35


This booklet was produced by the Science/AAAS Custom Publishing Office and sponsored by the State Key Laboratory of Medical Genetics in China. Materials that appear in this booklet were commissioned, edited, and published by the Science/AAAS Custom Publishing Office and were not reviewed or assessed by the Science Editorial staff. Articles in this booklet can be cited using the following format: [AUTHOR NAME(S)], [ARTICLE TITLE] in Pathways to Cures: Neurodegenerative Diseases in China, S. Sanders, Z. Zhang, B. Tang, Eds. (Science/AAAS, Washington, DC, 2013), pp. [xx-xx].
Editors: Sean Sanders, Ph.D.; Zhuohua Zhang, Ph.D.; Beisha Tang, M.D.Assistant Editor: Jia-Da Li, Ph.D.Proofreader/Copyeditor: Yuse Lajiminmuhip; Designer: Amy Hardcastle
© 2013 by The American Association for the Advancement of Science. All rights reserved. 20 December 2013
NEURODEGENERATION RESEARCH
4 Molecular Mechanisms Underlying Neuronal Death and Synaptic Dysfunction in Neurodegenerative Diseases Kwok-OnLaiandNancyY.Ip
7 The Role of Autophagy, Apoptosis, Neuroinflammation, and Mitochondrial Dysfunction in Parkinson’s Disease DongChen,HaigangRen,QingsongHu,FengGao, andGuanghuiWang
10 The Multiple Roles of Autophagy in Neural Function and Disease RuiShengandZheng-HongQin
13 Iron and Neurodegeneration YaKe,Ka-ChunWu,andZhong-MingQian
16 Novel Pathways Regulating Function and Metabolism of ß-Amyloid Precursor Protein in Alzheimer’s Disease Yun-WuZhang,GuojunBu,andHuaxiXu
18 Role of Tau Hyperphosphorylation in Alzheimer’s Disease-Associated Neurodegeneration Jian-ZhiWang,QingTian,andDiGao
22 Dopaminergic Modulation of Astrocyte Functions in the Pathogenesis of Parkinson’s Disease YingjunLiuandJiaweiZhou
25 Proteolytic Pathways in Parkinson’s Disease ChengyuanTang,TongmeiZhang,andZhuohuaZhang
27 Animal Models of Huntington’s Disease Xiao-JiangLiandShihuaLi
29 FMRP Regulates Microtubule Network Formation, Neurogenesis, and DNA Damage Response AiyuYaoandYongQ.Zhang
GENETIC AND CLINICAL STUDIES
32 Progress in Treating Hereditary Ataxia in Mainland China HongJiang,JunlingWang,JuanDu,RanhuiDuan, JiadaLi,andBeishaTang
35 Genetic Etiology of Parkinson’s Disease in China JifengGuo,XinxiangYan,DanlingWang, QianXu,BeishaTang,andZhuohuaZhang
37 PKD and PRRT2-related Paroxysmal Diseases in China Jun-LingWang,NanLi,HongJiang, LuShen,KunXia,andBeishaTang
41 Clinical and Molecular Biological Studies of Amyotrophic Lateral Sclerosis in China ZhangyuZouandLiyingCui
43 Brainnetome Studies of Alzheimer’s Disease Using Neuroimaging TianziJiang,YongLiu,andBingLiu
46 Fragile X Syndrome in China: A Clinical Review RanhuiDuan
CONTENTS
THERAPEUTIC STRATEGIES
48 Deep-Brain Stimulation Research in China BominSunandWeiLiu
50 Neural Progenitors by Direct Reprogramming: Strategies for the Treatment of Parkinson’s and Alzheimer’s Diseases ChanghaiTianandJialinC.Zheng
53 Potential Neuroprotective Therapies for Parkinson’s Disease JunLiuandSheng-DiChen
56 Exploring Therapeutic Mechanisms of Traditional Chinese Medicine Extracts and Electroacupuncture in Parkinson’s Disease YanZheng,JunJia,andXiao-MinWang
59 Novel Therapeutic Strategies for Amyotrophic Lateral Sclerosis WeidongLeandXiaojieZhang
62 Novel Neuroprotective Strategy for Stroke— Activating Inherent Neuronal Survival Mechanisms MingChen,Bo-XingLi,andTian-MingGao
Produced by the Science/AAAS Custom Publishing Office
About the Cover: The ink and wash style of painting (Shui-mo) was developed about 1,500 years ago in China. In this painting, the artist has used a withered tree to signify degener-ating neurons. The red characters (in ancient Chinese calligraphy, Zhuan-shu) translate as “neurodegenera-tive diseases.” Credit: Zaihao Hu
INTRODUCTIONS 2 Understanding the Aging Brain By Alan Leshner 3 Neurodegenerative Research in China: Promise and Challenge By Xiong-Li Yang, Zhuohua Zhang, and Beisha Tang
Cosponsors of this publication
1

The population of China is aging. An ever-increasing num-ber of Chinese are afflicted with neurodegenerative dis-eases that result from the gradual and progressive loss of neural cells, leading to nervous system dysfunction. De-spite decades of basic and clinical endeavors, understand-ing of the etiology of these diseases and finding cures re-mains a great challenge.
It is well accepted that genetic factors contribute to the pathogenesis of neurodegenerative diseases. Genetic studies have defined the incidence of known disease-related genes in the Chinese population and identified a number of new causative genes. These findings lay the foundation for the design of genetic diagnoses of neurode-generative diseases and the basis for dissecting and un-derstanding the pathological mechanisms. Unfortunately, neurodegenerative diseases are most often genetically complex, with only a few being monogenic. The real dif-ficulty therefore is defining the genetic background of the sporadic cases. Unbiased genome-wide association stud-ies have identified a number of novel loci with associa-tions to Alzheimer’s and Parkinson’s diseases, providing a possible explanation for the heritability of certain diseases. Whole genome and exome analysis through next genera-tion sequencing will likely generate further insights into the molecular pathways.
Results from human genetic studies offer an entry point to de-fine the molecular etiology of neu-rodegenerative diseases. Func-tional investigations suggest that the causative genes in many neu-rodegenerative diseases appear to be distinct, but are in fact function-ally linked, indicating a potential disease mechanism. In Alzheim-er’s disease, multiple causative and susceptibility genes regulate β-amyloid metabolism, emphasiz-ing the important role of this protein in pathogenesis. Genes found to be related to Parkinson’s disease play roles in maintaining mitochon-drial homeostasis and modulating protein degradation, highlighting the involvement of these essential cellular pathways in the pathogen-
first section provides a sampling of the promising basic re-search coming out of China, highlighting potential targets for deeper investigation and for possible development of therapeutic interventions. Section two covers progress in understanding the genetic basis of neurodegenerative disorders as well as reviewing some recent clinical stud-ies that are helping researchers understand which treat-ments might be most effective and why. The third and final section of this booklet highlights some promising thera-pies for a number of neurodegenerative disorders, includ-ing deep brain stimulation and certain traditional Chinese medicines.
It is apparent that a few broad conclusions can be drawn from the work contained in this booklet. First, it is clear that although the brains from autopsied disease sufferers provide many clues, they are a poor substitute for being able to observe the disease progress in real-time. The development of better, noninvasive or minimally invasive monitoring technologies is therefore a priority, as is the creation of more physiologically and biologically accurate animal models. It is clear that in China, progress is already being made in both areas. Second, researchers need to look more broadly for both targets and cures. Autophagy, a process more recently accepted as playing a key role in neurodegeneration, promises to provide a number of potential drug targets. Traditional Chinese medicine seems to be providing some additional promis-ing potential therapeutics that war-rant deeper investigation, with a number having been used to treat the symptoms of these diseases for centuries, apparently with good effect.
It is likely that the research world will follow with interest the prog-ress in China over the coming de-cades as the country aggressively pushes its research agenda, deter-mined to make its mark in the field of neurodegenerative diseases, amongst others.
Alan Leshner, Ph.D.CEO, AAASExecutive Publisher, Science
2
Produced by the Science/AAAS Custom Publishing Office
3
Neurodegenerative Research in China: Promise and ChallengeUnderstanding the Aging Brain
In most cultures around the world, the brain is recognized to be the center of our minds and our personality, defining who, and even what, we are. We also depend on our brains to help us navigate a complex world of so-cial interactions, physical obsta-cles, and emotional challenges. It is therefore understandable that diseases affecting our cognitive ability are regarded as among the most devastating and debili-tating as well as being frightening on a personal level.
Standards of health care are changing rapidly across the globe and probably nowhere more quickly than in China. As treatments improve, survival to old age becomes more common, bringing about a shift from dis-eases of youth to diseases of old age, including a spectrum of neu-rodegenerative diseases such as Parkinson’s, Alzheimer’s, amyo-trophic lateral sclerosis, and Hun-tington’s. For this reason, there is some urgency among research-ers to gain a better understand-ing of the etiology and biological basis of these disorders in order to affect preventative measures or even possible cures as well as find ways to improve the qual-ity of life for current and future sufferers.
This booklet looks at the cur-rent state of neurodegenera-tive research in China, outlining recent progress in the field and future prospects. It has been divided into three sections that mirror the progress of research from discovery in the laboratory to clinical testing to cure. The
As treatments
improve,
survival to old
age becomes
more common,
bringing about a
shift from dis-
eases of youth
to diseases of
old age.
Results from
human genetic
studies offer
an entry point
to define the
molecular
etiology of
neurodegenera-
tive diseases.
esis of this disease. Recently, neurodegenerative diseases have been shown to involve not only neurons, but also the supporting cells such as astrocytes. These studies open new avenues of re-search into disease etiology and provide potential targets for treat-ment.
The eventual goal of neurode-generative disease research is to develop effective therapies. Different strategies are currently being pursued by Chinese scien-tists, ranging from identification of neuroprotective molecules to the generation of cells for neuro-nal replacement. Notably, many novel compounds from Chinese herbal medicines appear to show promise in, for example, improving dementia caused by Alzheimer’s disease and reliev-ing the symptoms of Parkinson’s disease.
There are still many questions to be answered before neurodegen-erative disease can be regarded as fully treatable. Nevertheless, incremental scientific advances over the coming years will likely continue to challenge our previ-ous thinking and shift the para-digm of current treatments. The pursuit of a deeper understand-ing of the cellular and genetic processes involved in neurode-generative diseases will undoubt-edly prove beneficial to patients in China and worldwide.
Xiong-Li Yang1
Zhuohua Zhang2
Beisha Tang2,3
1Institute of Neurobiology, Institutes of Brain Science and State Key Laboratory of Medical Neurobiology, Fudan University, Shanghai, China2State Key Laboratory of Medical Genetics, Xiangya Medical School, Central South University, Changsha, Hunan, China3Department of Neurology, Xiangya Hospital, Central South University, Changsha, Hunan, China

Produced by the Science/AAAS Custom Publishing Office
5
NEURODEGENERATION RESEARCH
Molecular Mechanisms Underlying Neuronal Death and Synaptic Dysfunction in Neurodegenerative Diseases
Kwok-On Lai and Nancy Y. Ip*
The number of people afflicted with neurodegenerative diseases will increase as the population ages. The National Institute of Aging at the U.S. National institutes of Health estimated that 524 million people were aged 65 or older in 2010 (8% of the world’s population), and the aging population is projected to grow to nearly two billion by 2050 (16% of the world’s population), with most of the increase in developing countries (www.nia.nih.gov/research/publication/global-health-and-aging/humanitys-aging). The implications of an aging population are profound: as this segment of the population lives long enough to suffer serious age-related chronic diseases, it will spur a massive demand for health care.
Among the brain diseases and disorders in the elderly, neu-rodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) represent a leading cause of mortality. Although therapeutic drugs for these diseases are available, they merely alleviate the symptoms and, in some cases, are only ef-fective in the early stages of the disease. The current obstacle to developing effective treatments for these neurodegenerative dis-eases is the lack of understanding of disease pathophysiology. Delineating the complex biological processes at the molecular level within the diseased brain is therefore crucial to uncovering new drug targets against which more effective therapeutic drugs can be developed. This article summarizes our efforts to eluci-date the molecular mechanisms that underlie the neuronal death and synaptic dysfunction in these diseases, with particular em-phasis on the role of a key serine/threonine kinase in the brain: cyclin-dependent kinase 5 (Cdk5).
Alzheimer’s DiseAseAD is the most common form of dementia, characterized by loss of cognitive function and memory. The disease generally affects individuals over the age of 60, and almost half of the popula-tion over the age of 85 eventually succumbs to it. AD is clinically characterized by progressive cognitive impairment and memory loss (1). The key pathologies of the patient’s postmortem brains include extensive neuronal loss, disposition of senile plaques that consist of the accumulation of b-amyloid peptides (Ab), and
appearance of neurofibrillary tangles, which are composed of hyperphosphorylated tau. Emerging evidence indicates that dys-regulation of Cdk5 activity is one of the major culprits responsible for neuronal death and loss of synapses in AD (2).
role of CDk5 in TAu hyperphosphorylATion AnD neuronAl DeAThThe catalytic activity of Cdk5 requires its binding to either one of the two activators: p35 or p39. Calpain-mediated cleavage of p35 and p39 to the more stable p25 and p29, respectively, is proposed to account for the aberrant activity of Cdk5 in AD (3,4). Hyperactivity of Cdk5 is involved in multiple aspects of AD patho-genesis. Cdk5 can directly phosphorylate tau, which results in destabilization of microtubules and is therefore believed to exert toxic effects through disruption of axonal transport in neurons (5). Indeed, knockdown of Cdk5 expression by RNAi reduces tau phosphorylation and decreases the number of neurofibrillary tangles in triple transgenic (3x-Tg) AD mice (6), suggesting that Cdk5 is the main kinase causing tau hyperphosphorylation in AD.
In addition to tau pathology, Cdk5 also mediates neuronal death induced by Ab, since inhibition of Cdk5-p25 activity pre-vents Ab-induced apoptosis in cortical neurons (7). Mechanisti-cally, Cdk5 might regulate neuronal death by activating down-stream kinases such as p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) (8,9). In addition, our laboratory and others have found that Cdk5 can promote AD pathogenesis through the regulation of gene transcription. The transcription factor STAT3 can be phosphorylated by Cdk5 at Ser-727 both in vitro and in vivo in the brains of rodent models, and the transcriptional activity of STAT3 is enhanced upon this phosphorylation event (10). This leads to increased transcription of b-secretase 1 mRNA, which encodes the enzyme b-secretase that participates in the amyloidogenic cleavage of amyloid pre-cursor protein (APP) to the Ab peptide. Consistent with the notion that Cdk5 is involved in Ab generation, enhanced Ab production is observed in mice overexpressing p25, and the increased APP processing can be reversed by a Cdk5 inhibitor (11). STAT3 is also involved in Ab-induced neuronal death. Enhanced tyrosine phosphorylation of STAT3, which indicates activation of the tran-scription factor, is observed in cultured cortical neurons upon Ab treatment or in the hippocampi of AD patients. Suppression of STAT3—either by RNAi or using a peptide that blocks STAT3 dimerization—can abolish Ab-induced caspase activity and neu-ronal apoptosis (12). These findings underscore that STAT3 and
the transcription of target genes, such as iNOS and TRAIL, are crucial mediators of neuronal loss in response to Ab.
While the aforementioned studies demonstrate that Cdk5 is required for neuronal apoptosis in AD, it should be empha-sized that Cdk5 also paradoxically promotes neuronal survival during brain development. We have previously shown that the anti-apoptotic protein Bcl-2 can be phosphorylated by Cdk5 at Ser-70. Notably, expression of a phosphodeficient S70A Bcl-2 mutant increases nuclear fragmentation (a sign of apoptosis) in cultured retinal neurons, but it does not further exacerbate apoptosis in Cdk5-/- neurons, indicating that the absence of Bcl-2 phosphorylation at Ser-70 contributes to the neuronal death of neurons lacking Cdk5 (13). Other studies also found that Cdk5 in the nucleus promotes neuronal survival by preventing cell cycle reentry (14,15). It therefore appears that an intricate balance in Cdk5 activity is critical to neuronal survival: either too much or too little Cdk5 activity can lead to neuronal death through distinct signaling pathways.
role of CDk5 in synApTiC DysfunCTionEmerging evidence indicates that loss of synapses and impaired synaptic plasticity in the hippocampus precedes neuronal death in AD, and the memory deficit seen is largely attributable to the synaptic abnormalities. Natural accumulation of soluble Ab oligomers accounts for the progressive loss of synapses of hip-pocampal neurons (16). Precise synaptic insertion and removal of the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-
methyl-4-isoxazolepropionic acid (AMPA) subtypes of glutamate receptors is essential for synaptic transmission and plastic-ity, and hence higher cognitive functions such as learning and memory (17). Ab affects neurotransmission and synaptic plas-ticity by inducing the internalization and removal of NMDA and AMPA receptors from synapses (18,19). Synaptic strength can also be modulated by regulating the number and morphology of dendritic spines—specialized structures on the neuronal pro-cesses of postsynaptic neurons where excitatory synapses are localized. While a typical mature spine contains a bulbous mush-room-shaped head separated from the dendritic shaft by a short spine neck, treatment using Ab peptides leads to elimination of dendritic spines, and the remaining spines become abnormally long and irregular (20). It is generally believed that the reduction of synaptic AMPA and NMDA receptors, and the loss of mature dendritic spines, are central to the cognitive impairment in AD.
Many synaptic proteins have recently been identified to be substrates of Cdk5 (21). Similar to the situation in neuronal sur-vival and death discussed above, a balance in Cdk5 activity is critical to normal synapse function. On the one hand, Cdk5 is required for synaptic plasticity and memory formation through phosphorylation of downstream targets such as the brain-de-rived neurotrophic factor receptor TrkB (22) or the regulation of the cyclic AMP pathway (23). On the other hand, conditional knockout mice with reduced Cdk5 expression show enhanced memory as a result of reduced degradation of the NMDA re-ceptor subunit GluN2 (24). Indeed, hyperactivity of Cdk5 in AD
Division of Life Science, Molecular Neuroscience Center and State Key Laboratory of Molecular Neuroscience, The Hong Kong University of Science and Technology, Hong Kong*Corresponding Author: [email protected]
Figure 1. Schematic diagram illustrating the multiple roles of Cdk5 in the pathogenesis of AD and PD. Cdk5 phosphorylates multiple targets in diverse cellular processes, including neuronal death, microtubule disassembly, Ab production, and synaptic loss. EndoB1, endophilin B1; UVRAG, UV radiation resistance-associated gene protein; JNK, c-Jun N-terminal kinase; STAT3, signal transducer and activator of transcription 3; BACE1, b-secretase; APP, amyloid precursor protein; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; NR2B, N-methyl D-aspartate receptor subtype 2B; EphA4, Eph receptor A4.
4

Produced by the Science/AAAS Custom Publishing Office
7
NEURODEGENERATION RESEARCH
6
might not only contribute to neuronal death and toxicity through tau hyperphosphorylation, but is also likely responsible for the impaired synapse function in the early stages of the disease. For example, phosphorylation of postsynaptic scaffold proteins such as PSD-95 and GKAP by Cdk5 might account for the loss of AMPA receptors induced by Aß (25,26). Moreover, a previ-ous study demonstrated that Cdk5 promotes spine elimination through the regulation of the actin cytoskeleton. Upon activation of the receptor tyrosine kinase EphA4 by the membrane-bound ligand ephrinA1, Cdk5 is activated, which in turn phosphory-lates and activates the guanine nucleotide exchange factor ephexin1. This leads to activation of the small GTPase RhoA, and triggers spine retraction (27). We also found that activation of EphA4 leads to degradation of AMPA receptors by the prote-asome (28). These studies indicate that EphA4 is an important negative regulator of excitatory neurotransmission. It would be interesting to further investigate whether the spine elimination and loss of AMPA receptors mediated by EphA4 signaling might contribute to the loss of synapses in AD.
pArkinson’s DiseAsePD is a progressive neurodegenerative disorder that is associ-ated with cognitive, emotional, and movement disorders, includ-ing resting tremor, rigidity, bradykinesia, and postural instability. The characteristic pathology of PD includes loss of dopaminer-gic neurons in the substantia nigra pars compacta and the pres-ence of intracytoplasmic inclusions known as Lewy bodies. The etiology and the pathogenesis of PD are not completely under-stood but genetic factors might account for the progression of the disease (29). In particular, two missense point mutations in the α-synuclein protein (A53T and A30P) are linked to the early-onset of the familial PD (30,31).
Deregulation of autophagy is an emerging cellular mecha-nism that underlies the pathophysiology of PD (32). Autoph-agy is a homeostatic pathway involved in protein degrada-tion and organelle turnover through the lysosomal machinery. The autophagic pathway is responsible for the clearance of α-synuclein, which is the major constituent of Lewy bodies. Moreover, the mutations of α-synuclein observed in familial PD attenuate its degradation by chaperone-mediated autophagy (33). While these findings suggest that impaired autophagy might contribute to the aberrant accumulation of α-synuclein in Lewy bodies observed in PD, hyperactivation of the au-tophagic pathway is associated with neuronal death in PD models (34, 35). Understanding the regulation of autophagy in PD will therefore provide insights into potential targets for drug screening that could ameliorate PD symptoms or disease progression.
We recently delineated an unexpected role for Cdk5 in regu-lating neuronal autophagy through the protein endophilin B1 (EndoB1). A protein exhibiting lipid-binding properties, EndoB1
is required for autophagy induction and cell death regulation in fi-broblasts (36). We found that EndoB1 is phosphorylated by Cdk5 at Thr-145. Knockdown of EnboB1 in cultured cortical neurons by RNAi abolished starvation-induced autophagy, as indicated by the formation of microtubule-associated protein 1 light chain 3 (LC3)-II-positive autophagosomes. Notably, the impaired au-tophagy could be rescued by coexpression of the wild type, but not the phosphodeficient T145A mutant form of EndoB1 (37). Phosphorylation by Cdk5 promotes EndoB1 dimerization, which then triggers formation of autophagosomes through the recruit-ment of the UV radiation resistance-associated gene protein and Beclin 1. Interestingly, increased Thr-145 phosphorylation of En-doB1 is detected in mice injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and also in α-synucleinA53T mice, two well-established animal models of PD. Furthermore, knockdown of either EndoB1 or Cdk5, or overexpression of the T145A mu-tant of EndoB1, abolished the α-synucleinA53T mutant-induced autophagy and neuronal death (37). Our findings therefore es-tablish the involvement of Cdk5-mediated phosphorylation of En-doB1 and autophagy deregulation in the pathophysiology of PD.
In conclusion, we and others have demonstrated a complex but essential role for Cdk5 in the pathogenesis of AD and PD. Aberrant Cdk5 activity contributes to different aspects of disease progression, including neuronal death and synaptic dysfunction, through phosphorylation of distinct substrates. The identifica-tion of small molecule inhibitors of Cdk5 would be a promising strategy for drug discovery targeting these neurodegenerative diseases.
REFERENCES 1. D. M. Walsh, D. J. Selkoe, Neuron 44, 181 (2004). 2. Z. H. Cheung, N. Y. Ip, Trends Cell Biol. 22, 169 (2012). 3. G. N. Patrick et al., Nature 402, 615 (1999). 4. H. Patzke, L. H. Tsai, J. Biol. Chem. 277, 8054 (2002). 5. D. B. Evans et al., J. Biol. Chem. 275, 24977 (2000). 6. D. Piedrahita et al., J. Neurosci. 30, 13966 (2010). 7. Y. L. Zheng et al., J. Biol. Chem. 285, 34202 (2010). 8. K. H. Sun, H. G. Lee, M. A. Smith, K. Shah, Mol. Biol. Cell 20, 4611 (2009). 9. K. H. Chang et al., J. Neurochem. 113, 1221 (2010).10. A. K. Y. Fu et al., Proc. Natl. Acad. Sci. U.S.A. 101, 6728 (2004).11. Y. Wen et al., Neuron 57, 680 (2008).12. J. Wan et al., J. Neurosci. 30, 6873 (2010).13. Z. H. Cheung, K. Gong, N. Y. Ip, J. Neurosci. 28, 4872 (2008).14. J. Zhang, K. Herrup, Cell Cycle 7, 3487 (2008).15. J. Zhang et al., J. Neurosci. 30, 5219 (2010).16. G. A. Krafft, W. L. Klein, Neuropharmacology 59, 230 (2010).17. J. D. Shepherd, R. L. Huganir, Annu. Rev. Cell Dev. Biol. 23, 613 (2007).18. H. Hsieh et al., Neuron 52, 831 (2006).
The Role of Autophagy, Apoptosis, Neuroinflammation, and Mitochondrial Dysfunction in Parkinson’s Disease
Dong Chen, Haigang Ren, Qingsong Hu, Feng Gao, and Guanghui Wang*
Parkinson’s disease (PD) is the second most common neuro-degenerative disease, characterized by the loss of dopaminegic (DA) neurons in the substantia nigra pars compacta (SNpc) and the presence of Lewy bodies in the surviving neurons, accom-panied by the formation of dystrophic neurites (1–2). Although the etiology of PD remains unknown, it is apparent that the in-teraction between environmental and genetic factors is critical. Mitochondrial dysfunction plays an important role in PD patho-genesis; damaged mitochondria induce many cellular and patho-logical processes in the diseased brain, including the induction of apoptosis and the production of reactive oxygen species (ROS). ROS formation may result in oxidative stress and downstream signaling that promotes neuroinflammation.
Mitophagy is a cellular process that clears damaged mitochon-dria. A number of PD-related gene products appear to be tightly associated mitophagy, including PINK1 and parkin (3). Mutations in these genes can result in a failure to clear damaged mitochon-
dria, leading to an accumulation of dysfunctional mitochondria (4–5). Many factors involved in PD, including the genetic fac-tors—parkin, DJ-1, Omi—and environmental factors—rotenone and 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP)—play roles in the regulation of mitochondria. Loss of protective func-tion or gain of toxic function in these factors is thought to induce oxidative stress, apoptosis, and neuroinflammation in associa-tion with PD (6–9). This review summarizes and provides context for these findings.
AuTophAgy AnD pDThere is growing evidence showing that autophagy is associated with the pathogenesis of PD (10–12). The postmortem brains of PD patients present with autophagic vacuoles in the SNpc, indicating an involvement of autophagy (13). Additionally, the loss of autophagy-related genes appears to result in neurode-generation. For example, a conditional deletion of Atg7 in the central nervous system or SNpc selectively impairs autophagy and leads to a progressive loss of DA neurons, indicating the protective role of basal levels of autophagy against neurodegen-eration (14–15). Interestingly, many PD-related proteins such as LRRK2, α-synuclein, DJ-1, PINK1, parkin, and Omi are involved in the autophagy process (16–21). The protease Omi activates
Laboratory of Molecular Neuropathology, Department of Pharmacology, Soochow University College of Pharmaceutical Sciences, Suzhou, Jiangsu, China*Corresponding Author: [email protected]
19. P. Kurup et al., J. Neurosci. 30, 5948 (2010).20. P. N. Lacor, J. Neurosci. 24, 796 (2007).21. K. O. Lai, N. Y. Ip, Biochim. Biophys. Acta 1792, 741 (2009).22. K. O. Lai et al., Nat. Neurosci. 15, 1506 (2012).23. J. S. Guan et al., PLOS ONE 6, e25735 (2011).24. A. H. Hawasli et al., Nat. Neurosci. 10, 880 (2007).25. F. Roselli, P. Livrea, O. F. Almeida, PLOS ONE 6, e23097 (2011).26. F. Roselli et al., J. Neurosci. 25, 11061 (2005).27. W. Y. Fu et al., Nat. Neurosci. 10, 67 (2007).28. A. K. Fu et al., Nat. Neurosci. 14, 181 (2011).29. E. Broussolle, S. Thobois, Rev. Neurol. 158, 11 (2002).30. R. Kruger et al., Nat. Genet. 18, 106 (1998).31. M. H. Polymeropoulos et al., Science 276, 2045 (1997).32. T. Pan et al., Neurobiol. Dis. 32, 16 (2008).33. A. M. Cuervo, L. Stefanis, R. Fredenburg, P. T. Lansbury, D. Sulzer,
Science 305, 1292 (2004).34. E. H. Baehrecke, Nat. Rev. Mol. Cell Biol. 6, 505 (2005).35. M. C. Maiuri, E. Zalckvar, A. Kimchi, G. Kroemer, Nat. Rev. Mol. Cell Biol. 8, 741 (2007).36. Y. Takahashi et al., Nat. Cell Biol. 9, 1142 (2007).37. A. S. Wong et al., Nat. Cell Biol. 13, 568 (2011).
Acknowledgments: We thank all members of the Ip laboratory and are grateful to Ka-Chun Lok for preparing the figure in this review. The studies were supported in part by the Research Grants Council of Hong Kong (661109, 661309, 660810, 661010, 661111, and 661212), and the Theme-based Research Scheme (T13-607/12R), as well as the National Key Basic Research Program of China (2013CB530900), the Innovation and Technology Fund for State Key Laboratory (ITCPT/17-9), the Shenzhen Peacock Plan, and the SH Ho Foundation.

8
Produced by the Science/AAAS Custom Publishing Office
9
NEURODEGENERATION RESEARCH
autophagy through its cleavage of Hax-1 as part of the Beclin 1-dependent autophagic pathway. In vitro data showed that Omi-induced autophagy promotes the degradation of neurode-generative proteins such as pathogenic A53T α-synuclein and truncated polyglutamine (polyQ)-expanded huntingtin, whereas knockdown of Omi decreases the basal level of autophagy and increases the level of A53T α-synuclein and polyQ-expanded huntingtin. Further, S276C Omi, a protease-defective mutant present in the brains of mnd2 (motor neuron degeneration 2) mice, is unable to regulate autophagy. These results indicate that Omi is a regulator of autophagy and provide evidence that Omi-associated autophagy may provide an essential means for protein quality control in neurodegenerative diseases (21).
In addition, two other PD-related proteins, DJ-1 and parkin, also regulate autophagy. Knockdown of DJ-1 induces autophagy through activation of JNK and upregulation of Beclin 1 transcrip-tion. Inhibition of the JNK pathway was found to block autophagy activation and p62 degradation induced by DJ-1 knockdown, suggesting that autophagy regulation by DJ-1 is JNK-dependent (18).
Parkin is an E3 ubiquitin ligase that has been shown to reg-ulate mitophagy. Interestingly, in the cytosol, parkin represses autophagy. Parkin, but not its E3 ligase-deficient mutants, mono-ubiquitinates Bcl-2, leading to a stabilization of this protein. The resulting effective increase in Bcl-2 levels enhances the interac-tion between Bcl-2 and Beclin 1, thereby repressing autophagy. In parkin-overexpressed cells, LC3 conversion is decreased, whereas it is increased in parkin-knockdown cells, further sug-gesting that parkin represses autophagy. In contrast to cytosolic parkin, mitochondrial parkin, after its recruitment to mitochondria upon carbonyl cyanide m-chlorophenylhydrazone treatment, in-creases mitophagy, suggesting a differential role of cytosolic and mitochondrial parkin in autophagy (19).
miToChonDriA AnD ApopTosisRecent studies have shown that the selective degradation of damaged mitochondria by autophagy through the PINK1/parkin pathway is an important quality control mechanism in PD patho-genesis (4). Loss of function of PINK1 or parkin causes an ac-cumulation of abnormal mitochondria, which leads to increased oxidative stress. Mitochondria have an integral role in the apop-totic cell death pathway by releasing Bax and cytochrome c into the cytosol to induce apoptosis. Oxidative stress can lead to the collapse of mitochondrial membrane potential, which may induce a translocation of Bax to activate apoptotic pathways (22). Our previous studies showed that DJ-1 has anti-apoptotic effects that act through a mitochondria-associated pathway. DJ-1 ex-erts its cytoprotection through inhibition of the p53/Bax/caspase pathway: it interacts with and represses p53 in the nucleus to downregulate the expression of Bax and thereby inhibit caspase activation. However, a K130R mutant of DJ-1 has been shown
to lose its inhibitory effects on p53 due to failed translocation of the mutant protein to the nucleus (23–24). DJ-1 also functions in the TNF-related apoptosis-inducing ligand (TRAIL) pathway by blocking death-inducing signaling complex (DISC) formation and inhibiting procaspase-8 activation (25). DJ-1 inhibits cas-pase-8 activation by its interaction with Fas-associated protein death domain (FADD) to inhibit the formation of DISC. DJ-1, but not its pathogenic mutant L166P, inhibits procaspase-8 activa-tion by competing with it to bind to the death effector domain of FADD. Decreased binding of procaspase-8 to DISC results in lower activation of caspase-8 and the repression of apopto-sis (25). Apart from the anti-apoptotic effect of DJ-1 exerted at the cell membrane, it also has an effect on mitochondria. DJ-1 translocates to the mitochondria and binds to Bcl-XL following oxidative stress. The binding of DJ-1 stabilizes Bcl-XL by inhibit-ing its ubiquitination and degradation through the ubiquitin-pro-teasome system. Knockdown of DJ-1 decreases Bcl-XL levels, subsequently leading to mitochondrial Bax enrichment, cas-pase-3 activation, and cell death in response to oxidative stress (26). Interestingly, the pathogenic DJ-1 mutant L166P shows increased mitochondrial distribution and affinity for Bcl-XL. So, unlike wild-type DJ-1, DJ-1(L166P) does not influence Bcl-XL levels, but rather disrupts mitochondrial Bcl-XL/Bax heterodimer-ization, thereby releasing Bax from Bcl-XL/Bax heterodimers. The released Bax oligomerizes in the outer mitochondrial mem-brane and induces cell death under oxidative stress. Thus, both DJ-1 and its L166P mutant have direct effects on mitochondrial Bcl-XL, but apparently induce mitochondria-related cell death in different ways.
ros, neuroinflAmmATion, AnD pDNeuroinflammation presents in both animal models and the post-mortem brains of PD patients (6,27,28). It has been proposed that mitochondrial dysfunction may exert a crucial role in neuro-inflammation and pathogenesis of PD (29). Injury of mitochon-dria induces the production of ROS in both neuronal and glial cells, and causes DA neuronal cell death. Microglia, the main cell type of the innate immune system in the brain, can be acti-vated to produce inflammatory factors, promoting DA neuronal cell death (30,31). Rotenone, a mitochondrial complex I inhibitor, is well known as an environmental factor that can induce ROS production and oxidative damage in DA neurons. We found re-cently that rotenone induces ROS production in microglia. This does not cause microglial cell death, but activates the nuclear factor kappa B (NF-κB) signaling pathway, thereby inducing a significantly increased expression of inflammatory cytokines, ac-tivation of caspase-1, and maturation of IL-1β (32). Treatment of BV2 cells (a microglial cell line) with rotenone activates p38 mitogen-activated protein kinase (MAPK), whereas removal of ROS with N-acetylcysteine (NAC), a ROS scavenger, reduces p38 and NF-κB activation in BV2 cells, suggesting that this en-
vironmental toxin activates microglia through the p38 MAPK pathway (32). Most research has shown that those PD-related genetic factors that have been studied cause DA neuronal cell death directly or result in DA neurons sensitive to environmental stimuli. It is well accepted that damage to DA neurons induces neuroinflammation (30), but little is known about whether any gene products related to PD pathogeneses are able to activate microglia. Recently, we found that in mnd2 mice, which harbor a protease-deficient Omi (S276C Omi) and show neurodegen-eration, the activation of microglia may be a direct result of the protease deficient Omi. Omi cleaves MEK1, which is upstream of the extracellular signal-regulated kinase 1 and 2 (ERK1/2), thereby repressing ERK1/2 activation. Loss of Omi protease activity in mnd2 mice and knockdown of Omi in BV2 cells acti-vates NF-κB and results in the production of inflammatory fac-tors, including tumor necrosis factor-α and inducible nitric oxide synthase. By contrast, inhibition of MEK1 blocks the loss of Omi activity-induced NF-κB activation. These data suggest that the PD genetic factor Omi is able to activate microglia directly (9), hinting at a fundamental link between neuroinflammation, ROS, and mitochondrial function in both environmentally and geneti-cally induced PD.
In summary, mitochondrial dysfunction caused by genetic and environmental factors—either by loss of protection or toxic in-sult—plays a crucial role in PD pathogenesis. Impairment of mi-tochondrial function triggers multiple cellular processes, includ-ing impaired autophagy, increased sensitivity to oxidative stress, and induction of apoptosis associated with both neurodegenera-tion and neuroinflammation.
REFERENCES 1 J. M. Savitt, V. L. Dawson, T. M. Dawson, J. Clin. Invest. 116, 1744 (2006). 2. C. Liu et al., J. Biol. Chem. 282, 14558 (2007). 3. S. M. Jin, R. J. Youle, J. Cell Sci. 125, 795 (2012). 4. D. Narendra, A. Tanaka, D. F. Suen, R. J. Youle, J. Cell Biol. 183, 795 (2008). 5. N. Matsuda et al., J. Cell Biol. 189, 211 (2010). 6. T. C. Frank-Cannon et al., J. Neurosci. 28, 10825 (2008). 7. C. Zhou, Y. Huang, S. Przedborski, Ann. N.Y. Acad. Sci. 1147, 93
(2008). 8. J. Waak et al., FASEB J. 23, 2478 (2009). 9. Q. Hu et al., Sci. Signal. 5, ra61 (2012).10. B. Levine, G. Kroemer, Cell 132, 27 (2008).11. Z. H. Cheung, N. Y. Ip, Mol. Brain 2, 29 (2009).12. P. A. Jaeger, T. Wyss-Coray, Mol. Neurodegener. 4, 16 (2009).13. P. Anglade et al., Histol. Histopathol. 12, 25 (1997).14. M. Komatsu et al., Nature 441, 880 (2006).15. I. Ahmed et al., J. Neurosci. 32, 16503 (2012).16. E. D. Plowey, S. J. Cherra, 3rd, Y. J. Liu, C. T. Chu, J. Neurochem. 105, 1048 (2008).17. T. Vogiatzi, M. Xilouri, K. Vekrellis, L. Stefanis, J. Biol. Chem. 283, 23542 (2008).18. H. Ren et al., Cancer Lett. 297, 101 (2010).19. D. Chen et al., J. Biol. Chem. 285, 38214 (2010).17. T. Vogiatzi, M. Xilouri, K. Vekrellis, L. Stefanis, J. Biol. Chem. 283, 23542 (2008).18. H. Ren et al., Cancer Lett. 297, 101 (2010).19. D. Chen et al., J. Biol. Chem. 285, 38214 (2010).20. S. Michiorri et al., Cell Death Differ. 17, 962 (2010).21. B. Li et al., Cell Death Differ. 17, 1773 (2010).22. C. Henchcliffe, M. F. Beal, Nat. Clin. Pract. Neurol. 4, 600 (2008).23. J. Fan et al., J. Biol. Chem. 283, 4022 (2008).24. J. Fan et al., FEBS Lett. 582, 1151 (2008).25. K. Fu et al., Oncogene 31, 1311 (2012).26. H. Ren, K. Fu, D. Wang, C. Mu, G. Wang, J. Biol. Chem. 286, 35308 (2011).27. H. M. Gao et al., J. Neurosci. 28 7687 (2008).28. P. L. McGeer, S. Itagaki, B. E. Boyes, E. G. McGeer, Neurology 38, 1285 (1988).29. M. E. Witte, J. J. Geurts, H. E. de Vries, P. van der Valk, J. van Horssen, Mitochondrion 10, 411 (2010).30. M. L. Block, J. S. Hong, Biochem. Soc. Trans. 35, 1127 (2007).31. W. G. Kim et al., J. Neurosci. 20, 6309 (2000).32. F. Gao, D. Chen, Q. Hu, G. Wang, PLOS ONE 8, e72046 (2013).
Acknowledgments: This work was supported in part by the National High-Tech Research and Development Program of China (“973” Program, 2011CB504102), and the National Natural Sciences Foundation of China (911327723).

10
Produced by the Science/AAAS Custom Publishing Office
11
NEURODEGENERATION RESEARCH
Autophagy is an evolutionarily conserved pathway that involves the sequestration and delivery of cytoplasmic materials to lyso-somes, where proteins, lipids, and organelles are degraded and recycled. Generally, autophagy is divided into three categories: macroautophagy, microautophagy, and chaperone-mediated au-tophagy (CMA). Autophagy is also involved in many fundamental cellular activities and impacts numerous cellular regulatory path-ways involved in diverse processes, including tumorigenesis, longevity, immunity, organelle turnover, ER stress, and apopto-sis. Important progress has been made in defining the role of autophagy in human disease, enabling the development of better treatments for cancer, neurological diseases, and cardiovascular diseases.
Starting four decades ago, autophagy research expanded from a relatively minor area to one of the most exciting and impor-tant topics in cell biology. The total number of autophagy papers published in peer review journals worldwide has increased 15 fold in past 10 years (1). A number of autophagy-related genes and pathways have been identified and innovative molecular tools developed for genetic manipulation and chemical targeting of pathway components (2). In recent decades, autophagy has evolved into an active research field in China. Chinese scientists have contributed a great deal to basic and clinical autophagy research, their journal papers accounting for 10% of worldwide publications on this topic.
In the Laboratory of Aging and Nervous Diseases at Soochow University, one of the oldest domestic institutions carrying out autophagy research, we have been working on the role of au-tophagy in the regulation of cell metabolism and cell survival in the central nervous system (CNS), including neurodegenerative diseases, neuronal excitotoxicity, cerebral ischemia, and isch-emic preconditioning. In particular, our researches focus on the degradation of pathogenic proteins, excitotoxicity, precondition-ing, and the crosstalk between autophagy and apoptosis. This work has elucidated some of the pathogenic mechanisms of ner-vous system diseases and suggested novel targets for therapies. Below is a summary of some of the research recently performed in our laboratory.
AuTophAgy in meTAbolism of misfolDeD proTeinsAccumulation and aggregation of misfolded proteins is the hall-mark of several neurodegenerative diseases. Examples include the huntingtin (Htt) protein in Huntington’s disease (HD) and α-synuclein in Parkinson’s disease (PD).
HD is caused by an expansion of the polyglutamine (polyQ) tract in the HTT protein. This induces selective degeneration of striatal projection neurons and cortical pyramidal neurons due to the accumulation of N-terminal mutant HTT and intranulear inclu-sion formation in HD brains. Our studies suggest that autophagy and lysosomal cathepsins play critical roles in the degradation of N-terminal HTT. Altered processing of mutant HTT by autophagy and cathepsin D contributes to HD pathogenesis (3–5). In mac-roautophagy, Beclin 1 has been shown to be involved in the deg-radation of HTT (6). Additionally, we have shown that HTT frag-ments can be degraded by CMA. The CMA components—heat shock protein cognate 70 (Hsc70) and lysosome-associated protein 2A (LAMP-2A)—play essential roles in the clearance of HTT (7).
The pathological hallmark of PD is neuronal inclusions termed Lewy bodies that are composed mainly of the misfolded protein α-synuclein. Our studies have shown that autophagy plays a role in the degradation of α-synuclein (8). Lysosomal enzymes ca-thepsin L and B are involved in the regulation of autophagy and degradation of α-synuclein (9). Abnormal distribution of cathep-sin L is thought to contribute to neuronal death; thus drugs inhib-iting cathepsin L nuclear translocation might be neuroprotective against PD (10, 11). Recent unpublished work has shown that impaired autophagy flux is involved in PD, resulting in the accu-mulation and aggregation of α-synuclein protein and damage of lysosomal function in dopamine neurons.
AuTophAgy in neuronAl exCiToToxiCiTyExcitotoxicity is thought to play an important role in the patho-genesis of a number of neurological disorders, including HD, PD, and Alzheimer’s disease (AD) (12). In the late 1990s, we and other laboratories provided morphological and biochemical evi-dence demonstrating that an apoptotic mechanism was involved in excitotoxic neuronal death (13–15). In 2008, we reported that excitatory amino acid receptor agonists activate autophagy in animal models (16). Both kainic acid (KA) receptor agonist KA and N-methyl-D-aspartate (NMDA) receptor agonist quinolinic acid (QA) induced activation of autophagy, accompanied by downregulation of Bcl-2, and upregulation of Bax, tumor sup-
pressor protein p53 (TP53), and TP53-upregulated modulator of apoptosis (PUMA). Autophagy inhibitors and cathepsin inhibitors markedly inhibited autophagy activation and the mitochondria-mediated apoptotic pathway, suggesting that the autophagy-lys-osmal pathway plays an important role in excitotoxic neuronal in-jury (17,18). TP53 might be the key factor mediating autophagy activation and mitochondrial dysfunction in neuronal excitotoxic-ity. TP53 inhibitor pifithrin-α not only inhibited excitotoxic neuro-nal injury, but also suppressed KA- or QA-induced autophagy activation and mitochondrial dysfunction (17,18). These studies have advanced the understanding of the molecular mechanisms of excitotoxicity, which involve necrosis, apoptosis, and autopha-gic cell death (19).
DifferenT roles of AuTophAgy in CerebrAl isChemiA AnD isChemiC preConDiTioningStroke is a global health problem and one of the leading causes of adult disability in developed countries, while the phenomenon of ischemic preconditioning (IPC) has been recognized as one of the most potent mechanisms to protect against ischemic stroke (20). Morphological and biochemical evidence obtained from a rodent permanent focal ischemia model suggests that ischemic stroke induced autophagy activation. Using pharmacological ap-proaches, we demonstrated that the autophagy-lysosomal path-way is involved in the neuronal death induced by permanent fo-cal ischemia in adult rodents (21). Activation of autophagy and lysosomal proteases also occurred in astrocytes and contributed to the ischemia-induced glial death in both in vivo and in vitro ischemia models (22). However, we found that IPC also activat-ed autophagy and a blockade of autophagy activation during IPC abolished the neuroprotective effects of IPC (23).
The studies above clearly demonstrated that autophagy could play different roles under different pathological conditions. In an effort to better elucidate the mechanisms involved, we showed that both IPC and lethal cerebral ischemia induced activation of autophagy, but the extent and persistence of this activation were variable. The condition of permanent cerebral ischemia briefly activated autophagy, while IPC itself mildly triggered autophagy and this activation persisted longer during a second ischemic at-tack (24). The divergent effects of autophagy during ischemia and preconditioning were also found to be related to endoplas-mic reticulum (ER) stress (24,25), which induced ER-associated degradation (ERAD), a process that involves both the protea-somal and autophagic pathways. We found that pre-activation of autophagy by IPC can upregulate molecular chaperones and hence reduce excessive ER-stress–dependent apoptosis during fatal ischemia. Our current studies have focused on the contri-bution of ER chaperone GRP78 to IPC-induced autophagy and have found indications that GRP78 is a key mediator of autopha-gy activation during preconditioning (unpublished observations). This work also suggests that induction of autophagy and ER
stress may provide a strong justification for the introduction of IPC to treat cerebral ischemia (23).
CrossTAlk beTween AuTophAgy AnD ApopTosisAt the cellular level, the involvement of autophagy in the cell death and cell survival processes appears to be complex. In QA- and KA-induced apoptosis, the downregulation of pro-survival protein Bcl-2, was partially blocked by the autophagy inhibitor 3-methyladenine (3-MA) (16, 17). Our studies also revealed that mitochondrial inhibitor 3-nitropropionic acid (3-NP)-induced neuronal death involved both apoptotic and autophagic mech-anisms. TP53 was found to induce a number of downstream genes, including Bax, PUMA, and damage-regulated autophagy modulator 1 (DRAM1). Importantly, neuronal injury induced by 3-NP was inhibited by the TP53 specific inhibitor pifithrin-α, the autophagy inhibitor 3-MA, and by knockdown of DRAM1 (26,27). DRAM1 apparently affects autophagy through augmentation of lysosomal acidification, fusion of lysosomes with autophago-somes, and clearance of autophagosomes (27). Also, 3-NP–in-duced autophagy seemed to contribute to the degradation of Bcl-2 (28). We thus investigated the crosstalk between autophagy and apoptosis. It is well defined that autophagy activation under nutritional stress conditions benefits cell survival (29). We found that autophagy activation induced by serum deprivation was ac-companied by an upregulation of Bcl-2. Inhibition of Bcl-2 func-tion or downregulation of Bcl-2 expression amplified autophagy activation and resulted in apoptosis, while overexpression of Bcl-2 limited autophagy activation and blocked cell death in re-sponse to serum deprivation (30). Bcl-2 thus plays an important role in limiting autophagy activation and preventing initiation of programmed cell death during serum starvation. These studies have added significant information on a classical role of autoph-agy in nutritional stress.
Recently, we defined a novel pathway for crosstalk between autophagy and apoptosis involving Bax and DRAM1. We found that pro-apoptotic protein Bax was degraded through autophagy under basal condition, keeping its levels low. However, DRAM1-mediated activation of autophagy resulted in upregulation of Bax due to protein-protein interaction between DRAM1 and Bax. In-creased Bax was recruited to lysosomes by DRAM1, and Bax lysosomal translocation permeabilized lysosomal membranes. Apoptosis was then initiated by lysosomal protease cathepsin B-mediated Bid cleavage, cytochrome c release, and caspase 3 activation (unpublished observations).
AuTophAgy unDer oTher physiologiCAl AnD pAThologiCAl ConDiTionsIt is well known that regular endurance exercise is required to maintain muscle mass and body fitness in mammals. Our studies showed that long-term regular exercise increased autophagy activity in both adult and aged animals (31).
The Multiple Roles of Autophagy in Neural Function and Disease
Rui Sheng and Zheng-Hong Qin*
Department of Pharmacology and Laboratory of Aging and Nervous Diseases, Soochow University School of Pharmaceutical Science, Suzhou, China*Corresponding Author: [email protected]

12
Produced by the Science/AAAS Custom Publishing Office
13
NEURODEGENERATION RESEARCH
Iron is an essential element in many physiological processes in the ner-vous system, including DNA synthe-sis, oxygen transport, mitochondrial respiration, neurotransmitter synthe-sis and myelination (1). Brain iron lev-els increase with age, especially in those regions associated with motor functions, such as the globus pallidus and substantia nigra. Age-related iron accumulation in regions associated with cognitive function, such as frontal grey matter, has also been reported (2). Recently, abnormal brain iron ac-cumulation was reported in a wide spectrum of neurodegenerative dis-orders, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), suggesting a role for iron accumula-tion in neurodegeneration (1). In this review, we discuss iron metabolism in the brain and the toxic effects of iron overloading in neurodegenera-tive disorders as well as briefly intro-duce some treatment strategies and provide insights based on our recent findings.
brAin iron TrAnsporT AnD meTAbolismThe brain needs a stable environment in order to function normally. Both a deficiency and an excess of iron can cause severe damage, so it is important that iron levels in the brain are tightly regulated. The blood-brain barrier (BBB) forms a frontline boundary and maintains appropriate iron levels by regulating iron influx and efflux (3). Additionally, iron exchange can take place in the blood-cerebrospinal fluid barrier that separates circulating blood from the brain’s extracellular fluid (3). Inside the brain, glial
cells act cooperatively to maintain appropriate iron levels to meet the need of both neurons and the brain as a whole. Astrocytes act as a bridge between neurons and BBB, and are responsible for regulating iron transport within the brain. Microglia serve as a brain iron capacitor by quickly accumulating and releasing iron, and thus contain variable iron content depending on conditions (4,5).
Similar to cells outside the brain, brain cells such as neurons, microglia, and astrocytes express numerous iron-related pro-teins (2). We have previously described in detail the metabolic pathways for cellular iron in the brain (3,6). Briefly, transferrin (Tf), the transferrin receptor (TfR), and the divalent metal trans-porter 1 (DMT1) are involved in iron uptake. Tf carrying ferric iron (Fe3+) binds to TfR on the cell membrane, inducing internaliza-tion of the whole complex. With the help of endosomal metallore-ductase STEAP3, Fe3+ is converted to ferrous iron (Fe2+), which
Exercise training increased autophagy activity, but reduced apoptosis of muscle cells by modulating IGF-1 and its receptors, the Akt/mTOR and Akt/FOXO3a signaling pathways in skeletal muscles. Autophagy activation appeared to inhibit chloroquine-induced muscle damage through removal of abnormal mitochon-dria; it thus plays a positive role in muscle establishment and fitness (unpublished data).
Traumatic brain injury (TBI) also activates autophagy and ly-sosomal proteases in neurons. Inhibition of this autophagy-lys-osome axis may help attenuate traumatic damage and improve functional recovery (32 ).
In summary, our research demonstrates that autophagy is coor-dinated with other cellular activities such as apoptosis, ER stress, and mitochondrial function to maintain cell homeostasis. Autoph-agy plays different roles in neurodegenerative diseases, neuronal excitotoxicity, cerebral ischemia, and ischemic preconditioning. The multifunctional roles of autophagy are explained by its ability to interact with distinct key components in various signaling path-ways involving TP53, DRAM1, Bcl-2, and GRP78. We expect that a better understanding of the roles of autophagy and its regulation will bring deeper insight into pathogenic mechanisms of CNS dis-eases and provide useful information leading to the development of novel therapies.
REFERENCES 1. W. D. Le, Z. H. Qin, Acta Pharmacol. Sin. 34, 583 (2013). 2. Y. P. Yang, Z. Q. Liang, Z. L. Gu, Z. H. Qin, Acta Pharmacol. Sin. 26, 1421 (2005). 3. Z. H. Qin et al., Hum. Mol. Genet. 12, 3231 (2003). 4. Z. H. Qin et al., J. Neurosci. 24, 269 (2004). 5. L. L. Chen et al., Acta Pharmacol. Sin. 33, 385 (2012). 6. J. C. Wu et al., Acta Pharmacol. Sin. 33, 743 (2012). 7. L. Qi et al., PLOS ONE 7, e46834 (2012).
8. F. Yang et al., Neurosci. Lett. 454, 203 (2009). 9. B. Xiang et al., Brain Res. 1387, 29 (2011).10. L. Li et al., Neurosci. Lett. 489, 62 (2011).11. X. F. Fei et al., Brain Res. 1264, 85 (2009).12. X. X. Dong, Y. Wang, Z. H. Qin, Acta Pharmacol. Sin. 30, 379 (2009).13. Z. H. Qin, Y. Wang, M. Nakai, T. N. Chase, Mol. Pharmacol. 53, 33 (1998).14. Z. H. Qin, Y. Wang, T. N. Chase, Brain Res. 725, 166 (1996).15. Z. H. Qin et al., J. Neurosci. 19, 4023 (1999).16. Y. Wang et al., Autophagy 4, 214 (2008).17. Y. Wang et al., Eur. J. Neurosci. 30, 2258 (2009).18. X. X. Dong et al., Neuroscience 207, 52 (2012).19. Y. Wang, Z. H. Qin, Apoptosis 15, 1382 (2010).20. X. Q. Liu, R. Sheng, Z. H. Qin, Acta Pharmacol. Sin. 30, 1071 (2009).21. Y. D. Wen et al., Autophagy 4, 762 (2008).22. A. P. Qin et al., Autophagy 6, 738 (2010).23. R. Sheng et al., Autophagy 6, 482 (2010).24. R. Sheng et al., Autophagy 8, 310 (2012).25. B. Gao et al., Acta Pharmacol. Sin. 34, 657 (2013).26. X. D. Zhang et al., Autophagy 5, 339 (2009).27. X. D. Zhang, L. Qi, J. C. Wu, Z. H. Qin, PLOS ONE 8, e63245 (2013).28. X. D. Zhang et al., J. Neurosci. Res. 87, 3600 (2009).29. J. D. Rabinowitz, E. White, Science 330, 1344 (2010).30. H. D. Xu et al., PLOS ONE 8, e63232 (2013).31. L. Luo et al., Exp. Gerontol. 48, 427 (2013).32. C. L. Luo et al., Neuroscience 184, 54 (2011).
Acknowledgments: This work was supported by grants from the Natural Science Foundation of China (30930035 and 81271459) and the Ministry of Science and Technology (“973”) Program (2011CB510003).
Iron and Neurodegeneration
Ya Ke1*, Ka-Chun Wu1, and Zhong-Ming Qian2*
1School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong2Laboratory of Neuropharmacology, Fudan University School of Pharmacy, Shanghai, China*Corresponding Authors: [email protected] (Y. K.) and [email protected] (Z. M. Q.)
Figure 1. Brain iron metabolism and neurodegeneration. DMT1 and Lf/TfR, responsible for iron uptake, may be upregulated (in Parkinson’s disease), while APP-MTP1 coupling underlying iron release may be reduced (in Al-zheimer’s disease), facilitating iron accumulation. The resulting iron-induced increased Ab and α-synuclein synthesis may facilitate pathological aggregation. Moreover, abnormally high iron levels can promote free radical production via the Fenton reaction, leading to widespread oxidative damage. These processes likely contribute to the initia-tion of neurodegeneration. Ab, b-amyloid; MTP1, metal transporter protein-1 (ferroportin); APP, amyloid precursor protein; Hp, hephaestin; CP, ceruloplasmin; ROS, reactive oxygen species; DMT1, divalent metal transporter 1; Lf, lactoferrin; LfR, lactoferrin receptor; Tf, transferrin; TfR1, transferrin receptor 1.

14
Produced by the Science/AAAS Custom Publishing Office
15
NEURODEGENERATION RESEARCH
can then pass through the endosomal membrane and enter the cytosol with the aid of DMT1. Alternatively, extracellular Fe2+ that is not bound to transferrin may pass through the cell membrane directly, via DMT1. After entering the cytosol, Fe2+ ions bind to chaperone proteins that transfer it to target proteins, or it enters the mitochondria, an iron-laden organelle, via mitoferrin 1 and 2.
Ferritin, a large, iron storage protein, is composed of two sub-units: ferritin heavy chain (Ft-H)—a ferroxidase—and ferritin light chain (Ft-L). Excess intracellular Fe2+ can be converted to Fe3+ by Ft-H and sequestered by Ft-L for storage. Iron regulatory pro-tein 1 and 2 are responsible for regulating expression of proteins that are sensitive to iron content, i.e., those that carry an iron-responsive element in their mRNA transcript. Finally, ferroportin (or metal transporter protein-1, MTP1) functions to release cellu-lar ferrous iron, which is then converted to Fe3+ by ceruloplasmin (CP) or hephaestin (Hp). In the Fe3+ form, iron can then bind to transferrin again for recirculation. However, the expression of iron-related proteins is not ubiquitous in the brain (2), with cer-tain iron-related proteins being differentially expressed, revealing specialization of iron metabolism within different cells. For ex-ample, transferrin is most highly expressed in oligodendrocytes while astrocytes specialize in synthesizing CP (1,2), suggesting that different cell types in the brain work cooperatively to main-tain iron homeostasis.
iron ACCumulATion in The brAinDue to the pivotal role of iron, the brain has developed iron con-servation mechanisms, as evidenced by the minimal reduction of iron in the brain during systemic iron deficiency (7). Interestingly, iron is found to accumulate with age (1), a process that some suggest is passive and caused by a reduction in the efficiency of the BBB and cells that control iron levels, while others argue that a more active process is at work. A number of iron-related genes have been found to be associated with neurodegenerative disor-ders including AD and PD (8), suggesting that abnormal iron ac-cumulation may be a cause of accelerated neurodegeneration.
iron ToxiCiTy AnD neuroDegenerATionThe toxicity of iron can be explained by the fact that it is highly redoxactive (9). Under normal circumstances, most cellular iron is sequestered by proteins. However, when the level of iron ex-ceeds the capacity of iron-binding proteins, labile iron results. Hydrogen peroxide, which is constantly generated by mitochon-drial electron transport, can be converted to extremely toxic hy-droxyl free radicals in the presence of labile iron via the Fenton and Haber-Weiss reactions (9). These free radicals may elicit an array of cellular damage, including protein carbonylation and lipid peroxidation, and eventually evoke neuronal death. Since neurons normally have a high metabolic demand, they tend to be more susceptible to iron-induced oxidative damage. Moreover, iron has also been found to facilitate abnormal protein aggrega-
tion, which contributes to the pathogenesis of many neurodegen-erative disorders (10–12). However, the molecular mechanism of iron accumulation in neurodegenerative diseases such as AD and PD remains elusive.
Alzheimer’s DiseAseAD is the most common form of dementia (13). Symptoms include impairment of memory and disturbances in reasoning, planning, and perception. This disease is characterized by the pathological hallmarks of amyloid plaques and neurofibrillary tangles (NFTs) that contain hyperphosphorylated tau aggregates, found in the hippocampus, cortex, and other brain regions. The close rela-tionship between iron and the pathogenesis of AD is supported by several lines of evidence (14). First, iron accumulates in the same brain regions that exhibit β-amyloid (Aβ) accumulation, in-cluding the parietal cortex and hippocampus, and most amyloid plaques are found to contain iron. Second, amyloid precursor protein (APP) mRNA carries an iron-responsive element (IRE) in its 5’-untranslated region (5’-UTR). Third, iron can downregu-late the endoprotease, furin. This can lead to the suppression of α-secretase, and hence altered processing of APP and elevated Aβ production. Fourth, iron accumulation is associated with NFT formation, as iron binding to tau protein is found to precede the aggregation of hyperphosphorylated tau and the subsequent formation of NFTs. The cause of iron accumulation in AD still remains largely unknown. The presence of the C2 allele of trans-ferrin and the C282Y allele of the haemochromatosis gene have been found to be associated with increased risk of developing AD (15). Furthermore, deficiency of functional tau has also been found to impair APP trafficking to the cell surface and hence iron release mediated by APP-MTP1 coupling (16).
pArkinson’s DiseAsePD is the second most common neurodegenerative disorder (17). It manifests as resting tremor, bradykinesia, rigidity, and postural instability, and is caused by the selective loss of the dopaminergic neurons in the substantia nigra pars compacta (SNpc). Abnormal accumulation of α-synuclein in the form of Lewy bodies in SNpc is the major pathological hallmark (18). There are numerous reports showing an increased level of iron in the SNpc of PD patients (19). Studies indicate that accumu-lated iron in the SNpc is a critical factor in the degeneration of these neurons in PD, and that the level of iron accumulation in the SNpc is associated with the severity of PD symptoms (19–22). Recent studies showed that both increased non-transferrin–bound iron (NTBI) uptake via DMT1 and lactoferrin/lactoferrin re-ceptor (Lf/LfR)-mediated processes, as well as transferrin-bound iron (TBI) uptake into mitochondria mediated by Tf/TfR2, may contribute to iron accumulation in PD (20, 23). The exact role of iron in the pathogenic process of PD is not fully understood (9). Apart from free radicals production, iron may also promote
auto-oxidation of dopamine in SNpc neurons and hence induce oxidative stress. On the other hand, there is evidence suggest-ing that iron is related to α-synuclein toxicity and aggregation. For example, α-synuclein aggregation can be facilitated by ex-posure to iron and iron-induced reactive oxygen species (ROS). Additionally, the 5’-UTR of α-synuclein contains an IRE, imply-ing that elevated iron levels could increase posttranscriptional α-synuclein expression, facilitating its aggregation. Furthermore, the toxicity induced by α-synuclein in the presence of iron results in apoptosis, which is thought to be partly responsible for dopa-minergic neuronal death. α-synuclein has also been reported to be a ferrireductase that can generate highly active Fe2+, facili-tating free radical generation. Since mitochondria are known to use and store iron, sustained or enhanced iron import into mito-chondria in the presence of defective complex I assembly has been hypothesized to cause oxidative stress and contribute to PD (23).
TreATmenT sTrATegiesA number of iron chelators have been suggested as possible treatments for iron-related neurodegeneration. Deferoxamine (DFO), a natural iron scavenger isolated from Streptomycespilosus, was among the first-generation iron chelation drugs for treating iron overload. In one clinical trial, DFO significantly slowed AD progression (24), but its poor BBB penetration and short half-life makes it a less than ideal therapy. Deferiprone (DFP), which can pass through BBB, has been shown to be ther-apeutically effective in different animal and cell culture models of neurodegeneration, including PD. Clinical trials are ongoing to assess its treatment efficacy (25). Currently, most available iron chelators lack cellular specificity and can lead to a number of undesirable side-effects. The search for better iron restriction methods is therefore ongoing.
Hepcidin is a newly discovered short peptide found to pos-sess antimicrobial and iron-regulatory functions. It can bind to MTP1 to regulate iron levels, and inflammation and elevated iron levels can induce hepcidin expression (26). We have shown that hepcidin is also expressed throughout the central nervous system and its expression increases with age. The widespread distribution of hepcidin in the brain and its presence in the pe-ripheral organs implies that the peptide may also play a cen-tral role in brain iron homeostasis. We have also demonstrated that hepcidin may have unique functions in the brain, not only inhibiting iron release but also reducing uptake by downregulat-ing DMT1 and TfR1 expression (27,28). Our data also suggest a reduced net iron uptake in response to hepcidin treatment, which may help relieve iron overloading (27). It is not clear if hepcidin levels are altered in neurodegeneration characterized by brain iron accumulation. Further investigation is needed to assess the potential role of hepcidin in modifying iron-induced pathology.
ConClusionsDespite the indispensable role of iron in normal brain function, iron overload is known to cause oxidative damage and neuro-degeneration. However, we currently lack specific and effec-tive means of iron chelation that can slow down or reverse the progression of the resulting neurodegeneration. Understanding the molecular mechanisms of iron accumulation in the brain will hopefully lead to the identification of disease-modifying strate-gies that target the source of iron abnormality and the treatment of iron-related neurodegeneration.
REFERENCES 1. L. Zecca, M. B. Youdim, P. Riederer, J. R. Connor, R. R. Crichton, Nat. Rev. Neurosci. 5, 863 (2004). 2. T. A. Rouault, Nat. Rev. Neurosci. 14, 551 (2013). 3. Y. Ke, Z. M. Qian, Prog. Neurobiol. 83, 149 (2007). 4. P. Dusek, J. Jankovic, W. Le, Neurobiol. Dis. 46, 1 (2012). 5. R. Dringen, G. M. Bishop, M. Koeppe, T. N. Dang, S. R. Robinson, Neurochem. Res. 32, 1884 (2007). 6. Y. Ke, Z. M. Qian, Lancet Neurol. 2, 246 (2003). 7. T. A. Rouault, S. Cooperman, Semin. Pediatr. Neurol. 13, 142 (2006). 8. C. Borie et al., J. Neurol. 249, 801 (2002). 9. J. Sian-Hulsmann, S. Mandel, M. B. Youdim, P. Riederer, J. Neurochem. 118, 939 (2011).10. M. Matsuzaki et al., Brain Res. 1004, 83 (2004).11. A. Yamamoto et al., J. Neurochem. 82, 1137 (2002).12. P. W. Mantyh et al., J. Neurochem. 61, 1171 (1993).13. C. Qiu, M. Kivipelto, E. von Strauss, Dialogues Clin. Neurosci. 11, 111 (2009).14. S. Altamura, M. U. Muckenthaler, J. Alzheimers Dis. 16, 879 (2009).15. A. Lleo et al., J. Neurol. Neurosurg. Psychiatry 72, 820 (2002).16. P. Lei et al., Nat. Med. 18, 291 (2012).17. L. M. de Lau, M. M. Breteler, Lancet Neurol. 5, 525 (2006).18. A. G. Kazantsev, A. M. Kolchinsky, Arch. Neurol. 65, 1577 (2008).19. S. L. Rhodes, B. Ritz, Neurobiol. Dis. 32, 183 (2008).20. J. Salazar et al., Proc. Natl. Acad. Sci. U.S.A. 105, 18578 (2008).21. W. R. Martin, M. Wieler, M. Gee, Neurology 70, 1411 (2008).22. G. Bartzokis et al., Magn. Reson. Imaging 17, 213 (1999).23. P. G. Mastroberardino et al., Neurobiol. Dis. 34, 417 (2009).24. M. P. Cuajungco, K. Y. Faget, X. Huang, R. E. Tanzi, A. I. Bush, Ann. N.Y. Acad. Sci. 920, 292 (2000).25. R. B. Mounsey, P. Teismann, Int. J. Cell. Biol. 2012, 983245 (2012).26. Q. Wang et al., Endocrinology 149, 3920 (2008).27. F. Du et al., J. Nutr. Biochem. 23, 1694 (2012).28. F. Du et al., Glia 59, 936 (2011).
Acknowledgments: This work was supported by the grants from the National Basic Research “973” Program of China (2011CB510004), the National Natural Science Foundation of China (31271132, 31371092, and 31330035), and the Hong Kong RGC (CUHK 466713).

16
Produced by the Science/AAAS Custom Publishing Office
17
NEURODEGENERATION RESEARCH
Alzheimer’s disease (AD) is the most common neurodegenera-tive disorder worldwide, defined by two classical hallmark pa-thologies: extracellular senile plaques and intraneuronal neu-rofibrillary tangles (NFTs) (1, 2). NFTs are composed of the hyperphosphorylated microtubule-associated protein tau that is abnormally phosphorylated primarily by glycogen synthase ki-nase-3 (GSK-3) and cyclin D kinase 5 (Cdk5) (2). Senile plaques are composed of heterogeneous small peptides collectively called β-amyloid (Aβ), derived from the β-amyloid precursor pro-tein (APP) through sequential cleavage by β- and γ-secretases. APP is synthesized in the endoplasmic reticulum (ER) and trans-ported through the Golgi/trans-Golgi network (TGN) to the plasma membrane, where it can be cleaved by α-secretase to produce sAPPα. Non-cleaved APP is re-internalized and is subjected to amyloidogenic processing for Aβ generation (1). Multiple lines of evidence suggest that overproduction/aggregation of Aβ in the brain is the primary cause of AD: Aβ is highly toxic to neurons and can trigger a cascade of pathogenic events leading to cell death. Therefore, detailed delineation of the function, process-ing, and regulated trafficking of APP is crucial for understanding the mechanism underlying AD pathogenesis and for developing AD therapeutic strategies.
App proCessing TowArDs Aß generATionFull-length APP is a type I transmembrane protein transported through the constitutive secretory pathway. During its endocytic trafficking, APP can first be cleaved by β-secretase to release a soluble APP extracellular domain called sAPPβ. The remain-ing membrane-associated APP carboxyl-terminal fragment-β (β CTF) can then be cleaved by γ-secretase to generate Aβ and an APP intracellular domain (AICD).
The type I transmembrane aspartyl protease beta-site APP-cleaving enzyme (BACE1) is the primary β-secretase species. Optimal enzymatic BACE1 activity requires acidic environments such as those found in the TGN and endosomes where BACE1 is present in abundance (3). Mechanisms regulating BACE1 traf-ficking and activity have not been fully elucidated. Sorting nexin (SNX) family members contain a conserved lipid-binding PX do-
main and play important roles in membrane trafficking and pro-tein sorting (4). We recently found that a member of the SNX family, SNX12, interacts with BACE1. Downregulation of SNX12 accelerates BACE1 endocytosis, thus increasing Aβ production, whereas overexpression of SNX12 has the opposite effect. In ad-dition, SNX12 levels are decreased in AD brains, suggesting that changes in SNX12 levels may contribute to AD pathology (5). We also found that the human CUTA protein, another novel protein that interacts with BACE1, regulates its intracellular trafficking. Downregulation of CUTA can decelerate intracellular trafficking of BACE1 from the Golgi/TGN to the cell surface and increase BACE1-mediated APP processing/Aβ generation (6).
In addition to its function as a β-secretase for APP, we recently found that BACE1 may also contribute to memory and cogni-tive deficits associated with AD through an Aβ-independent mechanism: BACE1 interacts with adenylate cyclases via its transmembrane domain, resulting in a reduction in cellular cAMP levels and thus decreased protein kinase A (PKA) activity and CREB phosphorylation (7). Interestingly, during our search for new genes that regulate Aβ generation, we identified a new gene family, designated Rps23rg, whose encoded proteins also inter-act with adenylate cyclases via their transmembrane domains. However, RPS23RG proteins increase cellular cAMP levels to activate PKA, causing increased CREB phosphorylation and GSK-3 phosphorylation. Phosphorylation of GSK-3 inhibits its activity, resulting in reduced Aβ generation and tau phosphoryla-tion (8,9).
γ-cleavage is the last step in APP processing to generate Aβ peptides. In addition to cleaving APP, the high molecular mass, membrane-bound γ-secretase complex cleaves many other sub-strates such as Notch, Cadherin, and ErbB4. γ-secretase con-sists of four essential components: presenilin (PS, PS1, or PS2), nicastrin, anterior pharynx-defective-1 (APH-1), and presenilin enhancer-2 (PEN-2). We and others have shown that deficiency in any one of these may dramatically affect the stability and in-tracellular trafficking of other components and impair γ-secretase activity (1).
PS1 is the catalytic component of the γ-secretase complex. In addition to cleaving γ-secretase substrates, PS1 has been shown to have other functions, some of which are independent of γ-enzymatic activity. For example, we and others show that PS1 can reciprocally regulate the intracellular trafficking of APP (see next page) as well as several other membrane proteins (1,10).
funCTionAl roles for App AnD iTs meTAboliTesSince its identification as the precursor of Aβ, APP has been studied extensively. However, the physiological function of APP remains largely undetermined. APP is proteolyzed into various fragments during intracellular trafficking and these APP metabo-lites mediate various and sometimes opposing functions. The net effect of APP on cellular activity may be determined by the rela-tive amounts of APP itself and its various metabolites.
In cells and brains deficient in APP, we observed an elevation of Cdk5 activity where tau phosphorylation can be inhibited by re-expressing APP or sAPPα. In addition, APP-deficient neurons exhibit reduced metabolism and survival rates and are more sus-ceptible to excitotoxic glutamate-induced apoptosis through a mechanism involving Cdk5 activation. Our results define a novel neuroprotective function for APP, specifically the extracellular APPα domain, in preventing tau hyperphosphorylation through the suppression of Cdk5 overactivation (11).
APP undergoes rapid anterograde transport in neurons. Dur-ing its transport, APP interacts with kinesin-I and functions as a membrane-associated kinesin-I receptor to mediate axonal transport of β-secretase and PS1 (12,13). We find that APP can regulate cell surface delivery of the PS1/γ-secretase; APP de-ficiency accelerates transport of PS1 from the TGN to the cell surface and increases cell surface levels of PS1, which can be reversed by restoring APP levels (14). APP dosage also mark-edly decreases retrograde transport of nerve growth factor (15). Moreover, APP interacts with the choline transporter and affects its endocytosis (16). Together, these findings suggest that APP plays a critical role in regulating protein trafficking.
Using AICD as bait, we identified a mitochondrial carrier protein as an APP-interacting protein and designated it as appoptosin. We found that elevated appoptosin-mediated heme biosynthesis induced the release of reactive oxygen species and activated intrinsic caspase-dependent apoptosis. Importantly, appopotosin levels were upregulated in neurons treated with Aβ and in AD brains, whereas downregulation of appoptosin prevents cell death and caspase activation caused by Aβ insult, thereby implicating a novel appoptosin-dependent mechanism underlying Aβ neurotoxicity. Moreover, we found that although APP interacts with appoptosin through the AICD domain, AICD was unable to rescue appoptosin-induced cell death. These results suggest that membrane-associated domains in the full-length APP and APP CTFs are required to inhibit appoptosin-induced apoptosis. Hence, membrane-anchored APP may interact with and retain a certain amount of appoptosin in the cytosol, thus keeping the level of appoptosin in mitochondria from being elevated for more heme production in the presence of cellular insults or under pathological conditions. Since membrane-dissociated AICD has little effect on appoptosin-induced caspase activation, this could imply that membrane-associated APP/appoptosin complexes can be released and transported to mitochondria upon
γ-cleavage to increase heme synthesis and apoptosis. These results demonstrate a function of APP in mediating trafficking of nascent appoptosin from the cytosol to mitochondria (17).
Cytosolic AICD within the cell may translocate into the nucle-us to regulate the transcription of several genes such as APP, GSK-3β, BACE1, and low density lipoprotein receptor-related protein 1 (LRP1) (18). We also find that AICD can bind to the promoter region of the epidermal growth factor receptor (EGFR) gene and regulate its expression. Consistent with the notion that dysregulation of EGFR expression and activation is involved in cancers, we found that PS1/γ-secretase deficient mice have in-creased EGFR levels and increased tumorigenesis, in particular skin cancer. As AICD is generated concurrently with Aβ, which is elevated in AD, our results imply that there is a negative cor-relation between AD and cancer incidence and that the strategy for inhibiting PS1/γ-secretase activity to treat AD may increase the risk of tumorigenesis. Both implications are supported by recent findings that epidemiological studies have identified an inverse association between cancer and AD (19), while AD clini-cal trials of the γ-secretase inhibitor semagacestat from Eli Lilly have demonstrated that patients receiving the drug have an in-creased risk of skin cancer compared with those who received a placebo.
regulATion of App inTrACellulAr TrAffiCkingSubcellular APP trafficking to divergent sAPPα or Aβ cleavage pathways is critical to neurodegenerative onset, and mecha-nisms underlying APP trafficking are therefore integral to deter-mining neuropathological AD outcome. We found that SNX17 can interact with APP in the early endosome and that downregu-lation of SNX17 leads to reduced APP levels with a concomitant increase in Aβ (20). In addition, we and others have demonstrat-ed that members of the low-density lipoprotein (LDL) receptor family, including LRP1, LRP1B, SorLA/LR11, and apolipoprotein E receptor 2, interact with APP and regulate its endocytic traf-ficking and Ab generation. Moreover, we have shown that one of the γ-secretase components, PS1, also regulates APP traffick-ing where loss of PS1 results in increased budding/generation of APP-containing vesicles from both the ER and TGN, along with a concomitant increase in cell surface localization of APP. More-over, familial AD-linked PS1 variants are significantly impaired in vesicle budding, thereby attenuating cell surface delivery of APP (10). We also found that PS1 interacts with phospholipase D1 (PLD1), a phospholipid-modifying enzyme regulating membrane trafficking events, and recruits PLD1 to the Golgi/TGN, thus po-tentially altering APP trafficking. Indeed, PLD1 overexpression promotes budding of APP vesicles from the TGN, concomitant-ly increasing cell surface levels of APP (21,22).
perspeCTiveOverproduction and aggregation of Aβ in the brain are key patho-
Novel Pathways Regulating Function and Metabolism of ß-Amyloid Precursor Protein in Alzheimer’s Disease
Yun-Wu Zhang, Guojun Bu, and Huaxi Xu*
Fujian Provincial Key Laboratory of Neurodegenerative Dis-ease and Aging Research, Institute of Neuroscience, College of Medicine, Xiamen University, Xiamen, Fujian, China*Corresponding Author: [email protected]

18
Produced by the Science/AAAS Custom Publishing Office
19
NEURODEGENERATION RESEARCH
causes tau hyperphosphorylation at several sites related to impairment of spatial memory in AD (8). Mul-tiple factors can cause tau hyper-phosphorylation through GSK-3β activation, including peroxide nitrite (9), advanced glycation end prod-ucts (10,11), persistent light illumi-nation (12), endoplasmic reticulum stress (13,14), Aβ (8,15), and pro-teasome dysfunction (16,17). While activating GSK-3β inhibits long-term potentiation (LTP), inhibiting it pro-motes LTP through mechanisms involving the presynaptic release of neurotransmitter (18, 19). Inhibiting GSK-3β significantly attenuates tau hyperphosphorylation and improves memory (20,21), while overexpres-sion of neuroprotective neuroglobin decreases tau hyperphosphorylation by inhibiting GSK-3β (22,23).
In the brains of rats, inhibition of phosphatidylinositol 3-kinase (PI3K) alone induces a transient activation of GSK-3β, while simultaneous inhibition of PI3K and protein ki-nase C (PKC) results in a sustained activation of GSK-3β, lead-ing to prolonged tau hyperphosphorylation and spatial memory deficits with reduction of acetylcholine (8,24,25). Phosphoryla-tion of GSK-3β at serine-9 is recognized to be negatively corre-lated with GSK-3β activation, while cleavage of GSK-3β by cal-pain counteracts the inhibitory effect of serine-9 phosphorylation on GSK-3β activity induced by hydrogen peroxide (26).
Phosphorylation of tau by protein kinase A (PKA) primes tau, making it a better substrate for GSK-3β, and at least partially explaining why the GSK-3β-preferred sites on tau can be hyper-phosphorylated even after transient activation by PKA (27,28). Interestingly, we demonstrated that tau hyperphosphorylation by GSK-3β seemed to be required for hippocampal neurogenesis in the dentate gyrus. Further, tau phosphorylation and GSK-3β activation are essential for the adult neurogenesis in the subven-tricular zone (SVZ), another niche of neurogenesis in the adult brains (29, 30), suggesting that the neurogenesis in the brain may be tightly regulated by local microenvironments.
Due to its role in tau phosphorylation, GSK-3β has been con-sidered as a drug target for inhibiting neurodegeneration. How-ever, GSK-3β has other functions in the cell, so complete inhibi-tion would likely be detrimental. Recent studies that attempted spatiotemporal targeting of abnormal GSK-3β activation found that pathologies and memory deficits in an AD mouse model could be effectively attenuated (31). Further studies are needed
Role of Tau Hyperphosphorylation in Alzheimer’s Disease-Associated Neurodegeneration
Jian-Zhi Wang*, Qing Tian, and Di Gao
Tau proteins play an important role in maintaining the stability of the neuronal cytoskeleton system. In Alzheimer’s disease (AD), tau is abnormally hyperphosphorylated and aggregates into paired helical filaments (PHFs) forming neurofibrillary tangles (NFTs) in neurons (1). Clinical investigations have shown that the intracellular accumulation of NFTs is positively correlated with the severity of dementia. The transmission of abnormal tau or NFTs from the entorhinal cortex to the hippocampus and cere-bral cortex matches the clinical manifestation, which is the inter-national gold standard at present for assessing AD progression (Braak grading) (2). Recent studies suggest that the toxicity of b-amyloid protein (Ab, another pathogenic factor in AD) needs
Department of Pathology and Pathophysiology, Key Laboratory of the Ministry of Education of China for Neurological Disorders, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China*Corresponding Author: [email protected]
Figure 1. Schematic summarizing our current understanding of the various signaling pathways through which tau acts within the cell.
to clearly define the conditions, but this work shows promise.
role of pp2A in TAu hyperphosphorylATionTau proteins are dephosphorylated by protein phosphatases such as PP2A; therefore, inactivation of phosphatases results in tau hyperphosphorylation. PP2A is the most effective at de-phosphorylating abnormally hyperphosphorylated tau isolated from the AD brains (32, 33). In vitro, PP2A can dephosphory-late abnormal tau at multiple sites and thus restore its biologi-cal activity (34). Inhibiting PP2A in vivo by injection of okadaic acid or homocystine into the brain, or in vitro by incubating cells with PP2A inhibitors, induces AD-like tau hyperphosphoryla-tion, intracellular accumulation, axoplasmic transport deficits, and learning/memory dysfunction (35, 36). PP2A is activated in the astrocytes of tg2567 mice—a widely used amyloido-genic model of AD—and activation of PP2A stimulates the migration of astrocytes to the amyloid plaques through p38 MAPK inhibition, indicating that PP2A deficits observed in AD brains may cause Aβ accumulation by hindering astrocyte migration (37).
Since PP2A activity is significantly inhibited in AD brains, upreg-ulating PP2A seems a promising therapeutic strategy. However, currently no specific activator of PP2A exists, making the search for an upstream regulator of PP2A a critical mission. We have demonstrated that GSK-3β activation can inhibit PP2A by upreg-ulating protein tyrosine phosphatase 1Β, which phosphorylates
genic events in AD. Studies from our group and others have re-vealed novel pathways by which APP function and processing are regulated. Further studies investigating the function and regula-tion of APP in AD will not only help to elucidate the mechanism underlying disease pathogenesis, but also to identify new targets for disease treatment.
REFERENCES 1. Y. W. Zhang, R. Thompson, H. Zhang, H. Xu, Mol. Brain 4, 3 (2011). 2. V. M. Lee, M. Goedert, J. Q. Trojanowski, Annu. Rev. Neurosci. 24, 1121 (2001). 3. R. Yan, P. Han, H. Miao, P. Greengard, H. Xu, J. Biol. Chem. 276, 36788 (2001). 4. R. D. Teasdale, B. M. Collins, Biochem. J. 441, 39 (2012). 5. Y. Zhao et al., Mol. Neurodegener. 7, 30 (2012). 6. Y. Zhao, Y. Wang, J. Hu, X. Zhang, Y. W. Zhang, J. Biol. Chem. 287, 11141 (2012). 7. Y. Chen et al., J. Neurosci. 32, 11390 (2012). 8. X. Huang et al., Hum. Mol. Genet. 19, 3835 (2010). 9. Y. W. Zhang et al., Neuron 64, 328 (2009).10. D. Cai et al., J. Biol. Chem. 278, 3446 (2003).
11. P. Han et al., J. Neurosci. 25, 11542 (2005).12. A. Kamal, G. B. Stokin, Z. Yang, C. H. Xia, L. S. Goldstein, Neuron 28, 449 (2000).13. A. Kamal, A. Almenar-Queralt, J. F. LeBlanc, E. A. Roberts, L. S. Goldstein, Nature 414, 643 (2001).14. Y. Liu et al., J. Biol. Chem. 284, 12145 (2009).15. A. Salehi et al., Neuron 51, 29 (2006).16. B. Wang, L. Yang, Z. Wang, H. Zheng, Proc. Natl. Acad. Sci. U.S.A. 104, 14140 (2007).17. H. Zhang et al., J. Neurosci. 32, 15565 (2012).18. Q. Liu et al., Neuron 56, 66 (2007).19. J. A. Driver et al., B.M.J. 344, e1442 (2012).20. J. Lee et al., J. Biol. Chem. 283, 11501 (2008).21. D. Cai et al., Proc. Natl. Acad. Sci. U.S.A. 103, 1941 (2006).22. D. Cai et al., Proc. Natl. Acad. Sci. U.S.A. 103, 1936 (2006).
Acknowledgments: This work was supported by grants from National Natural Science Foundation of China (81225008 and 81161120496), Natural Science Foundation of Fujian Province of China (2009J06022 and 2010J01233), the Fundamental Research Funds for the Central Universities of China, and the Fok Ying Tung Education Foundation.
the tau protein as a mediator (3). These data together suggest that abnormal tau plays an important role in the onset and evolu-tion of neurodegeneration and the learning/memory deficits in AD patients.
To date, no tau gene mutation has been found in the AD pa-tients. The main focus of tau research therefore has been on posttranslational modifications, of which hyperphosphorylation is the most extensively studied. The imbalance of protein kinases (generally upregulation) and protein phosphatases (generally downregulation) is the direct cause for abnormal tau hyperphos-phorylation. Among various protein kinases and protein phos-phatases, glycogen synthase kinase-3β (GSK-3β) and protein phosphatase-2A (PP2A) are most heavily involved in AD-like tau hyperphosphorylation (4–6).
role of gsk-3ß in TAu hyperphosphorylATionGSK-3β was the first tau kinase to be identified; it can phosphor-ylate tau at multiple sitesin vitro (7). In vivo activation of GSK-3β

20
Produced by the Science/AAAS Custom Publishing Office
21
NEURODEGENERATION RESEARCH
PP2A at tyrosine-307 (38,39), suggesting that activation of GSK-3β and inhibition of PP2A may create a positive feedback loop that promotes tau hyperphosphorylation. Recent studies suggest that PP2A activity is regulated by an endogenous nuclear pro-tein inhibitor called inhibitor 2 of PP2A (I2PP2A) that is increased in AD brains. By generating a phosphorylation site-specific an-tibody, we showed that I2PP2A is phosphorylated at serine-9, resulting in cytoplasmic accumulation, as seen in AD brains (40 ). We have also shown that zinc induces PP2A inactivation and tau hyperphosphorylation through Src-dependent tyrosine-307 phosphorylation of PP2A (41). In addition, agricultural pesticides can also inhibit PP2A and activate GSK-3β (42). Conversely, ad-ministration of betaine or nicotinamide mononucleotide adenylyl-transferase 2 (the latter being a key enzyme involved in energy metabolism that shows decreased expression in the AD brain), can attenuate tau phosphorylation through activation of PP2A and improve the memory deficits in AD mice (43,44).
TAu glyCosylATion AnD phosphorylATionIn the AD brain, abnormally phosphorylated tau protein is also highly glycosylated through N-linked glycosidic bonds. Deglyco-sylation releases functional tau proteins from NFTs isolated from the AD brains (45). O-glycosylation of tau has been found both in normal and AD brains, and seems to inhibit phosphorylation of tau proteins, as evidenced by the negative correlation seen between tau O-glycosylation and phosphorylation in a rat model of starvation (46).
CellulAr effeCTs of TAu hyperphosphorylATionTo better understand how abnormal tau hyperphosphorylation causes neurodegeneration, we expressed exogenous tau pro-teins in HEK293 (a human embryonic kidney cell line that does not express endogenous tau) or N2a (a mouse neuroblastoma cell line that expresses very low levels of endogenous tau), and treated these cells, plus primary hippocampal neurons, with different pro-apoptotic factors. We measured cell viability and apoptotic markers after treatment as a proxy for neurodegen-eration. We found that tau phosphorylation was significantly increased upon treatment with pro-apoptotic factors and, unex-pectedly, the intracellular accumulation of hyperphosphorylated tau led the neurons to abort a chemically induced apoptosis pro-gram, both in vitro and in vivo, through pathways involving the survival factor β-catenin (47). Further studies demonstrated that overexpression of tau could antagonize apoptosis promoted by Aβ and other pro-apoptotic factors, resulting in reduced p53 lev-els, decreased release of cytochrome c from mitochondria, and inhibition of caspase-9/caspase-3 (48,49), while dephosphoryla-tion of tau induced apoptosis through failed dephosphorylation of Bcl-2 (50). We also showed that elevation of I2PP2A or inhibition of PP2A caused tau hyperphosphorylation with concomitant up-regulation of p53 and Akt, a key serine/threonine protein kinase
(51). Since p53 and Akt serve as pro- and anti-apoptotic factors, respectively, we speculate that the anti-apoptotic effects of Akt may overwhelm the pro-apoptotic role of p53, leading the cells to abort apoptosis and partially clarifying the role of hyperphos-phorylated tau in blocking apoptosis. Taken together, our data strongly suggest that tau hyperphosphorylation plays a dual role in leading neurons to escape apoptosis while at the same time promoting chronic neurodegeneration.
poTenTiAl meAsuremenTs for CliniCAl DiAgnosis of AD The early clinical diagnosis of AD is still a challenge. Since ce-rebrospinal fluid (CSF) surrounds the brain tissue, biochemical changes in the brain can be reflected in CSF. Therefore, quan-titative measurement of the phosphorylated tau protein at spe-cific sites in CSF may help to follow the progression of neuro-degeneration. Since the level of phosphorylated tau in the form of PHFs in CSF is too low to be detected by the conventional enzyme-linked immunosorbent assay (ELISA), we have estab-lished an ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay by linking the conventional ELISA (with high specificity) with a bienzyme substrate cycle (with high sensitivity). Using this method, we found that phosphorylation of tau at the PHF-1 monoclonal antibody epitope was significantly increased, as was the level of phosphorylated neurofilament pro-teins in CSF of AD patients (52,53).
Using quantitative magnetic resonance phase-corrected im-aging, we detected iron deposition in specific brain regions of AD patients. The iron concentration in the parietal cortex was positively correlated with the severity of cognitive impairment in AD patients (54), which may represent a potential biomarker to evaluate the progression of AD.
Blood oxygenation level dependent (BOLD) functional mag-netic resonance imaging (fMRI) was used to analyze the rela-tionship between the altered resting T2 fMRI signal and memory capacity/tau phosphorylation in rats. We observed that tau hy-perphosphorylation and memory deficit induced by activating GSK-3β was positively correlated with a reduced BOLD signal in rats (55), suggesting that the resting BOLD signal may serve as a noninvasive quantitative marker for predicting AD-like spatial memory deficits and tau hyperphosphorylation.
The level of acetylcholine, an important excitatory neurotrans-mitter, is decreased in the early stages of AD. Measurement of acetylcholine in the brain may therefore help with the clinical di-agnosis of AD. However, current methods for brain acetylcholine measurement are invasive and not feasible to use in the clinic. To address this problem, we employed both noninvasive proton magnetic resonance spectroscopy (MRS), to measure choline-containing compounds, and high-performance liquid chroma-tography to measure the extracellular levels of acetylcholine by microdialysis of samples from different brain regions. Analysis of
the data suggested that the choline signal intensity can be used as a noninvasive indicator of the acetylcholine level in rat brains (56), a procedure that could be applied in humans.
In summary (see Fig. 1), we have found that (1) GSK-3β and PP2A can actively and reversely regulate tau phosphorylation; (2) the activity of GSK-3β and PP2A can be modulated by mul-tiple factors; (3) priming phosphorylation of tau by PKA makes tau a better substrate for GSK-3β; (4) O-glycosylation inhibits tau phosphorylation, while N-glycosylation and nitration may pro-mote tau phosphorylation; and (5) hyperphosphorylation of tau enables cells to block apoptosis through competitively reducing the phosphorylation of β-catenin, and also inhibits proteasome activity, promotes tau aggregation, damages axonal transport, and impairs synaptic functions, all of which can trigger chronic neurodegeneration.
REFERENCES 1. I. Grundke-Iqbal et al., Biol. Chem. 261, 6084 (1986). 2. I. Alafuzoff et al., Brain Pathol. 18, 484 (2008). 3. L. M. Ittner, J. Götz, Nature Rev. Neurosci. 12, 65 (2011). 4. J. Z. Wang, F. Liu, Prog. Neurobiol. 85, 148 (2008). 5. Q. Tian, J. Z. Wang, Neurosignals 11, 262 (2002). 6. J. Z. Wang, Y. Y. Xia, I. Grundke-Iqbal, K. Iqbal, J. Alzheimers Dis. 33 Suppl. 1, S123 (2012). 7. J. Z. Wang, I. Grundke-Iqbal, K. Iqbal, Eur. J. Neurosci. 25, 59 (2007). 8. S. J. Liu et al., J. Neurochem. 87, 1333 (2003). 9. Y. J. Zhang, Y. F. Xu, Y. H. Liu, J. Yin, J. Z. Wang, FEBS Lett. 579, 6230 (2005).10. X. H. Li et al., Cell Death Dis. 4, e673 (2013).11. X. H. Li, et al., Neurobiol. Aging 33, 1400 (2012).12. Z. Q. Ling et al., J. Alzheimers Dis. 16, 287 (2009).13. Z. C. Liu et al., J. Alzheimers Dis. 29, 727 (2012).14. Z. Q. Fu et al., J. Alzheimers Dis. 21, 1107 (2010).15. Z. F. Wang et al., J. Neurochem. 98, 1167 (2006).16. Q. G. Ren, X. M. Liao, Z. F. Wang, Z. S. Qu, J. Z. Wang, FEBS Lett. 580, 2503 (2006).17. Y. H. Liu et al., Neurobiol. Aging 30, 1949 (2009).18. L. Q. Zhu et al., J. Neurosci. 27, 12211 (2007).19. L. Q. Zhu et al., J. Neurosci. 30, 3624 (2010).20. Y. S. Xiong et al., CNS Neurol. Disord.-DR 12, 436 (2013).21. J. Yin et al., J. Pineal Res. 41, 124 (2006).22. P. Zhou et al., Rejuv. Res. 14, 669 (2011).23. L. M. Chen et al., J. Neurochem. 120, 157 (2012).
24. G. G. Xu, Y. Q. Deng, S. J. Liu, H. L. Li, J. Z. Wang, Acta Bioch. Bioph. Sin. 37, 349 (2005).25. Y. Wang et al., J. Neurochem. 106, 2364 (2008).26. Y. Feng et al., J. Neurochem. 126, 234 (2013).27. S. J. Liu et al., J. Biol. Chem. 279, 50078 (2004).28. Y. Zhang, H. L. Li, D. L. Wang, S. J. Liu, J. Z. Wang, J. Neural Transm. 113, 1487 (2006).29. X. P. Hong et al., Hippocampus 20, 1339 (2010).30. X. P. Hong et al., Neurochem. Res. 36, 288 (2011).31. C. X. Peng et al., Neurobiol. Aging 34, 1555 (2013).32. J. Z. Wang, C. X. Gong, T. Zaidi, I. Grundke-Iqbal, K. Iqbal, J. Biol. Chem. 270, 4854 (1995).33. J. Z. Wang, I. Grundke-Iqbal, K. Iqbal, Brain Res. Mol. Brain Res. 38, 200 (1996).34. J. Z. Wang, I. Grundke-Iqbal, K. Iqbal, Eur. J. Neurosci. 25, 59 (2007).35. L. Sun et al., Neuroscience 118, 1175 (2003).36. Y. Yang et al., J. Neurochem. 102, 878 (2007).37. X. P. Liu et al., Glia 60, 1279 (2012).38. X. Q. Yao et al., Biochem. J. 437, 335 (2011).39. G. P. Liu et al., Neurobiol. Aging 29, 1348 (2008).40. G. Yu et al., Neurobiol. Aging 34, 1748 (2013).41. Y. Xiong et al., Neurobiol. Aging 34, 745 (2013).42. N. N. Chen et al., J. Alzheimers Dis. 30, 585 (2012).43. G. S. Chai et al., J. Neurochem. 124, 388 (2013).44. X. S. Cheng et al., J. Alzheimers Dis. 36, 185 (2013).45. J. Z. Wang, I. Grundke-Iqbal, K. Iqbal, Nat. Med. 2, 871 (1996).46. X. Li, F. Lu, J. Z. Wang, C. X. Gong, Eur. J. Neurosci. 23, 2078 (2006).47. H. L. Li et al., Proc. Natl. Acad. Sci. U.S.A. 104, 3591 (2007).48. Z. F. Wang et al., J. Alzheimers Dis. 20, 145 (2010).49. H. H. Wang et al., J. Alzheimers Dis. 21, 167 (2010).50. X. A. Liu et al., J. Alzheimers Dis. 19, 953 (2010).51. G. P. Liu et al., Neurobiol. Aging 33, 254 (2012).52. Y. Y. Hu et al., Am. J. Pathol. 160, 1269 (2002).53. Y. Y. Hu et al., Neurosci. Lett. 320, 156 (2002).54. W. Z. Zhu et al., Radiology 253, 497 (2009).55. Z. H. Hu et al., Acta Bioch. Bioph. Sin. 36, 803 (2004).56. X. C. Wang, X. X. Du, Q. Tian, J. Z. Wang, Neurochem. Res. 33, 814 (2008).
Acknowledgments: This work was supported by the Natural Science Foundation of China (81171195 and 91132305) and the Ministry of Science and Technology (2013DFG32670).

22
Produced by the Science/AAAS Custom Publishing Office
23
NEURODEGENERATION RESEARCH
Dopaminergic Modulation of Astrocyte Functions in the Pathogenesis of Parkinson’s Disease
Yingjun Liu and Jiawei Zhou*
Dopamine is one of the most ver-satile neurotransmitters in the central nervous system (CNS). It is involved in multiple brain func-tions including movement coordi-nation, reward, learning, and mem-ory. Dopamine functions through five G protein-coupled dopamine receptors (D1-5) that are highly expressed in many subtypes of neurons throughout the CNS. In-terestingly, accumulating evidence has demonstrated that dopamine receptors are also expressed in glial cells, although expression is much lower than in neuronal cells (1–4). Exactly how dopamine regu-lates glial cell functions in health and disease through glial dopa-mine receptors is still not fully un-derstood. Glial cells are the most abundant components of the CNS, constituting 65% to 90% of the total cells in the mammalian CNS. They are associated with many aspects of normal brain function and are important players in neurode-generative disease (5–8). Here, we summarize our recent data on the dopaminergic modulation of astrocyte functions, especial-ly in the context of Parkinson’s disease (PD).
DopAmine signAling ConTrols AsTroCyTiC fgf-2 biosynThesis AnD seCreTionPD is a movement disorders in which dopaminergic neurons in the substantia nigra pars compacta selectively degenerate. It is believed that gradual dopamine loss in the striatal brain tissues of PD patients occurs with disease progression and is the princi-pal mechanism underlying the cardinal clinical symptoms of PD. Dopamine receptor agonists are among the main therapeutic in-
terventions for symptomatic relief of PD, although there are still no available treatments that stop or reverse the progression of the disease (9). Receptor agonists can mimic dopamine func-tions, directly restoring the dopamine signaling in postsynaptic neuronsin vivo. They may also exert neuroprotective effects by enhancing the expression and secretion of neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) (10), which are known to be produced primarily in neuronal cells. GDNF is predominantly expressed in parvalbumin-positive neostriatal interneurons in normal and injured rodents (11), even though it has been previ-ously thought to be largely produced in glial cells (12). Converse-ly, fibroblast growth factor-2 (FGF-2), an important regulator of cell growth and differentiation for nigral DA neurons, is predomi-nantly synthesized by astrocytes, while other members of the FGF family are primarily synthesized by neurons (13). Whether the production of astrocyte-derived FGF-2 is influenced by dopa-mine receptors, and the biological significance of its regulation in
Institute of Neuroscience, State Key Laboratory of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China *Corresponding Author: [email protected]
Figure 1. Schematic showing signal transduction pathways involved in dopamine D1, D2 receptors, or PI-linked D1-like receptor-modulated biosynthesis and release of FGF-2 in striatal astrocytes. Enhanced release of high molecule weight (HMW) and low molecule weight (LMW) FGF-2 leads to increased survival of DA neurons. DA, do-pamine; D1, dopamine D1 receptor; D2, dopamine D2 receptor; PI-linked D1LR, phosphatidylinositol-linked D1-like receptor; GluR, glutamate receptor; cAMP, cyclic adenosine monophosphate; p-PKC, phosphorylated protein kinase C; IP3, inositol 1,4,5-trisphosphate; p-PKA, phosphorylated protein kinase A; p-MAPK, phosphorylated mitogen-acti-vated protein kinase; Ca, calcium; CaM, calmodulin; MZF-1, myeloid zinc finger-1; FGF-2, fibroblast growth factor-2.
the context of PD, remain elusive. We demonstrated that clas-sical dopamine D1 and D2 receptors potently regulated astro-cytic FGF-2 biosynthesis and secretion (14). FGF-2 synthesis and release were markedly upregulated in primary cultured as-trocytes exposed to apomorphine (APO), a non-selective dopa-mine receptor agonist, in a dose- and time-dependent manner. Conditioned medium from APO-treated astrocytes (APO-CM) dramatically enhanced the survival of dopaminergic (DA) neu-rons in vitro. Interestingly, neutralization of FGF-2 in APO-CM by using FGF-specific antibodies abrogated the survival-promoting effect of the conditioned medium upon DA neurons in culture, indicating that FGF-2 is the major effector component. Further studies showed that activation of the D1 receptor preferentially increased protein kinase A (PKA) activity, whereas activation of the D2 receptor only promoted phosphorylation of mitogen-activated protein kinase (MAPK). The D1 receptor-associated pathway had an inhibitory effect on the D2 receptor pathway via PKA and MAPK, but not on cyclic adenosine monophosphate (cAMP), suggesting a delicate system of regulation for control of FGF-2 expression in astrocytes (Fig. 1). Importantly, APO-mod-ulated FGF-2 expression was independent of Akt/phosphoinosit-ide 3-kinase signaling (14), which is known to mediate classical dopaminergic signaling in neurons. Furthermore, APO-induced enhancement of the biosynthesis and release of FGF-2 was me-diated by the activation of transcription factor myeloid zinc finger protein 1 (MZF-1) in striatal astrocytes (15).
Besides dopamine receptors D1 and D2, the phosphatidylino-sitol (PI)-linked D1-like receptor is also involved in modulation of FGF-2 expression in astrocytes (16). SKF83959, a selective PI-linked D1-like receptor agonist, induced a remarkable increase in the levels of FGF-2 in cultured striatal astrocytes in a classi-cal dopamine D1 and D2 receptor-independent manner. In vivo administration of SKF83959 reversed neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropypridine (MPTP)-induced reduction in FGF-2 expression in the nigrostriatum and resulted in marked protection of dopaminergic neurons from MPTP-induced neu-rotoxicity. Interestingly, inositol 1,4,5-trisphosphate (IP3)-depen-dent calcium oscillations were essential for SKF83959-mediated FGF-2 induction in astrocytes. Collectively, our data suggest that dopamine signaling plays crucial roles in the regulation of biosynthesis and secretion of growth factor FGF-2 in astrocytes via both well-characterized and uncharted intracellular signaling pathways (Fig. 1). However, we should bear in mind that most of these pharmacological studies were performed using dopamine receptor agonists or antagonists. Although these compounds have been widely used in the field, the possibility that they may have multiple targets and complex biological consequences can-not be entirely excluded. For example, recent studies showed that SKF83959 and some of its analogs are also potent allosteric modulators of the sigma-1 receptor, which modulates calcium signaling through IP3 receptors (17,18). Future investigation is
required to determine the underlying mechanisms of SKF83959-induced FGF-2 expression.
AsTroCyTiC DopAmine D2 reCepTor ACTivATion normAlly suppresses neuroinflAmmATion in The CnsIt has been widely recognized that neuroinflammation primar-ily mediated by microglia and astrocytes is a common feature of neurodegenerative diseases including PD and may play im-portant roles in the initiation and progression of neurodegenera-tive diseases. It has thus been proposed that anti-inflammation treatment is a promising therapeutic approach in patients with neurodegenerative disease. However, the molecular properties and regulation of neuroinflammation in the CNS is poorly under-stood, hampering attempts at anti-inflammatory treatments. Pre-vious results have demonstrated that dopamine receptors were expressed in immune-relevant cells, such as macrophages, and modulated their immune responses in the periphery tissue and organs, suggesting the possible involvement of dopamine in im-mune regulation (19,20). Our own recent studies have shown that dopamine D2 receptor (Drd2) negatively modulated the ex-pression of inflammatory cytokines in astrocytes, but not microg-lia (21). Drd2-deficient astrocytes showed hyper-responsiveness
Figure 2. Astrocytic Drd2 functions to suppress inflammatory responses specifically in astrocytes. Model for communications among astrocytes, microg-lial, and dopaminergic neurons.

24
Produced by the Science/AAAS Custom Publishing Office
25
NEURODEGENERATION RESEARCH
both in primary astrocytic cultures and specific astrocytic Drd2 conditional knockout (cKO) mice in response to immune stimuli. Moreover, global Drd2 KO mice and astrocytic Drd2 cKO mice were more vulnerable to MPTP-induced neurotoxicity, suggest-ing a potentially clinically relevant role for Drd2. Furthermore, we identified alpha-B crystalline (CRYAB), a small heat shock protein with anti-inflammatory and neuroprotective activities, that was responsible for astrocytic Drd2-mediated suppression of neuroinflammation. Deletion of Drd2 resulted in a drastic de-crease in the levels of CRYAB expression in cultured astrocytes and brain tissues, while over-expression of CRYAB specifically targeting astrocytes dramatically inhibited the upregulation of inflammatory cytokines in Drd2-null mice. These results indi-cate that CRYAB levels are tightly controlled by Drd2 and are responsible for dopamine-mediated immune inhibition (Fig. 2). However, how dopamine regulates CRYAB expression and how CRYAB controls immune responses of astrocytes are still open questions.
ConCluDing remArks Traditionally, as a neurotransmitter, dopamine is deemed to be a critical mediator of neuron-neuron communication through syn-apses. However, new evidence elucidating the functional roles of glial dopamine receptors under both physiological and pathologi-cal conditions is challenging the old dogma. Based on observa-tions from our group and others, it is very likely that dopamine is also involved in neuron-glia interactions. Our studies clearly demonstrate that dopamine signaling is critical for the regulation of astrocyte function under both normal and pathologic condi-tions. These observations also highlight the important roles of astrocytes in neurodegenerative diseases. Further investigation is required to unravel the complex and intricate biological func-tions of dopamine in CNS cell-cell communication.
REFERENCES 1. Z. U. Khan, P. Koulen, M. Rubinstein, D. K. Grandy, P. S. Goldman-
Rakic, Proc. Natl. Acad. Sci. U.S.A. 98, 1964 (2001). 2. K. M. Lucin, T. Wyss-Coray, Neuron 64, 110 (2009). 3. I. Miyazaki, M. Asanuma, F. J. Diaz-Corrales, K. Miyoshi, N. Ogawa, Brain Res. 1029, 120 (2004). 4. M. Mladinov et al., Translat. Neurosci. 1, 238 (2010). 5. N. J. Maragakis, J. D. Rothstein, Nat. Clin. Pract. Neurol. 2, 679 (2006). 6. B. Liu, J. S. Hong, J. Pharmacol. Exp. Ther. 304, 1 (2003). 7. W. J. Streit, Glia 40, 133 (2002). 8. C. S. Lobsiger, D. W. Cleveland, Nat. Neurosci. 10, 1355 (2007). 9. O. Rascol, C. Goetz, W. Koller, W. Poewe, C. Sampaio, Lancet 359, 1589 (2002).10. H. Guo et al., Eur. J. Neurosci. 16, 1861 (2002).11. M. Hidalgo-Figueroa, S. Bonilla, F. Gutierrez, A. Pascual, J. Lopez- Barneo, J. Neurosci. 32, 864 (2012).12. M. Ohta et al., Biochem. Biophys. Res. Commun. 272, 18 (2000).13. B. Reuss, O. von Bohlen und Halbach, Cell Tissue Res. 313, 139 (2003).14. A. Li et al., FASEB J. 20, 1263 (2006).15. X. Luo, X. Zhang, W. Shao, Y. Yin, J. Zhou, J. Neurochem. 108, 952 (2009).16. X. Zhang et al., J. Neurosci. 29, 7766 (2009).17. L. Guo et al., Mol. Pharmacol. 83, 577 (2013).18. T. Hayashi, T. P. Su, Cell 131, 596 (2007).19. P. J. Gaskill, L. Carvallo, E. A. Eugenin, J. W. Berman, J. Neuroinflamm. 9, 203 (2012).20. G. Hasko, C. Szabo, Z. H. Nemeth, E. A. Deitch, J. Neuroimmunol. 122, 34 (2002).21. W. Shao et al., Nature 494, 90 (2013).
Acknowledgments: This work was supported by grants from the Chinese Academy of Sciences, National Basic Research Program of China (2011CBA00408 and 2011CB504102), the Natural Science Foundation of China (31021063 and 31123002), and the Shanghai Metropolitan Fund for Research and Development.
State Key Laboratory of Medical Genetics, Xiangya Medical School, Central South University, Changsha, Hunan, China*Corresponding Author: [email protected]
genes associated with PD, the products of parkin, PINK1, DJ-1, uchL1, and FBXO7 are involved in the ubiquitin pathway (6–11), at least three of which have E3 ligase activity: uchl1, FBXO7 as part of the SKP1/cullin/F-box (SCF) complex, and the parkin/PINK1/DJ-1 (PPD) complex. UchL1 functions as both a ubiquitin E3 ligase and ubiquitin hydrolase (10). FBXO7 is known to regu-late cyclin D via its E3 ligase activity (12). The PPD E3 ligase complex uses parkin as a catalytic unit and PINK1 and DJ-1 as the regulatory units (Fig. 1) (8). Nevertheless, how impairment of these E3 ligases affects PD pathogenesis remains unknown.
The substrates, particularly those related to PD pathogen-esis, have yet to be defined for these previously characterized PD-related E3 ligases. However, it has been shown that UchL1
Figure 1. Schematic illustration of PPD E3 ligase complex. In the complex, PINK1 and DJ-1 function as regulatory units (Reg) while parkin serves as a catalytic domain (Cat). In the presence of ubiquitin conjugat-ing enzyme (E2) and ubiquitin (Ub), PPD promotes poly-ubiquitination of substrate (S).
Parkinson’s disease (PD) is the most common neurodegenera-tive movement disorder and is currently incurable. The pathologi-cal hallmarks of the disease include loss of dopaminergic (DA) neurons in the substantia nigra pars compacta and the formation of intracellular Lewy bodies in surviving neurons (1). Lewy bod-ies are formed by the accumulation of a number of unfolded/misfolded proteins, including synaptic protein α-synuclein. The role of α-synuclein in selective neurodegeneration in PD remains unknown. Human studies have demonstrated the critical role of genetic background in PD pathogenesis, identifying mutations in at least 14 genes associated with the disease (2–4). Neu-rons, as postmitotic cells, rely heavily on the cellular quality control system to remove newly synthesized or stress-induced proteins that are misfolded. The ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP) are two major proteolytic quality control pathways. UPS degrades mostly short-lived, soluble proteins while ALP removes damaged intracellular components, such as proteins and organelles, through lysosomes. Recent studies strongly indicate that impairment of both UPS and ALP contributes to PD pathogenesis.
ubiquiTin-proTeAsome sysTemUPS plays critical role in cellular quality con-trol by degrading soluble intracellular proteins. The first step in the UPS degradation process is ATP-dependent covalent linkage of a chain of activated ubiquitin to lysine residues on the tar-get substrates. This process is accomplished by the sequential action of ubiquitin-activating enzymes (E1), ubiq-uitin-conjugating enzymes (E2), and ubiquitin ligases (E3). The ubiquitin chain serves as a signal to target the substrate to the proteasome for degradation. In this process, E3 ubiquitin ligases are critical for the specificity of substrates to be degraded. Many neurodegenerative diseases, including PD, are the consequence of dysfunctional folding and trafficking of proteins (5). Compo-nents of the ubiquitin proteasomal pathway as well as chap-erones have been detected in Lewy bodies. Of the 14 known
Proteolytic Pathways in Parkinson’s Disease
Chengyuan Tang, Tongmei Zhang, and Zhuohua Zhang*
Figure 2. Neddylation stabilizes 55 kDa PINK1 cleavage product in cells. (A) Representative im-munoblot for FLAG-tagged PINK1 and NEDD8 upon cycloheximide treatment. Duration of treatment is shown above gel. (B) Quantitation of PINK1 and its cleavage product after cycloheximide treatment. Results are average from two independent experiments. Values represent mean ± standard error. 55kDa PINK1, the 55kDa PINK1 fragment; Control, empty vector. *, P=0.0314 at 6hr treatment time point between 55kDa PINK1+NEDD8 and 55kDa PINK1+Control using unpaired, two-tailed t test. Adapted from (14).

26
Produced by the Science/AAAS Custom Publishing Office
27
NEURODEGENERATION RESEARCH
promotes K63 ubiquitination of α-synuclein in vitro and that two PD-associated mutations have been shown to reduce E3 ligase activity (10). FBXO7 interacts with a number of cell cycle regulatory proteins, but does not promote UPS-mediated deg-radation of these proteins. A recent study suggests that FBXO7 regulates ubiquitination of mitofusin-1, and thereby mitophagy, via direct interaction with parkin and PINK1 (13). Likewise, the substrates of the PPD E3 ligase complex include those of pre-viously identified parkin substrates. Overexpression of parkin substrates or heat shock treatment resulted in parkin substrate accumulation in PINK1-deficient cells (8). One explanation for this observation is that the PPD complex functions as a qual-ity control system to degrade misfolded parkin substrates in-duced by oxidative stress in neurons. Pathogenic mutations that reduce PPD activity likely disrupt UPS, leading to increased susceptibility to oxidative stress and accumulation of misfold-ed proteins in cells, and eventual Lewy body formation and neuronal death.
The regulation of the PD-associated E3 ligases is not yet well understood. FBXO7 is reported to regulate parkin E3 ligase ac-tivity (13). Nedd8 modification of both parkin and PINK1 is likely another way to modulate PPD E3 ligase activity. In this case, PINK1 neddylation results in increased stability of its 55KDa pro-teolytic fragment, which forms a complex with parkin in the cyto-sol (Fig. 2). Furthermore, PD associated neurotoxin 1-methyl-4-phenylpyridinium (MPP+) suppresses the PPD E3 ligase activity via, at least partially, inhibiting neddylation of parkin and PINK1 (14). These results suggest a PD pathogenic mechanism of ge-netic and environmental interaction.
AuTophAgy-lysosomAl pAThwAyAutophagy maintains cellular homeostasis and integrity by recy-cling metabolic precursors and clearing subcellular debris. In re-sponse to environmental cues, the autophagic machinery—prod-ucts of the autophagy-related genes (Atg)—initiates a series of membrane inclusion processes that packages the unnecessary or dysfunctional cellular components and delivers them to the lysosome for degradation (15). PD-associated genes have only recently been shown to be involved in regulating autophagy, de-spite the fact that autophagy has been observed in DA neurons of the substantia nigra of PD patients for many years.
Parkin and PINK1 have been shown to mediate mitophagic degradation of depolarized mitochondria (16, 17). Upon de-polarization, PINK1 recruits parkin and promotes parkin’s E3 ubiquitin ligase activity resulting in the ubiquitination of outer membrane proteins and ultimately leading to mitochondrial au-tophagy. FBXO7 regulates parkin translocation to mitochondria and parkin-mediated ubiquitination of mitofusin-1, resulting in mitophagy (13). A protein encoded by the PD-related leucine-rich repeat kinase 2 (LRRK2) gene likely plays a role in the late stages of autophagy by altering overall cellular autophagy flux
through modulation of lysosome acidification (18). These studies linked abnormalities in autophagy to PD pathogenesis, but did not show a causal relationship. However, two independent stud-ies recently demonstrated that inactivation of autophagy-related protein 7 in mouse brain resulted in loss of dopaminergic neu-rons, formation of ubiquitinated protein aggregates, and accumu-lation of α-synuclein and LRRK2 proteins (19,20), strongly sug-gesting an important role for autophagy in PD pathogenesis. It remains to be seen whether mitophagy impairment results in the accumulation of damaged mitochondria in DA neurons in these mouse models. It will also be important to demonstrate in vivo that PD-gene–regulated autophagy is involved in the pathologi-cal changes seen in the disease.
Evidence has accumulated from in vitro studies indicating that both UPS and ALP protein degradation mechanisms play critical roles in PD pathogenesis. In vivo evidence shows that UPS and ALP components are associated with PD pathology, providing additional strong support. The two pathways likely functionally interact and are under tight regulatory control in neurons. The actual mode of action of UPS and ALP in PD pathogenesis in vivo remains to be elucidated.
REFERENCES 1. D. W. Dickson, Cold Spring Harb. Perspect. Med. 2, 1 (2012). 2. C. E. Krebs et al., Hum. Mutat. 34, 1200 (2013). 3. M. Quadri et al., Hum. Mutat. 34, 1208 (2013). 4. A. B. Singleton, M. J. Farrer, V. Bonifati, Mov. Disord. 28, 14 (2013). 5. M. Jucker, L. C. Walker, Ann. Neurol. 70, 532 (2011). 6. Y. Imai, M. Soda, R. Takahashi, J. Biol. Chem. 275, 35661 (2000). 7. H. Shimura et al., Nat. Genet. 25, 302 (2000). 8. H. Xiong et al., J. Clin. Invest. 119, 650 (2009). 9. Y. Zhang et al., Proc. Natl. Acad. Sci. U.S.A. 97, 13354 (2000).10. Y. Liu, L. Fallon, H. A. Lashuel, Z. Liu, P. T. Lansbury, Jr., Cell 111, 209 (2002).11. S. Shojaee et al., Am. J. Hum. Genet. 82, 1375 (2008).12. H. Laman et al., EMBO J. 24, 3104 (2005).13. V. S. Burchell et al., Nat. Neurosci. 16, 1257 (2013).14. Y. S. Choo et al., Hum. Mol. Genet. 21, 2514 (2012).15. M. A. Lynch-Day, K. Mao, K. Wang, M. Zhao, D. J. Klionsky, Cold Spring Harb. Perspect. Med. 2, a009357 (2012).16. D. Narendra, A. Tanaka, D. F. Suen, R. J. Youle, J. Cell Biol. 183, 795 (2008).17. D. P. Narendra et al., PLOS Biol. 8, e1000298 (2010).18. P. Gomez-Suaga et al., Hum. Mol. Genet. 21, 511 (2012).19. I. Ahmed et al., J. Neurosci. 32, 16503 (2012).20. L. G. Friedman et al., J. Neurosci. 32, 7585 (2012).
Acknowledgments: The study was supported by grants from the National Basic Research Program of China (2011CB510002), the Natural Science Foundation of China (81161120498 and 31330031), and the Program of Introducing Talents of Discipline to Universities in China (B13036).
Animal Models of Huntington’s Disease
Xiao-Jiang Li1,2* and Shihua Li1
1Department of Human Genetics, Emory University School of Medicine, Atlanta, GA2Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China *Corresponding Author: [email protected]
A variety of transgenic animal models have been established for uncovering the pathogenesis and therapeutic targets of Hunting-ton’s disease (HD). However, most genetic mouse models lack the striking neurodegeneration found in the brains of HD victims. Large animal models of HD may demonstrate pathological fea-tures that are more similar to those in patients and thereby help uncover more effective therapeutic targets. This review focuses on the differential neuropathology seen in mouse and large ani-mal models of HD and discusses the use of large animal models to investigate the pathogenesis of the disease.
inTroDuCTionAlzheimer’s disease (AD), Parkinson’s disease (PD), Hunting-ton’s disease (HD), and several other neurodegenerative dis-eases share the common pathological feature of age-dependent, selective neurodegeneration. These diseases are all caused by misfolded proteins that form aggregates in the brains of affected patients. Consistently, the age-dependent formation of these ag-gregates in patient brains is correlated with the progression of the neurological symptoms of AD, PD, and HD.
HD shares common pathological changes with other neurode-generative diseases, but is caused by an autosomal dominant genetic mutation, making it an ideal model to study how protein misfolding leads to selective neurodegeneration. The mechanis-tic insight obtained from studying HD should be very helpful for understanding other neurodegenerative diseases resulting from protein misfolding.
hD AnD polygluTAmine expAnsionHD is characterized by motor dysfunction, cognitive decline, and psychological dysfunction. HD displays selective neurodegener-ation that occurs preferentially in the brain striatum. The genetic cause of HD is the expansion of a CAG repeat (>36 CAGs) in exon 1 of the HD gene (HTT). The CAG repeat expansion results in an expanded polyglutamine (polyQ) tract in the N-terminal re-gion of the large (3,144 amino acid) huntingtin protein (Htt) (1,2).
While the primary function of Htt has yet to be determined, it is
known to be essential for early development and probably plays a role in cellular trafficking as a scaffold protein (1,2). It is evident that only N-terminal mutant fragments of Htt are able to form aggregates and are in fact more toxic than full-length mutant Htt (mHtt) (3,4). This has prompted a search for proteolytic cleavage sites mediated by various proteases, including calpains, aspartyl proteases, and caspases capable of cleaving mHtt to generate N-terminal mHtt fragments (5). These fragments can accumulate in the nucleus, whereas the majority of full-length mHtt remains in the cytoplasm (3, 6). The nuclear localization of N-terminal mHtt can lead to abnormal binding of the fragments to various transcription factors, affecting gene expression (1,2).
mouse moDels of hDVarious HD mouse models have been established, including transgenic mice (R6/2, N171-82Q) expressing N-terminal mHtt (7,8), full-length mHtt transgenic mice (using YACs and BACs) (9,10), and HD repeat knock-in (KI) mice (11–13). Characteriza-tion of various HD mouse models provides clear evidence that small N-terminal Htt fragments with expanded polyQ tracts be-come misfolded and form aggregates. Despite the milder pheno-types of HD mice that express full-length mHtt, such as YAC128 and HD KI mice, these mice show accumulation of mHtt in the striatum, consistent with the preferential loss of the medium spiny neurons in the striatum of HD patients. Thus, the accumulation of mHtt in neuronal cells is clearly a prerequisite for neuronal dysfunction and degeneration.
Since mHtt is ubiquitously expressed in various types of cells including neurons and male germ cells, and localized in different subcellular regions including the nucleus and synapses, it seems important to investigate how mHtt localization might contribute to disease progression. Expression of N-terminal mHtt in astro-cytes is sufficient to cause age-dependent neurological symp-toms (14). Recently, transgenic mice that selectively expressed N-terminal mHtt in presynaptic terminals were established and also showed severe neurological phenotypes, providing convinc-ing evidence for the critical role of synaptic mHtt in the disease (15).
Although HD mouse models have been used widely to un-cover the pathogenesis of HD and to develop treatments, most of these mouse models do not show overt neurodegeneration. Similarly, in other polyQ expansion mouse models, the lack of striking neurodegeneration is a noteworthy phenomenon. These facts highlight the fact that, although mouse models are used

28
Produced by the Science/AAAS Custom Publishing Office
29
NEURODEGENERATION RESEARCH
11. V. C. Wheeler et al., Hum. Mol. Genet. 9, 503 (2000).12. C. H. Lin et al., Hum. Mol. Genet. 10, 137 (2001).13. L. B. Menalled et al., J. Neurosci. 22, 8266 (2002).14. J. Bradford et al., Proc. Natl. Acad. Sci. U.S.A. 106, 22480 (2009).15. Q. Q. Xu et al., J. Cell Biol. 202, 1123 (2003).16. A. W. Chan et al., Science 291, 309 (2001).17. S. H. Yang et al., Nature 453, 921 (2008).18. E. Sasaki et al., Nature 459, 523 (2009).19. Y. Niu et al., Proc. Natl. Acad. Sci. U.S.A. 107, 17663 (2010).20. P. H. Cheng et al., Am. J. Hum. Genet. 93, 306 (2013).21. D. Yang et al., Hum. Mol. Genet. 19, 3983 (2010).22. J. C. Jacobsen et al., Hum. Mol. Genet. 19, 1873 (2010).23. L. Lai, R. S. Prather, Cloning Stem Cells 5, 233 (2003).24. B. Shen et al., Cell Res. 23, 720 (2013).25. H. Wang et al., Cell 153, 910 (2013).
Acknowledgments: This work was supported by the U.S. National Institutes of Health grants NS036232, AG019206, and NS041669 for X.J.L. and AG031153 for S.H.L.
widely to investigate the pathogenesis of HD and other neuro-logical diseases, they have their limitations and do not replicate the full range of neurological phenotypes seen in related human diseases. Such limitations reflect the importance of species dif-ferences in the etiology of neurodegeneration.
neuropAThology of lArge TrAnsgeniC AnimAl moDels of hD The insertion of a foreign gene into the monkey genome in 2001 (16) demonstrated that nonhuman primate models expressing transgenes was possible, including a rhesus monkey express-ing exon 1 mHtt with an 84 unit polyQ tract under the control of the human ubiquitin promoter (17–19). Unlike transgenic mice, which can survive after birth when expressing the same exon 1 mHtt with an even longer polyQ repeat (150Q) (7, 20), HD transgenic monkeys with 84Q died postnatally and this early death was directly related to the mHtt transgene (17). Despite their early death, some transgenic monkeys developed key clini-cal HD features including dystonia, chorea, and seizure (17). Like the brains of HD mouse models and patients, the HD mon-key brains also show abundant Htt aggregates in neuronal nu-clei and processes. More importantly, transgenic HD monkeys display the degeneration of axons and neuronal processes in the absence of obvious cell body degeneration, suggesting that neuronal degeneration in HD may initiate at the neuronal processes.
In collaboration with Dr. Liangxue Lai at the Chinese Acad-emy of Sciences’ Guangzhou Institutes of Biomedicine and Health, we created transgenic HD pigs that expressed N-ter-minal mHtt consisting of the first 208 amino acids and includ-ing 105Q (N208-105Q) (21). Like transgenic monkey models of HD, most of these transgenic piglets died postnatally, with some showing a severe chorea phenotype before death, suggesting that mHtt is more toxic to larger animals than to mice. More im-portantly, in all transgenic pig brains examined, apoptotic cells were seen (21), something that had not been reported in any HD mouse models.
In contrast to the above results, transgenic HD sheep express-ing full-length mHtt with a 73Q tract live normally and show only a decrease in the expression of the medium spiny neuron marker DARPP-32 (22). Thus, as with HD mouse models, the expression of N-terminal mHtt can result in robust neurological phenotypes and pathological changes in large animals. These studies also suggest that the protein context and the length of Htt fragments may determine the nature of the neuropathology. It is possible that neurodegeneration in large animals only oc-curs when a sufficient level of degraded N-terminal fragments have accumulated in old animals. Thus, expressing N-terminal mHtt fragments alone can speed the progression of the dis-ease, resulting in the early postnatal death in transgenic HD pigs and monkeys.
insighTs from lArge TrAnsgeniC AnimAl moDels of hDIt is clear that species differences play a critical role in the neuro-logical phenotype of HD in small and large animal models. There are considerable differences in development, life span, physiol-ogy, genetics, and anatomy between small and large animals. Of greater interest is elucidating the mechanisms behind these differences. The short lifespan of mice is often believed to be responsible for the failure of HD mouse models to develop overt neurodegeneration. It is also possible that the misfolded form of N-terminal mHtt is more toxic to the neuronal cells of pigs and monkeys than to rodent neurons. Also, because the brain cir-cuitry in pigs and monkeys is more complex than in mice, this may render neurons in large animals more vulnerable to mis-folded mHtt. Finally, the cellular ability to cope with misfolded proteins during development and adulthood may differ between species and the rapid maturation of rodent neurons during ear-ly brain development may reduce their sensitivity to misfolded proteins, potentially explaining why mouse models survive to adulthood even when they express the same mHtt N-terminal fragment.
fuTure sTuDies wiTh lArge AnimAl moDelsThe generation of genetic models using large animals is sig-nificantly more challenging than with rodents. Most work done on transgenic monkeys involved the use of lentiviral vector in-fection of fertilized oocytes followed by embryo transplantation (17–19), requiring a considerable number of donor and surro-gate monkeys. Successful generation of transgenic pigs can be achieved via nuclear transfer, a cloning strategy that has a low success rate (<2%) for transferred pig embryos developing to term (23). In addition, the costs of maintaining and breeding large animals, as well as the ethical concerns and strict regula-tions for use, also make biomedical research with these animals more difficult.
Recent advances in gene targeting have opened new avenues for generating large animal models. New technology using bac-terially derived transcription activator-like effector nucleases (TALENs) and clusters of regularly interspaced short palindromic repeats (CRISPR)/cas9 enable very specific gene targeting, even eliminating the need for using embryonic stem cells (24,25). This approach can be applied to one-cell fertilized embryos to generate null mutations in specific genes.
Although rodent models of neurodegenerative diseases will remain an important modeling system for investigating a vari-ety of diseases, in large part because of the difficulty and ex-pense of generating and characterizing large animals, large animal models provide a more rigorous system for validating the relevance of critical findings from small animal models. Transgenic models using higher mammalian species to model important neurodegenerative diseases will give us deeper in-
sight into the pathogenesis of neurodegenerative diseases. In addition, given the frequent failures when it comes to clini-cal trials of drugs based on successful testing in small ani-mal models, large transgenic animals could yield a more reli-able system for verifying therapeutic efficacy before moving to clinical trials.
REFERENCES 1. P. Harjes, E. E. Wanker, Trends. Biochem. Sci. 28, 425 (2003). 2. S. H. Li, X. J. Li. Trends Genet. 20, 146 (2004). 3. C. A. Gutekunst et al., J. Neurosci. 19, 2522 (1999). 4. H. Zhou et al., J. Cell. Biol. 163, 109 (2003). 5. Z. H. Qin, Z. L. Gu, Acta Pharmacol. Sin. 25, 1243 (2004). 6. M. DiFiglia et al., Science 277, 1990 (1997). 7. S. W. Davies et al., Cell 90, 537 (1997). 8. G. Schilling et al., Hum. Mol. Genet. 8, 397 (1999). 9. E. J. Slow et al., Hum. Mol. Genet. 12, 1555 (2003).10. M. Gray et al., J. Neurosci. 28, 6182 (2008).
FMRP Regulates Microtubule Network Formation, Neurogenesis, and DNA Damage Response
Aiyu Yao and Yong Q. Zhang*
Fragile X syndrome (FXS) is the most common form of inher-ited mental retardation worldwide (1) and is caused by the loss of function of the Fragile X mental retardation protein (FMRP; encoded by the FMR1 gene), a multi-domain RNA-binding pro-tein believed to regulate synaptic development and plasticity by controlling the localization and translation of specific mRNA tar-gets. Though the gene mutated in FXS was identified over two decades ago, exciting new discoveries on the functions of FMRP continue to emerge from in vivostudies. In this review, important advances in our current understanding of FMRP’s role in micro-tubule regulation, neurogenesis, and DNA damage response are discussed. The reader is referred to two excellent reviews for general information on the neuronal functions of FMRP (1) and the pathophysiology of FXS (2).
fmrp regulATes miCroTubule formATionThe first line of evidence implicating FMRP in microtubule regula-tion came from the discovery that FMRP binds and suppresses
the translation of mRNA for the microtubule-stabilizing microtu-bule-associated protein 1B (MAP1B) (3,4) and its homolog Fut-sch in Drosophila (5). In the neonatal mouse brain, FMRP sup-presses the expression of MAP1B during synaptogenesis, and the elevated expression of MAP1B in FMR1 knockout mice leads to increased microtubule stability (6). Together, these data es-tablish a key role for FMRP in microtubule regulation. However, direct in vivo evidence linking the loss of FMRP to microtubule abnormalities has been scarce.Spastin was identified through a genetic screen in Drosophila
as a dominant suppressor of the rough eye phenotype caused by dfmr1 (the Drosophilahomolog of FMR1) over-expression (7). Spastin encodes a microtubule-severing protein, and a mutation in this gene can lead to neurodegenerative hereditary spastic paraplegia. Muscle cells mutant for dfmr1 have more perinuclear but fewer distal microtubules, and the distal microtubules have short, bent, and tangled fibers. Conversely, dfmr1 overexpres-sion produces thick, parallel microtubule bundles, along with in-

30
Produced by the Science/AAAS Custom Publishing Office
31
NEURODEGENERATION RESEARCH
Key Laboratory of Molecular and Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China*Corresponding Author: [email protected]
creased levels of α-tubulin protein and acetylated microtubules (7). Genetic analysis revealed that dfmr1 acts upstream of or in parallel with spas-tin in multiple processes, including neuromuscu-lar junction growth and microtubule formation. The mechanism by which dFMRP regulates the microtubule network remains unclear. However, based on the observed genetic interaction between dfmr1 and spastin (7), the two proteins may regu-late microtubules through a similar mechanism (Fig. 1).
A recent study confirmed the regulation of micro-tubules by dFMRP at the neuromuscular junction, and further demonstrated that the FMRP-MAP1B/Futsch microtubule pathway is regulated by bone morphogenetic protein signaling (8).
fmrp regulATes neurogenesisA significant recent discovery is the involvement of FMRP in adult neurogenesis. In FMRP-deficient mice, both proliferation and fate specification of adult neural progenitor cells (aNPCs) were altered in the subgranular zone of the dentate gyrus of the hippocampus through dysregulation of the Wnt/β-catenin signal-ing pathway. A reduction in Wnt signaling activity changed the expression of neurogenin 1, and consequently, aNPC fate speci-fication (9). It was later shown that targeted ablation of FMR1 in aNPCs via inducible gene recombination decreased hippo-campal neurogenesis, and significantly impaired hippocampus-dependent learning in mice, while restoring FMR1 expression rescued the learning deficits (10). Based on these results, the authors proposed that defective adult neurogenesis contributes to the learning impairment exhibited by FXS patients, and that these can be alleviated by a restoration of FMRP function spe-cifically in aNPCs, suggesting new therapeutic strategies for the treatment of FXS.
In addition to adult neurogenesis, FMRP also regulates em-bryonic and post-embryonic neurogenesis in mammals and Dro-sophila (11–13). Intriguingly, studies using mammalian embry-onic NPCs versus aNPCs reveal opposite roles for FMRP. In the embryonic brain, loss of FMRP leads to the overproduction of neurons in the cortex (12,13), whereas in aNPCs, fewer neurons and more glia are generated in the hippocampus (9,10), imply-ing that the role of FMRP in mammalian neurogenesis is context-specific. In Drosophila, dFMRP is required to control NPC prolif-
eration during larval brain development (11). Loss of dfmr1 leads to a significant increase in the number of mitotic neuroblasts (NBs) in the early larval brain. dFMRP also inhibits neuroblast exit from quiescence in larval brains, as evidenced by the aber-rant expression of cyclin E, a critical regulator of the G1/S tran-sition of the cell cycle. Clonal analyses in the developing brain further revealed that dfmr1-deficient NBs generated significantly more neuronal progeny than wild-type NBs (11). Consistent with an essential role in a wide range of developmental processes, dFMRP also regulates germline proliferation and maintenance, as well as cell cycle progression during oogenesis (14,15). It is unclear whether FMRP has similar functions in the mammalian germline.
fmrp regulATes DnA DAmAge response by suppressing CyClin b expressionThe changes in differentiation and proliferation of germline cells and NPCs in both mammals and flies resulting from (d)FMRP deficiency point to an important role for FMRP in cell cycle reg-ulation. The molecular pathways for cell cycle control, together with those involved in DNA repair and apoptosis, are activated in response to DNA damage (16). It was observed that dfmr1 mutants were hypersensitive to DNA damage (17). Loss of dfmr1 compromised the G2/M DNA damage checkpoint, lead-ing to increased mitotic activity accompanied by elevated levels of cyclin B (CycB), a major regulator of the G2/M transition. In addition, dfmr1 mutants had excessive apoptosis, more DNA strand breaks, and greater genomic instability. Taken together,
Figure 1. Fragile X mental retardation protein (FMRP) regulates microtubules to control synapse growth and axonal transport by suppressing the expression of the microtubule-associated protein, MAP1B/Futsch. FMRP also regulates synapse growth in parallel with the microtubule-severing pro-tein, spastin, mutations of which lead to hereditary spastic paraplegia.
these data strongly argue that dFMRP regulates the DNA dam-age response by suppressing CycB expression and inhibiting apoptosis (17).
At present, it is unknown whether FXS patients and FMR1 knockout mice also have a defective DNA repair pathway. In Drosophila, there is a single FMRP homolog, compared to three in mammals. It is thus possible that functional redundancy between these three family members can mask the phenotype of a single mutant. It would be interesting to test whether double or triple mutants of FMR1 exhibit DNA damage hypersensitivity, as in the case of dfmr1 mutants. If confirmed, it would suggest that the DNA repair pathway could be exploited as a new target for intervention in FXS pathogenesis.
TrAnslATionAl impliCATionsFXS patients display a range of neurobehavioral abnormalities such as learning and memory deficits, attention deficit, hyperac-tivity, sleep disturbance, and autistic features. FMRP participates in many processes, including microtubule network regulation, neurogenesis, and DNA damage response, which are cellular context-dependent and developmental stage-specific (Fig. 2). At present, there is no unifying mechanism that connects the di-verse in vivo functions of FMRP. Drugs targeting specific FMRP-mediated pathways are currently in clinical trials, but effective interventions for FXS may require a combinatorial approach that targets multiple pathways, rather than a single pathway regu-lated by FMRP.
REFERENCES 1. O. Penagarikano, J. G. Mulle, S. T. Warren, Annu. Rev. Genomics Hum. Genet. 8, 109 (2007). 2. C. L. Gatto, K. Broadie, Curr. Opin. Neurobiol. 21, 834 (2011). 3. V. Brown et al., Cell 107, 477 (2001). 4. J. C. Darnell et al., Cell 107, 489 (2001). 5. Y. Q. Zhang et al., Cell 107, 591 (2001). 6. R. Lu et al., Proc. Natl. Acad. Sci. U.S.A. 101, 15201 (2004). 7. A. Yao et al., Hum. Mol. Genet. 20, 51 (2011). 8. M. Nahm et al., Neuron 77, 680 (2013). 9. Y. Luo et al., PLOS Genet. 6, e1000898 (2010).10. W. Guo et al., Nat. Med. 17, 559 (2011).11. M. A. Callan et al., Hum. Mol. Genet. 19, 3068 (2010).12. M. Castren et al., Proc. Natl. Acad. Sci. U.S.A. 102, 17834 (2005).13. T. A. Tervonen et al., Neurobiol. Dis. 33, 250 (2009).14. A. M. Epstein, C. R. Bauer, A. Ho, G. Bosco, D. C. Zarnescu, Dev. Biol. 330, 83 (2009).15. Y. Yang et al., PLOS Genet. 5, e1000444 (2009).16. J. W. Harper, S. J. Elledge, Mol. Cell. 28, 739 (2007).17. W. Liu, F. Jiang, X. Bi, Y. Q. Zhang, Hum. Mol. Genet. 21, 4655 (2012).
Acknowledgments: This work was supported by grants from the Chinese Academy of Sciences Strategic Project B (XDB02020400 and KSCX1-YW-R-69) and the National Science Foundation of China (NSFC, 30430250 and 30525015) to Y. Q. Z. and NSFC (31271121) to A. Y.
Figure 2. In mammals (Mamm.), FMRP regulates proliferation and fate specification of an adult neural progenitor cell (aNPC) by inhibiting the expression of cyclin D1 (CycD1) and the Wnt signal-ing pathway component glycogen synthase kinase 3 β (GSK3β), respectively. In Drosophila (Dros.), dFMRP controls proliferation of larval neuroblasts and the female germline by suppressing the ex-pression of cyclin E (CycE). dFMRP also suppresses cyclin B (CycB) expression to regulate cell cycle progression in embryos and to control DNA damage response in other stages of the life cycle.
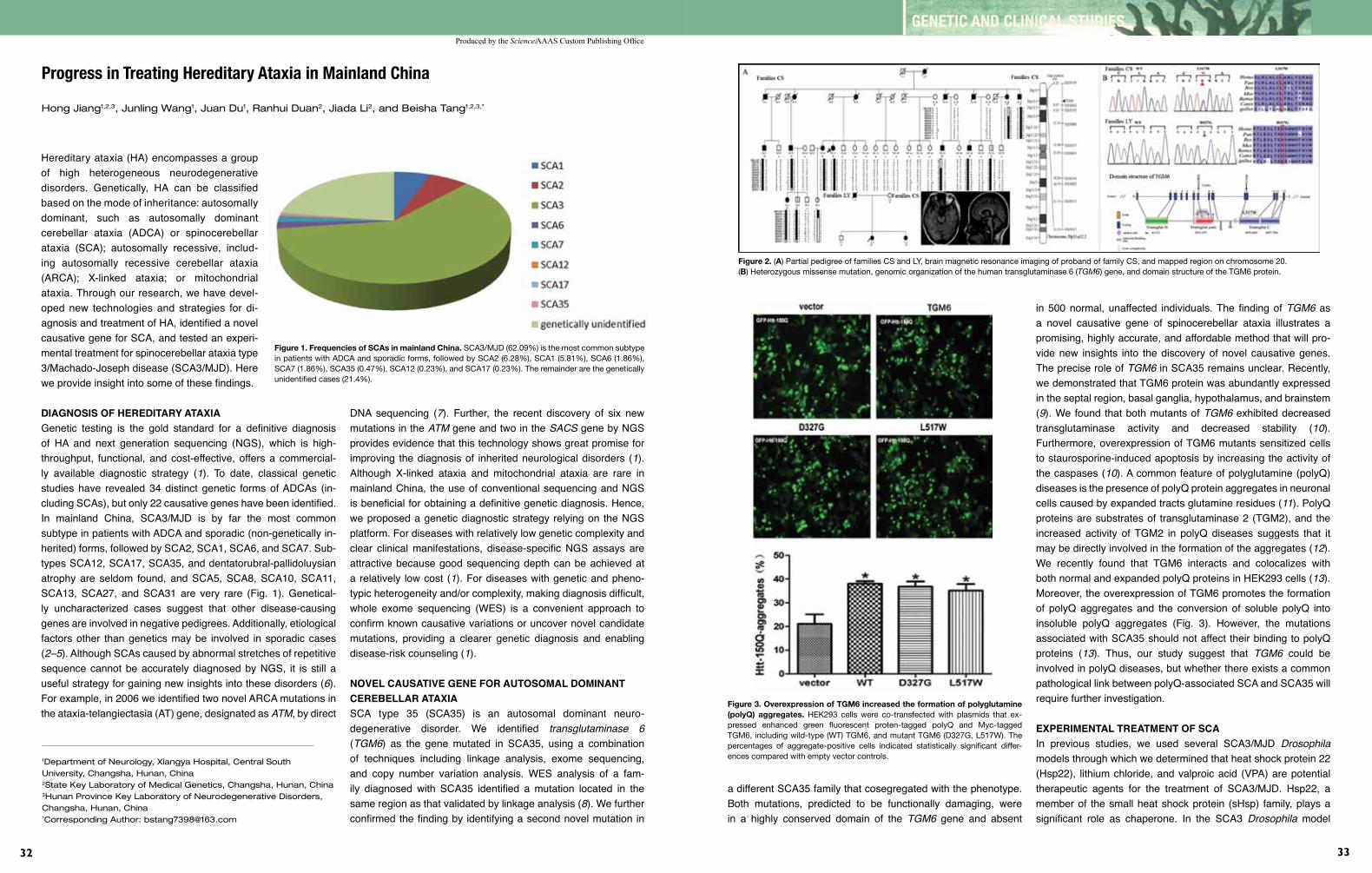
Produced by the Science/AAAS Custom Publishing Office
32
GENETIC AND CLINICAL STUDIES
33
Hereditary ataxia (HA) encompasses a group of high heterogeneous neurodegenerative disorders. Genetically, HA can be classified based on the mode of inheritance: autosomally dominant, such as autosomally dominant cerebellar ataxia (ADCA) or spinocerebellar ataxia (SCA); autosomally recessive, includ-ing autosomally recessive cerebellar ataxia (ARCA); X-linked ataxia; or mitochondrial ataxia. Through our research, we have devel-oped new technologies and strategies for di-agnosis and treatment of HA, identified a novel causative gene for SCA, and tested an experi-mental treatment for spinocerebellar ataxia type 3/Machado-Joseph disease (SCA3/MJD). Here we provide insight into some of these findings.
DiAgnosis of hereDiTAry ATAxiAGenetic testing is the gold standard for a definitive diagnosis of HA and next generation sequencing (NGS), which is high-throughput, functional, and cost-effective, offers a commercial-ly available diagnostic strategy (1). To date, classical genetic studies have revealed 34 distinct genetic forms of ADCAs (in-cluding SCAs), but only 22 causative genes have been identified. In mainland China, SCA3/MJD is by far the most common subtype in patients with ADCA and sporadic (non-genetically in-herited) forms, followed by SCA2, SCA1, SCA6, and SCA7. Sub-types SCA12, SCA17, SCA35, and dentatorubral-pallidoluysian atrophy are seldom found, and SCA5, SCA8, SCA10, SCA11, SCA13, SCA27, and SCA31 are very rare (Fig. 1). Genetical-ly uncharacterized cases suggest that other disease-causing genes are involved in negative pedigrees. Additionally, etiological factors other than genetics may be involved in sporadic cases (2–5). Although SCAs caused by abnormal stretches of repetitive sequence cannot be accurately diagnosed by NGS, it is still a useful strategy for gaining new insights into these disorders (6). For example, in 2006 we identified two novel ARCA mutations in the ataxia-telangiectasia (AT) gene, designated as ATM, by direct
Progress in Treating Hereditary Ataxia in Mainland China
Hong Jiang1,2,3, Junling Wang1, Juan Du1, Ranhui Duan2, Jiada Li2, and Beisha Tang1,2,3,*
DNA sequencing (7). Further, the recent discovery of six new mutations in the ATM gene and two in the SACS gene by NGS provides evidence that this technology shows great promise for improving the diagnosis of inherited neurological disorders (1). Although X-linked ataxia and mitochondrial ataxia are rare in mainland China, the use of conventional sequencing and NGS is beneficial for obtaining a definitive genetic diagnosis. Hence, we proposed a genetic diagnostic strategy relying on the NGS platform. For diseases with relatively low genetic complexity and clear clinical manifestations, disease-specific NGS assays are attractive because good sequencing depth can be achieved at a relatively low cost (1). For diseases with genetic and pheno-typic heterogeneity and/or complexity, making diagnosis difficult, whole exome sequencing (WES) is a convenient approach to confirm known causative variations or uncover novel candidate mutations, providing a clearer genetic diagnosis and enabling disease-risk counseling (1).
novel CAusATive gene for AuTosomAl DominAnT CerebellAr ATAxiASCA type 35 (SCA35) is an autosomal dominant neuro-degenerative disorder. We identified transglutaminase 6 (TGM6) as the gene mutated in SCA35, using a combination of techniques including linkage analysis, exome sequencing, and copy number variation analysis. WES analysis of a fam-ily diagnosed with SCA35 identified a mutation located in the same region as that validated by linkage analysis (8). We further confirmed the finding by identifying a second novel mutation in
1Department of Neurology, Xiangya Hospital, Central South University, Changsha, Hunan, China2State Key Laboratory of Medical Genetics, Changsha, Hunan, China3Hunan Province Key Laboratory of Neurodegenerative Disorders, Changsha, Hunan, China*Corresponding Author: [email protected]
Figure 1. Frequencies of SCAs in mainland China. SCA3/MJD (62.09%) is the most common subtype in patients with ADCA and sporadic forms, followed by SCA2 (6.28%), SCA1 (5.81%), SCA6 (1.86%), SCA7 (1.86%), SCA35 (0.47%), SCA12 (0.23%), and SCA17 (0.23%). The remainder are the genetically unidentified cases (21.4%).
a different SCA35 family that cosegregated with the phenotype. Both mutations, predicted to be functionally damaging, were in a highly conserved domain of the TGM6 gene and absent
in 500 normal, unaffected individuals. The finding of TGM6 as a novel causative gene of spinocerebellar ataxia illustrates a promising, highly accurate, and affordable method that will pro-vide new insights into the discovery of novel causative genes. The precise role of TGM6 in SCA35 remains unclear. Recently, we demonstrated that TGM6 protein was abundantly expressed in the septal region, basal ganglia, hypothalamus, and brainstem (9). We found that both mutants of TGM6 exhibited decreased transglutaminase activity and decreased stability (10). Furthermore, overexpression of TGM6 mutants sensitized cells to staurosporine-induced apoptosis by increasing the activity of the caspases (10). A common feature of polyglutamine (polyQ) diseases is the presence of polyQ protein aggregates in neuronal cells caused by expanded tracts glutamine residues (11). PolyQ proteins are substrates of transglutaminase 2 (TGM2), and the increased activity of TGM2 in polyQ diseases suggests that it may be directly involved in the formation of the aggregates (12). We recently found that TGM6 interacts and colocalizes with both normal and expanded polyQ proteins in HEK293 cells (13). Moreover, the overexpression of TGM6 promotes the formation of polyQ aggregates and the conversion of soluble polyQ into insoluble polyQ aggregates (Fig. 3). However, the mutations associated with SCA35 should not affect their binding to polyQ proteins (13). Thus, our study suggest that TGM6 could be involved in polyQ diseases, but whether there exists a common pathological link between polyQ-associated SCA and SCA35 will require further investigation.
experimenTAl TreATmenT of sCAIn previous studies, we used several SCA3/MJD Drosophila models through which we determined that heat shock protein 22 (Hsp22), lithium chloride, and valproic acid (VPA) are potential therapeutic agents for the treatment of SCA3/MJD. Hsp22, a member of the small heat shock protein (sHsp) family, plays a significant role as chaperone. In the SCA3 Drosophila model
Figure 2. (A) Partial pedigree of families CS and LY, brain magnetic resonance imaging of proband of family CS, and mapped region on chromosome 20. (B) Heterozygous missense mutation, genomic organization of the human transglutaminase 6 (TGM6) gene, and domain structure of the TGM6 protein.
Figure 3. Overexpression of TGM6 increased the formation of polyglutamine (polyQ) aggregates. HEK293 cells were co-transfected with plasmids that ex-pressed enhanced green fluorescent proten-tagged polyQ and Myc-tagged TGM6, including wild-type (WT) TGM6, and mutant TGM6 (D327G, L517W). The percentages of aggregate-positive cells indicated statistically significant differ-ences compared with empty vector controls.

GENETIC AND CLINICAL STUDIES
35
Produced by the Science/AAAS Custom Publishing Office
34
Figure 4. Gmr-SCA3tr-78Q is a strain of transgenic Dro-sophila that expresses expanded polyQ tracts in fly com-pound eyes. Overexpression of Hsp22, lithium chloride, and valproic acid (VPA) prevents polyQ-induced eye depigmenta-tion in an SCA3 Drosophila model. Paired dissecting micro-scope (Magnification: A, ×65; B, ×115; C, ×80) and scanning electron microscope (×1000) images of adult fly eyes are shown. (A) Hsp22, (B) VPA, (C) lithium chloride.
(gmr-GAL4/+; elav-GAL4/+; UAS-MJDtr-Q78/+; UAS-HSP22/+), expression of the MJDtr-Q78 transgene—containing an expanded polygluta-mine tract—showed a greater loss of cell integri-ty, and the pigmentation of adult flies was faded and showed black, point-like necrosis. Drosoph-ila co-expressing an expanded polyQ protein and either one or two copies of the HSP22 gene underwent heat shock, resulting in induction of expression of the HSP22 gene to differing levels depending on the number of copies introduced. The strains car-rying the HSP22 gene were identical, except for the number of gene copies. Findings showed that Hsp22 expression impacted eye depigmentation (Fig. 4), growth restriction, ability for eclo-sion, and average lifespan (14). Previous studies have suggest-ed that an imbalance in histone acetylation may play a key role in transcriptional dysregulation in polyQ diseases. We examined the effect of VPA, a promising therapeutic HDAC inhibitor, in a Drosophila SCA3 model (elav-MJDtr-Q78; gmr-MJDtr-Q78). We expressed the MJDtr-Q78 transgene both in the developing eyes and in neurons. Expression of the MJDtr-Q78 protein produced deleterious phenotypes including faded eye pigmentation, im-paired climbing ability, and decreased mean lifespan, similar to the characteristics of human SCA3. To test the therapeutic po-tential of VPA in vivo, a series of daily doses of VPA were admin-istered to SCA3 flies before cross-breeding. Results showed that long-term use of VPA at an optimal dose partly prevented eye de-pigmentation (Fig. 4), alleviated climbing disability, and extended the average lifespan of SCA3/MJD transgenic flies (15).
Lithium (Li) is known to be neuroprotective in various models of neurodegenerative disease and can reduce mutant protein ag-gregation by inducing autophagy. In an SCA3 Drosophila model (gmr-MJDtr-Q78; nrv2-MJDtr-Q78), expression of MJDtr-78Q caused faded eye pigmentation, decreased locomotor ability, and reduced lifespans in adult flies. These flies also showed a late-onset, progressive, neurodegenerative phenotype with similar features to SCA3/MJD patients. To study the effects of Li treatment on SCA3 pathogenesis in vivo, we administered daily doses of Li chloride (LiCl) at different levels to SCA3 flies prior to cross-breeding. Results confirmed that chronic treatment with LiCl prevented eye depigmentation (Fig. 4), alleviated locomotor disability, and extended the average lifespan (16).
In summary, DNA-based technologies have provided new di-agnostic strategies, the ability to identify causative genes, and
the opportunity to find new therapeutic interventions for HA, as well as provided new insights in pathogenic mechanisms. In the future, individuals suspected of suffering from HA can be genetically evaluated to gain a quicker and more complete diag-nosis as well as a personalized disease-risk profile and improved genetic counseling.
REFERENCES 1. Z. Chen et al., Neurobiol. Aging 34, 2411 (2013). 2. J. Wang et al., Zhong Nan Da Xue Xue Bao Yi Xue Ban 36, 482 (2011). 3. B. Tang et al., Arch. Neurol. 57, 540 (2000). 4. H. Jiang et al., Chin. Med. J. (Engl.) 118, 837 (2005). 5. H. Jiang et al., J. Neurol. Sci. 236, 25 (2005). 6. A. Sailer et al., Neurology 79, 127 (2012). 7. H. Jiang et al., J. Neurol. Sci. 241, 1 (2006). 8. J. L. Wang et al., Brain 133, 3510 (2010). 9. Y. T. Liu et al., Anat. Rec. 296, 1576 (2013).10. W. J. Guan et al., Biochem. Biophys. Res. Commun. 430, 780 (2013).11. A. Michalik, C. Van Broeckhoven, Hum. Mol. Genet. 12 Spec. No. 2, R173 (2003).12. C. A. Ross, M. A. Poirier, Nat. Med. 10 Suppl., S10 (2004).13. W. J. Guan et al., Biochem. Biophys. Res. Commun. 437, 94 (2013).14. Q.H. Li et al., Prog. Biochem. Biophys. 35, 1430 (2008).15. J. Yi et al., PLOS ONE 8, e54792 (2013).16. D. D. Jia et al., Cerebellum 12, 892 (2013).
Acknowledgments: These studies were supported by grants from the Major State Basic Research Development Program of China (“973” Program) (2012CB944601, 2012CB517902, and 2011CB510002), a New Century Excellent Talents in University grant from the Department of Education (NCET-10-0836), and the National Natural Science Foundation of China (81130021, 81271260, 30971585, 30871354, 30710303061, and 30400262).
Genetic Etiology of Parkinson’s Disease in China
Jifeng Guo1,2,3, Xinxiang Yan2,3, Danling Wang1, Qian Xu2,3, Beisha Tang1,2,3, and Zhuohua Zhang1*
Parkinson’s disease (PD) is generally considered to be a multi-factorial neurodegenerative movement disorder commonly char-acterized by bradykinesia, resting tremor, rigidity, and postural instability. Approximately 10% to 15% of patients with PD have a family history of the disease, but the majority of PD is sporadic in origin. Although the pathogenesis of PD remains unclear, it is generally accepted that genetic factors, environmental insult, and gene-environment interactions contribute to PD etiology. Ex-ploration the genetics of PD may provide significant insights into the disease mechanism, particularly since the SNCA gene, cod-ing for α-synuclein, was reported to be a causative factor in PD. However, the genetic heterogeneity inherent in PD means that there are still much work to be done understanding the genetic basis of this disease.
AuTosomAl DominAnT pDThe SNCA (PARK1/4), LRRK2 (PARK8), UCHL1 (PARK5), VPS35, and EIF4G1 genes have been identified as being as-sociated with autosomal dominant PD (ADPD). However, only rarely have mutations in these genes been found in Chinese fa-milial ADPD cases, suggesting that they are not the main genetic cause of PD in China (1–3).
Wang etal. (4,5) reported identification of two novelvariants in Chinese familial PD: c.A1847G (p.K616R) in a conserved residue within a domain of unknown function in the leucine-rich repeat kinase 2 gene (LRRK2), and c.G77A (p.G26E) in a non-conserved position in the high temperature requirement protein A (HtrA) serine peptidase 2 (HtrA2) gene. However, other family members have not yet been tested and cosegre-gation analysis was not performed. Further genetic and func-tional studies are therefore required to verify whether these two variants are causative. Similarly, no pathogenic mutations have been found in Chinese families with ADPD in the growth factor receptor-bound protein 10 (GRB10) interacting GYF pro-tein 2 (GIGYF2) gene. However, eight heterozygous and one homozygous sequence variants were identified in sporadic PD that do not present in 300 control individuals (6). Nevertheless, polymorphic alleles p.Ala99Ala and p.Pro460Thr in GIGYF2
have been suggested to be associated with increased risk of sporadic PD (7).
CAG triplet repeat expansions in the SCA3 gene have been found in patients with PD. In China, mutations in SCA2, SCA3, and SCA17arelikely linked to both familial and sporadic forms of PD (8, 9). Patients carrying these mutations show typical PD features with a bilateral reduction in uptake of a dopamine trans-porter tracer in PET scans. Moreover, some had mild symptoms of ataxia. The mutation frequency in SCA2andSCA3 is 1.5% and 3%, respectively, in familial cases, and 0.5% and 0.8%, respectively, in sporadic cases (8). Thus, it is suggested that SCA2, SCA3, and SCA17 be screened for mutations in order to diagnose ADPD in the ethnic Chinese population.
AuTosomAl reCessive pDMutations in parkin (PARK2), PINK1 (PARK6), DJ-1 (PARK7), FBXO7(PARK15),ATP13A2(PARK9), and PLA2G6(PARK14) are known to be involved in autosomal recessive PD (ARPD). In contrast to those genes known to be associated with ADPD, mutations in these ARPD-associated genes are well-represented in cases of both familial autosomal recessive early-onset Parkin-sonism (AREP) (10,11) and sporadic early onset Parkinsonism (EOP) in Chinese families (12–14). Parkinmutations are the most commonly identified genetic variations in Chinese patients with AREP (48.3%) or EOP (12.6%). Consistent with findings in other ethnic groups, exonic rearrangements within parkin are the most common types of mutations in Chinese PD cases with AREP. Mu-tations in the PINK1, DJ-1, and PLA2G6genes were also found in cases of AREP (6.9%, 3.4%, 3.4%, respectively) and sporadic EOP (3.1%, 2.4%, 0.79%, respectively). Thus far, no pathogenic mutations have been detected in the ATP13A2 and FBXO7 genes in the Chinese population. A list of PD-related genes is shown in Figure 1.
Mutations in PLA2G6 at the PARK14 locus have previous-ly been shown to be linked to infantile neuroaxonal dystrophy (INAD), neurodegeneration with brain iron accumulation (NBIA), and dystonia-parkinsonism. In the mainland Chinese population, a homozygous PLA2G6 missense mutation c.G991T (p.D331Y) was identified in PD patients in a consanguineous family (11) (Fig. 2). In this family, the proband showed typical PD clinical features with no extrapyramidal symptoms, while a PET study indicated a substantial reduction in dopamine transporter (DAT) binding in the brain. A slight reduction of DAT binding was also found in his younger sister who showed no PD symptoms. Fur-ther studies indicated that the mutant protein had only 30% phos-
1State Key Laboratory of Medical Genetics, Changsha, Hunan, China2Department of Neurology, Xiangya Hospital, Central South Univer-sity, Changsha, Hunan, China3Hunan Province Key Laboratory of Neurodegenerative Disorders, Changsha, Hunan, China*Corresponding Author: [email protected]

Produced by the Science/AAAS Custom Publishing Office
36
GENETIC AND CLINICAL STUDIES
37
pholipase enzyme activity comparing to its wild-type counterpart. Based on this data, we proposed AREP without complex pheno-types as the fourth subtype of PLA2G6-related neurodegenera-tive disorders. Partial loss of PLA2G6 enzyme activity likely con-tributes to the PD pathogenesis in the Han Chinese population. This is further supported by a novel PLA2G6 mutation found in a case of sporadic EOP (14).
In contrast to monogenic diseases in which Mendelian inheri-tance can adequately explain pathogenesis, PD is a complex dis-order, likely involving a combination of multiple genetic factors. Identification of a family with EOP harboring heterozygous mis-sense mutations in both PINK1 and DJ-1genes indicates a digen-ic inheritance of PD, suggesting a new genetic mechanism for PD pathogenesis (15).
oTher loCi AnD snps in pDSusceptibility genes play important roles in the pathogenesis of sporadic PD in many different populations. The following genes have been studied in the Han Chinese population.
SNCASNCA, which codes for α-synuclein, has been shown to modu-late susceptibility of sporadic PD using both a candidate gene approach and genome wide association studies. The single nucleotide polymorphism (SNP) rs11931074, located in 3’ region of SNCA, is hypothesized to modify expression of α-synuclein in certain cases of sporadic PD in China (16); other SNPs in SNCA have also been reported to contribute to sporadic PD (17).
GBAMutations in theGBA (glucosidase, beta, acid) gene was first re-ported in consanguineous relatives with Gaucher disease (GD), a lysosomal storage disorder. Heterozygous mutations GBA have been linked to PD pathogenesis: in the Han Chinese popu-lation, the L444P mutation in GBA is linked with susceptibility to sporadic PD, with an odds ratio of 24.29 (18). By contrast, the F213I, R353W, and N370S mutations—the most frequent muta-tions in non-Jewish populations after L444P—are rare in Han Chinese (18).
Figure 1. PD-related Genes in Chinese Population. (A) Frequency of mutations in familial autosomal recessive early-on-set Parkinsonism (AREP). (B) Frequency of mutations in sporadic early onset Par-kinsonism (EOP).
Figure 2. Family Carrying PLA2G6 Mutation (A) Pedigree of consanguineous family with PLA2G6 p.D331Y mutation. [PET], PET scan performed; m/m, homozygous mutation carrier; m/wt, heterozygous mutation carrier; wt/wt, homozygous wild-type. (B) Representative results of enzyme activity assay from three independent experiments. The mock group represented endogenous phospholipases enzyme activity of 0.37nmol/min/mg and was subtracted from each of the other groups. (C) PET images of the family members with the p.D331Y PLA2G6 mutation. Proband (II-2) shows significiant reduction in dopa-mine transporter (DAT) binding. The homozygous mutation carrier (II-4) shows slight reduction in DAT binding in the right posterior putamen. The heterozygous D331Y mutation carriers (I-2 and II-3) and the normal genotype member (II-1) show no abnormalities in this assay.
PITX3The gene for pituitary homeobox 3 (PITX3) is reported to be as-sociated with PD in several populations (19). The rs3758549 SNP within PITX3 has been associated with PD in China, es-pecially late onset PD. Further meta-analysis of PITX3 has provided strong evidence to support its role in susceptibility in Chinese populations (19). However, evidence of the involvement of rs2281983, rs4919621, and other SNPs in PITX3 in Chinese patients has not been found.
oTher genesA large number of susceptibility genes/loci related to sporadic PD have been identified in various populations by GWAS, in-cluding LRRK2, GAK, BST1, and PARK16. Recently, large-scale studies revealed that PARK16 (rs823128, rs823156, and rs16856139), BST1 (rs11724635),GAK (rs1564282), and LRRK2 (G2385R and R1268P) were associated with increased risk of developing sporadic PD in the Chinese population. Consider-ing that genetic heterogeneity is common in complex diseases such as PD and the genetic basis for the majority of sporadic PD cases remains unclear, a comprehensive and large-scale study focusing on genome level analysis of sporadic PD in China is recommended.
REFERENCES 1. J. Y. Tian et al., Neurosci. Lett. 516, 207 (2012). 2. J. F. Guo et al., Parkinsonism Relat. Disord. 18, 983 (2012). 3. K. Li et al., BMC Neurol. 13, 38 (2013). 4. L. Wang et al., Neurosci. Lett. 468, 198 (2010). 5. C. Y. Wang et al., Brain Res. 1385, 293 (2011). 6. L. Wang et al., Neurosci Lett. 473, 131 (2010). 7. L. Wang et al., J. Clin. Neurosci. 18, 1699 (2011). 8. J. L. Wang et al., Mov. Disord. 24, 2007 (2009). 9. Q. Xu et al., Parkinsonism Relat. Disord. 16, 700 (2010).10. J. F. Guo et al., Mov. Disord. 23, 2074 (2008).11. C. H. Shi et al., Neurology 77, 75 (2011).12. J. F. Guo et al., J. Neurol. 257, 1170 (2010).13. L. Z. Luo et al., Neurosci. Lett. 482, 86 (2010).14. J. Y. Tian et al., Neurosci. Lett. 514, 156 (2012).15. B. Tang et al., Hum. Mol. Genet. 15, 1816 (2006).16. Y. Hu et al., J. Neurol. 259, 497 (2012).17. C. Wang et al., Parkinsonism Relat. Disord. 18, 958 (2012).18. Q. Y. Sun et al., Mov. Disord. 25, 1005 (2010).19. J. Liu et al., Brain Res. 1392, 116 (2011).
Acknowledgments: This work was supported by grants from the Major State Basic Research Development Program of China (“973” Program) (2011CB510001) and the National Natural Science Foundation of China (81130021, 81171198, and 81371405).
PKD and PRRT2-Related Paroxysmal Diseases in China
Jun-Ling Wang1,2,3, Nan Li1,2,3, Hong Jiang1,2,3, Lu Shen1,2,3, Kun Xia2, and Beisha Tang1,2,3*
tury. PKD is characterized by childhood or adolescent onset of brief but recurrent attacks of the involuntary movements triggered by sudden voluntary movements. The episodes usually present with dystonia, chorea, athetosis, ballism, or their combination, with no loss or alteration of consciousness. The attacks usually last for under 15 seconds and the frequency of attacks can range from more than 100 per day to less than one per month. Other characteristic features include excellent control of attacks with carbamazepine or phenytoin, a history of benign infantile convul-sions in some patients, and a normal neurological examination. Most idiopathic PKD patients have a family history, usually in an
1Department of Neurology, Xiangya Hospital, Central South University, Changsha, Hunan, China2State Key Laboratory of Medical Genetics, Changsha, Hunan, China3Human Province Key Laboratory of Neurodegenerative Disorders, Changsha, Hunan, China*Corresponding Author: [email protected]
Paroxysmal kinesigenic dyskinesia (PKD) (OMIM# 128200), also known as episodic kinesigenic dyskinesia (EKD), is a rare and remarkable hereditary neurological disorder that was first de-scribed in 1892 and more precisely defined during the 20th cen-

Produced by the Science/AAAS Custom Publishing Office
38
GENETIC AND CLINICAL STUDIES
39
Figure 1. Schematic diagram of PRRT2 gene and wild type and mutant PRRT2 protein. The three mutations identified are indicated with red arrows (top). Schematic representation of chro-mosome 16, shows the overlapping loci for PKD (EKD2), ICCA, and BFIS, and the position of the PPRT2 gene within the 16p11.2 region (bottom).
autosomal dominant mode, although sporadic cases are also observed.
Linkage studies with a variety of ethnicities confirmed a critical region at 16p11.2–q12.1 that was named PKD1, was about 12.4cM long, and encompassed over 157 candidate genes. Two other regions that did not overlap with PKD1 were also identified and denoted PKD2 (16q12.1–q21) and PKD3 (precise loca-tion not known). Over the last decade, a con-siderable amount of work has been done on the identification of the genetic basis for PKD using traditional positional cloning strategies. Never-theless, no causative gene could be identified. Using a cloning strategy as a primary means for disease gene identification is limited, especially in circumstances such as small pedigrees, locus heterogeneity, substantially reduced reproduc-tive fitness, and an abundance of candidate genes present in the mapped region. Recently, whole exome sequencing has been shown to be a powerful approach to identify causative genes underlying a number of Mendelian disorders. For example, in 2010, we used a combined strategy of exome sequencing and linkage analysis to identify a novel spinocerebellar ataxia causative gene, TGM6(1). This strategy considerably re-duces the number of variants potentially impli-cated, making it a highly accurate and affordable method for identifying a causative gene using only a very small number of samples.
In 2011, our group utilized the above strategy to identify proline-rich transmembrane protein 2 (PRRT2) as the first causative gene of PKD (2). We performed genetic linkage mapping with eleven markers that encompassed the pericen-tromeric region of chromosome 16 in 27 mem-bers of two families with autosomal dominant PKD. We then performed whole exome se-quencing in three patients from these two fami-lies. By combining the sequencing and linkage data (region 16p12.1–q12.1), we identified a c.649dupC (p.P217fsX7) mutation in one family and a nonsense c.487C>T (p.Q163X) mutation in the other. Both mutations created premature
termination codons, indicating either a truncated protein or deg-radation of the mutated messenger RNA by nonsense-mediated decay. To confirm these findings, we sequenced the exons and flanking introns of PRRT2 in another three PKD families. The c.649dupC (p.P217fsX7) mutation was found in two families, and a missense mutation, c.796C>T (R266W), was identified in the other (2). This finding was rapidly followed by many simi-lar reports of mutations in the same gene in other families with PKD in China, and PRRT2 was identified as the major causative gene for PKD by independent groups in China. Studies of differ-ent Chinese PKD cohorts have identified PRRT2 mutations in 37% of cases, including 40 out of 58 familial cases (69%) and 26 out of 120 sporadic cases (22%) (3–10). The fact that the remaining 63% of typical PKD patients do not harbor mutations in PRRT2 suggests at least one other causative gene, or other types of PRRT2 gene disruptions that are difficult to detect by gene sequencing. In total, 26 mutations have been reported, with the c.649dupC (p.P217fsX7) mutation being the most com-mon (~80% of PKD sufferers), making a good case for priority screening for this mutation in Chinese PKD patients. Notably, the c.649dupC mutation is also observed as a de novo mutation in some sporadic PKD cases and is adjacent to nine consecutive cytosine (C) bases, indicating that the region is potentially ge-netically unstable (5,11).
oxysmal dyskinesia. Family studies of PKD and ICCA showed linkage to the same pericentromeric region on chromosome 16 (Fig. 1). It has long been proposed that ICCA and PKD are two different expressions of the same disorder. In our study, al-though most patients with a PRRT2 mutation exhibited the pure forms of PKD, there were two members separately diagnosed with ICCA syndrome in infancy from two unrelated families. This suggested that PRRT2may also be the causative gene for ICCA (12).
BFIS without PKD was also linked to the same locus on chro-mosome 16p12-q12 (Fig. 1). Despite distinct clinical features, we hypothesized that mutations in PRRT2might cause multiple clinical phenotypes including pure forms of PKD, pure forms of BFIS, or a combination of both (ICCA). To test this, we col-lected samples from two BFIS families of Han Chinese origin. Linkage analysis showed that the BFIS-causing locus mapped to 16p12.1–q12.2, the location of PRRT2. We then performed mu-tational analysis of PRRT2 by direct sequencing and identified a c.649dupC (p.P217fsX7) mutation in all BFIS patients (12). Inter-estingly, in the subsequent studies in China, PRRT2 gene muta-tions were also found be implicated in other paroxysmal disor-ders, including paroxysmal exercise-induced dyskinesia (PED), paroxysmal non-kinesigenic dyskinesia (PNKD), febrile convul-sions (FC), infantile non-convulsive seizures (INCS), nocturnal
Disease Mutation Type China World
pkD c.649dupC, c.649 delC, and others + +
iCCA c.649dupC, c.649 delC, and others + +
bfis c.649dupC, and others + +
pnkD c.649dupC + +
peD c.649dupC + -
fC c.649dupC, and others + -
nC c.649dupC + -
inCs c.649dupC,c.510dupT, and others + -
episodic ataxia (eA) c.649dupC - +
hemiplegic migraine (hm) c.649dupC, and others - +
infantile focal epilepsy c.649dupC - +
Table 1. Summary of PRRT2 screening in paroxysmal disease patient. “+” = observed; “-”= not observed
Some PKD patients, or their first and second degree relatives, presented with infantile convulsions (IC) or infantile convulsion and choreoathetosis (ICCA). ICCA is inherited in an autosomal dominant fashion and is characterized by benign familial infantile seizures (BFIS) that start within the first year of life and usu-ally stop by three years of age, variably associated with par-
convulsions (NC), and one case representing a phenotypic over-lap among different subtypes of paroxysmal dyskinesis (PxDs) that did not fit into the traditional classification groups (Table 1) (4,9,13).
We also noticed that diseases such as BFIS, PKD, and ICCA with PRRT2 mutations share some characteristics, such as

Produced by the Science/AAAS Custom Publishing Office
40
GENETIC AND CLINICAL STUDIES
41
responding well to anti-epileptic treatment and attacks being paroxysmal. Therefore, considering the fact that this group of PRRT2-related diseases share the same causative gene (pos-sibly even the same mutation), that these disorders show over-lapping occurrence, and that they respond to the same treatment method, we suggest naming this spectrum of disorders “PRRT2-related paroxysmal diseases,” or PRPDs, to simplify clinical clas-sification. PRPDs should be regarded as the general terminology currently associated with BFIS, PKD, PNKD, PED, ICCA, and certain types of epilepsies, and in which paroxysmal attacks and mutations in PRRT2 are characteristic features. This nosologi-cal criterion is meant to serve several purposes. It highlights the use of anticonvulsants to treat this spectrum of diseases and en-larges the disease spectrum of PxDs, especially for those PxDs cases that are difficult to fit into current the classifications based on triggers.
Although the PKD causative gene has been identified, the pathophysiology of PRRT2 mutations remains largely unknown. Whether PKD has a subcortical or cortical origin is controver-sial. A resting-state functional magnetic resonance imaging study performed in ten Chinese PKD patients suggested that in-creased spontaneous brain activity in the cortical-basal ganglia circuitry, especially in the motor preparation areas, is a common pathology of PKD, indicating a lack of readiness for movement in PKD. It was also found that the differences in the spatial pat-terns of increased amplitude of low-frequency fluctuation (ALFF) between PKD patients with and without the p.P217fsX7 mutation might reflect the distinct pathological mechanism resulting from the PRRT2 mutation (14). In addition, a genotype/phenotype re-lationship study in China revealed that PRRT2-PKD has differ-ent phenotypes and drug response compared with non-PRRT2–PKD. These findings provide new insights into the neural system effects of the PRRT2 gene in PKD (15).
In conclusion, we used a strategy combining exome sequenc-ing and classical genome-wide linkage studies to successfully
identify PRRT2 as the causative gene for PKD. This gene also appears to be responsible for other similar disorders including BFIS, ICCA, PNKD, PED, FC, INCS, NC, and a series of parox-ysmal disorders. For convenience in both clinical practice and research, we suggest naming this group of paroxysmal diseases harboring PRRT2 mutations, PRPDs. It is conceivable that PRP-Ds will provide a new research area in the field of neuroscience and provide new insights into the causes of epilepsy and move-ment disorders.
REFERENCES 1. J. L. Wang et al., Brain 133, 3510 (2010). 2. J. L. Wang et al., Brain 134, 3493 (2011). 3. W. J. Chen et al., Nat. Genet. 43, 1252 (2011). 4. J. Li et al., J. Med. Genet. 49, 76 (2012). 5. L. Cao et al., Parkinsonism Relat. Disord. 18, 704 (2012). 6. Q. Liu et al., J. Med. Genet. 49, 79 (2012). 7. Y. P. Chen et al., Eur. J. Neurol., Epub ahead of print (2013) doi: 10.1111/ene.12122. 8. C. H. Shi et al., Mov. Disord. 28, 1313 (2013). 9. X. R. Liu et al., Genes Brain Behav. 12, 234 (2013).10. X. Y. Jing et al., Parkinsonism Relat. Disord. 19, 639 (2013).11. H. F. Li, W. Ni, Z. Q. Xiong, J. Xu, Z. Y. Wu, CNS Neurosci. Ther. 19, 61 (2013).12. J. L. Wang et al., Neurosci. Lett. 552, 40 (2013).13. K. Wang et al., Brain Dev. 35, 664 (2013).14. C. Luo et al., Neurol. Sci., Epub ahead of print (2013) doi: 10.1007/ s10072-013-1408-7.15. H. F. Li et al., Neurology 80, 1534 (2013).
Acknowledgments: This work was funded by projects in the Major State Basic Research Development Program of China (“973” Program) (2011CB510001), the State Key Program of the National Natural Science Foundation of China (81130021), and the National Natural Science Foundation of China (81300981).
Clinical and Molecular Biological Studies of Amyotrophic Lateral Sclerosis in China
Zhangyu Zou1 and Liying Cui2*
1Department of Neurology, Affiliated Union Hospital of Fujian Medical University, Fuzhou, China2Department of Neurological, Peking Union Medical College Hospital, Beijing, China*Corresponding Author: [email protected]
Amyotrophic lateral sclerosis (ALS) is a syndrome characterized by progressive muscle weakness and atrophy resulting from loss of motor neurons in the corticospinal tract, brainstem, and ante-rior cells of the spinal cord. More than 60% of patients die within three years of onset (1). In the past fifteen years, epidemiological, genetic, and electrophysiological studies, as well as research into the treatment of ALS, have been performed at the Peking Union Medical College Hospital of China. Here we summarize some of the findings.
epiDemiologiCAl sTuDiesA registry-based study of ALS was conducted from March 1, 2009 to August 31, 2009 at 10 centers overseen by the Chinese ALS Association. Data from 455 patients yielded the following statistics: the male to female ratio was 1.63:1; sites of onset in-cluded the bulbar region (30.9%), the upper limbs (54.1%), the thoracic region (0.7%), the lower limbs (20.7%), and unspecified regions (3.7%); and the mean age at onset in China was 52.4 years, which was earlier than that reported in many other coun-tries, including Japan, Uruguay, Germany, France, and the United States (mean age at onset range 55.7–66.0 years) (2). The data showed an earlier age at onset for manual (blue collar) workers (50.7±11.4 years) than that of professional (white collar) workers (56.2±12.9 years) (P < 0.001). The most common age at onset (45 to 49 years for females and 55 to 59 years for males) was also earlier than that of patients from Europe and the Unit-ed States. The earlier mean and peak age at onset for Chinese ALS patients suggests a potential role for genetic susceptibility and exogenous factors, such as environmental pollution, in the pathogenesis of ALS (2).
geneTiC sTuDiesApproximately 5%–10% of patients have familial ALS (FALS) and show a Mendelian pattern of inheritance; the remaining 90%–95% carry sporadic ALS (SALS) mutations (3). Sixteen genes and loci have been identified in FALS: SOD1, ALS2, SETX,
SPG11, FUS, VAPB, ANG, TARDBP, FIG4, OPTN, ATXN2, VCP, UBQLN2, C9ORF72, and PFN1 (1,4). Mutations in SOD1, ANG, TARDBP, FUS, VCP, C9orf72, and PFN1have been screened in ALS patients of Chinese origin.
In a Chinese cohort of 142 SALS patients and seven FALS pedigrees, three missense mutations in the SOD1 gene—p.H46R, p.G72C, and p.E133V—were identified in three FALS patients; another three missense mutations—p.V29A, p.V47A, and p.N86I—were identified in three SALS cases. Phenotypic analysis revealed that all four patients within the p.H46R mutation FALS pedigree showed lower limb onset and very slow progression, with a survival of more than ten years (5,6). The clinical features of a p.H46R mutation pedigree in our study were similar to those previously reported in p.H46R pedigrees analyzed in studies of Japanese patients (7, 8). These findings suggest that the p.H46R mutation is more com-mon in the Asian population, and associated with a specific phenotype of lower limb onset, slow progression, and long survival (5,6).
In 202 SALS patients and 10 FALS pedigrees of Chinese origin, a novel missense mutation—ANG c.379G>A (p.V103I)—was identified in one SALS patient. This was the first ANGmutation identified in an Asian population. The prevalence of ANG mutations in SOD1-, FUS-, and TARDBP-mutation–negative ALS patients in Chinese population was 0.5% (9). Although an association of ALS with the G variant of the ANG rs11701 (T/G) single nucleotide polymorphism (SNP) was reported in Italian, Irish, and Scottish populations, no such association was demonstrated in the Chinese population (9).
In a Chinese cohort of 13 FALS pedigrees and 321 SALS patients, two heterozygous missense mutations in the TARDBP gene—c.875G>A (p.S292N) and c.1043G>T (p.G348V)—were identified in three SALS patients. In the Chinese population, the estimated frequency of TARDBP mutations in SALS patients (0.73%) is high-er than Japanese and lower than Caucasian populations, where-as the estimated mutation frequency in SOD1-negative FALS patients (15.2%) is higher than both Japanese and Caucasian populations (10).
Screening for mutations in the FUS gene was performed in 320 ALS patients and 16 FALS pedigrees identified two missense mutations—c.1562G>T (p.R521L) and c.1561C>G (p.R521G)—in one FALS proband. Two additional missense mutations—c.1562G>A (p.R521H) and c.1574C>T (p.P525L)—as well as a

Produced by the Science/AAAS Custom Publishing Office
42
GENETIC AND CLINICAL STUDIES
43
two-base pair deletion c.1509_1510delAG (p.G504Wfs*12) and a nonsense mutation c.1483C>T (p.R495X), were identified in five SALS patients (11,12). In the Chinese population, the fre-quency of FUSmutation in FALS is 10.0%, similar to the Japa-nese (10%), but higher than in Caucasians (4.9%). The frequen-cy of FUS mutation in SALS patients is 1.5%, which is similar to Korean sufferers (1.6%), but higher than in Caucasians (0.6%) (11,12).
No mutations in the VCP or PFN1 genes (13,14) and no ab-normal GGGGCC hexanucleotide repeat expansions in the non-coding region of the C9orf72gene (15) were identified in either FALS or SALS patients of Chinese origin, which suggests that mutations in VCP, PFN1, and C9orf72 are rare in this population.
The SNP rs10260404 in the DPP6 gene is strongly associated with susceptibility to ALS across different populations of Europe-an and American ancestry. However, it was not associated with ALS susceptibility in Chinese SALS patients (16).
Our studies suggest that mutations in SOD1 and FUSare the most common mutations in ALS patients of Chinese origin (5,11,12), whereas mutations in C9orf72are rare (15), highlighting another difference between Chinese and Caucasian populations in the genetic background of ALS.
eleCTrophysiologiCAl sTuDiesALS affects both upper motor neurons (UMN) and lower motor neurons (LMN), however, in the early stages of the disease patients may have focal involvement of only UMN or LMN. Therefore, it can be challenging to diagnose ALS in the early stages by clinical examination alone (17). A registry-based study showed that the mean duration from onset to diagnosis of ALS was 13.8 months in a Chinese cohort, longer than that reported in western countries (9 to 12 months) (2, 18). Electrodiagnos-tic evaluation is valuable in the differential diagnosis of ALS. It extends a physical examination and is able to detect LMN abnormalities early in the disease. It is also key to enabling doc-tors to exclude other diseases that mimic ALS and to establish some diagnostic certainty once other disease processes have been excluded (17).
Nerve conduction studies are an essential part of the electro-diagnostic evaluation of patients with suspected ALS. A study of nerve conduction in 205 ALS patients identified common abnormalities including prolonged distal motor latency (20.6%), absent compound muscle action potentials (CMAPs) or CMAPs with decreased amplitude (52.4%), decreased F-wave frequency (68.9%), and slowed conduction velocity consistent with axon loss (19). A strong association was found between decreased CMAP amplitudes and Medical Research Council (MRC) Scale strength and disease duration. The amplitudes of CMAP decreased as muscle strength decreased and the disease progressed. F-wave persistence was abnormal in 122 out of 177 patients with ALS and decreased as MRC grade strength decreased (19).
Needle electromyography (EMG) is the most important compo-nent of the electrodiagnostic evaluation in ALS. Since the clinical and electrophysiological identification of LMN abnormalities in the same body region have equal diagnostic weigh, needle EMG can enable earlier diagnosis and treatment before clinically obvi-ous symptoms arise (17). Needle EMG in ALS patients should reveal decreased motor unit recruitment and/or large-amplitude, long-duration motor unit potentials in combination with abnormal spontaneous activity, including positive sharp waves, fibrillations, and/or fasciculation potentials (17,20).
For a definitive diagnosis of ALS, LMN abnormalities must be documented in at least three of the four anatomical regions usually examined. Needle EMG evaluation should therefore include the cervical and lumbar regions, as well as the muscles innervated by the cranial nerves and the thoracic region (17). The tongue is traditionally sampled to evaluate LMN involvement, but it is difficult to achieve relaxation and obtain accurate readings. The sternocleidomastoid muscle (in the neck) is innervated by the cranial nerve and upper cervical roots, so EMG changes in this muscle could be regarded as evidence of LMN involvement in the bulbar region (21). Paraspinal muscles between the T8-T10 root levels are not innervated by the lower cervical or upper lumbar segments and could therefore be regarded as thoracic region muscles for the purposes of EMG testing (22).
UMN involvement in patients with ALS can be physiologi-cally assessed using transcranial magnetic stimulation (TMS). An ALS study performed with 40 patients, eight with pure LMN involvement and 34 health controls, suggested that TMS-motor evoked potential (MEP) is not a good measure for early diagno-sis of ALS. However, in patients with so-called clinically prob-able, laboratory-supported ALS or possible ALS (measured based on the number of body regions involved), TMS-MEP may help to demonstrate UMN involvement in these patients and facilitate early diagnosis (23). Fifty ALS patients and 22 healthy controls were examined using conventional TMS and the triple stimulation technique (TST) at the foot muscle, abductor digiti minimi. Central motor conduction time (CMCT), MEP, and rest motor threshold (RMT) were assessed. The TST amplitude ratio was significantly decreased in ALS patients with symptoms of UMN involvement compared to those without involvement or the controls (both P<0.001). Results using TST correlated well with those assessed on the Modified Ashworth Scale (r=-0.772, P<0.001). These findings suggest that TST may be a more ac-curate and sensitive measure of detecting UMN abnormality in ALS patients than previously used techniques, and could reveal subclinical UMN impairment in ALS patients. It may also help to assess the UMN loss in ALS and serve as an objective parameter for monitoring disease progression (24).
reseArCh inTo TreATmenTsCurrently, there are no effective treatments or drugs for this fatal
disease. In vivo studies have demonstrated neuroprotective ef-fects of L-3-n-butylphthalide (L-NBP), extracted from the seeds of Apiumgraveolensvar. Linn. (also known as Chinese celery), in ischemic, vascular dementia, and b-amyloid–infused animal models (25). DL-3-n-butylphthalide (DL-NBP), an analog of L-NBP, has been chemically synthesized and was approved in 2002 by the Chinese State Food and Drug Administration (SFDA) for clinical use in stroke patients. In vivo studies have shown that, following the presentation of ALS-like symptoms in transgenic SOD1-G93A mice, oral administration of DL-NBP significantly decreased the progression of motor deficits, suppressed body weight loss, attenuated motor neuron loss, delayed motor unit reduction, and extended the lifespan, compared with vehicle controls. DL-NBP treatment also caused a significant reduction in immune reactivity of CD11b and glial fibrillary acidic protein, markers for microglia and astrocytes, respectively. Additionally, downregulation of nuclear factor кB (NF-кB), p65, and tumor necrosis factor-α protein levels, and a slight upregulation of NF-E2–related factor 2 (Nrf2) and heme oxygenase-1 (HO-1) were found in the spinal cords of SOD1-G93A mice treated with DL-NBP. These results suggest that DL-NBP could be a promising therapeutic agent for ALS (26,27).
REFERENCES 1. O. Hardiman, L. H. van den Berg, M. C. Kiernan, Nature Rev. Neurol. 7, 639 (2011). 2. M. S. Liu, L. Y. Cui, D. S. Fan, Acta Neurol. Scand. Epub ahead of print (2013) doi: 10.1111/ane.12157. 3. A. Chio et al., Neurology 70, 533 (2008). 4. C. H. Wu et al., Nature 488, 499 (2012). 5. X. G. Li et al., Zhonghua Shen Jing Ke Za Zhi 43, 686 (2010). 6. X. G. Li et al., Xie He Yi Xue Zha Zhi 3, 337 (2012).
7. K. Abe et al., J. Neurol. Sci. 136, 108 (1996). 8. T. Arisato et al., Acta Neuropathol. 106, 561 (2003). 9. Z. Y. Zou et al., Amyotroph Lateral Scler. 13, 270 (2012).10. Z. Y. Zou et al., Neurobiol. Aging 33, 2229.e11 (2012).11. Z. Y. Zou et al., Neurobiol. Aging 34, 1312.e1 (2013).12. Z. Y. Zou et al., Eur. J. Neurol. 19, 977 (2012).13. Z. Y. Zou, M. S. Liu, X. G. Li, L. Y. Cui, Neurobiol. Aging 34, 1519. e3 (2013).14. Z. Y. Zou, Q. Sun, M. S. Liu, X. G. Li, L. Y. Cui, Neurobiol. Aging 34, 1713.e5 (2013).15. Z. Y. Zou, X. G. Li, M. S. Liu, L. Y. Cui, Neurobiol. Aging 34, 1710. e5 (2013).16. X. G. Li et al., Chinese Med. J. 122, 2989 (2009).17. L. Y. Cui, Zhong Guo Sheng Jin Mian Yi Xue He Sheng Jin Bing Xue Zha Zhi 19, 247 (2012).18. A. Al-Chalabi, O. Hardiman, Nature Rev. Neurol. Epub ahead of print (2013) doi: 10.1038/nrneurol.2013.203.19. X. H. Feng et al., Zhong Hua Sheng Jin Ke Zha Zhi 44, 178 (2011).20. M. S. Liu et al., Zhong Hua Sheng Jin Ke Zha Zhi 43, 204 (2010).21. M. S. Liu, L. Y. Cui, X. F. Tang, B. H. Li, H. Du, Zhong Hua Sheng Jin Ke Zha Zhi 35, 361 (2002).22. X. F. Tang, H. Pan, B. H. Li, H. Du, Zhong Hua Sheng Jin Ke Zha Zhi 36, 176 (2003).23. F. Jian et al., Nao Yu Shen Jin Ji Bin Zha Zhi 14, 345 (2006).24. Y. Wang, L. Y. Cui, H. Wang, Zhong Hua Sheng Jin Ke Zha Zhi 43, 562 (2010).25. Y. Peng et al., J. Alzheimers Dis. 29, 379 (2012).26. X. Feng, Y. Peng, M. Liu, L. Cui, Neuropharmacology 62, 1004 (2012).27. X. H. Feng, W. Yuan, Y. Peng, M. S. Liu, L. Y. Cui, Chin. Med. J. 125, 1760 (2012).
Brainnetome Studies of Alzheimer’s Disease Using Neuroimaging
Tianzi Jiang1,2,3,4*, Yong Liu1,2, and Bing Liu1,2
The human brain has been modeled as a hierarchy of complex networks on different temporal and spatial scales. The brain-netome (brain-net-ome), a new “ome” in which the brain net-work is the basic research unit used to investigate the structural and functional organization of the human brain, from genes and neuronal circuits to behaviors (1). Alzheimer’s disease (AD), the
most common form of dementia, is a neurodegenerative disease with typically but not exclusively memory deficits that affect daily cognitive function. The symptoms of AD are not thought to be due to a single, regionally specific pathophysiology, but rather re-sult from abnormal interactions between different brain regions. Therefore, the malfunctioning of connections and brain networks

Produced by the Science/AAAS Custom Publishing Office
44
GENETIC AND CLINICAL STUDIES
45
in cognitive function impairment, while the increased con-nectivity could be interpreted as a compensatory recruit-ment of cognitive resources to maintain task performance in AD patients.
AbnormAliTies of speCifiC brAin neTworks in ADUsing independent component analysis, Song and colleagues found severity-related decreased functional connectivity in the several cognitive-related brain networks in AD, including the bi-lateral precuneus of the precuneus network, the posterior cin-gulate cortex and left precuneus of the posterior default mode network, and the left superior parietal lobule of the left fronto-parietal network (3). In addition, we found that the functional con-nectivity pattern of the default mode network and its anticorrela-tion network are disrupted, and that these connections can be used as features to distinguish AD patients from healthy subjects (13). These results suggest that the brain networks supporting complex cognitive processes are specifically and progressively impaired over the course of AD.
AbnormAliTies of whole brAin neTworks in ADWe carried out a study in both AD patients and healthy elderly subjects in which we virtually divided the entire brain into regions using an anatomically labeled template and paired every region with every other region, calculating the correlation coefficients between each pair of regions (14). Our results demonstrated that most of the decreased functional connectivity occurred be-tween frontal and parietal lobes. This abnormal brain network was confirmed by another independent study in which we found that patients with AD had weaker functional connectivity between regions that were separated by a greater physical distance, es-pecially in several regions previously described as components of the default mode network. More importantly, the impaired functional connectivity in severe AD was consistent with results from studies of mild AD and amnestic mild cognitive impairment (4). The more severe the impairment, as measured by the mini-mental state examination (MMSE), the greater the attenuation of functional connectivity, especially in brain regions functionally connected over a long distance (4).
AbnormAl TopologiCAl ChArACTerisTiCs of The brAin neTwork in ADThe human brain is a complex network that reflects a balance between local processing and global integration of information. Two of our independent studies demonstrated that the topologi-cal characteristics of the brain network are disrupted in AD (4,8). We found that the altered brain regions were mainly located in the default mode network, the temporal lobe, and certain subcor-tical regions that are closely associated with neuropathological changes in AD (4,8). In patients with severe AD, as mentioned above, functional connectivity was particularly attenuated be-
may underlie some of the manifestations of AD. Here we review advances in the brainnetome studies of AD using neuroimaging techniques including magnetic resonance imaging (MRI), func-tional MRI (fMRI), and diffusion MRI. We have demonstrated that the patients with AD have brain atrophy (2–5), and lower ampli-tude and reduced regional homogeneity (4,6,7) in brain activity. Emerging evidence has shown that AD is also associated with impaired functional integration among spatially distinct brain re-gions (4,8,9).
DisrupTeD brAin neTworks in ADThe human brain can generate and integrate information with high efficiency and maintain a perfect balance between local and global interactions. Studies have demonstrated that the symp-toms of AD result from distortions of functional and anatomical networks.
AlTereD brAin neTworks bAseD on region-of-inTeresT sTuDiesRegion-of-interest analysis is the most common method for in-vestigating the functional connectivity pattern of a specific re-gion. A region is selected as a ‘seed’ and the correlation map between this region and every other voxel of the brain is then evaluated. Applying this method to AD patients, disrupted func-tional connectivity between the hippocampus and a set of brain regions in the default mode network was found, as well as in-creased functional connectivity between the left hippocampus and the right lateral prefrontal cortex (10). Zhou and colleagues, selecting the thalamus as the region of interest, found that al-terations in connectivity between the thalamo-default mode network and thalamo-cortical network were related to the se-verity of AD symptoms (11). Additionally, using fMRI on AD sufferers, researchers found impaired functional connectivity (compared to normal controls) to be located mainly between the amygdala and regions that are encompassed by the de-fault mode (i.e., medial posterior parietal cortex and dorsal medial prefrontal cortex), context conditioning, and extinction networks (12).
These disease-related decreases in functional connectiv-ity support the hypothesis of network disconnection reflected
tween regions separated by a greater physical distance, and loss of long distance connectivity was associated with a less efficient global and nodal network topology. Another independent struc-tural network study also confirmed that the AD brain network is distorted, showing a longer path length (9) and suggesting a loss of network efficiency in AD. The results also showed greater at-tenuation in those patients with more severe cognitive impair-ment, hinting at the potential for brainnetome measures to be used as biomarkers or predictors of disease progression in AD.
brAin neTworks Are moDulATeD by AD risk genes Recent pedigree studies have demonstrated that brain networks disrupted in AD are affected by genetic factors. For example, data from a resting state fMRI study showed that the heritabil-ity of default network functional connectivity was about 42% (15). Also, the information transfer efficiency of functional brain networks has been reported to be under genetic control. Previ-ous genetic and bioinformatics studies have demonstrated that AD was affected by interactions between multiple genetic vari-ants and environments (16). Therefore, genetic imaging studies, which investigate the influence of specific genetic variations on disruptions in brain networks in AD, are attracting increased in-terest from Alzheimer’s researchers.
There is particular interest in the genetic aspects of the default mode network, which is consistently reported to be disrupted in AD. The KIBRA gene, which is associated with memory perfor-mance and AD, has been found to impact synchronization within the default-mode in healthy, young populations (17). Studies have also reported that distinct patterns of resting brain activ-ity within the default network in young and older carriers of the apolipoprotein (Apo) E-ε4 allele can be detected before any neurophysiological expression of neurodegenerative processes. An additional resting state fMRI study reported that the ApoE genotype affects brain network properties, especially in AD pa-tients (8). Moreover, Liu etal. (18) reported that the catechol-O-methyltransferase (COMT) val158met polymorphism, which plays a unique role in regulating the prefrontal dopamine level, significantly modulates prefrontal-related functional connectiv-ity within the default mode network. All of these studies suggest that default network activity and connectivity are affected by AD-related genetic variants.
Besides functional imaging, structural brain connectivity and neural integrity markers are also attracting increasing attention in imaging the genetics of AD. Studies have reported that the spe-cific polymorphism and haplotypes of the COMT gene modulate the integrity of important white matter tracts (19,20). A diffusion MRI study found that the global information transfer efficiency of the human brain anatomical network was affected by a disruption in the schizophrenia 1 gene (21). Studies also reported that AD-related cortical thickness and gray matter volume was modulated by ApoE genetic variants (22) and specific long noncoding RNAs
(23). However, recent studies found that certain specific brain connectivity or brain networks were modulated by risk genes for AD (8,17,18,20,24), and thus systematically investigating the genetic control of neural connectivity within AD-related brain sys-tems will likely be an important research direction in the near future.
ConClusionsAs discussed, impairment of the functional and anatomical network architecture of the brain in AD has been verified us-ing neuroimaging. Moreover, studies have demonstrated that genes associated with AD risk play a role. These findings in-dicate that abnormalities in brain networks may be biomark-ers of disease progression in AD. The brainnetome research may therefore open up new avenues for understanding and treating AD.
REFERENCES 1. T. Jiang, Neuroimage 80C, 263 (2013). 2. S. Li et al., Acta Radiol. 49, 84 (2008). 3. J. Song et al., PLOS ONE 8, e63727 (2013). 4. Y. Liu et al., Cereb. Cortex. Epub ahead of print (2013) doi: 10.1093/ cercor/bhs410. 5. S. Li et al., Am. J. Neuroradiol. 28, 1339 (2007). 6. Y. He et al., Neuroimage 35, 488 (2007). 7. Z. Zhang et al., Neuroimage 59, 1429 (2012). 8. X. Zhao et al., PLOS ONE 7, e33540 (2012). 9. Z. Yao et al., PLOS Comput Biol. 6, e1001006 (2010).10. L. Wang et al., Neuroimage 31, 496 (2006).11. B. Zhou et al., Curr. Alzheimer Res. 10, 754 (2013).12. H. Yao et al., Eur. J. Radiol. 82, 1531 (2013).13. K. Wang et al., Med. Image Comput. Comput. Assist. Interv. 9, 340 (2006).14. K. Wang et al., Hum. Brain Mapp. 28, 967 (2007).15. D. C. Glahn et al., Proc. Natl. Acad. Sci. U.S.A. 107, 1223 (2010).16. B. Liu et al., Biochem. Biophys. Res. Commun. 349, 1308 (2006).17. D. Wang et al., Neuroimage 69, 213 (2013).18. B. Liu et al., J. Neurosci. 30, 64 (2010).19. J. Li et al., Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 375 (2009).20. B. Liu et al., Neuroimage 50, 243 (2010).21. Y. Li et al., Cereb. Cortex. 23, 1715 (2013).22. M. Fan et al., Neurosci. Lett. 479, 332 (2010).23. G. Chen et al., Hum. Mutat. 34, 338 (2013).24. A. J. Trachtenberg et al., Neuroimage 59, 565 (2012).
Acknowledgments: This work was partially supported by the National Key Basic Research and Development “973” Program (2011CB707800), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB02030300), and the Natural Science Foundation of China (91132301 and 81270020).
1Brainnetome Center, Institute of Automation, Chinese Academy of Sciences, Beijing, China2National Laboratory of Pattern Recognition, Institute of Automation, Chinese Academy of Sciences, Beijing, China
3The Queensland Brain Institute, The University of Queensland, Brisbane, Australia4Key Laboratory for NeuroInformation of Ministry of Education, School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, China*Corresponding Author: [email protected]

Produced by the Science/AAAS Custom Publishing Office
46
GENETIC AND CLINICAL STUDIES
47
Fragile X syndrome (FXS) (MIM#300624) is the most common cause of inherited intellectual disability, affecting 1 in 4,000 to 1 in 6,000 males and 1 in 8,000 females (1). Since expansion of a trinucleotide repeat in the fragile X mental retardation gene (FMR1) was first identified to cause FXS in 1991, basic science and clinical researchers have been working to find ways to im-prove the disease outcome in FXS families. Chinese scientists and health care professionals have made significant contribu-tions to this work. This review describes advances in the diag-nosis, genetic testing, and counseling of FXS sufferers in China.
inTroDuCTionJ. Purdon Martin and Julia Bell firstly described a pedigree of X-linked mental retardation in 1943 that we now know as FXS. The vast majority of cases are caused by expansion of the tri-nucleotide CGG repeat in the 5ʹ-untranslated region of FMR1. Normal alleles contain between six and 44 repeats, intermedi-ate alleles between 45 and 54 repeats, and premutation alleles in the range of 55 to 200 unmethylated repeats. During either oogenesis or early post-zygotic events, premutations may un-dergo further expansion, and methylation, to generate over 200 repeats. Subsequent hypermethylation and epigenetic silencing of FMR1, resulting in the loss of its protein product—fragile X mental retardation protein (FMRP)—causes FXS (2). Moreover, 40% of males (over 50 years old) with premutations will develop fragile X-associated tremor/ataxia (FXTAS), a progressive neu-rodegenerative disorder, and approximately 20% of females with premutations are at increased risk for premature ovarian insuf-ficiency (3,4).
A survey of the parents of children with special needs dem-onstrated that 95% of them were not aware of FXS (5). An in-vestigation among medical school students showed more than two-thirds had never heard of FXS before attending the medical genetic classes (6). The first cases of FXS in China were report-ed 30 years ago (by Xu Bizhen and Xue Jinglun) (7), but, despite the basic and clinical research undertaken since then, there is still much to be done to improve diagnosis and treatment of FXS patients. Here, we briefly review the clinical aspects of FXS in China and provide recommendations to assist Chinese health care professionals in the future.
CliniCAl DiAgnosisEpidemiological studies indicated a lower premutation frequency among Chinese and other east Asians compared with Europe-ans and Africans; the prevalence of the full fragile X mutation in China remains unknown (8).
Both boys and girls with FXS have certain physical, behavioral, and intellectual phenotypes. Screening checklists have been ap-plied to evaluate these physical characteristics for appropriate referrals to genetic testing, while some of them have also been validated to reduce unnecessary testing (9). A six-item screening checklist for children that includes characteristics such as men-tal retardation, family history of mental retardation, protruding ears, elongated face, attention deficit hyperactivity disorder, and autistic-like behavior has proved to be a reliable and effective diagnostic tool in two studies in China of groups with suspected fragile X or developmental delays (10,11). Similar characteristics were in fact noted in a large Chinese population survey in 1986 (12). The checklist designed by Nolin and Laing has been dem-onstrated to not be applicable for use in China due to the age and ethnic differences of patients used to develop the checklist (13).
Based on our clinical records, individuals affected by FXS are most likely to visit a doctor under the following circumstances: (1) around one to two years old when obvious developmen-tal delay is observed, (2) between seven and nine years old, when learning difficulties are noticed at school, (3) as teenag-ers who have been misdiagnosed or under-diagnosed, (4) ages 16 and over, when family members or relatives identify a problem.
moleCulAr TesTingThe FMR1gene is located on the X chromosome at Xq27.3. Ap-proximately 95–99% of fragile X cases are caused by an increase in the number of CGG repeats of the gene. A cytogenetic method to count the number of fragile sites in cultured folate-deficient cells was the first assay to be developed for diagnosis of FXS. It has been gradually replaced by a combination of molecular methods: CG-rich polymerase chain reaction (PCR) for accurate sizing, together with Southern blotting to determine methylation status (14). If standard molecular tests indicate a normal CGG tract, sequencing of FMR1 to assess deletion and point mutation may be performed upon request. Additional detection methods have been developed that may be adopted as clinical tests in the future, including methylation-sensitive PCR, methylation-specific melting curve analysis, and methylation-specific multiplex liga-
Fragile X Syndrome in China: A Clinical Review
Ranhui Duan
State Key Laboratory of Medical Genetics, Central South University, Changsha, Hunan, China. Corresponding Author: [email protected]
tion-dependent probe amplification (15,16). Certain large hospi-tals in China provide genetic testing services for FXS, but not all can perform high-CG–content PCR and southern blot analysis in parallel due to difficulties amplifying CG-rich sequences. As a result, female carriers and those showing genetic mosaicism may be misdiagnosed. Some commercial FXS tests have been promoted in China such as the AmplideX™ FMR1 PCR Kit from Asuragen, a fragile X PCR test from Abbott, and a trial product from PerkinElmer. The cost, instrument requirements, and train-ing needs impede the widespread use of these technologies in the laboratory or clinic for adult and prenatal diagnosis.
Physician referrals and networks formed by FXS families are the main channels for FXS patients and their families to advo-cate for testing. As of the end of 2012, most counties and cities in China had joined the national medical insurance program, but the cost of genetic testing for FXS is still not covered by the major health care insurance plans. Furthermore, the cost of the test is $300 to $600, unaffordable for some low-income families (17).
geneTiC CounselingUnlike in Western countries, genetic counseling guidelines in China have only recently been developed. In China, genetic counselors must have completed a medical genetics specialty and have a Master’s or doctoral degree in medicine. Physi-cians mainly focus on the diagnostic and clinical aspects of the disease, but may overlook ethical issues and essential emotional support for the patients, areas covered by genetic counselors (18). China has some unique challenges that re-quire careful handling, including newborn screening, carrier screening, confidentiality issues, and reproductive options for FXS patients.
In China, children with mild intellectual disability are eligible for the standard nine-year compulsory education, while children with moderate to severe intellectual disability must make use of special mental health services. Since drug development for FXS is still in the research phase, Chinese physicians currently follow western conventions by treating the symptoms of the disease; a diagnosis of FXS generally doesn’t influence the therapies prescribed. The most common symptoms seen include attention deficit problems, anxiety, and autism (19). Traditional Chinese medicine offers some options for improving various developmental and behavioral aspects in children with autism (20), which may be exploited for use with FXS sufferers.
We propose that a committee of multidisciplinary experts should be established in China to formulate standards and guidelines regarding diagnosis, genetic testing, and counsel-ing for FXS. Doctors of obstetrics and gynecology specialized in clinical genetics should be provided with periodic training in the diagnosis and care of individuals with FXS. In addition, we suggest that a publicly funded network should be provided to educate and assist patients as well as provide resources to sup-port clinics carrying out genetic testing.
REFERENCES 1. F. J. Song, P. Barton, V. Sleightholme, G. L. Yao, A. Fry-Smith, Health. Technol. Assess. 7, 1 (2003). 2. T. Wang, S. M. Bray, S. T. Warren, Curr. Opin. Genet. Dev. 22, 256 (2012). 3. S. Jacquemont et al., JAMA 291, 460 (2004). 4. D. J. Allingham-Hawkins et al., Am. J. Med. Genet. 83, 322 (1999). 5. J. L. Sun, C. Y. Ji, N. N. Xiong, Y. Zhang, J. Wang, Chin. J. Public Health 23, 140 (2007). 6. J. Li, W. Huang, S. Y. Luo, Y. T. Lin, R. H. Duan, J. Genet. Couns. Epub ahead of print (2013) doi: 10.1007/s10897-013-9634-y. 7. J. L. Xue et al., Journal of Fudan University 23, 319 (1984). 8. M. K. Hill, A. D. Archibald, J. Cohen, S. A. Metcalfe, Genet. Med. 12, 396 (2010). 9. V. A. Johnson, J. Cult. Divers. 15, 117 (2008).10. Y. Z. Guo et al., Acta Acad. Med. Sin. 22, 85 (2000).11. Z. W. Zhu et al., paper presented at the JiangZheHu Pediatric Academic Conference, Jiaxing, China, 1 November 2012.12. X. T. Zhou et al., Acta Genet. Sin. 13, 310 (1986).13. Y. H. Yi, X. F. Lu, W. P. Liao, Z. Chen, X. F. Xia, J. Clin. Neurol. 15, 167 (2002).14. A. McConkie-Rosell et al., J. Genet. Couns. 14, 249 (2005).15. X. Y. Guo, J. Liao, F. H. Lan, Chin. J. Med. Genet. 29, 296 (2012).16. L. Q. Wu et al., Hereditas (Beijing) 25, 123 (2003).17. T. J. Musci, A. B. Caughey, Am. J. Obstet. Gynecol. 192, 1905 (2005).18. S. L. Sui, Chin. Med. Ethics 26, 252 (2013).19. J. H. Hersh, R. A. Saul, Pediatrics 127, 994 (2011).20. V. C. Wong, J. G. Sun, J. Altern. Complement. Med. 16, 545 (2010).
Acknowledgments: This work was funded in part by grants from the National Natural Science Foundation of China (81071028 and 81172513), the National Basic Research “973” Program (2011CB510000 and 2012CB944600), and the Program for New Century Excellent Talents (7603230006).

Produced by the Science/AAAS Custom Publishing Office
48
THERAPEUTIC STRATEGIES
49
China has the largest single population in the world, at 1.3 billion, accounting for 20% of the total global population. In the past 10 years, China has been advancing universal health care cover-age, and a basic social medical insurance system has been de-veloped and is expanding rapidly (1). However, due to inequity between the urban and rural areas, bad health service utilization, and escalation of the costs, the financial burden of health care has rapidly increased and affordability of care for the rural popu-lation remains a challenge (2,3).
Following the pioneering work of Benabid etal., (4) deep-brain stimulation (DBS) has been shown to be an effective treatment for a variety of neurological and psychotic disorders refractory to normal treatments, including Parkinson’s disease (PD), dystonia, tremors, and obsessive-compulsive disorder (OCD) (5–8). In the United States, the application of DBS in essential tremor (ET), PD, primary generalized and segmental dystonia, and OCD was approved by the U.S. Food and Drug Administration in 1997, 2002, 2003, and 2009, respectively, while in Europe, DBS has been approved to treat epilepsy and OCD. Many researchers in other countries such as Japan, Korea, Brazil, and China, have also studied the application of DBS in neurological and psychotic disorders (9–13). In China, the first article introducing DBS was published in Chinese in 1995 (14), and the first DBS surgery was performed on two PD patients in 1998 (15). Since then, DBS has rapidly gained acceptance in China as an effective treatment for patients with certain refractory movement disorders. However, due to the high cost of implantable hardware, this technology is still not affordable for the majority of patients, especially those in the rural areas. Here we review the development of DBS in China over the past decade, and discuss the ongoing challenges and possible solutions for the future.
The DevelopmenT of Dbs in ChinAIn the decade after the first DBS surgery was performed, its use in China underwent a period of rapid development. During this time, more than 200 articles on DBS were published, in Chinese as well as English, and more than 2,000 patients underwent DBS implantations. Most of the medical centers providing DBS treatment were located in the more developed eastern part of China, such as Tiantan and Xuanwu Hospitals in Beijing, Ruijin and Changhai Hospitals in Shanghai, Anhui Provincial Hospital
Department of Functional Neurosurgery, Shanghai Jiao Tong University School of Medicine, Rui Jin Hospital, Shanghai, China*Corresponding Author: [email protected]
in Anhui, and Zhujiang Hospital in Guangdong. Similar to other countries, PD patients account for the largest DBS treatment population, followed by dystonia and ET patients. Although Chi-na lagged behind western countries in DBS treatment, Chinese neurosurgeons make important contributions to the develop-ment of DBS technology and application. For example, Sun etal. introduced subthalamic nucleus (STN) stimulation in primary dystonia, a new target of DBS, at the 2003 quadrennial meeting hosted by the American Society for Stereotactic and Functional Neurosurgery (16). In 2006, Zhang et al. reported on six sec-ondary dystonia patients who received STN DBS treatment (17). Besides movement disorders, new indications such as anorexia nervosa (18–20) and drug psychological dependence (21, 22) have been investigated by Chinese researchers.
AppliCATion of Dbs in ChinAParkinson’sDisease(PD)As mentioned above, the largest treatment group for DBS is PD sufferers. The targets chosen for the first two patients were the ventrolateral thalamus (Vim) and STN. Both of the patients showed clear improvement relative to before the surgery (15). Although Vim DBS has been effective in controlling parkinsonian tremors, the lack of reliable effects on other motor symptoms has limited Vim DBS for the treatment of PD. Therefore, STN as well as the internal globus pallidus (GPi) have been the most com-monly used targets in PD patients in China. Since publication of the first DBS study, many neurosurgeons in medical centers across China have performed the DBS procedure in PD patients, with satisfactory outcomes. Authors have not only explored the mechanisms of DBS treatment, but have also shared their ex-periences including complications encountered, changes seen using positron emission tomography imaging, and postoperative management of DBS (23–25).
DystoniaIn the last century, stereotactic GPi ablation was the most com-monly used method for the treatment of dystonia in China (26). Successful application of DBS in PD patients led Chinese neuro-surgeons to start using DBS for the treatment of dystonia. STN, rather than the more commonly used GPi, is the most frequent stimulation target for dystonia in China and outcomes are report-ed to be satisfactory. In 2006, Sun etal. described 15 primary dystonia patients who received STN DBS treatment; over 76% of their symptoms improved by the six month follow-up (27). In the same year, Zhang etal. published on six cases of secondary
dystonia treated with STN DBS, obtaining similar results (17). In 2009, another group in China reported on 17 dystonia patients treated with STN DBS in which the Burke-Fahn-Marsden scale improved from 22% to 95.8% (28). More recently, Cao etal. (29) published a long-term follow-up study that proposed the sub-thalamus as an alternative target to GPi for DBS to treat primary dystonia.
EssentialTremorOnly a few journal papers have been published from China involving DBS treatment in ET patients. Hu et al. reported that two ET patients demonstrated significant tremor control after STN DBS (30), while Meng et al. treated five ET pa-tients with Vim DBS and demonstrated satisfactory tremor control (31). Despite some success, the outcomes in ET pa-tients treated with DBS are relatively poor compare to those with PD.
OtherDisordersApart from movement disorders, Chinese neurosurgeons have been investigating new indications for DBS. For example, in a long-term follow-up study Sun etal. reported an average of 65% increase in body weight in four severe and refractory patients with anorexia nervosa following a DBS procedure. To the best of our knowledge, this is the first such report of DBS treatment in anorexia patients.
Before the introduction of DBS, capsulotomy was the most frequently used method for the treatment of medically refractory OCD in China (32). DBS has now mostly replaced capsulotomy because it has fewer complications and is reversibility. Chen etal. reported that of six refractory OCD patients who underwent nucleus accumbens (NAc) DBS on the right side of their brain and anterior capsulotomy on the left side, five patients showed obvious improved while one patient exhibited mild improvement (33). Zhou etal. investigated white matter abnormalities in pa-tients with treatment-resistant OCD by comparing diffusion ten-sor imaging (DTI) data before and after DBS procedures, and concluded that the mechanism of DBS action in refractory OCD may be related to ablation of the function of fibers in the frontal lobes and cingulates fasciculus (34).
Xu et al. presented the first case report in China of NAc DBS for the treatment of psychological opiate drug depen-dence (21). At the end of a three month follow-up, there is no evidence of relapse. We obtained similar results in our medical center and believe that a bright future exists for DBS treatment of addiction in patients (35). NAc is now consid-ered a promising DBS target for the treatment of addiction (22).
The application of DBS in chronic pain and epilepsy are still in the early stages, with only a few studies published in China (36,37).
problems AnD sTrATegiesDBS has been actively used in China for more than 10 years, but there are still some challenges associated with its broader ap-plication. First, the high cost of DBS might be the chief obstacle in China. Of the over 1.7 million people aged 55 years or older suffering from PD in mainland China (38), many will become eli-gible for the surgical intervention because of intense levodopa usage. However, only very few will receive DBS treatment due to high out-of-pocket expenses. With the advent of new, cheaper devices (39), it is hoped that more Chinese patients will be able to afford this treatment in the future.
Second, inclusion criteria for patients need to be strictly en-forced. We were pleased to see an expert consensus statement on DBS treatment in PD patients in China published in 2012 (40), which has greatly enhanced the development of DBS for PD pa-tients. However, expert consensus is also needed for other dis-orders such as dystonia, OCD, and depression. The potentially positive effects of DBS in the treatment of psychiatric disease have not yet been recognized by most psychiatrists in China. Therefore, greater communication and collaboration should be encouraged between neurosurgeons and psychiatrists in the fu-ture. Finally, strict inclusion criteria for psychotic patients should be required to ensure that excessive and unnecessary opera-tions are not performed.
REFERENCES 1. C. Li, X. Yu, J. R. Butler, V. Yiengprugsawan, M. Yu, Soc. Sci. Med. 73, 359 (2011). 2. S. Hu et al., Lancet 372, 1846 (2008). 3. Q. Long, L. Xu, H. Bekedam, S. Tang, Int. J. Equity Health 12, 40 (2013). 4. A. L. Benabid, P. Pollak, A. Louveau, S. Henry, J. de Rougemont, Appl. Neurophysiol. 50, 344 (1987). 5. G. Deuschl et al., N. Engl. J. Med. 355, 896 (2006). 6. J. Mueller et al., Mov. Disord. 23, 131 (2008). 7. D. O’Sullivan, M. Pell, Brain Res. Bull. 78, 119 (2009). 8. W. K. Goodman, R. L. Alterman, Annu. Rev. Med. 63, 511 (2011). 9. P. R. Arantes et al., Mov. Disord. 21, 1154 (2006).10. T. Kaido et al., Neuromodulation 14, 123 (2011).11. S. H. Paek et al., J. Neurol. Sci. 327, 25 (2013).12. B. Sun, S. Chen, S. Zhan, W. Le, S. E. Krahl, Acta Neurochir. Suppl. 97, 207 (2007).13. J. Zhang et al., Acta Neurochir. Suppl. 99, 43 (2006).14. P. Wei, Foreign Med. Sci. (Section on Neurology & Neurosurgery) (Chin.) 22, 299 (1995).15. X. Guan, G. Luan, B, Zhang, Mod. Rehabilit. (Chin.) 5, 33 (2001).16. B. Sun et al., paper presented at the 2003 Quadrennial Meeting held by the American Society for Stereotactic and Functional Neurosurgery, New York City, May 18–21, 2003.17. J. Zhang, K. Zhang, Z. Wang, M. Ge, Y. Ma, Chin. Med. J. (Engl.) 119, 2069 (2006).
Deep-Brain Stimulation Research in China
Bomin Sun* and Wei Liu

Produced by the Science/AAAS Custom Publishing Office
50
THERAPEUTIC STRATEGIES
51
Age-related neurodegenerative diseases, such as Parkinson’s (PD) and Alzheimer′s (AD) disease, are seriously threatening the health of Chinese citizens, with PD and AD patient populations increasing at a rate of 100,000 and 300,000 per year, respec-tively (1,2). Although there is no cure for AD or PD, a variety of medications can provide relief from some of the symptoms. Recently, the development of direct reprogramming technolo-gies has shed light on promising strategies for stem cell-based therapies for AD and PD. This review will focus on neural pro-genitor cells and induced neural progenitor cells studied in our laboratory, and discuss their potential applications in AD and PD treatment.
Neural Progenitors by Direct Reprogramming: Strategies for the Treatment of Parkinson’s and Alzheimer’s Diseases
Changhai Tian1,2 and Jialin C. Zheng1,2,3*
1Center for Translational Neurodegeneration and Regenerative Therapy, Shanghai Tenth People’s Hospital affiliated to Tongji University School of Medicine, Shanghai, China2Department of Pharmacology & Experimental Neuroscience, and 3Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, Nebraska*Corresponding Author: [email protected]
18. C. Wu, Y. Pan, F. Li, B. Sun, Med. Recapi. (Chin.) 16, 2290 (2010).19. C. Wu et al., World Neurosurg. 80, 29 (2012).20. C. Wu et al., paper presented at INS – 9th World Congress, Seoul, Korea, September, 2009.21. J. Xu et al., Chin. J. Stereotact. Funct. Neurosurg. (Chin.) 18, 140 (2005).22. B. Sun , D. Li , C. Wu, Neuromodulation (Academic Press, London, UK, 2009).23. B. Sun et al., Chin. J. Nerv. Ment. Dis. 29, 410 (2003).24. Y. Ma et al., Chin. J. Neurosurg. (Chin.) 23, 452 (2007).25. C. Zuo et al., Chin. J. Nucl. Med. 27, 335 (2007).26. Y. Li et al., Chin. J. Neurosurg. (Chin.) 17, 350 (2001).27. B. Sun et al., Chin. J. Neurosurg. (Chin.) 22, 717 (2006).28. Y. Li et al., Chin. J. Neurosurg. (Chin.) 25, 605 (2009).29. C. Cao et al., Mov. Disord. 12, 1877 (2013). 30. X. Hu et al., Shanghai Med. J. 29, 353 (2006).31. F. Meng et al., Nat. Med. J. China 92, 1037 (2012).32. B. Sun et al., Chin. J. Nerv. Ment. Dis. 29, 81 (2003).
neurAl progeniTors from feTAl Tissues Multipotent neural progenitor cells (NPCs) exist in the mamma-lian developing and adult nervous system, and are capable of migrating toward the specific sites and giving rise to the main components of the nervous system. Transplantation of NPCs is a promising therapy for various neurodegenerative diseases and brain injuries (3–5). It has been reported that the transplantation of NPCs into the brain contributes to the improvement of cog-nitive impairment in animal models of PD and AD. This occurs through cell replacement, the release of specific neurotransmit-ters, and the production of neurotrophic factors that protect in-jured neurons and promote neuronal growth (6–10). The trans-plantation of NPCs derived from fetal brain raises serious ethical concerns, particularly in the acquisition of the fetal tissue, but the understanding gained from these in vitro experiments will be useful for the future application of NPCs obtained from non-fetal sources. Our laboratory has been working on human NPCs for many years and has revealed that C-X-C motif chemokine 12 (CXCL12, also known as stromal cell-derived factor 1) plays an important role not only in increasing cell proliferation through the Akt/Foxo3a signaling pathway, but also in protecting against
33. B. Sun et al., CMINSJ 13, 58 (2008).34. Y. Zhou, J. Zhu, L. Li, J. Xu, Chin. J. Imaging Technol. 22, 971 (2006).35. B. Sun, W. Liu, Surgical Treatments for Drug Addictions in Humans. Deep Brain Stimulation (Springer-Verlag, Berlin/Heidelberg, 2012).36. S. Liang, Chin. J. NeuRosurg. (Chin.) 28, 802 (2012).37. L. Wang et al., Chin. J. Stereotact. Funct. Neurosurg. (Chin.) 15, 166 (2002).38. Z. Zhang et al., Lancet 365, 595 (2005).39. Y. Ma et al., Chin. J. Neruosurg. (Chin.) 29, 415 (2013).40. B. Chen et al., Chin. J. Neruosurg. (Chin.) 28, 855 (2012).
Acknowledgments: We thank Shikun Zhan, Dianyou Li, Sijian Pan, and Chunyan Cao for their valuable advice and assistance in the preparation of this manuscript. These studies were supported by the National Natural Science Foundation of China (81271518) and the Natural Science Foundation of Shanghai, China (11ZR1422600).
Figure 1. Strategies to Generate iNPCs from Somatic Cells. iNPCs can be induced by conceptually different mechanisms. (A) Ectopic expression of Yamanaka factors converts somatic cells to induced pluripotent (iPS) cells that can then be differentiated into primitive iNPCs, and subse-quently into different subtypes of neurons based on differ-entiation conditions. (B) Ectopic expression of Yamanaka factors converts somatic cells to primitive iNPCs through an intermediate unstable pluripotent stage. (C) Direct re-programming of primitive iNPCs by expression of lineage-specific transcription factors. (D) Direct reprogramming of region-specific iNPCs by expression of lineage-specific transcription factors and location-specific determinants. (E) Generation of neuronal subtypes through direct conversion of cells in vitro and in vivo. (F) Generation of neuronal pro-genitor subtypes through direct conversion of somatic cells using sets of defined transcription factors.
NPC apoptosis through chemokine receptor CXCR7- and CX-CR4-mediated endocytotic signaling pathways (11, 12). These data provide insight into the essential role for CXCL12 in neuro-genesis and also suggests a novel role for CXCR7 in NPC sur-vival contributing to neurogenesis, as well as potentially offering some theoretical guidance for NPC-based therapy.
neurAl progeniTor Cells generATeD by DireCT reprogrAmmingSelective degeneration of functional neurons is associated with the pathogenesis of neurodegenerative disorders, such as de-generation of midbrain dopaminergic neurons in PD (13) and forebrain cholinergic neurons in AD (14). How to achieve suf-ficient cell replacement to halt PD or AD progression, or pos-sibly even provide a cure, is the main challenge. The discovery of induced pluripotent stem (iPS) cells has facilitated the deri-vation of stem cells from adult somatic cells for the personal-ized treatment of PD and AD without depending on fetal tis-sues. However, ethical and safety concerns still exist. Recently, neuronal subtypes including dopaminergic and cholinergic neurons have been generated successfully through direct re-programming of somatic cells by expression of developmental genes (15–17). The low yield of neurons from this method has,
however, limited its broad application in cell transplantation. In addition to NPCs obtained from iPS cell differentiation (Fig. 1, path A) and neuronal subtypes induced by direct reprogram-ming (Fig. 1, path E), the direct reprogramming of NPCs from differentiated, non-neuronal somatic cells (Fig. 1, paths B and C) (18–21) will not only provide an alternative to fetal tissue or pluripotent cells as precursors, but also furnish a potentially un-limited source of neurons. Our laboratory was among the first to successfully convert fibroblasts into induced NPCs (iNPCs) by ectopic expression of transcription factors. iNPCs share many characteristics with primary NPCs and are able to differentiate into neurons (Fig. 2). Recently, the direct reprogramming of iN-PCs has also been achieved by forced expression of a single factor (22) or by 3-D sphere culture of fibroblasts on low attach-ment surfaces (23). These findings suggest that neural progeni-tor cell fate can be reprogrammed by modifying intrinsic and extrinsic cues.
Stem cell-based therapy for AD and PD is essentially based on the regeneration of different neuronal subtypes, such as do-paminergic neurons in PD and forebrain cholinergic neurons in AD. Recently, we have been working on the direct reprogram-ming of somatic cells into region-specific iNPCs (Fig. 1, path D) as well as subtype-specific iNPCs (Fig. 1, path F) by over-

Produced by the Science/AAAS Custom Publishing Office
52
THERAPEUTIC STRATEGIES
53
Figure 2. Direct Conversion of Fibro-blasts into NPCs by a Novel Com-bination of Transcription Factors. (A) Schematic showing direct repro-gramming of fibroblasts derived from E/Nestin:EGFP transgenic mice into NPCs using defined transcription fac-tors including Brn2, Sox2, Bmi1, TLX, and c-Myc. (B) Differentiation of iNPCs into neurons (panel a; MAP-2, red), astrocytes (panel b; GFAP, red), oligo-dendrocytes (panel c; O4, red). Panel d shows synapse formation between neurons (β-III Tubulin, green; Synap-tophysin, red), with magnification of white box in d shown in panel e. Nu-clei are stained with DAPI (blue). EGFP, enhanced green fluorescent protein; MAP-2, microtubule-associated pro-tein 2; GFAP, glial fibrillary acidic pro-tein; O4, marker for oligodendrocytes.
expression of defined growth factors. Both of these pathways show promise for AD and PD therapies. Direct in vivo conver-sion of somatic cells, such as fibroblasts and astrocytes, into functional neurons has also been achieved (24), providing proof of principle that it may be feasible to convert somatic cells—such as activated astrocytes—into region- or subtype-specific iNPCs in the brains of AD and PD patients. In the future, the further development of this technology will undoubtedly pro-vide promising strategies for the effective treatment of AD and PD.
REFERENCES 1. J. Xian, Public Health (Chin.) 36, 25 (2013). 2. J. Wang, Asia-Pacific Traditional Medicine (Chin.) 7, 157 (2011). 3. A. Bjorklund, O. Lindvall, Nat. Neurosci. 3, 537 (2000). 4. O. Lindvall, Z. Kokaia, Nature 441, 1094 (2006). 5. J. K. Ryu, T. Cho, Y. T. Wang, J. G. McLarnon, J. Neuroinflammation 6, 39 (2009). 6. M. Blurton-Jones et al., Proc. Natl. Acad. Sci. U.S.A. 106, 13594 (2009). 7. D. E. Redmond, Jr. et al., Proc. Natl. Acad. Sci. U.S.A. 104, 12175 (2007). 8. S. Wu et al., Pathobiology 75, 186 (2008). 9. T. Yasuhara et al., J. Neurosci. 26, 12497 (2006).10. L. Madhavan, B. F. Daley, K. L. Paumier, T. J. Collier, J. Comp.
Neurol. 515, 102 (2009).11. Y. Wu et al., J. Neurochem. 109, 1157 (2009).12. B. Zhu et al., Stem Cells 30, 2571 (2012).13. W. Dauer, S. Przedborski, Neuron 39, 889 (2003).14. D. S. Auld, T. J. Kornecook, S. Bastianetto, R. Quirion, Prog. Neurobiol. 68, 209 (2002).15. M. Caiazzo et al., Nature 476, 224 (2011).16. U. Pfisterer et al., Proc. Natl. Acad. Sci. U.S.A. 108, 10343 (2011).17. M. L. Liu et al., Nat. Commun. 4, 2183 (2013).18. C. Tian et al., Curr. Mol. Med. 12, 126 (2012).19. E. Lujan, S. Chanda, H. Ahlenius, T. C. Sudhof, M. Wernig, Proc. Natl. Acad. Sci. U.S.A. 109, 2527 (2012).20. M. Thier et al., Cell Stem Cell 10, 473 (2012).21. D. W. Han et al., Cell Stem Cell 10, 465 (2012).22. K. L. Ring et al., Cell Stem Cell 11, 100 (2012).23. G. Su et al., Biomaterials 34, 5897 (2013).24. O. Torper et al., Proc. Natl. Acad. Sci. U.S.A. 110, 7038 (2013).
Acknowledgments: This work was supported in part by research grants from the National Institutes of Health (R01 NS 41858-01 and R01 NS 061642-01), the State of Nebraska (DHHS-LB606 Stem Cell 2009-10 to J.Z. and LB606 Stem Cell 2010-10 to S.D. and C.T.), the China National Science and Technology major project (2014CB965001), and the National Natural Science Foundation of China (81028007 and 81329002 to J.Z., and 81271419 to C.T.).
Parkinson’s disease (PD), the second most common neurode-generative disease, is characterized by progressive loss of do-paminergic neurons in the substantia nigra pars compacta and formation of intracytoplasmic Lewy inclusion bodies (1). Its car-dinal clinical features are resting tremor, rigidity, bradykinesia, and postural instability. The most effective approach currently for managing PD patients is utilizing a dopamine replacement strategy. Even though they provide considerable benefit during the early stages of the disease, these treatments exhibit draw-backs with long-term use. Most critically, they are thought to be only symptomatically effective, not disease-modifying. We have therefore explored four avenues to provide favorable changes in dopaminergic cell loss and neurologic symptoms in PD mod-els: traditional Chinese herbs, small peptides, synthesized com-pounds, and stem cell therapies. Results have been encourag-ing, showing neuroprotective effects in vitro and in vivo through a variety of different molecular pathways. It is hoped that these agents may provide promising new drug candidates for PD inter-vention by blocking the ongoing degeneration of dopaminergic neurons and slowing the disease progression.
TrADiTionAl herb-bAseD TreATmenTs
1. CurCumin Curcumin, isolated from the roots of the turmeric plant (Curcumalonga), is a natural polyphenol and the primary active component of turmeric, a widely used spice and the major constituent of curry powder in South and Southeast Asian cuisine, particularly in Indi-an. Over the past several years, curcumin has been extensively studied for its neuroprotective effects and therapeutic potential in both in vitro and in vivo models of PD. Our laboratory and other investigators have demonstrated that curcumin can modulate multiple processes, including antioxidation, antiapoptosis, anti-neuroinflammation, and antiaggregation of proteins (2).
InVitroStudiesThe antioxidant and anti-inflammatory effects of curcumin in 6-hydroxydopamine (6-OHDA)-treated MES23.5 cells have
been reported (3). Curcumin counteracts oxidative stress by not only downregulating reactive oxygen species (ROS), but also by upregulating the antioxidant enzyme superoxide dismutase (SOD). It alleviates inflammation and mitochondrial dynamic dysfunction by suppressing the activation and nuclear trans-location of nuclear factor кB (NF-кB) or inhibiting extracellular signal-regulated kinase (ERK) (4). Moreover, Chen etal. demon-strated that curcumin possesses antiapoptotic properties, reduc-ing 1-methyl-4-phenylpyridinium ions (MPP+)-induced PC12 cell death mediated via the Bcl-2/mitochondrial/ROS/iNOS (inducible nitric oxide synthase) pathways (5). Our study demonstrated the neuroprotective effects of curcumin in SH-SY5Y cells exposed to α-synuclein and its efficient inhibition of α-synuclein fibril ac-cumulation through downregulation of mTOR/p70S6K signaling, which is a classical suppressive pathway of autophagy and re-covery of macroautophagy (6).
InVivoStudiesSome studies have evaluated the neuroprotective value of cur-cumin against both 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)- and 6-OHDA-induced dopaminergic degeneration in rodents. Recent experiments indicated that intraperitoneally ad-ministered curcumin could protect dopaminergic neurons from apoptosis in a mouse model of PD (7). Our studies showed that curcumin inhibited MPTP-induced hyperphosphorylation of c-Jun N terminal Kinase (JNK), especially JNK3, and its downstream molecules, which contributed to the prevention of mitochondria-mediated apoptosis (8). In addition, our laboratory has provided evidence that a reduction in ROS levels and the inhibition of mi-croglial activation play important roles in curcumin’s neuropro-tective effect in MPTP-treated PD mice (9). Furthermore, others have demonstrated that intravenous and oral curcumin exhibited protective effects against dopaminergic neurotoxicity induced by MPTP- or 6-OHDA in mice (10,11). Dietary supplementation with curcumin could protect dopaminergic neurons against MPTP-in-duced neurodegeneration through inhibition of protein nitration, elevation of glutathione levels, and by boosting mitochondrial complex I activity (12). Recent studies showed that curcumin im-proved motor function in a mouse model of PD (13).
2. sAliDrosiDe Salidroside, a remedy used in Tibet, is a phenylpropanoid glyco-side isolated from RhodiolaroseaL. A recent study indicated that
Department of Neurology & Institute of Neurology, Rui Jin Hospital affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China*Corresponding Author: [email protected]
Potential Neuroprotective Therapies for Parkinson’s Disease
Jun Liu and Sheng-Di Chen*

Produced by the Science/AAAS Custom Publishing Office
54
THERAPEUTIC STRATEGIES
55
salidroside played antioxidant and antiapoptotic roles, protect-ing PC12 cells in culture against MPP+ toxicity by inhibiting the nitric oxide pathway (14). Our work has shown that salidroside could partially block the loss of dopaminergic neurons induced by MPTP in a PD mouse model. The mechanism of action in-volved activation of the PI3K/Akt and PI3K/protein kinase B signaling pathways (15) and may be related to the elevated se-cretion of endogenous glial cell line-derived neurotrophic factor (GDNF) (16).
3. TripChloroliDe Tripchlorolide (TW397), an analogue of triptolide (T10) and one of the active ingredients from TripterygiumwilfordiiHookF.—a traditional Chinese herb widely used in the amelioration of a host of inflammatory and autoimmune diseases—possesses potent anti-inflammatory and immunosuppressive properties (17). Li etal. reported that TW397 exerted a neuroprotective effect against dopaminergic lesions and restored their function after neurotox-icity induced by MPP+ in vitro (18). Our own study demonstrated a neuroprotective action of TW397 in vivo in the MPTP-lesioned PD mouse model by suppression of the astroglial response with-in the striatum. We found that intraperitoneal injection of TW397 not only dramatically attenuated the death of tyrosine hydrox-ylase-immunoreactive (TH-IR) neurons in the substantia nigra pars compacta and TH-IR fibers in the striatum after MPTP treat-ment, but also significantly improved the level of dopamine in the substantia nigra and striatum. In addition, intriguingly, in MPTP-treated animals the locomotive performance of those treated with TW397 improved significantly (19).
ArTifiCiAl neuroproTeCTive CompounDsOur previous studies have indicated that apoptosis mediated by the JNK signaling pathway plays an important role in animal mod-els with MPTP- or 6-OHDA–induced PD. We have since reported the inhibitory effect of a synthesized compound, K252a, on JNKs activation, and also generated a novel small peptide, Tat-JBD, comprised of the membrane transduction domain (Tat) of human immunodeficiency virus-type 1 (HIV-1) plus the sequence of an 11-amino acid peptide corresponds to the JNK-binding domain (JBD) found in JNK-interacting protein-1 (JIP-1) (20). Tat-JBD interfered with JIP-1-JNK complex formation, suppressed the ac-tivation of JNK induced by MPTP, and inhibited the downstream apoptosis cascade.
Overall, the loss of dopaminergic neurons in the substantia nigra and their axons in the striatum was significantly minimized by administration of Tat-JBD in MPTP-treated mice.
K252a has numerous modes of action, including inhibition of Trk, MLK3, and apoptosis-inducing kinase 1 (ASK1), as well as activation of mitogen-activated protein kinase signaling pathways through its interaction with multiple molecular targets. It has also been shown that K252a protects dopaminergic neurons against
6-OHDA-induced cell death, and partially improved the behav-ioral asymmetry in 6-OHDA-treated rats (20,21). Moreover, we found that the antioxidant N-acetylcysteine and the N-methyl-D-aspartate receptor antagonist ketamine also exerted a preven-tive effect on MPTP-induced loss of dopaminergic neurons by suppressing the nuclear translocation and activation of JNK3, the only neural-specific isoform of JNK (22).
In summary, these findings suggest that Tat-JBD and K252a possess neuroprotective properties following MPTP or 6-OHDA insult through inhibition of the JNK signaling pathway.
sTem Cell TherApy In previous work, we have explored the curative potential of neu-ral stem cells (NSCs) and bone marrow stromal cells (BMSCs) in PD mouse models (23,24). Genetically engineered neural stem cells (NSCs) have been found to be a promising vector for deliv-ery of neurotrophic factors, which has been used to treat various nervous system disorders (25). We genetically engineered NSCs to delivery neurturin (NTN), a GDNF family member, into the brains of a rat model of PD and demonstrated that it promoted the survival of embryonic dopaminergic neurons. These special-ized NSCs not only engrafted and integrated in the host striatum successfully, but also differentiated into neurons, astrocytes, and oligodendrocytes with stable, high-level NTN expression. Our study revealed that NTN expression decreased the loss of do-paminergic neurons in the substantia nigra and improved motor symptoms for at least four months after the NSC graft. These findings suggested that transplantation of NTN-secreting NSCs exerted a protective effect in a PD rat model (23).
BMSCs, non-hemopoietic stem cells found in adult mammals or in human bone marrow, possess several advantages over stem cells—abundant availability, and ease of preparation, iso-lation, purification, and culture—as well as trans-differentiation properties that allow BMSCs to differentiate into various types of cells under certain conditions, including neuron- and glia-like cells (26,27), providing a new and reliable source for cell sub-stitution and gene transfection. One study revealed that differ-entiated BMSCs contributed to higher dopamine levels in the le-sioned striatum and significant behavioral improvement over five months (28). BMSCs containing NTN also significantly attenuat-ed behavioral symptoms in 6-OHDA–lesioned rats. Interestingly, in contrast to NSCs transfected with NTN genes, the mechanism of action of NTN-expressing BMSCs appeared to involve func-tional enhancement of residual dopaminergic neurons by NTN, rather than an increase in the numbers of existing dopaminergic neurons (29). These results may help us to further evaluate the neuroprotective value of BMSC transplantation in PD treatment.
Moreover, it is well known that regulation of the immune re-sponse after grafting is a critical issue in PD transplantation (30). Our work found that NSCs transfected with the interleu-kin-10 gene, an important regulator of the immune system,
could downregulate intracerebral cellular and humoral immune responses in 6-OHDA-lesioned PD rats (31). Furthermore, sel-enite—the source of the essential micronutrient selenium—in-traperitoneally injected into 6-OHDA–lesioned rats before the transplantation of DA neurons derived from embryonic stem cells, could inhibit the expression of proinflammatory factors such as TNFα and iNOS. It also improved the survival of im-planted neurons and the rotational behavior in the recipient animals (32).
In summary, the agents above have been shown to be neuropro-tective in both in vitro and in vivo experiments, and may slow down disease progression and provide new therapeutic approaches for PD. However, some challenges still lie ahead. More precise knowledge is needed of the signaling pathways responsible for neuronal death and the mechanisms underlying the protective effects. More critically, the effectiveness of these potential thera-pies will need to be confirmed in large scale clinical trials with PD patients.
REFERENCES 1. K. Jellinger, Movement Disord. 2, 124 (1987). 2. A. Jitoe-Masuda, A. Fujimoto, T. Masuda, Curr. Pharm. Des. 19, 2084 (2013). 3. J. Wang, X. X. Du, H. Jiang, J. X. Xie, Biochem. Pharmacol. 78, 178 (2009). 4. Y. X. Gui et al., Neurobiol. Aging 33, 2841 (2012). 5. J. Chen et al., Apoptosis 11, 943 (2006). 6. T. F. Jiang et al., J. Neuroimmune Pharmacol. 8, 356 (2013). 7. Z. Mansouri, M. Sabetkasaei, F. Moradi, F. Masoudnia, A. Ataie, J. Mol. Neurosci. 47, 234 (2012). 8. J. Pan et al., Translational Neurodegeneration 1, 16 (2012). 9. J. Pan, J. Q. Ding, S. D. Chen, Chin. J. Contemp. Neurol. Neurosurg. 7, 421 (2007).10. O. Vajragupta et al., Free Radic. Biol. Med. 35, 1632 (2003).
11. V. Zbarsky et al., Free Radic. Res. 39, 1119 (2005).12. R. B. Mythri, J. Veena, G. Harish, R. B. S. Shankaranarayana, B. M. M. Srinivas, Br. J. Nutr. 106, 63 (2011).13. W. Tripanichkul, E. O. Jaroensuppaperch, Int. J. Neurosci. 122, 263 (2012).14. X. Li et al., Brain Res. 1382, 9 (2011).15. Y. H. Zhang et al., J. Clin. Neurol. 21, 133 (2008). 16. Y. H. Zhang et al., Chin. J. Neurol. 39, 540 (2006). 17. W. Z. Gu, S. R. Brandwein, Int. J. Immunopharmacol. 20, 389 (1998).18. F. Q. Li et al., Exp. Neurol. 179, 28 (2003).19. Z. Hong et al., Eur. J. Neurosci. 26,1500 (2007).20. J. Pan et al., Lab. Invest. 90, 156 (2010).21. J. Pan et al., Mol. Pharmacol. 72, 1607 (2007). 22. J. Pan et al., Neurochem. Int. 54, 418 (2009).23. W. G. Liu, G. Q. Lu, B. Li, S. D. Chen, Parkinsonism Relat. Disord. 13, 77 (2007).24. M. Ye et al., Parkinsonism Relat. Disord. 13, 44 (2007).25. C. Chen, Y. Wang, G. Y. Yang, Curr. Drug Targets 14, 81 (2013).26. B. A. Horger et al., J. Neurosci. 18, 4929 (1998).27. J. Sanchez-Ramos, F. Cardozo-Pelaez, S. Song, Mov. Disord. 13S, 122 (1998).28. J. Sanchez-Ramos et al., Exp. Neurol. 164, 247 (2000).29. M. Ye et al., Brain Res. 1142, 206 (2007).30. K. E. Soderstrom et al., Neurobiol. Dis. 32, 229 (2008). 31. X. J. Wang, W. G. Liu, Y. H. Zhang, G. Q. Lu, S. D. Chen, Neurosci. Lett. 423, 95 (2007). 32. L. P. Tian et al., Curr. Mol. Med. 12, 1005 (2012).
Acknowledgments: These studies were supported by the National Program of Basic Research of China (2010CB945200 and 2011CB504104), the Natural Science Fund (30971031, 81129018, and 81371407), the Shanghai Key Project of Basic Science Research (10411954500), and the Program for Outstanding Medical Academic Leader (LJ 06003).

Produced by the Science/AAAS Custom Publishing Office
56
THERAPEUTIC STRATEGIES
57
inTroDuCTion Parkinson’s disease (PD) is a common neurodegenerative disor-der of the central nervous system (CNS), found particularly in the elderly and affecting approximately two million people in China (1,2). It is characterized by the loss of dopaminergic (DAergic) neurons in the substantia nigra of the midbrain and the subse-quent decrease in dopamine (DA) levels in the striatum. The imbalance in the substantia nigra-striatum basal ganglia neural signaling resulting from DA insufficiency can be corrected with DA replacement treatments such as levodopa (L-dopa). Howev-er, severe side effects result from long-term administration. The complicated pathogenesis of PD makes the development of rem-edies difficult, driving investigators to continually look for novel treatments. China is fortunate to be able to draw on abundant traditional Chinese medicine (TCM) resources, including herbal medicines and acupuncture, which are increasingly being recog-nized as providing potentially effective treatments. Our research into natural extracts and electroacupuncture (EA) targeting PD pathogenesis have shown therapeutic potency. However, their mechanisms of action are not fully understood, creating a bar-rier to the popular acceptance of TCM treatments. For this rea-son, we investigated the therapeutic mechanisms and efficacy of these therapies.
nATurAl TrADiTionAl Chinese meDiCine exTrACTsDiverse factors are involved in the loss of DAergic neurons in PD, among which inflammation and oxidative stress play criti-cal roles. They interact with each other to activate and drive the course of neuronal death. Three natural TCM extracts have been discovered that target these pathways: triptolide, fucoidan, and tenuigenin (Chinese patents ZL00107779.1, ZL200710099008.8, and ZL 201010200579.8, respectively). The therapeutic and neu-roprotective effects of these compounds have been confirmed in vitro and in rodent PD models, including an oxidative stress model induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
Exploring Therapeutic Mechanisms of Traditional Chinese Medicine Extracts and Electroacupuncture in Parkinson’s Disease
Yan Zheng, Jun Jia, and Xiao-Min Wang*
(MPTP)/1-methyl-4-phenylpyridinium ion (MPP+) or 6-hydroxy-dopamine (6-OHDA), and an inflammation model simulated by microglia activation using lipopolysaccharides (LPS).
TripToliDeTriptolide (T10) is the major active component extracted from Tripterygiumwilfordii Hook. F. (TWHF) and has been used to treat inflammatory diseases for centuries in China. Our data showed for the first time that T10 and tripchlorolide, an analogue of T10, prevented the death of DAergic neurons in neurotoxic le-sions induced by MPP+ or LPS (3,4). In primary microglial cul-tures, T10 significantly suppressed the activation of microglia and the release of interleukin-1b (IL-1b), tumor necrosis factor α (TNFα), and nitric oxide (NO) from LPS-activated microglia (5), indicating that T10 exhibited neuroprotective effects by re-ducing inflammation. As expected, in LPS- or MPP+-induced rodent PD models, the protective effect of T10 on DAergic neu-rons and the inhibition of microglial activation were detected simultaneously, contributing to behavioral improvement (6, 7). Two parallel signaling pathways may underlie the suppression effect of T10 on microglial activation: T10 inhibited prostaglandin E2 (PGE2) expression by blocking the phosphorylation of the c-jun NH2-terminal kinase (JNK); T10 reduced the transcriptional activity of NF-kB, which in turn down-regulated cyclooxygen-ase-2 (COX2) expression and PGE2 release (8). In addition to impacting microglia, T10 promoted the synthesis and release of nerve growth factor (NGF) from primary astrocyte cultures. Surprisingly, it had no effect on brain-derived neurotrophic fac-tor (BDNF) and glial cell-derived neurotrophic factor (GDNF) levels (9). Tripchlorolide, however, promoted BDNF gene ex-pression in astrocytes and stimulated the neurite growth of DAergic neurons (3). This evidence suggests that T10 and its derivatives exert their neuroprotective function in a number of ways, especially through anti-inflammatory and neurotrophic effects (10).
fuCoiDAn AnD TenuigeninFucoidan is a sulfated polysaccharide from edible brown sea-weeds, while Tenuigenin (TEN) is an active compound found in polygalatenuifoliaroot extracts. Our group first showed their value as a potential PD therapies by demonstrating that they appeared to prevent motor deficits, alleviate apomorphine-
Department of Neurobiology, Department of Physiology, Capital Medical University, Key Laboratory for Neurodegenerative Disorders of the Ministry of Education, Beijing Institute for Brain Disorders, Beijing, China*Corresponding Author: [email protected]
induced abnormal rotation, and attenuate DAergic neuron loss and decreases in DA levels in an MPTP-induced mouse model and LPS-induced rat model of PD (11–13). The results indicate that these compounds likely have an anti-inflammatory mode of action in addition to the anti-oxidative effects reported previously. In vitro studies revealed that fucoi-dan blocked NO synthesis (14) and TNFα release (12)—both essential to the inflammatory re-sponse—and, interestingly, also dramatically reduced the produc-tion of reactive oxygen species (ROS) by activated microglia (12), implying that fucoidan might impede the self-propagating cy-cle of neurodegenerative prog-ress in PD since ROS acts as a bridge between inflammation and oxidative stress. As for TEN, it stabilized mitochondrial mem-brane potential and decreased neurotoxic damage through increased antioxidant ex-pression (15). Recently, it has been observed that TEN at-tenuated phosphorylation of α-synuclein, a major com-ponent of Lewy bodies in degenerating DAergic neurons, thereby protecting neurons from toxicity induced by α-synuclein overexpression (16).
Taken together, these promising PD treatments exhibited protective effects through multiple mechanisms including anti-inflammation, anti-oxidation, neurotrophy, and impacting post-translational protein modifications (Fig. 1).
eleCTroACupunCTure for sympTom AlleviATion in pArkinson’s DiseAseAcupuncture is an ancient TCM technology that has been ap-plied to alleviate the symptoms of PD patients and improve their quality of life (17,18); however, its therapeutic mode of ac-tion remains unconfirmed. To clarify these issues, electroacu-puncture (EA) was developed by adding a pulsed electric current through the needles in place of manual manipulation (2). One of the big advantages is that the stimulation param-eters—such as frequency, pulse width, intensity, and period of treatment can be controlled precisely. It has been demon-strated that high-frequency EA (100 Hz) can significantly miti-gate motor deficits in a mechanically-damaged (by medial
Figure 1. Possible therapeutic mechanisms of traditional Chinese medicine extracts in Parkinson’s disease. Neu-rotoxins (MPP+/LPS/6-OHDA) activate microglia to release pro-inflammatory factors (PGE2/IL-1b/TNFα) and ROS, and also inhibit neurotrophic activity of astrocytes, both of which result in DAergic neuron degeneration. Some of these neu-rotoxins can also be transported into the cytosol through a dopamine transporter (DAT) located on the membrane of DAe-rgic neurons, initiating oxidative stress. TCM extracts may inhibit neuroinflammation and oxidative stress (1), stimulate the production and release of neurotrophic factors from astrocytes (2), prevent neuronal damage from neurotoxicity (3), and downregulate α-synuclein hyperphosphorylation (4). DA, dopamine; MPP+, 1-methyl-4-phenylpyridinium ion; LPS, lipopolysaccharide; 6-OHDA, 6-hydroxydopamine; PGE2, prostaglandin E2; IL-1b, interleukin-1b; TNFα, tumor necrosis factor α; ROS, reactive oxygen species.
forebrain bundles (MFBs) axotomy) rodent PD model (19–22) and in rodents suffering 6-OHDA- (23, 24) or MPTP-induced damage (25).
neuroproTeCTive effeCTs of eleCTroACupunCTurePrevious investigations to elucidate acupuncture mechanisms of action have hinted at a neuroprotective role in animal mod-els of PD that functions through various pathways in the sub-stantia nigra and striatum (26). Similarly, in rodents suffering MPTP (27, 28), 6-OHDA (24), or MFB insult (19, 21), EA in-hibited the loss of DAergic neurons, possibly attributable to EA-enhanced expression of GDNF (20), BDNF (29), or tyro-sine kinase B (the BDNF receptor) (24, 30). It has been also determined that EA significantly attenuated the activation of microglia, reduced the levels of TNFα and IL-1b mRNA in ven-tral midbrains (20), and limited the upregulation of COX-2 and inducible nitric oxide synthase (iNOS) (28). Additionally, EA ef-fectively inhibited the oxidative stress response and increased the activity of antioxidants in the striatum (27). These results in-dicate that EA could serve as a neuroprotective intervention by inhibiting the inflammatory response and restoring the oxidant- antioxidant balance.

Produced by the Science/AAAS Custom Publishing Office
58
THERAPEUTIC STRATEGIES
59
non-DopAmine ApproACh for TherApeuTiC effeCTs of eleCTroACupunCTureContrary to current thinking that increasing DA is crucial for PD therapy, data from our group and others (31) have shown that the efficacy of EA was independent of DA levels in the striatum (21,22,29,32). This implies that a non-DA–dependent neuroprotec-tive pathway exists. EA was able to modulate neurotransmitter stability and reverse the neurotransmitter imbalance caused by DA depletion. EA impacted many neurotransmitters in basal gan-glia circuitry, including neurotrophic factors (19,29), substance P (21), g-aminobutyric acid (GABA) (22), glutamate, and ace-tylcholine (32), and also affected synaptic plasticity (31). Taken together, these studies suggest that restoration of homeostasis in the basal ganglion circuitry was the predominant therapeutic effect generated by EA.
Genome-wide analysis was used to study the gene expres-sion of those molecules regulated by EA. In a mouse model of PD caused by MPTP, EA significantly shifted gene expression profiles, and comparative analyses suggested that EA improved motor abnormalities by correcting imbalances in the expression of functional gene clusters in both the cortex and striatum (24).
In summary, high-frequency EA stimulation could effectively al-leviate behavioral disorders in PD animal models. Further work to elucidate EA mechanisms will focus on understanding how neuronal network balance can be restored in the basal ganglion circuitry through the application of high throughput assays and functional imaging technologies. TCM extracts and EA are clear-ly promising prospects for PD therapy, even though details of their therapeutic mechanism of action remain to be clarified. It is prudent to acknowledge that they may not completely replace treatments such as L-dopa, but rather may be useful for prolong-ing the effect of current therapies and alleviating the side effects of those treatments.
REFERENCES 1. Z. X. Zhang et al., Lancet 365, 959 (2005). 2. X. Wang et al., Neurochem. Res. 33, 1956 (2008). 3. F. Q. Li et al., Exp. Neurol. 179, 28 (2003).
4. F. Q. Li et al., J. Neuroimmunol. 148, 24 (2004). 5. H. F. Zhou et al., NeuroReport 14, 1091 (2003). 6. H. F. Zhou et al., Neurobiol. Dis. 18, 441 (2005). 7. J. P. Gao, S. Sun, W. W. Li, Y. P. Chen, D. F. Cai, Neurosci. Bull. 24, 133 (2008). 8. Y. Gong, B. Xue, J. Jiao, L. Jing, X. Wang, J. Neurochem. 107, 779 (2008). 9. B. Xue et al., Neurochem. Res. 32, 1113 (2007).10. Y. Zheng, W. J. Zhang, X. M. Wang, CNS Neurosci. Ther. 19, 76 (2013).11. D. Luo et al., Eur. J. Pharmacol. 617, 33 (2009).12. Y. Q. Cui, Y. J. Jia, T. Zhang, Q. B. Zhang, X. M. Wang, CNS Neurosci. Ther. 18, 827 (2012).13. H. L. Yuan et al., CNS Neurosci. Ther. 18, 584 (2012).14. Y. Q. Cui et al., Clin. Exp. Pharmacol. Physiol. 37, 422 (2010).15. Z. Liang et al., Neurosci. Lett. 497, 104 (2011).16. J. X. Zhou et al., CNS Neurosci. Ther. 19, 688 (2013).17. A. A. Rabinstein, L. M. Shulman, Neurologist 9, 137 (2003).18. L. M. Shulman et al., Mov. Disord. 17, 799 (2002).19. X. B. Liang et al., NeuroReport 14, 1177 (2003).20. X. Y. Liu et al., Exp. Neurol. 189, 189 (2004).21. J. Jia et al., Behav. Brain Res. 205, 214 (2009).22. J. Jia et al., Behav. Neurosci. 124, 305 (2010).23. H. J. Park et al., Exp. Neurol. 180, 93 (2003).24. L. R. Huo et al., Evid. Based Complement Alternat. Med. 2012, 908439 (2012).25. H. Wang et al., PLOS ONE 8, e64403 (2013).26. T. H. Joh, H. J. Park, S. N. Kim, H. Lee, Neurol. Res. 32, 5 (2010).27. H. Wang et al., PLOS ONE 6, e19790 (2011).28. J. M. Kang et al., Brain Res. 1131, 211 (2007).29. X. B. Liang et al., Mol. Brain Res. 108, 51 (2002).30. Y. K. Kim et al., Neurosci. Lett. 384, 133 (2005).31. S. N. Kim et al., PLOS ONE 6, e27566 (2011).32. Z. L. Sun et al., Neurosci. Lett. 520, 32 (2012).
Acknowledgments: This work was supported by the National Basic Research “973” Program (2006CB500700 and 2011CB504100) and the National Natural Science Foundation of China (81030062 and 81072858).
Novel Therapeutic Strategies for Amyotrophic Lateral Sclerosis
Weidong Le1* and Xiaojie Zhang2
Amyotrophic lateral sclerosis (ALS) is a devastating, adult-onset neuro-degenerative disease. The patho-logical hallmark of ALS is the selec-tive death of motor neurons (MNs) in the spinal cord and brain (1,2). The only drug approved by the U.S. Food and Drug Administration (FDA) for ALS treatment is riluzole, but it has limited impact on disease symptoms and patients survival (3). Although the underlying causes of ALS have not been fully defined, progress has been made in recent years, which has led to the development of several neuro-protective agents and potential thera-peutic strategies for treatment. Our recent work has shown that selective degeneration of MNs is usually asso-ciated with impairment of the autoph-agy pathway, which appears to play a critical role in the pathogenic process leading to neuron degeneration (4,5). We have also reported on a range of biological effects using mammalian target of rapamycin (mTOR)-depen-dent and independent autophagic activators in ALS mice (4, 5). Fur-thermore, we and others have found that ALS patients have higher levels of homocysteine (Hcy) in their blood and spinal fluid, suggesting that Hcy may be involved in the pathogenesis of ALS and that Hcy-lowering treat-ments might have therapeutic po-tential (6,7). We present hereby our findings of autophagy and Hcy in ALS (summarized in Fig. 1).
11st Affiliated Hospital, Dalian Medical University, Dalian, China2Ruijin Hospital, Shanghai 2nd Medical University, Shanghai, China*Corresponding Author: [email protected]
AuTophAgy As TherApeuTiC TArgeT for AlsProtein aggregation and inclusion body formation have been rec-ognized as common cellular and molecular processes in many neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and ALS (8). Postmortem neuropathological studies have revealed the increased number of autophagosomes (AVs) in MNs of sporadic ALS (SALS) and familial ALS (FALS) patients (4, 9). In mammals, the predominant route for the degradation of long-
Figure 1. Novel findings and possible mechanisms of action of autophagy and Hcy in ALS. Small molecule autophagy enhancers induce autophagy activation via mTOR-dependent or independent pathways in motor neurons (MNs). If the autophagic flux is intact, the activated autophagy shows neuroprotective effects by enhancing the degradation of toxic protein aggregates. However, if the autophagic flux is defective, the enhanced autophagy might be harmful to the MNs due to accumulated toxic protein aggregates. Rescue of the impaired autophagic flux can induce protein degradation and improve MNs survival. Low levels of folic acid and 5-methyltetrahydrofolate (5-MTHF), as well as other unknown factors, may contribute to elevated homocysteine (Hcy) levels in ALS. Hcy-lowering therapies such as folic acid, B12, or GOS-rich prebiotic yogurt show neuroprotective effects mediated by lowering Hcy in ALS mice.

Produced by the Science/AAAS Custom Publishing Office
60
THERAPEUTIC STRATEGIES
61
lived proteins, aggregated proteins, and injured organelles is through the autophagosome-lysosome pathway. We investigat-ed the activation status of autophagy in transgenic SOD1-G93A mice (hereafter referred to as ALS mice, which carry a G93A mutation in the superoxide dismutase 1 gene), demonstrating for the first time that the level of autophagy marker microtubule-associated protein 1 light chain 3-II (LC3-II) is markedly and spe-cifically increased in the spinal cord of these mice (4,5). Electron microscopy and immunochemistry studies have shown that AVs are significantly accumulated in the dystrophic axons of spinal cord MNs in ALS mice. These changes appeared by the pre-symptomatic stage (90 days) and became more severe at the end stage of the disease (120 days) (5).
Several previous studies suggest that the activation of autoph-agy may be neuroprotective in neurodegenerative diseases by enhancing the removal of toxic protein aggregates (10). Specific ablation of autophagy-related genes Atg 5 or Atg 7 in mouse brain results in a neurodegenerative phenotype and the forma-tion of protein aggregates (11). Recent findings indicate that increased autophagy can protect Caenorhabditis elegans mo-tor neurons against the toxicity of mutant SOD1 (12). Based on these findings, we propose that autophagy is a potential target for the development of antineurotoxicity treatments for ALS. The classical pathway for autophagy control is through the serine/threonine kinase, mTOR, which is a negative regulator of the pro-cess (13). Several pathways are thought to be involved, includ-ing insulin/growth factor pathways, the Ca2+-related pathway, as well as other as-yet unknown pathways, all classified as mTOR-independent pathways.
Rapamycin is a widely used mTOR-dependent autophagic en-hancer that shows neuroprotective effects in HD and PD models through autophagy-mediated protein degradation (14). Our own studies have demonstrated that rapamycin unexpectedly accel-erates disease progression in ALS mice, causing accumulation of AVs, but fails to reduce the level of mutant SOD1 protein ag-gregates even though these are a known target of autophagy-mediated degradation (5). In addition, rapamycin treatment re-sults in severe mitochondrial impairment, higher Bax levels, and increased caspase-3 activation in ALS mice (14). Another study reported that rapamycin had no effect on the disease progres-sion in SOD1-H46R/H48Q mutant mice (15). Since the mTOR pathway touches a broad variety of important cellular processes including cell growth, mRNA translation, metabolism, and in-flammation (13), rapamycin may not be a safe drug to treat ALS patients. We therefore chose to investigate mTOR independent autophagic inducers in ALS.
It is known that autophagy can be induced by lowering intracel-lular inositol 1,4,5-trisphosphate (IP3) levels. IP3-lowering agents, such as lithium, carbamazepine, and sodium valproate, have been demonstrated to induce autophagy in neurons (14). There are contradictory data on lithium treatment in the disease pro-
gression of ALS (16,17). Lithium has been reported to increase survival and attenuate the disease progression in patients and mouse models, partially due to induction of autophagy (16). However, using the same strain of ALS mice and similar treat-ment methods, two other independent investigators did not find neuroprotective effects of lithium (17). On the contrary, lithium may cause an earlier disease onset and a reduction of survival time in ALS mice (17). Moreover, concerns about the safety of lithium carbonate as a therapy for ALS patients have been raised after the halting of a trail that found serious side effects in its participants (18). The efficacy of other IP3-lowering agents has not been determined.
Trehalose is a non-reducing natural disaccharide with chemical chaperone activity that induces autophagy in an mTOR-indepen-dent pathway by an unknown mechanism. A recent study found that trehalose administration can significantly delay disease on-set and prolong lifespan, and that such effects are associated with activation of autophagy and improvement of autophagic flux in the MNs of ALS mice (unpublished data). These results are consistent with a report from Castillo and colleagues (19).
AuTophAgiC flux DefeCTs AnD AlsAutophagy is a highly dynamic and complex process, hence the term “autophagic flux,” that can be regulated at multiple steps. Early studies have demonstrated the accumulation of AVs in a diverse range of neurodegenerative diseases. However, it is difficult to determine whether this change is the result of induc-tion of autophagy or impairment of autophagic flux (20). It has been reported that the autophagic flux defect observed in ALS could be caused by ALS-related mutant genes such as the dy-nein/dynactin complex, chargedmultivesicularbodyprotein-2B (CHMP2B), and ubiquilin2(21,22). Our previous study showed that rapamycin administration could cause further accumulation of AVs, but failed to reduce the level of mutant SOD1 aggrega-tion in the spinal cords of ALS mice (5); moreover, we found that p62/SQSTM1 accumulates in the spinal cord, indicating a defec-tive autophagy flux (5). These results suggest the possibility that autophagy activity or flux may be impaired in SOD1 mutant ALS mice.
In our present study using the mTOR-independent autophagy activator trehalose, we documented that it can decrease SOD1 and p62 aggregation, and reduce the accumulation of ubiqui-tinated proteins in MNs of ALS mice. Furthermore, electron mi-croscopy revealed that the reduction of protein aggregates by trehalose is associated with improved autophagic flux in ALS mice (unpublished data). These studies highlight the possibil-ity that autophagy flux may be altered in SOD1-mediated ALS, and enhancing autophagy with rapamycin may exaggerate the degeneration of MNs. Instead, the use of multifunctional agents that regulate autophagy at multiple points, or combinatorial therapies to repair the autophagic flux defect and enhance ac-
tivation of the process, may have a more beneficial impact and improve MNs function and survival.
elevATeD homoCysTin (hCy) levels As biomArker for AlsThe identification of ALS-specific biomarkers may provide op-portunities to improve early disease diagnosis, monitor disease progression, and possibly delay time to morbidity in clinical pa-tients. Until now, no biomarkers have been identified that meet the desired criteria for the diagnosis of ALS. Hcy is a sulfhydryl-containing amino acid that is produced by demethylation of me-thionine. High plasma levels of Hcy have been reported to be related to PD, AD, and other neurodegenerative disorders (24,25). Using the ALS mice model, we detected high levels of Hcy at early and late stages (90 and 120 days old) as compared with the age-matched wild-type mice (6,7). Further clinical study has verified that Hcy levels are significantly higher in patients with ALS compared to age- and sex-matched controls (26).
Folic acid and its metabolite, 5-methyltetrahydrofolate (5-MTHF), play a key role in one-carbon metabolism, a process that includes the remethylation of Hcy. We have documented that the levels of folic acid and 5-MTHF are significantly lower in ALS mice (7) and lower folic acid levels have also been reported in ALS patients (26). Most interestingly, the decreased level of 5-MTHF is found at a young ages (30 days), indicating that the transmethylation cycle might be altered very early, leading to greater accumulation of Hcy by the later stages (7). Methylene tetrahydrofolate reductase (MTHFR) catalyzes the conversion of folic acid to 5-MTHF. A cross-sectional study has reported a significant increase in ALS risk among clinical subjects with the C677T mutation in the MTHFR gene (27).
The increase in plasma Hcy levels have been reported in pa-tients with AD, PD, mild cognitive impairment, and ALS, so moni-toring Hcy levels alone is not sufficient to diagnose ALS and track disease progression. However, tracking the changes in folic acid and 5-MTHF might help in early diagnosis, and preliminary evi-dence indicates that early intervention to reduce Hcy levels may modify disease progression and even extend the lifespan of ALS patients.
hCy-lowering TherApy in AlsTo investigate the efficacy of Hcy-lowering therapies, we first tested folic acid and vitamin B12 (B12) in ALS mice to determine their biological effects on Hcy levels and neuroprotection. We found that a combination treatment with both folic acid and B12 can reduce the Hcy levels by up to 69% and, more importantly, this regimen significantly delayed disease onset, prolonged lifes-pan, and enhanced MNs survival in ALS mice (6). A recent study also demonstrated the neuroprotective effects of galactooligo-saccharide (GOS)-rich prebiotic yogurt in ALS mice (28). GOSs are oligomeric, non-digestible carbohydrates that help probiot-
ics in the colon improve the absorption and synthesis of B vita-mins. In ALS mice, administration of GOS-rich prebiotic yogurt increased the levels of folate and B12, and reduced the level of Hcy (28). Furthermore, chronic use of GOS-rich prebiotic yo-gurt attenuated MNs loss, improved mitochondrial activity, and reduced atrophy of myocytes (28). These results suggest that GOS-rich prebiotic yogurt may be an effective adjunct nutritional therapy to lower Hcy in ALS sufferers.
A double-blind clinical trial conducted on ALS patients reported that short-term high-dosage administration of methylcobalamin, a form of vitamin B12, is effective in improving motor action of patients, potentially through the reduction in Hcy (29). A clinical review of 12 studies has confirmed that accumulation of Hcy in-creases the risk and progression of motoneuronal degeneration in ALS patients (30).
While our studies indicate that altered autophagy status and possibly an autophagic flux defect play a role in the pa-thology of ALS in our mouse model, the exact role of au-tophagy in ALS has not yet been fully elucidated. Additional studies are required to more directly manipulate autophagy in ALS model systems and detect the impact in disease pro-gression and pathogenesis. Further, animal and clinical stud-ies have provided growing evidence that elevated Hcy levels impact ALS pathogenesis, but clearly more work is needed to evaluate the efficacy and safety of directly targeting Hcy in ALS patients.
REFERENCES 1. J. A. Rumfeldt, J. R. Lepock, E. M. Meiering, J. Mol. Biol. 385, 278 (2009). 2. P. Pasinelli, R. H. Brown, Nat. Rev. Neurosci. 7, 710 (2006). 3. M. C. Bellingham, CNS Neurosci. Ther. 17, 4 (2011). 4. L. Li, X. Zhang, W. Le, Autophagy 4, 290 (2008). 5. X. Zhang et al., Autophagy 7, 412 (2011). 6. X. Zhang, S. Chen, L. Li, Q. Wang, W. Le, Neuropharmacology 54, 1112 (2008). 7. X. Zhang, S. Chen, L. Li, Q. Wang, W. Le, J. Neurol. Sci. 293, 102 (2010). 8. C. A. Ross, M. A. Poirier, Nat. Med. 10 Suppl, S10 (2004). 9. S. Sasaki, J. Neuropathol. Exp. Neurol. 70, 349 (2011).10. Z. H. Cheung, N. Y. Ip, J. Neurochem. 118, 317 (2011).11. M. Komatsu et al., Nature 441, 880 (2006).12. J. Li, K. X. Huang, W. D. Le, Acta Pharmacol. Sin. 34, 644 (2013).13. D. C. Rubinsztein, J. E. Gestwicki, L. O. Murphy, D. J. Klionsky, Nat. Rev. Drug Discov. 6, 304 (2007).14. S. Sarkar, B. Ravikumar, R. A. Floto, D. C. Rubinsztein, Cell. Death Differ. 16, 46 (2009).15. A. Bhattacharya et al., Neurobiol. Aging 33, 1829 (2012).16. F. Fornai et al., Proc. Natl. Acad. Sci. U.S.A. 105, 2052 (2008).17. A. Gill, J. Kidd, F. Vieira, K. Thompson, S. Perrin, PLOS ONE 4, e6489 (2009).

Produced by the Science/AAAS Custom Publishing Office
62
THERAPEUTIC STRATEGIES
63
Stroke is a leading cause of death, long-term disability, and so-cioeconomic cost worldwide. Thrombolysis is the only therapy for acute ischemic stroke, but its use is limited by a narrow therapeutic window. Although progress has been made in un-derstanding the pathophysiology of stroke, clinically effective neuroprotectants have remained elusive until now. Over the past 30 years, scientists worldwide had conducted many studies on the neuronal death pathways induced by cerebral ischemia, and have developed neuroprotective drugs targeting the key mol-ecules mediating cell death; but thus far, all clinical trials have failed (1). For example, clinical trials using N-methyl-D-aspartate (NMDA) receptor antagonists—developed based on the calcium (Ca2+) overload theory, the dominate theory in ischemic neuronal cell death (2)—failed because of the short therapeutic time win-dow and severe side effects of the drugs. Developing novel neu-roprotective targets with a longer therapeutic window and fewer side effects has become an urgent issue. The survival of neurons after cerebral ischemia is determined not only by the activation of cell death pathways, but also by the robustness of inherent neuronal survival mechanisms. Activating these inherent survival mechanisms is an attractive, novel neuroprotective strategy for stroke. The survival of neurons depends on them experiencing
Novel Neuroprotective Strategy for Stroke— Activating Inherent Neuronal Survival Mechanisms
Ming Chen, Bo-Xing Li, and Tian-Ming Gao*
Department of Neurobiology, Key Laboratory of Neuroplasticity of Guangdong Higher Education Institutes, Southern Medical University, Guangzhou, China*Corresponding Author: [email protected]
physiological levels of electrical activity. This activity-dependent survival depends on L-type Ca2+ channel-mediated Ca2+ influx and the induction of neuronal survival-promoting gene expres-sion (3–5). Here, we review our recent findings on the roles of ion channels, which mediate Ca2+ influx and are required for neuronal survival in ischemic neuronal death, and the protection against ischemic cerebral injury afforded by neuregulin-1 (NRG1), the expression of which is regulated by L-type Ca2+ channel activity (Fig. 1).
open l-Type CA2+ ChAnnels—new neuroproTeCTive sTrATegy wiTh long TherApeuTiC winDowTransient forebrain ischemia induces delayed, selective neuronal death in the CA1 region of the hippocampus. The induction of Ca2+ overload mediated by glutamate receptors is the dominant theory in delayed neuronal death. Our previous work has shown that the increase in L-type Ca2+ current 30 minutes after reperfusion might be one of the influx pathways leading to acute postischemic Ca2+ overload during this early period (6). Maintaining the proper intracellular Ca2+ levels (Ca2+ set point) is the key to the survival of neurons; too high or too low Ca2+ levels is not conducive to neuronal survival. Studies have reported that although intracellular Ca2+ concentration increases in CA1 pyramidal neurons during the period of ischemia and early reperfusion (30 minutes), at the late stage of reperfusion the resting Ca2+ levels are below normal in hippocampal CA1 neurons. We therefore hypothesize that Ca2+ overload at the time of ischemia and immediately after reperfusion is only a trigger,
but the later persistent lack of Ca2+ may be one of the executors of delayed neuronal death. Using patch-clamp techniques, we found that in vulnerable CA1 neurons, L-type Ca2+ channel activity was transiently upregulated and then persistently downregulated after ischemic insult, whereas in invulnerable CA3 neurons, no such change occurred. The lack of change in protein expression levels of the L-type Ca2+ channel suggests that only functional changes occurred after ischemia (7). Inhibition of the L-type, but not the N-type or the P/Q-type Ca2+ channels, significantly suppressed survival in hippocampal neurons in culture. Treatment with a L-type Ca2+ channel agonist after 1 to 12 hours of reoxygenation or as late as 24 hours after reperfusion significantly reduced cell death induced by oxygen/glucose deprivation (OGD) and ischemia-reperfusion injury in an animal model (7,8). This neuroprotection was mediated by the extracellular-signal-regulated kinase (ERK) pathway (8). These results suggest that the later, persistent L-type Ca2+ channel hypoactivity may be responsible for the delayed neuronal death. These findings also develop and correct the traditional Ca2+ overload theory and suggest a novel neuroprotective target with a long therapeutic time window, namely the L-type Ca2+ channel.
We further elucidated the mechanisms underlying the ac-tivity changes of the L-type Ca2+ channel after ischemia. Nitric oxide is an important signaling molecule mediat-ing ischemic neuronal death (9–11). At the early stage of ischemia, elevated NO production enhances hippocampal L-type Ca2+ channels by S-nitrosylation during the initial phase of hypoxia and OGD (10,11). During late stage of reperfusion, oxidation modulation may be responsible for the L-type Ca2+channel hypoactivity (7). These results sug-gest a new direction for drug design based on the disease state, such as an effective agonist of the L-type Ca2+ chan-nel in its oxidative state.
bloCking lArge ConDuCTAnCe CA2+-ACTivATeD poTAssium ChAnnels—AlTernATive new sTrATegy for neuroproTeCTionIn addition to regulation by L-type Ca2+ channels in the plasma membrane, Ca2+ influx is also regulated by membrane excitabil-ity; a reduction in membrane excitability can inhibit L-type Ca2+ channel activation. By comparing the membrane properties of ischemia-vulnerable CA1 pyramidal neurons and ischemic-invul-nerable CA3 pyramidal neurons after mild and severe ischemia with intracellular recording and staining techniques in vivo, we found that only after severe ischemia did CA1 pyramidal neurons show persistently decreased membrane excitability and a sus-tained increase in the magnitude of hyperpolarization mediated by large conductance Ca2+-activated potassium (BKCa) channels.
By contrast, in ischemic-invulnerable neurons, no changes were detected (12,13). Therefore, the hyperactivity of BKCa channels might persistently decrease membrane excitability and thereby inhibit L-type Ca2+ channel activation, leading to ischemic neuro-nal death. This notion is supported by studies showing that BKCa channel activity increased significantly in cultured hippocampal neurons after hypoxia/reoxygenation and in CA1 neurons iso-lated from animals after forebrain ischemia/reperfusion (14,15). Administration of a BKCa channel-specific opener, NS1619, or in vitro overexpression of BKCa channel genes induced apoptosis in hippocampal neuronal cultures (15). Specific inhibition of BKCa
Figure 1. (A) Pathways of neuronal death following brain ischemia/reperfusion. Calcium (Ca2+) overload occurs at early stage of reperfusion and may trigger oxidative modifi-cations of ion channels. Later, persistent L-type Ca2+channel hypoactivity may be re-sponsible for delayed neuronal death. In addition, BKCa channel hyperactivity decreased membrane excitability, thereby further inhibiting L-type Ca2+ channel function. (B) Novel neuroprotective strategies for stroke by activation of inherent neuronal survival mecha-nisms. Activation of L-type Ca2+ channels, blockade of BKCa channels, and enhance-ment of ErbB4 signaling are all candidate protective strategies. NMDAR, N-methyl-D-aspartate receptor; BKCa, channel, large conductance Ca2+-activated potassium channel; ERK, extracellular signal-regulated kinase; NRG1, neuregulin 1; ErbB4, v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4; GABA, γ-aminobutyric acid; K+, po-tassium ion.
18. A. Chio et al., Neurology 75, 619 (2010).19. K. Castillo et al., Autophagy 9, 1308 (2013). 20. E. Wong, A. M. Cuervo, Nat. Neurosci. 13, 805 (2010).21. S. Chen, X. Zhang, L. Song, W. Le, Brain Pathol. 22, 110 (2012).22. X. J. Zhang, S. Chen, K. X. Huang, W. D. Le, Acta Pharmacol. Sin. 34, 595 (2013).23. M. Nassif, C. Hetz, Autophagy 7, 450 (2011).24. M. S. Morris, Lancet Neurol. 2, 425 (2003).25. P. Quadri et al., Am. J. Clin. Nutr. 80, 114 (2004).26. S. Zoccolella et al., Neurology 70, 222 (2008).
27. P. Kuhnlein et al., Amyotroph. Lateral. Scler. 12, 136 (2011).28. L. Song, Y. Gao, X. Zhang, W. Le, Neuroscience 246, 281 (2013).29. R. Kaji et al., Muscle Nerve 21, 1775 (1998).30. S. Zoccolella, C. Bendotti, E. Beghi, G. Logroscino, Amyotroph. Lateral. Scler. 11, 140 (2010).
Acknowledgments: This study was funded by research grants from the National Nature Science Foundation of China (81000541and 81171201) and by the National Basic “973” Research Program (2011CB510003).

Produced by the Science/AAAS Custom Publishing Office
64
channels was neuroprotective in neurons subjected to hypoxic/reoxygenation or OGD/reoxygenation, or in animals subjected to forebrain ischemia/reperfusion (15,16). Together, these results indicate that BKCa channel hyperactivity mediates hippocampal neuronal death after ischemia. The mechanism leading to cell death may be due to decreased membrane excitability and inhi-bition of L-type Ca2+ channel function. Therefore, blocking BKCa channels might provide a novel neuroprotective strategy.
We carried out a patch-clamp study to clarify the mechanisms underlying the hyperactivity of BKCa channels after severe isch-emia and reperfusion. Single-channel analysis revealed a sus-tained increase in open probability of BKCa channels in ischemic CA1 pyramidal neurons, predominantly due to the increased Ca2+ sensitivity (17). Redox regulation of BKCa channels was investi-gated by comparing the sensitivity of BKCa channels to oxidants and reductants in normal and ischemic neurons. We found that more BKCa channels were in a reduced state in normal neurons, while in ischemic neurons, more were in their oxidized state. This suggests that oxidative modification may mediate the enhanced BKCa channel activity following cerebral ischemia (17,18).
erbb4, novel TArgeT for neuroproTeCTionNRG1 is a trophic factor that acts by stimulating ErbB receptor kinases. The expression of NRG1 is regulated by L-type Ca2+ channel activity. Using transgenic mice expressing green fluo-rescent protein under the control of glutamic acid decarboxyl-ase promoter (directs specific expression in γ-aminobutyric acid (GABA) interneurons), we found that ErbB4 (v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4) was local-ized in GABAergic presynaptic terminals (19). NRG1 enhanced both the amplitude of evoked inhibitory postsynaptic currents (eIPSCs) and the depolarization-induced release of GABA in a dose-dependent manner (19). Further study showed that NRG1 significantly reduced the paired-pulse ratios (PPRs) of eIPSCs in response to two successive stimuli, indicating that NRG1-en-hanced GABAergic transmission works directly through the pre-synaptic terminals and not via postsynaptic mechanisms (19). To verify that endogenous NRG1 regulates GABA release, we gen-erated ecto-ErbB4, a neutralizing peptide containing the entire extracellular region of ErbB4 that can specifically block NRG1 activation of ErbB kinases. Treatment with ecto-ErbB4 reduced depolarization-induced GABA release and eIPSCs, indicating a role for endogenous NRG1 in regulating these processes (19,20). Two subtypes of ErbB receptors are expressed in neurons, but only ErbB4 plays a role in the regulation of GABAergic trans-mission by NRG1 (19,20). These findings identify a novel func-tion of NRG1. More recently, we found that NRG1 modulation of GABAergic transmission via ErbB4 occurred mainly in parval-bumin-positive interneurons, but not in pyramidal neurons (20).
We further studied whether NRG1 plays a neuroprotective role through ErbB4 and GABAergic pathways in an OGD model. The
results showed that neurons in which ErbB4 had been knocked out were more sensitive to OGD-induced injury, and both ecto-ErbB4 and GABA receptor antagonists could block the neuropro-tective effect of NRG1. There was no synergism in the neuropro-tective effects of NRG1 and GABA receptor agonists, suggesting that the ErbB4 and GABAergic pathways are involved in the protective effects of NRG1 (21). Our results reveal not only a new mechanism underlying NRG1 neuroprotection against ischemic brain injury, but also provide an experimental basis for developing novel targets for neuroprotection such as the ErbB4 receptor.
In summary, activating inherent neuronal survival pathways is a novel neuroprotective strategy. L-type Ca2+ channels, BKCa
channels, and ErbB4 provide potentially exciting targets for treat-ing ischemic stroke.
REFERENCES 1. S. I. Savitz, M. Fisher, Ann. Neurol. 61, 396 (2007). 2. D. W. Choi, Nature 433, 696 (2005). 3. P. L. Greer, M. E. Greenberg, Neuron 59, 846 (2008). 4. S. J. Zhang et al., PLOS Genet. 5, e1000604 (2009). 5. F. Leveille et al., J. Neurosci. 30, 2623 (2010). 6. X. M. Li et al., Prog. Biochem. Biophys. 30, 755 (2003). 7. X. M. Li et al., J. Neurosci. 27, 5249 (2007). 8. H. H. Hu et al., Mol. Neurobiol. 47, 280 (2013). 9. M. Chen et al., NeuroSignals 17, 162 (2009).10. K. Jian et al., Biochem. Biophys. Res. Commun. 359, 481 (2007).11. Y. W. Tjong et al., Free Radic. Biol. Med. 42, 52 (2007).12. T. M. Gao, E. M. Howard, Z. C. Xu, J. Neurophysiol. 80, 2860 (1998).13. T. M. Gao, W. A. Pulsinelli, Z. C. Xu, Neuroscience 90, 771 (1999).14. L. Gong, T. M. Gao, X. Li, H. Huang, Z. Tong, Brain Res. 884, 147 (2000).15. M. Chen et al., Mol. Neurobiol. Epub ahead of print (2013) doi: 10.1007/ s12035-013-8467-x.16. H. Huang, T. M. Gao, L. Gong, Z. Zhuang, X. Li, Neurosci. Lett. 305, 83 (2001).17. L. Gong, T. M. Gao, H. Huang, Z. Tong, Eur. J. Neurosci. 15, 779 (2002).18. L. Gong, T. M. Gao, H. Huang, Z. Tong, Neurosci. Lett. 286, 191 (2000).19. R. S. Woo et al., Neuron 54, 599 (2007).20. Y. J. Chen et al., Proc. Natl. Acad. Sci. U.S.A. 107, 21818 (2010). 21. C. Y. Wu et al., Procedings of the Chinese Society for Neuroscience Ninth National Academic Conference and the Fifth Congress p.135 (2011).
Acknowledgments: This work was supported by grants from the National Natural Science Foundation of China (81030022, 81070983, 81329003, 31371146, and U1201225), the Key Project of Guangdong Province (9351051501000003 and CXZD1018), the Guangzhou Science and Technology Project (7411802013939), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1142).
Produced by the Science/AAAS Custom Publishing Office


















