Page 1
Accepted Manuscript
Molecular structure, vibrational and electronic properties of 4-Phenyl-3H-1,3-thiazol-2-ol using density functional theory and comparison of drug efficacy ofketo and enol forms by QSAR Analysis
Alok K. Sachan, Shilendra K. Pathak, Satish Chand, Ruchi Srivastava, OnkarPrasad, Salah Belaidi, Leena Sinha
PII: S1386-1425(14)00768-9DOI: http://dx.doi.org/10.1016/j.saa.2014.05.011Reference: SAA 12159
To appear in: Spectrochimica Acta Part A: Molecular and Biomo-lecular Spectroscopy
Received Date: 14 March 2014Revised Date: 23 April 2014Accepted Date: 2 May 2014
Please cite this article as: A.K. Sachan, S.K. Pathak, S. Chand, R. Srivastava, O. Prasad, S. Belaidi, L. Sinha,Molecular structure, vibrational and electronic properties of 4-Phenyl-3H-1,3-thiazol-2-ol using density functionaltheory and comparison of drug efficacy of keto and enol forms by QSAR Analysis, Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy (2014), doi: http://dx.doi.org/10.1016/j.saa.2014.05.011
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customerswe are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, andreview of the resulting proof before it is published in its final form. Please note that during the production processerrors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Page 2
Molecular structure, vibrational and electronic properties of 4-Phenyl-3H-1,3-thiazol-2-ol using density functional theory and comparison of drug efficacy of keto and enol forms
by QSAR Analysis
Alok K. Sachana, Shilendra K. Pathaka, Satish Chanda, Ruchi Srivastavaa, Onkar Prasada, Salah Belaidib
and Leena Sinhaa,*
aDepartment of Physics, University of Lucknow, 226007, Lucknow,India bGroup of Computational and pharmaceutical Chemistry, LMCE Laboratory,
Faculty of sciences, University of Biskra, 07000, Biskra, Algeria
* Corresponding author. Tel.: +91 9415313779.
E-mail address: [email protected] .
Page 3
Abstract
4-Phenyl-3H-1,3-thiazol-2-ol can exist in two tautomeric forms – keto and enol. Comprehensive
investigation of molecular geometry and electronic structure in ground as well as in the first excited
state of 4-Phenyl-3H-1,3-thiazol-2-ol (enol) has been carried out. To determine lowest-energy
molecular conformation of the title molecule, the selected torsion angles were varied in steps of 10º
and molecular energy profile was calculated from -180º to +180º. Experimental FT-IR and FT-Raman
spectra of title compound were compared with the spectral data obtained by DFT/B3LYP method.
Dipole moment, polarizability, first static hyperpolarizability and molecular electrostatic potential
surface map have been calculated to get a better insight of the properties of title molecule. Natural
bond orbital (NBO) analysis has been done to study the stability of the molecule arising from charge
delocalization. UV–Vis spectrum of the title compound was also recorded and electronic properties
such as frontier orbitals and band gap energies were calculated by TD-DFT approach. To compare
the drug efficacy of enolic and keto forms, QSAR properties of both forms have also been computed
and discussed.
Keywords: Vibrational analysis, FT-IR, FT-Raman and UV-vis spectra, NBO analysis, QSAR
Page 4
1.Introduction
Thiazoles exhibit a variety of biological activity namely antibacterial, antifungal, anti-HIV, anti-
hypertension, anti-inflammatory, anticancer, anticonvulsant and antidepressant [1-6], hence are valuable
structural components in the field of medicinal chemistry. In fact Thiazole moiety appears commonly in
structures of various natural products and biologically active compounds, like thiamine (vitamin-B) and
also is an integral part of most of the available antibiotics drugs such as penicillin, micrococcin which
have revolutionized the therapy of bacterial diseases [7]. Phenyl and substituted phenyl-thiazoles are also
common structures of a wide range of biologically active natural products [8]. Recently it has been found
that phenyl-thiazole ring system provides a template for the design and synthesis of antiviral agents which
inhibit the flavi-viruses by targeting their E-protein [9]. Pharmaceutical importance of thiazoles and their
derivatives drove us to investigate the molecular structural properties, vibrational and energetic data of 4-
Phenyl-3H-1,3-thiazol-2-ol (4P3HT) with a long-term objective to achieve a better understanding of the
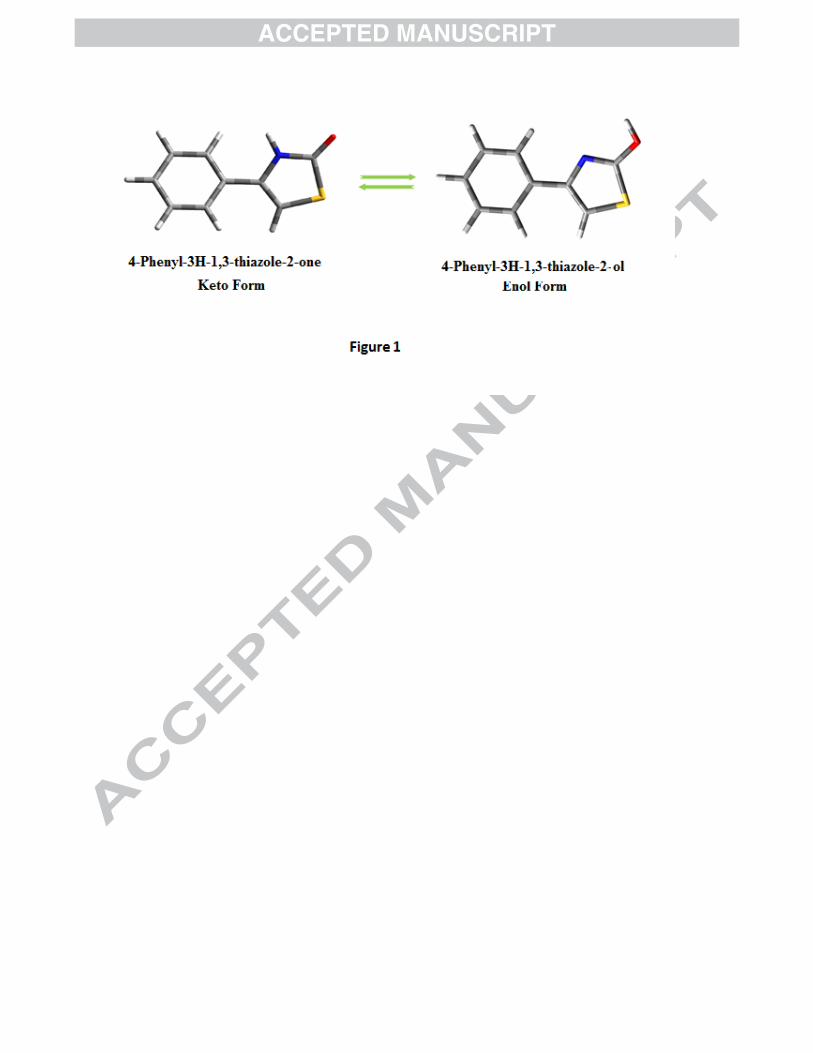
properties of such derivatives. 4P3HT can exist in two tautomeric forms – keto and enol (Fig. 1).
DFT/B3LYP/6-311++G(d,p) calculations show that the keto form (Ground state energy -875.50601 a.u.)
is more stable than enol form (Ground state energy -875.48940 a.u.). K. Pihlaja et. al. [10] have reported
geometric and electronic properties of 4-phenylthiazol-2(3H)-one (keto form), at the most elementary HF
level of theory which does not take into account the electronic correlation effects. The work reported in
the present communication deals with the comprehensive investigation of geometrical and electronic
structure of enolic form of 4P3HT in ground as well as in the first excited state. The significance of enol
form lies in the fact that this form ionizes into the enolate form under physiological conditions and
increases the interaction of the drug with the vis-à-vis receptors, functional proteins or enzymes. To
compare the drug efficacy of enolic and keto forms, QSAR properties of both forms have also been
computed and discussed. Experimentally observed spectral data (FT-TR and FT-Raman) of the title
compound is compared with the spectral data obtained by DFT/B3LYP method. The molecular properties
like dipole moment, polarizability, first static hyperpolarizability and molecular electrostatic potential
surface, contour map have been calculated to get a better understanding of the properties of the title
molecule. Natural bond orbital (NBO) analysis has been applied to study the stability of the molecule
arising from charge delocalization. UV–Vis spectrum of the title compound was also recorded and
Page 5
electronic properties, such as frontier orbitals and band gap energies were calculated by TD-DFT
approach.
2. Experimental and computational methods
2.1 Sample and instrumentation
The pure 4-Phenyl-3H-1,3-thiazol-2-ol (4P3HT) of spectral grade was purchased from M/s Aldrich
Chemical Co., as a white crystalline solid and was used as such without any further purification. The
sample was used to record FT-Raman and FT-IR spectra. FT-IR and FT-Raman spectra were recorded on
a Varian 7000 series spectrometer in the region 4000–400 cm-1 with a spectral resolution of 0.5 cm-1 at
AIRF, Jawaharlal Nehru University, New Delhi. For Raman Spectra the 1064 nm laser line of Nd:YAG
laser was used as the exciting wavelength with an output power of about 2 mW at the sample position.
The spectrum was recorded in the range of 4000–100 cm-1 with a scanning speed of 10 cm-1min-1 and the
spectral resolution of 4.0 cm-1. UV absorption spectra of 4P3HT were recorded in methanol and
chloroform using the Shimadzu 1800 UV–Vis recording spectrometer in the spectral region of 200–500
nm.
2.2 Computational details
Density functional theory [11] treated according to hybrid Becke’s three parameter and the Lee–
Yang–Parr functional (B3LYP) [12–14] supplemented with polarized triple-zeta 6-311++G(d,p)basis sets
was used to study 4P3HT, as this quantum chemical method provides a very good overall description of
medium-sized molecules. It has also been used to calculate the dipole moment, mean polarizability and
first static hyperpolarizability based on the finite field approach. All calculations in this study have been
performed with the Gaussian 09 program package [15] and results were analysed with the Gaussview 5.0
molecular visualization program [16]. The most stable geometry of the molecule has been determined
from the potential energy scan by varying the S17-C15-O18-H19 and N16-C12-C3-C4 dihedral angles at
B3LYP/6-311++G(d,p) level of theory. 3-dimensional Potential energy surface showing the variation of
dihedral angles and their corresponding energies are given in Fig. 2(a) and 2(b) and thus obtained stable
conformers of the title molecule are shown in Fig. 2(c). Geometrical structure corresponding to the lowest
minima in the potential energy surface (represented as conformer A in fig. 2(c)) has been used for the
calculation of molecular properties and for the calculation of vibrational wavenumbers. Optimized
Page 6
parameters of the title molecule are very close to the experimental values reported by J. Garbarczyk et. al.
[17] for N-phenylthioamide thiazole-2. Positive value of all the calculated wavenumbers confirms the
stability of optimized geometry. An empirical uniform scaling factor of 0.983 up to 1700 cm-1 and 0.958
for greater than 1700 cm-1 [18,19] was used to offset the systematic errors caused by basis set
incompleteness, neglect of electron correlation and vibrational anharmonicity [20]. Theoretical
vibrational assignment of the title compound using percentage potential energy distribution (PED) has
been done with the MOLVIB program (version V7.0-G77) written by T. Sundius [21-23]. The theoretical
UV–Vis spectrum has been computed by TD-DFT method with 6-311++G(d,p) basis set for gas phase
and solvent effect also has been taken into consideration by implementing IEFPCM model at the same
level of theory.
Natural bonding orbital (NBO) calculations [24] were performed using Gaussian 09 package in
order to understand various second order interactions between the filled orbitals of one subsystem and
vacant orbitals of another subsystem which is a measure of the intermolecular delocalization or hyper
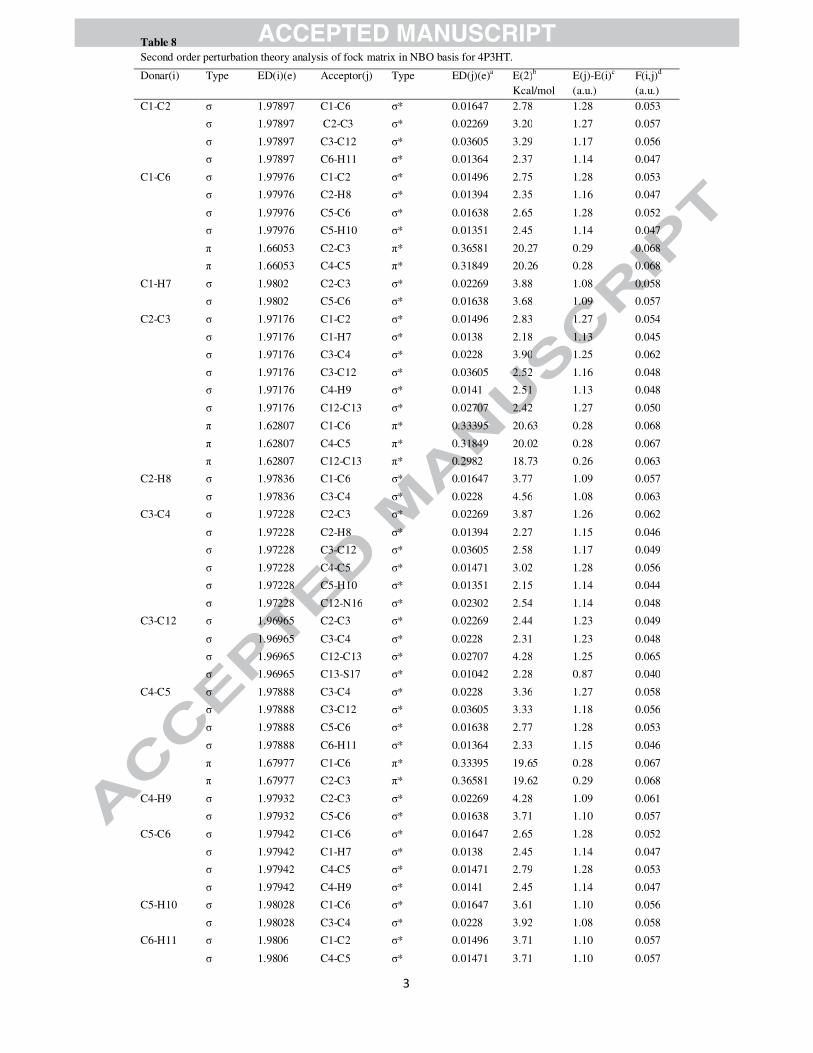
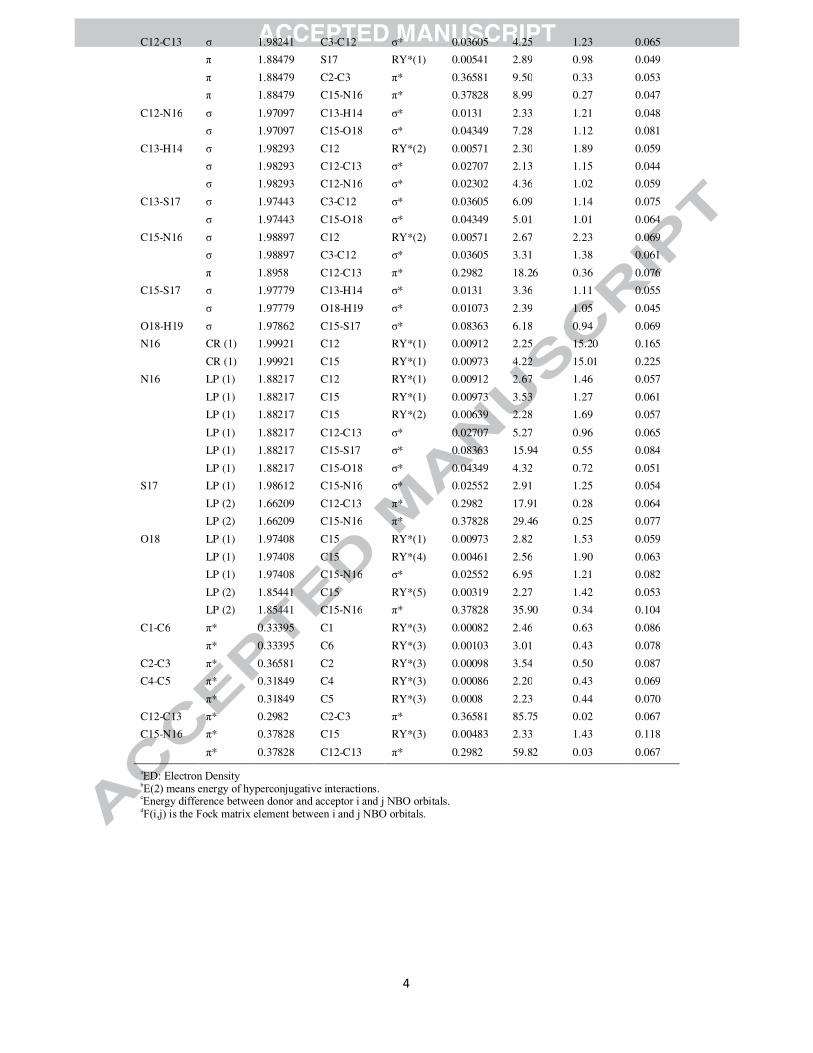
conjugation. The second order perturbation theory analysis of Fock matrix in NBO basis of 4P3HT was
carried out to evaluate the donor-acceptor interactions. The interactions result in a loss of occupancy from
the localized NBO of the idealized Lewis structure into an empty non-Lewis orbital. For each donor (i)
and acceptor (j), the stabilization energy �� associated with the delocalization
i → j is estimated as
�(�) = ∆��� = ��(�,�)�
� ��� (1)
Where �� is the donor orbital occupancy, �� and �� are diagonal elements and F(i,j) is the off diagonal
NBO Fock matrix element. Natural bond orbital analysis provides an efficient method for studying intra
and intermolecular bonding as well as interaction among bonds. It also provides a useful basis for
investigating charge transfer or conjugative interaction in molecular systems. The QSAR parameters of
keto and enolic form of 4P3HT have been calculated employing Hyperchem 8.0 software [25].
2.3 Prediction of Raman intensities
Page 7
The Raman activities (��) calculated with the Gaussian 09 program were subsequently converted to
relative Raman intensities (��) using the following relationship derived from the basic theory of Raman
scattering [26-27].
�� = [�(�� − ��)���] [⁄ ��{1 − exp(−ℎ!�� "#)}]⁄ (2)
Where �� is the exciting frequency in cm-1, �� the vibrational wave number of the ith normal mode, h, c
and " are the fundamental constants and � is a suitably chosen common normalisation factor for all the
peak intensities. The calculated Raman and IR spectra were plotted using the pure Lorentzian band shape
with a band width of FWHM of 5 cm-1.
3. Results and discussion
3.1 Molecular geometry and PES scan studies
To calculate the minimum energy structure of the molecule, potential energy surface (PES) scan were
performed at DFT/B3LYP/6-311++G(d,p) level of theory by varying dihedral angles S17-C15-O18-H19
and N16-C12-C3-C4 in steps of 10o from -180o to 180o and all the geometrical parameters were
simultaneously relaxed during the scan except the two selected dihedral angles. Dihedral angle N16-C12-
C3-C4 and S17-C15-O18-H19 are the relevant torsional angles to gauge conformational flexibility within
the title molecule. The torsional profiles of PES scan are shown in Fig. 2 (a) and (b). Stable conformers
(A, B, C, and D) corresponding to the minima on potential energy surface are shown in Fig. 2(c) with
their respective ground state energies. Eigen values obtained from scan output reveals that, the structure
(A) positioning the dihedral N16-C12-C3-C4/S17-C15-O18-H19 at 170°/180°, possesses minimum
(least) energy at -875.489360 Hartree while the three other minima at B, C and D at 20o/180°, 170o/0o
and 10o/0o correspond to -875.489357, -875.482521 and -875.482520 Hartree respectively. The
optimized bond lengths, bond angles and dihedral angles are listed in Table 1. Since the crystal structure
of the title molecule is not available, the optimized structure was compared with other similar system
[17]. In the six-membered ring all the C-C and C-H bond distances are in the range 1.391-1.403 Å and
1.082-1.084 Å respectively. In the hetero ring, S17-C15 bond length is the longest (1.749 Å) while C15-
N16 is the shortest (1.288 Å). The longest distance attributes the pure single bond character. The S17-C15
and C13-S17 bond lengths are 1.749 Å and 1.744 Å respectively, in between the standard bond lengths
for a C-S (1.820 Å) bond and for C=S (1.61 Å) bond. With the electron donating substituents on the
Page 8
benzene ring, the symmetry of the ring is distorted, yielding ring angles smaller than 120o at the point of
substitution and slightly larger than 120o at the ortho and meta positions [28]. More distortion in bond
parameters has been observed in the hetero ring than in the benzene ring. The variation in bond angle
depends on the electro negativity of the central atom, the presence of lone pair of electrons and the
conjugation of the double bonds. If the electronegativity of the central atom decreases, the bond angle
decreases. Thus the difference in the bond angle C12-N16-C15 (111.0°) as compared to C13-S17-C15
(87.6°) is due to higher electro-negativity of nitrogen than sulphur. The structure of title molecule
deviates significantly from planar structure because the phenyl and hetero rings are rotated around the C3-
C12 axis to give a C4-C3-C12-N16 torsion angle of 165.5°.
3.2 Vibrational analysis
The 4P3HT molecule consists of 19 atoms, which undergo 51 normal modes of vibrations. The molecule
possesses C1 symmetry. Vibrational spectral assignments were performed at the B3LYP level with the
triple split valence basis set 6-311++G(d,p). A detailed vibrational description can be given by means of
normal coordinate analysis. The specific assignment to each wavenumber is attempted through potential
energy distribution (PED). For this purpose the full set of internal coordinates are defined and given in
Table 2. The local symmetry coordinates for 4P3HT were defined as recommended by Fogarasi and
Pulay [29] and are presented in Table 3. The method is useful for determining the mixing of other modes,
but the maximum contribution is accepted to be the most significant mode. Observed FT-IR and FT-
Raman bands with their relative intensities and calculated wave numbers and assignments are given in
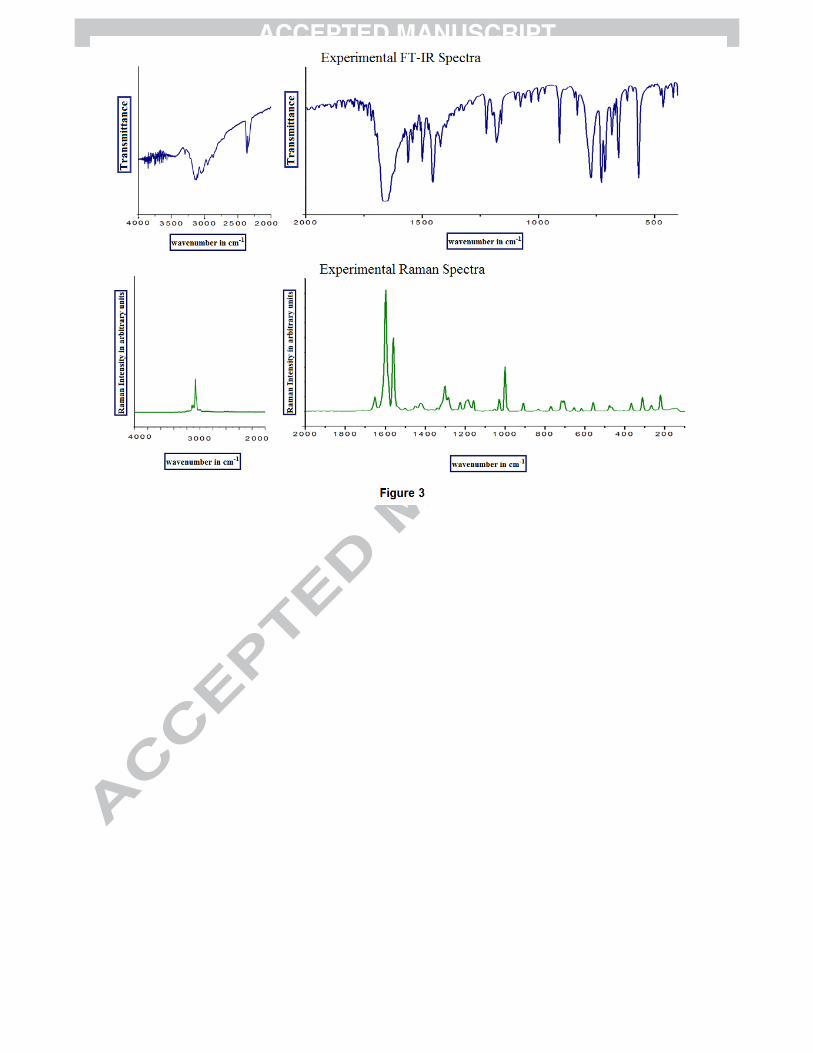
Table 4. The experimental FT-Raman and FT-IR spectra of 4P3HT have been presented in Fig. 3 while
calculated (simulated) spectra are given in Fig. S1. The title compound 4P3HT consists of a thiazole ring
substituted with phenyl ring and a hydroxyl group hence the vibrational modes are discussed under three
heads:
(i) Thiazole ring vibrations (ii) Phenyl ring vibrations (iii) O-H group
3.2.1 Thiazole ring vibrations
As the key moiety in 4P3HT is the thiazole moiety having the conjugated -C=C-N=C system and two
hetero atoms, vibrations of these hetero atoms are themselves influenced and modified. It is worth here to
discuss the C-S, C-N and C=N, C=C vibrations under this head. The C-S stretching vibration cannot be
Page 9
identified easily as it results in weak infrared bands, which is susceptible to coupling effects and is also of
variable intensity. In general C-S stretching vibration occurs in the region 700-600 cm-1. The theoretically
computed values in case of 4P3HT are at 821, and 698 cm-1 which are matched with the FT-IR bands at
832 and 683 cm-1. The shifting of this wavenumber to the higher side can be explained on the basis of
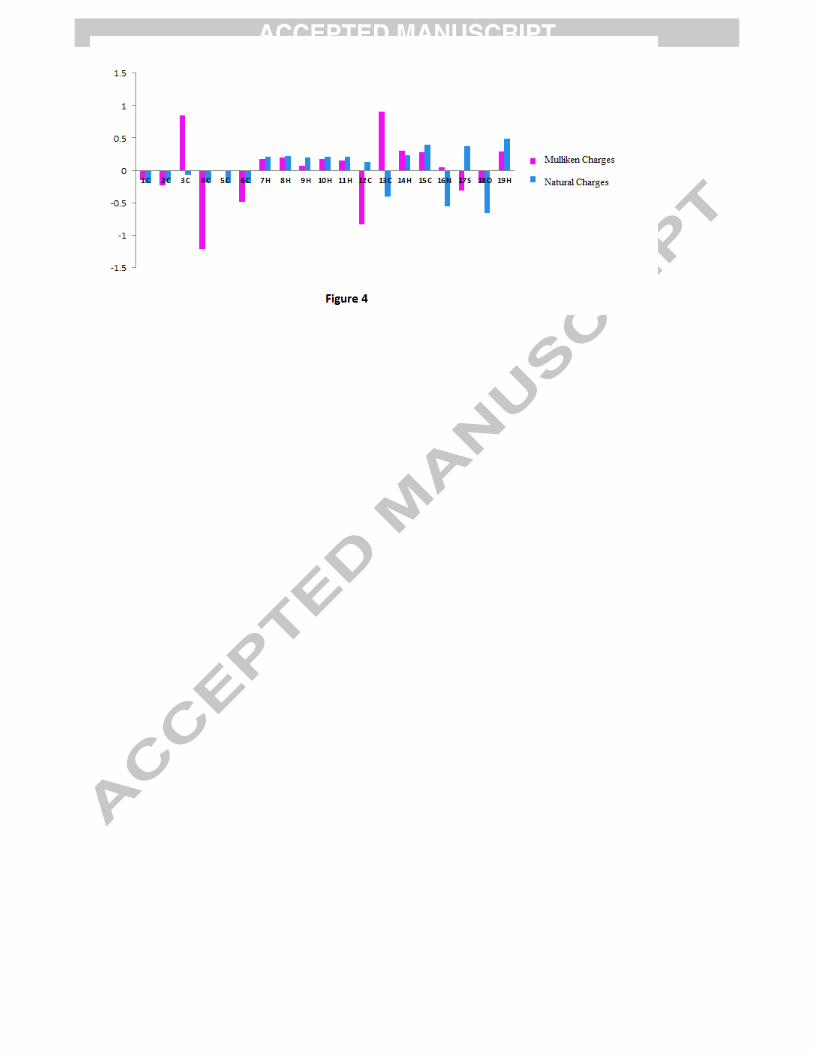
Mulliken Population analysis (MPA) (refer to Fig. 4). According to MPA the positive charge is
concentrated on sulphur atom and negative charge is concentrated on nitrogen atom on the heterocyclic
ring, consequently there is a strong attraction in thiazole ring. NPA charges also show strong attraction
due to opposite charges on sulphur and nitrogen atoms. This results in reduction of bond length and thus
shifting up of vibrational wavenumbers of heterocyclic ring. The band occurring at 569/559 in FT-IR/FT-
Raman is assigned to C-S-C bending vibration; the calculated value for this mode is at 571 cm-1. V.
Arjunan et.al. have observed this bending vibration at 526 cm-1 for 2-amino-4-methylbenzothiazole [30].
Another important vibration in thiazole ring is the C-N stretching vibration. Identification of C-N
vibrations is a very difficult task because of the mixing of several bands in this region. Silverstein et. al.
[31] assigned C=N and C-N stretching vibrations in the range 1382-1266 cm−1and 1250-1020 cm−1
respectively. However, molecular simulation program (Gauss View 5.0) and normal mode analysis of the
molecule 4P3HT helped us to define the C-N vibrations correctly. A very strong band observed at 1559
and 1560 cm-1 in FT-IR and FT-Raman spectra respectively has been assigned to C=N stretching
vibration (64% P.E.D.). The mode calculated at 1284 cm-1 is the C-N stretching mode (23% P.E.D.)
which is in good agreement with experimental value. It is a mixed mode having contribution from C-C
stretch and C-H bending vibrations. The C=C-N in-plane bending vibration is calculated as a mixed mode
at 698 cm-1.
3.2.2 Phenyl Ring vibrations:
The phenyl ring spectral region predominantly involves the C-H, C-C and C=C stretching, and C-C-C as
well as H-C-C bending vibrations. The ring stretching vibrations are very prominent, in the vibrational
spectra of benzene and its derivatives. Usually the carbon hydrogen stretching vibrations give rise to
bands in the region of 3100-3000 cm-1 in all aromatic compounds [32, 33]. In the present study, the bands
in the region 3121- 3029 cm-1 have been assigned to the ring C-H stretching vibrations with more than
90% potential energy contribution. The C–H in-plane and out-of-plane bending vibrations generally lies
Page 10
in the range 1300–1000 cm-1 and 1000–675 cm-1 [34–37], respectively. In this work, vibrations involving
C-H in plane bending are found in the region 1488-1053 cm-1. The computed wavenumbers at 999 cm-1 is
identified as the trigonal ring bending mode and is in complete agreement with FT-IR/FT-Raman peak at
998/999 cm-1. The wavenumber calculated at 682 cm-1 is assigned to the ring puckering mode. A good
agreement between the calculated and experimentally observed wavenumbers has allowed us to establish
a detailed and precise assignment of normal mode wavenumbers in the entire spectral region.
3.2.3 O-H vibrations
A free hydroxyl group or a non-hydrogen bonded hydroxyl group absorbs in the range 3700–3500 cm-1.
In hydrogen bonded structure, the O-H stretching results in a broad band in the region 3300–2500 cm-1
[38]. In the FT-IR spectra of 4P3HT, there is a broad band in the region 3300–2600 cm-1 containing the
wavenumbers due to the motion of O-H stretching and phenyl ring stretching vibrations. The scaled
wavenumber calculated at 3618 cm-1 in case of 4P3HT are identified as O-H stretching with 100%
contribution to P.E.D. The O-H group vibrations being the most sensitive to the environment show
marked shifts in the spectra of the hydrogen bonded species. Several bands between 2400 and 2300 cm−1
found in the FT-IR spectrum of 4P3HT are also characteristic of the hydrogen bonds. Present calculations
showed that there was a marked wavenumber downshift of O-H stretching vibration which must be due to
the presence of intermolecular interaction. The bands identified at 1368, and 1159 cm-1 in the Raman
spectrum are assigned to in-plane O-H bending vibrations while the out-of-plane bending vibration is
calculated at 390 cm-1. The characteristics band due to out-of-plane bending observed in the range 450-
350 cm-1 indicates the presence of hydrogen bonding [39]. Although the crystal structure of 4P3HT is not
available but above discussion asserts the existence of hydrogen bonding in 4P3HT.
3.3 Electric moments
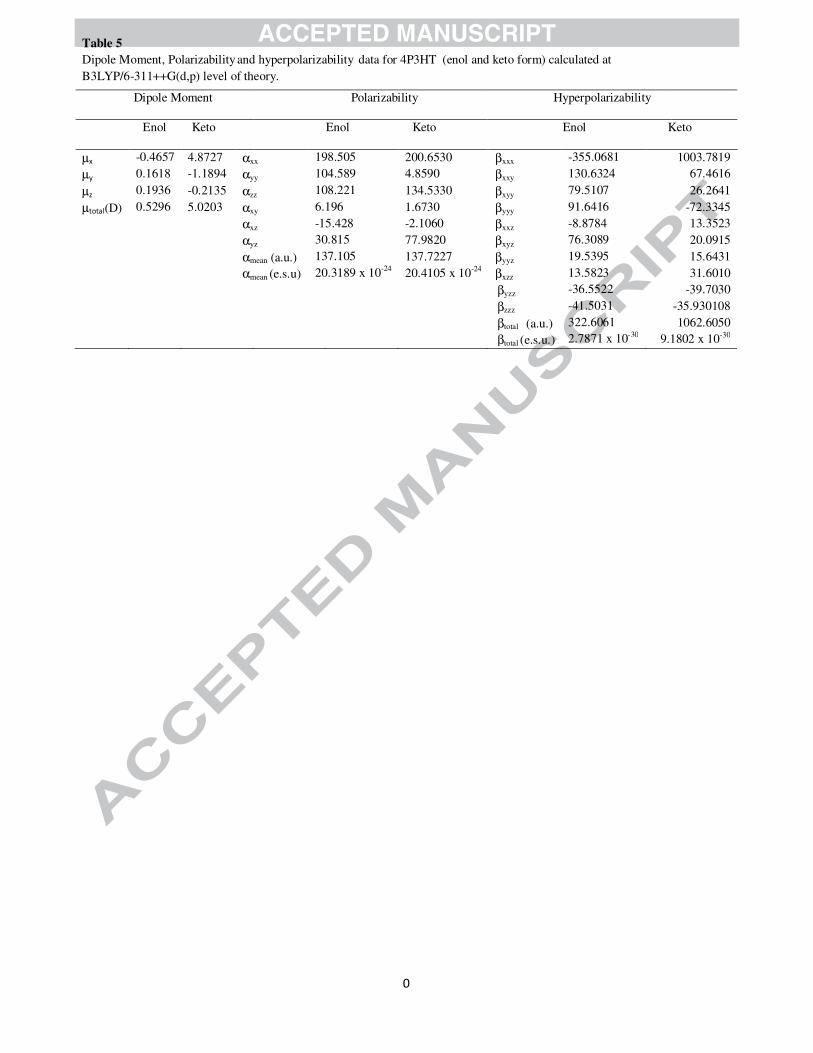
The B3LYP results of electronic dipole moment µ, polarizability α and first order hyperpolarizability β
are listed in Table 5. The polarizability and first hyperpolarizability (β) calculated for 4P3HT is based on
the finite-field approach. In presence of an applied electric field, the energy of a system is a function of
the electric field. The first hyperpolarizability is a third rank tensor that can be described by a 3 × 3 × 3
matrix. The 27 components of the matrix can be reduced to 10 components due to the Kleinman
Page 11
symmetry [40]. The components of β are defined as the coefficients in the Taylor series expansion of the
energy in the external electric field. When the electric field is weak and homogeneous, this expansion
becomes
E = E0– µiFi− 1/2 αijFiFj− 1/6 βijkFiFjFk+ . . . (3)
where E0 is the energy of the unperturbed molecules, Fi is the field at the origin µi, αij and βijk are the
components of dipole moment, polarizability, and the first hyperpolarizability, respectively. The total
electric dipole moment (µ), the mean polarizability <α>, and the total first order hyperpolarizability
(βtotal), have been calculated using the x, y, and z components of these electric moments. The calculated
value of mean polarizability and first hyperpolarizability are 137.105 a.u. or 20.3189×10-24 e.s.u. and βtotal
=2.7871 x 10-30 e.s.u. respectively. Urea is one of the prototypical molecules used in the study of the NLO
properties of molecular systems. Therefore it is used frequently as a threshold value for comparative
purposes. The calculated value of β for the title compound is nearly fourteen times higher than that of
Urea and thus the 4P3HT molecule possesses considerable non-linear optical properties. Theoretically
calculated value of dipole moment is 0.5296 Debye.
Electric moments of keto form (4-phenyl-3H-1,3-thiazol-2-one) at DFT/B3LYP/6-311++G(d,p) have also
been calculated. Theoretically calculated values of mean polarizability of both keto and enol forms are
found to be nearly same but the dipole moment (5.0203 Debye) and first static hyperpolarizability
(βtotal=9.1802 x 10-30 e.s.u.) of keto form are appreciably higher than enolic form.
3.4 Electronic properties and UV-spectral analysis
The Frontier orbitals, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular
orbital (LUMO) are important factors in quantum chemistry [41] as these determine the way the molecule
interacts with other species. The frontier orbital gap helps characterize the chemical reactivity and kinetic
stability of the molecule. A molecule with a small frontier orbital gap is more polarizable and is generally
associated with a high chemical reactivity, low kinetic stability and is also termed as soft molecule [42].
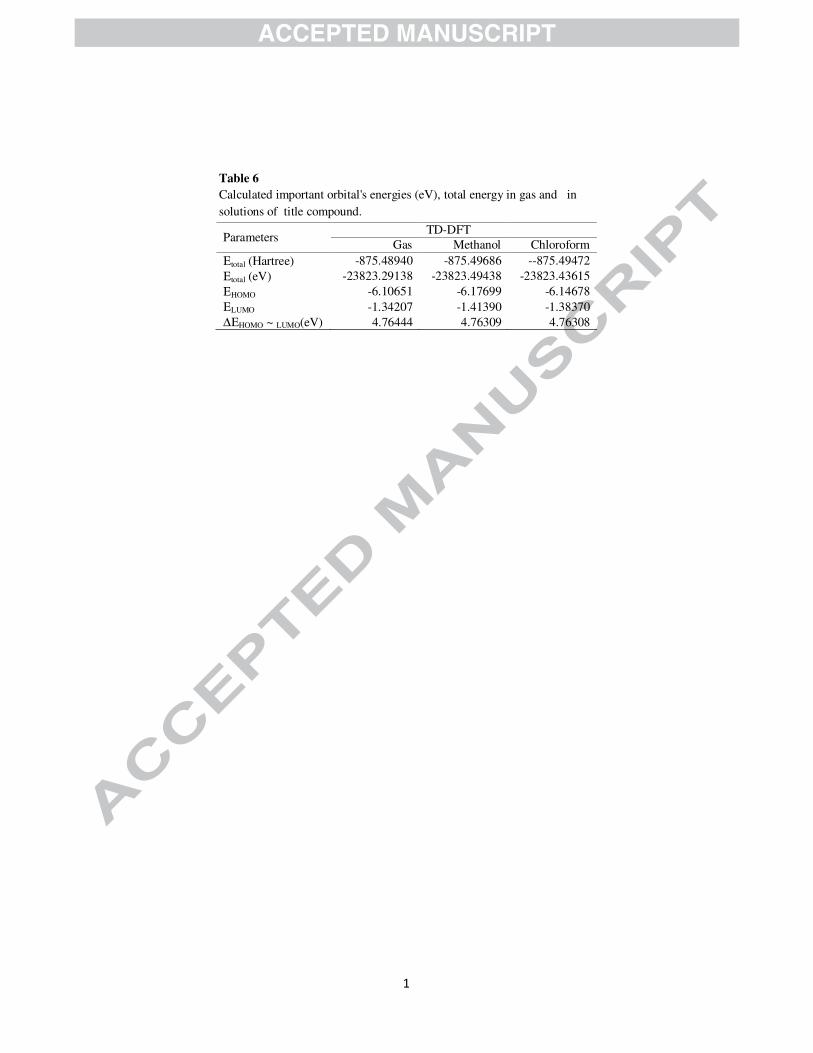
Fully optimized ground-state structure has been used to determine energies (Table 6) and 3D plots (Fig.
S2) of HOMO, LUMO and other MOs involved in the UV transitions of 4P3HT at TD-DFT/B3LYP-
6311++G(d,p) level of theory. Gauss-Sum 2.2 Program [43] was used to calculate the character of the
molecular orbitals (HOMO and LUMO) and prepare the total density of the states (TDOS) and Partial
Page 12
Density of states (PDOS) plots as shown in Fig. 5. DOS plot shows population analysis per orbital and
demonstrates a clear view of the makeup of the molecular orbitals in a certain energy range while PDOS
plot shows percentage contribution of a group to each molecular orbital. It can be seen from figure that
HOMO and LUMO both are spread over the entire molecule having contribution from both phenyl ring
and heterocyclic ring but LUMO has more anti-bonding character than HOMO.
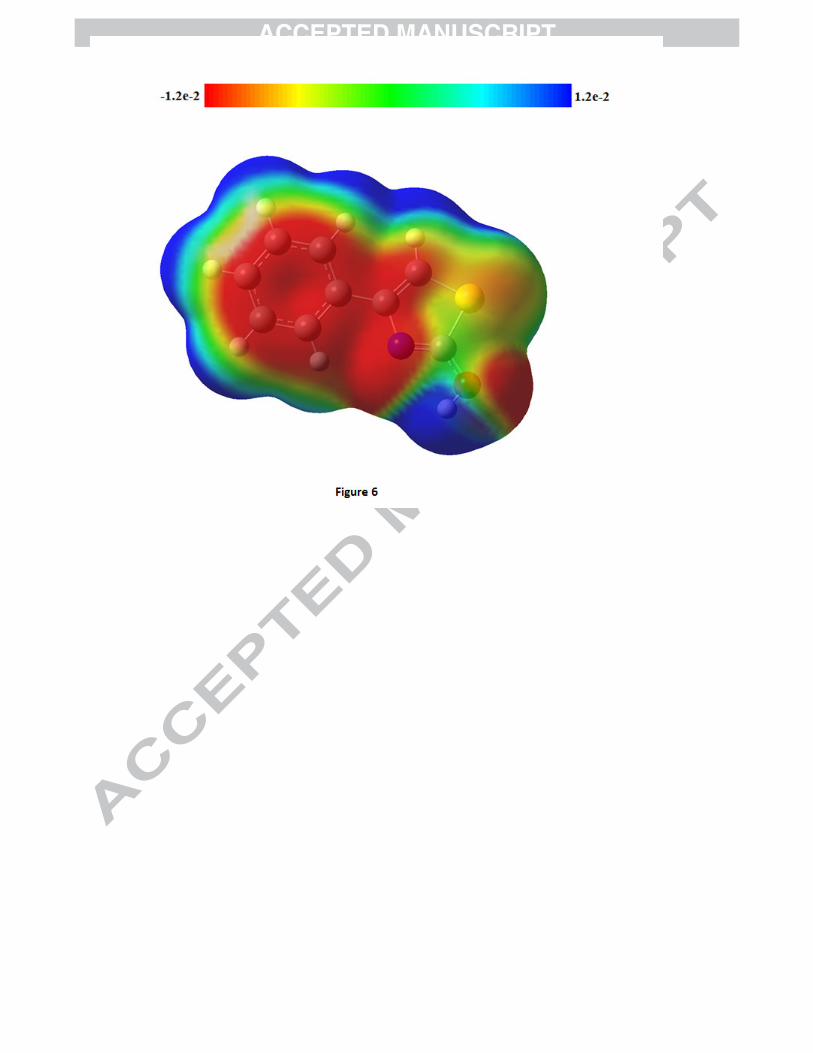
MESP may be employed to distinguish regions on the surface which are electron rich (subject to
electrophilic attack) from those which are electron poor (subject to nucleophilic attack) and has been
found to be a very convenient tool in exploration of correlation between molecular structure and the
physiochemical property relationship of molecules including bio molecules and drugs [44-49]. The MESP
map of 4P3HT (Fig. 6) clearly suggests that the electron rich (red) region is spread around carbon atoms
in benzene ring, bridge carbon atoms, most part of the thiazole ring as well as oxygen atom of O-H group
whereas the hydrogen atoms shows the maximum burnt of positive charge (blue). Ultraviolet spectral
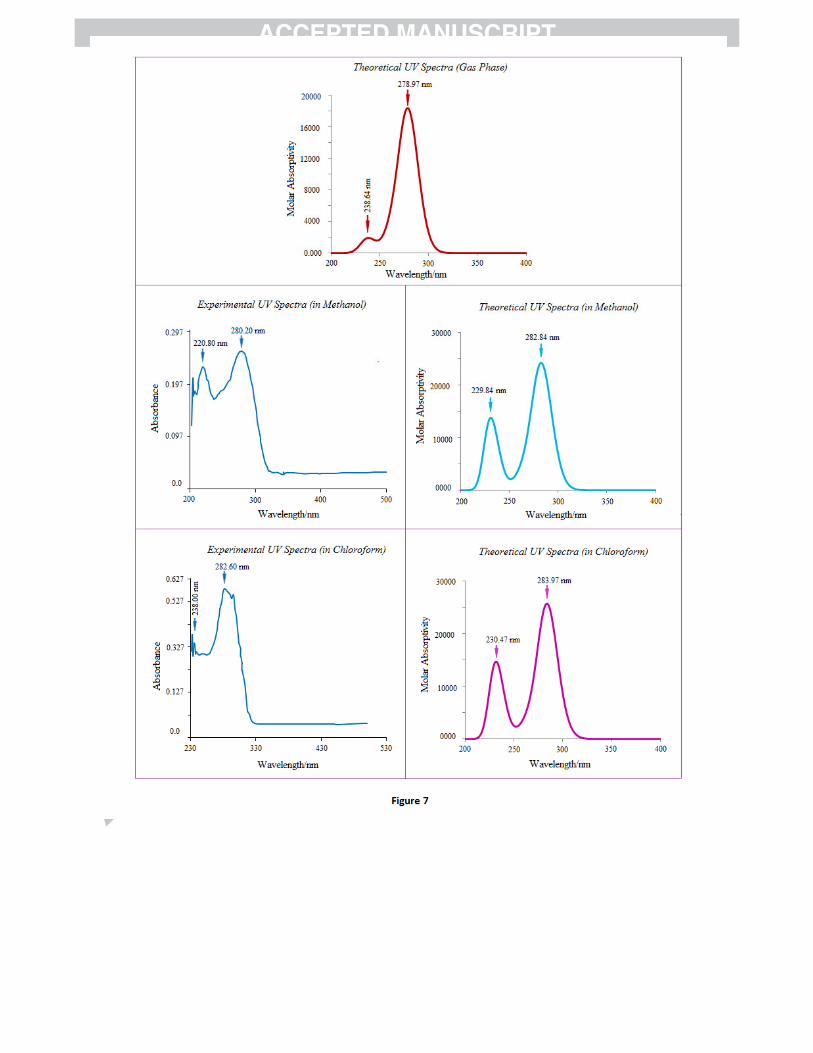
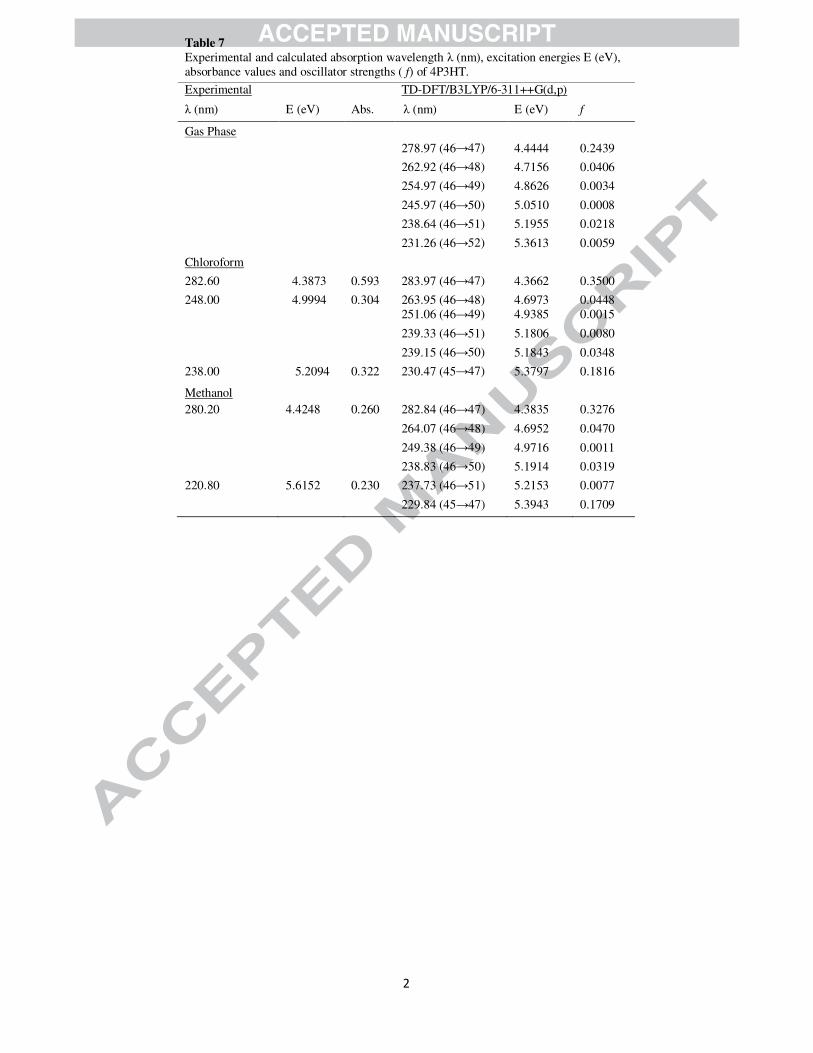
analyses of 4P3HT have been made by experimental as well as theoretical calculations (Fig. 7). In order
to understand electronic transitions of compound, time-dependent DFT (TD-DFT) calculations on
electronic absorption spectra in gas phase and solvent (methanol and chloroform) were performed. The
calculated absorption wavelengths (%), oscillator strengths (f) and vertical excitation energies (E) for gas
phase and solvent (methanol and chloroform) were carried out and compared with experimental values
(Table 7). The calculated absorption maxima values have been found to be 278.97 and 238.64 nm for gas
phase , 282.84 and 229.84 nm for methanol solution and 283.97 and 230.47 nm for chloroform solution
at DFT/B3LYP/6-311++G(d,p) method. The intense electronic transition at 278.97 nm with an oscillator
strength f = 0.2439, is in good agreement with the measured experimental data (λ = 280.20, in methanol
and 282.60 nm in chloroform). This electronic absorption corresponds to the transition from the
molecular orbital HOMO (46) to the LUMO (47) excited state, is a π → π* transition. The weak band at
220.80/238.00 nm in methanol/chloroform in experimental UV spectra of title molecule is also a π → π*
electronic transition, and shows blue shift in more polar solvent.
3.5 NBO analysis
The calculation pertaining to delocalization of the electron density between occupied Lewis type (bond
(or) lone pair) NBO orbitals and formally unoccupied (anti-bond (or) Rydberg) non-Lewis NBO orbitals
Page 13
corresponding to a stabilizing donor–acceptor interactions, have been performed at B3LYP/6-
311++G(d,p) basis set. The energy of these interactions can be estimated by the second order perturbation
theory [50]. Table 8 lists the calculated second-order interaction energies (E(2)) between the donor–
acceptor orbitals in 4P3HT. The larger E(2) (energy of hyper-conjugative interaction) value, the more
intensive is the interaction between electron donors and acceptors i.e., the more donation tendency from
electron donors to electron acceptors and the greater the extent of conjugation of the whole system.
The intra-molecular interaction formed by the orbital overlap between bonding (C-C) and (C-C)
anti-bonding orbital results in intra-molecular charge transfer (ICT) causing stabilization of the system.
These interactions are observed as increase in electron density (ED) in C-C anti-bonding orbital that
weakens the respective bonds. Table 8 clearly shows that the strong intra-molecular hyper conjugative
interaction of π electrons of (C1-C6) with π*(C2-C3) and π*(C4-C5), of π (C2-C3) with π*(C1-C6) and
π*(C4-C5) and of π(C4-C5) with π*(C1-C6) and π*(C2-C3) of the ring. On the other hand, the π(C2-C3)
of phenyl ring conjugate to the anti-bonding π orbital (C12- C13) of thiazole ring and π(C15-N16) to
the π*(C12- C13) with energies 18.73 kcal/mol and 18.26 kcal/mol respectively, resulting in strong
delocalization. A pair of interactions in the title molecule involving the lone pairs LP S17(2) and LP
O18(2),with that of anti-bonding π (C15-N16) results in the stabilization of 29.46 kcal/mol and 35.90
kcal/mol, respectively. The π*(C12- C13) NBO conjugated with π*(C2-C3) and π*(C15- N16) with
π*(C12- C13) also results in an enormous stabilization of 85.75 kcal/mol and 59.82 kcal/mol
respectively. Several other types of valuable data, such as directionality, hybridization, and partial
charges, have been analysed from the NBO results. The direction of the line of centres between the two
nuclei is compared with the hybrid direction to determine the bending of the bond, expressed as the
deviation angle (Dev.) between these two directions. The hybrid directionality and bond bending analysis
of natural hybrid orbitals (NHOs) offer indications of the substituent effect and steric effect. It is evident
from Table 9 that the C12 and C13 NHOs of σ (C12-C13) are away from the line of centres by ~ 3°. In
σ(C12-N16) and σ(C15-N16), N16 NHOs show deviation of 4.9° and 3.9° with C12 and C15, the sulphur
(S17) NHOs in σ (C13-S17) and σ (C15-S17) show very large deviations of 9.7° and 9.5° with line of
nuclear centres whereas C13 and C15 show deviation of 2.8° and 2.7° respectively. These deviations
provide a strong charge transfer path within the molecule.
Page 14
3.6 Quantitative structure activity relationship (QSAR) properties: Keto and enol form
QSAR [51] is the quantitative association of the biological activity to the structure of chemical
compounds [52-53] which permits the prediction of drug efficacy of a structurally related compound.
QSAR properties allow calculation and estimation of a variety of molecular descriptors. In this paper
QSAR properties like surface area, volume, log P, hydration energy, refractivity, polorizability, mass and
total energy etc. of enol and keto forms of 4P3HT were determined by HyperChem software and collected
in Table 10. Partition coefficient Log P is a vital factor used in medicinal chemistry to gauge the drug-
likeness of a given molecule, and used to calculate lipophilic ligand efficiency (LipE). LipE is an
imperative parameter to normalize potency relative to lipophilicity. LiPE is used to compare compounds
of different potencies (pIC50s) and lipophilicities (LogP). For a given compound lipophilic efficiency is
defined as pIC50 (or pEC50) of interest minus Log P of the compound [54-55]. For a drug to be orally
absorbed, it normally must first pass through lipid bilayers in the intestinal epithelium. For efficient
transport, the drug must be hydrophobic enough to partition into the lipid bilayer, but not so hydrophobic,
that once it is in the bilayer, it will not partition out again [56]. Likewise, hydrophobicity plays a major
role in determining where drugs are distributed within the body after absorption and as a consequence in
how rapidly they are metabolized and excreted. For good oral bioavailability of any compound, the log P
must be greater than zero and less than 3. Both tautomers of title compound have optimal values of log P.
Higher value of log P of the enol form (1.54) predicts that it is more orally absorbent product than keto
form (log P=0.50) and have important capacity to be dependent on plasmatic proteins. The absolute value
of hydration energy is also found to be larger in enol form (12.11Kcal/mol) than in keto form (5.61
Kcal/mol) of 4P3HT. This establishes the efficacy of enol form of the studied title compound under
physiological conditions and hence predicts its enhanced interaction with the vis-à-vis receptors,
functional proteins or enzymes.
4. Conclusions
In the present study, we have carried out the experimental and theoretical spectroscopic analysis of
4P3HT for the first time, using FT-IR, FT-Raman and UV–Vis techniques and implements derived from
the density functional theory. In general, a good agreement between experimental and calculated normal
modes of vibrations has been observed. The molecular geometry, vibrational frequencies, infrared and
Page 15
Raman intensities of the molecules have been calculated by using DFT (B3LYP) method with 6-
311++G(d,p) basis sets. The MESP plot provides the visual representation of the chemically active sites
and comparative reactivity of atoms. NBO analysis shows that the most important interactions in the title
molecule having lone pairs LP S17(2) and LP O18(2), with that of anti-bonding π (C15-N16) resulting in
the stabilization of 29.46 kcal/mol and 35.90 kcal/mol, respectively. NLO behaviour of the title molecule
has been investigated by the dipole moment, polarizability and first hyperpolarizability. Theoretically
calculated values of mean polarizability of both keto and enol forms are found to be nearly same but the
dipole moment (5.0203 Debye) and first static hyperpolarizability (βtotal=9.1802 x 10-30 e.s.u.) of keto
form are appreciably higher than enolic form (0.5296 Debye, βtotal = 2.7871× 10-30 e.s.u.). The calculated
electronic properties show good correlation with the experimental UV–Vis spectrum. QSAR analysis of
both keto and enol form establishes the efficacy of enol form of the studied title compound under
physiological conditions and hence predicts its enhanced interaction with the vis-à-vis receptors,
functional proteins or enzymes.
Acknowledgements
The authors are thankful to Dr. Manoj Singh for running the FT-IR and FT-Raman spectra at AIRF,
Jawaharlal Nehru University, New Delhi and SAIF Central Drug Research Institute, Lucknow for
recording UV-Vis spectra. Prof. T. Sundius is also gratefully acknowledged for his MOLVIB program.
One of the authors (Shilendra K. Pathak) is grateful to the UGC (India) for the financial assistance.
References
[1] N. Ulusoy, M. Kiraz, O. Kucukbasmaci, Monatsh. Chem. 133 (2002) 1305-1315.
[2] Z. A. Kaplancikli, G. T. Zitouni, G. Revial, K. Guven, Arch. pharm. Res. 27 (2004) 1081-1085.
[3] M. S. Al-Saddi, H. M. Faidallah, S. A. F. Rostom, Arch. Pharm. Chem. Life Sci. 341 (2008) 424-434.
[4] K. A. Karpov, A. V. Nazarenko, B. V. Pekarevskii, V. M. Potekhin, Russ. J. Appl. chem. 74 (2001)
998-1001.
[5] T. Baselt, K. Rehse, Arch. der pharmazie. 24 (2008) 645-654.
[6] H. N. Karade, B. N. Acharya, S. Manisha, M. P. Kaushik, Med. Chem. Res. 17 (2008) 19-29.
[7] M. J. Rogers, E. Cundliffe, T. F. Mccutchan, Antimicrob. Agents Chemother. 42 (1998) 715-716.
[8] M. C. Bagley, J. W. Dale, E. A. Merritt, X. Xiong, Chem. Rev. 105 (2005) 685-714.
Page 16
[9] A. S. Mayhoub, M. Khaliq, C. Botting, Z. Li, R. J. Kuhn, M. Cushman, Bioorg. Med. Chem. 19
(2011) 3845–3854.
[10] K. Pihlaja, V. Ovcharenko, E. Kolehmainen, K. Laihia, W. M. F. Fabian, H. Dehne, A. Perjéssy, M.
Kleist, J. Teller and Z. Susteková, J. Chem. Soc., Perkin Trans. 2 (2002) 329–336.
[11]W. Kohn, L.J. Sham, Phys. Rev. 140 (1965) A1133–A1138.
[12] A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652.
[13] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1998) 785–789.
[14] B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett. 157 (1989) 200–206.
[15] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani,
V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F.
Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J.
Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery,
Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R.
Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M.
Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo,
R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L.
Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich,
A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian Inc.,
Wallingford, CT, 2009.
[16] E. Frisch, H.P. Hratchian, R.D. Dennington II, T.A. Keith, J. Millam with B. Nielsen, A.J. Holder, J.
Hiscocks. Gaussian, Inc. GaussView Version 5.0.8, 2009.
[17] J. Garbarczyk, G. Kamyszek, R. Boese, J. Mol. Struc. 479 (1999) 21-30.
[18] M. Karabacak, M. Kurt, M. Cinar, A. Coruh, Mol. Phys. 107 (2009) 253–264.
[19] N. Sundaraganesan, S. Ilakiamani, H. Saleem, P.M. Wojciechowski, D. Michalska, Spectrochim.
Acta A 61 (2005) 2995–3001.
[20] J. B. Foresman, A. Frisch, Exploring Chemistry with Electronic Structure Methods, second ed.,
Gaussian Inc., Pittsburgh, PA, 1996.
[21] T. Sundius, J. Mol. Spectrosc. 82 (1980) 138-151.
[22] T. Sundius, J. Mol. Struct. 218 (1990) 321-336.
[23] T. Sundius, Vib. Spectrosc. 29 (2002) 89-95.
[24] E.D. Glendening, C.R. Landis, F. Weinhold, WIREs Comput. Mol. Sci. 2 (2011) 1–42.
[25] Hypercube, Inc. HyperChem molecular system, USA, 2007.
[26] G. Keresztury, S. Holly, J. Varga, G. Besenyei, A.Y. Wang, J. R. Durig, Spectrochem. Acta 49
(1993) 2007–2017.
[27] G. Keresztury, Raman spectroscopy: theory, in: J.M. Chalmers, P.R. Griffith (Eds.), Handbook of
Vibrational Spectroscopy, John Wiley & Sons, New York, 2002.
[28] Y. Wang, S. Saebo, C. U. Pittman Jr., J. Mol. Struct. (Theochem.) 281 (1993) 91-98.
[29] P. Pulay, G. Fogarasi, F. Pang, and J. E. Boggs, J. Am. Chem. Soc. 101 (1979) 2550-2560.
Page 17
[30] V. Arjunan, S. Sakiladevi, T Rani, C.V. Mythili, S. Mohan, Spectrochim. Acta A 88 (2012) 220-231.
[31] R. M. Silverstein, F. X. Webstor, Spectrometric Identification of Organic Compounds, 6th edition,
Wiley, New York, 1998.
[32] V. K. Rastogi, M. A. Palafox, R. P. Tanwar, L. Mittal, Spectrochim. Acta A 58 (2002) 1987-2004.
[33] M. Silverstein, G. C. Basseler, C. Morill, Spectrometric Identification of Organic Compounds,
Wiley, New York, 1981.
[34] V. Krishnakumar, N. Prabavathi, Spectrochim. Acta A 71 (2008) 449–457.
[35] A. Altun, K. Golcuk, M. Kumru, J. Mol. Struct. (Theochem) 155 (2003) 637–639.
[36] V. Krishnakumar, R.J. Xavier, Spectrochim. Acta A 61 (2005) 253–258.
[37] Y. Sun, Q. L. Hao, Z. X. Yu, W. J. Jiang, L. D. Lu, X. Wang, Spectrochim. Acta A 73 (2009) 892–
901.
[38] B. Stuart, Infrared Spectroscopy: Fundamentals and Applications, Wiley India Ed., 2010.
[39] Y Erdogdu, M. T. Gulluoglu, Spectrochim. Acta A 74 (2009) 162-167.
[40] D. A. Kleinman, Phys. Rev. 126 (1962) 1977-1979.
[41] J. M. Seminario, Recent Developments and Applications of Modern Density Functional Theory,
Elsevier, Amsterdam, 1996.
[42] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, John Wiley and Sons, New York,
1976.
[43] N. M. O’ Boyle, A. L. Tenderholt, K. M. Langer, J. Comput. Chem. 29 (2008) 839–845.
[44] I. Alkorta, J. J. Perez, Int. J. Quantum. Chem. 57 (1996) 123–135.
[45] E. Scrocco, J. Tomasi, in: P. Lowdin (Ed.), Advances in Quantum Chemistry, Academic Press, New
York, 1978.
[46] F. J. Luque, M. Orozco, P. K. Bhadane, S.R. Gadre, J. Phys. Chem. 97 (1993) 9380–9384.
[47] J. S. Murray, K. Sen, Molecular Electrostatic Potentials, Concepts and Applications, Elsevier,
Amsterdam, 1996.
[48] R. K. Pathak, S. R. Gadre, J. Chem. Phys. 93 (1990) 1770–1774.
[49] S. R. Gadre, I. H. Shrivastava, J. Chem. Phys. 94 (1991) 4384–4390.
[50] I. Hubert Joe, I. Kostova, C. Ravikumar, M. Amalanathan, S. C. Pinzaru, J. Raman Spectrosc. 40
(2009) 1033–1038.
[51] C. Hansch, A. Leo and D.H. Hoekman, Exploring QSAR fundamentals and applications in chemistry
and Biology, American Chemical Society, Washington DC, USA, 1995.
[52] P. V. Khadikar, S. Karmarkar, V. K. Agrawal, M. Mandloi, and S. Joshi, Natl. Acad. Sci. Lett. 3-4
vol. 23 no. 3-4 (2000) 50–56.
[53] Min Li, Dongbin Wei, Huimin Zhao, Yuguo Du, Chemosphere 95 (2014) 220–226.
[54] Edwards MP, Price DA, Annual Reports in Medicinal Chemistry 45 (2010) 381–391.
[55] Leeson PD, Springthorpe B, Nat Rev Drug Discov 6 (2007) 881–90.
[56] Kubinyi H, Farmaco [Sci] 34 (3) (1979) 248–76.
Page 18
Figure Captions
Figure 1: Tautomeric forms (keto and enol) of 4P3HT Figure 2(a): The potential energy surface (PES) scan of 4P3HT along the S17-C15-O18-H19 and N16-C12-C3-C4 dihedral angles. Figure 2(b): PES projection showing the position of stable conformers (minima’s) of 4P3HT. Figure 2(c): Stable conformers of 4P3HT at DFT/B3LYP/6-311++G(d,p) along with their energies. Figure 3: Experimental (FT-IR and FT-Raman) vibrational spectra of 4P3HT. Figure 4: Mulliken and Natural charges of 4P3HT. Figure 5: DOS and PDOS plots of 4P3HT. Figure 6: The MESP map of 4P3HT. Figure 7: Experimental and simulated UV absorption spectra of 4P3HT Figure S1: Theoretical vibrational spectra of 4P3HT
Figure S2: HOMO, LUMO and other significant molecular orbitals calculated at the TD- DFT/B3LYP/6-311++G(d,p) level in gas phase.
Page 26
Table 1
The optimized geometric parameters of 4P3HT, with bond lengths in angstrom (Aº), bond angles and selected dihedral angles in degrees (º).
Bond Length
Calculated Value
Bond Angle
Calculated Value
Dihedral Angles
Calculated Value
C1-C2 1.392 C2-C1-C6 120.4 C6-C1-C2-C3 -0.2 C1-C6 1.394 C2-C1-H7 119.6 C6-C1-C2-H8 179.9 C1-H7 1.084 C6-C1-H7 120.0 H7-C1-C2-C3 179.8 C2-C3 1.402 C1-C2-C3 120.7 H7-C1-C2-H8 -0.1 C2-H8 1.082 C1-C2-H8 120.4 C2-C1-C6-C5 -0.2 C3-C4 1.403 C3-C2-H8 118.9 C2-C1-C6-H11 -179.9 C3-C12 1.475 C2-C3-C4 118.5 H7-C1-C6-C5 179.8 C4-C5 1.391 C2-C3-C12 120.1 H7-C1-C6-H11 0.1 C4-H9 1.084 C4-C3-C12 121.5 C1-C2-C3-C4 0.7 C5-C6 1.395 C3-C4-C5 120.8 C1-C2-C3-C12 -179.1 C5-H10 1.084 C3-C4-H9 120.0 H8-C2-C3-C4 -179.4 C6-H11 1.084 C5-C4-H9 119.2 H8-C2-C3-C12 0.8 C12-C13 1.367 C4-C5-C6 120.3 C2-C3-C4-C5 -0.8 C12-N16 1.392 C4-C5-H10 119.6 C2-C3-C4-H9 178.2 C13-H14 1.078 C6-C5-H10 120.1 C12-C3-C4-C5 179.1 C13-S17 1.744 C1-C6-C5 119.4 C12-C3-C4-H9 -2.0 C15-N16 1.288 C1-C6-H11 120.3 C2-C3-C12-C13 164.4 C15-S17 1.749 C5-C6-H11 120.2 C2-C3-C12-N16 -14.8 C15-O18 1.342 C3-C12-C13 126.4 C4-C3-C12-C13 -15.4 O18-H19 0.968 C3-C12-N16 119.2 C4-C3-C12-N16 165.5
C13-C12-N16 114.4 C3-C4-C5-C6 0.3 C12-C13-H14 129.1 C3-C4-C5-H10 179.8 C12-C13-S17 110.9 H9-C4-C5-C6 -178.7 H14-C13-S17 120.0 H9-C4-C5-H10 0.8 N16-C15-S17 116.1 C4-C5-C6-C1 0.2 N16-C15-O18 125.2 C4-C5-C6-H11 179.8 S17-C15-O18 118.7 H10-C5-C6-C1 -179.3 C12-N16-C15 111.0 H10-C5-C6-H11 0.4 C13-S17-C15 87.6 C3-C12-C13-H14 -1.3 C15-O18-H19 107.2 C3-C12-C13-S17 -179.7
N16-C12-C13-H14 177.9 N16-C12-C13-S17 -0.5 C3-C12-N16-C15 179.7 C13-C12-N16-C15 0.5 C12-C13-S17-C15 0.3 H14-C13-S17-C15 -178.3 S17-C15-N16-C12 -0.3 O18-C15-N16-C12 179.4 N16-C15-S17-C13 0.0 O18-C15-S17-C13 -179.7 N16-C15-O18-H19 -0.5
S17-C15-O18-H19 179.1
Page 27
Table 2
Definition of internal coordinates of 4P3HT at B3LYP/6-311++G(d,p) level of theory. I.C.No. Symbol Type Definitions Stretching
1-5 ri C-H(R1) C1-H7, C2-H8, C4-H9, C5-H10, C6-H11. 6 ri C-H(R2) C13-H14. 7-12 ri C-C(R1) C1-C2, C2-C3, C3-C4, C4-C5, C5-C6, C6-C1. 13 ri C-C(R2) C12-C13. 14-15 ri C-S(R2) C13-S17, S17-C15. 16-17 ri C-N(R2) C15-N16,N16-C12 18 pi C-C(brd) C3-C12. 19 pi C-O C15-O18. 20 pi O-H O18-H19. In-plane bending
21-30 αi CCH(R1) C6-C1-H7,C2-C1-H7,C1-C2-H8,C3-C2-H8,C3-C4-H9,C5-C4-H9,C4-C5-H10,C6-C5-H10, C5-C6-H11, C1-C6-H11.
31-32 αi CCH(R2) C12-C13-H14, S17-C13-H14. 33-34 αi CCC(brd) C2-C3-C12, C4-C3-C12. 35 αi NCO N16-C15-O18. 36 αi SCO S17-C15-O18. 37-38 αi NCC(brd) N16-C12-C3, C13-C12-C3. 39 αi COH C15-O18-H19. 40-45 αi R1 C6-C1-C2,C1-C2-C3,C2-C3-C4,C3-C4-C5,C4-C5-C6,C5-C6-C1. 46-50 αi R2 N16-C12-C13, C12-C13-S17, C13-S17-C15, S17-C15-N16, C15-N16-C12. Out of plane bending
51-55 ψi CH(R1) H7-C1-C6-C2, H8-C2-C1-C3, H9-C4-C3-C5, H10-C5-C4-C6, H11-C6-C5-C1. 56 ψi CH(R2) H14-C13-C12-S17. 57 ψi CC(brd) C12-C3-C2-C4. 58 ψi CO O18-C15-N16-S17. 59 ψi CC(brd) C3-C12-N16-C13. Torsion
60-65 ti R1 C6-C1-C2-C3, C1-C2-C3-C4, C2-C3-C4-C5, C3-C4-C5-C6, C4-C5-C6-C1, C5-C6-C1-C2. 66-70 ti R2 N16-C12-C13-S17, C12-C13-S17-C15,C13-S17-C15-N16,S17-C15-N16-C12,C15-N16-C12-C13. 71-74 ti C-C(brd) C2-C3-C12-N16,C2-C3-C12-C13,C4-C3-C12-C13,C4-C3-C12-N16. 75-76 ti C-O N16-C15-O18-H19, S17-C15-O18-H19.
Page 28
Table 3
Local symmetry coordinates of 4P3HT at B3LYP/6-311++G(d,p) level of theory.
No. Symbol Definitions No. Symbol Definitions 1 ν(C1-H) r1 30 β(O-H) α39 2 ν(C2-H) r2 31 δtrig(R1) (α40- α41+ α42-α43+α44- α45)/√6 3 ν(C4-H) r3 32 δs(R1) (2α40- α41- α42+2α43-α44- α45)/√12 4 ν(C5-H) r4 33 δas(R1) (α41- α42+α44- α45)/√4 5 ν(C6-H) r5 34 δs(R2) α46+a( α47+ α50)+b(α48+α49) 6 ν(C13-H) r6 35 δas(R2) (a-b)( α47- α50)+(1-a)( α48- α49) 7-12 νCC(R1) r7, r8, r9, r10, r11, r12 36 γ(C1-H) Ψ51
13 νCC(R2) r13 37 γ(C2-H) Ψ52 14-15 νCS(R2) r14, r15 38 γ(C4-H) Ψ53 16-17 νCN(R2) r16, r17 39 γ(C5-H) Ψ54 18 νCC(brd) r18 40 γ(C6-H) Ψ55 19 νCO r19 41 γ(C13-H) Ψ56 20 νOH r20 42 γ(C3-C12) Ψ57 21 β(C1-H) (α21- α22)/√2 43 γ (C-O) Ψ58 22 β(C2-H) (α23- α24)/√2 44 γ(C12-C3) Ψ59 23 β(C4-H) (α25- α26)/√2 45 τR1puck. (t60-t61+t62-t63+t64-t65)/√6 24 β(C5-H) (α27- α28)/√2 46 τR1s (t60-t62+t63-t65)/√4 25 β(C6-H) (α29- α30)/√2 47 τR1as (-t60+2t61-t62-t63+2t64-t65)/√12 26 β(C13-H) (α31-α32)/√2 48 τ1R2 b(t66+t70)+a(t67+t69)+t68 27 β(C3-C12) (α33- α34)/√2 49 τ2R2 (a-b) (t69-t67)+(1-a)(t70-t66) 28 β(C-O) (α35- α36)/√2 50 τC-C(brd) (t71+t72+t73+t74)/√4 29 β(C12-C3) (α37- α38)/√2 51 τC-O (t75+t76)/√2
Page 29
Table 4 FT-IR, FT-Raman spectral data and computed vibrational wavenumbers along with the assignments of vibrational modes based on TED results.
S. No.
Calculated Wavenumbers
Experimental Wavenumber
IIRa IRa
a Assignment of dominant modes in order of decreasing potential energy distribution (PED) Unscaled in cm-1
Scaled in cm-1
FTIR in cm-1
Raman in cm-1
1 3777 3618 3145 - 95.89 3.85 ν(O-H)(100)
2 3258 3121 3127 3124 w 3.33 3.13 ν(C-H)R2(98)
3 3204 3069 3055 bb 3067 s 3.25 5.48 ν(C-H)R1(98)
4 3190 3056 - 3053 sh 18.18 12.70 ν(C-H)R1(96)
5 3180 3046 3047 - 23.22 3.20 ν(C-H)R1(97)
6 3170 3037 - - 4.71 6.26 ν(C-H)R1(97)
7 3162 3029 3022 - 2.21 1.84 ν(C-H)R1(98)
8 1643 1615 1657 vs 1654 m 17.32 86.66 ν(C-C)R1(64) + δas(R1)(8) + β(C4-H)(7) + β(C4-H)(7)
9 1620 1592 1588 vw 1598 vs 12.60 9.61 ν(C-C)R1(57) + β(C6-H)(8) + δs(R1)(7) + δas(R2)(5) + ν(C-N)R2(5)
10 1584 1557 1559 s 1560 s 337.12 5.66 ν(C-N)R2(64) + δas(R2)(17) + ν(C-O)(11)
11 1557 1531 1542 m - 8.20 111.65 ν(C-C)R2(40) + δas(R2)(26) + ν(C-C)brd(11) + ν(C-C)R1(8)
12 1514 1488 1491 s 1499 vw 16.95 12.09 ν(C-C)R2(26) + ν(C-C)R1(19) + β(C5-H)(13) + β(C2-H)(12) + δas(R2)(8) + β(C1-H)(6)
13 1474 1449 1454 s 1451 w 9.90 11.07 ν(C-C)R1(30) + β(C6-H)(19) + β(C1-H)(17) + β(C5-H)(7) + ν(C-C)R2(7) + δas(R2)(6)
14 1390 1366 1362 vw 1368 w 35.39 2.79 β(O-H)(32) + ν(C-S)R2(13) + ν(C-N)R2(11) + ν(C-O)(11) + δs(R2)(10) + β(C-O)(6)
15 1357 1334 1340 w 1339 w 0.81 4.91 β(C4-H)(26) + ν(C-C)R1(23) + β(C2-H)(21) + β(C6-H)(10)
16 1335 1312 1322 w 1301 s 18.80 14.71 ν(C-C)R1(60) + ν(C-N)R2(8)
17 1306 1284 1284 w 1282 m 11.02 17.83 ν(C-C)R1(29) + ν(C-N)R2(23) + ν(C-C)R2(15) + ν(C-C)brd(9) + β(C2-H)(6)
18 1220 1199 1196 w 1197 10.31 35.93 β(C13-H)(43) + ν(C-C)brd(15) + ν(C-C)R1(10) + δtrig(R1)(7) + ν(C-N)R2(5)
19 1205 1185 1180 s 1186 1.41 8.08 β(C5-H)(23) + β(C4-H)(20) + ν(C-C)R1(20) + β(C2-H)(17) + β(C1-H)(15)
20 1183 1163 - - 1.84 2.72 β(C6-H)(35) + β(C1-H)(21) + ν(C-C)R1(18) + β(C5-H)(17)
21 1175 1155 1158 m 1159 w 205.80 1.59 β(O-H)(27) + ν(C-O)(25) + ν(C-N)R2(15) + δas(R2)(9) + β(C13-H)(9)
22 1102 1083 1075 m - 16.09 0.63 ν(C-C)R1(51) + β(C6-H)(14) + β(C2-H)(12) + β(C4-H)(8)
23 1071 1053 1056 m 1055 w 59.52 2.28 ν(C-N)R2(22) + β(C13-H)(22) + ν(C-C)R1(15) + ν(C-C)R2(9) + β(O-H)(6) + ν(C-N)R2(5)
24 1047 1029 1031 m 1028 m 19.59 7.27 ν(C-C)R1(57) + δtrig(R1)(15)
25 1016 999 998 m 999 s 0.15 40.43 δtrig(R1)(62) + ν(C-C)R1(37)
Page 30
26 995 978 972 w - 0.33 0.16 γ(C1-H)(37) + γ(C2-H)(21) + γ(C6-H)(19) + τR1(puck.)(13) + γ(C5-H)(7)
27 982 965 - - 0.17 0.02 γ(C5-H)(41) + γ(C4-H)(21) + γ(C2-H)(16) + γ(C6-H)(7) + γ(C1-H)(5)
28 933 917 909 s - 2.90 0.15 γ(C4-H)(28) + γ(C2-H)(25) + γ(C6-H)(24) + γ(C3-C12)(6)
29 919 903 882 vw 908 w 1.28 4.44 δas(R2)(24) + ν(C-C)R1(17) + ν(C-S)R2(11) + δtrig(R1)(10) + δs(R2)(9) + ν(C-N)R2(8)
30 853 838 844 w 831 vw 0.10 0.71 γ(C4-H)(30) + γ(C1-H)(26) + γ(C2-H)(20) + γ(C5-H)(20)
31 835 821 832 m - 19.892 1.63 ν(C-S)R2(62) + δas(R2)(18)
32 785 772 773 s 772 w 18.84 1.84 τ1R2(20) + τR1(puck.)(20) + γ(C6-H)(15) + γ(C3-C12)(13) + τ2R2(9) + γ(C12-C3)(8)
33 721 709 713 s 705 m 94.74 0.45 γ(C13-H)(56) + γ(C1-H)(9) + τ1R2as(8) + γ(C5-H)(8)
34 710 698 683 s - 27.66 27.86 ν(C-S)R2(43) + δas(R2)(33) + β(C-O)(8)
35 694 682 - - 6.97 1.99 τR1(puck.)(57) + γ(C13-H)(13) + γ(C5-H)(11) + γ(C1-H)(10) + γ(C3-C12)(5)
36 682 670 669 m - 8.40 0.25 τ1R2(49) + τ2R2(36) + γ(C12-C3)(6)
37 669 658 654 s 654 w 18.20 6.02 δas(R1)(34) + δs(R1)(18) + δas(R2)(14) + ν(C-S)R2(5)
38 634 623 618 m 617 w 0.11 4.14 δs(R1)(54) + δas(R1)(29) + ν(C-C)R1(5)
39 585 575 592 w - 13.46 4.39 τ2R2(39) + γ(C-O)(22) + δs(R2)(13) + τ1R2(11)
40 581 571 569 vs 559 m 8.31 5.23 τ2R2(34) + γ(C-O)(20) + δs(R2)(18) + τ1R2(8)
41 492 484 474 w 475 w 8.40 0.48 τR1as(24) + τ1R2(23) + γ(C3-C12)(18) + τR1s(8) + γ(C6-H)(5) + τ2R2(5)
42 445 437 449 w 465 w 11.35 2.42 β(C-O)(16) + β(C12-C3)(13) + τ1R2(12) + ν(C-S)R2(12) + β(C3-C12)(9) + δas(R2)(9)
43 411 404 419 m - 2.84 0.43 τR1s(62) + τR1as(20)
44 397 390 - - 91.35 2.65 γ(O-H)(68)+ τ2R2(17) + τ(C-O)(7)
45 349 343 - 363 m 7.27 6.39 β(C-O)(28) + β(C3-C12)(20) + δas(R2)(11) + ν(C-S)R2(6) + ν(C-S)R2(5) + β(C12-C3)(5)
46 303 298 - 310 m 0.22 11.97 ν(C-C)brd(21) + δas(R2)(20) + δas(R1)(14) + β(C-O)(10)
47 274 269 - 267 w 0.54 3.02 τ2R2(44) + γ(C-O)(15) + γ(C13-H)(11) + τR1as(11) + τ1R2(6) + τR1s(5)
48 243 239 - 222 m 0.87 9.59 τ1R2(35) + τ2R2(33) + τR1as(13) + τR1s(5)
49 131 129 - - 0.06 7.53 β(C12-C3)(39) + β(C3-C12)(23) + δas(R2)(6)
50 91 89 - - 0.83 21.25 γ(C12-C3)(26) + γ(C3-C12)(23) + τ1R2(21) + τ2R2(10) + τR1as(6)
51 35 34 - - 0.04 222.44 τ(C-C)brd(76) + τ1R2(5)
Abbreviations: R1: benzene ring; R2: five-membered ring; s: symmetric; as: asymmetric; ν: stretching; β: in-plane bending; γ: out-plane bending; δ: deformation; τ: torsion (τ1 & τ2 defined in table 3); brd: bridge; a = cos(1440) and b = cos(720). aIIR and IRa, IR and Raman Intensity (kmmol-1);
Page 31
0
Table 5
Dipole Moment, Polarizability and hyperpolarizability data for 4P3HT (enol and keto form) calculated at B3LYP/6-311++G(d,p) level of theory.
Dipole Moment Polarizability Hyperpolarizability
Enol Keto Enol Keto Enol Keto
µx -0.4657 4.8727 αxx 198.505 200.6530 βxxx -355.0681 1003.7819 µy 0.1618 -1.1894 αyy 104.589 4.8590 βxxy 130.6324 67.4616
µz 0.1936 -0.2135 αzz 108.221 134.5330 βxyy 79.5107 26.2641 µtotal(D) 0.5296 5.0203 αxy 6.196 1.6730 βyyy 91.6416 -72.3345 αxz -15.428 -2.1060 βxxz -8.8784 13.3523 αyz 30.815 77.9820 βxyz 76.3089 20.0915 αmean (a.u.) 137.105 137.7227 βyyz 19.5395 15.6431 αmean (e.s.u) 20.3189 x 10-24 20.4105 x 10-24 βxzz 13.5823 31.6010 βyzz -36.5522 -39.7030 βzzz -41.5031 -35.930108 βtotal (a.u.) 322.6061 1062.6050 βtotal (e.s.u.) 2.7871 x 10-30
9.1802 x 10-30
Page 32
1
Table 6
Calculated important orbital's energies (eV), total energy in gas and in solutions of title compound.
Parameters TD-DFT
Gas Methanol Chloroform Εtotal (Hartree) -875.48940 -875.49686 --875.49472 Εtotal (eV) -23823.29138 -23823.49438 -23823.43615 ΕHOMO -6.10651 -6.17699 -6.14678 ΕLUMO -1.34207 -1.41390 -1.38370 ∆ΕHOMO ~ LUMO(eV) 4.76444 4.76309 4.76308
Page 33
2
Table 7
Experimental and calculated absorption wavelength λ (nm), excitation energies E (eV), absorbance values and oscillator strengths ( f) of 4P3HT.
Experimental TD-DFT/B3LYP/6-311++G(d,p)
λ (nm) E (eV) Abs. λ (nm) E (eV) f
Gas Phase 278.97 (46→47) 4.4444 0.2439
262.92 (46→48) 4.7156 0.0406
254.97 (46→49) 4.8626 0.0034
245.97 (46→50) 5.0510 0.0008
238.64 (46→51) 5.1955 0.0218
231.26 (46→52) 5.3613 0.0059
Chloroform
282.60 4.3873 0.593 283.97 (46→47) 4.3662 0.3500
248.00 4.9994 0.304 263.95 (46→48) 4.6973 0.0448 251.06 (46→49) 4.9385 0.0015
239.33 (46→51) 5.1806 0.0080
239.15 (46→50) 5.1843 0.0348
238.00 5.2094 0.322 230.47 (45→47) 5.3797 0.1816
Methanol 280.20 4.4248 0.260 282.84 (46→47) 4.3835 0.3276
264.07 (46→48) 4.6952 0.0470
249.38 (46→49) 4.9716 0.0011
238.83 (46→50) 5.1914 0.0319
220.80 5.6152 0.230 237.73 (46→51) 5.2153 0.0077
229.84 (45→47) 5.3943 0.1709
Page 34
3
Table 8 Second order perturbation theory analysis of fock matrix in NBO basis for 4P3HT.
Donar(i) Type ED(i)(e) Acceptor(j) Type ED(j)(e)a E(2)b Kcal/mol
E(j)-E(i)c
(a.u.) F(i,j)d
(a.u.) C1-C2 σ 1.97897 C1-C6 σ* 0.01647 2.78 1.28 0.053
σ 1.97897 C2-C3 σ* 0.02269 3.20 1.27 0.057
σ 1.97897 C3-C12 σ* 0.03605 3.29 1.17 0.056
σ 1.97897 C6-H11 σ* 0.01364 2.37 1.14 0.047
C1-C6 σ 1.97976 C1-C2 σ* 0.01496 2.75 1.28 0.053
σ 1.97976 C2-H8 σ* 0.01394 2.35 1.16 0.047
σ 1.97976 C5-C6 σ* 0.01638 2.65 1.28 0.052
σ 1.97976 C5-H10 σ* 0.01351 2.45 1.14 0.047
π 1.66053 C2-C3 π* 0.36581 20.27 0.29 0.068
π 1.66053 C4-C5 π* 0.31849 20.26 0.28 0.068
C1-H7 σ 1.9802 C2-C3 σ* 0.02269 3.88 1.08 0.058
σ 1.9802 C5-C6 σ* 0.01638 3.68 1.09 0.057
C2-C3 σ 1.97176 C1-C2 σ* 0.01496 2.83 1.27 0.054
σ 1.97176 C1-H7 σ* 0.0138 2.18 1.13 0.045
σ 1.97176 C3-C4 σ* 0.0228 3.90 1.25 0.062
σ 1.97176 C3-C12 σ* 0.03605 2.52 1.16 0.048
σ 1.97176 C4-H9 σ* 0.0141 2.51 1.13 0.048
σ 1.97176 C12-C13 σ* 0.02707 2.42 1.27 0.050
π 1.62807 C1-C6 π* 0.33395 20.63 0.28 0.068
π 1.62807 C4-C5 π* 0.31849 20.02 0.28 0.067
π 1.62807 C12-C13 π* 0.2982 18.73 0.26 0.063
C2-H8 σ 1.97836 C1-C6 σ* 0.01647 3.77 1.09 0.057
σ 1.97836 C3-C4 σ* 0.0228 4.56 1.08 0.063
C3-C4 σ 1.97228 C2-C3 σ* 0.02269 3.87 1.26 0.062
σ 1.97228 C2-H8 σ* 0.01394 2.27 1.15 0.046
σ 1.97228 C3-C12 σ* 0.03605 2.58 1.17 0.049
σ 1.97228 C4-C5 σ* 0.01471 3.02 1.28 0.056
σ 1.97228 C5-H10 σ* 0.01351 2.15 1.14 0.044
σ 1.97228 C12-N16 σ* 0.02302 2.54 1.14 0.048
C3-C12 σ 1.96965 C2-C3 σ* 0.02269 2.44 1.23 0.049
σ 1.96965 C3-C4 σ* 0.0228 2.31 1.23 0.048
σ 1.96965 C12-C13 σ* 0.02707 4.28 1.25 0.065
σ 1.96965 C13-S17 σ* 0.01042 2.28 0.87 0.040
C4-C5 σ 1.97888 C3-C4 σ* 0.0228 3.36 1.27 0.058
σ 1.97888 C3-C12 σ* 0.03605 3.33 1.18 0.056
σ 1.97888 C5-C6 σ* 0.01638 2.77 1.28 0.053
σ 1.97888 C6-H11 σ* 0.01364 2.33 1.15 0.046
π 1.67977 C1-C6 π* 0.33395 19.65 0.28 0.067
π 1.67977 C2-C3 π* 0.36581 19.62 0.29 0.068
C4-H9 σ 1.97932 C2-C3 σ* 0.02269 4.28 1.09 0.061
σ 1.97932 C5-C6 σ* 0.01638 3.71 1.10 0.057
C5-C6 σ 1.97942 C1-C6 σ* 0.01647 2.65 1.28 0.052
σ 1.97942 C1-H7 σ* 0.0138 2.45 1.14 0.047
σ 1.97942 C4-C5 σ* 0.01471 2.79 1.28 0.053
σ 1.97942 C4-H9 σ* 0.0141 2.45 1.14 0.047
C5-H10 σ 1.98028 C1-C6 σ* 0.01647 3.61 1.10 0.056
σ 1.98028 C3-C4 σ* 0.0228 3.92 1.08 0.058
C6-H11 σ 1.9806 C1-C2 σ* 0.01496 3.71 1.10 0.057
σ 1.9806 C4-C5 σ* 0.01471 3.71 1.10 0.057
Page 35
4
C12-C13 σ 1.98241 C3-C12 σ* 0.03605 4.25 1.23 0.065
π 1.88479 S17 RY*(1) 0.00541 2.89 0.98 0.049
π 1.88479 C2-C3 π* 0.36581 9.50 0.33 0.053
π 1.88479 C15-N16 π* 0.37828 8.99 0.27 0.047
C12-N16 σ 1.97097 C13-H14 σ* 0.0131 2.33 1.21 0.048
σ 1.97097 C15-O18 σ* 0.04349 7.28 1.12 0.081
C13-H14 σ 1.98293 C12 RY*(2) 0.00571 2.30 1.89 0.059
σ 1.98293 C12-C13 σ* 0.02707 2.13 1.15 0.044
σ 1.98293 C12-N16 σ* 0.02302 4.36 1.02 0.059
C13-S17 σ 1.97443 C3-C12 σ* 0.03605 6.09 1.14 0.075
σ 1.97443 C15-O18 σ* 0.04349 5.01 1.01 0.064
C15-N16 σ 1.98897 C12 RY*(2) 0.00571 2.67 2.23 0.069
σ 1.98897 C3-C12 σ* 0.03605 3.31 1.38 0.061
π 1.8958 C12-C13 π* 0.2982 18.26 0.36 0.076
C15-S17 σ 1.97779 C13-H14 σ* 0.0131 3.36 1.11 0.055
σ 1.97779 O18-H19 σ* 0.01073 2.39 1.05 0.045
O18-H19 σ 1.97862 C15-S17 σ* 0.08363 6.18 0.94 0.069
N16 CR (1) 1.99921 C12 RY*(1) 0.00912 2.25 15.20 0.165
CR (1) 1.99921 C15 RY*(1) 0.00973 4.22 15.01 0.225
N16 LP (1) 1.88217 C12 RY*(1) 0.00912 2.67 1.46 0.057
LP (1) 1.88217 C15 RY*(1) 0.00973 3.53 1.27 0.061
LP (1) 1.88217 C15 RY*(2) 0.00639 2.28 1.69 0.057
LP (1) 1.88217 C12-C13 σ* 0.02707 5.27 0.96 0.065
LP (1) 1.88217 C15-S17 σ* 0.08363 15.94 0.55 0.084
LP (1) 1.88217 C15-O18 σ* 0.04349 4.32 0.72 0.051
S17 LP (1) 1.98612 C15-N16 σ* 0.02552 2.91 1.25 0.054
LP (2) 1.66209 C12-C13 π* 0.2982 17.91 0.28 0.064
LP (2) 1.66209 C15-N16 π* 0.37828 29.46 0.25 0.077
O18 LP (1) 1.97408 C15 RY*(1) 0.00973 2.82 1.53 0.059
LP (1) 1.97408 C15 RY*(4) 0.00461 2.56 1.90 0.063
LP (1) 1.97408 C15-N16 σ* 0.02552 6.95 1.21 0.082
LP (2) 1.85441 C15 RY*(5) 0.00319 2.27 1.42 0.053
LP (2) 1.85441 C15-N16 π* 0.37828 35.90 0.34 0.104
C1-C6 π* 0.33395 C1 RY*(3) 0.00082 2.46 0.63 0.086
π* 0.33395 C6 RY*(3) 0.00103 3.01 0.43 0.078
C2-C3 π* 0.36581 C2 RY*(3) 0.00098 3.54 0.50 0.087
C4-C5 π* 0.31849 C4 RY*(3) 0.00086 2.20 0.43 0.069
π* 0.31849 C5 RY*(3) 0.0008 2.23 0.44 0.070
C12-C13 π* 0.2982 C2-C3 π* 0.36581 85.75 0.02 0.067
C15-N16 π* 0.37828 C15 RY*(3) 0.00483 2.33 1.43 0.118
π* 0.37828 C12-C13 π* 0.2982 59.82 0.03 0.067
aED: Electron Density
bE(2) means energy of hyperconjugative interactions.
cEnergy difference between donor and acceptor i and j NBO orbitals.
dF(i,j) is the Fock matrix element between i and j NBO orbitals.
Page 36
5
Table 9
NHO directionality and ''bond bending'' (deviations from line of nuclear centres).
Bond (A-B) Deviation at A (°) Deviation at B (°)
C1-C2 1.5 1.1 C1-C6 1.1 --- C3-C4 1.1 --- C4-C5 --- 1.5 C4-H9 1.2 --- C5-C6 1.1 --- C12-C13 2.6 2.4 C12-N16 --- 4.9 C13-S17 2.8 9.7 C15-N16 --- 3.9 C15-S17 2.7 9.5 C15-O18 2.3 1.0 O18-H19 2.8 ---
Table 10
Comparison of QSAR properties of 4P3HT molecule in enol and keto form S. no. Parameters Enol form Keto Form 1. Molecular Surface Area( Grid)(Å2) 342.65 340.57 2. Molecular Volume(Å3) 524.49 524.21 3. Hydration Energy (Kcal/mol) -12.11 -5.61 4. Log(P) 1.54 0.50 5. Refractivity (Å3) 49.79 49.55 6. Molecular Mass (amu) 177.22 177.22
Page 37
6
Research Highlights
� FT-Raman, FT-IR and UV-vis spectroscopic analysis of 4-Phenyl-3H-1,3-thiazol-2-ol has been done for the first time. � The mean polarizability and first hyperpolarizability calculated to be 20.3189×10-24 e.s.u. and 2.7871 x 10-30 e.s.u. respectively. � DOS and PDOS plots have been drawn to show the make–up of the molecular orbitals. � QSAR analysis of both the keto and enol form of the title compound establishes the efficacy of enol
form.
Page 38
7
Graphical abstract Molecular structure, vibrational and electronic properties of 4-Phenyl-3H-1,3-thiazol-2-ol using
density functional theory and comparison of drug efficacy of keto and enol forms by QSAR
Analysis
Alok K. Sachana, Shilendra K. Pathaka, Satish Chanda, Ruchi Srivastavaa, Onkar Prasada, Salah Belaidib and Leena Sinhaa,*
aDepartment of Physics, University of Lucknow, 226007, Lucknow,India bScientific Council of the Faculty of Science, University of Biskra, Algeria