n ilola& and ce War w e Molecular and Cellular Endocrinology 107 (1995) 29-40 ELSEVIER Multiple intracellular signallings are involved in thyrotropin-releasing hormone (TRH)-induced c-fos and jun B mRNA levels in clonal prolactin cells Emmanuelle Passegue*, Jean-Lx Richard, Genevikve Boulla, Danielle Gourdji Groupe de Biologic de la Cellule Neuraendocrine, CNRS URA I1 15. Coll?ge de France, I I place Marcelin Berthelor, 75231 Paris Cedex 05, France Received 8 August 1994; accepted 8 October 1994 Abstract In mammosomatotropes GH3B6 cells, one of the primary responses to thyrotropin-releasing hormone (TRH) is the parallel induction of two proto-oncogenes, C--OS andjun B, which code for constituents of API transcription factor. To better understand the mode of action of TRH and to look for possible functions of c-fos and jun B in these cells, we have investigated the role of different intracellular signals in the induction of each proto-oncogene on the one hand, and on prolactin (PRL) release and PRL gene expression on the other hand. Northern and dot-blot analyses revealed that the activation of protein kinase C (PKC)-, Ca*+- or adenylyl cyclase-dependent pathways acutely increased both c-fos and jun B transcripts. However, a gene specific responsiveness was revealed using phorbol 12-myristate 13- acetate (TPA) and several combined treatments. The simultaneous activation of PKC and Ca*+-dependent pathways resulted in synergistic stimulations of c-j& mRNA levels only. Consistently, ionomycin plus low doses of TPA solely reproduced the potent effect of TRH on c- fos transcripts. Data collected from TRH and TPA down-regulated cells indicated that TRH probably recruits TPA-dependent PKC isoforms for stimulating c-fos but not jun B transcripts. On the contrary, the TRH-induced stimulation of either proto-oncogene likely involves Ca*‘-dependent mechanisms because calcium agonists and the peptide exert non-additive effects. Finally, the synergistic stimulations observed in response to TRH combined with forskolin, indicate that adenylyl cyclase-dependent mechanisms are jnterconnected with TRH-induced proto-oncogene expression. The overall study also reveals that among the agonists tested, the dihydropyridine Bay K 8644 and forskolin only were capable to induce a long-lasting stimulation of c-fos and jun B mRNA levels, concomitant to increased levels of PRL transcripts, as does TRH. Considering that APl is assumed to be involved in signal transmission from the cell surface to the nucleus, it might be thus proposed that a common stimulation of c-fos and jun B gene expression is possibly involved in the activation of the PRL gene. On the other hand, the systematic coincidence between acute PRL release and proto-oncogenes expression suggest a role for c-jiis and jun B in the control of genes involved in the secretory process. Keywords: Proto-oncogenes; Prolactin; Rat pituitary cells; Gene expression; Intracellular signalling 1. Introduction It is now admitted that a number of nuclear proto- oncogenes, such as fos and jun, could play a role in the cascade of events coupling membrane receptors to the nucleus (see reviews in Sheng and Greenberg, 1990; Cur- ran et al., 1993; Lin et al., 1993). Thefos and jun genes are classified into two families, each comprising several members. All the corresponding proteins contain a leucine zipper motif allowing the formation of diverse *Corresponding author, Tel.: +33 I 44 27 15 88; Fax: +33 1 44 27 10 84. dimers, constituting the transcription factor API. This diversity helped to suggest the hypothesis that fosljun could play a role of master switch at the nuclear level in the case of pleiotropic responses triggered by external signals (see reviews in Diestel and Spiegelman, 1990; Vogt and Boss, 1990). This prompted us, and others, to look for a possible role of such genes in the mode of ac- tion of thyrotropin-releasing hormone (TRH) (Weisman et al., 1987; Carr et al., 1993; Passegut? et al., 1994), a hypophysiotropic neuropeptide that primarily acts on thy- rotropes and lactotropes (see review in Metcalf and Jack- son, 1989). TRH was shown to exert multiple actions in rat pituitary prolactin (PRL) clonal cell lines namely GH3 0303-7207/95/$09.50 0 1995 Elsevier Science Ireland Ltd. All rights reserved SSDI 0303-7207(94)03417-R
Transcript
n ilola& and
ce War
w e Molecular and Cellular Endocrinology 107 (1995) 29-40 ELSEVIER
Multiple intracellular signallings are involved in thyrotropin-releasing hormone (TRH)-induced c-fos and jun B mRNA levels
Groupe de Biologic de la Cellule Neuraendocrine, CNRS URA I1 15. Coll?ge de France, I I place Marcelin Berthelor, 75231 Paris Cedex 05, France
Received 8 August 1994; accepted 8 October 1994
Abstract
In mammosomatotropes GH3B6 cells, one of the primary responses to thyrotropin-releasing hormone (TRH) is the parallel induction of
two proto-oncogenes, C--OS andjun B, which code for constituents of API transcription factor. To better understand the mode of action of TRH and to look for possible functions of c-fos and jun B in these cells, we have investigated the role of different intracellular signals in the induction of each proto-oncogene on the one hand, and on prolactin (PRL) release and PRL gene expression on the other hand. Northern and dot-blot analyses revealed that the activation of protein kinase C (PKC)-, Ca*+- or adenylyl cyclase-dependent pathways acutely increased both c-fos and jun B transcripts. However, a gene specific responsiveness was revealed using phorbol 12-myristate 13- acetate (TPA) and several combined treatments. The simultaneous activation of PKC and Ca*+-dependent pathways resulted in synergistic stimulations of c-j& mRNA levels only. Consistently, ionomycin plus low doses of TPA solely reproduced the potent effect of TRH on c- fos transcripts. Data collected from TRH and TPA down-regulated cells indicated that TRH probably recruits TPA-dependent PKC isoforms for stimulating c-fos but not jun B transcripts. On the contrary, the TRH-induced stimulation of either proto-oncogene likely
involves Ca*‘-dependent mechanisms because calcium agonists and the peptide exert non-additive effects. Finally, the synergistic stimulations observed in response to TRH combined with forskolin, indicate that adenylyl cyclase-dependent mechanisms are jnterconnected with TRH-induced proto-oncogene expression. The overall study also reveals that among the agonists tested, the dihydropyridine Bay K 8644 and forskolin only were capable to induce a long-lasting stimulation of c-fos and jun B mRNA levels,
concomitant to increased levels of PRL transcripts, as does TRH. Considering that APl is assumed to be involved in signal transmission
from the cell surface to the nucleus, it might be thus proposed that a common stimulation of c-fos and jun B gene expression is possibly involved in the activation of the PRL gene. On the other hand, the systematic coincidence between acute PRL release and proto-oncogenes expression suggest a role for c-jiis and jun B in the control of genes involved in the secretory process.
Keywords: Proto-oncogenes; Prolactin; Rat pituitary cells; Gene expression; Intracellular signalling
1. Introduction
It is now admitted that a number of nuclear proto- oncogenes, such as fos and jun, could play a role in the cascade of events coupling membrane receptors to the
nucleus (see reviews in Sheng and Greenberg, 1990; Cur- ran et al., 1993; Lin et al., 1993). Thefos and jun genes
are classified into two families, each comprising several members. All the corresponding proteins contain a leucine zipper motif allowing the formation of diverse
dimers, constituting the transcription factor API. This
diversity helped to suggest the hypothesis that fosljun could play a role of master switch at the nuclear level in the case of pleiotropic responses triggered by external signals (see reviews in Diestel and Spiegelman, 1990;
Vogt and Boss, 1990). This prompted us, and others, to look for a possible role of such genes in the mode of ac- tion of thyrotropin-releasing hormone (TRH) (Weisman et al., 1987; Carr et al., 1993; Passegut? et al., 1994), a hypophysiotropic neuropeptide that primarily acts on thy- rotropes and lactotropes (see review in Metcalf and Jack- son, 1989). TRH was shown to exert multiple actions in rat pituitary prolactin (PRL) clonal cell lines namely GH3
0303-7207/95/$09.50 0 1995 Elsevier Science Ireland Ltd. All rights reserved SSDI 0303-7207(94)03417-R
30 E. Passegue er al. I Molecular and Cellular Endocrinology 107 (1995) 29-40
or GH4C1, collectively termed GH cells thereafter. The response primarily includes the acute stimulation of pre- synthesised hormone release, as well as the activation of PRL gene transcription and ensuing long term PRL se- cretion, concomitantly with morphological changes (see reviews in Gourdji et al., 1982; Laverriere et al., 1983;
Murdoch et al., 1983).
In agreement with our working hypothesis, TRH was shown to stimulate c-fos,jun B and c-&n mRNA levels in GH3B6 cells (Weisman et al., 1987, PasseguC et al.,
1994). The stimulation of c-fos and jun B is potent and displays a similar kinetic pattern: following an early peak, a long-lasting stimulation was observed, an unusual find- ing concerning immediate early genes activated by a pep- tide. The increase in c-jun transcripts was by contrast limited and transient. On the other hand, Carr et al. (1993) reported an almost parallel triphasic stimulation of c-fos
and c-jun mRNA accumulation in GH3 cells. The control of fos and jun gene expression by second
messengers systems is firmly established in a number of models, including some endocrine cells and neurones (see
reviews in Sheng and Greenberg, 1990; Curran et al., 1993; Lin et al., 1993). The survey of the literature actu- ally reveals a cellular specificity. In this respect, very little is known regarding the glandular cells of the anterior pituitary and namely PRL secreting cells. It is no longer disputable that TRH primarily acts on GH pituitary cells via membrane receptors stimulating the catabolism of phosphatidylinositol phosphate through the activation of a G aq/a,l GTP-binding protein (Hsieh and Martin, 1992). It
is also admitted that the resulting mobilisation of intracel- lular Ca2+ stores, activation of protein kinase C (PKC)
and influx of extracellular Ca*+ (see review in Gershen- gorn, 1989) account for TRH-induced acute release of
stored PRL (Gershengorn, 1989) as well as for the activa- tion of rPRL gene transcription (Laverriere et al., 1988). On the contrary, the mechanisms by which TRH induced fos and jun expression in GH cells were not found. Fur- thermore, among the few observations reporting the effect calcium or PKC agonists, several do not coincide (Gourdji et al., 1988; Bandyopadhyay and Bancroft, 1989; Duchemin et al., 1992; Carr et al., 1993). An in-
creasing number of recent studies indicates that TRH may also act through adenylyl cyclase-dependent mechanisms
(Gordelaze et al., 1989; Paulssen et al. 1992; Gollash et al., 1993). This further questions the mechanisms respon- sible for the stimulation of fos and ;un gene expression elicited by TRH.
Finally, the physiological relevance of the activation of these nuclear proto-oncogenes in PRL secreting cells has so far been ignored. Nevertheless, in the case of TRH, this activation and the secretory response display common dose-dependency and kinetic coincidences (Passegue et al., 1994). The early and transient fodjun induction im- mediately succeeds in the first phase of TRH action on PRL release and levels off after the achievement of the
second phase. This prompted us and others to postulate that these genes could play a role in the complex and multistep adaptive processes linked to increased hormone
exocytosis (Weisman et al., 1987; Carr et al., 1993; Passegue et al., 1994). On the other hand, the early c- fosljun B induction precedes TRH-induced PRL gene
transcription whereas the sustained plateau phase accom-
panies the long-term stimulation of PRL mRNA levels and PRL secretion. This questions a possible involvement of APl (c-fos/jun B) in the regulation of the PRL gene,
which does not contain a canonical APl site but displays several APl-like sequences (see Passegue et al., 1994). Thus, in the present work, we have addressed two ques- tions: (1) does TRH stimulates c-fos and jun B expression via identical mechanisms, and are these proto-oncogenes regulated in parallel whatever the type of second messen-
ger activated; (2) is the induction of c-fos and/or jun B systematically associated with PRL release and/or PRL gene activation? To this aim, cells were submitted to
separate and combined treatments with agents activating PKC-, calcium- or adenylyl cyclase-dependent intracellu- lar mechanisms and with TRH. Changes in c-fos and jun
B transcripts were analysed by dot and Northern blotting, and systematically related to medium PRL and PRL mRNA levels. Thereby, similarities, but also noticeable differences, were detected in the intracellular pathways regulating c-fos and jun B mRNA levels, including a re- sponse to TRH that appears to act, at least in part, through mechanisms interconnected with adenylyl cyclase- dependent events. On the other hand, acute PRL release
systematically accompanied the early induction of c-fos and jun B, but increased PRL gene expression appeared
preferentially associated with stimuli that elicit sustained
proto-oncogene responses.
2. Materials and methods
2.1. Materials
Phorbol 1Zmyristate 13-acetate (TPA), forskolin, ionomycin and staurosporine were purchased from Sigma Chemical Co. (St Louis, MO, USA) and TRH from Cal- biochem (San Diego, CA, USA). [3H]Phorbol 12,13-
dibutyrate ([3H]PDBu, 42 Ci/mmol) was obtained from Amersham-France (Les Ulis, France). Bay K 8644 was
kindly provided by Dr. Franckowiak (Bayer, Wupertal- Eberfeld, Germany). All other chemicals were of analyti- cal grade and obtained from Merck (Darmstadt, Ger- many) or Prolabo (Paris, France). Oligonucleotides were synthesised with the Gene-Assembler-Plus (Pharmacia, LKB), purified by electrophoresis in a 8 M urea/8% poly- acrylamide gel, eluted and frozen at -80°C. The 40-mer antisense oligonucleotides rat c-fos and mouse jun B probes were derived from sequences located in the first exon of each gene (Curran et al., 1987; Ryder et al., 1988). The rat PRL 800 cDNA (Cooke et al., 1980) in- serted in pSPT18 plasmid and the rat cyclophilin 650
E. Passegue et al. I Molecular and Cellular Endocrinology 107 (1995) 2940 31
cDNA (Danielson et al., 1987) inserted in pSP65 plasmid
were kindly provided by Dr J.A. Martial (Universitt de Liege, Belgium) and by Dr R. Counis (Gif-sur-Yvette,
France), respectively. [y-32P]dATP (>4000 Ci/mmol) and [a-32P]dATP (>3000 Ci/mmol,) were obtained from ICN
Biomedicals France (Orsay, France).
2.2. Cell cultures GH3B6 cells, subcloned from GH3 tumour-derived rat
pituitary cells, were routinely grown as monolayers in Ham’s F12 medium supplemented with 15% heat-
inactivated horse serum (ATGC Biotechnologie, France) and 2.5% fetal calf serum (Gibco, Ireland) (Gourdji et al.,
1982). The cells used were between their seventh and
fifteenth passages. The experiments designed for mRNA and PRL secre-
tion analysis were performed as previously described (Passegut et al., 1994). In brief, 0.5 x lo6 cells were plated in 60-mm culture dishes and grown to 50-70%
confluence for 5 days in the routine serum-supplemented medium, with a medium renewal after 2 or 3 days. Then, because serum-starvation was earlier shown to enhance
the responsiveness of both proto-oncogenes to TRH (Passegut et al., 1994), cells were switched for 35 h to a
minimum serum-free medium (SF = Ham’s F12 supple- mented with 3 x 10m8 M sodium selenite, 5 pg/ml trans- ferrin, 5 U/ml penicillin and 5 @ml streptomycin)
(Weisman et al., 1987). SF medium was thereafter re- newed to perform the different treatments stated in the figure legends. 1000x stock solutions were prepared in dimethyl-sulfoxide for TPA, forskolin, staurosporine and Bay K 8644, in ethanol for ionomycin and in water for TRH. Vehicle was added in untreated groups. At the end of the treatments, cells and culture media were collected and submitted to mRNA analysis and rPRL assay, re-
spectively. In some experiments, cells were exposed to TPA
(1.6pM), TRH (50 nM) or to the PKC inhibitor staurosporine (10 nM) during the 35-h serum starvation in
SF medium. Cell monolayers were then rinsed twice with Ham’s F12 medium containing 0.1% BSA, and incubated
for 30 min at 36.5”C in SF medium supplemented with
0.1% BSA before being treated as above or exposed to [3H]PDBu (see below).
2.3. RNA analysis RNA analysis was performed using total RNA,
extracted by a guanidium thiocyanate/lithium chloride method (Laverrike et al., 1988; Passegue et al., 1994). The concentration of total RNA per sample was measured in triplicate by ethidium bromide fluorescence. C-fos and jun B mRNAs were analysed by dot and Northern blotting. For dot blotting analysis, duplicate aliquots of 5,ug per RNA sample were denaturated according to Cheley and Anderson (1984), filtered under vacuum on nitrocellulose filters and baked overnight at 60°C.
Hybridisations were performed at 65°C using single
strand c-fos orjun B oligonucleotide probes y-32P-labelled
by T4 polynucleotide kinase to a specific activity of
(l-3) x lo8 cpm&g. The hybridised dots were punched out and bound radioactivity was measured by liquid scintillation counting. Northern blotting analysis was
performed on glyoxalated RNA samples, submitted to 1% agarose gel electrophoresis and transferred onto Gene Screen membrane (NEN Research Products, Boston, MA, USA). Hybridisations with c-fos and jun B probes were performed under the conditions described above and analysed by autoradiography. RNA loading was monitored by re-hybridisation of the mem-
branes with a cyclophilin cDNA probe a-32P-labelled by random priming (Stratagene, Prime-IT@11 kit, CA,
USA).
PRL mRNAs were quantified by dot blotting, using
triplicate aliquots of 1.4 pg per RNA sample. PRL mRNAs, transcribed in vitro using SP6 polymerase, were included in each filter allowing the number of copies of PRL mRNA per dot to be determine. Pre-hybridisation and hybridisation with PRL cDNA a-32P-labelled by
nick-translation ((2-5) x lo8 cpm/pg) were performed as previously described (Laverriere et al., 1988).
2.4. Medium PRL enzyme-immunoassay PRL was quantitated in a competitive enzyme-
immunoassay (Duhau et al., 1992) performed on dupli- cate samples, using a rPRL antiserum prepared in the laboratory and rPRL conjugated to acetyl-cholinesterase
as a tracer (CEN Saclay, France). Results were expressed in nanogram-equivalents of the rat PRL-RP3 standard provided by the NIDDK.
2.5. Binding of rH]PDBu Cells were precultured as described above, excepted
that they were seeded in 24-well plates (40 000 cells per
well), and the pretreatment in SF medium was for 48 h.
[3H]PDBu binding by intact monolayers was measured according to a protocol slightly modified from Jaken et al.
(1981). SF medium (200~1) containing 7.4 pmol of [3H]PDBu (37 nM) were added per well and incubated for
1 h at 36.5”C. At the end of this period, the plates were placed on ice and the media collected. Each plate was then rinsed rapidly in four beakers containing ice-cold
0.9% NaCl. Cells were scraped, lysed and collected in a total volume of 600~1 of 0.03 N NaOH. Then 400~1 of the lysate were submitted to scintillation counting and total proteins were determined in 50-100~~1 aliquots (Bradford, 1976). Binding determined in the presence of a
500-fold excess of unlabelled PDBu was subtracted to calculate specific binding. Data obtained from four ex- periments performed in triplicate showed that a TPA pre- treatment (1.6pM) resulted in a 74% loss of [‘H]PDBu binding (40 + 7 versus 156 + 17 dpm bound per ,ug of total cell proteins).
32 E. Passegue et ul. I Molecular and Cellulur Endocrinchgy 107 (1995) 2940
2.6. Nuclear extracts and gel mobility shif assay Nuclear extracts were prepared from 35-h serum-
starved GH3B6 cells as described by Dignam et al. (1983). Nuclear proteins were quantitated (Bradford, 1976) and stored at -80°C. Fifteen micrograms of nuclear
extract were mixed on ice with 2,ug of poly(dIdC) in binding buffer (12 mM HEPES, pH 7.9, 4 mM Tris-HCl
pH 7.9, 60 mM KCl, 0.8 mM EDTA, 1 mM DTT, 12% glycerol and 3OO~giml bovine serum albumin). After
15 min at 0-4”C, 0.2 ng of double strand oligonucleotide
A 4 hours
probe y-32P-labelled by T4 polynucleotide kinase (10 000-20 000 cpm) were added alone or with competi- tors. Incubation was performed at room temperature for
30 min. DNA-protein complexes were resolved on a 5% polyacrylamide non-denaturing gel in lx TBE (90 mM Tris-borate, 1 mM EDTA), electrophoresed at 80 V,
dried and autoradiographed.
2.7. Data analysis Each treatment was performed using triplicate culture
B 8
c-fos mRNA l
1
6- z .- iij 5 .- : 4-
0 IL”
o kL_ /. .
4;ours
...... . ........ . ........ . h ..... .
0 1 IO 100 1000 10000
TPA (nM)
3
jun B mRNA *
i
30 min
n
4 hours
0 1 10 100 1000 10000
TPA (nM)
Fig. I. Dose-dependent effect of TPA on the levels of c7@ mRNA and jun B mRNA. GH3B6 cells were cultured for 5 days in serum-containing me-
dium and were serum-starved for 35 h as described in Section 2. Cells were then treated for 30 min and 4 h in fresh serum-free medium containing
TRH (50 nM) or increasing concentrations of TPA (I .6 nM to 1.6pM) as indicated. Total RNA was extracted and analysed by Northern (A) and dot
blotting (B). (A) Northern blot analysis: 1Opg of glyoxalated total RNA were loaded per lane, fractionated on 1% agarose gel, transferred onto nylon
membrane and hybridised with 32 P-labelled ~~f0.r or jun B oligonucleotide probe. After completion of the autoradiographic analysis, each membrane
was de-hybridised for 10 min in boiling water and re-hybridised with the 32P-labelled cyclophilin cDNA probe (cycle). (B) Dot-blot analysis: 5yg of total RNA were loaded per dot on nitro-cellulose membrane and hybridised with 32P-labelled c,fos or jun B oligonucleotide probe. The bound radio- activity was measured by liquid scintillation counting from duplicate dots. Results, expressed as treated/control ratios, are the means * SD (bars) of two independent experiments each performed using triplicate culture dishes per treatment (control values: c-f0.r mRNA = 64 f 8 cpm/dot; jun B
mRNA = 76 + 2 cpm/dot). The continuous lines are theoretical and were obtained by non-linear regression analysis of the experimental values as de-
scribed in Section 2. Asterisks (*) indicate differences versus control (P < 0.01). Bars when not visible are included in the symbols.
E. Passegue et al. I Molecular and Cellular Endocrinology 107 (1995) 2940 33
dishes and experiments were repeated as indicated. The
values displayed are means f SD. Each effect was ex- pressed as fold stimulations, i.e. the treated value/control
value ratio, except when indicated. Statistical compari- sons between means were done by one-way analysis of
variance using Fisher’s test. Combined treatments were performed to discriminate interacting from independent cellular pathways. To this aim, the theoretical additivity
(theoretical sum) was compared to the observed experi- mental value. Theoretical sum of a treatment combining A and B = [(A-induced fold-stimulation) + (B-induced fold-stimulation) - 11. The concentrations inducing the E&c were determined by non-linear regression analysis (Wilkinson, 1961).
3. Results
3.1. Pharmacological activation of protein kinase C-, calcium- or adenylyl cyclase-dependent mechanisms: comparison with TRH
Levels of c-fos and jun B mRNAs, as well as PRL se- cretion, were measured in cells exposed to pharmacologi- cal activators of the major second messenger-dependent mechanisms. The efficiency of each treatment was com-
pared to that of TRH, separately and by means of com- bined treatments. Two durations, at least, were tested:
30 min in order to investigate the early response, and 4 h
which corresponds to the plateau phase of TRH action (Passegue et al., 1994).
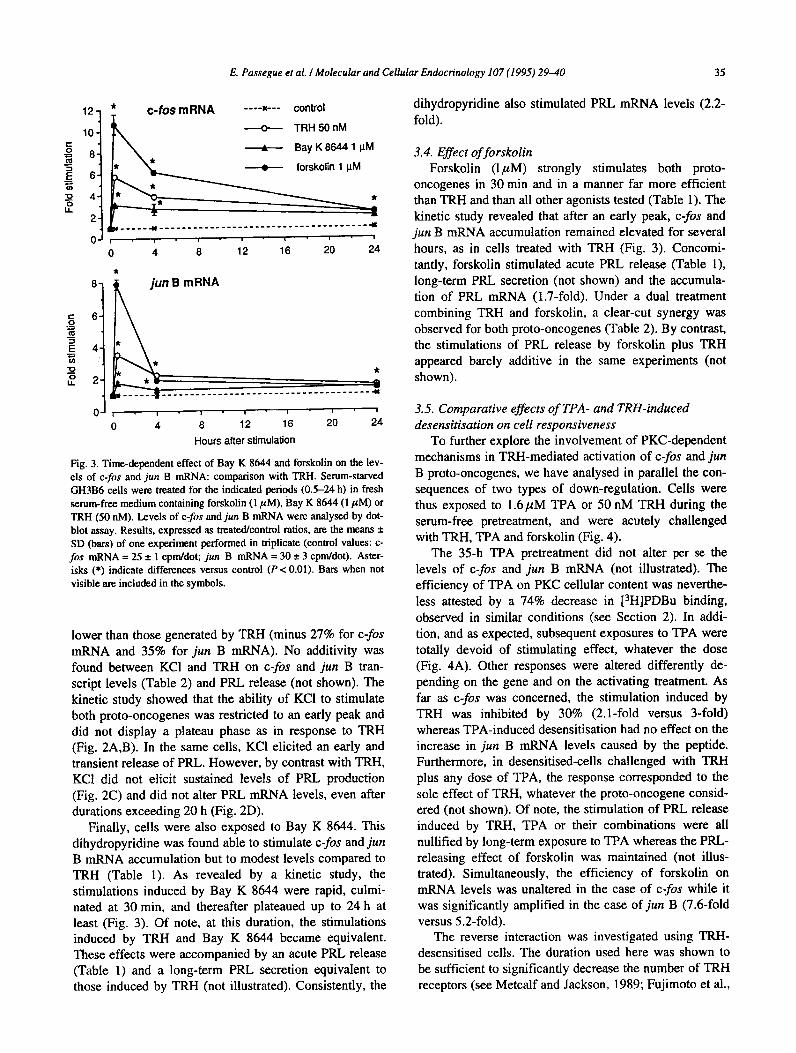
3.2. Effects of TPA A dose-dependent study (1.6 nM to 1.6,~M) was first
performed at 30 min and 4 h. In cells treated for 30 min, both proto-oncogene mRNA levels displayed a concen- tration-dependent stimulation as shown in Fig. 1. The ECso was estimated as 14 + 5 nM for c-fos and
24 + 16 nM for jun B. The maximal increases were in- duced in both cases by 1.6,uM TPA, the highest dose
tested. After a 4-h exposure, the response to TPA was dependent on the gene considered: a dose-dependent de- crease of c-fos mRNA accumulation was observed, down
Table 1
to 25% of control levels, whereas jun B mRNA levels
remained unaltered whatever the TPA concentration (Fig. 1B). Indeed, when cells were exposed for 4 h to 1.6pM
TPA, the accumulation of c-fos transcripts was consis- tently and significantly lower than in control cells (0.3- fold + 0.2) whereas jun B mRNA levels ranged control values (l.l-fold + 0. l), among seven independent ex- periments. A kinetic study, performed from 30 min to 24 h, confirmed that the ability of a high concentration of
TPA to stimulate both proto-oncogenes was restricted to an early peak culminating at 30 min (not illustrated). This study also indicated that, following the inhibition ob- served at 4 h, c-fos mRNA returned to control levels at 24 h. Concomitantly and as expected, TPA strongly in- creased acute PRL release (Table l), and elicited a mod-
erate long-term PRL secretion and PRL mRNA accumu- lation (not shown).
Compared to TRH, 16 nM of TPA was found to be less efficient for stimulating the expression of both proto- oncogenes and medium PRL accumulation in 30 min. This concentration reproduced 53%, 37% and 56% of TRH-induced c-fos and jun B mRNA accumulation and acute PRL release, respectively (Table 1). At 1 .6pM, the phorbol ester was more efficient than TRH regarding c-
fos transcript (165%) and PRL release (152%) but, still less efficient regarding jun B transcript (75%). The stimulations induced by 16 nM or 1.6pM TPA were also
found to be additive to that of TRH on proto-oncogene expression (Table 2) and PRL release (not shown).
3.3. Effects of treatments that increase intracellular calcium levels
Increases in cytosolic calcium were obtained via two different mechanisms: mobilisation of intracellular Ca2+
stores by exposure to the ionophore ionomycin (100 nM) (Albert and Tashjian, 1986) and increased Ca2+ influx
through voltage-dependent calcium channels by exposure to depolarising concentration of KC1 (30 mM) or to the dihydropyridine Bay K 8644 (1 PM).
Ionomycin promoted to similar levels the expression of C--OS and jun B mRNA within 30 min (Table 1). Com-
Effects of TRH, TPA, ionomycin, KCI, Bay K 8644 and forskolin on the levels of c-for mRNA, jun B mRNA and PRL release
TRH
50 nM
(n = 9)
TPA
16nM
(n = 7)
TPA
1.6/lM
(n = 7)
ionomycin
100 nM
(n = 3)
KCL
30 mM
(n = 3)
Bay K 8644 Forskolin
lpM l@M (n= I) (n = 2)
c-for mRNA 6.0 + 0.3a 3.2 f O.la 9.9 f 0.4a 2.3 + 0.2a 5.0 + 0.1” 3.0*0.1” 10.8 f 0.4a
jun B mRNA 4.3 *0.2a 1.6 + 0.3a 3.2 f 0.3a 2.4 f 0.1” 2.8 f 0.1” 1.8 + 0.1” 12.0 f 0.9”
PRL release 2.7 f 0.4a 1.5 +O.lh 4.1 f 0.3a 2.7 i 0.2a 2.4ztO.l” 3.4kO.l” 2.0 *0.3a
Serum-starved GH3B6 cells were treated for 30 min in fresh serum-free medium with the indicated agents. Levels of c-fos and jun B mRNA were
determined by dot blot assay and medium PRL accumulation by enzyme-immunoassay as described in Section 2 and in the legend to Fig. 1. Results,
expressed as treated/control ratios, are the means & SD of n independent experiments, each performed using triplicate culture dishes per treatment. All
treatments resulted in values different from their respective control. aP IO.01; bP 50.05) (c-fos mRNA = 23 + 5 cpm/dot, jun B mRNA =
39 + 8 cpm/dot, medium PRL = 75 + 11 @ml).
34 E. Pussepe et ~1. I Molecuiur und Cellular Endocrinnloy 107 (1995) 2940
Table 2
Effects of TRH in combination with TPA, ionomycin, KCL and forskolin on the levels of c&s andjun B mRNA
TRH 50 nM +
TPA
16nM
(II = 4)
TPA
1.6/lM
(n = 4)
ionomycin
100 nM
(n = 3)
KCL
30 mM
(n = 3)
Forskolin
tpM (n = 2)
c$os mRNA Experimental values
Theoretical sum
9.0 * 0.2 IS.1 kO.1 5.1 f 0.2 5.8 + 0.2 22.9 r I .9
Serum-starved GH3B6 cells were treated for 30 min in fresh serum-free medium with the indicated agents either alone or in combination. Levels of c-
fns and jun B mRNA were determined by dot blot assay. Results, expressed as treated/control ratios, are the means & SD of n independent experiments,
each performed in triplicate. Values observed in response to separate treatments arc included in Table I. The ‘theoretical sum’ was calculated as de-
scribed in Section 2 from the separate effect of each agent, displayed in Table I.
pared to TRH-mediated stimulations, this effect (-2.3- fold control values) ranged 38% and 56% of c-for and jurz B mRNA accumulation, respectively. When cells were
treated simultaneously with ionomycin and TRH, the stimulation elicited was lower than the theoretical sum of the separate effects. This was also true concerning PRL
release, which was actually stimulated to a similar extent
c-fos mRNA ___*__. control
d KCI 30 mM
-O- TRH50nM
* 0
0 4 8 12 16 20 24 0 4 8 12 16 20 24
B 8
1
jun B mRNA
*
O-J, . I . I , I . I ’ D . I
0 4 0 12 16 20 24
by each agent (2.7-fold). Long-term kinetics studies were not performed due to the cytotoxic action of ionomycin for durations exceeding 4 h.
Exposure of the cells to KC1 resulted in the stimulation of both proto-oncogenes, c-fos being increased to a higher extent than jun B (Table 1, Fig. 2A,B). These effects,
important compared to those induced by ionomycin, were
c 31 PRL release P4r $3
6 $2 $
'3 * 91 & 0.0
0 6 12 18 24 * *
IL” 1
* D 41 PRL mRNA T
$ , _______~____________________________________~ I- O’, . I . I . I . , , 1 (
0 4 8 12 16 20 24
Hours after stimulation
Fig. 2. Time-dependent effect of KCI on the levels of cfd.r mRNA, jun I3 mRNA, medium PRL accumulation and PRL mRNA: comparison with TRH.
Serum-starved GH3B6 cells were treated for the indicated periods (0.5-24 h) in fresh serum-free medium containing TRH (50 nM) or KC1 (30 mM).
Total RNA was extracted and analysed by dot-blot as described in Section 2 and in the legend to Fig. 1, (A) [email protected] mRNA; (B) jun B mRNA. (D) PRL mRNA. Medium PRL accumulation (C) was determined by enzyme-immunoassay. Results, expressed as treated/control ratios, except for the insert in
(C), are the means + SD (bars) of three independent experiments each performed in triplicate (control values: c,fi,s mRNA = 25 + 3 cpm/dot; jun B
mRNA = 44 f 4 cpm/dot; PRL mRNA = 1826 * I4 Mcopies/dot; medium PRL = 69 k 5 ng/ml). Asterisks (*) indicate differences versus control (P < 0.01). Bars when not visible are included in the symbols.
36
c-fos mRNA
0 no pretreatment
F%Zd TPA pretreatment
TRH TPA TPA forskolin
50 nM 16nM 1.6pM 1 PM
0 no pretreatment
LZZd TRH pretreatment
8 t
TRH TPA TPA forskolin
50 nM 16nM 1.6pM 1 PM
Fig. 4. Comparative effect of TPA and TRH pretreatment on the level5 of c-fos and jun H mRNA induced by TRH. TPA and forskolin. TPA 1.6pM (dark shaded bars), TRH SO nM (light shaded bars) or vehicle (open bars) was added during the 35 h of starvation in strum-free medium. Following
washings to eliminate TPA or TRH. cells were treated for 30 min in fresh serum-free medium as described in Section 2. Levels of C+JS and jun B
mRNA were analysed by dot blot assay. Results, expressed as trcatedicontrol ratios, are the means r SD (bars) of n independent experiments each
performed in triplicate. Asterisks (*) indicate differences versus non-pretreated cells (I’ < 0.01). (A) TPA pretreatment (n = 3). Control values: c-fi,s
mRNA = 12 + I cpm/dot; jun B mRNA = 9 t I cpm/dot. (B) TRH pretreatment (II = I). Control values: C+U mRNA = 21 + 2 cpm/dot; jun R
mRNA = I9 + 2 q&dot.
1991) and PKC activity (Kiley et al., 1990). Such cells displayed unaltered levels of c-fos and jz~z B mRNA compared to untreated cells whereas the efficiency of the peptide was attested by an increased spontaneous PRL release (1.9-fold; not illustrated). As expected, all the TRH-induced stimulations were repressed: by about 50- 55% for the expression of the proto-oncogenes (Fig. 4B) and by 35% for PRL release (not shown). The ability of
the same ceiis to respond to other stimuii was aitered dif- ferently. TPA-induced responses were either attenuated, namely for jun B mRNA levels (minus 19% for 16 nM and minus 37% for I .6pM TPA) or unaltered for c-fn.~ RNA accumulation. When TRH and TPA were added together, the stimulation was inhibited to levels corre- sponding to the sum of the separate effects observed in TRH down-regulated cells (not illustrated). Finally, TRH pretreatment inhibited by 33% the stimulation of I PM forskolin on jun B mRNA (7.5fold versus 1 l.l-fold) but did not compromise the effect of forskolin on cYfos mRNA.
3.6. Effects of staurosporine A third approach was carried out to analyse PKC-
dependent mechanisms involved in TRH action by means of staurosporine, a specific PKC inhibitor at low concen-
trations (Riiegg and Burgess, 1989). GH3B6 cells were treated with 10 nM staurosporine during the 35 h of se- rum starvation and were exposed to TRH or TPA (Fig. 5). This pretreatment did not modify, per se, the levels of c- fos and jun B mRNAs or spontaneous PRL release (not illustrated). C-fos and jurl B mRNA responses to 1.6pM
TPA feii to about 75% of controi ieveis. Staurosporine repressed all the TRH-induced stimulations studied here: the c-fos and jun B gene responses by 44% and 58%, and PRL release by more than 60%.
3.7. Efects @treatments combining a Ca2+ agonist and TPA
It was previously shown that elevated intracellular calcium levels plus TPA-dependent events reproduce TRH action on PRL release and PRL gene transcription (Laverrikre et al., 1988). Thus, GH3B6 cells were ex- posed for 30 min to TPA and ionomycin or KC1 (Table 3) and results compared to TRH-treated cells shown in Table I. Treatments combining 100 nM ionomycin plus 16 nM TPA resulted in c-fos mRNA values exceeding the theo-
E. PasseRue et ul. I Molecular and Cellular Endocrinology 107 (1995) 29-40 31
0 no pretreatment
staurospofine pretreatment
‘4 I- c-fos mRNA
8 r “” B mRTNA
6 10 6
';i 48 E 4 .- 6 ti
; 4 2
2
TRH TPA TRH TPA 50 nM 1.6kM 50 nM 1.6pM
Fig. 5. Effect of staurosporine on the levels of c-fi,s and jun B mRNA
induced by TRH and TPA. Staurosporine 10 nM (shaded bars) or vehi-
cle (open bars) was added during the 35 h of starvation in serum-free
medium. Following washings to eliminate staurosporine, cells were
treated for 30 min in fresh serum-free medium as described in Section
2. Levels of c-fk and jun B mRNA were analysed by dot blot assay.
Results, expressed as treated/control ratios, are the means + SD (bars)
of two independent experiments each performed in triplicate (control
values: c-fos mRNA = 15 -e 3 cpm/dot; jun B mRNA = 19 f 4 cpm/
dot). Asterisks (*) indicate differences versus staurosporine non-
pretnzated cells (P < 0.01).
retical sum of the separate effects, indicating a co-
operative stimulation. Of note, this mimicked the effect of TRH (6.4-fold versus 6.0-fold). By contrast, the same
treatment induced a level of jun B mRNA and medium PRL accumulations lower than those elicited by the pep-
tide and lower than the theoretical sum of separate effects. On the other hand, the treatment combining ionomycin and the high dose of TPA (1.6pM) resulted in a co- operative stimulation of both C-$X and jun B mRNA lev- els, but only in a partial additivity (-67%) on induced
PRL release. Finally, the effects of TPA (16 nM or 1.6,~M) were found strictly additive to that of KC1
(30 mM) on the level of both mRNA species as well as on
PRL release (not illustrated).
Altogether, the data presented validated the search for nuclear target(s) of APl or API-related proteins in
GH3B6 cells. This was initiated by investigating the overall API binding activity by the means of gel retarda- tion assays.
3.8. Binding of GH3B6 nuclear proteins to API sites In the presence of GH3B6 nuclear extracts, a major
retarded complex was revealed using an oligonucleotide probe encompassing the consensus APl site of the human
collagenase promoter [TGACTCA] (Angel et al., 1987) (Fig. 6). A minor and less retarded complex was also de- tected. Homologous competitions with unlabelled oli- gonucleotides resulted in a dose-dependent decrease in the major band (not illustrated), which eventually disap- peared (lane 10). In an attempt to relate this API binding
activity to the regulation of PRL gene transcription, we have performed competing experiments using a 32-bp fragment of the rat PRL gene. This sequence, located at positions -106/-75 from the transcription start site, was shown to contain cis elements important for basal and hormonally regulated PRL gene expression (Jackson et
al., 1992). As illustrated in Fig. 6, incubation with in- creasing amounts of this competitor abolished the major
and minor retarded complexes. This indicates that ele- ments included in the -106/-75 rPRL sequence compete
for the binding of protein(s) to the consensus APl site, although this oligonucleotide does not comprise consen-
sus API sites.
4. Discussion
A pharmacological study was carried out to identify the intracellular pathways controlling c-fos and jun B mRNA accumulation in GH3B6 cells, particularly in re- sponse to the neuropeptide TRH. It demonstrates that
both transcripts are augmented in response to treatments
Table 3
Combined effects of calcium activating agents and TPA on the levels of c+s and jun B mRNA
Cells were treated and RNA analysed as stated in the legend to Table 2. Results, expressed as treated/control ratios, are the means rt SD of two inde-
pendent experiments each performed in triplicate. Values observed in response to sepamte treatments are included in Table I. The ‘theoretical sum’
was calculated as described in Section 2 from the separate effect of each agent, displayed in Table I.
38 E. Pussegue et ~1. I Molecular und Cellular Endocrinolqqy IO7 (I 99s) 2940
2Pj i 1P TATA box
-1061-75 rPRL oligonucleotide
B
competitor: -106/-75 rPRL APl
I
1 2 3 4 5 6 7 0 910
Fig. 6. API-binding activity in GH3B6 cells. (A) Schematic represen-
tation of the S’-flanking region of the rat PRL gene illustrating the
localisation of the sequence corresponding to the -106/-75 rPRL oli-
gonucleotide probe. IP and 2P correspond to Pit-l binding sites (grey
boxes). Numbers are in base pairs relative to the transcription initiation
site (arrow). (B.) Serum-starved GH3B6 cell nuclear extracts were
incubated as indicated in Section 2 with 25mer 32P-lahelled oligonu-
cleotide probes (0.2 ng) containing the consensus APl site of the hu-
man collagenase promoter [TGACTCA] (Angel et al., 1987). Competi-
tions were performed with the indicated unlabelled oligonucleotides
(competitor): Lanes 2-9, 0. I -. I-, S-, IO-. SO-. IOO-, 500- and I OOO-fold
excess of unlabelled -106/-75 rPRL; lane IO, IOOO-fold excess of
unlabelled AP 1. DNA-protein complexes were resolved on a 5% poly-
acrylamide non-denaturing gel. Arrows indicate the free probe
(arrowhead) and complexes (arrow).
increasing specifically intracellular Ca*+ levels or activat-
ing protein kinase C or adenylyl cyclase. This is in accor- dance with the identification of cis responsive elements allowing transcriptional activation of the c-fos and jun B genes by PKC-dependent and Ca2+/cAMP-dependent mechanisms (Velcich and Ziff, 1990; Perez-Albuerne et al., 1993). The overall study, however, reveals that the response varies, in amplitude and/or time-course, with the signalling system on the one hand, and between the two proto-oncogenes, on the other hand. Namely, a majority of treatments stimulate c-fos transcript levels with a higher efficiency than jun B transcript. The overall study also reveals that the occurrence of persistent stimulations of c-fos and jun B mRNA levels depends on the stimulus. In turn, the time course of the responses to KCL or micro- molar concentrations of TPA argue against a former pro- posal, from us and others, suggesting that the mechanisms involved in shutting of the c-fos gene were not efficient in
GH pituitary cells (Weisman et al., 1987; Bandyopadhyay and Bancroft, 1989; Carr et al., 1993).
As far as Ca2+-evoked stimulations are concerned, the responses depend on the calcium activating treatment. In terms of acute efficiency, KC1 is the more potent and only ionomycin is capable of activating both proto-oncogenes
to the same, although modest, level. From a kinetic point of view, only Bay K 8644 is able to elicit a long-lasting stimulation. The findings concerning c-fos are consistent
with our preliminary observations (Gourdji et al., 1988) and with the ionomycin-induced stimulation observed by Carr et al. (1993). They contrast, however, with the ab-
sence of effect or with the inconsistent increase reported by Duchemin et al (1992) in response to KCL or Bay K 8644. As compared to TRH effect, the increases elicited
by any calcium agonist were lower, and when combined to the peptide no additivity was revealed. This is in ac- cordance with the idea that TRH induces c-fir and jun B gene expression at least in part via calcium-dependent mechanisms, as for PRL release and PRL gene expression (see in Gershengorn, 1989; Laverribe et al., 1988).
The positive control of c-fos and jun B gene expression by protein kinase C-dependent mechanisms is shown here by the direct dose-dependent stimulations in cells exposed to TPA for 30 min. Furthermore, the partial inhibition of TRH-induced stimulation by staurosporine indicates that
PKC-dependent mechanisms likely subserve TRH effects. Nonetheless, the PKC- dependent pathways triggered by
TRH are not totally clear since the stimulations elicited by TPA on c-fos and jun B transcripts were found to be fully additive to that of TRH in 30 min. To conciliate these findings with the unquestionable capacity of TRH to acti-
vate PKC (see review in Gershengorn, 1987), one may speculate that by analogy with other GH cell lines, mul- tiple PKC isoforms are probably expressed in GH3B6
cells. It is clearly established that these isozymes are not similarly activated and down-regulated by TRH and phorbol esters. Namely, TRH selectively down-regulates
E-PKC only whereas TPA desensitises several isoforms
(a-, /?-, 6- and E-PKC isozymes) (Akita et al., 1990; Kiley et al., 1990, 1992). These observations might also explain that only the TRH-mediated stimulation of C--OS gene was partially inhibited in TPA-desensitised cells, whereas in TRH-desensitised cells only the TPA-mediated stimula- tion of jun B gene was attenuated. Apart from differences between the mode of action of TPA and TRH, these ob- servations illustrate that the mechanisms controlling both proto-oncogenes are not identical; the stimulation of the c-for gene by TRH appears partially dependent on TPA- sensitive PKC isozymes whereas TRH-induced stimula- tion of jun B seems to involve TPA-insensitive PKC isozymes. Such differential control regarding PKC- dependent pathways is further substantiated by the re- sponses to TPA after the initial early peak; when meas- ured after 4 h, increasing doses of TPA actually result in a dose-dependent decrease in c-for mRNA levels whereas
E. Passegue et al. I Molecular and Cellular Endocrinology 107 (1995) 2940 39
expression of the jun B gene meets control values. In agreement, a kinetic study (not shown) revealed that fol-
lowing the early and transient peak elicited by 1.6pM TPA, jun B transcripts returned directly to control levels within 24 h. Meanwhile, the amounts of c-fos mRNA decreased under control levels and then slowly increased to reach control values in 24 h
The fact that TRH stimulates C-$X and jun B gene ex- pression through partially distinct intracellular mecha- nisms is also supported by data acquired using cells si- multaneously exposed to ionomycin and TPA. This treatment results in a synergistic stimulation of c-fos mRNA levels, in agreement with preliminary observa- tions (Gourdji et al., 1988) and with reports from
Duchemin et al. (1992) and Carr et al. (1993). Such recip- rocal co-operation between Caz+- atid PKC-dependent
pathways is not clear-cut regarding jun B transcripts. Of note, the occurrence of this cross-talk is conditioned by the mechanism by which cytosolic calcium is increased; treatments combining KC1 and TPA, yielded complete additive stimulations on all the parameters studied here.
Finally, apart from these mechanisms classically asso- ciated with the TRH receptor, several observations indi- cate that the TRH signalling pathway that activates c-fos and jun B expression is probably interconnected with adenylyl cyclase-dependent mechanisms. The first indi- cation was that forskolin, a very potent stimulus per se, interacts positively with TRH in stimulating c-fos and jun
B mRNA levels. Furthermore, in TPA down-regulated and TRH-desensitised cells, the stimulating effect of forskolin on the levels of jun B transcripts was modified, positively in the first case and negatively in the second. In
contrast, changes in C-$X transcript levels or PRL release were unaltered. Cross-talks between adenylyl cyclase- and phospholipase C-dependent signalling pathways,
have already been reported in GH cells (Summers and Cronin, 1988). They are consistent with the present con-
cept of an integrated network versus a linear cascade of signal transmission (see review in De Vivo and Iyengar, 1994). An increasing number of reports now indicate that the TRH receptor may be linked to divergent intracellular pathways. This includes a coupling to adenylyl cyclase via the activation of the Gs stimulatory GTP-binding protein (Paulssen et al., 1992), the involvement of the
Gaiz and Gai3 (Gollash et al., 1993), and the activation of tyrosine phosphorylation (Ohmichi et al., 1994).
The second question asked in this work was whether
the induction of C-$X and/or jun B was systematically associated with an increased PRL release and/or PRL gene activation. The data show that such coincidence ex- ists regarding acute PRL release under separate and combined treatments. Nevertheless, no strict relationship was observed between the respective amplitudes of the induced release and the stimulation of either proto- oncogene. Thus, the hypothesis for an involvement of c- fos and/or jun B proto-oncogenes in TRH-induced acute
secretion requires additional experimentation. Regarding a possible physiological link between c-fosljun B induc-
tion and PRL gene expression, indirect arguments only were obtained, as expected from the experiments per- formed. Examination of the results actually reveals that increases in PRL mRNA and long-term PRL secretion were not systematically observed and, when present, were associated with the induction of a sustained plateau phase as in response to TRH, Bay K 8644 and forskolin, all previously shown to increase PRL gene transcription. In contrast, KC1 is unable to evoke a sustained elevation of proto-oncogene mRNA levels and to induce the accumu- lation of PRL mRNA. Thus, the hypothesis that c-Fos and or Jun B might play a role in the activation of PRL gene transcription still deserves consideration. In this respect, it is valuable to note that the binding of GH3B6 nuclear extracts to a canonical API site is inhibited, in a dose- dependent manner, by a DNA probe derived from the sequence of the rat PRL proximal promoter, which is de- void of a canonical APl site. This sequence, located be- tween two Pit-l binding sites (1P and 2P), confers re-
sponsiveness to TRH and TPA (Jacken et al., 1992) as well as to CAMP-induced stimulations (Liang et al., 1992). Interestingly, Kim et al. (1993) have recently shown that an APl-like factor binds to the promoter of the human thyrotropin p gene, and that this binding is necessary together with the binding of Pit-l to permit the trans-activation induced by TRH in transient assays.
This study reveals that TRH triggers calcium, PKC but also adenylyl cyclase-dependent signallings for stimulat- ing c-fos and jun B genes in GH3B6 prolactin cells. Nev- ertheless, a gene specific responsiveness is also revealed concerning the mode of action of TRH, which is probably
due to a differential control of both genes by PKC- dependent pathways. This allows us to speculate that c-fos and jun B could be involved in common but also in dis- tinct cellular functions in these cells. Their identification should be facilitated by the knowledge of targets for c- Fos and Jun B oncoproteins in this model.
Acknowledgements
E. Passegut is supported by a fellowship from the
Minis&e de la Recherche et de la Technologie. We thank C. Pennarun for the preparation of half-tone illustrations
and Drs J.-N. Laverribre, E. Vila-Porcine and V. Ngo for reviewing the manuscript. This work was supported by
the Centre National de la Recherche Scientifique (URA 1115) and a grant from Association de la Recherche con- tre le Cancer (ARC-600084).
References
Akita, Y., Ohno, S., Yajima, Y. and Suzuki, K. (1990) Biochem. Bio-
phys. Res. Commun. 172, 184-189.
Albert, P.R. and Tashjian, Jr., A.H. (1986) Am. J. Physiol. 251, (387.
40 E. Passepe et al. I Moleculur und Cellular Endocrinology IO7 (1995) 29-40
Angel, P., Imagawa, M., Chiu, R., Stein, B., Imbra, R.J., Rahmsdorf, Jackson, S.M., Keech, C.A., Williamson, D.J. and Gutiertez-Hartmann.
H.J., Jonat, C., Herrlich, P. and Karim, M. (1987) Cell 49.729-739. A. (1992) Mol. Cell. Biol. 12,2708-2719.
Bandyopadhyay, S.K. and Bancroft, C. (1989) 1. Biol. Chem. 264, Jaken, S., Tashjian, Jr., A.H. and Blumberg, P.M (1981) Cancer Res.