Page 1
Bachelor Thesis Scheikunde
Multicyclic peptides via CLIPS and oxime ligation
Synthesis of acid labile amino-oxy protection groups
door
Filip Aleksic
14-07-2017
Studentnummer
10754830
Onderzoeksinstituut
Van ’t Hoff Institute for Molecular Sciences
(HIMS)
Onderzoeksgroep
Synthetic Organic Chemistry (SOC)
Verantwoordelijk docent
Prof. Dr. J. H. van Maarseveen
Begeleider
Dieuwertje Streefkerk MSc.
Page 3
3
Abstract
The synthesis of multi-cyclic peptide chains has proven to be important for possible applications in the
antibody design field. CLIPS technology has shown to be an effective way to constrain peptide chains
into cyclic structures. However, when tricyclic structures are attempted to be synthesized using CLIPS
regioisomers are rapidly formed. To avoid the formation of these regioisomers, a second orthogonal
coupling reaction was used: oxime ligation. For a sequential orthogonal coupling of CLIPS and oxime
ligation the pH has to be mediated to fit the reaction conditions of both coupling methods. CLIPS is
performed at pH = 8, after which the pH is lowered to <1 to remove the acid labile protection group
(Boc) on the amino-oxy moiety. The pH is then increased to pH = 4 to meet the optimal oxime ligation
conditions. The large volumes of acid and base needed are not practical to work with when applying
this method to screening a large peptide library. It was envisioned that through the use of a highly acid
labile protection group a pH buffer could simply be used to mediate the reactions and deprotect the
acid labile protection group, thereby eliminating the need for large volumes of acid and base. The aim
of this project was to find a highly acid labile protection group for an amino-oxy moiety. Two acid
labile protection groups were tested; Mtt and t-Bumeoc. It was found that the free amino-oxy rapidly
reacts with trace amounts of acetone, thereby nullifying the reactivity of the amino-oxy. The removal
of the acetone adduct was successful by allowing the acetone adduct to react with methoxyamine. The
Mtt protected amino-oxy was synthesized. However, the high acid lability of the compound resulted in
rapid degradation on acidic silica when purification was attempted using column chromatography.
Crystallisation was additionally attempted in Et2O and pentane with DCM as a co-solvent, however
neither showed any product. The tertiary alcohol of t-Bumeoc was synthesized. However, the
instability of the chloroformate, and imidazole carbamate of t-Bumeoc lead to the inability to couple
the protection group to an amino-oxy.
Page 4
4
Samenvatting
Het nabootsen van de in het immuunsysteem aanwezige moleculen om deze als medicijn toe te dienen
is een zeer effectieve manier bij bepaalde ziektes die het immuunsysteem aantasten. HIV en influenza
zijn bijvoorbeeld twee virussen die op deze manier bestreden kunnen worden. Echter is het maken van
dit soort mimicry moleculen geen simpele taak omdat processen in ons lichaam met een hoge
specificiteit plaatsvinden. In het geval van het immuunsysteem reageert een antilichaam en een antigen
met elkaar met behulp van het sleutel-slot-principe. Als men een antigen synthetisch wil namaken
moet de ‘sleutel’ van dit molecuul (die selectief in het slot van een antilichaam past) zo goed mogelijk
lijken op het door het lichaam gemaakte antigen. Antigenen hebben echter vaak een complexe
structuur met veel verschillende gebogen bindingsplekken waar een keten aminozuren in de vorm van
meerdere lussen op past.
Het maken van deze lussen kan bereikt worden door de keten aminozuren vast te maken aan de
buitenkant van een rigide steiger molecuul. Bij het koppelen van de keten aan de steiger wordt gebruik
gemaakt van twee verschillende koppelingsreacties die individueel plaatsvinden bij verschillende
condities, gehete CLIPS (Chemical Linkage of Peptides onto Scafolds) en oxime ligatie. De CLIPS
reactie vindt eerst plaats, waarna oxime ligatie wordt geïnitieerd (Figuur 1).
Figuur 1: Een schematische weergave van de twee koppelings reacties. Links is een T4 rigide steiger
molecuul te zien met 2 rode CLIPS bindingsplekken en 2 groene oxime ligatie bindingsplekken. Op de
lineaire aminozuurketen zitten ook 2 CLIPS en oxime ligatie bindingsplekken in rood en blauw
respectievelijk. Allereerst wordt een CLIPS reactie uitgevoerd gevolgd door oxime ligatie.
De koppelingsreacties vinden plaats bij verschillende condities. De CLIPS koppelings reactie wordt
geïnitieerd bij een licht basische pH van rond de 8, terwijl oxime ligatie het snelst is bij licht zure
condities plaatsvindt van een pH waarde rond de 4. Men wil echter uiteraard niet dat de koppelings
reacties door elkaar heen kunnen plaatsvinden.
Page 5
5
Daarom wordt gebruik gemaakt van een speciale groep die wordt vastgeplakt aan de reactieve plek
van oxime ligatie aan het rigide steiger molecuul die de reactiviteit van deze plek compleet tenietdoet.
Zo een speciale groep wordt ook wel een beschermgroep genoemd (immers beschermt men de oxime
ligatie reactieve plek zodat deze niet meer kan reageren).
In dit geval kan deze beschermgroep weer worden verwijdert met behulp van geconcentreerd zuur. De
ontscherming vindt plaats bij een pH waarde lager dan 1, dit is te laag voor oxime ligatie en de pH
moet vervolgens weer omhoog gebracht worden met een base. Deze grote hoeveelheden zuur en base
die moeten worden toegevoegd zijn praktisch niet handig wanneer op kleine schaal veel verschillende
aminozuurketens worden getest. De buisjes waarin deze testjes worden gedaan zijn zo klein dat ze
simpelweg overvloeien nadat al het zuur en base is toegevoegd.
Een oplossing voor dit probleem is het gebruik maken van een beschermgroep die onder minder zure
condities er al af gaat. Dit zou het benodigde volume zuur verminderen en het gebruik van een base
zou dan niet meer nodig zijn (Figuur 2).
Figuur 2: Schema van toevoegingen zuur en base die nodig zijn. Bij een beschermgroep die er onder
minder zure condities al af gaat is geen base nodig om oxime ligatie te initiëren.
In dit project zijn twee beschermgroepen gebruikt: Mtt en t-Bumeoc. De Mtt beschermde oxime ligatie
bindingsplek was gesynthetiseerd, echter was het zuiveren van dit molecuul tot dusver niet gelukt. Het
zuiveringsproces dat gebruikt werd vindt plaats onder licht zure condities en dit was al genoeg om de
beschermgroep van het molecuul af te splitsen. De tweede beschermgroep t-Bumeoc was ook
gesynthetiseerd. De volgende stap zou zijn om de beschermgroep te koppelen aan de oxime ligatie
bindingsplek, echter is dit niet gelukt. De beschermgroep bleek na meerdere verschillende manieren
geprobeerd te hebben om het te koppelen simpelweg niet geschikt voor dit project.
Page 6
6
List of abbreviations
Adpoc 1-(1-Adamantyl)-1-methylethoxycarbonyl carbamate
Boc Tert-butoxycarbonyl
Cbz Carboxybenzyl
CLIPS Chemical Linkage of Peptides onto Scaffolds
CDI 1,1-Carbonyldiimidazole
d Doublet (by NMR)
DCM Dichloromethane
DIAD Diisopropyl azordicarboxylate
DIPEA N,N-Diisopropylethylamine
DMSO Dimethylsulfoxide
Equiv Equivalents
EtOAc Ethyl acetate
EtOH Ethanol
IR Infrared spectroscopy
m Multiplet (by NMR)
Mtt 4-Methyltrityl
NBS N-Bromosuccinimide
NMR Nuclear Magnetic Resonance
OSu N-Hydroxysuccinimide
pAcF p-Acetylphenylalanine
Pd/C Palladium on activated carbon
PE Petroleum ether (40-60)
PG Protection Group
phth Phthalimide
py Pyridine
q Quartet (by NMR)
s Singlet (by NMR)
t-Bumeoc 1-(3,5-Di-t-butylphenyl)-1-methylethyl carbamate
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TLC Thin layer chromatography
Tol Toluene
Page 7
7
Table of contents
Abstract. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Samenvatting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
List of abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Table of contents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.1 Chemical Linkage of Peptides onto Scaffolds (CLIPS) . . . . . . . . . . . . . . . . . . . 9
1.2 Orthogonal coupling reaction: Oxime ligation . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.3 Results in the group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
1.4 Aim of the project. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2 Experimental. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.1 Synthesis of scaffold: CLIPS half. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.2 Synthesis of scaffold: Protected amino-oxy. . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.2.1 Acetone adduct formation and prevention . . . . . . . . . . . . . . . . . . . . . 16
2.2.2 Purification of the Mtt protected amino-oxy. . . . . . . . . . . . . . . . . . . . 18
2.3 Alternative protection group: t-Bumeoc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.3.1 Mechanistic discussion of the Friedel-Crafts alkylation . . . . . . . . . . 19
2.3.2 Synthesis towards the chloroformate. . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.3.3 Coupling t-Bumeoc to an amine: Literature approach . . . . . . . . . . . 22
2.3.4 Coupling t-Bumeoc to an amine: The use of CDI. . . . . . . . . . . . . . . . 23
3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4 Future prospects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
5 Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
6 References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
7 Experimental. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
7.1 General remarks. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
8 Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
Page 8
8
1. Introduction Immunotherapy is widely regarded as one of the most promising ways to cure patients from chronic
diseases which target the immune system.1 Therefore, understanding the complex peptide interactions
of the immune system can be tremendously beneficial to the design of new immunotherapy drugs.
Typically, immunotherapy drugs consist of artificial antibodies which replace the antibodies that have
been destroyed by a debilitating disease. Antigens contain binding sites (epitopes) to which an
antibody can bind with high affinity and selectively with regions on the antibody, called paratropes
(Figure 1).
Figure 1: Schematic view of an antigen and antibody. The paratrope and epitope bind to each other
with remarkable specificity due to their pairing 3D-structure.
There are two different types of epitopes: continuous and discontinuous. Continuous epitopes are
linear peptide chains which interact with the paratrope on an antibody, whereas discontinuous epitopes
consist of multiple peptide chains (Figure 2).
Figure 2: Left: Continuous epitope. A single loop on a linear peptide forms the binding site for the
paratrope. Right: Discontinuous epitope. Multiple loops on different parts of the epitope form the
binding site.2
In vitro experiments have been performed to synthesize peptide loops which can effectively mimic
continuous epitopes with the use of a peptide which was constrainted into 1 loop.3 However,
discontinuous epitopes have proven to be particularly difficult to emulate. It has also been reported
that the vast majority, if not all, epitopes are discontinuous to some extent.4
Page 9
9
Therefore, an extensive amount of research has been conducted to find ways to efficiently synthesize
peptides into a multi-loop structure. Various methods to constrain linear peptides into the correct
conformation have since been developed, such as CLIPS (Chemical Linkage of Peptides onto
Scaffolds) technology, oxime ligation, Click-chemistry, and disulfide formation.5, 6, 7, 8
1.1 Chemical Linkage of Peptides onto Scaffolds (CLIPS)
CLIPS was found to be particularly promising due to the fact that it is a remarkably clean and fast
reaction under mild reaction conditions (Figure 3).9, 10, 11
As the name suggests the main synthetic
strategy is the constraining of the linear peptide chain onto a scaffold by substituting the bromine on
the scaffold with the thiol of a cysteine residu of the peptide chain.
Figure 3: Schematic view of CLIPS technology.5
The potential of CLIPS was explored by Heinis et al. who showed that the binding affinity of a
bicyclic system to an enzyme dramatically increased relative to the mono-cyclic and linear systems.12
Page 10
10
This raises the question about whether or not a tricyclic, tetracyclic, or even a pentacyclic peptide
system would show an even better activity. However, this is where the limitations of CLIPS
technology come into view. When a tricyclic (or more) system is synthesized using CLIPS
regioisomers are rapidly formed (Figure 4).
Figure 4: Regioisomer formation for CLIPS reaction on a scaffold with 4 binding sites. After the first
CLIPS couples, the next thiol on the peptide can react with any of the 3 remaining bromines on the
scaffold, thereby forming regioisomers.
1.2 Orthogonal coupling reaction: Oxime ligation
For potential applications of CLIPS in the pharmaceutical industry it is crucial to have a process which
is able to produce a product without regioisomers to avoid costly large scale separation procedures (if
the separation is even possible at all). Combining CLIPS with another orthogonal coupling reaction
with each only having 2 reactive sites on the scaffold could prevent the formation of regioisomers. In
our group prevention of regioisomer formation is explored by combining CLIPS technology with an
orthogonal coupling reaction; oxime ligation (Figure 5).
Figure 5: Schematic view of the process of an orthogonal CLIPS and oxime ligation reaction with a 4
functional group scaffold forming a peptide chain suspended into 3 loops.
Oxime ligation is the reaction of an amino-oxy with a ketone resulting in the formation of an oxime
with the removal of H2O (Figure 6). The reaction is typically executed in acidic conditions of a pH
value at around 4. Performing the reaction at an even more acidic pH would lower the reaction rate
significantly, as the free amino-oxy would become protonated which would reduce its nucleophilic
strength.
Page 11
11
Figure 6: Oxime ligation mechanism in an acidic environment. An amino-oxy group performs a
nucleophilic attack on a ketone in an acidic environment. After elimination of water the oxime is
formed.
When a primary amine reacts with a ketone forming an imine, the equilibrium heavily favours the
ketone. However α-effect nitrogens, such as amino-oxy groups, favour the formation of the oxime
(Figure 7).13
The high reactivity and selectivity, coupled with the mild reaction conditions makes
oxime ligation a highly effective orthogonal coupling reaction.
Figure 7: Above: Formation of an imine. The equilibrium is favoured for the ketone. Below: Oxime
ligation. The equilibrium is favoured for the oxime. Both E- and Z-isomers are formed.
By adding an amino-oxy functional group to the scaffold, which can react with a ketone on the side
chain of a peptide, oxime ligation can be used as an orthogonal coupling reaction. However, no natural
amino acids contain a ketone group. Therefore the synthetic amino acid p-acetylphenylalanine (pAcF)
was used in the synthesis of the linear peptide chain to allow for the coupling reaction (Figure 8).
Figure 8: Structure of p-acetylphenylalanine (pAcF)
Page 12
12
1.3 Results in the group
The 4 functional group scaffold T4-N3 was tested in the group showing promising results (Figure 9).
The linear peptide chain with the amino acid sequence AcC-E-pAcF-A-pAcF-A-K-CNH2 (C=
Cysteine, E = Glutamate, A = Alanine, K = Lysine) coupled with high efficiency to the scaffold for the
CLIPS reaction. However, following the oxime ligation it was found that both E- and Z-isomers are
formed of the oxime bond. The problem of E- and Z- isomers forming was not in the scope of this
project and will therefore not be discussed in more detail.
Figure 9: The orthogonal coupling of the linear peptide chain AcC-E-pAcF-A-pAcF-A-K-CNH2 onto
the T4-N3 scaffold. The CLIPS reaction was performed under basic conditions, after which the pH
was reduced to remove the Boc group. The pH was then increased slightly for an efficient oxime
ligation reaction.
A problem arose with the Boc PG used to protect the amino-oxy, as the conditions to remove this PG
are relatively harsh. Typically concentrated HCl or pure trifluoroacetic acid (TFA) is used. These
conditions are not practical to work with when screening a large number of different linear peptide
chains in peptide library format. These screening reactions are done in microwell plates which fill up
rapidly and can even overflow due to the large volumes of acid and base needed to mediate the
conditions for the different coupling, and deprotection reactions.
Therefore a new, more acid labile amino-oxy PG is desired. The research question of this project is:
can a scaffold be synthesized with a highly acid labile PG on the amino-oxy substituent for use in an
orthogonal CLIPS and oxime ligation?
Page 13
13
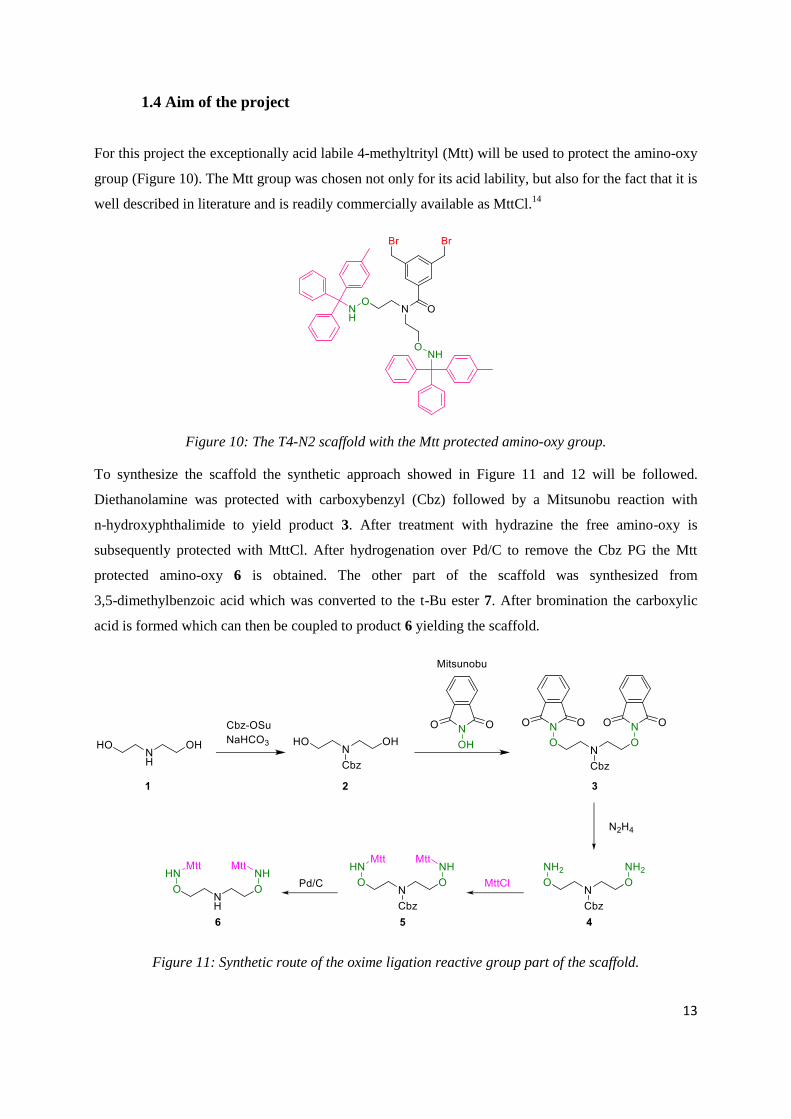
1.4 Aim of the project
For this project the exceptionally acid labile 4-methyltrityl (Mtt) will be used to protect the amino-oxy
group (Figure 10). The Mtt group was chosen not only for its acid lability, but also for the fact that it is
well described in literature and is readily commercially available as MttCl.14
Figure 10: The T4-N2 scaffold with the Mtt protected amino-oxy group.
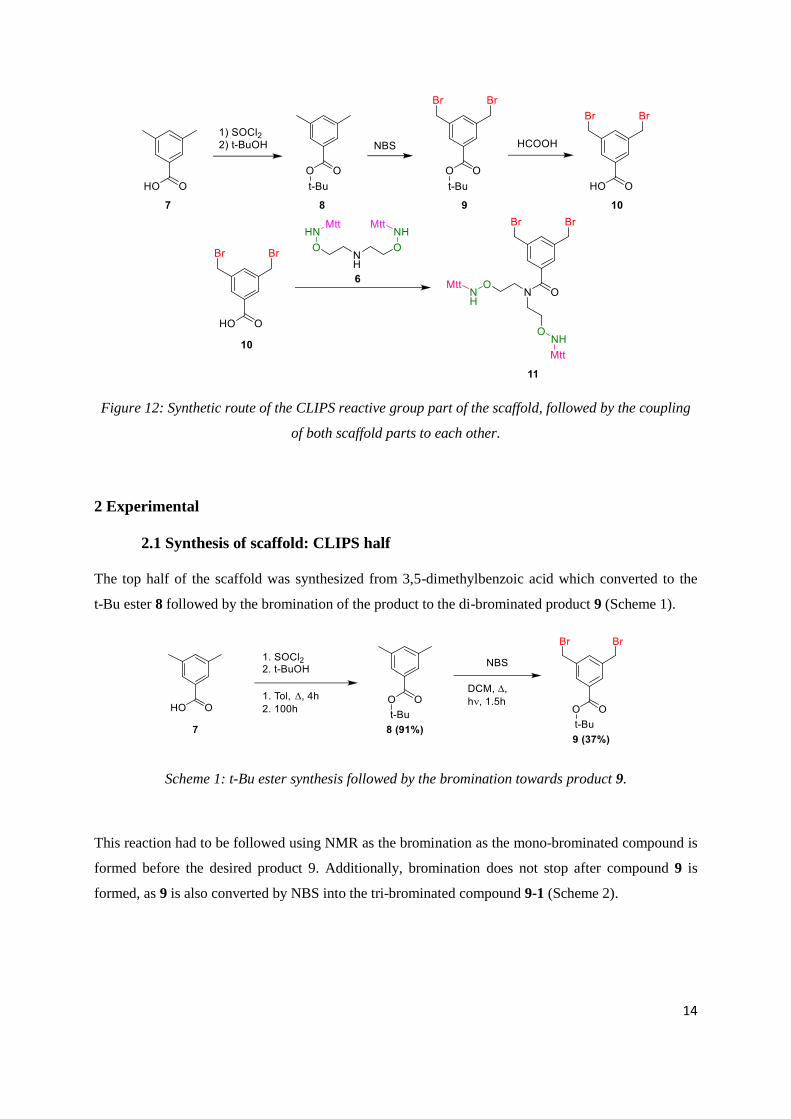
To synthesize the scaffold the synthetic approach showed in Figure 11 and 12 will be followed.
Diethanolamine was protected with carboxybenzyl (Cbz) followed by a Mitsunobu reaction with
n-hydroxyphthalimide to yield product 3. After treatment with hydrazine the free amino-oxy is
subsequently protected with MttCl. After hydrogenation over Pd/C to remove the Cbz PG the Mtt
protected amino-oxy 6 is obtained. The other part of the scaffold was synthesized from
3,5-dimethylbenzoic acid which was converted to the t-Bu ester 7. After bromination the carboxylic
acid is formed which can then be coupled to product 6 yielding the scaffold.
Figure 11: Synthetic route of the oxime ligation reactive group part of the scaffold.
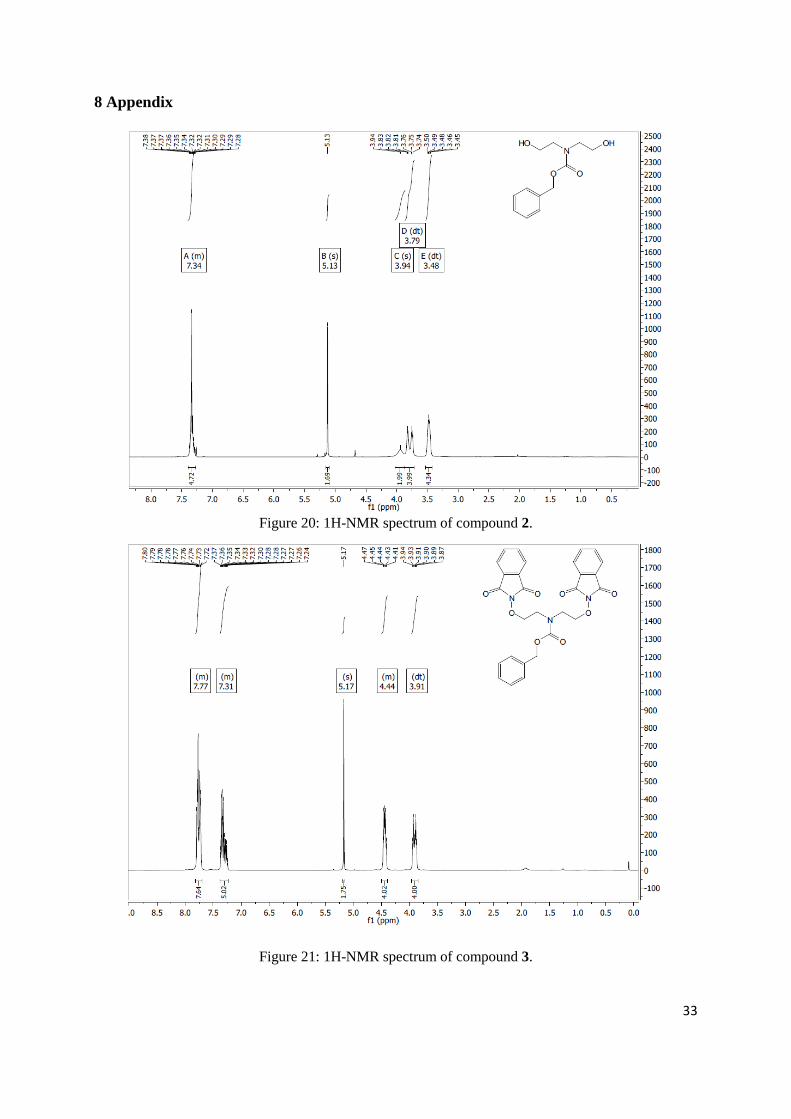
Page 14
14
Figure 12: Synthetic route of the CLIPS reactive group part of the scaffold, followed by the coupling
of both scaffold parts to each other.
2 Experimental
2.1 Synthesis of scaffold: CLIPS half
The top half of the scaffold was synthesized from 3,5-dimethylbenzoic acid which converted to the
t-Bu ester 8 followed by the bromination of the product to the di-brominated product 9 (Scheme 1).
Scheme 1: t-Bu ester synthesis followed by the bromination towards product 9.
This reaction had to be followed using NMR as the bromination as the mono-brominated compound is
formed before the desired product 9. Additionally, bromination does not stop after compound 9 is
formed, as 9 is also converted by NBS into the tri-brominated compound 9-1 (Scheme 2).
Page 15
15
Scheme 2: Bromination of 9 towards the tri-, and tetra-brominated products 9-1 and 9-2.
An equilibrium is desired where most of the starting material 8 is converted to the product 9 whereby
most of 9-1 is converted and the concentration of 9-2 is kept at a minimum. Typically crystallization in
n-hexane is carried out to purify the product 9. However, after 3 attempts no crystallization was
observed. Co-crystallization with left over product 9 from previous experiments was also
unsuccessful. Therefore, it was decided to continue the synthesis of the scaffold with the crude
product. Following the bromination the t-Bu ester was converted to the carboxylic acid 10 using
formic acid in dichloromethane (DCM) (Scheme 3).
Scheme 3: Carboxylic acid synthesis from the t-Bu ester.
The next step of the synthesis is the coupling of the carboxylic acid to the free secondary amine of the
Mtt protected amino-oxy. The synthesis of this compound will be discussed next.
2.2 Synthesis of scaffold: Protected amino-oxy
The sysnthesis was started with a straightforward protection of the secondary amine of diethanolamine
to yield product 2 followed by a Mitsunobu reaction with n-hydroxyphthalimide to introduce the
amino-oxy moiety, which yielded the diphthalimide product 3 (Scheme 4).
Page 16
16
Scheme 4: Cbz protection of diethanolamine.
Afterwards, the phth groups were removed with hydrazine to yield the free amino-oxy 4 which allows
us to add the desired acid labile PG. However, after analysis of the product it became evident that no
free amino-oxy was present, instead the acetone adduct 4-1 was formed (Scheme 5).
Scheme 5: Removal of phth groups with hydrazine followed by the unwanted side reaction with trace
amounts of acetone towards the oxime 4-1.
2.2.1 Acetone adduct formation and prevention
Although no acetone was used in any steps of the synthesis trace amounts appeared to be present in the
reaction mixture, either from cleaning glassware, vapours which entered the flask during the use of the
rotary evaporator, or simply trace amounts present in the solvents used. Ethanol specifically
commonly has ketone and aldehyde impurities which react with the free amino-oxy. The rapid
formation of the acetone adduct showed the strength of the oxime ligation reaction, however the
formed oxime nullifies the reactivity of the scaffold towards future oxime ligation, therefore it is a
highly undesired product.
Page 17
17
In an attempt to recover the free amino-oxy from the acetone adduct methoxyamine was added in a
large excess and after 2 days at room temperature the free amino-oxy 4 was obtained in poor yield of
9% (Scheme 6).
Scheme 6: Recovering the free amino-oxy from the acetone adduct with the use of methoxyamine
The low yield could be a result of low solubility of the protonated free amino-oxy into the water phase
during the acid-base extraction which was performed. The large, apolar Cbz was likely too difficult to
solute in water despite the highly polar protonated amino-oxy groups. However, the aim of this
reaction was to recover some of the lost product, therefore a low yield was acceptable.
Avoiding the formation of the acetone adduct was a pressing issue and several special measures were
taken to make sure no acetone was present in reactions involving free amino-oxy groups. All
glassware which was used was either flame dried or kept in an oven at 150°C before use. Additionally,
distilled tetrahydrofuran (THF) was chosen as a solvent for future reactions, as it has been shown in
our group to be an effective solvent when working with free amino-oxy containing compounds.
Further prevention of the acetone adduct formation was successfully attempted by performing a
one-pot deprotection - protection sequence from the di-phth compound 3 to the Mtt protected amino-
oxy 5. Previously hydrazine soluted in water was used for the removal of the phth groups. However,
this resulted in a 2 phase system with THF, which was not ideal. Methylhydrazine was instead used to
avoid this problem. Additionally, the use of methylhydrazine resulted in reaching full conversion
towards the free amino-oxy after only 5 to 10 min. Afterwards, MttCl was added alongside N,N-
diisopropylethylamine (DIPEA) in 10 and 5 equiv respectively and left to stir overnight to yield the
Mtt protected amino-oxy 5 (Scheme 7).
Scheme 7: One-pot synthesis of Mtt protected amino-oxy using methylhydrazine in THF.
Page 18
18
Analysis of the product was performed with NMR whereby three signals were used to determine
conversion of the product (Figure 13).
Figure 13: 1H-NRM spectrum of the crude product from Scheme 10.
The 1H-NMR signals of the hydrogens marked with 1 and 2 did not only shift to a lower ppm value
compared to the phth protected compound from 4.44 ppm to 3.63 ppm and from 3.91 ppm to 3.18
ppm respectively, but the splitting pattern also changed. Due to hindered rotation around carbamate
Cbz bond the phth protected compound showed doublet of triplet-like structures for signals 1 and 2.
After the Mtt protection signal 1 changed into a broad singlet, and 2 into two separate broad signals.
This change indicated a more rotationally hindered compound, which is what is expected given the
more bulky Mtt PG.
2.2.2 Purification of the Mtt protected amino-oxy
To purify the Mtt protected compound column chromatography was not the preferred method given
that the acid lability of the product could result in rapid degradation on the acidic silica. It was argued
that due to the large aromatic structure of the Mtt PG the isolated product could be crystalline.
Therefore, crystallization was firstly attempted in Et2O, and pentane with DCM as a co-solvent.
However, neither showed any sign of product after multiple attempts.
Page 19
19
NEt3 treated silica was therefore used despite the possible degradation problems. After two sequential
attempts to isolate the product using column chromatography no product could be isolated due to the
similarity in polarity of the by-products and main product. The structure of the compounds which are
formed as by-products has not been determined. However, due to the large signals in the aromatic
region of the 1H-NMR spectrum in Figure 13 it was assumed that the by-products are likely Mtt-
containing compounds. Following the second column it also became evident that the mixture became
more complicated than before, as more spots appeared on TLC even closer together than previously
observed after the first column. This change likely occurred due to degradation of the Mtt protected
compound.
2.3 Alternative protection group: t-Bumeoc
Following the difficulties to isolate the Mtt protected compound alternative acid labile PGs were
investigated. Different carbamates were explored as alternatives, as they would likely be more robust
towards acidic degradation on silica, which would allow for the purification of the protected product.
1-(3,5-Di-t-butylphenyl)-1-methylethyl carbamate (t-Bumeoc) (Figure 14) was chosen as an
alternative, as it has been shown to be removed from an amine in an 80% AcOH/H2O solution 4000
times faster than Boc.15, 20
The synthesis of t-Bumeoc will now be discussed in more detail.
Figure 14: t-Bumeoc protection group coupled to a generic amino-oxy RONH2.
2.3.1 Mechanistic discussion of the Friedel-Crafts alkylation
The first step of the t-Bumeoc synthesis is a Friedel-Crafts alkylation of toluene with tert-butyl
chloride (t-BuCl) (Scheme 8).
Scheme 8: Friedel-Crafts alkylation of toluene with t-BuCl to yield 3,5-di-tert-butyltoluene.
Page 20
20
Interestingly the t-Bu groups are added at the meta positions relative to the methyl of toluene, which is
odd because typically methyl substituents are ortho-para direction groups. The exact mechanism of
this reaction was not found in literature. However, two different mechanisms will be proposed. The
first mechanism relies on the addition and removal of t-Bu groups being in equilibrium with each
other. Due to steric hindrance between the t-Bu and methyl group the ortho addition is rare. On the
contrary, the meta addition of a t-Bu group results in no steric interaction with the methyl group. This
results in the reaction eventually moving towards the 3,5-di-tert-butylated compound which can be
seen as a thermodynamic product (Figure 15).
Figure 15: First proposal of the mechanism of the Friedel-Crafts alkylation of toluene with t-BuCl.
A second mechanistic explanation can be a methyl shift from the 2,4-di-tert-butylated product towards
the 3,5- substituted product.
Figure 16: Second proposal of the mechanism of the Friedel-Crafts alkylation of toluene with t-BuCl.
Page 21
21
To determine which of these mechanistic explanations is correct two experiments could be performed.
Firstly, the Friedel-Crafts alkylation of 4-tert-Butyltoluene can be performed to see if any 3,5- product
is formed, which would indicate a methyl shift. Additionally, the alkylation of toluene can be
performed using deuterated toluene to see if any deuterium scrambling takes place. Deuterium
scrambling would occur when the t-Bu groups detach and re-attach from the toluene repeatedly and
would therefore indicate the presence of the thermodynamic product 3,5-di-tert-Butyltoluene.
2.3.2 Synthesis towards the chloroformate
3,5-Di-tert-butyltoluene was oxidized to the carboxylic acid 15 using potassium permanganate. The
low yield for this reaction could be contributed to incomplete oxidation of 14. However, a large
portion of the product was likely also lost during the difficult workup due to the formed MnO2, which
stained the glassware that was used, and made it difficult to see a phase separation during extractions.
Afterwards, the methyl ester 16 was formed which was reacted with the Grignard reagent MeMgBr to
yield the tertiary alcohol 17 (Scheme 9).
Scheme 9: Synthesis of the Me ester, followed by a Grignard reaction to yield the tertiary alcohol 17.
The next step of the synthesis was to convert the tertiary alcohol into an activated compound which
could then be coupled to the amino-oxy 4. Firstly, the chloroformate was attempted to be synthesized
using triphosgene (Scheme 10).
Scheme 10: Synthesis of the chloroformate using triphosgene.
Page 22
22
However, no conversion towards the chloroformate was observed. It was not clear what had happened,
as the only difference in the 1H-NMR spectrum was a shift in ppm of all the signals. IR and
13C-NMR
were also not conclusive about what had happened.
2.3.3 Coupling t-Bumeoc to an amine: Literature approach
In literature the tertiary alcohol of t-Bumeoc is coupled to an amine using the fluoroformate (Figure
17).
Figure 17: Fluoroformate of t-Bumeoc.
Theoretically it is expected that the fluoroformate is more stable than the chloroformate due to
electronic effects. In the article where t-Bumeoc was synthesized and used it was reported that the
fluoroformate is only stable at -18°C, which would explain why no conversion could be seen when
attempting to synthesize the chloroformate, as it likely decomposes instantly upon formation at room
temperature. The fluoroformate is synthesized using carbonic chloride fluoride which was formed
from reacting anhydrous sulphuric acid with trichlorofluoromethane (Figure 18).16
Figure 18: Synthesis of the fluoroformate from the tertiary alcohol using carbonic chloride fluoride.
Due to the highly hazardous reagents and the practically difficult procedure it was chosen not to
follow this synthetic route.
Page 23
23
2.3.4 Coupling t-Bumeoc to an amine: The use of CDI
Another method which was attempted to form an imidazole carbamate with the use of 1,1-
carbonyldiimadazole (CDI), as it has been shown in literature that CDI can be used to couple tertiary
alcohols to amines.17
The reaction was followed with TLC which indicated full conversion to the
elimination product 19-1 (Scheme 11).
Scheme 11: Synthesis of the CDI activated ester and the following elimination. After elimination of
CO2 the highly stabilised carbocation of t-Bumeoc is formed, after which a hydrogen is abstracted by
the imidazole anion to yield the alkene elimination product.
3 Conclusion
The goal of this project was to find highly acid labile amino-oxy PG for the use in orthogonal CLIPS
and oxime ligation. Two amino-oxy protection groups were attempted to be synthesized with these
applications in mind: Mtt and t-Bumeoc. The Mtt protected amino-oxy was synthesized, however
purification of the product was so far unsuccessful due to similar polarities of the by-products and
product, alongside difficulties with product degradation on the acidic silica.
The tertiary alcohol of t-Bumeoc was synthesized. However, coupling of the PG to an amino-oxy
using a chloroformate, or by using CDI was unsuccessful. The fluoroformate was used in literature to
couple the tertiary alcohol to an amine, though this synthesis deals with highly hazardous reagents and
was therefore not attempted.
Page 24
24
4 Future prospects
Using an Mtt PG for orthogonal CLIPS and oxime ligation reactions might still be an attractive option
despite difficulties with isolation of the product. By limiting the amount of by-products formed by
optimizing the coupling reaction separation might become more straightforward. It is envisioned that
the main reaction condition which should be changed is the amount of MttCl added. The products
which were attempted to isolate were all synthesized with 10 equiv MttCl. A coupling reaction with
2.1 equiv of MttCl can be attempted to analyse the conversion to the desired protected amino-oxy.
Although t-Bumeoc turned out not to be a usable protection group other carbamates could potentially
still be used due to them being more robust towards acidic degradation compared to the Mtt PG. For
example 1-(1-Adamantyl)-1-methylethoxycarbonyl carbamate (Adpoc) is another carbamate PG
which is highly acid labile and could serve at a potential alternative to Mtt (Figure 19).15
Figure 19: Adpoc PG coupled to a generic amino-oxy R-ONH2.
The adamantane carboxylic acid can be purchased commercially after which it is chlorinated and
reacted with MeMgI to obtain the tertiary alcohol.18
The coupling of the tertiary alcohol to an amine is
once again performed in literature using the carbonic chloride fluoride reagent.19
If it is decided that
Adpoc is to be researched an alternative to the fluoroformate should first be looked for.
Page 25
25
5 Acknowledgements
Firstly I would like to thank Prof. Dr. H. Hiemstra, Prof. Dr. J. H. van Maarseveen and Dr. S.
Ingemann Jorgensen for allowing me to do this project in the Synthetic Organic Chemistry group.
Additionally I would like to thank Prof. Dr. A. M. Brouwer for being the second reviewer of this
project. I would also like to express my deepest gratitude and appreciation to my daily supervisor
Dieuwertje Streefkerk. She has taken a vast amount of time out of her busy schedule to guide me
through any difficulties I may have had. Not only has she helped me tremendously in the lab by
teaching me how to perform experimental work both safely and efficiently, but she was also more than
willing to guide me with the work outside the lab. I have learned a great deal during this project
because of her willingness to actively help me with anything I may have needed help with.
Lastly I would like to thank everyone in the Synthetic Organic Chemistry group for creating a
wonderful atmosphere which made the long days in the lab more enjoyable.
Page 26
26
6 References
1: Waldmann, T. A. Nat. Med. 2003, 9 (3), 269–277.
2: Werkhoven, P. R.; Elwakiel, M.; Meuleman, T. J.; Quarles, H. C.; Quarles van Ufford, H. C.;
Kruijtzer, J. A. W.; Liskamp, R. M. J. Org. Biomol. Chem., 2016, 14, 701–710.
3: Scott, J. K.; Smith, G. P. Science 1990, 249, 386–390.
4: Barlow, D. J.; Edwards, M. S.; Thornton, J. M. Nature 1986, 322, 747–748.
5: Timmerman, P.; Beld, J.; Puijk, W. C.; Meloen, R. H. ChemBioChem 2005, 6, 821–824.
6: Tang, L.; Yin, Q.; Xu, Y.; Zhou, Q.; Cai, K.; Yen, J.; Dobrucki, L. W.; Cheng, J. Chem. Sci.
2015, 6, 2182–2186.
7: Angell, Y. L.; Burgess, K. Chem. Soc. Rev. 2007, 36, 1674–1689.
8: Zeng, W.; Ghosh, S.; Macris, M.; Pagnon, J.; Jackson, D. C. Vaccine 2001, 19, 3843–3852.
9: Timmerman, P.; Puijk, W. C.; Meloen, R. H. J. Mol. Recognit. 2007, 20, 283–299.
10: Smeenk, L. E. J.; Dailly, N.; Hiemstra, H.; van Maarseveen, J. H.; Timmerman, P. Org. Lett.
2012, 14 (5), 1194–1197.
11: Timmerman, P.; Puijk, W. C.; Boshuizen, R. S.; van Dijken, P.; Slootstra, J. W.; Beurskens, F. J.;
Parren, P. W. H. I.; Huber, A.; Bachmann, M. F.; Meloen, R. H. Open Vaccine J. 2009, 2, 56–67.
12: Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Nat. Chem. Biol. 2009, 5 (7), 502–507.
13: Dirksen, A.; Hackeng, T. M.; Dawson, P. E. Angew. Chem. Int. Ed. 2006, 45, 7581–7584.
14: Liu, F.; Thomas, J.; Burke Jr., T. R. Synthesis 2008, 15, 2432–2438.
15: Wuts, P. G. M. Greene’s Protective Groups In Organic Synthesis, 5th ed.; Wiley: Michigan, USA.
16: Voelter, W.; Müller, J. Liebigs Ann. Chem., 1983, 248–260.
17: Stoddart, A.; Feast, W. J.; Rannard, S. P. Soft Matter 2012, 8, 1096–1108.
18: Kolocouris, A.; Busath, D. D.; Johnson, B. Antiviral compounds for treating amantadine resistance
influenza A. PCT Int. Appl. 2014121170, 2014.
19: Kalbacher, H.; Voelter, W. J. Chem. Soc., Chem. Commun. 1980, 72, 1265–1266.
20: Voelter, W.; Müller, J. Liebigs Ann. Chem. 1983, 248–260.
Page 27
27
7 Experimental
7.1 General remarks
Reagents which were purchased were used as supplied. Unless stated otherwise, reactions were
performed at room temperature without any special precautions such as drying or N2 atmosphere.
Dried solvents were obtained by distillation with sodium. Reactions were followed with thin layer
chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60F-254). TLC analysis of
acid labile compounds such as the Mtt protected amino-oxy were performed by first hanging the TLC
plate in a NEt3 vapour rich atmosphere for several seconds and adding 1-10% of NEt3 to the liquid
phase. SilaFlash® P60 (particle size 40-63μm) was used for column chromatography. NMR spectra
were recorded on Bruker DRX-400, and 300 MHz instruments and calibrated on residual undeuterated
solvent signals as internal standard. IR spectra were recorded on a Bruker Alpha FTIR.
benzyl bis(2-hydroxyethyl)carbamate
Diethanolamine (4.8 mL, 50 mmol, 1 equiv), 1,4-dioxane (85 mL) and sat. NaHCO3 (aq., 100 mL)
were mixed and cooled with an ice bath. To the white suspension a solution of Cbz-OSu (13.08 g, 52.5
mmol, 1.05 equiv) dissolved in acetone (60 mL) was added and the flask was stirred at rt overnight.
The volatiles were removed until ca. 100 mL solvent remained. The solution was extracted three times
with ethyl acetate, which was subsequently washed six times with KHSO4 (aq., 1M), and brine. After
drying over MgSO4, the volatiles were removed under reduced pressure yielding the colourless oil
(9.11 g, 38.1 mmol, 76.2%). 1H NMR (400 MHz, Chloroform-d) δ 7.39 – 7.31 (m, 5H), 5.15 (s, 2H),
3.96 (s, 2H), 3.84 (s, 2H), 3.77 (s, 2H), 3.57 – 3.42 (m, 4H)
benzyl bis(2-((1,3-dioxoisoindolin-2-yl)oxy)ethyl)carbamate
Cbz-diethanolamine (5.73 g, 23.97 mmol, 1.0 equiv), PPh3 (13.19 g, 50.34 mmol, 2.1 equiv) and N-
hydroxyphthalimide (8.19 g, 50.34 mmol, 2.1 equiv) were dissolved in distilled THF (370 mL) in a
flame dried flask.
Page 28
28
At 0°C DIAD (9.86 mL, 50.34 mmol, 2.1 equiv) was added dropwise and left to stir for 45 minutes
after which the reaction mixture was allowed to warm to rt. The reaction mixture was left to stir
overnight after which the volatiles were removed under reduced atmosphere and the yellow oil crude
product was purified by column chromatography (silica powder, 2:1 PE: EtOAc) to yield an off white
solid (6.62 g, 12.50 mmol, 53%). 1H NMR (400 MHz, Chloroform-d) δ 7.77 (m, J = 19.3, 5.5, 2.9 Hz,
8H), 7.42 – 7.23 (m, 5H), 5.18 (s, 2H), 4.45 (dt, J = 11.9, 5.4 Hz, 4H), 3.92 (dt, J = 16.3, 5.5 Hz, 4H);
13C NMR (101 MHz, Chloroform-d) δ 163.22, 155.89, 136.28, 134.34, 128.75, 128.36, 127.85,
127.69, 123.38, 77.28, 76.96, 76.64, 67.35, 47.73, 47.14; IR (cm-1
): 1733, 903, 878, 723, 700, 648.
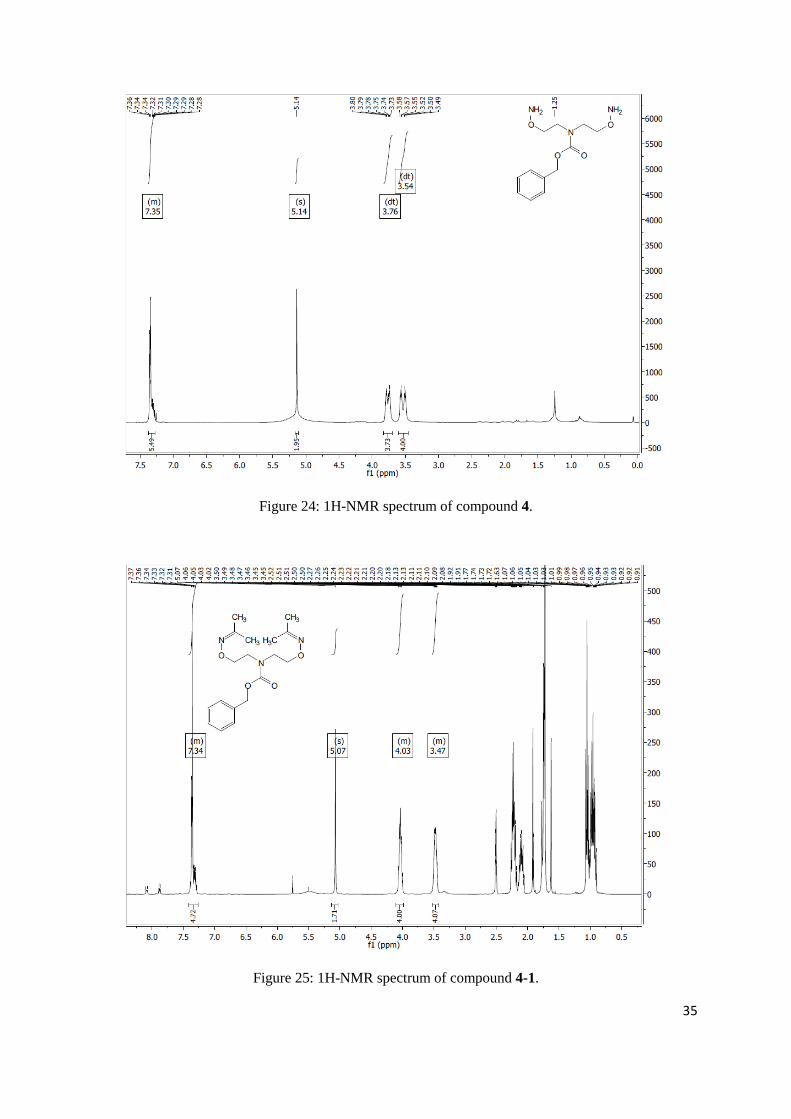
benzyl bis(2-(aminooxy)ethyl)carbamate
Acetone derivate oxime 4-1 (3.74 g, 13.9 mmol, 1.0 equiv) was suspended in distilled THF (80 mL),
after which methoxyamine (25-35 wt% HCl salt) (21 mL, 69.12 mmol, 4.97 equiv) was added. The
solution was left to stir at r.t. for 48h. The organic layer was extracted with strongly acidic water. The
water layer was subsequently made strongly basic with NaHCO3. The water layer was then extracted
with EtOAc. The organic layer was dried with MgSO4 and the volatiles were removed to yield the
product as a colourless oil (310 mg, 1.15 mmol, 8.29%). 1H NMR (400 MHz, Chloroform-d) δ 7.38 –
7.27 (m, 6H), 5.14 (s, 2H), 3.76 (dt, J = 20.0, 5.2 Hz, 4H), 3.54 (dt, J = 25.8, 5.4 Hz, 4H).
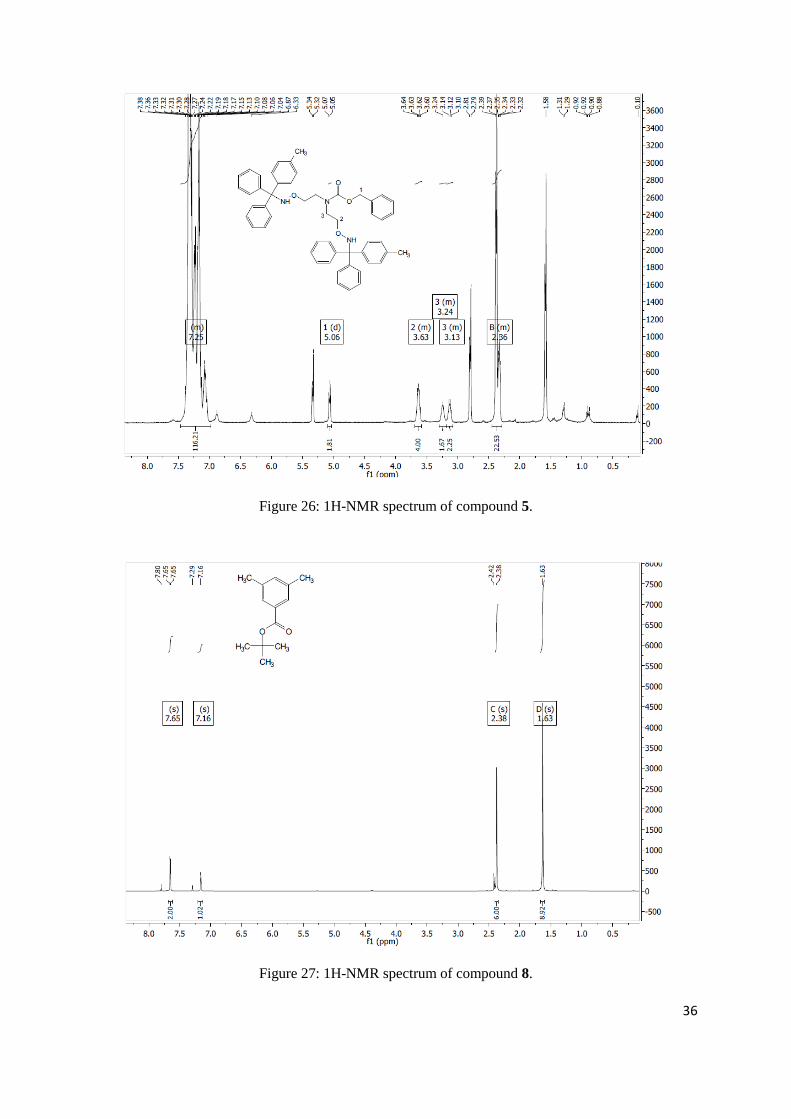
benzyl bis(2-(((diphenyl(p-tolyl)methyl)amino)oxy)ethyl)carbamate
In a flame dried flask phth protected compound 3 (1.50 g, 2.84 mmol, 1 equiv) was dissolved in
distilled THF (45 mL) after which methylhydrazine (0.18 mL, 3.41 mmol, 1.2 equiv) was added. After
2 hours of stirring a white solid had formed and DIPEA (4.9 mL, 14.2 mmol, 5.0 equiv) and MttCl
(4.15 g, 28.4 mmol, 10 equiv) were added and the reaction mixture was left to stir overnight.
The reaction mixture was washed twice with H2O and brine. After drying with K2SO4 the volatiles
were removed under reduced atmosphere to yield a yellow oil crude product which was attempted to
purify by column chromatography (silica powder pre-treated with 10% NEt3, 6:1 PE:EtOAc).
Page 29
29
No pure product was obtained, however NMR indicated product formation. 1H NMR (300 MHz,
Chloroform-d) δ 7.47 – 6.98 (m, 116H), 5.06 (d, J = 5.8 Hz, 2H), 3.69 – 3.58 (m, 4H), 3.30 – 3.19 (m,
2H), 3.19 – 3.08 (m, 2H), 2.45 – 2.29 (m, 23H).
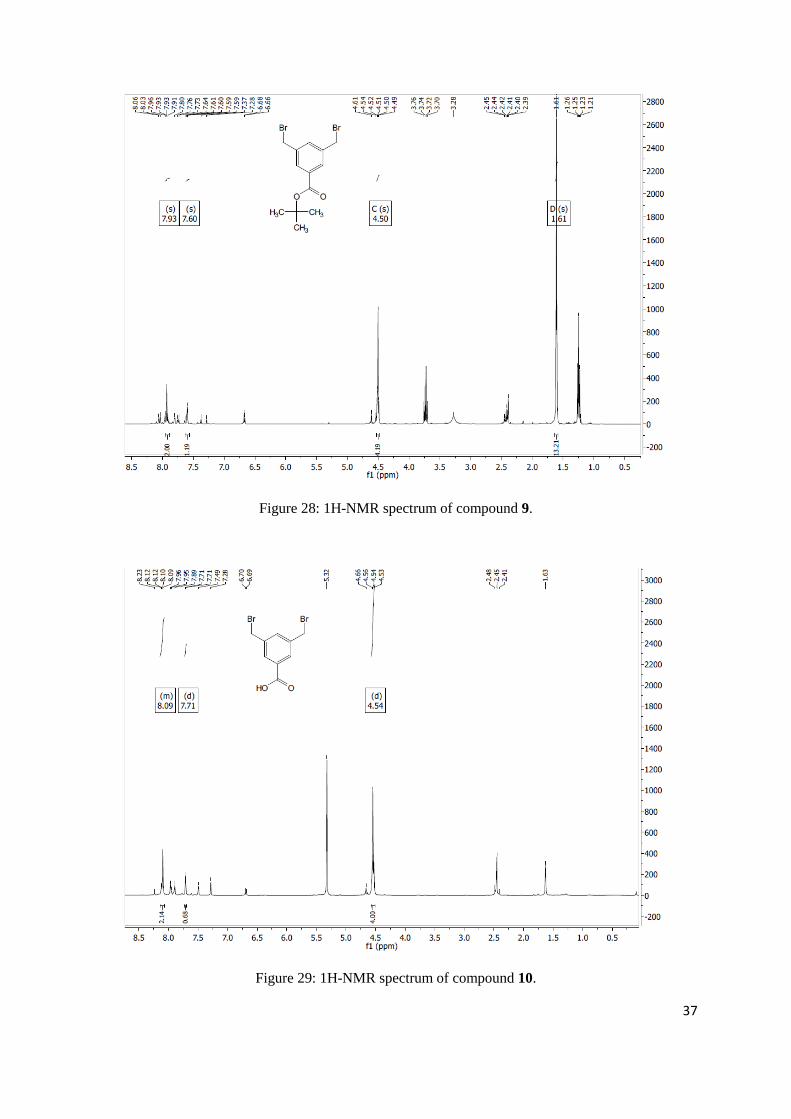
tert-butyl 3,5-dimethylbenzoate
3,5-dimethyl benzoic acid (7.25g, 48.25 mmol, 1.00 equiv) was suspended in toluene (6 mL) and
thionyl chloride (7.23 mL, 77.20 mmol, 1.60 equiv) was added and the solution was stirred at reflux
for 4 hours. The volatiles were removed and t-BuOH (9.3 mL, 96.5 mmol, 2.00 equiv) and pyridine
(4.12 mL, 51.1 mmol, 1.1 equiv) were added. After 100 hours the solution was washed with HCl
(aq., 4M), water, NaOH (aq., 2M) and water. After drying with K2CO3 the volitiles were removed to
yield a yellow oil (9.06 g, 43.92 mmol, 91%). 1H NMR (400 MHz, Chloroform-d) δ 7.65 (s, 2H), 7.16
(s, 1H), 2.38 (s, 6H), 1.63 (s, 9H).
tert-butyl 3,5-bis(bromomethyl)benzoate
To a flame dried flask under N2 atmosphere tBu ester 8 (8.90 g, 43.20 mmol, 1.00 equiv) was
dissolved in DCM (200 mL) and NBS (16.50 g, 90.76 mmol, 2.1 equiv) was added. The yellow
suspension was irradiated with a 500W lamp to reflux and the reaction was monitored using 1H-NMR.
After 1.5h it was determined the reaction was complete and the mixture was diluted with DCM and
washed with water. After drying with Na2SO4 the volatiles were removed to yield a yellow oil (5.82 g,
15.98 mmol, 31%). 1H NMR (400 MHz, Chloroform-d) δ 7.93 (s, 2H), 7.60 (s, 1H), 4.50 (s, 4H), 1.61
(s, 9H).
Page 30
30
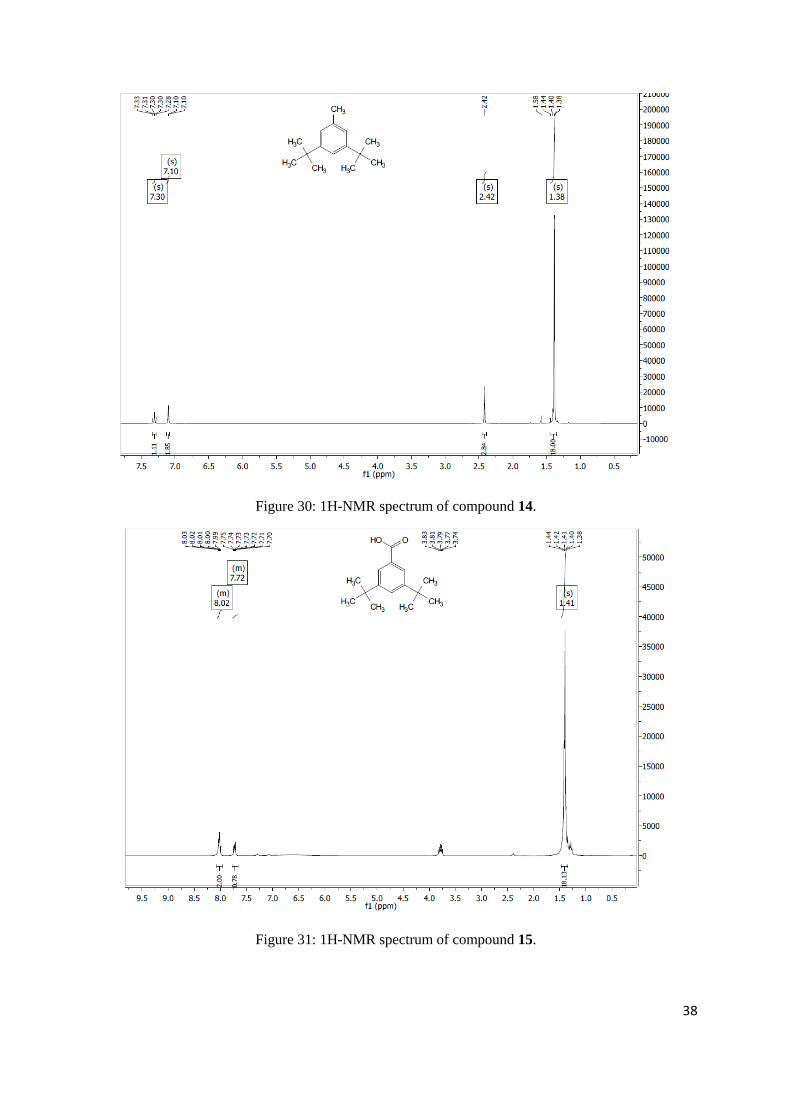
3,5-bis(bromomethyl)benzoic acid
t-Bu ester 9 (610 mg,1.68 mol, 1.00 equiv) was dissolved in DCM (6 mL) and formic acid (6 mL) was
added. The solution was stirred overnight after which the volatiles were removed to yield the product
as an off white solid (516 mg, 1.67 mmol, 99%). 1H NMR (400 MHz, Chloroform-d) δ 8.14 – 8.07 (s,
2H), 7.71 (s, 1H), 4.54 (s, 4H).
1,3-di-tert-butyl-5-methylbenzene
t-BuCl (143 mL, 1.3 mol, 2.6 equiv) and toluene (53 mL, 0.5 mol, 1.0 equiv) were added to a flame
dried 3-neck flask fitted with an oil bubbler. AlCl3 (6.07 g, 0.046 mol, 0.09 equiv) was added in small
batches and the reaction was left to stir overnight. Water was added to quench the reaction and the
product was extracted in Et2O. The crude mixture was distilled at reduced pressure to yield the product
as a colourless liquid (37.3 g, 0.18 mol, 37%). 1H NMR (300 MHz, Chloroform-d) δ 7.30 (s, 1H), 7.10
(s, 2H), 2.42 (s, 3H), 1.38 (s, 18H).
3,5-di-tert-butylbenzoic acid
In a three neck flask fitted with a reflux condenser 14 (35.99 g, 0.176 mol, 1.0 equiv) was added to a
solution of KOH (15.8 g, 0.282 mol, 1.6 equiv), pyridine (130 mL) and H2O (35 mL) and the solution
was heated to 95°C under vigorous mechanical stirring. KMnO4 (70.0 g, 0.443 mol, 2.52 equiv) was
added in portions and the suspension was left to stir overnight. The reaction mixture was then filtrated
and washed with NaOH (aq., 1M). After removal of ca. 200 mL of volatiles the mixture was made
strongly acidic with HCl (aq., 6M) and EtOAc (100 mL) was added.
Page 31
31
The organic phase was separated and the water phase was washed with EtOAc. The organic phases
were combined and washed with water until neutral. After drying over Na2SO4 the volatiles were
removed to yield a white solid which was washed with EtOH to yield the product 15 as a white crystal
(8.36 g, 0.00357 mol, 20%). 1H NMR (300 MHz, Chloroform-d) δ 8.02 (m, 2H), 7.72 (m, 1H), 1.41 (s,
18H).
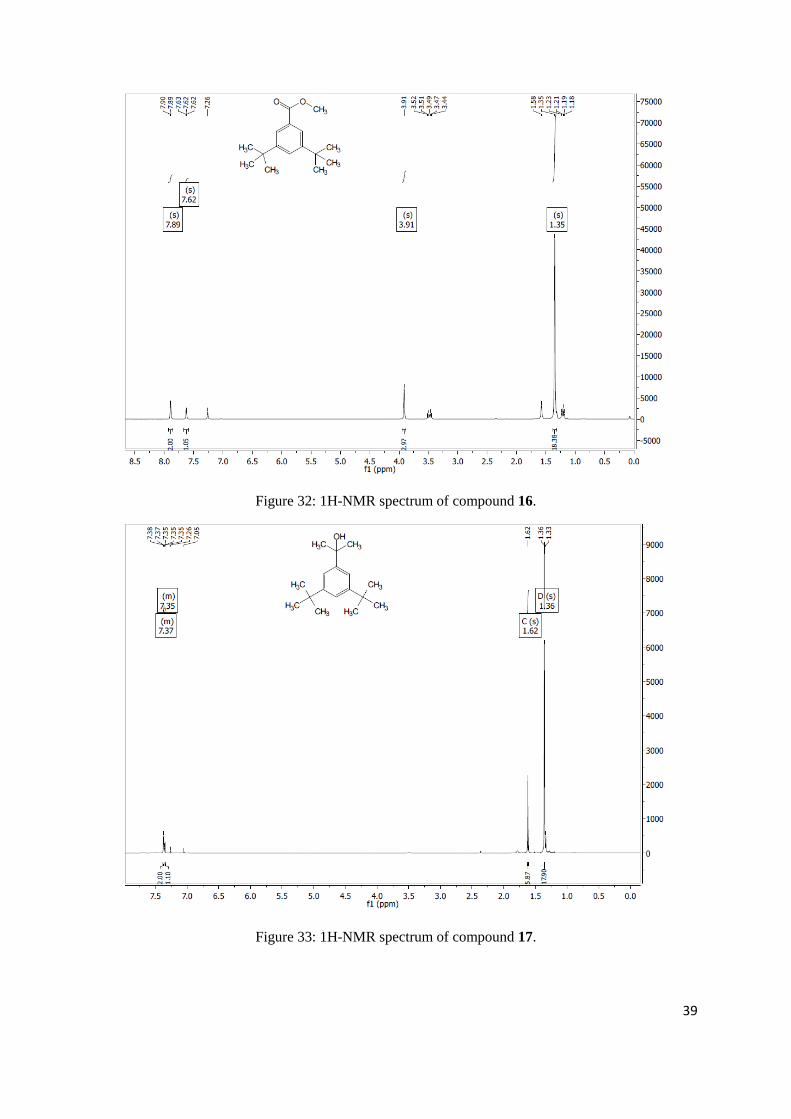
methyl 3,5-di-tert-butylbenzoate
Benzoic acid 15 (8.36 g, 35.67 mmol, 1.0 equiv) was suspended in MeOH (17 mL) and SOCl2 (1.5
mL, 20.56 mmol, 0.58 equiv) was added and the solution was heated to reflux overnight. The reaction
mixture was diluted with Et2O and washed with H2O, Na2CO3 and H2O. After drying with MgSO4 the
volatiles were removed to yield 16 as white crystals (8.16 g, 32.86 mmol, 92%). 1H NMR (300 MHz,
Chloroform-d) δ 7.89 (s, 2H), 7.62 (s, 1H), 3.91 (s, 3H), 1.35 (s, 18H).
2-(3,5-di-tert-butylphenyl)propan-2-ol
In a flame dried flask methyl-ester 16 (7.19 g, 28.95 mmol, 1.0 equiv) in distilled Et2O (25 mL) was
slowly added to MeMgBr (Et2O, 3M) (29.0 mL, 86.85 mmol, 3.0 equiv). The solution was then heated
to 35°C overnight, after which the solution was poured over ice and made strongly acidic with KHSO4
1M. The organic layer was separated and the water layer was washed with Et2O. After combining the
organic layers they were washed with Na2CO3 and H2O and dried with MgSO4. The volatiles were
removed to yield the tertiary alcohol 17 as a yellow oil (6.83 g, 27.49, 95%). 1H NMR (400 MHz,
Chloroform-d) δ 7.37 (m, 2H), 7.35 (m, 1H), 1.62 (s, 6H), 1.36 (s, 18H).
Page 32
32
2-(3,5-di-tert-butylphenyl)propan-2-yl carbonochloridate
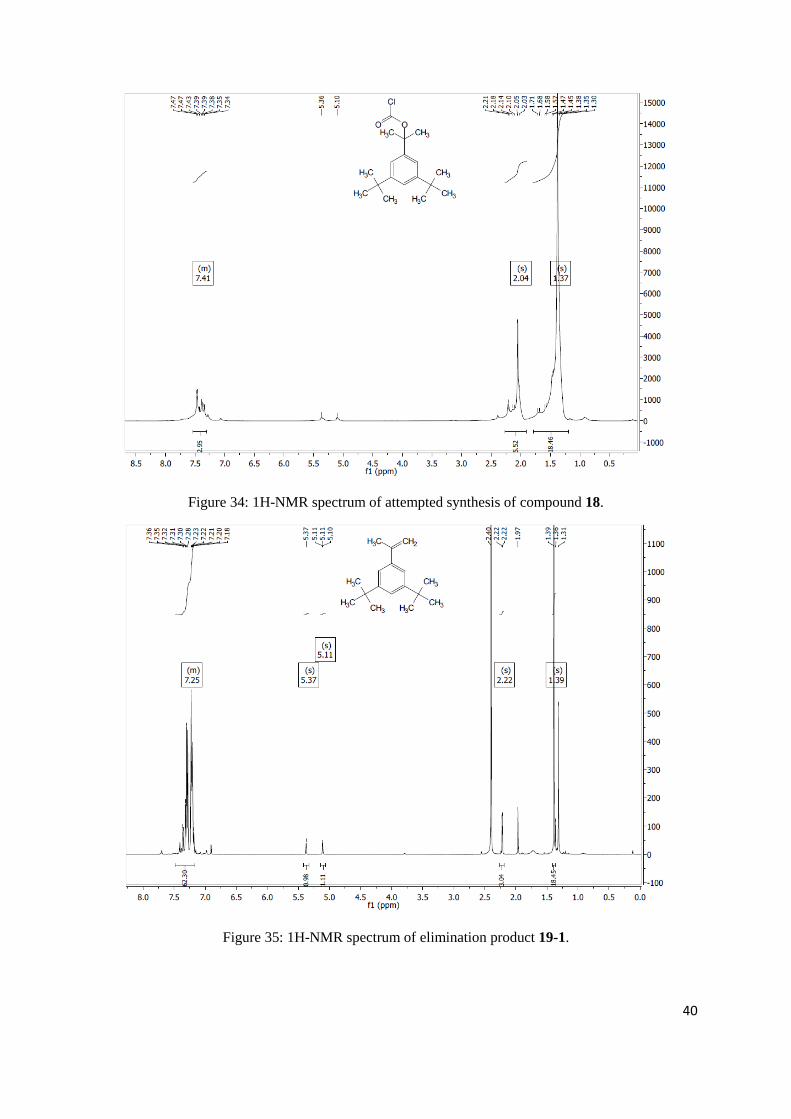
At 0°C pyridine (0.78 mL, 9.66 mmol, 1.2 equiv) and triphosgene (1.00 g, 3.36 mmol, 0.4 equiv) were
added to a solution of 17 (2.09 g, 8.40 mmol, 1.0 equiv) in DCM (50 mL). The solution was allowed
to come up to r.t. and was left overnight. IR showed no product was formed. 1H NMR (300 MHz,
Chloroform-d) δ 7.54 – 7.30 (m, 3H), 2.04 (s, 6H), 1.37 (s, 18H).
2-(3,5-di-tert-butylphenyl)propan-2-yl 1H-imidazole-1-carboxylate
To a solution of 17 (200 mg, 0.80 mmol, 1.0 equiv) in toluene (2 mL) was added CDI (182 mg, 1.14
mmol, 1.43 equiv) and the suspension was heated to 60°C overnight. Full conversion to the
elimination product 19-1 was observed. 1H NMR (400 MHz, Chloroform-d) δ 7.48 – 7.17 (m, 62H),
5.37 (s, 1H), 5.11 (s, 1H), 2.22 (s, 3H), 1.39 (s, 18H).
Page 33
33
8 Appendix
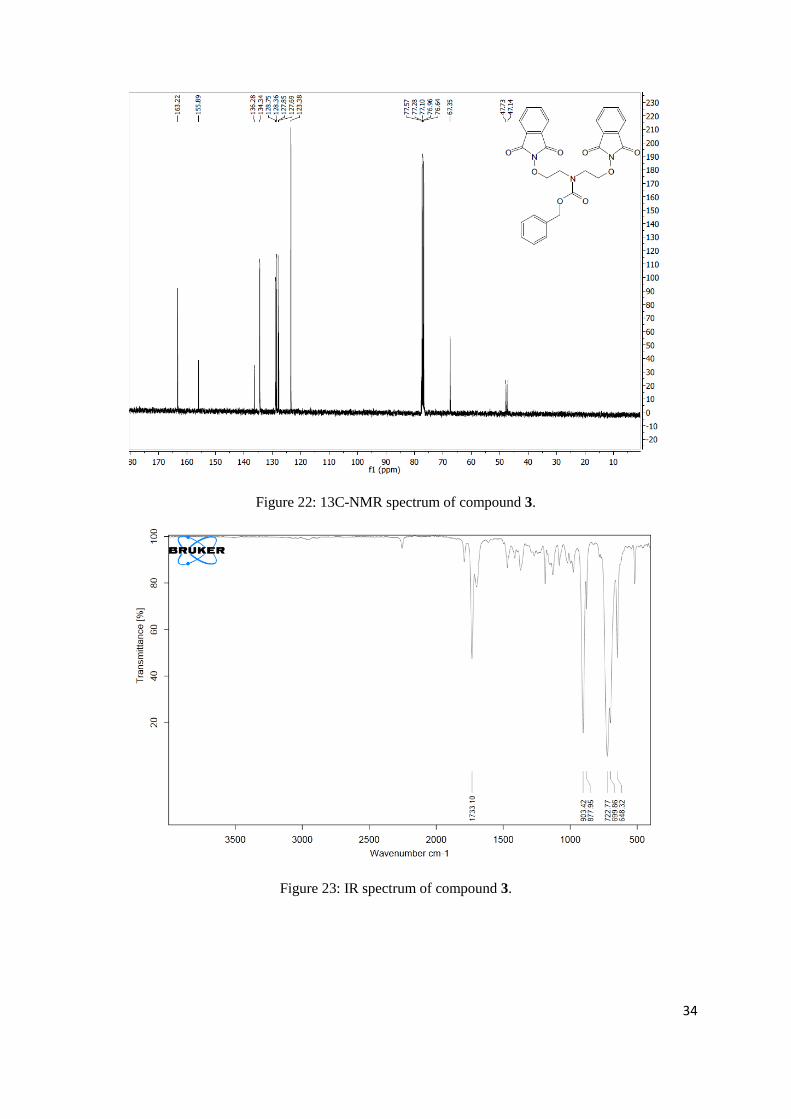
Figure 20: 1H-NMR spectrum of compound 2.
Figure 21: 1H-NMR spectrum of compound 3.
Page 34
34
Figure 22: 13C-NMR spectrum of compound 3.
Figure 23: IR spectrum of compound 3.
Page 35
35
Figure 24: 1H-NMR spectrum of compound 4.
Figure 25: 1H-NMR spectrum of compound 4-1.
Page 36
36
Figure 26: 1H-NMR spectrum of compound 5.
Figure 27: 1H-NMR spectrum of compound 8.
Page 37
37
Figure 28: 1H-NMR spectrum of compound 9.
Figure 29: 1H-NMR spectrum of compound 10.
Page 38
38
Figure 30: 1H-NMR spectrum of compound 14.
Figure 31: 1H-NMR spectrum of compound 15.
Page 39
39
Figure 32: 1H-NMR spectrum of compound 16.
Figure 33: 1H-NMR spectrum of compound 17.
Page 40
40
Figure 34: 1H-NMR spectrum of attempted synthesis of compound 18.
Figure 35: 1H-NMR spectrum of elimination product 19-1.

































![Synthesis and Antibacterial Activity of Oxime Ester Derivatives … · 2016. 7. 31. · Gram-positive bacteria only [25-28]. The oxime ester derivatives containing acrylpimaryl group](https://static.documents.pub/doc/80x56/6098c12840631e79f03687e7/synthesis-and-antibacterial-activity-of-oxime-ester-derivatives-2016-7-31-gram-positive.jpg)













