INVITED REVIEW ARTICLE Molecular and diagnostic aspects of genetic skin fragility Cristina Has * , Leena Bruckner-Tuderman Department of Dermatology, University of Freiburg, Hauptstr. 7, 79104 Freiburg, Germany Received 6 July 2006; received in revised form 8 August 2006; accepted 9 August 2006 Contents 1. Introduction ............................................................ 130 2. Molecular and structural basis of adhesion and stability of skin compartments .................. 132 2.1. Cytoskeleton and cell—cell adhesions ........................................ 132 2.2. The hemidesmosomes and the dermal—epidermal junction .......................... 134 3. Inherited skin fragility disorders ............................................... 135 3.1. Keratin defects ...................................................... 135 3.1.1. Epidermolytic hyperkeratosis (syn. bullous ichthyosiform erythroderma) ............. 135 3.1.2. Ichthyosis bullosa Siemens .......................................... 135 3.1.3. Epidermolysis bullosa simplex ........................................ 137 Journal of Dermatological Science (2006) 44, 129—144 www.intl.elsevierhealth.com/journals/jods KEYWORDS Skin fragility; Cytoskeleton; Keratin; Desmosomes; Hemidesmosomes; Collagen; Laminin Summary Genetic syndromes with skin fragility represent a heterogeneous group of very rare disorders caused by mutations in genes encoding proteins or protein subunits important for the mechanical resistance of keratinocytes and for cell—cell or cell— extracellular matrix adhesion. The common symptoms are skin blistering or peeling, with various degrees of severity and distribution, ranging from localized to generalized forms. Associated features include involvement of skin annexes, mucous membranes, teeth, muscles or the digestive tract. Morphological investigation of skin samples provides evidence for the tissue level of blister formation, while immunostainings may reveal defective proteins, providing clues concerning the genetic origin of the disease. Extensive mutation analysis and subsequent identification of new gene defects provide accurate diagnostics, and lead to better understanding of the functions of the respective proteins, with the potential for new therapeutic strategies. # 2006 Japanese Society for Investigative Dermatology. Published by Elsevier Ireland Ltd. All rights reserved. Abbreviations: EB, epidermolysis bullosa; HD, hemidesmosome; IF, intermediate filaments; TG, transglutaminase; ECM, extra- cellular matrix; EM, electron microscopy; DEJ, dermal—epidermal junction; NC, non-collagenous; EHK, epidermolytic hyperkeratosis; IBS, ichthyosis bullosa Siemens; IIF, indirect immunofluorescence; PSS, peeling skin syndrome; KS, Kindler syndrome; PTC, premature termination codon; JEB, junctional epidermolysis bullosa * Corresponding author. Tel.: +49 761 270 6785; fax: +49 761 270 6720. E-mail address: [email protected](C. Has). 0923-1811/$30.00 # 2006 Japanese Society for Investigative Dermatology. Published by Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.jdermsci.2006.08.003
Transcript
Journal of Dermatological Science (2006) 44, 129—144
www.intl.elsevierhealth.com/journals/jods
INVITED REVIEW ARTICLE
Molecular and diagnostic aspects of genetic skinfragility
Cristina Has *, Leena Bruckner-Tuderman
Department of Dermatology, University of Freiburg, Hauptstr. 7, 79104 Freiburg, Germany
Received 6 July 2006; received in revised form 8 August 2006; accepted 9 August 2006
Summary Genetic syndromes with skin fragility represent a heterogeneous group ofvery rare disorders caused by mutations in genes encoding proteins or protein subunitsimportant for the mechanical resistance of keratinocytes and for cell—cell or cell—extracellular matrix adhesion. The common symptoms are skin blistering or peeling,with various degrees of severity and distribution, ranging from localized to generalizedforms. Associated features include involvement of skin annexes, mucous membranes,teeth, muscles or the digestive tract. Morphological investigation of skin samplesprovides evidence for the tissue level of blister formation, while immunostainingsmay reveal defective proteins, providing clues concerning the genetic origin of thedisease. Extensivemutation analysis and subsequent identification of new gene defectsprovide accurate diagnostics, and lead to better understanding of the functions of therespective proteins, with the potential for new therapeutic strategies.# 2006 Japanese Society for Investigative Dermatology. Published by Elsevier IrelandLtd. All rights reserved.
0923-1811/$30.00 # 2006 Japanese Society for Investigative Dermatology. Published by Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.jdermsci.2006.08.003
The major role of the skin is to separate and protectthe inner environment of the organism from theexternal milieu and its physical, chemical and biolo-gical insults. To achieve these complex functions,numerous structural proteins andmolecule networksin the epidermis corroborate to assure the cohesionof keratinocytes and their adhesion to the basementmembrane.Thedermisalso cushions thebodyagainstmechanical injury by conferring elasticity and plas-ticity to the skin. Genodermatoses with skin fragilityare caused by mutations in genes encoding proteinsthat contribute to cutaneous resistance.
Epidermolysis bullosa (EB) hereditaria, the pro-totype of the skin fragility syndromes, represents aclinically and genetically heterogeneous group ofgenodermatoses characterized by skin blisteringafter minor trauma. Mutations in 12 distinct genesare responsible for the disease subtypes (Table 1)that are classified into three main categories based
Table 1 Genetic diseases with skin fragility, underlying mo
on the level of skin splitting (Fig. 1) and furthersubtyped on the basis of the clinical severity, whichusually reflects the degree of the protein defect[1,2]. In the different EB forms, the phenotypesrange from very mild symptoms, such as nail dys-trophy to extensive muco-cutaneous blistering lead-ing to a multiorgan disease. For example, skinfragility is minimal in EB simplex localisataWeber—Cockayne and even vigorous rubbing ofthe skin can be well tolerated, consequently, thequality of life is onlyminimally affected. In contrast,at the other end of the clinical spectrum, Herlitzjunctional EB is characterized by extreme fragilityof skin and mucous membranes and lethality. Thevery recently recognized acantholytic epidermolysisbullosa is lethal in the neonatal period due toextensive transcutaneous fluid loss [3].
For many years, EB subtypes were strictly sepa-rated on the basis of ultrastructural defects and,subsequently on the basis of protein abnormalities.Extensive mutation analysis in the last years has
Molecular and diagnostic aspects of genetic skin fragility 131
Fig. 1 Schematic representation of the epidermis and upper dermis. On the right, the genetic skin fragility syndromesare noted and the level of separation of the skin indicated. Abbreviations: APSS, acral peeling skin syndrome; IBS,ichthyosis bullosa of Siemens; SFEDS, skin fragility–—ectodermal dysplasia syndrome; SFWHS, skin fragility–—wooly hairsyndrome; EB, epidermolysis bullosa; EHK, epidermolytic hyperkeratosis.
revealed the classical EB categories to be less dis-tinct than previously considered. Nevertheless, themorphological classification has been maintainedfor practical purposes. A new classification basedsolely on gene mutations would be too detailed, andtherefore difficult to implement in clinical practise.For example, it would be complicated to definedifferent forms of EB simplex, such as EB simplex
Table 2 Other genodermatoses characterized by blistering
Disease (MIM) Gene Protein
AEC syndrome(MIM 106260)
p63 p63
Incontinentiapigmenti(MIM 300248)
NEMO NF-kB essentialmodulator
Porphyriacutanea tarda(MIM 176100)
UROD Uroporphyrinogendecarboxylase
Ehlers—Danlossyndrome type I(MIM 130000)
COL5A1,COL5A2,COL1A1
Collagen V,collagen I
Ehlers—Danlossyndrome type II(MIM 130010)
COL5A1,COL5A2
Collagen V
Hailey—Haileydisease(MIM 16960)
ATP2C1 Humanhomologueof an CaATP-ase pump
with intraepidermal blistering and a COL17A1muta-tion affecting the intracellular domain of collagenXVII [4], as opposed to lethal EB simplex with pyloricatresia caused by plectin deficiency [5].
Recently, the molecular basis of skin fragilitysyndromes, such as skin fragility–—ectodermal dys-plasia syndrome [6] and Kindler syndrome [7], havebeen elucidated, adding new real ‘‘epidermolysis’’
Similar toEhlers—Danlostype I but milderRecurrent blisteringpredominantly atintertriginous areas
Generalized lesions,warty papules
132 C. Has, L. Bruckner-Tuderman
members to the EB disease family. In addition,tetraspanin CD151 has been implicated in the cor-rect assembly of human basement membranes inkidney and skin: two siblings with pretibial EB,hereditary nephritis, sensorineural deafness andb-thalassemia minor were shown to be homozygousfor a CD151 single-nucleotide insertion mutationaltering the integrin-binding domain of CD151 [8].However, mice lacking CD151 have normal skin mor-phology including normal hemidesmosome (HD)structure, which may be due to compensation bya closely related tetraspanin [9]. Molecular charac-terization of further patients will be necessary toconfirm the identity of this entity.
Several other genetic multiorgan diseases exhibitan associated skin fragility symptom (Table 2).These disorders are heterogenous and must be con-sidered in the differential diagnosis of skin fragility.
2. Molecular and structural basis ofadhesion and stability of skincompartments
2.1. Cytoskeleton and cell—cell adhesions
Keratins are major components of the intermediatefilament (IF) cytoskeletal network of epithelial cells(Fig. 1). Their primary function is to protect cellsfrom mechanical and non-mechanical stress. Morethan 50 keratin genes are specifically expressed indifferent epithelia [10]. According to their biochem-ical properties, keratins are divided into two types,the acidic type I keratins (K9-K20) and the neutral-basic type II keratins (K1—K8). Pairs of types I and IIkeratins are synthesized together. Basal keratino-
Fig. 2 Schematic represen
cytes express K14 and K5 when in contact with abasal lamina, although these keratins persist in thecells at biochemically detectable levels also in theupper epidermal layers. In the process of cornifyingdifferentiation, suprabasal keratinocytes express K1and K10. Keratin 2A (or keratin 2E) is located in theupper spinous and granular layers and associateswithK10 at specific thickened sites of the skin. As in othertypesof IFs, keratinmonomers consist of a central roddomain in four a-helical segments (1A, 1B, 2A, 2B)connected by non-helical linker stretches (L1, L12,L2). Theconservedhelixboundarypeptides, thehelixinitiation peptide and the helix termination peptideare located at either end of the rod domain and areflanked by non-helical end domains, the amino-term-inal head domain and the carboxyl-terminal taildomain. The rod domain, in particular its N- and C-termini, is essential for anti-parallel hetero-dimer-ization of specific pairs of types I and II keratins,whilethe globular domains interact with cytolinker pro-teins of the plakin family. A filament of around 8—10 nm in diameter is composed by repeated coiled-coiling of a single polypeptide chain [11].
The barrier to the environment is constituted byterminally differentiating keratinocytes,whichmovefrom the proliferating basal layer to the granularlayer where the cornified envelope is formed andfinally to the cornified layer, an association of flat-tened, dead cell remnants [12]. The resistance andinsolubility of the cornified envelope is based on theformation of very stable covalent bonds betweentype II keratins, catalysed by the transglutaminases(TG), TG1, TG3 and TG5. This results in the formationand stabilization of the keratin IF filaggrin network inthe cornified cell. TG5 is widely expressed in tissuesand strongly expressed in theepidermis, especially at
tation of desmosomes.
Molecular and diagnostic aspects of genetic skin fragility 133
the junction between stratum granulosum and stra-tum corneum [13]. Mutations in the gene encodingTG1 cause lamellar ichthyosis [14].
Desmosomes are key adhesion complexes in mostepithelia, forming circular domains 0.1—0.5 mm indiameter at the plasma membrane (Fig. 2). Theyconsist of a number of adhesive proteins and glyco-proteins that link keratinocytes to one another andconnect the cell membrane to the cytoplasmic IFnetwork. The main components of desmosomes arethe transmembrane cadherins, desmogleins 1—4 anddesmocollins 1—3. The plakins represent a family oflarge cytolinker proteins that havemultiple functionsin the cross-talk of cytoskeletal networks by cross-linking actin microfilaments, microtubules and IFs.They are central components in desmosomes, HDsand focal adhesions, forming a link to the cytoskele-ton. Desmoplakin is the most prominent desmosomalplaque protein and is required for assembly of des-mosomes and their association with IF [15]. The N-terminal plakin domain peptide of desmoplakin isessential for targeting to desmosomal plaques byinteractions with plakoglobin and plakophilin-1,while the C-terminal domain is composed of threeplakin repeats. Furthermore, desmoplakin can bindto itself, to desmocollin and to plectin. Plakophilin 1is an armadillo protein, which is not only a cell type-specific component of stratifying epithelial desmo-somes, but also a nuclear component of many celltypes, including epithelial cells, fibroblasts andhematopoietic cells. It comprises a unique N-term-inal headdomain and a C-terminal domain containinga characteristic 9.2 armadillo repeat unit [16]. Thehead domainsmediate interactionswith desmosomal
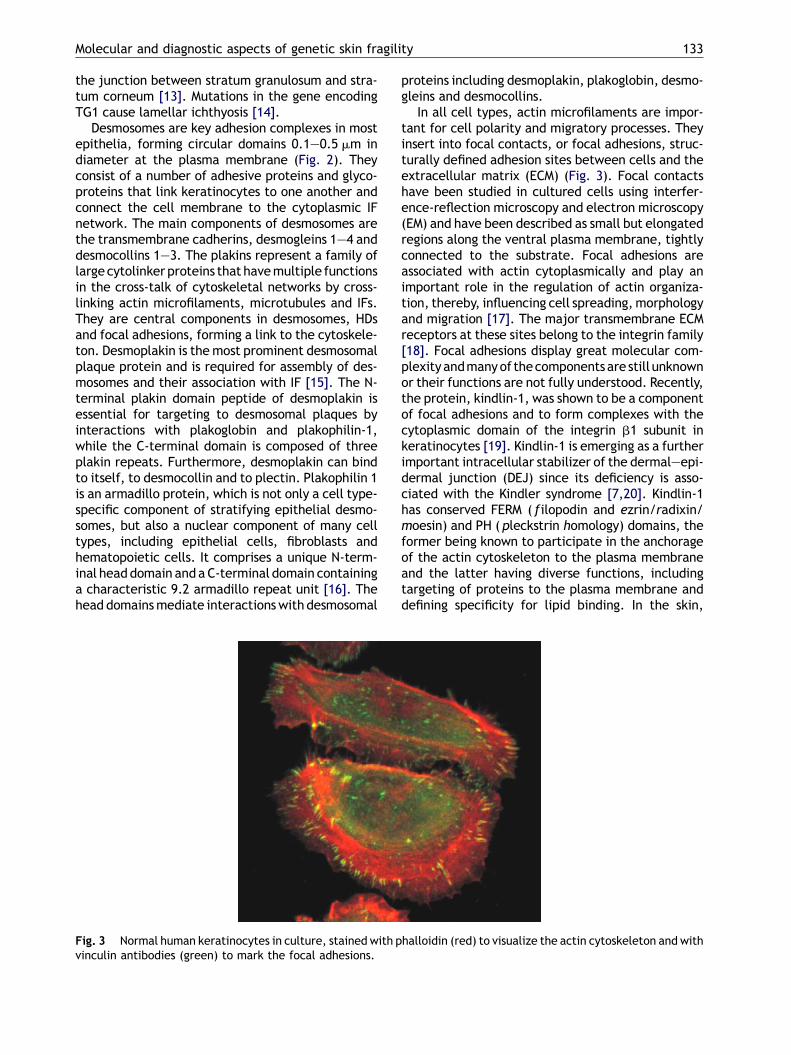
Fig. 3 Normal human keratinocytes in culture, stained with pvinculin antibodies (green) to mark the focal adhesions.
proteins including desmoplakin, plakoglobin, desmo-gleins and desmocollins.
In all cell types, actin microfilaments are impor-tant for cell polarity and migratory processes. Theyinsert into focal contacts, or focal adhesions, struc-turally defined adhesion sites between cells and theextracellular matrix (ECM) (Fig. 3). Focal contactshave been studied in cultured cells using interfer-ence-reflection microscopy and electron microscopy(EM) and have been described as small but elongatedregions along the ventral plasma membrane, tightlyconnected to the substrate. Focal adhesions areassociated with actin cytoplasmically and play animportant role in the regulation of actin organiza-tion, thereby, influencing cell spreading,morphologyand migration [17]. The major transmembrane ECMreceptors at these sites belong to the integrin family[18]. Focal adhesions display great molecular com-plexity andmanyof thecomponents are still unknownor their functions are not fully understood. Recently,the protein, kindlin-1, was shown to be a componentof focal adhesions and to form complexes with thecytoplasmic domain of the integrin b1 subunit inkeratinocytes [19]. Kindlin-1 is emerging as a furtherimportant intracellular stabilizer of the dermal—epi-dermal junction (DEJ) since its deficiency is asso-ciated with the Kindler syndrome [7,20]. Kindlin-1has conserved FERM ( filopodin and ezrin/radixin/moesin) and PH (pleckstrin homology) domains, theformer being known to participate in the anchorageof the actin cytoskeleton to the plasma membraneand the latter having diverse functions, includingtargeting of proteins to the plasma membrane anddefining specificity for lipid binding. In the skin,
halloidin (red) to visualize the actin cytoskeleton and with
134 C. Has, L. Bruckner-Tuderman
kindlin-1 is expressedwithin basal keratinocyteswitha predominant localization at their basal surfacealong the DEJ [20]. In cultivated epithelial cells,kindlin-1 co-localizes with vinculin and with actinmicrofilaments [19,20].
2.2. The hemidesmosomes and thedermal—epidermal junction
The DEJ is essential for adhesion and signalingbetween the epidermis and dermis. Its complexstructure is based on molecular networks that con-nect the basal keratinocytes to the ECM. HDs ultra-structurally resemble a desmosome, but have acompletely different molecular composition(Fig. 4). The 230-kDa BPAG1 (bullous pemphigoidantigen-1 or BP230) contains a plakin domain, acoiled-coil rod domain and two PLEC repeats. Itresides in the inner cytoplasmic plaque of HD, whereit contributes to the tethering of the keratin IFs viaits C-terminal domain [15]. In a transgenic mousemodel, deletions in bpag1 gene have been shown tocause mechanical fragility of basal keratinocytes,reminiscent of EB simplex. However, no mutations inthe human BPAG1 gene have been reported, to date.Plectin, a huge plakin protein of 500 kDa, is loca-lized to HD and focal adhesion structures in basalkeratinocytes and myogenic tissues, including the Z-bands of striated muscle, dense plaques of smoothmuscle, and intercalated discs of cardiac muscle. Inthe HD, plectin directly links IF to the cytoplasmicdomains of the b4 integrin and collagen XVII [15].HDs also contain CD151, a member of the tetraspa-nin protein superfamily, characterized by four trans-membrane domains, one small and one largeextracellular loop and short cytoplasmic N- and C-terminal domains. CD151 is believed to play a role inthe stabilization of HD by regulating the spatial
Fig. 4 Schematic repre
organization and lateral interactions of hemidesmo-somal proteins [15].
An important and specific component of the DEJ iscollagen XVII, also known as the 180-kDa bullouspemphigoid antigen-2, or BP180. Collagen XVII isthe largest transmembrane collagen known and con-sists of three identical a-chains, which form a homo-trimer inserted into the plasmamembrane in a type IIorientation [21]. The intracellular portion of thehomotrimer is located in the cytoplasmic plaque ofHDs where it co-localizes and interacts with theintracellular tail of the integrin b4 subunit, BPAG1and plectin. The extracellular domain of human col-lagen XVII, which contains 15 collagenous and 16 non-collagenous segments, forms the anchoring filamentstogether with laminin 5/332 [21,22].
The integrins comprise a large family of adhesionreceptors. They are hetero-dimers of a and b sub-units that contain a large extracellular domainresponsible for ligand binding, a transmembraneand a cytoplasmic domain [18]. At least 24 integrinsare known in humans. In addition to their roles inadhesion to ECM ligands or to counter receptors onadjacent cells, integrins serve as transmembranemechanical links between the extracellular matrixand the intracellular cytoskeleton. The integrinsalso regulate and modulate the architecture ofcytoskeletal networks [23]. All integrins, excepta6b4, link to actin microfilaments. a6b4 integrinis involved in anchoring the keratin IF network to theHD. However, it is not only present in tissues thatcontain HD, but also in various cells that apparentlylack HD, like Schwann cells, perineural fibroblasts,endothelial cells and immature thymocytes. In theskin, the extracellular domain of a6b4 integrin bindsto laminin 5/332 [15].
Laminins are the major extracellular glycopro-teins located in the basement membrane and are
sentation of the DEJ.
Molecular and diagnostic aspects of genetic skin fragility 135
heterotrimers formed by a, b and g subunits. Todate, five a, three b and three g chains have beenidentified that can combine to form up to 15 differ-ent isoforms [22]. In the skin and mucosae, laminina3A and a3B chains associate with b3 and g2 chainsto form laminin 3A32 and 3B32 (previously known aslaminin 5), or with the b1 and g1 chains to formlaminin 311 (previously synonymous laminin 6).Together with the ectodomain of collagen XVII theyconstitute the anchoring filaments of the skin. TheC-terminus of the laminin a3 chain interacts witha3b1 and a6b4 integrins while the N-terminus oflaminin 332 binds to collagen VII, the major compo-nent of the anchoring fibrils located in the sub-lamina densa resulting in a direct connectionbetween anchoring filaments and fibrils [24].
Collagen IV is a ubiquitous and major componentin all basement membranes. Individual type IV col-lagen molecules are heterotrimers of differenta(IV)-chains. Each molecule has a long triple helicaldomain and a large globular domain at the N-termi-nus (NC1). The self-polymerization of collagen IVinvolves dimerization of the NC1 domains and tetra-mer formation by interactions between the C-term-inal regions. The association is stabilized bycovalent bonds. These end-to-end interactionsresult in an extended two-dimensional network thatis the basis of basement membrane organization andto which other matrix proteins, like laminins, per-lecan and nidogens, bind. Six genetically distincta(IV) chains exist: a1(IV) and a2(IV) chains are foundin all basement membranes, whereas the a3(IV),a4(IV), a5(IV) and a6(IV) chains have a morerestricted tissue localization. At the DEJ, thea1(IV) and a2(IV) containing network dominates,but an a1(IV)/a2(IV)/a5(IV)/a6(IV) containing net-work is also present. Mutations in the genes encod-ing the different a(IV) chains have not beenidentified in skin diseases, but are associated withAlport syndrome or leiomyomatosis associated withAlport syndrome [25]. Recently, a COL4A1 mutationwas identified in a family with small-vessel disease,which often underlies ischemic strokes and intra-cerebral hemorrhages [26].
Collagen VII, the main component of the anchor-ing fibrils, is the longest collagen and a homotrimerof three a1(VII) chains. Each a-chain consists of acentral collagenous region flanked by NC terminalsequences. The large N-terminal domain, NC1, iscomposed of two von Willebrandt A modules andnine fibronectin type III repeats. It interacts with thebasement membrane components, collagen IV andlaminin 332, and plays a critical role in connectingthe lamina densa to the papillary dermis. CollagenVII also interacts with fibronectin via the collage-nous triple helix. Collagen VII is secreted as a homo-
trimeric molecule, which aggregates extracellularlyinto anti-parallel dimers. After proteolytic cleavageof the NC-2 domains, the dimers associate laterallyto form a tightly packed fibrous structure, theanchoring fibril [27].
3. Inherited skin fragility disorders
3.1. Keratin defects
3.1.1. Epidermolytic hyperkeratosis (syn.bullous ichthyosiform erythroderma)Epidermolytic hyperkeratosis (EHK) is an autosomaldominant disorder with an incidence of 1 in200 000—300 000 newborns, with up to 50% of thecases being sporadic. The disease manifests at birth,frequently with erythroderma, blistering and peel-ing (Fig. 5A). There is notable perinatal and child-hood mortality and morbidity resulting fromepidermal erosions and infections. Erythrodermaand blistering diminish during the first year of lifeand hyperkeratosis develops predominantly overthe flexural areas of the extremities. Histopatholo-gical investigation reveals orthohyperkeratosis anda huge stratum corneum. The granular layer isbroadened and degenerated and contains coarsekeratohyalin granula. Suprabasal keratinocytesshow perinuclear vacuolar degeneration and lysis,which is the basis for the name epidermolytic hyper-keratosis (Fig. 5B), and there are slight to moderateperivascular infiltrates in the corium.
EHK is caused by mutations in the KRT1 or KRT10genes coding for keratin 1 or 10. Amino acid substitu-tions have dominant negative effects. An unusualvariant of EHK with late onset is caused by a hetero-zygous single-nucleotide insertion (c.1752insG) inKRT1, predicted to lead to the replacement of thevariableK1enddomainbyanaberrant sequence [28].Recently, the first case of recessive inheritance ofEHK was described and sequence analysis revealed ahomozygous nonsense mutation of the KRT10 gene inseverely affected individuals [29].
3.1.2. Ichthyosis bullosa SiemensIchthyosis bullosa Siemens (IBS) is a rare autosomaldominant disease, which manifests from birth withwidespread blistering. Later, the patients developlarge, dark grey hyperkeratosis, with a lichenoidappearance on the flexural areas of the arms andlegs. Bullae occur after minor mechanical trauma,especially in summer. Skin fragility is superficial,with shedding of the stratum corneum, and leads tolocalized denuded areas. This phenomenon is oftenobserved on the backs of the hands and feet, andwas described as ‘‘Mauserung’’ or a ‘‘molting
136 C. Has, L. Bruckner-Tuderman
effect’’ by Siemens in 1937, when he first describedthis phenotype. A progressive amelioration of symp-toms with age is reported. Clinical and histopatho-logical differentiation from EHK is difficult [30],since histology of IBS reveals several features ofepidermolytic hyperkeratosis. However, vacuoliza-tion of the keratinocytes is less dominant and moresuperficial than that observed in EHK. Biopsies fromfresh blisters show splitting in both granular andhorny layers. Ichthyosis exfoliativa was initially
Fig. 5 Clinical and morphological phenotypes in genetic skinepidermolytic hyperkeratosis. (B) Histologic features of epideEB simplex Weber—Cockayne with blisters and keratosis on thewas identified in the patient and her affected mother. (D) Sevehomozygous KRT14 deletion mutation. (E) Two-year-old girl wwho is homozygous for a KIND1 duplication mutation. (F) IIF odiscontinuous staining pattern. (G) Blistering in a 6-year-old gNon-Hallopeau—Siemens dystrophic EB in a newborn who is coand a nonsense mutation in COL7A1.
described as a distinctive entity [31], but was sub-sequently shown to be a form of IBS [32].
IBS is caused by mutations in the keratin 2A gene(KRT2A or KRT2E) on chromosome 12q11 in the typeII keratin gene cluster. So far, 13 different missensemutations have been described (HGMD, http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html). Themutation p.E493K, located at the end of the roddomain, is by far the most frequent mutation in thisdisorder [33].
fragility disorders. (A) Clinical phenotype in newborn withrmolytic hyperkeratosis. (C) Twenty-year-old patient withsoles of the feet. A heterozygous KRT5 deletion mutationrely affected newborn with recessive EB simplex due to aith Kindler syndrome showing blisters on the extremities,f KS skin with antibodies against collagen XVII showing airl with JEB non-Herlitz caused by COL17A1mutations. (H)mpound heterozygous for a glycine substitution mutation
Molecular and diagnostic aspects of genetic skin fragility 137
3.1.3. Epidermolysis bullosa simplexEB simplex is themost common EB subtype, account-ing for one half of all cases [34]. It is clinicallycharacterized by non-scarring blisters of the skincaused by minimal trauma and morphologically byintraepidermalblistering. Theclinical spectrumofEBsimplex ranges from mild blistering of the hands andfeet in EB simplex Weber—Cockayne (Fig. 5C), tomore generalized blistering in EB simplex Kobner,EB simplex Dowling—Meara and EB simplex withmottled pigmentation. EM and indirect immunofluor-escence (IIF) staining of skin sections with antibodiesto keratins and proteins of the DEJ have localized thelevel of splitting to the stratum basale.
The major subtypes of EB simplex result frommutations in either thekeratin 5 (KRT5) or thekeratin14 (KRT14) gene. Mutations in the gene for plectin(PLEC1) cause the rare forms, EB simplex with mus-cular dystrophy and EB simplex Ogna, which will bediscussed below. EB simplex, as most keratin disor-ders identified in humans, is caused by dominantmissense mutations. The position of the mutationin the protein correlates with the phenotypic sever-ity. For example, Dowling—Meara EB simplex is asso-ciated with mutations in the helix boundary motifs,while the more mild Kobner and Weber—Cockaynephenotypes are associated with mutations in the roddomainoutside thehelixboundary peptides or innon-helical domains such as the linker regions or the H1domain in the head [11]. Patients with recessive EBsimplex (Fig. 5D) represent about 5% of all EB simplexkeratin mutations [11,35].
3.2. Defects in enzymes involved interminal differentiation of keratinocytes
3.2.1. Acral peeling skin syndromePeeling skin syndrome (PSS) is a heterogeneousgroup of skin disorders characterized by the con-tinuous shedding of the outer epidermis layers ([13]and citations herein). Generalized non-inflamma-tory (type A) and inflammatory (type B), as wellas localized forms, have been described [36]. In theacral form the dorsal surfaces of the hands and feetare predominantly affected and exhibit asympto-matic erythematous and thin-scaling patches with asharply demarcated border. The peeling seems tooccur spontaneously and manual removal of hornystrips is easy and painless. The skin lesions areexacerbated by elevated ambient temperatureand humidity [37—39,13].
In both generalized and acral PSS, histological andultrastructural analyses reveal tissue separation atthe junction between the granular layer and thestratum corneum. In two Dutch kindreds with acralPSS, a homozygous mutation, p.G113C, has been
identified in the TGM5 gene, which encodes trans-glutaminase 5 [13]. The mutation changes glycine113, a residue conserved in all members of the TGfamily in many species, and completely abolishesenzymatic activity. To date, no TGM5mutations havebeen identified in patients with generalized PSS.
3.3. Defects of desmosomal proteins
3.3.1. Skin fragility–—ectodermal dysplasiasyndrome (syn. McGrath syndrome)The skin fragility–—ectodermal dysplasia syndrome,a very rare autosomal recessive skin disorder, com-prises trauma-induced skin fragility and ectodermaldysplasia affecting hair, nails and sweat glands[6,40]. Patients have red, raw skin at birth, followedby loss of hair (scalp, eyebrows and eyelashes),painful palmoplantar fissuring and keratoderma,nail dystrophy, reduced sweating and perioral ero-sions [41]. Sometimes, late-onset painful plantarhyperkeratosis causes the patient to become wheel-chair-bound. Skin fragility manifests as trauma-induced tearing and blisters. The histologic hall-marks of this syndrome are widened intercellularspaces and dysadhesion in the suprabasal layers ofthe epidermis, with resulting intraepidermal clefts,blisters and a few acantholytic keratinocytes in thelower epidermis, or with a complete detachment ofthe epidermis above the upper spinous layers,accounting for the skin fragility and tendency toblistering. EM reveals loss of keratinocyte—kerati-nocyte adhesion and small desmosomes that arereduced in number. These changes are most promi-nent in the lower suprabasal layers. The keratin IFnetwork association with the cell membrane is alsodisrupted, particularly in mid-spinous layers. Inmost suprabasal keratinocytes, tonofilamentsappear condensed and compacted in a perinucleardistribution and lack connections to desmosomes[40,42]. Immunohistology of skin sections with anti-bodies against plakophilin 1 is a helpful diagnostictool, since lack of staining is the consequence ofmost mutations.
Compound heterozygous or homozygous muta-tions in the gene PKP1 encoding plakophilin 1 havebeen identified in patients with the epidermal dys-plasia skin fragility syndrome. This phenotypedemonstrates the importance of plakophilin 1 instabilizing components of the desmosomal plaqueand in linking desmosomes to the cytoskeleton. ThePKP1 mutations described so far are nonsense, fra-meshift and splice site mutations, all causing loss offunction of the protein. Hamada et al. [41] have alsoreported a patient with a milder clinical phenotypeand a homozygous splice site mutation: RT-PCRprovided evidence for low levels of the full length
138 C. Has, L. Bruckner-Tuderman
PKP1 transcript and additional near full-length tran-scripts while immunohistology revealed positivestaining of the skin with an anti plakophilin 1 anti-body recognizing the head domain of the molecule.
3.3.2. Skin fragility–—woolly hair syndromeTwo patients with the autosomal recessive skinfragility–—woolly hair syndrome have beendescribed [43]. They show neonatal skin fragility,focal and diffuse palmoplantar keratoderma,hyperkeratotic plaques on the trunk and limbs,and woolly hair with varying degrees of alopecia.The clinical severity did not improve with age andthe patients became disabled. Ultrastructural ana-lysis of the skin demonstrated acantholysis through-out all layers of the epidermis, focal detachment ofdesmosomes, and perinuclear condensation of thesuprabasal keratin IF network. Immunohistochem-istry of skin biopsies revealed that desmoplakin waslocated not only at the cell periphery, but also in thecytoplasm while genetic analysis revealed com-pound heterozygosity for a nonsense/missensecombination of mutations in the desmoplakin gene,DSP [43]. However, heterozygous carriers displayedno phenotypic abnormalities. Mutations in DSPmaycause various phenotypes. A very similar autosomalrecessive disorder, designated dilated cardiomyo-pathy with woolly hair and keratoderma has alsobeen shown to be due to a missense DSP mutation[44]. In some cases of the autosomal dominantdisorder, striate palmoplantar keratoderma, aDSP nonsense mutation is associated with haploin-sufficiency [45].
3.3.3. Lethal acantolythic epidermolysisbullosaA single case of a newly recognized syndrome, lethalacantholythic EB, was reported [3]. The newbornshowed severe fragility of the skin and mucousmembranes, with epidermolysis commencing duringdelivery, and progressing to 90% of the body surfacearea within the first 5 days of life, leaving extensiveeroded areas. The phenotype also comprised uni-versal alopecia, neonatal teeth and nail loss. Thepatient died neonatally due to immense transcuta-neous fluid loss. Histology revealed suprabasal cleft-ing and acantholysis throughout the spinous layer,while EM revealed disconnection of keratin IFs fromdesmosomes. IIF staining for desmoplakin showed adistinct punctuate intercellular pattern in thepatient’s skin.
Mutation detection in the DSP gene disclosedcompound heterozygosity for a nonsense mutationleading to p.R1934X and a deletion c.6370delTT.Aberrant mRNA transcripts were detected by RT-PCR, which predict premature termination of trans-
lation with loss of the IF binding subdomains in thedesmoplakin tail, while protein analysis revealedexpression of truncated polypeptides. Thus, in thiscase, the dramatic phenotype was caused by loss ofthe desmoplakin tail, emphasizing its essential rolein epidermal integrity.
3.4. Defects in proteins associated withthe actin cytoskeleton
3.4.1. Kindler syndromeKindler syndrome (KS) is a rare autosomal recessivegenodermatosis first described in 1954. Since then,around 100 cases have been published, and thecausative genetic defect was described in 2003[7]. The clinical features of KS include congenitalskin blistering (Fig. 5E) and mild photosensitivityboth of which improve with age, and an early,generalized, progressive poikiloderma with exten-sive atrophy. The presence of palmoplantar kerato-derma and nail abnormalities is variable; webbing ofthe fingers and contractures may also occur. Gingivalfragility, poor dentition, as well as early and rapidlyprogressive periodontitis occur in some patients.Other mucous membranes, i.e. urethral, anal, oeso-phageal and genital mucosae, may be involved andthe lesions can lead to stenosis. Squamous cellcarcinomas have been reported as complicationsof KS [46]. EM shows reduplications of the epidermalbasement membrane and different levels of blisterformation. IIF staining with antibodies against lami-nin 332, a6b4 integrin, and collagens XVII and VIIdemonstrate abnormal and discontinuous stainingpatterns (Fig. 5F).
Mutations in the KIND1 gene, encoding kindlin-1,have been identified in patients with the KS [7,20],with more than 20 loss-of-function mutations iden-tified to date [47]. Recently, a recurrent Alu-mediated large deletion spanning from KIND1introns 9 to 11 was described in several Italian KSpatients. Predictive analysis of repetitive elementsrevealed that KIND1 is rich in repetitive sequences,such as the Alu-repeats. Hence, complex genomicrearrangements may explain why mutations couldnot be identified by common PCR-based mutationscreening strategies in several KS patients [48].
3.5. Defects in proteins of thehemidesmosomes and anchoring filaments
3.5.1. Epidermolysis bullosa simplex causedby plectin defectsMutations in the gene encoding plectin (PLEC1) leadto EB simplex with muscular dystrophy and EB sim-plex Ogna. Recently, PLEC1 mutations were alsoreported in the novel clinical subtype, EB simplex
Molecular and diagnostic aspects of genetic skin fragility 139
with pyloric atresia [49,50]. Furthermore, a homo-zygous plectin mutation has been shown to be asso-ciated with a lethal form of recessive EB [51]. Thisnew variant is characterized by generalized blister-ing, loss of skin from the extremities, particularlythe limbs (aplasia cutis), complex developmentalanomalies and rapid demise after birth.
3.5.1.1. EB simplex with muscular dystrophy. EBsimplex with muscular dystrophy manifests as con-genital, generalized blistering associated with late-onset progressive muscular dystrophy. Healing of theskin lesions occurs without scarring, but with atrophyand nail abnormalities. Mild palmoplantar keratosis,dental anomalies and corneal involvement, aswell asextensive mucosal erosions and urethral stricturesare described in some patients. The onset and extentof the muscle symptoms vary greatly, and in mostcases, eventually lead to the patients’ becomingwheelchair-bound and to premature death. EMreveals blistering low in basal cells, HDs with hypo-plastic attachment plates and impaired keratin fila-ment insertion into the inner hemidesmosomalplaque. IIF with plectin antibodies shows absenceof this protein. This rare autosomal recessive diseaseis caused by nonsense mutations, small deletions orinsertions in the PLEC1 gene, which lead to prema-ture termination codons (PTCs) and to absence ofplectin both in the skin and in the muscle.
3.5.1.2. EB simplex Ogna. EB simplex Ogna is anautosomal dominant disease described in only onelarge Norwegian family. The patients exhibited sea-sonal acral blistering with onset in infancy andgeneralized tendency to skin bruising, as well asonychogryphosis. A heterozygous missense PLEC1mutation (p.R2110W) has been documented in thisfamily [52].
3.5.1.3. EB simplex with pyloric atresia. So far, sixrelated patients with the newly identified EB sim-plex with pyloric atresia have been described[49,50]. All of the patients exhibited severe gener-alized congenital blistering and pyloric atresia, anddied from complications of the disease shortly afterbirth. EM showed cleavage within the lamina lucida[49] or at the base of the basal keratinocytes [50],and HDs were reduced in frequency and hypoplastic.Immunostaining for plectin was markedly attenu-ated or absent. Homozygous nonsense, splice siteand deletion mutations in PLEC1 were identified inthe affected neonates [50,49]. Due to similarities inthe symptoms of EB simplex with pyloric atresia withthose of junctional EB with pyloric atresia, causedby mutations in the gene for b4 integrin, Nakamuraet al. [49,50] have postulated that the development
of pyloric atresia must be associated with defects ininteractions between plectin and the b4 integrin.
3.5.2. Junctional epidermolysis bullosaThe term junctional epidermolysis bullosa (JEB)comprises a heterogenous group of disorders, whichshare mechanically induced blistering, with a skinsplit level within the lamina lucida of the DEJ. Theclinical phenotypes range from very severe lethalforms to milder forms in which skin, its annexes andmucosae are involved. The genetic background isalso heterogenous; mutations in six genes areresponsible for JEB.
3.5.2.1. JEB Herlitz. JEB Herlitz (syn. JEB gravis) isone of the most severe EB subtypes and usually leadsto death within the first years of life [53]. The dis-order is characterized by widespread erosions andblistering of skin and mucosae from birth. Extremi-ties, the scalp and the face are usually affectedearly,together with oral and respiratory mucous mem-branes. The skin is extremely fragile and normalhandling of an affected baby may cause extensivedetachment of the epidermis. Typically, laryngealmucosal blistering leads to hoarseness. Blisters healwithout scars, but atrophy develops after recurrenterosions. At some stage, however, the healing of theskin blisters ceases, and large areas of chronic ero-sions and granulation tissue result. Nails are lost andparonychia-like lesions are characteristic. Toothenamel is defective, but dental growth is normal.Eventually, fluidandprotein loss, combinedwithpoorfeeding, cause systemic complications, like severeanemia, growth retardation, chronic infections and,finally, organ failure.
EM reveals rudimentary HDs and absence ofanchoring filaments. Immunostaining of skin sectionsis a very useful tool for rapid diagnosis of Herlitz JEB,with characteristic absence or significant reductionin staining with antibodies to laminin 332. In accor-dance with the concept that receptor—ligand inter-actions are essential for the formation of a functionalHD-anchoring filament complex, absence of laminin332 is frequently associated with reduced depositionof other HD components. Rare exceptions exists ofneonates with JEB Herlitz but positive staining forlaminin 332 and for HD proteins, which may beexplained by the existence of splice site mutationsthat result in milder phenotypes [54,2].
Homozygous or compound heterozygous nullmutations in the genes encoding the three chainsof laminin 332, a3, b3 and g2 (LAMA3, LAMB3,LAMC2) are responsible for Herlitz JEB. Nonsensemutations, small insertions or deletions result inframeshift, PTCs and in nonsense mediated mRNAdecay, and drastic reduction or complete loss of the
140 C. Has, L. Bruckner-Tuderman
polypeptide. LAMB3 mutations account for 80% ofthe laminin 332 defects [54]. This is mainly due tothe occurrence of the hotspot mutation p.R635X,which represents 45% of all mutated LAMB3 allelesin Caucasian Herlitz JEB patients, but not in theMiddle Eastern population [55].
3.5.2.2. JEB non-Herlitz. The JEB non-Herlitz sub-type comprises all non-lethal JEB forms that are notassociated with pyloric atresia. The clinical spec-trum ranges from localized to generalized pheno-types with onset at birth or later in life. Typically,JEB non-Herlitz manifests as generalized, life-longblistering of skin and mucous membranes (Fig. 5G),skin atrophy, alopecia, nail dystrophy and dentalabnormalities [56]. Blister formation occurs at thelevel of the lamina lucida, and immature HDs arepresent. Immunostaining for laminin 332 is positive,although frequently attenuated, while collagen XVIIstaining may be reduced or absent.
JEB non-Herlitz is genetically heterogeneous:mutations in either the laminin 332 genes, LAMA3,LAMB3 or LAMC2, or in the COL17A1 gene encodingcollagen XVII can result in this phenotype. Patientswith laminin 332 defects are often compound het-erozygous, with combinations of one nonsense andone missense or splice site mutation. Amino acidsubstitutions do not abolish expression but result ina conformation of laminin 332, which alters itsadhesive function [54]. A few patients with JEBnon-Herlitz carrying two PTC mutations have beendescribed; in these cases in-frame skipping of exonscontaining the mutations was the explanation forthe presence of altered but partially functionallaminin 332 polypeptides [54].
Most COL17A1 defects are nonsensemutations, orsmall insertions or deletions, which result in a PTCand absence of collagen XVII in the skin. Missensemutations are more rare and associated with milderphenotypes, including moderate skin blistering anddystrophy of nails and teeth [57,58]. In some cases,a particular mutational constellation may providevaluable new knowledge on biological functions ofcollagen XVII. One example is a large genomicCOL17A1 deletion that results in a truncated col-lagen XVII with a very short intracellular domain ofonly 76 amino acids, rather than the normal 466residues. This results in perturbed interactions ofcollagen type XVII with the intracellular HD compo-nents, b4 integrin, BPAG-1 and plectin, and cytolysisof keratinocytes as also observed in EB simplex [4].
3.5.2.3. JEB with pyloric atresia. JEB with pyloricatresia is a rare EB subtype, characterized by skinblistering with pyloric atresia. In severe cases, thecondition is perinatally lethal and affected children
die of systemic complications despite surgical cor-rection of the intestinal defect. Mild JEB with pylo-ric atresia has a favorable prognosis after surgery tocorrect pyloric atresia. In such cases, skin involve-ment may be minimal, sometimes with late onset ofacral blistering and nail dystrophy. Enamel hypopla-sia may be associated with the disease.
Mutations in the genes coding for a6 and b4 integ-rin (ITGA6 and ITGB4) subunits cause JEBwith pyloricatresia, with the majority of mutations involving theb4 subunit. As a general rule, lethal JEB with pyloricatresia results from nonsense mutations in ITGA6 orITGB4, whereas mild phenotypes occur mainly inpatients with missense mutations in ITGB4 [59].
3.6. Defects in dermal collagens
3.6.1. Dystrophic epidermolysis bullosaDystrophic epidermolysis bullosa is a clinically andgenetically heterogeneous group. It is characterizedby blistering of the skin and mucous membranesfollowingminor trauma,with abroad rangeof clinicalseverity [2,60]. Dermal blisteringand scarringare thehallmarks of dystrophic EB. It can be inherited byautosomal dominant or recessive transmission [2].Collagen VII, the major component of the anchoringfibrils, is reduced or absent in the skin of dystrophicEB patients and, correspondingly, the anchoringfibrils are morphologically altered or absent.
3.6.1.1. Dominant dystrophic EB. Patients withdominant dystrophic EB have a mild clinical pheno-type, mostly limited to the skin. Blistering usuallyoccurs at birth or shortly thereafter, and there is apredilection for the extremities. Patients often havedystrophy and/or loss of nails. IIF of the skin showsnormal or reduced collagen VII staining at the roof ofa trauma-induced blister.
3.6.1.2. Non-Hallopeau—Siemens recessive dys-trophic EB. Non-Hallopeau—Siemens recessive dys-trophic EB is characterized by generalized blisteringat birth (Fig. 5H), mucosal involvement, scarring,dystrophy of teeth, and dystrophy and/or loss ofnails. However, mutilations or pseudosyndactylies ofhands and feet do not develop. Immunostaining ofthe skin demonstrates a reduced collagen VII signalat the blister roof.
3.6.1.3. Hallopeau—Siemens recessive dystrophicEB. Hallopeau—Siemens recessive dystrophic EB isone of the most severe EB subtypes. Generalizedblistering is present at birth and increases progres-sively. Poorly healing ulcerations and massive scar-ring are typical. The synecchia and mutilationsof hands and feet, which are characteristic of
Molecular and diagnostic aspects of genetic skin fragility 141
Fig. 6 Diagnostic algorithm for genetic diseases with skin fragility.
Hallopeau—Siemens recessive dystrophic EB, usuallydevelop early in life. Oral and gastrointestinal invol-vement leads to malnutrition, which in, combina-tion with protein loss through ulcerations, results ingrowth retardation and anaemia. IIF of the skinshows absence or significant reduction of collagenVII.
All forms of dystrophic EB are caused by muta-tions in COL7A1, the gene coding for type VII col-lagen. Glycine substitution mutations in the regioncoding for the triple helical domain of the proteinare characteristic for dominant dystrophic EB [27],and cause the disease through dominant negativeinterference. The genetic background of non-Hallo-peau—Siemens recessive dystrophic EB is heteroge-neous, including missense or splice site mutationsresulting in in-frame exon skipping in at least oneallele. Frequently, non-Hallopeau—Siemens reces-sive dystrophic EB patients are compound hetero-zygous and the second mutation often causes a PTC.In contrast, in the majority of Hallopeau—Siemensrecessive dystrophic EB cases, both the mutationscause PTCs, which lead to nonsense mediated mRNAdecay or truncated collagen VII polypeptides thatare degraded within the cell. Hence, absence ofcollagen VII is a hallmark of Hallopeau—Siemensrecessive dystrophic EB.
Around 300 different COL7A1 mutations havebeen reported in dystrophic EB, which are distrib-uted throughout the gene, and novel mutationscontinue to be identified at a high rate [61]. Thegenotype—phenotype correlations are complex and,with the exception of null mutations, it has not beenpossible to predict phenotypes from gene defects.For example, an apparent missense mutation,p.G114V, was shown to induce a cryptic splice site[62], while in a recessive dystrophic EB patient witha gene deletion an additional missense mutation,p.M2415I was shown to result in complex effects on
splicing [63]. Therefore, although mutation detec-tion in COL7A1 remains technically difficult, timeconsuming and expensive, it is essential for precisediagnosis, including prenatal diagnosis, and prog-nosis of the disease, as well as for genetic counseling[61].
4. Diagnostic algorithm for geneticskin fragility syndromes
Clinical diagnosis of skin fragility syndromes mightbe obvious in children and adults in whom the fullclinical phenotype and complications have devel-oped, but can be very difficult postnatally becauseof the absence of typical secondary symptoms. In allcases, careful investigation of the family historymust be accompanied by a systematic clinical exam-ination of the entire skin surface, nails, hair, denti-tion and mucosal surfaces (Fig. 6). The involvementof other organs, e.g. digestive, urogenital, or mus-culoskeletal systems, must also be considered. Mor-phological/immunohistochemical analysis of skinbiopsies is an essential step in the diagnostic algo-rithm, as it reveals the level of tissue separation andcan often indicate the defective cellular structureor protein. Microbiological tests should be used toexclude primary or secondary infections as thecause of skin blisters or erosions. If other syndromesthat are associated with skin fragility as an auxiliarysymptom (Table 2) are suspected, investigation ofinternal organs is necessary. Mutation detection canbe performed if the defective gene has been iden-tified and if genetic counseling and prenatal diag-nosis are requested for future pregnancies. Inaddition, a precise diagnosis and correct informa-tion on the disease prognosis are often very helpfulfor a family dealing with the enormous psychologicaland general burden of skin fragility.
142 C. Has, L. Bruckner-Tuderman
Acknowledgements
The work of the authors is supported by: EU ProgramGENESKIN, the Network Epidermolysis bullosa sup-ported by the Federal Ministry for Education andResearch (BMBF) and the German Research Council(DFG).
References
[1] Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuder-man L, Christiano A, et al. Revised classification system forinherited epidermolysis bullosa: report of the second inter-national consensus meeting on diagnosis and classification ofepidermolysis bullosa. JAmAcadDermatol 2000;42:1051—66.
[2] Bruckner-Tuderman L. Epidermolysis bullosa. In: Royce PM,Steinmann B, editors. Connective tissue and its heritabledisorders. Molecular, genetic and medical aspects. 2nd ed.,New York: Wiley—Liss; 2002. p. 689—725.
[3] Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, TerHorst HJ, Timmer A, et al. Loss of desmoplakin tail causeslethal acantholytic epidermolysis bullosa. Am J Hum Genet2005;77:653—60.
[4] Fontao L, Tasanen K, Huber M, Hohl D, Koster J, Bruckner-Tuderman L, et al. Molecular consequences of deletion ofthe cytoplasmic domain of bullous pemphigoid 180 in apatient with predominant features of epidermolysis bullosasimplex. J Invest Dermatol 2004;122:65—72.
[5] Pfendner E, Rouan F, Uitto J. Progress in epidermolysisbullosa: the phenotypic spectrum of plectin mutations.Exp Dermatol 2005;14:241—9.
[6] McGrath JA, McMillan JR, Shemanko CS, Runswick SK, LeighIM, Lane EB, et al. Mutations in the plakophilin 1 gene resultin ectodermal dysplasia/skin fragility syndrome. Nat Genet1997;17:240—4.
[7] Jobard F, Bouadjar B, Caux F, Hadj-Rabia S, Has C, Matsuda F,et al. Identification of mutations in a new gene encoding aFERM family protein with a pleckstrin homology domain inKindler syndrome. Hum Mol Genet 2003;12:925—35.
[8] Karamatic CV, Burton N, Kagan A, Green CA, Levene C,Flinter F, et al. CD151, the first member of the tetraspanin(TM4) superfamily detected on erythrocytes, is essential forthe correct assembly of human basement membranes inkidney and skin. Blood 2004;104:2217—23.
[9] Wright MD, Geary SM, Fitter S, Moseley GW, Lau LM, ShengKC, et al. Characterization of mice lacking the tetraspaninsuperfamily member CD151. Mol Cell Biol 2004;24:5978—88.
[10] Coulombe PA, Omary MB. ‘Hard’ and ‘soft’ principles defin-ing the structure, function and regulation of keratin inter-mediate filaments. Curr Opin Cell Biol 2002;14:110—22.
[11] Porter RM, Lane EB. Phenotypes, genotypes and their con-tribution to understanding keratin function. Trends Genet2003;19:278—85.
[12] CandiE, SchmidtR,MelinoG.Thecornifiedenvelope: amodelof cell death in the skin. Nat RevMol Cell Biol 2005;6:328—40.
[13] Cassidy AJ, van Steensel MA, Steijlen PM, van Geel M, van dV, Morley SM, et al. A homozygous missense mutation inTGM5 abolishes epidermal transglutaminase 5 activity andcauses acral peeling skin syndrome. Am J Hum Genet2005;77:909—17.
[14] Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, HashemN, Compton JG, et al. Mutations in the gene for transglu-taminase 1 in autosomal recessive lamellar ichthyosis. NatGenet 1995;9:279—83.
[15] Koster J, Borradori L, Sonnenberg A. Hemidesmosomes:molecular organization and their importance for cell adhe-sion and disease. In: Beissert T, Nelson CF, editors. Hand-book of experimental pharmacology, vol. 165. Berlin:Springer; 2004. p. 245—67.
[16] Getsios S, Huen AC, Green KJ. Working out the strengthand flexibility of desmosomes. Nat Rev Mol Cell Biol 2004;5:271—81.
[17] Zamir E, Geiger B. Molecular complexity and dynamics ofcell—matrix adhesions. J Cell Sci 2001;114:3583—90.
[18] Brakebusch C, Fassler R. The integrin—actin connection, aneternal love affair. EMBO J 2003;22:2324—33.
[19] Kloeker S, Major MB, Calderwood DA, Ginsberg MH, JonesDA, Beckerle MC. The Kindler syndrome protein is regulatedby transforming growth factor-beta and involved in integrin-mediated adhesion. J Biol Chem 2004;279:6824—33.
[20] Siegel DH, Ashton GH, Penagos HG, Lee JV, Feiler HS,Wilhelmsen KC, et al. Loss of kindlin-1, a human homologof the Caenorhabditis elegans actin—extracellular-matrixlinker protein UNC-112, causes Kindler syndrome. Am JHum Genet 2003;73:174—87.
[21] Franzke CW, Bruckner P, Bruckner-Tuderman L. Collagenoustransmembrane proteins: recent insights into biology andpathology. J Biol Chem 2005;280:4005—8.
[22] Aumailley M, Bruckner-Tuderman L, Carter WG, DeutzmannR, Edgar D, Ekblom P, et al. A simplified laminin nomencla-ture. Matrix Biol 2005;24:326—32.
[24] Aumailley M, El Khal A, Knoss N, Tunggal L. Laminin 5processing and its integration into the ECM. Matrix Biol2003;22:49—54.
[25] Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG.Alport’s syndrome, Goodpasture’s syndrome, and type IVcollagen. N Engl J Med 2003;348:2543—56.
[26] Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K,Massin P, et al. Role of COL4A1 in small-vessel disease andhemorrhagic stroke. N Engl J Med 2006;354:1489—96.
[27] Bruckner-Tuderman L, Hopfner B, Hammami-Hauasli N. Biol-ogy of anchoring fibrils: lessons from dystrophic epidermo-lysis bullosa. Matrix Biol 1999;18:43—54.
[28] Sprecher E, Yosipovitch G, Bergman R, Ciubutaro D, Indel-man M, Pfendner E, et al. Epidermolytic hyperkeratosis andepidermolysis bullosa simplex caused by frameshift muta-tions altering the v2 tail domains of keratin 1 and keratin 5. JInvest Dermatol 2003;120:623—6.
[29] Muller FB, Huber M, Kinaciyan T, Hausser I, Schaffrath C,Krieg T, et al. A human keratin 10 knockout causes recessiveepidermolytic hyperkeratosis. Hum Mol Genet 2006;15:1133—41.
[30] Akiyama M, Tsuji-Abe Y, Yanagihara M, Nakajima K, KodamaH, Yaosaka M, et al. Ichthyosis bullosa of Siemens: its correctdiagnosis facilitated by molecular genetic testing. Br JDermatol 2005;152:1353—6.
[32] Kremer H, Zeeuwen P, McLean WH, Mariman EC, Lane EB,van de Kerkhof CM, et al. Ichthyosis bullosa of Siemens iscaused by mutations in the keratin 2e gene. J Invest Der-matol 1994;103:286—9.
[33] Basarab T, Smith FJ, Jolliffe VM, McLean WH, Neill S, RustinMH, et al. Ichthyosis bullosa of Siemens: report of a familywith evidence of a keratin 2e mutation, and a review of theliterature. Br J Dermatol 1999;140:689—95.
[34] Pfendner EG, Sadowski SG, Uitto J. Epidermolysis bullosasimplex: recurrent and de novo mutations in the KRT5and KRT14 genes, phenotype/genotype correlations, and
Molecular and diagnostic aspects of genetic skin fragility 143
implications for genetic counseling and prenatal diagnosis.J Invest Dermatol 2005;125:239—43.
[35] Has C, Chang YR, Volz A, Hoeping D, Kohlhase J, Bruckner-Tuderman L. Novel keratin 14 mutations in patients withsevere recessive epidermolysis bullosa simplex. J InvestDermatol 2006.
[36] Traupe H. The ichthyoses Berlin: Springer; 1989.[37] Shwayder T, Conn S, Lowe L. Acral peeling skin syndrome.
Arch Dermatol 1997;133:535—6.[38] Brusasco A, Veraldi S, Tadini G, Caputo R. Localized peeling
skin syndrome: case report with ultrastructural study. Br JDermatol 1998;139:492—5.
[39] Hashimoto K, Hamzavi I, Tanaka K, Shwayder T. Acral peelingskin syndrome. J Am Acad Dermatol 2000;43:1112—9.
[40] McGrath JA, Hoeger PH, Christiano AM, McMillan JR, MellerioJE, Ashton GH, et al. Skin fragility and hypohidrotic ecto-dermal dysplasia resulting from ablation of plakophilin 1. BrJ Dermatol 1999;140:297—307.
[41] Hamada T, South AP, Mitsuhashi Y, Kinebuchi T, Bleck O,Ashton GH, et al. Genotype—phenotype correlation inskin fragility–—ectodermal dysplasia syndrome resultingfrom mutations in plakophilin 1. Exp Dermatol 2002;11:107—14.
[42] McMillan JR, Haftek M, Akiyama M, South AP, Perrot H,McGrath JA, et al. Alterations in desmosome size and numbercoincide with the loss of keratinocyte cohesion in skinwith homozygous and heterozygous defects in the desmoso-mal protein plakophilin 1. J Invest Dermatol 2003;121:96—103.
[43] Whittock NV,Wan H, Morley SM, GarzonMC, Kristal L, Hyde P,et al. Compound heterozygosity for non-sense and mis-sensemutations in desmoplakin underlies skin fragility/woolly hairsyndrome. J Invest Dermatol 2002;118:232—8.
[47] Ashton GH, McLean WH, South AP, Oyama N, Smith FJ, AlSuwaid R, et al. Recurrent mutations in kindlin-1, a novelkeratinocyte focal contact protein, in the autosomal reces-sive skin fragility and photosensitivity disorder, Kindler syn-drome. J Invest Dermatol 2004;122:78—83.
[48] Has C, Wessagowit V, Pascucci M, Baer C, Didona B, WilhelmC, et al. Molecular basis of Kindler syndrome in Italy: noveland recurrent Alu/Alu recombination, splice site, nonsense,and frameshift mutations in the KIND1 gene. J Invest Der-matol 2006.
[49] Pfendner E, Uitto J. Plectin gene mutations can causeepidermolysis bullosa with pyloric atresia. J Invest Dermatol2005;124:111—5.
[50] Nakamura H, Sawamura D, Goto M, Nakamura H, McMillanJR, Park S, et al. Epidermolysis bullosa simplex associatedwith pyloric atresia is a novel clinical subtype caused bymutations in the plectin gene (PLEC1). J Mol Diagn2005;7:28—35.
[51] Charlesworth A, Gagnoux-Palacios L, Bonduelle M, OrtonneJP, De Raeve L, Meneguzzi G. Identification of a lethal formof epidermolysis bullosa simplex associated with a homo-zygous genetic mutation in plectin. J Invest Dermatol2003;121:1344—8.
[52] Koss-Harnes D, Hoyheim B, Anton-Lamprecht I, Gjesti A,Jorgensen RS, Jahnsen FL, et al. A site-specific plectinmutation causes dominant epidermolysis bullosa simplexOgna: two identical de novo mutations. J Invest Dermatol2002;118:87—93.
[53] Muhle C, Jiang QJ, Charlesworth A, Bruckner-Tuderman L,Meneguzzi G, Schneider H. Novel and recurrent mutations inthe laminin-5 genes causing lethal junctional epidermolysisbullosa: molecular basis and clinical course of Herlitz dis-ease. Hum Genet 2005;116:33—42.
[54] Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuder-man L, Pfendner E, et al. Laminin 5 mutations in junctionalepidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet 2002;110:41—51.
[55] Abu SJ, Indelman M, Pfendner E, Falik-Zaccai TC, Mizrachi-Koren M, Shalev S, et al. Molecular epidemiology of heredi-tary epidermolysis bullosa in a Middle Eastern population. JInvest Dermatol 2006;126:777—81.
[56] Floeth M, Bruckner-Tuderman L. Digenic junctional epider-molysis bullosa: mutations in COL17A1 and LAMB3 genes. AmJ Hum Genet 1999;65:1530—7.
[57] Vaisanen L, Has C, Franzke C, Hurskainen T, Tuomi ML,Bruckner-Tuderman L, et al. Molecular mechanisms of junc-tional epidermolysis bullosa: Col 15 domain mutationsdecrease the thermal stability of collagen XVII. J InvestDermatol 2005;125:1112—8.
[58] Tasanen K, Floeth M, Schumann H, Bruckner-Tuderman L.Hemizygosity for a glycine substitution in collagen XVII:unfolding and degradation of the ectodomain. J InvestDermatol 2000;115:207—12.
[59] Ashton GH, Sorelli P, Mellerio JE, Keane FM, Eady RA,McGrath JA. Alpha 6 beta 4 integrin abnormalities in junc-tional epidermolysis bullosa with pyloric atresia. Br J Der-matol 2001;144:408—14.
[60] Uitto J. Epidermolysis bullosa: the expanding mutationdatabase. J Invest Dermatol 2004;123:xii—i.
[61] Kern JS, Kohlhase J, Bruckner-Tuderman L, Has C. Expandingthe COL7A1 mutation database: novel and recurrent muta-tions and unusual genotype—phenotype constellations in 41patients with dystrophic epidermolysis bullosa. J InvestDermatol 2006;126:1006—12.
[62] Wessagowit V, Kim SC, Woong OS, McGrath JA. Genotype—phenotype correlation in recessive dystrophic epidermolysisbullosa: when missense doesn’t make sense. J Invest Der-matol 2005;124:863—6.
[63] Titeux M, Mejia JE, Mejlumian L, Bourthoumieu S, Mirval S,Tonasso L, et al. Recessive dystrophic epidermolysis bullosacaused by COL7A1 hemizygosity and a missense mutationwith complex effects on splicing. Hum Mutat 2006;27:291—2.
Cristina Has studied medicine during1985—1991 at the Medical UniversityCluj-Napoca, Romania. She is specia-lised in dermatology and venerologyfrom the University Hospital Cluj-Napoca during 1992—1995 and workedas a dermatologist at the Departmentof Dermatology in the same hospitalduring 1995—1998. In 2000, shereceived her doctorate from the Uni-versity of Cluj-Napoca. From 2001 to
2003 she was a postdoctoral fellow at the University of Munster,Germany and the National Centre of Genotyping Evry, France.Since March 2003, she is working at the Department of Dermato-logy at the University of Freiburg, Germany. Her scientific andclinical specialties are: blistering skin diseases, genodermatoses,and molecular genetics of the skin.
144 C. Has, L. Bruckner-Tuderman
Leena Bruckner-Tuderman is specia-lised in dermatology and venerologyfrom the University Hospital Zurich,Switzerland during 1984—1987 andworked as a senior staff dermatologistat the Department of Dermatology atthe University Hospital Zurich from 1988to 1993. In 1989, she received her habili-tation from the University of Zurich andworked as a docent for dermatology andvenerology. In 1995, she became a specia-
list for dermatology and venerology in Germany and the EU. At theUniversity of Muenster, she became Professor of Dermatology in1995. Since February 2003, she is the Chair of the Department ofDermatology at the University of Freiburg, Germany. Her scientificand clinical specialties are: blistering skin diseases, genoderma-toses, autoimmune diseases, connective tissue diseases, woundhealing, molecular genetics, and gene therapy of the skin. LeenaBruckner-Tuderman is a member of numerous dermatological andresearch societies and coordinates the German national networkof excellence ‘‘Epidermolysis bullosa.’’She has over 150 originalpublications in international peer reviewed journals.