUT2-positive (41), and GSIS hasvirtually disappeared (9,13) (Table 3, 12-week-old rats). Asthe diabetes progresses, <20% of p-cells remain GLUT2-
positive and GLUT2 mRNA declines to -25% of normal (13).Similar observations have been made in all models of rodentNIDDM thus far studied, including the GK rat (15), the db/dbmouse (14), and the Chinese hamster (42).Role of GLUT2 loss in glucose incompetence of P-cells.The remarkable correlation between the reduction inGLUT2-positive P-cells and glucose incompetence (13)seemed to suggest that a defect in glucose transport inP-cells was the underlying cause of the functional impair-ment (9). But subsequent evidence of defects distal toGLUT2 (43,44) has cast doubt on the role of impaired p-cellglucose transport in the loss of GSIS, the report of atransport-defective GLUT2 mutation in a patient withNIDDM (45) notwithstanding. Ironically, however, there arereasons to suspect that GLUT2 in p-cells may nevertheless berequired for glucose responsiveness, albeit through functionsother than glucose transport. First, expression of GLUT2antisense mRNA in p-cells results in NIDDM in transgenicmice (46). Second, GLUT2 expression confers glucose re-sponsiveness in GLUT2-deficient glucose-unresponsive insu-lin-secreting cell lines (47,48), but transfer of a GLUT2 cDNAinto GLUT2-deficient insulinoma cells is associated withenhanced glucokinase activity (48). Consistent with thisevidence of the functional versatility of GLUT2 is the factthat transfection of insulin-secreting AtT-20 cells to expresseither GLUT2 or GLUTl results in a similar enhancement ofglucose transport and yet confers glucose responsivenessonly to the GLUT2-expressing cells (49). These findingssuggest that GLUT2 may serve as the proximal anchor of asignaling pathway for high Km glucose metabolism (47-49).Lipotoxicity as cause of glucose incompetence andGLUT2 loss. The original assumption that loss of GLUT2was the cause rather than the consequence of the associatedmetabolic abnormalities of diabetes (9) was based on thefact that normalization of glycemia did not prevent the loss(41). But Thorens et al. (14) subsequently found that GLUT2disappeared from normal P-cells when transplanted intostreptozotocin-induced diabetic mice and, conversely, thatGLUT2 reappeared on GLUT2-deficient p-cells of diabeticdb/db mice when transplanted to nondiabetic recipients (14).
TABLE 2FFA effects on
(mmol/1)
012
glucose usage by cultured islets
n
333
Glucose usage (pmol
2.8 mmol/1
11.42 ± 1.2623.28 ± 1.7432.28 ± 1.74
•h"1- islet-1)
5.6 mmol/1
16.86 ± 3.5335.81 ± 2.8446.80 ± 1.52
Glucose usage (pmol • h l
2.8 mmol/1
1.63 ± 0.183.33 ± 0.254.59 ± 0.21
•ngDNA"1)
5.6 mmol/1
2.41 ± 0.505.12 ± 0.416.69 ± 0.21
Data are means ± SE. From Milburn et al. (35).
DIABETES, VOL. 44, AUGUST 1995 865
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
LIPOTOXICITY OF OBESITY-DEPENDENT NIDDM
FA +
FIG. 3. Effect of FFA on BrdU incorporation by islet cells. Islets isolated from 6-week-old Wistar rats were cultured for 7 days with (FA+ ) orwithout (FA-) a 2 mmol/1 palmitate-oleate mixture. Adjacent sections were stained with either anti-insulin (upper panels) or anti-BrdU antisera(lower panels) to determine if BrdU incorporation is in p-cells. From Milburn et al. (35).
This proved conclusively that loss of P-cell GLUT2 requireda metabolic abnormality that was present in diabetic mice.Since hyperglycemia had already been excluded as themetabolic cause of glucose incompetence (50) and of GLUT2loss in (3-cells (41,51), a lipid abnormality was suspected,particularly since acute inhibitory effects of FFAs on GSIS(52,53), glucokinase (54), phosphofructokinase (55), glucoseoxidation (52,53), and glucose-stimulated insulin biosynthe-sis (33) had previously been established. We therefore mea-sured plasma FFA and triglyceride (TG) levels longitudinally
in obese male ZDF rats before and after the onset ofhyperglycemia, using nonobese male and obese nonpredia-betic female ZDF littermates as controls (39) (Fig. 4). PlasmaTG levels were increased in both prediabetic and nonpredia-betic obese groups (Fig. 4B). Plasma FFA levels were highonly in the prediabetic rats (Fig. 4(7). The hyperlipacidemiabegan at 7 weeks of age, 2 weeks before the onset ofdiabetes, and peaked at 2 mmol/1 at 9 weeks, at which timehyperglycemia first appeared. Every hyperglycemic rat hadan FFA level above 1.5 mmol/1 (Fig. 5) despite a high basal
TABLE 3Insulin levels during pancreas perfusion at 5.6 mmol/1 glucose (baseline) and at 20 mmol/1 glucose (incremental), p-cell volumefractions and percentage of GLUT2-positive p-cells in 6- and 12-week-old Wistar and Zucker rats
Animals
6-week-old Wistarmale
ZDF (fa/+) maleZDF (fa/fa) femaleZDF (fa/fa) male
12-week-old Wistarmale
ZDF (/a/+) maleZDF(fa/fa) femaleZDF (fa/fa) male
Immunoreactive
Baseline
6.9 ± 0.911.8 ± 1.627.5 ± 2.523.6 ± 1.9
3.4 ± 0.39.6 ± 1.6
119.2 ± 12.1145.5 ± 12.2
insulin (u,U/ml)
Incremental
168.8 ± 14.4169.6 ± 15.4
207 ± 34.2226 ± 35
231.8 ± 12.7304.3 ± 15.4138.3 ± 13.2
4.7 ± 23.4
P-cell volumefraction (%)
0.43 ± 0.10.40 ± 0.071.01 ± 0.171.32 ± 0.34
0.45 ± 0.090.45 ± 0.141.52 ± 0.341.78 ± 0.35
p-cellGLUT2 (%)
-10096 ± 0.5
98.5 ± 1.6100.3 ± 2.1
-10098.5 ± 2.598.5 ± 1.625.1 ± 6.4
Islet TG(jjLg/islet)
—0.02 ± 0.0090.13 ± 0.020.17 ± 0.03
—0.06 ± 0.010.24 ± 0.50.99 ± 0.3
Data are means ± SE. From Ohneda et al. (58).
866 DIABETES, VOL. 44, AUGUST 1995
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
R.H. UNGER
8ooS
(0UJS
(0Q.
E,•ou
30
20
10
0
30
20
10
0
3.0
2.0
B
f 1X
3
I
0.0
1
0.8
0.6
0.4
0.2
0
..I-a T
8 9 10 11
AGE (weeks)
12 13 14
FIG. 4. Longitudinal studies of blood glucose (A), plasma TG (£), FFA(C), and TG content of islets (D) in lean male ZDF rats (fa/+) (O);obese female ZDF rats (fa/fa) (A), which do not develop diabetes; andobese male ZDF rats (fa/fa) ( • ) , which develop diabetes between theages of 8 and 10 weeks. From Lee et al. (39).
insulin secretion (Table 3, 12-week-old rats). While thetiming and the magnitude of the FFA elevations were con-sistent with a role in the pathogenesis of the p-cell malfunc-tion, increased tissue levels of fatty acyl CoA would berequired to impair p-cell function. If, in fact, islet fatty acylCoA is high, the glycerol 3-phosphate generated by glucosemetabolism would provide a substrate for TG formation. Wetherefore measured the islet TG content of the three groupsfrom 5 to 14 weeks of age as an index of substrate overload.Compared with lean controls islet TG content was increasedin islets of both prediabetic and nonprediabetic obese rats
600^
500-
400-
300
200-
100-
PLASMA GLUCOSE(mg/100 ml)
R = 0.873P = .001
0.5 1 1.5PLASMA FFA(mM)
2.5
FIG. 5. The relationship between plasma FFA and plasma glucose levelsin obese male ZDF rats in specimens obtained between 6 and 14 weeks ofage (39).
beginning at 6 weeks of age. However, at the onset of overtdiabetes at 9 weeks of age the TG content of islets in theprediabetic group had risen to 0.6 (xg/islet, more than twicethat of obese nonprediabetic controls and 10 times that oflean controls (Fig. AD). At present we consider the highTG content of diabetic islets to be the passive conse-quence of chronically elevated fatty acyl CoA in islet cells,rather than a cause of the p-cell abnormalities. (TGformation and lipolysis in islets is probably not rapidenough to create a quantitative significant diversion ofglycerol 3-phosphate from its postulated role in glucosesignaling (56,57].)
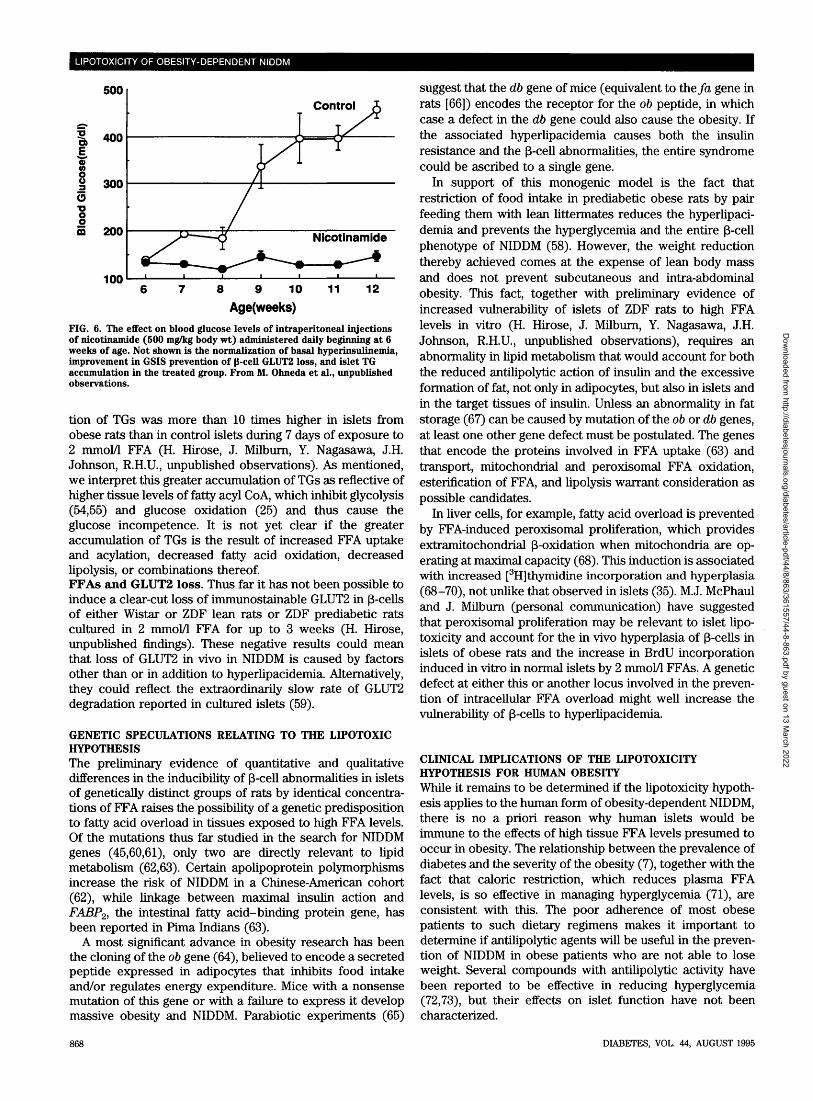
Chronic excesses of fat in islets could, however, have latesequelae. Late in the course of diabetes in ZDF rats, theincreased p-cell volume noted at onset declines by almost75% and fibrosis appears (58); conceivably the chronicallyhigh fat content in these islets plays a role in the cellulardepletion and associated fibrosis analogous to steatonecro-sis in liver.Effects of reducing FFAs on NIDDM p-cell phenotype. Ifincreased islet FFA levels do in fact cause the p-cell changesof NIDDM, these abnormalities should be preventable bymeasures that reduce the substrate overload. Two suchmaneuvers, caloric restriction by pair-feeding to lean litter-mates (58) and nicotinamide treatment (M. Ohneda, Y.Nagasawa, Y. Lee, J. Milburn, R.H.U., unpublished data),reduce plasma FFA levels to <1.5 mmol/1 and prevent theaccumulation of islet TGs. They also prevent the hypergly-cemia (Fig. 6), the basal hyperinsulinemia, the loss ofGLUT2, and the glucose incompetence (58) (Table 3). How-ever, the fact that these interventions have actions other thanreduction of plasma and tissue FFA levels makes it impos-sible to ascertain the mechanism of these dramatic effects.Can FFAs induce the p-cell phenotype in nondiabeticislets? High concentrations of free fatty acids are known toimpair p-cell function. Sako and Grill (52) observed a 50%reduction in GSIS by the perfused pancreas of normal ratsafter 48 h of a TG emulsion infusion, while Elks (53) noted amore substantial loss of GSIS in perifused islets exposed to1 mmol/1 palmitate. However, Zhou and Grill (33) observed areduction of only 30-50% in GSIS in islets isolated fromnormal rats and cultured for 48 h in the presence oflong-chain FFAs. Clearly, in vitro suppression of GSIS byFFAs is less complete than that which occurs at the onset ofNIDDM in intact ZDF rats.
To determine if this difference reflected a greater vulner-ability of islets from ZDF rats to the lipotoxic effects of highFFAs, we compared their effects on islets isolated fromprediabetic obese male ZDF rats, from nonprediabetic obesefemale ZDF rats, and from normal Wistar rats. All islets werecultured for 7 days in 2 mmol/1 FFAs and 2% bovine serumalbumin. Evidence of qualitative and quantitative differencesin FFA effects on the different groups were observed (H.Hirose, J. Milburn, Y. Nagasawa, J.H. Johnson, R.H.U., un-published observations). In normal islets from Wistar rats, 1and 2 mmol/1 FFAs induced a progressive increase in basalinsulin secretion (at 3 mmol/1 glucose). In islets from bothlean (heterozygous) and obese (homozygous) ZDF rats, aprogressive decrease in basal insulin secretion was induced.In islets from Wistar rats, 2 mmol/1 FFAs lowered the insulinresponse to 23 mmol/1 glucose by 57% in Wistar islets,compared with 91 and 98% in islets from lean ZDF and obeseprediabetic rats, respectively. Interestingly, the accumula-
DIABETES, VOL. 44, AUGUST 1995 867
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
LIPOTOXICITY OF OBESITY-DEPENDENT NIDDM
500
"3T
soom 200
1008 9 10
Age(weeks)12
FIG. 6. The effect on blood glucose levels of intraperitoneal injectionsof nicotinamide (500 mg/kg body wt) administered daily beginning at 6weeks of age. Not shown is the normalization of basal hyperinsulinemia,improvement in GSIS prevention of 0-cell GLUT2 loss, and islet TGaccumulation in the treated group. From M. Ohneda et al., unpublishedobservations.
tion of TGs was more than 10 times higher in islets fromobese rats than in control islets during 7 days of exposure to2 mmol/1 FFA (H. Hirose, J. Milburn, Y. Nagasawa, J.H.Johnson, R.H.U., unpublished observations). As mentioned,we interpret this greater accumulation of TGs as reflective ofhigher tissue levels of fatty acyl CoA, which inhibit glycolysis(54,55) and glucose oxidation (25) and thus cause theglucose incompetence. It is not yet clear if the greateraccumulation of TGs is the result of increased FFA uptakeand acylation, decreased fatty acid oxidation, decreasedlipolysis, or combinations thereof.FFAs and GLUT2 loss. Thus far it has not been possible toinduce a clear-cut loss of immunostainable GLUT2 in (3-cellsof either Wistar or ZDF lean rats or ZDF prediabetic ratscultured in 2 mmol/1 FFA for up to 3 weeks (H. Hirose,unpublished findings). These negative results could meanthat loss of GLUT2 in vivo in NIDDM is caused by factorsother than or in addition to hyperlipacidemia. Alternatively,they could reflect the extraordinarily slow rate of GLUT2degradation reported in cultured islets (59).
GENETIC SPECULATIONS RELATING TO THE LIPOTOXICHYPOTHESISThe preliminary evidence of quantitative and qualitativedifferences in the inducibility of p-cell abnormalities in isletsof genetically distinct groups of rats by identical concentra-tions of FFA raises the possibility of a genetic predispositionto fatty acid overload in tissues exposed to high FFA levels.Of the mutations thus far studied in the search for NIDDMgenes (45,60,61), only two are directly relevant to lipidmetabolism (62,63). Certain apolipoprotein polymorphismsincrease the risk of NIDDM in a Chinese-American cohort(62), while linkage between maximal insulin action andFABP2, the intestinal fatty acid-binding protein gene, hasbeen reported in Pima Indians (63).
A most significant advance in obesity research has beenthe cloning of the ob gene (64), believed to encode a secretedpeptide expressed in adipocytes that inhibits food intakeand/or regulates energy expenditure. Mice with a nonsensemutation of this gene or with a failure to express it developmassive obesity and NIDDM. Parabiotic experiments (65)
suggest that the db gene of mice (equivalent to the fa gene inrats [66]) encodes the receptor for the ob peptide, in whichcase a defect in the db gene could also cause the obesity. Ifthe associated hyperlipacidemia causes both the insulinresistance and the p-cell abnormalities, the entire syndromecould be ascribed to a single gene.
In support of this monogenic model is the fact thatrestriction of food intake in prediabetic obese rats by pairfeeding them with lean littermates reduces the hyperlipaci-demia and prevents the hyperglycemia and the entire p-cellphenotype of NIDDM (58). However, the weight reductionthereby achieved comes at the expense of lean body massand does not prevent subcutaneous and intra-abdominalobesity. This fact, together with preliminary evidence ofincreased vulnerability of islets of ZDF rats to high FFAlevels in vitro (H. Hirose, J. Milburn, Y. Nagasawa, J.H.Johnson, R.H.U., unpublished observations), requires anabnormality in lipid metabolism that would account for boththe reduced antilipolytic action of insulin and the excessiveformation of fat, not only in adipocytes, but also in islets andin the target tissues of insulin. Unless an abnormality in fatstorage (67) can be caused by mutation of the ob or db genes,at least one other gene defect must be postulated. The genesthat encode the proteins involved in FFA uptake (63) andtransport, mitochondria! and peroxisomal FFA oxidation,esterification of FFA, and lipolysis warrant consideration aspossible candidates.
In liver cells, for example, fatty acid overload is preventedby FFA-induced peroxisomal proliferation, which providesextramitochondrial 3-oxidation when mitochondria are op-erating at maximal capacity (68). This induction is associatedwith increased [3H]thymidine incorporation and hyperplasia(68-70), not unlike that observed in islets (35). M.J. McPhauland J. Milburn (personal communication) have suggestedthat peroxisomal proliferation may be relevant to islet lipo-toxicity and account for the in vivo hyperplasia of (3-cells inislets of obese rats and the increase in BrdU incorporationinduced in vitro in normal islets by 2 mmol/1 FFAs. A geneticdefect at either this or another locus involved in the preven-tion of intracellular FFA overload might well increase thevulnerability of (3-cells to hyperlipacidemia.
CLINICAL IMPLICATIONS OF THE LIPOTOXICITYHYPOTHESIS FOR HUMAN OBESITYWhile it remains to be determined if the lipotoxicity hypoth-esis applies to the human form of obesity-dependent NIDDM,there is no a priori reason why human islets would beimmune to the effects of high tissue FFA levels presumed tooccur in obesity. The relationship between the prevalence ofdiabetes and the severity of the obesity (7), together with thefact that caloric restriction, which reduces plasma FFAlevels, is so effective in managing hyperglycemia (71), areconsistent with this. The poor adherence of most obesepatients to such dietary regimens makes it important todetermine if antilipolytic agents will be useful in the preven-tion of NIDDM in obese patients who are not able to loseweight. Several compounds with antilipolytic activity havebeen reported to be effective in reducing hyperglycemia(72,73), but their effects on islet function have not beencharacterized.
868 DIABETES, VOL. 44, AUGUST 1995
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
R.H. UNGER
ACKNOWLEDGMENTSThis work was supported by National Institutes of HealthGrants DK-02700 and 1-P01-DK42582 and Veterans Adminis-trations Research Support Grant 549-8000.
We wish to thank Drs. J. Denis McGarry, Daniel W. Foster,Christopher B. Newgard, and Helen Hobbs for their con-structive criticisms of this manuscript and Teresa Autrey foroutstanding secretarial work.
REFERENCES1. National Center for Health Statistics cited in "An epidemic of obesity."
Newsweek, 1 Aug 1994: p. 622. Harris MI: Impaired glucose tolerance in the US population. Diabetes
Care 12:464-474, 19893. Harris MI: Undiagnosed NIDDM: clinical and public health issues.
Diabetes Care 16:642-652, 19934. DeFronzo RA: The triumvirate: 3-cell, muscle, liver: a collusion respon-
sible for NIDDM. Diabetes 37:667-687, 19885. Reaven GM: The fourth musketeer: from Alexandre Dumas to Claude
Bernard. Diabetologia 38:3-13, 19956. Unger RH, Foster DW: Diabetes mellitus. In William's Textbook of
Endocrinology. Wilson JD, Foster DW, Eds. Philadelphia, Saunders, 1992,p. 1255-1333
7. Warram JH, Rich SS, Krolewski AS: Epidemiology and genetics ofdiabetes mellitus. In Joslin's Diabetes Mellitus. 13th ed. Kahn CR, WeirGC, Eds. Media, PA, Lea and Febiger, 1994, p. 208-211
8. Weir GC, Leahy JL: Pathogenesis of non-insulin-dependent (type II)diabetes mellitus. In Joslin's Diabetes Mellitus. 13th ed. Kahn CR, WeirGC, Eds. Media, PA, Lea and Febiger, 1994, p. 256-257
9. Unger, RH: Diabetic hyperglycemia: link to impaired high K,,, glucosetransport in pancreatic fi-cells. Science 251:1200-1205, 1991
10. Ogawa A, Johnson JH, Ohneda M, McAllister CT, Inman L, Alam T, UngerRH: Roles of insulin resistance and 3-cell dysfunction in dexamethasone-induced diabetes. J Clin Invest 90:497-504, 1992
11. Palmer JP, Benson JW, Walter RM, Ensinck JW: Arginine-stimulatedacute phase of insulin and glucagon secretion in diabetic subjects. J ClinInvest 58:565-570, 1976
12. Pfeiffer MA, Halter JB, Porte D Jr: Insulin secretion in diabetes mellitus.AmJMed 70:579-588, 1981
13. Johnson JH, Ogawa A, Chen L, Orci L, Newgard CB, Alam T, Unger RH:Underexpression of 3-cell high Km glucose transporters in noninsulin-dependent diabetes. Science 250:546-549, 1990
14. Thorens B, Wu YJ, Leahy JL, Weir GC: The loss of GLUT2 expression byglucose-unresponsive 3-cells of db/db mice is reversible and is inducedby the diabetic environment. J Clin Invest 90:77-85, 1992
15. Ohneda M, Johnson JH, Inman LR, Chen L, Suzuki K-I, Goto Y, Alam T,Orci L, Unger RH: GLUT-2 expression and function in p-cells of GK ratswith NIDDM: dissociation between reductions in glucose transport andglucose-stimulated insulin secretion. Diabetes 42:1065-1072, 1993
16. Brunzell JD, Robertson RP, Lerner RL, Hazzard WR, Ensinck JW,Bierman EL, Porte D Jr: Relationships between fasting plasma glucoselevels and insulin secretion during intravenous glucose tolerance tests. JClin Endocrinol Metab 42:222-229, 1976
17. Gottlieb MS: Diabetes in offspring and siblings of juvenile and maturity-onset-type diabetics. J Chronic Dis 33:331-339, 1980
18. Cahill GF Jr: Starvation in man. N Engl J Med 282:668-675, 197019. Greenough WB, Crespin, SR, Steinberg D: Hypoglycemia and hyperinsuline-
mia in response to raised free-fatty acid levels. Lancet 2:1334-1336, 196720. Madison LL, Seyffert WA, Unger RH, Barker B: Effect of plasma free fatty
acids on plasma glucagon and serum insulin concentrations. Metabolism17:301-304, 1968
22. Gorden ES: Non-esterified fatty acids in blood of obese and lean subjects.Am J Clin Nutr 8:740-747, 1960
23. Chen YDI, Golay A, Swislocki ALM, Reaven GM: Resistance to insulinsuppression of plasma free fatty acids concentrations and insulin stimula-tion of glucose uptake in NIDDM. J Clin Endocrinol Metab 64:17-21, 1987
24. Groop LC, Bonadonna RC, Simonson DC, Petrides AS, Shank M, DeFronzoRA: Effect of insulin on oxidative and nonoxidative pathway of free fattyacid metabolism in human obesity. AmJPhysiol 263:E79-E84, 1992
25. Randle PJ, Garland PB, Hales CN, Newsholme FA: The glucose fatty-acidcycle: its role in insulin sensitivity and the metabolic disturbances ofdiabetes mellitus. Lancet 1:785-789, 1963
26. Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA: Effect of fatty acids onglucose production and utilization in man. J Clin Invest 72:1737-1747, 1983
27. Groop LC, Saloranta C, Shank M, Bonadonna RC, Ferrannini E, DeFronzoRA: The role of free fatty acid metabolism in the pathogenesis of insulinresistance in obesity and noninsulin-dependent diabetes mellitus. J ClinEndocrinol Metab 72:96-107, 1991
28. McGarry JD: What if Minkowski had been ageusic? An alternative angleon diabetes. Science 258:766-774, 1992
29. McGarry JD: Disordered metabolism in diabetes: have we underempha-sized the fat cell component? J Cell Biochem 555:29-38, 1994
30. Jensen MD, Haymond MW, Rizza RA, Cryer PE, Miles JM: Influence ofbody fat distribution on free fatty acid metabolism in obesity. J ClinInvest 83:1168-1173, 1989
31. Martin ML, Jensen MD: Effects of body fat distribution on regionallipolysis in obesity. J Clin Invest 88:609-613, 1991
33. Zhou Y-P, Grill VE: Long-term exposure of rat pancreatic islets to fattyacids inhibits glucose-induced insulin secretion and biosynthesis througha glucose fatty acid cycle. J Clin Invest 93:870-876, 1994
34. Campbell PJ, Carlson MG, Jurjhan N: Fat metabolism in human obesity.AmJPhysiol 266:E600-E604, 1994
35. Milburn JL, Hirose H, Lee YH, Nagasawa Y, Ogawa A, Ohneda M,BeltrandelRio H, Newgard C, Johnson JH, Unger RH: Pancreatic 3-cellsin obesity: evidence for induction of functional, morphologic and meta-bolic abnormalities by increased long-chain fatty acids. J Biol Chem270:1295-1299, 1995
36. Matschinsky FM, Ellerman JE: Metabolism of glucose in the islets ofLangerhans. J Biol Chem 243:2730-2736, 1968
37. Malaisse WJ, Sener A, Herchuelz A, Hutton JC: Insulin release: the fuelhypothesis. Metabolism 28:373-386, 1979
38. Meglassson MD, Matschinsky FM: Pancreatic islet metabolism and theregulation of insulin secretion. Diabetes Metab Rev 2:163-214, 1986
39. Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH: 3-celllipotoxicity in the pathogenesis of non-insulin-dependent diabetes melli-tus of obese rats: impairment in adipocyte-p-cell relationships. Proc NatlAcad Sci USA 91:10878-10882, 1994
40. Peterson RG, Shaw WN, Neel M, Little LA, Eichberg J: Zucker diabeticfatty rat as a model for non-insulin-dependent diabetes mellitus. ILARNews 32:16-19, 1990
41. Orci L, Ravazzola M, Baetens D, Inman L, Amherdt M, Peterson RG,Newgard CB, Johnson JH, Unger RH: Evidence that down-regulation of3-cell glucose transporters in non-insulin-dependent diabetes may be thecause of diabetic hyperglycemia. Proc Natl Acad Sci USA 87:9953-9957,1990
42. Kohnert K, Kloting I, Hehmke B, KeBler J, Sickel E, Besch W: Impairedbeta-cell function and GLUT-2 expression in the new Chinese-HamsterChig/Han subline are not reversible by long-term insulin-treatment.Diabetologia 37:Al 13(439), 1994
43. Giroix M-H, Baetens D, Rasschaert J, Leclercq-Meyer V, Sener A, PorthaB, Malaisse WJ: Enzymic and metabolic anomalies in islets of diabeticrats: relationship to 3-cell mass. Endocrinology 130:2634-2640, 1992
44. Ohneda M, Johnson JH, Lee Y, Nagasawa Y, Unger RH: Post-GLUT-2defects in p-cells of non-insulin-dependent diabetic obese rats. Am JPhysiol 267:E968-E974, 1994
45. Mueckler M, Kruse M, Strube M, Riggs AC, Chiu KC, Permutt MA: Amutation in GLUT-2 glucose transporter gene of a diabetic patientabolishes transport activity. J Biol Chem 269:17765-17767, 1994
46. Valera A, Salanes G, Fernandez-Alvarez J, Pujol A, Ferrer J, Asins G,Gomis R, Bosch F: Expression of GLUT-2 antisense RNA in 3-cells oftransgenic mice leads to diabetes. J Biol Chem 269:28543-28546, 1994
47. Hughes SD, Johnson JH, Quaade C, Newgard CB: Engineering of glucose-stimulated insulin secretion and biosynthesis in non-islet cells. Pmc NatlAcad Sci USA 89:688-692, 1992
48. Ferber S, BeltrandelRio H, Johnson JH, Noel RJ, Cassidy LE, Clark S,Becker TC, Hughes SD, Newgard CB: GLUT-2 gene transfer into insuli-noma cells confers both high and low affinity glucose-stimulated insulinrelease: relationship to glucokinase activity. J Biol Chem 269:11523-11529, 1994
49. Hughes SD, Quaade C, Johnson JH, Ferber S, Newgard CB: Transfectionof AtT-20ins cells with GLUT-2 but not GLUT-1 confers glucose-stimu-lated insulin secretion: relationship to glucose metabolism. J Biol Chem268:15205-15212, 1993
50. Komiya I, Baetens D, Kumajima M, Orci L, Unger RH: Compensatorycapabilities of islets of BB/Wor rats exposed to sustained hyperglycemia.Metabolism 39:614-618, 1990
51. Chen L, Alam T, Johnson JH, Hughes S, Newgard CB, Unger RH:Regulation of 3-cell glucose transporter gene expression. Proc Natl AcadSci USA 87:4088-4092, 1990
52. Sako Y, Grill VE: A 48-hour lipid infusion in the rat time-dependentlyinhibits glucose-induced insulin secretion and 3-cell oxidation through aprocess likely coupled to fatty acid oxidation. Endocrinology 127:1580-1589, 1990
53. Elks, ML: Chronic perifusion of rat islets with palmitate suppressesglucose-stimulated insulin release. Endocrinology 133:208-214, 1993
56. MacDonald MJ: High content of mitochondrial glycerol-3-phosphatedehydrogenase in pancreatic islets and its inhibition by diazoxide. J BiolChem 256:8287-8290, 1981
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M
arch 2022
LIPOTOXICITY OF OBESITY-DEPENDENT NIDDM
Worley JF III: Dependence on NADH produced during glycolysis forP-cell glucose signalling. J Biol Chem 269:10979-10982, 1994
58. Ohneda M, Inman LR, Unger RH: Caloric restriction in obese prediabeticrats prevents beta-cell depletion, loss of beta-cell GLUT-2 and glucoseincompetence. Diabetologia 38:173-179, 1995
59. Thorens B, Gerard N, Deriaz N: GLUT-2 surface expression and intracel-lular transport via the constitutive pathway in pancreatic |3-cells andinsulinoma: evidence for a block in trans-Golgi network exit by brefeldinA. J Cell Biol 123:1687-1694, 1993
60. Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, Lesage S,Stoffel M, Takeda J, Passa P, Permutt A, Beckman JS, Bell GI, Cohen D:Familial hyperglycemia due to mutations on glucokinase. N Engl J Med328:697-702, 1993
61. Bell GI, Xiang K-S, Newman MV, Wu SH, Wright LG, Fajans SS, SpielmanRS, Cox NJ: Genes for non-insulin-dependent diabetes mellitus (maturityonset diabetes of the young) is linked to DNA polymerization on humanchromosome 20. Proc Natl Acad Sci USA 88:1484-1488, 1991
62. Xiang K-S, Cox NJ, Sanz N, Huang P, Karam JH, Bell GI: Insulin-receptorand apolipoprotein genes contribute to development of NIDDM inChinese Americans. Diabetes 38:17-23, 1989
63. Prochazka M, Lillioja S, Tait JF, Knowler WC, Mott DM, Spraul M,Bennett PH, Bogardus C: Linkage of chromosomal markers on 4q with aputative gene determining maximal insulin action in Pima Indians.Diabetes 42:514-519, 1993
64. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM:Positional cloning of the mouse obese gene and its human homologue.Nature 372:425-432, 1994
65. Coleman DL: Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 14:141-148, 1978
66. Truett GE, Bahary N, Friedman JM, Leibel RL: Rat obesity gene fatty (fa)maps to chromosome 5: evidence for homology with the mouse genediabetes (db). Proc Natl Acad Sci USA 88:7806-7809, 1991
67. Briquet-Laugier V, Dugail I, Ardouin B, Le Liepvre X, Lavau M, QuignardBA: Evidence for a sustained genetic effect on fat storage capacity incultured adipose cells from Zucker rats. Am J Physiol 30:E439-E446,1994
68. Lock EA, Mitchell AM, Elcombe CR: Biochemical mechanisms of induc-tion of hepatic peroxisome proliferation. Annu Rev Pharmacol Toxicol29:145-163, 1989
69. Autwerx J: Regulation of gene expression by fatty acids and fibric acidderivatives: an integrative role for peroxisome proliferator activatedreceptors. Horm Res 38:269-277, 1992
70. Motojima K: Peroxisome proliferator-activated receptor (PPAR): struc-ture, mechanisms of activation and diverse functions. Cell Struct Fund18:267-277, 1993
71. Newburgh LH: Control of hyperglycemia of obese diabetes by weightreduction. Ann Intern Med 17:935-942, 1942
72. Fulcher GR, Walker M, Catalano C, Aquis L, Alberti KG: Metabolicsuppression of nonesterified fatty acid levels with acipimox in obeseNIDDM patients. Diabetes 41:1400-1408, 1992
73. Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, KraegenEW. A new antidiabetic agent, BRL 49653, reduces lipid availability andimproves insulin action and glucoregulation in the rat. Diabetes 43:1203-1210, 1994
870 DIABETES, VOL. 44, AUGUST 1995
Dow
nloaded from http://diabetesjournals.org/diabetes/article-pdf/44/8/863/361557/44-8-863.pdf by guest on 13 M