Mechanisms Regulating theSecretion of the PromalignancyChemokine CCL5 by Breast TumorCells: CCL5’s 40s Loop andIntracellular Glycosaminoglycans1,2
Gali Soria*, Yaeli Lebel-Haziv*,3, Marcelo Ehrlich*,3,Tsipi Meshel*, Adva Suez†, Edward Avezov*,Perri Rozenberg* and Adit Ben-Baruch*
*Department of Cell Research and Immunology, George S.Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv,Israel; †Bioinformatics Unit, George S. Wise Faculty of LifeSciences, Tel Aviv University, Tel Aviv, Israel
AbstractThe chemokine CCL5 (RANTES) plays active promalignancy roles in breast malignancy. The secretion of CCL5 bybreast tumor cells is an important step in its tumor-promoting activities; therefore, inhibition of CCL5 secretionmay have antitumorigenic effects. We demonstrate that, in breast tumor cells, CCL5 secretion necessitated thetrafficking of CCL5-containing vesicles on microtubules from the endoplasmic reticulum (ER) to the post-Golgi stage,and CCL5 release was regulated by the rigidity of the actin cytoskeleton. Focusing on the 40s loop of CCL5, we foundthat the 43TRKN46 sequence of CCL5 was indispensable for its inclusion in motile vesicles, and for its secretion. TheTRKN-mutated chemokine reached the Golgi, but trafficked along the ER-to-post-Golgi route differently than thewild-type (WT) chemokine. Based on the studies showing that the 40s loop of CCL5 mediates its binding to glyco-saminoglycans (GAG), we analyzed the roles of GAG in regulating CCL5 secretion. TRKN-mutated CCL5 had lowerpropensity for colocalization with GAG in the Golgi compared to the WT chemokine. Secretion of WT CCL5 wassignificantly reduced in CHO mutant cells deficient in GAG synthesis, and the WT chemokine acquired an ER-likedistribution in these cells, similar to that of TRKN-mutated CCL5 in GAG-expressing cells. The release of WT CCL5was also reduced after inhibition of GAG presence/synthesis by intracellular expression of heparanase, inhibition ofGAG sulfation, and sulfate deprivation. The need for a 43TRKN46 motif and for a GAG-mediated process in CCL5secretion may enable the future design of modalities that prevent CCL5 release by breast tumor cells.
Neoplasia (2012) 14, 1–19
IntroductionThe inflammatory milieu plays a key role in regulating tumor growthand progression [1–3]. A growing number of studies suggest that theinflammatory CC chemokine CCL5 (also known as RANTES) hasmajor tumor-supporting activities in several cancer diseases [4,5].CCL5 was extensively studied in breast cancer, where it was shownto causatively promote malignancy [4,5]. The chemotactic propertiesof CCL5 lead to elevated levels of deleterious tumor-associated macro-phages in breast tumors, and it was suggested that this chemokine recruitsinflammatory TH17 cells to the tumor site [6–9]. In parallel, the chemo-kine promotes the release of matrix-degrading enzymes by the tumorcells [7,10] and induces their migration and invasion [10–19]. Particu-larly, the chemokine was shown to promote the invasiveness of cellshaving the CD44+/CD24− phenotype of tumor-initiating cells [19].The importance of CCL5 in breast cancer is reinforced by the fact
that its inhibition has led to reduced malignancy in animal modelsystems of breast cancer, indicating that the chemokine has a causative
role in promoting breast cancer [6,8,13,20–22]. In line with the above,CCL5 was intimately linked with advanced and aggressive disease inpatients and with lymph node involvement and was suggested as a
Abbreviations: BFA, brefeldin A; ER, endoplasmic reticulum; ERAD, ER-associatedprotein degradation; HMEC, human mammary normal epithelial cell; GAG, glycos-aminoglycan; mAb, monoclonal antibody; MW, molecular weight; WT, wild-typeAddress all correspondence to: Adit Ben-Baruch, PhD, Ela Kodesz Institute for Researchon Cancer Development and Prevention, Department of Cell Research and Immuno-logy, George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv 69978,Israel. E-mail: [email protected] study was supported by Israel Science Foundation and by Federico Foundation.All authors declare that they have no conflict of interest.2This article refers to supplementary materials, which are designated by Videos W1 toW6 and Figures W1 and W2 and are available online at www.neoplasia.com.3These authors equally contributed to this article.Received 7 August 2011; Revised 5 December 2011; Accepted 12 December 2011
potential prognostic factor predicting progression in stage II breastcancer patients [19,23–26].In biopsies of breast cancer patients, the most important source for
CCL5 is the cancer cells themselves [5,9,19,23–30]. Recent studiesindicate that the expression of the procancerous chemokine CCL5 isacquired in the course of malignant transformation, and its release bythe tumor cells enables its paracrine and autocrine activities on cells ofthe tumor microenvironment and on the tumor cells, respectively[4,5,19,27,31]. Therefore, the secretion of CCL5 by breast cancer cellsis a key regulatory step whose inhibition may lead to a significant re-duction in the tumor-promoting activities induced by this chemokine.The aim of the present study was to characterize the mechanisms
that control the secretion of CCL5 by breast tumor cells. Specifically,we wished to identify chemokine domains that are required for CCL5secretion and cellular components that regulate the release of thischemokine by breast tumor cells. The findings of this study indicatethat the chemokine is mobilized in well-organized vesicles on micro-tubules from the endoplasmic reticulum (ER) to the post-Golgi stageand that its release by the tumor cells is an actin-regulated process.Furthermore, by using a mutated CCL5, we have identified a four-amino-acid motif in the 40s region of CCL5, 43TRKN46, that isessential for its inclusion in motile vesicles and for its secretion bybreast cancer cells. We have also shown that glycosaminoglycans(GAG) play an important regulatory role, although partial, in mediat-ing CCL5 release by the tumor cells.The above results indicate that the 43TRKN46 sequence of CCL5
and intracellular GAG are essential for the secretion of CCL5. Whenthese results are considered with additional findings provided in thisstudy, and in the literature, we suggest that one of the mechanisms thatmediate the secretion of CCL5 by breast tumor cells is based on theassociation of the 40s loop of CCL5 with intracellular GAG that maketheir way to the cell surface or to the cell exterior. The identification of a43TRKN46-mediated and GAG-mediated process of CCL5 secretionmay set the basis for the future design of inhibitors that would re-duce the secretion of CCL5 by breast tumor cells and thus may limitCCL5-dependent processes that are involved in breast malignancy.
Materials and Methods
Cell CulturesThe MCF-7 and T47D human breast carcinoma cell lines were
grown in Dulbecco modified Eagle medium (DMEM) as describedpreviously [24]. MDA-MB-231 cells were cultured in DMEM asabove. MCF-10A cells were cultured in DMEM/F12 supplementedwith 5% horse serum, 10 μg/ml insulin, 20 ng/ml epidermal growthfactor (all purchased from Biological Industries, Beit Haemek, Israel),100 ng/ml cholera toxin (Sigma-Aldrich, St Louis, MO), and 0.5 μg/mlhydrocortisone (Sigma). WI-38 cells (kindly provided by Prof Rotter,Weizmann Institute of Science, Rehovot, Israel) were cultured in min-imum essential medium (Sigma) supplemented with 10% fetal calfserum and 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mlpenicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin(Biological Industries). Human mammary normal epithelial cells(HMECs; kindly provided by Dr Berger, Sheba Medical Center,Tel-Hashomer, Israel) were cultured in MEGM Ready Medium(Lonza, Walkersville, MD). CHO-K1 and CHO-pgsA-745 cells(kindly provided by Prof Vlodavsky, Technion, Haifa, Israel) were cul-tured in DMEM and RPMI 1640medium, respectively, supplementedwith 10% fetal calf serum, 2 mM L-glutamine, 1 mM sodium pyru-
vate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 250 ng/mlamphotericin (Biological Industries).Before the different experimental procedures, the various cell lines were
transferred to their corresponding serum-free media, except for MCF-10A cells that were transferred to LPM medium (Biological Industries).
Chemokine SequencesThe vector used in most analyses in this study was pEGFP-N1, ex-
pressing WT or 43TRKN46-mutated CCL5 (Table 1). The constructsof WT and mutated CCL5 were produced by polymerase chain re-action, and the sequences of both chemokines were validated byfull-length sequencing. In parallel, we used the pcDNA3.1-zeo(−)vector, expressing HA-tagged WT CCL5, whose sequence was vali-dated by full-length sequencing.
Bioinformatics Analysis of Chemokine StructureModels were built using Rosetta 3.1 AbinitioRelax protocol, with
protein structure prediction using Rosetta [32]. The sequence of theTRKN-mutated CCL5 [named CCL5(TRKN–)], SPYSSDTTPCC-FAYIARPLPRAHIKEYFYTSGKCSNPAVVFVAAAARQVCAN-PEKKWVREYINSLEMS, was superimposed on the x-ray structureof WT CCL5 (PDB 1U4M [33]; the modeling started at amino acid14 because the mutated chemokine was modeled as monomer, whereasthe x-ray structure of WT CCL5 was of a dimer, in which the N′ isdifferently organized compared to the monomer). To predict the ap-propriate folding of the mutated CCL5 protein, an ab initio protocolwas applied, which predicts the structure of a protein based on its se-quence. The final model presented was chosen out of 10,000 decoys,based on their lowest score. The model was further assessed usingthe MolProbity Web server [34], which examines psi and phi angles,C-beta deviations, atom clashes, and rotamers.
Transfection and Determination of Transfection YieldsMCF-7 and T47D cells were transfected by electroporation as de-
scribed previously [35]. The other cell types were transfected by ICAFectin 441 DNA transfection reagent (MedProbe, Oslo, Norway)according to the manufacturer’s instructions.Transfection outcome was evaluated by flow cytometry analyses
(fluorescence-activated cell sorter [FACS]). The cells were washed withphosphate-buffered saline supplemented with 0.02% sodium azide,and the expression of GFP was determined with a Becton DickinsonFACSort (Mountain View, CA) using the CellQuest software.
Determination of CCL5 Secretion by ELISAThe different cell types were grown in serum-free medium for 24 to
48 hours. In specific cases, the cells were treated by brefeldin A (BFA;Sigma; in two experiments, we used 5 μg/ml BFA for 2 hours, and inone additional experiment, we used 25 μg/ml for 5 hours; the two con-ditions yielded similar results), by latrunculin (5 μg/ml; Sigma), or
Table 1. Sequences of GFP-CCL5(WT) and GFP-CCL5(TRKN–) Used in the Study.
Chemokine Carboxyl Terminus Sequence
GFP-CCL5(WT) N′-CSNPAVVFVTRKNRQVC…-GFP43
GFP-CCL5(TRKN–) N′-CSNPAVVFVAAAARQVC…-GFP43
The sequences of the GFP-CCL5(WT) and GFP-CCL5(TRKN–) are presented starting at their34th amino acid. The sequence that was mutated to alanines (43TRKN46) is underlined.
2 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
by jasplakinolide [10−6 M; Alexis, Farmingdale, NY]) for 2 hours at37°C. In other cases, the cells were treated by BFA for 20 minutes atroom temperature.When the cells were treated by nocodazole (15 μg/ml;Sigma), they were incubated as follows: 15 minutes at 4°C without thedrug; 15 minutes at 4°C with the drug; 1.5 hours at 37°C with the drug.Treatment with sodium chlorate (30 mM; Sigma) was performed for48 hours at 37°C. In all cases, control treatments included incubationof the cells with the relevant diluents of the reagents for similar periods.The inhibitors did not affect cell viability.When sulfate deprivation was induced, the cells were grown for
48 hours with magnesium sulfate–deprived medium (Biological Indus-tries; to replenish the magnesium, a similar quantity of magnesiumchloride was added to the medium). Control cells were grown withregular DMEM.In other experiments, MCF-7 cells were transiently or stably trans-
fected with a myc-tagged pSecTag vector-expressing heparanase or withan empty vector as control (kindly provided by Prof I. Vlodavsky,Technion, Haifa, Israel). In parallel, the cells were transiently trans-fected with GFP-CCL5(WT).Chemokine levels in cell supernatants were determined by ELISA,
using standard curves with recombinant human CCL5 at the linearrange of absorbance. The ELISA analyses were performed in thefollowing manners: Procedure 1. Unless otherwise indicated, the ELISAstudies were performed with antibodies against CCL5: coating—mousemonoclonal antibodies (mAbs) against human CCL5 (PeproTech,Rocky Hill, NJ); detection—biotinylated polyclonal goat anti-humanCCL5 antibodies (R&D Systems, Minneapolis, MN). Procedure 2.Coating—mouse mAb against GFP (Covance, Princeton, NJ; MBLInternational, Woburn, MA); detection—biotinylated polyclonal rab-bit anti-GFP antibodies (Sigma). Procedure 3. Coating—mouse mAbagainst GFP (Covance); detection—biotinylated polyclonal goatanti-human CCL5 antibodies (PeproTech). Procedure 4. Coating—polyclonal rabbit antibodies against human CCL5 (PeproTech);detection—biotinylated polyclonal goat anti-human CCL5 antibodies(PeproTech). P values were calculated by Student’s t test.
Determination of CCL5 Expression by Western Blot AnalysisMCF-7 cells were transiently transfected with vectors expressing
GFP-CCL5(WT), GFP-CCL5(TRKN–) or GFP only. The proteinswere immunoprecipitated from cell lysates by mouse mAb againstGFP (Covance), and Western blot for GFP was performed by mousemAb against GFP (Covance).
Confocal AnalysesCells transiently transfected with vectors expressingGFP-CCL5(WT),
GFP-CCL5(TRKN–), GFP only, or HA-CCL5(WT) were grown ingrowth medium on coverslips for 24 hours and then in serum-free me-dium for another 24 hours at 37°C. Actin polymerization was detectedby phalloidin (40 minutes; Molecular Probes, PoortGebouw, theNetherlands) after cell fixation and permeabilization [36].To determine the localization of CCL5 in the ER, staining was
performed with rabbit antibodies against calnexin (Sigma), thenwith DyLight 549–conjugated AffiniPure goat anti-rabbit antibodies( Jackson Immunoresearch Laboratories, West Grove, PA). To de-termine the localization of CCL5 in the Golgi, a vector expressingthe Golgi marker α mannosidase IB, tagged by HA, was expressedby transfection in the cells. HA was detected by rabbit antibodiesagainst HA (Santa Cruz Biotechnology, Santa Cruz, CA), then stainedby DyLight 549–conjugated AffiniPure goat anti-rabbit antibodies
(Jackson Immunoresearch Laboratories). Negative controls includedcell staining by secondary antibodies only, as well as by an isotype-matched nonrelevant antibody (data not shown). In this set of ex-periments, stained cells were imaged by Zeiss LSM-510 confocalmicroscope (Zeiss, Thornwood, NY). Determination of colocalizationof GFP-CCL5(WT) or GFP-CCL5(TRKN–) with markers of the ERor of the Golgi was performed by Slidebook software (Slidebook,Denver, CO), applied on a large number of cells.In parallel, experiments were performed to determine the colocaliza-
tion of GFP-CCL5(WT) or GFP-CCL5(TRKN–) with GAG in theGolgi. In these experiments, MCF-7 cells were transiently transfectedby constructs expressing GFP-CCL5(WT) or GFP-CCL5(TRKN–)and simultaneously with the Golgi marker α mannosidase IB, taggedby HA. The cells were stained with Texas Red–labeled LycopersiconEsculentum (Tomato) Lectin (Vector Laboratories, Burlingame, CA)and with rabbit antibodies against HA (Santa Cruz), followed by AlexaFluor 647 goat anti-rabbit IgG (H + L) highly cross-adsorbed anti-bodies (Invitrogen, Carlsbad, CA). Stained cells were imaged withZeiss LSM-510 confocal microscope and with spinning disk confocalmicroscope, carried out with Axiovert 200M microscope (Zeiss) cou-pled to a Yokogawa CSU-22 spinning disk confocal head (Yokogawa,Sugar Land, TX). Slidebook software was used for determination ofcolocalization of GFP-CCL5(WT) or GFP-CCL5(TRKN–) withGAG in the Golgi.In additional experiments, HA-CCL5(WT) was detected in the
cells, stained by antibodies against HA (as above), and then withDyLight 549–conjugated AffiniPure goat anti-rabbit IgG (JacksonImmunoresearch Laboratories). Analysis was performed with ZeissLSM-510 confocal microscopy, as above.
Live Cell ImagingMCF-7 cells were transiently transfected by vectors expressing GFP-
CCL5(WT) or GFP-CCL5(TRKN–) and, 48 hours later, were imagedby live cell spinning disk confocal microscopy. Two-dimensional time-lapse series were acquired with a 3-second interval. To visualize thepaths taken by moving vesicles containing GFP-CCL5(WT), imageswere projected in two dimensions using the maximum-value-per-pixelalgorithm of Slidebook. The two dimensional algorithm was pseudo-colored in red and expanded through the entire time-lapse series.
Results
Vesicles Containing CCL5 Are Shuttled from the ER tothe Post-Golgi Stage, Leading to CCL5 Secretion byBreast Tumor CellsIn the present study, we wished to identify CCL5 domains and intra-
cellular components regulating the secretion of the chemokine by breasttumor cells and to detect intracellular regulatory determinants of CCL5secretion. To this end, we have expressed in the tumor cells GFP-taggedhuman wild-type (WT) CCL5, namely GFP-CCL5(WT). At first, wewished to guarantee that the GFP-tagged CCL5 acts in similar mannersto those characterized for endogenous CCL5 that is constitutively pro-duced by the same tumor cells [27] and to further extend our understand-ing of basic mechanisms involved in CCL5 secretion by the tumor cells.Being a secretory protein expressing a signal peptide, we have shown
in our published study that endogenous CCL5 is organized in vesiclesin MCF-7 breast tumor cells and is secreted by breast tumor cells in anER-to-Golgi–dependent process [27]. In the present study, we showsimilar characteristics for the GFP-tagged CCL5 that we have produced,
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 3
analyzed in MCF-7 cells (Figures 1 and 2). The results in Figure 1Ademonstrate that the GFP-tagged chemokine had a pronounced vesicularorganization, with high propensity to Golgi localization, as was con-firmed in colocalization analyses described below. This pattern of intra-cellular localization also applied to CCL5 labeled by a smaller tag, whenwe used HA-tagged CCL5(WT), showing vesicular organization anddistribution typical of Golgi (Figure W1).Using the GFP-tagged CCL5 in our further analyses, we found that
CCL5-containing vesicles had a very dynamic motility in the cells(Video W1). As expected, owing to the transfection with GFP-CCL5(WT) (Figure 1B1), high levels of the chemokine were detected in thesupernatants of the cells (Figure 1B2). The transfection of the cells byGFP-tagged empty vector (named GFP) allowed us to determine thebackground levels of endogenous CCL5 released by the cells. In mostanalyses included in the study (see below), it was difficult to detectthe endogenous chemokine after transfection of the cells by GFP (asis the case in Figure 1B2). Please note that the MCF-7 cells used in this
analysis, before their transfection by vector expressing GFP-CCL5(WT), release endogenous CCL5 [24,27,31], but in lower levels thanthe nanogram amounts released after the expression of the transfectedWT chemokine. Therefore, the endogenous CCL5 is hardly detectedunder the current experimental conditions, designed to provide reliabledetection curves of high nanogram CCL5 levels.After these analyses that have verified the secretion of the GFP-
tagged CCL5, we asked if the chemokine is released by the tumor cellsin an ER-to-Golgi–dependent process, as was shown to be the case inour previous study of endogenous CCL5 in breast tumor cells. In thatpublished investigation [27], we have shown that BFA, a drug thatinduces the collapse of the Golgi apparatus and blocks the transportof proteins from the ER to the trans-Golgi network [37,38], has led toa pronounced inhibition of secretion of endogenous CCL5 by thetumor cells. In line with those findings on the endogenous chemokine,when GFP-CCL5(WT)–transfected tumor cells were treated withBFA (2-5 hours; see Materials and Methods), GFP-CCL5(WT)
Figure 1. CCL5 is organized in vesicles and is secreted by breast tumor cells. Human MCF-7 breast tumor cells were transiently trans-fected by a vector expressing GFP-CCL5(WT) or by a control vector expressing GFP only (=GFP). (A) GFP-CCL5(WT) acquires a vesiculardistribution in the tumor cells, as determined by confocal analysis (similar localization pattern was observed for HA-tagged WT CCL5, asshown in Figure W1). This vesicular distribution is similar to that of endogenous CCL5 produced by the cells [27]. Live cell imaging ofmotility of GFP-CCL5(WT)–containing vesicles is demonstrated in Video W1. The control empty vector expressing GFP had a diffusenonorganized distribution in the cells (data not shown). (B) Determination of transfection yields and of CCL5 secretion by MCF-7 cellstransfected with the GFP-CCL5(WT) vector and by the control GFP vector. (B1) Transfection yields based on GFP expression in FACSanalysis. (B2) CCL5 secretion to the cell supernatants, determined by ELISA assays with antibodies against human CCL5, as described inprocedure 1 in Materials and Methods. In each part of the figure, the results are representatives of at least n = 3.
4 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
assumed a reticulate pattern with an enhanced localization to thenuclear membrane, typical of ER localization (Figure 2A1), and almostcomplete inhibition of CCL5 secretion was obtained (Figure 2A2).This observation indicated that the secretion of the GFP-taggedCCL5 that we have produced, similarly to that of the endogenousCCL5 [27], requires ER-to-Golgi trafficking.Also, in our published study, we have shown that endogenously ex-
pressed CCL5 is secreted from premade vesicles, localized to the cellperiphery [27]. To confirm the presence of such a secretion-readyvesicle population and to extend our understanding of the mechanismsinvolved in CCL5 secretion, we used a relatively short BFA treatment(20 minutes) designed to induce the collapse of the Golgi withoutaffecting the vesicle population that has already reached the post-Golgistage close to the cell periphery. Video W2, provided as static picturesin Figure 2, B1 and B2, shows by live cell imaging that despite thecollapse of the Golgi, CCL5 was still localized to motile vesicles, inaccord with its localization to a post-Golgi secretory compartmentadjacent to the cell membrane.
Overall, the above findings validate the suitability of the GFP-taggedCCL5 for further research and are in line with our published results[27]. In addition, these results provide additional insights into basicevents taking place in the course of CCL5 secretion, showing thatCCL5-containing vesicles mobilize in a dynamic manner from theER to the post-Golgi stage and give rise to a productive process ofCCL5 secretion to the cell exterior.
Vesicles Containing CCL5 Move on Structured MicrotubuleTracks, and the Release of the Chemokine Is Regulated bythe Rigidity of the Actin CytoskeletonThe observations obtained in the previous part of the study demon-
strated a dynamic behavior of vesicles containing GFP-CCL5(WT),along well-structured paths in the cells (Video W1). To envision thetracks undertaken by the vesicles and to understand if the samepaths were reused over time, we projected and pseudocolored in redthe maximum intensity of the GFP signal of individual frames andsuperimposed this signal on the original time-lapse series. Video W3,
Figure 2. Vesicles containing CCL5 are shuttled from the ER to the post-Golgi stage. The effects of treatment by BFA on the intracellularorganization of vesicles containing GFP-CCL5(WT) and on CCL5 secretion. Control cells were treated by the solubilizer of the drug. (A)Human MCF-7 breast tumor cells were transiently transfected by a vector expressing GFP-CCL5(WT), and the pool of cells was split tocells treated by BFA or to control cells treated by the solubilizer of the drug. The drug did not affect cell viability (data not shown). (A1)Confocal analysis showing cells treated by BFA for 2 hours. Before BFA addition, the localization of GFP-CCL5(WT) was as in Figure 1A(vesicular and punctuate; data not shown). The picture is a representative of multiple cells analyzed in n = 2. (B) ELISA analysis of CCL5amounts in supernatants of cells untreated or treated by BFA (for 2-5 hours, as described in Materials and Methods). The results aresimilar to those obtained for the endogenous CCL5 produced by breast tumor cells, shown to be mobilized toward secretion in an ER-to-Golgi–dependent manner [27]. CCL5 secretion to the cell supernatants was determined by ELISA assays with antibodies against humanCCL5, as described in procedure 1 in Materials and Methods. (B) The effect of short-term treatment by BFA (20 minutes) on motility ofCCL5-containing vesicles. Human MCF-7 cells were transiently transfected by GFP-CCL5(WT), and the motility of CCL5-containingvesicles was determined by live cell imaging in spinning disk confocal microscope after the treatment by BFA. The figure provides staticpictures of Video W2. (B1) At the beginning of the BFA treatment, prominent localization of CCL5 was detected in the Golgi, and thechemokine was also found in peripheral vesicles. (B2) At advanced stages after this short treatment by BFA, there was almost an entirecollapse of the Golgi, CCL5 was minimally detected in the Golgi, but it was still vastly localized in peripheral vesicles. The pictures arerepresentatives of multiple cells analyzed in n ≥ 2.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 5
provided as a static picture in Figure 3A, clearly shows that the vesiclescontaining GFP-CCL5(WT) (yellow, owing to the superimposition ofthe red signal of the tracks and the green signal of GFP-CCL5(WT))traffic along structured, defined, and extended routes, which stretchthroughout the cells (shown in red).Because vesicles containing GFP-CCL5(WT) had apparent
directional motility and they reused defined paths, we tested the
possibility that vesicles carrying the chemokine move along cyto-skeleton filaments. Depolymerization of microtubules by nocoda-zole [39] reduced considerably the motility of CCL5-containingvesicles, and decelerated their movement along well-defined paths(Video W4: before nocodazole treatment; Video W5: after nocodazoletreatment). In parallel, depolymerization of the microtubules sig-nificantly inhibited CCL5 secretion by the tumor cells (Figure 3B),
Figure 3. The trafficking and secretion of CCL5 are regulated by cytoskeleton elements. The characteristics of CCL5 trafficking and secretionwere determined in human MCF-7 breast tumor cells, transiently transfected by a vector expressing GFP-CCL5(WT). (A) GFP-CCL5(WT)–containing vesicles traffic on structured cellular tracks. The cells were imaged by live cell imaging in confocal microscopy. The figure showsa static picture of Video W3. To visualize the paths taken by moving GFP-CCL5(WT)–containing vesicles, images were projected in twodimensions using the maximum-value-per-pixel algorithm of Slidebook. The two-dimensonal algorithm was pseudocolored in red andexpended through the entire time-lapse series. The tracks used by GFP-CCL5(WT)–containing vesicles are demonstrated in red, and thevesicles containing GFP-CCL5(WT) (green) are shown in yellow, owing to the superimposition of red and green signals. The pictures arerepresentatives ofmultiple cells analyzed in n=2. (B, C, D) The roles ofmicrotubules (B) and of actin filaments (C, D) in regulating themotilityof CCL5-containing vesicles and the secretion of CCL5were determined in MCF-7 cells. After transient transfection with a vector expressingGFP-CCL5(WT), the pool of cells was split to cells treated with (B) nocodazole (microtubule depolymerizing), (C) latrunculin (actin depoly-merizing), or (D) jasplakinolide (actin polymerizing) and to control cells that were treated by the solubilizers of the drugs. The drugs did notaffect cell viability (data not shown). (B) CCL5 secretion after treatment by nocodazole was determined as indicated below. In parallel,the motility of vesicles containing GFP-CCL5(WT) was followed by live cell imaging in confocal microscopy, without (Video W4) and follow-ing nocodazole treatment (Video W5). (C) The effects of latrunculin on the organization of actin filaments were determined by staining con-trol cells (C1) or latrunculin-treated cells (C2) with phalloidin. (C3) CCL5 secretion after latrunculin treatment was determined as indicatedbelow. (D) The effects of jasplakinolide on the shape and contour of the cells were determined by light microscope in control cells (D1) and injasplakinolide-treated cells (D2). (D3) CCL5 secretion after jasplakinolide treatment was determined as indicated below. (B, C3, D3) CCL5secretion to the cell supernatants was determined by ELISA analyses with antibodies against human CCL5, as described in procedure 1 inthe Materials and Methods. In all parts of the figure, the ELISA analyses are of a representative experiment of n = 3, and the pictures arerepresentatives of multiple cells analyzed in n = 3 (except for the live cell imaging in B, where n = 2).
6 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
being in line with the above findings showing that the inclusion ofCCL5 into motile vesicles is important for its secretion (Video W1and Figure 2).In addition, we have analyzed the roles of the actin cytoskeleton in
regulating the secretion of CCL5 by the tumor cells. After depolymeri-zation of the actin filaments by latrunculin [40] (Figure 3, C1 and C2),the secretion of CCL5 was promoted (Figure 3C3), suggesting that themembrane-proximal actin cortex acts as a partial barrier that preventsmaximal vesicle fusion and secretion of the chemokine. This possibilitywas supported by taking the opposite approach, in which the cells weretreated by the actin polymerizing agent jasplakinolide [41]. This drughas led to change in cell shape and contour that are consistent withincreased rigidity of the actin cortex (Figure 3, D1 and D2) and inhib-ited the secretion of CCL5 by the tumor cells (Figure 3D3).Together, the above observations indicate that microtubules serve
as structured tracks along which the chemokine-containing vesiclesshuttle toward secretion, whereas the actin cytoskeleton controlsthe extent of chemokine release to the extracellular milieu of the cells.
The 43TRKN46 Sequence of CCL5 Is Essential for Its Secretionby Breast Tumor Cells and for Its Inclusion in Motile VesiclesNext, we asked what are the chemokine motifs that are essential for
its release by breast tumor cells and chose to focus on the 43TRKN46
sequence located in the 40s loop of CCL5. The rationale for focusingon this motif was based on the following two observations: 1) A recentstudy by El Golli et al. [42] has shown that the 45LKNG48 sequenceof CXCL4 facilitates the targeting of this chemokine to granules inplatelets. That research has suggested that the 45LKNG48 sequenceof CXCL4 exhibits the same surface-exposed hydrophilic turn/loopfeatures as the 43TRKN46 sequence of CCL5, thus motivating us toask whether the 43TRKN46 sequence of CCL5 regulates its secretionby breast tumor cells. 2) The 43TRKN46 sequence is found in theexposed 40s loop of CCL5, known to be important for CCL5 bindingto GAG and to the CCL5 receptors CCR1 and CCR3 [33,43,44].Therefore, we speculated that such a region may mediate the inter-actions of the chemokine with intracellular components that shuttleit toward secretion.On the basis of the above, we asked if the 43TRKN46 motif of CCL5
is required for the secretion of the chemokine by breast tumor cells.To this end, we generated a CCL5(TRKN–) variant in which the43TRKN46 motif was mutated to alanines. To guarantee that theTRKN-mutated chemokine is correctly folded, we have performedthe following two analyses: 1) We determined the predicted three-dimensional structure of the TRKN-mutated chemokine by super-imposing it on the x-ray structure of WT CCL5 [33]. This analysishas shown that the TRKN-mutated CCL5 is correctly folded (Fig-ure 4A), and this conclusion is supported by past studies showing thatCCL5 mutated at 44RKNR47 (a sequence shifted one amino acid fromour 43TRKN46 motif ) had similar x-ray characteristics to WT CCL5[33]. 2) Further below in the article, we describe studies determiningthe intracellular localization of GFP-CCL5(WT) and GFP-CCL5(TRKN–). In those experiments (see below), we found that the mu-tated CCL5 exited from the ER to the Golgi, a process that couldnot have taken place if the mutated chemokine was misfolded owingto ER-associated protein degradation (ERAD) that ensures that onlyproperly folded and assembled proteins proceed to the Golgi for furtherprocessing and secretion [45–47].We now asked if the vectors expressing GFP-CCL5(WT) and GFP-
CCL5(TRKN–) generate proteins at the correct molecular weights
(MWs). Western blot analyses performed on cell lysates of cells trans-fected with GFP-CCL5(WT) and with GFP-CCL5(TRKN–) haveshown that the two proteins had a similar MW, and they both assumedthe expected MW of 35 kDa, as expected for CCL5 tagged by GFP(CCL5, ∼8 kDa; GFP, ∼27 kDa) (Figure 4B).After these analyses, we have determined the secretion of GFP-CCL5
(WT) and GFP-CCL5(TRKN–) by the tumor cells. Figure 4C showsthat the transfection yields of GFP-CCL5(TRKN–) were very similarto those of GFP-CCL5(WT) in the tumor cells. However, whereasthe WT chemokine was highly secreted and was detected in high levelsin the cell supernatants, very substantial inhibition of secretion wasobtained for the mutated GFP-CCL5(TRKN–) (Figure 4, D1 andD2). This was indicated by ELISAs performed with antibodies againstCCL5 (Figure 4D1) and with antibodies against the GFP tag(Figure 4D2). Very prominent reduction in the secretion of the GFP-CCL5(TRKN–) was obtained also by two additional ELISA analysesperformed with other combinations of antibodies (Figure W2).In line with the perturbed secretion of the mutated GFP-
CCL5(TRKN–) by the tumor cells, we found that the mutated chemo-kine had a diffuse/reticulate distribution, with no definite vesicularorganization (Figures 5 and 6). Live cell imaging experiments have shownthat the motility of the mutated chemokine was limited, random, andnondirectional (Video W6), in contrast to the dynamic and directionalmotility of vesicles containing GFP-CCL5(WT) (Video W1). Thesefindings, showing lack of vesicular localization ofGFP-CCL5(TRKN–),support fully the prominent reduction observed in secretion of themutated chemokine by the tumor cells.
The TRKN-Mutated Chemokine Reaches the Golgi, butTraffics along the ER-to-Post-Golgi Route in a DifferentManner than the WT ChemokineThe above results indicate that the 43TRKN46 motif of CCL5 is
essential for the vesicular organization and for the secretion of thechemokine by breast tumor cells. To provide further insights tothe mechanisms responsible for reduced secretion of the TRKN-mutated CCL5, we have determined its intracellular localizationalong the ER-to-Golgi trafficking process, relative to the WT che-mokine. To this end, we studied the colocalization of the WTCCL5 or mutated CCL5 chemokines with markers of the ER(calnexin) and of the Golgi (α mannosidase IB). The extent ofcolocalization was also determined quantitatively, as described inMaterials and Methods.The results in Figures 5 and 6 indicate that the GFP-CCL5
(TRKN–) exited from the ER to the Golgi, however, with modifiedproportions compared to the WT chemokine. The TRKN-mutatedchemokine had higher propensity for ER localization than the WTchemokine (Figure 5), and in parallel, its expression in the Golgi wassomewhat reduced (Figure 6). These results indicate that: 1) The43TRKN46 motif regulates the trafficking of CCL5 at two points alongthe mobilization process, where the first stage takes place at the exitfrom the ER to the trans-Golgi network and the second is in traffickingof CCL5, in motile vesicles at the post-Golgi stage, a stage that isrequired for completion of secretion. These findings agree well withthe reduced secretion of the TRKN-mutated CCL5 by the tumor cells.2) GFP-CCL5(TRKN–) exited from the ER and reached the Golgi,indicating that it is correctly folded because its inappropriate foldingwould have led to activation of the ERAD process [45–47] that wouldhave completely prevented its exit from the ER.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 7
The 43TRKN46 Sequence Is a Ubiquitous Motif Required forSecretion of CCL5 in Many Different Cell TypesIn our search for the mechanisms that may be involved in the
43TRKN46-mediated process of CCL5 secretion, we first asked if thedependence on the 43TRKN46 motif is a general phenomenon shared
by many cell types or whether it is specific to breast tumor cells in gen-eral, or to MCF-7 cells in particular. To answer this question, we haveexpressed GFP-CCL5(WT) and GFP-CCL5(TRKN–), which werecompared with control GFP vector, in several cell types. In each ofthe cell types, we validated that the expression levels of GFP-CCL5
Figure 4. The 43TRKN46 motif of CCL5 is essential for its secretion by breast tumor cells. (A) The predicted three-dimensional structureof 43TRKN46-mutated CCL5, superimposed on the three-dimensional structure of WT CCL5 obtained by x-ray analyses [33]. Of note,this article also showed similar x-ray structures for WT CCL5 and the 44RKNR47 CCL5 mutant. (B) Human MCF-7 breast tumor cellswere transiently transfected by vectors expressing GFP-CCL5(WT), GFP-CCL5(TRKN–), or GFP alone (GFP), followed by determinationof chemokine expression in cell lysates. The chemokines were immunoprecipitated by mAb against GFP, and Western blot analysis wasperformed with mAb against GFP. The results are of a representative experiment of n > 3. (C, D) The effects of the 43TRKN46 mutationon CCL5 secretion. Human MCF-7 breast tumor cells were transiently transfected by vectors expressing GFP-CCL5(WT) or GFP-CCL5(TRKN–), followed by determination of chemokine secretion. (C) FACS analyses showing the transfection yields of vectors expressingGFP-CCL5(WT) or GFP-CCL5(TRKN–). (D) Determination of CCL5 secretion to cell supernatants, performed by ELISA assays. (D1) ELISAassays with antibodies against human CCL5, as described in procedure 1 in Materials and Methods. (D2) ELISA assays with antibodiesagainst human GFP, as described in procedure 2 in Materials and Methods. The results in all parts of the figure are of a representativeexperiment of n > 3. Additional ELISA analyses with other combinations of antibodies, showing the reduced secretion of GFP-CCL5(TRKN–), are provided in Figure W2.
8 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
(WT) were similar to those of GFP-CCL5(TRKN–) (Figure 7, A1-D1)and determined the release of the WT or the mutated CCL5 by the cells(Figure 7, A2-D2; please note that the levels of chemokines are presentedin different scales in the various panels of the figure).We began this analysis by studying the T47D human breast tumor
cells that, like MCF-7 cells are tumorigenic but not metastatic, and theMDA-MB-231 cells that are highly metastatic human breast tumorcells [35]. In both cell types, the patterns of CCL5 release were similarto those of MCF-7 cells: GFP-CCL5(WT) was highly secreted, whereasthe release of mutated CCL5 was prominently inhibited by the muta-tion of the 43TRKN46 motif (Figure 7, A2 and B2). This has indicatedthat the 43TRKN46 motif is involved in CCL5 trafficking in breasttumor cells, independently of their metastatic potential.We then asked if the same is correct for nontransformed human cells
of the breast (HMEC) and for normal human lung fibroblasts (WI-38)(Figure 7, C2 and D2). We found that also in these two cell types, the
secretion of the mutated GFP-CCL5(TRKN–) chemokine was muchreduced when compared with the release of GFP-CCL5(WT) bythe cells. In general, reduced secretion of GFP-CCL5(TRKN–) wasalso detected in the MCF-10A nontransformed breast epithelial cells(data are not presented because, in several of the experiments, thetransfection procedure has led to very high release of endogenousCCL5 by the cells, making the expression of GFP-CCL5(WT) ineffi-cient. In these specific cases, it was difficult to interpret the exact patternof secretion of the mutated chemokine and to correctly appreciate whatseemed to be a dominant negative effect induced by GFP-CCL5(TRKN–)).Taken together, the above results indicate that the 43TRKN46-
dependent mechanism of CCL5 secretion is shared by many cell typesand is probably ubiquitous. These observations have led us to searchfor a general mechanism, by which the 43TRKN46 motif is requiredfor the secretion of CCL5.
Figure 5. GFP-CCL5(TRKN–) is found in the ER in higher propensity than GFP-CCL5(WT). Confocal pictures showing the localization pat-tern of GFP-CCL5(WT) and of GFP-CCL5(TRKN–) with the ER marker calnexin. (A) Human MCF-7 breast tumor cells were transiently trans-fected by vectors expressing GFP-CCL5(WT) or GFP-CCL5(TRKN–). The colocalization of CCL5 (green) with an ER marker (calnexin, red)was determined by confocal analysis and shown in orange/yellow. The pictures also show that, in contrast to the vesicular organization ofGFP-CCL5(WT), the GFP-CCL5(TRKN–) had a diffuse/reticulate organization. The nondirectional and limited motility of GFP-CCL5(TRKN–)in the tumor cells is shown in Video W6. The pictures are representatives of multiple cells analyzed in n > 3. (B) Quantitative analysis ofthe colocalization of the mutated and GFP-CCL5(WT) molecules with calnexin, performed on a large number of cells. The graph showsthe mean ± SD of the normalized values obtained in n = 3. P values were obtained from actual values of the computational analysisbefore normalization.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 9
The Process of CCL5 Secretion Is Partly Regulated by GAGIn search for a general mechanism which leads to CCL5 secretion
and is shared by many cell types, we investigated the possibility thatCCL5 interacts with intracellular GAG on its trafficking route towardrelease from the cells. This approach was based on published studiesshowing that GAG-decorated proteoglycans participate in the packag-ing of positively charged proteins in cytotoxic lymphocytes—such asgranzymes and perforin [48,49]—and that supernatants of activatedhuman immunodeficiency virus 1–specific cytotoxic T cells includeCC chemokines complexed with proteoglycans [50]. The latter studydid not address the involvement of GAG in the intracellular mobiliza-tion of the chemokines toward secretion, but it motivated us to de-termine the possibility that GAG regulate the intracellular traffickingand release of the CC chemokine CCL5 in breast tumor cells. Thispossibility was reinforced by the fact that the CCL5-secretory motifof 43TRKN46 is only one amino acid shifted from the 44RKNR47 se-quence of CCL5, known to be essential for CCL5 binding to GAG
[33,43,44]. Importantly, both 43TRKN46 and 44RKNR47 express posi-tively charged amino acids that are part of the sequence required forGAG binding [33,43,44]. Therefore, if indeed intracellular GAG arerequired for CCL5 secretion, it is possible that they interact with the43TRKN46 motif of CCL5, which is essential for the release of thischemokine by the tumor cells.On the basis of the above, our primary goal in this part of the study
was to determine whether the secretion of CCL5 by breast tumorcells was regulated by intracellular GAG. To achieve this aim, we tookseveral approaches in which we determined the effects of GAG inhibi-tion on the secretion of WT CCL5. In the framework of this study,we have also addressed indirectly the possibility that the 43TRKN46
motif associates with such intracellular GAG. This point was notaddressed by direct measures because of two restrictions: 1) We foundthat, because of technical reasons, it is impossible to isolate fromthe transfected cells the actual molecules of GFP-CCL5(WT) and ofGFP-CCL5(TRKN–) in a manner that will enable determination of their
Figure 6. GFP-CCL5(TRKN–) reaches the Golgi apparatus but in a lower propensity than GFP-CCL5(WT). Confocal pictures showing thelocalization pattern of GFP-CCL5(WT) and of GFP-CCL5(TRKN–) with the Golgi marker αmannosidase IB. (A) Human MCF-7 breast tumorcells were transiently transfected by vectors expressing GFP-CCL5(WT) or GFP-CCL5(TRKN–). The colocalization of CCL5 (green) with aGolgi marker (α mannosidase IB, red) was determined by confocal analysis and is shown in orange/yellow. The pictures also show that,in contrast to the vesicular organization of GFP-CCL5(WT), the GFP-CCL5(TRKN–) had a diffuse/reticulate organization. The nondirectionaland limited motility of GFP-CCL5(TRKN–) in the tumor cells is shown in Video W6. The pictures are representatives of multiple cellsanalyzed in n > 3. (B) Quantitative analysis of the colocalization of the mutated and GFP-CCL5(WT) molecules with α mannosidase IB,performed on a large number of cells. The graph shows the mean ± SD of the normalized values obtained in n = 3. P values wereobtained from actual values of the computational analysis before normalization.
10 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
binding to GAG on solid surfaces (plates or beads). 2) The approach ofCCL5 immunoprecipitation with GAGwas expected to be problematicowing to the weak nature of the chemical bonds between them.Below we provide data on the analyses that we have performed to
address the roles of intracellular GAG in CCL5 secretion and themeasures taken to address the possible involvement of the CCL5
43TRKN46 sequence in such a GAG-mediated process. The findingsprovided below indicate that intracellular GAG are indeed involvedin CCL5 secretion. In parallel, our findings are suggestive of a processin which the 43TRKN46 motif of CCL5 participates in a mechanismmediated by GAG, and this possibility is supported by additionalfindings in the literature, as detailed in the Discussion section.
Figure 7. The 43TRKN46 sequence is required for CCL5 secretion in many cell types. Different cell types were transiently transfected byvectors expressing GFP-CCL5(WT), GFP-CCL5(TRKN–), or GFP alone (GFP) (please note the different scales used for the various cell typesin the ELISA analyses presented). (A1-D1) FACS analyses showing the transfection yields of GFP-CCL5(WT) and GFP-CCL5(TRKN–) in thedifferent cell types. (A2-D2) The secretion of CCL5 was determined by ELISA assays, performed on cell supernatants of the different celltypes, with antibodies against human CCL5, as described in procedure 1 in the Materials and Methods. (A1, A2) Human T47D nonaggres-sive breast carcinoma cells. (B1, B2) Human MDA-MB-231 metastatic breast carcinoma cells. (C1, C2) Human mammary normal epithelialcells. (D1, D2) Human WI-38 normal lung fibroblasts. In all parts of the figure, the results are of a representative experiment of n = 3.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 11
First, we asked if the TRKN-mutated CCL5 diverges from the WTchemokine in its ability to associate with GAG in the Golgi. Thisapproach was based on our findings showing that the secretion ofWTCCL5 requires positioning of the chemokine in theGolgi (Figure 2and [27]) and that, in the Golgi, saccharides are appended to proteincores by glycosyltransferases, leading to formation of proteoglycans
[51]. By performing triple-dye analyses, we have shown that GFP-CCL5(WT) was highly colocalized with GAG in the Golgi (Figure 8),whereas the mutated GFP-CCL5(TRKN–) had a considerably re-duced colocalization with GAG in the Golgi (Figure 9). These datashow that CCL5 is highly localized in secretion-related organelles(Golgi) that are enriched with GAG and that this localization is
Figure 8. The localization of GFP-CCL5(WT) with GAG in the Golgi. Human MCF-7 breast tumor cells were transiently transfected by avector expressing GFP-CCL5(WT). The colocalization of CCL5 (green) with GAG (red), and with a Golgi marker (αmannosidase IB, purple/blue) was determined by confocal analysis. (A) The pictures show each of the proteins alone, as well as combinations of the following:GFP-CCL5(WT) + GAG, GFP-CCL5(WT) + Golgi, or GFP-CCL5(WT) + GAG + Golgi. The colocalization of GFP CCL5(WT) + GAG + Golgiis demonstrated in white. (B) Higher magnification of the colocalization of GFP-CCL5(WT) with GAG and Golgi, demonstrated in brightwhite. The percentage of GFP-CCL5(WT) that was colocalized with GAG and Golgi is indicated in the figure. The pictures are represen-tatives of multiple cells analyzed in n = 3.
12 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
associated with the expression of the positively charged 43TRKN46
motif of CCL5.Next, we asked if GAG are required for CCL5 secretion, and to
what extent. To address this issue, we have used a highly specificapproach, in which we measured the degree of GFP-CCL5(WT)
secretion by mutated CHO cells that have substantially reduced levelsof GAG synthesis due to deficiency in xylosyltransferase, which ini-tiates GAG biosynthesis [52]. The secretion of GFP-CCL5(WT) bythese “CHO-deficient GAG” cells (original name CHO-pgsA-745cells) was compared to “CHO-GAG+++” cells that expressed normal
Figure 9. The localization of GFP-CCL5(TRKN–) with GAG in the Golgi. Human MCF-7 breast tumor cells were transiently transfected by avector expressing GFP-CCL5(TRKN–). The colocalization of CCL5 (green) with GAG (red), and with a Golgi marker (α mannosidase IB,purple/blue) was determined by confocal analysis. (A) The pictures show each of the proteins alone, as well as combinations of thefollowing: GFP-CCL5(TRKN–) + GAG, GFP-CCL5(TRKN–) + Golgi, or GFP-CCL5(TRKN–) + GAG + Golgi. The colocalization of GFP-CCL5(TRKN–) + GAG + Golgi is demonstrated in white. (B) Higher magnification of the colocalization of GFP-CCL5(TRKN–) with GAG andGolgi, demonstrated in bright white. The percentage of GFP-CCL5(TRKN–) that was colocalized with GAG and Golgi is indicated in thefigure. The pictures are representatives of multiple cells analyzed in n = 3.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 13
Figure 10. The secretion of GFP-CCL5(WT) and its vesicular organization are perturbed in CHO-deficient GAG cells. CHO cells were trans-fected by vectors expressing GFP-CCL5(WT), GFP-CCL5(TRKN–), or GFP alone (GFP), followed by determination of secretion and intracellularorganization of CCL5. The analyses were performed in cells that expressed normal GAG levels, termed herein CHO-GAG+++ cells (=CHO-K1 cells), compared to CHO cells deficient in GAG expression, termed herein CHO-deficient GAG cells (=CHO-pgsA-745 cells). (A1, A2)FACS analyses showing the transfection yields of GFP-CCL5(WT) and GFP-CCL5(TRKN–) in CHO-GAG+++ cells and in CHO-deficientGAG cells. The results are of a representative experiment of n= 2-3. (B) CCL5 secretion, determined by ELISA assays performed on super-natants of the different cell types, with antibodies against human CCL5, as described in procedure 1 in Materials and Methods. (B1) Secre-tion of WT CCL5 in CHO-GAG+++ cells, transfected with vectors expressing GFP-CCL5(WT), GFP-CCL5(TRKN–), or GFP vector only(=GFP). The results are of a representative experiment of n = 3. (B2) Secretion of CCL5 by CHO-GAG+++ cells and by CHO-deficientGAG cells, transfected with GFP-CCL5(WT). The results are mean ± SD of normalized values of CCL5 secretion in n = 3. P values wereobtained from actual values of the computational analysis before normalization. (C, D) Intracellular localization of GFP-CCL5(WT) (C1, D1) andof GFP-CCL5(TRKN–) (C2, D2) in CHO-GAG+++ cells (C1, C2) and in CHO-deficient GAG cells (D1, D2). In C and D, the results are of arepresentative experiment of n = 2, with multiple cells analyzed in each experiment.
14 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
GAG levels (original name CHO-K1 cells). Here, it is important toindicate that 1) we ensured that the CHO-deficient GAG cells indeeddid not express GAG (data not shown) and that 2) the CHO-deficientGAG cells do not have a general defect in secretion [53]; therefore,they were valid for our studies.This analysis was begun by validating that similarly to MCF-7 cells,
CHO cells that express normal levels of GAG mobilize CCL5 throughthe 43TRKN46 motif. Indeed, Figure 10, A and B1, shows that, despitesimilar transfection yields of GFP-CCL5(WT) and GFP-CCL5(TRKN–) in the CHO-GAG+++ cells, the mutated GFP-CCL5(TRKN–) chemokine was not secreted by the cells. Furthermore, themutated chemokine assumed a reticulate intracellular localization inthe CHO-GAG+++ cells (Figure 10C), as was the case in MCF-7 cells(Figures 5 and 6).Then, we compared the release of GFP-CCL5(WT) in CHO-
GAG+++ cells to its secretion by CHO-deficient GAG cells. The re-sults in Figure 10B2 show that the control CHO-GAG+++ cellsreleased high levels of GFP-CCL5(WT); however, the secretion ofGFP-CCL5(WT) by CHO-deficient GAG cells was prominentlyimpaired, although only partly. The CHO-deficient GAG cells alsofailed to release the mutated CCL5, as was the case in the other celllines that were investigated (data not shown).Further analyses that were performed in the CHO-deficient GAG
cells have indicated that GAG are important not only for the secretionof GFP-CCL5(WT) but also for its vesicular localization. Figure 10C1shows that in CHO-GAG+++ cells, GFP-CCL5(WT) was localized invesicles that had a very definite punctuate distribution. In contrast, inCHO-deficient GAG cells, the punctuate distribution of GFP-CCL5(WT) was perturbed (Figure 10D1), and the WT chemokine waslargely localized in the ER and acquired a phenotype which was in gen-eral similar to that of the TRKN-mutated CCL5, which was expressedin normal, GAG-expressing cells (Figures 10C2, 5, and 6).The roles of GAG in regulating the secretion of GFP-CCL5(WT)
was further supported by three additional methods taken to inhibitGAG presence or synthesis in the cells. These experiments were per-formed in the original MCF-7 breast tumor cells, in which the secre-tion of the GFP-CCL5(WT) was analyzed with and without differentGAG inhibitory measures. We did not analyze the effects of thesemeasures on the mutated chemokine because it was not secreted bythe MCF-7 cells, as shown in Figure 4. In all three types of analysis,we have ensured similar expression of the GFP-CCL5(WT) in cellsnot treated, compared with cells treated by the different inhibitorymeasures (see the relevant figures, as indicated herein).The following three methods were applied to inhibit GAG func-
tions in the tumor cells: 1) We have expressed in breast tumor cellsheparanase (Figure 11), an enzyme that degrades heparane sulfate,which is one of the GAG that bind CCL5 [44,54–56]. The resultsin Figure 11A show that heparanase was expressed intracellularly inthe tumor cells, at the expected MW for the intracellularly expressedenzyme, tagged by myc (∼65 kDa of heparanase [57] + the myctag). In cells expressing the enzyme intracellularly, the secretion ofGFP-CCL5(WT) was significantly inhibited, compared to control cellstransfected by vector only. However, the reduction in CCL5 secretionafter expression of heparanase was only partial (Figure 11, B and C ). 2)In view of the importance of GAG sulfation for binding of chemokines,including of CCL5 [33,58–60], we treated the cells with sodium chlo-rate, a competitive inhibitor of ATP-sulfurylase that inhibits the sulfa-tion process of GAG and is used routinely as a measure for reduction ofGAG sulfation, including in aspects related to chemokine activities
[61–63]. This treatment has also led to significant, but partial, reduc-tion in secretion of CCL5 by the tumor cells (Figure 12A). 3) We havedeprived sulfate out of the growth medium of the cells. As shown inFigure 12B, this measure has also led to significant inhibition ofGFP-CCL5(WT) secretion by the cells. Here again, the reduction
Figure 11. Intracellular expression of heparanase leads to reducedsecretion of GFP-CCL5(WT) by breast tumor cells. Human MCF-7breast tumor cells were transiently transfected by a vector express-ing GFP-CCL5(WT). In parallel, the cells were transfected with amyc-tagged vector expressing heparanase or by a control myc-tagged empty vector. (A) Western blot showing the expression ofheparanase in cells transfected with heparanase-containing vectorbut not in cells transfected with the control vector. The analysis wasperformed with antibodies againstmyc. The MW of the heparanaseis the one expected for the intracellularly expressed enzyme, taggedby myc (∼65 kDa of heparanase + the myc tag). No signal was de-tected in the control cells transfected with the myc-tagged vectoronly because of its small MW (in the conditions used for appropriatedetection of the heparanase, the myc protein run out of the gel).(B) FACS analysis showing the transfection yields of GFP-CCL5(WT)in cells transfected with the heparanase vector and those transfectedwith control vector. (C) CCL5 secretion to the cell supernatants de-termined by ELISA assays with antibodies against human CCL5, asdescribed in procedure 1 in Materials and Methods. In all parts ofthe figure, the results are of a representative experiment of n > 3.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 15
was partial. The combination of the sodium chlorate treatment + sulfatedeprivation did not yield additive inhibitory effects on the secretion ofGFP-CCL5(WT) by the tumor cells, and the inhibition level remainedpartial (data not shown). These results indicate that the involvement ofGAG activities has reached a saturation point, with respect to their rolesin regulating CCL5 secretion.To conclude, by taking four different inhibitory approaches, we
have shown that GAG play a significant and an important role inmobilizing GFP-CCL5(WT) toward secretion. However, theseGAG-inhibitory measures have led to only partial reduction in secre-tion of GFP-CCL5(WT), suggesting that other mechanisms regulateCCL5 release as well. Our results show that GFP-CCL5(TRKN–) hadreduced localization with GAG in the Golgi and that GFP-CCL5(WT) had an ER-like organization in GAG-deficient cells, which wassimilar to the organization of the TRKN-mutated chemokine in GAG-expressing cells. Together with supporting findings in the literature (seeDiscussion), it is possible that one of the mechanisms mediating CCL5secretion is a process involving 43TRKN46-GAG associations.
DiscussionThe results of this study provide novel information on the molecularmechanisms involved in the secretion of CCL5. They are the first toidentify a role for the 40s loop of CCL5 and for GAG in traffickingand secretion of this chemokine. These findings contribute to ourunderstanding of basic processes controlling the expression of pro-malignancy chemokines in tumor cells and may have potentialtherapeutic implications, as follows:
The 43TRKN 46 Motif Is Essential for CCL5 Secretion andfor Its Inclusion in Motile VesiclesThe results of this study indicate that the secretion of a TRKN-
mutated CCL5 by breast tumor cells is prominently reduced and thatthe 43TRKN46 sequence is a ubiquitous motif required for the secretionof the chemokine by many cell types. Additional analyses that we haveperformed (article in preparation) indicate that the TRKN sequencerequires the backbone of CCL5 to act as a secretion-determining motifbecause it did not support the secretion of a different chemokine when
Figure 12. The secretion of GFP-CCL5(WT) by breast tumor cells is inhibited by reduced sulfation of GAG. The effects of GAG undersulfationon the secretion of CCL5were determined in MCF-7 cells, transfected with GFP-CCL5(WT). (A) Treatment by sodium chlorate, a competitiveinhibitor of ATP-sulfurylase that inhibits the sulfation process of GAG (30mM, 48 hours). (B) The cells were exposed to sulfate deprivation bygrowth in sulfate-deficient medium (48 hours). (A1, B1) FACS analyses showing the transfection yields of GFP-CCL5(WT) in control cells andin cells in which undersulfation was induced by (A1) sodium chlorate or (B1) sulfate deprivation. (A2, B2) The secretion of CCL5 was deter-mined by ELISA assays, performed on supernatants of control cells and of cells in which undersulfation was induced, performed with anti-bodies against human CCL5, as described in procedure 1 in Materials and Methods. (A2) Sodium chlorate. (B2) Sulfate deprivation. In allparts of the figure, the results are of a representative experiment of n = 3.
16 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
it was positioned at its 40s domain (data not shown). The need forthe 40s domain of a chemokine for its secretion is supported by recentfindings showing that regions of the 40s loop regulate the sortingof the chemokine CXCL4 (45LKNG48) into α granules in plateletsand of CXCL8 (44DSG46) into Weibel-Palade bodies in endothelialcells [42,64].Our confocal analyses identified a defect in the intracellular organi-
zation and in the ability of the mutated chemokine to use the appro-priate routes on its way toward secretion. Whereas the WT chemokinewas mobilized from the ER to the post-Golgi stage and was organizedin vesicles that moved on definite microtubule tracks, the TRKN-mutated chemokine was loosely organized in diffuse vesicles that hadrandom, unstructured, and limited motility in the cells.Furthermore, our findings indicate that the 43TRKN46 motif of
CCL5 regulates the trafficking of the chemokine at two points of theER-to-post-Golgi path: Some of the molecules of the TRKN-mutatedchemokine could not find their way to the Golgi, and those thatdid reach the Golgi could not give rise to a productive release of thechemokine to the cell exterior. The fact that a considerable amountof GFP-CCL5(TRKN–) molecules did exit from the ER to the Golgiindicates that the folding of the mutated chemokine is correct; other-wise, the ERAD process [45–47] would have led to complete arrestof their exit from the ER.
Secretion of CCL5 Is Partly Regulated by aGAG-Dependent ProcessWe have shown by four different approaches that GAG are necessary
and important for CCL5 secretion. These approaches included threehighly specific methods: one based on GAG-mutated CHO cells, thesecond on the use of the enzyme heparanase that degrades heparanesulfate, and the third on perturbation of GAG sulfation by a com-petitive inhibitor of ATP-sulfurylase. The results of these studieswere reinforced by sulfate deprivation. Furthermore, our results withheparanase provided a partial clue to the identity of the GAG involvedin this process, suggesting that heparane sulfate participates in therelease of CCL5 by the cells.On the basis of the above, we propose that one of the mechanisms
mediating CCL5 secretion is based on the ability of CCL5 to “hitch-hike” on GAG, possibly on GAG-decorated proteoglycans, thatmake their way to the cell membrane or to the cell exterior. Support-ing this mechanism are two recent studies, one by Meen et al. [65]that suggested that a proteoglycan-mediated process was involved inthe secretion of the chemokine CXCL1 by endothelial cells and thesecond on FGF-2, showing that extracellular heparane sulfate proteo-glycans formed a molecular trap that translocated the protein acrossthe plasma membrane [66].It is interesting to note that all four measures that were taken in our
study to reduce GAG activities/expression were in good agreementwith each other, all yielding similar inhibition levels of CCL5 secre-tion, between 40% and 60%. It is possible that the presence orsynthesis of GAG was not completely shut down by any of thesemeasures; however, as indicated in the Results section, when sulfatedeprivation was combined with sodium chlorate, there was no additiveinhibitory effect, and the inhibition of CCL5 secretion remainedpartial (data not shown).Therefore, it is very likely that the partial reduction in CCL5 secre-
tion, which was obtained by the different measures of GAG inhibition,actually reflects the existence of alternative mechanisms that are in-volved in CCL5 secretion. Because the absence of the 43TRKN46 motif
has led to complete inhibition of CCL5 secretion whereas the degreeof GAG involvement in CCL5 secretion was only partial, such addi-tional mechanisms possibly also regulate the secretion of CCL5 in a43TRKN46-dependent manner. On the basis of the literature, we spec-ulate that one such additional mechanism may be based on CCR1and CCR3, at least in breast tumor cells, because these cells expresssuch receptors [7,19]. This possibility is supported by the fact thatthe 44RKNR47 sequence mediates the binding of CCL5 to CCR1and CCR3 [43,44]. These findings stand in the basis of our plans toinvestigate whether CCR1 and CCR3 regulate the release of CCL5 inbreast tumor cells.Taking our novel findings a step further and considering them with
additional results provided in our study and in the literature, it is logicalto assume that the secretion of CCL5 by breast tumor cells is regulatedby associations formed through its 43TRKN46 motif with GAG. Thispossibility is supported by the following observations: 1) In contrast toWT CCL5 that had a definite colocalization with GAG in the Golgi,the TRKN-mutated chemokine had much lower propensity to suchcolocalization. These findings indicate that the existence of the posi-tively charged 43TRKN46 motif of CCL5 is associated with the locali-zation of the chemokine in GAG-enriched secretion-related organelles(Golgi) and supports the role of 43TRKN46-GAG associations in thesecretion of CCL5. 2) Our confocal analyses have shown that WTCCL5 acquired a reticulate localization phenotype, which is typicalof ER, in CHO-deficient GAG cells, similar to the one detected forthe TRKN-mutated chemokine in normal GAG expressing cells. Thisobservation suggests that the organization of CCL5 in the Golgi ismediated through the 43TRKN46 motif. 3) Our secretion-regulating43TRKN46 sequence carries two of the three basic residues found inthe 44RKNR47 motif, that is essential for CCL5 binding to GAG[33,43,44,60]. 4) Being a CC chemokine, CCL5 was found to be ex-pressed in supernatants of human immunodeficiency virus–specific cyto-lytic T cells in complexes with proteoglycans [50]. Our study suggeststhat the reason forCCL5 associationwith proteoglycans in the extracellularmilieu of those cells is that the chemokine associated with them intra-cellularly on its path toward secretion and that this process involveda 43TRKN46-GAG–mediated mechanism.From the therapeutic point of view, the identification of the compo-
nents that regulate the secretion of CCL5 may have potential implica-tions, as they may pave the way toward the future design of modalitiesthat inhibit 43TRKN46-mediated or GAG-mediated mechanisms, lead-ing thereafter to reduced release of CCL5 by breast tumor cells. More-over, the fact that the 43TRKN46 motif is required for the secretion ofCCL5 bymany cell types (Figure 7) suggests that the different measurestaken to reduce the 43TRKN46-mediated process of CCL5 secretionmay inhibit the release of the chemokine also by tumor-promoting hostcells that are found at the tumor microenvironment, like leukocytesor mesenchymal stem cells, that contribute to tumor growth throughCCL5 release [13,24].Although such an approach may lead to inhibition of CCL5 secre-
tion also at inflammatory sites, it is not expected to impose a threaton the immune integrity of the host because the immune activities ofCCL5 are backed up by other chemokines [67,68]. One additionalaspect of inhibitory modalities that target the 43TRKN46-mediatedor GAG-mediated mechanism is the question whether they wouldlead also to reduced secretion of other chemokines or to interferencewith their binding to GAG in other cell types. The interactionsof many chemokines with GAG are mediated by basic amino acidsof the chemokines and negative charges of the GAG molecules.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 17
However, the different chemokines diverge in the sequences throughwhich they bind to GAG and in the positions of such domains inthe chemokine sequence. Hence, it is possible that the secretion ofother chemokines would not be affected when the 43TRKN46-mediatedor GAG-mediated trafficking of CCL5 is targeted and inhibited.To conclude, our study has provided novel findings on the regula-
tory processes involved in the secretion of CCL5 by breast tumor cellsand are the first to identify the important roles played by the 40s loopof CCL5 and of GAG in the secretion of this chemokine. In our futurestudies, we will aim at targeting the 43TRKN46-mediated or GAG-mediated mechanisms that lead CCL5 toward secretion and will de-termine the effects of such inhibitory measures on the malignancyphenotype of breast tumor cells.
AcknowledgmentsThe authors thank Prof Vlodavsky and Dr Ilan from the Technion(Haifa, Israel) for providing the CHO cells and the heparanase con-structs used in the study and Dr Barbul (Tel Aviv University) for hisassistance with the confocal analyses.
References[1] Hanahan D and Weinberg RA (2011). Hallmarks of cancer: the next generation.
Cell 144, 646–674.[2] Colotta F, Allavena P, Sica A, Garlanda C, and Mantovani A (2009). Cancer-
related inflammation, the seventh hallmark of cancer: links to genetic instability.Carcinogenesis 30, 1073–1081.
[3] Hagemann T, Balkwill F, and Lawrence T (2007). Inflammation and cancer:a double-edged sword. Cancer Cell 12, 300–301.
[4] Soria G and Ben-Baruch A (2009).The CCL5/CCR5 Axis in Cancer. Humana Press,New York, NY.
[5] Soria G and Ben-Baruch A (2008). The inflammatory chemokines CCL2 andCCL5 in breast cancer. Cancer Lett 267, 271–285.
[6] Adler EP, Lemken CA, Katchen NS, and Kurt RA (2003). A dual role for tumor-derived chemokine RANTES (CCL5). Immunol Lett 90, 187–194.
[7] Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M,Wigler N, Keydar I, and Ben-Baruch A (2002). The CC chemokine RANTESin breast carcinoma progression: regulation of expression and potential mecha-nisms of promalignant activity. Cancer Res 62, 1093–1102.
[8] Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, and BalkwillFR (2003). A chemokine receptor antagonist inhibits experimental breast tumorgrowth. Cancer Res 63, 8360–8365.
[9] Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, and Peng G (2010). Tumor micro-environments direct the recruitment and expansion of human TH17 cells. J Immunol184, 1630–1641.
[10] Kim JE, Kim HS, Shin YJ, Lee CS, Won C, Lee SA, Lee JW, Kim Y, Kang JS,Ye SK, et al. (2008). LYR71, a derivative of trimeric resveratrol, inhibits tumori-genesis by blocking STAT3-mediated matrix metalloproteinase 9 expression.Exp Mol Med 40, 514–522.
[11] Cappellen D, Schlange T, Bauer M, Maurer F, and Hynes NE (2007). Novelc-MYC target genes mediate differential effects on cell proliferation and migra-tion. EMBO Rep 8, 70–76.
[12] Jiao X, Katiyar S, Willmarth NE, Liu M, Ma X, Flomenberg N, Lisanti MP,and Pestell RG (2010). c-Jun induces mammary epithelial cellular invasion andbreast cancer stem cell expansion. J Biol Chem 285, 8218–8226.
[13] Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, RichardsonAL, Polyak K, Tubo R, and Weinberg RA (2007). Mesenchymal stem cells withintumour stroma promote breast cancer metastasis. Nature 449, 557–563.
[14] Mira E, Lacalle RA, Gonzalez MA, Gomez-Mouton C, Abad JL, Bernad A,Martinez AC, and Manes S (2001). A role for chemokine receptor transactivationin growth factor signaling. EMBO Rep 2, 151–156.
[15] Pinilla S, Alt E, Abdul Khalek FJ, Jotzu C, Muehlberg F, Beckmann C, and SongYH (2009). Tissue resident stem cells produce CCL5 under the influence of cancercells and thereby promote breast cancer cell invasion. Cancer Lett 284, 80–85.
[16] Prest SJ, Rees RC, Murdoch C, Marshall JF, Cooper PA, Bibby M, Li G, andAli SA (1999). Chemokines induce the cellular migration of MCF-7 human
breast carcinoma cells: subpopulations of tumour cells display positive and nega-tive chemotaxis and differential in vivo growth potentials. Clin Exp Metastasis17, 389–396.
[17] Youngs SJ, Ali SA, Taub DD, and Rees RC (1997). Chemokines induce migra-tional responses in human breast carcinoma cell lines. Int J Cancer 71, 257–266.
[18] ZabouoG, Imbert AM, Jacquemier J, Finetti P,Moreau T, Esterni B, BirnbaumD,Bertucci F, and Chabannon C (2009). CD146 expression is associated with a poorprognosis in human breast tumors and with enhanced motility in breast cancer celllines. Breast Cancer Res 11, R1.
[19] Zhang Y, Yao F, Yao X, Yi C, Tan C, Wei L, and Sun S (2009). Role of CCL5in invasion, proliferation and proportion of CD44+/CD24− phenotype of MCF-7cells and correlation of CCL5 and CCR5 expression with breast cancer progres-sion. Oncol Rep 21, 1113–1121.
[20] Forst B, Hansen MT, Klingelhofer J, Moller HD, Nielsen GH, Grum-SchwensenB, Ambartsumian N, Lukanidin E, and Grigorian M (2010). Metastasis-inducingS100A4 and RANTES cooperate in promoting tumor progression in mice. PLoSOne 5, e10374.
[21] Manes S, Mira E, Colomer R, Montero S, Real LM, Gomez-Mouton C, Jimenez-Baranda S, Garzon A, Lacalle RA, Harshman K, et al. (2003). CCR5 expressioninfluences the progression of human breast cancer in a p53-dependent manner.J Exp Med 198, 1381–1389.
[22] Stormes KA, Lemken CA, Lepre JV, Marinucci MN, and Kurt RA (2005).Inhibition of metastasis by inhibition of tumor-derived CCL5. Breast CancerRes Treat 89, 209–212.
[23] Bieche I, Lerebours F, Tozlu S, Espie M, Marty M, and Lidereau R (2004).Molecular profiling of inflammatory breast cancer: identification of a poor-prognosisgene expression signature. Clin Cancer Res 10, 6789–6795.
[24] Luboshits G, Shina S, Kaplan O, Engelberg S, Nass D, Lifshitz-Mercer B,Chaitchik S, Keydar I, and Ben-Baruch A (1999). Elevated expression of theCC chemokine regulated on activation, normal T cell expressed and secreted(RANTES) in advanced breast carcinoma. Cancer Res 59, 4681–4687.
[25] Sauer G, Schneiderhan-Marra N, Kazmaier C, Hutzel K, Koretz K, Muche R,Kreienberg R, Joos T, and Deissler H (2008). Prediction of nodal involvementin breast cancer based on multiparametric protein analyses from preoperativecore needle biopsies of the primary lesion. Clin Cancer Res 14, 3345–3353.
[26] Yaal-Hahoshen N, Shina S, Leider-Trejo L, Barnea I, Shabtai EL, Azenshtein E,Greenberg I, Keydar I, and Ben-Baruch A (2006). The chemokine CCL5 as apotential prognostic factor predicting disease progression in stage II breast cancerpatients. Clin Cancer Res 12, 4474–4480.
[27] Soria G, Yaal-Hahoshen N, Azenshtein E, Shina S, Leider-Trejo L, Ryvo L,Cohen-Hillel E, Shtabsky A, Ehrlich M, and Meshel T (2008). Concomitantexpression of the chemokines RANTES and MCP-1 in human breast cancer:a basis for tumor-promoting interactions. Cytokine 44, 191–200.
[28] Dupre SA, Redelman D, and Hunter KW Jr (2007). The mouse mammarycarcinoma 4T1: characterization of the cellular landscape of primary tumoursand metastatic tumour foci. Int J Exp Pathol 88, 351–360.
[29] Niwa Y, Akamatsu H, Niwa H, Sumi H, Ozaki Y, and Abe A (2001). Correla-tion of tissue and plasma RANTES levels with disease course in patients withbreast or cervical cancer. Clin Cancer Res 7, 285–289.
[30] Tedla N, Palladinetti P, Wakefield D, and Lloyd A (1999). Abundant ex-pression of chemokines in malignant and infective human lymphadenopathies.Cytokine 11, 531–540.
[31] Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Weitzenfeld P, Meshel T, Shabtai E, Gutman M, et al. (2011). Aninflammatory network in breast cancer: coordinated expression of TNFα &IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition.BMC Cancer 11, 130–149.
[32] Rohl CA, Strauss CE, Misura KM, and Baker D (2004). Protein structure predic-tion using Rosetta. Methods Enzymol 383, 66–93.
[33] Shaw JP, Johnson Z, Borlat F, Zwahlen C, Kungl A, Roulin K, Harrenga A,Wells TN, and Proudfoot AE (2004). The x-ray structure of RANTES: heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure12, 2081–2093.
[34] Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, MurrayLW, Arendall WB III, Snoeyink J, Richardson JS, et al. (2007). MolProbity:all-atom contacts and structure validation for proteins and nucleic acids. NucleicAcids Res 35, W375–W383.
[35] Soria G, Meshel T, and Ben-Baruch A (2009). Chemokines in human breasttumor cells: modifying their expression levels and determining their effects onthe malignancy phenotype. Methods Enzymol 460, 3–16.
18 Regulation of CCL5 Secretion in Breast Cancer Soria et al. Neoplasia Vol. 14, No. 1, 2012
[36] Feniger-Barish R, Yron I, Meshel T, Matityahu E, and Ben-Baruch A (2003).IL-8–induced migratory responses through CXCR1 and CXCR2: association withphosphorylation and cellular redistribution of focal adhesion kinase. Biochemistry42, 2874–2886.
[37] Strous GJ, van Kerkhof P, van Meer G, Rijnboutt S, and Stoorvogel W (1993).Differential effects of brefeldin A on transport of secretory and lysosomal proteins.J Biol Chem 268, 2341–2347.
[38] Miller SG, Carnell L, and Moore HH (1992). Post-Golgi membrane traffic:brefeldin A inhibits export from distal Golgi compartments to the cell surfacebut not recycling. J Cell Biol 118, 267–283.
[39] Williams RC Jr, Caplow M, and McIntosh JR (1986). Cytoskeleton. Dynamicmicrotubule dynamics. Nature 324, 106–107.
[40] Coue M, Brenner SL, Spector I, and Korn ED (1987). Inhibition of actinpolymerization by latrunculin A. FEBS Lett 213, 316–318.
[41] Cingolani LA and Goda Y (2008). Actin in action: the interplay between theactin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9, 344–356.
[42] El Golli N, Issertial O, Rosa JP, and Briquet-Laugier V (2005). Evidencefor a granule targeting sequence within platelet factor 4. J Biol Chem 280,30329–30335.
[43] Proudfoot AE, Fritchley S, Borlat F, Shaw JP, Vilbois F, Zwahlen C, Trkola A,Marchant D, Clapham PR, and Wells TN (2001). The BBXB motif ofRANTES is the principal site for heparin binding and controls receptor selec-tivity. J Biol Chem 276, 10620–10626.
[44] Martin L, Blanpain C, Garnier P, Wittamer V, Parmentier M, and Vita C(2001). Structural and functional analysis of the RANTES-glycosaminoglycansinteractions. Biochemistry 40, 6303–6318.
[45] Boelens J, Lust S, Offner F, Bracke ME, and Vanhoecke BW (2007). Review.The endoplasmic reticulum: a target for new anticancer drugs. In Vivo 21,215–226.
[46] Ellgaard L, Molinari M, and Helenius A (1999). Setting the standards: qualitycontrol in the secretory pathway. Science 286, 1882–1888.
[47] Tsai YC and Weissman AM (2010). The unfolded protein response, degrada-tion from endoplasmic reticulum and cancer. Genes Cancer 1, 764–778.
[48] MacDermott RP, Schmidt RE, Caulfield JP, Hein A, Bartley GT, Ritz J,Schlossman SF, Austen KF, and Stevens RL (1985). Proteoglycans in cell-mediated cytotoxicity. Identification, localization, and exocytosis of a chondroitinsulfate proteoglycan from human cloned natural killer cells during target cell lysis.J Exp Med 162, 1771–1787.
[49] Stevens RL, Otsu K, Weis JH, Tantravahi RV, Austen KF, Henkart PA, GalliMC, and Reynolds CW (1987). Co-sedimentation of chondroitin sulfate Aglycosaminoglycans and proteoglycans with the cytolytic secretory granules ofrat large granular lymphocyte (LGL) tumor cells, and identification of a mRNAin normal and transformed LGL that encodes proteoglycans. J Immunol 139,863–868.
[50] Wagner L, Yang OO, Garcia-Zepeda EA, Ge Y, Kalams SA, Walker BD,Pasternack MS, and Luster AD (1998). β-Chemokines are released fromHIV-1–specific cytolytic T-cell granules complexed to proteoglycans. Nature391, 908–911.
[51] Handel TM, Johnson Z, Crown SE, Lau EK, and Proudfoot AE (2005). Regula-tion of protein function by glycosaminoglycans—as exemplified by chemokines.Annu Rev Biochem 74, 385–410.
[52] Esko JD, Stewart TE, and Taylor WH (1985). Animal cell mutants defective inglycosaminoglycan biosynthesis. Proc Natl Acad Sci USA 82, 3197–3201.
[53] Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F,Bashenko Y, Flugelman MY, Vlodavsky I, and Ilan N (2004). Heparanaseuptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem279, 44084–44092.
[54] Rabenstein DL (2002). Heparin and heparan sulfate: structure and function.Nat Prod Rep 19, 312–331.
[55] Zcharia E, Metzger S, Chajek-Shaul T, Friedmann Y, Pappo O, Aviv A, Elkin M,Pecker I, Peretz T, and Vlodavsky I (2001). Molecular properties and involve-ment of heparanase in cancer progression and mammary gland morphogenesis.J Mammary Gland Biol Neoplasia 6, 311–322.
[56] Kuschert GS, Coulin F, Power CA, Proudfoot AE, Hubbard RE, Hoogewerf AJ,andWells TN (1999). Glycosaminoglycans interact selectively with chemokines andmodulate receptor binding and cellular responses. Biochemistry 38, 12959–12968.
[57] Goldshmidt O, Nadav L, Aingorn H, Irit C, Feinstein N, Ilan N, Zamir E,Geiger B, Vlodavsky I, and Katz BZ (2002). Human heparanase is localizedwithin lysosomes in a stable form. Exp Cell Res 281, 50–62.
[58] Sweeney MD, Yu Y, and Leary JA (2006). Effects of sulfate position on heparinoctasaccharide binding to CCL2 examined by tandem mass spectrometry. J AmSoc Mass Spectrom 17, 1114–1119.
[59] Meissen JK, Sweeney MD, Girardi M, Lawrence R, Esko JD, and Leary JA(2009). Differentiation of 3-O-sulfated heparin disaccharide isomers: identifica-tion of structural aspects of the heparin CCL2 binding motif. J Am Soc MassSpectrom 20, 652–657.
[61] Farley JR, Nakayama G, Cryns D, and Segel IH (1978). Adenosine triphosphatesulfurylase from Penicillium chrysogenum equilibrium binding, substrate hydro-lysis, and isotope exchange studies. Arch Biochem Biophys 185, 376–390.
[62] Dubrac A, Quemener C, Lacazette E, Lopez F, Zanibellato C, Wu WG,Bikfalvi A, and Prats H (2010). Functional divergence between 2 chemokinesis conferred by single amino acid change. Blood 116, 4703–4711.
[63] Culley FJ, Fadlon EJ, Kirchem A, Williams TJ, Jose PJ, and Pease JE (2003).Proteoglycans are potent modulators of the biological responses of eosinophils tochemokines. Eur J Immunol 33, 1302–1310.
[64] Hol J, Kuchler AM, Johansen FE, Dalhus B, Haraldsen G, and Oynebraten I(2009). Molecular requirements for sorting of the chemokine IL-8/CXCL8 toendothelial Weibel-Palade bodies. J Biol Chem 284, 23532–23539.
[65] Meen AJ, Oynebraten I, Reine TM, Duelli A, Svennevig K, Pejler G, Jenssen T,and Kolset SO (2011). Serglycin is a major proteoglycan in polarized humanendothelial cells and is implicated in the secretion of the chemokine GROα/CXCL1. J Biol Chem 286, 2636–2647.
[66] Zehe C, Engling A, Wegehingel S, Schafer T, and Nickel W (2006). Cell-surfaceheparan sulfate proteoglycans are essential components of the unconventionalexport machinery of FGF-2. Proc Natl Acad Sci USA 103, 15479–15484.
[67] Devalaraja MN and Richmond A (1999). Multiple chemotactic factors: finecontrol or redundancy? Trends Pharmacol Sci 20, 151–156.
[68] Mantovani A (1999). The chemokine system: redundancy for robust outputs.Immunol Today 20, 254–257.
Neoplasia Vol. 14, No. 1, 2012 Regulation of CCL5 Secretion in Breast Cancer Soria et al. 19
Figure W1. Intracellular distribution of HA-CCL5(WT). HumanMCF-7breast tumor cells were transiently transfected by a vector express-ing HA-CCL5(WT). Confocal analysis has shown vesicular distributionand Golgi localization of the chemokine, similarly to the intracellularphenotype obtained for GFP-CCL5(WT) (Figs. 1, 6, and 8) and toendogenous CCL5 in breast tumor cells [27].
Video W1. Vesicles containing GFP-CCL5(WT) have dynamic motil-ity in MCF-7 human breast tumor cells. The control empty vectorexpressing GFP had a diffuse nonorganized distribution in the cells(data not shown).
Video W2. After short BFA treatment of MCF-7 cells, the Golgicollapsed but vesicles containing GFP-CCL5(WT) were still apparentclose to the cell periphery, indicating that they have reached thepost-Golgi stage.
Video W3. Vesicles containing GFP-CCL5(WT) moved along direc-tional and structured tracks in MCF-7 human breast tumor cells.
Video W5. The movie follows the same cells that were shown inVideo W4, but now after nocodazole treatment, demonstratingreduced motility of CCL5-containing vesicles, and their deceleratedmovement along well defined paths.
Figure W2. Further evidence to prominent reduction in secretion of GFP-CCL5(TRKN–) compared to GFP-CCL5(WT) by breast tumorcells. Human MCF-7 breast tumor cells were transiently transfected by vectors expressing GFP-CCL5(WT) or GFP-CCL5(TRKN–), fol-lowed by determination of chemokine expression in cell supernatants. (A) ELISAs with mAb to GFP and polyclonal detecting antibodiesto CCL5 (“Procedure 3” in “Materials and methods”). (B) ELISAs with polyclonal coating antibodies to CCL5 and polyclonal detectingantibodies to CCL5 (“Procedure 4” in “Materials and methods”).
Video W4. MCF-7 cells expressing GFP-CCL5(WT) before treatmentwith nocodazole showing dynamic motility of vesicles. The samecells after nocodazole treatment are shown in Video W5.
Video W6. In transfected MCF-7 cells, GFP-CCL5(TRKN–) is lessorganized in vesicles than GFP-CCL5(WT) (Video W1), and thosevesicles that contain GFP-CCL5(TRKN–) have nondirectional andlimited motility.




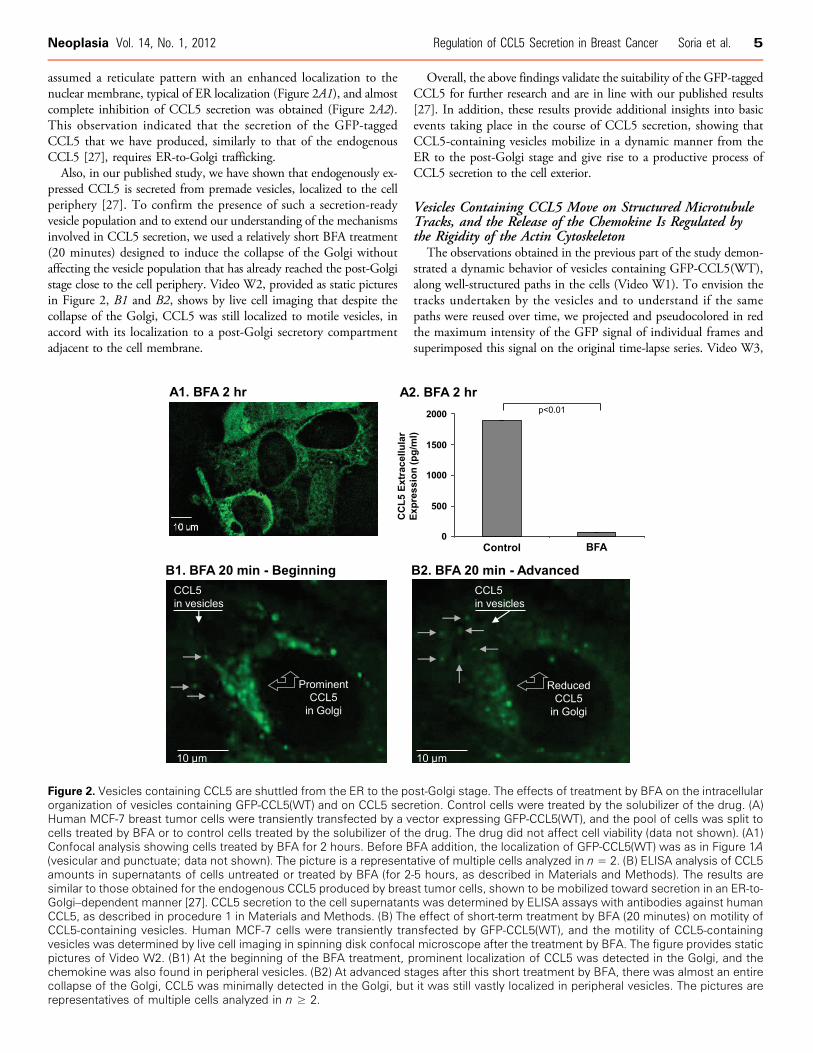


































![Regulation of the intracellular Ca2+. Regulation of intracellular [H]:](https://static.documents.pub/doc/80x56/5a4d1b717f8b9ab0599b56a5/regulation-of-the-intracellular-ca2-regulation-of-intracellular-h.jpg)