Materials Science and Engineering C 33 (2013) 1091–1101
Contents lists available at SciVerse ScienceDirect
Materials Science and Engineering C
j ourna l homepage: www.e lsev ie r .com/ locate /msec
A new hydroxyapatite-based biocomposite for bone replacement
Devis Bellucci a,⁎, Antonella Sola a, Matteo Gazzarri b, Federica Chiellini b, Valeria Cannillo a
a Department of Engineering “Enzo Ferrari”, University of Modena and Reggio Emilia, Via Vignolese 905, 41125 Modena, Italyb Laboratory of Bioactive Polymeric Materials for Biomedical and Environmental Applications (BIOlab) & UdR INSTM, Department of Chemistry & Industrial Chemistry, University of Pisa,Via Vecchia Livornese 1291, 56122S. Piero a Grado, Pisa, Italy
Since the 1970s, various types of ceramic, glass and glass–ceramic materials have been proposed and used toreplace damaged bone in many clinical applications. Among them, hydroxyapatite (HA) has been successfullyemployed thanks to its excellent biocompatibility. On the other hand, the bioactivity of HA and its reactivitywith bone can be improved through the addition of proper amounts of bioactive glasses, thus obtainingHA-based composites. Unfortunately, high temperature treatments (1200 °C÷1300 °C) are usually requiredin order to sinter these systems, causing the bioactive glass to crystallize into a glass–ceramic and henceinhibiting the bioactivity of the resulting composite. In the present study novel HA-based composites are re-alized and discussed. The samples can be sintered at a relatively low temperature (800 °C), thanks to the em-ployment of a new glass (BG_Ca) with a reduced tendency to crystallize compared to the widely used 45S5Bioglass®. The rich glassy phase, which can be preserved during the thermal treatment, has excellent effectsin terms of in vitro bioactivity; moreover, compared to composites based on 45S5 Bioglass® having the sameHA/glass proportions, the samples based on BG_Ca displayed an earlier response in terms of cell proliferation.
The loss of an organ or tissue due to cancer, disease or trauma is acritical problem in human health care. An attractive and promisingapproach to address such issue is to create biological or hybrid sub-stitutes for implantation into the body, exploiting the self-healingpotential of the body itself, as proposed in the framework of theemerging tissue engineering [1–4]. Tissue engineering, following theprinciples of cell transplantation and materials science, seeks toregenerate healthy biological tissues, as opposed to the traditionalapproach of synthetic implants and organ transplantation. Amongthe many tissues in the body, the regeneration of bone with pre-determined shapes for orthopaedic surgery applications is of primaryinterest, since “there are roughly 1 million cases of skeletal defects ayear that require bone-graft procedures to achieve union” [5]. Fur-thermore, bone is a dynamic tissue, in constant resorption and forma-tion, and has the highest potential for regeneration [6].
Biomaterials play a critical role in the success of tissue engineer-ing, since they provide mechanical stability to the self-healing tissuesand drive their shape and structure [7]. Moreover, they can controland stimulate the regeneration of the living tissue itself by activatingspecific genes through their dissolution or, if required, releasinggrowth factors and drugs [8–10].
+39 059 2056243.ci).
rights reserved.
Among biomaterials for bone tissue engineering, hydroxyapatite(HA) has raised great interest for many applications in both dentistryand orthopaedics, due to its close chemical and crystal resemblance tothe mineral phase of bone, which results in an excellent biocompati-bility [11,12]. In particular, HA has been widely employed in recentyears in dental devices and hard tissue surgery thanks to its abilityto form a bond with the surrounding bone tissue after implantation[13–15]. As an alternative to HA, bioactive glasses [16,17] offer re-markable advantages due to their higher bioactivity index. Amongthem 45S5 Bioglass®, whose proportions are 45 wt.% SiO2, 24.5 wt.%CaO, 24.5 wt.% Na2O and 6 wt.% P2O5, is the most bioactive glass,since it is able to bond to soft tissues as well as to hard ones [18,19].
Unfortunately, the use of bulk HA and bioactive glasses has beenlimited so far to non-load-bearing applications due to their relativelypoor mechanical properties; moreover, even if the HA biocompatibil-ity is excellent, its close similarity to the mineral component of boneresults in the lack of HA biodegradation in the body [20,21]. In fact, al-though its degradation rate increases with porosity [21], HA has alimited in vitro reactivity, and in vivo assays have shown low forma-tion of osseous tissue. For these reasons, HA is expected to remainin the body for long periods of time, with no resorption [22–25]. Usu-ally this is an undesirable feature for many applications, in particularfor the realization of scaffolds, one of the key ingredients of tissue en-gineering [26–28].
Recently there have beenmany attempts to reinforce and combineHA with other ceramics [29], polymers [30] and bioactive glasses,
1092 D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
aiming to obtain composite materials with improved biological prop-erties, not achievable by any of the elementalmaterials acting alone. Inparticular the possibility ofmixing HA and bioactive glasses, which aremuch more reactive than HA, looks rather promising and may lead tothe development of new generation composites with tailored biologi-cal properties. The glass composition and volume fraction have a largeeffect on the phase assembly, mechanical properties and bioactivityof the resulting composites. Many glasses belonging to the Na2O\CaO\P2O5 or CaO\P2O5\SiO2 systems have been tested as secondphase in a HA-based composite [31–37]. A very important characteris-tic of silica-based glasses is that they release critical concentrationsof ions (e.g. Si, Ca, P) during their dissolution, whichmay induce intra-cellular and extracellular responses, such as gene activation in osteo-blasts, and stimulate neo-vascularisation and angiogenesis [8,9,38,39].In addition, silicate-based glasses offer additional advantages with re-spect to phosphate-based ones when they are used in HA-containingcomposites, as structural and chemical analyses have demonstratedthat favourable ionic substitutions may occur in the HA lattice [40].These ionic substitutions, which include Na+ for Ca2+ and, in particular,SiO4
4− for PO43−, strongly affect the stability of HA [41,42] and its surface-
structure and charge, which in turn influence the bioactivity of the com-posite system.
The main disadvantage of using bioactive glasses as second phasein HA-based composites is probably that high-temperature treat-ments (1200 °C÷1300 °C) are usually required in order to sinterthese systems [43], causing the glass to crystallize with possible nega-tive effects on its bioactivity [44]. In fact, as proved for 45S5 Bioglass®-derived glass-ceramics, although crystallization does not inhibit the invitro development of a HA surface layer, the onset time for HA forma-tion is increased up to three to four times with respect to the corre-sponding parent glass [45,46]. Since 45S5 Bioglass® is already proneto crystallize at temperatures as low as 610 °C [47], alternative glasscompositions with a reduced tendency to crystallize are expected toopen interesting scenarios for applications in HA-based composites.Additional negative side effects may be caused by high-temperaturethermal treatments. First of all, thermal treatments around 1200 °Cmay elicit reactions between glass and HA, with the subsequent for-mation of new phases, which in turn may alter the biodegradabilityof the final system. Moreover, at about 1200 °C, the HA itself can de-compose, resulting in the formation of tricalcium phosphate [48,49]or CaO [43]. In order to avoid the degradation of the constituentphases, it is mandatory to define new processing routes to obtain bio-active glass-HA composite materials.
Recently another glass (BG_Ca), whose composition is 47.3 mol%SiO2, 45.6 mol% CaO, 4.6 mol% Na2O and 2.6 mol% P2O5 [50], hasbeen employed in a preliminary study to realize HA-based compositeswith relatively low HA contents (50 wt.%) [51]. This glass shows a re-duced tendency to crystallize with respect to the widely used 45S5Bioglass®,which belongs to the sameNa2O\CaO\P2O5\SiO2 system,due to its relatively high CaO-to-Na2O ratio. In this work, this glasscomposition was applied to realize HA-based composites with variousHA/glass proportions, up to 80 wt.% of HA. The novel samples couldbe sintered at a lower temperature (800 °C) compared to HA/45S5Bioglass® composites with the sameHA/glass ratio (Tsintering~1150 °C).This fact greatly helped to preserve the amorphous nature of the glass inthe HA/BG_Ca composites, thus preventing the HA decomposition,which typically occurs at higher temperatures, and limited the reactionsbetween HA and glass, with excellent effects in terms of bioactivity.
In view of a potential application for bone tissue engineering, apreliminary evaluation of the composites biocompatibility and bio-activity was carried out. As a model the mouse calvaria-derived pre-osteoblastic cell line MC3T3-E1 was selected. This cell line mimicsosteoblast progenitors by expressing markers associated with differ-entiation into a mineralizing phenotype [52]. In particular, samplesbased on HA/BG_Ca displayed an earlier response in terms of cell pro-liferation in comparison to HA/45S5 Bioglass®.
2. Materials and methods
2.1. Composites preparation
The BG_Ca glass powders were prepared by melting the raw pow-der materials (commercial SiO2, CaCO3, Ca3(PO4)2, Na2CO3 by CarloErba Reagenti, Italy) in a platinum crucible at 1450 °C. Then the meltwas rapidly quenched in water in order to obtain a frit that was subse-quently dried overnight in a furnace at 110 °C, ball-milled and finallysieved to a grain size below 38 μm.
BG_Ca and 45S5 Bioglass® powders were added to HA powderswith the aim of producing different composites. The glass-to-HA ra-tios selected for further investigations and the corresponding labelsare reported in Table 1.
Commercial HA (CAPTAL® Hydroxylapatite, Plasma Biotal Ltd,UK), with an average particle size below 25 μm, was used. Glass/HApowders were mixed for 6 h in a plastic bottle using a rolls shaker.Subsequently the mixture was used to produce green bodies by uni-axial pressing at 140 MPa for 10 s using propanol as a liquid binder.The pressed samples (shaped in form of disks: 4 cm of nominal diam-eter and 0.8 cm of thickness) were then heat-treated in a kiln for 3 h.Several sintering temperatures and thermal treatments were investi-gated in order to obtain samples with adequate compactness andlow crystallization of the glassy phase; in particular, the densificationof the composites was monitored by measuring the volume shrink-age (in percent) Δ%, which was calculated according to the followingequation:
Δ% ¼ d0−ded0
⋅100 ð1Þ
where d0 is the nominal diameter of the press (4 cm) and de is themea-sured diameter of the sample; five samples were considered for eachcomposition.
The thermal treatment was set at a final temperature of 1150 °C forHA/45S5BG and 800o C for HA/BG_Ca, respectively. Both HA/45S5BGandHA/BG_Cawere heat-treated for 3 h. Theheating ratewas 10 °C/min.
2.2. Microstructural characterization and assessment of in vitrobioactivity
The microstructure of the composites was investigated by meansof a scanning electron microscope, SEM (ESEM Quanta 200, FEI Co.,Eindhoven, The Netherland). Moreover, a local chemical analysis wasperformed by X-ray energy dispersion spectroscopy, EDS (Inca, OxfordInstruments, UK). The SEMwas operated in low-vacuummode with apressure of 0.5 Torr.
The composites were also studied by means of X-ray diffraction,XRD (PANalytical X'pertPRO diffractometer), employing a Cu Ka radi-ation. Data were collected in the angular range 10–70o 2θ with stepsof 0.02o and 5 s/step.
The in vitro bioactivity of the obtained composites was studied bysoaking them in an acellular simulated body fluid (SBF pH 7.4), withion concentrations approximately equal to those of human blood plas-ma [53,54]. In fact, it is generally believed that a biocompatible mate-rial able to form an apatite layer on its surface in SBF can develop sucha layer also in the living body, therefore the in vitro bioactivity is usu-ally considered as a pre-requirement for in vivo bioactivity. The SBF
Table 2The Effect of Temperature on Sample Density and Volume Shrinkage.
Sample Code T (°C) Volume shrinkage (%)
20% BG_Ca 700 0.34±0.03800 0.88±0.02900 0.76±0.04
1000 0.45±0.0440% BGCa 700 0.55±0.03
800 1.22±0.08900 0.88±0.04
1000 0.55±0.07850 0.31±0.05950 0.48±0.07
1050 0.78±0.081150 1.53±0.12
40% 45S5 BG 850 0.71±0.07950 0.94±0.09
1050 1.83±0.161150 2.63±0.17
1093D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
solution was prepared according to the protocol developed by Kokuboand co-workers [53,54]. Each sample was immersed in a polyethyleneflask containing an excess of SBF (20 ml) calculated on the basis of theequation Vs=Sa/10, where Vs is the volume of SBF (ml) and Sa is theapparent surface area of the specimen (mm2) [53,54]. The sampleswere maintained at 37 °C and the SBF was refreshed every 48 h. Thesamples were then extracted from the solution after given times of3, 7 and 14 days. Subsequently, the SEM investigation was repeatedwith the aim to evaluate the amount and morphology of the precipi-tated HA.
In order to further investigate the chemistry of the precipitated HA,Raman spectroscopywas performed using a Jobin-Yvon RamanMicro-scope spectrometer (Horiba Jobin-Yvon Inc., Edison, NJ). The sampleswere studied after immersion in SBF for 3, 7 and 14 days. With thisaim, a 632.8 diode laser was employed (output power: 20 mW). Pho-tons scattered by the samples were collected on a CCD camera and thecollection optic was set at 100 X ULWD objective. A spectra collectionof 10 acquisitions of 60 s each was employed.
2.3. Biological evaluation
2.3.1. Preparation, sterilization and neutralizationComposites were cut into pieces of about 0.5 g and sterilized in dry
heat at 180 °C for 3 h [55]. Samples were then pre-treated in SBF, pre-pared according to the Kokubo protocol [53,54] and kept at 37 °C for19 days. Each sample was soaked in 20 ml of SBF. The solution wasrefreshed every 24 h to simulate the recirculation of physiologicalfluid and the consequent formation of HA aggregates on the glass sur-face [52]. During soaking in SBF, pH measurements were performedon each sample to monitor the pH variations due to ion exchange pro-cess between bioactive composites and the surrounding fluids. At theend of the incubation time with SBF, samples were rapidly soaked incomplete alpha-Minimum Essential Medium (α-MEM) [Sigma] for3 h at 37 °C 5% CO2 prior to cell seeding.
2.3.2. Cell seeding and culturingTo investigate the ability of the prepared composite samples to
support cell growth for bone tissue regeneration mouse calvaria-derived pre-osteoblastic MC3T3-E1 (CRL 2594) cell line from AmericanType Culture Collection [ATCC] was selected. Cells were propagatedas indicated by the supplier using α-MEM containing ribonucleo-sides, deoxyribonucleosides, sodium bicarbonate and supplementedwith 2 mM of L-glutamine, 1% of penicillin:streptomycin solution(10,000 U/ml:10 mg/ml), 10% of fetal bovine serum and antimycotic(complete α-MEM). Cells were allowed to proliferate for 24 h priorto the incubation with osteogenic medium, prepared by adding tothe complete α-MEM ascorbic acid γ-irradiated [50 μg/ml] andβ-glycerolphosphate disodium salt hydrate [10 mM] [56].
2.3.3. Cell adhesion and proliferation assayA preliminary biological evaluation of the suitability of the pre-
pared composites to sustain cell adhesion and proliferation was car-ried out as follows: samples (pieces of 0.5 g) were placed in 24 wellplates and cells were seeded directly onto the scaffold's surface at aconcentration of 2.5×104 per sample in a final volume of 0.8 ml, andwere then allowed to proliferate for 15 days. After 24 h from theseeding samples were transferred in a new plate, in order to evaluatethe proliferation of only the cells grown onto their surfaces. Growthmedium was refreshed every 48 h and the proliferation rate wasmeasured at day 2, 7 and 14 after conditioning in osteogenic medium,by using the Alamar-Blue® assay [Invitrogen]. Briefly, the Alamar-Blue® reagent, diluted 1:10, was added to the culture and incubatedfor 24 h. Supernatants were then re-plated in 96 well culture platesand analyzed with a Biorad microplate reader. Measurements of reso-rufin dye absorbance were carried out at 565 nm, with the referencewavelength at 595 nm. Cell proliferation was expressed as percentage
with respect to the value obtained for cells grown on tissue culturepolystyrene.
2.3.4. Alkaline phosphatase activityAlkaline phosphatase (ALP) activity was determined in cultured
MC3T3-E1-sample constructs on days 2, 7 and 14 after conditioningin osteogenic medium. The measurement was assessed with a colori-metric method that is based on the conversion of p-nitrophenylphosphate into p-nitrophenol by the ALP enzymatic activity. The sam-ples were washed two times with PBS and then placed into 1 ml of alysis buffer, containing Triton X-100 (0.2%), magnesium chloride[5 mM] and Trizma Base [10 mM] at pH 10. Samples underwentfreezing–thawing cycles by keeping at –20 °C and subsequently atroom temperature (RT) [57]. This process was repeated three timesin order to extract the intracellular ALP [58]. Following this step, avolume of 20 μl of supernatant was taken from the samples andadded into 100 μl of p-nitrophenyl phosphate substrate (Sigma). Astandard calibration, prepared by dissolving alkaline phosphatasefrom bovine kidney (Sigma) in the same lysis buffer, was added tothe substrate and the reaction was left to take place at 37 °C for30 min. The reaction was stopped by adding 50 μl of 2 M NaOH solu-tion and after 5 min waiting absorbance was measured at 405 nm in aspectrophotometer. The molar concentration of alkaline phosphataseactivity was normalized with the total protein content of each sam-ple, which was measured using micro BCA™ protein assay (Pierce).The amount of the proteins was calculated against a standard curveof serum bovine albumine. The results for alkaline phosphatase activ-ity assay were reported as nano-moles (nmol) of substrate convertedinto product⁎(mg of protein⁎min)−1.
2.3.5. Morphological observation of cultured cellsMorphological analysis ofMC3T3-E1 cultured on composite samples
was carried out at day 21 after osteogenic conditioning. After removal ofthe culture medium, each cell-cultured samples was rinsed twice withPBS, and the cells were then fixed with 2% glutaraldehyde solution,which was diluted from a 25% glutaraldehyde solution (Sigma) withPBS 1X, at 1.5 ml/well. After 1 hour of incubation, it was rinsed againwith PBS and then treated with 1.5 ml/well of sodium cacodylate[0.1 M] pH 7.4 for approximately 1 minute. After cell fixation, the spec-imen was dehydrated in ethanol solution of varying concentration(i.e. 10, 30, 50, 70, 90, and 100%, respectively) for 15 min at each con-centration. It was then dried in 100% of tetramethylsilan to removeanywater traces. The fixed samplewasmounted on a Scanning ElectronMicroscopy (SEM) stub, coated with gold, and observed by SEM.
Fig. 1. (a) XRD analysis of a BG_Ca sample treated at 800 °C for 3 h; (b) XRD analysis ofthe 20%-BG_Ca composite treated at 800 °C for 3 h.
1094 D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
2.3.6. Statistical analysisThe in vitro biological tests were performed on triplicate samples for
each material, and the data are represented as mean±standard devia-tion. Statistical difference was analyzed using one-way analysis of vari-ance (ANOVA) [59], and a p value of b0.05 was considered significant.
3. Results and discussion
3.1. Microstructural characterization
Preliminary sintering tests were performed in order to identifyadequate sintering conditions for each composition. Table 2 reports thevolume shrinkage of the sintered samples as a function of temperature.
Fig. 2. (a) XRD analysis of the 40%-BG_Ca composite treated at 800 °C for 3 h; (b)
Several works in the literature report an increased density and volumeshrinkage with increasing temperature and glass content for HA-basedcomposites [48,60]. However, these systems are usually sintered athigher temperatures (1200 °C÷1300 °C) compared to the ones em-ployed in the present contribution, causing a wide devetrification ofthe glassy phase, non-trivial reactions between glass and HA, the devel-opment of new phases and the alteration of the lattice parameters of theresidual HA. For these reasons, the interpretation of volume shrinkagewith increasing temperature is not straightforward. In Table 2 it is possi-ble to observe that the volume shrinkage increasedwith temperature forboth 20%- and 40%-45S5BG composites, although the glass is likely tocrystallize at temperatures exceeding600 °C [61] and sintering and crys-tallization are competing mechanisms. Since the data in Table 2 suggestthat thedensification of 45S5-derived samples improved as the sinteringtemperature increased but higher temperatures would cause the abovementioned undesired phenomena, the heat treatment was performedat 1150 °C for 3 h. On the contrary, in BG_Ca-based composites the vol-ume shrinkage reached a peak at 800 °C both for 20% and 40% samples.For higher temperatures, the shrinkage stopped, probably due to a par-tial crystallization of the glasswhich inhibited any further densifications.The BG_Ca glass, in fact, has been characterized in a previous work [51],where a differential thermal analysis reported a crystallization onsettemperature at about 850 °C.
The BG_Ca ability to maintain its amorphous nature up to very hightemperatures is confirmed by the XRD analysis (Fig. 1(a)) performed ona BG_Ca sample (pressed powder) treated at 800 °C for 3 h,which is thesame temperature employed to sinter the BG_Ca-based composites. TheXRD spectrum shows a broad halo, thus confirming that the glass is stillamorphous.
The XRD investigation of the HA/BG_Ca sintered samples beforesoaking in SBF is reported in Figs. 1(b) and 2(a). The XRD spectralook rather similar, since all the main peaks are associated to HA, in-dependently of the BG_Ca amount in the composite. This suggeststhat the low-temperature sintering cycle minimizes both the glassdevetrification process and the reaction between glass and HA. In-stead the literature concerning HA/glass composites often reportsa complete crystallization of the glassy phase and/or a reactionbetween the original constituent phases, resulting in a reductionof the HA amount and the formation of additional phases such asα- and β-tricalcium phosphate (TCP). Tancred et al., for example, re-ported that glass additions as low as 2.5 and 5 wt.% promoted thedevelopment of α-TCP or β-TCP in the HA matrix [48]. Generallyspeaking, the results obtained by Tancred et al. suggest that thefinal HA/TCP ratio is strongly influenced by the glass amount intro-duced in the composite. Moreover, these authors observed thatthe addition of glass delays the composite's densification to highertemperatures, thus negatively affecting the sintering process. The
XRD analysis of the 40%-45S5 Bioglass® composite treated at 1150 °C for 3 h.
Fig. 3. (a) Micrograph of the 40%-BG_Ca surface specimen and (b) EDS results of the analysis carried out on the area reported in (a).
1095D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
analysis of the fracture surfaces of the samples investigated byTancred et al. (25 and 50 wt.% glass addictions) also revealed theformation of progressively larger pores as the sintering tempera-ture increases which is a consequence of the reaction betweenbioglass and HA. Göller et al. reported the transformation of HAinto silicocarnotite (Ca5(PO4)2SiO4) and Ca2P2O7·4H2O in samplessintered at 1200 °C and originally composed of HA with a 10 wt.%
Fig. 4. Micrographs of the 40%-45S5 Bioglass® surface specimen at different magnification d
of Bioglass® [62]; a complete transformation of HA in silicocarnotiteand α-TCP was also observed by Santos and co-workers for HA/5 wt.%Bioglass® sintered at 1350 °C [33]. The presence of these phases canlead to non trivial consequences in terms of mechanical stability andbioactivity of the resulting composites.
In the present work, 45S5 Bioglass®-HA composites were pro-duced as a term of comparison with respect to BG_Ca-based ones.
egrees (a, b) and (c) EDS results of the analysis carried out on the area reported in (a).
1096 D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
As previously described (Table 2), in order to obtain fully densematerials, the sintering temperature was fixed at 1150 °C, which ismuch higher than the sintering temperature of the BG_Ca-basedcounterparts. The XRD analysis of a 40%-45S5Bioglass® sample(sintered at 1150 °C) before soaking in SBF is reported in Fig. 2(b).The XRD pattern reveals that crystallization had occurred extensively;at least two phases can be identified: a sodium calcium silicate(wollastonite, CaSiO3) and rhenanite (NaCaPO4). Presumably theoriginal bioactivity of the constituent phases (HA and Bioglass®)was modified by the sintering process. Nevertheless, the new crystal-line phases in the composite materials are still bioactive. In factNaCaPO4 has been shown to support cellular proliferation and to pos-sess high ability to enhance osteogenesis [63,64]. Also NaCaPO4-containing glass ceramics may be bioactive [65]. On the other hand,the bioactivity and biocompatibility of wollastonite is well demon-strated in the literature [66,67].
Fig. 3 reports a micrograph of the surface of the 40%-BG_Ca speci-men after heat treatment at 800 °C for 3 h. The constituent phasesare homogeneously distributed and the composite is well consolidat-ed. In particular, the 40%-BG_Ca surface looks rather similar to the40%-45S5 Bioglass® sample reported in Fig. 4. This is an interesting re-sult, since a higher temperature treatment was required for 40%-45S5Bioglass® to obtain comparable compactness. Fig. 3(b) presents theresults of the EDS analysis performed on the 40%-BG_Ca surface. Itshould be noted the higher Ca/Na ratio compared to the EDS spectrain Fig. 4(c), which refers to a 40%-45S5 Bioglass® sample.
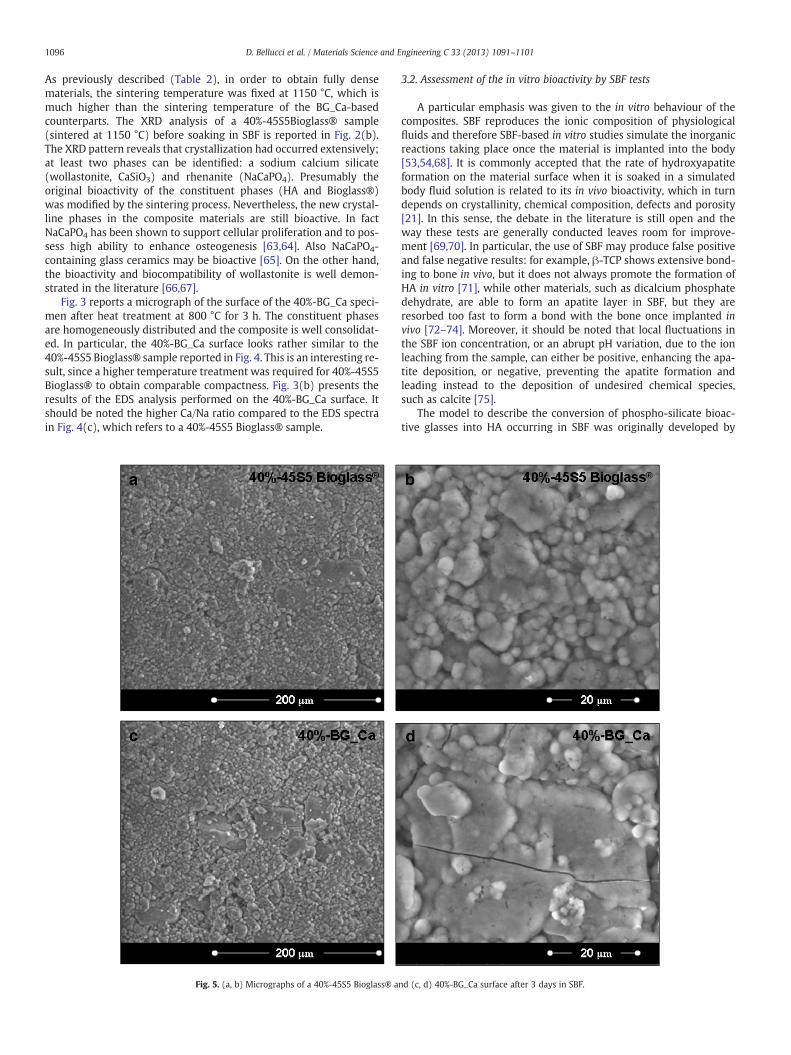
Fig. 5. (a, b) Micrographs of a 40%-45S5 Bioglass® a
3.2. Assessment of the in vitro bioactivity by SBF tests
A particular emphasis was given to the in vitro behaviour of thecomposites. SBF reproduces the ionic composition of physiologicalfluids and therefore SBF-based in vitro studies simulate the inorganicreactions taking place once the material is implanted into the body[53,54,68]. It is commonly accepted that the rate of hydroxyapatiteformation on the material surface when it is soaked in a simulatedbody fluid solution is related to its in vivo bioactivity, which in turndepends on crystallinity, chemical composition, defects and porosity[21]. In this sense, the debate in the literature is still open and theway these tests are generally conducted leaves room for improve-ment [69,70]. In particular, the use of SBF may produce false positiveand false negative results: for example, β-TCP shows extensive bond-ing to bone in vivo, but it does not always promote the formation ofHA in vitro [71], while other materials, such as dicalcium phosphatedehydrate, are able to form an apatite layer in SBF, but they areresorbed too fast to form a bond with the bone once implanted invivo [72–74]. Moreover, it should be noted that local fluctuations inthe SBF ion concentration, or an abrupt pH variation, due to the ionleaching from the sample, can either be positive, enhancing the apa-tite deposition, or negative, preventing the apatite formation andleading instead to the deposition of undesired chemical species,such as calcite [75].
The model to describe the conversion of phospho-silicate bioac-tive glasses into HA occurring in SBF was originally developed by
1097D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
Hench and co-workers in the 70s [17,18]. In the sequence of interfa-cial reactions occurring on the surface of bioactive glasses soaked inphysiological fluids, the glass initially exchanges alkaline oralkaline-earth cations (which are present as network modifiers inthe glasses) with protons from the solution, while the interfacial pHincreases. At the same time, as the Si\O\Si bonds of the glass net-work break, a SiO2 rich layer (silica gel) forms on the glass surfaceand an amorphous calcium phosphate is formed from the silica gel. Fi-nally, microcrystalline apatite nucleates and grows from the calciumphosphate film. The silica gel and apatite film will provide adsorptionsites for the cellular growth factors produced by stem cells andmacro-phages in vivo, thus promoting the formation of new bone tissue.
This model can be applied to explain the bioactivity of BG_Ca com-posites, whose glassy phase was preserved after the thermal treatment.As far as the 45S5 Bioglass® composites are concerned, different reac-tion mechanisms in SBF are involved, since the glass experienced awide crystallization during the heat treatment to consolidate the com-posite. A few years ago Boccaccini at al. extensively studied the reac-tivity of bioactive glass-ceramics during immersion in SBF, with aparticular emphasis on 45S5 Bioglass®-derived crystalline phases,such as Na2Ca2Si3O9 crystallites [44,76]. In particular, they suggestedthat the general idea of the model proposed by Hench could be appliedto 45S5-derived glass ceramics as well, provided that the crystallinephases undergo a preliminary amorphization in SBF. In fact, the initialion leaching results in amorphization of the crystalline network by theformation of point defects and the interfacial reactions occur at a slowerrate than on a glass.
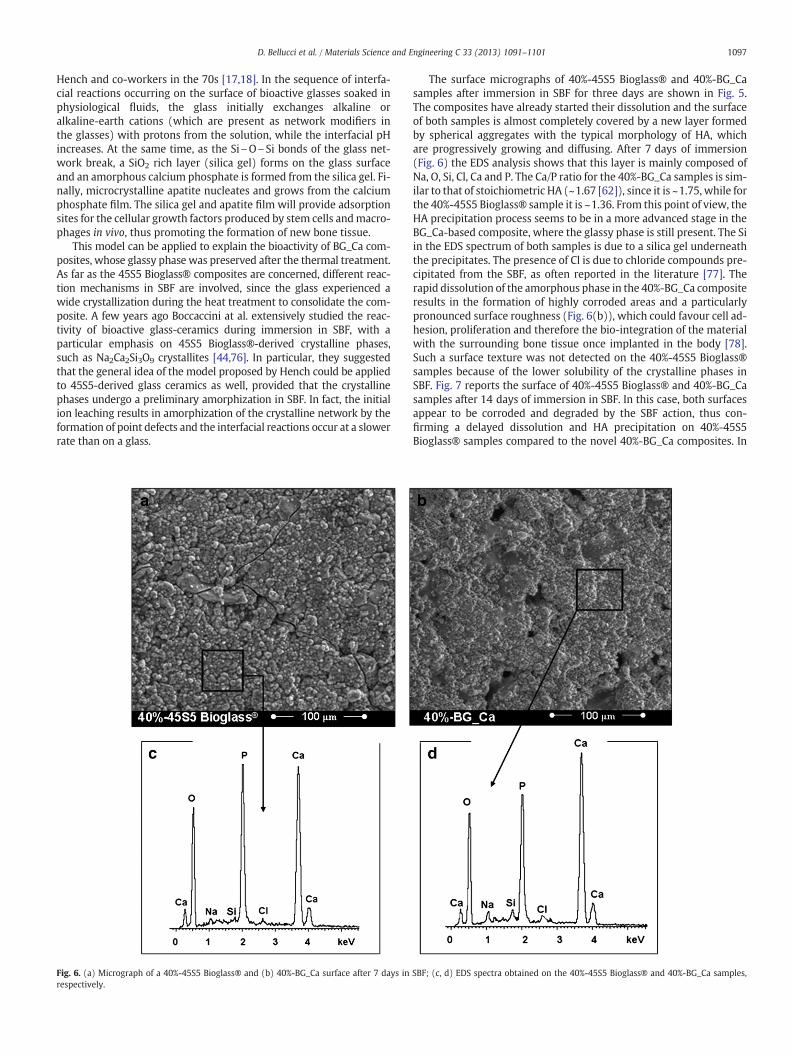
Fig. 6. (a) Micrograph of a 40%-45S5 Bioglass® and (b) 40%-BG_Ca surface after 7 days inrespectively.
The surface micrographs of 40%-45S5 Bioglass® and 40%-BG_Casamples after immersion in SBF for three days are shown in Fig. 5.The composites have already started their dissolution and the surfaceof both samples is almost completely covered by a new layer formedby spherical aggregates with the typical morphology of HA, whichare progressively growing and diffusing. After 7 days of immersion(Fig. 6) the EDS analysis shows that this layer is mainly composed ofNa, O, Si, Cl, Ca and P. The Ca/P ratio for the 40%-BG_Ca samples is sim-ilar to that of stoichiometric HA (~1.67 [62]), since it is ~1.75, while forthe 40%-45S5 Bioglass® sample it is ~1.36. From this point of view, theHA precipitation process seems to be in a more advanced stage in theBG_Ca-based composite, where the glassy phase is still present. The Siin the EDS spectrum of both samples is due to a silica gel underneaththe precipitates. The presence of Cl is due to chloride compounds pre-cipitated from the SBF, as often reported in the literature [77]. Therapid dissolution of the amorphous phase in the 40%-BG_Ca compositeresults in the formation of highly corroded areas and a particularlypronounced surface roughness (Fig. 6(b)), which could favour cell ad-hesion, proliferation and therefore the bio-integration of the materialwith the surrounding bone tissue once implanted in the body [78].Such a surface texture was not detected on the 40%-45S5 Bioglass®samples because of the lower solubility of the crystalline phases inSBF. Fig. 7 reports the surface of 40%-45S5 Bioglass® and 40%-BG_Casamples after 14 days of immersion in SBF. In this case, both surfacesappear to be corroded and degraded by the SBF action, thus con-firming a delayed dissolution and HA precipitation on 40%-45S5Bioglass® samples compared to the novel 40%-BG_Ca composites. In
SBF; (c, d) EDS spectra obtained on the 40%-45S5 Bioglass® and 40%-BG_Ca samples,
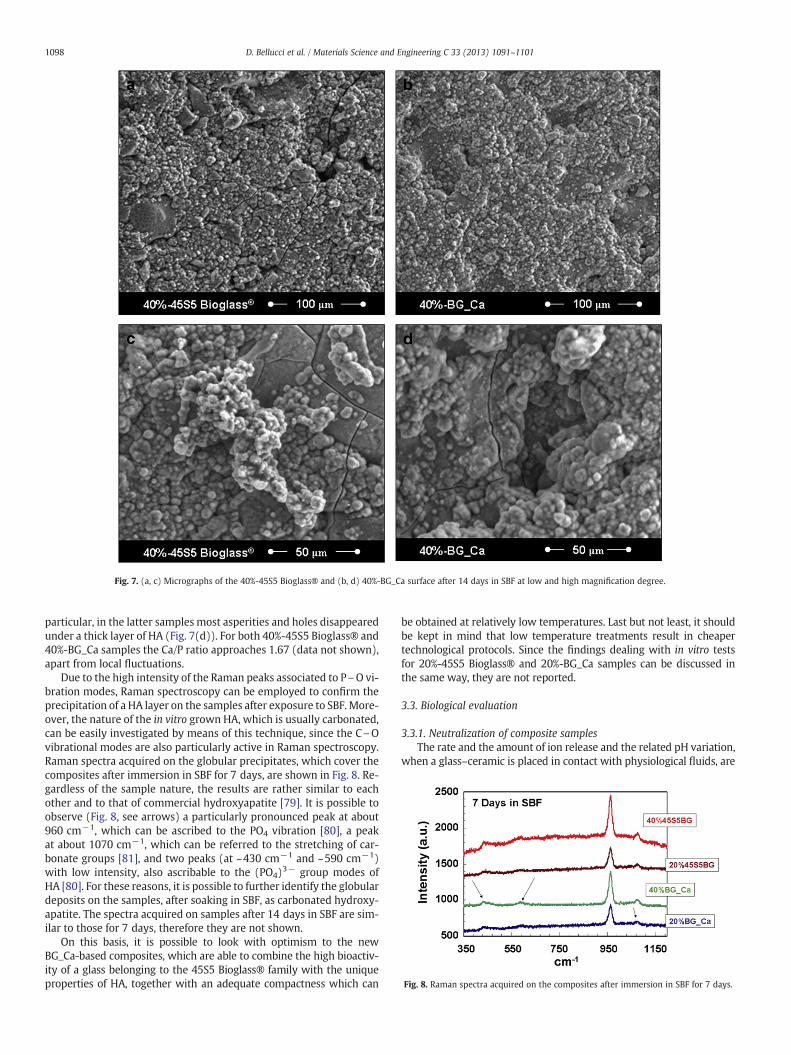
Fig. 7. (a, c) Micrographs of the 40%-45S5 Bioglass® and (b, d) 40%-BG_Ca surface after 14 days in SBF at low and high magnification degree.
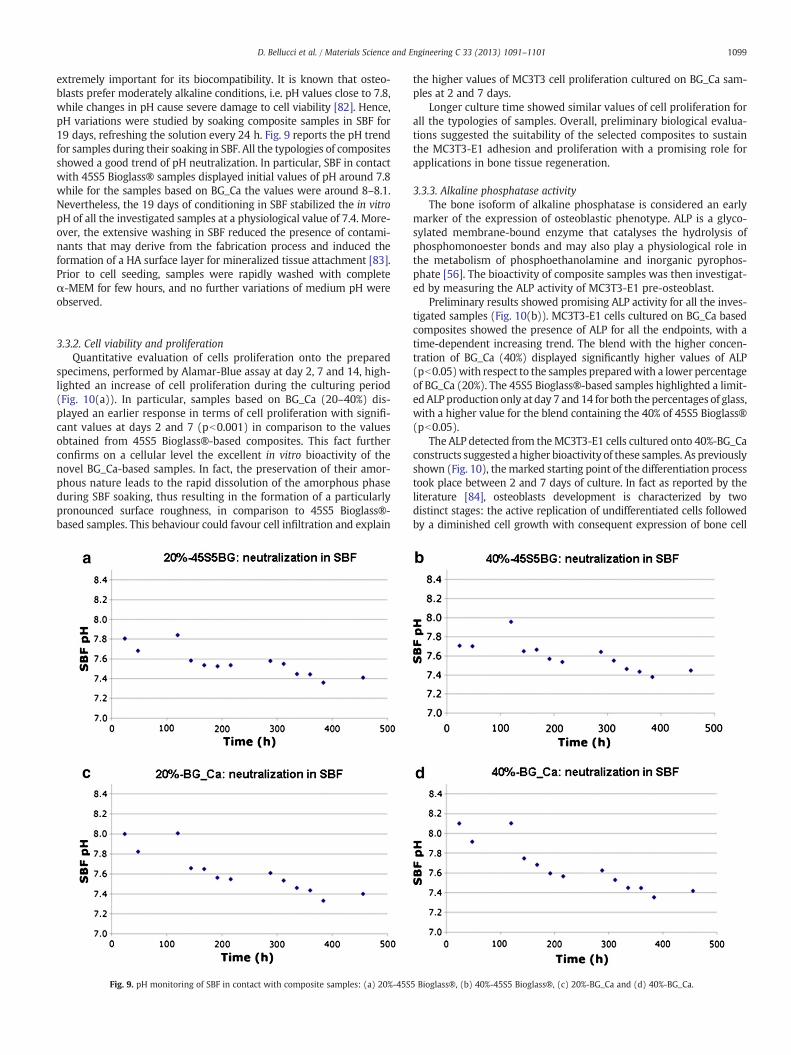
Fig. 8. Raman spectra acquired on the composites after immersion in SBF for 7 days.
1098 D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
particular, in the latter samples most asperities and holes disappearedunder a thick layer of HA (Fig. 7(d)). For both 40%-45S5 Bioglass® and40%-BG_Ca samples the Ca/P ratio approaches 1.67 (data not shown),apart from local fluctuations.
Due to the high intensity of the Raman peaks associated to P\O vi-bration modes, Raman spectroscopy can be employed to confirm theprecipitation of a HA layer on the samples after exposure to SBF.More-over, the nature of the in vitro grown HA, which is usually carbonated,can be easily investigated by means of this technique, since the C\Ovibrational modes are also particularly active in Raman spectroscopy.Raman spectra acquired on the globular precipitates, which cover thecomposites after immersion in SBF for 7 days, are shown in Fig. 8. Re-gardless of the sample nature, the results are rather similar to eachother and to that of commercial hydroxyapatite [79]. It is possible toobserve (Fig. 8, see arrows) a particularly pronounced peak at about960 cm−1, which can be ascribed to the PO4 vibration [80], a peakat about 1070 cm−1, which can be referred to the stretching of car-bonate groups [81], and two peaks (at ~430 cm−1 and ~590 cm−1)with low intensity, also ascribable to the (PO4)3− group modes ofHA [80]. For these reasons, it is possible to further identify the globulardeposits on the samples, after soaking in SBF, as carbonated hydroxy-apatite. The spectra acquired on samples after 14 days in SBF are sim-ilar to those for 7 days, therefore they are not shown.
On this basis, it is possible to look with optimism to the newBG_Ca-based composites, which are able to combine the high bioactiv-ity of a glass belonging to the 45S5 Bioglass® family with the uniqueproperties of HA, together with an adequate compactness which can
be obtained at relatively low temperatures. Last but not least, it shouldbe kept in mind that low temperature treatments result in cheapertechnological protocols. Since the findings dealing with in vitro testsfor 20%-45S5 Bioglass® and 20%-BG_Ca samples can be discussed inthe same way, they are not reported.
3.3. Biological evaluation
3.3.1. Neutralization of composite samplesThe rate and the amount of ion release and the related pH variation,
when a glass–ceramic is placed in contact with physiological fluids, are
1099D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
extremely important for its biocompatibility. It is known that osteo-blasts prefer moderately alkaline conditions, i.e. pH values close to 7.8,while changes in pH cause severe damage to cell viability [82]. Hence,pH variations were studied by soaking composite samples in SBF for19 days, refreshing the solution every 24 h. Fig. 9 reports the pH trendfor samples during their soaking in SBF. All the typologies of compositesshowed a good trend of pH neutralization. In particular, SBF in contactwith 45S5 Bioglass® samples displayed initial values of pH around 7.8while for the samples based on BG_Ca the values were around 8–8.1.Nevertheless, the 19 days of conditioning in SBF stabilized the in vitropH of all the investigated samples at a physiological value of 7.4. More-over, the extensive washing in SBF reduced the presence of contami-nants that may derive from the fabrication process and induced theformation of a HA surface layer for mineralized tissue attachment [83].Prior to cell seeding, samples were rapidly washed with completeα-MEM for few hours, and no further variations of medium pH wereobserved.
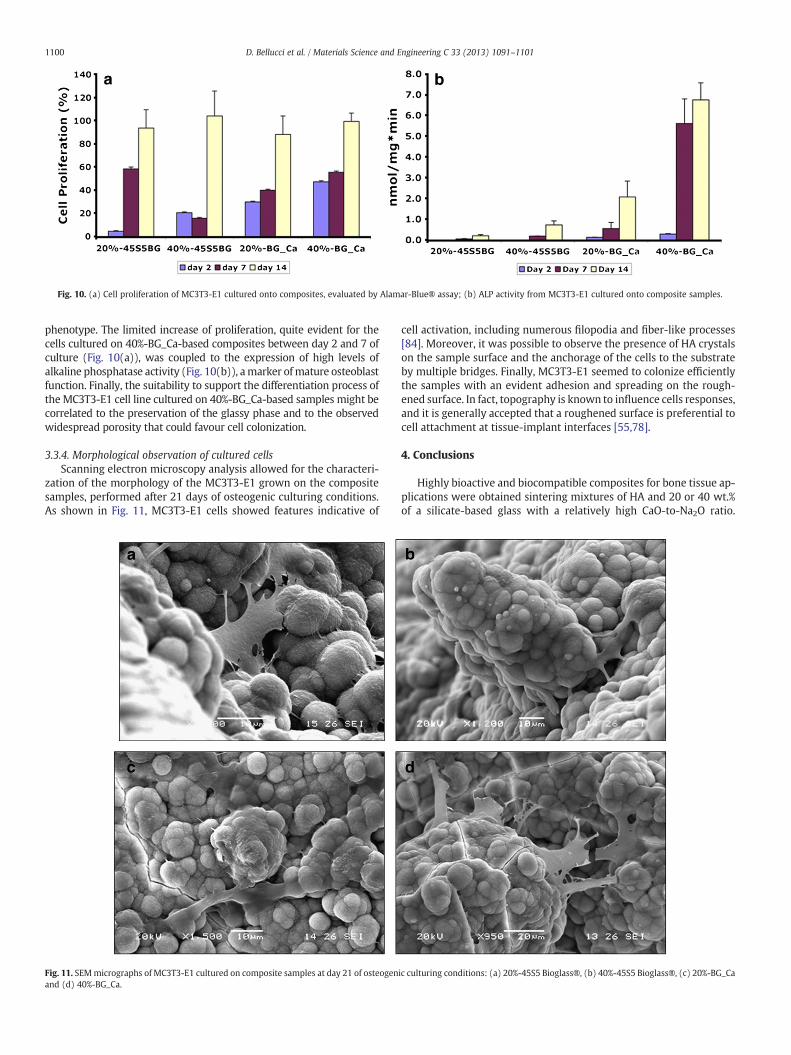
3.3.2. Cell viability and proliferationQuantitative evaluation of cells proliferation onto the prepared
specimens, performed by Alamar-Blue assay at day 2, 7 and 14, high-lighted an increase of cell proliferation during the culturing period(Fig. 10(a)). In particular, samples based on BG_Ca (20–40%) dis-played an earlier response in terms of cell proliferation with signifi-cant values at days 2 and 7 (pb0.001) in comparison to the valuesobtained from 45S5 Bioglass®-based composites. This fact furtherconfirms on a cellular level the excellent in vitro bioactivity of thenovel BG_Ca-based samples. In fact, the preservation of their amor-phous nature leads to the rapid dissolution of the amorphous phaseduring SBF soaking, thus resulting in the formation of a particularlypronounced surface roughness, in comparison to 45S5 Bioglass®-based samples. This behaviour could favour cell infiltration and explain
Fig. 9. pH monitoring of SBF in contact with composite samples: (a) 20%-45S
the higher values of MC3T3 cell proliferation cultured on BG_Ca sam-ples at 2 and 7 days.
Longer culture time showed similar values of cell proliferation forall the typologies of samples. Overall, preliminary biological evalua-tions suggested the suitability of the selected composites to sustainthe MC3T3-E1 adhesion and proliferation with a promising role forapplications in bone tissue regeneration.
3.3.3. Alkaline phosphatase activityThe bone isoform of alkaline phosphatase is considered an early
marker of the expression of osteoblastic phenotype. ALP is a glyco-sylated membrane-bound enzyme that catalyses the hydrolysis ofphosphomonoester bonds and may also play a physiological role inthe metabolism of phosphoethanolamine and inorganic pyrophos-phate [56]. The bioactivity of composite samples was then investigat-ed by measuring the ALP activity of MC3T3-E1 pre-osteoblast.
Preliminary results showed promising ALP activity for all the inves-tigated samples (Fig. 10(b)). MC3T3-E1 cells cultured on BG_Ca basedcomposites showed the presence of ALP for all the endpoints, with atime-dependent increasing trend. The blend with the higher concen-tration of BG_Ca (40%) displayed significantly higher values of ALP(pb0.05)with respect to the samples preparedwith a lower percentageof BG_Ca (20%). The 45S5 Bioglass®-based samples highlighted a limit-ed ALP production only at day 7 and 14 for both the percentages of glass,with a higher value for the blend containing the 40% of 45S5 Bioglass®(pb0.05).
The ALP detected from theMC3T3-E1 cells cultured onto 40%-BG_Caconstructs suggested a higher bioactivity of these samples. As previouslyshown (Fig. 10), themarked starting point of the differentiation processtook place between 2 and 7 days of culture. In fact as reported by theliterature [84], osteoblasts development is characterized by twodistinct stages: the active replication of undifferentiated cells followedby a diminished cell growth with consequent expression of bone cell
5 Bioglass®, (b) 40%-45S5 Bioglass®, (c) 20%-BG_Ca and (d) 40%-BG_Ca.
Fig. 10. (a) Cell proliferation of MC3T3-E1 cultured onto composites, evaluated by Alamar-Blue® assay; (b) ALP activity from MC3T3-E1 cultured onto composite samples.
1100 D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
phenotype. The limited increase of proliferation, quite evident for thecells cultured on 40%-BG_Ca-based composites between day 2 and 7 ofculture (Fig. 10(a)), was coupled to the expression of high levels ofalkaline phosphatase activity (Fig. 10(b)), amarker ofmature osteoblastfunction. Finally, the suitability to support the differentiation process ofthe MC3T3-E1 cell line cultured on 40%-BG_Ca-based samples might becorrelated to the preservation of the glassy phase and to the observedwidespread porosity that could favour cell colonization.
3.3.4. Morphological observation of cultured cellsScanning electron microscopy analysis allowed for the characteri-
zation of the morphology of the MC3T3-E1 grown on the compositesamples, performed after 21 days of osteogenic culturing conditions.As shown in Fig. 11, MC3T3-E1 cells showed features indicative of
Fig. 11. SEMmicrographs of MC3T3-E1 cultured on composite samples at day 21 of osteogenand (d) 40%-BG_Ca.
cell activation, including numerous filopodia and fiber-like processes[84]. Moreover, it was possible to observe the presence of HA crystalson the sample surface and the anchorage of the cells to the substrateby multiple bridges. Finally, MC3T3-E1 seemed to colonize efficientlythe samples with an evident adhesion and spreading on the rough-ened surface. In fact, topography is known to influence cells responses,and it is generally accepted that a roughened surface is preferential tocell attachment at tissue-implant interfaces [55,78].
4. Conclusions
Highly bioactive and biocompatible composites for bone tissue ap-plications were obtained sintering mixtures of HA and 20 or 40 wt.%of a silicate-based glass with a relatively high CaO-to-Na2O ratio.
1101D. Bellucci et al. / Materials Science and Engineering C 33 (2013) 1091–1101
The employed glass, named BG_Ca, shows a reduced tendency to crys-tallizewith respect to thewidely used 45S5 Bioglass®, therefore it waspossible to sinter the composites at a relatively low temperature, thuspreserving the glassy phase in the composite during sintering, withexcellent effects in terms of bioactivity. Additionally, it was possibleto avoid reactions between glass and HA or the decomposition of theHA itself, which typically occurs at higher temperatures. The realizedsamples were used as three-dimensional supports for the culture ofmouse calvaria-derived pre-osteoblastic cells MC3T3-E1. The samplesdemonstrated to be able to support cell adhesion and proliferation anda promising initialmechanism of differentiation towards an osteoblas-tic phenotype. In particular, compared to composites based on 45S5Bioglass® with the same HA/glass proportions, samples based onBG_Ca (20–40%) displayed an earlier response in terms of cell prolifer-ation, probably due to an increased surface roughness of the con-structs that promotes cell colonization. This fact further confirms ona cellular level the excellent in vitro bioactivity of the novel composi-tions. Future studies will be devoted to perform additional investiga-tions of osteoblast differentiation process by assessing the collagenproduction and later markers as extracellular matrix mineralizationand osteopontin.
Acknowledgements
The authors would like to thank Ms. Silvia Volpi for her help andsupport during experiments. Dr. Aura Bonaretti is kindly acknowl-edged for recording SEM images of biological samples.
References
[1] R.M. Nerem, Ann. Biomed. Eng. 19 (1991) 529–545.[2] L.G. Griffith, G. Naughton, Science 295 (2002) 1009–1014.[3] R. Lanza, R. Langer, J. Vacanti, Principles of Tissue Engineering, third ed. Academic
Press, San Diego, CA, USA, 2007.[4] R. Langer, J.P. Vacanti, Science 260 (1993) 920–926.[5] M.J. Yaszemski, J.B. Oldham, L. Lu, B.L. Currier, in: J.E. Davis (Ed.), Bone Engineer-
ing, Em Squared, Toronto, 2000, pp. 541–547.[6] J.P. Fisher, A.H. Reddi, in: N. Ashammakhi, P. Ferretti (Eds.), Topics in Tissue Engi-
[9] R.M. Day, Tissue Eng. 11 (2005) 768–777.[10] K. Anselme, Biomaterials 21 (2000) 667–681.[11] H. Aoki, Science and Medical Applications of Hydroxyapatite, Japanese Associa-
tion of Apatite Layer, Tokyo, 1991.[12] W. Suchanek, M. Yoshimura, J. Mater. Res. 13 (1998) 94–117.[13] V.P. Orlovskii, V.S. Komlev, S.M. Barinov, Inorg. Mater. 38 (2002) 973–984.[14] R.V. Silva, J.A. Camilli, C.A. Bertran, N.H. Moreira, Int. J. Oral Maxillofac. Surg. 10
(2004) 1–7.[15] L.L. Hench, J. Wilson, An Introduction to Bioceramics, World Scientific Inc., 1993.[16] M.N. Rahaman, D.E. Day, B.S. Bal, Q. Fu, S.B. Jung, L.F. Bonewald, A.P. Tomsia, Acta
117–141.[19] L.L. Hench, J. Mater. Sci: Mater. Med. 17 (2006) 967–978.[20] M. Marcacci, E. Kon, V. Moukhachev, A. Lavroukov, S. Kutepov, R. Quarto, M.
Mastrogiacomo, R. Cancedda, Tissue Eng. 13 (2007) 947–955.[21] S.V. Dorozhkin, Biomaterials 31 (2010) 1465–1485.[22] P. Ducheyne, S. Radin, L. King, J. Biomed. Mater. Res. 27 (1993) 25–34.[23] E. Schepers, M. Declercq, P. Ducheyne, J. Oral. Rehab. 18 (1991) 439–452.[24] H. Oonishi, L.L. Hench, J. Wilson, F. Sugihara, E. Tsuji, M. Matsuura, S. Kin, T.
Yamamoto, S. Mizokawa, J. Biomed. Mater. Res. 51 (2000) 37–46.[25] J. Zhong, D.C. Greenspan, J. Biomed. Mater. Res. (Appl. Biomat.) 53 (2000) 694–701.[26] P.X. Ma, Mater. Today 7 (2004) 30–40.[27] D.W. Hutmacher, Biomaterials 21 (2000) 2529–2543.[28] V. Karageorgiou, D. Kaplan, Biomaterials 26 (2005) 5474–5491.[29] J. Li, B. Fartash, L. Hermannsson, Biomaterials 16 (1995) 417–422.[30] W. Bonfield, in: L.L. Hench, J. Wilson (Eds.), An Introduction to Bioceramics, World
(1994) 5–10.[34] G. Georgiou, J.C. Knowles, Biomaterials 22 (2001) 2811–2815.[35] J.D. Santos, F.J. Monteiro, J.C. Knowles, J. Mater. Sci: Mater. Med. 6 (1995) 348–352.[36] R. Ravarian, F. Moztarzadeh, M. Solati Hashjin, S.M. Rabiee, P. Khoshakhlagh, M.
Tahriri, Ceram. Int. 36 (2010) 291–297.[37] J.C. Knowles, W. Bonfield, J. Biomed. Mater. Res. 27 (1993) 1591–1598.[38] E.M. Carlisle, Science 167 (1970) 179–180.[39] S. Hu, J. Chang, M. Liu, C. Ning, J. Mater. Sci. Mater. Med. 20 (2009) 281–286.[40] G. Evans, J. Behiri, J. Currey, W. Bonfield, J. Mater. Sci: Mater. Med. 1 (1990) 38–43.[41] J.C. Knowles, Br. Ceram. Trans. 93 (1994) 100–103.[42] M. Vallet-Regi, J.M. Gonzales-Calbet, Prog. Solid. State. Chem. 32 (2004) 1–31.[43] W. Suchanek, M. Yashima, M. Kakihana, M. Yoshimura, Biomaterials 18 (1997)
923–933.[44] Q.Z. Chen, I.D. Thompson, A.R. Boccaccini, Biomaterials 27 (2006) 2414–2425.[45] O. Peitl, E.D. Canotto, J.J. Hench, J. Non-Cryst. Solids 292 (2001) 115–126.[46] O.P. Filho, G.P. LaTorre, L.L. Hench, J. Biomed. Mater. Res. 30 (1996) 509–514.[47] L. Lefebvre, J. Chevalier, L. Gremillard, R. Zenati, G. Thollet, D. Bernache-Assolant,
A. Govin, Acta Mater 55 (2007) 3305–3313.[48] D.C. Tancred, A.J. Carr, B.A.O. McCormack, J. Mater. Sci: Mater. Med. 12 (2001)
81–93.[49] C.P.A.T. Klein, A.A. Driessen, K. de Groot, A. Van den Hooff, J. Biomed. Mater. Res.
17 (1983) 769–784.[50] M.G.W. Lockyer, D. Holland, R. Dupree, J. Non-Cryst. Solids 188 (1995) 207–219.[51] D. Bellucci, V. Cannillo, A. Sola, Materials 4 (2011) 339–354.[52] V.G. Varanasi, E. Saiz, P.M. Loomer, B. Ancheta, N. Uritani, S.P. Ho, A.P. Tomsia, S.J.
Marshall, G.W. Marshall, Acta Biomater 5 (2009) 3536–3547.[53] T. Kokubo, H. Takadama, Biomaterials 27 (2006) 2907–2915.[54] T. Kokubo, H. Kushitani, S. Sakka, T. Kitsugi, T. Yamamuro, J. Biomed. Mater. Res.
24 (1990) 721–734.[55] Q.Z. Chen, A. Efthymiou, V. Salih, A.R. Boccaccini, J. Biomed. Mater. Res. A 84
(2008) 1049–1060.[56] G.R. Beck, E.C. Sullivan, E. Moran, B. Zerler, J. Cell. Biochem. 68 (1998) 269–280.[57] P. Wutticharoenmongkol, P. Pavasant, P. Supaphol, Biomacromolecules 8 (2007)
2602–2610.[58] J. Wang, X. Yu, Acta Biomater. 6 (2010) 3004–3012.[59] D.S. Soper, Analysis of Variance (ANOVA) Calculator - One-Way ANOVA from
Summary Data (Online Software), http://www.danielsoper.com/statcalc3 2012.[60] J.C. Knowles, S. Talal, J.D. Santos, Biomaterials 17 (1996) 1437–1442.[61] D.C. Clupper, L.L. Hench, J. Non-Cryst. Solids 318 (2003) 43–48.[62] G. Göller, H. Demirkiran, F.N. Oktar, E. Demirkesen, Ceram. Int. 29 (2003) 721–724.[63] M.M.A. Ramselaar, P.J. van Mullem, W. Kalk, J.R. de Wijn, A.L.H. Stols, F.C.M.
Driessens, J. Mater. Sci: Mater. Med. 4 (1993) 311–317.[64] M.M.A. Ramselaar, F.C.M. Driessens, W. Kalk, J.R. de Wijn, P.J. van Mullem,
J. Mater. Sci: Mater. Med. 2 (1991) 63–70.[65] A.R. El-Ghannam, J. Biomed. Mater. Res. A 69 (2004) 490–501.[66] X. Liua, C. Dinga, P.K. Chub, Biomaterials 25 (2004) 1755–1761.[67] V. Cannillo, F. Pierli, S. Sampath, C. Siligardi, J. Eur. Ceram. Soc. 29 (2009) 611–619.[68] S. Aryal, S.R. Bhattarai, K.C. R. Bahadur, M.S. Khil, D.R. Lee, H.Y. Kim, Mater. Sci.
Eng., A 426 (2006) 202–207.[69] M. Bohner, J. Lemaître, Biomaterials 30 (2009) 2175–2179.[70] T.Y. Juliana, J.T.Y. Lee, Y. Leng, K.L. Chow, F. Ren, X. Ge, K. Wang, X. Lu, Acta
Biomater. 7 (2011) 2615–2622.[71] S. Kotani, Y. Fujita, T. Kitsugi, T. Nakamura, T. Yamamuro, C. Ohtsuki, et al.,
J. Biomed. Mater. Res. 25 (1991) 1303–1315.[72] W.R. Walsh, P. Morberg, Y. Yu, J.L. Yang, W. Haggard, P.C. Sheath, et al., Clin.
Orthop. 406 (2003) 228–236.[73] H. Chan, D. Mijares, J.L. Ricci, in: Transactions - seventh world biomaterials con-
gress, 2004. Sydney, 2004, p. 627.[74] D. Apelt, F. Theiss, A.O. El-Warrak, K. Zlinszky, R. Bettschart-Wolfisberger, M.
Bohner, et al., Biomaterials 25 (2004) 1439–1445.[75] J.R. Jones, P. Sepulveda, L.L. Hench, J. Biomed. Mater. Res. 58 (2001) 720–726.[76] A.R. Boccaccini, Q. Chen, L. Lefebvre, L. Gremillard, J. Chevalier, Faraday Discuss.
136 (2007) 27–44.[77] S. Padilla, J. Román, S. Sánchez-Salcedo, M. Vallet-Regí, Acta Biomater 2 (2006)
87–96.[79] D. Bellucci, G. Bolelli, V. Cannillo, A. Cattini, A. Sola, Mater. Charact. 62 (2011)
1021–1028.[80] S. Koutsopoulos, J. Biomed. Mat. Res. 62 (2002) 600–612.[81] A. Awonusi, M.D. Morris, M.M.J. Tecklenburg, Calcif. Tissue Int. 81 (2007) 46–52.[82] I.A. Silver, J. Deas, M. Erecinska, Biomaterials 22 (2000) 175–185.[83] D. Bellucci, V. Cannillo, A. Sola, F. Chiellini, M. Gazzarri, C. Migone, Ceram. Int. 37
(2011) 1575–1585.[84] L.D. Quarles, D.A. Yohay, L.W. Lever, R. Caton, R.J. Wenstrup, J. Bone Miner. Res. 7