This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys. Cite this: DOI: 10.1039/c3cp44617d Mediated electron transfer in glucose oxidising enzyme electrodes for application to biofuel cells: recent progress and perspectives Paul Kavanagh and Do ´nal Leech Glucose oxidising enzyme electrodes have long been studied for their application to biosensors and, more recently, anodes in biofuel cells. At a fundamental level, insight into enzyme electron transfer and oxidation current generation at enzyme electrodes can be gained by systematic studies on integration of surfaces, biocatalysts, and artificial substrates (mediators). In this perspective, we present an overview of methods to aid the development of glucose oxidising enzyme electrodes based on mediated electron transfer for application to continuous-use anodes in a biofuel cell. Focus is placed on the rational design of mediators, based on osmium redox complexes, and screening of the activity of such complexes as mediators for glucose oxidising enzymes. An overview of the performance of enzyme electrodes, focused predominantly on crosslinked films of redox polymers and glucose oxidase, for glucose oxidation, is presented and approaches to improve both current output and stability of such enzyme electrodes are discussed. 1. Introduction Enzymes are naturally occurring molecules which catalyse chemical reactions of biological importance. A particular class of enzyme of interest to electrochemists are the oxidoreductases which catalyse the transfer of electrons from one molecule (the electron donor) to another (the electron acceptor). Glucose oxidase (GOx), which catalyses the oxidation of glucose to gluconolactone coupled to the reduction of O 2 to H 2 O 2 , is a well known example, because of its significance to electrochemical sensing technology. The first glucose enzyme electrode was demons- trated by Clark and Lyons 1 using a thin layer of GOx entrapped on an oxygen electrode by a semi-permeable membrane. School of Chemistry & Ryan Institute, National University of Ireland Galway, Galway, Ireland. E-mail: [email protected]Paul Kavanagh Paul Kavanagh received his BSc in Pure and Applied Chemistry from Dublin City University, Dublin, Ireland in 2002. He obtained a PhD (2006) in chemistry from National Univer- sity of Ireland Galway, focusing on the synthesis and characterisa- tion of redox polymer films for application to DNA biosensors and biofuel cells. His research activities include application of electrochemistry for enzyme and microbial biofilm studies, bio- analysis, microfluidics and transi tion metal complex syntheses. Do´nalLeech Professor Do ´nal Leech is Director of the Biomolecular Electronics Research Laboratory at the National University of Ireland Galway. He obtained his PhD from Dublin City University, and held positions at the University of Hawaii and Universite ´ de Montre ´al, prior to his appointment to a lectureship in NUIG in 1997. His research interests span the broad themes of bioelectrochemistry, electroanalytical chemistry and electrocatalysis. Received 20th December 2012, Accepted 19th February 2013 DOI: 10.1039/c3cp44617d www.rsc.org/pccp PCCP PERSPECTIVE Downloaded by University of Illinois - Urbana on 05 March 2013 Published on 19 February 2013 on http://pubs.rsc.org | doi:10.1039/C3CP44617D View Article Online View Journal
Transcript
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
Cite this: DOI: 10.1039/c3cp44617d
Mediated electron transfer in glucose oxidisingenzyme electrodes for application to biofuel cells:recent progress and perspectives
Paul Kavanagh and Donal Leech
Glucose oxidising enzyme electrodes have long been studied for their application to biosensors and,
more recently, anodes in biofuel cells. At a fundamental level, insight into enzyme electron transfer and
oxidation current generation at enzyme electrodes can be gained by systematic studies on integration
of surfaces, biocatalysts, and artificial substrates (mediators). In this perspective, we present an overview
of methods to aid the development of glucose oxidising enzyme electrodes based on mediated
electron transfer for application to continuous-use anodes in a biofuel cell. Focus is placed on the
rational design of mediators, based on osmium redox complexes, and screening of the activity of such
complexes as mediators for glucose oxidising enzymes. An overview of the performance of enzyme
electrodes, focused predominantly on crosslinked films of redox polymers and glucose oxidase, for
glucose oxidation, is presented and approaches to improve both current output and stability of such
enzyme electrodes are discussed.
1. Introduction
Enzymes are naturally occurring molecules which catalysechemical reactions of biological importance. A particular classof enzyme of interest to electrochemists are the oxidoreductases
which catalyse the transfer of electrons from one molecule (theelectron donor) to another (the electron acceptor). Glucoseoxidase (GOx), which catalyses the oxidation of glucose togluconolactone coupled to the reduction of O2 to H2O2, is awell known example, because of its significance to electrochemicalsensing technology. The first glucose enzyme electrode was demons-trated by Clark and Lyons1 using a thin layer of GOx entrappedon an oxygen electrode by a semi-permeable membrane.
School of Chemistry & Ryan Institute, National University of Ireland Galway,
Paul Kavanagh received his BScin Pure and Applied Chemistryfrom Dublin City University,Dublin, Ireland in 2002. Heobtained a PhD (2006) inchemistry from National Univer-sity of Ireland Galway, focusingon the synthesis and characterisa-tion of redox polymer films forapplication to DNA biosensorsand biofuel cells. His researchactivities include application ofelectrochemistry for enzyme andmicrobial biofilm studies, bio-analysis, microfluidics and transition metal complex syntheses.
Donal Leech
Professor Donal Leech is Directorof the Biomolecular ElectronicsResearch Laboratory at theNational University of IrelandGalway. He obtained his PhDfrom Dublin City University, andheld positions at the University ofHawaii and Universite de Montreal,prior to his appointment to alectureship in NUIG in 1997. Hisresearch interests span the broadthemes of bioelectrochemistry,electroanalytical chemistry andelectrocatalysis.
Received 20th December 2012,Accepted 19th February 2013
Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
Glucose concentration was measured indirectly by monitoringthe O2 consumed by GOx during the enzyme catalysed reaction.Further improvements on this design2–4 led to the developmentof the first practical glucose biosensors, measuring the amountof H2O2 produced.4 However, the measurement of H2O2 using aconventional platinum electrode requires a high overpotential(B0.6 V vs. Ag/AgCl) leading to risk of interference fromelectroactive species, such as ascorbic and uric acids, for glucosetesting of blood samples.5 The 1980s saw the emergence of‘second generation’ glucose sensors, which relied on usingsmall, synthetic electron transfer mediators, in place of O2 (thenatural co-substrate of GOx), as electron acceptors to shuttleelectrons between enzyme active site and electrode surface, in amechanism termed mediated electron transfer (MET). Utilisa-tion of redox mediators allowed detection of glucose at loweroverpotentials resulting in improved sensitivity and accuracy indetermining blood glucose levels. Fig. 1 depicts a scheme forthe MET process in which the enzyme takes part in the firstredox reaction with the substrate and is re-oxidised or reducedby the mediator, which in turn is regenerated, with electronstransported through a combination of mediator physical diffusionand electron self-exchange, at the electrode surface. Themediator circulates continuously between the enzyme and theelectrode, cycled between its oxidised and reduced forms,producing catalytic current.
Advances in MET based enzyme electrodes throughout the1990’s led to a renewed interest in their potential as fueloxidising and/or oxidant reducing electrodes for biofuel cell (BFC)applications. This resurgence followed a seminal publication byPalmore et al.6 describing the complete oxidation of methanolto CO2 using a combination of solution-phase enzymes and aredox mediator at a graphite electrode. Focus soon shiftedtowards examining glucose as a potential fuel source, presentat relatively high concentrations (B5.5 mM) at normal bloodsugar levels, with the long-term aim of developing self-poweredimplantable or semi-implantable biomedical devices.7 This isnot a new concept: it had been explored in the 1960’s, buthad largely been abandoned when it became apparent that
sufficient power to drive pacemaker devices, becoming avail-able at that time, could not be generated using in vivo glucoseas the sole fuel source.8,9 However, advances over the last twodecades in both MET based enzyme electrode design anddevelopment of ever smaller and lower power demandingmicroelectronic devices make feasible the concept of using aBFC to power a miniature biomedical device. A BFC configurationcould therefore consist of a fuel oxidising anode and an oxygenreducing cathode connected with all catalytic components(i.e. enzyme and mediator) confined to the electrode surface,thus eliminating the need for a separating membrane andaiding miniaturisation of the device.
Here we provide a perspective on the optimisation andperformance of mediated enzyme electrodes for the catalyticoxidation of glucose. While GOx remains the most widelystudied enzyme for this purpose, a range of dehydrogenases,which do not donate electrons to O2, capable of oxidisingglucose have also been studied.10 It is worthwhile to note thatattempts have been made to connect (or ‘wire’) the active site ofGOx directly to the electrode surface, mainly through enzymedeglycosylation11 or by addition of carbon nanotubes (CNTs)into the biofilm matrix,12 whilst maintaining the catalyticactivity of the enzyme and thereby eliminating the need foran electron transfer mediator. Where catalytic current isobserved, and attributed to direct electron transfer (DET), it isnot clear if this is due to true electrical connection of the activesite to the electrode or to diffusion of loosely bound FADco-factor to the electrode surface.13 In any case, the level ofcurrents observed tend be significantly lower than that of theirMET-based counterparts given that the rate of electron transferis related exponentially to the distance of closest approachbetween an electron donor and acceptor.14 Negligible rates ofelectron transfer pertain for distances beyond 2 nm, meaningthat DET can only take place when an electrode is placed withinthis distance to an enzyme cofactor in the active site. Correctorientation of each enzyme at the electrode surface is requiredto maintain the active site within this distance of closestapproach and even when the electrode and the enzyme activesite are sufficiently close to permit electron transfer, the electrodeitself can block access to the active site by the co-substrate/substrate,thereby inactivating the catalytic property of the enzyme.10 Suchlimitations are avoided by the use of redox mediators as artificialco-substrates, which permit the ‘wiring’ of multi-layers of enzymesirrespective of their orientation. One drawback, however, is that theuse of redox mediators can result in lower open circuit, andoperating, cell voltages of BFCs, although open circuit voltages(OCV) in excess of 0.5 V for BFCs employing MET based bioanodeand biocathode electrodes are frequently reported.15–18
Demonstrator BFCs for both implanted power and portablepower generation provide useful benchmarks to target improve-ments in device configuration and performance. For example,Sakai et al.17 describe a two-compartment BFC, using vitaminK3, diaphorase, NADH and glucose dehydrogenase (GDH)deposited on carbon fibre sheets, for glucose oxidation (0.4 Mconcentration) at the anode. When coupled to a ferricyanidemediated bilirubin oxidase (BOD) oxygen-reducing cathode, the
Fig. 1 Simplified schematic depiction of mediated electron transfer (MET) forsubstrate oxidation, with electron transfer to the electrode by self-exchangebetween reduced, Med(Red), and oxidised, Med(Ox), forms of the mediator.
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
assembled BFC delivered a maximum power density of1.45 mW cm�2 at 0.3 V. Stacking of four such cells providedsufficient power (>100 mW) to operate a toy car or portablemusic player for at least 2 h. Recent reports also describe thetransition from in vitro to in vivo testing of BFCs, implanted indiverse living organisms.19 Cinquin et al.20 report on theoperation of a BFC implanted in the retroperitoneal space ofa rat. The BFC, based on a mediated GOx enzyme electrode forglucose oxidation at the anode, generated a maximum powerdensity of 24.4 mW cm�3 at 0.13 V, with production of 2 mWpower observed over several hours for the implanted system.Katz and co-workers have demonstrated operation of BFCsimplanted in snails,21 clams22 and lobsters.23 In a recent study,an OCV of up to 1.2 V was recorded from two lobster-implantedBFCs connected in series, and this configuration was used topower an electronic watch.23 In addition, stacking of five BFCsin series, operating on human serum in an in vitro fluidic system,generated the 3 V necessary to power a cardiac pacemaker. Liuet al.24 highlight the feasibility of using a subcutaneouslyimplanted miniature BFC-powered sensor system for continuousblood glucose monitoring of a diabetic patient. The performanceof the self-powered sensor matched that of a conventional battery-powered amperometric sensor over the 5 day trial period.
In this perspective, we pay particular attention to the develop-ment of enzyme-based glucose oxidising anodes for applicationto BFCs, giving a brief overview of MET systems and methods toidentify appropriate mediators for each enzyme. Approaches todetermine the conditions for optimum current and poweroutput are also examined. We finish by discussing the challengesthat we consider to be significant for research on developmentand future applications of BFCs.
2. Selection of mediator and enzymecombination
Judicious choice of enzyme and mediator combination at anodesand cathodes is essential to maximise BFC power output. Desirablecharacteristics for redox mediators include: rapid heterogeneous,enzyme–mediator and self-exchange electron transfer rates, fordelivery of electrons; and chemical stability in oxidised and reducedforms, to ensure stability of catalytic turnover of enzyme reaction.When selecting a mediator for each enzyme, two factors shouldtherefore be considered. Firstly: the redox potential of themediator, which should be thermodynamically favourable withrespect to the enzyme redox potential, i.e. more positive foroxidative biocatalysis or more negative for reductive biocatalysis,to drive electron transfer. Secondly: the mediator–enzymeelectron transfer rate, which should be sufficiently high so asto generate high currents.
Initial studies of MET in BFCs focused on addition ofmediators to electrolyte solutions, which necessitates inclusionof a separating membrane between half-cell compartments toprevent cross reactions between enzymes and mediators at eachelectrode short-circuiting the cell. Immobilisation of mediatorand enzyme at the electrode surface facilitates BFC miniaturisation,
as it permits removal of any separating membrane. Althoughsolution-phase mediation may not be desirable for an ‘endproduct’ BFC, studies of such systems can aid in the design andselection of appropriate mediators for a particular enzyme, ininitial stages of BFC development. The following sectionsdescribe the use of solution phase studies to probe mediator–enzyme interaction, and subsequent immobilisation of enzymeand mediator at electrode surfaces.
2.1 Solution-phase studies
Homogeneous mediation represents the simplest configurationfor bioelectrocatalysis and provides an elegant starting pointfor probing enzyme–mediator interactions. Cyclic voltammetry(CV) can be used to determine the redox potential (E10) of themediator and reversibility of the redox couple, and the rate ofelectron transfer between enzyme and mediator. As the redoxpotential of the mediator is shifted further from the redoxpotential of the enzyme active site, the rate of enzyme–mediatorelectron transfer is predicted to increase exponentially, due tothe increased driving force.14,25 However an increase in thedriving force at each electrode may result in a decrease inoverall cell voltage (as cell voltage is strongly influenced by thedifference in redox potential between anodic and cathodicmediators7). Consequently, a compromise between catalyticrate and cell voltage is necessary for maximum power output.For example, it has been proposed that a mediator should havean E10 value B50 mV downhill of the enzyme active site togenerate current with minimal loss in cell voltage.26
The chemical structure of the mediator may also influencethe rate of mediator–enzyme electron transfer, as the mediatorand enzyme active site need to be in close proximity to permitefficient transfer of electrons. In the case of GOx, for example, itis postulated that steric hindrance of the mediator during theapproach to the flavin prosthetic group in the active site of GOxcan significantly slow down the rate of electron transfer.27,28
Based on theory developed by Nicholson and Shain,29 kineticparameters, such as the pseudo first-order and second orderrate constant can be evaluated for mediator–enzyme inter-actions, under defined conditions. Such studies are normallyperformed in homogenous solution-phase at low mediatorconcentration with a fixed concentration of active enzymeand a high substrate concentration, permitting estimation ofa second order rate constant, k, for enzyme–mediator reaction.Many solution phase studies have focused on GOx, utilisingdifferent series of structurally similar mediators e.g. ferroceneand its derivatives, osmium and ruthenium polypyridyl complexes,and quinones. A simplified reaction scheme for the oxidation ofGOx by a one-electron mediator, eqn (1), presumes the FADH2
active site of GOx is oxidised to FAD by transfer of electrons tomediator species.
Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
of ferrocene derivative structure on GOx mediation in aqueoussolution, reporting second order rate constant values inthe range of 105 M�1 s�1, leading to selection of an optimal1,10-dimethyl-3-(2-amino-1-hydroxyethyl)ferrocene mediator fora commercial glucose enzyme electrode test. However, no clearrelationship between rate constant and redox potential wasapparent, as the method of varying redox potential relied uponvariation in structure, which also influences the value estimatedfor the rate constant through steric interactions in the active site.Indeed the simplified reaction in eqn (1) has been expanded totake account of sequential electron and proton transfer, of theacid dissociation equilibrium for the flavin species, and of theformation of a precursor affinity complex between enzyme andmediator,31 with results using selected ferrocene derivatives asmediators again demonstrating that rate constants do not trendwith driving force alone, providing evidence for the formation ofsuch affinity complexes.
Ferrocene derivatives display relatively high redox potentials(B0.35 V vs. Ag/AgCl), contributing to cell voltage losses if usedas anode mediators in BFC assemblies. In addition, ferrocenein its oxidised form, ferricenium, is unstable in aqueoussolution as it undergoes hydrolysis32 and is not readily solublein its reduced form in aqueous solvents, which may lead todifficulties for enzyme electrode fabrication. Zakeeruddinet al.33 focused on use of a range of tris(4,40-substituted-2,2 0-bipyridine) complexes of iron, ruthenium and osmium asmediators for GOx, with the complexes displaying redox poten-tials spanning a wider range to the ferrocene derivatives. Anattempt was made to correlate rate constants, through Marcustheory, to driving force, with however significant deviationsfrom the model and a range of simplifying assumptions used.Overall, for a driving force above 0.3 V between GOx and osmiumcomplexes, rate constant values in the range 105–106 M�1 s�1 areobserved, indicating that such complexes are useful for enzymeelectrode studies.
Extraction of kinetic parameters from dynamic electro-chemical experiments is not a straightforward task, however,given the interplay between the factors that can contributeto controlling the catalytic current during a transient cyclicvoltammetric experiment. In most cases kinetic parameters areestimated under conditions of high substrate and low mediatorconcentrations, where an enzyme-limited reaction rate isexpected. Such conditions, however, only represent one of arange of rate-limiting cases for the homogeneous system, with avariety of factors influencing the overall reaction, representedby the schematic in Fig. 2.31,34,35 Also, certain parameters, suchas enzyme activity, normally assumed to remain constant,29 maychange throughout the timeframe of the experiment. Recentreports have highlighted, based on use of a case diagram forsolution-phase mediation of enzymes, substrate depletion, enzymeinactivation, low solubility of redox mediator and relativity betweenmass transport and kinetic control contributing to difficultiesin extracting reliable kinetic parameters.36
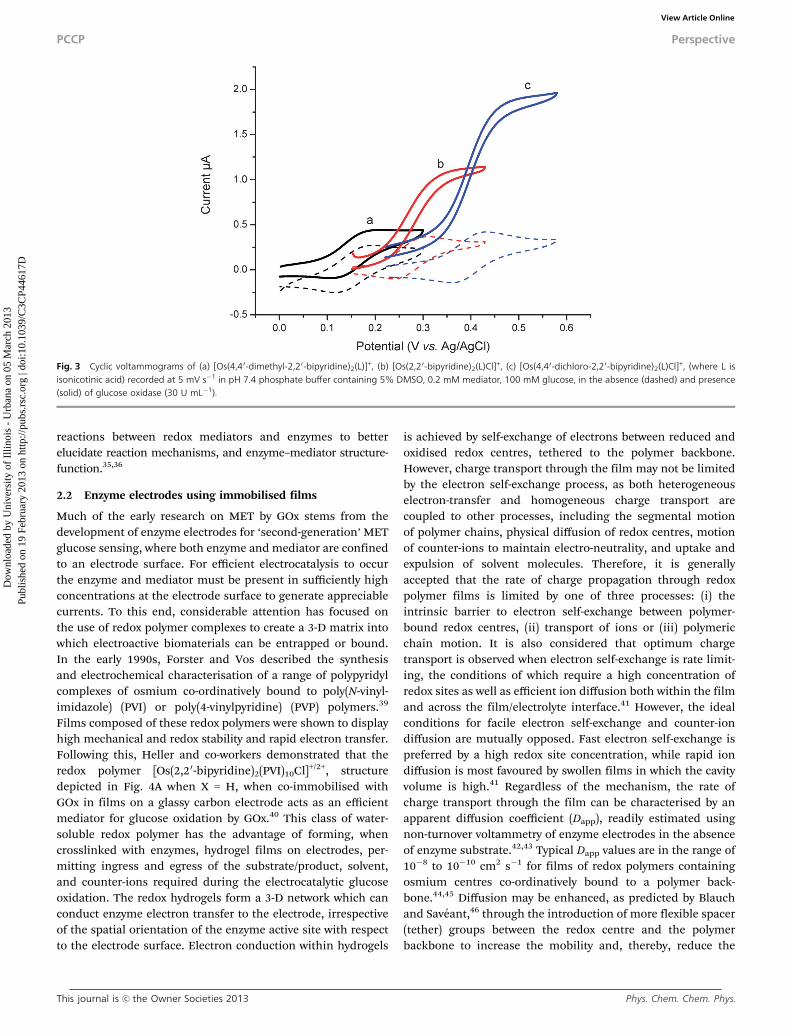
From our perspective, a rapid yet effective comparison of thecapability of each redox compound to act as a mediator can beundertaken by recording, under identical conditions, voltammetry
with selection of either mediator or enzyme as the singlevariable. We have applied this approach for the screening of aseries of osmium complexes of general formula [Os(4,40-sub-stituted-2,20-bipyridine)2(L)Cl]+, where L is Cl or an amino- orcarboxylic acid-terminated 4-pyridine ligand, as redox media-tors for a range of glucose oxidising enzymes. For example,Fig. 3 shows CVs recorded for a series of [Os(4,40-XX-2,20-bipyridine)2(isonicotinic acid)Cl]+ complexes (where X = CH3,H and Cl) as mediators of glucose oxidation by GOx (30 U mL�1)at glassy carbon electrodes. The ‘tuning’ of the redox potentialof the osmium metal centre, by introduction of electronwithdrawing/donating groups on the bipyridine, can be pre-dicted using an empirical approach developed by Lever andcoworkers,37 and offers a means to probe the influence ofstructure and thermodynamic driving force on current genera-tion. Variation in mediator selection, present in relativelylow concentration, 0.2 mM, compared to that of the glucosesubstrate, results in higher catalytic currents for mediators ofmore positive oxidation potentials, supporting the case thatthermodynamic driving force can influence electron transferkinetics in solution.25
It should be noted, though, that significant differences existbetween systems where the mediator and enzyme are free todiffuse in solution (homogeneous catalysis) to when they areconfined to an electrode surface (heterogeneous catalysis). Forexample, when mediators are adsorbed on electrodes, theirlocal concentrations can become elevated, leading to difficultyin comparison between solution phase and immobilised situa-tions. Also, the kinetics and/or affinity of reactions involvingimmobilised enzymes are normally different from those of thesame enzyme freely present in solution.38 Therefore, enzymeand mediator combinations effective in solution-phase studiesfor current generation may not necessarily have their effective-ness translated to immobilised systems. Nonetheless there isscope for expanding detailed solution-phase characterisation of
Fig. 2 Schematic representation of homogeneous enzyme mediated oxidationas considered by Albery et al.34 and Flexer et al.36 Mox and Mred are the oxidisedand reduced forms of the mediator. Eox and Ered are the oxidised and reducedenzyme. S and P are the substrate and product of the enzymatic reaction. DE andDM are the diffusion coefficients of enzyme and mediator respectively; thecoefficients of the oxidised and reduced forms are assumed to be equal. Fromref. 36, with permission granted by Elsevier.
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
reactions between redox mediators and enzymes to betterelucidate reaction mechanisms, and enzyme–mediator structure-function.35,36
2.2 Enzyme electrodes using immobilised films
Much of the early research on MET by GOx stems from thedevelopment of enzyme electrodes for ‘second-generation’ METglucose sensing, where both enzyme and mediator are confinedto an electrode surface. For efficient electrocatalysis to occurthe enzyme and mediator must be present in sufficiently highconcentrations at the electrode surface to generate appreciablecurrents. To this end, considerable attention has focused onthe use of redox polymer complexes to create a 3-D matrix intowhich electroactive biomaterials can be entrapped or bound.In the early 1990s, Forster and Vos described the synthesisand electrochemical characterisation of a range of polypyridylcomplexes of osmium co-ordinatively bound to poly(N-vinyl-imidazole) (PVI) or poly(4-vinylpyridine) (PVP) polymers.39
Films composed of these redox polymers were shown to displayhigh mechanical and redox stability and rapid electron transfer.Following this, Heller and co-workers demonstrated that theredox polymer [Os(2,20-bipyridine)2(PVI)10Cl]+/2+, structuredepicted in Fig. 4A when X = H, when co-immobilised withGOx in films on a glassy carbon electrode acts as an efficientmediator for glucose oxidation by GOx.40 This class of water-soluble redox polymer has the advantage of forming, whencrosslinked with enzymes, hydrogel films on electrodes, per-mitting ingress and egress of the substrate/product, solvent,and counter-ions required during the electrocatalytic glucoseoxidation. The redox hydrogels form a 3-D network which canconduct enzyme electron transfer to the electrode, irrespectiveof the spatial orientation of the enzyme active site with respectto the electrode surface. Electron conduction within hydrogels
is achieved by self-exchange of electrons between reduced andoxidised redox centres, tethered to the polymer backbone.However, charge transport through the film may not be limitedby the electron self-exchange process, as both heterogeneouselectron-transfer and homogeneous charge transport arecoupled to other processes, including the segmental motionof polymer chains, physical diffusion of redox centres, motionof counter-ions to maintain electro-neutrality, and uptake andexpulsion of solvent molecules. Therefore, it is generallyaccepted that the rate of charge propagation through redoxpolymer films is limited by one of three processes: (i) theintrinsic barrier to electron self-exchange between polymer-bound redox centres, (ii) transport of ions or (iii) polymericchain motion. It is also considered that optimum chargetransport is observed when electron self-exchange is rate limit-ing, the conditions of which require a high concentration ofredox sites as well as efficient ion diffusion both within the filmand across the film/electrolyte interface.41 However, the idealconditions for facile electron self-exchange and counter-iondiffusion are mutually opposed. Fast electron self-exchange ispreferred by a high redox site concentration, while rapid iondiffusion is most favoured by swollen films in which the cavityvolume is high.41 Regardless of the mechanism, the rate ofcharge transport through the film can be characterised by anapparent diffusion coefficient (Dapp), readily estimated usingnon-turnover voltammetry of enzyme electrodes in the absenceof enzyme substrate.42,43 Typical Dapp values are in the range of10�8 to 10�10 cm2 s�1 for films of redox polymers containingosmium centres co-ordinatively bound to a polymer back-bone.44,45 Diffusion may be enhanced, as predicted by Blauchand Saveant,46 through the introduction of more flexible spacer(tether) groups between the redox centre and the polymerbackbone to increase the mobility and, thereby, reduce the
Fig. 3 Cyclic voltammograms of (a) [Os(4,4 0-dimethyl-2,20-bipyridine)2(L)]+, (b) [Os(2,2 0-bipyridine)2(L)Cl]+, (c) [Os(4,40-dichloro-2,2 0-bipyridine)2(L)Cl]+, (where L isisonicotinic acid) recorded at 5 mV s�1 in pH 7.4 phosphate buffer containing 5% DMSO, 0.2 mM mediator, 100 mM glucose, in the absence (dashed) and presence(solid) of glucose oxidase (30 U mL�1).
Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
mean distance between redox centres. For example, Dapp for filmsof a redox polymer bearing a 13-atom-long flexible spacer armsituated between a PVP backbone and an alkyl functional group ofan [Os(N,N0-dialkylated-2,20-biimidazole)3]2+/3+ complex (Fig. 4B)was significantly higher than that for films of redox polymer thatdid not contain the spacer arm.47,48 It should be noted that lowercharge transport rates were reported for films of the long tethersystem upon either crosslinking48 or operation in perchlorate saltsolutions.49 As a result of the enhanced charge transport, films ofGOx and the chloride salt of the long tether redox polymer, withan optimised ratio of Os centres to polymer repeat unit, on carbonfibre electrodes generate substantial current density, 1.5 mA cm�2
at�0.1 V vs. Ag/AgCl, in pH 7.2 phosphate buffered saline at 37 1Ccontaining 15 mM glucose,50 although some concerns have beenraised about the possible oxidation of the Os centres by oxygen atthese low redox potentials.51
Research based on use of the range of [Os(4,40-XX-2,20-bipyridine)2(PVI)10Cl]+/2+ polymers, depicted in Fig. 4A, as, mediatingfilms on electrodes, relies upon preparation of redox polymers byligand substitution of the more labile Cl, of a Os(4,40-XX-2,20-bipyridine)2Cl2 monomeric complex, by an imidazole from the PVIpolymer, at reflux in a suitable solvent.39 Substitution of the Clligand by the more strongly co-ordinating imidazole results in a+0.2 V shift in redox potential of the osmium complex. Furtherheating at reflux can result in substitution of the second Clligand by an additional imidazole from PVI, resulting in afurther shift in osmium complex redox potential. In this case,the metal complex can act as a crosslinking agent betweenadjacent polymer chains, which may lower the solubility of theproduct in aqueous media and alter the charge transportcharacteristics of films of the redox polymer.
Forster et al.52 examined the effect of electroactive siteconcentration, electrolyte composition and concentration, and
polymer structure on charge transport through osmium redoxpolymer films, suggesting that ion diffusion is rate limiting atlow electrolyte concentration and high redox complex loadingcombinations. Ohara et al.40 established a 10 : 1 ratio of poly-mer monomeric unit to osmium redox complex co-ordinationas optimum for efficient charge transport through [Os(2,20-bipyridine)2(PVI)nCl]+/2+ polymer films containing GOx, althoughthe 10 : 1 ratio quoted is based upon relative molar ratios ofstarting materials, with an assumption of complete reaction.Recently, Barton and co-workers have highlighted the impor-tance of optimisation of redox complex loading,53–55 and ofredox potential,55 for optimisation of oxygen reduction currentby films of osmium-based redox polymers and a Trametesversicolor laccase on carbon electrodes. For example, variationin final osmium complex ratios within PVI redox polymers canoccur39,53–55 as the osmium site loading is synthetically difficultto control, can vary between batches and ligand substitutionreactivity is dependent on the structure of Os complex selected.Moreover, variation in the average molecular weight of the PVI,usually prepared by bulk free radical polymerisation,56 canaffect redox polymer solubility and lead to batch-to-batchvariations in enzyme electrode responses, although a recentreport on controlled radical polymerisation of N-vinylimidazoleto produce well-defined, mono-disperse homo-polymers,provides a route for alleviation of this issue.57
To gain better control over redox polymer composition,Poller et al.58 have described a synthetic route in which analready fully co-ordinated (so-called N6) osmium complex withone of the ligands containing a suitable functional group isattached to a pre-synthesised polymer via epoxide ring openingreaction. Redox polymers prepared by this route have shownpotential to act as mediators for Trametes hirsuta laccase for thereduction of oxygen at low overpotential. We recently reported
Fig. 4 Structures of redox polymers based on co-ordination of osmium complexes directly to a N-vinylimidazole polymer (A) or via a flexible spacer to a4-vinylpyridine polymer (B).
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
on high current densities (0.2–0.5 mA cm�2) for glucose oxida-tion by films of analogues of these osmium redox polymersco-immobilised with carbon nanotubes (CNTs) and GOx orglucose dehydrogenase (FAD dependent) at graphite electro-des.59 In a similar approach, Boland et al.60 describe a rationalroute to synthesis and characterisation of a library of osmiumpolypyridyl redox complexes bearing, for example, a 4-amino-methyl pyridine (4AMP) ligand, and the coupling of suchcomplexes, and GOx, to carboxymethyl dextran polymersupports at functionalised electrode surfaces. However, in theabsence of a bi-functional cross-linker, film thickness is limitedto the length of the surface-bound polymer chain which, inturn, can limit the amount of osmium mediator and GOx at theelectrode surface. In view of this, O Conghaile et al.61 describethe preparation of films prepared by cross-linking (using adi-epoxide reagent) [Os(2,20-bipyridine)2(4-AMP)Cl]+, a polyally-lamine polymer support and GOx on graphite electrodes,reporting a glucose oxidation current density, in 5 mM glucose,of 120 mA cm�2 at 0.45 V vs. Ag/AgCl, in pH 7.4 phosphatebuffered saline. Replacement of the 2,20-bipyridine of thecomplex with 4,40-dimethoxy-2,20-bipyridine, or 4,40-dimethyl-2,20-bipyridine ligands, provides mediators of lower redoxpotential, resulting in current densities of 30 and 70 mA cm�2
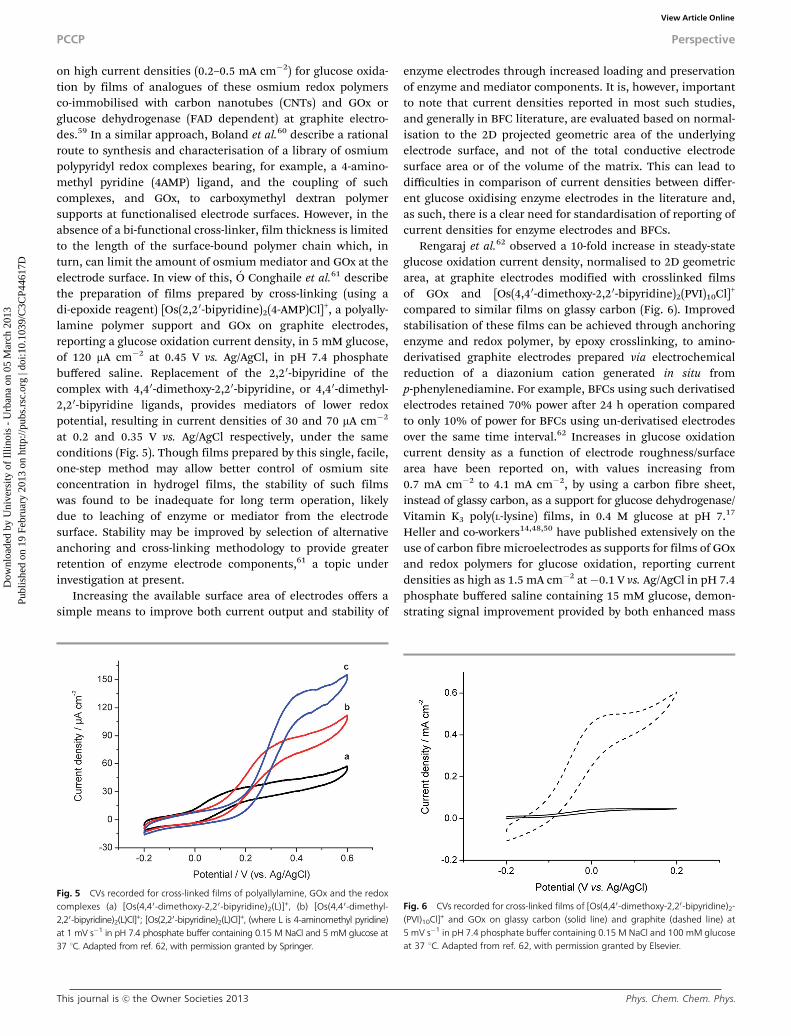
at 0.2 and 0.35 V vs. Ag/AgCl respectively, under the sameconditions (Fig. 5). Though films prepared by this single, facile,one-step method may allow better control of osmium siteconcentration in hydrogel films, the stability of such filmswas found to be inadequate for long term operation, likelydue to leaching of enzyme or mediator from the electrodesurface. Stability may be improved by selection of alternativeanchoring and cross-linking methodology to provide greaterretention of enzyme electrode components,61 a topic underinvestigation at present.
Increasing the available surface area of electrodes offers asimple means to improve both current output and stability of
enzyme electrodes through increased loading and preservationof enzyme and mediator components. It is, however, importantto note that current densities reported in most such studies,and generally in BFC literature, are evaluated based on normal-isation to the 2D projected geometric area of the underlyingelectrode surface, and not of the total conductive electrodesurface area or of the volume of the matrix. This can lead todifficulties in comparison of current densities between differ-ent glucose oxidising enzyme electrodes in the literature and,as such, there is a clear need for standardisation of reporting ofcurrent densities for enzyme electrodes and BFCs.
Rengaraj et al.62 observed a 10-fold increase in steady-stateglucose oxidation current density, normalised to 2D geometricarea, at graphite electrodes modified with crosslinked filmsof GOx and [Os(4,40-dimethoxy-2,2 0-bipyridine)2(PVI)10Cl]+
compared to similar films on glassy carbon (Fig. 6). Improvedstabilisation of these films can be achieved through anchoringenzyme and redox polymer, by epoxy crosslinking, to amino-derivatised graphite electrodes prepared via electrochemicalreduction of a diazonium cation generated in situ fromp-phenylenediamine. For example, BFCs using such derivatisedelectrodes retained 70% power after 24 h operation comparedto only 10% of power for BFCs using un-derivatised electrodesover the same time interval.62 Increases in glucose oxidationcurrent density as a function of electrode roughness/surfacearea have been reported on, with values increasing from0.7 mA cm�2 to 4.1 mA cm�2, by using a carbon fibre sheet,instead of glassy carbon, as a support for glucose dehydrogenase/Vitamin K3 poly(L-lysine) films, in 0.4 M glucose at pH 7.17
Heller and co-workers14,48,50 have published extensively on theuse of carbon fibre microelectrodes as supports for films of GOxand redox polymers for glucose oxidation, reporting currentdensities as high as 1.5 mA cm�2 at�0.1 V vs. Ag/AgCl in pH 7.4phosphate buffered saline containing 15 mM glucose, demon-strating signal improvement provided by both enhanced mass
Fig. 5 CVs recorded for cross-linked films of polyallylamine, GOx and the redoxcomplexes (a) [Os(4,40-dimethoxy-2,20-bipyridine)2(L)]+, (b) [Os(4,40-dimethyl-2,20-bipyridine)2(L)Cl]+; [Os(2,20-bipyridine)2(L)Cl]+, (where L is 4-aminomethyl pyridine)at 1 mV s�1 in pH 7.4 phosphate buffer containing 0.15 M NaCl and 5 mM glucose at37 1C. Adapted from ref. 62, with permission granted by Springer.
Fig. 6 CVs recorded for cross-linked films of [Os(4,40-dimethoxy-2,20-bipyridine)2-(PVI)10Cl]+ and GOx on glassy carbon (solid line) and graphite (dashed line) at5 mV s�1 in pH 7.4 phosphate buffer containing 0.15 M NaCl and 100 mM glucoseat 37 1C. Adapted from ref. 62, with permission granted by Elsevier.
Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
transport and available surface area at microelectrodes com-pared to macroelectrodes.48
The addition of conductive supports into film matrices hasshown potential to improve the performance of mediatedenzyme electrodes. Binyamin et al.63 ascribe production ofmechanically strong composite films of GOx and redox hydrogelon vitreous carbon, yielding a maximum current density of1.6 mA cm�2 for glucose oxidation, to addition of graphiteparticles to the films. Cinquin et al.20 use mechanical compres-sion of graphite particles, GOx, catalase and ubiquinone toprovide a stable composite graphite disc enzyme electrode forglucose oxidation in a BFC assembly, subsequently implanted inthe retroperitoneal space of a rat. Nishizawa and co-workers64,65
show that glucose oxidation current improves by addition ofKetjen black (KB) particles into a film matrix composed ofVitamin K3, poly(allylamine), diaphorase and glucose dehydro-genase on a GC electrode. In addition, Kamitaka et al.66 demon-strate that KB particles bound to carbon paper electrodespromote DET to adsorbed fructose dehydrogenase (FDH),generating a current density of 4 mA cm�2 in a continuouslystirred pH 5 buffered solution containing 0.2 M fructose.
The co-immobilisation of CNTs with enzymes at electrodesurfaces for application to BFCs has received considerableattention in recent years.12 CNTs, which display excellentconductivity, provide a high specific surface area for immobi-lisation of enzymes and mediators compared to 2D surfaces.Notable increases to both current magnitude and stability wereobserved for mediated systems incorporating CNTs, reportedmostly for applications related to biosensors67 and morerecently to BFCs.12 In a recent study, incorporation of single-walled carbon nanotubes (SWCNTs) into GOx and ferroceneredox polymer hydrogel films on electrodes, yields glucose(saturated solution) oxidation current density of 3 mA cm�2
compared to 0.6 mA cm�2 for films without SWCNTs addition.68
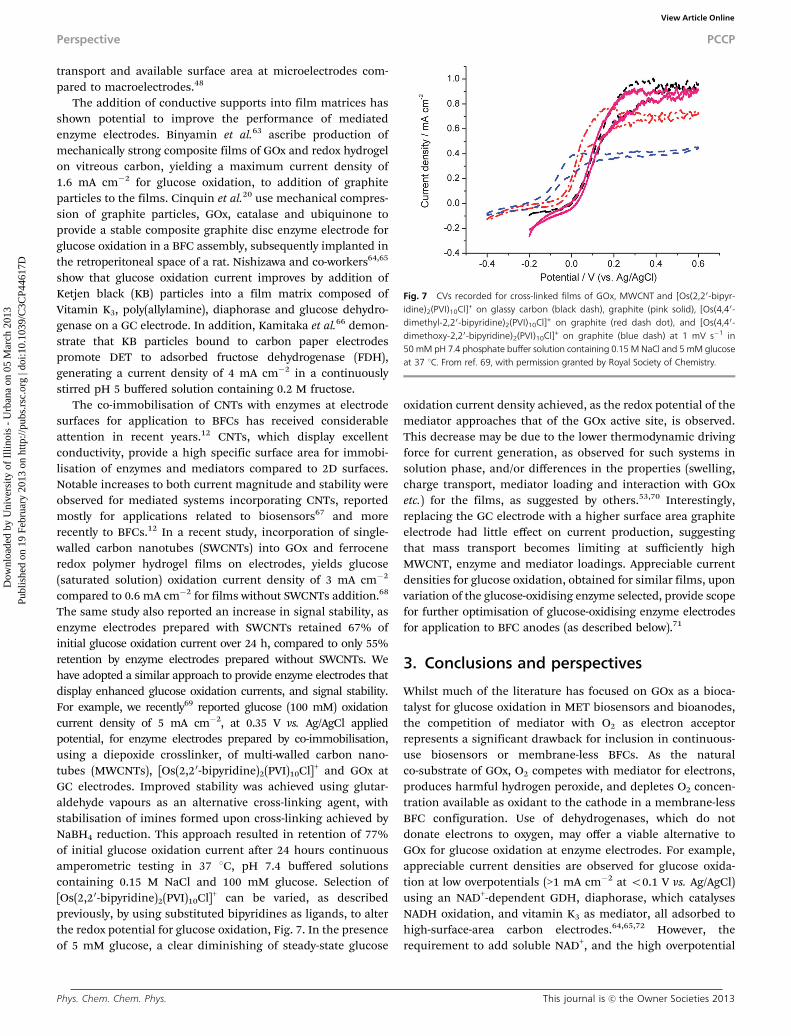
The same study also reported an increase in signal stability, asenzyme electrodes prepared with SWCNTs retained 67% ofinitial glucose oxidation current over 24 h, compared to only 55%retention by enzyme electrodes prepared without SWCNTs. Wehave adopted a similar approach to provide enzyme electrodes thatdisplay enhanced glucose oxidation currents, and signal stability.For example, we recently69 reported glucose (100 mM) oxidationcurrent density of 5 mA cm�2, at 0.35 V vs. Ag/AgCl appliedpotential, for enzyme electrodes prepared by co-immobilisation,using a diepoxide crosslinker, of multi-walled carbon nano-tubes (MWCNTs), [Os(2,20-bipyridine)2(PVI)10Cl]+ and GOx atGC electrodes. Improved stability was achieved using glutar-aldehyde vapours as an alternative cross-linking agent, withstabilisation of imines formed upon cross-linking achieved byNaBH4 reduction. This approach resulted in retention of 77%of initial glucose oxidation current after 24 hours continuousamperometric testing in 37 1C, pH 7.4 buffered solutionscontaining 0.15 M NaCl and 100 mM glucose. Selection of[Os(2,20-bipyridine)2(PVI)10Cl]+ can be varied, as describedpreviously, by using substituted bipyridines as ligands, to alterthe redox potential for glucose oxidation, Fig. 7. In the presenceof 5 mM glucose, a clear diminishing of steady-state glucose
oxidation current density achieved, as the redox potential of themediator approaches that of the GOx active site, is observed.This decrease may be due to the lower thermodynamic drivingforce for current generation, as observed for such systems insolution phase, and/or differences in the properties (swelling,charge transport, mediator loading and interaction with GOxetc.) for the films, as suggested by others.53,70 Interestingly,replacing the GC electrode with a higher surface area graphiteelectrode had little effect on current production, suggestingthat mass transport becomes limiting at sufficiently highMWCNT, enzyme and mediator loadings. Appreciable currentdensities for glucose oxidation, obtained for similar films, uponvariation of the glucose-oxidising enzyme selected, provide scopefor further optimisation of glucose-oxidising enzyme electrodesfor application to BFC anodes (as described below).71
3. Conclusions and perspectives
Whilst much of the literature has focused on GOx as a bioca-talyst for glucose oxidation in MET biosensors and bioanodes,the competition of mediator with O2 as electron acceptorrepresents a significant drawback for inclusion in continuous-use biosensors or membrane-less BFCs. As the naturalco-substrate of GOx, O2 competes with mediator for electrons,produces harmful hydrogen peroxide, and depletes O2 concen-tration available as oxidant to the cathode in a membrane-lessBFC configuration. Use of dehydrogenases, which do notdonate electrons to oxygen, may offer a viable alternative toGOx for glucose oxidation at enzyme electrodes. For example,appreciable current densities are observed for glucose oxida-tion at low overpotentials (>1 mA cm�2 at o0.1 V vs. Ag/AgCl)using an NAD+-dependent GDH, diaphorase, which catalysesNADH oxidation, and vitamin K3 as mediator, all adsorbed tohigh-surface-area carbon electrodes.64,65,72 However, therequirement to add soluble NAD+, and the high overpotential
Fig. 7 CVs recorded for cross-linked films of GOx, MWCNT and [Os(2,20-bipyr-idine)2(PVI)10Cl]+ on glassy carbon (black dash), graphite (pink solid), [Os(4,40-dimethyl-2,20-bipyridine)2(PVI)10Cl]+ on graphite (red dash dot), and [Os(4,40-dimethoxy-2,20-bipyridine)2(PVI)10Cl]+ on graphite (blue dash) at 1 mV s�1 in50 mM pH 7.4 phosphate buffer solution containing 0.15 M NaCl and 5 mM glucoseat 37 1C. From ref. 69, with permission granted by Royal Society of Chemistry.
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
for NADH oxidation at solid electrodes, makes use of NAD+-dependent dehydrogenases less promising for continuous-usesystems. The PQQ-dependent GDHs, which contain pyrrolo-quinoline quinone (PQQ) as a tightly bound cofactor, may offera more practical alternative, although adoption of their use islimited due to the inherent instability of the wild-typeenzyme.73 Generation of PQQ-dependent GDH mutants canenhance the stability73 and improve properties for DET73–76
by the enzyme for use in anodes of BFCs operating underphysiological conditions. Recent reports focus on exploringproperties of FAD-dependent GDHs for glucose oxidation inbiosensors and BFCs, offering advantages of high turnoverrates, substrate selectivity, good stability, and a lower redoxpotential than that of PQQ-GDHs.77 For example, films pre-pared by co-immobilisation of a novel FAD-GDH, isolated fromGlomerella cingulata, with [Os(4,40-dimethyl-2,2 0-bipyridine)2-(PVI)10Cl]+ on graphite electrodes yield glucose oxidation currentdensity of 0.49 mA cm�2 in pH 7.4 phosphate buffered solutioncontaining 30 mM glucose.77 Alternatively, Gorton and co-workershave reported extensively on the use of a range of cellobiosedehydrogenase (CDH) variants for application as enzyme electro-des in biosensors and as putative bioanodes in BFCs.78 The CDHs,which have active sites consisting of both a flavin and hemedomain, connected by a flexible linker region, are capable of bothDET and MET to electrodes, via the heme and flavin domains,respectively.79 Recent research has focused on production of CDHvariants that can efficiently oxidise glucose, instead of cellobioseor lactose, under physiological conditions, for application asglucose-oxidising bioanodes in BFC configurations.71,80,81
Power output of BFC assemblies can also be improved uponby improving anode coulombic efficiencies, through the combi-nation of rationally selected enzymes to provide for moreextensive (deeper) oxidation of the biofuel. This was firstdemonstrated in a BFC anode by Palmore et al.,6 providingfor complete oxidation of methanol to CO2 using NAD+/NADHregeneration by diaphorase and benzylviologen as mediator,coupled to three dehydrogenases. Subsequently, multiple-enzyme cascades for full or partial oxidation of ethanol,82,83
pyruvate,84 and glycerol85 in BFC assemblies has been demon-strated to improve power output. Enzyme electrodes preparedusing only a combination of CDH and pyranose dehydrogenase(PDH) co-immobilised with SWCNTs and an osmium redoxpolymer on pyrolytic graphite electrode have been used toincrease oxidation currents of glucose-oxidising anodes,extracting up to six electrons from one molecule of glucose.86
There is further scope for improving our understanding ofthe role each component plays in BFCs, by comparison ofperformance of enzyme electrodes to models. Such studiescan, in particular, enable better determination of limitingfactors in enzyme electrode reaction kinetics, mass transport,and system design. Bartlett and Pratt87 propose a general modelto describe diffusion and kinetic effects in substrate oxidisingenzyme electrodes, where enzyme and mediator are entrappedwithin the film. Excellent correlation between experimentalobservations and the theoretical model was demonstratedunder varying conditions of film thickness, enzyme loading,
mediator loading and substrate concentration, showingpotential for wider application of the model to glucose oxidisingenzyme electrodes. In order to compare such kinetic models toexperimental results it is important to accurately determine andreproducibly control the concentrations of enzyme and mediatorimmobilised at the electrode surface, a task which, as statedpreviously, is often difficult to achieve. To this end, Flexeret al.88,89 employed a layer-by-layer self-assembly approach fordeposition of layers of alternately charged GOx and osmiumredox polymer onto pre-treated (using self-assembly of thiols)gold electrodes, allowing precise control over the film thickness,enzyme loading and osmium concentration within the biofilm.Enzyme electrodes prepared in this manner were used togenerate an extensive range of data for glucose oxidationby dynamic electrochemistry, to which digital simulationscompared. Excellent agreement between both experiment andsimulations was achieved, facilitating the rational approach todesign of glucose oxidising enzyme electrodes if adoptedby others.
The adoption of a standardised methodology for BFC testingand reporting of data, as has been recommended for thecomparable field of microbial fuel cells,90,91 to allow bettercomparison and benchmarking between BFC electrodes,mediators and configurations is long overdue. Comparisonof reported half-cell current densities, onset potentials andpotentials for maximum current, as well as figures of meritfor assembled BFCs, is difficult as there is, to date, pooradherence to normalisation of such data to standard or acceptedconditions. The variability in experimental approaches includesdifferences, often unspecified, in specific surface areas of theelectrodes used, the control of mass transport of substrate to thesurface, the coverage, thickness, density or activity of the enzyme,mediator or support on the surface, the stability of the system, theoperational (and optimal) conditions (pH, ionic strength, bufferand chloride ion concentration, and temperature), and themethod used to estimate current density (amperometry at fixedpotential, cyclic voltammetry at various scan rates or in a BFCcircuit) or power density (fixed load, potentiostatic or galvano-static control). In addition, the conditions under which stability ofelectrodes are evaluated are often ill-defined i.e. if stability ismeasured intermittently, in which case storage conditionsbetween measurements should be described, or whether it ismeasured under (defined) continuous operational conditions.
In conclusion, many challenges exist for the future develop-ment of glucose oxidising enzyme electrodes for application asanodes in BFCs, most notably in terms of stability and currentgeneration whilst configuring systems to provide a cell voltagesufficient to power microelectronic devices. For example, METbased BFCs typically display optimum power output in therange of 0.2–0.5 V cell voltages, requiring stacking of cells toachieve the necessary voltage to power devices. Voltage losseswithin the cell may be minimised by astute selection of enzymeand mediator combinations to provide glucose oxidisingcurrents at low overpotentials. Electrode stability and currentoutput can be further enhanced through surface modifications,e.g. by addition of conductive supports to the biofilm matrix,
Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
which can facilitate high enzyme and mediator loadings andgreater retention of components. Moreover, the application oftheoretical models as an approach to aid enzyme electrodedesign is presently under-utilised and merits more rigorousinvestigation, although achieving precise control of the level ofeach component in a 3D matrix remains challenging. To this end,adoption of methodology for preparation of chemically function-alised surfaces, and deposition of films of controlled componentconcentration,58–61 provide scope for testing of such models, andfor delivery of enzyme electrodes capable of providing stablesignals of high currents at low redox potentials for applicationto continuous-use biosensors and anodes in biofuel cells.
Acknowledgements
Financial support by a Charles Parsons Energy Research Awardthrough Science Foundation Ireland is gratefully acknowledged.We also thank Brenda Egan and Todd Silverstein for usefuldiscussions. We thank Mike Lyons and Edmond Magner for theinvitation to present at the Electrochem 2012 meeting in Dublin,and to subsequently contribute this paper to PCCP.
References
1 L. C. Clark and C. Lyons, Ann. N.Y. Acad. Sci., 1962, 102,29–45.
2 S. J. Updike and G. P. Hicks, Science, 1967, 158, 270–272.3 S. J. Updike and G. P. Hicks, Nature, 1967, 214, 986–988.4 G. G. Guilbault and J. G. Montalvo, J. Am. Chem. Soc., 1969,
91, 2164–2165.5 J. Wang, Chem. Rev., 2008, 108, 814–825.6 G. T. R. Palmore, H. Bertschy, S. H. Bergens and
G. M. Whitesides, J. Electroanal. Chem., 1998, 443, 155–161.7 S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004,
104, 4867–4886.8 A. T. Yahiro, S. M. Lee and D. O. Kimble, Biochim. Biophys.
Acta, 1964, 88, 375–383.9 A. Heller, Phys. Chem. Chem. Phys., 2004, 6, 209–216 and
references therein.10 P. Kavanagh and D. Leech, Enzymatic fuel cells, in Bio-
electrochemistry: Fundamentals, Applications and Recent Develop-ments, Advances in Electrochemical Science and Engineering,ed. R. C. Alkire, D. M. Kolb and J. Lipkowski, Wiley, 2011,vol. 13, p. 229.
11 O. Courjean, F. Gao and N. Mano, Angew. Chem., Int. Ed.,2009, 48, 5897–5899.
12 M. Holzinger, A. L. Goff and S. Cosnier, Electrochim. Acta,2012, 82, 179–190 and references therein.
13 Y. Wang and Y. Yao, Microchim. Acta, 2012, 176, 271–277.14 R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811,
265–322.15 N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2003, 125,
6588–6594.16 F. Barriere, P. Kavanagh and D. Leech, Electrochim. Acta,
2006, 51, 5187–5192.
17 H. Sakai, T. Nakagawa, Y. Tokita, T. Hatazawa, T. Ikeda,S. Tsujimura and K. Kano, Energy Environ. Sci., 2009, 2,133–138.
18 M. T. Meredith, D. Y. Kao, D. Hickey, D. W. Schmidtke andD. T. Glatzhofera, J. Electrochem. Soc., 2011, 158, B166–B174.
20 P. Cinquin, C. Gondran, F. Giroud, S. Mazabrard,A. Pellissier, F. Boucher, J. P. Alcaraz, K. Gorgy,F. Lenouvel, S. Mathe, P. Porcu and S. Cosnier, PLoS One,2010, 5, e10476.
21 L. Halamkova, J. Halamek, V. Bocharova, A. Szczupak,L. Alfonta and E. Katz, J. Am. Chem. Soc., 2012, 134,5040–5043.
22 A. Szczupak, J. Halamek, L. Halamkova, V. Bocharova,L. Alfontac and E. Katz, Energy Environ. Sci., 2012, 5,8891–8895.
23 K. MacVittie, J. Halamek, L. Halamkova, M. Southcott,W. D. Jemison, R. Lobel and E. Katz, Energy Environ. Sci.,2013, 6, 81–86.
24 Z. Liu, B. Cho, T. Ouyang and B. Feldman, Anal. Chem.,2012, 84, 3403–3409.
25 T. P. Silverstein, J. Chem. Educ., 2012, 89, 1159–1167.26 A. Heller, AIChE J., 2005, 51, 1054–1066.27 P. Alzari, N. Anicet, C. Bourdillon, J. Moiroux and
J. M. Saveant, J. Am. Chem. Soc., 1996, 118, 6788–6789.28 N. J. Forrow, G. S. Sanghera and S. J. Walters, J. Chem. Soc.,
Dalton Trans., 2002, 3187–3194.29 R. S. Nicholson and I. Shain, Anal. Chem., 1965, 37, 178–190.30 A. E. G. Cass, G. Davis, G. D. Francis, H. A. O. Hill,
W. J. Aston, I. J. Higgins, E. V. Plotkin, L. D. L. Scott andA. P. F. Turner, Anal. Chem., 1984, 56, 667–671.
31 C. Bourdillon, C. Demaille, J. Moiroux and J. M. Saveant,J. Am. Chem. Soc., 1993, 115, 2–10.
32 P. Yeh and T. Kuwana, J. Electroanal. Chem., 1976, 123,1334–1339.
33 S. M. Zakeeruddin, D. M. Fraser, M. K. Nazeeruddin andM. Gratzel, J. Electroanal. Chem., 1992, 337, 253–283.
34 W. J. Albery, P. N. Bartlett, B. J. Driscoll and R. B. Lennox,J. Electroanal. Chem., 1992, 323, 77–102.
35 B. Limoges, J. Moiroux and J. M. Saveant, J. Electroanal.Chem., 2002, 521, 1–7.
36 V. Flexer, M. V. Ielminia, E. J. Calvo and P. N. Bartlett,Bioelectrochemistry, 2008, 74, 201–209.
37 A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285.38 B. Limoges and J. M. Saveant, J. Electroanal. Chem., 2003,
549, 61–70.39 R. J. Forster and J. G. Vos, Macromolecules, 1990, 23,
4372–4377.40 T. J. Ohara, R. Rajagopalan and A. Heller, Anal. Chem., 1993,
65, 3512–3517.41 G. Inzelt, Electrochim. Acta, 1989, 34, 83–91.42 P. Daum, J. R. Lenhard, D. Rolison and R. W. Murray, J. Am.
Chem. Soc., 1980, 102, 4649–4653.43 P. Daum and R. W. Murray, J. Phys. Chem., 1981, 85,
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
44 R. J. Forster and J. G. Vos, Langmuir, 1994, 10, 4330–4338.45 A. Aoki and A. Heller, J. Phys. Chem., 1993, 97, 11014–11019.46 D. N. Blauch and J. M. Saveant, J. Am. Chem. Soc., 1992, 114,
3323–3332.47 N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2002, 124,
2962–12963.48 F. Mao, N. Mano and A. Heller, J. Am. Chem. Soc., 2003, 125,
4951–4957.49 R. J. Forster, D. A. Walsh, N. Mano, F. Mao and A. Heller,
Langmuir, 2004, 20, 862–868.50 N. Mano, F. Mao and A. Heller, Chem. Commun., 2004,
2116–2117.51 A. Prevoteau and N. Mano, Electrochim. Acta, 2012, 68,
128–133.52 R. J. Forster and J. G. Vos, Electrochim. Acta, 1992, 37,
159–167.53 J. W. Gallaway and S. A. C. Barton, J. Electroanal. Chem.,
2009, 626, 149–155.54 N. S. Hudak, J. W. Gallaway and S. C. Barton, J. Electroanal.
Chem., 2009, 629, 57–62.55 J. W. Gallaway and S. A. C. Barton, J. Am. Chem. Soc., 2008,
130, 8527–8536.56 B. B. Dambatta and J. R. Ebdon, Eur. Polym. J., 1986, 22,
783–786.57 M. H. Allen, S. T. Hemp, A. E. Smith and T. E. Long,
Macromolecules, 2012, 45, 3669–3676.58 S. Poller, Y. Beyl, J. Vivekananthan, D. A. Guschin and
W. Schuhmann, Bioelectrochemistry, 2012, 87, 178–184.59 P. O Conghaile, S. Poller, D. MacAodha, W. Schuhmann and
D. Leech, Biosens. Bioelectron., 2013, 43, 30–37.60 S. Boland, P. Kavanagh and D. Leech, ECS Trans., 2008, 13,
77–87.61 P. O Conghaile, S. Kamireddy, D. MacAodha, P. Kavanagh
and D. Leech, Anal. Bioanal. Chem., 2013, DOI: 10.1007/s00216-012-6628-9.
62 S. Rengaraj, P. Kavanagh and D. Leech, Biosens. Bioelectron.,2011, 30, 294–299.
63 G. Binyamin, J. Cole and A. Heller, J. Electrochem. Soc., 2000,147, 2780–2783.
64 F. Sato, M. Togo, M. K. Islam, T. Matsue, J. Kosuge,N. Fukasaku, S. Kurosawa and M. Nishizawa, Electrochem.Commun., 2005, 7, 643–647.
65 M. Togo, A. Takamura, T. Asai, H. Kaji and M. Nishizawa,Electrochim. Acta, 2007, 52, 4669–4674.
66 Y. Kamitaka, S. Tsujimura, N. Setoyama, T. Kajinob andK. Kano, Phys. Chem. Chem. Phys., 2007, 9, 1793–1801.
67 J. Wang, Electroanalysis, 2005, 17, 7–14.68 T. O. Tran, E. G. Lammert, J. Chen, S. A. Merchant,
D. B. Brunski, J. C. Keay, M. B. Johnson, D. T. Glatzhoferand D. W. Schmidtke, Langmuir, 2011, 27, 6201–6210.
69 D. MacAodha, M. L. Ferrer, P. O Conghaile, P. Kavanagh andD. Leech, Phys. Chem. Chem. Phys., 2012, 14, 14667–14672.
70 M. N. Zafar, X. Wang, C. Sygmund, R. Ludwig, D. Leech andL. Gorton, Anal. Chem., 2012, 84, 334–341.
71 D. MacAodha, P. O Conghaile, B. Egan, P. Kavanagh,C. Sygmund, R. Ludwig and D. Leech, Electroanalysis,2013, 25, 94–100.
72 A. Sato, K. Kano and T. Ikeda, Chem. Lett., 2003, 32,880–881.
73 N. Yuhashi, M. Tomiyama, J. Okuda, S. Igarashi,K. Ikebukuro and K. Sode, Biosens. Bioelectron., 2005, 20,2145–2150.
74 J. Okuda-Shimazaki, N. Kakehi, T. Yamazaki, M. Tomiyamaand K. Sode, Biotechnol. Lett., 2008, 30, 1753–1758.
75 F. Durand, C. Stines-Chaumeil, V. Flexer, I. Andre andN. Mano, Biochem. Biophys. Res. Commun., 2010, 402,750–754.
76 V. Flexer, F. Durand, S. Tsujimura and N. Mano, Anal.Chem., 2011, 83, 5721–5727.
77 M. N. Zafar, X. Wang, C. Sygmund, R. Ludwig, D. Leech andL. Gorton, Anal. Chem., 2012, 84, 334–341.
78 R. Ludwig, W. Harreither, F. Tasca and L. Gorton, Chem-PhysChem, 2010, 11, 2674–2697 and references therein.
79 F. Tasca, L. Gorton, W. Harreither, D. Haltrich, R. Ludwigand G. Noll, Anal. Chem., 2009, 81, 2791–2798.
80 W. Harreither, C. Sygmund, M. Augustin, M. Narciso,M. L. Rabinovich, L. Gorton, D. Haltrich and R. Ludwig,Appl. Environ. Microbiol., 2011, 77, 1804–1815.
81 W. Harreither, A. K. G. Felice, R. Paukner, L. Gorton,R. Ludwig and C. Sygmund, Biotechnol. J., 2012, 7,1359–1366.
82 N. L. Akers, C. M. Moore and S. D. Minteer, Electrochim.Acta, 2005, 50, 2521–2525.
83 S. Topcagic and S. D. Minteer, Electrochim. Acta, 2006, 51,2168–2172.
84 D. Sokic-Lazic and S. D. Minteer, Electrochem. Solid-StateLett., 2009, 12, F26–F28.
85 R. L. Arechederra and S. D. Minteer, Fuel Cells, 2009, 9,63–69.
86 F. Tasca, L. Gorton, M. Kujawa, I. Patel, W. Harreither,K. P. Clemens, R. Ludwig and G. Noll, Biosens. Bioelectron.,2010, 25, 1710–1716.
87 P. N. Bartlett and K. F. E. Pratt, J. Electroanal. Chem., 1995,397, 61–78.
88 V. Flexer, K. F. E. Pratt, F. Garay, P. N. Bartlett andE. J. Calvo, J. Electroanal. Chem., 2008, 616, 87–98.
89 V. Flexer, E. J. Calvo and P. N. Bartlett, J. Electroanal. Chem.,2010, 646, 24–32.
90 J. Menicucci, H. Beyenal, E. Marsili, R. Angathevar, G. Demirand Z. Lewandowski, Environ. Sci. Technol., 2006, 40,1062–1068.
91 B. E. Logan, B. Hamelers, R. Rozendal, U. Schroder, J. Keller,S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey,Environ. Sci. Technol., 2006, 40, 5181–5192.