Research paper Myocardial ER chaperone activation and protein degradation occurs due to synergistic, not individual, cold and hypoxic stress Kanika Jain, Geetha Suryakumar * , Rajendra Prasad, Som Nath Singh, Lilly Ganju Cellular Biochemistry Group, Defence Institute of Physiology and Allied Sciences, Lucknow Road, Timarpur, Delhi 110054, India article info Article history: Received 19 October 2012 Accepted 19 June 2013 Available online 29 June 2013 Keywords: Heart Hypobaric hypoxia Cold Chaperones ER stress abstract Environmental stress at high altitude affects the myocardium at the physiological and molecular level. Characterized by hypobaric hypoxia and low temperatures, the cumulative impact of these stressors on the protein folding homeostasis in the heart is yet unexplored. The present study evaluates the collective effect of cold and hypoxia on the myocardial protein oxidation and activation of the endoplasmic re- ticulum (ER) stress response. Adult rats were exposed to either a singular acute stress of cold (10 C; C), hypobaric hypoxia (7620 m; H) or simultaneously to both cold and hypobaric hypoxia (CH) for 6 h. Hypoxic stress amplified the free radical generation in H and CH groups, leading to enhanced HIF-1a expression. Coupled to cold stress, reduced oxygen availability caused substantial protein oxidative modifications, as well as cardiac tissue injury and matrix remodeling, evident in the histological staining. Presence of oxidized proteins caused a significant upregulation in expression of ER chaperones GRP78 and PDI in the cold hypoxia exposed animals. Enhanced proteolytic activity signaled the removal of misfolded proteins. Linked intricately to cellular stress response, cell survival kinases were expressed higher in CH group; however apoptotic CHOP (C/EBP homologous protein) expression remained unal- tered. Administration of ER stress inducer, tunicamycin along with cold hypoxic stress, caused a discernible increase in protein oxidation and GRP78 expression, along with a significant elevation in proteasome and apoptotic activity. Highlighting the significance of a synergistic, rather than individual, effect of low oxygen and temperature on the protein folding machinery, our study provides evidence for the activation of ER stress response in the myocardium under acute high altitude stress. Ó 2013 Elsevier Masson SAS. All rights reserved. 1. Introduction High altitude conditions are emphasized by diminished ambient oxygen pressure, decreased temperature, lower humidity, and increased ultraviolet radiation. These hostile environmental con- ditions require a high degree of physiological and metabolic ad- aptations to preserve function and ensure long term survival; indeed prolonged exposure may have adverse medical conse- quences for those inhabiting high altitude regions. Hypoxia induces adaptive changes in the myocardium at systemic and cellular levels, thereby having a profound effect on the morphology and function of the cardiopulmonary system [1]. Physiologically, this leads to an increase in heart rate, myocardial contractility, and cardiac output. Chronic high altitude exposure can cause an in- crease in hypoxic pulmonary vasoconstriction, with the remodeling of the pulmonary arterioles, leading to pulmonary hypertension and right ventricular hypertrophy [2]. Growing evidence has shown the exacerbated production of reactive oxygen species (ROS) generation under acute hypoxic condition leading to oxidative damage to proteins, lipids and nucleic acids [3e5]. The fall in temperature in the high altitude milieu can also disrupt the physiological systems in the body. Although cold exposure decreases the metabolic rate, it also alters the microcir- culation and affects the oxygen supply to the tissues. While mild hypothermia has been shown to be protective against myocardial ischemic injury and acute hypoxic stress [6e10], chronic low tem- peratures have been shown to have a marked impact on the cellular redox homeostasis [11,12]. Despite the evident decrease in tem- peratures as one ascends to high altitude, the cumulative effect of hypoxia and cold has been unexplored in most studies elaborating the impact of such environmental conditions on the mammalian physiology. A study by Templeman et al., in 2010 identified the cardiac ventricular remodeling that occurs in response to chronic hypoxia and cold [13]. They found that the cardiac muscles respond * Corresponding author. Tel.: þ91 11 23883311; fax: þ91 11 23914790. E-mail address: [email protected](G. Suryakumar). Contents lists available at SciVerse ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi 0300-9084/$ e see front matter Ó 2013 Elsevier Masson SAS. All rights reserved. http://dx.doi.org/10.1016/j.biochi.2013.06.018 Biochimie 95 (2013) 1897e1908
Transcript
at SciVerse ScienceDirect
Biochimie 95 (2013) 1897e1908
Contents lists available
Biochimie
journal homepage: www.elsevier .com/locate/biochi
Research paper
Myocardial ER chaperone activation and protein degradation occursdue to synergistic, not individual, cold and hypoxic stress
Kanika Jain, Geetha Suryakumar*, Rajendra Prasad, Som Nath Singh, Lilly GanjuCellular Biochemistry Group, Defence Institute of Physiology and Allied Sciences, Lucknow Road, Timarpur, Delhi 110054, India
a r t i c l e i n f o
Article history:Received 19 October 2012Accepted 19 June 2013Available online 29 June 2013
0300-9084/$ e see front matter � 2013 Elsevier Mashttp://dx.doi.org/10.1016/j.biochi.2013.06.018
a b s t r a c t
Environmental stress at high altitude affects the myocardium at the physiological and molecular level.Characterized by hypobaric hypoxia and low temperatures, the cumulative impact of these stressors onthe protein folding homeostasis in the heart is yet unexplored. The present study evaluates the collectiveeffect of cold and hypoxia on the myocardial protein oxidation and activation of the endoplasmic re-ticulum (ER) stress response. Adult rats were exposed to either a singular acute stress of cold (10 �C; C),hypobaric hypoxia (7620 m; H) or simultaneously to both cold and hypobaric hypoxia (CH) for 6 h.Hypoxic stress amplified the free radical generation in H and CH groups, leading to enhanced HIF-1aexpression. Coupled to cold stress, reduced oxygen availability caused substantial protein oxidativemodifications, as well as cardiac tissue injury and matrix remodeling, evident in the histological staining.Presence of oxidized proteins caused a significant upregulation in expression of ER chaperones GRP78and PDI in the cold hypoxia exposed animals. Enhanced proteolytic activity signaled the removal ofmisfolded proteins. Linked intricately to cellular stress response, cell survival kinases were expressedhigher in CH group; however apoptotic CHOP (C/EBP homologous protein) expression remained unal-tered. Administration of ER stress inducer, tunicamycin along with cold hypoxic stress, caused adiscernible increase in protein oxidation and GRP78 expression, along with a significant elevation inproteasome and apoptotic activity. Highlighting the significance of a synergistic, rather than individual,effect of low oxygen and temperature on the protein folding machinery, our study provides evidence forthe activation of ER stress response in the myocardium under acute high altitude stress.
� 2013 Elsevier Masson SAS. All rights reserved.
1. Introduction
High altitude conditions are emphasized by diminished ambientoxygen pressure, decreased temperature, lower humidity, andincreased ultraviolet radiation. These hostile environmental con-ditions require a high degree of physiological and metabolic ad-aptations to preserve function and ensure long term survival;indeed prolonged exposure may have adverse medical conse-quences for those inhabiting high altitude regions. Hypoxia inducesadaptive changes in the myocardium at systemic and cellularlevels, thereby having a profound effect on the morphology andfunction of the cardiopulmonary system [1]. Physiologically, thisleads to an increase in heart rate, myocardial contractility, andcardiac output. Chronic high altitude exposure can cause an in-crease in hypoxic pulmonary vasoconstriction, with the remodeling
: þ91 11 23914790.. Suryakumar).
son SAS. All rights reserved.
of the pulmonary arterioles, leading to pulmonary hypertensionand right ventricular hypertrophy [2].
Growing evidence has shown the exacerbated production ofreactive oxygen species (ROS) generation under acute hypoxiccondition leading to oxidative damage to proteins, lipids andnucleicacids [3e5]. The fall in temperature in the high altitude milieu canalso disrupt the physiological systems in the body. Although coldexposure decreases the metabolic rate, it also alters the microcir-culation and affects the oxygen supply to the tissues. While mildhypothermia has been shown to be protective against myocardialischemic injury and acute hypoxic stress [6e10], chronic low tem-peratures have been shown to have amarked impact on the cellularredox homeostasis [11,12]. Despite the evident decrease in tem-peratures as one ascends to high altitude, the cumulative effect ofhypoxia and cold has been unexplored in most studies elaboratingthe impact of such environmental conditions on the mammalianphysiology. A study by Templeman et al., in 2010 identified thecardiac ventricular remodeling that occurs in response to chronichypoxia and cold [13]. They found that the cardiac muscles respond
K. Jain et al. / Biochimie 95 (2013) 1897e19081898
to the two stressors simultaneously but not additively, whenexposed to environmentally realistic hypoxic cold.
Enhanced free radical generation orchestrates a shift in theredox status and alteration in the protein homeostasis in thecardiac tissue. In response, there occurs a reprogramming inthe protein content of the heart, involving the upregulation ofspecific proteins and a general down-regulation of myocardialprotein synthesis [14e16]. Stadtman in 1986 [17] proposed thatpartial oxygen pressure influences the rate of protein turnover andoxidative modifications of amino acids, having a considerableimpact on cellular homeostasis. High altitude training increased thelevels of protein carbonyl derivatives, known indicators of oxidativemodifications of proteins, in skeletal muscle of rats [18]. Alteredskeletal muscle protein turnover under chronic hypobaric hypoxia,accompanied by an upregulation in the ubiquitineproteasomesystem and calpain activity, was recently shown by our group [19].
In the endoplasmic reticulum (ER), the site for protein folding,there are adaptive programs to detect the misfolding of protein andmediate comprehensive defense signals to remove such aberrantproteins and if needed, increase the folding capacity of the cell.Such sophisticated cellular mechanisms, known collectively asprotein quality control, are highly sensitive to even minor pertur-bations in the redox state of the cell [20]. An accumulation ofmisfolded proteins in the ER lumen results in activation of theunfolded protein response (UPR) [21]. Having been implicated inmyocardial ischemia, hypertrophy, cardiomyopathy and heart fail-ure, the UPR causes simultaneous activation of both adaptive andpro-apoptotic pathways to deal with the load of improper folding ina number of cardiac pathophysiologies [22e25]. Recent evidencehas indicated that this highly ubiquitous response is activated inpulmonary hypertension, associated with high altitude exposure,and its attenuation may be a novel therapeutic target for thetreatment of high altitude pathologies [26,27].
Although each stress paradigm at high altitude affects themyocardium, low oxygen tension and temperature, singularly andin a synergistic manner, may cause a significant disruption in themyocardial proteostasis. The present study was performed toidentify the effect of such simulated stress conditions of cold andhypobaric hypoxia, individually and in combination, on the proteinoxidation within the heart and the subsequent activation of thecytoprotective responses to alleviate the misfolded protein load.We show that the protein homeostasis is most substantially alteredon exposure to simultaneous cold hypoxic stress and the interfer-ence in ER function leads to the activation of ER stress response inthe heart under such conditions.
2. Materials and methods
2.1. Chemicals and reagents
All chemicals, including tunicamycin, were obtained from Sigma(St. Louis, MO, USA) and were at least analytical grade. All anti-bodies were from Sigma (St. Louis, MO, USA) and Santa CruzBiotechnology (Santa Cruz, Ca, USA). Nitrocellulose membrane wasfrom Millipore (Millipore, Billerica, USA). X-ray films were pur-chased from Kodak (Kodak, Rochester, NY, USA).
2.2. Study design
2.2.1. Ethical clearance for animal studyMale SpragueeDawley rats (150 � 15 g) were used for all ex-
periments. Animals were maintained under a 12-h lightedark cycleat temperature 24� 2 �C in the Institute’s animal house facility. Thestudy was approved by the Animal Ethical Committee of theinstitute in accordance with Committee for the Purpose of Control
and Supervision on Experiments on Animals (CPCSEA) of theGovernment of India.
2.2.2. Experimental designThe rats were randomly divided into four groups with n ¼ 8 in
each group e Group 1: unexposed normoxic (N), Group II: exposedto cold (C, 10 �C), Group III: exposed to hypobaric hypoxia (H,7,620 m) and Group IV: exposed to cold & hypobaric hypoxia (CH,10 �C & 7,620 m), each for 6 h duration. Simulated high altitudeexposure was performed in an animal decompression chambermaintained at pressure of 282 torr (equivalent to an altitude of7620 m, 8% oxygen), coupled to mercury barometer, at 25 �C forhypoxic group and at 10 �C for cold hypoxic group (Decibel In-struments, India). The airflow in the chamber was 2 L/min withrelative humidity maintained at 50e55%. Control group rats weremaintained in the normoxic condition within the same laboratory.
To assess the contribution of ER stress response to simultaneouscold hypoxic stress in the myocardium, we administered tunica-mycin (Tm, 0.3 mg/kg bw) intraperitoneally, freshly prepared as a0.05 mg/ml suspension in 150 mM dextrose [28]. Animals werecategorized randomly as control, only tunicamycin (Tm only), coldhypoxia and exposure to cold hypoxic stress after tunicamycinadministration (Tm þ cold hypoxia), with n ¼ 6 animals per group.
2.3. Oxidative stress markers
After exposure, animals were anaesthetized using sodiumpentobarbital. Hearts from control and exposed animals wererapidly excised and used fresh or snap frozen in liquid nitrogen andstored at �80 �C for further use. For biochemical estimations thetissue were homogenized in 0.154 M KCleEDTA buffer.
2.3.1. ROS measurementROS levels were measured with a nonfluorescent lipophilic dye,
dichlorofluorescein diacetate (DCFH-DA). DCFH-DA passively dif-fuses through cellular membranes where it is cleaved into 2, 7-dichlorofluorescein (DCF) by the action of intracellular esterasesand thus fluoresces in the presence of ROS. The production of freeradicals was determined as described earlier [29]. Briefly, 150 ml ofheart homogenatewas incubatedwith 10 ml of 100 mMDCFH-DA for30 min in dark. Fluorescence was read using a fluorimeter (PerkinElmer, UK) with excitation at 485 nm and emission at 535 nm.Readings were obtained in arbitrary fluorometric units and resultsexpressed as fold change in free radical generation.
2.3.2. Lipid peroxidationLipid peroxides are unstable indicators of oxidative stress in cells
that decompose to form more complex and reactive compoundssuch as Malondialdehyde (MDA). Lipid peroxidation was measuredby direct estimation of MDA using the modified method of Buegeand Aust [30]. In brief, 100mg tissuewas homogenized in 15% (w/v)TCA. 0.355% (w/v) TBA was added to supernatant and incubated inboiling water bath for 30 min. Following which, the absorbancewas read at 535 nm using UVevis spectrophotometer (BioRad,USA). The malondialdehyde levels were expressed as mmol/ml.
2.4. Histopathological studies
For histological studies, after the hypoxia and cold exposure,heart was excised and immediately fixed in 10% buffered neutralformalin solution. The fixed tissues were embedded in paraffin andserial sections (4 mm thick) were cut using microtome (Leica RM2125, Germany). Each section was stained with hematoxylin andeosin (H and E). The sections were examined under the light mi-croscope (Nikon Tokyo, Japan) and photomicrographs were taken.
K. Jain et al. / Biochimie 95 (2013) 1897e1908 1899
2.5. Measurement of MMP activity by gelatin zymography
Gelatin zymography is mainly used for the detection of thegelatinases, MMP-2 and MMP-9, respectively [31]. Briefly, frozencardiac tissue was homogenized in Tris buffer (saline 0.9%, Tris0.05 M, Triton X-100 0.25% and CaCl2 0.02 M) and centrifuged at4500 g for 30 min. Tissue homogenate (75 mg protein) was sub-jected to 10% SDS-PAGE containing 0.1% SDS and 1 g/L gelatin undernon-reducing conditions without prior boiling. After electropho-resis, gels were washed in 2.5% Triton X-100 for 30 min to removeSDS and allow protein renaturation. Gels were then subsequentlyimmersed in activity buffer (50 mM TriseHCl, 5 mM CaCl2, 0.2 MNaCl, 0.02% NaN3) for 16 h at 37 �C. Following incubation, gels werestained with 0.25% Coomassie brilliant blue (CBB R250) in meth-anol, acetic acid and water (4:1:5) followed by destaining withmethanol, acetic acid and water (4:1:5). Enzymatic activities weredetected as clear bands of gelatin lysis against a blue background.
2.6. Oxidative protein modifications
2.6.1. Protein carbonyl contentOxidative modifications of amino acid residues include deriva-
tization of amino acid residues such as proline, arginine, and lysineto reactive carbonyl derivatives. Briefly, 2,4-dinitrophenylhydrazinereacts with protein carbonyl forming a Schiff base to produce thecorresponding hydrazone, which can be analyzed spectrophoto-metrically [32]. Heart tissue was homogenized in ice cold 50 mMphosphate buffer (pH 7.2) containing 1 mM EDTA and proteinaseinhibitor cocktail. The homogenate was centrifuged at 10,000 g for15 min and supernatant was checked for presence of nucleic acids.To 200 ml of sample, 600 ml 10 mM 2,4-dinitrophenylhydrazine(DNPH) in 2 N HCl was added, or 600 ml of 2 N HCl was added asa blank control. The mixture was incubated for 1 h at room tem-perature. The proteinwas precipitated with an equal volume of 20%trichloroacetic acid and was washed three times with ethanol/ethylacetate (1:1 v/v). The final precipitate was dissolved in 400 ml of 6Mguanidine hydrochloride (pH 2.3), and insoluble debris wasremoved by centrifugation. The absorbance of the DNPH de-rivatives was measured at 360 nm. The concentration of carbonylgroups was calculated by using an absorbance coefficient 22 nM/cmand expressed as nanomole carbonyl per milligram of protein.
2.6.2. Advanced oxidation protein products (AOPPs)AOPPs are considered as markers to evaluate oxidant induced
protein damage. The determination of AOPP levels in heart wascarried out by spectrophotometric detection according to Witko-Sarsat et al. [33] modified for heart tissue. Briefly, heart tissue ho-mogenates, prepared in 0.154 M KCleEDTA, were diluted 1:5 withphosphate-buffered saline (PBS), pH 7.4. For standard curve, 200 mlof chloramine T (0e100 mmol/L) and 200 ml of PBS as blank wereapplied on a microtiter plate. Similarly 200 ml of diluted sampleswere applied. 10 ml of 1.16 M potassium iodide and 20 ml of aceticacid were added to each well and absorbance at 340 nm measuredimmediately. Concentration of AOPPs were obtained in chloramineunits and expressed as mmol chloramine/mg protein.
2.6.3. Fluorescence measurement of protein oxidationIsolated frozen hearts were thawed and homogenized in 0.3 M
sucrose, 10 mM HEPES with pH 7.4. Homogenate was centrifuged(1200 � g for 10 min) to remove the debris (total membrane frac-tion). Protein concentration was determined by the Bradfordmethod [34]. Fluorescence measurements were performed in so-lution containing 50 mg of homogenate protein per ml, 10 mMHEPES, 100 mM KCl, pH 7.0 at 25 �C on a PerkineElmerspectrofluorometer.
Fluorescence emission spectra (300e450 nm, slit width 2 nm) ofTrp were measured with excitation at 295 nm (slit width 2 nm)[35]. Fluorescence emission spectra of bityrosine, product of tyro-sine oxidation, were recorded in range 380e440 nm (slit width2 nm), at excitation wavelength 325 nm (slit width 2 nm) [36].
2.7. Protein degradation pathways
2.7.1. Protein degradation rateProtein degradation rate was determined by the release of
tyrosine over a period of 2 h as described previously [37]. Sincetyrosine is neither synthesized nor degraded in tissue, its releasefrom cardiac tissue section, into the incubation medium, reflectsthe net protein degradation rate. In brief, 100 mg of tissue wasplaced in 2 ml Krebs-Henseleit buffer and incubated for 2 h.Following incubation, the supernatant was collect and fluorescencewas measured in a PerkineElmer fluorimeter (LS-45) at excitation465 nm, emission 535 nm. The rate of protein degradation wasexpressed as nmoles of tyrosine released per mg protein per min.
2.7.2. 20 S Proteasome activityDegradation via the proteasome pathway was studied by
assaying the chymotrypsin-like enzyme activity of 20 SProteasome, as described earlier [38]. The fluorogenic peptidesuccinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC) served as substrate for the chymotrypsin-like activity. Smallportion (w50 mg) of tissue was homogenized in buffer containing50 mM TriseHCl, 1 mM dithiothreitol (DTT), 5 mM MgCl2, 5%glycerol and 5 mM ATP. The homogenates were incubated for30min at 37 �C in 50 ml of a buffer containing 100mM TriseHCl (pH8.0), 1 mM DTT, 5 mM MgCl2, 1 mM Suc-LLVY-AMC, 2 mg/mlovalbumin, and 0.07% SDS. The reaction was terminated by 25 mlof 10% SDS and diluted by 2 ml of 0.1 M TriseHCl (pH 9.0). Fluo-rescence of the liberated amidomethylcoumarin was monitored ina PerkineElmer fluorimeter at excitation 380 nm and emission460 nm. Chymotrypsin-like enzyme activity was expressed asarbitrary units per minute per milligram of protein.
2.7.3. Calpain assayActivity of calpains, calcium activated proteases, was measured
in the homogenate using N-succinyl-Leu-Tyr-7-amido-4-methylcoumarin (SLY-AMC) as a substrate [39]. A stock solution of50 mM SLY-AMC was prepared in dimethyl sulfoxide and storedat �20 �C. Hearts were homogenized in buffer having 50 mM TriseHCl, 150 mM NaCl, 10 mM NaH2PO4, 1% Nonidet P-40, and 0.4 mMsodium orthovanadate. Homogenate was then centrifuged at13,000 g for 15 min at 4 �C. The following procedure was used formeasuring calpain activity in tissue extracts: 30ml heart tissue extractwas incubated for 60 min at 37 �C in a buffer solution (pH 7.4) con-taining 25 mMHEPES, 0.1% CHAPS, 10% sucrose, 10 mM DTT, 0.1 mg/ml bovine serum albumin. After addition of 5 ml of the substrate so-lution, buffer was added to adjust the volume of the assay to 2 ml.Fluorescence of the liberated AMCwasmonitored in a PerkineElmerfluorimeter (LS-45) at excitation 380 nm, emission 460 nm. Calpainactivity, Caþ2 dependent cleavage of SLY-AMC, was expressed asmicromole arbitrary units of AMC released per milligram of protein.
2.8. Protein expression analysis
2.8.1. Preparation of nuclear and cytosolic extractFor nuclear and cytosolic fractionation, frozen heart tissue was
homogenized in an ice-cold buffer A (0.5 M sucrose, 10 mMHEPES, 10 mM KCl, 1.5 mM MgCl2, 10% glycerol, 1 mM EDTA,1 mM DTT, 1 mM PMSF) fortified with protease inhibitors. Ho-mogenates were kept on ice for 15 min and 0.6% Nonidet P-40
K. Jain et al. / Biochimie 95 (2013) 1897e19081900
was added, and then centrifuged for 10 min at 2000 g at 4 �C. Thesupernatant with cytosolic fraction was collected, stored and thepellet was dissolved in cold buffer B (20 mM HEPES, 1.5 mMMgCl2, 0.3 mM NaCl, 0.2 mM EDTA, 20% glycerol, 0.5 mM DTT,0.5 mM PMSF and cocktail of protease inhibitors) for nuclearfraction. It was incubated for 30 min on ice followed by centri-fugation at 20,000 g at 4 �C for 15 min. The supernatant con-taining the nuclear fraction was aliquoted and stored at �80 �Cfor further analysis. Total protein concentrations were deter-mined using the Bradford protein method [34].
2.8.2. Western blottingProtein expression of HO-1, HSP70, HSP60, HSP90, glucose
regulated proteins, GRP78 and GRP94, PDI, CHOP, mTOR, Akt, p-Aktand VEGF, were quantified in cytosolic extract, whereas HIF-1awasanalyzed in the nuclear extract by Western blot analysis. Protein(50 mg) was separated by 10% SDS-polyacrylamide gel electropho-resis and transferred onto a nitrocellulose membrane. The mem-branes were blocked with 3% bovine serum albumin in PBScontaining 0.1% Tween 20, washed and probed with respectivemouse/rabbit monoclonal antibodies. The membranes were thenincubated with anti-mouse/rabbit-IgG HRP conjugate. The mem-branewas washed and incubated with chemiluminescent substrateand the bands were developed using X-ray films. Quantificationwas performed by densitometry using ImageJ software.
2.9. Statistical analysis
All the experiments were performed on three different occa-sions, and data are presented as mean � Standard Error Mean(SEM). One-way analysis of variance with post-hoc Bonferroni
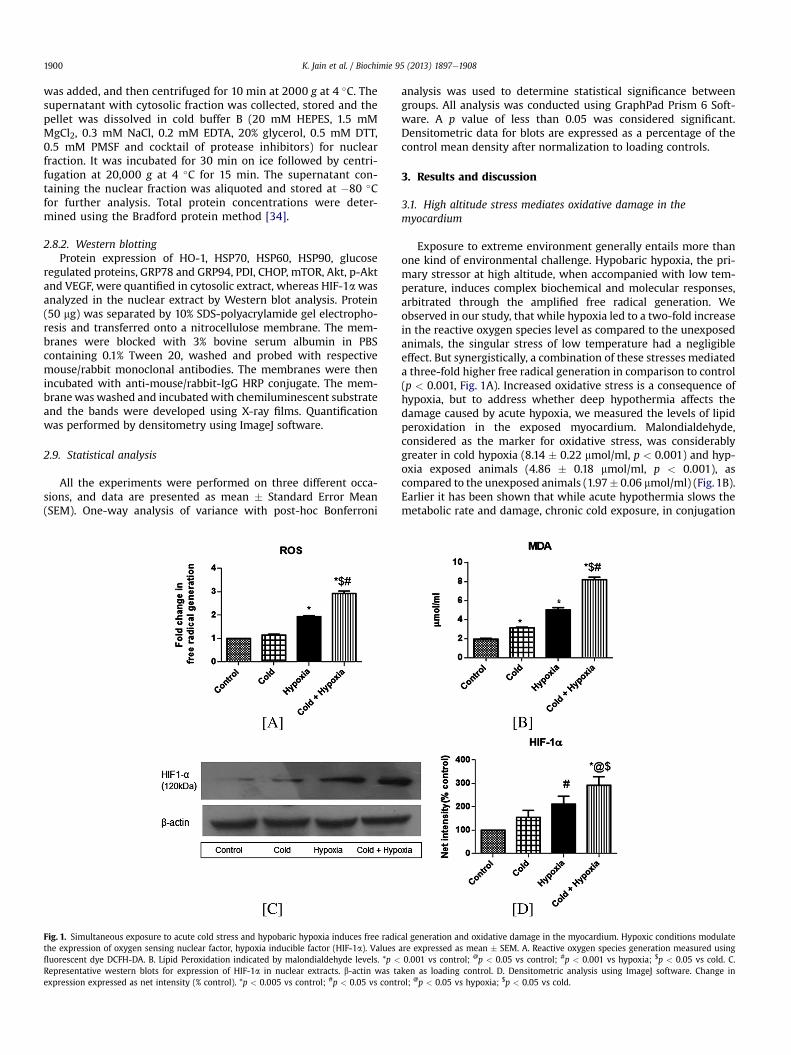
Fig. 1. Simultaneous exposure to acute cold stress and hypobaric hypoxia induces free radicthe expression of oxygen sensing nuclear factor, hypoxia inducible factor (HIF-1a). Valuesfluorescent dye DCFH-DA. B. Lipid Peroxidation indicated by malondialdehyde levels. *p <
Representative western blots for expression of HIF-1a in nuclear extracts. b-actin was taexpression expressed as net intensity (% control). *p < 0.005 vs control; #p < 0.05 vs contr
analysis was used to determine statistical significance betweengroups. All analysis was conducted using GraphPad Prism 6 Soft-ware. A p value of less than 0.05 was considered significant.Densitometric data for blots are expressed as a percentage of thecontrol mean density after normalization to loading controls.
3. Results and discussion
3.1. High altitude stress mediates oxidative damage in themyocardium
Exposure to extreme environment generally entails more thanone kind of environmental challenge. Hypobaric hypoxia, the pri-mary stressor at high altitude, when accompanied with low tem-perature, induces complex biochemical and molecular responses,arbitrated through the amplified free radical generation. Weobserved in our study, that while hypoxia led to a two-fold increasein the reactive oxygen species level as compared to the unexposedanimals, the singular stress of low temperature had a negligibleeffect. But synergistically, a combination of these stresses mediateda three-fold higher free radical generation in comparison to control(p < 0.001, Fig. 1A). Increased oxidative stress is a consequence ofhypoxia, but to address whether deep hypothermia affects thedamage caused by acute hypoxia, we measured the levels of lipidperoxidation in the exposed myocardium. Malondialdehyde,considered as the marker for oxidative stress, was considerablygreater in cold hypoxia (8.14 � 0.22 mmol/ml, p < 0.001) and hyp-oxia exposed animals (4.86 � 0.18 mmol/ml, p < 0.001), ascompared to the unexposed animals (1.97� 0.06 mmol/ml) (Fig.1B).Earlier it has been shown that while acute hypothermia slows themetabolic rate and damage, chronic cold exposure, in conjugation
al generation and oxidative damage in the myocardium. Hypoxic conditions modulateare expressed as mean � SEM. A. Reactive oxygen species generation measured using0.001 vs control; @p < 0.05 vs control; #p < 0.001 vs hypoxia; $p < 0.05 vs cold. C.ken as loading control. D. Densitometric analysis using ImageJ software. Change inol; @p < 0.05 vs hypoxia; $p < 0.05 vs cold.
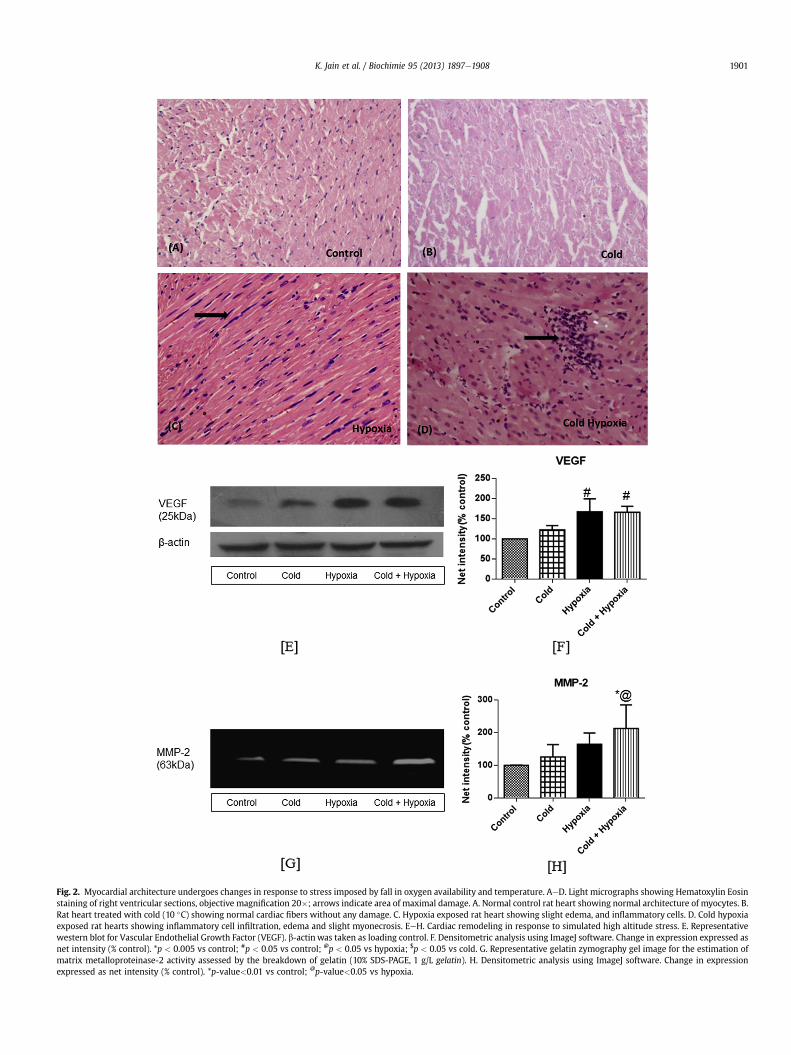
Fig. 2. Myocardial architecture undergoes changes in response to stress imposed by fall in oxygen availability and temperature. AeD. Light micrographs showing Hematoxylin Eosinstaining of right ventricular sections, objective magnification 20�; arrows indicate area of maximal damage. A. Normal control rat heart showing normal architecture of myocytes. B.Rat heart treated with cold (10 �C) showing normal cardiac fibers without any damage. C. Hypoxia exposed rat heart showing slight edema, and inflammatory cells. D. Cold hypoxiaexposed rat hearts showing inflammatory cell infiltration, edema and slight myonecrosis. EeH. Cardiac remodeling in response to simulated high altitude stress. E. Representativewestern blot for Vascular Endothelial Growth Factor (VEGF). b-actin was taken as loading control. F. Densitometric analysis using ImageJ software. Change in expression expressed asnet intensity (% control). *p < 0.005 vs control; #p < 0.05 vs control; @p < 0.05 vs hypoxia; $p < 0.05 vs cold. G. Representative gelatin zymography gel image for the estimation ofmatrix metalloproteinase-2 activity assessed by the breakdown of gelatin (10% SDS-PAGE, 1 g/L gelatin). H. Densitometric analysis using ImageJ software. Change in expressionexpressed as net intensity (% control). *p-value<0.01 vs control; @p-value<0.05 vs hypoxia.
K. Jain et al. / Biochimie 95 (2013) 1897e1908 1901
K. Jain et al. / Biochimie 95 (2013) 1897e19081902
to hypoxia at high altitude, is liable to mediate free radical gener-ation and tissue injury [7,10e12]. Our study demonstrates thatacute cold conditions (10 �C), coupledwith hypoxia, can causemoreoxidative damage than either of these stressors alone.
Hypoxia inducible factor (HIF-1a) has long been identified as acritical component of cellular systemic response to hypoxia,regulating a number of physiological responses to enhance tissueoxygen delivery [40]. HIF-1a stabilization has been observedunder both chronic and acute exposure to hypobaric hypoxia inmammalian systems indicating its central role in oxygen sensingand physiological response to low oxygen tensions [5,40]. In ourstudy, only hypobaric hypoxia singularly caused a significant in-crease in HIF-1a expression from control (p < 0.05), while cold hadno discernible effect on its stabilization (Fig. 1C and D). EarlierTanaka et al. showed that persistent hypothermia suppressed theexpression of HIF-1a and its inducible genes under hypoxic con-ditions [41]. In contrast, we find in our study, a significant increasein the expression of this nuclear factor under the simultaneouseffect of environmentally realistic cold and hypobaric hypoxia(p < 0.005). Our results indicate that collectively, acute hypother-mia and hypoxia significantly intensified the free radical mediatedphysiological response in the myocardium.
3.2. Acute exposure to hypoxia and hypothermia affects themyocardial tissue architecture
To identify the extent of damage in themyocardium at the tissuelevel, we stained the right ventricular sections of the myocardium,known to be more susceptible to hypoxic stress [42], with standard
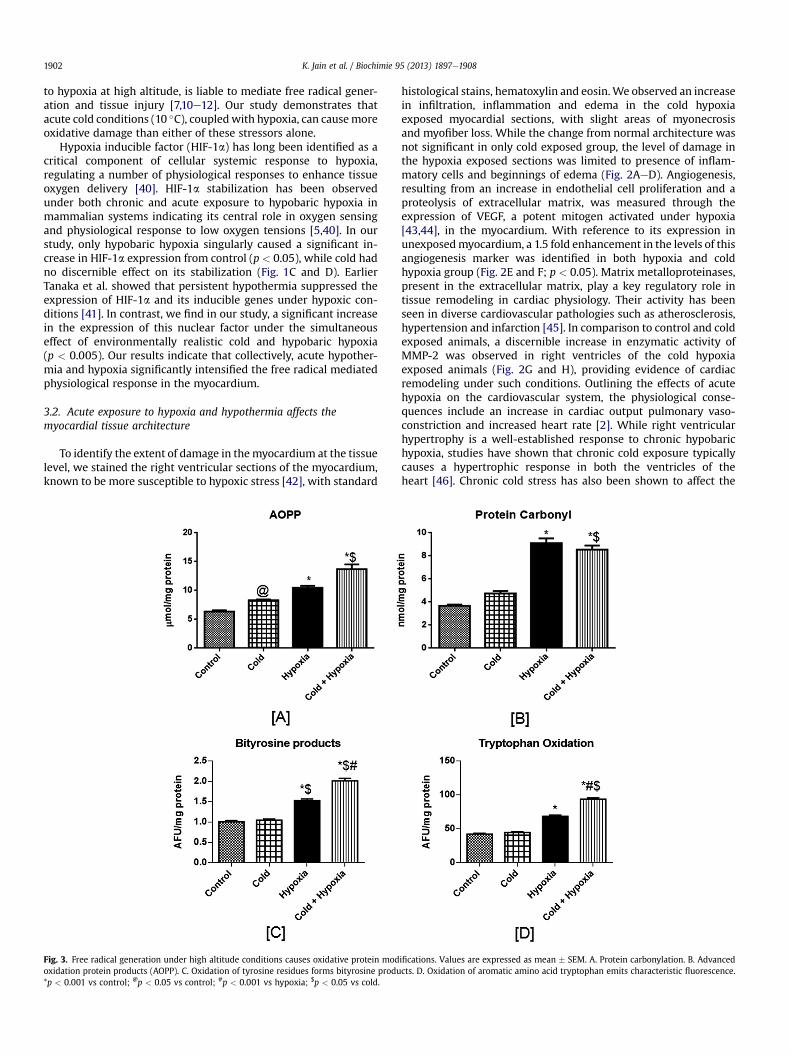
Fig. 3. Free radical generation under high altitude conditions causes oxidative protein modoxidation protein products (AOPP). C. Oxidation of tyrosine residues forms bityrosine produ*p < 0.001 vs control; @p < 0.05 vs control; #p < 0.001 vs hypoxia; $p < 0.05 vs cold.
histological stains, hematoxylin and eosin.We observed an increasein infiltration, inflammation and edema in the cold hypoxiaexposed myocardial sections, with slight areas of myonecrosisand myofiber loss. While the change from normal architecture wasnot significant in only cold exposed group, the level of damage inthe hypoxia exposed sections was limited to presence of inflam-matory cells and beginnings of edema (Fig. 2AeD). Angiogenesis,resulting from an increase in endothelial cell proliferation and aproteolysis of extracellular matrix, was measured through theexpression of VEGF, a potent mitogen activated under hypoxia[43,44], in the myocardium. With reference to its expression inunexposedmyocardium, a 1.5 fold enhancement in the levels of thisangiogenesis marker was identified in both hypoxia and coldhypoxia group (Fig. 2E and F; p < 0.05). Matrix metalloproteinases,present in the extracellular matrix, play a key regulatory role intissue remodeling in cardiac physiology. Their activity has beenseen in diverse cardiovascular pathologies such as atherosclerosis,hypertension and infarction [45]. In comparison to control and coldexposed animals, a discernible increase in enzymatic activity ofMMP-2 was observed in right ventricles of the cold hypoxiaexposed animals (Fig. 2G and H), providing evidence of cardiacremodeling under such conditions. Outlining the effects of acutehypoxia on the cardiovascular system, the physiological conse-quences include an increase in cardiac output pulmonary vaso-constriction and increased heart rate [2]. While right ventricularhypertrophy is a well-established response to chronic hypobarichypoxia, studies have shown that chronic cold exposure typicallycauses a hypertrophic response in both the ventricles of theheart [46]. Chronic cold stress has also been shown to affect the
ifications. Values are expressed as mean � SEM. A. Protein carbonylation. B. Advancedcts. D. Oxidation of aromatic amino acid tryptophan emits characteristic fluorescence.
K. Jain et al. / Biochimie 95 (2013) 1897e1908 1903
hemodynamic function of the heart, also causing cardiac fibrosisand remodeling, and subsequently triggering cardiovascular com-plications [11,47,48]. In response to synergistic low temperatureand hypoxic stress, Templemen et al. [13] showed an increase inmetabolic remodeling in right and left ventricles. At the tissue level,our study demonstrates that while hypothermia of 10 �C failed toinduce a significant degree of damage to the tissue; concomitant tohypoxia; it enhanced the degree of injury. Our findings thus showthat the effects of high altitude stress on natives and sojournersalike, can only be completely identified taking into account theinjurious consequences of not just hypobaric hypoxia but also lowtemperature.
3.3. Enhanced ROS levels induce oxidative protein modifications
Oxidative modification of proteins due to free radical generationat high altitude affects their physiological activity, making themhighly sensitive to proteolysis. The levels of protein carbonyl de-rivatives in the hypoxia group and the cold hypoxia group(9.08 � 0.42 and 8.55 � 0.35 nmol/mg protein respectively,p < 0.001, Fig. 3A), were found to be more than two fold higher ascompared to the level in control animals (3.67 � 0.10 nmol/mgprotein).
Advanced oxidation protein products are considered as relevantmarkers for oxidant induced protein damage. In our study (Fig. 3B),we observed a 1.5 times higher AOPP formation in the hypoxicgroup (10.45 � 0.34 mmol/mg protein, p < 0.001) and a 2.5 foldincrease in cold hypoxic group (13.69 � 0.82 mmol/mg protein,
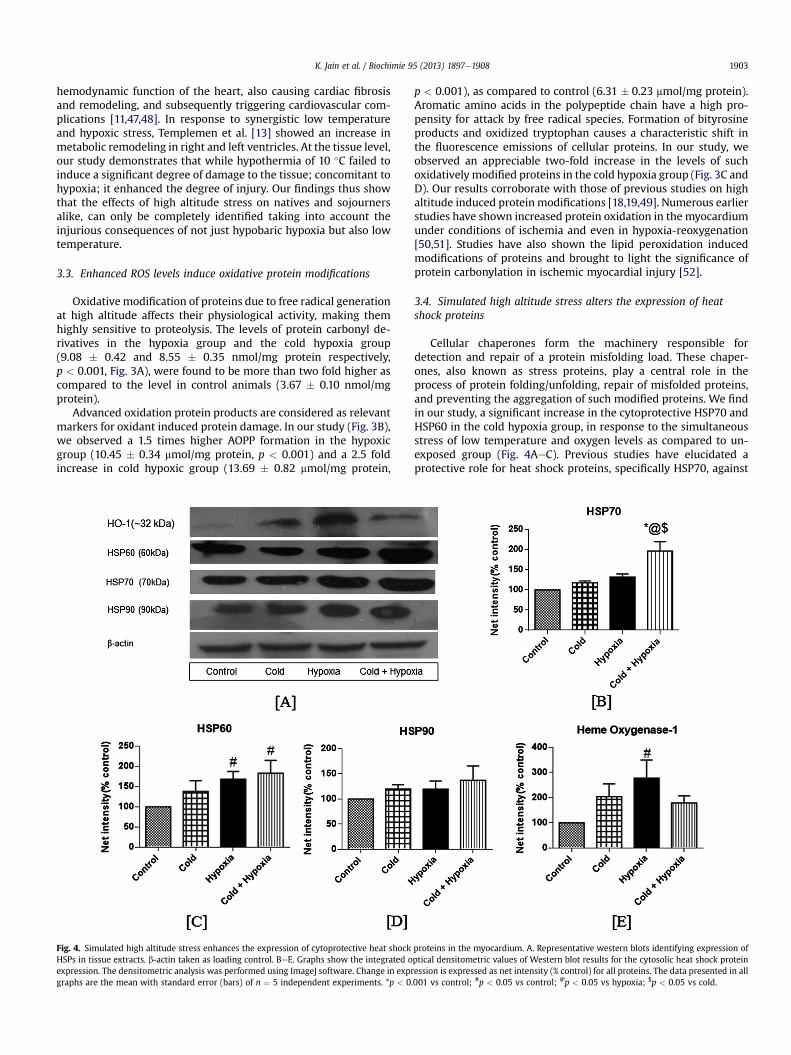
Fig. 4. Simulated high altitude stress enhances the expression of cytoprotective heat shockHSPs in tissue extracts. b-actin taken as loading control. BeE. Graphs show the integrated oexpression. The densitometric analysis was performed using ImageJ software. Change in exprgraphs are the mean with standard error (bars) of n ¼ 5 independent experiments. *p < 0
p < 0.001), as compared to control (6.31 � 0.23 mmol/mg protein).Aromatic amino acids in the polypeptide chain have a high pro-pensity for attack by free radical species. Formation of bityrosineproducts and oxidized tryptophan causes a characteristic shift inthe fluorescence emissions of cellular proteins. In our study, weobserved an appreciable two-fold increase in the levels of suchoxidatively modified proteins in the cold hypoxia group (Fig. 3C andD). Our results corroborate with those of previous studies on highaltitude induced protein modifications [18,19,49]. Numerous earlierstudies have shown increased protein oxidation in the myocardiumunder conditions of ischemia and even in hypoxia-reoxygenation[50,51]. Studies have also shown the lipid peroxidation inducedmodifications of proteins and brought to light the significance ofprotein carbonylation in ischemic myocardial injury [52].
3.4. Simulated high altitude stress alters the expression of heatshock proteins
Cellular chaperones form the machinery responsible fordetection and repair of a protein misfolding load. These chaper-ones, also known as stress proteins, play a central role in theprocess of protein folding/unfolding, repair of misfolded proteins,and preventing the aggregation of such modified proteins. We findin our study, a significant increase in the cytoprotective HSP70 andHSP60 in the cold hypoxia group, in response to the simultaneousstress of low temperature and oxygen levels as compared to un-exposed group (Fig. 4AeC). Previous studies have elucidated aprotective role for heat shock proteins, specifically HSP70, against
proteins in the myocardium. A. Representative western blots identifying expression ofptical densitometric values of Western blot results for the cytosolic heat shock proteinession is expressed as net intensity (% control) for all proteins. The data presented in all.001 vs control; #p < 0.05 vs control; @p < 0.05 vs hypoxia; $p < 0.05 vs cold.
K. Jain et al. / Biochimie 95 (2013) 1897e19081904
hypoxic insult [53,54]. Hypothermia induced increase in HSP70levels has been shown in the ischemic tissue, thus identifying akey role for the chaperone in the protective mechanism of hypo-thermia [8]. The combined stress of hypothermia and fall in oxy-gen availability also elevated the expression of HSP90 (Fig. 4D).Heme oxygenase-1, known as HSP32, plays a protective role incardiac cells under redox stress [53]. Hypoxia alone led to anappreciable two fold increase in its expression from control, whileHO-1 expression showed a similar pattern of elevation on expo-sure to only cold and cold hypoxia (Fig. 4E). The elevatedexpression of these chaperones under the concurrent stress of coldand hypobaric hypoxia is indicative of the altered protein load inthe cardiac tissue.
3.5. Low oxygen and hypoxic stress activates the ER chaperoneresponse
Bip/Grp78 and Grp94, the best characterized amongst theER chaperones, interact intricately to prevent aggregation ofmisfolded proteins, their refolding and eventually targetingsuch proteins for degradation by proteasomes [55]. While indi-vidually these stressors did not cause any significant change, thesynergistic effect of these factors caused a two fold increase inexpression of ER stress marker, BiP/GRP78 (p < 0.001 vs control,Fig. 5A and B). GRP94, another hallmark of the ER stress response,showed a marked increase in the only hypoxia and cold hypoxiagroup as compared to unexposed control group (Fig. 5C). High-lighting the significance of the GRPs in the ER stress mediatedadaptive and maladaptive pathways, earlier studies have shownthat overexpression of BiP/GRP78 protects the cell against thedeleterious consequences of pathophysiologies [27,56]. Higher
Fig. 5. Simultaneous stress of low temperature and hypobaric hypoxia led to an increasmyocardium. A. Representative western blots for expression of ER chaperones in myocardialof the ER chaperone expression is presented in the graphs. The densitometric analysis is sh*p < 0.001 vs control; #p < 0.05 vs control; @p < 0.05 vs hypoxia; $p < 0.01 vs cold.
GRP78 expression is also being implicated in the hypothermiamediated protection against ischemic injury [57]. In context ofenvironmental pathophysiology, a recent study by Drompariset al. [27] indicated attenuation of ER stress using chemicalchaperones as a novel therapeutic target against pulmonaryarterial hypertension. Within the ER, an altered redox potentialcauses the disruption of the normal protein folding milieu andactivation of the UPR to both combat the deleterious effects toprotein function and prevent further damage. This involves afinely tuned interaction between the ER chaperones to ensurethat only proteins that are properly assembled and folded leavethe ER compartment and thus alleviate the threat of cellulardysfunction.
The oxidizing environment within the ER is critical for the di-sulfide bond formation mediated by oxidoreductases such as pro-tein disulfide isomerases and oxidoreductins. In our study, we findan equivalent increase in the protein disulfide isomerase in boththe hypoxia and cold hypoxia group, while no significant changewas seen in the cold exposed group (p < 0.05, Fig. 5D). As disulfidebond formation requires oxygen and is compromised by reactiveoxygen species, the enzymatic redox function of PDI may be criticalin its role in imparting a protective tolerance to environmentalstress including hypoxia and cold, thereby ensuring functionalprotein viability [58].
3.6. Elevated levels of oxidized proteins modulate cellulardegradation, survival and apoptotic machinery
Various studies have highlighted that mild oxidation of globular,soluble proteins increases their susceptibility for proteasomaldegradation [59] and the degradation of such abnormal proteins
e in expression of canonical hallmarks of the ubiquitous ER stress response in thecytosolic extracts. b-actin considered the loading control. B-D. Semiquantitative analysisown as mean with standard error (bars) performed in n ¼ 5 independent experiments.
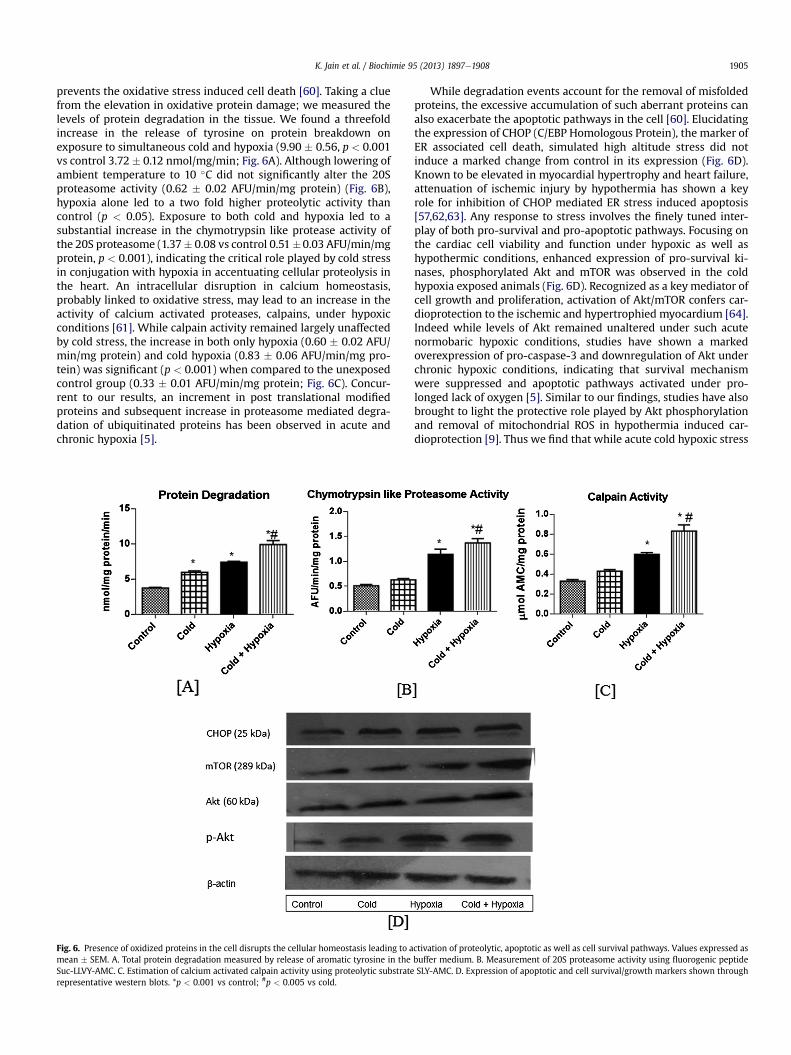
K. Jain et al. / Biochimie 95 (2013) 1897e1908 1905
prevents the oxidative stress induced cell death [60]. Taking a cluefrom the elevation in oxidative protein damage; we measured thelevels of protein degradation in the tissue. We found a threefoldincrease in the release of tyrosine on protein breakdown onexposure to simultaneous cold and hypoxia (9.90 � 0.56, p < 0.001vs control 3.72 � 0.12 nmol/mg/min; Fig. 6A). Although lowering ofambient temperature to 10 �C did not significantly alter the 20Sproteasome activity (0.62 � 0.02 AFU/min/mg protein) (Fig. 6B),hypoxia alone led to a two fold higher proteolytic activity thancontrol (p < 0.05). Exposure to both cold and hypoxia led to asubstantial increase in the chymotrypsin like protease activity ofthe 20S proteasome (1.37� 0.08 vs control 0.51�0.03 AFU/min/mgprotein, p < 0.001), indicating the critical role played by cold stressin conjugation with hypoxia in accentuating cellular proteolysis inthe heart. An intracellular disruption in calcium homeostasis,probably linked to oxidative stress, may lead to an increase in theactivity of calcium activated proteases, calpains, under hypoxicconditions [61]. While calpain activity remained largely unaffectedby cold stress, the increase in both only hypoxia (0.60 � 0.02 AFU/min/mg protein) and cold hypoxia (0.83 � 0.06 AFU/min/mg pro-tein) was significant (p < 0.001) when compared to the unexposedcontrol group (0.33 � 0.01 AFU/min/mg protein; Fig. 6C). Concur-rent to our results, an increment in post translational modifiedproteins and subsequent increase in proteasome mediated degra-dation of ubiquitinated proteins has been observed in acute andchronic hypoxia [5].
Fig. 6. Presence of oxidized proteins in the cell disrupts the cellular homeostasis leading to amean � SEM. A. Total protein degradation measured by release of aromatic tyrosine in theSuc-LLVY-AMC. C. Estimation of calcium activated calpain activity using proteolytic substraterepresentative western blots. *p < 0.001 vs control; #p < 0.005 vs cold.
While degradation events account for the removal of misfoldedproteins, the excessive accumulation of such aberrant proteins canalso exacerbate the apoptotic pathways in the cell [60]. Elucidatingthe expression of CHOP (C/EBP Homologous Protein), the marker ofER associated cell death, simulated high altitude stress did notinduce a marked change from control in its expression (Fig. 6D).Known to be elevated in myocardial hypertrophy and heart failure,attenuation of ischemic injury by hypothermia has shown a keyrole for inhibition of CHOP mediated ER stress induced apoptosis[57,62,63]. Any response to stress involves the finely tuned inter-play of both pro-survival and pro-apoptotic pathways. Focusing onthe cardiac cell viability and function under hypoxic as well ashypothermic conditions, enhanced expression of pro-survival ki-nases, phosphorylated Akt and mTOR was observed in the coldhypoxia exposed animals (Fig. 6D). Recognized as a key mediator ofcell growth and proliferation, activation of Akt/mTOR confers car-dioprotection to the ischemic and hypertrophied myocardium [64].Indeed while levels of Akt remained unaltered under such acutenormobaric hypoxic conditions, studies have shown a markedoverexpression of pro-caspase-3 and downregulation of Akt underchronic hypoxic conditions, indicating that survival mechanismwere suppressed and apoptotic pathways activated under pro-longed lack of oxygen [5]. Similar to our findings, studies have alsobrought to light the protective role played by Akt phosphorylationand removal of mitochondrial ROS in hypothermia induced car-dioprotection [9]. Thus we find that while acute cold hypoxic stress
ctivation of proteolytic, apoptotic as well as cell survival pathways. Values expressed asbuffer medium. B. Measurement of 20S proteasome activity using fluorogenic peptideSLY-AMC. D. Expression of apoptotic and cell survival/growth markers shown through
K. Jain et al. / Biochimie 95 (2013) 1897e19081906
did not affect the apoptotic pathways in the cell, an upregulation inthe cell survival kinases may mediate the pro-survival response.
Collectively, our results indicate that the synergistic effect ofacute hypothermia and hypoxia led to an appreciable increase inthe levels of misfolded proteins, with a concomitant elevation inthe proteolytic and pro-survival pathway to sustain proper proteinfunction under high altitude conditions. Alleviation of the oxidizedprotein load through the adaptive UPR response, involving chap-erones as well as degradative pathways for removal of such pro-teins, prevented the activation of the pro-apoptotic machinery ofthe myocardium under stress conditions.
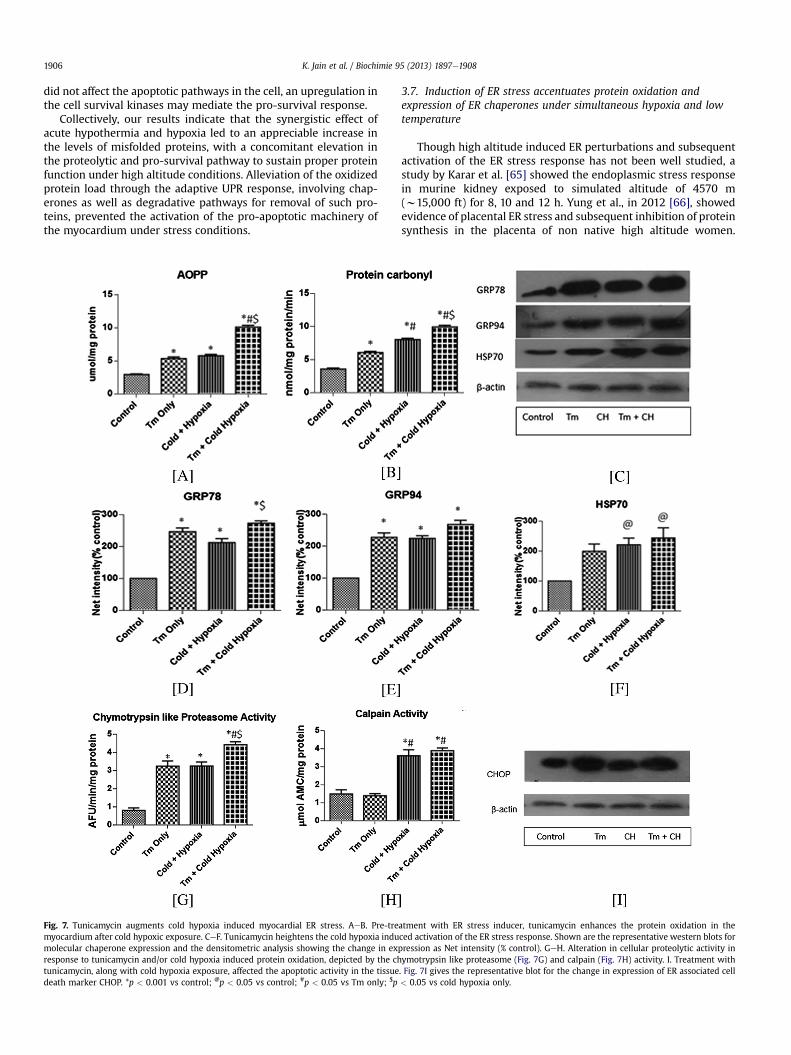
Fig. 7. Tunicamycin augments cold hypoxia induced myocardial ER stress. AeB. Pre-tremyocardium after cold hypoxic exposure. CeF. Tunicamycin heightens the cold hypoxia indumolecular chaperone expression and the densitometric analysis showing the change in exresponse to tunicamycin and/or cold hypoxia induced protein oxidation, depicted by the chtunicamycin, along with cold hypoxia exposure, affected the apoptotic activity in the tissuedeath marker CHOP. *p < 0.001 vs control; @p < 0.05 vs control; #p < 0.05 vs Tm only; $p
3.7. Induction of ER stress accentuates protein oxidation andexpression of ER chaperones under simultaneous hypoxia and lowtemperature
Though high altitude induced ER perturbations and subsequentactivation of the ER stress response has not been well studied, astudy by Karar et al. [65] showed the endoplasmic stress responsein murine kidney exposed to simulated altitude of 4570 m(w15,000 ft) for 8, 10 and 12 h. Yung et al., in 2012 [66], showedevidence of placental ER stress and subsequent inhibition of proteinsynthesis in the placenta of non native high altitude women.
atment with ER stress inducer, tunicamycin enhances the protein oxidation in theced activation of the ER stress response. Shown are the representative western blots forpression as Net intensity (% control). GeH. Alteration in cellular proteolytic activity inymotrypsin like proteasome (Fig. 7G) and calpain (Fig. 7H) activity. I. Treatment with. Fig. 7I gives the representative blot for the change in expression of ER associated cell< 0.05 vs cold hypoxia only.
K. Jain et al. / Biochimie 95 (2013) 1897e1908 1907
Recent studies have explored the activation of the unfolded proteinresponse in the context of pulmonary hypertension, induced bychronic hypoxia or monocrotaline [26,27]. Within the myocardium,the studies on role of ER stress has largely remained focussed on theischemic and hypertrophied heart; the effect of high altitude on thecardiac protein quality control has been not widely investigated. Toclearly elucidate whether the collective effect of low temperaturesand hypobaric hypoxia activates the ER stress response within themyocardium, we administered tunicamycin, at a dose of 0.3 mg/kgbw to the animals before subjecting them to cold hypoxic stress.Tunicamycin, a well-known inducer of ER stress, inhibits the N-linked glycosylation of nascent proteins, leading to the activation ofUPR in the mammalian system [28]. Measuring the AOPP levels, weobserved that treatment with this drug before simulated highaltitude stress, mediated a significant 2-fold elevation from bothonly Tm and cold hypoxia group, which showed similar increasewith reference to control (p < 0.05, Fig. 7A). Protein carbonyl levelsincreased in only Tm group as compared to control animals(p < 0.05, Fig. 7B). In contrast to the only cold hypoxia group, thelevels of carbonylationwere higher in the Tmþ cold hypoxia group.Identifying the effect of tunicamycin in conjugation with the lowoxygen and temperature stress on ER stress marker GRP78, therewas a significant increase in its expression in the Tmþ cold hypoxiagroup from only cold hypoxia (p < 0.05, Fig. 7C and D). AlthoughGRP94 was elevated in the cold hypoxia group, presence of tuni-camycinwith cold hypoxic stress augmented its expression (Fig. 7C,E). While tunicamycin is a known inducer of ER stress chaperones,it also led to an elevation of the heat shock protein HSP70 onexposure to cold and hypoxia (Fig. 7F). In conjugation with coldhypoxic stress, tunicamycin mediated activation of the ER stressresponse amplified the protein misfolding load on the cell.Expounding on the proteolytic response of the heart under suchconditions, we observed that the elevation in the proteasome ac-tivity was maximal in the Tm þ cold hypoxia group as compared toonly Tm or only cold hypoxia groups (p < 0.05, Fig. 7G). On theother hand, the increase in calpain activity was equivalent in thecold hypoxia group, with or without Tm administration (Fig. 7H).Tm þ cold hypoxia group showed a considerable enhancedexpression of CHOP, as compared to control and only cold hypoxiagroup (Fig. 7I). We find that while cold hypoxia alone was unable toinduce ER stress associated cell death in the myocardium, pre-treatment with tunicamycin greatly heightened the apoptoticresponse in the exposed myocardium.
Our results show that tunicamycin induced alteration in theprotein folding homeostasis, intensifies the activation of ER stressresponse under high altitude condition, as evident by the distinctupregulation of the chaperones, the cellular proteolysis andapoptotic activity of heart. Thus our study highlights the congruenteffect of acute cold and fall in oxygen tensions on the proteinfolding and ER homeostasis and how these stressors differentiallymodulate the chaperone and degradation machinery within thecardiac tissue.
4. Conclusion
In summary, our study presents compelling evidence that underthe synergistic effect of acute cold and hypobaric hypoxia, themyocardium faces the maximal degree of protein oxidation andtissue injury. While the disruption in cardiac proteome functionwithin a short period is restricted by the concomitant activation ofprotein degradation and chaperone machinery, prolonged highaltitude exposure may lead to activation of both adaptive andmaladaptive effectors of the UPR. As demonstrated by the priorinduction of ER stress by tunicamycin, severe or extensive activa-tion of the ER stress response may be detrimental to the cell,
leading to activation of apoptosis if the cell is unable to cope withthemisfolding load. Our study finds relevance in understanding therole of an altered protein folding milieu in the manifestation ofcardiac pathologies including myocardial infarction, right ventric-ular hypertrophy and pulmonary hypertension prevalent underextreme environmental conditions.
Acknowledgment
The study was supported by Defence Research and Develop-ment Organisation, Ministry of Defence, Government of India. Thefirst author is a Council of Scientific and Industrial Research SeniorResearch Fellow.
References
[1] R. Bohuslavova, F. Kolar, L. Kuthanova, J. Neckar, A. Tichopad, G. Pavlinkova,Gene expression profiling of sex differences in HIF1-dependent adaptivecardiac responses to chronic hypoxia, J. Appl. Physiol. 109 (2010) 1195e1202.
[2] P. Bartsch, J.S. Gibbs, The effect of altitude on the heart and lungs, Circulation116 (2007) 2191e2202.
[3] J.A. Jefferson, J. Simoni, E. Escudero, M.E. Hurtado, E.R. Swenson, D.E. Wesson,G.F. Schreiner, R.B. Schoene, R.J. Johnson, A. Hurtado, Increased oxidativestress following acute and chronic high altitude exposure, High Alt. Med. Biol.5 (2004) 61e69.
[4] P. Maiti, S.B. Singh, A.K. Sharma, S. Muthuraju, P.K. Banerjee, G. Ilavazhagan,Hypobaric hypoxia induces oxidative stress in rat brain, Neurochem. Int. 49(2006) 709e716.
[5] A. Viganò, M. Vasso, A. Caretti, V. Bravatà, L. Terraneo, C. Fania, D. Capitanio,M. Samaja, C. Gelfi, Protein modulation in mouse heart under acute andchronic hypoxia, Proteomics 11 (2011) 4202e4217.
[6] S.L. Hale, R.A. Kloner, Myocardial temperature reduction attenuates necrosisafter prolonged ischemia in rabbits, Cardiovasc. Res. 40 (1998) 502e507.
[7] C. Gordon, The therapeutic potential of regulated hypothermia, Emerg. Med. J.18 (2001) 81e89.
[8] Y. Terao, S. Miyamoto, K. Hirai, H. Kamiguchi, H. Ohta, M. Shimojo, Y. Kiyota,S. Asahi, Y. Sakura, Y. Shintani, Hypothermia enhances heat-shock protein 70production in ischemic brains, Neuroreport 20 (2009) 745e749.
[9] Z.H. Shao, W.W. Sharp, K.R. Wojcik, C.Q. Li, M. Han, W.T. Chang,S. Ramachandran, J. Li, K.J. Hamann, T.L. Vanden Hoek, Therapeutic hypo-thermia cardioprotection via Akt- and nitric oxide-mediated attenuation ofmitochondrial oxidants, Am. J. Physiol. Heart Circ. Physiol. 298 (2010) H2164eH2173.
[10] N. Alva, D. Azuara, J. Palomeque, T. Carbonell, Deep hypothermia protectsagainst acute hypoxia in vivo in rats: a mechanism related to the attenuationof oxidative stress, Exp. Physiol. 98 (2013) 1115e1124.
[11] S.S. Purkayastha, R.P. Sharma, G. Ilavazahagan, K. Siridharan, S. Ranganathan,W. Selvamurthy, Effect of vitamin C and E in modulating peripheral vascularresponse to local cold stimulus in man at high altitude, Jpn. J. Physiol. 49(1999) 159e167.
[12] E. Sahin, S. Gümüslü, Cold-stress-induced modulation of antioxidant defence:role of stressed conditions in tissue injury followed by protein oxidation andlipid peroxidation, Int. J. Biometeorol. 48 (2004) 165e171.
[13] N.M. Templeman, J.L. Beaudry, C.M. Le Moine, G.B. McClelland, Chronic hyp-oxia- and cold-induced changes in cardiac enzyme and gene expression inCD-1 mice, Biochim. Biophys. Acta 1800 (2010) 1248e1255.
[14] A.J. Ouellette, R.K. Watson, K. Billmire, M.K. Dygert, J.S. Ingwall, Protein syn-thesis in the cultured fetal mouse heart: effects of deprivation of oxygen andoxidizable substrate, Biochemistry 22 (1983) 1201e1207.
[15] V.R. Preedy, D.M. Smith, P.H. Sugden, The effects of 6 hours of hypoxia onprotein synthesis in rat tissues in vivo and in vitro, Biochem. J. 228 (1985)179e185.
[16] L. Piacentini, J.S. Karliner, Altered gene expression during hypoxia and reox-ygenation of the heart, Pharmacol. Ther. 83 (1999) 21e37.
[17] E.R. Stadtman, Oxidation of proteins by mixed-function oxidation system:implication in protein turnover, aging and neutrophil function, Trends Bio-chem. Sci. 11 (1986) 11e12.
[18] Z. Radak, K. Asano, K. Lee, H. Ohno, A. Nakamura, H. Nakamoto, S. Goto, High-altitude training increases reactive carbonyl derivatives but not lipid peroxi-dation in skeletal muscle of rats, Free Radic. Biol. Med. 22 (1997) 1109e1114.
[19] P. Chaudhary, G. Suryakumar, R. Prasad, S.N. Singh, S. Ali, G. Ilavazhagan,Chronic hypobaric hypoxia mediated skeletal muscle atrophy: role ofubiquitin-proteasome pathway and calpains, Mol. Cell. Biochem. 364 (2012)101e113.
[20] D.T. Rutkowski, R.J. Kaufman, A trip to the ER: coping with stress, Trends CellBiol. 14 (2004) 20e28.
[21] D. Ron, P. Walter, Signal integration in the endoplasmic reticulum unfoldedprotein response, Nat. Rev. Mol. Cell Biol. 8 (2007) 519e529.
[22] K. Okada, T. Minamino, Y. Tsukamoto, Y. Liao, O. Tsukamoto, S. Takashima,A. Hirata, M. Fujita, Y. Nagamachi, T. Nakatani, C. Yutani, K. Ozawa, S. Ogawa,
K. Jain et al. / Biochimie 95 (2013) 1897e19081908
H. Tomoike, M. Hori, M. Kitakaze, Prolonged endoplasmic reticulum stress inhypertrophic and failing heart after aortic constriction: possible contributionof endoplasmic reticulum stress to cardiac myocyte apoptosis, Circulation 110(2004) 705e712.
[23] E. Szegezdi, A. Duffy, M.E. O’Mahoney, S.E. Logue, L.A. Mylotte, T. O’Brien,A. Samali, ER stress contributes to ischemia-induced cardiomyocyte apoptosis,Biochem. Biophys. Res. Commun. 349 (2006) 1406e1411.
[24] D.J. Thuerauf, M. Marcinko, N. Gude, M. Rubio, M.A. Sussman, C.C. Glembotski,Activation of the unfolded protein response in infarcted mouse heart andhypoxic cultured cardiac myocytes, Circ. Res. 99 (2006) 275e282.
[25] H. Yoshida, ER stress and diseases, FEBS J. 274 (2007) 630e658.[26] M.E. Yeager, M.B. Reddy, C.M. Nguyen, K.L. Colvin, D.D. Ivy, K.R. Stenmark,
Activation of the unfolded protein response is associated with pulmonaryhypertension, Pulm. Circ. 2 (2012) 229e240.
[27] P. Dromparis, R. Paulin, T.H. Stenson, A. Haromy, G. Sutendra, E.D. Michelakis,Attenuating endoplasmic reticulum stress as a novel therapeutic strategy inpulmonary hypertension, Circulation 127 (2013) 115e125.
[28] G. Petrovski, S. Das, B. Juhasz, A. Kertesz, A. Tosaki, D.K. Das, Cardioprotectionby endoplasmic reticulum stress-induced autophagy, Antioxid. Redox Signal.14 (2011) 2191e2200.
[29] R. Cathcart, E. Schwiers, B.N. Ames, Detection of pico mole levels of hyder-operoxides using fluorescent dichlorofluoroscein assay, Anal. Biochem. 134(1983) 111e116.
[31] P.Y. Cheung, G. Sawicki, M. Wozniak, W. Wang, M.W. Radomski, R. Schulz,Matrix metalloproteinase-2 contributes to ischemiaereperfusion injury in theheart, Circulation 101 (2000) 1833e1839.
[32] R.L. Levine, D. Garland, C.N. Oliver, A. Amici, I. Climent, A. Lenz, B. Ahn,S. Shaltiel, E.R. Stadtman, Determination of carbonyl content in oxidativelymodified proteins, Methods Enzymol. 186 (1990) 464e478.
[33] V. Witko-Sarsat, M. Friedlander, C. Capeillere-Blandin, T. Nguyen-Khoa,A.T. Nguyen, J. Zingraff, P. Jungers, B. Descamps-Latscha, Advanced oxidationprotein products as a novel marker of oxidative stress in uremia, Kidney Int.49 (1996) 1304e1313.
[34] M.M. Bradford, Rapid and sensitive method for the quantitation of microgramquantities of protein utilizing the principle of protein-dye binding, Anal.Biochem. 72 (1976) 248e254.
[35] N. Dousset, G. Ferretti, M. Taus, P. Valdiguie, G. Curatola, Fluorescence analysisof lipoprotein peroxidation, Methods Enzymol. 233 (1994) 459e469.
[36] C. Giulivi, K.J.A. Davies, Dityrosine: a marker for oxidatively modified proteinsand selective proteolysis, Methods Enzymol. 233 (1994) 363e371.
[37] T.P. Waalkes, S. Udenfriend, A fluorimetric method for the estimation oftyrosine in plasma and tissues, J. Lab. Clin. Med. 50 (1957) 733e736.
[38] R.T. Hepple, M. Qin, H. Nakamoto, S. Goto, Caloric restriction optimizes theproteasome activity, Am. J. Physiol. Regul. Integr. Comp. Physiol. 295 (2008)R1231eR1237.
[39] R. Mastrocola, P. Reffo, F. Penna, C.E. Tomasinelli, G. Boccuzzi, F.M. Baccino,M. Aragno, P. Costelli, Muscle wasting in diabetic and in tumor bearing rats:role of oxidative stress, Free Radic. Biol. Med. 44 (2008) 584e593.
[40] G.L. Semenza, O2 regulated gene expression: transcriptional control ofcardiorespiratory physiology by HIF-1, J. Appl. Physiol. 96 (2004) 1173e1177.
[41] T. Tanaka, T. Wakamatsu, H. Daijo, S. Oda, S. Kai, T. Adachi, S. Kizaka-Kondoh,K. Fukuda, K. Hirota, Persisting mild hypothermia suppresses hypoxia-inducible factor-1alpha protein synthesis and hypoxia-inducible factor-1-mediated gene expression, Am. J. Physiol. Regul. Integr. Comp. Physiol. 298(2010) R661eR671.
[42] A. Genovese, A. Accinni, G. Spadaro, S. Quattrin, M. Condorelli, Myocardialhypertrophy in rats exposed to simulated high altitude, Arch. Int. Physiol.Biochim. 93 (1985) 331e338.
[43] P. Carmeliet, Y.S. Ng, D. Nuyens, G. Theilmeier, K. Brusselmans, I. Cornelissen,E. Ehler, V.V. Kakkar, I. Stalmans, V. Mattot, J.C. Perriard, M. Dewerchin,W. Flameng, A. Nagy, F. Lupu, L. Moons, D. Collen, P.A. D’Amore, D.T. Shima,Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice
[44] O.J. Birot, A. Peinnequin, N. Simler, H. van Cuyck Gandre, R. Hamel, X.A. Bigard,Vascular endothelial growth factor expression in heart of rats exposed tohypobaric hypoxia: differential response between mRNA and protein, J. Cell.Physiol. 200 (2004) 107e115.
[45] F.G. Spinale, Myocardial matrix remodeling and the matrix metal-loproteinases: influence on cardiac form and function, Physiol. Rev. 87 (2007)1285e1342.
[46] S.R. Kayar, N. Banchero, Volume overload hypertrophy elicited by cold and itseffects on myocardial capillarity, Respir. Physiol. 59 (1985) 1e14.
[47] Z. Sun, Cardiovascular responses to cold exposure, Front. Biosci. (Elite Ed.) 2(2010) 495e503.
[48] Y. Zhang, N. Hu, Y. Hua, K.L. Richmond, F. Dong, J. Ren, Cardiac overexpressionof metallothionein rescues cold exposure-induced myocardial contractiledysfunction through attenuation of cardiac fibrosis despite cardiomyocytemechanical anomalies, Free Radic. Biol. Med. 53 (2012) 194e207.
[49] Z. Radak, A. Nakamura, H. Nakamoto, K. Asano, H. Ohno, S. Goto, A period ofanaerobic exercise increases the accumulation of reactive carbonyl derivativesin the lungs of rats, Pflugers Arch. 435 (1998) 439e444.
[50] Y. Park, S. Kanekal, J.P. Kehrer, Oxidative changes in hypoxic rat heart tissue,Am. J. Physiol. 260 (1991) H1395eH1405.
[51] Z. Tatarková, P. Kaplán, M. Matejovi�cová, J. Lehotský, D. Dobrota, W. Flameng,Effect of ischemia and reperfusion on protein oxidation in isolated rabbithearts, Physiol. Res. 54 (2005) 185e191.
[52] P. Eaton, D.J. Hearse, M.J. Shattock, Lipid hydroperoxide modification of pro-teins during myocardial ischaemia, Cardiovasc. Res. 51 (2001) 294e303.
[53] W.H. Dillmann, R. Mestril, Heat shock proteins in myocardial stress, Z. Kardiol.84 (1995) 87e90.
[54] R.M. Mohan, S. Golding, D.J. Paterson, Intermittent hypoxia improves atrialtolerance to subsequent anoxia and reduces stress protein expression, ActaPhysiol. Scand. 172 (2001) 89e95.
[55] M.J. Gething, Role and regulation of the ER chaperone BiP, Semin. Cell Dev.Biol. 10 (1999) 465e472.
[56] J.D. Malhotra, H. Miao, K. Zhang, A. Wolfson, S. Pennathur, S.W. Pipe,R.J. Kaufman, Antioxidants reduce endoplasmic reticulum stress and improveprotein secretion, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 18525e18530.
[57] X. Liu, M. Wang, H. Chen, Y. Guo, F. Ma, F. Shi, Y. Bi, Y. Li, Hypothermia protectsthe brain from transient global ischemia/reperfusion by attenuating endo-plasmic reticulum response-induced apoptosis through CHOP, PLoS One 8(2013) e53431.
[58] S. Tanaka, T. Uehara, Y. Nomura, Up-regulation of protein-disulfide isomerasein response to hypoxia/brain ischemia and its protective effect againstapoptotic cell death, J. Biol. Chem. 275 (2000) 10388e10393.
[59] K.J.A. Davies, Degradation of oxidized proteins by the 20S proteasome,Biochimie 83 (2001) 301e310.
[61] K. Iizuka, H. Kawaguchi, H. Yasuda, Calpain is activated during hypoxicmyocardial cell injury, Biochem. Med. Metab. Biol. 46 (1991) 427e431.
[62] S.J. Marciniak, C.Y. Yun, S. Oyadomari, I. Novoa, Y. Zhang, R. Jungreis,K. Nagata, H.P. Harding, D. Ron, CHOP induces death by promoting proteinsynthesis and oxidation in the stressed endoplasmic reticulum, Genes Dev. 18(2004) 3066e3077.
[63] S. Oyadomari, M. Mori, Roles of CHOP/GADD153 in endoplasmic reticulumstress, Cell Death Differ. 11 (2004) 381e389.
[64] S. Balasubramanian, R.K. Johnston, P.C. Moschella, S.K. Mani, W.J. Tuxworth Jr.,D. Kuppuswamy, mTOR in growth and protection of hypertrophyingmyocardium, Cardiovasc. Hematol. Agents Med. Chem. 7 (2009) 52e63.
[65] J. Karar, K.S. Dolt, M.A. Qadar Pasha, Endoplasmic reticulum stress response inmurine kidney exposed to acute hypobaric hypoxia, FEBS Lett. 582 (2008)2521e2526.
[66] H.W. Yung, M. .Cox, M. Tissot van Patot, G.J. Burton, Evidence of endoplasmicreticulum stress and protein synthesis inhibition in the placenta of non-nativewomen at high altitude, FASEB J. 26 (2012) 1970e1981.