JOURNAL OF RAMAN SPECTROSCOPY J. Raman Spectrosc. 2002; 33: 359–380 Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/jrs.866 Molecular structures of supported metal oxide catalysts under different environments M. A. Ba ˜ nares 1∗ and I. E. Wachs 2 1 Instituto de Catalisis y Petroleoquimica, CSIC, Campus UAM-Cantoblanco, E-28049 Madrid, Spain 2 Zettlemoyer Center for Surface Studies and Department of Chemical Engineering, Lehigh University, Bethlehem, Pennsylvania 18015, USA Received 3 August 2001; Accepted 15 February 2002 The use of in situ Raman spectroscopy to study the molecular structures of supported metal oxide catalysts under different environments is reviewed. The molecular structures under ambient (hydrated) and dehydrated conditions are presented. The effect of moisture at elevated temperatures is also presented and discussed with regard to its implications for catalytic phenomena. The molecular structural transformations during C 2 –C 4 lower alkane (LPG) oxidation, methane oxidation, methanol oxidation and selective catalytic reduction of NO with NH 3 reaction conditions are presented. In situ spectroscopy during catalytic reaction with simultaneous activity/selectivity measurement (‘operando’ spectroscopy) is emphasized owing to its contribution to the fundamental understanding of catalytic performance. The reducibility of the different surface metal oxide species, the relevance of surface coverage (surface monomeric vs polymeric species) and the specific oxide support are discussed when LPG, methane, methanol or hydrogen is the reducing agent. In situ Raman spectroscopy provides molecular-level information about the surface metal oxide species: structures, stability and transformations under different environments. In many cases, the use of complementary spectroscopic techniques results in a more complete understanding of the molecular structure–activity/selectivity relationships for supported metal oxide catalysts. Copyright 2002 John Wiley & Sons, Ltd. INTRODUCTION Raman spectroscopy is an extremely powerful catalyst characterization technique because it can provide funda- mental information about catalytic molecular structures and surface reaction intermediates. Furthermore, Raman spectroscopy can provide this information under in situ conditions (temperature, partial pressure of gas phase com- ponents, etc.). 1 Consequently, Raman spectroscopy has been used to examine essentially all types of catalytic materi- als: bulk and supported metals, bulk mixed metal oxides, supported metal oxides, bulk and supported metal sulfides, zeolites and molecular sieves, heteropolyoxo anions and clays. 2 The combination of fundamental molecular struc- tural information and in situ capabilities has resulted in a powerful tool for catalysis science, which allows for the development of a molecular-level understanding of struc- ture–activity/selectivity relationships for catalytic reactions. Ł Correspondence to: M. A. Ba ˜ nares, Instituto de Catalisis y Petroleoquimica, CSIC, Campus UAM-Cantoblanco, E-28049 Madrid, Spain. E-mail: [email protected]Contract/grant sponsor: National Science Foundation; Contract/grant number: CTS-9901643. Contract/grant sponsor: US Department of Energy, Basic Energy Sciences; Contract/grant number: DE-FG02-93ER14350. Contract/grant sponsor: CICYT; Contract/grant numbers: QUI98-0784; IN96-0053. Several publications have extensively reviewed the contribu- tions of Raman studies in catalysis. 3–17 This paper, however, must only review the Raman studies of catalysts performed over the past decade at Lehigh University (USA) and the Institute of Catalysis and Petroleum Chemistry (Spain). In particular, the use of Raman spectroscopy to study supported metal oxides under the following environments is presented: ambient conditions (hydrated), dehydrated conditions, wet oxidation reaction conditions, oxidation reaction conditions (alkane oxidation, methanol oxidation and selective catalytic reduction of NO x ) and under reducing conditions (by alka- nes or by hydrogen, with subsequent reoxidation). The role of the environment (dry air, humid air, reaction conditions, etc.) on the structural transformations of supported metal oxides is also discussed. The combination of fundamental molecular structural information and in situ capabilities has resulted in an explosion of Raman spectroscopic character- ization studies in the catalysis literature that began in the 1970s, from three publications in 1970 to ¾140 publications in 1999. 2,9 Among those published in 1999, about 30 corre- spond to Raman studies of supported metal oxides. 2 In recent years, there has been an emphasis on the molecular charac- terization of catalysts under reaction conditions since such fundamental information should lead to the development of molecular structure–activity/selectivity relationships and Copyright 2002 John Wiley & Sons, Ltd.
Transcript
JOURNAL OF RAMAN SPECTROSCOPYJ. Raman Spectrosc. 2002; 33: 359–380Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/jrs.866
Molecular structures of supported metal oxidecatalysts under different environments
M. A. Banares1∗ and I. E. Wachs2
1 Instituto de Catalisis y Petroleoquimica, CSIC, Campus UAM-Cantoblanco, E-28049 Madrid, Spain2 Zettlemoyer Center for Surface Studies and Department of Chemical Engineering, Lehigh University, Bethlehem, Pennsylvania 18015, USA
Received 3 August 2001; Accepted 15 February 2002
The use of in situ Raman spectroscopy to study the molecular structures of supported metal oxide catalystsunder different environments is reviewed. The molecular structures under ambient (hydrated) anddehydrated conditions are presented. The effect of moisture at elevated temperatures is also presented anddiscussed with regard to its implications for catalytic phenomena. The molecular structural transformationsduring C2 –C4 lower alkane (LPG) oxidation, methane oxidation, methanol oxidation and selective catalyticreduction of NO with NH3 reaction conditions are presented. In situ spectroscopy during catalytic reactionwith simultaneous activity/selectivity measurement (‘operando’ spectroscopy) is emphasized owing to itscontribution to the fundamental understanding of catalytic performance. The reducibility of the differentsurface metal oxide species, the relevance of surface coverage (surface monomeric vs polymeric species)and the specific oxide support are discussed when LPG, methane, methanol or hydrogen is the reducingagent. In situ Raman spectroscopy provides molecular-level information about the surface metal oxidespecies: structures, stability and transformations under different environments. In many cases, the useof complementary spectroscopic techniques results in a more complete understanding of the molecularstructure–activity/selectivity relationships for supported metal oxide catalysts. Copyright 2002 JohnWiley & Sons, Ltd.
INTRODUCTION
Raman spectroscopy is an extremely powerful catalystcharacterization technique because it can provide funda-mental information about catalytic molecular structuresand surface reaction intermediates. Furthermore, Ramanspectroscopy can provide this information under in situconditions (temperature, partial pressure of gas phase com-ponents, etc.).1 Consequently, Raman spectroscopy has beenused to examine essentially all types of catalytic materi-als: bulk and supported metals, bulk mixed metal oxides,supported metal oxides, bulk and supported metal sulfides,zeolites and molecular sieves, heteropolyoxo anions andclays.2 The combination of fundamental molecular struc-tural information and in situ capabilities has resulted in apowerful tool for catalysis science, which allows for thedevelopment of a molecular-level understanding of struc-ture–activity/selectivity relationships for catalytic reactions.
ŁCorrespondence to: M. A. Banares, Instituto de Catalisis yPetroleoquimica, CSIC, Campus UAM-Cantoblanco, E-28049Madrid, Spain. E-mail: [email protected]/grant sponsor: National Science Foundation;Contract/grant number: CTS-9901643.Contract/grant sponsor: US Department of Energy, Basic EnergySciences; Contract/grant number: DE-FG02-93ER14350.Contract/grant sponsor: CICYT; Contract/grantnumbers: QUI98-0784; IN96-0053.
Several publications have extensively reviewed the contribu-tions of Raman studies in catalysis.3 – 17 This paper, however,must only review the Raman studies of catalysts performedover the past decade at Lehigh University (USA) and theInstitute of Catalysis and Petroleum Chemistry (Spain). Inparticular, the use of Raman spectroscopy to study supportedmetal oxides under the following environments is presented:ambient conditions (hydrated), dehydrated conditions, wetoxidation reaction conditions, oxidation reaction conditions(alkane oxidation, methanol oxidation and selective catalyticreduction of NOx) and under reducing conditions (by alka-nes or by hydrogen, with subsequent reoxidation). The roleof the environment (dry air, humid air, reaction conditions,etc.) on the structural transformations of supported metaloxides is also discussed. The combination of fundamentalmolecular structural information and in situ capabilities hasresulted in an explosion of Raman spectroscopic character-ization studies in the catalysis literature that began in the1970s, from three publications in 1970 to ¾140 publicationsin 1999.2,9 Among those published in 1999, about 30 corre-spond to Raman studies of supported metal oxides.2 In recentyears, there has been an emphasis on the molecular charac-terization of catalysts under reaction conditions since suchfundamental information should lead to the developmentof molecular structure–activity/selectivity relationships and
Copyright 2002 John Wiley & Sons, Ltd.
360 M. A. Banares and I. E. Wachs
the molecular engineering of improved catalytic materials.At present, this is resulting in spectroscopic studies of cat-alysts under real reaction conditions, where structure andactivity/selectivity are measured simultaneously. This ‘real’reaction in situ spectroscopy is given the tentative name‘operando,’ by one of the authors (M.A.B.), to provide a sim-ple word which underlines the simultaneous evaluation ofboth structure and activity/selectivity. This name has beenborrowed from the Latin gerund ‘operando,’ which meansworking or operating, since the spectra are of an ‘operating’catalyst. Operando spectroscopy is poised to become a pow-erful tool in catalysis research.16 These developments mayalso be of use in other fields where environmental (temper-ature, pressure and atmosphere) spectroscopy will providefundamental information about the molecular structure andperformance of various materials.2
Supported metal oxide catalysts generally consist of two-dimensional surface metal oxide overlayers on an oxidesupport (e.g. alumina, titania, zirconia, silica). The oxidesupport are typically of ca 10–300 m2 g�1. In addition tothe two-dimensional surface metal oxide overlayer, smallmetal oxide crystallites may also be present. The metal oxidecrystallites tend to form when (a) the precursor salt is poorlydistributed over the support in the catalyst synthesis step,(b) there is a weak interaction between the deposited metaloxide and the underlying oxide support (e.g. metal oxideson silica) or (c) monolayer coverage has been exceeded.
Detailed fundamental surface information on a molecularlevel can be obtained from model supported metal oxidecatalysts containing the two-dimensional overlayers ofsurface metal oxides.4,7,11 A unique feature of supportedmetal oxide catalysts is that the active component isexclusively present as a surface phase, 100% dispersion,below monolayer coverage and there is no spectroscopiccomplication from the co-existence of bulk crystalline phases.The only bulk crystalline phase present below monolayercoverage is due to the oxide supports, which tend to give riseto weak Raman signals (e.g. Al2O3, SiO2� or Raman signalsthat generally occur at much lower wavenumbers than theactive supported metal oxide species (e.g. CeO2, ZrO2, TiO2�.
Supported metal oxide catalysts are widely employedin industrial applications: alkane dehydrogenation,olefin polymerization, olefin metathesis, selectiveoxidation/ammoxidation/reduction of organic moleculesand inorganic emissions as well as precursors to supportedHDS and metallic hydrotreating catalysts.7,11,18 – 20 The use ofsupported metal oxide catalysts for oxidation reactions hasgrown significantly over the past few decades owing to theirexcellent catalytic properties in the manufacture of manychemical intermediates and pollution control strategies.21
Supported metal oxide catalysts are ideally suited forRaman spectroscopic studies because the dispersed metaloxide phases generally give rise to strong Raman signalsand the oxide support tends to possess weak Raman sig-nals or strong Raman signals that occur at much lower
wavenumbers. Consequently, Raman spectroscopy providesfundamental information about the surface metal oxidesspecies present in supported metal oxide catalysts: spe-cific location, surface coverage, molecular structure andits potential transformation in different environments, theparticipation of specific metal oxygen bonds in catalytic oxi-dation reactions (e.g. with the aid of isotopic tracer studies)and surface acidity/basicity. Raman spectroscopy also pro-vides direct fundamental surface information about the ratioof isolated and polymerized surface metal oxide species,terminal M O and bridging M—O—M bonds, extent ofreduction of the surface oxide species during reaction con-ditions, influence of the oxide support ligands, influenceof acidic/basic metal oxide additives (promoters/poisons)and participation of specific M—O bonds in catalysis (e.g.with the aid of oxygen-18-labeled isotope experiments). Thebridging M—O—support bonds possess enough of a cova-lent character that should result in weak Raman bands.7 Theintensity of the bridging M—O—support band dependson the covalency of that bond and is, therefore, directlyrelated to the electronegativity of the cations involved. Forexample, the intensity of the bridging V—O—support bondin supported vanadium oxide catalysts should depend onthe electronegativity of the cation of the oxide support(Si4C > Al3C > Ti4C > Zr4C). Thus, in the series V2O5/SiO2,V2O5/Al2O3, V2O5/TiO2, V2O5/ZrO2, the polarizability ofthe bridging V—O—support bond increases, and the Ramansignal of the bridging V—O—support bond increases. Thus,bridging V—O—support bonds have not been observedfor V2O5/SiO2, V2O5 or Al2O3 owing to the higher elec-tronegativity of Si and Al cations in silica22 and alumina,23
so they must be very weak or possibly inactive in Raman.For V2O5/TiO2, the bridging V—O—Ti bond should exhibitweak Raman bands near 638 and 248 cm�1 that cannot bedetected owing to the very intense Raman bands of the tita-nia support. However, they are visible on a system wherevanadium oxide is supported on a substrate where titania ishighly dispersed as a surface species on silica.24 Similarly, thebridging V—O—Zr bond also exhibits weak Raman bandsthat are visible when vanadium oxide is supported on asystem where zirconia is highly dispersed on silica.25 Ramanspectroscopy also provides structural information about thepresence of microcrystallites (<40 A), XRD amorphous par-ticles and surface reaction intermediates.
The results presented in this paper are from investi-gations performed at Lehigh University and Instituto deCatalisis y Petroleoquımica, CSIC. The Raman system usedat Lehigh University is a triple-grating spectrometer (Spex,Model 1877), a CCD detector and an argon ion laser. The insitu Raman studies were run in a quartz cell with a sampleholder made of metal alloy (Hastalloy C), and a 100–200 mgsample disc is held by the cap of the sample holder. The sam-ple holder is mounted on a ceramic shaft that is rotated by a115 V CD motor at a speed of 1000–2000 rpm. A cylindricalheating coil surrounding the quartz cell is used to heat the
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 361
cell. The quartz cell is capable of operating up to 873 K andflowing gas is introduced into the cell at a rate of 50–100ml min�1 at atmospheric pressure. An on-line mass spec-trometer is used to monitor the conversion and selectivityof the specific reaction–catalyst system. The Raman systemused at the CSIC institute is a Renishaw Micro-Raman System1000 equipped with a cooled CCD detector (200 K), a holo-graphic super-Notch filter and a 1800 grooves mm�1 grating.The powdered samples were excited with an argon ion laser.In situ Raman spectra were run in a Linkam TS-1500 hotstage that allows heating of the powdered sample to 1773 Kunder a controlled atmosphere or stream of gases. OperandoRaman–GC spectra were run with a laboratory-made reac-tion cell that consists of a fixed-bed quartz microreactorcontained by quartz-wool plugs on both ends; the catalyst(ca 170 mg) is in powder form. The reaction feed is controlledby mass flow controllers and the reactor outlet is connectedon-line with a gas chromatograph (HP 5890 Series II). Themicro-reactor walls have optical quality and no appreciabledifferences can be observed between the Raman reaction celland a fixed-bed microreactor. The laser power on the sam-ple was kept below 5 mW to prevent local heating. Specificdetails of the experimental procedures in the work discussedbelow can be found in the original papers cited.
AMBIENT CONDITIONS (HYDRATED)
Under ambient conditions, moisture adsorbs on the surfaceof supported metal oxide catalytic systems and there isextensive solvation of the surface metal oxides (equivalentto ¾20–40 monolayers of water). Therefore, the surfacechemistry of supported oxides (V, Nb, Ta, Cr, Mo, Wand Re) corresponds to that of aqueous solutions. As inaqueous solution, the molecular structure of the metal oxidedepends on the concentration and pH of the aqueous system.Many publications have shown that Raman spectroscopycan follow the hydrated molecular structures of thesesupported metal oxides as a function of oxide loading andthe nature of the oxide support.20,21,26 – 29 For instance, in
the case of hydrated supported vanadium oxide catalysts,Raman spectroscopy can efficiently discriminate betweenthe different kind of vanadium oxides present as a functionof the specific oxide support and vanadia loading. It wasfound that the molecular structures of supported transitionmetal oxide overlayers on different oxide supports (MgO,Al2O3, ZrO2, TiO2 and SiO2� are determined by the net pHat the point of zero charge (PZC). Gil-Llambias et al.30 haveshown that for the V2O5/TiO2 and V2O5/Al2O3 systems,the pH at PZC of the samples decreases with increasingvanadium oxide loadings (with surface vanadium oxidecoverage). The pH at PZC of the supported vanadium oxidecatalytic systems is expected to be much lower than thepH at PZC of the oxide support at high surface vanadiumoxide coverage and close to that of the oxide support at lowsurface coverage because the pH at PZC of V2O5 is ¾0.5.The surface vanadium oxide species depends on the net pHat PZC of the surface and can be predicted from the knownvanadium oxide aqueous phase diagram as shown in Table 1for low and high surface vanadium oxide loadings. At lowsurface vanadium oxide coverage, the net pH at PZC shouldclosely reflect the specific oxide support. At high surfacevanadium oxide coverage, the pH at PZC is significantlylowered owing to the presence of the acidic vanadium oxideoverlayer. The qualitative agreement between the predictedhydrated surface vanadium oxide species and the observedhydrated surface vanadium oxide species on the differentoxide supports are shown in Table 1 and underlines theinfluence of the net pH at PZC of each oxide support upon thesurface structure of the hydrated surface vanadium species.
For the silica-supported vanadium oxide system,UV–Vis31 and EXAFS/XANES32 studies indicate thepresence of VO5 and octahedral coordinated surfacevanadium oxide under ambient conditions. The partialhydration of the dehydrated sample changes the color fromwhite to light yellow and the sample becomes deep redwhen fully hydrated. For the partially hydrated sample,the broad Raman bands observed are very different fromthe bands observed for the fully hydrated and dehydrated
Table 1. Predicted and observed hydrated surface vanadium oxide species on different oxide supports28,29
Net pH at PZC of Hydrated structures
Oxide support oxide support Coverage Predicted Observed
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
362 M. A. Banares and I. E. Wachs
samples.22 Therefore, the molecular structure of the surfacevanadium oxide species on silica is also dependent onthe degree of hydration. A combination of in situ Raman,UV–Vis–NIR DRS (Diffuse Reflectance Spectroscopy) andXANES spectroscopy also revealed that the structure ofhydrated surface vanadium oxide species is dependent onthe degree of hydration.22 Fully hydrated surface vanadiumoxide species on SiO2 closely resemble a V2O5ÐnH2O gel,which is distinct from V2O5 crystallites.
DEHYDRATED CONDITIONS
The molecular structure of supported transition metal oxidecatalysts under dehydrated conditions has been under debatein the literature for almost two decades. In particular, itwas not clear how many terminal M O bonds are presentin the surface molecular entities. Transition metal oxidesmay possibly be present as mono-, di- or trioxo species.The combination of in situ Raman spectroscopy and oxygenisotopic labeling studies provides fundamental informationabout the number of terminal M O bonds and, thus,discriminates between mono-, di- and trioxo species. Thisis illustrated schematically in Fig. 1. A monooxo species(structure A) should give rise to two strong Raman bands(M 16O and M 18O), a dioxo species (structure B) shouldhave three strong Raman bands (16O M 16O, 16O M 18Oand 18O M 18O) and a trioxo species (structure C)
should give rise to four strong Raman bands (16O3 M,16O2 M 18O, 16O M 18O2, 18O3 M).33,34
Isotopic oxygen labeling studies have allowed the deter-mination of the molecular structures of zirconia-supportedV(V), Nb(V), Cr(VI), Mo(VI), W(VI) and Re(VII) oxides.35
After several reduction and reoxidation cycles, a signifi-cant extent of isotopic exchange is achieved and Table 2shows the Raman bands of representative catalysts aftercalcinations with 16O2 and after partial isotopic exchangewith oxygen-18. The Raman bands of the dehydrated sup-ported transition metal oxides after calcination in 16O2 at823 K in the 980–1030 cm�1 range corresponds to the shortterminal M O bond (�s[M O]) and the Raman bandsin the 750–950 cm�1 range correspond to either the anti-symmetric stretch of M—O—M bonds or the symmetricstretch of (—O—M—O—)n bonds (�as [M—O—M] and�s[(—O—M—O—)n], respectively). Isotopic substitutionby oxygen–18 results in significant Raman shifts of thebands positions owing to the greater mass of oxygen-18than oxygen-16. Both the number of the new Raman bandsformed, only one additional band, and the shift to lowerwavenumber values are dependent on the molecular struc-ture of the dehydrated supported transition metal oxides(see Fig. 1) and correspond very well with the calculatedvalues for monooxo dehydrated surface metal oxide species(structure A corresponding to one terminal bond).
Additional evidence for the monooxo structure of thesupported transition metal oxides can be found from the
16O
16O
16O
16O
O16
16O 16O 18O 18O 18O
18O|| M
\\ //M
||
\\ //M
\\ //M
M
|| M
MonooxoStructure A
DioxoStructure B
TrioxoStructure C
==18O
18O
18O||M ==16O
18O
18O||M ==16O
16O
18O||M ==
Figure 1. Schematic representation of possible structures of surface metal oxide species.
Table 2. Observed Raman bands (wavenumbers in cm�1) before and after partialexchange with 18O2
35
Experimentally observed wavenumber/cm�1
Catalyst After calcination with 16O2 After partial exchange with 18O2
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 363
comparison of the Raman and IR bands obtained for thesame catalyst under dehydrated conditions. The presence ofonly one vibration at the same wavenumber from Ramanand IR indicates a monooxo species because a diatomicoscillating system has only one stretching mode (�s) sincefor a dioxo species two bands due to the symmetric (�s)and antisymmetric (�as) stretching modes will be visible.33,34
These wavenumbers should be separated by ca 20 cm�1.Furthermore, for dioxo species the symmetric mode will bemore intense in Raman, while the antisymmetric mode willdominate in IR. A more complex situation should occur fora trioxo species. Based on the coincident values of the IRand Raman wavenumbers it appears that the supportedtransition metal oxides are present as surface monooxospecies.36
The dehydrated monooxo species can be present as eithersurface isolated or polymerized structures, where dehy-drated polymeric species also possesses bridging M—O—Mbonds that can be detected at ¾900 (antisymmetric stretch),600 (symmetric stretch) and 200 (bending mode) cm�1. Thus,molybdena on zirconia is present as a surface polymericmonooxo species because of the Raman vibrations at 996(Mo O symmetric stretch) and 850 cm�1 (Mo—O—Moantisymmetric stretch). The structural information can befurther complemented by other techniques, such as XANES,that reveal that Mo has a formal coordination of five, whichis close to a distorted octahedral coordination.27 Similarstructures can be envisioned also for the dehydrated sur-face tungsten oxide and niobia species. A surface polymericspecies occurs for surface vanadia species on ZrO2, whichpossesses a pseudo-tetrahedral coordination, giving rise toRaman vibrations near 1030 (terminal V O) and 920 (bridg-ing V—O—V) cm�1.35 The coordination model for rheniumoxide on zirconia is proposed as a surface polymeric species,in which at least two octahedron-like coordinated Re7C atomsare linked via an oxygen atom, one terminal Re O bond andfour bridging Re—O—Zr bonds.35
The Raman study of the dehydrated catalysts does notprovide any indication as to which of the Raman bandsof the terminal M O bond corresponds to the surfaceisolated or polymeric species. However, in situ reactivitystudies under reducing conditions37 have shown that theRaman bands due to the surface isolated and polymericspecies can be discriminated by their different reductionactivity. For example, supported polymeric chromium oxidespecies are more reducible than the isolated species andreveal that the terminal M O bond of dehydrated isolatedmonooxo species are characterized by a Raman band at ahigher wavenumber (1030 cm�1) than that of the dehydratedpolymeric monooxo species (1010 cm�1). In the case ofthe silica support, only dehydrated monochromate surfacespecies are observed.37 For vanadium oxide supported onCeO2, the terminal V O mode of the dehydrated isolatedsurface vanadium oxide occurs at 1034 cm�1 and that of thedehydrated polymeric vanadium oxide species is observed
at 1017 cm�1.38 Similar observations have also been madefor molybdenum oxide, tungsten oxide and rhenium oxidesupported on CeO2.39 In the case of the silica support,only dehydrated isolated surface metal oxide species areobserved.
The terminal M O bond and the bridging M—O—Mbonds are detectable by Raman spectroscopy; however,the bridging M—O—support bonds are not detectableby Raman spectroscopy since they are too ionic to beRaman active. The study of the bridging M—O—supportbonds cannot be done directly by Raman spectroscopy.For V2O5/TiO2/SiO2, however, where a bilayer of surfacevanadia layer on a surface titania layer on silica is present, thebridging V—O—Ti bonds have been directly detected withRaman spectroscopy.24 Other characterization techniquescan provide additional structural information about thelocal environment of the different metal cations, solid-stateNMR, XANES/EXAFS and UV–Vis revealed that both thecoordination of the surface vanadia species and the surfacetitania species are altered by the formation of the bridgingV—O—Ti bond between the two layers.24
WET OXIDATION
Supported metal oxide catalysts under ambient conditionsare extensively hydrated owing to the presence of significantamounts of adsorbed moisture at room temperature. Theadsorbed moisture has a pronounced effect on the molecularstructures of the surface metal oxide phases and the situationin aqueous environments has already been described (seeabove). Under in situ dehydrated conditions, the adsorbedmoisture desorbs upon heating and the surface metal oxidespecies becomes dehydrated. The molecular structures of thedehydrated supported metal oxides have also been describedabove. However, water vapor is usually present in the feedgas or as a reaction product in many catalytic reactions atelevated temperatures. The presence of significant amountsof water vapor can affect the extent of surface hydroxylation,the ratio of Brønsted-to-Lewis surface acid sites or themolecular structures of the surface metal oxide species,which can have a significant effect on many reactions oversupported metal oxide catalysts.
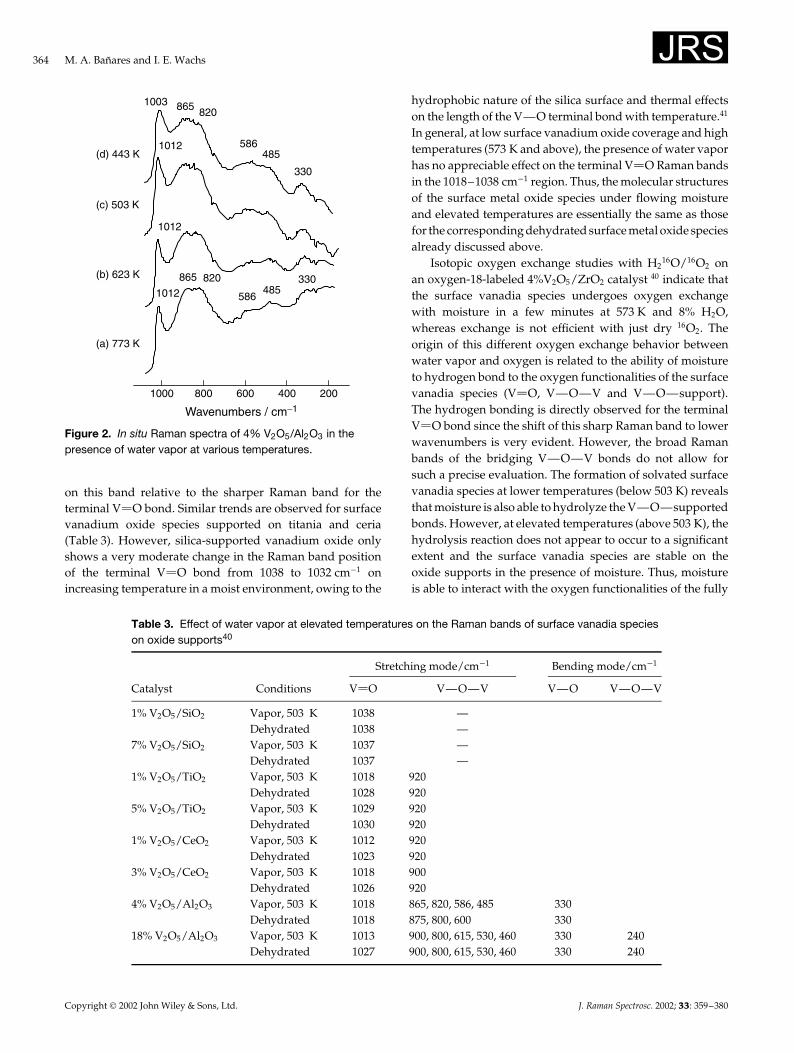
A detailed fundamental study on the nature of supportedvanadium oxide catalysts at high temperature in the presenceof water vapor was recently reported.40 This is illustratedin Fig. 2 for surface vanadium oxide species on an Al2O3
support. For 4% V2O5/Al2O3, the Raman band of theterminal V O bond of the surface vanadium oxide speciesshifts from 1012 to 1003 cm�1 on decreasing the catalysttemperature from 773 to 443 K and simultaneously becomesbroad in the presence of moisture. However, the bridgingV—O—V functionality of the surface vanadia species onalumina (900–800, 620–450 and 350–200 cm�1� appears tobe only modestly influenced by the presence of adsorbedwater vapor, which is primarily related to the broadness
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
364 M. A. Banares and I. E. Wachs
2004006008001000
Wavenumbers / cm−1
1003 865
586485
330
1012
865
586485
330
1012
1012
820
820
(a) 773 K
(b) 623 K
(c) 503 K
(d) 443 K
Figure 2. In situ Raman spectra of 4% V2O5/Al2O3 in thepresence of water vapor at various temperatures.
on this band relative to the sharper Raman band for theterminal V O bond. Similar trends are observed for surfacevanadium oxide species supported on titania and ceria(Table 3). However, silica-supported vanadium oxide onlyshows a very moderate change in the Raman band positionof the terminal V O bond from 1038 to 1032 cm�1 onincreasing temperature in a moist environment, owing to the
hydrophobic nature of the silica surface and thermal effectson the length of the V—O terminal bond with temperature.41
In general, at low surface vanadium oxide coverage and hightemperatures (573 K and above), the presence of water vaporhas no appreciable effect on the terminal V O Raman bandsin the 1018–1038 cm�1 region. Thus, the molecular structuresof the surface metal oxide species under flowing moistureand elevated temperatures are essentially the same as thosefor the corresponding dehydrated surface metal oxide speciesalready discussed above.
Isotopic oxygen exchange studies with H216O/16O2 on
an oxygen-18-labeled 4%V2O5/ZrO2 catalyst 40 indicate thatthe surface vanadia species undergoes oxygen exchangewith moisture in a few minutes at 573 K and 8% H2O,whereas exchange is not efficient with just dry 16O2. Theorigin of this different oxygen exchange behavior betweenwater vapor and oxygen is related to the ability of moistureto hydrogen bond to the oxygen functionalities of the surfacevanadia species (V O, V—O—V and V—O—support).The hydrogen bonding is directly observed for the terminalV O bond since the shift of this sharp Raman band to lowerwavenumbers is very evident. However, the broad Ramanbands of the bridging V—O—V bonds do not allow forsuch a precise evaluation. The formation of solvated surfacevanadia species at lower temperatures (below 503 K) revealsthat moisture is also able to hydrolyze the V—O—supportedbonds. However, at elevated temperatures (above 503 K), thehydrolysis reaction does not appear to occur to a significantextent and the surface vanadia species are stable on theoxide supports in the presence of moisture. Thus, moistureis able to interact with the oxygen functionalities of the fully
Table 3. Effect of water vapor at elevated temperatures on the Raman bands of surface vanadia specieson oxide supports40
Stretching mode/cm�1 Bending mode/cm�1
Catalyst Conditions V O V—O—V V—O V—O—V
1% V2O5/SiO2 Vapor, 503 K 1038 —Dehydrated 1038 —
7% V2O5/SiO2 Vapor, 503 K 1037 —Dehydrated 1037 —
1% V2O5/TiO2 Vapor, 503 K 1018 920Dehydrated 1028 920
5% V2O5/TiO2 Vapor, 503 K 1029 920Dehydrated 1030 920
1% V2O5/CeO2 Vapor, 503 K 1012 920Dehydrated 1023 920
3% V2O5/CeO2 Vapor, 503 K 1018 900Dehydrated 1026 920
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 365
oxidized surface vanadia species via hydrogen bonding, butthe structural transformations in the presence of moistureare only observed at low temperatures (below 503 K).
The ability of moisture to interact with the surface vana-dia species at elevated temperatures implies that moisturecompetitively adsorbs with the reactants on such surfaceadsorption sites during oxidation reactions. Thus, the rate ofoxidation reactions over supported vanadia catalysts shouldgenerally decrease in the presence of moisture and this effectshould increase with decreasing reaction temperature. Thisis in line with literature studies on the effect of moistureupon the selective catalytic reduction of NOx with NH3 overV2O5/TiO2 catalysts. All of these studies demonstrate thatthe presence of moisture decreases the conversion of NOand that the effect of moisture becomes more pronouncedat lower reaction temperatures.42 In addition, the selectiv-ity towards undesirable N2O formation is suppressed bythe presence of moisture, which has been attributed to sur-face hydroxylation.42 – 44 In fact, in situ IR studies during SCR(Selective Catalytic Reduction) over vanadia/titania catalystsshow that moisture adsorbs via hydrogen bonds, increasesthe surface hydroxyl concentration and coordinates to thesurface vanadia species.43
OXIDATION REACTIONS CONDITIONS
C2 –C4 alkanes (LPG) oxidation reaction conditionsButane oxidation to maleic anhydride was investigatedover supported vanadia/alumina.45 The Raman spectrumof the fully oxidized, dehydrated system exhibits bandsat 920, 800, 600 and 550 cm�1 due to the presence ofbridging V—O—V bonds and 1028 cm�1 due to the terminalV O bonds. During n-butane oxidation, the polymericV—O—V functionalities are preferentially reduced relativeto the terminal V O bonds. This trend becomes moreevident as the reaction temperature increases. In the absenceof oxygen, both the terminal V O and the bridgingV—O—V bonds are absent from the in situ Raman spectrumowing to extensive reduction of the surface vanadia sites.Unfortunately, the reduced surface vanadia species doesnot give rise to new Raman bands owing to its veryweak or inactive Raman signal (most likely because of theabsence of the V O functionality, which gives rise to astrong Raman signal). Exposure to an oxygen environmentreoxidizes the surface vanadia species and yields the initialRaman spectrum of the fresh catalysts.45 Essentially thesame trends were observed with vanadia supported ontitania, zirconia, niobia, ceria and binary oxide supportssuch as P2O5/TiO2. Thus, the surface vanadia species onoxide supports reduces partially during n-butane oxidationand the bridging V—O—V functionality is preferentiallyreduced relative to the terminal V O bond.45,46
The extent of reduction of supported vanadium oxidespecies depends on the reactivity of the hydrocarbon. Thus,reduction is less evident during the selective oxidation of
propane and ethane for the same catalytic systems; only4% V2O5/ZrO2 shows some reduction of the polymericV—O—V functionalities.47 – 49 Other in situ techniquesprovide complementary information about the states of thesupported vanadium oxide species under reaction conditionsfor butane and ethane selective oxidation reactions.47,50 Thecomplementary study by in situ UV–Vis DRS 50 on supportedvanadium oxide catalysts shows that the extent of reductionof supported vanadium oxide is higher under n-butaneoxidation than under ethane oxidation. In particular, theextents of reduction of V(V) sites determined by in situUV–Vis DRS for 1% V2O5/ZrO2 and 4% V2O5/ZrO2 areca 2.8 and 3.6% under ethane oxidation, ca 1.8 and 8.2%under propane oxidation and ca 8.0 and 9.6% under butaneoxidation.50 This is consistent with the expected reactivity ofthe C2 –C4 alkanes, determined by the weakest C—H bond,and the trends observed by in situ Raman reaction studieswith the same C/O atomic ratio in the hydrocarbon/O2/Hefeed.44 – 50
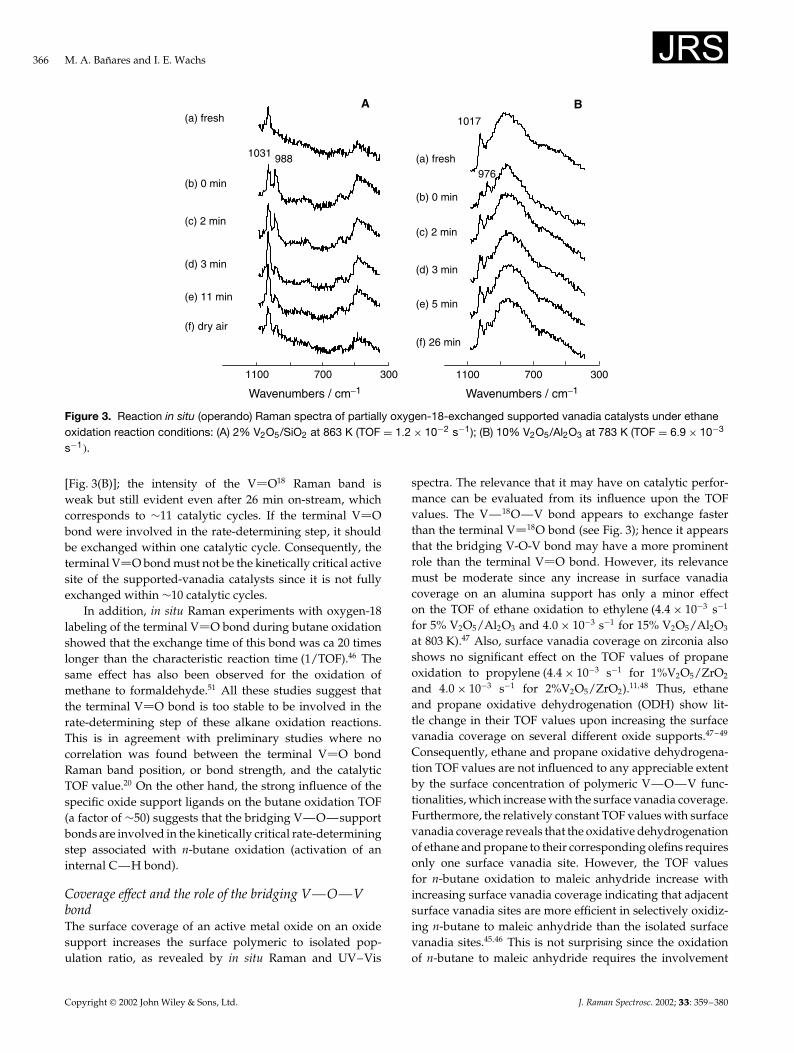
Role of the terminal V O bondThe butane turnover frequency (TOF) is a strong functionof the specific oxide support and varies by a factor of ¾50(titania > ceria > zirconia > niobia > alumina > silica),but does not correspond with the changes in the positionof the terminal V O bond.45 A lack of such a correlationwas also found for ethane oxidation on supported-vanadiacatalysts.20 The in situ Raman spectra of the oxygen-18exchanged 2% V2O5/SiO2 and 10% V2O5/Al2O3 duringethane oxidation reaction conditions are illustrated inFig. 3. Each supported-vanadia catalyst initially shows aRaman band at 1031 or 1017 cm�1 that corresponds tothe terminal V 16O vibrations of the dehydrated surfacevanadia on silica and alumina, respectively. New Ramanbands appear near 988 and 976 cm�1 upon exchange with18O2 that correspond to the terminal V 18O vibrations onsilica and alumina, respectively. These wavenumber shiftsquantitatively correspond with the predicted values from thechange in the reduced mass of the V—O bond when oxygen-16 is replaced with oxygen-18. Alumina-supported vanadiaexhibits a very broad Raman band at ¾860 cm�1, due to thebridging V—O—V bond. A weak shoulder at ¾900 cm�1
is evident after oxygen-18 exchange, which corresponds tothe vibration of the exchanged V—18O—V bond. The insitu Raman spectra in Fig. 3 for V2O5/Al2O3 and V2O5/SiO2
show that the supported vanadia species is essentially notreduced during ethane oxidation. The Raman intensities ofthe 988 and 1031 cm�1 bands of the 2% V2O5/SiO2 catalystare monitored as a function of time-on-stream during ethaneoxidation with 16O2 [Fig. 3(A)]. The Raman band of theterminal V 18O bond decreases rather fast during the firstminutes under reaction conditions at 863 K; however, it isnot fully exchanged, even after more than 11 min time-on-stream, which corresponds to about eight catalytic cycles.The exchanged 10% V2O5/Al2O3shows a similar behavior
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
366 M. A. Banares and I. E. Wachs
Wavenumbers / cm−1
3007001100
B1017
976(a) fresh
(b) 0 min
(c) 2 min
(d) 3 min
(e) 5 min
(f) 26 min
1100
Wavenumbers / cm−1
A
1031 988
(a) fresh
(b) 0 min
(c) 2 min
(d) 3 min
(e) 11 min
(f) dry air
700 300
Figure 3. Reaction in situ (operando) Raman spectra of partially oxygen-18-exchanged supported vanadia catalysts under ethaneoxidation reaction conditions: (A) 2% V2O5/SiO2 at 863 K (TOF D 1.2 ð 10�2 s�1); (B) 10% V2O5/Al2O3 at 783 K (TOF D 6.9 ð 10�3
s�1�.
[Fig. 3(B)]; the intensity of the V O18 Raman band isweak but still evident even after 26 min on-stream, whichcorresponds to ¾11 catalytic cycles. If the terminal V Obond were involved in the rate-determining step, it shouldbe exchanged within one catalytic cycle. Consequently, theterminal V O bond must not be the kinetically critical activesite of the supported-vanadia catalysts since it is not fullyexchanged within ¾10 catalytic cycles.
In addition, in situ Raman experiments with oxygen-18labeling of the terminal V O bond during butane oxidationshowed that the exchange time of this bond was ca 20 timeslonger than the characteristic reaction time (1/TOF).46 Thesame effect has also been observed for the oxidation ofmethane to formaldehyde.51 All these studies suggest thatthe terminal V O bond is too stable to be involved in therate-determining step of these alkane oxidation reactions.This is in agreement with preliminary studies where nocorrelation was found between the terminal V O bondRaman band position, or bond strength, and the catalyticTOF value.20 On the other hand, the strong influence of thespecific oxide support ligands on the butane oxidation TOF(a factor of ¾50) suggests that the bridging V—O—supportbonds are involved in the kinetically critical rate-determiningstep associated with n-butane oxidation (activation of aninternal C—H bond).
Coverage effect and the role of the bridging V—O—VbondThe surface coverage of an active metal oxide on an oxidesupport increases the surface polymeric to isolated pop-ulation ratio, as revealed by in situ Raman and UV–Vis
spectra. The relevance that it may have on catalytic perfor-mance can be evaluated from its influence upon the TOFvalues. The V—18O—V bond appears to exchange fasterthan the terminal V 18O bond (see Fig. 3); hence it appearsthat the bridging V-O-V bond may have a more prominentrole than the terminal V O bond. However, its relevancemust be moderate since any increase in surface vanadiacoverage on an alumina support has only a minor effecton the TOF of ethane oxidation to ethylene (4.4 ð 10�3 s�1
for 5% V2O5/Al2O3 and 4.0 ð 10�3 s�1 for 15% V2O5/Al2O3
at 803 K).47 Also, surface vanadia coverage on zirconia alsoshows no significant effect on the TOF values of propaneoxidation to propylene (4.4 ð 10�3 s�1 for 1%V2O5/ZrO2
and 4.0 ð 10�3 s�1 for 2%V2O5/ZrO2).11,48 Thus, ethaneand propane oxidative dehydrogenation (ODH) show lit-tle change in their TOF values upon increasing the surfacevanadia coverage on several different oxide supports.47 – 49
Consequently, ethane and propane oxidative dehydrogena-tion TOF values are not influenced to any appreciable extentby the surface concentration of polymeric V—O—V func-tionalities, which increase with the surface vanadia coverage.Furthermore, the relatively constant TOF values with surfacevanadia coverage reveals that the oxidative dehydrogenationof ethane and propane to their corresponding olefins requiresonly one surface vanadia site. However, the TOF valuesfor n-butane oxidation to maleic anhydride increase withincreasing surface vanadia coverage indicating that adjacentsurface vanadia sites are more efficient in selectively oxidiz-ing n-butane to maleic anhydride than the isolated surfacevanadia sites.45,46 This is not surprising since the oxidationof n-butane to maleic anhydride requires the involvement
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 367
of four oxygen atoms [each surface vanadia site can onlycontribute one oxygen atom since the maximum reductionis from V(V) to V(III), assuming that gas-phase oxygen doesnot directly participate (Mars–van Krevelen mechanism)].Therefore, the specific requirements of the reactant and itcorresponding product, rather than the ratio of polymeric toisolated surface vanadia species, determine the relevance ofthe surface metal oxide coverage.
Support effectThe selectivity to maleic anhydride during n-butane oxi-dation depends strongly on the specific oxide support:Al2O3 > Nb2O5 > TiO2 > SiO2 > ZrO2 > CeO2. This trendparallels the strength of the Lewis acidity of the oxide sup-port. Alumina has the strongest Lewis acid sites, followedby niobia, and the other supports have weak (titania andzirconia) or no Lewis acid sites (silica).52 This observation isconsistent with the critical reaction step involving the bridg-ing V—O—support bond, which is in immediate vicinity ofthe Lewis acid sites of the oxide support.
The TOF values for n-butane oxidation to maleicanhydride45,46 and for the ethane ODH (Oxidative Dehy-drogenation) to ethylene over supported vanadium oxidecatalysts20 also depend strongly on the specific oxide sup-port. Ethane ODH TOF values change by more than an orderof magnitude (TiO2 > ZrO2 >> Al2O3 > Nb2O5 > CeO2 >SiO2) and those for n-butane oxidation change by a factor of50 (TiO2 > CeO2 > ZrO2 > Nb2O5 > Al2O3 > SiO2). Withthe exception of the vanadia/ceria catalytic system, which
undergoes solid-state reaction and decreases the number ofsurface vanadia active sites, the TOF values for n-butaneoxidation increase with decreasing electronegativity of theoxide support (the more basic bridging V—O—S bondsare more efficient in activating the internal C—H bonds).The dependence of the TOF values for n-butane oxidationalso parallels the reducibility trend of the surface vanadiumoxide species on these supports, which is most probably justcoincidental. However, the ethane ODH TOF values showsome deviation from this trend owing to solid-state reactionsbetween the surface vanadia and the ceria and niobia sup-ports at the much higher reaction temperatures required forethane activation. This issue will be discussed further in thesection below on structural transformations under differentenvironments.
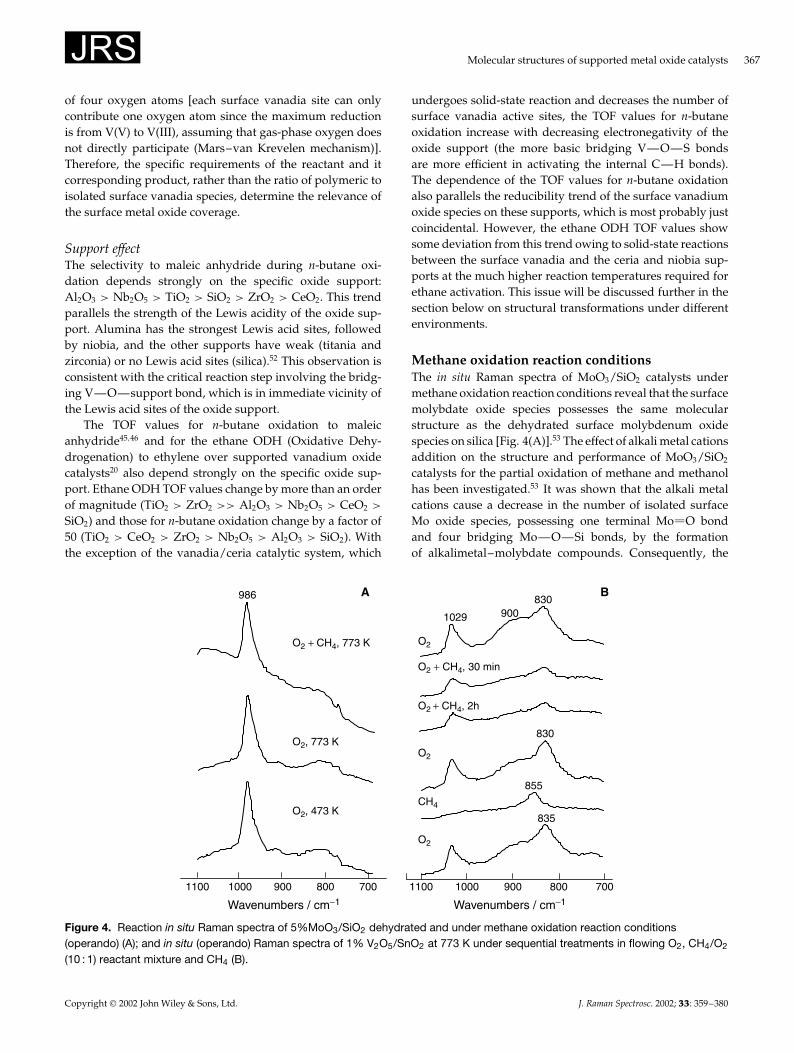
Methane oxidation reaction conditionsThe in situ Raman spectra of MoO3/SiO2 catalysts undermethane oxidation reaction conditions reveal that the surfacemolybdate oxide species possesses the same molecularstructure as the dehydrated surface molybdenum oxidespecies on silica [Fig. 4(A)].53 The effect of alkali metal cationsaddition on the structure and performance of MoO3/SiO2
catalysts for the partial oxidation of methane and methanolhas been investigated.53 It was shown that the alkali metalcations cause a decrease in the number of isolated surfaceMo oxide species, possessing one terminal Mo O bondand four bridging Mo—O—Si bonds, by the formationof alkalimetal–molybdate compounds. Consequently, the
1100 1000 900 800 700
Wavenumbers / cm−1
O2, 773 K
O2, 473 K
O2 + CH4, 773 K
A986
1100 1000 900 800 700
8309001029
Wavenumbers / cm−1
CH4
O2
O2
O2 + CH4, 2h
O2 + CH4, 30 min
O2
B
855
830
835
Figure 4. Reaction in situ Raman spectra of 5%MoO3/SiO2 dehydrated and under methane oxidation reaction conditions(operando) (A); and in situ (operando) Raman spectra of 1% V2O5/SnO2 at 773 K under sequential treatments in flowing O2, CH4/O2
(10 : 1) reactant mixture and CH4 (B).
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
368 M. A. Banares and I. E. Wachs
catalytic activity for methane oxidation to formaldehydeover the MoO3/SiO2 catalyst is decreased since the numberof isolated surface Mo oxide active species is reduced.When both surface vanadium and molybdenum oxidesare supported on silica, both species are isolated and co-exist over the silica support without any interactions andthe catalytic performance is dominated by the more activesurface vanadium oxide species.54
In situ Raman studies of V2O5/SiO2, V2O5/TiO2 andV2O5/SnO2 and V2O5/TiO2/SiO2 demonstrate that thedehydrated surface vanadia species exhibits a sharp Ramanband at ¾1027–1034 cm�1 from the terminal V O bond,while the surface bridging V—O—V polymeric speciesexhibit a broad Raman band ¾900 cm�1 on the SnO2 andTiO2 supports. In addition, the SnO2 system shows a newband near 830 cm�1, associated with a bulk V—Sn—Ocompound.54 Under methane oxidation reaction conditions,the Raman intensities of the surface vanadium oxide speciesdecreases for the supported V2O5/TiO2 and V2O5/SnO2
catalytic systems owing to the reduction of the surfacevanadium oxide species under the reducing methane oxida-tion environment, but no significant changes were observedfor the V2O5/SiO2 and V2O5/TiO2/SiO2 systems. In thecase of the SnO2-supported vanadia catalyst, reductionresulted in a reduced surface V(IV) or V(III) phase char-acterized by a weak and broad Raman band at 855 cm�1
[Fig. 4(B)].Similar to the oxidation of the C2 –C4 alkanes, the
oxygen in the terminal V O bond does not appear tobe the critical site for methane oxidation. Oxygen isotopicstudies show that the terminal V O bond in the V2O5/SiO2
catalyst is too stable under the reaction conditions to bethe kinetically critical active site for activating the C—Hbond in methane.51 However, the catalytic data do showthat the overall methane oxidation activity is dependenton the specific oxide support. The V—O—support bondstrength must vary with the support and is most probablyresponsible for the different catalytic TOF values observedover these supported vanadium oxide catalytic systems.It is also possible that some surface V(V) species will bereduced and provide sites for oxygen adsorption to formelectrophilic oxygen species.55 The population of the reducedsurface vanadia species depend on the specific catalystsupport since the in situ Raman spectra show that thereduced species are not present over the SiO2 support andare much more prevalent over titania and SnO2 supportsunder the reaction conditions employed. The extent ofreduction of the surface vanadium species oxide follows thetrend SnO2 > TiO2 > SiO2, which correlates with the CO2
selectivity and inversely with the formaldehyde selectivity.The CO product arises form the direct decomposition ofthe formaldehyde intermediate product and CO2 arises fromthe direct combustion of methane and further oxidationof CO.
SCR (NO and NH3/ reaction conditionsThe in situ Raman spectra of the supported vanadium oxidecatalysts for the selective catalytic reduction (SCR) of NOwith NH3 reveal that increasing the surface vanadia coverageincreases the ratio of polymerized to isolated surfacevanadia species, the same Raman spectra are obtained underdehydrated and SCR reaction conditions. Corresponding IRstudies, employing NH3 adsorption, demonstrated that thenumber of surface Brønsted acid sites also increases withincreasing surface vanadia coverage. The SCR DeNOx TOFvalues were found to increase with surface vanadia coverage,but the specific redox properties of the surface vanadiaspecies were not affected. The increasing SCR TOF withsurface vanadia coverage reveals that the bimolecular NOand NH3 SCR reaction requires more than one surface site.This suggests that the immediate environment of the surfaceredox site is critical. This could originate from the need fortwo adjacent surface redox sites, a surface redox site adjacentto an acid site or to the higher activity of polymerizedsurface vanadia species relative to that of isolated surfacevanadia species. However, it is not straightforward todiscriminate among these different possibilities from thesurface vanadia coverage effect on the SCR reaction. Theaddition of surface tungsten oxide and niobium oxide speciesto a 1% V2O5/TiO2 catalyst increases the SCR TOF by upto an order of magnitude, with the surface tungsten oxidesomewhat more efficient than the surface niobium oxidespecies. Surface NbOx (Lewis acid) and WOx (Brønsted acid)do not possess redox properties. Thus, the DeNOx reactiondoes not necessarily require two adjacent surface redox sites(isolated or polymeric). Nor does the introduction of theseacidic promoters alter the ratio of polymerized to isolatedsurface vanadia species to any appreciable extent, but theSCR TOF values increase with the introduction of the acidicsurface additives. The presence of surface WOx or NbOx
promoters increases the presence of surface Brønsted orLewis acid sites, respectively. Ammonia initially adsorbson the surface acid sites and reduces the adjacent surfacevanadia species from V(V) to V(III), which creates a vacancyfor the adsorption of NOx [NOx does not adsorb on surfaceV(V) sites]. The adjacent chemisorbed NOx and NHx speciesthen combine to form the desired N2 and H2O reactionproducts. The reduced surface vanadia sites are then rapidlyreoxidized by gas-phase oxygen. Thus, the SCR reaction ismost efficient in the presence of adjacent surface redox andsurface acid sites (WOx > VOx > NbOx).56
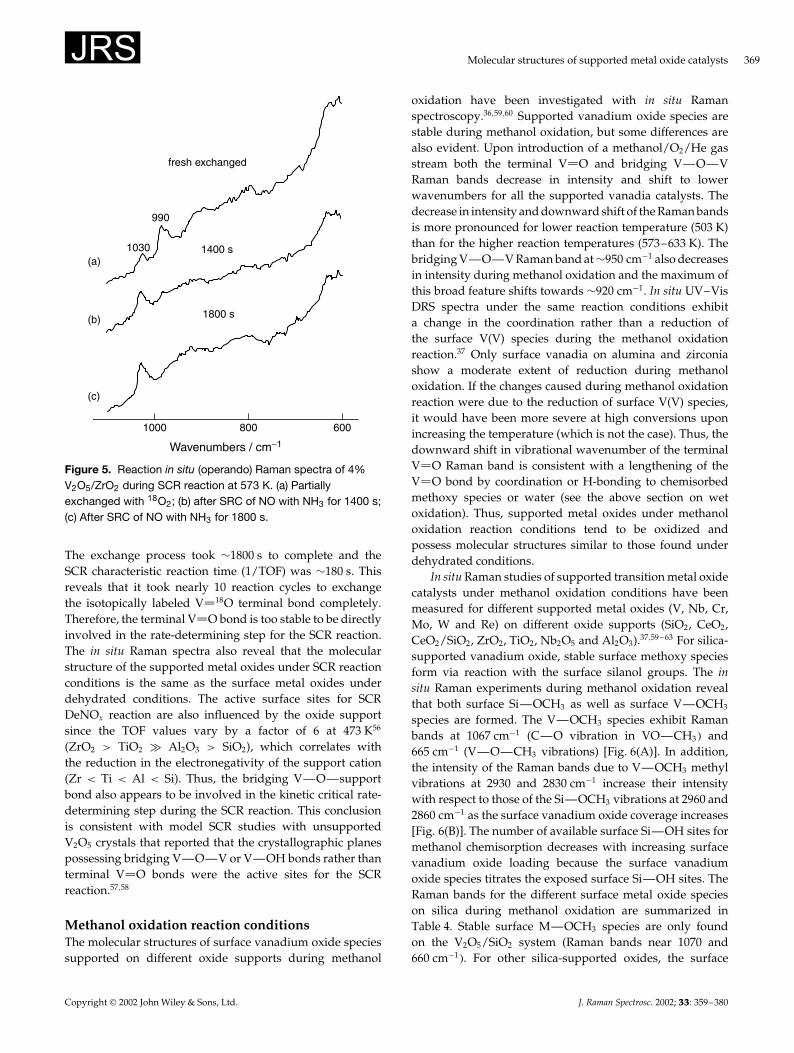
The reaction in situ Raman spectra of 4% V2O5/ZrO2
during the SCR reaction at 573 K are presented in Fig. 5.The catalyst was partially exchanged with oxygen-18 via aseries of successive n-butane reduction and 18O2 oxidationcycles. The heavier mass of oxygen-18 relative to oxygen-16shifts the Raman band from ¾1030 to ¾990 cm�1 (Fig. 5).The time required to exchange the terminal V 18O bond toV 16O during the SCR reaction (containing only 16O2, N16Oand NH3� was monitored by in situ Raman spectroscopy.
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 369
1000 800 600
Wavenumbers / cm−1
fresh exchanged
1400 s
1800 s
1030
990
(a)
(b)
(c)
Figure 5. Reaction in situ (operando) Raman spectra of 4%V2O5/ZrO2 during SCR reaction at 573 K. (a) Partiallyexchanged with 18O2; (b) after SRC of NO with NH3 for 1400 s;(c) After SRC of NO with NH3 for 1800 s.
The exchange process took ¾1800 s to complete and theSCR characteristic reaction time (1/TOF) was ¾180 s. Thisreveals that it took nearly 10 reaction cycles to exchangethe isotopically labeled V 18O terminal bond completely.Therefore, the terminal V O bond is too stable to be directlyinvolved in the rate-determining step for the SCR reaction.The in situ Raman spectra also reveal that the molecularstructure of the supported metal oxides under SCR reactionconditions is the same as the surface metal oxides underdehydrated conditions. The active surface sites for SCRDeNOx reaction are also influenced by the oxide supportsince the TOF values vary by a factor of 6 at 473 K56
(ZrO2 > TiO2 × Al2O3 > SiO2), which correlates withthe reduction in the electronegativity of the support cation(Zr < Ti < Al < Si). Thus, the bridging V—O—supportbond also appears to be involved in the kinetic critical rate-determining step during the SCR reaction. This conclusionis consistent with model SCR studies with unsupportedV2O5 crystals that reported that the crystallographic planespossessing bridging V—O—V or V—OH bonds rather thanterminal V O bonds were the active sites for the SCRreaction.57,58
Methanol oxidation reaction conditionsThe molecular structures of surface vanadium oxide speciessupported on different oxide supports during methanol
oxidation have been investigated with in situ Ramanspectroscopy.36,59,60 Supported vanadium oxide species arestable during methanol oxidation, but some differences arealso evident. Upon introduction of a methanol/O2/He gasstream both the terminal V O and bridging V—O—VRaman bands decrease in intensity and shift to lowerwavenumbers for all the supported vanadia catalysts. Thedecrease in intensity and downward shift of the Raman bandsis more pronounced for lower reaction temperature (503 K)than for the higher reaction temperatures (573–633 K). Thebridging V—O—V Raman band at ¾950 cm�1 also decreasesin intensity during methanol oxidation and the maximum ofthis broad feature shifts towards ¾920 cm�1. In situ UV–VisDRS spectra under the same reaction conditions exhibita change in the coordination rather than a reduction ofthe surface V(V) species during the methanol oxidationreaction.37 Only surface vanadia on alumina and zirconiashow a moderate extent of reduction during methanoloxidation. If the changes caused during methanol oxidationreaction were due to the reduction of surface V(V) species,it would have been more severe at high conversions uponincreasing the temperature (which is not the case). Thus, thedownward shift in vibrational wavenumber of the terminalV O Raman band is consistent with a lengthening of theV O bond by coordination or H-bonding to chemisorbedmethoxy species or water (see the above section on wetoxidation). Thus, supported metal oxides under methanoloxidation reaction conditions tend to be oxidized andpossess molecular structures similar to those found underdehydrated conditions.
In situ Raman studies of supported transition metal oxidecatalysts under methanol oxidation conditions have beenmeasured for different supported metal oxides (V, Nb, Cr,Mo, W and Re) on different oxide supports (SiO2, CeO2,CeO2/SiO2, ZrO2, TiO2, Nb2O5 and Al2O3).37,59 – 63 For silica-supported vanadium oxide, stable surface methoxy speciesform via reaction with the surface silanol groups. The insitu Raman experiments during methanol oxidation revealthat both surface Si—OCH3 as well as surface V—OCH3
species are formed. The V—OCH3 species exhibit Ramanbands at 1067 cm�1 (C—O vibration in VO—CH3� and665 cm�1 (V—O—CH3 vibrations) [Fig. 6(A)]. In addition,the intensity of the Raman bands due to V—OCH3 methylvibrations at 2930 and 2830 cm�1 increase their intensitywith respect to those of the Si—OCH3 vibrations at 2960 and2860 cm�1 as the surface vanadium oxide coverage increases[Fig. 6(B)]. The number of available surface Si—OH sites formethanol chemisorption decreases with increasing surfacevanadium oxide loading because the surface vanadiumoxide species titrates the exposed surface Si—OH sites. TheRaman bands for the different surface metal oxide specieson silica during methanol oxidation are summarized inTable 4. Stable surface M—OCH3 species are only foundon the V2O5/SiO2 system (Raman bands near 1070 and660 cm�1�. For other silica-supported oxides, the surface
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
370 M. A. Banares and I. E. Wachs
A B
633 K (O2/He)
573 K (O2/He)
923 K (O2/He)
1100 900 700
1029
1034
1028
1035
1027
1033
1067
3% V2O5/SiO2
2960
2930
2860
2830
3000 2900 2800
Si-OCH3
V-OCH3
Wavenumbers / cm−1 Wavenumbers / cm−1
665
(O2/CH3OH/He)
(O2/CH3OH/He)
(O2/CH3OH/He)
(d)
(c)
(b)
(a)
x% V2O5/SiO2
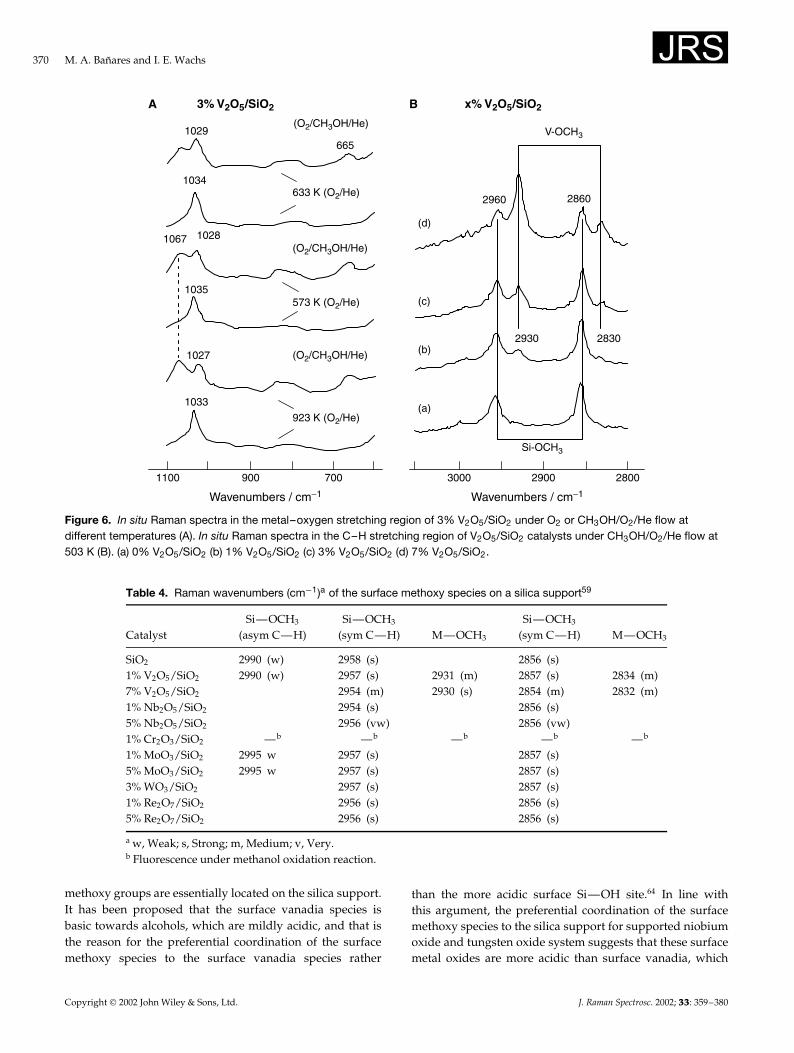
Figure 6. In situ Raman spectra in the metal–oxygen stretching region of 3% V2O5/SiO2 under O2 or CH3OH/O2/He flow atdifferent temperatures (A). In situ Raman spectra in the C–H stretching region of V2O5/SiO2 catalysts under CH3OH/O2/He flow at503 K (B). (a) 0% V2O5/SiO2 (b) 1% V2O5/SiO2 (c) 3% V2O5/SiO2 (d) 7% V2O5/SiO2.
Table 4. Raman wavenumbers (cm�1)a of the surface methoxy species on a silica support59
SiO2 2990 (w) 2958 (s) 2856 (s)1% V2O5/SiO2 2990 (w) 2957 (s) 2931 (m) 2857 (s) 2834 (m)7% V2O5/SiO2 2954 (m) 2930 (s) 2854 (m) 2832 (m)1% Nb2O5/SiO2 2954 (s) 2856 (s)5% Nb2O5/SiO2 2956 (vw) 2856 (vw)1% Cr2O3/SiO2 —b —b —b —b —b
1% MoO3/SiO2 2995 w 2957 (s) 2857 (s)5% MoO3/SiO2 2995 w 2957 (s) 2857 (s)3% WO3/SiO2 2957 (s) 2857 (s)1% Re2O7/SiO2 2956 (s) 2856 (s)5% Re2O7/SiO2 2956 (s) 2856 (s)
a w, Weak; s, Strong; m, Medium; v, Very.b Fluorescence under methanol oxidation reaction.
methoxy groups are essentially located on the silica support.It has been proposed that the surface vanadia species isbasic towards alcohols, which are mildly acidic, and that isthe reason for the preferential coordination of the surfacemethoxy species to the surface vanadia species rather
than the more acidic surface Si—OH site.64 In line withthis argument, the preferential coordination of the surfacemethoxy species to the silica support for supported niobiumoxide and tungsten oxide system suggests that these surfacemetal oxides are more acidic than surface vanadia, which
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 371
agrees with the acidic character of these metal oxides.Furthermore, transient Raman studies have demonstratedthat the surface Si—OCH3 species are not reactive under thecurrent methanol oxidation reaction conditions and are justspectator species.
For V2O5/TiO2, The Ti—OCH3 Raman bands decreasewith increasing surface vanadia coverage and essentiallydisappear at monolayer coverage of surface vanadia specieson titania,65,66 indicating that no V—OCH3 species aredetected in the V2O5/TiO2 system by Raman spectroscopy.NbOx/SiO2 and WOx/SiO2 show the same trend.59 TheZr—OMe Raman vibrations for vanadia/zirconia cata-lysts behave similarly to those of the V2O5/TiO2 catalysts.No information was available for the V2O5/Al2O3 andCr2O3/SiO2 catalysts since the surface methoxy vibrationregion was obscured by fluorescence.36,37,59 However, com-plementary IR spectroscopic measurements demonstratedthe existence of adsorbed V—OCH3 species by the IR bandsat ¾2932 and ¾2832 cm�1.36 Moreover, the surface vanadiamonolayers of TiO2, CeO2, ZrO2 and Al2O3 exhibit onlyV—OCH3 bands and methanol appears to have no accessto exposed support cation sites for the formation of sup-port—OCH3 species, consistent with the formation of acomplete monolayer of surface vanadia on these oxide sup-ports. In contrast, the 10% V2O5/SiO2 catalyst contains bothsurface V—OCH3 and Si—OCH3 bands in the IR spectrumowing to the presence of exposed silica surface sites. There-fore, Raman spectroscopy presents some limitations for the
detection of surface methoxy species since it only detectssurface methoxy bands on silica, V2O5/SiO2, TiO2 and ZrO2
surfaces, with no V—OCH3 bands being detected in theV2O5/TiO2 or V2O5/ZrO2 catalysts. This phenomenon isattributed to a support-induced effect upon the electronicstructure of the surface V—OCH3 species that reduces theRaman scattering cross-section in these catalysts. Conse-quently, in situ IR spectroscopy is a better method for thedetection and quantification of chemisorbed surface methoxyintermediates on the active surface V2O5 sites during steady-state methanol oxidation and in situ Raman is a better methodto study the molecular structural changes of the surface metaloxide species present in supported metal oxide catalysts.
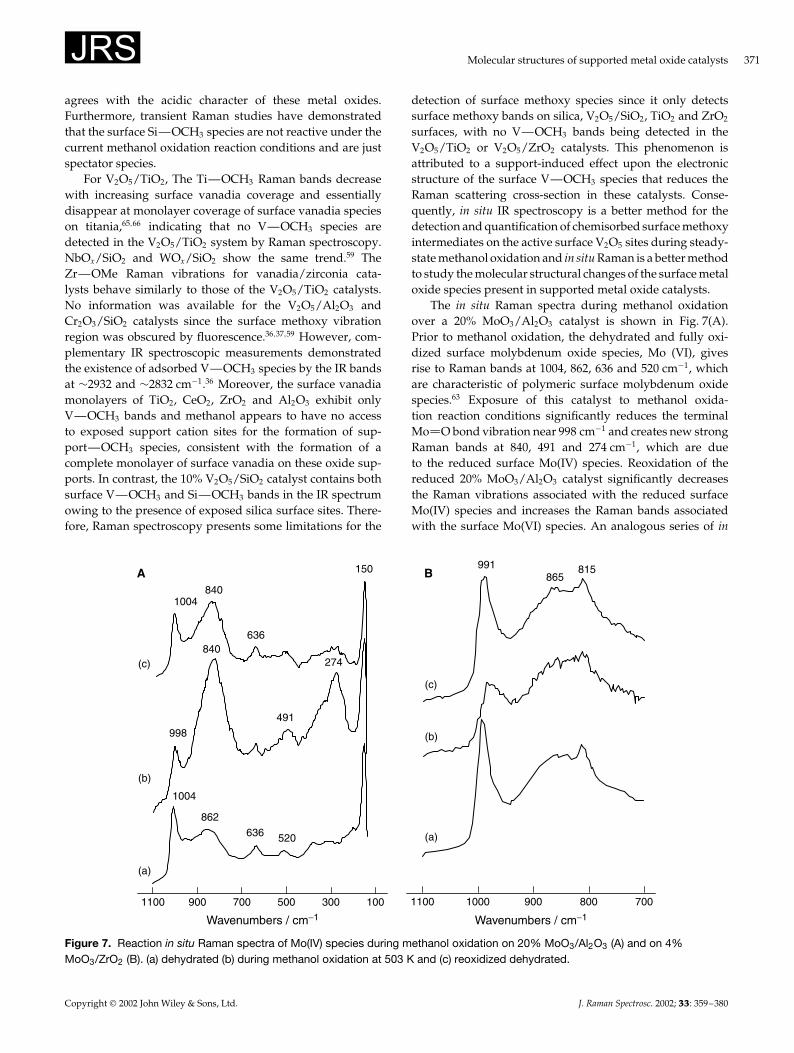
The in situ Raman spectra during methanol oxidationover a 20% MoO3/Al2O3 catalyst is shown in Fig. 7(A).Prior to methanol oxidation, the dehydrated and fully oxi-dized surface molybdenum oxide species, Mo (VI), givesrise to Raman bands at 1004, 862, 636 and 520 cm�1, whichare characteristic of polymeric surface molybdenum oxidespecies.63 Exposure of this catalyst to methanol oxida-tion reaction conditions significantly reduces the terminalMo O bond vibration near 998 cm�1 and creates new strongRaman bands at 840, 491 and 274 cm�1, which are dueto the reduced surface Mo(IV) species. Reoxidation of thereduced 20% MoO3/Al2O3 catalyst significantly decreasesthe Raman vibrations associated with the reduced surfaceMo(IV) species and increases the Raman bands associatedwith the surface Mo(VI) species. An analogous series of in
1100 900 700 500 300 100
1004
862
636 520
150A
998
840
491
274
1004840
636
(a)
(b)
(c)
Wavenumbers / cm−1
1100 1000 900 800 700
B991
865815
(a)
(b)
(c)
Wavenumbers / cm−1
Figure 7. Reaction in situ Raman spectra of Mo(IV) species during methanol oxidation on 20% MoO3/Al2O3 (A) and on 4%MoO3/ZrO2 (B). (a) dehydrated (b) during methanol oxidation at 503 K and (c) reoxidized dehydrated.
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
372 M. A. Banares and I. E. Wachs
situ studies over 4% MoO3/ZrO2 did not show Raman bandscharacteristic of reduced surface molybdenum oxide species[Fig. 7(B)].63 For ZrO2- and TiO2-supported Cr2O3 catalysts,the surface chromate Raman signals disappear upon intro-ducing a reducing CH3OH/He gas feed due to reduction ofCr(VI) to Cr(III). In the case of SiO2- or Al2O3-supported sur-face Cr2O3 species, no Raman bands are observed undera reducing CH3OH/He gas feed owing to fluorescencefrom reduced surface chromium oxide species [especiallythe Cr(III) sites that are known to cause fluorescence].37
REDUCTION CONDITIONS
Exposure of supported metal oxides (V, Cr, Mo) to reducingenvironments or during oxidation reaction conditions maylead to the formation of reduced surface metal oxidespecies that may be directly monitored by in situ Ramanspectroscopy. However, as shown above for methanoloxidation and reduction, it appears that it is generallydifficult to obtain good Raman signals for reduced surfacemetal oxide species of supported metal oxide catalysts. Incontrast, the oxide supports do not appear to be reducedduring the reduction of surface vanadia species. Whilethe average oxidation state of reduced surface vanadiaspecies can be quantitatively determined by temperature-programmed reduction (TPR) and gravimetric measurement,more specific information about the distribution of oxidationstates can be obtained with in situ UV–Vis DRS studies,which are also informative about the chemical environmentof the surface metal oxide sites. However, molecularinformation on the structures of the reduced surface oxide
species is not readily available, but may be obtained by insitu XANES/EXAFS measurements (especially if only oneoxidation state is present under specific conditions).
Reduction by C2 –C4 alkanesIn situ Raman studies of supported chromium oxide speciesunder reducing conditions show that the specific oxidesupport has a pronounced effect. Silica-supported chro-mia catalysts show reduction under n-butane oxidation thatleads to fluorescence and no visible Raman features canbe detected. Zirconia-supported chromia exhibits Ramanbands at 1030 cm�1, characteristic of the terminal Cr Obond of isolated surface chromate species, and the bandsat 1010 and 880 cm�1, characteristic of the terminal Cr Oand bridging Cr—O—Cr bonds of the surface polychro-mate species. Under n-butane oxidation reaction conditions,surface chromium oxide species reduce. The relative inten-sity of the bands at 880 and 1010 cm�1 exhibit a paralleldecrease in intensity, whereas the 1030 cm�1 Raman bandintensity decreases independently and at a slower rate. Itis evident that each surface chromate species possesses dif-ferent reduction properties. Therefore, polymeric surfacechromium oxide species reduce more readily than the iso-lated surface chromium oxide species.
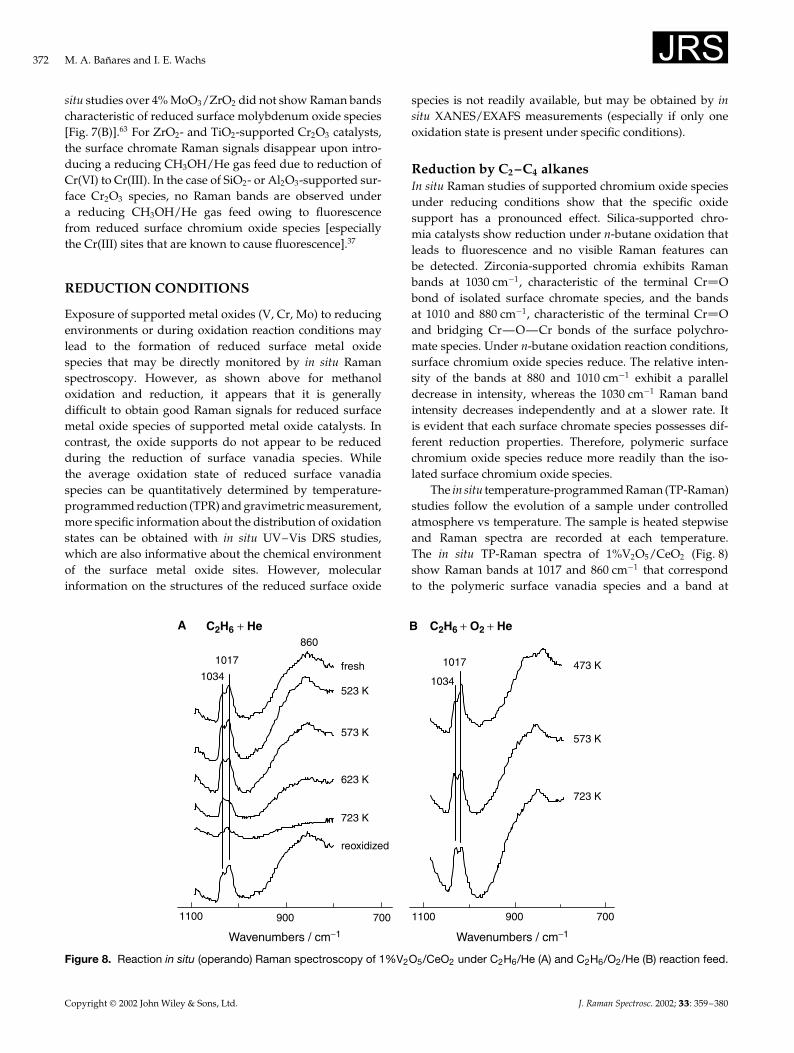
The in situ temperature-programmed Raman (TP-Raman)studies follow the evolution of a sample under controlledatmosphere vs temperature. The sample is heated stepwiseand Raman spectra are recorded at each temperature.The in situ TP-Raman spectra of 1%V2O5/CeO2 (Fig. 8)show Raman bands at 1017 and 860 cm�1 that correspondto the polymeric surface vanadia species and a band at
7009001100
473 K
573 K
723 K
C2H6 + O2 + HeB
1017
1034
Wavenumbers / cm−1
7009001100
reoxidized
523 K
573 K
623 K
723 K
fresh
C2H6 + HeA
1017
1034
860
Wavenumbers / cm−1
Figure 8. Reaction in situ (operando) Raman spectroscopy of 1%V2O5/CeO2 under C2H6/He (A) and C2H6/O2/He (B) reaction feed.
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 373
1034 cm�1 corresponding to isolated surface vanadia sites.Under a reducing atmosphere (C2H6 C He), the polymericsurface vanadia species are more easily reduced thanthe isolated surface vanadium sites, as reflected by thepreferential decrease of the two Raman bands characteristicof polymeric surface vanadia species during the TP-Raman experiment [Fig. 8(A)]. Under reaction conditions(C2H6 C O2 C He), however, only a small fraction of thesurface vanadia sites were reduced since at 723 K the Ramanspectrum remains very similar to the Raman spectrum ofthe fresh, dehydrated sample [Fig. 8(B)]. In situ UV–VisDRS spectroscopy measures no significant reduction forthe silica- and alumina-supported vanadia catalysts duringethane oxidation.47 The 1% V2O5/ZrO2 catalyst shows aslight vanadia reduction during ethane oxidation and only5.5% reduction is observed during ethane reduction. Thus,the average oxidation state under steady-state oxidationconditions does not rely on the reducibility of the catalyst,but on the equilibrium of the reduction and reoxidationrates of the catalytic cycle. UV–Vis DRS spectroscopy is alsosensitive to the degree of polymerization of surface vanadiaspecies.50 During ethane oxidation, the edge energy valuesare essentially unchanged on silica- or alumina-supportedvanadia, but increase on more reducible supports likezirconia, which indicates preferential reduction of the surfacepolymerized species. The polymerization degree of thesurface V(V) cations increases from the 1% to 4% V2O5/ZrO2
catalysts owing to the higher surface vanadia density.47 Thepolymerized surface vanadia species on 4% V2O5/ZrO2 aremore easily reduced than the 1% V2O5/ZrO2 under ethanereduction conditions at 723 K (23.0% vs 5.5% reduction). Thein situ TP-Raman spectra of 1% V2O5/CeO2 provide furtherevidence for the preferential reduction of polymeric surfacevanadia species over the isolated surface vanadia species.
The extent of reduction of supported metal oxidecatalysts is dependent on the hydrocarbon reactivity. In situtemperature-programmed reduction Raman (TPR-Raman)studies consist of a stepwise temperature-programmedreduction and Raman spectra are acquired at every step. Insitu TPR-Raman experiments with 17.5% V2O5/Al2O3 underreducing C4H10/He feed shows the complete disappearanceof the Raman bands of the surface vanadium oxide speciesat 623 K.45 However, 15% V2O5/Al2O3 under a reducingC2H6/He gas feed shows a very strong decrease in the Ramanbands at 803 K, but not complete reduction.47 However, itis important to stress that even when surface polymericmetal oxide species are more reducible than the isolatedsurface metal oxide species, there is not much differencebetween the extent of reduction of surface metal oxidemonomers and polymers during steady-state hydrocarbonoxidation reactions. The average oxidation state duringcatalytic operation depends on the balance between thereduction by the hydrocarbon molecule and the reoxidationby gas-phase molecular oxygen, and it does appear thatreoxidation is faster than reduction. Therefore, supported
metal oxide catalysts are essentially oxidized during catalyticoperation and exhibit a zero-order dependence on the oxygenpartial pressure. Furthermore, oxygen isotopic switchingexperiments67 and comparative temperature-programmedreaction spectroscopy (TPRS) experiments with and withoutoxygen48 demonstrate that the surface lattice oxygen, and notgas-phase oxygen, is involved in these oxidation reactionsvia a Mars–van Krevelen mechanism.
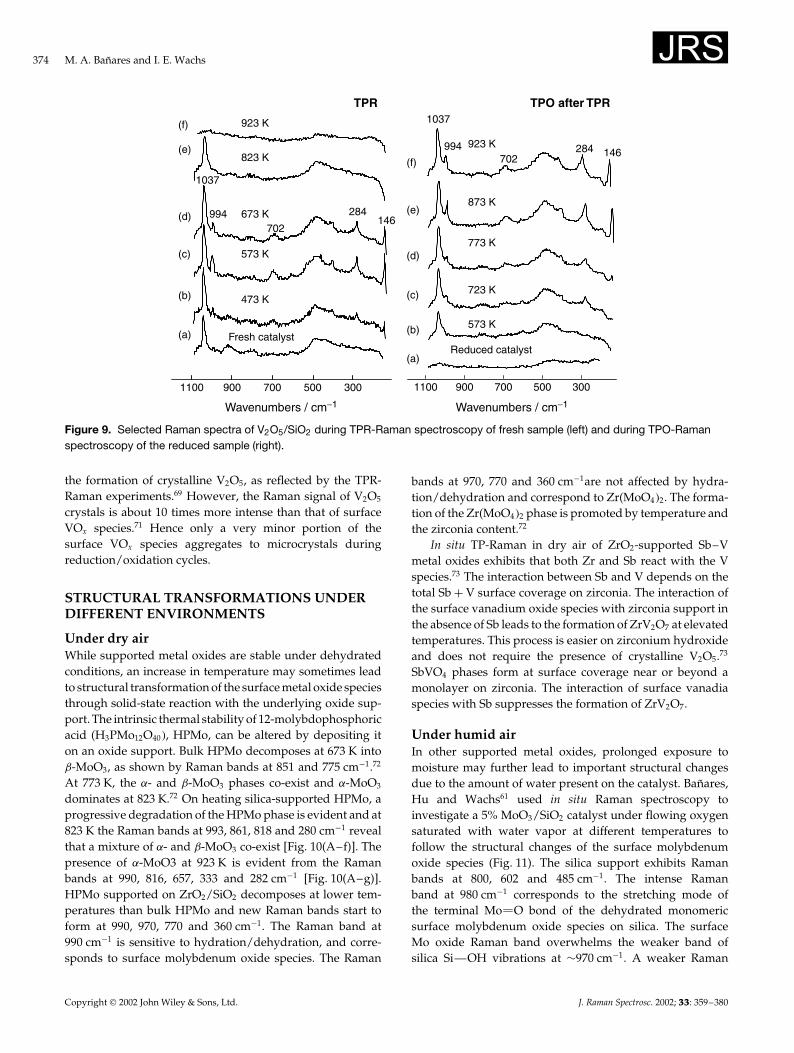
Reduction in hydrogen and reoxidationIn general, it appears that supported vanadia and chromiacatalysts show no new Raman bands for their reducedsurface metal oxide species.58,68 However, the structuralchanges taking place upon reduction of supported metaloxides may influence the structure of the reoxidized metaloxide phase. Thus, in situ TPR-Raman studies69 demonstratethe dynamic states of silica-supported vanadium oxidespecies. During the TPR of 0.9% V2O5/SiO2 (¾30% ofmaximum surface coverage for this specific silica), the Ramanintensity of the terminal V O bond of the surface vanadiumoxide species at 1037 cm�1 decreases monotonically with H2
consumption during the experiments. Upon reoxidation, the0.9% V2O5/SiO2 catalyst exhibits only the Raman band of theoriginal surface vanadia species.69 However, a sample with2.4% V2O5/SiO2, close to the maximum coverage of surfacevanadium oxide for this specific silica, undergoes structuralmodification upon hydrogen reduction and reoxidation(Fig. 9). At 473 K, during H2-TPR, new Raman bands at994, 702, 284 and 146 cm�1 appear which are characteristicof crystalline V2O5. As the temperature increases during theTPR, the Raman bands of crystalline V2O5 become moreevident and then completely disappear owing to reductionof V2O5 to V2O3 (black phase with extremely low Ramanscattering signal). The Raman band of the isolated surfacevanadium oxide species at 1037 cm�1 persists up to 823 K,which reflects its stability as surface V(V) species. TheTPO (Temperature Programmed Oxidation)-Raman spectrademonstrate the formation of both crystalline V2O5 andisolated surface vanadium oxide species. Therefore, near themaximum surface coverage, the V2O5/SiO2 catalyst doesnot completely undergo a reversible change modificationduring the redox cycles. Under reducing conditions, thesurface vanadium oxide species reduce and produce no newRaman signal, and therefore no information is available aboutthe reduced surface vanadium oxide species [quantitativeH2 consumption demonstrate that it is reduced to surfaceV(III) species]. However, as the surface vanadia coverageincreases, it is more likely to compensate for O removalby sharing oxygen sites among the surface vanadiumoxide species. Raman, XANES and 51V-NMR studies70 showthat surface vanadia on silica may exist in two extremeconfigurations under dehydrated conditions, isolated surfacevanadia species and crystalline V2O5, but not as polymericsurface vanadia species. Thus, any effect that promotesinteraction among surface vanadia species must lead to
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
374 M. A. Banares and I. E. Wachs
3005007009001100
573 K
723 K
773 K
873 K
923 K
Reduced catalyst
TPO after TPR
3005007009001100
Wavenumbers / cm−1
923 K
823 K
673 K
573 K
473 K
Fresh catalyst
TPR
(a)
(b)
(c)
(d)
(e)
(f)
(a)
(b)
(c)
(d)
(e)
(f)
1037
994702
284146
1037
994702
284 146
Wavenumbers / cm−1
Figure 9. Selected Raman spectra of V2O5/SiO2 during TPR-Raman spectroscopy of fresh sample (left) and during TPO-Ramanspectroscopy of the reduced sample (right).
the formation of crystalline V2O5, as reflected by the TPR-Raman experiments.69 However, the Raman signal of V2O5
crystals is about 10 times more intense than that of surfaceVOx species.71 Hence only a very minor portion of thesurface VOx species aggregates to microcrystals duringreduction/oxidation cycles.
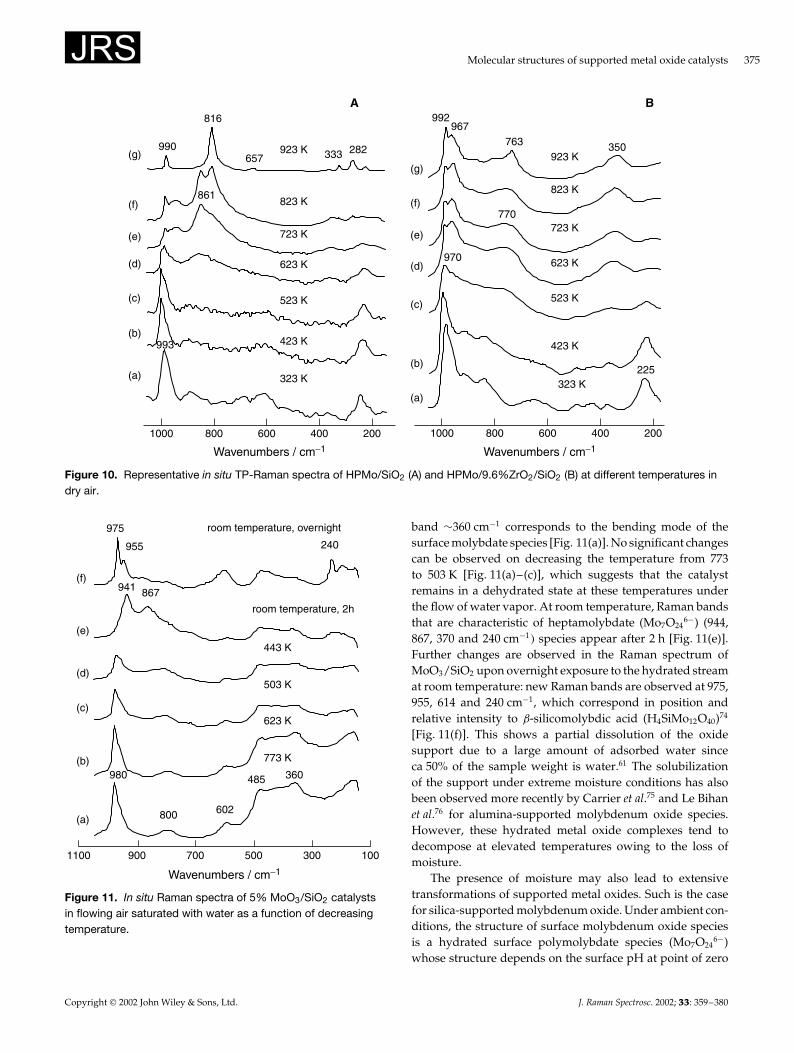
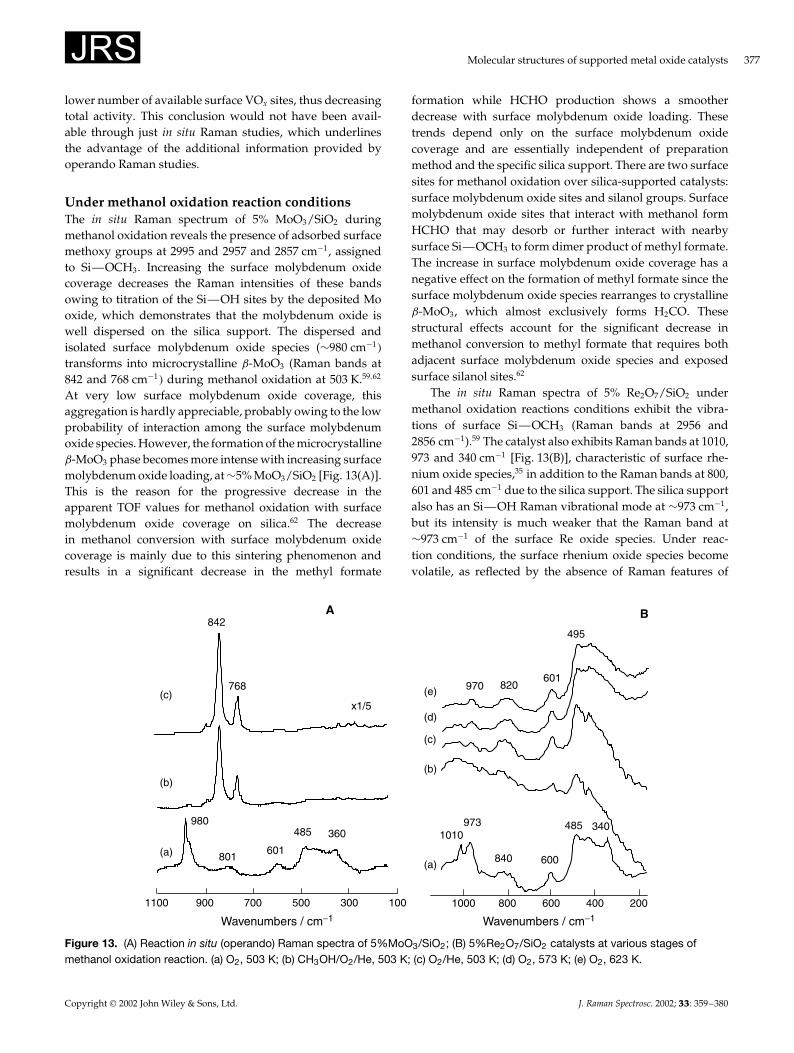
Under dry airWhile supported metal oxides are stable under dehydratedconditions, an increase in temperature may sometimes leadto structural transformation of the surface metal oxide speciesthrough solid-state reaction with the underlying oxide sup-port. The intrinsic thermal stability of 12-molybdophosphoricacid (H3PMo12O40�, HPMo, can be altered by depositing iton an oxide support. Bulk HPMo decomposes at 673 K intoˇ-MoO3, as shown by Raman bands at 851 and 775 cm�1.72
At 773 K, the ˛- and ˇ-MoO3 phases co-exist and ˛-MoO3
dominates at 823 K.72 On heating silica-supported HPMo, aprogressive degradation of the HPMo phase is evident and at823 K the Raman bands at 993, 861, 818 and 280 cm�1 revealthat a mixture of ˛- and ˇ-MoO3 co-exist [Fig. 10(A–f)]. Thepresence of ˛-MoO3 at 923 K is evident from the Ramanbands at 990, 816, 657, 333 and 282 cm�1 [Fig. 10(A–g)].HPMo supported on ZrO2/SiO2 decomposes at lower tem-peratures than bulk HPMo and new Raman bands start toform at 990, 970, 770 and 360 cm�1. The Raman band at990 cm�1 is sensitive to hydration/dehydration, and corre-sponds to surface molybdenum oxide species. The Raman
bands at 970, 770 and 360 cm�1are not affected by hydra-tion/dehydration and correspond to Zr(MoO4�2. The forma-tion of the Zr(MoO4�2 phase is promoted by temperature andthe zirconia content.72
In situ TP-Raman in dry air of ZrO2-supported Sb–Vmetal oxides exhibits that both Zr and Sb react with the Vspecies.73 The interaction between Sb and V depends on thetotal Sb C V surface coverage on zirconia. The interaction ofthe surface vanadium oxide species with zirconia support inthe absence of Sb leads to the formation of ZrV2O7 at elevatedtemperatures. This process is easier on zirconium hydroxideand does not require the presence of crystalline V2O5.73
SbVO4 phases form at surface coverage near or beyond amonolayer on zirconia. The interaction of surface vanadiaspecies with Sb suppresses the formation of ZrV2O7.
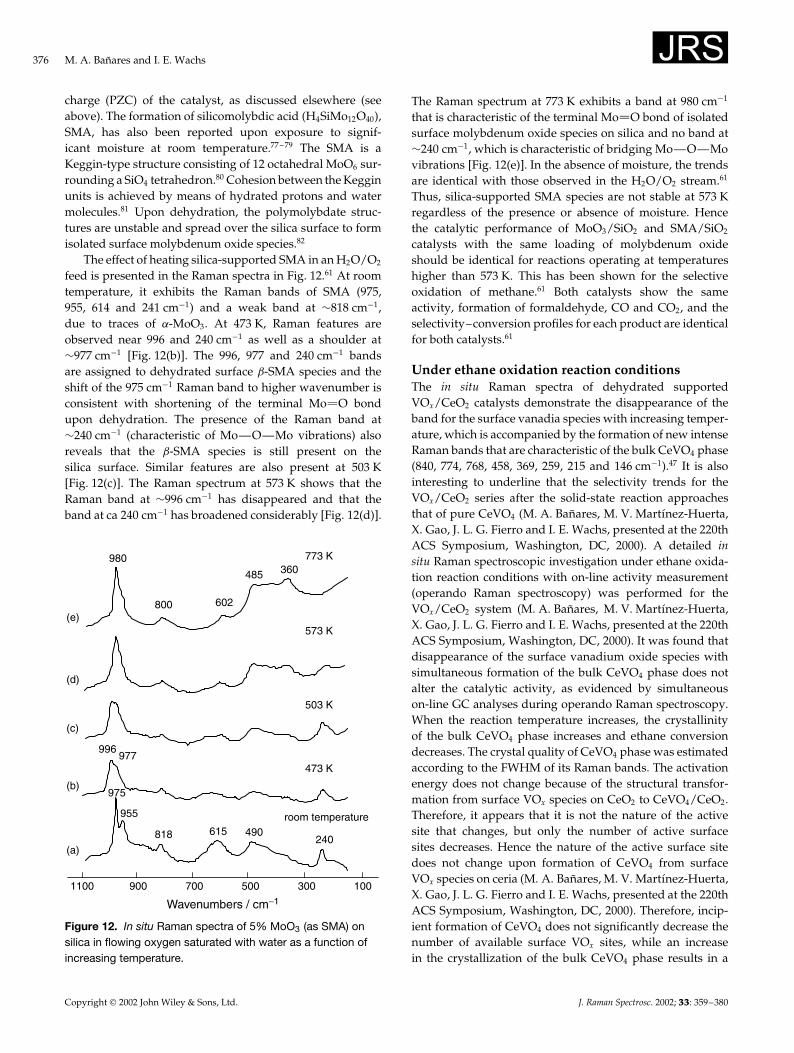
Under humid airIn other supported metal oxides, prolonged exposure tomoisture may further lead to important structural changesdue to the amount of water present on the catalyst. Banares,Hu and Wachs61 used in situ Raman spectroscopy toinvestigate a 5% MoO3/SiO2 catalyst under flowing oxygensaturated with water vapor at different temperatures tofollow the structural changes of the surface molybdenumoxide species (Fig. 11). The silica support exhibits Ramanbands at 800, 602 and 485 cm�1. The intense Ramanband at 980 cm�1 corresponds to the stretching mode ofthe terminal Mo O bond of the dehydrated monomericsurface molybdenum oxide species on silica. The surfaceMo oxide Raman band overwhelms the weaker band ofsilica Si—OH vibrations at ¾970 cm�1. A weaker Raman
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 375
A B
323 K 323 K
423 K423 K
523 K 523 K
623 K623 K
723 K723 K
823 K823 K
923 K923 K
Wavenumbers / cm−1 Wavenumbers / cm−1
(g)
(f)
(e)
(d)
(c)
(b)
(a)
(g)
(f)
(e)
(d)
(c)
(b)
(a)
1000 800 600 400 200 1000 800 600 400 200
990
816
657282333
861
993
970
770
992967
763 350
225
Figure 10. Representative in situ TP-Raman spectra of HPMo/SiO2 (A) and HPMo/9.6%ZrO2/SiO2 (B) at different temperatures indry air.
975
955
602800
240
867941
980
1100 700 500 300 100900
Wavenumbers / cm−1
773 K
623 K
503 K
443 K
room temperature, 2h
(f)
(e)
(d)
(c)
(b)
(a)
485 360
room temperature, overnight
Figure 11. In situ Raman spectra of 5% MoO3/SiO2 catalystsin flowing air saturated with water as a function of decreasingtemperature.
band ¾360 cm�1 corresponds to the bending mode of thesurface molybdate species [Fig. 11(a)]. No significant changescan be observed on decreasing the temperature from 773to 503 K [Fig. 11(a)–(c)], which suggests that the catalystremains in a dehydrated state at these temperatures underthe flow of water vapor. At room temperature, Raman bandsthat are characteristic of heptamolybdate (Mo7O24
6�) (944,867, 370 and 240 cm�1� species appear after 2 h [Fig. 11(e)].Further changes are observed in the Raman spectrum ofMoO3/SiO2 upon overnight exposure to the hydrated streamat room temperature: new Raman bands are observed at 975,955, 614 and 240 cm�1, which correspond in position andrelative intensity to ˇ-silicomolybdic acid (H4SiMo12O40)74
[Fig. 11(f)]. This shows a partial dissolution of the oxidesupport due to a large amount of adsorbed water sinceca 50% of the sample weight is water.61 The solubilizationof the support under extreme moisture conditions has alsobeen observed more recently by Carrier et al.75 and Le Bihanet al.76 for alumina-supported molybdenum oxide species.However, these hydrated metal oxide complexes tend todecompose at elevated temperatures owing to the loss ofmoisture.
The presence of moisture may also lead to extensivetransformations of supported metal oxides. Such is the casefor silica-supported molybdenum oxide. Under ambient con-ditions, the structure of surface molybdenum oxide speciesis a hydrated surface polymolybdate species (Mo7O24
6�)whose structure depends on the surface pH at point of zero
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
376 M. A. Banares and I. E. Wachs
charge (PZC) of the catalyst, as discussed elsewhere (seeabove). The formation of silicomolybdic acid (H4SiMo12O40),SMA, has also been reported upon exposure to signif-icant moisture at room temperature.77 – 79 The SMA is aKeggin-type structure consisting of 12 octahedral MoO6 sur-rounding a SiO4 tetrahedron.80 Cohesion between the Kegginunits is achieved by means of hydrated protons and watermolecules.81 Upon dehydration, the polymolybdate struc-tures are unstable and spread over the silica surface to formisolated surface molybdenum oxide species.82
The effect of heating silica-supported SMA in an H2O/O2
feed is presented in the Raman spectra in Fig. 12.61 At roomtemperature, it exhibits the Raman bands of SMA (975,955, 614 and 241 cm�1) and a weak band at ¾818 cm�1,due to traces of ˛-MoO3. At 473 K, Raman features areobserved near 996 and 240 cm�1 as well as a shoulder at¾977 cm�1 [Fig. 12(b)]. The 996, 977 and 240 cm�1 bandsare assigned to dehydrated surface ˇ-SMA species and theshift of the 975 cm�1 Raman band to higher wavenumber isconsistent with shortening of the terminal Mo O bondupon dehydration. The presence of the Raman band at¾240 cm�1 (characteristic of Mo—O—Mo vibrations) alsoreveals that the ˇ-SMA species is still present on thesilica surface. Similar features are also present at 503 K[Fig. 12(c)]. The Raman spectrum at 573 K shows that theRaman band at ¾996 cm�1 has disappeared and that theband at ca 240 cm�1 has broadened considerably [Fig. 12(d)].
1100 900 700 500 300 100
Wavenumbers / cm−1
room temperature
473 K
503 K
573 K
773 K
975
955
818 615 490240
996977
980
602
485 360
(e)
(d)
(c)
(b)
(a)
800
Figure 12. In situ Raman spectra of 5% MoO3 (as SMA) onsilica in flowing oxygen saturated with water as a function ofincreasing temperature.
The Raman spectrum at 773 K exhibits a band at 980 cm�1
that is characteristic of the terminal Mo O bond of isolatedsurface molybdenum oxide species on silica and no band at¾240 cm�1, which is characteristic of bridging Mo—O—Movibrations [Fig. 12(e)]. In the absence of moisture, the trendsare identical with those observed in the H2O/O2 stream.61
Thus, silica-supported SMA species are not stable at 573 Kregardless of the presence or absence of moisture. Hencethe catalytic performance of MoO3/SiO2 and SMA/SiO2
catalysts with the same loading of molybdenum oxideshould be identical for reactions operating at temperatureshigher than 573 K. This has been shown for the selectiveoxidation of methane.61 Both catalysts show the sameactivity, formation of formaldehyde, CO and CO2, and theselectivity–conversion profiles for each product are identicalfor both catalysts.61
Under ethane oxidation reaction conditionsThe in situ Raman spectra of dehydrated supportedVOx/CeO2 catalysts demonstrate the disappearance of theband for the surface vanadia species with increasing temper-ature, which is accompanied by the formation of new intenseRaman bands that are characteristic of the bulk CeVO4 phase(840, 774, 768, 458, 369, 259, 215 and 146 cm�1).47 It is alsointeresting to underline that the selectivity trends for theVOx/CeO2 series after the solid-state reaction approachesthat of pure CeVO4 (M. A. Banares, M. V. Martınez-Huerta,X. Gao, J. L. G. Fierro and I. E. Wachs, presented at the 220thACS Symposium, Washington, DC, 2000). A detailed insitu Raman spectroscopic investigation under ethane oxida-tion reaction conditions with on-line activity measurement(operando Raman spectroscopy) was performed for theVOx/CeO2 system (M. A. Banares, M. V. Martınez-Huerta,X. Gao, J. L. G. Fierro and I. E. Wachs, presented at the 220thACS Symposium, Washington, DC, 2000). It was found thatdisappearance of the surface vanadium oxide species withsimultaneous formation of the bulk CeVO4 phase does notalter the catalytic activity, as evidenced by simultaneouson-line GC analyses during operando Raman spectroscopy.When the reaction temperature increases, the crystallinityof the bulk CeVO4 phase increases and ethane conversiondecreases. The crystal quality of CeVO4 phase was estimatedaccording to the FWHM of its Raman bands. The activationenergy does not change because of the structural transfor-mation from surface VOx species on CeO2 to CeVO4/CeO2.Therefore, it appears that it is not the nature of the activesite that changes, but only the number of active surfacesites decreases. Hence the nature of the active surface sitedoes not change upon formation of CeVO4 from surfaceVOx species on ceria (M. A. Banares, M. V. Martınez-Huerta,X. Gao, J. L. G. Fierro and I. E. Wachs, presented at the 220thACS Symposium, Washington, DC, 2000). Therefore, incip-ient formation of CeVO4 does not significantly decrease thenumber of available surface VOx sites, while an increasein the crystallization of the bulk CeVO4 phase results in a
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 377
lower number of available surface VOx sites, thus decreasingtotal activity. This conclusion would not have been avail-able through just in situ Raman studies, which underlinesthe advantage of the additional information provided byoperando Raman studies.
Under methanol oxidation reaction conditionsThe in situ Raman spectrum of 5% MoO3/SiO2 duringmethanol oxidation reveals the presence of adsorbed surfacemethoxy groups at 2995 and 2957 and 2857 cm�1, assignedto Si—OCH3. Increasing the surface molybdenum oxidecoverage decreases the Raman intensities of these bandsowing to titration of the Si—OH sites by the deposited Mooxide, which demonstrates that the molybdenum oxide iswell dispersed on the silica support. The dispersed andisolated surface molybdenum oxide species (¾980 cm�1�transforms into microcrystalline ˇ-MoO3 (Raman bands at842 and 768 cm�1� during methanol oxidation at 503 K.59,62
At very low surface molybdenum oxide coverage, thisaggregation is hardly appreciable, probably owing to the lowprobability of interaction among the surface molybdenumoxide species. However, the formation of the microcrystallineˇ-MoO3 phase becomes more intense with increasing surfacemolybdenum oxide loading, at ¾5% MoO3/SiO2 [Fig. 13(A)].This is the reason for the progressive decrease in theapparent TOF values for methanol oxidation with surfacemolybdenum oxide coverage on silica.62 The decreasein methanol conversion with surface molybdenum oxidecoverage is mainly due to this sintering phenomenon andresults in a significant decrease in the methyl formate
formation while HCHO production shows a smootherdecrease with surface molybdenum oxide loading. Thesetrends depend only on the surface molybdenum oxidecoverage and are essentially independent of preparationmethod and the specific silica support. There are two surfacesites for methanol oxidation over silica-supported catalysts:surface molybdenum oxide sites and silanol groups. Surfacemolybdenum oxide sites that interact with methanol formHCHO that may desorb or further interact with nearbysurface Si—OCH3 to form dimer product of methyl formate.The increase in surface molybdenum oxide coverage has anegative effect on the formation of methyl formate since thesurface molybdenum oxide species rearranges to crystallineˇ-MoO3, which almost exclusively forms H2CO. Thesestructural effects account for the significant decrease inmethanol conversion to methyl formate that requires bothadjacent surface molybdenum oxide species and exposedsurface silanol sites.62
The in situ Raman spectra of 5% Re2O7/SiO2 undermethanol oxidation reactions conditions exhibit the vibra-tions of surface Si—OCH3 (Raman bands at 2956 and2856 cm�1).59 The catalyst also exhibits Raman bands at 1010,973 and 340 cm�1 [Fig. 13(B)], characteristic of surface rhe-nium oxide species,35 in addition to the Raman bands at 800,601 and 485 cm�1 due to the silica support. The silica supportalso has an Si—OH Raman vibrational mode at ¾973 cm�1,but its intensity is much weaker that the Raman band at¾973 cm�1 of the surface Re oxide species. Under reac-tion conditions, the surface rhenium oxide species becomevolatile, as reflected by the absence of Raman features of
1000 800 600
Wavenumbers / cm−1
400 200
1010973
600840
485 340
970 820
(a)
(b)
(c)
(d)
(e)
B
601
495
1003005007009001100
x1/5
842
768
980
801
485
A
(c)
(b)
(a) 601
360
Wavenumbers / cm−1
Figure 13. (A) Reaction in situ (operando) Raman spectra of 5%MoO3/SiO2; (B) 5%Re2O7/SiO2 catalysts at various stages ofmethanol oxidation reaction. (a) O2, 503 K; (b) CH3OH/O2/He, 503 K; (c) O2/He, 503 K; (d) O2, 573 K; (e) O2, 623 K.
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
378 M. A. Banares and I. E. Wachs
the surface rhenium oxide species after reoxidation of thesamples [Fig. 13(B-c)].59
The mobility and volatility of surface Mo and Re oxidesare enhanced by the formation of metal oxide–methoxycomplexes. The Raman bands for the terminal M O bondsdecrease in intensity and shift to lower wavenumbersduring methanol oxidation.35,59 The mobility of the surfacemolybdenum oxide and the volatility of the rhenium oxidespecies on the silica support are attributed to the formationof M—OCH3 complexes. These complexes result in thevolatilization of the surface rhenium oxide species and thecrystallization of the surface molybdenum oxide species. TheMo—OCH3 species were also somewhat volatile becauseMo deposits were detected on the glass portion of thereactor exit.62 The strong interaction of surface molybdenumoxide species with methanol that leads to its structuralrearrangement into microcrystalline ˇ-MoO3 also affects thestability of silica-supported silicomolybdic acid, which at573 K under methanol oxidation reaction conditions formsˇ-MoO3.61
CONCLUSIONS
Raman spectroscopy is a very versatile molecular charac-terization technique for studying supported metal oxidecatalysts under different environments. Dehydrated sup-ported metal oxide species consist of metal cations sur-rounded by oxygen ions, one of which is a terminalM O bond and the others bridge the cations to the oxidesupport (M—O—support) or to adjacent surface cations(M—O—M). Exposure to moisture leads to coordination ofwater, which under reaction conditions may compete withreacting molecules. At high temperature, moisture read-ily exchanges oxygens in the supported metal oxides. Theinteraction with moisture becomes more pronounced as thetemperature decreases. At ambient temperature, this inter-action leads to the solvation and even the dissolution of thesupported oxides. Under ambient conditions, the molecu-lar structures of the hydrated supported metal oxides aredetermined by their chemistry in aqueous solutions, whichdepends on their concentration and solution pH. The pHvalue corresponds to the PZC (point of zero charge). ThepH at PZC value is determined by (1) the specific oxidesupport, (2) the specific active supported metal oxide and(3) the surface coverage of the active supported metal oxide.The presence of extensive water may also lead to a partialdissolution of the oxide support cation.61,75,76
Dehydrated polymeric surface metal oxide species arepreferentially reduced over the corresponding dehydratedsurface monomeric species. Under reaction conditions,however, the average oxidation state does not depend onthe reducibility of the surface oxide but on the balancebetween reduction and reoxidation rates during the catalyticprocess. The catalytic reactivity of the supported metal oxidecatalysts does not appear to be associated with the terminal
M O bond, which is too stable to be directly involved inthe kinetically critical rate-determining step. The bridgingoxygen sites among surface metal oxide cations, M—O—M,do not appear to have a significant role since the TOF valuesare not affected by the coverage of the active surface metaloxide species. Several independent factors strongly suggestthat the V—O—support bridging oxygen is the kineticallycritical active site during oxidation reactions since changingthe oxide support ligand can influence the TOF values byorders of magnitude. In some cases, as in the oxidation ofn-butane to maleic anhydride, surface coverage does havea strong effect and the TOF values increase, but this isrelated to a geometric requirement of several sites in closevicinity for an efficient activation of the n-butane moleculeand its oxidation by four oxygen atoms to yield maleicanhydride. Similarly, the bimolecular reaction between NOx
and NH3 during SCR requires two adjacent surface acidand redox sites and the TOF value increases with surfacemetal oxide coverage. However, for unimolecular oxidationreactions that require only the consumption of one surfaceoxygen atom, only one surface active metal oxide site appearsto be required (methanol to formaldehyde,59,60,63 methaneto formaldehyde,53,54 ethane to ethylene,20,38,47 propane topropylene,48,49 1-butene to butadiene83 and SO2 to SO3
84).A complete fundamental understanding of the structural
changes of supported metal oxides under different environ-ments cannot always be fully understood just through in situRaman spectroscopy, but requires the application of comple-mentary spectroscopic characterization techniques. Also, theincorporation of simultaneous catalytic activity and selec-tivity measurement with the spectroscopic studies underreaction conditions (operando spectroscopy) provides keyinformation to be able to understand more fully the struc-ture–activity/selectivity relationship at a molecular level forsupported metal oxide catalysts.
The performance of bulk mixed-metal oxides must bedetermined by the nature of the outermost surface layer,directly exposed to reacting molecules rather than by thebulk structure. Recent studies have revealed that the surfacesof bulk mixed molybdates are covered by monolayers ofsurface Mo oxide.85,86 Therefore, we are now at the dawnof a new approach to understanding fundamentally themolecular structure–activity/selectivity relationship of bulkmixed-metal oxide catalysts, since the knowledge developedfor supported metal oxides in the past decade can be directlyapplied to the molecular-level understanding of bulk mixed-metal oxide catalysts.
AcknowledgementsThe financial support of the US National Science Foundation (NSF),grant CTS-9901643, US Department of Energy (DOE), Basic EnergySciences, grant DE-FG02-93ER14350, and CICYT, Spain (ProjectsQUI98-0784 and IN96-0053) for this collective work is greatlyappreciated.
The authors also thank all the individuals in their laboratoriesat Lehigh University and the Institute of Catalysis and Petroleum
Copyright 2002 John Wiley & Sons, Ltd. J. Raman Spectrosc. 2002; 33: 359–380
Molecular structures of supported metal oxide catalysts 379
Chemistry who have made significant contributions to the advance-ment of Raman spectroscopy of catalysis; without their contributionswe would not possess all the fundamental knowledge that we havetoday: Frank D. Hardcastle, Jih-Mirn Jehng, Goutam Deo, Dou-soung Kim, Michael A. Vuurman, Andrzej M. Turek, HangchunHu, Xingtao Gao, Bert Weckhuysen, Vadim Guliants, Loyd Bur-cham, Chuan-Bao Wang, Yeping Cai, M. Victoria Martınez-Huertaand M. Olga Guerrero-Perez.
REFERENCES1. Thomas JM, Thomas WJ. Principles and Practice of Heterogeneous
Catalysis. VCH: New York, 1997.2. Wachs IE. In Handbook of Spectroscopy, Lewis IR, Edwards HGM
(eds). Marcel Dekker: New York, 2001; chapt. 20, 799–833.3. Bartlett JR, Cooney RP. In Spectroscopy of Inorganic-Based
Materials, Clark RJH, Hester RE (eds). Wiley: New York, 1987;187.
4. Stencel JM. Raman Spectroscopy for Catalysis. Van Nostrand: NewYork, 1990.
5. Mehicic M, Grasselli JG. In Analytical Raman Spectroscopy,Grasselli JG, Bulkin BJ (eds). Wiley: New York, 1991.
6. Wachs IE, Hardcastle FD. In Catalysis—A Specialist PeriodicalReport, vol. 10, Royal Society of Chemistry: Cambridge, 1993;102.
Benziger JB, Sundaresan S. J. Catal. 1997; 170: 75.47. Banares MA, Martınez-Huerta MV, Gao X, Fierro JLG, Wachs
IE. Catal. Today 2000; 61: 295.48. Gao X, Jehng J-M, Wachs IE. J. Catal. in press.49. Watling TC, Deo G, Seshan K, Wachs IE, Lercher JA. Catal. Today
1996; 28: 139.50. Gao X, Banares MA, Wachs IE. J. Catal. 1999; 188: 325.51. Cardoso JH. PhD Dissertation, Universidade Federal de Sao
Carlos, Sao Paulo, 1998.52. Datka J, Turek AM, Jehng J-M, Wachs IE. J. Catal. 1992; 135: 186.53. Banares MA, Spencer ND, Jones MD, Wachs IE. J. Catal. 1994;
146: 204.54. Sun Q, Jehng J-M, Hu H, Herman RG, Wachs IE, Klier K. J. Catal.
1997; 165: 101.55. Bielanski A, Haber J. Catal. Rev. Sci. Eng. 1979; 19: 1.56. Wachs IE, Deo G, Weckhuysen BM, Andreini A, Vuurman MA,
de Boer M, Amiridis MD. J. Catal. 1996; 161: 211.57. Ozkan US, Cai Y, Kumthekar M. Appl. Catal. A 1993; 96: 365.58. Gasior M, Haber J, Machej T, Czeppe T. J. Mol. Catal. 1988; 43:
359.59. Jehng J-M, Hu H, Gao X, Wachs IE. Catal. Today 1996; 28: 335.60. Jehng J-M. J. Phys. Chem. B 1998; 102: 5816.61. Banares MA, Hu H, Wachs IE. J. Catal. 1995; 155: 249.62. Banares MA, Hu H, Wachs IE. J. Catal. 1994; 150: 407.63. Hu H, Wachs IE. J. Phys. Chem. 1995; 99: 10 911.64. Oyama ST. Chem. Intermed. 1991; 15: 165.65. Busca G, Elmi AS, Forzatti P. J. Phys. Chem. 1987; 91: 5263.66. Busca G. Catal. Today 1996; 27: 457.67. Chen K, Khodakov A, Yang J, Bell AT, Iglesia E. J. Catal. 1999;