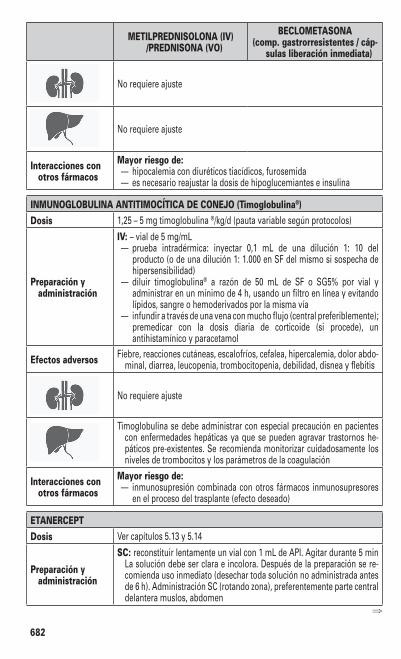
Manual de Trasplante Hematopoyético 5. a edición ENRIC CARRERAS I PONS Director Médico del Registro de Donantes de Médula Ósea (REDMO). Fundación Josep Carreras contra la Leucemia. Barcelona. Médico Consultor Senior. Servicio de Hematología. Hospital Clínic Universitari (Barcelona). Institut de Recerca Josep Carreras / Campus Clínic-Universitat de Barcelona. MONTSERRAT ROVIRA I TARRATS Responsable del Programa de Trasplante Hematopoyético. Médico Consultor. Servicio de Hematología. Hospital Clínic Universitari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Colaboradora Docente. Universitat de Barcelona. IZASKUN ZEBERIO ECHECHIPIA Médico Adjunto. Servicio Hematología - Hospital de Día. Hospital Donostia (Gipuzkoa). Biodonostia. Profesora Asociada Hematología. Euskal Herriko Unibertsitatea. DAVID VALCÁRCEL FERREIRAS Director del Programa Trasplante Hematopoyético. Servicio Hematología. Hospital Vall d´Hebron (Barcelona). Profesor Asociado. Universitat Autònoma de Barcelona.
Transcript
Manualde Trasplante
Hematopoyético5.a edición
ENRIC CARRERAS I PONSDirector Médico del Registro de Donantes de Médula Ósea (REDMO).
Fundación Josep Carreras contra la Leucemia. Barcelona.Médico Consultor Senior. Servicio de Hematología.
Hospital Clínic Universitari (Barcelona).Institut de Recerca Josep Carreras / Campus Clínic-Universitat de Barcelona.
MONTSERRAT ROVIRA I TARRATSResponsable del Programa de Trasplante Hematopoyético.
Médico Consultor. Servicio de Hematología.Hospital Clínic Universitari (Barcelona).
Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS).Colaboradora Docente. Universitat de Barcelona.
Advertencia. Los autores han hecho todos los esfuerzos posibles para asegurarse de que las indicaciones y las dosis que figuran en este Manual son las correctas y las generalmente recomen-dadas por las Autoridades Sanitarias, las Organizaciones Nacionales e Internacionales de Trasplante Hematopoyético y la literatura médica, en el momento de la publicación del libro.
Sin embargo, queremos advertir a los lectores que deben consultar las recomendaciones y las informaciones que, de forma periódica, proporcionan las Autoridades Sanitarias, las mencionadas Or-ganizaciones y los fabricantes de los productos, y que no podemos hacernos responsables de las con-secuencias que pudieran derivarse de cualquier error en el texto que haya podido pasar inadvertido.
Finalmente, cuando para el manejo o tratamiento de una determinada situación haya más de una opción admitida, las recomendaciones del libro expresan exclusivamente la preferencia de los auto-res, sin que ello indique que otras opciones no puedan ser eficaces o recomendables.
«Cualquier forma de reproducción, distribución, comunicación pública o transformación de esta obra sólo puede ser realizada con la autorización de sus titulares, salvo excepción prevista por la ley. Di-ríjase a CEDRO (Centro Español de Derechos Reprográficos, www.cedro.org) si necesita fotocopiar o escanear algún fragmento de esta obra (www.conlicencia.com; 91 702 19 70 / 93 272 04 47)».
PAU ABRISQUETA COSTA. Médico Adjunto. Servicio de Hematología. Hospital Universitari Vall d’Hebron (Barcelona). Universitat Autònoma de Barcelona.
LAURA ALONSO GARCÍA. Médico adjunto. Servicio de Oncología y Hematología Pediátricas. Hos-pital Universitari Vall d’Hebron (Barcelona). Universitat Autònoma de Barcelona.
MARTA AYMERICH GREGORIO. Hematóloga Consultora. Unidad de Hematopatología. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
ISABEL BADELL SERRA. Directora Unidad. Unidad Pediátrica de TPH. Hospital de la Santa Creu i Sant Pau y Hospital Sant Joan de Deu (Barcelona). Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau. Profesora Titular de Pediatría. Universitat Autònoma de Barcelona.
PASCUAL BALSALOBRE LÓPEZ. Coordinador de Trasplante Hematopoyético. Servicio de Hema-tología-Unidad de Trasplante Hematopoyético. Hospital General Universitario Gregorio Marañón (Madrid). Instituto de Investigación Sanitaria Gregorio Marañón (IISGM). Universidad Complutense de Madrid.
PERE BARBA SUÑOL. Médico adjunto. Servicio de Hematología. Hospital Universitari Vall d’Hebron (Barcelona). Profesor Asociado. Universitat Autònoma de Barcelona.
MONTSERRAT BATLLE MASSANA. Facultativo Especialista. Servicio Hematología Clínica. ICO-Hospital Universitari Germans Trias i Pujol (Badalona). Universitat Autònoma de Barcelona.
GUIOMAR BAUTISTA CARRASCOSA. Médico Adjunto. Hematología-Hemoterapia. Hospital Uni-versitario Puerta de Hierro-Majadahonda (Madrid). Instituto de Investigación Sanitaria Puerta de Hierro. Colaborador Clínico Docente. Universidad Autónoma de Madrid.
ARANCHA BERMÚDEZ RODRÍGUEZ. Médico Adjunto. Servicio de Hematología /Sección Clínica. Hospital Universitario Marqués de Valdecilla (Santander). Fundación Instituto de Investigación Mar-qués de Valdecilla (IDIVAL). Profesora Asociada de Hematología. Universidad de Cantabria.
JOAN BLADÉ I CREIXENTÍ. Consultor Senior y Jefe de Unidad. Servicio de Hematología / Unidad de Amiloidosis y Mieloma. Hospital Clínic Universitari (Barcelona). Institut d’Investigacions Biomè-diques August Pi i Sunyer (IDIBAPS). Universitat de Barcelona.
FRANCESC BOSCH ALBAREDA. Jefe de Servicio de Hematología. Servicio de Hematología. Hospi-tal Universitari Vall d’Hebron (Barcelona). Universitat Autònoma de Barcelona.
SALUT BRUNET MAURI. Directora de Unidad de Hematología Clínica. Servicio de Hematología. Hospital de la Santa Creu i Sant Pau (Barcelona). Profesora Asociada Hematología. Universitat Au-tònoma de Barcelona.
MARÍA DOLORES CABALLERO BARRIGÓN. Jefe de Sección. Servicio de Hematología Hospital Universitario (Salamanca). Instituto Biosanitario de Salamanca (IBSAL). Profesor Titular de Hemato-logía. Universidad de Salamanca.
JOSÉ RAFAEL CABRERA MARÍN. Jefe de Servicio. Hematología-Hemoterapia. Hospital Universi-tario Puerta de Hierro-Majadahonda (Madrid). Instituto de Investigación Sanitaria Puerta de Hierro. Profesor Titular Hematología. Universidad Autónoma de Madrid.
MÓNICA CABRERO CALVO. Médico Adjunto. Servicio de Hematología. Hospital Universitario (Sa-lamanca). Instituto de Investigación Biomédica de Salamanca (IBSAL). Universidad de Salamanca.
ESTHER CARCELERO SANMARTÍN. Facultativo Especialista. Servicio de Farmacia. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
VI
CELIA CARDOZO ESPINOLA. Médico Becario. Servicio de Infecciones. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
FRANCISCO CERVANTES REQUENA. Médico Consultor Senior. Servicio de Hematología. Hospital Clínic Universitari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Profesor Asociado. Universitat de Barcelona.
M.a TERESA CIBEIRA LÓPEZ. Médico Adjunto. Servicio de Hematología / Unidad de Amiloidosis y Mieloma. Hospital Clínic Universitari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Universitat de Barcelona.
NATALIA CREUS BARÓ. Facultativo Especialista Sénior. Servicio de Farmacia. Hospital Clínic Uni-versitari (Barcelona). Universitat de Barcelona.
FELIPE DE ARRIBA DE LA FUENTE. Médico Especialista en Hematología. Servicio de Hematología y Oncología Médica. Hospital General Universitario Morales Meseguer (Murcia). Instituto Murciano de Investigación Biosanitaria (IMIB). Profesor Asociado de Medicina. Universidad de Murcia.
JOSÉ LUIS DE LA CALLE REVIRIEGO. Coordinador. Unidad para el Estudio y Tratamiento del Do-lor. Hospital Universitario Ramón y Cajal (Madrid). Universidad de Alcalá de Henares.
RAFAEL DE LA CÁMARA DE LLANZA. Médico Adjunto. Servicio de Hematología. Hospital de la Princesa (Madrid). Universidad Autònoma de Madrid.
M.a CONSUELO DEL CAÑIZO FERNÁNDEZ-ROLDÁN. Jefe de Servicio. Departamento de He-matología. Hospital Clínico (Salamanca). Instituto Biosanitario de Salamanca (IBSAL) Catedrática. Universidad de Salamanca.
CRISTINA DÍAZ DE HEREDIA RUBIO. Jefe de Sección. Servicio de Oncología y Hematología Pediá-tricas. Hospital Universitari Vall d’Hebron (Barcelona). Universitat Autònoma de Barcelona.
MIGUEL ANGEL DÍAZ PÉREZ. Jefe de Sección. Servicio de Onco-Hematología y Trasplante Hema-topoyético. Hospital Infantil Universitario Niño Jesús (Madrid).
MARIBEL DÍAZ RICART. Consultor. Servicio de Hemoterapia-Hemostasia. Hospital Clínic Univer-sitari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) Universitat de Barcelona.
MARÍA DÍEZ CAMPELO. Médico Adjunto. Servicio de Hematología. Hospital Universitario (Sala-manca). Instituto de Investigación Biomédica de Salamanca (IBSAL). Universidad de Salamanca.
JOSÉ LUIS DÍEZ MARTÍN. Jefe de Servicio. Servicio de Hematología. Hospital General Universitario Gregorio Marañón (Madrid). Instituto de Investigación Sanitaria Gregorio Marañón (IISGM). Profe-sor Asociado. Universidad Complutense de Madrid.
EVA DOMINGO DOMÈNECH. Coordinador Asistencial. Servicio Hematología Clínica. Institut Ca-talà d’Oncologia. Hospital Duran i Reynals (L’Hospitalet de Llobregat, Barcelona). Universitat de Barcelona.
IZASKUN ELORZA ÁLVAREZ. Médico Adjunto, Especialista en Hematología y Oncología Pediátri-cas. Unidad Pediátrica de Trasplante de Progenitores Hematopoyéticos. Hospital Universitari Vall d´Hebron (Barcelona). Vall d´Hebron Institut de Reserca. Universitat Autònoma de Barcelona.
GUADALUPE ERCILLA GONZÁLEZ. Consultor Sénior. Servicio de Inmunología. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
FRANCISCO FERNÁNDEZ AVILÉS. Hematólogo Consultor. Unidad de Trasplante Hematopoyético. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
MARÍA LAURA FOX. Servicio Hematología. Hospital Universitari Vall d’Hebron (Barcelona). Univer-sitat Autònoma de Barcelona.
CRISTINA FUSTÉ GIRÓ. Coordinadora Técnica. Registro de Donantes de Médula Ósea. Fundació Josep Carreras (Barcelona).
VII
DAVID GALLARDO GIRALT. Director Asistencial. Jefe de Servicio. Servicio de Hematología. Institut Catalá d’Oncologia (Girona). lnstitut d’Investigació Biomèdica de Girona (IDIBGI). Profesor Asocia-do. Universitat de Girona.
EVA GÁLVEZ DE LA VILLA. FEA Hematología y Hemoterapia. Servicio de Hemato-Oncología Pe-diátrica. Servicio de Transfusión. Hospital Infantil Universitario Niño Jesús. Fundación Investigación Biomédica. Hospital Infantil Universitario Niño Jesús. Grupo Clínico Vinculado CIBERER.
IRENE GARCÍA CADENAS. Médico adjunto. Servicio de Hematología. Hospital de la Santa Creu i Sant Pau (Barcelona). Universitat Autònoma de Barcelona.
JORGE GAYOSO CRUZ. Jefe de Sección Hematología Clínica-UTMO. Servicio de Hematología-Uni-dad de Trasplante Hematopoyético. HGU Gregorio Marañón (Madrid). Instituto de Investigación Sanitaria Gregorio Marañón. Universidad Complutense de Madrid.
MONTSERRAT GIMÉNEZ PÉREZ. Facultativo Especialista. Servicio de Microbiologia. Hospital Uni-versitari Germans Trias i Pujol (Badalona). Profesor Asociado Microbiologia. Grado de Medicina y Biomedicina. Universitat Autònoma de Barcelona.
MARTA GONZÁLEZ VICENT. Médico Adjunto. Servicio de Onco-Hematología y Trasplante Hemato-poyético. Hospital Infantil Universitario Niño Jesús (Madrid).
FRANCESC GRAUS RIBAS. Jefe de Servicio. Servicio de Neurología. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
GONZALO GUTIÉRREZ GARCÍA. Hematólogo Especialista. Unidad de Trasplante Hematopoyético. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
JUAN CARLOS HERNÁNDEZ BOLUDA. Facultativo Especialista. Servicio de Hematología. Hos-pital Clínico Universitario (Valencia). Instituto de Investigación Sanitaria (INCLIVA). Universitat de València.
MI KWON. Médico Adjunto. Servicio de Hematología. Hospital General Universitario Gregorio Mara-ñón (Madrid). Instituto de Investigación Sanitaria Gregorio Marañón (IISGM). Profesor Colaborador. Universidad Complutense de Madrid.
FRANCESCO LO COCO. Jefe del Laboratorio de Diagnóstico Hematológico. Departamento de Bio-patología. Profesor Titular. Universidad Tor Vergata (Roma).
JAVIER LÓPEZ JIMÉNEZ. Jefe de Servicio. Servicio de Hematología y Hemoterapia. Hospital Ramón y Cajal (Madrid). Profesor Asociado del Dpto. de Medicina. Universidad de Alcalá de Henares.
ESTAFANÍA LÓPEZ PÉREZ. Médico Adjunto. Servicio de Hematología. Hospital Universitario (Sa-lamanca). Instituto de Investigación Biomédica de Salamanca (IBSAL).Universidad de Salamanca.
OLGA LÓPEZ VILLAR. Médico Adjunto. Servicio de Hematología y Hemoterapia. Hospital Universi-tario (Salamanca). Universidad de Salamanca.
NÚRIA MARIEGES VIA. Coordinadora General. Registro de Donantes de Médula Ósea. Fundació Josep Carreras (Barcelona).
ALEJANDRO MARTÍN GARCÍA-SANCHO. Médico Adjunto. Servicio de Hematología. Hospital Universitario (Salamanca). Instituto Biosanitario de Salamanca (IBSAL). Universidad de Salamanca.
CARMEN MARTÍNEZ MUÑOZ. Hematólogo Consultor. Unidad de Trasplante Hematopoyético. Hospital Clínic Universitari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Colaboradora Docente. Universitat de Barcelona.
RODRIGO MARTINO BOFARULL. Médico Adjunto. Servicio de Hematología / Unidad de Trasplante Hematopoyético. Hospital Santa Creu i Sant Pau (Barcelona). Universitat Autònoma de Barcelona.
JAUME MARTORELL PONS. Consultor Sénior. Servicio de Inmunología. Hospital Clínic Universi-tari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Universitat de Barcelona.
VIII
MARÍA-VICTORIA MATEOS. Médico Adjunto. Servicio de Hematología. Complejo Asistencial Uni-versitario (Salamanca). Instituto Biosanitario de Salamanca (IBSAL). Profesor Asociado de Hemato-logía. Universidad de Salamanca.
EOIN McGRATH. JACIE Operations Manager. JACIE Accreditation Office (Barcelona). European So-ciety for Blood & Marrrow Transplantation (EBMT).
LAURA MEDINA MARRERO. Médico adjunto. Banc de Sang i Teixits / Unitat d’afèresi i de processa-ment cel·lular. Hospital Universitari de la Santa Creu i Sant Pau (Barcelona). Universitat Autònoma de Barcelona.
JOSEP MENSA PUEYO. Médico Consultor. Servicio de Infecciones. Hospital Clinic Universitari (Bar-celona). Universitat de Barcelona.
M.a TERESA MIANA MENA. Farmacéutica Especialista en farmacia hospitalaria. Servicio de Farma-cia. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
ANNA MILLÁN ÁLVAREZ. Médico Adjunto. Banc de Sang i Teixits Girona. Hospital Universitari Dr. Josep Trueta (Girona). Profesora asociada de Hematología. Universitat de Girona.
PAU MONTESINOS FERNÁNDEZ. Médico adjunto. Servicio de Hematología. Hospital Universitari i Politècnic La Fe (Valencia). Universitat de València.
SILVIA MONTOTO ALMIRALL. Médico adjunto. Departamento de Hemato-Oncología. St Bartholo-mew’s Hospital, Barts Health NHS Trust, Londres, RU. Queen Mary University of London.
JOSÉ M.a MORALEDA JIMÉNEZ. Jefe de Servicio. Servicio de Hematología y Hemoterapia. Unidad de TPH. Hospital Clínico Universitario Virgen de la Arrixaca (Murcia). Instituto Murciano de Investi-gación Biosanitaria (IMIB). Catedrático de Medicina. Universidad de Murcia. .
FEDERICO MOSCARDÓ GARCÍA. Médico adjunto. Servicio de Hematología. Hospital Universitari i Politècnic La Fe (Valencia). Universitat de València.
NÚRIA MUNDÓ ROSELL. DUE Dietista Nutricionista. Unidad de Dietética. Servicio de Endocrinolo-gía y Nutrición. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
TERESA OLIVÉ OLIVERAS. Médico Adjunto, Especialista en Hematología y Hematología y On-cología Pediátricas. Unidad Pediátrica de Trasplante de Progenitores Hematopoyéticos. Hospital Universitari Vall d´Hebron (Barcelona). Vall d´Hebron Institut de Reserca. Universitat Autònoma de Barcelona.
SANDRA ORTEGA SÁNCHEZ. Médico adjunto. Banc de Sang i Teixits / Unitat d’afèresi. Hospital Universitario de Bellvitge (L’Hospitalet de Llobregat).
NURIA PARDO GARCÍA. Jefe Clínico, Directora de Hospitalización y Hospital de día. Servicio de Pediatría. Hospital de la Santa Creu i Sant Pau (Barcelona). Institut de Recerca Hospital Santa Creu i Sant Pau. Profesor asociado. Universitat Autònoma de Barcelona.
ROCÍO PARODY PORRAS. Médico Adjunto. Servicio de Hematología/ Unidad de Trasplante Hema-topoyético. Institut Catalá d’Oncologia, Duran i Reynals (Barcelona).
LUIS PINTOR PÉREZ. Psiquiatra Consultor-1. Servicio de Psiquiatría. Unidad de Psiquiatría de Enlace Hospitalaria. Hospital Clínic Universitari (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Profesor Clínico de Psiquiatría. Universidad de Barcelona.
JOSÉ LUIS PIÑANA SÁNCHEZ. Facultativo especialista en Hematología. Unidad de Trasplante He-matopoyético. Hospital Clínico Universitario (Valencia). Universidad de Valencia.
FELIPE PROSPER CARDOSO. Co-Director de Hematología. Servicio de Hematología. Clínica Uni-versidad (Navarra). Centro de investigación Médica Aplicada (CIMA). Acreditado Catedrático de He-matología. Universidad de Navarra.
IX
LLUIS PUIG ROVIRA. Director Asistencial. Banc de Sang i Teixits (Barcelona).SERGIO QUEROL GINER. Director. Servicio de Terapia Celular. Banc de Sang i Teixits (Barcelona).JOSEP M.a RIBERA SANTASUSANA. Jefe del Servicio de Hematologia Clínica. Institut Català d’On-
cologia. Hospital Germans Trias i Pujol (Badalona). Institut de Recerca Josep Carreras / Campus HGTiP (Badalona). Profesor Titular de Medicina-Hematología. Universitat Autònoma de Barcelona.
JOSÉ RIFÓN ROCA. Consultor Clínico. Hematología y Área de Terapia Celular. Clínica Universidad de Navarra (Pamplona). Instituto de Investigación Sanitaria de Navarra (IDISNA). Profesor Asociado de Hematología. Universidad de Navarra .
MAR RIVEIRO-BARCIELA. Médico Adjunto. Servicio de Hepatología-Medicina Interna. Hospital Uni-versitari Vall d’Hebron (Barcelona). CIBERehd. Universitat Autònoma de Barcelona.
SUSANA RIVES SOLÀ. Médico Adjunto Consultor. Servicio de Hematología y Oncología pediátri-cas. Hospital Sant Joan de Déu de Barcelona (Barcelona). Fundació Sant Joan de Déu-Hospital Sant Joan de Déu. Universitat de Barcelona.
MARTA RODRIGUEZ ALIBERAS. Médico Adjunto, Especialista en Hematología. Banc de Sang i Teixits- Centre Vall d’Hebron (Barcelona).
LAURA ROSIÑOL DACHS. Médico adjunto. Servicio de Hematología. Hospital Clínic Universita-ri (Barcelona). Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Universitat de Barcelona.
ALBERT SAIZ HINAREJOS. Consultor Senior. Servicio de Neurologia/Unidad Neuroinmunologia. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
NURIA SÁNCHEZ ABAD. Psicóloga Especialista Senior. Servicio de Psiquiatría. Unidad de Psico-On-cología. Hospital Clínic Universitari (Barcelona). Universitat de Barcelona.
FERMÍN SÁNCHEZ-GUIJO MARTÍN. Médico Adjunto. Servicio de Hematología. Hospital Universi-tario (Salamanca). Instituto Biosanitario de Salamanca (IBSAL). Profesor Asociado de Ciencias de la Salud. Universidad de Salamanca.
ANDRÉS SÁNCHEZ SALINAS. Médico Adjunto. FEA. Servicio de Hematología y Hemoterapia. Uni-dad de TPH. Hospital Clínico Universitario Virgen de la Arrixaca (Murcia). Instituto Murciano de Investigación Biosanitaria (IMIB). Universidad de Murcia.
JUAN MANUEL SANCHO CIA. Coordinador Asistencial. Servicio de Hematología Clínica. Institut Català d’Oncologia. Hospital Germans Trias i Pujol (Badalona, Barcelona). Institut de Recerca Josep Carreras / Campus HGTiP (Badalona). Universitat Autònoma de Barcelona.
MIGUEL A. SANZ ALONSO. Jefe de Servicio. Servicio de Hematología. Hospital Universitari i Poli-tècnic La Fe (Valencia). Profesor Titular. Universitat de València.
JAIME SANZ CABALLER. Médico Adjunto. Servicio de Hematología. Hospital Universitario i Politèc-nic La Fe (Valencia). Profesor Asistencial. Universitat de València.
GUILLERMO F. SANZ SANTILLANA. Jefe de Sección. Servicio de Hematología. Hospital Universi-tari i Politècnic La Fe (Valencia). Profesor Asistencial. Universidad de València.
DAVID SERRANO SIMONNEAU. Médico Adjunto. Servicio de Hematología. Hospital General Universitario Gregorio Marañón (Madrid). Instituto de Investigación Sanitaria Gregorio Marañón (IISGM). Profesor Colaborador. Universidad Complutense de Madrid.
JULIÁN SEVILLA NAVARRO. FEA Hematología y Hemoterapia. Servicio de Hemato-Oncología Pe-diátrica. Servicio de Transfusión. Hospital Infantil Universitario Niño Jesús (Madrid). Fundación In-vestigación Biomédica Hospital Infantil Universitario Niño Jesús. Grupo Clínico Vinculado CIBERER.
CARLOS SOLANO VERCET. Jefe de Servicio. Servicio de Hematología y Hemoterapia. Hospital Clínico Universitario (Valencia). Instituto de Investigación Sanitaria (INCLIVA). Profesor Titular de Medicina. Universidad de Valencia.
X
MARIA SUAREZ-LLEDÓ GRANDE. Médico Adjunto. Servicio de Hematología / Unidad de TPH. Hospital Clínic Universitari (Barcelona). Institut de Recerca Josep Carreras/Campus Clínic-Univer-sitat de Barcelona.
ANNA SUREDA BALARÍ. Jefe de Sección. Servicio de Hematología Clínica. Institut Català d’On-cologia. Hospital Duran i Reynals (L’Hospitalet de Llobregat, Barcelona). Universitat de Barcelona.
MONTSERRAT TORRENT ESPAÑOL. Médico Adjunto. Servicio de Pediatría. Hospital de la Santa Creu i Sant Pau (Barcelona). Universitat Autònoma de Barcelona.
JUAN CARLOS VALLEJO LLAMAS. Jefe de Sección. Unidad de Trasplante Hematopoyético. Hos-pital Universitario de Donostia (San Sebastián). Universidad del País Vasco.
M.a LOURDES VÁZQUEZ LÓPEZ. Médico Adjunto. Servicio de Hematología / Unidad de Trasplante Hematopoyético. Hospital Universitario (Salamanca). Instituto de Investigación Biomédica de Sala-manca (IBSAL). Colaborador en Prácticas. Universidad de Salamanca.
SUSANA VIVES POLO. Médico Adjunto. Servicio de Hematología Clínica. Institut Català d’Oncolo-gia. Hospital Germans Trias i Pujol (Badalona). Institut de Recerca Josep Carreras / Campus HGTiP (Badalona).
LUCRECIA YÁÑEZ SANSEGUNDO. Médico Adjunto. Servicio de Hematología / Sección Clínica. Hospital Universitario Marqués de Valdecilla (Santander). Fundación Instituto de Investigación Mar-qués de Valdecilla (IDIVAL). Profesora Asociada de Hematología. Universidad de Cantabria.
XI
Al Biel, el meu primer nét, en el dia del seu 5è aniversari
PRÓLOGO A LA QUINTA EDICIÓN
¡Todo llega! Parece que fue ayer cuando, en abril de 2010, escribí, como frase final de la cuarta edición del Manual Práctico de Trasplante Hemopoyético “… he empezado a tomar notas para mejorar la próxima edición que espero poder realizar en 2016 coincidiendo con mi 65 aniversario”.
Mis deseos se han visto cumplidos y, a pocos días de dicho aniversario, estoy escribiendo el prólogo de la 5.a edición de un Manual que se ha convertido en un principal libro de consulta en el día a día de centenares de profesionales de habla hispana dedicados al Trasplante Hematopoyético.
No sé si llegaré a los 70 ni si, de llegar, aún tendré ánimos (o neuronas suficientes) para realizar una sexta edición, pero en cualquier caso estoy muy satisfecho de haber podido asegurar su continuidad gracias a los excepcionales co-editores de esta edición, que se han comprometido a seguir actualizando el Manual, y al apoyo incondicional de nuestros patrocinadores.
La principal novedad de esta 5.a edición es que, si bien se seguirá realizando una versión en papel, su contenido se ha elaborado pensando en el desarrollo de una aplicación para sistemas Android e iOS, que podrá descargarse en los próximos meses y permitirá varias mejoras fundamentales. En primer lugar, la fácil corrección de las erratas que suelen descubrirse al poco tiempo de manejar el Manual. En segundo lugar, la posibilidad de ir introduciendo las novedades destacables que nos hagan llegar los autores sin necesidad de esperar a una nueva edición. Estas modificaciones se irán actualizando automáticamente en todos los dispositivos. En tercer lugar, la agilidad de consulta, gracias a los hiperenlaces entre capítulos, la posibilidad de descargar la bibliografía de acceso gratuito referenciada, y el acceso a páginas web con material de utilidad, como las calculadoras de índices online. Finalmente, la no limitación de espacio (aspecto importante si tenemos en cuenta que la versión en papel excede las 700 páginas) y la posibilidad de incluir imágenes en color. En resumen, una serie de ventajas inimaginables hace unos pocos años.
No podría concluir este prólogo sin hacer mención específica a: — Los doctores Montserrat Rovira, Izaskun Zeberio y David Valcarcel, por su ayuda en la elaboración del índice y selección de autores, por la realización de gran parte de los capítulos, y por su compromiso de futuro.
— Los 99 autores que han colaborado desinteresadamente en la realización de los 90 capítulos del Manual y que han soportado mi bombardeo de preguntas, sugerencias, requerimientos y galeradas.
— Los alma mater de la edición en papel, la Dra. Carme Escofet (Ed. Antares), y de la versión electrónica, el Dr. Juan Pablo Burgués Gasión (Go-Space).
— Mis hijos, Enric y Josep, por su esfuerzo en la elaboración de la versión electrónica y de la portada, respectivamente.
— Mi esposa, Maria, por soportarme, una vez más, durante los más de 6 meses en que he estado prácticamente desaparecido para poder completar el Manual en los plazos previstos.
A todos ellos muchas gracias.
ENRIC CARRERASLa Vall d’en Bas, Girona
5 de agosto de 2016
PRÓLOGO A LA PRIMERA EDICIÓN
El trasplante hemopoyético constituye un procedimiento terapéutico de introducción relativamente re-ciente en la práctica clínica diaria. Este es probablemente el motivo, por el cual sigue en plena evolución. En efecto, tras las primeras experiencias con el trasplante de medula ósea realizadas por E. Donnall Thomas en las décadas de 1950-1960, comienza su tímida expansión mundial en la década de 1970, para experimentar un espectacular desarrollo en los años 1980. Pero aún en la década actual (y previsiblemente ello va a ocurrir también en el futuro), los cambios han sido tan numerosos que el trasplante de medula ósea, de un solo procedimiento se ha convertido en un amplio grupo de modalidades terapéuticas. Así, por ejemplo, si en los inicios las células progenitoras de la hemopoyesis se obtenían siempre a partir de la medula ósea, actual-mente se pueden emplear como fuente del material a trasplantar la sangre periférica o la sangre de cordón umbilical. En efecto, hoy en día disponemos de diversas técnicas que nos permiten movilizar las células progenitoras hacia la sangre periférica y recogerlas mediante un proceso de leucaféresis. Por otro lado, el número de progenitores hemopoyéticos contenidos en la sangre del cordón umbilical y de la placenta es suficiente como para garantizar un buen implante, por lo menos en niños o adultos de peso inferior a 40 kg. El hecho de que dispongamos de fuentes tan diversas, nos ha obligado incluso a un cambio terminológico. En efecto, ya no es idóneo –por restrictivo– hablar de trasplante de medula ósea, sino que es obligado referirnos en este contexto al trasplante de células progenitoras de la hemopoyesis o, simplemente, tras-plante hemopoyético. Otro factor que ha conducido a la ampliación de modalidades terapéuticas, es el tipo de donante. En los inicios, el trasplante habitual era el alogénico, realizado a partir de un donante familiar HLA-compatible (amén de raros trasplantes isogénicos, es decir, entre gemelos univitelinos). En la década de los 80 se generalizan dos modalidades adicionales. Por un lado, se introduce con frecuencia creciente el trasplante autogénico, es decir, la posibilidad de obtener células progenitoras a partir del propio paciente para reimplantarlas más tarde. Y por otro, se comienza a practicar el trasplante alogénico a partir de donan-tes no emparentados. Ello ha sido posible gracias a la creación de registros de donantes voluntarios y de bancos de cordón umbilical en numerosos países del mundo civilizado. Actualmente se dispone ya de más de 5 millones de donantes no familiares y más de 15.000 unidades de progenitores de cordón umbilical. Por último, la importancia asistencial del trasplante hemopoyético se ha acrecentado en los años recientes al haberse incorporado nuevas indicaciones para su empleo. Junto a las ya clásicas aplicaciones en aplasia medular, hemopatías malignas, inmunodeficiencias congénitas y otros defectos genéticos, se ha ampliado el uso del trasplante a las neoplasias sólidas (posibilitando megaterapias en dosis supraletales, seguidas de rescate hemopoyético) y, muy recientemente, al interesantísimo campo de la enfermedades autoinmunes. La posibilidad de curar pacientes con esclerosis en placas, dermatomiositis, esclerodermia o lupus eritematoso sistémico constituye un reto apasionante.
Teniendo en cuenta la creciente importancia clínica del trasplante hemopoyético, no es de extrañar que en la bibliografía médica crezca de forma muy intensa el número de artículos de investigación, revisiones, monografías y libros sobre la materia. En este sentido me es particularmente grato introducir la aparición del presente «Manual de Trasplante Hemopoyético» que a no dudarlo va a rellenar un hueco entre las fuentes bibliográficas del mundo hispano, en el que carecíamos de un texto dedicado a este campus. La excelente sistematización de las diversas materias, la gran calidad de las numerosas aportaciones basadas en la expe-riencia propia y lo atractivo de su presentación convierten este texto en enormemente interesante.
El Dr. Enric Carreras, director del Manual, los co-directores Dres. Salut Brunet, Montserrat Rovira, Jorge Sierra y Álvaro Urbano-Ispizua, así como la mayor parte de los colaboradores, pertenecen a lo que podría-mos denominar la segunda generación de profesionales dedicados a esta materia. Para los de primera gene-ración –entre los que me cuento– el trasplante hemopoyético era un procedimiento extraordinario, heroico o casi milagroso, por lo cual nuestra actitud al respecto era, con frecuencia, excesivamente personalista. Por contra, los directores de este texto pertenecen a la mencionada segunda generación, ya muy numerosa en España, para la cual este procedimiento constituye una actividad profesional normal u ordinaria, parangona-ble al cultivo de cualquier otra especialidad médica. Ello hace que sus miembros se caractericen por una gran capacidad de colaboración, ayuda mutua y coexistencia civilizada. Este grupo de excelentes profesionales ha recogido con notable eficacia el testigo de los pioneros elevando de modo admirable el nivel asistencial, docente e investigador en el terreno que nos ocupa.
A mi juicio, el presente «Manual de Trasplante Hemopoyético» va a constituir un notable éxito editorial y seguirá apareciendo con regularidad en el futuro. Este es, por lo menos, mi ferviente deseo.
CIRIL ROZMAN
PRÓLOGO A LA SEGUNDA EDICIÓN
The transplantation of bone marrow was used in the 1970s and 1980s to treat patients with leukemia with marrow from an HLA matched sibling. As the success of this modality increased, additional subjects with a variety of hematopoietic diseases were considered. In the 1980s and 1990s bone marrow transplantations were carried out increasingly using peripheral blood or cord blood as the source of donor stem cells. For this reason the field became known as hematopoietic cell transplantation rather than bone marrow trans-plantation.
During the 1970s and 1980s there was an increasing knowledge of HLA, the major histocompatibility systems in man, and greatly improved technology resulted in recognition of the important components of the HLA system. During the 1980s and 1990s it became possible to identify HLA matched donors unrelated to the patient. This made it possible to recruit and HLA-type large panels of volunteer unrelated donors for patients without a suitable family donor.
Donor registries were created in England and the United States and now include many countries. The Ca-rreras Foundation played a major role in establishing the marrow registry in Spain. At the present time, there are some 6 million volunteer donors worldwide, the majority of whom are Caucasians. There is still a great need for volunteers among other ethnic groups, including Spanish-speaking individuals in the Americas.
In the 1990s and early 2000s two major developments are being exploited. The first is specific adoptive immunotherapy through identifying a single cell of desired reactivity, and cloning and growing the cell with the aid of interleukin-2. The second is the achievement of hematopoietic cell grafts without the use of myeloa-blative regimens, socalled minitransplants. These transplants are currently being explored for hematopoietic diseases and autoimmune diseases.
These developments in the field of hematopoeitic cell transplantation mean that increasing thousands of transplants are being done worldwide each year with a particularly rapid growth in the Spanish speaking countries.
The first edition of the «Manual de Trasplante Hemopoyético» met the need for a condensed manual in Spanish. The second edition has been expanded to include the most recent developments while retaining the format of a manual for maximal utility by Spanish-speaking physicians, nurses and technicians. It provides an easily accessible reference guide for those involved in dealing with the complexities of the rapidly changing field of hematopoietic cell transplantation.
E. DONNALL THOMAS
PRÓLOGO A LA TERCERA EDICIÓN
It gives us enormous pleasure to be invited to contribute an introduction to the third edition of this ma-nual of haemopoietic stem cell transplantation. We have watched the development of this project with great interest and have been very impressed with the vision, clarity of thought and hard work of the editors. This latest edition builds on the work presented in the previous versions, incorporating additional information on transplant indications, up-dating practice according to the latest research and providing important recent references. The original editors have been joined by co-workers from other major transplant units thus giving a wide perspective of management based on a huge collective experience.
The management of patients by transplantation is an exacting art. It requires not only understanding and experience of the underlying haematological problem, but also the practice of modern internal medicine and great attention to detail. For the haematology trainee the decision making process is challenging and complex. The ability to refer to an up to date manual of common transplant indications, complications and outcomes is invaluable in their daily work. The size and tabular format are designed to make the information readily accessible. The authors are themselves highly competent practitioners of transplantation and convey their knowledge in a highly informative and readable fashion. The European Group for Blood and Marrow Transplant (EBMT) is composed of transplant physicians throughout the world who have constantly demons-trated their willingness to share their knowledge, support their colleagues and develop the field. There has been a long-standing commitment to the ongoing education of both experienced and less experienced hae-matologists. In this context we are delighted with this valuable contribution from our Spanish colleagues and congratulate them on their achievement!.
JANE APPERLEY ELIANE GLUCKMAN
ALOIS GRATWOHLJanuary 2004
El pasado es un prólogo (William Shakespeare)PRÓLOGO A LA CUARTA EDICIÓN
Jamás hubiera podido imaginar cuando me propuse editar un pequeño Manual de bolsillo dedicado al Trasplante Hemopoyético, que 14 años después estaría escribiendo el prólogo de la cuarta edición gracias al éxito alcanzado por las tres anteriores. Mi intención inicial era resumir los conocimientos prácticos adqui-ridos a lo largo de 20 años de mis maestros (Dr. Albert Grañena y Profesor Ciril Rozman), de centenares de horas leyendo artículos, de muchos días asistiendo a congresos y, muy en especial, de los 900 trasplantes realizados desde que empecé a dedicarme de pleno a esta modalidad terapéutica en 1978. Pensaba en hacer algo similar a las Guías Clínicas que elaboré en 1980 para nuestra Unidad pero en formato de libro de bolsillo, para poder disponer de él en cualquier momento del día a día asistencial, enfocado a los aspectos prácticos del trasplante, y con poco texto y muchas tablas que facilitaran una rápida localización y lectura. Al elaborar el índice de capítulos se hizo patente que mi objetivo era inalcanzable si no contaba con la ayuda de mis colegas dedicados al Trasplante Hemopoyético en los Hospitales Clínic, Sant Pau y Materno-Infantil de la Vall de Hebron. Afortunadamente, todos ellos aceptaron participar en el proyecto sin vacilar.
Para esta primera edición conté, además, con la inestimable ayuda de mi compañero de ambulatorio en épocas de juventud, el Dr. Ramon Limiñana, en aquel entonces Director de AMGEN España quien, sin dudarlo ni un momento, aceptó sufragar la primera edición, con la experiencia en la elaboración de libros de bolsillo de mi buen amigo el Dr. Josep Mensa, y con el buen hacer de la Dra. Carmen Escofet, inagotable editora de todos sus manuales y a partir de entonces de todos los míos.
Aquel primer Manual de 1998, que contaba con 30 colaboradores y 400 páginas, y que amablemente aceptó prologar el Profesor Ciril Rozman, dejaba mucho que desear. Además de una portada horrible, di-señada por mí, tenía evidentes deficiencias y carencias. Con todo, fue tal su éxito que en tan sólo 2 años ya habíamos preparado una segunda edición revisada y ampliada a 75 capítulos con 550 páginas y más de 40 colaboradores, con una portada digna (diseñada por mi hijo Josep) y con un prólogo de excepción al haber sido realizado por el Profesor Donald Thomas, padre espiritual de todos los trasplantólogos de nuestra generación.
A pesar de que la segunda edición se agotó de nuevo en pocos meses, esperamos hasta el 2004 para reali-zar la tercera ya que durante aquellos años no se habían producidos avances lo suficientemente importantes como para justificar una nueva edición. En ella seguimos un formato similar al anterior pero contamos ya con 50 colaboradores (destacando la incorporación de los compañeros del Hospital Germans Trias y Pujol de Badalona), casi 600 páginas y un prólogo de lujo firmado por tres de los grades del Trasplante Hemopoyético en Europa, los Dres. Jane Apperley, Eliane Gluckman y Alois Gratwohl.
Coincidiendo con aquella edición tuve el honor de ser nombrado editor del “Handbook on Haema to-poietic Stem Cell Transplantation” del European Group for Blood and Marrow Transplantation (EBMT). La experiencia adquirida en la cuarta y quinta edición de dicho Manual me sirvió para ver que su apertura a quien más supiera sobre cada tema, independientemente de su lugar de trabajo, si bien hacía mucho más compleja la edición, enriquecía mucho su contenido. Por ello, cuando me planteé esta nueva edición, decidí ampliar los límites originales de Barcelona a todo el país. Con placer puede observar que todos los colegas a quienes se lo ofrecí aceptaron participar de forma inmediata y con gran entusiasmo. Ahora, con el Manual 2010 a punto de entrar en imprenta, con más de 670 páginas, 86 capítulos y 80 colaboradores tan sólo la-mento no haber podido incluir, ante el riesgo de dejar de tener un libro de bolsillo, a todos los compañeros que, destacando en un tema en concreto, hubieran merecido participar en algunas de las secciones; a todos ellos pido disculpas. Además, esta nueva etapa del Manual me obligó a reestructurar el consejo editorial y a distribuir la coordinación de las distintas secciones entre la Dra. Montserrat Rovira, mi brazo derecho desde que se incorporó al equipo de trasplante hace 21 años, y la Dra. Carmen Martínez, mi brazo izquierdo desde que me hice cargo de la dirección del Programa de Trasplante del Clínic hace 15 años; sin su apoyo y buen hacer esta nueva edición no habría sido posible.
Como colofón a este largo prólogo quiero hacer llegar mi agradecimiento a:— los pacientes y familiares de los casi 2000 trasplantes realizados a lo largo de más de 32 años, por la
oportunidad única que me han brindado para ir aprendiendo, día a día, sobre las peculiaridadesde esta modalidad terapéutica,— todos los colaboradores de esta cuarta edición, por los excelentes capítulos realizados y su disponibi-
lidad para ajustarse a los cortos plazos de entrega dados,Hasta tal punto ha sido para mi un placer la experiencia y satisfactorio el resultado alcanzado, que ya he
empezado a tomar notas para mejorar la próxima edición que espero poder realizar en 2016 coincidiendo con mi 65º aniversario.
ENRIC CARRERASAbril 2010
El médico sabe por los libros y demuestra por la experiencia (Ramon Llull)
XV
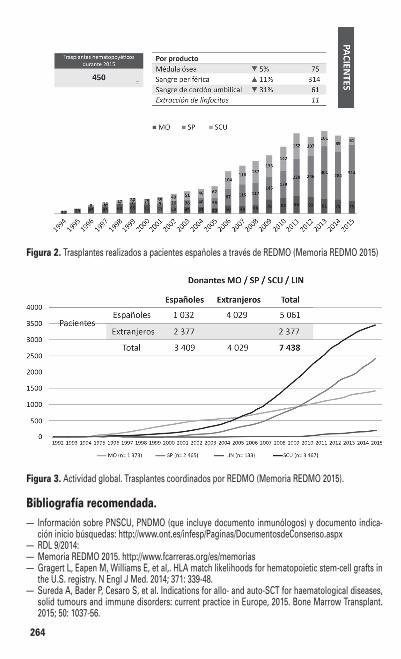
6MP 6-mercaptopurinaa añosAC arabinósido de citosinaAc anticuerposAcMo anticuerpo(s) monoclonal(es)ADAC altas dosis de ACADR adriamicinaAF anemia de FanconiAg antígeno(s) AgCMV antigenemia CMVAIR acondicionamiento de intensidad
reducidaalo- alogénicoAM aplasia medularAMA acondicionamiento mieloablativoAMO aspirado de medula óseaAnB anfotericina Baprox. aproximadamenteATB antibiótico(s)ATG globulina antitimocíticaauto- autogénico/autólogoAZA azacitidinaBGN bacilo gram negativoBMDW Bone Marrow Donor WorldwideBMO biopsia de medula óseaBMT Bone Marrow TransplantationBOS síndrome de bronquiolitis obliteranteBu busulfán cap. capítuloCB crisis blástica cGy centigreyCIBMTR Center for International Blood &
Marrow Transplant ResearchClcr clearence creatinina (mL/min)CMV citomegalovirusCMN celularidad mononucleada CNT celularidad nucleada total crom. cromosomaCsA ciclosporina ACSF factor estimulante de coloniasCVC catéter venoso centralCy ciclofosfamidaCyPT ciclofosfamida posTPHd día(s)DE donate emparentadoDLT eliminación de linfocitos TDMSO dimetilsulfóxidoDNB daunoblastinaDnE donante no emparentadoDR locus DR del sistema HLAEAI enfermedad(es) autoinmune(s)EBMT European Group for Blood and Marrow
EVOH enfermedad veno-oclusiva hepáticaFA fase aceleradaFAB clasificación Franco-Américo-Británica
de las leucemiasFac. factoresFBS fibrobroncoscopiaFC fase crónicaFG filtrado glomerular (mL/min) Fluda fludarabinafrac. fraccionadaG-CSF factor estimulante de colonias granulo-
cíticasGETH Grupo Español de Trasplante Hemato-
poyético y terapia celularGI gastrointestinalGM-CSF factor estimulante de colonias granulo-
cito-macrofágicasGy grey (unidad de radiación)h hora(s)HBsAg antígeno AustraliaHdD Hospital de DíaHEPA high-efficiency particulate arresting
filtersHLA antígenos leucocitarios humanosHTLV human T leukemia virusICC insuficiencia cardíaca congestivaICN inhibidor de la calcineurinaICT irradiación corporal total id. idénticoIDA idarrubicinaIFI infección fúngica invasivaIFN interferónIg inmunoglobulinasIH insuficiencia hepáticaIL interleucinaILD infusión de linfocitos del donanteIM intramuscularINT irradiación nodal totalIPS síndrome de neumonía idiopáticaIR insuficiencia renalIS inmunodepresióniso- isogénicoIT intratecalITA irradiación tóraco abdominalITK inhibidores de la tirosinkinasaIV intravenosoIKS índice de KarnofskyLA leucemia agudaLAF habitación con flujo laminar de aire
LNH linfoma no-hodgkinianoLT linfocitos Tm mes(es)MA mieloablativoMAT microangiopatía trombótica MEL melfalánmetilPDN metilprednisolonaMFP mielofibrosis primaria Mg magnesiomin minutosMM mieloma múltipleMMF micofenolato de mofetiloMO medula óseaMRT mortalidad relacionada con el trasplan-
teMTX metotrexatoNE nutrición enteralNK natural killerNMA no mieloablativoNMDP National Marrow Donor ProgramNP nutrición parenteralONT Organización Nacional de Trasplantespac. paciente(s)PCR reacción en cadena de la polimerasaPDN prednisonaPF perfusión continuaPFR pruebas funcionales respiratoriasPh cromosoma FiladelfiaPH progenitores hemopoyéticosPJ Pneumocystis jiroveciiPL punción lumbarPMO progenitores de medula óseaPSCU progenitores de sangre de cordón
umbilicalPSP progenitores de sangre periféricaPVC presión venosa centralQT quimioterapiaR rituximabRC remisión completaRC ≥2 segunda o posteriores remisiones
completasRC1 primera remisión completaREC recidiva/recaídaREDMO Registro de Donantes de Médula ÓseaRM (o RMN) resonancia magnéticaRP remisión parcial
RT radioterapiaRx radiografíaSC subcutáneoSCU sangre de cordón umbilical sem semana(s)SF suero fisiológico SG5% suero glucosado 5% SGS suero glucosalino síndr. síndromeSL sublingualSLE supervivencia libre de enfermedadSLP síndromes linfoproliferativos SMD síndromes mielodisplásicos SNC sistema nervioso centralSOS síndrome de obstrucción sinusoidalSP sangre periféricaSRV supervivencia Tacro tacrolimust-PA activador del plaminógeno tisularTA temperatura ambienteTAMO trasplante autogénico de medula óseaTAPH trasplante autogénico de progenitores
hemopoyéticosTASP trasplante autogénico de progenitores
de sangre periféricaTC ( o TAC) tomografía computarizadaTHS tratamiento hormonal sustitutivoTIS tratamiento inmunosupresorTMO trasplante de medula óseaTNF tumor necrosis factorTPH trasplante de progenitores hemopoyé-
ticosTPSP trasplante de progenitores de sangre
periféricaTT tiotepatto. tratamientoVEB virus de Epstein-Barr VHB virus de la hepatitis B VHC virus de la hepatitis C VHS virus del herpes simpleVIH virus de la inmunodeficiencia humanaVO vía oralVP (VP-16) etopósidoVRS virus respiratorio sincitialVVZ virus varicela zóster
XVII
ÍNDICE DE CAPÍTULOS
Capítulo 1. GENERALIDADES.1.1 Fundamento y modalidades de TPH (E. Carreras) . . . . . . . . . . . . . . . . . . . . . . . . . 11.2 La unidad de TPH (E. Carreras, M. Rovira) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.3 Acreditación de Centros de Trasplante (F. de Arriba, E. McGrath) . . . . . . . . . . . 13
Capítulo 2. INDICACIONES Y RESULTADOS. 2.1 Indicaciones del TPH (A. Sureda, E. Carreras) . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.2 Leucemia mieloblástica aguda (S. Brunet) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 232.3 Leucemia promielocítica aguda (M. A. Sanz, P. Montesinos, F. Lo Coco) . . . . . 372.4 Leucemia mieloblástica aguda del niño (M. González-Vicent, M. A. Díaz) . . . . 452.5 Leucemia linfoblástica aguda del adulto (J. M. Ribera, S. Vives) . . . . . . . . . . . . 532.6 Leucemia linfoblástica aguda del niño (S. Rives, I. Badell) . . . . . . . . . . . . . . . . . 632.7 Síndromes mielodisplásicos y síndromes mielodisplásicos/mielopro-
Capítulo 7. OTROS ASPECTOS DE INTERÉS.7.1 Impacto de los polimorfismos génicos en el TPH (D. Gallardo) . . . . . . . . . . . . 6557.2 Terapia celular en el TPH y en otras aplicaciones (F. Prosper, F. Sánchez-Guijo,
C. del Cañizo) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6597.3 Papel del microbioma en el TPH (A. Bermúdez, L. Yañez) . . . . . . . . . . . . . . . . 6677.4 Farmacos más empleados en el TPH (N. Creus, C. Carcelero) . . . . . . . . . . . . . 673
ACREDITACIÓN DE CENTROS DE TRASPLANTE(F. de Arriba, E. McGrath) 1.3
1. INTRODUCCIÓN. — JACIE (Joint Accreditation Committee of ISCT and EBMT) es un comité conjunto creado por la colaboración del EBMT y la International Society For Cellular Therapy (ISCT), dos organizaciones profesionales sin ánimo de lucro, cuya finalidad es promover la calidad y seguridad en los diferentes procesos implicados en el TPH.
— JACIE emite un certificado a aquellos centros que, de forma voluntaria, se someten a su valoración y consiguen demostrar que su actividad, en el ámbito del TPH, se ajusta a todos los requisitos (conocidos como estándares) que JACIE tiene establecido.
— La certificación JACIE no es imprescindible para que un centro tenga un programa de TPH que funciona correctamente, aunque si hay países donde la acreditación ya es un requisito administrativo (http://www.jacie.org/about/national-regulations). Sin embargo, la implantación de un sistema de gestión de calidad que se ajuste a los estándares JACIE, además de garantizar que el programa de TPH se desarrolla con criterios internacionales de excelencia, aporta algunos beneficios adicionales, puesto que la certificación:
1) Es reconocida y avalada por la comunidad científica y las autoridades.2) Aborda todos los aspectos relacionados con el TPH. 3) Mejora de resultados clínicos e impacta positivamente en los aspectos operacionales. 4) Permite la difusión de la cultura de calidad entre los empleados que participan en el programa. — El principal inconveniente de la implantación de un sistema de gestión de calidad exigente, como puede ser JACIE, estriba en la necesidad de dedicar recursos materiales y humanos adicionales a la actividad asistencial propia de un programa de TPH.
— Desde el 1 de Marzo de 2015, los centros que deseen contar con la certificación JACIE deben aplicar la 6.a edición de los estándares http://www.jacie.org/standards/6th-edition-2015) y, para ello, deben tener en cuenta que:
– Cuando los estándares y la legislación aborden aspectos similares, se deberá aplicar la norma más exigente
– El centro que solicita la certificación es el encargado de demostrar la adherencia a cada uno de los estándares, así como justificar aquellos que no aplican para su organización
– La redacción de los estándares es lo suficientemente genérica para que se pueda aplicar en los distintos países, con sistemas de salud diferentes, y a diferentes centros, con esquemas de organización dispares. No obstante, dependiendo de la exigencia requerida para cada estándar, hay diferentes grados de permisividad: “shall” (cumplimiento estricto), “should” (recomendado, pero se admiten alternativas) o “may” (más laxo)
– Los estándares están divididos en 5 partes y varios apéndices. En este capítulo nos referiremos fundamentalmente a la Parte B (Programa clínico), y a aquellos estándares de la Parte CM (Obtención de MO) y Parte C (Obtención de PHSP) que habitualmente son responsabilidad del área clínica. La parte D, que cubre las actividades relacionadas con el procesamiento de PH, queda fuera del alcance de este capítulo
– Cada requisito está definido por una(s) letra(s) (A, B, CM, C, D) que remite a cada una de las 5 partes en que se han dividido los estándares, y una sucesión de números. En este capítulo la referencia a los estándares se realiza siguiendo esta numeración, aunque siempre refiriéndonos a los requisitos más genéricos
14
2. IMPACTO DE LA CERTIFICACIÓN JACIE EN LA UNIDAD DE TRASPLANTE. — Aunque hay pocos estudios que evalúen este aspecto, los puntos más relevantes que podemos destacar son:
– Un estudio observacional de Gratwohl A, 2011 y actualizado recientemente (Gratwohl A, 2014), constata un incremento en la SG entre el 11 – 14% en los pacientes sometidos a trasplante en centros que cuentan con la certificación JACIE si se compara con aquellos no acreditados
– Impacto menor en el contexto del trasplante autólogo, debido a la menor complejidad del procedimiento y a que el resultado final está condicionado de forma fundamental por el riesgo de la enfermedad de base
– Mejora de la seguridad de los donantes de PH familiares con la implantación de los estándares JACIE (5.a edición) (Anthias, 2016)
3. IMPLANTACIÓN DE UN SISTEMA DE GESTIÓN DE CALIDAD EN UNA UNIDAD DE TPH. — El sistema de gestión de calidad es la estructura organizativa que permite dar cumplimiento a los diferentes requisitos relacionados con el TPH. Su diseño debe ser: 1) Global para cubrir todos los aspectos del programa. 2) Flexible para adaptarse a los avances científicos y los cambios en la legislación. 3) Capaz de generar dinámicas de mejora contínua.
— En primer lugar, para que la Unidad Clínica pueda alcanzar la certificación JACIE, debe contar con: 1) Autorización de los responsables sanitarios competentes (B1.3). 2) Acuerdos con terceros (B4.6) que garantice el cumplimiento de los estándares en actividades
no cubiertas por el programa de TPH: obtención, procesamiento de PH (B1.2) y distribución de PH (B7.6), inmunología e histocompatibilidad (B2.11), farmacia (B3.8; B7.4), oncología radiote-rápica (B7.5), estudios de quimerismo (B2.12), fotoaféresis extracorpórea (B7.9).
3) Cumplir la normativa relativa tanto a la investigación clínica (B8), como a los registros clínicos o del programa de TPH (B10) y
4) Cumplimentación de los registros MED-A requeridos por el EBMT (B9).
4. ESTRUCTURA CLÁSICA DE UN SISTEMA DE GESTIÓN DE CALIDAD [1].
1.– Planificar 5.– Revisión
3.– Registros
4.– Identificar tendencias negativas2.– Acción según protocolosMEJORA
CONTINUA
15
PlanificarDefine la estructura del sistema de gestión de calidad (Manual de Calidad), de los procedimientos
generales (controlan los aspectos relacionados con la gestión de calidad) y los procedimientos normalizados de trabajo (PNTs) — Manual de Calidad (B4.1; B4.2) — Organigrama (B4.3; B4.4) — Control documental (B4.5); — Otros: Trazabilidad (B4.11); Interrupción de las operaciones del programa clínico (B4.12) — PNTs (B5; B3.3.3; B3.3.4; B3.7.3, B3.7.4), Evaluación del donante (B6), Cuidados del receptor (B7)
Acción según los protocolosPara poder desarrollar la actividad según lo planificado, el centro debe contar con las instalaciones
adecuadas y el personal adiestrado para el desempeño de las funciones que tiene asignadas: — Instalaciones y Servicios de apoyo requeridos (B2); Acuerdos con terceros (B4.6); — Personal (B3): a) Director del Programa (B3.1), facultativos (B3.2) y su formación requerida (B3.3) b) Médicos en formación (B3.4); c) Enfermeros (B3.7); d) Farmacéutico (B3.8); e) Otros: Facultativos especialistas (B3.9), Coordinador de Calidad (B3.10), Personal de apoyo (B3.11)
— Seguridad y Salud Laboral (B2.14, B2.15)Registros
Dejan constancia tanto de la adherencia de la actividad con lo planificado como de las desviaciones que hayan podido ocurrir — Análisis de resultados clínicos (B4.7) — Auditorías internas de calidad (B4.8) — Productos de terapia celular con cultivo microbiológico positivo (B4.9) — Incidencias (errores, accidentes, efectos adversos) (B4.10) — Indicadores de calidad — Validación del proceso de obtención de PHMO (B4.13) — Otros: Fichas de formación, actas, informes de calidad, informes de validación, etc….
Identificación de las tendencias negativasPermite poner en evidencia las desviaciones y estrategias para corregirlas.
— Medidas Correctoras (B4.10.5)Revisión
Análisis periódico del funcionamiento de los diferentes elementos del sistema de gestión de calidad — Revisión anual por el Director (B4.1.2) — Revisión cuatrimestral del sistema de Gestión de Calidad (B4.2.2) (indicadores de calidad, acciones correctoras, incidencias, etc…)
— Revisión de PNTs bienal (B5.3.8) — Revisión de acuerdos con terceros (B4.6.2)
PlanificaciónEn respuesta a la hallazgos obtenidos en la revisión, se elabora la planificación del siguiente ejerci-
cio y, de esta forma, se completa la estructura cíclica que genera la dinámica de mejora continua
[1] En los programas de TPH, con frecuencia es el área clínica donde se asume el proceso de obtención de PHMO (Parte CM) y, en ocasiones, también las aféresis de PHSP (Parte C). En estos casos, puede recaer sobre el área clínica la responsabilidad del cumplimiento de los estándares relativos a la codi-ficación y etiquetaje del producto (CM7 ó C7), el control del proceso (CM8 ó C8), almacenamiento del producto (CM9 ó C9) o su trasporte (CM10 ó C10).
16
5. EL PROCESO DE CERTIFICACIÓN JACIE. — La oficina de JACIE coordina y promueve la acreditación de centros y, entre sus actividades, mantiene una página web (www.jacie.org) con documentos e información que puede resultar de utilidad a los centros que quieren implantar un sistema de gestión de calidad. Desde JACIE también se organizan cursos que abordan los diferentes aspectos de la implantación y certificación de las unidades de trasplante.
5.1 Diseño y puesta en marcha del sistema de gestión de calidadEl centro debe diseñar los diferentes elementos que formarán parte del sistema de gestión de ca-
lidad. En la página web de JACIE, además de los estándares vigentes http://www.jacie.org/stan-dards/6th-edition-2015), está accesible un Manual en el que se da una información más detallada de cada uno de los estándares, y estrategias para su adecuada implantación (Manual-ed-6.pdf)
5.2 Inspección JACIE — El centro debe remitir a la oficina JACIE la solicitud de inspección de su programa de trasplante y transferir el pago de las tasas. Las tarifas se pueden consultar en http://www.jacie.org/applicants/fees
— Una vez que se ha formalizado el contrato con JACIE, el centro solicitante dispone de 30 días para remitir la documentación solicitada y, debidamente cumplimentado, un documento en formato Excel proporcionado desde la oficina de JACIE (CH-001-08-Inspection Checklist.xlsx)
— Durante la visita de inspección, los inspectores designados revisaran como el centro cumple con cada uno de los estándares y emitirán un informe que será remitido a la oficina JACIE
5.3 CertificaciónUna vez acreditado el cumplimiento con todos los estándares, la oficina de JACIE emite un certi-
ficado con una vigencia de 4 años. No obstante, al cabo de dos años, se va a solicitar al centro información específica con la intención de demostrar el funcionamiento del sistema de gestión de calidad y la adecuada adherencia a los estándares
Bibliografía recomendada.— Gratwohl A, Brand R, Niederwieser D, et al. Introduction of a Quality Management System and
Outcome After Hematopoietic Stem-Cell Transplantation. J Clin Oncol. 2011; 29: 1980-6.— Piccirillo N, Ausoni G, Chiusolo P, et al. Twenty years of unrestricted hematopoietic stem cell co-
llection and storage: impact of Joint Accreditation Committee International Society for Cellular Therapy Europe standards implementation on stem cell storage policy and resource utilization. Cytotherapy. 2013; 15: 519-21.
— Gratwohl A, Brand R, McGrath E, et al. Use of the quality management system “JACIE” and outco-me after hematopoietic stem cell transplantation. Haematologica. 2014; 99: 908-15.
— Jiménez M-J, Ferra C, García O, et al. The commitment and evaluation of the quality management plan by professionals from accredited stem cell transplant centers in Spain. Bone Marrow Trans-plant. 2014; 49: 990-2
— Anthias C, O’Donnell P V., Kiefer DM, et al. EBMT transplant centers with FACT-JACIE accreditation have significantly better compliance with related donor care standards. Biol Blood Marrow Trans-plant. 2016; 22: 514-9.
111
ANEMIA DE FANCONI Y OTRAS INSUFICIENCIAS MEDULARES (J. Sevilla, E. Gálvez) 2.10
ANEMIA DE FANCONI (AF).
1. ASPECTOS BÁSICOS. — La AF es un síndrome complejo que se caracteriza principalmente por insuficiencia medular progresiva, anomalías somáticas constitucionales, hipersensibilidad a agentes que producen entrecruzamientos en las cadenas del ADN y predisposición a desarrollar LMA-SMD y tumores sólidos.
— La prevalencia de AF es de 1 – 5 pacientes por millón, con una frecuencia de portadores heterocigotos de entre 0,3 – 1%, y se distribuyen en todas las razas y grupos étnicos.
2. DIAGNÓSTICO. — Las características clínicas y las alteraciones hematológicas son en la mayoría de los casos orientativas del diagnóstico. En ausencia de malformaciones, la orientación diagnóstica de una hipoplasia medular no es tan evidente.
Test de fragilidad cromosómica — Es la prueba diagnóstica básica de la AF. Se realiza en cultivos de linfocitos de SP y se basa en el análisis de roturas cromosómicas y cromatídicas, inducidas por un agente que produce entrecruzamientos en el DNA, tales como mitomicina C (MMC) y diepoxibutano (DEB)
— En caso de que el test fuera dudoso o negativo en SP y hubiera alta sospecha clínica, es recomendable realizar el test diagnóstico en fibroblastos de la piel
— Las pruebas de fragilidad cromosómica deben realizarse en todos los niños y jóvenes con aplasia medular, fallos medulares congénitos y SMD por las repercusiones que su diagnóstico implica en el tratamiento, y de manera muy especial en el TPH
— En casos de LMA y, en algunos tumores en pacientes con malformaciones también debe valorarse el realizar esta prueba
Estudios genéticosDesde el punto de vista genético, se conocen 18 grupos de complementación asociados a mu-
taciones en los correspondientes genes. La herencia es autosómica recesiva, excepto en el caso de los pacientes del grupo de complementación B, puesto que el gen FANCB está localizado en el cromosoma X. Más del 50% de los pacientes con AF poseen mutaciones en el gen FANCA
3. TRATAMIENTO DE PRIMERA LÍNEA. — El TPH es el único tratamiento que puede restaurar una hematopoyesis normal, pero no previene el desarrollo de tumores sólidos. El resto de manifestaciones de la enfermedad requiere su tratamiento específico (cirugía de las malformaciones, o tratamiento de los tumores).
112
3.1 Tratamiento de soporteLos pacientes que carecen de donante compatible para el TPH pueden recibir tratamiento sustitutivo
en espera de encontrar donante1. Soporte transfusional: cuando la anemia y trombocitopenia son intensas y sintomáticas2. Derivados androgénicos:
— Oximetolona: dosis de 2 – 4 mg/kg/día. Elevación de la Hb en el 75% de los pacientes, lo que previene la realización de un elevado número de transfusiones
— Danazol: dosis de 5 mg/kg/día. Su uso se está extendiendo por asociar menos efectos secun-darios virilizantes, pero su verdadera eficacia está por determinar
En ambos casos debe vigilarse la aparición de efectos secundarios (virilización, aceleración del cre-cimiento, disfunción hepática, peliosis hepatis y tumores hepáticos)
3. Factores de crecimiento: el G-CSF puede tener cierta eficacia en pacientes con neutropenia intensa e infecciones recurrentes, aunque actualmente su indicación es limitada
3.2 Terapia génicaSe ha demostrado eficaz en modelos murinos para el tratamiento del fallo medular. Los primeros
ensayos de terapia génica con vectores lentivirales en humanos con Anemia de Fanconi de grupo A están en marcha
3.3 TPH alogénico.
Indicación• El TPH debe realizarse sin dilación si el paciente entra en situación de pancitopenia grave (Hb <8
g/dL, neutrófilos <0,5 x10^9/L o plaquetas <20 x10^9/L)• El TPH en AF en fase de SMD o LMA tiene peor pronóstico, siendo muy debatido el uso de QT
citorreductora previa al TPHTipo de TPH preferibleHermano HLA idéntico > DnE 9 – 10/10 > SCU 6/6 (más de 5 x10^7 CNT/kg) > Haplo (muy escasa
experiencia)Estudios pre-TPH específicos
• En todos los casos de AM debe excluirse el diagnóstico de AF con un test de fragilidad cromosómica por su implicación para el acondicionamiento
• Realizar estudio de fragilidad cromosómica en donantes familiares• Debe descartarse SMD y leucemia aguda (modificación del acondicionamiento)
Acondicionamiento• El empleo de AMA clásicos está claramente contraindicado• En los TPH a partir de donantes familiares, el empleo de AIR con fludarabina y DLT del inóculo
ha generado excelentes resultados, con una incidencia de EICR mínima. Se ha abandonado el uso de ICT en el acondicionamiento. Esto no sólo ha mejorado los resultados en cuanto a SRV global, si no que probablemente también ha disminuido la incidencia de neoplasia secundarias, muy frecuentemente relacionadas con la aparición de EICR crónica
• Régimen recomendado: Cy 5 mg/kg/d IV x 4 días y Fluda 35 mg/kg/d IV x 4 días (días –5 a –2) y ATG conejo 3 mg/kg/d IV x 5 días (días –5 a –1) [1]
• En TPH-DnE se aconsejan regímenes con Fluda; el uso de ICT es controvertido• Régimen recomendado:
– ICT (300 cGy) ( –6), Cy 10 mg/kg/d IV x 4 días y Fluda 35 mg/kg/d IV x 4 días (días –5 a –2) [2]– Fluda 30 mg/m^2/d IV (días −9 a −4), Bu 1 mg/kg/d en dos dosis VO (días −5 y −4), Cy 20 mg/
kg/d IV (días −3 y −2), y Alemtuzumab 5 mg/m^2/d (día −7) seguido por 10 mg/m^2/d IV (días −6 a −4) [3]
⇒
113
Fuente de progenitores• TPH familiares HLA idénticos: se aconseja el uso de MO o SP con DLT, no obstante, la fuente
de PH es aún hoy debatida. Se rechaza la SP no DLT• TPH con SCU ha mejorado sus resultados al disminuir los fallos de injerto. La experiencia
reportada por Eurocord-Netcord y el EBMT alcanza una SRV global del 50% en los casos con identidad HLA, buena celularidad y empleando Fluda en el acondicionamiento
• El desarrollo del diagnóstico pre-implantacional permite seleccionar embriones no afectos de AF y, al mismo tiempo, HLA-compatibles con el paciente. Esto facilita la utilización de unidades de SCU para un TPH entre hermanos. Es un proceso complejo, difícil y caro, pero con el que se han obtenido algunos éxitos terapéuticos en diferentes enfermedades monogénicas, la primera la AF
• El TPH con donantes haploidénticos ha ampliado las posibilidades de alcanzar el TPH para algunos pacientes
Profilaxis EICR recomendadaCsA + esteroides (debido a la tendencia a la hiperglucemia se encuentra en estudio el uso de com-
binaciones CsA + MMF)Estudios a largo plazo recomendadosDespistaje tumores cabeza y cuello y genitales. Seguimiento de complicaciones endocrinológicasFactores pronósticosDel resultado del TPH: compatibilidad HLA donante/receptor, transfusiones recibidas, historia previa
de infecciones oportunistas, y edad >10 añosResultados del TPH
• La mayoría de las series actuales comunican SRV en torno al 95% de los casos en el caso de TPH de donante familiar HLA idéntico
• En el caso de DnE la SRV está en torno al 70% en la última serie del grupo europeo, aunque bastantes Centros, en pacientes seleccionados y de manera aislada, reportan resultados iguales a los del trasplante familiar (SRV 95%) (Chao, 2015; MacMillan, 2015)
• Recientemente se han comunicado resultados esperanzadores con donante haploidénticos con SRV 83% a 5 años (Zecca, 2014). Sin embargo, la experiencia general aún es muy escasa
[1] Protocolo GETMON basado en protocolo Minnesota. [2] Protocolo Minnesota. [3] Protocolo Grupo Alemán.
114
4. ALGORITMO TERAPÉUTICO.
Obse
rvac
ión
Recu
ento
hem
atol
ógic
o af
ecta
do
sin n
eces
idad
de
trans
fusió
nRe
cuen
to h
emat
ológ
ico
afec
tado
*
Fam
iliar
HLA
idén
tico
Tras
plan
te h
emat
opoy
étic
o
*Hab
itualm
ente
: Hb
<8 g
/dL, P
laque
tas <
50 x1
0^9/L
; Gr
anul
ocito
s <0,
5 x1
0^9/L
Tipa
je H
LA d
e pa
cien
tes y
fam
iliar
esM
ielo
disp
lasia
Diag
nóst
ico
Reco
gida
pro
geni
tore
s par
a te
rapi
a gé
nica
Búsq
ueda
don
ante
alte
rnat
ivo
No d
onan
teSi
don
ante
Trat
amie
nto
sopo
rte: D
anaz
ol, A
ndró
geno
s, Tr
ansf
usio
nes,
G-CS
F
NoSí
115
DISQUERATOSIS CONGÉNITA (DC).
1. ASPECTOS BÁSICOS. — Síndrome hereditario caracterizado por alteración en la pigmentación cutánea, distrofia ungueal, leucoplaquia oral, fallo medular y aumento del riesgo de tumores.
— Caracterizado por alteración de la reparación del telómero, se reserva el término DC para el cuadro clásico, si bien se han descrito otras enfermedades (telomeropatías) con esta misma base patogénica, pero que constituyen cuadros clínicos heterogéneos (fibrosis pulmonar, hepática, síndrome de Revesz, …).
— La incidencia anual es inferior a un caso entre un millón de recién nacidos.
2. DIAGNÓSTICO. — Las características clínicas son fundamentales para el diagnóstico. — El diagnóstico se basa en la medida de la longitud del telómero, que demuestra un telómero significativamente más corto (<primer percentil) que la media para la edad del paciente.
— Desde el punto de vista genético se han descrito mutaciones hasta día de hoy en DKC1, TERC, TINF2, NOP10, TERT, NPH2, TCAB1, RTEL1, CTC1, NHEJ11 y C16orf57. La confirmación del gen afectado sólo se consigue en un 60% de los casos.
3. TRATAMIENTO DE PRIMERA LÍNEA. — El TPH es el único tratamiento que puede restaurar una hematopoyesis normal (Gadalla, 2013).
3.1 Tratamiento de soporteLos pacientes que carecen de donante compatible para el TPH pueden recibir tratamiento sustitutivo
igual que los pacientes diagnosticados de AF• El tratamiento con derivados androgénicos como la oximetolona a dosis de 2 – 4 mg/kg/
día, o el uso de danazol a dosis incluso menores de 5 mg/kg/día, consigue mejores resultados que en la AF
3.2 TPH alogénico.
IndicaciónEl TPH debe realizarse sin dilación si el paciente entra en situación de pancitopenia graveTipo de TPH preferibleHermano HLA idéntico > DnEEstudios pre-TPH específicosEvaluar función hepática y pulmonar por mayor toxicidad a estos nivelesDescartar genéticamente la enfermedad en donantes familiaresAcondicionamiento
• El empleo de regímenes AMA clásicos está contraindicado• No existe un régimen de acond. que haya demostrado clara superioridad, ya que son pocos
los casos reportados• Régimen recomendado: Cy 200 mg/kg; o Cy 120 mg/kg + Fluda
Fuente de progenitoresEscasa experiencia, imposible comparar fuentes de progenitores
⇒
116
Estudios a largo plazo recomendadosDespistaje tumores secundarios. Seguimiento de complicaciones pulmonares, fibrosisResultados del TPH
• Son pocos los estudios que estudian estos pacientes de manera aislada. Los más recientes datos del CIBMTR estiman la supervivencia en un 57% a 5 años, con mejor SRV en el grupo trasplantado tras el año 2000 (65%)
• La principal causa de muerte es el fallo de implante y las complicaciones pulmonares. Otras series comunican supervivencias similiares en el grupo con donante familiar (71%), pero claramente inferiores con donante alternativo (31% a dos años) (Alter, 2009)
ANEMIA DE BLACKFAN DIAMOND (ABD).
1. ASPECTOS BÁSICOS. — La ABD constituye el síndrome congénito más frecuente en el que tiene lugar un fallo único de células eritroides. Se manifiesta mediante anemia macrocítica con retículocitopenia, presencia de hemoglobina F y elevación de adenosina desaminasa en los eritrocitos. Algunos pacientes también presentan neutropenia, trombocitosis o trombocitopenia. Los pacientes con ABD también poseen predisposición para el desarrollo de cáncer, principalmente SMD y LMA, siendo el riesgo de LMA a lo largo de la vida del 25%.
— En un 20% de los pacientes se produce resolución espontánea de la enfermedad. — Se trata de una enfermedad con un patrón de herencia mixta, aunque la mayor parte de estos pacientes poseen mutaciones esporádicas que en caso de transmitirse lo harán de modo autosómico dominante.
— La incidencia estimada es de 5 – 7 casos por millón de nacimientos vivos.
2. DIAGNÓSTICO. — La mayor parte de estos pacientes son diagnosticados durante el primer año de vida. El signo guía para el diagnóstico de la enfermedad es la anemia macrocítica con eritroblastopenia. Los niveles elevados de adenosina desaminasa eritrocitaria se han propuesto como bastante específicos de la enfermedad; sin embargo, el 16% de estos pacientes tienen valores normales de esta enzima.
— La confirmación diagnóstica se basa en la demostración de las mutaciones específicas en los genes que codifican proteínas ribosomales (RPS17, RPS19, RPS24, RPS7, RPS10, RPS26, RPS27, RPS28, RPS29, RPL5, RPL11, RPL15, RPL19, RPL26, RPL27, RPL31, RPL35A y TSR2). Recientemente se ha implicado también el gen GATA1. Hasta la fecha, el diagnóstico genético sólo se consigue en el 65% de los pacientes estudiados.
3. TRATAMIENTO DE PRIMERA LÍNEA. — El tratamiento inicial es prednisona (o prednisolona) a dosis de 2 mg/kg. En los pacientes que responden, se irá disminuyendo progresivamente hasta alcanzar una dosis a días alternos capaz de mantener los niveles de hemoglobina. Aproximadamente el 79% de los pacientes responden inicialmente a los corticoides, pero sólo un 60 – 70%, adquieren una independencia transfusional.
— En aquellos pacientes no respondedores a esteroides, y sin donante para trasplante hematopoyético, el tratamiento será de soporte con transfusiones de hematíes y terapia quelante (Vlachos, 2010).
117
3.1 TPH alogénico.
IndicaciónEl TPH debe realizarse en aquellos pacientes no respondedores a esteroides y transfusión depen-
dientes, con donante HLA idénticoTipo de TPH preferibleHermano HLA idéntico > DnE (mejores resultados desde el año 2000, aunque aún discutido) Estudios pre-TPH específicosDescartar genéticamente la enfermedad en donantes familiaresAcondicionamiento
• Los AMA clásicos (Bu-Cy) estarían indicados en niños entre 3 – 9 años• También se emplean regímenes: Bu (16 mg/kg) + Tiotepa 6 – 10 mg/kg) + Fluda (120 – 160 mg/m^2)• El empleo de AIR, que también ha demostrado utilidad, se reserva para adultos y niños con
toxicidades moderadasFuente de progenitoresAunque la experiencia es escasa, en general se recomienda el uso de MO como principal fuente de
progenitores seguido de SCU. No se recomienda el uso de PHSP por el aumento de EICR crónicaEstudios a largo plazo recomendadosDespistaje tumores secundarios. Seguimiento de complicaciones relacionadas con la sobrecarga de
hierro y alteraciones endocrinológicasResultados del TPH
• Escasos estudios con gran número de casos. Los datos del registro americano (Vlachos, 2010; Narla, 2011) demuestran que los TPH de antes del año 2000 tenían peores SRV que los trasplantados con posterioridad (47,1% vs. 72,4%)
• Además, se demuestra que los casos con donante alternativo han mejorado claramente sus resultados después del año 2000: 23,1% vs. 85,7%. Por este motivo, cada vez más centros opinan que los pacientes no respondedores a esteroides deberían proceder a trasplante en caso de tener cualquier donante compatible, aunque sigue siendo un tema discutido
• El registro italiano (Fagioli, 2014) reporta sus resultados recientemente: 86,6% en aquellos trasplantados tras el año 2000. No encuentran diferencias significativas según el tipo de donante (familiar – 80,4% – vs. no relacionado – 69,9% – ). De nuevo la edad es el factor más importante (100% menores de 10 años vs. 29,6% mayores de 10 años)
SÍNDROME DE SHWACHMAN-DIAMOND (SSD).
1. ASPECTOS BÁSICOS. — La alteración hematológica más frecuente de los pacientes con SSD es la neutropenia (85%). La anemia con reticulocitopenia (42 – 82%) y la trombocitopenia (24 – 88%) son otras manifestaciones de la enfermedad hematológica.
— Disfunción pancreática exocrina y en un 50% de los pacientes se producen alteraciones esqueléticas, con retraso en el crecimiento, baja estatura, anomalías torácicas y disóstosis metafisaria.
— Estos pacientes también muestran elevada predisposición al desarrollo de SMD y LMA, con un riesgo estimado del 19% a los 19 años y del 36% a los 30 años (Donadieu, 2013).
— La incidencia estimada es 1/76.500.
118
2. DIAGNÓSTICO. — Se basa en la sospecha clínica con alteraciones hematológicas tempranas del paciente, hipocelularidad en los aspirados de MO, y presencia de heces grasas. Se demuestran bajos niveles de tripsinógeno e isoamilasa pancreática.
— El diagnóstico genético confirma la enfermedad. El 90% de los casos tienen mutaciones en el gen Shwachman-Bodian-Diamond (SBDS) localizado en el cromosoma 7q11.
3. TRATAMIENTO DE PRIMERA LÍNEA. — La administración de enzimas pancreáticas y de vitaminas liposolubles son la primera opción para la insuficiencia pancreática. Este tratamiento no es necesario que sea de por vida, porque en cerca del 50% de los pacientes, la esteatorrea se resuelve espontáneamente con la edad.
— Cuando existe neutropenia grave y persistente existiendo, por tanto, riesgo de infección está indicado la administración de G-CSF. La dosis será la más baja que permita mantener unos niveles aceptables de neutrófilos.
3.1 TPH alogénico.
Indicación• El TPH debe realizarse en aquellos pacientes con donante HLA idéntico no respondedores a
G-CSF, en aquellos que evolucionan a fallo medular o desarrollan SMD/LAComentariosEl TPH es el único tratamiento curativo de la enfermedad hematológica. La experiencia en este gru-
po de enfermos, es escasa y se limita fundamentalmente al “French Neutropenia Registry” y al EBMT que comunican una supervivencia alrededor del 60%, con mejores resultados en aquellos pacientes con fracaso medular que en aquellos con SMD/LMA (Cesaro, 2005). Cuestiones tales como la fuente de progenitores o tipo de acondicionamiento, están todavía por esclarecer
OTROS FALLOS MEDULARES.
1. NEUTROPENIA CONGÉNITA.
Aspectos básicos — Grupo heterogéneo de enf. caracterizadas por neutropenia crónica grave — Prevalencia 1 – 1,7 / 333.300 habitantes. Incidencia anual 1 / 250.000 nacimientos
Indicaciones de trasplanteAusencia de respuesta a G-CSF (más de 8 µg/kg/día), o evolución a SMD o LA, o mutación Gly185Arg
en el gen ELANEAcondicionamientoSe han empleado AMA clásicos (Bu-Cy) y AIR, con similares resultados (92 vs. 87%)Fuente de progenitores y donantes
— Los mejores resultados se han comunciado con donantes familiares HLA idénticos, pero en los últimos años los trasplantes con donantes alternativos han mejorado significativamente sus resultados
— Se describen casos con MO y con SCU. Imposible analizar diferencias⇒
119
ResultadosEl análisis combinado de los casos reportados arroja unos resultados excelentes: SRV 89%, SLE
75%. Estos resultados empeoran en caso de indicación por SMD o LA
2. TROMBOPENIA AMEGACARIOCITICA (TAMC).
Aspectos básicos — Suele presentarse como trombopenia al nacimiento que progresa a fallo medular completo. En algunos pacientes también se ha observado una evolución maligna hacia SMD y LMA
— En la literatura se han reportado 100 casos con TAMC — Se produce por mutaciones en el gen c-MPL
Indicaciones de trasplanteTrombocitopenia grave, evolución a fallo medular, SMD o LMAAcondicionamiento y donantes
— Aunque la experiencia es escasa, los resultados con donantes familiares son mejores. — El uso de DnE se está extendiendo a pesar de mayor toxicidad y fallo de implante con regímenes mieloablativos. Sin embargo éstos resultados han mejorado los datos de toxicidad orgánica con el uso de AIR: alemtuzumab + Fluda + MEL (Words, 2014)
Bibliografía recomendada.— Cesaro S, Oneto R, Messina C, et al; EBMT Severe Aplastic Anaemia and Paediatric Diseases Wor-
king Party. Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation. Br J Haematol. 2005; 131: 231-6.
— Tan PL, Wagner JE, Auerbach AD, et al. Successful engraftment without radiation after fludarabi-ne-based regimen in Fanconi anemia patients undergoing genotypically identical donor hemato-poietic cell transplantation. Pediatr Blood Cancer. 2006; 46: 630-6.
— Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009; 113: 6549-57.
— Pasquini R, Carreras J, Pasquini MC, et al. HLA-matched sibling hematopoietic stem cell transplan-tation for fanconi anemia: comparison of irradiation and nonirradiation containing conditioning regimens. Biol Blood Marrow Transplant. 2008; 14: 1141-7.
— MacMillan ML1, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia - when and how? Br J Haematol. 2010; 149: 14-21.
— Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010; 116: 3715-23. — Connelly JA, Choi SW, Levine JE. Hematopoietic stem cell transplantation for severe congenital
neutropenia. Curr Opin Hematol. 2012; 19: 44-51.— Donadieu J, Fenneteau O, Beaupain B, et al; Associated investigators of the French Severe Chronic
Neutropenia Registry*. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012; 97: 1312-9.
— Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplan-tation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013; 19: 1238-43.
— Fagioli F, Quarello P, Zecca M, et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol. 2014; 165: 673-81.
— Narla A, Vlachos A, Nathan DG. Diamond Blackfan anemia treatment: past, present, and future. Semin Hematol. 2011; 48: 117-23
— Zecca M, Strocchio L, Pagliara D, et al. HLA-haploidentical T cell-depleted allogeneic hematopoietic
259
LA BÚSQUEDA DE DONANTES NO EMPARENTADOS. (E. Carreras, N. Marieges) 3.4
1. SISTEMÁTICA PARA EL INICIO DE UNA BÚSQUEDA INTERNACIONAL DE DnE. — Valorar riesgos y beneficios de un TPH de DnE frente a otras opciones de menor riesgo (auto-TPH, QT) (ver indicaciones EBMT en cap. 2.1).
— Confirmar que el paciente tiene una de las indicaciones establecidas por la Subcomisión de TPH de la Comisión Interterritorial de Trasplantes para el inicio de una búsqueda (ver apartado 1.1). En caso contrario deberá solicitarse la valoración del caso por el Comité de Expertos de dicha Subcomisión.
— Confirmar que no existe donante compatible entre los familiares de primer grado (ver apartado 1.1).
— Confirmar que la edad del paciente no supera el máximo autorizado para iniciar una búsqueda. En caso contrario deberá solicitarse la valoración del caso por el Comité de Expertos de dicha Subcomisión.
— Antes de iniciar una búsqueda es recomendable valorar las posibilidades de localización de donante con los inmunólogos del centro.
— Si la búsqueda se inicia desde un centro no autorizado para realizar TPH de DnE, deberá solicitar la aceptación del caso en un centro autorizado.
— Una vez recibida la aceptación del centro de trasplante (si procede), solicitar el visto bueno del Coordinador de Trasplantes de la CA (si procede; en algunas CCAA función delegada en la Gerencia de los Hospitales).
— A continuación, enviar a REDMO la siguiente documentación:> Impreso de solicitud de inicio de búsqueda con todos los datos cumplimentados:
* Datos pacientes, datos del centro solicitante, diagnóstico y fase de la enfermedad.* Datos centro de trasplante, tipo de búsqueda (indicativo del producto deseado), fecha
deseable para la realización de TPH y grado de compatibilidad aceptable.> Autorización inicio búsqueda (Coordinador Autonómico de Trasplantes y/o Gerencia Hos-
pital).> Dictamen laboratorio de tipaje HLA del paciente (HLA-A, B, C, DRB1 y DQB1 por técnicas
de alta resolución, 4 dígitos)> Dictamen laboratorio de tipaje HLA de padres y hermanos con resolución suficiente para
demostrar que presentan ≥2 diferencias HLA con el paciente. Si no están disponibles o presentan alguna comorbilidad que impida la donación, no es necesario realizar el tipaje, es suficiente con indicarlo en el formulario y/o en el informe)
> Breve historia clínica (incluir características del paciente y justificación de la búsqueda).> En caso de considerarlo oportuno solicitar la Selección de Cordón por parte de la Oficina
de SCU de REDMO mediante la cumplimentación del formulario oportuno.
1.1 Requisitos para el inicio de una búsqueda internacional.
Condiciones generales — Edad ≤55 años. Para pacientes entre 55 y 65 años se acepta el criterio del centro de trasplante — Ausencia donante familiar compatible aceptable (hermano o progenitor compatible o con una única diferencia HLA en los loci HLA-A, B, C, DRB1, DQB1)
— En enfermedades congénitas, ausencia de alteraciones neurológicas — Resultados esperables superiores a otras terapias
⇒
260
Indicaciones: Enfermedades malignas [1]En el momento del diagnóstico
— SMD con IPSS intermedio-2 ó alto en adultos y en todos los casos en niños — LMA con alteraciones citogenéticas o moleculares de alto riesgo — LLA con alteraciones citogenéticas o moleculares de alto riesgo — Leucemia aguda postmielodisplasia — Leucemia aguda secundaria
Durante la inducción a la remisión — LA y necesidad >1 ciclo de QT para alcanzar una RP (<25% blastos MO) o RC — LLA con enfermedad significativa al final de la inducción, consolidación o ambas — LLA con respuesta lenta con valor pronóstico adverso respecto al protocolo empleado, durante el tratamiento de inducción
A lo largo de su evolución — LMC: Pacientes en FA, FC2, CB, FC1 con fracaso a ITK de 1.a – 2.a línea, mutación T315i — LLC: Factores mal pronóstico (deleción 17p/mutaciones TP53) sin respuesta a R-FC o R-FCM — Leucemias agudas en segunda remisión completa o posterior
En recaída — LA en 1.a REC (en LLA si RC1 duró menos de 12 meses) — Leucemia aguda en segunda recaída
Síndromes linfoproliferativos — LNH alto riesgo en REC quimiosensible con indicación de TPH de hermano HLA-idéntico según el EBMT
— Mieloma múltipleIndicaciones: Enfermedades no malignas [1]
— AM al diagnóstico o que no responda al TIS — HPN con aplasia medular o complicaciones graves que no responden a otros ttos. — Anemia de Fanconi y otras IMC (disqueratosis congénita, Blackfand-Diamond, disgenesia reticular; Shwachman-Diamond, etc)
— Inmunodeficiencias congénitas o adquiridas grave — Alt. congénitas de los macrófagos y neutrófilos (Linfohistiocitosis hemofagocítica, Kostman, Chediak-Higashi, Enfermedad granulomatosa crónica, etc.)
— Errores congénitos del metabolismo. (Adrenoleucodistrofia, Hurler, Enfermedad de Gaucher, etc.)
— Osteopetrosis
[1] Se podrán contemplar otras indicaciones previa valoración por la Comisión de Expertos de la Organización Nacional de Trasplantes.
1.2 Requisitos para la realización del trasplante.
En los pacientes en que se disponga de un donante compatible, o de una unidad de SCU idónea, el trasplante tan sólo debería llevarse a cabo si se cumple, en el momento de cursar el “work-up” (ver apartado 3), alguna de las indicaciones reconocidas como una práctica “estándar” u “opción clínica” en el documento de indicaciones del EBMT (2015) [1] (ver capítulo 2.1)
[1] Sureda, 2015.
261
2. LA SELECCIÓN DEL MEJOR DONANTE O UNIDAD DE SCU (ver capítulo 3.3).
3. CONDUCTA A SEGUIR UNA VEZ IDENTIFICADO EL DONANTE IDÓNEO
Solicitud formal del TPHUna vez seleccionado al donante ideal, activar la solicitud formal de donación (“work-up”) a través
del REDMO, empleando el formulario específico del registro al que pertenezca el donante, en el que debemos indicar: — Fechas aproximadas para realizar el TPH (dar varias opciones) — Muestras requeridas para los estudios confirmatorios (normalmente 2.a muestra HLA [1], grupo sanguíneo, Ac. irregulares, serologías víricas y estudio del quimerismo)
— Fecha máxima para recibir la confirmación de disponibilidad y buen estado físico/emocional del donante (clearance). Recordar que hasta la recepción del mismo no debe iniciarse el tratamiento de acondicionamiento
— Tipo de progenitores (MO/SP) y celularidad solicitada [2], anticoagulante — y temperatura de conservación preferida — Necesidad de courier [3]
[1] EFI recomienda realizar una segunda confirmación de tipaje (por baja resolución) antes de realizar el TPH. [2] La celularidad infundida influye significativamente en los resultados del TPH. Intentar que sea >3 x10^8/CNT/kg en los TMO y entre 4 – 5 x10^6 CD34+/kg en los TPH de SP. En lugar de indicar una cifra fija es recomendable indicar un rango aceptable (p. ej. 2 – 4 x10^6 CD34+/kg), de este modo en algunos casos es posible evitar la realización de una segunda aféresis al donante. [3] Desde 2008 los centros españoles que lo solicitan disponen de un servicio de mensajería profesionalizado para estos transportes (NOPC).
Recogida, transporte y administración de los PHLos courier de aquellos centros que no utilicen el servicio de NOPC deben recordar:
— La recogida de los PHSP normalmente se realiza a primera hora de la mañana del día siguiente a la 1.a aféresis (si una sola aféresis) o al mediodía (si 2 aféresis) [1]
— La Recogida de los PHMO se realiza a mediodía del día de la extracción [2] — Temperatura de transporte:
MO: 2 – 24 ºC en viajes <12 h; si >12 h, 2 – 10 oC SP: 2 – 24 ºC en viajes <4 h; si >4 h, 2 – 10 oC
— Se requiere una autorización del Ministerio de Sanidad y del organismo equivalente del país de origen para el paso de fronteras
— En los controles de aduanas no someter los PH a irradiación — Efectuar nuevos recuentos y pruebas cruzadas antes de infundir — Infundir como se indica en el capítulo 3.12
Sobre la congelación o destrucción de los PH no empleados — Ocasionalmente puede ocurrir que, por problemas de programación previsibles, el CT se plantee criopreservar el producto para su utilización unos días después y así no tener que volver a repetir todos los estudios serológicos del donante, con una validez limitada a 30 días. Debe saberse que la mayoría de registros exigen que se les consulte específicamente y que en muchas ocasiones prefieren modificar las fechas
— Puede ocurrir que estos problemas de programación sean imprevisibles (p. ej. avería irradiador) y ocurran una vez ya ha empezado la movilización del donante, en estos supuestos la única alternativa válida es criopreservar el producto a su llegada e informar a REDMO, y por añadidura al registro de origen del donante, de la demora en su administración
⇒
262
Sobre la congelación o destrucción de los PH no empleados (Cont.) — Algo muy distinto es que la demora sea debida a las condiciones del paciente (infección intercurrente, recaída, etc.). En dicho supuesto NUNCA debe de procederse a solicitar la criopreservación, es obligado posponer la donación hasta que sea seguro que podrá realizarse el trasplante
— En ocasiones excepcionales puede ocurrir que un producto no pueda ser infundido por fallecimiento del paciente durante el acondicionamiento. En dicho supuesto el producto debe de ser destruido. En España, con la entrada en vigor de los nuevos modelos de CI, los donantes de reciente incorporación autorizan dicha destrucción. No siempre ocurre lo mismo con productos procedentes de terceros países si bien la política seguida por la mayoría de países es proceder a la destrucción, guardando siempre la documentación del procedimiento, informando a REDMO, y por añadidura al registro de origen del donante
[1] Estos aspectos deben de ser pactados con anterioridad con el centro de obtención que informará unos días antes sobre la hora y el lugar de recogida. De forma excepcional y aceptando un sobrecoste en los casos de una sola aféresis puede plantearse su recogida el mismo día del procedimiento. [2] Algunos centros exigen que el courier notifique su llegada a la ciudad la noche anterior de la colecta de MO para iniciar la misma.
4. CONDUCTA A SEGUIR UNA VEZ IDENTIFICADA UNA UNIDAD DE SCU IDÓNEA.
Selección inicialLa selección de unidades de SCU se basa principalmente en la celularidad de la unidad y su tipaje
HLA (ver capítulo 3.3). Dicha selección debe realizarla el facultativo responsable del centro de trasplante (CT), en base a los listados remitidos por REDMO, que contienen el código de identifi-cación de la unidad, su HLA (A, B, DR por baja resolución y en ocasiones C y/o DRB1 por alta), y su celularidad (celularidad nucleada total y, casi siempre, CD34+). Esta selección también puede realizarla la Oficina de SCU de REDMO si así lo solicita el CT mediante la cumplimentación de un formulario creado a tal efecto
Informe preliminar de la unidad — Si la selección no la realiza REDMO, una vez escogidas las “mejores” unidades debe solicitarse el “Unit report” (UR) de todas aquellas que se consideren de interés. Este informe incluye en la mayor parte de casos los siguientes datos: banco de origen, fecha de colecta, volumen criopreservado, celularidad nucleada y CD34+ (si disponible), número de bolsas, grupo sanguíneo, sexo, marcadores serológicos y estudios de viabilidad precongelación. Si la unidad está incluida en EMDISCord y dispone de la información básica el UR se facilita desde REDMO de forma inmediata. Si no se dispone de toda la información, ésta debe ser solicitada a origen y puede estar disponible en 24 – 48 h (12 h si es española)
— Si la Selección de las “mejores” unidades de SCU se realiza a través de la oficina REDMO, junto con el informe de selección se remiten todos los UR de las unidades seleccionadas
Estudios adicionales solicitables — Ampliación de tipaje no disponible en origen [1] [2] — Envío de muestras de ADN, suero y plasma materno o del cordón, o segmento adjunto a la bolsa [3]
— Ampliación de marcadores serológicos, estudio de hemoglobinopatías, etc. — Reserva de la unidad durante 2 – 3 meses. Conducta muy variable según bancos, desde una simple reserva hasta la realización de una serie de comprobaciones de la unidad [4]
⇒
263
Envío, transporte y recepción de la unidadUna vez escogida la unidad que se considere más idónea procede solicitar su envíoPara ello se deberá:
— Rellenar el formulario requerido por cada BSCU/Registro — Indicar fecha aproximada para el envío (recordar que, si no se ha reservado con anterioridad, los estudios a realizar en la unidad previos a la salida de ésta pueden demorar hasta 15 días su envío, dado que es recomendable disponer de la cuantificación de CFU)
— Fecha estimada de inicio del tto. de acondicionamiento y de infusión (recordar que nunca debe iniciarse el tto. de acond. hasta no haber confirmado la correcta recepción de la unidad)
— Persona encargada de su recepción y descripción exacta del lugar donde debe ser entregada la unidad
— El courier encargado del trasporte suele ser el servicio de mensajería profesional escogido por el Banco de SCU
— Si a su recepción, se observa cualquier anomalía en el contenedor (o en la bolsa) es recomendable documentarlo fotográficamente en vistas a posibles reclamaciones posteriores, y valorar cuidadosamente si se debe infundir o no dicha unidad
— Una vez en destino y efectuados los obligados controles de calidad (cap. 3.11 y 3.12) los PSCU pueden ser remitidos a la unidad de TPH para su descongelación y administración o bien mantenerse criopreservados hasta su posterior utilización
[1] Algunos centros prefieren disponer de A, B, C, DRB1 por alta resolución, si bien ningún estudio ha demostrado que con ello se mejoren los resultados, haciendo más compleja la localización de unida-des compatibles. [2] Por contra, el estudio del locus C es cada vez más frecuente por su impacto en los resultados. [3] Es cada vez más frecuente que los bancos de cordón se nieguen a mandar segmentos de cordón dado su limitado número. En las recomendaciones actuales de EFI se acepta que la reconfir-mación de tipaje se realice en el banco de origen. [4] Estos estudios comportan unos gastos que serán facturados aún en el caso de que finalmente no se solicite el envío de la unidad.
5. ACTIVIDAD GLOBAL DE REDMO (figuras 1, 2 y 3)
Figura 1. Búsquedas iniciadas REDMO a lo largo de los años (memoria REDMO 2015).
264
Figura 2. Trasplantes realizados a pacientes españoles a través de REDMO (Memoria REDMO 2015)
Figura 3. Actividad global. Trasplantes coordinados por REDMO (Memoria REDMO 2015).
Bibliografía recomendada.— Información sobre PNSCU, PNDMO (que incluye documento inmunólogos) y documento indica-
ción inicio búsquedas: http://www.ont.es/infesp/Paginas/DocumentosdeConsenso.aspx— RDL 9/2014:— Memoria REDMO 2015. http://www.fcarreras.org/es/memorias— Gragert L, Eapen M, Williams E, et al,. HLA match likelihoods for hematopoietic stem-cell grafts in
the U.S. registry. N Engl J Med. 2014; 371: 339-48.— Sureda A, Bader P, Cesaro S, et al. Indications for allo- and auto-SCT for haematological diseases,
solid tumours and immune disorders: current practice in Europe, 2015. Bone Marrow Transplant. 2015; 50: 1037-56.
673
FÁRMACOS MÁS EMPLEADOS EN EL TPH(N. Creus, E. Carcelero) 7.4
QUIMIOTERÁPICOS UTILIZADOS EN LOS TRATAMIENTOS DE ACONDI-CIONAMIENTO
BUSULFAN
Dosis [1] VO: 1 mg/kg/dosis c/6 h (x 4 días)IV: 0,8 mg/kg/dosis c/6 h (x 4 días)
Preparación y administración
VO: para facilitar la administración se puede realizar una suspensión ex-temporánea de los comprimidos de busulfano con agua
IV: 10 veces volumen busulfan de SF o SG5% (aprox 0,5 mg/mL)Infusión: 2 hEstabilidad: 12 h N. + 3 h TA o (8 h TA)
Toxicidad limitante de dosis (no he-matológica) [2]
HepatotoxicidadMucositisConvulsiones (toxicidad característica pero no limitante de dosis) en un
10% de los pacientes. Se realiza de forma rutinaria profilaxis con anticon-vulsivantes (fenitoína, benzodiacepinas o levetiracetam)
No requiere ajuste
No requiere ajuste
Interacciones con otros fármacos
Evitar paracetamol y metronidazol 72 h antes y después de busulfano (au-mentan niveles busulfano)
Itraconazol, voriconazol, posaconazol pueden aumentar niveles busulfano (los estudios PK se hicieron con fluconazol)
Induce el metabolismo de fenitoína y viceversa
CARBOPLATINODosis IV: 700 mg/m^2/d (x3 días)
Preparación y administración
IV: 250 – 500 mL SG5% (no compatible con SF)Infusión: 30 min – 1 hEstabilidad: 28 d N
Toxicidad limitante de dosis (no hematológica)
Toxicidad renalMucositisNeuropatía periféricaOtotoxicidad (toxicidad característica pero no limitante de dosis)
⇒
674
CARBOPLATINONo requiere ajuste si se dosifica por AUCSi dosificación por mg/m^2 se sugiere:
Administración concomitante de aminoglucósidos aumenta el riesgo de ototoxicidad
CARMUSTINADosis IV: 300 mg/m^2 (x1 día)
Preparación y administración
IV: 250 – 500 mL SF o SG5% (no PVC)Infusión: 1 – 2 hEstabilidad: 48 h N. Proteger de la luz
Toxicidad limitante de dosis (no hematológica)
Toxicidad pulmonarLa administración de dosis altas (>150 mg/m2) se asocia con dolor de cabe-
za, parestesias periorales y enrojecimiento debido al diluyente alcohóli-co utilizado en su reconstitución. Se recomienda administrar lentamente en 2 h y premedicar con paracetamol, morfina o benzodiacepinas
No administrar si Clcr <10 mL/min
Podría ser necesario el ajuste en pacientes con alteración hepática. No existen guías específicas
Interacciones con otros fármacos
Evitar inhibidores de la aldehído deshidrogenasa-2 o fármacos que indu-cen reacción disulfiram-like debido al contenido en alcohol del diluyente
IV: 250 – 500 mL SF o SG5% Infusión: 30 min – 2 hEstabilidad: 21 d N
Toxicidad limitante de dosis (no hematológica)
CardiotoxicidadCistitis hemorrágica (toxicidad característica de ciclofosfamida a dosis
altas pero no limitante de dosis). Causada por el metabolito acroleína. Se recomienda la administración de mesna y diuresis forzada para su prevención
⇒
678
glucosado 5%. SF = suero fisiológico. min = minutos. TA = temperatura ambiente. VO = vía oral. [1] Dosificación más frecuente en régimen de acondicionamiento. [2] Toxicidad no hematológica limi-tante de dosis. También se citan los principales efectos adversos distintos a los habituales en toda quimioterapia
FÁRMACOS INMUNOSUPRESORES. — A continuación se describen los principales fármacos inmunosupresores utilizados en el contexto del trasplante hematopoyético para la prevención y tratamiento de la enfermedad del injerto contra el huésped.
CICLOSPORINADosis Ver capítulos 4.4, 5.13 y 5.14
Preparación y administración
IV: diluir cada ampolla en 20 – 100 mL de SF o SG5% en envase de polieti-leno o vidrio (no utilizar PVC)
Est. Dil.: en SF 12 h TA y en SG5% 24 h TAAdministrar en 2 – 6 hVO: con o sin alimentos, pero siempre en la misma condición. Se reco-
mienda no modificar el contenido de grasas de las comidas o el líquido empleado para tragar el fármaco para evitar cambios en la biodisponibi-lidad. La solución oral debe diluirse preferentemente en zumo de naranja o manzana (no zumo de pomelo). Administrar en vaso de vidrio. Evitar que la jeringa entre en contacto con el diluyente. No enjuagar la jerin-ga, limpiar el exterior con un pañuelo de papel seco. Desechar 2 meses después apertura
Efectos adversos
Nefrotoxicidad, hipertensión, hipomagnesemia, temblores de las extremi-dades, neurotoxicidad, hipercalemia, hiperglicemia, hipertrigliceridemia, hirsutismo, microangiopatía trombótica, hiperplasia gingival, reacciones alérgicas (por excipiente)
Normalmente se ajustará dosis en función de los niveles plasmáticos Ajuste de dosis recomendado en función de nefrotoxicidad: Si Cr 2 – 2,9 x basal → reducir dosis en mínimo un 25%Si Cr >3 x basal → reducir dosis en mínimo un 50%
Podría ser necesario el ajuste en pacientes con alteración hepática. No existen recomendaciones específicas
Interacciones con otros fármacos
Es sustrato de CYP3A4 y la glicoproteína P. Precaución cuando se adminis-tra concomitantemente con fármacos que inducen o inhiben CYP3A4 o la glicoproteína P
⇑ niveles (inhibidores CYP3A4): p. ej. voriconazol, posaconazol (antifúngi-cos azólicos), zumo de pomelo
⇓ niveles (inductores CYP3A4): p. ej. carbamacepina, fenitoína, hierba de San Juan
Mayor riesgo de: — nefrotoxicidad con fármacos nefrotóxicos sistémicos (AINEs, aminoglucósidos.)
— hiperpotasemia con suplementos de potasio, diuréticos ahorradores de potasio, IECA
682
METILPREDNISOLONA (IV) /PREDNISONA (VO)
BECLOMETASONA (comp. gastrorresistentes / cáp-
sulas liberación inmediata)
No requiere ajuste
No requiere ajuste
Interacciones con otros fármacos
Mayor riesgo de: — hipocalemia con diuréticos tiacídicos, furosemida — es necesario reajustar la dosis de hipoglucemiantes e insulina
INMUNOGLOBULINA ANTITIMOCÍTICA DE CONEJO (Timoglobulina®)Dosis 1,25 – 5 mg timoglobulina ®/kg/d (pauta variable según protocolos)
Preparación y administración
IV: – vial de 5 mg/mL — prueba intradérmica: inyectar 0,1 mL de una dilución 1: 10 del producto (o de una dilución 1: 1.000 en SF del mismo si sospecha de hipersensibilidad)
— diluir timoglobulina® a razón de 50 mL de SF o SG5% por vial y administrar en un mínimo de 4 h, usando un filtro en línea y evitando lípidos, sangre o hemoderivados por la misma vía
— infundir a través de una vena con mucho flujo (central preferiblemente); premedicar con la dosis diaria de corticoide (si procede), un antihistamínico y paracetamol
Timoglobulina se debe administrar con especial precaución en pacientes con enfermedades hepáticas ya que se pueden agravar trastornos he-páticos pre-existentes. Se recomienda monitorizar cuidadosamente los niveles de trombocitos y los parámetros de la coagulación
Interacciones con otros fármacos
Mayor riesgo de: — inmunosupresión combinada con otros fármacos inmunosupresores en el proceso del trasplante (efecto deseado)
ETANERCEPTDosis Ver capítulos 5.13 y 5.14
Preparación y administración
SC: reconstituir lentamente un vial con 1 mL de API. Agitar durante 5 min La solución debe ser clara e incolora. Después de la preparación se re-comienda uso inmediato (desechar toda solución no administrada antes de 6 h). Administración SC (rotando zona), preferentemente parte central delantera muslos, abdomen
⇒
683
Efectos adversosReacción local en punto de inyección que dura 3 – 5 días. Cefalea, rinitis,
mareo, molestias gastrointestinales, dolor abdominal. Aumento riesgo de infecciones. Reacciones de hipersensibilidad
No requiere ajuste
No requiere ajuste
Interacciones con otros fármacos
Mayor riesgo de: — infecciones si administración concomitante con otros fármacos inmunosupresores (anakinra o abatacept)
Adm = administración. API = agua para inyección. Clcr = aclaramiento de creatinina. Cr = creatinina. d = días. GI = gastrointestinales. IV = vía intravenosa. h = horas. N. = nevera. SC = subcutánea. SNC = sistema nervioso central. SG5% = suero glucosado 5%. SF = suero fisiológico. min = minutos. TA = temperatura ambiente. VO = vía oral.