Brain Research Bulletin, Vol. 24, pp. 611-686. 0 Pergamon Press plc, 1990. Printed in the U.S.A. 0361-9230/90 $3.00 + .OO Microangiopathy , the Vascular Basement Membrane and Alzheimer’s Disease: A Review LYNN S. PE~M~R*’ AND HELENA CNANG CHUI~ University of Southern California School of Medicine, *Department of Neurology, Los Angeles, CA 90033 and ~Al~i~r’s Disease Diagnostic and Treatment Center Ranch0 Los Amigos Medical Center, Downey, CA 93242 PERLMUTITR, L. S. AND H. C. CHUI. Microangiopathy, the vascular basement membrane and Alzheimer’s disease: A review. BRAIN RES BULL 24(5) 677-686, 1990.-Tbe present review focuses on the vascular basement membrane (VRM) and its relationship to the lesions of Alzbeimer’s disease (AD). Examination of the fine structure of the microvasculature reveals AD-associated VBM alterations, which include both thickening and vacuolization. ~un~~he~s~ confirms that all three intrinsic VBM components [collagen type IV, laminin, and heparan sulfate proteoglycan (HSPG)] outline the capillary bed, which is pathologically altered in AD patients (microangiopathy). Ultrastructural analyses of AD tissue samples demonstrate that HSPG’s normal staining pattern is disrupted on the endothelial surface of the VBM in brain regions affected by Alzbeimer lesions. Similarly altered VBM is reported to occur in the kidney of patients with diabetes mellitus, where it is associated with a leakage of protein. All three VBM components i~~ola~l capillaries, amyloid and pl~ue-~s~ia~d glial processes, suggesting a link between microangiopathy and senile plaque formation. In addition, the consistent colocaliration of HSPG with several forms of amyloid implies an involvement in amyloidogenesis. Finally, the neurotrophic effects of P-amyloid, combined with neurite-promoting effects of laminin and HSPG, could create a strong focus for an aberrant sprouting response. Such a response is postulated to result in plaque-associated degenerating net&es. Thus, VBM components could serve as a nidus for plaque formation, playing a role in the development of neuritic as well as amyloidotic elements. Heparan sulfa& proteoglycan Collagen type IV Laminin Blood-brain barrier Amyloid Glia ALZHEIMER’S DISEASE basement membrane (VBM), in the disease process. Alzheimer’s disease (AD), the most common cause of progres- ‘Ihe pathogenetic mechanisms involved in the progression of sive dementia, results in atrophy of the brain and neuronal loss. In AD are unknown. Each of the three characteristic lesions can addition, three classic lesions show a characteristic regional occur inde~ndent of the other two: NFI ate found in Postenceph- distribution (10,7’S), preferentially affecting the hippocampus and alitic Parkinsonism and Parkinson-Dementia Complex of Guam association cortices. Primary sensory and motor cortices, as well (25); CAA is observed in familial congophilic angiopathy (10); as the cerebellum, are relatively spared. One of these lesions senile plaques are associated with normal aging in a number of affects the neuron, where accumulations of paired helical fila- mammalian species (70). Thus, while several lesions are charac- ments form ne~o~b~ll~ tangles (NFI). A second, known as teristic, no single lesion can be considered pa~o~omonic for the cerebral amyloid angiopathy (CAA), involves the deposition of disease. amyloid in the walls of the cerebral vasculature (10,lS). The third Amyloid, on the other hand, is associated with all three of the typical lesion, the senile plaque, is a more heterogeneous struc- ture. It is comprised of a central core of amyloid around which are classic Alzheimer lesions. The term amyloid refers to an abnormal dispersed amyloid fib&, dystrophic net&es, capillaries, and fibrillar substance with specific conformational (&pleated sheet) reactive astroglia and microglia (48, 82-84). This lesion is of and staining (Congo red and thioflavine) characteristics (18,19). particular interest, as it suggests interrelationships between neu- Amyloids are found in several systemic and cerebral disorders, ritic, vascular and glial elements in AD. Thus, AD affects virtually and apparently result from the pathologic processing of chemically every component of the central nervous system-but in a very unrelated proteins (18,19). The composition of the amyloid specific, regional fashion. The present report is a review of the contained in NFT is ~~0~; the amyloid found in plaques and possible role of the vasculature, and specifically the vascular cerebral vessels, however, has been sequenced and found to be virtually identical (20,SS). The presence of this p- (also known as ‘Requests for reprints should be addressed to Lynn S. Perlmutter, University of Southern California School of Medicine, Department of Neurology, 2025 Zonal Avenue, RMR-407, Los Angeles, CA 90033 677
Transcript
Brain Research Bulletin, Vol. 24, pp. 611-686. 0 Pergamon Press plc, 1990. Printed in the U.S.A. 0361-9230/90 $3.00 + .OO
Microangiopathy , the Vascular Basement Membrane and
Alzheimer’s Disease: A Review
LYNN S. PE~M~R*’ AND HELENA CNANG CHUI~
University of Southern California School of Medicine, *Department of Neurology, Los Angeles, CA 90033 and ~Al~i~r’s Disease Diagnostic and Treatment Center
Ranch0 Los Amigos Medical Center, Downey, CA 93242
PERLMUTITR, L. S. AND H. C. CHUI. Microangiopathy, the vascular basement membrane and Alzheimer’s disease: A review. BRAIN RES BULL 24(5) 677-686, 1990.-Tbe present review focuses on the vascular basement membrane (VRM) and its relationship to the lesions of Alzbeimer’s disease (AD). Examination of the fine structure of the microvasculature reveals AD-associated VBM alterations, which include both thickening and vacuolization. ~un~~he~s~ confirms that all three intrinsic VBM components [collagen type IV, laminin, and heparan sulfate proteoglycan (HSPG)] outline the capillary bed, which is pathologically altered in AD patients (microangiopathy). Ultrastructural analyses of AD tissue samples demonstrate that HSPG’s normal staining pattern is disrupted on the endothelial surface of the VBM in brain regions affected by Alzbeimer lesions. Similarly altered VBM is reported to occur in the kidney of patients with diabetes mellitus, where it is associated with a leakage of protein. All three VBM components i~~ola~l capillaries, amyloid and pl~ue-~s~ia~d glial processes, suggesting a link between microangiopathy and senile plaque formation. In addition, the consistent colocaliration of HSPG with several forms of amyloid implies an involvement in amyloidogenesis. Finally, the neurotrophic effects of P-amyloid, combined with neurite-promoting effects of laminin and HSPG, could create a strong focus for an aberrant sprouting response. Such a response is postulated to result in plaque-associated degenerating net&es. Thus, VBM components could serve as a nidus for plaque formation, playing a role in the development of neuritic as well as amyloidotic elements.
Heparan sulfa& proteoglycan Collagen type IV Laminin Blood-brain barrier Amyloid Glia
ALZHEIMER’S DISEASE basement membrane (VBM), in the disease process. Alzheimer’s disease (AD), the most common cause of progres- ‘Ihe pathogenetic mechanisms involved in the progression of
sive dementia, results in atrophy of the brain and neuronal loss. In AD are unknown. Each of the three characteristic lesions can addition, three classic lesions show a characteristic regional occur inde~ndent of the other two: NFI ate found in Postenceph- distribution (10,7’S), preferentially affecting the hippocampus and alitic Parkinsonism and Parkinson-Dementia Complex of Guam association cortices. Primary sensory and motor cortices, as well (25); CAA is observed in familial congophilic angiopathy (10); as the cerebellum, are relatively spared. One of these lesions senile plaques are associated with normal aging in a number of affects the neuron, where accumulations of paired helical fila- mammalian species (70). Thus, while several lesions are charac- ments form ne~o~b~ll~ tangles (NFI). A second, known as teristic, no single lesion can be considered pa~o~omonic for the cerebral amyloid angiopathy (CAA), involves the deposition of disease. amyloid in the walls of the cerebral vasculature (10,lS). The third Amyloid, on the other hand, is associated with all three of the typical lesion, the senile plaque, is a more heterogeneous struc- ture. It is comprised of a central core of amyloid around which are
classic Alzheimer lesions. The term amyloid refers to an abnormal
dispersed amyloid fib&, dystrophic net&es, capillaries, and fibrillar substance with specific conformational (&pleated sheet)
reactive astroglia and microglia (48, 82-84). This lesion is of and staining (Congo red and thioflavine) characteristics (18,19).
particular interest, as it suggests interrelationships between neu- Amyloids are found in several systemic and cerebral disorders,
ritic, vascular and glial elements in AD. Thus, AD affects virtually and apparently result from the pathologic processing of chemically
every component of the central nervous system-but in a very unrelated proteins (18,19). The composition of the amyloid
specific, regional fashion. The present report is a review of the contained in NFT is ~~0~; the amyloid found in plaques and
possible role of the vasculature, and specifically the vascular cerebral vessels, however, has been sequenced and found to be virtually identical (20,SS). The presence of this p- (also known as
‘Requests for reprints should be addressed to Lynn S. Perlmutter, University of Southern California School of Medicine, Department of Neurology, 2025 Zonal Avenue, RMR-407, Los Angeles, CA 90033
677
PEKLMI: I”ft:K .iNL) (‘til:i
FIG. 1. The cerebral rn~cro~~as~ui~[ure. Red blood cells (KBC) fill the lumen trr this capillary from the cerebellum of a G-year-old male Pick‘s d&se patient ~p(~strno~~m delay = 4 hours). The endatheii~i ceil nucleus (N) is present. and tight junctions can be seen (arrowheads). A pericyte (PI abuts the capillary. and the vascular basement membrane (arroos) can be traced around the cndothelial cell and around the pericyte. Astrocytic endfect I*) wrround the capillary. Final magnification, X500 Y
A4) amyloid in both lesions has suggested a pathogenetic relation- ship between the fo~ation of the senile plaque and atnyloid-laden vasculature.
AD-associated pathology of the microvasculature has long been described (27). Capillaries appear fragmented and irregular (27,65,66, 87), and develop thickened and reduplicated basement membranes (41,55). Recently, the severity of these changes has been correlated with neuronal loss (87). Furthermore. components of the capillary vascular basement membrane (VBM) have been colocalized with senile plaques. The present review focuses on the VBM and its relationship to other characteristic lesions of AD. First, the normal anatomy and function of the microvasculature and the VBM will be described. Second, clinical and experimental VBM pathology in the brain and elsewhere will be discussed. Finally, the relationship between VBM pathology and the classic lesions of AD will be examined. It is suggested that the VBM may play an active role in the pathogenesis of this disease.
THE CEREBRAL MICROVASCULATURE
The cerebral microvasculature is comprised of three cell types (Fig. 1). The first type is the specialized endothelial cells of the brain which form the bl~-brain barrier. They contain no fenes- trations, few pinocytotic vesicles, and epithelial-like tight junc- tions (5, 21, 52, 53). Second are the pericytes, which appear as flattened cells whose processes encompass the capillary wall. They share antigenic determinants with macrophages (23), and are thought to transform into microglia (23, 32, 49,58). Pericytes also inhibit endothelial cell growth in cutture (2.64) and may regulate capillary tone (60). The third cell type is the astrocyte. A sheath of astrocytic endfeet surround the endothelium. While their function is unclear, they are thought to influence endothelial cell expression of brain-specific features (e.g., tight junctions) (5, 21, 3.5). The basement membranes produced by these cells fuse to form a
trilaminar structure similar to that seen in the renal glomerulus (76). This flocculent-appearing “membrane” provides physical support. controls cellular migration, selectively filters macromol- ecules. and may influence endothelial cell function (2 I, 22, 76). The VBM. along with the astrocytic endfeet, may also provide the brain with additional protection against extravasated protein (77).
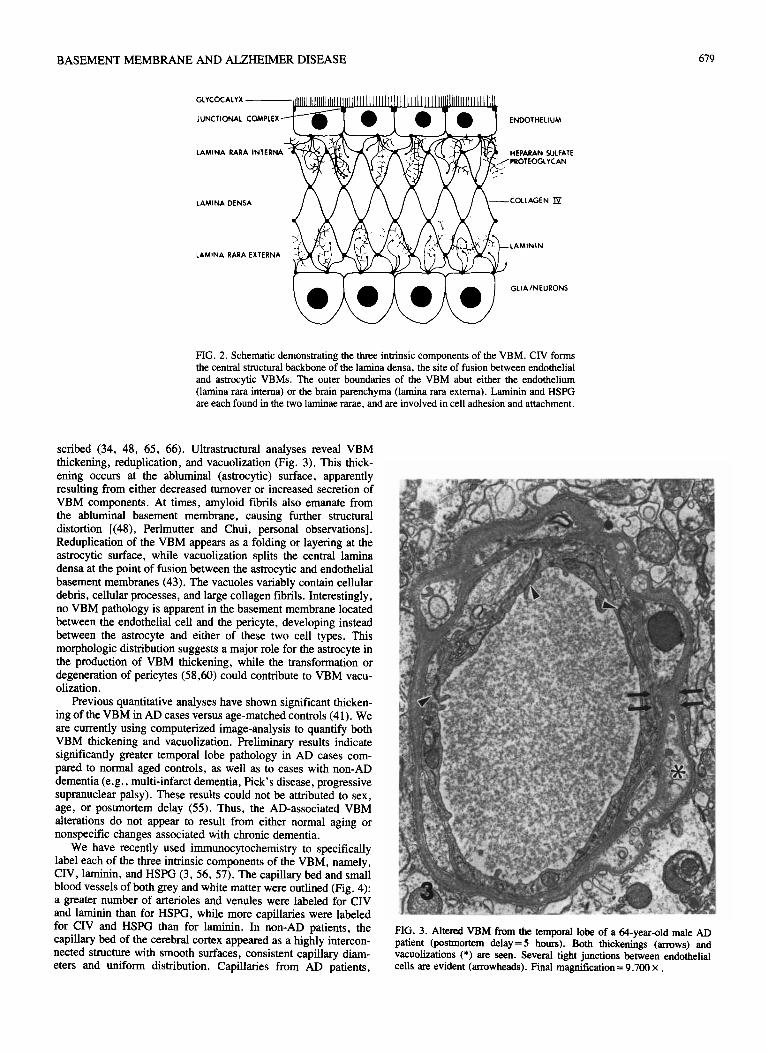
The three intrinsic components of the VBM are collagen type 1V (CIV), laminin, and heparan sulfate proteoglycan (HSPG) (Fig. 2). CIV forms the central structural backbone of the lamina densa. the site of fusion between endothelial and astrocytic VBMs (43). Laminin and HSPG are each found at the endothelial (lamina rara interna) and the astrocytic (lamina rara externa) VBM surfaces (15.43). They are involved in cell adhesion and attachment ( 15.43), and are potent neurite-promoting factors (8, 9, 37-39). In addition, the negatively charged HSPG molecule can filter anionic and neutral proteins f 15). Charge is irn~~nt in the maintenance of the renal glomerular barrier. and may be important for the BBB as well (12. 26. 51. 59).
HSPG has been associated with neuronal cell surfaces and cholinergic synaptic vesicles as well as the VBM (24,74). CIV and laminin, on the other hand, are found exclusively in the basement membranes of healthy tissues (43, 61, 86). Moreover, astrocytes respond to injury with de novo production of all three VBM components (40,47j, and basement membrane-lined astrocyte lacunae form during revascularization (47). Thus, endothelial cells, pericytes and astrocytes produce all three intrinsic VBM components; this fact may prove important in understanding cerebral VBM pathologies.
PATHOLOGIC ALTER.~TIONS OF THE VASCULAR
BASEMENT MEMBRANE
Al-heher ‘s Di.wu.~e .,
AD-associated alterations of the VBM have long been de-
BASEMENT MEMBRANE AND ALZHEIMER DISEASE 619
GLYCOCALYX
JUNCTIONAL COMPLEX
LAMINA RARA INTERNA
ENDOTHELIUM
HEPARAN SULFATE PROTEOGLVCAN
LAMINA DENSA COLLAGEN IS!
LAMININ
LAMINA RARA EXTERNA
GLIAINEURONS
FIG. 2. Schematic demonstrating the three intrinsic components of the VBM. CIV forms the central structural backbone of the lamina densa, the site of fusion between endothelial and astrocytic VBMs. The outer boundaries of the VBM abut either the endothelium (lamina rara intema) or the brain parenchyma (lamina rara extema). Laminin and HSPG are each found in the two laminae rarae. and are involved in cell adhesion and attachment.
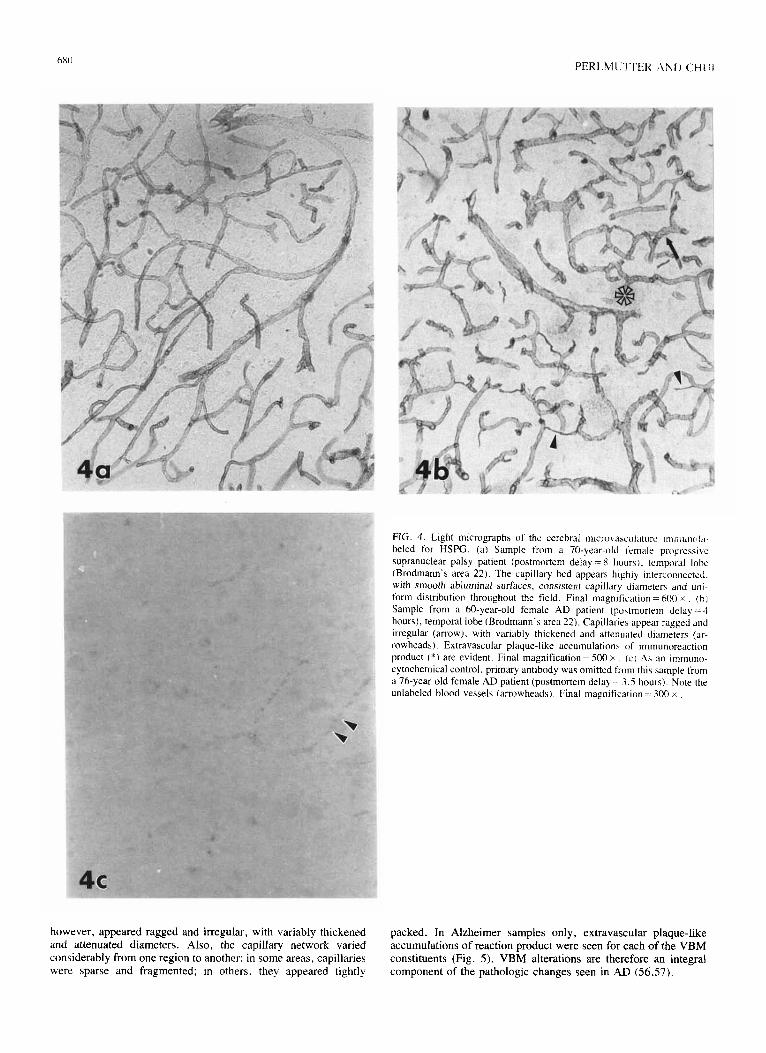
scribed (34, 48, 65, 66). Ultrastructural analyses reveal VBM thickening, reduplication, and vacuolization (Fig. 3). This thick- ening occurs at the abluminal (astrocytic) surface, apparently resulting from either decreased turnover or increased secretion of VBM components. At times, amyloid fibrils also emanate from the abluminal basement membrane, causing further structural distortion [(48), Perlmutter and Chui, personal observations]. Reduplication of the VBM appears as a folding or layering at the astrocytic surface, while vacuolization splits the central lamina densa at the point of fusion between the astrocytic and endothelial basement membranes (43). The vacuoles variably contain cellular debris, cellular processes, and large collagen fibrils. Interestingly, no VBM pathology is apparent in the basement membrane located between the endothelial cell and the pericyte, developing instead between the astrocyte and either of these two cell types. This morphologic distribution suggests a major role for the astrocyte in the production of VBM thickening, while the transformation or degeneration of pericytes (5860) could contribute to VBM vacu- olization.
Previous quantitative analyses have shown significant thicken- ing of the VBM in AD cases versus age-matched controls (41). We are currently using computerized image-analysis to quantify both VBM thickening and vacuolization. Preliminary results indicate significantly greater temporal lobe pathology in AD cases com- pared to normal aged controls, as well as to cases with non-AD dementia (e.g., multi-infarct dementia, Pick’s disease, progressive supranuclear palsy). These results could not be attributed to sex, age, or postmortem delay (55). Thus, the AD-associated VBM alterations do not appear to result from either normal aging or nonspecific changes associated with chronic dementia.
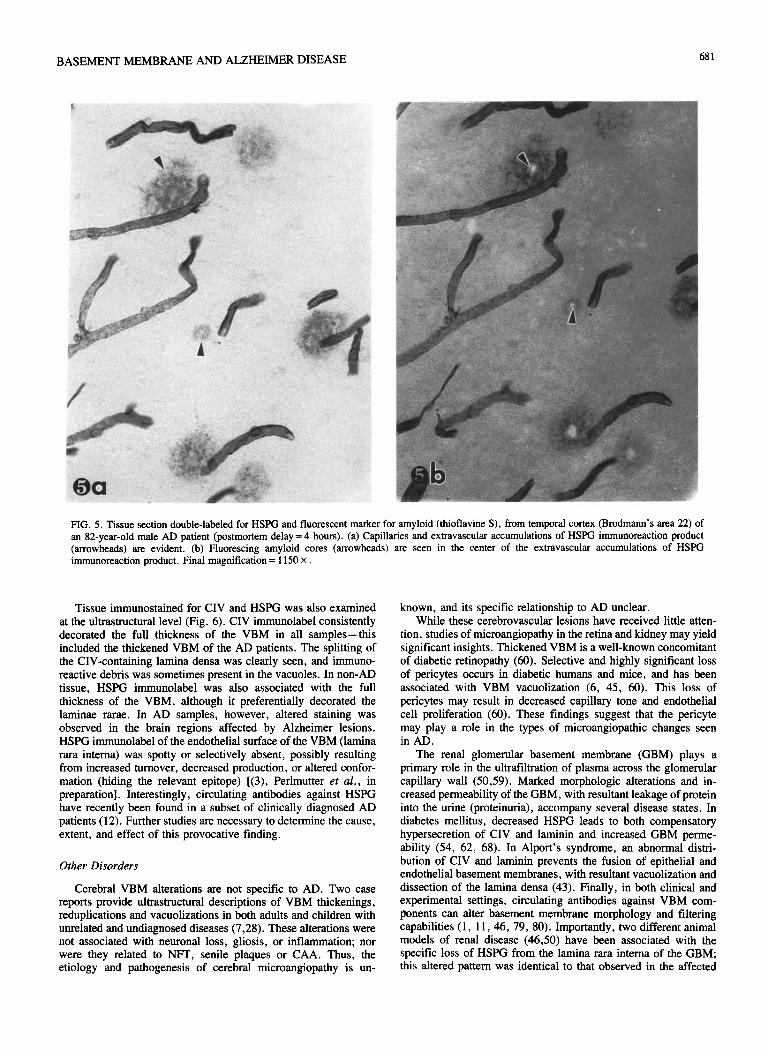
We have recently used immunocytochemistry to specifically label each of the three intrinsic components of the VBM, namely, CIV, laminin, and HSPG (3.56, 57). The capillary bed and small blood vessels of both grey and white matter were outlined (Fig. 4): a greater number of arterioles and venules were labeled for CIV and laminin than for HSPG, while more capillaries were labeled for CIV and HSPG than for laminin. In non-AD patients, the capillary bed of the cerebral cortex appeared as a highly intercon-
nected structure with smooth surfaces, consistent capillary diam- eters and uniform distribution. Capillaries from AD patients,
FIG. 3. Altered VBM from the temporal lobe of a f+year-old mate AD patient (postmortem &lay=5 hours). Both thickenings (arrows) and vacuolizations (*) are seen. Several tight junctions between endothelial cells are evident (arrowheads). Final magnification = 9,700 X .
FIG. 1. Light micrographs of the cerebral mtcrovasculature ~mmuool,~. beled for HSPG. (a) Sample from a 70-year-old female progresstcc supranuclear palsy patient (postmortem delay = 8 hour\). temporal lobe (Brodmann’s area 22). The capillary bed appears highly interconnected. with smooth abluminal surfaces, consistent capillary diameters and unr- form distribution throughout the field. Final magnification = 600 x (hi Sample from a 60-year-old female AD patient (postmortem delay-4 hours), temporal lobe (Brodmann’s area 22). Capillaties appear ragged and irregular (arrow). with variably thickened and attenuated diameters (ar- rowheads). Extravascular plaque-like accumulations of immunoreaction product t*) are evident. Final magnification = 500 x (c-1 As an immuno- cytochemical control, primary antibody was omitted from thts sample from a 76-year-old female AD patient (postmortem delay = 3.5 hours). Kate the unlabeled blood vessels (arrowheads). Final magnification = 300 x
however, appeared ragged and irregular, with variably thickened and attenuated diameters. Also, the capillary network varied considerably from one region to another: in some areas, capillaries were sparse and fragmented: in others. they appeared tightly component of the pathologic changes seen in AD (56.57)
packed. In Alzheimer samples only, extravascular plaque-like accumulations of reaction product were seen for each of the VBM constituents (Fig. 5). VBM alterations are therefore an integral
BASEMENT MEMBRANE AND ALZHEIMER DISEASE
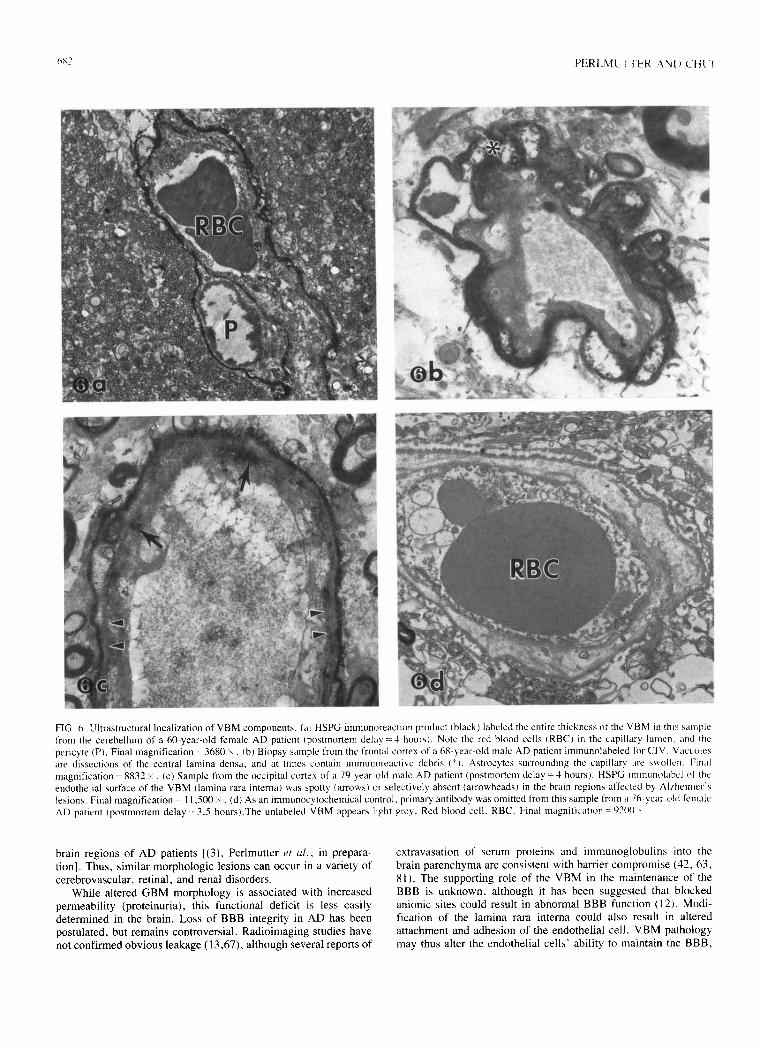
PIG. 5. Tissue section double-labeled for HSPG and fluorescent marker for amyloid (thioflavine S), from temporal cortex (Brodmann’s area 22) of an 82-year-old male AD patient (postmortem delay = 4 hours). (a) Capillaries and extravascular accumulations of HSFG immunoreaction product (arrowheads) are evident. (b) Fluorescing amyloid cores (arrowheads) are seen in the center of the extravascular accumulations of HSPG immunoreaction product. Final magnification = I 150 X .
Tissue immunostained for CIV and HSPG was also examined at the ultrastructural level (Fig. 6). CIV immunolabel consistently decorated the full thickness of the VBM in all samples-this included the thickened VBM of the AD patients. The splitting of the CIV-containing lamina densa was clearly seen, and immuno- reactive debris was sometimes present in the vacuoles. In non-AD tissue, HSPG immunolabel was also associated with the full thickness of the VBM, although it preferentially decorated the laminae rarae. In AD samples, however, altered staining was observed in the brain regions affected by Alzheimer lesions. HSPG immunolabel of the endothelial surface of the VBM (lamina rara intema) was spotty or selectively absent, possibly resulting from increased turnover, decreased production, or altered confor- mation (hiding the relevant epitope) [(3), Perlmutter er al., in preparation]. Interestingly, circulating antibodies against HSPG have recently been found in a subset of clinically diagnosed AD patients (12). Further studies are necessary to determine the cause, extent, and effect of this provocative finding.
Other Disorders
Cerebral VBM alterations are not specific to AD. Two case reports provide ultrastmcturaI descriptions of VBM thickenings, reduplications and vacuolizations in both adults and children with unrelated and undiagnosed diseases (7,28). These alterations were not associated with neuronal loss, gliosis, or inflammation; nor were they related to NFT, senile plaques or CAA. Thus, the etiology and pathogenesis of cerebral microangiopathy is un-
known, and its specific relationship to AD unclear. While these cerebrovascular lesions have received little atten-
tion, studies of microangiopathy in the retina and kidney may yield significant insights. Thickened VBM is a well-known concomitant of diabetic retinopathy (60). Selective and highly significant loss of pericytes occurs in diabetic humans and mice, and has been associated with VBM vacuolization (6, 45, 60). This loss of pericytes may result in decreased capillary tone and endothelial cell proliferation (60). These findings suggest that the pericyte may play a role in the types of microangiopathic changes seen in AD.
The renal glomerular basement membrane (GBM) plays a primary role in the ultrafiltration of plasma across the glomerular capillary wall (50,59). Marked morphologic alterations and in- creased permeability of the GBM, with resultant leakage of protein into the urine (proteinuria), accompany several disease states. In diabetes mellitus, decreased HSPG leads to both compensatory hypersecretion of CIV and laminin and increased GBM perme- ability (54, 62, 68). In Alport’s syndrome, an abnormal distri- bution of CIV and laminin prevents the fusion of epithelial and endothelial basement membranes, with resultant vacuolization and dissection of the lamina densa (43). Finally, in both clinical and experimental settings, circulating antibodies against VBM com- ponents can alter basement membrane morphology and filtering capabilities (1, 11,46, 79, 80). Importantly, two different animal models of renal disease (46,50) have been associated with the specific loss of HSPG from the lamina rara intema of the GBM; this altered pattern was identical to that observed in the affected
f’tiRI_MI. I-‘I’E+! .4tl) (‘HI :
FIG. 6. Illtrastructural localization of VBM components. la) HSPG immunoreaction product (black) labeled the entire thickness ot the LBM 111 thl\ ~rrple from the cerebellum of a ho-year-old female AD patient (postmortem delay :=J hours). Note the red blood cells rRBC) in the capillary lumen. and the pericyte (P). Final magnification = 3680 x (b) Biopsy sample from the frontal cortex of a h&year-old male AD patient immunolabeled for CIV. Vacuoles are dissections of the central lamina densa. and at times contain immunoreacti$e debris (“l. Astrocyte\ surrounding the capillar! are svollcn Final magnification = 8832 )i (c) Sample from the occipital cortex of a 70.year-old male AD patient (postmortem delay = 4 hours). HSPG ~mmunolabcl of the endothelial surface of the VBM (lamina rara interna) was spotty (arrows) or selectively absent (arrowheads) in the brain regions affected b] Alrheimer’\ lesions. Final magnification = 11,500 x (d) As an immunocytochemical control. primary antibody wah omitted from this sample from a 76-year-old female AD patient (postmortem delay = 3.5 hours).The unlabeled VBM appears light grey. Red blood cell. RBC. Final magnification = W)(l .
brain regions of AD patients [(3), Perlmutter et u/. , in prepara- tion]. Thus, similar morphologic lesions can occur in a variety of cerebrovascular. retinal, and renal disorders.
While altered GBM morphology is associated with increased permeability (proteinuria), this functional deficit is less easily determined in the brain. Loss of BBB integrity in AD has been postulated, but remains controversial. Radioimaging studies have not confirmed obvious leakage (13,67). although several reports of
extravasation of serum proteins and immunoglobulins into the brain parenchyma are consistent with barrier compromise (42, 63, 81). The supporting role of the VBM in the maintenance of the BBB is unknown, although it has been suggested that blocked anionic sites could result in abnormal BBB function (12). Modi- fication of the lamina rara interna could also result in altered attachment and adhesion of the endothelial cell. VBM pathology may thus alter the endothelial cells’ ability to maintain the BBB.
BASEMENT MEMBRANE AND ALZHEIMER DISEASE 683
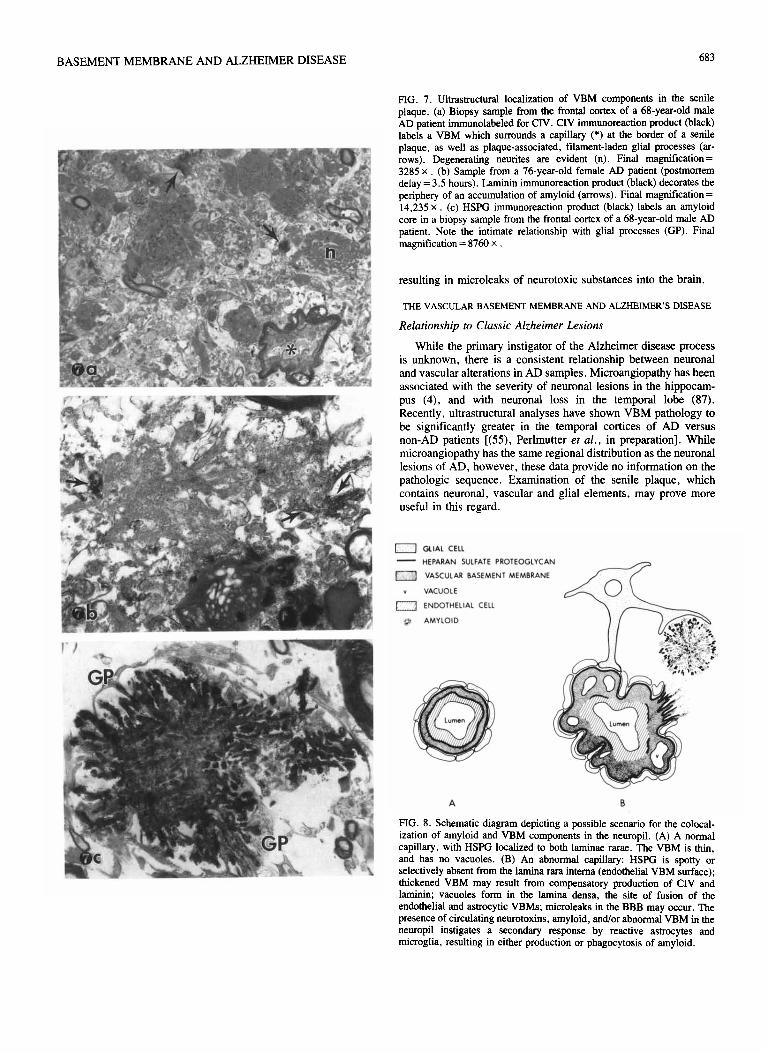
FIG. 7. Ultrastructural localization of VBM components in the senile plaque. (a) Biopsy sample from the frontal cortex of a 68-year-old male AD uatient immunolabeled for ClV. CIV immunoreaction product (black) lab& a VBM which surrounds a capillary (*) at the border of a senile plaque, as well as plaque-associated, filament-laden glial processes (ar- rows). Degenerating neurites are evident (n). Final magnification = 3285 x (b) Sample from a 76-year-old female AD patient (postmortem delay = 3.5 hours). Laminin immunoreaction product (black) decorates the periphery of an accumulation of amyloid (arrows). Final magnification = 14,235 x (c) HSPG immunoreaction product (black) labels an amyloid core in a biopsy sample from the frontal cortex of a 68-year-old male AD patient. Note the intimate relationship with glial processes (GP). Final magnification = 8760 x
resulting in microleaks of neurotoxic substances into the brain.
THE VASCULAR BASEMENT MEMBRANE AND ALZBEIMER’S DISEASE
Relationship to Classic Alzheimer Lesions
While the primary instigator of the Alzheimer disease process is unknown, there is a consistent relationship between neuronal and vascular alterations in AD samples. Microangiopathy has been associated with the severity of neuronal lesions in the hippocam- pus (4), and with neuronal loss in the temporal lobe (87). Recently, ultrastructural analyses have shown VBM pathology to be significantly greater in the temporal cortices of AD versus non-AD patients [(55), Perlmutter et al., in preparation]. While microangiopathy has the same regional distribution as the neuronal lesions of AD, however, these data provide no information on the pathologic sequence. Examination of the senile plaque, which contains neuronal, vascular and glial elements, may prove more useful in this regard.
n GLIAL CELL
- HEPARAN SULFATE PROTEOGLYCAN
m VASCULAR BASEMENT MEMBRANE
Y VACUOLE
m ENDOTHELIAL CELL
@ AMYLOID
A B
FIG. 8. Schematic diagram depicting a possible scenario for the colocal- ization of amyloid and VBM components in the neuropil. (A) A normal capillary, with HSPG localized to both laminae rarae. The VBM is thin, and has no vacuoles. (B) An abnormal capillary: HSPG is spotty or selectively absent from the lamina rara intema (endothelial VBM surface); thickened VBM may result from compensatory production of CIV and laminin; vacuoles form in the lamina densa, the site of fusion of the endothelial and astrocytic VBMs; microleaks in the BBB may occur. The presence of circulating neurotoxins, amyloid, and/or abnormal VBM in the neuropil instigates a secondary response by reactive astrocytes and microglia, resulting in either production or phagocytosis of amyloid.
hK4 PERLMI:‘~“l%K r\Nlf CHI /
S~VSII lines of evidence link the cerebral vasculature and the senile plaque. Amyloid fibrils emanate from the capillary base- ment membrane toward the plaque [(48t. Perlmutter and Chut. personal observations], seriai section ultrast~~tural analyses IY- veal close anatomic associations between capillaries and plaques (48). and the amyloid proteins deposited in the cerebrovasculature and the plaque are biochemically virtually identical (20.85). These findings suggest an intimate relationship between elements of the vasculature and the plaque. but leave the role of the VBM unaddressed.
We have used imlnunocyt~hemist~ to explore the relation- ship between specific VBM components and the plaque (3, 56. 57). At the light microscopic level, plaque-like extravascular accumulations of all three VBM components were seen in brain tissue from AD patients. These were always in the vicinity of capillaries. commonly colocalized with amyloid (Fig. 5), and never found in tissue from non-AD patients (56.57). At the ultrastructural level, HSPG, CIV and laminin immunoreaction product decorated amyloid fibrils in the core and corona of the plaque L(57.72): Fig. 7b. c]. While it is possible that HSPG is deposited in the plaque by degenerating cholinergic neurites (24.73). CIV and laminin are made exclusively by endothelial cells. pericytes and astrocytes (43,861. Thus. the combined ~olocalization of all three VBM components in senile plaques suggests a nonneuronal and possibly glial origin.
Reactive glia have long been recognized as an integral plaque constituent (83). Recently, we have localized all three VBM components within processes morphologically identified as astro- glial [(57) Perlmutter et al., submitted]. While phagocytosis of degenerating capillaries is possible, astrocytes also produce VBM components in response to injury (33.40.47). Thus, the intra-glial localization of CIV. laminin and HSPG could represent either phagocytosis or de novo production of the VBM.
Microglia are also an inherent component of the senile plaque. Recent ultrastructural analyses have revealed microglial cell pro- cesses enveloping amyloid stars (57a,84). Amyloid fibrils appear to be emanating from &sterna within the microglial cell, although phagocytosis of extracellular fibrils ia also possible. Several researchers (23.32.49) suggest that micro&a may be transformed pericytes. This scenario could account for the vacuolization of the VBM as well as the colocalization of VBM components with the amyloid.
Despite their close anatomical association. a pathogenetic relationship between the VBM and amyloid remains to be deter- mined. As was seen in the kidney, pathology of the VBM could be associated with impaired filtering capabilities, resulting in leakage of toxic plasma substances or circulating amyloid precursor into the brain (18, 25, 3 1, 48). Moreover. HSPG has been consistently colocalized with several types of cerebral and systemic amyloid (72). HSPG may influence the biochemical processing of various
amyloid precursor proteins, perhaps by virtue of its high negative charge (36. 69, 73). Thus, one of the intrinsic components of the VBM may be actively involved in amyloidogenesis.
Aizheimer-associated lesions could result from multiple. tntlepen- dent mechanisms (30). parsimony would dictate a single pathogc- netic cascade. For example. a primary neuronal l~~etaboli~ defect ( 13.44) could lead to I ) the forjnation of paired helical filaments and NFT , 2) neuronal and neuritic degeneration, and 3) release and subsequent processing of abnormal neuronal filaments into extraneuronal amyloid. Alternately, an initial dysfunction of the BBB (18,48) could result in 1) influx and accumulation of amyloid , 2) leakage of circulating neurotoxins into the neuropil, and 3) neurotic and neuronal death. Finally, a primary metabolic defect or immunologic insult could induce a pathology of the capillary itself, as was seen in the kidney. This could result in I ) loss of HSPG from the luminal surface of the VBM. 2) compen- satory hypersecretion of CIV and laminin, and 3 I altered metabolic or BBB functions of the endothelial cell resulting in increased BBS permeability.
Regardless of the initiating sequence, howevet-. the presence of either degenerating neurites, amyloid, neurotoxins. or abnormal VBM in the neuropil could instigate a secondary response by reactive astrocytes and microglia, resulting in either phagocytosis or production (82,84) of VBM and amyloid (Fig. 8). The recent discovery that P-amyloid is deposited in tissues other than brain (31 I increases the likelihood that it is either derived from a circulating substance or has t~lultiple sites of local pr~u~t~[~n, Vascular involvement in such a process is ccrtainlg worthy of study.
The source of plaque-associated dystrophic neurites is the subject of intense investigation. On the one hand, they may result from the development of NET in the neuronal somata (29); on the other hand, they might represent an aberrant sprouting response (16.17). P-Amyloid has recently been shown to exhibit neuro- trophic effect (78), and laminin and HSPG are known to be potent neurite-promoting factors (8, 9. 37-39). The collective presence of P-amyloid. laminin, and HSPG in the neuropil could initially create a strong neurotrophic focus. Further processing of this abnormal extravascular/extracelluIar mass could result in the loss of trophic effect and subsequent degeneration of the sprouting fibers. Thus, VBM components could serve as a nidus for plaque formation, playing a role in the development of both neuritic and amyloidotic elements.
The VBM is a vascular support structure whose role in neuropathologic processes has long been overlooked. A disorder of the VBM could result in compromise of the BBB. alteration of endothelial, pericytic, and astrocytic functioning. as well as abnormal deposition of neurite-promoting substances into the neuropil. Alterations of the VBM have been consistently associ- ated with Alzheimer’s lesions; future studies should elucidate its role in the pathogenesis of this disease.
This research was supported by AG-07127 and John Douglas French Foundation (L.S.P.). NIH 1P50-AGO5142 (H.C.C.). and AG-07624 (H.C.C.. L.S.P. ). We thank Jonathan Lin, Emesto Barron, Martha Myers. Chris Zarow and Dr. David Saperia for excellent technical assistance. Some of the tissue specimens used were obtained from the National Neurological Research Bank (VAMC Wadsworth Division, Los Angeles, CA 90073). which is sponsored by N~CDS/N~H, NMSS, HD Foun- d&ion, Comprehensive Epilepsy Program, Tourette’s Syndrome Associa- tion. Dystonia Research Foundation, and the Veteran’s Administration. Other specimens were obtained from the Southern California Alzheimer’s Research Consortium. NIH 1 P50-AGO5 142.
VBM Lesions and the Pathogenesis of Akheimer’s Disease
The lack of a specific AD-related lesion hampers the determi- nation of the cause and course of this disease. NFT, CAA and senile plaques are independently found in a variety of neurode- generative disorders, and VBM alterations also accompany other cerebral and systemic diseases (7. 28, 43, 54, 68. 71). While
BASEMENT MEMBRANE AND ALZHEIMER DISEASE 685
1.
2.
3.
4.
9.
10.
11.
12.
13.
14.
15.
16.
17.
IS.
19.
20.
21.
22.
23.
REFERENCES
Abrahamson, D. R.; Caulfield, J. P. Proteinuria and structural alterations in rat glomerular basement membranes induced by intra- venously injected anti-laminin immtmoglobulin G. J. Exp. Med. 156:128-14.5; 1982. ~tonelli-~lidge, A.; Saunders, K. B.; Smith, S. R.; D’Amore, P. A. An activated form of ~ansfo~ng growth factor B is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. USA 86:4544-4548; 1989. Athanikar, J.; Perlmutter, L. 8; Saperia, D.; Chui, H. C. Alteration of basement membrane components in dementia. Sot. Neurosci. Abstr. 14:638: 1988. Bell, M.; Ball, M. J. Morphometric comparison of hippocampal ~c~v~ulam~ in ageing and demented people: Diameters and densities. Acta Neuropathol. (Be&) 53:299-318; 1981. Bradbury, M. W. B. The structure and function of the blood-brain barrier. Fed. Proc. 43:186-H@; 1984. Cogan, D. G.; Kuwarbara, T. The mural cell in perspective. Arch. Gphthalmol. 78:133-139; 1967. Donahue, S.; Zeman, W.; Watanabe, I. Alterations of basement membranes of cerebral capillaries. J. Neuropathol. 26:397-411; 1976. Dow, K. E.; Mirski, S. E, L.; Roder, J. C.; Riopelle, R. J. Neuronal proteoglycans: biosynthesis and functional interaction with neurons in vim. J. Neurosci. 8:3278-3289; 1988. Edgar, D.; Timpl, R.; Theonen, H. The heparin-binding domain of laminin is responsible for its effects on neurite outgrowth and neuronal survival. EMBO J. 3:1463-1468; 1984. Esiri, M. M.; Wilcock, G. K. Cerebral amyloid angiopathy in dementia and old age. J. Neural. Neurosurg. Psychiatry 491221- 1226; 1986. Filiit, H. M.; Damle, S. P.; Gregory, J. D.; Volin, C.; Poon-King, T.; Zabriskie, J. Sera from patients with poststreptococcal glomerulone- phritis contain antibodies to glomerular heparan sulfate proteoglycan. J. Exp. Med. 161:277-289; 1985. Fillit, H. M.; Kemeny, E.; Luine, V.; Weksler, M. E.; Zabrislcie, J. B. Antivascular antibodies in the sera of patients with senile dementia of the Alzheimer’s type. J. Gerontol. 42180-184; 1987, Friedland, R. P.; Yano, Y.; Budinger, T. F.; Ganz, E.; Huesman, R. H.; Derenzo, S. E.; Knittel, B. Quantitative evaluation of blood brain integrity in Alzheimer-type dementia: Positron emission tomographic studies with rubidium-82. Eur. Neural. 22(Su~ul. 2):19: 1983. Gambetti, P.; Perry, G.; Autilio-Gambem, r. Paired helical fila- ments: Do they contain neurofilament epitopes? Neurobiol. Aging 7:451-452; 1986. Garbisa, S.; Negro A. Mac~molecul~ organization and functional architecture of basement membranes. Appl. Pathol. 2:217-222; 1984. Geddes, J. W.; Monaghan, D. T.; Cotman, C. W.; Lo& I. T.; Kim, R. C.; Chui, H. C. Plasticity of hippocampal circuitry in Alzheimer’s disease. Science 230:1179-1181; 1985. Geddes, J. W.; Anderson, K. J.; Cotman, C. W. Senile plaques as aberrant sprout-stimulating structures. Exp. Neurol. 94:767-776; 1986.
24.
25.
26.
Greif, K. F.; Reichardt, L. F. Appearance and distribution of neuronal cell surface and synaptic vesicle antigens in the developing rat superior cervical ganglion. J. Neurosci. 2:843-852; 1982. Hardy, J. A.; Mann, D. M. A.; Webster, P.; Winblad, B. An integrative hypothesis concerning the patbogenesis and progression of Alzheimer’s disease. Neurobiol. Aging 7:489-502; 1986. Hart, M. N.; VanDyk, L.; Moore, S. A.; Shasby, D. M.; Canciila, P. A. Differential opening of the brain endothelial barrier following neutralization of the endothelial luminal anionic charge in vitro. J. Neuropathol. Exp. Neurol. 46:141-153; 1987.
Glenner, G. G. Congop~lic ~cm~giopa~y in the pathogenesis of Alzheimer’s syndrome (Presenile Dementia). Med. Hypotheses 5: 1231-1236; 1979. Glenner, G. G.; Eanes, E. D.; Bladen, H. A.; Lit&e, R. P.; Tennine, I. D. B-pleated sheet librils. A comparison of native amyloid with synthetic protein fibrils. J. His&hem. Cytochem. 22:1141-l 158; 1974. Glenner, G. G.; Wong, C. W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. B&hem. Biophys. Res. Commun. 120:885-890; 1984. Goldstein, G. W.; Betz, A. L. Recent advances in understanding brain capillary function. Ann. Neural. 14:389-395; 1983. Goldstein, G. W.; Betz, A. L.; Bowman, P. D.; Dorovini-Zis, K. In v&o studies of the blood-brain barrier using isolated brain capillaries and cultured endo~elial cells. Ann. NY Acad. Sci. 481:202-213; 1986. Graeber, M. B.; Streit, W. J.; Kreutzberg, G. W. Identity of ED2positive perivascular cells in rat brain. J. Neurosci. Res. 22: 103-106; 1989.
27.
28.
29.
30.
31.
Hassle;, 0. Vascular changes in senile brains: A micro-angiographic study. Acta Neuropathol. (Berl.) 5:40-53; 1965. Hughes, C. P.; Myers, F. K.; Smith, D.: Torack, R. M. Nosologic problems in dementia. Neurology 23:344-351; 1973. Hyman, B. T.; Kromer, L. J.; Van Hoesen, G. W. Reinnervation of the hippocampal perforant pathway zone in Alzheimer’s disease. Ann. Neurol. 21:259-267; 1987. Iqbal, K. Biology of Alzheimer’s Disease through biochemistry of taneles and ulaaues. Neurobiol. Aeina 7:434-437: 1986. Joaihim, C.*L.;‘Mori, H.; Selkoe, D. z Amyloid B-protein deposition in tissue other than brain in Alzheim~r’s disease. Nature 341:226-230; 1989.
32.
33.
34.
35.
36.
Jordan, F. L.; Thomas, W. E. Brain macrophages: questions of origin and interrelationship. Brain Res. Rev. 13:165-178; 1988. Kefalides, N, A.; Alper, R.; Clark, C. C. Biochemistry and metab- olism of basement membranes. Int. Rev. Cytol. 61:167-228; 1979. Kidd, M. Alzheimer’s disease-an electron microscopical study. Brain 87:307-320; 1964.
37.
38.
39.
40.
41.
42.
43.
44.
Kimelberg, H. K.; Norenberg, M. D. Astrocytes. Sci. Am. 260: 66-76; 1989. Kisilevsky, R. From arthritis to Alzheimer’s disease: Current concepts on the pathogenesis of amyloidosis. Can. J. Physiol. Pharmacol. 65:1805-1815; 1987. Lander, A. D.: Fujii, D. K.; Gospodarowicz, D.; Reichardt. L. F. Characterization of a factor that promotes neurite outgrowth: evidence linking activity to heparan sulfate proteoglycan. I. Cell Biol. 94: 574-585; 1982. Lander, A. D.; Fujii, D. K.; Reichardt, L. F. Purification of a factor that promotes neurite outgrowth: isolation of laminin and associated molecules. J. Cell Biol. 101:898-913; 1985. Lander, A. D.; Fujii, D. K.; Reichardt, L. F. Laminin is associated with the “neurite outgrowth-promoting factors” found in conditioned media. Proc. Natl. Acad. Sci. USA 82:2183-2187; 1985. Liesi, P.; Kaakkola, S.; Dahl, D.; Vaheri, A. bin is induced in astrocytes of adult brain by injury. EMBO J. 3:683-686; 1984. Mancardi, G. L.; Perdelli, F.; Rivano. C.; Leonardi, A.; Bugiani, 0. Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol. (Berl.) 49:79-83; 1980. Mann, D. M. A.; Davies, J. S.; Yates, P. 0.; Hawkes, J. Immuno- histochemical staining of senile plaques. Neuropathol. Appl. Neuro- biol. 8:55-61; 1982. M~inez-Hemandez, A.; Amenta, P. The basement membrane in pathology. Lab. Invest. 48:656-677; 1983. Masters, C. L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R. N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: neurotibril- lary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 4:2757-2763; 1985.
45. Midena, E.; Segato, T,; Radin, S.; di Giorgio, G.; Meneghini, F.; Piermarocchi, S.; Belloni, A. S. Studies on the retina of the diabetic db/db mouse. 1. Endothelial cell-pericyte ratio. Ophthalmic Res. 21:106-111; 1989.
46. Miettinen, A.; Stow, J. L.; Mentone, S.; Farquhar, M. G, Antibodies to basement membrane heparan sulfate proteoglycans bind to the laminae rarae of the glomerular basement membrane (GBM) and induce subepi~eli~ GBM thickening. J. Exp. Med. 163:1064-1084: 1986.
47. Mitchell, J.; Weller, R. 0.; Evans, H. Capillary regeneration follow- ing thermal lesions in the mouse cerebral cortex. Acta Neuropathol. (Berl.) 44:167-171; 1978.
48. Miyakawa, T.; Shimoji, A.; Kuramoto, R.; Higuchi, Y. The relation-
hX6 PERLMCI”[‘ER ANI1 CHI’I
ship between senile plaques and cerebral blood \ea~cls m Alzheimer’, disease and senile dementia. Virchows Arch. 30:1?1--129; 19X2.
49. Mori, S.; Leblond. C. P. Identification of mlcroglia m light and electron microscopy. J. Comp. Neural. 135:57-80: 1969.
50. Mynderse, L. A.; Hassell, J. R.; Kleinman, H, K.: Martin, G. R.. Martinez-Hemander, A. Loss of heparan sulfate proteoglycan from plomerular basement membrane of nephrotic rats. Lah. Invest. 48: 292-302: 1983.
51. Nagy, Z.; Peters, H.; Hiittner. I. Charge-related alterations of the cerebral endothelium. Lab. Invest. 49:662-671: 1983.
52. Pardridge, W. M.; Oldendorf, W. H.; Cancilla, P.: Frank, H. J. L. Blood-brain harrier: interface between internal medicine and the brain. Ann. Intern. Med. 105:82-95: 1986.
53. Pardridge. W. M.; Yang, J.; Eisenberg, J.; Mietub. I_. J. Antibodies to blood-brain barrier bind selectively to brain capillary endothelial lateral membranes and to a 46K protein. J. Cereh. Blood Flow Metab. 6:203-21 I: 1986.
54. Parthasarathy, N.; Spiro, R. G. Effect of diabetes on the glycosami- noglycan component of the human glomerular basement membrane. Diabetes 3 I :738-74 I ; 1982.
55. Perlmutter. L. S.; Athanikar, J.; Zarow, C.; Chui. H. C. Basement membrane pathology in dementia. Sot. Neurosci. Abstr. 14:639: 198X.
56. Perlmutter. L. S.; Chui, H. C.; Saperia, D.; Athanikar, J. Microan- giopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 508: 13-19: 1990.
57. Perlmutter, L. S.; Saperia, D.; Wu. J.; Chui. H. C. Vascular basement membrane components associated with senile plaque amy- loid. Sot. Neurosci. Abstr. 15:1376; 1989.
57a Perlmutter. L. S.; Barron, E.; Chui, H. C. Morphologic associates between microglia and senile plaque amyloid in Alzheimer disease. Neurosci. Lett. Submitted.
58. Peters, A.; Palay, S. L.; Webster. H. deF. The fine structure of the nervous system: The neurons and supporting ceils. Philadelphia: W. B. Saunders Co.; 1976.
59. Rennke, H. G.; Cotran, R. S.; Venkatachalam, M. A. Role of molecular charge in glomerular permeability. J. Cell Biol. 67: 638-646; 1975.
60. Rohison, W. G., Jr. Prevention of diabetes-related retinal microangio- pathy with aldose reductase inhibitors. Adv. Exp. Med. Biol. 246: 365-372; 1988.
61. Roggendorf, W.; Opitz, H.; Schuppan, D. Altered expression of collagen type IV in brain vessels of patients with chronic hyperten- sion. Acta Neuropathol. (Berl.) 77:5560; 1988.
62. Rohrhach, D. H.; Hassell, J. R.; Kleinman, H. K.: Martin, G. R. Alterations in the basement membrane (heparan sulfate) proteoglycan in diabetic mice. Diabetes 31:185-188; 1982.
63. Saperia, D.; Perlmutter, L. S.: Athanikar, J.; Chui, H. C. Amyloid P component in dementia. Sot. Neurosci. Ahstr. 14:638; 1988.
64. Sato, Y.: Rifkin. D. B. Inhibition of endothelial cell movement by perlcytes and smooth muscle cells: activation of a latent transforming growth factor-PI-like molecule by plasmin during co-culture. J. Cell Biol. 109:309-315; 1989.
65. Scheibel, A. B. Changes in brain capillary structure in aging and dementia. In: Wertheimer, J.; Marois, M., eds. Senile dementia: Outlook for the future. New York: Alan R. Liss; 1984:137-149.
66. Scheihel, A. B.; Duong, T.; Tomiyasu, U. Denervation microangio- pathy in senile dementia, Alzheimer type. Alzheimer Dis. ASSOC. Disord. l:19-37; 1987.
67. Schlapeter. N. L.; Carson, R. E.; Rapoport, S. 1. Examination of
69. Schubert, D.: Schroeder. R.: LaCorbiere, M.: Saitoh, T.: Cole. (1. Amyloid B protein precursor is possibly a heparan rulfate proteoglc- can core protein. Science 241:223-241; 1988.
70. Selkoe. D. J.: Bell, D. S.: Podlisny. M. B.; Price, 1). L.: Cork. L. C. Conservation of bram angiopathy proteins in aged mammals and humans with Alzheimer’s disease. Science 235:873-877: 1987.
71. Smith, L. H.: Martin. D. W.: Watts, H. D. Diabetic microangiopa- thy-Medical Staff Conference, University of California. San Fran- cisco. West. J. Med. 121:404--112; 1974.
72. Snow, A. D.: Mar. H.: Nochlin. D.; Kimata. K: Kate. M.; Suzuki. S.: Hassell. J.. Wight. T. N. The presence rrf heparan sulfate proteoglycans in the neuritlc plaques and congophilic angiopathy in Alzheimer’\ disease. Am. J. Pathol. 133:456363; 1988.
73. Snow. A. D.: Willmer. J.; Kisilevsky. R. A clobe ultrastructural relationship between sulfated proteoglycans and AA amyloid fibrils. Lab. Invest. 57:687-698; 1987.
74. Stadler. H.; Dowe. G. H. C. identification oi a heparan sulphate- containing proteoglycan as a specific core component of cholinergic synaptic vesicles from ir,jrpedo murmornra. EMBO J. I : 13X I - 1384; 19x2.
75. Terry, R. D.; Davies. P. Dementia of the Alzheimer type. Annu. Rev. Neurosci. 3:77-95; 1980.
76. Timpl, R.; Dziadek. M. Structure. development, and molecular pathology of basement membranes. Int. Rev. Exp. Pathol. 29: I-l 12: 1986.
77. van Deurs. B. Observations on the blood-brain barrier in hypertensive rats, with particular reference to phagocytic pericytes. J. Ultrastruct. Res. 56165-77; 1976.
78. Whitson, J. S.; Selkoe. D. J.; Cotman. C. W. Amyloid p protein enhances the survival of hippocampal neurons in vitro. Science 243:1488-1490; 1989.
79. Wick, G. Muller. P. U.; Timpl. R. In viva localization and patholog- ical effects of passively transferred antibodies to type IV collagen and laminin in mice. Clin. Immunol. Immunopathol. 23:656-665; 1982.
80. Wilson. C. B.; Dixon, F. J. Immunopathology and glomerulonephri- tis. Annu. Rev. Med. 25:83-98; 1974.
XI. Wisniewski, H. M.; Kozlowski, P. B. Evidence of blood brain barrier changes in senile dementia of the Alzheimer type. Ann. NY Acad. Sci. 396:119-129; 1982.
82. Wisniewski, H. M.; Mere, G. S. Neuritic and amyloid plaques in senile dementia of the Alzheimer type. In: Katzman. R.. rd. Biolog- ical aspects of Alzheimer’s disease. New York: Cold Spring Harbor: 1983:145-153.
83. Wisniewski, H.; Terry, R. D. Reexamination of the pathogenesis of the senile plaque. Prog. Neuropathol. II:l-26; 1973.
84. Wisniewski, H. M.; Wegiel, J.; Wang, K. C.; Kujawa, M.; Lath, B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. J. Can. Sci. Neural. 16535-542; 1989.
85. Wong. C. W.; Quaranta, V.; Glenner, G. G. Neuntic plaques and cerehrovascular amyloid in Alzheimer disease are antigenically re- lated, Proc. Natl. Acad. Sci. USA 82:8729-8732; 1985.
86. Yamada, K. M. Cell surface interactions with extracellular materials. Annu. Rev. Biochem. 52:761-799; 1983.
87. Zarow, C.; Chui, H. C.; Hsu, E.; Perlmutter. L. S. Correlation between microangiopathy and neuronal loss in Alzheimer’s Disease. Sot. Neurosci. Abstr. 15:1376: 1989.