13
Max Bachour Jessica Chen
| Date post: | 17-Dec-2015 |
| Category: |
Documents |
| Upload: | philomena-gibson |
| View: | 214 times |
| Download: | 1 times |

Max Bachour
Jessica Chen

Shotgun or 454 sequencing
•High throughput sequencing technique that can collect a large amount of data at a fast rate.
•Works by partially digesting a genome or big strand of DNA into small overlapping fragments
•These small fragments are sequenced and fragments that overlap are matched together.
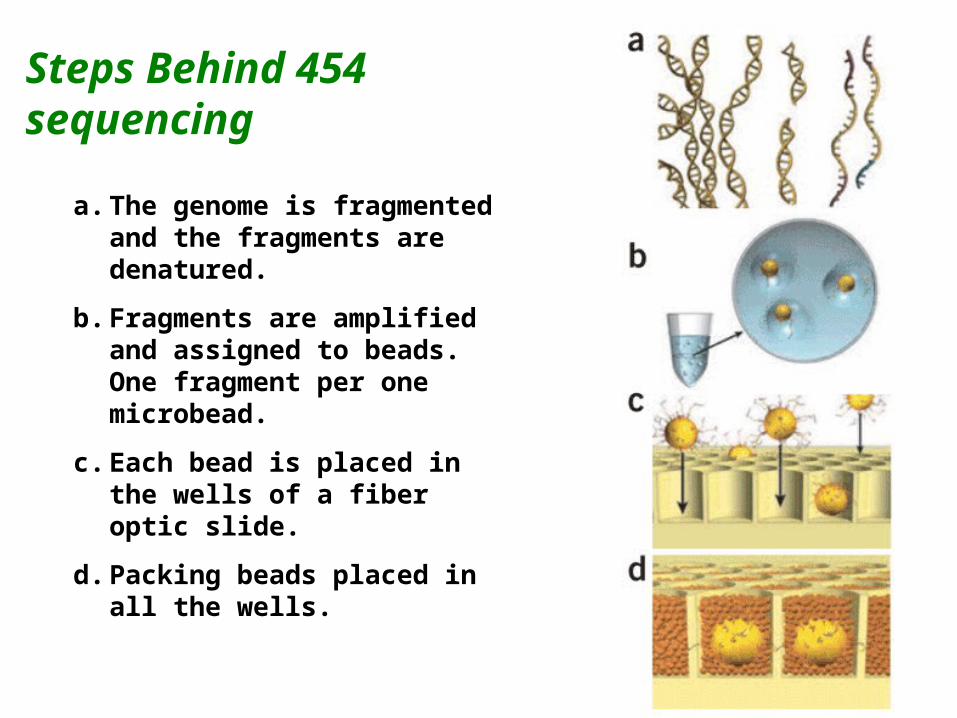
Steps Behind 454 sequencing
a. The genome is fragmented and the fragments are denatured.
b. Fragments are amplified and assigned to beads. One fragment per one microbead.
c. Each bead is placed in the wells of a fiber optic slide.
d. Packing beads placed in all the wells.

• Solution of one nucleoside is flooded onto tray.
• If base added is next in the sequence, it will be added to the single stranded DNA on the bead.
• When a nucleoside is added to DNA, 2 phosphates are given out
• Enzymes in packing beads convert phosphate groups to ATP and then the ATP to light energy.
Steps Behind 454 sequencing

• Computer and camera detect light in a certain well as a certain base is added to the tray.
• Base is washed off and process is repeated with another base.
• End product is large amount of fragments sequenced.
Steps Behind 454 sequencing

Genome Sequence Analysis
Contig Assembly
Identifying open reading frames (ORF) using gene prediction programs

What is the initial problem with assembly?
Sequenced fragmented
DNA
Incorrectly Assembled
DNA Sequence
CONTIG 1 CONTIG 2

How is this problem solved?
Sequenced fragmented
DNA
Masked DNA Sequence
CONTIG 1 CONTIG 3 CONTIG 5
CONTIG 2
CONTIG 4Assembled
DNA Sequence

How do we identify genes?
1) Use gene prediction programs (Fgenesh, Genscan, Genemark) to determine potential genes; also determine any repeat sequences
Enter contig
2) Which of the predicted genes are most likely existing genes?
Use BLAST

How do we use BLAST?
tblastn all predicted genes against an EST database (ESTDB)
Why ESTDB? – record of all known/identified mRNA (cDNA library)
Why tblastn? -- amino acid sequence more likely to be conserved
use blastn and blastp -blastp: determine expression of
gene

Analyzing BLAST data
• Critical data: – e-value– %match– EST
source
Gene 1:
Protein sequence: MFVVQYLGSSRSWTSCSHSSKPGVDSRGRAEPHLAVGRSSLLGRVQTGLKGGGMKDSDLT
GDSSLARANQSMGICKSEGTVDRRLKSQVSQLLLGLLLIRLEGLLATCMTGPHGDAGAGS
THK
>gb|FC457105.1| UCRVU04_CCNI646_g1 Cowpea 524B Mixed Tissue and Conditions cDNA
Library UCRVU04-1 Vigna unguiculata cDNA clone CCNI646, mRNA
sequence.
Length=807
Score = 215 bits (548), Expect(2) = 2e-55, Method: Compositional matrix adjust.
Identities = 110/112 (98%), Positives = 110/112 (98%), Gaps = 0/112 (0%)
Frame = -1
Query 12 SWTSCSHSSKPGVDSRGRAEPHLAVGRSSLLGRVQTGLKGGGMKDSDLTGDSSLARANQS 71
SWTSCSHS KPGVDSRGRAEPHLAVGRSSLLGRVQTGLKGGGMKDSDLTGDSSLARANQS
Sbjct 438 SWTSCSHS*KPGVDSRGRAEPHLAVGRSSLLGRVQTGLKGGGMKDSDLTGDSSLARANQS 259
Query 72 MGICKSEGTVDRRLKSQVSQLLLGLLLIRLEGLLATCMTGPHGDAGAGSTHK 123
MGICK EGTVDRRLKSQVSQLLLGLLLIRLEGLLATCMTGPHGDAGAGSTHK
Sbjct 258 MGICK*EGTVDRRLKSQVSQLLLGLLLIRLEGLLATCMTGPHGDAGAGSTHK 103

Advantages and Disadvantages
• Fast sequencing at a high volume• Cheap compared to other
methods• Much higher coverage protection• Repetitive sequences can disrupt
computer program into thinking that unrelated sequences are in fact connected.
• More prone to error and missing sequences

Drastically Drastically changed changed genomics in a genomics in a very short very short amount of amount of timetime















![Lander-Waterman Statistics for Shotgun Sequencing [10pt] Math …gptesler/283/slides/shotgun_f19-handout.pdf · Lander-Waterman Statistics for Shotgun Sequencing Math 283: Ewens &](https://static.documents.pub/doc/80x56/603f8be259e6e0059e20c3cf/lander-waterman-statistics-for-shotgun-sequencing-10pt-math-gptesler283slidesshotgunf19-.jpg)

