Mechanisms of Adoptive Immunotherapy: Improved Methods for in Vivo Trackingof Tumor-infiltrating Lymphocytes and Lymphokine-activated Killer Cells1
Paul K. Wallace,2 Larry D. Palmer, Donna Perry-Lalley, Ellen S. Bolton, Richard B. Alexander, Paul K. Horan,James C. Yang, and Katharine A. Muirhead3
Zynaxis Cell Science. Inc., Malvem. Pennsylvania ¡9355¡P.K. W., L D. P.. P. K. H.. K. A. M.¡.and Surgery Branch, Naliimal Cancer Institute. NIH. Bethesda. Maryland 20W2ID. P-L. E. S. B.. K. B. A.. J. C. Y.I
ABSTRACT
Adoptive immunotherapy with tumor-infiltrating lymphocytes (TIL)and lymphokine-activated killer cells has been demonstrated to mediate
regression of tumors in murine models and in selected patients with advanced cancer. Improved methods for monitoring immune cell traffic,particularly to sites of tumor, are needed to elucidate mechanisms ofantitumor activity and optimize treatment protocols. Traditional celltracking methods such as fluorescent protein labeling and radiolabelingusing '"In, I25I, or "Cr are limited by isotope half-life, leakage or transfer
of label from immune cells, and toxicity or altered cell function caused bythe labeling process. Labeling with genetic markers allows long-term cell
tracking but is laborious to perform and difficult to quantitate.We have used two recently described lipophilic cell tracking compounds
(PKH26 and 125I-PKH95) which stably partition into lipid regions of the
cell membrane to track immune cells in vivo. Concentrations of eachtracking compound which had no adverse effects were determined for avariety of murine TIL and lymphokine-activated killer cell functions.
Viability was unimpaired at labeling concentrations of up to 5 UM forPKH95 and 20 MMfor PKH26. TIL proliferation was unaltered by labelingwith up to 5 UMPKH95, 20 MMPKH26, or a combination of 15 MMPKH26and 5 MMPKH95. In vitro cytotoxic effector function and in vivo therapeutic efficacy of lymphokine-activated killer cells and TIL were alsounimpaired by labeling with 20 MMPKH26 or 1 MMI25I-PKH95. Subsequent studies in an adoptive transfer immunotherapy model used '-"I-
PKH95 to track the biodistribution of TIL in tumor and in non-tumor-
bearing animals and PKH26 fluorescence to monitor microdistributionwithin tissues and distinguish TIL from host T-cells. The results suggest
that differential accumulation, selective retention, or proliferation at thetumor site cannot account for the observed pattern of therapeutic efficacy.We hypothesize that a minimum number of TIL must reach the tumor sitein order to achieve a demonstrable therapeutic effect.
INTRODUCTION
Adoptive immunotherapy, a procedure in which immune cells withantitumor activity are transferred to a tumor-bearing host, is currently
under investigation in clinical trials for treatment of patients withmetastatic cancer (1, 2). These trials are based on animal modelswhich have demonstrated that syngeneic lymphocytes expanded andactivated in culture with cytokines can mediate tumor regression whenadoptively transferred into animals with experimentally induced tumors (3-5). Several different types of immune cells have been usedfor adoptive immunotherapy, including LAK4 cells and TIL (6-9).
Received 12/13/91; accepted 3/11/93.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1This work was supported in part by Small Business Innovative Research grant
number AI28600 awarded by the National Institutes of Allergy and Infectious Diseases.- Present address: Department of Microbiology and Immunology. Medical College of
Pennsylvania. Philadelphia. PA 191291To whom requests tor reprints should be addressed, at Zynaxis Cell Science. Inc.. 371
Phocnixville Pike. Mähern. PA 19355.4 The abbreviations used are: LAK ceils, lymphokine-activated killer cells: TIL. tumor-
infiltrating lymphocyte(s); rIL-2. recombinant interleukin-2; NK cell, natural killer cell;CM. complete medium: HBSS. Hanks' balanced salt solution: FITC. fluorescein isothio-
cyanate.
Different methods of preparation, cell phenotype, tumor target specificities, and in vivo potencies characterize each of these immune celltypes.
LAK activity is mediated by broadly reactive cytolytic cells that arenot major histocompatibility complex restricted and are capable oflysing fresh and cultured tumors, including both NK-sensitive andNK-resistant targets (9. 10). In humans the majority of LAK activityis associated primarily with cells of the CD3~CD56 ' phenotype (11),
the precursor of which is similar to NK cells (12, 13). A more potentpopulation of cells with antitumor activity can be derived from lymphocytes which have infiltrated the tumor after expansion with rIL-2
in vitro. In contrast to LAK, murine and human TIL demonstratecytolytic specificity against only the tumor from which they werederived or against closely related tumors (3, 14), and in murine systems TIL have proved to be 50 to 100 times more potent than LAKcells (3). Murine TIL are predominantly CD3 ^CD^CDS ', while
human TIL are phenotypically more heterogeneous, and perhaps forthis reason their in vitro cytotoxicity has not been a good predictor oftheir ability to cause in vivo tumor regression (1, 15). Both LAK andTIL mediate tumor regression, but only in selected patient groups, andthe mechanisms of this antitumor activity remain undefined.
Improved methods to monitor the distribution of LAK cells and TIL/«vivo should provide insight about interactions between immunecells and tumor, facilitating optimization of protocols for adoptiveimmunotherapy. Horan et al. (16) have developed a family of fluorescent and radiolabeled lipophilic compounds which bind stably andnontoxically to cell membranes, enabling long-term cell tracking invivo (17-23). For purposes of monitoring the in »'ivodistribution of
immune cells following adoptive transfer, methods are needed toassess both microdistribution within organs and macrodistributionamong organs and tissues. We have developed methods to label bothLAK cells and TIL with these lipophilic tracking compounds andexamined the feasibility of using these probes to study their migrationand homing patterns. Fluorescent tracking compounds have been usedto quantify total cell number recovered after organ disaggregation, andradiolabeled compound has been used to determine whole body bio-
distribution. In this communication, we demonstrate that measures ofseveral functions believed to be critical to their in vivo biologicalactivity were unaffected by labeling with either PKH26 or I25I-
Mice. Female C57BL/6 mice were obtained from the Small Animal Section, Veterinary Resource Branch. NIH (Bethesda. MD) and from CharlesRiver Laboratories (Wilmington. MA). All mice were maintained in pathogen-
free conditions and used in accordance with NIH guidelines for animal care.Mice were used at 10 weeks of age or older.
IMPROVER METHODS I-OR IN VIVO IMMUNE CEU. TRACKING
Tumors and Cell Lines. The murine colon adenocarcinoma MC-38. murine sarcomas MCA-205. and MCA-207 tumors were used in this study (3. 24).
The sarcomas were induced by injection of 0.1 ml of \7c 3-methylcholanthrene
in sesame oil and were passaged s.c. for up to 10 generations, at which time a
cryopreserved vial from the second or third generation was thawed and transplanted. The MC-38 adenocarcinoma was induced by I.2-dimethylhydra/ine
and passaged in vivn.YAC, a Moloney murine leukemia virus-induced T-cell lymphoma line, was
kept in continuous culture and maintained in CM consisting of RPMI 1640(Biofluids. Rockville. MDl supplemented with 10% heat-inactivated fetal calf
serum (Biofluids): O.I HIMnonessential amino acids. O.I imi sodium pyruvate.and 50 ug/ml gentamicin (M.A. Bioproducts. Walkersville, MD); 2 m\i freshglutamine. 100 units/ml penicillin, and KM)ug/ml streptomycin (NIH MediaUnit. Bethesda. MD): 5 x IQ-' M2-mercaptoethanol (Sigma Chemical Co., St.
Louis, MO); and 50 ug/ml amphotericin B (fungi/one; Gibco. Grand Island.NY).
Lymphocyte Preparation and LAK Cell Culture. Spleens from C57BL/6mice were aseptically excised and crashed into a single cell suspension withthe end of a syringe. The spleen cell suspension was filtered through 100-gauge
nylon mesh (Nytex. Advance Process Supply. Chicago. IL) and centrifuged.Erythrocytes were hypotonically lysed in a buffered ammonium chloride solution (ACK lysing buffer: Biofluids) at room temperature. The cells were thencentrifuged and washed twice with CM.
LAK cells were generated by culturing splenocytes at 1-3 x 10" cells/ml in
CM with 6000 lU/ml rIL-2 provided by the Cetus Corporation (Emeryville,CA). The flasks were incubated supine at 37°Cin 57c CO; for 3 or 4 days.
Cells were then harvested, passed over a discontinuous Ficoll gradient (Lym-pholyte-M; Cedarlane Laboratories. Ltd.. Hornby. Ontario, Canada) to remove
dead cells, and washed three times in HBSS (Biofluids) before use.TIL Cultures. Murine TIL were generated from s.c. tumors approximately
1-1.5 cm in diameter. Tumors were harvested aseptically, minced with scissorsinto fragments 1-2 mm in size, and stirred in a three-enzyme mixture con
taining 1500 units type I deoxyribonuclease. 50 mg type IV collagenase, and5 mg type V hyaluronidase (Sigma Chemical Co.) in a 50-ml volume of HBSSfor 2-3 h at room temperature. The dispersed tumor cells were collected,passed through Nytex. washed twice in HBSS. resuspended in CM at 1 X 10"
cells/ml, and placed on ice. When necessary, erythrocytes were removed byhypotonie lysis before washing in HBSS.
TIL were isolated from tumor digests as previously described (25, 26). Inbrief. anti-Thyl.2 monoclonal IgM (New England Nuclear, Boston. MA)-
coated paramagnetic beads (Dynal, Inc.. Great Neck, NY) were washed in CM.resuspended at I x 10* beads/ml, and combined with an equal amount of
dispersed tumor. This mixture was incubated on ice with periodic gentleagitation for 60 min. After incubation the tube was filled with 25 ml cold CM,slowly inverted to resuspend the pellet, and placed in a magnetic holder for 2min. The supernatant was discarded, and the remaining bead-cell conjugates
were gently resuspended in CM for one wash followed by magnetic collection.Bead-cell conjugates were resuspended in CM containing 120 lU/ml rIL-2 andincubated at 37°Cin 5% CO: in a 24-well plate (Costar, Cambridge. MA) ata density of 1-2 X IO7 beads/ml. After 18 h. the wells were vigorously
triturated with a Pasteur pipet to detach the beads, which were then removedby magnetic separation. Free cells were collected, replated at 1-2 X IO5
cells/ml, and stimulated with irradiated (5.000-10,000 cGy) autologous tumorcells at a density of 1-2 x IO5 cells/ml.
TIL were maintained ¡nCM with 120 lU/ml rIL-2. Every 3-5 days either
fresh medium was added to the cultures or the TIL were harvested, counted,and replated at a density of 1-2 X IO5cells/ml in 6- or 24-well plates. TIL were
routinely stimulated every 7-10 days with irradiated (3000 cGy) autologoustumor cells by adding 0.5-2 X 10' cells to each ml of TIL culture.
LAK Cell and TIL Labeling with PKH26 and PKH95. LAK cells andTIL were harvested, washed twice in HBSS. counted, and then stained withPKH26 or PKH95 (Zynaxis, Malvern, PA) according to the manufacturer's
instructions (16). In brief, washed cells were resuspended in Diluent C at twicethe final staining concentration and then immediately combined with an equalvolume of 2x PKH26 or PKH95 in Diluent C. Final staining conditions forPKH26 were 1 X IO7cells/ml in 5-20 UMPKH26; final staining conditions forPKH95 were 5-10 x ID7 cells/ml in 1-5 UMPKH95 ('-5I-PKH95 specific
activities, 5-30 mCi/umol). Rapid and complete mixing of the cells with the
label by vortex mixing or trituration was found to be important for reproducible
and homogeneous staining. After 2-2.5 min at room temperature an equal
volume of fetal calf serum was added to stop the staining reaction. Stained cells
were pelleted, transferred to a fresh centrifuge tube, and washed twice withCM. Viability was determined by trypan blue exclusion.
Proliferation Assays. To compare the in vitro proliferative capacity oflabeled TIL with that of unlabeled TIL. cells were stained and cultured in24-well plates as described above. At regular intervals TIL from several wells
were harvested and pooled. The number of viable cells in each pool (as
determined by trypan blue exclusion) was enumerated using a hemocytometer.Each proliferation experiment was performed a minimum of two times.
In Vitro Cytotoxicity Assay. LAK and TIL effector cells were prepared asdescribed above and plated at various effector: target cell ratios in 96-wellU-bottomed plates (Costar). YAC cells and fresh tumor obtained from enzyme
digests of s.c. tumors were used as targets. Targets were washed twice inHBSS. and the cells were resuspended in CM at 1 x IO7cells/ml. Two hundreduCi of 5lCr were added per ml of target cell suspension. The mixture wasincubated at 37°Cfor 2 h. then washed twice in HBSS, resuspended in CM.
and incubated an additional half-hour before washing a final time. Target cellswere resuspended in CM. and IO4 target cells plus varying numbers of effector
cells were added to each well. Targets and effectors were centrifuged at 50 Xf>for 5 min and then incubated at 37°Cfor 4 h. The supernatant from each well
was collected with filter frames (Skatron, Sterling, VA) and counted in agamma counter. The mean of triplicate samples was determined, and percentage lysis was calculated as follows:
X 100<7rlysis =' 5l
where 5lCr is measured in cpm, sample is the amount released when effector
and targets are combined experimentally, spontaneous is the backgroundamount released by targets alone, and maximum is the amount released by lysisof targets cells in l N HC1. Cytotoxicity experiments were performed a minimum of two times.
suspended in 0.5 ml HBSS. Three or six days after tumor cell injection, variousdoses of expanded TIL or LAK cells were injected i.v. in 0.5 ml HBSS.Commencing on the day of adoptive therapy and continuing twice daily for 5days, some mice received 60.000 IU rIL-2 in 0.5 ml HBSS i.p. Approximately
ml HBSS. Animals also received 60.000 IU rIl-2 in 0.5 ml HBSS i.p. com
mencing on the day of immune cell injection and continuing twice daily for 5days. Replicate aliquots of labeled cells were fixed in \'7< paratormaldehydeand stored at 4°Cfor use as counting standards. At various times after immune
cell injection, animals were sacrificed, major organs and tissues were harvestedinto preweighed tubes, and the tubes were reweighed and counted in a model1272 ClinGamma gamma counter (Wallac Pharmacia. Turku, Finland) alongwith the cellular counting standards. Decay-corrected results were expressed as
the adoptive immunotherapy model. Six days after tumor induction, mice weregiven i.v. injections of 0.5 ml HBSS containing 5 X IO11TIL labeled with 15
UMPKH26; animals were then maintained on rIL-2 as previously described. At
various times after immune cell injection animals were sacrificed, and lungs orother organs were harvested, rapidly frozen on dry ice, and stored at -70°C
prior to histological examination. Lungs were gently insufflated with OCTembedding medium (Miles Diagnostics. Elkhart. IN) prior to freezing to rnain-
IMPROVED METHODS I-OR /.V VIVO IMMUNE CELL TRACKING
tain good tissue architecture. Ten-um frozen sections were cut using a Reichert-
Jung histostat with a model 820 freezing microtome (Baxter Diagnostics.Scientific Products Division. McGaw Park. ID and were transferred to glassmicroscope slides. Fluorescence micrographs were taken immediately aftersectioning, using a calibrated microscope stage and recording the position ofthe field for each photomicrograph. Slides were then stained with hematoxylinand eosin and readjusted to the same stage position, and the field was repho-tographed. The relative densities of labeled immune cells in tumor-bearing and
normal regions of the lung were determined by comparing fluorescence andhematoxylin and eosin photomicrographs.
subsequently maintained on rIL-2 as previously described. At various times
after immune cell injection animals were sacrificed, lungs or other organs wereharvested, and a single cell suspension was prepared using the enzymaticdisaggregation procedure described for preparing TIL cultures. Erythrocyteswere removed by hypotonie lysis, and the cells were washed twice in coldstaining buffer (phosphate-buffered saline. pH 7.2. with \<7cfetal calf serumando.\'7r sodium a/ide) (Sigma Chemical Co.). counted, and adjusted toa finalconcentration of IO7 cells/ml. Immunofluorescence labeling was performed on
ice in a 96-well round-bottomed microtiter plate. Nonspecific binding of an
tibody to Fc receptors was blocked by preincubating 100 ul of cells with 10 ul(2(K) ug/ml) of clone 2.4G2 (generously provided by David Segal. Bethesda.MD). After 5 min. 20 ul of FITC-labeled anti-Thyl.2 or control antibody withirrelevant specificity (Leu-2: Becton Dickinson. Mountain View. CA) was
added. Cells were stained for 30 min and washed twice in staining bufferbefore flow cytometric analysis.
Labeled cells were analyzed on a FACScan or a FACStar Plus (BectonDickinson) in a dual laser configuration. Linear forward scatter versus logarithmic side scatter displays were used on both instruments to set a broad gatewhich eliminated small debris and large aggregates prior to collection of listmode data. On the FACScan. nonviable cells labeled with propidium iodide(Sigma Chemical Co.) were excluded from analysis by linear amplification inthe FL3 channel (650 longpass filter). FITC-labeled antibodies were detected
using logarithmic amplification in the FL1 channel (530/30 bandpass filter).PKH26 and PKH95 fluorescence were evaluated using logarithmic amplification in the FL2 channel (585/42 bandpass filter). FACScan research softwarewas used to collect and analyze flow cytometric data. On the FACStar. FITC-
labeled antibodies were excited with the first argon ion laser tuned at 488 nmand were delected by logarithmic amplification in the FL1 channel (530/30bandpass filter). PKH26 fluorescence was also excited at 488 nm and evaluatedwith logarithmic amplification in the FL2 channel (575/26 bandpass filter).Nonviable cells which incorporated propidium iodide were excited at 540 nmusing a dye head containing rhodamine 110 pumped with an argon ion laser,detected in the FL3 channel (640 longpass filter), and later eliminated fromanalysis by gating on FL3. Lysis-II (Becton Dickinson) software was used on
the FACStar to collect and analyze flow cytometric data.Statistical Analysis. Statistical differences in all in vitro experiments were
determined using a paired Student's t test analysis. All in vivo metastasis
measurements were compared using the nonparametric. Wilcoxon two-sample
Characterization of Probe Effects on Cell Function
Optimization of PKH26 and PKH95 Labeling. Initial experiments were designed to determine optimal labeling conditions withthe tracking compounds (i.e.. the maximum amount of PKH26 orPKH95 membrane incorporation which did not decrease cell viability)since we have found that these conditions vary for each cell type(16-19). Important variables include cell concentration and dye concentration. A cell concentration for labeling ranging from 1 X IO7 to10 X IO7 cells/ml was chosen based on the total number of cells
required for in vivo experiments (5 X IO7 to 2 X IO8) and the
requirement for maintaining a final staining volume of less than 5 ml.Fluorescence intensity is a function of the cell:dye ratio; higher cellconcentrations and/or lower dye concentration give decreased stainingintensity. We have found that relatively small staining volumes facilitate rapid mixing and give more homogeneous cell staining, whilelower dye concentrations give less homogeneous staining. Fig. \Aillustrates selection of optimal stain concentration for TIL withPKH26. Concentrations greater than 20 UM were toxic to TIL. Atlabeling concentrations of 5 and 20 UMPKH26 and 2 X IO7cells/ml,
TIL were, respectively, 100 and 500 times brighter than unlabeledcells. In Fig. Ißan overlay of flow cytometric histograms fromunlabeled and 20 MMPKH26-labeled TIL is illustrated; 20 UMwas
selected as the maximum concentration for further evaluation of effects on cell function because it gave maximum fluorescence andhomogeneous staining and had minimal effects on cell viability.
Similar experiments were carried out with PKH95. an iodinatedanalogue of PKH26 which is characterized by weaker fluorescencebut can be used as a radiotracer. The maximal nontoxic concentrationof PKH95 was found to be 5 UMat 5 X IO7cells/ml (data not shown).
The maximum nontoxic dye concentrations established for TIL werealso found to be nontoxic for LAK cells stained under the sameconditions (data not shown). Staining with a mixture of PKH26 andPKH95 was tested to determine whether a combination of dyes couldbe used for studies of whole body biodistribution using the radiolabeland histological microdistribution using the fluorescent label. A combination of 5 |JMPKH95 with 15 UMPKH26 was found to be nontoxicand to give fluorescent intensities 75 times greater than unlabeled cells(cell concentration, 5 X 107/ml).
5010 20 30
UMPKH2640
B400
300
io 200ÚJ
100
t¿r;
v..10" 101 10Z 103
Log Fluorescence Intensity (FL2)
10«
Fig. I. PKH26 staining optimization for murine TIL. A. MC-38 TIL were stained withthe indicated concentrations of PKH26 at a final cell concentration of I X IO7cells/ml asdescribed in "Materials and Methods." Viability (A) was determined by trypan blue
exclusion, and mean fluorescence intensity •¿�' was determined from the flow cylomelrichistograms. B, overlay of flow cytometric histograms from unstained MC-38 TIL ( —¿�;mean intensity 1.9) and MC-38 TIL stained with 20 JJMPKH26 ( : mean intensity662).
Fig. 2. Effect of PKH26 and PKH95 labeling on proliferation of murine TIL. Cultureswere maintained as described in "Materials and Methods." and multiple wells were pooled
for enumeration of viable cells at the indicated times. A. MC-38 TIL were treated with 20UMPKH26 (A). 5 UMPKH26 (•).diluent only (A), or medium only (O). Proliferation ofPKH26- and diluent-treated TIL was not significantly different (P > 0.20) from that ofTIL treated with medium only. B. MCA-205 TIL were treated with 5 UMPKH95 (A). IUM'-'1-PKH95 •¿�0.265 cpm/cell I, or medium only (O). Proliferation of PKH95 labeled
TIL was not significantly different (P > 0.12) from that of TIL treated with medium only.C. MCA-205 TIL were treated with IS M" PKH26 and 5 UMPKH95 (Al. or K) UMPKH26
and 5 UMPKH95 (•).or medium only (O). Proliferation of TIL labeled with both PKH26and PKH95 was not significantly different (P > 0.II) from that of TIL treated withmedium only.
In Vitro Proliferation of Labeled Cells. Although no acute tox-
icity was observed immediately following staining, it was also necessary to demonstrate that labeling did not affect other critical cellfunctions such as proliferation and ability to recognize and kill tumor.As shown in Fig. 2, PKH26 (Fig. 2A), PKH95 or I25I-PKH95 (Fig.
2B), or a combination of both PKH26 and PKH95 (Fig. 2C) had nosignificant effect on the rate of proliferation. Labeling different TILcultures with 1 UM I2SI-PKH95 of varying specific activities gave0.033-0.265 cpm/cell (1.8 X 10~x to 1.2 X I0~7 uCi/cell). an expo
sure well below the level causing radiotoxicity (27) but more thansufficient for radiotracking purposes. Data shown are for the firstweek in culture; however, no late effects were seen when cultureswere followed for up to 3 weeks (data not shown). This is as expected,because the label is partitioned between daughter cells and thereforethe amount of dye per cell progressively decreases with each celldivision.
In Vitro Cytotoxic Effector Function of Labeled Cells. Havingestablished that proliferation was not affected by labeling, we then
examined the effect of PKH26 or PKH95 labeling on the ability ofLAK cells and TIL to lyse appropriate tumor targets. Targets examinedincluded autologous tumor (tumor line from which the TIL werederived), nonautologous tumor (other syngeneic solid tumors), andYAC (a cell line known to be sensitive to NK and LAK cell cytotoxicmechanisms). Neither compound affected the cytotoxic activity ortarget specificity of either effector cell type; representative experiments are shown in Fig. 3. Labeled TIL were able to recogni/.e and killautologous tumor as effectively as unlabeled TIL (Fig. 3, A and B) butdid not lyse nonautologous tumors (data not shown) or YAC targets(Fig. 3. C and D). Labeled LAK cells were able to lyse all targets; asexpected, they exhibited lower potency than TIL directed againstautologous tumors. Therefore, we conclude that neither in vitro cytotoxic potency nor target specificity is altered by labeling with PKH26,PKH95, or l2il-PKH95.
unlabeled effector cells. No differences in gross physical appearanceor behavior were observed in mice receiving PKH26-. PKH95-, orl25I-PKH95-labeled effector cells as compared with mice receiving
unlabeled cells.
In Vivo Tracking of Labeled Immune Cells
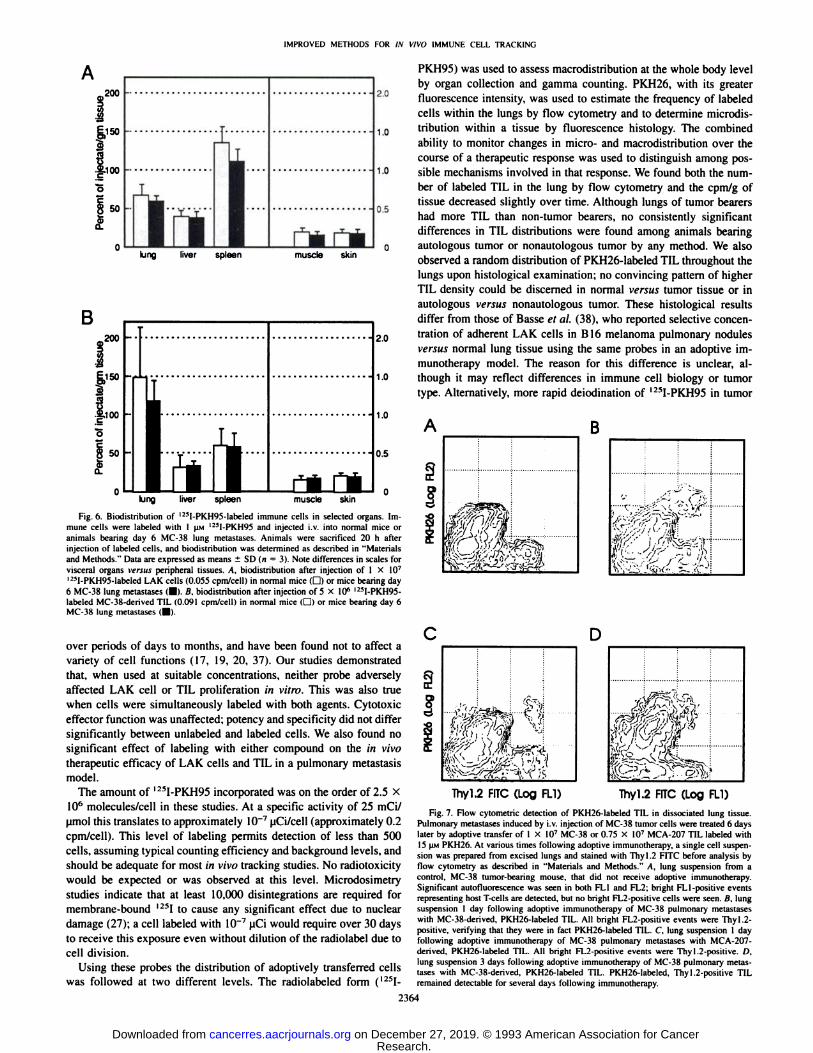
Utility of Fluorescent and Radiolabeled Probes for Tracking.Since neither in vitro nor in vivo effector functions of LAK cells andTIL were altered by labeling with PKH26 or PKH95. we evaluated theuse of these compounds for in vivo cell tracking. The presence oflabeled TIL in various organs was readily identifiable by gammacounting (Fig. 6). flow cytometry (Fig. 7). and fluorescence microscopy (Fig. 8).
Active mechanisms which may account for the therapeutic efficacyof immune cells include homing to. selective retention in. and/orproliferation of these cells within the autologous tumor. Alternatively,no active mechanisms may exist; instead a demonstrable therapeuticresponse may be achieved only when a minimum number of cellspassively accumulate within tumor deposits. To attempt to distinguishamong the various possible mechanisms, we used PKH26 and I25I-
PKH95 to track the in vivo micro- and macrodistribution of labeledTIL in the adoptive transfer model. A cross-over experimental design
IMPROVED METHODS FOR /,V VIVO IMMUNE CELL TRACKING
100
80
| 60
S«40
20
0100:1 20:1 4:1 0.8:1
100:1 20:1 4:1 0.8:1
EffectorJarget Ratio
B
D
100
80
60
40
20
O
100
80
60
40
20
100:1 40:1 16:1 6.4:1 1.6:1
100:1 40:1 16:1 6.4:1
Effector Target Ratio1.6:1
Fig. 3. In viim cytotoxiceffector functionof PKH26- and PKHu5-labeled murine LAK cells andTIL. TIL or LAK cells were labeledandcombinedwith autologoustumoror YACcell targetsin a standard4-h MCr-releasecytotoxicilyassayat the indicatedeffectorto targetratio (see"Materials andMethods"!.Means ±SD are shownfor unlabeledTIL (Al. labeled
TIL (A), unlabeled LAK cells (O). and labeled LAK cells •¿�A. LAK cells and MC-38-derived TIL were labeled with 20 UM PKH26 and tesled for lysis of MC-38 tumor.Effectortarget ratioswere 100:1. 20:1. 4:1. and0.8:1 for LAK cells andTIL. Cytotoxicity of PKH26-labeled TIL (P > 0.721 and LAK cells (P > 0.95l wasnoi significantlydifferentfrom their unlabeledanalogue.0. LAK cells and MCA-205-derived TIL were labeledwith l UM'-5I-PKHU5 andtestedfor lysisof MCA-205 lumor. Effectortarget ratioswere I(X):I.40:1. 16:1, and 6.4:1 for LAK cells and 50:1. 20:1. 8:1, and 3.2:1 for TIL. Cytotoxicity of PKH95-labeled TIL (P > 0.79) and LAK cells </>> 0.48) was not significantlydifferentfrom the Cytotoxicityof their unlabeledanalogue.C. LAK cells and MC-38-derived TIL were labeledwiih 20 UMPKH26 andtestedfor lysisof YAC cell targets.Effectontargetratioswere 100:1, 20:1, 4:1. and0.8:1 for LAK cellsandTIL. The Cytotoxicityof PKH26 labeledTIL (P > 0.48) andLAK cells (P > 0.51) wasnot significantlydifferent from thatof theirunlabeledanalogue.I), LAK cells and MCA-205-derived TIL were labeledwith l UM I:51-PKH95 and tesledagainstYAC cell targets.Effectortarget ratios were 100:1, 40:1. 16:1.and6.4:1 for LAK cells and50:1. 20:1, 8:1. and3.2:1 for TIL. The Cytotoxicityof PKH95-labeled TIL (P > 0.46) and LAK cells </>> 0.82) wasnot significantlydifferent from that
of their unlabeledanalogue.
selective homing of these cells to the tumor type from which theywere originally derived, but not to nonautologous tumor. To determineif there was preferential accumulation of TIL in organs bearing autologous tumor, TIL were labeled with I2SI-PKH95 and injected into
mice using a two-way cross-over design. Typically, the majority of
label was found in the liver, lungs, and spleen: no consistently significant differences were observed in any organ except the lungs: andalthough there was a trend toward greater accumulation of TIL intumor-bearing versux non-tumor-bearing lung, lungs from tumor-bear
ing animals receiving the autologous TIL did not consistently containmore label than lungs from animals receiving nonautologous TIL. Arepresentative experiment is shown in Table 1. In this experiment,significantly more label was found in the lungs of most tumor-bearingmice compared with those from non-tumor bearers. However, no
consistently greater accumulation was seen in lungs from animalsreceiving the autologous tumor/TIL combinations versus the nonautologous tumor/TIL combination.
These observations suggest that the increased amount of label foundin the tumor-bearing lungs, compared with normal lungs, was not the
result of any selective homing mechanism but was possibly due tomechanical trapping or nonspecific effects of tumor on pulmonaryendothelium and vasculature. An alternative explanation for the therapeutic efficacy of TIL against autologous tumor is that TIL arepreferentially retained at sites of autologous tumor. This hypothesispresumes that equal numbers of TIL arrive at both tumors but that TILrecognize autologous tumor and leave the circulation, while TIL encountering nonautologous tumor remain in the circulation. This hypothesis was addressed by examining the redistribution of I25I-
PKH95-labeled TIL over time in animals bearing no tumor,
autologous tumor, or nonautologous tumor. As shown in Table 1. thereis no evidence for any preferential retention over time; levels of probein the lung decrease over time for all treatments.
Flow Cytometric Quantitation of TIL Frequency in Tumor-bearing and Non-tumor-bearing Lungs. Although the distribution
of radiolabel clearly indicates that TIL did not selectively accumulateand were not preferentially retained within the autologous tumor, it ispossible that selective proliferation of autologous TIL or selectiverecruitment of host T-cells accounts for their therapeutic effect. These
possible mechanisms could not be assessed by examining the distribution of radiolabel but could be evaluated using flow cytometry bycomparing the frequency of TIL and host T-cells in lungs bearing
autologous versus nonautologous tumor. Results from a representativeexperiment are shown in Table 2. The number of PK.H264, Thy 1.2*
TIL did not increase significantly over time in lungs bearing autologous tumor, suggesting that there was no selective proliferation. Significantly greater numbers of PKH26", Thy 1.2* cells were noted in
MC-38-bearing lungs compared with non-tumor-bearing or MCA-207-bearing lungs. However, there was not a significantly greaternumber of host T-cells in lungs bearing autologous compared with
nonautologous tumor, suggesting that therapeutic efficacy was not dueto selective recruitment of host T-cells to autologous tumor.
Microdistribution of PKH-26-labeled TIL in Tumor-bearingLungs. Whole organ evaluation (Tables 1 and 2) showed no evidencefor a preferential accumulation or proliferation of TIL in lungs bearingautologous versus nonautologous tumor. However, such measurements cannot determine whether TIL are randomly distributed in
IMPROVED MKTHODS I OR l\ VIVO IMMUNE CELL TRACKING
normal and tumor tissue or whether they selectively concentrate intumor tissue. To address this question we examined frozen sections oflungs from mice receiving PKH26-labeled TIL in a cross-over exper
imental design. For all TIL/tumor combinations, labeled TIL wereobserved in normal tissue as well as in tumor deposits within anyindividual lung. Occasional sections could be found which appearedto have a greater concentration in tumor tissue than in surroundinglung tissue. However, after examining many sections, no convincingpattern of higher TIL density could be discerned in either tumor ornormal organ parenchyma or in autologous versus nonautologoustumor (data not shown).
DISCUSSION
Adoptive immunotherapy using TIL or LAK cells has been shownto be effective against some metastatic cancers refractory to standardtherapies. Although substantial tumor regression is observed only fora limited number of tumor histologies, durable complete responseshave been achieved in some patients ( I. 2. 7. 8). Further optimisationto increase efficacy and reduce toxicity is required to establish theutility of this modality. Many of the critical questions about TIL/LAKcell therapy revolve around issues of cell trafficking. What are thetissue and organ affinities of LAK cells and TIL. and what are thekinetics of their distribution? Is there a correlation between traffickingpattern and clinical efficacy? Do specific subpopulations mediate
clinical responses and is this via direct cytotoxic mechanisms orsecondary mediators?
These types of questions are under investigation in both animalmodels and clinical studies. Lotze el til. (28) reported no selectivelocalization of LAK cells to tumor. Selective accumulation of '"In-
labeled LAK cells was reported in one case study (29) and in one ofsix patients in another study (30). All of these investigations reportedaccumulation predominantly in the liver and spleen. Similar studiesusing '"In-labeled TIL from patients with melanoma have also re
ported accumulation predominantly in the liver and spleen (31. 32).However, in these studies, preferential localization of TIL at tumorsites was demonstrated. This localization was seen as early as 24 h andincreased over time. Most recently, localization of ' ' ' In-labeled TIL to
tumor in patients with melanoma was significantly associated withclinical responses to therapy*.
Use of '"In for cell tracking has significant limitations. These
include a relatively short half-life (which limits the length of study),
radiotoxicity to cells, and significant spontaneous release. The amountof label per cell must be limited in order to minimize effects on cellfunction (31 ), necessitating the use of large numbers of cells ( 10'°)per
study in order to detect localization at later time points. Therefore,recent studies have utilized genetic markers for cell trafficking (25,33-35). These types of markers are excellent for monitoring long-term
survival and the appearance of TIL at specific tumor sites after immunotherapy. However, with genetic markers quantification of wholebody distribution and degree of localization in tumor is difficult andat best semiquantitative.
We have demonstrated the feasibility of using two recently described probes to study the migration and homing patterns of adoptively transferred cells. PKH26 and '-5I-PKH95 are lipophilic cell-
labeling agents which provide potential solutions to some of thedifficulties in cell trafficking studies (16. 19. 36). These agents partition into the lipid region of cell membranes, are suitable for tracking
' B. A. Puckaj, R. Sherry. J. Wei. J. R. Yannelli. C. S. Carter. S. F Leitman. J. R.
Carrasquillo. D. E. White. S. M. Steinberg. S. A. Rosenberg, and J. C. Yang. Localizationof '"indium-labelled tumor infiltrating lymphocytes to tumor in patients receiving adop
tive ¡mmunotherapy: augmentation with cyclophosphamide and association with response,submitted for publication.
over periods of days to months, and have been found not to affect avariety of cell functions (17, 19, 20, 37). Our studies demonstratedthat, when used at suitable concentrations, neither probe adverselyaffected LAK cell or TIL proliferation in vitro. This was also truewhen cells were simultaneously labeled with both agents. Cytotoxiceffector function was unaffected; potency and specificity did not differsignificantly between unlabeled and labeled cells. We also found nosignificant effect of labeling with either compound on the in vivotherapeutic efficacy of LAK cells and TIL in a pulmonary metastasismodel.
The amount of I2SI-PKH95 incorporated was on the order of 2.5 XIO6 molecules/cell in these studies. At a specific activity of 25 mCi/umol this translates to approximately 10~7uCi/cell (approximately 0.2
cpm/cell). This level of labeling permits detection of less than 500cells, assuming typical counting efficiency and background levels, andshould be adequate for most in vivo tracking studies. No radiotoxicitywould be expected or was observed at this level. Microdosimetrystudies indicate that at least 10,000 disintegrations are required formembrane-bound I25I to cause any significant effect due to nucleardamage (27); a cell labeled with 10~7uCi would require over 30 days
to receive this exposure even without dilution of the radiolabel due tocell division.
Using these probes the distribution of adoptively transferred cellswas followed at two different levels. The radiolabeled form (I25I-
PKH95) was used to assess macrodistribution at the whole body levelby organ collection and gamma counting. PKH26, with its greaterfluorescence intensity, was used to estimate the frequency of labeledcells within the lungs by flow cytometry and to determine microdistribution within a tissue by fluorescence histology. The combinedability to monitor changes in micro- and macrodistribution over the
course of a therapeutic response was used to distinguish among possible mechanisms involved in that response. We found both the number of labeled TIL in the lung by flow cytometry and the cpm/g oftissue decreased slightly over time. Although lungs of tumor bearershad more TIL than non-tumor bearers, no consistently significant
differences in TIL distributions were found among animals bearingautologous tumor or nonautologous tumor by any method. We alsoobserved a random distribution of PKH26-labeled TIL throughout the
lungs upon histológica! examination: no convincing pattern of higherTIL density could be discerned in normal versus tumor tissue or inautologous versus nonautologous tumor. These histológica! resultsdiffer from those of Basse et al. (38), who reponed selective concentration of adherent LAK cells in B16 melanoma pulmonary nodulesversus normal lung tissue using the same probes in an adoptive im-
munotherapy model. The reason for this difference is unclear, although it may reflect differences in immune cell biology or tumortype. Alternatively, more rapid deiodination of I25I-PKH95 in tumor
B
Wirnm^-^^ÃÃ^'V-^^o-^ff.^-'?'•?•>•&=#•'
D
,i, : 1;:SÄ.
Thyl.2 FITC (Log FU) Thyl.2 FITC (Log FU)Fig. 7. Flow cytometric detection of PKH26-labeled TIL in dissociated lung tissue.
15 UMPKH26. At various times following adoptive immunotherapy. a single cell suspension was prepared from excised lungs and stained with Thyl.2 FITC before analysis byflow cytometry as described in "Materials and Methods." A, lung suspension from a
IMPROVED METHODS (OR /,V VIVO IMMUNE CELL TRACKING
hr ** •¿�'•±*:*,r -*•" -". - *-
Fig. 8. Light and fluorescence micrographs of lung and spleen showing tissue architecture and distribution of PKH26-labeled TIL. MC-38-derived TIL were labeled with 20 UMPKH26. and 5 X 10* were injected i.v. into normal mice. On day 3. the organs were harvested, and lungs were gently insufflated with embedding medium and frozen on dry ice.
Fluorescent micrographs were taken immediately after sectioning on a microtome cryostat. Sections were then stained with H&E. A. lung section stained with H&E. li. fluorescentmicrograph of lung section shown in A taken with a fluorescein filter combination. Bright yellow PKH26-stained TIL appear to be randomly distributed throughout: dim yellow greentissue autofluorescence is also visible. C fluorescent micrograph of lung section shown in A taken with a rhodamine filter combination, which provides more efficient excitation ofPKH26 than the fluorescein combination. Essentially no tissue autofluorescence is seen with this filler combination. 1), spleen section stained with H&E. /:'. fluorescent micrograph
of spleen section shown in D taken with fluorescein filter combination. Bright yellow PKH26-stained TIL appear to preferentially accumulate in the outer regions of the follicle.Considerable tissue auttifluurescence is also visible. F. fluorescent micrograph of spleen section shown in D taken with a rhodamine filter combination. Minimal tissue autofluorescenceis visible.
deposits as compared with normal lung tissue might explain the apparent lack of preferential accumulation of radioluhel in tumor bearinglung. We believe this explanation is unlikely for two reason: (a) thechemical linkage in I25I-PKH95 is much more stable than that
achieved by radiolabeling aromatic amino acids (36); and (i>)a similarlack of preferential accumulation was found with the fluorescentprobe, PKH26, where deiodination would not be a problem.
These results argue that in murine models TIL do not selectivelymigrate or home to tumor by an active mechanism. If continued influxof effector cells and net accumulation within tumor were occurring,one would expect to see an increased percentage of labeled cells overtime using flow cytometry, increasing values for cpm/g tissue bygamma counting, and an increasing concentration of fluorescence-
positive cells in tumor deposits upon histológica! examination. Local
Table I Biodistribution of l~'<il-PKH95-lubelt'd TIL in normal and tumor-hearing mice
Treatment"TIL38
3838207
207207TumorNone
38207None
20738n35
53
55Day
19.18(0.50)''
11.77(0.68)''16.81(1.67)''6.81
(1.14)9.20(1.20)7.39(0.89)TreatmentTIL38
3838207
207207TumorNone
38207None
20738n35
53
55Day
10.03(0.01)
0.03 (0.00)0.07(0.02)0.03
(0.00)0.02(0.01)0.02 (O.(X)ILungs'7Day
36.53(1.13)
10.30(1.25)13.90(1.56)''2.93(0.17)8.44(0.58)''
6.15(0.52)''MuscleDay
30.03(0.01)
0.02 (0.00)0.02(0.00)0.01
(0.01)0.01 (O.(X))0.02 (0.01)Day
54.52(0.16)
6.66(0.67)''11.67(0.52)'2.71
(0.35)6.67(0.56)''5.31(0.14)''Day50.01
(0.00)0.02 (0.00)0.02(0.01)0.02(0.01)
0.01 (0.00)0.01 (O.(X))Day
152.00(1.10)
43.52 (2.25)47.27(2.34)54.39
(0.99)41.80(2.70)44.33(3.35)Day
10.00
(0.00)0.00 (0.00)0.00(O.(X))0.57(0.13)
0.21 (0.02)/
0.29(0.05)LiverDay
344.84(1.52)
45.15(1.54)40.60(3.56)45.92(2.41)
43.89(2.02)40.53(1.69)KidneysDay
30.20(0.02)
0.20 (0.04)0.14(0.03)0.25
(0.02)0.11 (0.01)'
0.19(0.04)Day
540.38(0.32)
34.90(3.44)37.96(2.79)52.00(3.83)
40.18(2.96)41.45(3.12)Day
50.13(0.00)0.14(0.02)
0.17(0.05)0.18(0.02)
0.09(0.01)0.14(0.03)Day
11.98(0.06)
1.51 (0.11)1.84(0.24)2.32(0.05)
1.89(0.27)2.02(0.11)Day
10.33
(0.08)0.68(0.15)0.73(0.17)0.53(0.31)
0.78(0.19)0.08 (0.03)SpleenDay
31.77(0.09)
1.67(0.15)1.73(0.08)1.76(0.06)
2.19(0.25)2.43(0.52)UrineDay
30.04(0.02)
0.04(0.03)0.07(0.02)0.05
(0.03)0.10(0.03)0.07(0.03)Day
51.63(0.09)
1.37(0.13)1.72(0.22)2.15(0.19)
1.91 (0.33)1.84(0.24)Day
50.05
(0.02)0.02 (0.00)0.04(0.02)0.05
(0.00)0.06(0.03)0.04 (0.02)
" Irradiated C57BL/6 mice were innoculuted i.v. with either the MC-38 or MCA-207 tumor. Six days later these und other non-tumor-beuring mice were treated with either I x IO7|:SI-PKH95 labeled MC-38 or MCA-207 TIL. All mice were maintained on rlL-2 us described in "Materials and Methods."
''On the indicated duys, animuls were sacrificed, major organs were harvested into preweighed tubes and the tubes were reweighed and then counted.' Mean (SE); decay corrected results expressed as percentage of injected dose/g tissue.'' Significantly greater (P < 0.05) thun non-tumor bearer which received similur TIL.'' Significantly greater (P < 0.05) thun both the non-tumor-bearing control and the other tumor beurer which received similar TIL.
' Significantly less (P < 0.05) than non-tumor-bearing control which received similar TIL.
Tuble 2 Flow cytometric ÃfuunÃitationof PKH26-labeled TIL and Thyi-positive host T-cells from murine lungs
Treatment"TILNone
None38
3838207
207207Tumor38
207None
38207None
20738n2
22
442
44TIL(PKH26*.
THYI.2VDay
10.27(0.06)'
0.33(0.05)2.08
(0.04)3.74(0.71)1.95(0.64)0.80
(0.30)0.69(0.14)1.30(0.18)Day
30.19(0.07)
0.21(0.03)1.00(0.05)
1.60(0.20)0.85(0.23)0.13(0.06)
0.20(0.08)0.54 (0.09)Day
50.39(0.19)
0.09(0.00)1.06(0.47)
2.76(0.60)4.24(1.20)0.27
(0.03)0.18(0.05)1.90(0.28)''Host
T-cells (PKH26-,THY1.2+)Day
12.99
(0.89)1.71(0.00)0.95
(0.24)4.95(0.76)''
1.01(0.26)0.82(0.13)
0.94(0.37)2.53(0.24)''Day
32.59(0.85)
2.30(0.47)1.89(0.32)
4.46(1.11)1.42(0.44)0.98(0.31)
1.39(0.54)3.64(1.16)Day52.67
(0.99)1.75(0.30)1.84(0.39)
10.25(2.43)''
6.49 ( 1.80)2.39
(0.45)1.62(0.48)9.38(1.60)''
" Irradiated C57BL/6 mice were inoculated i.v. with either the MC-38 or MCA-207 tumor. Six days later some of these tumor hearers and other normal mice were treated with either1 x IO7 PKH26-labeled MC-38 TIL or 7.5 x IO6 labeled MCA-207 TIL. Animals were maintained on rIL-2 as described in "Materials and Methods."
'' On the indicated days, animals were sacrificed, lungs were harvested, and single cell suspensions were prepared. Total cell counts were determined before directly labeling the
suspensions with Thyl.2 (FITC) for flow cytometric analysis. Using forward and side scatter gates to eliminate small debris and propidium iodide to exclude dead cells, the numberof Thyl.2-positive. PKH26-positive TIL and the number of Thyl.2-positive, PKH26-negative host T-cells was determined.
' Absolute number of the cells recovered = % viable cells with the indicated phenotype determined by flow cytometric analysis x total number of viable cells recovered. Valuesshown are mean (SE) in units x l()?.
''Significantly greater (P < 0.05) than both the non-tumor-bearing control and the other tumor bearer receiving similar TIL.
redistribution of TIL to tumor deposits within the lung would not beevident from radiolabeling results but should be evident on fluorescence histological examination. Finally, if neither further influx norredistribution within organs was required but. rather, antigen-stimu
lated proliferation of those effector cells encountering tumor occurred,then one would see an increasing percentage of labeled cells by flowcytometry with a decreasing mean fluorescence intensity for thosecells. None of these changes were evident in these studies.
High levels of TIL localization in normal lung and variability fromanimal to animal may limit our ability to detect small amounts ofdifferential accumulation of TIL within lungs bearing autologous tumor. Background levels of TIL accumulation in normal skin andmuscle are much lower than in lung, and in a preliminary experiment,an increased accumulation of autologous TIL in small intradermaltumors was observed in comparison with the surrounding skin andmuscle tissues. This is similar to what has been observed in humanstudies (31, 32). However, the number of cells accumulating at thetumor site was much lower than in the lung (<1% versus 10-20%).
Unlike in patients with melanoma, the therapeutic efficacy of TIL forintradermal lesions has not been demonstrated in the mouse model. Inaddition, lymphocyte chemotactic factors and endothelial ligands maybe species specific, and animal models may not predict TIL behavior
in patients. These considerations may contribute to differences seenbetween patient studies and these preclinical murine studies.
The studies described here demonstrate that neither PKH26 norPKH95 alters the in vitro or in vivo biological characteristics of LAKcells and TIL and suggest that in order to achieve a therapeutic effect,a minimum number of cells must be directly delivered to the tumortissue. We believe that these probes will be useful in developing andtesting strategies designed to deliver the maximum number of effectorcells to tumor tissue. Analogues of these probes capable of binding yemitters for imaging may eventually permit extension of this approachto clinical studies.
ACKNOWLEDGMENTS
The authors wish to thank Mr. Thomas C. Schmitt for his expert assistancewith the preparation of frozen sections and with hematoxylin and eosin stainingand Mr. David Jones for his help with animal care and tumor maintenance.
REFERENCES
1. Rosenberg. S. A. Immunotherapy and gene therapy of cancer. Cancer Res., 51-(Suppl.): 5074s-5()79s. 1991.
2. Chang. A. E.. and Shu. S. Immunotherapy with sensitized lymphocytes. CancerInvest.. 10: 357-369. 1992.
IMPROVED METHODS FOR /.V VIVO IMMUNE CELL TRACKING
3. Spiess. P. J.. Yang. J. C.. and Rosenberg. S. A. In vivo antitumor activity of lumor-infillraling lymphocytes expanded in recomhinant interleukin-2. J. Nail. Cancer Inst..79: 1067-1075, 1987.
4. Cameron. R. B.. Spiess, P. J.. and Rosenberg. S. A. Synergistic anlitumor activity oftumor-infiltrating lymphocytes, interleukin-2. and local tumor irradiation. J. Exp.Med.. 171: 244-26.1. 1991)'
5. Sacchi. M.. Vitólo.D.. Sedlmayr. P.. Rabinowich. H.. Johnson. J. T. H.. and Whiteside.T. L. Induction of tumor regression in experimental model of human head and neckcancer by human A-LAK cells and IL-2. Int. J. Cancer. 47: 784-791, 1991.
6. Rosenberg. S. A.. Lotze. M. T.. Yang. J. C.. Aebersold. P. M.. Linehan. W. M.. Seipp.C. A., and White. D. E. Experience with the use of high-dose inlerleukin-2 in thetreatment of 652 cancer patients. Ann. Surg.. 210: 474-485. 1989.
7 Albenini. M. R.. Sosman. J. A.. Hank. J. A.. Moore. K. H.. Borchert. A.. Schell. K..Kohler. P. C.. Bechhoter. R.. StörenB., and Sondel. P. M. The influence of autologouslymphokine-acti\ated killer cell infusions on the toxicity and antitumor effect ofrepetitive cycles of interleukin-2. Cancer (Phila.l. 66: 2457-2464. 1990.
S. Butcher. J. P.. Gaynor. E. R.. Boldt. D. H.. Doroshow. J. H., Bar. M. H.. Sznol. M. M..Sparano. J.. Fisher. R. I.. Weiss. G.. Margolin. K.. Aronson. F. R.. Hawkins. M.. andAtkins, M. A phase II study of high-dose continuous infusion interleukin-2 withlymphokine-activated killer cells in patients with metastatic melanoma. J. Clin. On-col.. 9: 641-648. 1991
9. Grimm. E. A.. Ma/umder. A.. Zhang. A., and Rosenberg. S. A. The lymphokineactivated killer cell phenomenon: lysis of NK resistant fresh solid tumor cells by IL-2activated autologous human peripheral blood lymphocytes. J. Exp. Med.. 155: 1823-1841. 1982.
K). Melder. R. J.. Whiteside. T. L.. Vujanovic. N. L.. Hiserodt. J. V.. and Herherman. R.B. A new approach to generating antitumor effectors for adoptive immunotherapyusing human adherent lytnphokine-aclivated killer cells. Cancer Res.. 4X: 3461-3469.1988.
11. Savary, C. A., and Lotzova. E. Adhesion molecules on MHC-nonrestricted lymphocytes: high density expression and role in oncolysis. Lymphokine Cytokine Res.. //:149-156. 1992.
12. Herberman. R. B.. Hiserodt. J., Vujanovic. N., Balch, C.. Lotzova. E., Bolhuis. R..Golub. S.. Lanier. L. L.. Phillips. J. H.. Riccardi. C. Ritz. J., Santoni. A.. Schmidt. R.E.. and Uchida. A. Lymphokine activated killer cell activity. Characteristics of effector cells and their progenitors in blood and spleen. Immunol. Today. K: 178-181, 1987
13. Melder. R. J., Walker. E. R.. Herberman. R. B.. and Whiteside. T. L. Surface characteristics, morphology, and ultraslructure of human adherent lymphokine-activatedkiller cells. J. Leukocyte Biol.. 48: 163-173. 1990.
15. Topalian. S. L., Solomon, D.. and Rosenberg. S. A. Tumor-specific cytolysis bylymphocytes infiltrating human melanomas. J. Immunol.. 142: 3714-3725. 1989.
16. Horan, P. K.. Melnicoff. M. J., Jensen. B. D.. and Slezak. S. E. Fluorescent celllabeling for in vivo and in vitm cell tracking. Methods Cell Biol.. 33: 469-490. 1990.
17. Melnicoff, M. J.. Morahan. P. S.. Jensen. B. D., Breslin. E. W., and Horan. P. K. Invii1»labeling of resident peritoneal macrophages. J. Leukocyte Biol.. 43: 387-397,
1988.18. Slezak. S. E.. and Horan. P. K Fluorescent m viro tracking of hematopoietic cells.
Part I. Technical considerations. Blood. 74: 2172-2177. 1989.19. Jensen. B. D.. Schmitt, T. C.. and Slezak. S. E. Labeling of mammalian cells for in
vivo cell tracking by a fluorescence method. Prog. Clin. Biol. Res., 355: 199-207.1990.
20. Teare. G. F.. Horan. P. K., Slezak, S. E.. Smith, C.. and Hay. J. B. Long-term trackingof lymphocytes in vivo: the migration of PKH-labeled lymphocytes. Cell. Immunol..134: 157-170. 1991.
21. Hugo. P.. Kappler. J. W., Godfrey. D. I., and Marrack. P. A cell line that can inducethymocyte positive selection. Nature (Lond.t. 3ftu: 679-682. 1992.
22. Messina, L. M.. Podrazik. R. M.. Whitehill. T. A.. Ekhterae. D.. Brothers. T. E..Wilson. J. M., Burkel. W. E., and Stanley. J. C. Adhesion and incorporation of/«rZ-transduced endothelial cells into the intact capillary wall in the rat. Proc. Nati.Acad. Sei. USA. Ä9:I2018-I2022. 1992.
23. Rosenman. S. J.. Ganji, A. A.. Tedder. T. F.. and Gallatin. W. M. .Vv/i-capping ofhuman T lymphocyte adhesion/activation molecules and their redistribution duringinteraction with endothelial cells. J. Leukocyte Biol.. 53: 1-10. 1993.
25. Alexander. R. B.. and Rosenberg. S. A. Long term survival of adoptively transferredtumor-infiltrating lymphocytes in mice. J. Immuni)!.. 145: 1615-1620. 1990.
26. Yang. J. C.. Perry-Lalley. D.. and Rosenberg. S. A. An improved method for growingmurine tumor-infiltrating lymphocytes with in vivo antitumor activity. J. Biol. Response Modif.. 9: 149-159. 1990.
27. Hofer, K. G. Microdosimetry of labeled cells. In: C. F. Krüger(ed.). Blood Cells inNuclear Medicine. Pan II. pp. 224-243. The Hague: Martin N'ijhoff Publishers. 1984.
28. Lotze. M. T.. Line. B. R.. Mathisen. D. J.. and Rosenberg. S. A. The in vivo distribution of autologous human and murine lymphoid cells grown in T cell growth factor(TCGF): implications for the adoptive immunotherapy of tumors. J. Immunol.. 125:1487-1493. 1980.
29. Thakur. M. L. Radiolabelcd blood cells: perspectives and directions. NucÃ.Med. Biol..17: 41-47. 1990.
30. Mukherji. B.. Ambjarnarson. O.. Spilzangle. L. A.. Hoffman. J.. Ergin. M. T.. andSpencer. R. P. Biodislribution of In-111 labeled tumor sensitized autologous lymphocytes in cancer patients. Prog. Clin. Biol. Res.. 244: 325-334. 1987.
31. Fisher. B.. Packard. B. S.. Read. E. J.. Carrasquillo. J. A.. Carter, C. S., Topalian. S.L.. Yang. J. C.. Yoiles. P.. Larson, S. M.. and Rosenberg. S. A. Tumor localization ofadoptively transferred indium-Ill labeled tumor infiltrating lymphocytes in patientswith metastatic melanoma. J. Clin. Oncol.. 7: 250-261. 1989.
32. Griffith. K. D.. Read. E. J.. Carrasquillo, J. A., Carter, C. S., Yang. J. C.. Fisher. B..Aebersold. P.. Packard. B. S.. Yu. M. Y. and Rosenberg. S. A. In vivo distribution ofadoptively transferred indium-111 labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J. Nati. Cancer Inst..HI: 1709-1717. 1989.
33. Alexander. R. B.. and Rosenberg. S. A. Adoptively transferred tumor-infiltratinglymphocytes can cure established metastatic tumor in mice and persist long-term invivo as functional memory T lymphocytes. J. Immunolher.. 10: 389-397, 1991.
34. Culver. K.. Cornetta. K.. Morgan. R.. Morecki. S.. Aebersold. P.. Kasid. A. L.,Rosenberg, S. A., Anderson, W. F., and Blaese, R. M. Lymphocytes as cellularvehicles for gene therapy in mouse and man. Proc. Nati. Acad. Sci. USA. US:3155-3159. 1991.
35. Wong. R. A.. Alexander, R. B.. Puri. R. K.. and Rosenberg. S. A. In vivo proliferationof adoptively transferred tumor-infiltrating lymphocytes in mice. J. Immunolher.. lu:120-130, 1991.
36. Brooks. K.. Palmer. L.. Jensen. B.. Gavin. E.. and Muirhead. K. A radioiodinatedlipophilic probe for quantitative in vivo cell tracking. Cytometry Suppl.. 5: 86. 1991.
37. Yuan, Y.. and Fleming. B. P. A method for labeling rat neutrophils with a fluorescentdye for intravital microvascular studies. Microvasc. Res.. 40: 218-229. 1990.