28
Modeling Mesoscale Structure In Comb Polymer Materials for Anhydrous Proton Transport Applications Barry Husowitz Barry Husowitz Peter Monson Peter Monson
| Date post: | 14-Dec-2015 |
| Category: |
Documents |
| Upload: | sage-morison |
| View: | 224 times |
| Download: | 0 times |

Modeling Mesoscale Structure In Comb Polymer Materials for Anhydrous Proton Transport
Applications
Barry HusowitzBarry Husowitz
Peter MonsonPeter Monson

Proton Exchange Membrane (polyelectrolyte)Proton Exchange Membrane (polyelectrolyte)Membranes need to beMembranes need to beChemically stableChemically stableThermodynamically stableThermodynamically stableReasonable proton conductanceReasonable proton conductance
Important for Operation of Fuel Cell (Proton Exchange Membrane)
U.S. Department of Energy Hydrogen Program
Why is this important?

Water Assisted Proton Conductance
Nafion (comb polymer)Nafion (comb polymer)The conductance of water containing nafion is The conductance of water containing nafion is
highly dependent on the state of hydration of the highly dependent on the state of hydration of the membrane and structural characteristicsmembrane and structural characteristics
Temperature range of operation highly dependent Temperature range of operation highly dependent on solventon solvent
P. J. Brookman, J. W. Nichoson, "Developments in Ionic Polymers, vol 2" Elsevier, London, pp. 269-283 (1986)

Anhydrous proton conductance
Conductance does not depend on physical Conductance does not depend on physical properties of the solventproperties of the solvent
Investigating proton-conducting polymers Investigating proton-conducting polymers which do not rely on a solvent is a which do not rely on a solvent is a revolutionary approach to new hydrogen fuel revolutionary approach to new hydrogen fuel cellscells

Center for Chemical Innovations (CCI) : Candidates for Anhydrous Proton Conductance
NHN
NN
HN
Comb diblocksComb diblocks
Comb polymersComb polymers
ONH
O
n
O OC10H21 N
H
O
n
O
BrO
O
SS
O
O
Br
O
nm
NH
HN
N
O
Imidazole
Benzotriazole

NHN
NN
HN
Dendrimer-LinearDendrimer-Linear
PT
PT
Center for Chemical Innovations (CCI) : Candidates for Anhydrous Proton Conductance

ModelingPolymer Architecture (chemistry/structure) Predict
(model)
Mesoscale Structure (ordering)
What is the correlation between the nanostructureand proton conductance?

Coarse Grained Models
bond vibrations~ 10-15 s
diffusion ~ 10-9 -10-6 s
conformational rearrangements ~ 10-12 - 10-10 s
Number of Atoms Lumped into Effective Segment (interaction center) MC, MD, DPD
Want a coarse-grained model for polymers that captures relevant interactions, excluded volumes, repulsion between unlike atoms- Using softer potentials and reducing the degrees of freedom is an efficient technique for large dense systems (106 segments)
5
12
34
5
F. Muller-Plathe, CHEMPHYSCHEM 3, 754-769 (2002)

Multiscale Modeling: Force Fields
Quantum Calculations of energy surface
Force Field - Functional forms and Parameters to describe the potential energy of atoms or groups of atoms
Molecular Mechanics – Newtonian mechanics to model molecular system
Quantum Mechanics
Classical Physics
conformational rearrangements ~ 10-12 - 10-10 s
bond vibrations~ 10-15 s
diffusion ~ 10-9 -10-6 s
12
3
4
5
H. Sun, Macromolecules 28, 701-712 (1995)

Basic Model For Polymers
H=Hb + Hnb
Bonding interactions within the chains (Conformational states of individual polymers), connectivity
Interactions between other chains and non-bonded sites
Base Case Model Hamiltonian
Minimal coarse graining model that captures Minimal coarse graining model that captures relevant interactions, connectivity, excluded relevant interactions, connectivity, excluded volumesvolumes

Bonding Hamiltonian (Ideal Chain Models)
Connectivity (Molecular Architecture)Connectivity (Molecular Architecture)Freely joined chainFreely joined chainKey Parameter bKey Parameter b
Discrete Gaussian chainDiscrete Gaussian chainri(s) ri(s+1)
b
Fixed
ri(s)
ri(s+1)G. R. Strobl, Chapter 6 "The Physics of Polymers, 2'nd Ed." Springer, NY, (1997)
RReoeo=bN=bN1/21/2

Non-bonding Hamiltonian
{
{
L
L
ΦA,m
K. Daoulas and M. Muller, J. Chem. Phys. 125, 184904 (2006)
Essential Interactions described through simple Essential Interactions described through simple parameters, chain extension Rparameters, chain extension R
eoeo, Flory-Huggins , Flory-Huggins
( N( NABAB), compressibility (N), compressibility (N), acts like excluded ), acts like excluded
volumevolume Symmetric Diblock
-1 0 1
r-rc
RReoeo
No explicit volume exclusion, segments can overlap, enforce low compressibility on
length scale of interest Reo
L=0.17Reo

Metropolis Algorithm For Monte Carlo Simulation
The basic idea is that we assume each configuration of a system has a probability proportional to a Boltzmann factor or
Consider two configurations A and B, each of which occurs with probability proportional to the Boltzmann factor. Then
The nice thing about forming the ratio is that it converts relative probabilities involving an unknown proportionality constant (called the inverse of the partition function), into a pure number. We can achieve the relative probability of the last equation in a simulation by proceeding as follows:

Metropolis Algorithm For Monte Carlo Simulation
1. Start from configuration A, with know energy EA, make a change in the configuration to obtain a new (nearby) configuration B.2. Compute EB (typically a small change from EA, but not to small)3. If EB < EA, assume the new configuration , since it has lower energy (desirable thing, according to the boltzmann factor).4. If EB > EA, accept the new (higher energy) configuration with This means that when the temperature is
high, we don’t care if we take a step in the “wrong” direction, but as the temperature is lowered, we settle into the lowest configuration we can find in our neighborhood.

Metropolis Algorithm For Monte Carlo Simulation
If we follow these rules, then we will sample points in the space of all possible configurations with probability proportional to the Boltzmann factor, consistent with the theory of equilibrium statistical mechanics. We can compute average properties by summing them along the path we follow through possible configurations.
The hardest part about implementing the Metropolis algorithm is the first step: how to generate “useful” new configurations.
Canonical Partition function – Sum of all the different possible energy states of the system (However, maybe one of these configurations is very very low in energy and dominates the sum)

Monte Carlo Moves Incorporated
1. Moved single beads in the chain
2. Translated the whole chain
3. Reptation

Simulation Methods
Single Chain in Mean Field (SCMF)Single Chain in Mean Field (SCMF)Chains move in a field created by other chains Chains move in a field created by other chains
via model Hamiltonian via model Hamiltonian Update the fields periodically based on segment Update the fields periodically based on segment
distributiondistributionDirect Monte Carlo (MC) simulation of the model Direct Monte Carlo (MC) simulation of the model
HamiltonianHamiltonianConsider new and old fields for every MC moveConsider new and old fields for every MC moveUpdate fields after every accepted moveUpdate fields after every accepted move

Advantage of Model
Retains computational advantage of Self-consistent Retains computational advantage of Self-consistent Field theory (SCF), but includes fluctuations.Field theory (SCF), but includes fluctuations.
The evaluation of interactions via a grid and The evaluation of interactions via a grid and densities speeds up the energy calculation by about densities speeds up the energy calculation by about two order of magnitude compared to explicit two order of magnitude compared to explicit pairwise interactions.pairwise interactions.
Can easily implement different polymeric Can easily implement different polymeric structures (branched polymers, dendrimeric structures (branched polymers, dendrimeric polymers etc.) or architectures with this model.polymers etc.) or architectures with this model.

Experimental Results: Morphology of Comb Polymers by X-ray Scattering
q1
q2
q2/q1=√3
X-ray scattering clearly reveals nano-scale phase-segregation induced by alkyl chains
Chen et al, Nature Chemistry , 2010, 2, 503-508

X-ray scattering clearly reveals nano-scale phase-segregation induced by alkyl chains
Experimental Results: Morphology of Comb Polymers by X-ray Scattering
q1
q2
q2/q1=4
Chen et al, Nature Chemistry , 2010, 2, 503-508

Center for Chemical Innovations (CCI) Comb Polymers - Coarse Graining

Comparison of SCMF with Previous Self-Self-Consistent Field Theory (SCF)Consistent Field Theory (SCF) Study of Comb Study of Comb PolymersPolymers
Liangshun Zhang et al, J. Phys. Chem. B 2007, 111, 351-357
Changing the position of the graft points and number of branch points provides aroute to a cylinder morphology

Alkyl side chains gives rise to cylinder morphology
ΑΒ= 52.2
Monte Carlo simulations of coarse grained model
q2/q1=
q2
q1
Experimental Our Calculated Structure Factor

Disordered structure in this case
But, if we lower the interaction parameter to 40.6 then …
ΑΒ= 52.2
q2/q1=4
q1
q2

MC simulations of coarse grained model
ΑΒ= 52.2
Alkyl side chains give rise to lamellar morphology
q2
q1 q2/q1=4
Experimental Our Calculated Structure Factor
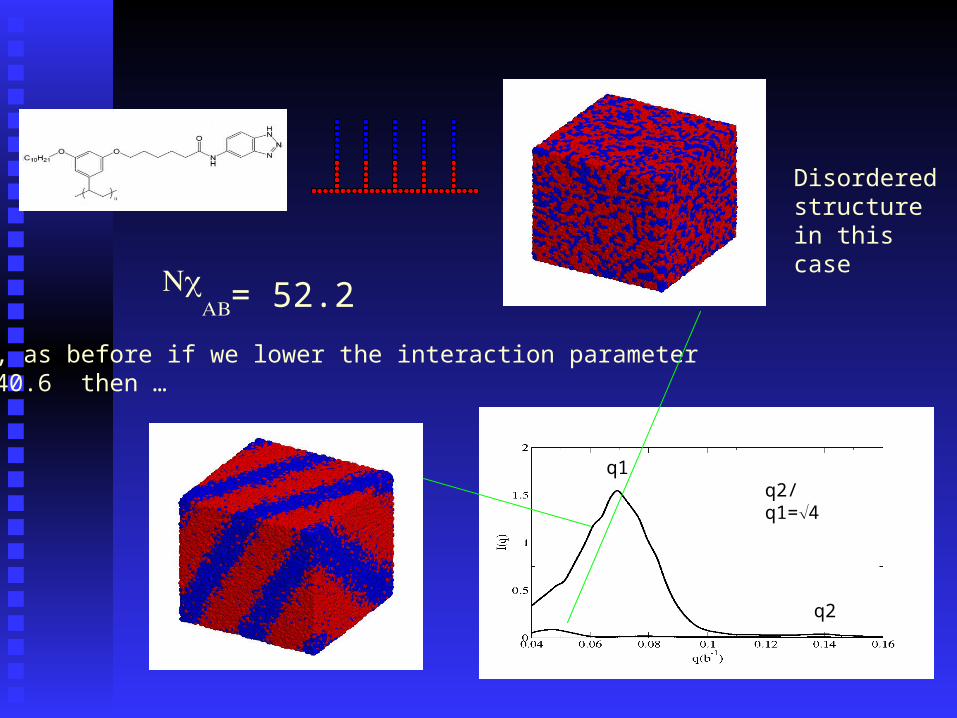
Disordered structure in this case
ΑΒ= 52.2
But, as before if we lower the interaction parameter To 40.6 then …
q2/q1=4q1
q2

Summary and Conclusions Coarse grained models of CCI polymers show lamellar, Coarse grained models of CCI polymers show lamellar,
cylindrical and disordered structures found in experimentscylindrical and disordered structures found in experiments Simulations may explain origins of disordered structures Simulations may explain origins of disordered structures
seen in some experimentsseen in some experimentsAddition of alkyl groups increases value of Addition of alkyl groups increases value of required for required for
ordering in the system (effect of chain branching on order-ordering in the system (effect of chain branching on order-disorder transitions)disorder transitions)
Simulations of comb polymer systems for large Simulations of comb polymer systems for large starting starting from a disordered state can lead to quenched disorderfrom a disordered state can lead to quenched disorder
Thus simulations of alkylated polymers can show ordered Thus simulations of alkylated polymers can show ordered structures when those of nonalkylated polymers structures when those of nonalkylated polymers at the same value of do not

Thank You Peter MonsonPeter Monson NSF NSF Center for Chemical Innovation Center for Chemical Innovation
(CCI) at the University of (CCI) at the University of Massachusetts, AmherstMassachusetts, Amherst
Everyone here todayEveryone here today

















