Page 1
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 1/8
3516 J ourna l
l inkage at C-9 in the real substrate (3) .
(16) (a) G. A. Berchtold and G. F. Uh lig, J. Org. Chem., 28, 1459 (1963); (b) K.
C. Brannock. R . D. Burpitt, and
J.
G. Thwe att, ibid., 28, 1462 (1963); (c)
C. F. Huebner,
L.
Dorfmann, M. M. Robinson, E. Donog hue, W. G. Pierson,
and P. Strachan , hid., 28, 3134 (1963); (d) K. C. Brannoc k,
R.
D. Burpitt,
V.
W .
Goodiett, and J. G. Thweatt, ibid., 29, 8 18 (1964).
(17)
R.
Ratcliffe and R . Rodehorst, J.
Org.
Chem., 35, 4000 (1970) .
(18) The trans-fused symmetrical ketone analogous to 32 has a C-2 axis.
Therefore, the m ethyl groups are equivalent and the trans ketone would
only exhibit seven signals. The cis-fused diketone
32,
on the other hand,
has no
C-2
axis and thus the methyl groups are diastereotopic.
(19)
The methyl peaks of
32
also overlapped in the 'H NMR. The Eu(fod)3
spectrum shifted them from 6
1
.O
o
two singlets at 6 3 .0 and 3.05.
(20) H. C. Brown and
S.
Krishnamurthy, J. Am. Chem. SOC., 94, 7159
(1972) .
(21)
G. Hofle and
W.
Steglich, Synthesis,
619 (1972),
and references cited
therein.
(22) N.
S.
hacca,J. E. Gurst, and D.
H.
Wil l iams, J. Am.
Chem. SOC.
7, 302
(1965) .
(23) if one assumes that isom ers 17 and 19 are normal , then the characteristic
shift for H in CDC13 should be (4.30+ 4.15) /2
6
4.22. The deshieldin
caused by the carbony l group is therefore 6 -
.53
for isomer
16
and
- .58 for isomer 18. In CsDs he n ormal position for Ha is at (4.45 +
4 .85 ) /2
6
4.65. The deshielding or isomer 16
is
therefore
6
- . 4 and
0.2 for isomer 18. The positive s olvent shift is thereby seen, in that the
deshielding of Ha caused by the carbo nyl group is less in C& than in CDC13
for each isom er. The gross CsD, deshielding of all isom ers is probably a
consequence of the polar benzenesulfonatesubstituen t. See D. H. W illiams
and N.S. Bhacca, Tetrahedron, 21, 2021 (1965)) or an extended discussion
of benzene solvent shifts with carbonyl compounds.
(24)
Dienone
49
could be isolated from s everal of the
17
.
5 1
reaction mix-
tures, but the major com ponent was alwa ys dimer 51.
(25) L. M. Jackm an and S. Sternhell, Applications of Nuclear Magnetic Res-
onance Spectrosco py in Organic Chem istry, Pergamon Press, Oxford,
1969, p 185.
(26) (a) See ref 25, p 225; (b) H. B. Kagan, Ed., Determ ination of Configurations
by Spectrom etric Methods, Vol. I.Georg Thieme Verlag, Stuttgart, 1977,
p 59.
of the American Chemical Socie ty / 101:13 / J u n e 20, 1979
(27) Hoffmann has done a m olecular orbital analysis of fragmentation reactions
[R.
Gleiter. W.
D.
Stohrer, and R . Hoffmann, Helv. Chim. Acta, 55, 893
(1972)] and finds that, for thos e cases where th e dihedral angle $J = Oo
cyclobutane formation can be preferred. The cases in point (41-fold,
53-fold) have J 60° , but the Hoffm ann calculations do not give results
other than for 4
0,
90, 180 .
(28) Technical assistance for some of the 360-MHz NMR experiments by
R.
E. Santini, J. Dallas, and N. Nowicki is greatly appreciated.
(29)
International Flavors and Fragrances, sold as transdecahydro-p-na-
phthol.
(30) W. G. Dauben,
R .
C. Tweit. and C. Mannersk antz,J. Am. Chem.
SOC.
76,
(31) H. C. Brow n, C. P. GarQ,and K. T. Liu,
J.
O r a Chem., 36, 387 (1971).
4420 (1954).
(32) A. Kandiah, J. Chem. Soc., 922 (1931).
(33) (a) K. Taguchi and F. H. Westheimer, J.
Org.
Chem., 36, 1571 (1971); (b)
-
H.
Weingarten and W. A. W hite,
bid.,
32, 213 (1967);
(c)
I.
Moretti and G.
Torre. Synthesis, 141 (1970).
(34) (a) H. Weingarten and W. A. W hite,
J.
Org. Chem., 32, 213 (1967); (b) I.
Moretti and G. Torre, Synthesis,
141 (1970).
(35) (a) A.
K.
Bose, G. Mina, M.
S.
Manhas, and E. Rzucidlo. Tetrahedron Lett.,
1467 (1963); (b) K. L. Brannock,
R. D.
Burpitt, U. W. G oodlett, and J. G.
Thweatt, J. Org. Chem.
29,81 8 (1964);
(c) C. F. Huebner, L. Dorfman. M.
M. Robison, E. Donoghue, W. G. Pierso n, and P. Strachan , ibid., 28 ,3134
(1963); (d) J. A. Hirs ch and F. J. Cross, ibid., 36,95 5 (1971); (e) A. J. Birch
and E. G. Hutchins on, J. Chem. SOC.
C 671 (1971);
(f)
E.
Yoshi and S.
Kimoto, Chem. Pharm.Bull., 17, 629 (1969) : (9) G. A. Berch told and G. F.
Uhlig,
J .
Org. Chem., 28, 1459 (1963); (h) D. W. Bo erth and F. A. Van-
Catledge,
ibid., 40,
3319 (1975).
(36)
R. Mozing, Organic Syntheses , Collect. Vol. Ill, Wiley, New York,
1955,
p 181.
(37) E. J. Corey and M. Chaykovsky,
J.
Am. Chem.
Soc.
87, 1345, 1353
(1965).
(38) (a)
H.
C. Brown, Organic Synthesis via Boranes , Wiley-lnterscienc e, New
York, 1975, (b) H. C. Brow n, A. K. M andai. and
S.
U.Kulkarni, J. Org. Chem.,
42, 1392 (1977); (c)
H.
C. Brown and N. Ravindran, ibid., 42, 2533
(1977) .
(39) L.
F.
Fieser and M. Fieser, Reagents for Organic Synthesis , Vol. 1, Wiley.
New York,
1967,
p
1180.
Mechanism of the Cannizzaro R e a ~ t i o r r l - ~
C.
Gardner Swain, Arnet L. P o ~ e l l , ~illiam A. Sheppard, and Charles
R .
Morgan
Contribution fr o m the Department o f Chem istry and the Laboratory f o r Nuclear Science,
Massachusetts Insti tute of Technology, Cambridge, Massachusetts
02139.
Receiced Nocember
13, 1978
.Abstract: A n ultimate technique for disqualifying compounds suspected of being intermediates is illustrated by the use of iso-
tope dilution to prove that benzyl benzoate is not an intermediate i n the Canniz zaro reaction of
0.5
M benzaldehyde-p-t (tri-
tium labeled) with 0.25
M
sodium hydroxide in
74%
methanolL26% water at 100 OC. Th e adduct from hydroxide ion and two
molecules of benzaldehyde th at was though t to rearrange to benzyl benzoate could alternatively rearrang e directly to the prod-
ucts. benzoate ion and benzyl alcohol. Houever, this mechanism also is disproved because methoxide
ion
acting instead
of
hy-
droxide ion should lead to benzyl methyl e the r, but less than 1 is found. Two other mechanisms involving a proton transfer
concerted Mith the hydride transfer a re disproved by the k ~ ~ ~ / k ~ ~ osotope effect of 1.9 . The rate-determining steps can be
rcprcsented by tivo hydride transfer reactions to C~ H S C H O .rom the adduct from HO- + ChHsC HO and from the adduct
from CH30-
+
ChHsCHO, or. equivalently. by t w o termolecular reactions. H O - + 2ChHsCHO and CH3O- +
2
c >ti
J C
H
0
T h e C a n n iz za r o r eac t io n b is t h e d i sp ro p o r t ion a t io n o f an
a ld ch y d c to an eq u im o la r m ix tu re o f p r im ary a l co h o l an d
ca rb o x y l ic sa l t . I t i s ch a rac t e r i s t i c o f a ld eh y d es th a t h av e n o
1
hydrogens, and therefo re canno t undergo aldo l condensat ion .
T h e r cac t io n i s u su a l ly b ro u g h t ab o u t i n a h o m o g en eo u s ,
s t rong ly basic so lu t ion o r in a heterogeneous sys tem consis t ing
o f an o rg an ic p h ase an d a s t ro n g ly b as i c aq u eo u s p h ase .
A
typ ical example is react ion o f benzaldehyde I) i t h co n cen -
t r a t ed so d iu m h y d ro x id e
i n
h o t aq u eo u s m e th an o l t o y i e ld
bcn7yl a lcohol (I I ) an d so d iu m b en zo a te ( I I ) . F o rm ald eh y d e
d i sp ro p o r t io n a tes i n ac id so lu tio n a lso .' T h e C an n izz a ro r e-
ac t io n w as co n s id e red o n e o f t h e m o s t im p o r t an t sy n th e t i c
reactio ns of organic chem istry prior to the discovery of LiAIH4
i n 1946, but has now been totally supplanted by metal hydrides
fo r l ab o ra to r y sy n th eses .
0002-7863/19/1501-3576$01
OO / O
C H 3 0 H
2 C b H 5 C H 0
+
N a O H
-
6 H 5 C H 2 0 H
I
I 1
+
C ~ H S C O ~ N ~
I I I
B en zy l b en zo a te (V I ) w as i so l a ted f ro m t h e r eac t io n o f I
w i t h N a O H
i n
w a t e r
or
i n h o m o g en eo u s aq u eo u s m e th an o l
solution w h en h ea t in g an d ex cess N a O H w ere av o id ed. 8 In
h eav y w a te r (DzO), t h e a l co h ol p ro d u ced f ro m th e r eac t io n
of
I
or fo rm ald eh y d e co n ta in s n o ca rb o n -b o u n d
D;9
th is ex -
c lu d es all mechan isms invo lv ing a hydride t ransfer f rom or to
o x y g en a to m s , for ex am p le , eq
A
or B.
T h e k in e t i c o rd e r w i th
I
and several der ivat ives
i n
w a t e r ,
m e t h a n o l , a q u e o u s m e t h a n o l , o r aq u eo u s d io x an e i s t h i rd :
979
America n Chemical Society
Page 2
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 2/8
S w a i n e t
al.
/ M e c h a n i s m of t h e C a n n i z z a r o R e a c t i o n
3517
R 0-
J
o=c
H
Q-0-C-R - RCH20-
+
HOCOR (A)
Ll
H
91
-
O r )
R
H-C=O QR
AJ
HO-H
I
OH
HO- + RCHZOH + RCOiH (B)
k 3
[
J 2 [ b a s e ] . o W i t h f u r f u r a l ' l a , b a n d f o r m a l d e h y d e ' t h e
orde r var ies f rom f i rs t to second in base, hence th i rd to four th
o v era l l d ep en d in g
on
t h e co n d i t i on s ; so d iu m m -fo rm y l -
b en zen es u l fo n a te a l so g ives a fo u r th -o rd e r r eac t io n , k4-
[R C H O J2 . [b ase l2 , u n d er ce r t a in co n d i t io n s . l I f T h e H am m e t t
r eac t io n co n s t an t fo r sev en a ro m at i c a ld eh y d es w i th so d iu m
h y d ro x id e
in
50 m e t h a n o l a t 100 "C is +3 .7 6 (correlat ion
coeff ic ien t 0.998). Ioc~12
Under typ ical Cann izzaro react ion cond i t ions, 0 .6 M I a n d
0.25
M
N a O H in 74% C H 3 0 H - 2 6 % w a te r a t 100 " C , t h e
reac t an t s ex i s t p red o m in an t ly a s f r ee
I
( n o t h y d r a t e or h em i -
ace t a l ) , N a+ , an d H O - , t h e r eac t io n is c lo se to seco n d o rd e r
i n I and f i rs t o rder in
HO-,
a n d t h e p r i nc i p al p r o d uc t s a r e I1
an d T h e p u rpo se of th is work was to invest igate the main
ro u te b y w h ich I 1 i s form ed u n d er t h ese co n d i t i o ns in o rd e r t o
ex c lu d e r ig o ro u s ly f iv e o f t h e s ix m ech an i sm s (1-6) pro-
posed .
Proposed
Mechanisms.
R a d i c a l - c h a i n m e c h a n i s m s h a v e
b een p ro p o sed , I3 b u t a r e ex c lu d ed u n d er h o m o g en eo u s co n -
ditions because radical initiators (benzoyl or sodium peroxide)
or i n h ib i to r s (h y d ro q u in o n e or d ip h en y lam in e) h av e no effect
on t h e r a t e . '
l a , 1 4
U n d e r h e t e ro g en eo u s co n d i t i o ns co n s i s t i n g
of a benzaldehyde phase and a s t rong ly alkal ine aqueous phase,
the react ion is a compos i te
of
two homogeneous react ions; the
reac t io n in t h e o rg an ic p h ase is ca t a ly zed b y I 1 produced by
the s lower react ion in the aqueous phase, bu t in bo th phases
th e r eac t io n ap p ea r s t o b e p 0 1 ar . I ~
S ev era l m ech an i sm s s t il l co n s i s t ent w i th t h e d a t a so f a r
p resen ted have been p roposed fo r the homogeneous Can n izza ro
reac t ion . T h e f i r ~ t ~ ~ , ~ . ~ ~ . ~ ~ ~ , ~ ~a s e q I as i t s r a t e -d e t e rm in in g
s t e p ( R
=
pheny l) .
0
0-
II fast
I
H-C + HO- -C-OH
R
I
R
I IV
0- 0- OH
I fast I I
I
0
II
I
R-C + H-C-OH - -C-0-C-R
H
I
H
~~
I IV V
0 H
slow
II
R-c-O-C-R -C-0-C-R
+
HO- (1)
1-4
H H
I
H
V VI
fast
R C H , O C O R H O -
-
C H , O H + R C 0 , -
VI I1 I11
T h is m ech an i sm in vo lves fo rm at io n o f VI as an in t e rm ed i -
a t e . S i n c e 18Q ex ch an g e b e tw een w a te r an d
I
is m u c h f a s t e r
t h a n t h e C a n n i z z a r o r e ac t i on , " t h e r a t e - d e t e r m i n i ng s t e p
can n o t b e fo rm at io n o f ad d u c t IV
or
t h e m ech an i s t i ca l ly
s im i l a r fo rm at io n o f ad d u c t
V,
b u t i t m ig h t b e th e r ea r r an g e-
m e n t ( e q 1) of V t o VI. E s te r h y d ro ly s is i s k n ow n t o b e f a s t
u n d e r C a n n i z z a r o
condition^.'^
A
seco nd m ech an i sm I8 i s eq
2
with a p r io r equ i l ib r ium fo r
I V a s i n m e c h a n i s m 1. T h is m ech an i sm in vo lves a r a t e -d e t e r -
H 0-1
H
0
1
n
slow
I
II
R-C H-C-R
-
-C-H + C-R
(2 )
I
OH
I + IV
I
OH
I
0-
VI1 vn1
fast
VI1 + VI11
-
C H , O H R C 0 , -
m in in g in t e rm o lecu la r h y d r id e sh i f t , f ol low ed b y f a s t p ro to n
t r an s fe r .
R e a r r a n g e m e n t s of
V
t h a t d o n o t l ead to fo rm a t io n o f V I
h av e a l so b een p ro p o sed, a s d esc r ib ed b y m ec h an i s m 319 or
m ech an i sm 4.20 In (3) , t h e s t a b l e p r o d u c t s (I1 a n d 111) a r e
I1 I11
ro-
OH
0
OH
Y
I
slow II I
R-C-0-C-R
-
-(2-0- + H-C-H
3 )
l
I
A H
I
R
V
I11 I1
0-
OH
0-
OH
I I slow I
I
R-C-0-C-R - -C-H + O=C-R(4)
I
H
I
H
V VI1 VI11
formed directly in the rate-de termin ing st ep, while in (4) a fast
p r o t on t r a n s f e r o c c u r s a f t e r t h e r e a r r a n g e m e n t t o f o r m t h e
s t ab l e p ro d u c ts .
A n o th e r r ea r r an g em en t o f V, m ech an i sm 5, involves a
pro ton t ransfer in the s low step to p roduce the s tab l e p roducts .
0
I L I
H H
I
H
V I1 I11
S im i l a r ly , t h e l a s t tw o s t ep s o f m ech an i sm
2
might be te le-
scoped to ( 6 ) .
OH
0
R-C
(p
0-C-R T -C-H
I
+
-0-C-R
II
(6)
H
b - I
H H
I + IV I1 I11
I n any so lu t ion o f IV a n d I , ther e wil l be some
V
a t eq u i l i b -
r iu m . I n m ech an i sm s
I ,
3 , 4 , a n d
5,
i t is conside red tha t
V,
in
sp i te of i t s low concen trat ion , reacts a t a fas ter ra te tha n more
a b u n d a n t r e a c t a n t s ( I a n d IV) because V h o ld s t h e m ig ra t in g
hydroge n in a favorab le posit ion fo r an in t r amo lecu lar rear-
r an g em en t . In m ech an i sm 2 or 6 the necessary in termolecu lar
hydride t ransfer canno t occur un less I an d
1V
happen to collide
w i th p rec i se ly co r rec t o r i en t a t io n s .
M e c h a n i s m s t h a t a r e s t il l s i m p l e r
i n
the sens e o f bypassing
IV can b e p ro p o sed . M ech an i sm 7 is a n e x a m p l e . H e r e IV is
Page 3
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 3/8
3578
Journal of t he Amer ican Chemical Soc ie t y
/ 101.13
/ June
20,
1979
0 0 0- 0
II
I1
slow
H-C
+
H-C +
HO--
R-C-H + C-OH i )
I
R R
I I
R
R
VI1 VI11
a r ev e r s ibly fo rm ed b y p ro d u c t i n eq u i l i b r iu m w i th th e r eac -
t an t s , b u t n o t an in t e rm ed ia t e a l o n g th e m a in r eac t io n p a th .
G en era l a rg u m en t s ag a in s t t e rm o lecu la r m ech an i sm s a re n ot
v a l id a t t h e h ig h co n cen t r a t io n s o rd in a r i l y u sed in t h e C an -
n izza ro r eac t io n . 2 ' T e rm o lecu la r m ech an i sm s h av e b een
dem onstra ted in var ious systems exper imental ly .22 Mec han is m
7 c o u l d h a v e t h e s a m e t r a n si t i on s t a t e a s m e c h a n i s m 2.
I so to pe e f f ec t s u s in g b en za ld eh y d e-a - t (1 . 2 4 -
I
.41)2a13a-23
a nd b e n ~ a l d e h y d e - c u - d ~ ~ , ~ ~ave been mea sured , and a re in the
n o rm al d i r ec t io n fo r p r im ary e f f ec t s ( t h o u g h sm al l e r t h a n
usual) . However , they do no t d is t ingu ish between mechan isms
1-7,
which al l invo lve a hydri de
or
h y d ro g en a to m t r an s fe r i n
t h e r a t e - d e t e r m i n i n g s t e p .
Isotope Dilution Applied
to
the Cannizzaro Reaction of
Benzaldehyde-p-t
in
74% Methanol-26% W ater at 1 0C.
Disproof of Mechanism 1 .
A
p ro m is in g t ech n iq u e fo r e l im i -
n a t in g a s a n i n t e r m e d i a t e a n y s u g g e s t e d c o m p o u n d C t h a t is
s t ab l e w h en i n p u re fo rm i s t o sh o w th a t i t s ac tu a l co n cen t r a -
tion i n the react ing system is less than would be requ ired i f i t
w e r e a n i n t e r m e d i a t e . T h i s r e q u i re s ( a ) m e a s u r i n g t h e r a t e o f
co n su m p t io n o f t h e su sp ec t ed in t e rm ed ia t e C u n d er r eac t io n
co n d i t i o n s s t a r t i n g w i th i t a s a r eac t an t . (b ) ca l cu l a t in g f ro m
t h i s t h e c o n c e n t r a t i o n s o f C t h a t s h o u l d be p r e s e n t a t v a r i o u s
t im es w h en n o n e i s ad d ed b u t o n th e a s su m p t io n th a t i t i s
fo rm ed as an e s sen t i a l i n t e rm ed ia t e a lo n g th e m a in r eac t io n
p a t h , a n d ( c ) s h ow i n g t h a t t h e m e a s u r e d c o n c e n t r a t i o n s o f C
ar e l e ss t h an th ese ca l cu la t ed v a lu es . S in ce l i k ely r eac t io n i n -
termedia tes a re o f ten very unstab le under react ion cond i t ions,
th i s u su a l ly r eq u i r es a n e x t r em ely sen s i t i ve an a lyt i ca l m e th o d
fo r m easu r in g th e co n cen t r a t io n o f C , e sp ec ia l ly w h en i t
o r ig in a t e s f rom th e u su a l r eac t an t s a lo n e ( a s i n p a r t c ) . T h e
m o s t sen s i t i v e g en era l an a ly t i ca l m e th o d k n o w n fo r h y d ro -
gen-con tain ing C compounds invo lves the use o f p r io r t r i t ium
lab e l in g of C ( in p a r t a ) o r r eac t a n t ( i n p a r t c ) , co u p led w i th
iso tope d i lu t ion by a larger measured amount o f un labeled C,
purification a nd analys is for radioactivity. Th e low cost of pure
t r i t i u m ( T l ) g a s ( c a .
$2
per cur ie) and the h igh sensi t iv i ty o f
r ad io ac t iv e co u n te r s m ak e it p rac t i ca l to m e a s u r e a c c u r a t e l y
concen trat ions o f t r i tia ted m ater ia l as low as M.24.25 h e
fo l lowing i l lust rates the use o f th is techn ique fo r p rov ing that
benzy l benzoate (VI) i s no t an in terme diate i n t h e C a n n i z z a r o
react ion o f I .
I n simul taneous base-catalyzed hydro lysis and m ethano lysis ,
m e th o x id e io n is a b o u t t h r e e t i m e s m o r e r e a c t i v e t h a n h y -
drox ide ion toward acety l L-pheny lalan ine methy l es ter in
80%
m e t h a n o l - 2 0 % w a t e r 2 6 dan d 4 5 t im es m o re r eac t iv e to w ard
p-n i t ropheny l acetate in dilute solutions of alcohol i n w'ater.2"b
W h e n t h e r a t e o f d i s a p p e a r a n c e o f 0.0625 M V I , p a r t i a ll y l a -
b e l ed w i th t r i t i u m i n t h e p a ra p o s i ti o n s of t h e r i n g s , w as d e -
t e r m i n e d u n d e r C a n n i z z a r o c o nd i ti o n s (0. 2 5 M to tal base
( m e t h o x i d e + h y d ro x id e ) , 7 4 % m eth an o l -2 6 % w ate r a t
IO0
"C) by isotope dilution for unchanged VI , i t was found tha t VI
disappears by two pdralk l f i rs t -order react ions, wi th methox ide
ion and with hydrox ide ion , wi th the combi ned f i rs t -o rd er ra te
constan t of 0 .37 s - l . F ro m th ese d a t a an d th e th i rd -o rde r r a t e
c o n s t a n t
k 3
= I . 8 6 X M-? S - I f o r t h e C a n n i r z a r o r e-
ac t io n o f
I ,
t h e co n cen t r a t io n o f VI t h a t s h o u l d b e f o r m e d a t
v a r io u s t im es
in
t h e C a n n i z z a r o r e a c t i o n , a s s u m i n g t h a t i t is
an es sen t i a l i n t e rm ed ia t e a s r eq u i r ed b y m ech an i sm I , w as
ca lcu la t ed b y ap p l i ca t io n o f t h e s t ead y - s t a t e ap p ro x im at io n .
Of co u r se , so m e
VI
m u st fo rm ev en i f m e c h a n i s m
2
o p e r a t e s ,
b ecau se th e ad d u c t o f p ro d u c t b en zy lo x id e io n
V I
I t o b en za l -
d eh y d e I sh o u ld b e ab o u t a s e f f ec t iv e a s IV as a h y d r id e d o n o r .
H o w ev er , IV i s no t fo rm ed in i t ia l ly if mec han is m 2 i s co rrect ,
w h ereas i t
is
a n e c e s s a r y i n t e r m e d i a t e f r o m t h e b e g i n n i n g
i n
m e c h a n i s m 1 T h ere fo re , i so to p e -di lu t io n m easu re m en t s w ere
rest r ic ted to the ear ly par t (6-19%) of the Cann izzaro react ion
of benzaldehyde-p-f . Th e calcu lated concen trat ions o f
VI
were
a t l e a s t 10-17.5 t i m e s t h e c o n c e n t r a t i o n s f o u n d . T h i s s h o w s
t h a t
VI
i s no t an essen t ial in terme diate a long the main reac t ion
p a t h a n d e x c l u d es m e c h a n i s m 1 .
Product Analysis. Disproof of Mechanisms 3 and 4. S i n c e
m eth o x id e io n i s m o re r eac t iv e th an h y d ro x id e ion to w ard es -
t e r s in m e th an o l - w a te r so lu t io n s ,2 6 h e re sh o u ld b e
a
consid -
e rab le am o u n t o f r eac t io n o f
I
w i t h C H 3 O - i n C a n n i z z a r o
reac t io n s ca r r i ed o u t in a lk a l in e m e th a n o l -w a te r so lu tio n s .
M e c h a n i s m s 1 an d 2 w o u ld y ie ld t h e sa m e p ro d u c t s w i th
C H 3 O - as w i th HO- since esters ar e rap id ly hydro lyzed under
t h e c o n d it i o ns . H o w e v e r , s u b s t i t u ti o n o f C H 3 0 - f o r
HO-
i n
m e c h a n i s m 3 requ ires fo rmat ion o f benzy l methy l e ther
( I X ) ,
w h i c h s h o u l d a c c u m u l a t e a s a s t a b l e p r o d u ct . IX ad d ed to a
Cann izza ro react ion m ix tu re in i t ia l ly (0.25 M) was still present
a t t h e e n d ( 0 . 2 4
M ) .
sh o w in g th a t t h i s e th e r i s no t d es t ro y ed
u n d er C an n izza ro co n d i ti o n s . Wh en n o n e w as ad d ed in it i a ll y ,
less than 1 was found a t t h e en d b y g as - l i qu id p a r t i t i o n
c h r o m a t o g r a p h y . T h is e x c l u d e s m e c h a n i s m 3.
M ec h an i s m 4 is fo rbid d en as a co n ce r t ed
or
o n e- s t ep r eac -
t ion by conservat ion o f o rb i ta l symmetry ru les . An equ ivalen t
two-step rearrangement v ia homolysis
to
a d i r ad i ca l
or
triplet
ca rb en e in t e rm ed ia t e h as b een p ro p o sed ?" b u t seem s ex c lu ded
b ecau se th e rm al en e rg y a lo n e i n the absence o f l igh t or f ree
rad ica l s sh o u ld n o t b reak th ese s t ro n g b o n d s th i s r ap id ly , an d
p ro d u c t s or rate shou ld be affected by rad ical inh ib i to rs
i f
rad icals were invo lved .
,4lso.
(4 ) p rov ides no exp lanat ion fo r
base catalysis s ince the homoly t ic re arran geme nt i n (4 ) shou ld
p ro ceed ab o u t a s w e ll w i th t h e n eu t r a l co n ju g a t e ac id o f
V
a s
w i th th e m o n o an io n .
Sovent Isotope Effect. Disproof of Mechanisms 5 and 6.
M e c h a n i s m s 5 a n d 6 a re ex c lu d ed b y
o u r
"solvation rule" ,
which s ta tes that a p ro ton being t ransferred between oxygens
(or o t h e r a t o m s w i th u n s h a r e d p a i r s ) i n t h e r a t e - d e t e r m i n i n g
s t ep o f an o rg an ic r eac t io n (o n e w i th b o n d ch an g e s o n ca rb o n
i n
the rate-determin ing s tep) shou ld l ie a t a po ten t ia l min im um
(ra th e r t h an m ax im u m ) a t t h e t r an s i t io n s t a t e . T h i s m ean s th a t
n o p r im ary k in e t i c i so to p e e f f ec t sh o u ld b e o bse rv ed fo r an y
such hydrogen because i t does no t lose zero -po in t v ib rat ional
e n e r g y f r om g r o u n d s t a t e to t r an s i t i o n s t a t e . S in ce i t s m o t io n
is not a c r i t i ca l p a r t o f t h e d eco m p o s i t i o n m o d e w e sh o u ld n o t
include an arrow or arrows for
its
t ransfer
in
this step. This docs
no t exclude the possib i l i ty o f a favorab le cycl ic or h q d ro g en -
bonded conformat ion fo r the t ransi t ion s ta te bu t does el iminate
th e m o re co n ce r t ed m ech an i sm s 5 a n d 6 .
E x p er im en ta l d a t a o n th e C a n n i rza ro r eac t ion , accu m u la ted
b e fo re t hi s ru le ~ l a sormulated and tes ted by o ther react ions,
l ead s to t h e s a m e c o n c l us i o n. T h e r e a c t a n t s o f m e c h a n i s m 2
i n
heavy water
D20)
r c
DO-
an d
two
molecules of aldehyde.
A t t h e t r an s i t i on s t a t e t h e DO- bond has been replaced bq a
DO bond (uncharged ) . Th is equ i l ib r ium is more favorab le u i th
DO-
i n D 2 0 t h a n w i t h H O -
i n
H 2 0 by a f a c t o r of a b o u t
2.0 .27 .2x n t h e o th e r h an d , i n m e c h a n i s m 5 or
6
th is shou ld
be more than o ffset by
a
prima ry iso tope effect
in
the opposi te
d i r ec t io n f ro m t r an s fe r
of
D r a t h e r t h a n H
i n
t h e r a t e - d e t e r -
m in in g s t ep . r e su l t i n g i n a n e q u a l or f a s t e r r a t e i n H 2 0 . T h e
observcd
k
[,?o/k i 2 s I
.90.
T h is ex c lu d es m ech an i sm s 5 a n d
6 a n d S ~ O W Sh a t t h e p ro to n i s t r an s fe r r ed a f t e r , r a th e r t h an
during , the rate-determin ing s tep . ' ' . ' "
Mechanisms
2
and 7. T h ese m ech an i sm s a r e b o th s t il l al -
lowed . We knovv th at the two molecu les o f rea cta n t
l
a re p re -
d o m in an t ly u n so lv a t ed an d u n asso c ia t ed w i th h y d ro x id e
or
m cth o x id e i n
our
so lu tio n s ,' w h ereas t h e b o n d in g o f t h c h q -
d ro x id c o x y g en to ca rb o n is c o m p l e t e o r n ear ly
so
a t t h e t r a n -
si t ion s ta te ( f rom the observed larg e values o f
k l ) ? ~ / k t i ~ ~
n d
Page 4
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 4/8
S w a i n
et
al . / Mechani sm of the Cannizzaro React ion
t h e H a m m e t t r eac t io n con s t an t
p .
W e c a n n o t s ay m u c h a b o u t
th e seq u en ce o f ev en ts b e tw een r e ac t an t s an d t r an s i t i on s t a t e ,
w h e th e r t h e r a t e -d e t e rm i n in g r eac t io n m o re u su a l ly in v olv es
two successive doub le co l l is ions o r on e t r ip le co l l is ion , because
t h e s a m e r a t e i s c a l c u l a t e d f o r t h e s a m e t r a n si t i on s t a t e w i th
e i th e r (2)
or 7 ) .
W e know of no operat ional way , exper imental
or
t h eo re t i ca l , t o d i s t i n g u i sh b e tw een (2) a n d
7 ) ,
a n d w e
th e re fo r e co n s ide r t h e m a s equ iv a l en t .
A r easo n ab le s t ru c tu re fo r t h e t r an s i t i o n s t a t e is i l lust rated
by
X .
T h e C - - H - - C b o n d m a y b e b e n t . 3 ' T h e c a r b o n y l
n 6 -
3579
X
o x y g en s of o n e or b o th a ld eh y d es a re l i ke ly to b e p o la r i zed b y
h y d ro g en b o n d in g to w a te r o r a l co h o l solv en t m o lecu les , an d
reasons have been g iven2 ' fo r bel ieving tha t such p ro tons have
n o rm al b o n d s w i th n o rm al ze ro -p o in t en e rg y a t t h e t r an s i t i o n
s t a t e .
T h e f o u r t h - o r d e r t e r m o b s e r v e d w i t h f o r m a l d e h y d e a n d
cer tain o ther a ldehydes may re presen t a t ransi t ion s ta te s imilar
to X excep t that the hydroxy l ic p ro ton has been t ransferred to
a seco n d h y d ro x id e
ion
t o f o r m a s e p a r a t e w a t e r m o l e cu l e .
Experimental
Section3
Toluene-p-t.
A Grignard reagent was prepared from 260
g
(1 .52
mol) ofp-brom otoluene and 40 g
( I
.65
mol) of
M g i n 370 mL of dry
ether. Tritium chloride,33 prepared from a heated m ixture (25 "C to
boiling point) of 1.0 g (0.1 1 equiv, 2.5 Ci) of tritiated water (see
"Inorganic Chemicals"), 5.0
g
(0.55 equiv) of HzO , and 250 gof re-
agent grade C,jH5COCI, was passed into the vigorously stirred Gri-
gnard mixture by a stream of purified N2. After th e ether had been
removed from the resulting mixture, the pressure was reduced to
I O
mm and the crude toluene-p-1 was collected in a dry ice-acetone
cooled flask. Distillation in a Vigreux column ( 1 6.5 cm X 2.5 cm 0.d.)
with a Claisen head gave 59
g
(97%) of partially tritium-labeled tol-
uene, bp 108-109 "C (lit.34 for toluene, bp l l l "C).
Benzaldehyde-p-t (I-p- t). This toluene (59 g, 0.64 mol) was pho-
tochlorinated by a Sylvania
RS
sunlamp
15
cm from the flask. Hy-
d r ~ l y s i s ~ ~f the resulting benzylidene chloride gave partially tri-
tium-labeled
I,
which was purified through the bisulfite addition
product35 and distilled under purified Nz in a semimicro column,36
bp 176- 177 "C, n z 5 ~.5424 (lit. for I, bp 179 0C,35 Z 0 ~.544637),
26.4
g
(39%).
Benzoic-p-t Acid and Benzyl-p-t Alcohol.
A Cannizz aro reaction
on this benzaldehyde (14 g, 0.13 mol)38gave 3.8 g (47% ) of partially
tritium-labeled V l l l after recrystallization from water, mp 121-122
"C (lit.39 for ChH5C 02H, 122.38 "C ), and 3.9 g (55%) of partially
tritium-labeled I 1 after distillation under z in a semim icro column,3h
bp 109-1
10 c
15 mm), f125D 1.5366 (/it ,4\ ' fo r 11, bp 104-105 c
(20 mm) , 1,5340).
Benzyl-p-t Benzoate-p-t
( V I - p , p - 12 ) was prepared by a Tis-
c ht sc he nk o r e a ~ t i o n . ~ 'he partially labeled I
(8.1
g, 0.076
mol)
was
treated with VI1 prepared from 0.033 g (0.001 4 mol) of N a and 0.70
g (0.0065 m ol) of the partially labeled
I I .
Th e pasty. gelatinous
mass
resulting was treated with 40 mL of H2O and 20 mL of ether
to
give
three layers (H 20 , ether, and an oil) which were separated. The H2O
and oil layers were each washed with 20 mL of ether. Th e combined
ether layers were dried over anhydrous NazSO4. After the ether had
been removed, the VI was distilled under K n a semimicro c0lumn.3~
5.1 g (63%), bp 133-134 " C (0.7 mm ), t i Z 5 ~.5664 (lit.I4 for V I . bp
133-1 35 "C (0.5 mm ), n 2 4 ~.5672).
Benzaldehyde-a-i I-a-t) . he Reissert compound, 1 -benzoyl-
I
2-dihydroquinaldonitrile as prepared from ChHsCO CI, quinoline,
and aqueous KCN and recrystallized twice from 95% ethanol as white
needles, mp 154- 154.8 "C (lit.42154- I55 "C) . Th is was hydrolyzed
i n 2.5 M H2S 04 by refluxing I2 g
i n 108 g
of
a
solution of 25.6 g of
96.5% H2S04 (0.25 mol of H2S04
+
0 .05 mol of H2 O) in 84 .6 g of
tritiated water (4.70 mol) of 1.37 mCi/mol activity for 2 h under N2.
Th e solid crystals disappea red within 30 min. The product was steam
distilled, extracted with ether, dried by azeotropic distillation with
15 mL of adde d C6H6, and distilled t hrough
a
semimicro column,36
3.0 g, bp 74.0-74.4 " C (20-2 I
mm.
The 2 4-dinitrophenylhydrazone
recrystallized twice from ethyl a ceta te, mp 241 .O-242.2 *C (l it j3 237
"C), had an activity
of 0.1 16
mCi/mol; the dimethone derivative,
recrystallized twice from CH 30 H, mp 196.5-197.7 "C (lit.44 194-195
C),had 0.120 mCi/m oJ; the semicarbazone, recrystallized twice from
50% ethanol, mp 220.2-221.1 "C (lit.43222
"C) ,
had
0.1
16 mCi/m ol
and changed less than 2% when it was dissolved in the minimum
amou nt of 50% ethanol, refluxed for I h, and recrystallized t o see i f
it exchanged with solvent; and V ll l from KM n0 4 oxidation45 of the
compd recrystallized twice from water, mp 122.8-1 23.5 "C, had no
significant tritium content (4.7
X
mCi/mol). I xchanges with
tritiated H zS 04 ess than I nder these conditions: 5.0 mL of I re-
fluxed with 52. 2 g of a solution of 12.6 g of 96.5% H2S0 4
in
42.3 g of
tritiated w ater of 3.54 mCi/m ol for 3 h under
N2
and worked up as
above gave 3.86 g, bp 66.8 " C
( I
5
mm,
pecific activity of semicar-
bazone 9.39
X
mCi/m ol, of
VI11
5.44
X
mCi/mol. These
tritium analyses reported for water and organic compounds were done
by the Mg46 and
Zn4'
reduction methods, respectively.
The isotope effect in this Reissert aldehyde synthesis is therefore
k H k T
= (1.37
X
4.70 mCi)/O.l16 mCi/mol
X
10.0 equiv of H in
Hz S04 solution = 5.6.
A
second synthesis with nine times t he activity
of the first gave 5.3.4s This isotope effect does not prove that the proton
transfe r occurs in the rate-determining step, because t here would be
a selective competition between proton a nd triton dono rs even in a fast
step unless it were diffusion controlled.
For the Cannizzaro reaction, the benzaldehyde-0-1 was converted
to semicarbazone, recrystallized, hydrolyzed back
to
aldehyde. and
freshly distilled under N 2 before use, bp 69.0-69.7 "C (17-18
mm).
Methanol-d (C H3 0D ) was prepared by decomposition of
Mg(OCH3)2 with DI O. ~'All the glasswa re was baked at 350 OC for
several hours, assembled rapidly while hot, and immediately a ttached
to Ascarite a nd Drierite towers to prevent introduction of CO2 and
moisture. AC S reagent grade CH 30 H was dried by the Mg method.50
Mg
(150
g) was added in small portions to 3.5 L of dry CH30H
without exposing the system to the atmosphere. After the Mg had all
dissolved and the solution had refluxed for 3 h, most of the CH30H
was distilled and the residue was heated at 150-200 "C ( I mm) for
24 h . Th e system was allowed to cool to 25 "C a nd then dr y, COz-free
air admitted. DzO (100 mL, >99.5%, degassed by bubbling purified
N2 through i t for 30 min) was added an d the resulting mixture was
refluxed. with frequent shaking, for 4 days. The flask containing t he
reaction mixture was equipped for trap-to-trap distillation. Dry,
COz-free air was admitted to the system while the distillation flask
was cooled by liquid
N2.
The system was then evacuated to 0.5 mm
and t he liquid
N 1
bath was moved from the distillation flask to the
receiver. After 24
h ,
dry, COz-free air was introduced and the distillate
was allowed to warm to 25 "C. Mg (2
g)
was added to the CH3 0D .
After the Mg had all dissolved. the solution was refluxed for 3 h and
the CH 30 D was distilled, bp 65 "C. 200 mL, GL C at 25 "C wi t h two
different columns (30% (by weight) 3-methyl-3-nitropimelonitrile
on 60- 100 mesh firebrick and 30% Carbowax 600 on SO- IO0 mesh
firebrick i n 8-m ni Pyrex tubes 190 cm long) indicated total im purities
of less than 0. . A determination of the D content by the falling-drop
method" gave 24.65 atom %exces s D (98.6% CH 3O D) .
Methanol was prepared i n the same manner as CH3O D for com-
parison of rates in light and heavy 74% methanol, bp 63.5-64.0 "C.
GL C indicated total impurities
o f
less than 0.1 CHj OH for all the
other
r u n s
was AC S reagent grade dried over Drierite.
Benzaldehyde
I ) ,
Eastman white label, was washed with 10%
a qu eo us N a ~ C 0 3 , ried over anhydrous N a2S 04 and freshly distilled
under N2
i n
a semim icro column3h before use, bp 176-177 "C,
t i Z 5 D
1.5432.
Benzyl benzoate (VI) and methyl benzoate XII ) , Eastman white
label, were redistilled under N2
i n
a Semimicro column:36 V I , bp
121-122 "C ( 0 . 5 mm), n 2 5 ~.5653; XI I , bp 7 5 "C ( I O mm,
n 2 5 0
1 . 5 1 2 2 ( l i t. 5 2 b p 8 3 C ( 1 2 m m ) , n 2 5 D . 51 55 ).
Benzyl benzoate
( V I )
used as the diluent for isotope dilution was
rccrystallized Eastm an white label grade . A CH 30 H solution of
V I
was cooled in an ice-salt water bath; water was added to th e cloud
point and a seed crystal was introduce d. Th e crystallized
V I
was col-
lected on an ice-jacketed fritted-glass funnel, washed with a sma ll
amount of ice-cold C H 30 H , and air dried. After three such recrys-
tallizations, it was recrystallized from a minimum of pure CH30H
Page 5
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 5/8
3580
Journal o f th e Amer ican Chemical Soc iet y /
101:13
/ June
20, 1979
Table
1 Reaction'of 0.5752 M I with 0.2875
M
Na OD in 74%
C H ?O D -D , O a t 9 9. 8
f
.1 "C in Teflon Tubes
Table IV. Kinetics of the Alkaline Hydrolysis of
0.1
84 M VI with
0. I77 M Na OH in 78% CH3OH at 99.4 "C
k3
X I O 4
time,
s [DO-1, M
reaction
M-2 s - I
k z a X I O 2 .
t ime,
s
[HO-1,
M
reaction
'
M-'
s - I
0 0.2872 0
3600 0.2 I64 24.7 3.21
7200 0. I779 38.1 3.38
I O 800
0.1599 44.9 3.12
1 8 000
0.1284 55.3 3.37
57 600 0.07 53 73.8 3.57
mean 3.33
320 0.0577 67.4 3.4
600 0.0246 86.1 5.0
900
0.0
145 91.8 5.6
I200
0.0109 93.9 5.5
I500
0.0092 94.8 5.0
1810
0.0089 94.9 4.2
mean 5
Table 11. Rate Constants for the Cannizzaro Reaction of I in Light
and Heavv 74% Methanol-26% Wa ter in Teflon Tubes
k3
X I O 4
temp, "C [NaOLIo, M
[ l ] ~ ,
M water,
L 2 0
M-*
S - I
-
99.4 0.2850
0.5702
DzO 3.76 f .07
99.4 0.2609
0.5216
H 2 0 1.79
f
0.10
99.8 0.2685
0.5370 H2O 1.86
0. I 1
99.8 0.2875
0.5752
D2O 3.33 0.13
Table
H I .
Cannizzaro Reaction of
1
in
Glass Tubes
solvent . C H IO H te m a " C k l X
IO4.
M-2 s-'
50
67
74
74
100
2.33 f .09O
IO0
2.22
f
.10b
98.3 3.31 f 1.03'
98.6 2.45 f .33d
E.
L. Molt;loa Tommilal°C reported a frequency factor of 4.66 X
I
O4
and a n activation ene rgy of 13.85 kcal for the Cannizza ro reaction
o r I
i n 50% CH3O H. Thes e gave a calculated k3 of 3.47 X M-*
5 - l at 100
"C.
K. B. Wiberg.23 W . A. Sheppard.2a
I-p-t; I
gave
2.36
f
.16
X
at 98.4 "C.
and dried over Anhydrone (MgC104) in a desiccator at
I O
"C, mp
19.3-20.0 "C (lit.s3 mp 19.4 "C),
n 2 5 ~
.5672.
Benzyl methyl ether
IX) , Eastman white label, was redistilled under
N2 in a s e m i m i c r o c ~ l u m n , ~ ~p 169-170 "C (lit.54bp 170.5 "C).
Inorganic Chemicals.
Purified nitrogen was prepurified N2 reed
of
CO2
and H20 by passage through a series of towers containing
Ascarite (NnOH on asbestos) and Drierite (Ca S04 ). Tritiated water
~ i i srepared from T I gas (AEC , Oa k Ridge) as described previous-
l y . H 2 0 was laboratory distilled water which was redistilled from
N aO H -K M n O a i n an all-Pyrex apparatus.
N a O D was prepared by the dropwise addition of 100 mL of de-
gassed D2O ( > 9 9 . 5 %) to 2.3 g (0. mol) of freshly cut reagent grade
N a under
a n
atmosphere of purified N2 and then standardized.
Analbsisil of the solvent of the K aO D solution gave 99.14% DzO.
NnOH was prepared i n the 5ame manner as NaOD for the runs
comparing rates in light and heavy 74% methanol . For the other runs,
i t
was prepared either bj the concentrated NaOH methodSSor by
diluting
I
M Acculute and standardizing. COz-free HzO was used
i n
a11
these preparations.
Kinetic Procedures.
The kinetics of the Canniz zaro reaction of I
i n 74% methanol was measured essentially by the method of Moltlod
and A l c ~ a n d c r . ' ~he procedure was the same in light and heavy
methanol. I (about 3 m L ) was transferred by means of a 5- mL syringe
and under a n atm osphere of purified N2 to a weighed 50-mL volu-
metric flask. The flask was reweighed a nd placed in a dryb ox, which
was
then tlushed with purified
Nz
or 20-30 min. Methanol
(25
mL)
was
added to the flask, and 12.87
mL
of
1 M
NaO H was added f rom
a buret with mixing by swirling. Th e volume was brought to the m ark
by addition of methanol. Af ter the solution had been thoroughly
mixed. i t was drawn into a 50-m L syringe and seven aliquots of ca.
6
mL each were injected into Teflon tubes of ca . 7 mL capacity pre-
viously fitted s nugly inside 18 X I50 mm Pyrex test tubes which had
been constricted about 2.5 cm above the top of the Teflon to 4-5 mm.
a Based on total base consumed
The tubes were protected from t he atmosphere by tight-fitting rubber
stoppers, removed from the drybox, cooled in an ice-water bath, sealed
at the constriction, and placed in the constant tempe rature ba th. After
the tubes had been in the bath for
10
min, one was withdra wn, cooled
in an ice-water bath, allowed to com e to 25 C, and opened.
A 5-mL
aliquot was pipetted into a known excess of 0.1 M sta ndar d HC I and
back-titra ted under purified N Z o the phenolphthalein end point with
0.1 M
Na OH . The constant was calculated from k3
=
x ( 2 a - x)/
8ta2 a- ) ~ , here
a
is the initial concentration of base a t I O min
( t = 0) and
x is
the concentration reacted
in
t ime t . Data for a typical
run a re given in Table I , and th e results for various runs in light and
heavy 74% CH 30 H in Table
11
Teflon tubes were used
in
the later phases of this work because rapid
attack of alkali on Pyrex tubes complicated the kinetics and gave
poorer reproducibility in the earlier runs. Teflon tubes a re convenient
to use as described above an d the solvent shows
no
tendency to distill
out of the Teflon tube into the small spa ce outside or under this tube
(between the Teflon liner and the outer Pyrex tube) because the
electrolyte (N aO H)
is
nonvolatile and keeps the vapor pressure below
tha t of salt-free solvent. A difference
in
height of liquid inside and
outside of over 30
m
would be required for gravity t o compensate this
osmotic difference. Without these inert reaction vessels about one-
quart er of the base was consumed by reaction with Pyrex under our
conditions. Kinetic results of investigations i n glass ar e reported i n
Table
1 1 1 .
I n 74% CH 30H -26 % H:O solutions at 100 "C CH3O- should
attack
V I
to form methyl benzoate
(XII)
more rapidly than HO-
attacks
V I
or
XI1
to form
1 1 1 . 2 6 . 2 7
Therefore the second-order rate
k.
C,H,COOCH,C,H, + CH,O- C,H,COO CH, C,H,CH,O-
k
HO
C , H , C O O H
+
C , H , C H , O -
k 1
HO
C , H , C O O H C H , O -
C , H , C O O - C , H , C H , O H
C , H , C O O -
+
CH,OH
k , > k , and
k ,
consta nt for hydrolysis of V I measured by the usual acid-base titration
procedure should be nearly the sam e as that for
XII.
Since these esters
hydrolyze rapidly even at 25 "C, the involved procedure for trans-
ferring samples under Nz, which was necessary for the Canniz zaro
rcaction runs, could not be used. Inst ead, as soon a s the reaction so-
lutions were prepared, samples were transferre d a s quickly as possible
by means of a syringe to 7-mL a mpules
( 1
5 X 125 mm Pyrex test tubes
constricted i n the middle to 4-6 mm ) which were cooled i n ice watcr
and which had been previously flushed with purified N l . The tubes
were sealed quickly and placed in the constant temper ature bath. A t
suitable intervals. one was withdrawn, cooled
in
ice water, and opened
and then a 5 - m L aliquot was pipetted into a known excess of 0. M
standard HCI and back-titrated with 0.1 M Na OH . The second-order
rate consta nt was calculated from t he equation
2.303 b ( a
- X )
k ? = -
log ~
a ( b
- x)
a - b )
where
a
and
h
are initial V I and NaOH conccntrations. respectively.
and .Y is the amount
of
reactant consumed in time
t .
The kinetic results
Page 6
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 6/8
Swa in et al.
/
Mechanism
of
the C annizzaro React ion
Table V. Kinetics of the Alkaline Hydrolysis of 0.244 M
XI1
with
0 .235 M NaOH i n 74% Methanol-26% Wat er a t 99.4 OC
k l a
X
IO 2 ,
[HO-1,
M
reactiona M-1 s - I
time, s
300
0.042 82.1 5.7
600
0.0121 94.8 9.4
900
0.0084 96.4 8 . 3
1200
0.0067 97.1
1 . 3
I500
0.0051 97.8 7.2
I800
0.0049 97.9 6.0
mean 7
358
1
Based
on
total base consumed
for the hydrolysis of V I and XI1 as determin ed by acid-base titration
are reported i n Tables
IV
and
V.
Despite the large errors inherent i n measuring the rates of these very
fast reactions, the close agreem ent of the second-order rate constant
for VI
( 5
X I O - ? M-I s - I ) with that for
XI1
(7 X
IO-*
M-' S - I ) in-
dicates that what is really being measured i n the ca se of V I by the ti-
tration method
is
the attack of base
on X I I .
The ra te of disappearance
of
VI is
very much faste r, as reported below under "Isotope Dilution
Technique".
The ra te of disappeara nce of
V I - p . p - t ?
under Cannizzaro condi-
tions was measured by determ ining the amou nt of unchanged ester
at various times
using
isotope dilution. All trans fers were m ade under
purified
I \r l
to glassware that had been flushed with purified
N2.
To
a 50-m L volumetric flask containing 1.3256
g
(0.006 25 mol) of V I
(with tracer-level p,p -r2 labeling) was added 25 mL of CH3OH.
COz-free water ( 1 2.50 mL ) was added from a buret. The temperature
rose I0 "C a nd some ester separated as fine droplets. After the mixture
had cooled to 25 O C . thevolum e was brought almost t o the mark with
CH IO H, and the flask was swirled gently
u n t i l
the ester had dissolved.
The volume wab brought to the m ark, and the resulting solution was
thoroughly mixed by shak ing. To a separat e 50 -mL volumetric flask
containing 25 mL of CH3OH was added
12.50
mL of 1.002
M
N aO H
from a b ure t. After t he solution had cooled to 25 OC, th e volume was
brought to the mark with C H 30 H . The resulting solution was thor-
oughly mixed by shaking. A constant-delivery automatic syringe was
used to deliver 3-mL aliquots of the 0.1250 M ester solution to one ar m
of inverted-Y-shaped Pyrex tubes. The tubes were stoppered with
rubber stoppers. Th e syringe was cleaned by repeated rinsing with
C H 30 H , dried, and then used to deliver
3 - m L
aliquots of the 0.2500
M Na OH solution to the other arm of the tubes. Eight tubes were
loaded in this manner. cooled
i n
ice water, and sealed
so
that the re-
sul t ing
third arm had about the same capacity
as
the other two.
(Measurement, after the run.
of
the total capacity of each
of
seven of
the tubes gave
17. 0.4
mL.) Some of the ester separ ated as fine
droplets when the solutions were cooled. One of the tube s, the zero
point, was opened and treated as described below u nder "Isotope
Dilution Technique". T o obtain each of the other points. a tube
was
placed in the constant temp erature bath, held upright for 4-5 min to
allow it to reach bath temperature, inverted (measurements
on
blanks
showed that there was no rise i n temperature when a 74% CH jO H
solution was mixed with an equal volume of 0.250
M
K a O H i n 74%
CH3OH), and shaken vigorously. At a suitable time, the tube was
removed from the bath and plunged into ice water. The time was
measured by means
of
a stopwatch from th e instant the tube was in-
verted to the moment it was immersed in ice water. After the tube had
been cooled, the sample was treated as described
i n
the next sec-
tion.
Isotope Dilution Technique. The 6- mL aliquots from the reaction
of tritium-labeled V I with base under C annizza ro conditions were
washed with C H 30 H solutions containing known am ounts of unla-
beled
VI.
The solution was thoroughly mixed and the ester was puri-
fied by recrystallization five times. For the first three recrystalliza-
tions, the CH 3 0 H solutions were cooled in an ice-salt-water bat h,
water was adde d to the cloud point. and a seed crystal was introduced.
The crystals were collected on an ice-jacketed fritted-glass funnel and
washed twice with ice-cold CH 30 H . Th e fourth and fifth recrystal-
lizations were from pure C H3O H: filtration was used to remove the
mother liquid and washings a fter the fourth. and decantation was used
aft er the fifth. After the ester had been dried over .Anhydrone i n a
desiccator kept at 10
"C,
melting points were ta ken. The ester was
liquefied and kept dr y by allowing the desiccator to wa rm to 25 OC.
Aliquots of the recovered ester were transferred to heighed counting
bottles. After the amount of ester had been determined, 20 ml. of
scintillation solution
( 1
5 mg of diphenylhexatriene and 4
g
of 2 , s -
diphenyloxazole/L of toluene) was added and the material & a s
counted
in
a Packard Tri-Carb liquid scintillation spectrometer at
I200 v .
The amount of VI (Ao) i n the aliquot of the reaction solution was
found from
A =
AS/So. where S is counts/min .g for recovered ester.
SOs counts/m in.g for undiluted VI . and A is g of untagged VI added
to the aliquot. Self-quenching by t he ester ma de
i t
necessarq to use
differ ent values of
So
for different amounts of ester coun ted. Corrected
So values were obtained from a plot of So vs. g of added
V I .
The ra te of consumption of
VI
under Cannizzaro conditions
is
slow
enough to be measurable,
as
shoun i n Table V I . Although the ratio
of total base (CH3O-
+
HO- ) to ester was
2:l
at 7ero time. the re-
action was treated as first order since
a t
7
s
the concentration of
VI
was 4.73 X M while the total strong base concentration was 6.72
X
IO-'
VI.
Accordingly.
a
plot
\ b a s
nude of the logarithm
of
the
concentration of V I
vs.
time. (Concentr ations used were those after
the fifth recrystallization.) The first-order rate constant
X
of 0.37
s-I obtained from the initial slope. which ~ 4 3 sonstant from 0 io ?0
s (four points), was interpreted as being that for the reaction? of
V I
wi t h HO- and CH 30- . The fractions of the initial V I left after 7.
12.
Table VI.
Reaction of Tritium-Labeled 0.0625
M V I
with 0.1250
M
N a O H i n 7 4% C H 3 0 H
;it
99.8
0.1
OC
sample counted,
S,
counts min-'
time.
s
A , g no. of recrystns mp. "C g sam ple coun ts",h '5-I
[V I ] , [ '
0 5.532 4 19.2- 19.7 0.0 108 278 857 71 7*
3.58 x
107 6 .30 X IO-'
4 6 4
x
10-3
5.490 4 19.0- 19.8 0.1001 I30
910 f 25*
1.31
x
106
3 .73
x
10-318.5-19. I 0.0999 I33 870 f 320* 1.34 X I O h
12 5.484 4 19.0- 19.7 0. I007 17 006 220* 1 .68
x
105 6.0 x lo-'
5 18.0- 18.5 0.1000 176 I67 1473 1.76
X
10'
6 .2
x
10-4
20 5.499 4
18.8- 19.8 0.0997
I3 833 I18 1.34 X I O 4 3 .7 x l o - '
5 18.0- 18.7 0. I000
I2
917 185
1.24
X I O J 4.4 x io-i
40 5.487
4 19.1-19.7 0.1001 9750 f 168 9.22
x
10' 3.8
x 10-5
5
18.0-18.5
0. I000 9465 162
8.99 x 103 3.2 x
l o - '
90 5.490 4 18.7- 19.5
0.1004 5683 58
5 . 1 4
x 103 1.8 x 10-5
5
18.2-18.7 0. I000 5065 64
4.59
x 103
1.6
x
10-5
300 5.487 4 19.0- 19.8 0.0996 1986
51
1.47 X
I O 3
5 . 2 x 10-6
5 18.2- 18.9
0. I002 I550 48
1.07
x
103 3.8
x
1 0 - 6
3630 5.485 4
18.8-19.7 0.0999
2086 f 3
I .-57 X 1 O3 _ .6 x 10-6
5 18.4- 19.0
0.1001 1551 5 3 6
1.07 X
I O 3
3.8
x
10-6
~~
['Astcrisked counts are in I min; ali others are i n 10 r n i n .
Mean value for ten trials wi th avera ge deviation from the iiiciin. .4lt cr subtrLicting
backgrou nd, which was 5 I8
f
2 count s/l 0 min for samples after the fourth recrystallization and 480
f
0 counts/
10
min for \amplea after
thc f i f t h recrystallimtion. (' alculated using So of I .75 X I O 9 counts/mi n.g for 7ero point and
SO
f I .2
I5
X IO' counts/niin.g for all othcr
points.
''
Calcula ted initial concentration was 0.0625 M.
Page 7
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 7/8
3 5 8 2
Journal of th e American Chemical Society /
101:13 /
June
20,
1979
Table VI I .
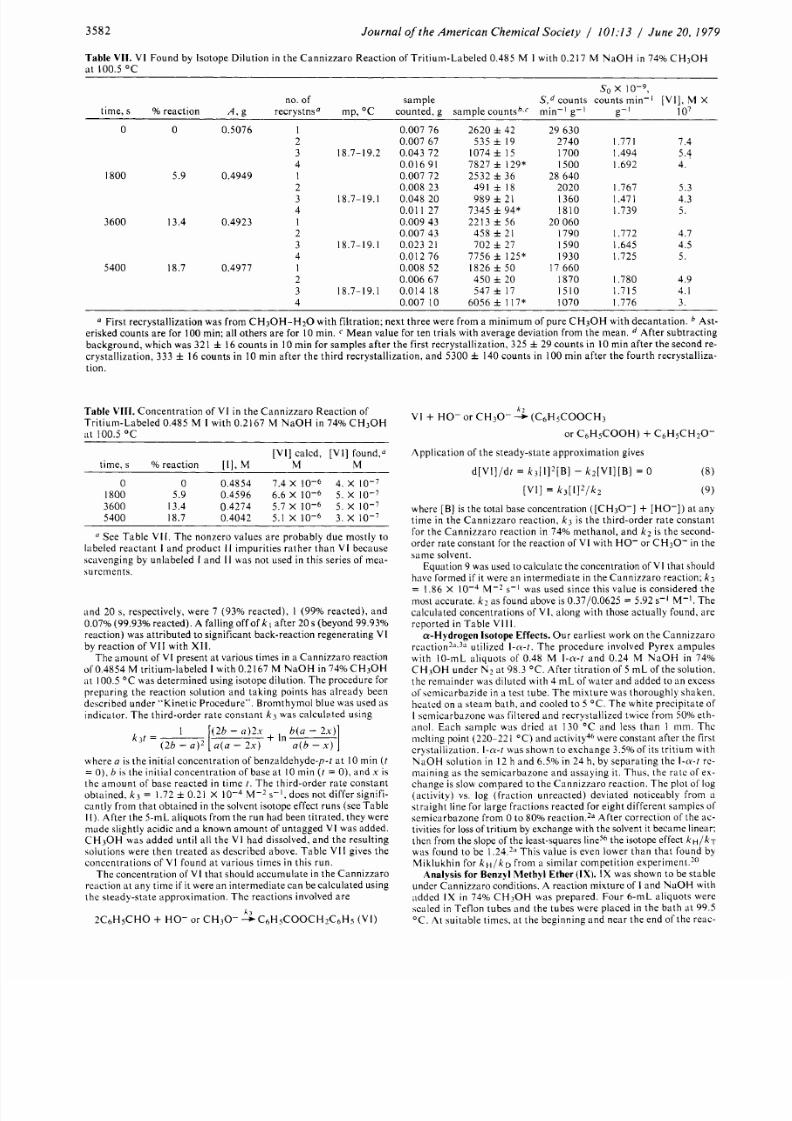
V I Found by Isotope Dilution in the Cannizz aro Re action of Tritium-Labe led 0.485
M I
with 0.217
M
N a O H i n 7 4% C H 3 0 H
at 100.5 "C
so
x 10-9,
no. of sam ple
S d
counts counts min-' [VI], M X
t ime, s reaction A g recr ystm u mp, "C counted , g sampl e countsb ,c min-' g-' g-' 107
0 0
0.5076
1
2
3
4
I800 5.9 0.4949 I
2
3
4
3600 13.4 0.4923
I
2
3
4
5400 18.7 0.4977
1
2
3
4
0.007 76
0.007 67
0.01 6 91
0.007 72
0.008 23
0.01
I
27
0.009 43
0.007 43
18.7-19.1 0.023 21
0.0
12 76
0.008 5 2
0.006 67
0.007 I O
18.7-19.2 0.043 72
18.7-19.1 0.04 8 20
18.7-19.1 0.014 18
2620
f
42
535 19
IO74
f 15
1827
f
129*
2532
f
36
491
f
18
989
f 1
7345
f
4*
2213
f
56
458
f 1
702 f 27
7756 f 125*
1826
f
50
450
f
20
547
f
17
6056 f 17*
29 630
2740
1700
1500
28 640
2020
1360
1810
20 060
I790
I590
I930
17 660
1870
1510
I070
I .77
1.494
1.692
1.767
1.47
1
1.739
1.772
1.645
1.725
1.780
1.715
1.776
7.4
4.
5.3
4.3
5.
4.7
4.5
5 .
4.9
4.
3.
5.4
First recrystallization was from CH 30 H -H 20 with filtration; next three were from a minimum of pure CH3O H with decan tation.
Ast-
erisked counts a re for 100 min; all others a re for 10min. C Mean value for ten trials with average deviation from the mean. After subtracting
background, which was 321 f 6 counts in 10 min for samples after the first recrystallization, 325 f 9 count s in
I O
min aft er the second re-
crystallization, 333 16 counts in 10 min after the third recrystallization, and 5300 140 counts in
100
min afte r the fourth recrystalliza-
tion
Table VI I I .
Concentration
of V I
i n the Cannizzaro Reaction of
Tritium-Labeled 0.485 M
I
with 0.2167
M
NaOH in 74% CH30H
at 100.5 "C
~ ~ ~ ~ ~ ~~~~~~
[VI ] calcd, [VI ] found,O
time.
s
%react ion 111 .
M
M
M
0 0 0.4854 7.4 X 4. X
I800 5.9 0.4596
6.6 X 5. X
3600
13.4 0,42 74 5.7 X 5. X
IO-7
5400 18.7 0.4042
5.1
X
3.
X
f 'See Tab le
V I I .
The nonzero values are probably due mostly to
labeled reactant
I
and product I I impurities rather than
V I
because
scavenging by unlabeled
I
and
I I
was not used in this series of mea -
surements.
and 20 s , respectively, were 7 (93% reacte d), I (99% reacted), and
0.07% (99.93% reacted). A falling off of k l after 20
s
(beyond 99.93%
reaction) was attributed to significant back-reaction regenerating VI
by reaction of VI1 with XII.
The amount of VI present at various times in a Cannizz aro reaction
of0.4854 M tritium-labeled
I
with0.2167 M N a O H i n 7 4 % C H 3 0 H
at 100.5 OC was determi ned using isotope dilution. The procedure for
preparing th e reaction solution and taking points has already been
described under "Kinetic Procedure". Bromthymol blue was used as
indicator. The third-order ra te constant k3 was calculated using
26 - Q ) ~ X
a ( a - 2x)
b (a - 2x)
a ( 6
- )
+
In
where
I
is the initial concentration
of
benzaldehyde-p-1 at 10 min ( t
= 0),
b
is the initial concentration of base a t I O min
r
= 0),and
x
is
Ihc amount of base reacted i n time t . The third-order rate constant
obtained , k3 = 1.72 * 0.21 X
M-' s-l ,
does not differ signifi-
cantly from that obtained
i n
the solvent isotope effect runs (see Table
I I ) .
After the 5 -mL aliquots from the run had been titrated, they were
made slightly acidic and a known amount of untagged V I was added.
CH3O H was added until all the V I had dissolved, and the resulting
solutions were then tre ated a s described above. Table
V I 1
gives the
concentrations of
V I
found at various times in this run.
The concentration of V I that should accumulate i n the Cannizzaro
reaction a t any time if it were an interm ediate can be calculated using
the steady-state approximation. The reactions involved ar e
2 C h H sC H O + H O - o r C H 3 0 - k C h H 5 C O O C H 2 C h H 5 (V I)
VI +
H O - o r C H 3 0 - C h H sC OO C H 3
or ChH5COOH)
+
C6HsCH'O-
Application of the steady-state approximation gives
d [ V l ] / d t
=
k3[I l2 [B] - kz[VI][B]
=
0
( 8 )
( 9 )
where [B] is the total base concentration ([CH3O-]
+
[ HO-1) at any
time i n the Canniz zaro reaction, k3 is the third-order rate consta nt
for the Cannizz aro reaction in 74% methanol, and kz is the second-
order rate constant for the reaction
of
VI with HO- or CH3O- in the
sam e solvent.
Equation 9 was used tocalcu late the concentration of VI that should
have formed if it were an intermediate
i n
the Cann izzaro reaction; k3
= I .86 X M-? s-' was used since this value is considered the
most accurate. k? as found above
is
0.37/0.0625 = 5.92
s-'
M-I. The
calculated concentrations of V I . along with those actually found, are
reported in Table
V I I I .
a-Hydro gen Isotope Effects.
Our earliest work on the Canniz zaro
r e i ~ c t i o n ~ ~ ~ ~ ~tilized
I-a-t.
The procedure involved Pyrex ampules
with IO-mL aliquots of 0.48
M
I-a-t and 0.24
M
N a O H i n 74%
CH iO H under Nz at 98.3 "C. After titration of
5
mL of the solution,
the remainder was diluted with 4
m L
of water and a dded to an excess
ol 'xmicarbazide i n a test tube. The mixture was thoroughly shake n,
heated on a ste am bath. and cooled to 5 OC. The white precipitate of
I scmic arbazo ne was filtered and recrystallized twice from 50% eth-
anol. Each sample w'as dried at 130 "C and less than
1
mm. The
melting point (220-221 "C ) and activitydhwereconstant after the first
crystallization.
I-a-t
was shown to exchange 3.5%of its tritium with
Na OH solution in I2 h and 6 .5% in 24 h, by sepa rating the I-a-t re-
maining as the semicarbazone and assaying it. Thus, the rate of ex-
change is slow com pared
to
the Can nizzaro reaction. The plot of log
(activity) vs. log (fraction unreacted) deviated noticeably from a
straight line for large fractions reacted for eight different samples of
scmic arbaz one from 0 to 80% reaction.'" After correction of the ac-
tivities for
loss
of tritium by exchange with the solvent it became linear;
then from the slope of the least-squares lineSh he isotope effect kH /kT
was found to be 1.24.':' Thi s value is even lower than t ha t found by
Miklukhin for kH /kD from a similar competition experiment.?"
Analysis for Benzyl Methyl Ether I X ) . IX was shown to be stable
under C annizza ro conditions. A reaction mixture of
I
and NaOH wi th
added IX i n 74% CH3OH was prepared. Four 6-mL aliquots were
scaled i n Teflon tubes and the tubes were placed in the bath a t 99.5
"C.
At suita ble times, at the beginning and near the end of the reac-
Page 8
7/26/2019 Mechanism of Cannizzaro Reaction
http://slidepdf.com/reader/full/mechanism-of-cannizzaro-reaction 8/8
Swa in e t a l . / Me c han i sm of the Cannizzaro React ion
t i on . tubes we re w i thd rawn , coo led to room tempe ra tu re , a nd opened .
A 5 -mL a l i q u o t w a s p i p e t t e d i n t o a 6 0 - m L s e p a r a t or y f u n n e l a n d
c x t ra c t e d w i t h t w o
IO-mL
p o r t i o n s o f p u r e p e n t a n e. T h e p e n t a n e
laye rs we re comb ined and evapo ra ted
to 2
mL. T h i s s a m p l e w as
w a s he d i n t o a 5 - m L v o l u m e t r i c f l as k a n d d i l u t e d t o t h e m a r k w i t h
p e n ta n e . T h e a m o u n t o f e t h e r w a s d e t e r m i n e d by GLC w i t h 30% (by
w e i g h t ) C a r b o w a x 600 o n
50-100
m e s h f i r e b r i c k a t 130 C. A r e a s
f o r t h r e e 1 5 - p L al i q uo t s of each samp le we re measu red by p l an ime te r
a numb er of t imes and averages used. Va lues for the areas for the thre e
15-pL
a l i quo ts o f a g i v e n s a m p le w e r e g e n e r a l ly w i t h i n 3% o f e a ch
o t h e r . F r o m k n o w n s ol u ti o ns o f 0 . 0 2 6 0 - 0 .2 8 0
M
IX i n p e n t an e , t h e
r a t i o o f a r e a t o c o n c e n t r a t io n f o r t h e c o l u m n w a s d e t e r m i n e d . This
v a l u e w a s t h e n u s e d t o c a l c u l a t e t h e c o n c e n t r at i o n o f IX i n s a m p le s
f r o m t h e C a n n i z r a r o r ea c t i o n. T h e r e w a s no s i g n i f ic a n t c h a n ge i n
c o n c e n t r a t i o n of IX u n d e r C a n n i z r a r o c o n d i t i o n s : c o n c e n t r a t io n s
found i n two samp les taken a f te r I O m i n a t 9 9 . 5 "C were 0 .26 and 0.25
M; a f t e r 3 3 4 a n d 4 9 9 h t h e y w e r e 0 . 2 5 a n d 0 . 2 4 M .
A f t e r t h e f i n al p o i n t f r o m o n e C a n n i r z a r o
run
( 2 6 2 h,
>90%
r e -
ac t i on ) ha d been t i t r a ted , the resu l t i ng so lu t i on was ex t rac ted w i th
t H o 2 5 - m L p o r t i o ns o f p u r e p e nt an e . T h e o r g a n ic l a ye r s u e r e c o m -
b i n e d a n d e v a p o r a te d t o 2 mL. This so lu t i on was washed i n to a 5-mL
v o l u m e t r i c f l a s k a n d d i l u t e d t o t h e m a r k w i t h p e n t a ne . G L C u s i n g
C a r b o w a x 6 0 0 s h o w e d no peak fo r IX. IX i n pen tane (0.0260 M , the
amoun t tha t wou ld have been p resen t i f
IO%
o f h e
I
h a d r e a c te d w i t h
CH30- i o n b y m e c h a n is m 3 ) w a s a c c u r a t e l y m e a s u ra b l e b y t h i s
t e c h n i q u e ( a r e a / c o n c e n tr a t i o n r a t i o w i t h i n I % o f t h a t f o r 0 .2 6 -0 . 28
M) .
The re fo re ce r ta i n l ) less t h a n I %
of
IX
was
present.
References and Notes
(1) Supported in part by the Off ice of Naval Research, the Atomic Energy
Commission, the National Science Foundation, and the National Institutes
of Health, and by a predoctoral NIH fellowship to C.R.M.
(2) For further experime ntal details, c f. (a) W A. Sheppard, Ph.D. Thesis,
Massachusetts nstitute of Technology, 1954; (b) C. R. Morgan, P h D Thesis,
Massachu setts Institute of Technology, 1963.
(3) (a) C. G. Swain and W. A . Sheppard, Abstracts, 127th National Meeting of
the American Chemical Society, Cincinnati, Ohio, April 195 5, p 40N; (b)
C . G. Swain, A. L. Powell, and C. R . Morgan, Abstracts, 144th National
Meeting of the American C hemical Society, Los Angeles, Calif.. April 1 963,
p 18M; (c) A. L. Powe ll, C. G. Swain, and C. R. Morgan in Tritium in the
Physical and Biological Science s , V ol. 1, International Atomic Energy
Agency Sym posium, Vienna, 1962, pp 153-160; C. G. Swain, A. L. Powell,
C.
R .
Morgan, T.
J.
Lynch.
S.
R. Alpha. and R. P. Dunlap, Abstracts, 166th
National Meeting of the American Chemical Society, Chicago, Ill., Aug
1973, No. ORGN -27.
(4)
C.
G. Swain, A. L. Powell, T.
J.
Lynch, S.
R.
Alpha, and
R .
P. Duniap, J. Am.
Chem.
Soc.
ollowing paper in this issue.
(5) Office o f Naval Resea rch, Boston, Mass. Guest of the Institute. 1956-
1979.
(6) (a) F. Wohler and J. Liebig, JUSIUSiebigs Ann. Chem ., 3, 252 (1832): (b)
S.
Cannizzaro,
ibid.
88, 129 (1853). For extensive discussions, see (c)
T. A. Geissm an, Org. React., 2, 94 (1 944); (d) E. R. Alexander, Principles
of Ionic Organic Reactions . Wiley, New York, 1950, p 168; (e)
J.
Hine,
Physical Organic Chemistry , 2nd ed., McGraw-Hill, New York, 1962, p
267.
3583
(7) M. S. Nemtsov and K. M . Trenke, Zh.
Obshch. Khim.,
22,415 (1952); Chem.
Abstr.. 46. 84 85 i (1952 ).
(8) A. Lachman,
J.
Am.
Chem.
SOC. 5, 2356 (1923).
(9) (a) H . Fredenhagen and K. F. Bonhoeffer,
2.
Phys. Chem., Abt. A, 181,379
(1938); (b) C. R. Hauser, P. J. Hamrick, J r., and A. T. S tewart, J. Org. Chem.,
21, 260 (1956).
(10) (a) E. L. Molt , R e d Trav. Chim. Pays-Bas, 56, 233 (1937); (b) A. Eitel and
G. Lock. Monatsh. Chem., 72, 392 (1 939): c) E. Tomm ila, Ann. Acad. Sci.
Fenn., Ser. A. 59 (a) , 3-69 (1942); Chem. Abstr., 38, 6175 (194 4)
(11) (a) K. H. Geib, Z. Phys. Chem., Abt. A, 169, 41 (1934); (b) A. Eitel, Monatsh.
Chem., 74, 124 (1942); (c) H. v. Euler and T. Lovgren,
Z.
Anorg. Chem.,
147, 123 (1925); (d)R. J. L. Martin,
Aus t .
J. Chem., 7, 335 (1954); (e) E.
Pfeil, Chem. Ber., 84, 229 (1951);
(f)
E. A. Shilov and G. I. Kudryavtsev,
Dokl. Akad. Nauk SSSR, 63, 681 (1948 ); Chem. Abstr., 43, 4547
(1949).
(12) H. H. Jaffe, Chem Rev., 53, 209 (1953).
(13) F. Haber and
R.
Willstatter. Ber., 64 , 2851 (1931); J. Weiss, Trans. Faraday
(14) E.
R.
Alexander, J. Am. Chem.
SOC.,
69, 289 (1947).
(15) M.
S.
Kharasch and
R.
H. Snyder, J. Org. Chem., 14, 819 (1949).
(16) L. Claisen,
Chem.
Ber., 20, 646 (1887); H. Meerwein and
R .
Schmidt, Justus
(17) M. Senkus and W. G. Brown,
J.
Org.Chem., 3,5 69 (193 8). Exchange occurs
Soc. 7, 782 (1941).
Liebigs Ann. Chem., 444, 2 21 (1925).
even at 2 5 OC with no base added.
(18) B. Eistert, Tautomerie und Mesom erie , F. Enke, Stuttgart, 1938, p 116;
ref lo b, p 410. L. P. Hamm ett, Physical Organic Chemistry , M cGraw-Hill,
New York, 1940, p 350, proposed this mechanism in slightly modified form
to explain the fourth-order term s; see also E. R. Alexander, J. Am. Chem.
SOC.,70, 259 2 (1948).
(19) A. E. Favorsky,
Zh.
Fiz. Khim., 27, 8 (1895): ref 16, p 230.
(20) G. P. Miklukhin and A. F. R ekasheva, J. Gen. Chem. USSR(€ngl. Trans/.),
25, 1099 (1955);
Zh.
Obshch.
Khim.,
25, 1146 (1955).
(21) K. J. Laidler. Che mica l Kinetics , 1st ed., McGraw -Hill, New York, 1950,
pp 96-101,2nd ed , 1965, pp 137-143
(22) Cf ref 25 in C G Swain and D
R
Crist,
J
Am Chem S o c , 94, 3199
(1972)
(23) K. B. Wiberg,
J.
Am. Chem. SOC., 78, 53 71 (1954): D. Luther and H. Koch.
Ber., 99, 2227 (1966).
(24) H, von Buttlar and W. F. Libby, J. lnorg. Nucl. Chem., 1, 75 (19 55). Tritium
contents of lo - ' ' in ordinary hydrogen can be me asured
to
f 5 % accu-
racy.
(25) C. G. Swain and A. J. Kresge, J. Am. Chem. Soc., 80, 5281 (1958).
(26) (a) M. L. Bender and W. A. Glasson, J. Am . Chem. Soc., 81, 1590 (1959);
(b) W. P. Jencks and M. Gilchrist. ibid., 84, 2 910 (196 2).
(27 ) C . G. Swain, D. A. Kuhn, and R. L. Schowen, J. Am. Chem. SOC. 87, 1553
(1965).
(28) C . G. Swain and E. R. Thornton, J. Am. Chem. SOC.,83, 3890 (1961).
However, exact agreement is not
to
be expected becau se the solvent is
74 % m ethanol-26% water at 100 OC instead of pure water at 25 C .
(29) Likewise J. Hine and H. W. Haworth, J. Am, Chem. SOC., 0, 2274 (1958),
found the rearrangement of benzil to benzilic acid
to
be 1.85 times as fast
in 67 % dioxane-33 % D20 as in 67 dioxane-33 H20 and concluded
that the phenyl migration precedes the proton transfer.
(30) It also excludes a cyclic m echanism for the Cannizzaro reaction in which
hydrogen is transferred between carbons as a proton rather than as a hy-
dride ion or hydrogen atom, an alternative suggested by G. A. Hamilton:
E. T. Ka iser and F. J. Kezdy, fr og . Bioorg. Chem ., 1 148-152 (1971).
(31) M. F. Hawthorne and E.
S.
Lewis, J. Am. Chem. Soc.. 80, 4296 (1958).
(32) Melting points and boiling points are uncorrected.
(33) The procedure used here was essentially that of H. C. Brown and
C.
Groot,
J. Am . Chem. Soc., 64, 2223 (194 2). for preparing deuterium chloride.
(34 ) D . M. Hughes and
J.
C. Reid, J.
Org.
Chem., 14, 524 (1949).
(35) L. Gattermann and H. Wieland, Laboratory Methods of Organic Chemistry ,
Macmillan, New York, 1938, p 209,
(36) C. W. Gould, Jr., G. Holzman, and C. Niemann, Anal. Chem., 20, 361
(1948).
(37) H. B. Haas and M . L. Bender, J. Am. Chem.
SOC.,
71, 17 68 (1949).
(38) Reference 35, p 220.
(39) F. W. Schwab and E. Wichers, J . Res. Natl. Bur. Stand., Sect. A, 34,333
(40) H. Adkins and H. R. Billica, J. Am. Chem. SOC.,70, 696 (1948).
(41)
0.
Kamm and W. F. Kamm, Organic Syntheses . Collect.
Vol.
I,Wiley,
(42) A. Reissert. Ber., 38, 1610 (1905).
(43)
R .
L. Shriner,
R.
C. Fuson, and D. Y. Curtin, Systematic identification of
(44) E. C. Horning and M. G. Horning, J. Org. Chem., 11, 97 (1946).
(45) Reference 43, p 218, procedure A.
(46) C. G. Swain, V. P. Kreiter, and W. A. Sheppard. Anal. Chem., 27, 1157
(1955).
(47) K. E. Wilzbach, L. Kapian, and W. G. Brown, S cience, 118, 522 (1953 ).
(48) These values were reported in 1955.3aDr. Clair J. Collins informed us in
1960 that on repeating this work he obtained a value of 5. 26.
(49)
0.
Redlich and F. Pordes. Monatsh. Chem., 67, 203 (1936).
(50)
H. Lund and J. Bjerrum, Ber., 64, 2 10 (1931); L. F. Fieser, Experiments
in Organic Chemistry , 3rd ed.. D. C. Heath, Boston, Mass., 1955, pp 289,
29.1
(1945).
New York, 1941, p 104.
Organic Com pounds , 4th ed., Wiley. New York, 19 56.
(53)
(54)
These analyses were performed by Mr. Josef Nameth. Urbana, 111
H. Rinderknecht and C. Niemann.
J.
Am. Chem. SOC.,70,26 05 (1948); R.
0.Clinton and
S.
C. Laskowski, ibid., 70, 3135 (1948).
J . Kendall and A. H. Wright, J. Am. Chem.
SOC.
2, 177 8 (1920).
W. T. Olson et al., J. Am. Chem. Soc. 69, 2451 (1947).
(55) H.Diehl and G. F. Smith, Quantitative Analysis, Elementary Principles and
Practices , Wiley, New York, 1952, p 207.
(56) A. M . Downes and G. M. Harris, J. Chem. Phys. 20, 196 (1952).