20
Molecular dynamics refinement and rescoring in WISDOM virtual screenings Gianluca Degliesposti University of Modena and Reggio Emilia Molecular Modelling & Drug Design Lab
| Date post: | 18-Dec-2015 |
| Category: |
Documents |
| Upload: | eugene-kennedy |
| View: | 214 times |
| Download: | 0 times |

Molecular dynamics refinement and rescoring in WISDOM virtual screenings
Gianluca Degliesposti
University of Modena and Reggio Emilia
Molecular Modelling &
Drug DesignLab

Protein Flexibility and Mobility in Structure-Based Drug Design
• Initially Structure-Based Drug Design was based on the “lock and key” model
• Many targets show significant flexibility upon ligand binding (e.g. local rearrangement of side-chains or small motions of loops)
• Effective methods are available for docking a flexible ligand into a rigid target
• Nowadays there are efforts to consider protein flexibility and mobility in drug design approaches
• Our approach is based on molecular dynamics

Why Molecular Dynamics?
• MD provide the “gold standard” when used to describe flexible biomolecular systems
• MD permit the simultaneus simulation of either target and/or ligands flexibility
• MD simulations can take in account solvent contribution to the system energy
• MD can be chosen to validate and refine the orientations of docked compounds and rescore them using a reliable scoring function
• For these reasons MD can be chosen to validate and refine the orientations of docked compounds and rescore them using a reliable scoring function

How to refine and rescore docking complexes using MD?
• A Refinement and Rescoring procedure based on MM/MD and MM-PBSA has been designed and then validated
• Multistep protocol:
• Minimization of ligand-target complexes
• MD simulation of minimized complexes
• Minimization of complexes after MD
• Free energy of binding (∆Gbind ) estimation with MM-PBSA and/or MM-GBSA
Ligand + Target Complex
∆ Gbinding = ∆ EMM + ∆ Gsolvent
∆EMM = interaction energy in vacuo∆Gsolvent = ∆Gsolv polar + ∆Gsolv non-polar

How to automate the refinement/rescoring procedure in a virtual screening?
Antechamber Leap Sander(MM)
Sander(MD)
Sander(MM) Ptraj
MM-PBSAMM-GBSA
DockedLigands(.mol2)
LigandTopology
ComplexTopology
Target structureand ligand
MMInput files
MinimizedStructure
MDStructure
MinimizedStructure
MMInput files
MMInput files
Update of 3Dcoordinates
OutputRMSD
Refined complexes
Output Freeenergy scores
Complex Preparation Complex Refinement Output retrieving and Rescoring

MD-Refinement/Rescore Procedure Validation
• Target: Aldose reductase
• Ligands: 28 known inhibitors with measured activities and known binding modes
• Predicted free energy of binding show good correlation with experimental data
• MM-PBSA estimated free energies of binding are the best correlating
R2=0.80 R2=0.73
G. Rastelli et al. Bioorganic & Medicinal Chemistry - 15, 2007 (7865-7877)

Molecular Dynamics in WISDOMA practical example
• Target: pf-DHFR
• Ligands: ZINC-DB
• Docking: FlexX
• MD-Refinement
• MM-PBSA rescoring

Docking Results Analysis
• Compounds ranking based on docking energy score
• Analysis of interactions established by each ligand with aminoacids of pf-DHFR binding site
• Selection of a subset of compounds to be refined using the MD-refinement procedure
Docking Results
Analysis of docking scores and interactions
with key compounds
SubsetSelection

Docking energy evaluation
• Analysis of Docking energy trend of first million ranked compounds
• Analysis of Docking energy trend with a cutoff of ~-8 Kcal mol-1
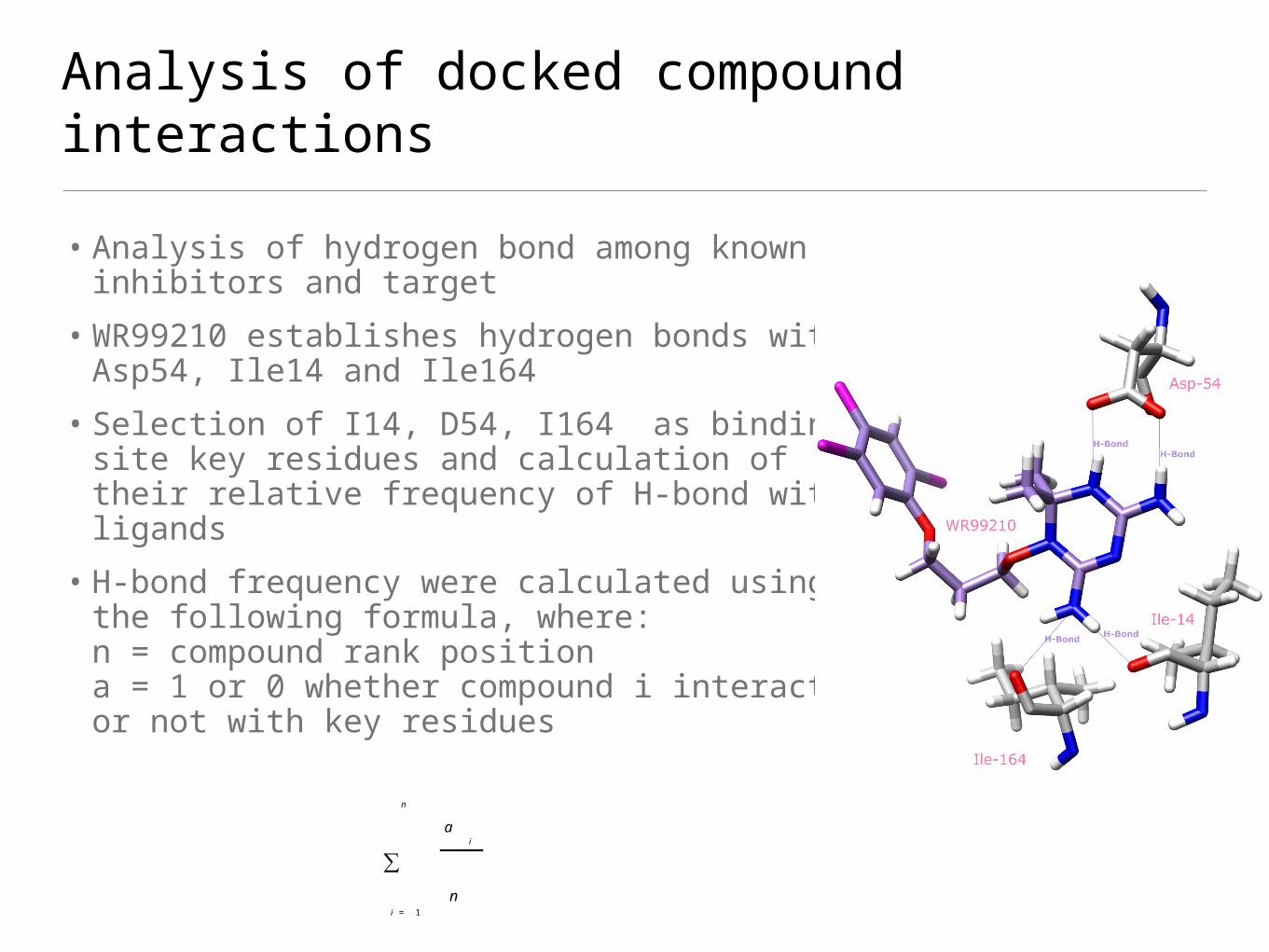
• Analysis of hydrogen bond among known inhibitors and target
• WR99210 establishes hydrogen bonds with Asp54, Ile14 and Ile164
• Selection of I14, D54, I164 as binding site key residues and calculation of their relative frequency of H-bond with ligands
• H-bond frequency were calculated using the following formula, where:n = compound rank positiona = 1 or 0 whether compound i interact or not with key residues
Analysis of docked compound interactions
€
ai
ni = 1
n
∑

Hydrogen bond frequency analysis

MD Refinement preparation
• Selection of first 15.000 compounds from the docking scores ranked list
• Compounds partial charges calculation
• Compounds separated by total charge (Insight)
• Partial atomic charges calculated with AM1-BCC (Antechamber)
• Creation of 300 packages containing 50 compounds
• Storage of created packages on SE
• Setting of input files

Analysis of MD Refinement Results
• Re-ranking of compounds according to MM-PBSA and evaluation of free energy of binding
• Analysis of interaction focusing on H-Bond among ligands and Asp54, Ile14 and Ile164
• Visualisation of best scoring compounds
• Evaluation of mobility after MD
• Evaluation of binding orientation, comparison with WR99210
• Evaluation of rank position difference between docking score and free energy of binding ranked lists

MM-PBSA Rescore Results
WR9
Pyr
Cyg

Interaction frequencies after MD-Refinement

Structure visualization analysis
• Complexes of first 100 compounds were visualized
• Comparison among WR99210 and compound orientations
• Analysis of compounds mobility from docking to post refinement complexes
• Analysis of interactions with binding site residues
• Main part of compounds interact similarly to WR99210
• Some compounds mobility among ligands in docking and after MD refinement complexes
Ile14
Ile164
Asp54
WR99210Cmpd1Cmpd2
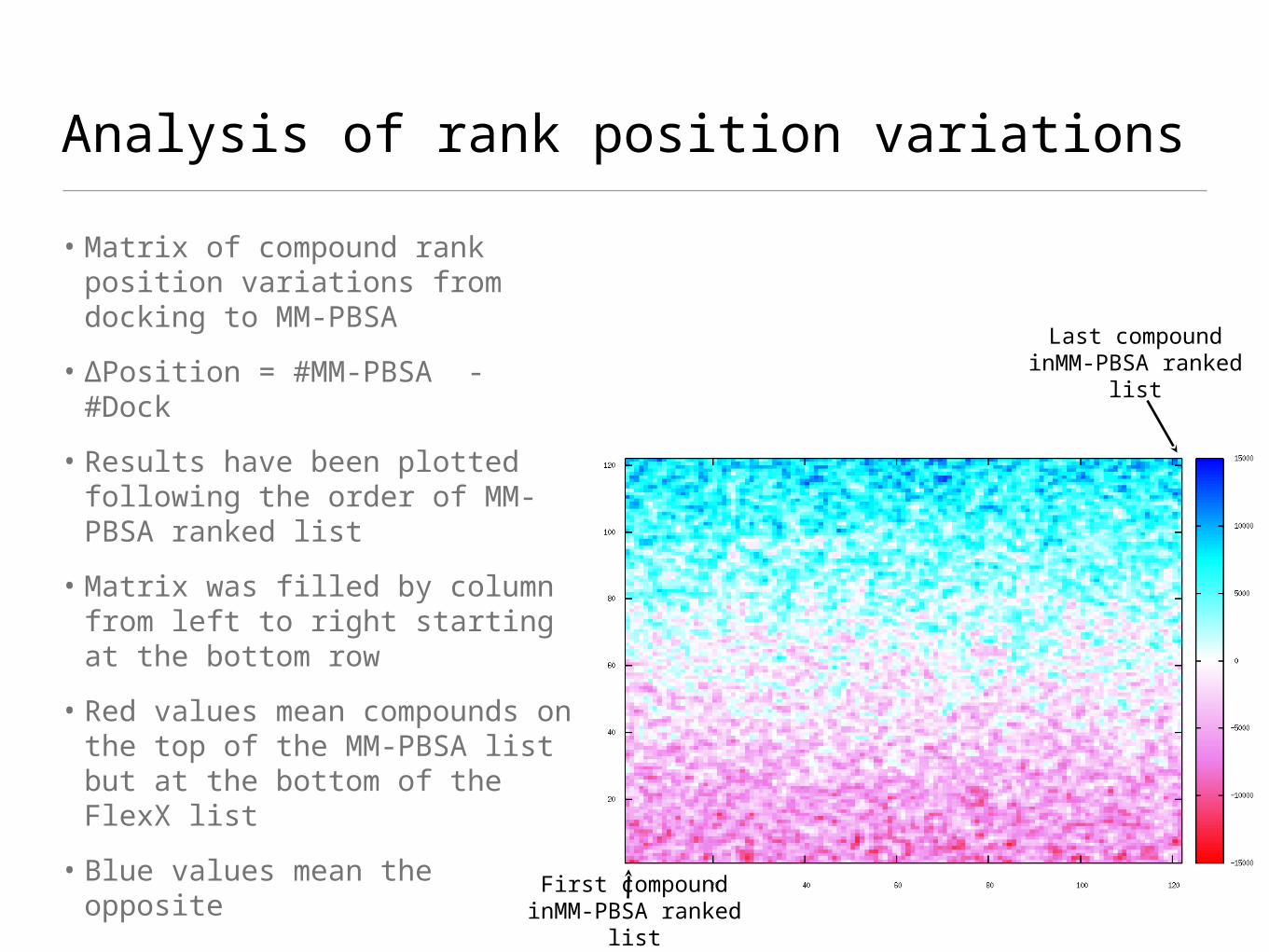
Analysis of rank position variations
• Matrix of compound rank position variations from docking to MM-PBSA
• ∆Position = #MM-PBSA - #Dock
• Results have been plotted following the order of MM-PBSA ranked list
• Matrix was filled by column from left to right starting at the bottom row
• Red values mean compounds on the top of the MM-PBSA list but at the bottom of the FlexX list
• Blue values mean the opposite
Last compound inMM-PBSA ranked
list
First compound inMM-PBSA ranked
list

Conclusions
• Molecular dynamics efficiently refined the orientation of docked compounds
• MM-PBSA rescoring permitted to estimate the free energy of binding of the docked compounds
• Applying the MD-refinement/rescoring procedure, some compounds were recovered from the tail of docking ranked list, while others moved from the head to the tail of MM-PBSA ranked list
• Interesting molecules were retrieved among the first 100 best scored compounds, and several of these will be selected for in vitro activity assays

Acknowledgement
• WISDOM Collaborators
• Worachart Sirawaraporn from Mahidol University (Thailand) for in-vitro test
• Prof. Giulio Rastelli
• Dott. Miriam Sgobba
• Dott. Anna Maria Ferrari
from Università di Modena e Reggio Emilia, Italy

Modena





![trechos.org … · Reggio Children. As cem linguagens em mini-histórias : contadas por professores e crianças de Reggio Emilia [recurso eletrônico] / Reggio Children, Escolas e](https://static.documents.pub/doc/80x56/609a5a68aad0cd466a1f568d/-reggio-children-as-cem-linguagens-em-mini-histrias-contadas-por-professores.jpg)



![1 2 3 Contextual Rescoring for Human Pose Estimationsergio/linked/posterbmvc2014_hupba.pdf · Contextual Rescoring for Human Pose Estimation [1] Y. Yang and D. Ramanan. “Articulated](https://static.documents.pub/doc/80x56/5f4c6ac1dbe0841ac119e076/1-2-3-contextual-rescoring-for-human-pose-sergiolinkedposterbmvc2014hupbapdf.jpg)

![[Paper Introduction] Efficient Lattice Rescoring Using Recurrent Neural Network Language Models](https://static.documents.pub/doc/80x56/587e29de1a28abb93e8b5b07/paper-introduction-efficient-lattice-rescoring-using-recurrent-neural-network.jpg)





