Page 1
1
Nitric Oxide disrupts H2O2-dependent activation of NF- B:
Role in Sensitization of human tumor cells to TNF- -induced cytotoxicity
Hermes J. Garbán and Benjamin Bonavida*
Department of Microbiology, Immunology and Molecular Genetics, and Jonsson Comprehensive
Cancer Center, UCLA School of Medicine, 10833 Le Conte Ave., Los Angeles CA 90095-1747,
USA.
*To whom proofs and requests for reprints should be addressed: Benjamin Bonavida, Ph.D.,
Department of Microbiology, Immunology, and Molecular Genetics, UCLA School of Medicine,
10833 Le Conte Avenue, Los Angeles, CA 90095-1747. Phone: (310) 825-2233; Fax: (310) 206-
3865; E-mail: [email protected] .
Running title: NO-mediated disruption of H2O2-dependent activation of NF-κB
Key words: nitric oxide, nitric oxide synthase, tumor necrosis factor, NF-kappa B, hydrogen
peroxide, apoptosis, ovarian carcinoma, tumor immunobiology, Chemo- and Immuno-
sensitization, Superoxide, Superoxide dismutase.
Copyright 2000 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on December 15, 2000 as Manuscript M008471200 by guest on February 18, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Page 2
2
Summary
Tumor Necrosis Factor alpha (TNF-α) exerts its effect by two distinct signaling
pathways. It can trigger cytotoxicity in sensitive target cells. TNF-α can also promote NF-κB
activity and regulate the expression of genes that interfere with apoptosis and thus conferring
resistance to several apoptotic stimuli. We have observed that interferon-γ (IFN-γ) sensitizes
human ovarian carcinoma cell lines to TNF-α-mediated apoptosis and further, IFN-γ induces the
expression of the inducible nitric oxide synthase (iNOS) and the generation of nitric oxide (NO).
This study examines the role of NO in the sensitization of the ovarian carcinoma cell line AD10
to TNF-α-mediated cytotoxicity. Treatment of AD10 cells with the NOS inhibitor L-NMA
blocked the IFN-γ-dependent sensitization whereas NO donors (SNAP) sensitized these cells to
TNF-α cytotoxicity. Analysis of the activation status of NF-κB upon treatment with NO donors
confirmed the inhibitory role of NO on both the NF-κB DNA-binding property and its activation.
Moreover, the inhibition of NF-κB nuclear translocation by NO donors directly correlated with
the intracellular concentration of H2O2 and was reversed by the addition of exogenous H2O2.
These findings show that NO might interfere with TNF-α-dependent NF-κB activation by
interacting with O2- and reducing the generation of H2O2, a potent NF-κB activator. Therefore,
NO-mediated disruption of NF-κB activation results in the removal of anti-apoptotic/resistance
signals and sensitizes tumor cells to cytotoxic cytokines like TNF-α.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 3
3
Introduction
The development of resistance to either the immune system or chemo-immunotherapeutic
strategies remains a disadvantage in the therapy of cancer, particularly in cases where recurrence
and/or relapses occurred. Apoptosis has been accepted as a distinct pathological mechanism in
tumors responding to anticancer therapies. Further, resistance to apoptosis in tumor cells has
been recognized as a common pathway to multiple drug resistance (1,2). Multiple lines of
evidence have implicated the activation of the transcription factor NF-κB as one of the primary
signals in the onset of resistance to many apoptotic stimuli, particularly TNF-α (3-5).
TNF-α is a proinflammatory cytokine that exerts a broad spectrum of biological effects
by its interaction with two distinct cell surface receptors, TNFR1 and TNFR2 (6). Most cytotoxic
effects of TNF-α are mediated by the TNFR1. It has been demonstrated that upon interaction
with TNF-α, trimerization of TNFR1 takes place and results in cellular signaling leading to the
recruitment of the TNFR1-associated death domain protein (TRADD) and the receptor-
interacting protein (RIP) to the receptor complex (7). TRADD interacts with the Fas-associated
death domain (FADD) to initiate the death pathway and engages several proteins such as the
TNFR-associated factor-1 (TRAF1), the TNFR-associated factor-2 (TRAF-2), and RIP to initiate
the TNF signaling pathways such as the activation of NF-κB (8).
Reactive oxygen species (ROS) have also been implicated in the signaling pathways
initiated by TNF-α. Stimulation of mammalian cells with TNF-α triggers the generation of
various ROS (9,10). Hence, the use of antioxidants results in the inhibition of various TNF-α-
related effects such as the activation of transcription factors, gene expression and cytotoxicity. In
addition, the use of exogenous ROS mimics the biological activity of TNF-α (11). These data
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 4
4
support the hypothesis that ROS function as second messengers for TNF-α-mediated signaling.
In biological systems the most important ROS generated upon TNF-α stimulation are the result
of enzymatic partial reduction of oxygen yielding superoxide (O•2-), which is either immediately
reduced by superoxide dismutase (SOD) to hydrogen peroxide (H2O2) or alternatively reacts
rapidly with nitric oxide (NO) to generate ONOO- (12-14). However, the regulatory role of NO
in TNF-α signaling via the disruption of ROS-dependent activation of NF-κB has not been
established.
Several lines of evidence showed that resistant tumors could be sensitized to TNF-α-
mediated cytotoxicity by various cytokines or pharmacological treatments (15-20). Recently, we
have reported that IFN-γ induced the sensitization of the human ovarian carcinoma AD10 cell
line to Fas-mediated apoptosis and the sensitization was due in part to the generation of nitric
oxide by the induction of iNOS in these cells (21). NO has been identified as a potential second
messenger based on its ability to chemically interact with a broad range of regulatory proteins.
Furthermore, NO can interact with metal clusters- and thiol-containing proteins (for review see
(22)) resulting in the modification of both the structures and functions of these proteins.
Although NO has been shown to react very rapidly with O•2-, the only biological effect to this
chemical reaction has been assigned to the generation of ONOO-, a proposed cytotoxic derivative
(23,24).
Herein, we hypothesize that NO is interfering with the TNF-α-mediated signaling by
chemically reacting with O•2-. Since O•
2- can serve as a precursor to H2O2, which is a proposed
activator of the anti-apoptotic transcription factor NF-κB, the reaction of O•2- with NO will
interfere with the activation of NF-κB and will result in the removal of anti-apoptotic signals and
sensitization of the tumor cells to TNF-α cytotoxicity. This study has been designed to test this
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 5
5
hypothesis and the following have been examined: a) the molecular mechanism by which IFN-γ
sensitizes the human ovarian carcinoma cell line to TNF-α-induced cytotoxicity b) the specific
role of NO in the disruption of TNF-α-mediated generation of H2O2 and subsequently c) the
mechanism by which NO can disrupt the TNF-α-dependent NF-κB activation.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 6
6
Experimental Procedures
Cell culture and reagents
The AD10 cell line is an Adriamycin resistant, MDR phenotype-expressing, subline
derived from the human ovarian carcinoma cell line A2780 and was obtained from Dr. Ozols
(Fox Chase Cancer Center, Philadelphia, PA). The PC-3 cell line is a metastatic bone-derived
human prostatic adenocarcinoma obtained from ATCC: CRL-1435 (American type culture
collection, Manassas. VA.). Cell cultures were maintained as monolayers on plastic dishes in
RPMI-1640 medium (MediaTech, Inc., Herndon, VA.), supplemented with 10% heat-inactivated
FBS (Gemini Bio-Products, Inc., Calabasas, CA), 1% L-glutamine (Life Technologies, Bethesda,
MD), 1% pyruvate (Life Technologies, Bethesda, MD), 1% nonessential amino acids (Life
Technologies, Bethesda, MD) and incubated at 37°C and 5% CO2. For every experimental
condition the cells were cultured in 1% FBS 24 hours prior to treatments. In cases where SNAP
(kindly provided and synthesized by Dr. Jon Fukuto at UCLA) was used, 500 µM of photo-
activated SNAP was added to the cultured cells 2 hours prior to stimulation with cytokines
unless otherwise indicated in the text. For iNOS induction, cultured cells were stimulated 18
hours with 100 U of human recombinant IFN-γ (PeproTech, Inc., Roky Hill, NJ). For Guanylate
Cyclase-related effects, cells were incubated in the presence of the cGMP analogue 8-bromo-
cGMP instead of SNAP or blocked using 300 µM of ODQ (1H-[1,2,4]oxadiazolo- [4,3-
a]quinoxalin-1-one) (Alexis Corporation, San Diego, CA).
Cytotoxicity assay
TNF-α-mediated cytotoxicity was assessed using recombinant TNF-α at the
concentrations of 0.01, 0.1 and 1ng/mL in a 24 h incubation assay. The lactate dehydrogenase
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 7
7
(LDH)-based CytoTox 96™ Assay (Promega, Madison, WI) was used to determine cytotoxicity
(25). Briefly, 1x104 cells/sample, in quadruplicate, were distributed into a 96-well flat-bottom
microtiter plate (Costar, Cambridge, MA) and cultured at low serum concentration (0.1% FBS)
18 hours prior to each treatment. After incubation for each different experimental condition,
released LDH into the culture supernatants was measured with a 30-minute coupled enzymatic
assay, which results in the conversion of a tetrazolium salt (INT) into a red formazan product
that is read at 490nm in an automated plate reader (Emax, Molecular Devices, Sunnyvale, CA).
Percentage cytotoxicity was calculated using the spontaneous release-corrected OD as follows:
% cytotoxicity = (OD of experimental well/OD of maximum release control well) x 100.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR).
Total RNA was extracted and purified from approximately 1x106 cells for each
experimental condition by a single step mono-phasic solution of Phenol and guanidine
isothiocyanate-chloroform using TRIZOL® reagent (Life Technologies, Bethesda, MD). 1 µg of
total RNA was reverse transcribed to first strand cDNA for 1 hour at 42°C with 200 U of
SuperScript™ II reverse transcriptase and 20µM random hexamer primers (Life Technologies,
Bethesda, MD). Amplification of 1/10 of these cDNA by PCR was performed using the
following gene-specific primers: TNF-α (F) (5-'AAG CCT GTA GCC CAT GTT GTA GC-3')
and TNF-α(R) (5-'GAA GAC CCC TCC CAG ATA GAT G-3') [342 bp expected product].
Internal control for equal cDNA loading in each reaction was assessed using the following gene-
specific glyceraldehyde-3-phosphate dehydrogenase (G-3-PDH) primers: G-3-PDH sense (5’-
GAA CAT CAT CCC TGC CTC TAC TG-3’), G-3-PDH antisense (5’-GTT GCT GTA GCC
AAA TTC GTT G-3’) [355 bp expected product]. PCR amplifications of each specific DNA
sequence were carried out using the "Hot Start" method using Platinum Taq™ polymerase (Life
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 8
8
Technologies, Bethesda, MD) followed by a two-steps thermal cycling incubations (95°C/15 sec;
60°C/30 sec for 30 cycles and a final extension at 72°C/10 min). The numbers of PCR cycles
were established based on preliminary titration of the relative amount of amplified product for
each gene representing the linear phase of the amplification process. The amplified products
were resolved on 1.5% agarose gel electrophoresis and their relative concentrations were
assessed by densitometric analysis of the digitized ethidium bromide (EtBr)-stained image,
performed on a Macintosh computer (Apple Computer Inc., Cupertino, CA.) using the public
domain NIH Image program (developed at the U.S. National Institutes of Health and available on
the Internet at http://rsb.info.nih.gov/nih-image/).
Nuclear extracts preparation
1x106 cultured cells treated under different experimental conditions were washed twice
with ice-cold DPBS (MediaTech, Inc., Herndon, VA.). P-40 lysis buffer (10 mM Tris.HCl, pH
7.4; 10 mM NaCl; 3 mM MgCl2; 0.5% NP-40; 0.1 mM EDTA) was added to the top of the
washed cells and incubated on ice for 5 minutes. Lysed cells were collected by gentle pipetting 3
to 4 times and transfer to a microcentrifuge tube. Nuclear pellets for each experimental condition
were generated by two consecutive centrifugation and washing steps at 1200 rpm. Nuclear
pellets were lysed in buffer C (20 mM HEPES pH 7.9; 25 % Glycerol; 0.42 M NaCl; 1.5 mM
MgCl2; 0.1 mM EDTA; 0.5 mM PMSF and 0.5 mM DTT). Total nuclear protein concentrations
were determined using the method of Bradford (26).
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear protein extracts (2 µg) were assayed for DNA interaction by EMSA as described
previously with modifications (27). The double-stranded NF-κB consensus binding sequence (5'-
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 9
9
AGT TGA GGG GAC TTT CCC AGG C-3') oligonucleotide was radiolabeled with [γ32P] ATP
(ICN Pharmaceuticals, Inc. Costa Mesa, CA.) by incubation with 10 U of T4 polynucleotide
kinase (New England Biolabs Inc. Bervely, MA.) and further purified using QIAquick nucleotide
removal kit (QIAGEN Inc., Valencia, CA.). After the DNA-binding reaction, the samples were
resolved on 4-15% Tris-HCl-polyacrylamide minigels (Bio-Rad, Richmond, CA), the gels were
dried and autoradiographed. Specificity of the DNA-binding reaction was determined by
competition assays performed with 100-fold excess of unlabeled NF-κB or unrelated
oligonucleotide (i.e. AP-1 : 5´-GAT CGA ACT GAC CGC CCG CGG CCC GT-3´). The relative
concentrations of specific NF-κB shifted bands were assessed by densitometric analysis of the
digitized autoradiographic images using the NIH Image program described above.
Determination of Intracellular H2O2 generation
1x106 cells were cultured in a 6-well plate for 18 hours in culture medium supplemented
with 1% FBS. In some instances, the minimal serum-cultured cells were treated with 500 µM of
photo-activated SNAP 2 hours prior to stimulation with 10 or 100 U/mL of TNF-α. Intracellular
H2O2 levels were evaluated using the fluorescent cell permeable probe, 2',7'-dichlorofluorescin
diacetate (H2DCFDA) (Molecular Probes, Inc. Eugene, OR). Then, the culture medium was
replaced with DPBS pH 7.4 containing 5 µM H2DCFDA. Fluorescence intensity was analyzed
on an EPICS® XL-MCL flow Cytometer (Beckman Coulter Inc. Fullerton, CA).
Transfections and Reporter gene system
The intracellular activation of NF-κB was determined by transient transfection of AD10
cells with the pNF-κB-d2EGFP reporter vector (CLONTECH laboratories Inc., Palo Alto, CA.).
7x106 cultured cells were transfected with 10 µg of DNA using 60 µL of Lipofectamine reagent
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 10
10
(Life Technologies, Bethesda, MD) according to the manufacturer’s recommendations.
Transfected cells were then distributed onto a 6-well culture plate and incubated under different
experimental conditions. The relative fluorescence intensity was analyzed on an EPICS® XL-
MCL flow Cytometer.
Statistical Analysis
The experimental values were expressed as the mean ± standard error of the mean (SEM)
for the number of separate experiments indicated in each case. One-way analysis of variance was
used to compare variances within groups and among them. Bartlett’s tests were used to establish
the homogeneity of variance on the basis of the differences among standard deviations (SDs).
Whenever needed, post hoc unpaired multiple comparison tests (Bonferroni’s test) and Student’s
t-test were used for comparison between two groups. Significant differences were considered for
those probabilities < 5% (P<0.05).
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 11
11
Results
IFN- -mediated sensitization of the human ovarian carcinoma AD10 to TNF- -induced
cytotoxicity is due, in part, to the generation of nitric oxide.
To investigate the role of nitric oxide on the sensitization of the human ovarian
carcinoma AD10 cell line to TNF-α-mediated cytotoxicity, we first stimulated quiescent AD10
cells with IFN-γ in the presence or absence of 1 mM of the potent NOS inhibitor L-NMA. The
sensitivity of AD10 cells to the cytotoxic effect of increasing concentrations of TNF-α (0.01, 0.1
and 1 ng/mL) was evaluated by the release of LDH into the culture medium after 24 hour of
incubation. Exposure of AD10 cells to IFN-γ (100 U/mL) for 18 hours sensitized the tumor cells
to TNF-α-mediated cytotoxicity and the degree of sensitization increased with increasing
concentrations of TNF-α. Sensitization by IFN-γ was significantly decreased in the presence of 1
mM of the NOS inhibitor L-NMA (Fig. 1A).
In order to confirm the specific role of nitric oxide in the sensitization of AD10 cells, we
assessed the cytotoxic effect of TNF-α in the presence of 10 and 100 µM of the nitric oxide
donor SNAP. After incubation for 18 hours with different concentrations of SNAP, we observed
a significant increase in the sensitivity of AD10 cells to TNF-α-mediated cytotoxicity in a 24 h
assay that directly correlated with the concentrations of SNAP (Fig. 1B).
Similarly, we have found that IFN-γ (100 U/ml) sensitized the prostatic adenocarcinoma
cell line PC3 to TNF-α-mediated cytotoxicity (1 ng/ml) from 5 ± 1.9 % to 37 ± 1.2 % and the
sensitization was blocked by the addition of L-NMA (1 mM) to 20 ± 2.1 %. Like IFN-γ, the use
of the NO donor SNAP (100 µM) sensitized PC-3 cells to TNF-α (1 ng/ml) cytotoxicity from 1.9
± 3 % to 58 ± 5 %.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 12
12
Nitric oxide and PDTC inhibit TNF- -induced expression of endogenous TNF- in AD10
cells.
The transcription factor NF-κ B has been demonstrated to tightly regulate the gene
expression of TNF-α, establishing a self-regulatory loop in tumor cells that secrete TNF-α that
in turn activates NF-κB(28). Furthermore, pyrrolidine-dithiocarbamate (PDTC) has been shown
to inhibit TNF-α-mediated activation of NF-κ B in several cell types and in macrophages (29).
In order to demonstrate the specific effect of nitric oxide on the NF-κ B-mediated expression of
TNF-α, we incubated AD10 cells with 1, 10, 100 and 500 µM of SNAP for 18 hours and then
stimulated the cells with 100 U/mL of TNF-α for 4 hours. The relative levels of endogenously
generated TNF-α were assessed by amplification of the specific TNF-α cDNA using RT-PCR.
The constitutive expression of TNF-α by AD10 cells was demonstrated and a significant
increased level was observed upon treatment with exogenous TNF-α. Moreover, this increased
level of TNF-α was blocked following treatment of the cells with SNAP (500 µM of the nitric
oxide donor) up to the complete disappearance of the amplified TNF-α mRNA (Fig. 2A). These
findings suggest that NO downregulates NF-κB and consequently downregulates TNF-α mRNA
expression. Similar results to those observed with AD10 cells were obtained with the human
prostatic adenocarcinoma cell line PC-3. The expression of TNF-α messenger RNA in PC-3 was
decreased approximately four to five folds upon treatment with 500 µM of SNAP suggesting the
role of nitric oxide in the NF-κB-dependent expression of TNF-α.
To confirm the control of NF-κB on TNF-α expression, we examined the relative levels
of expression of endogenous TNF-α mRNA after treatment of AD10 cells with 1, 10, 100 and
500 µM of PDTC for 18 hours followed with TNF-α (100 U/mL) stimulation for 4 hours.
Endogenous TNF-α gene expression of TNF-α-stimulated cells decreased in the presence of
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 13
13
PDTC but was never completely blocked as was observed above following treatment with SNAP
(Fig. 2B). These results confirm the role of ROS in the activation of the transcription factor NF-
κB and the subsequent expression of TNF-α.
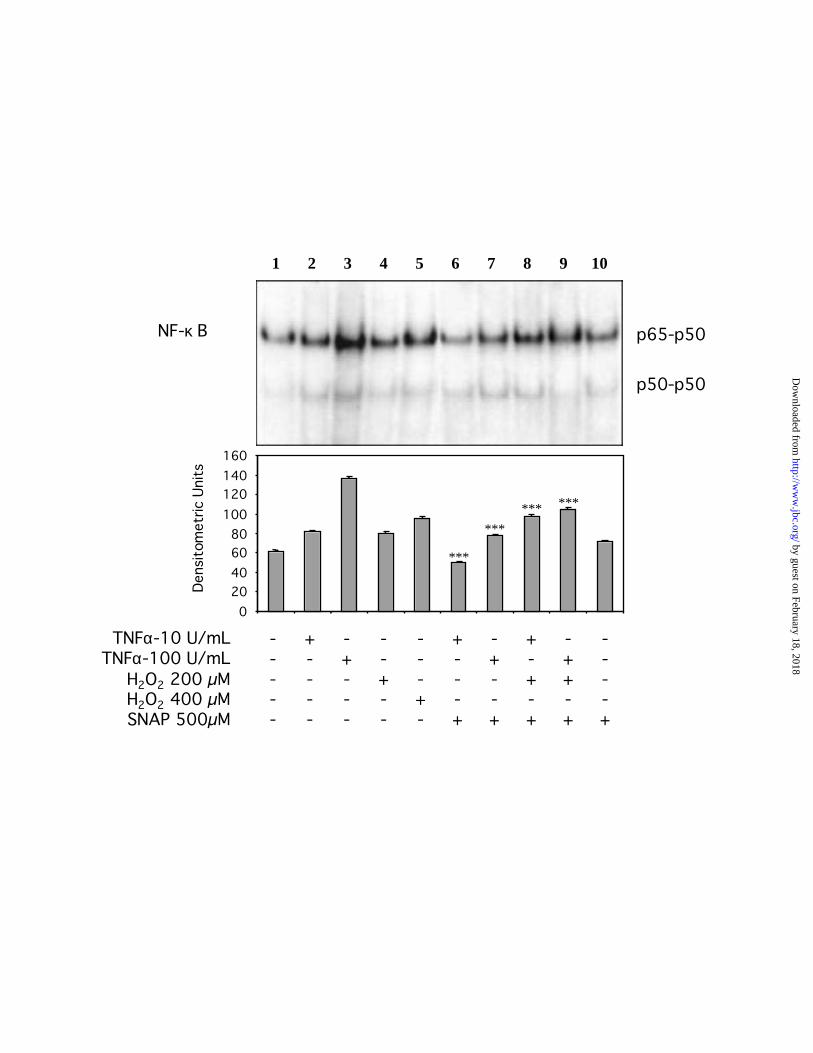
Nitric oxide disrupts the H2O2-dependent activation of NF- B in AD10 cells.
To determine whether nitric oxide could interfere with the TNF-α-mediated activation of
NF-κB, we examined the NF-κB DNA-binding activity by EMSA. As shown in Figure 3,
nuclear extracts from TNF-α-stimulated AD10 cells exhibited an increased binding activity
specific for the NF-κB heterodimer p65-p50. H2O2 also induced specific NF-κB binding activity
in AD10 cells after 30 min of incubation. Further, NF-κB binding activity was significantly
inhibited by the incubation of AD10 cells with 500 µM of SNAP for 2 hours prior to stimulation
with TNF-α for 30 min. The impaired NF-κB binding activity by SNAP was restored by the
addition of H2O2 to similar levels as those detected in the H2O2-stimulated AD10 cells. Thus,
these results suggest that the step at which nitric oxide interferes preceded the step at which H2O2
is generated after stimulation of AD10 cells with TNF-α.
Noteworthy, AD10 cells exhibit a constitutive level of NF-κB binding activity that is not
affected by nitric oxide (Fig. 3, lane 1 and 10), whereas in TNF-α-stimulated cells the NF-κB
binding activity decreased below the basal levels in the presence of nitric oxide (Fig. 3, lane 6).
Nitric oxide decreases TNF- -dependent generation of H2O2.
To examine whether nitric oxide affects the generation of H2O2 in AD10 cells stimulated
with TNF-α, we determined the intracellular generation of H2O2 using the fluorescent cell
permeable probe, 2',7'-dichlorofluorescin diacetate (H2DCFDA). AD10 cells were incubated in
the presence or absence of 500 µM of SNAP and then stimulated with 10 and 100 U/mL of TNF-
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 14
14
α, respectively, for 15 min. Fluorescence cytometric analysis of these experimental groups
revealed a significant increase in H2O2 levels generated by the TNF-α treatment. Incubation of
the TNF-α-stimulated cells in the presence of SNAP significantly reduced the relative amount of
H2O2 generated by these cells (Fig. 4). These data suggest that nitric oxide is affecting the intra
cellular bio-generation of H2O2 by SOD via its chemical interaction with TNF-α-induced O-2.
Exogenous H2O2 restored the nitric oxide-mediated blocking of the TNF- -dependent
activation of NF- B.
Nitric oxide has been shown to directly affect the structure of NF-κB and decrease its
DNA-binding ability due to thiol modification of critical amino acid residues (30). In order to
determine the direct effect of nitric oxide on the activation of NF-kB, we used an enhanced green
fluorescent protein-based reporter system driven by four tandem-repeated κB responsive
elements linked to the thymidine kinase minimal promoter (pNF-κB-d2EGFP). We transiently
transfected AD10 cells with the pNF-κB-d2EGFP reporter vector and then stimulated the cells in
the presence or absence of 500 µM of SNAP. Cytoflourometric analysis of these cells revealed a
significant activation of the reporter gene by TNF-α and H2O2 and the extent of activation was a
function of the concentrations used. The TNF-α-induced activation of the NF-κB-dependent
reporter gene was significantly decreased in the presence of 500 µM of SNAP (Fig. 5),
corroborating the findings obtained in the NF-κB binding assay. The inhibitory activity of SNAP
on the TNF-α-induced activation of the NF-κB-dependent reporter gene was significantly
rescued by stimulation with 200 µM of exogenous H2O2 (Fig. 5). These data confirm the
inhibitory effect of nitric oxide on the H2O2-dependent activation of NF-κB in TNF-α-treated
AD10 cells. We also noticed that untreated AD10 cells were able to maintain basal levels of NF-
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 15
15
κB activation that were not inhibited by treatment with nitric oxide, corroborating the findings
observed in the binding assay in Figure 3.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 16
16
Discussion
The activation of the transcription factor NF-κB by TNF-α and many other stimuli has
been implicated in the development of resistance of tumor cells to a variety of cytotoxic
molecules including TNF-α (3,5). NF-κB is an oxidative stress-responsive transcription factor
that has been shown to respond to small concentrations of exogenous H2O2 or to reactive oxygen
species endogenously generated as part of the signaling cascade triggered by many molecules
such as TNF-α (31-33). We have reported that the IFN-γ-induced sensitization of the human
ovarian carcinoma AD10 cell line to Fas-mediated apoptosis is due in part to the generation of
nitric oxide, or its reaction products, by iNOS in these cells (21). In the present study evidence is
presented for the first time that demonstrates that NO also sensitizes tumor cells to TNF-α-
mediated cytotoxicity. Further, we describe a novel molecular mechanism by which nitric oxide
disrupts the H2O2-dependent activation of NF-κB resulting in sensitization of the AD10 cells to
TNF-α cytotoxicity.
The specific role of nitric oxide in tumor biology is not established. A broad spectrum of
activities has been assigned to either the physiology or the patho-physiology of nitric oxide in
tumor cells (for a review, see ref. (34)). Low-output of nitric oxide has been correlated with
increased blood flow and new blood vessels feeding the tumor area (35). In addition, the
generation of nitric oxide by tumor cells may inhibit the activation and proliferation or may
increase the apoptosis of surrounding lymphocytes that can account for the immune suppression
accompanying tumor growth (unpublished data). Furthermore, high intratumoral-output of nitric
oxide could inhibit the activation of caspases and therefore antagonizes the pro-apoptotic signals
(36,37). However, the opposite effect has also been observed in many other systems whereby the
generation of high-output of nitric oxide, either by iNOS induction or by the use of NO donors,
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 17
17
inhibits tumor growth and metastasis (38). Therefore, the final outcome of NO mediated effects
would be determined by many factors including the local concentration and sources of nitric
oxide and the presence of reactive molecules that might redirect the redox status in the tumor
cell.
In the human ovarian carcinoma AD10 cell line stimulated with the pro-inflammatory
cytokine IFN-γ, we observed a markedly increased sensitivity of these tumor cells to the
cytotoxic effect of TNF-α. IFN-γ also induces iNOS expression in these cells (21). Sensitization
to TNF-α was antagonized by the use of the specific NOS inhibitor L-NMA and was mimicked
by the use of the NO donor SNAP, confirming the role of nitric oxide in the sensitization process
(Fig. 1). Frequently, IFN-γ treatment alone might not be sufficient to induce iNOS expression in
cultured cells. The participation of IFN-γ in the induction of iNOS is generally directed to the
potentiation of the activity of pro-inflammatory cytokines like TNF-α, IL-1 or bacterial LPS.
These cytokines and/or the bacterial LPS have been shown to activate the transcription factor
NF-κB, setting the basal threshold for the induction of the expression of iNOS that might be
enhanced by the action of IFN-γ (39). We observed that untreated AD10 cells (which
constitutively secrete TNF-α) display a constitutive level of activation of NF-κB (Fig. 3 and 5).
Therefore, the basal activation of NF-κB in AD10 cells could explain why the treatment with
IFN-γ alone was sufficient to induce iNOS and subsequently generate nitric oxide.
NF-κB has been shown to be a key transcription factor controlling TNF-α gene
expression in many cells, either as a major activator or synergistically in association with other
transcription factors (28). Thus, the significant basal activation of the NF-κB in AD10 cells
might explain the presence of a constitutive expression of TNF-α by these cells (Fig. 2A-B, last
lanes). Moreover, TNF-α has been implicated as a survival cytokine used by tumor cells either to
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 18
18
control anti-apoptotic mechanisms or promoting cellular proliferation (40-42). Therefore, the
maintenance of a self-regulated loop in which the expression of TNF-α is perpetuated by the
TNF-α-mediated basal activation of NF-κB could play a major role in the survival and/or
proliferation of tumor cells. Pyrrolidine- dithiocarbamate (PDTC) has been shown to be a potent
and specific inhibitor of the NF-κB-mediated expression of TNF-α (29,43). Untreated AD10
cells exhibited a basal expression of TNF-α, which was enhanced by stimulation with exogenous
TNF-α and subsequently inhibited by PDTC (Fig. 2B). Similarly, using the nitric oxide donor
SNAP, we were able to completely abrogate the expression of endogenous TNF-α (Fig. 2A). In
contrast, nitric oxide was unable to block the basal expression of endogenous TNF-α in the
absence of exogenous stimulation. These results strongly suggest the inhibitory role of nitric
oxide on TNF-α-induced activation of NF-κB and consequently resulting in the disruption of
TNF-α gene expression.
TNF-α induces the generation of reactive oxygen species (ROS) that may serve as second
messengers in the activation of divergent pathways related to the cell death process (44-46).
Stimulation of many cell types with TNF-α results in the generation of intracellular superoxide
(O•2-) (10). In biological systems, O•
2- is immediately reduced by SOD to H2O2 or rapidly reacts
with NO, generating ONOO- (13). Therefore, decreased amounts of TNF-α-generated O•2- will
result in a reduced generation of total H2O2. This could subsequently affect the H2O2-dependent
activation of NF-κB (47). Examining the endogenous generation of H2O2 in TNF-α stimulated
AD10 cells, we have found a significant reduction in the total amount of H2O2 being generated in
the presence of nitric oxide (Fig. 4). These results strongly suggest the scavenging effect of NO
on the O•2- being generated upon TNF-α treatment. Alternatively, NO can inhibits O•
2- production
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 19
19
by the modification of the activity of NADPH oxidase, the main enzyme that generates O•2-
within the cell (48,49).
Further, we have found that the addition of NO donors to TNF-α-stimulated AD10 cells
inhibited either the DNA binding activity of NF-κB (Fig. 3) or its activation (Fig. 5). This
inhibition was restored to the normal H2O2-stimulated level by treatment with exogenous H2O2.
In contrast, nitric oxide did not affect the NF-κB activation in untreated AD10 cells, confirming
the previous observation with TNF-α gene expression. These results suggest the presence of, at
least, two pathways in the activation of NF-κB in AD10 cells that may differ in their sensitivity
to H2O2 and the selectivity of nitric oxide to affect just one of these two pathways. The
inactivation of NF-κB upon NO treatment was not mediated by guanylate cyclase (GC)
activation since the cGMP analogue 8-bromo-cGMP had no effect on NF-κB and we could not
block the inhibitory effect of NO on NF-κB activation by the use of the GC blocker ODQ (data
not shown).
Previous reports have implicated the role of nitric oxide on the activation of NF-κB. NO
has been shown to increase the expression of the NF-κB inhibitory sub-unit IκB or affects its
cellular stability by inhibiting protein degradation (50). Due to the rapid generation of H2O2 upon
TNF-α treatment and the immediate activation NF-κB (less than 15 minutes) in AD10 cells, it is
highly unlikely that secondary regulatory factors like the induction of IκB interfered with the
rapid NF-κB activation. It is likely that in the long run, a combination of both mechanisms may
account for the total inhibitory role of nitric oxide on the TNF-α-induced activation of NF-κB.
An alternative proposed mechanism implicated in the inhibition of the NF-κB activity by
NO is via the alteration of critical thiol groups, resulting in the disruption of the NF-κB structure
and subsequently affecting its DNA-binding ability (30). However, the in vivo situation may be
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 20
20
much more complex due to the high concentrations of glutathione and other redox-active
proteins within the cell, which may prevent the modification of thiol groups.
In conclusion, our findings suggest that the mechanism by which NO sensitizes the
human ovarian carcinoma cell line to TNF-α-mediated apoptosis is due to the specific disruption
of the TNF-α-induced generation of H2O2 and the subsequent inhibition of the NF-κB-dependent
expression of anti-apoptotic genes. These results can be extended to other solid tumor cells as
observed with the human prostatic adenocarcinoma cell line PC-3. As shown in Figure 6, the
survival autocrine-paracrine loop involving the NF-κB-dependent expression of TNF-α could be
interrupted by the inhibitory activity that nitric oxide exerts on the TNF-α-induced activation of
NF-κB. Furthermore, in an in vivo situation, the exposure of tumor cells to pro-inflammatory
cytokines such as IFN-γ will promote the induction of iNOS by the tumor cells or neighboring
lymphocytes and which in turn will result in the generation of nitric oxide. Hence, the
endogenously generated or the exogenously provided NO would scavenge the TNF-α-generated
O•2- and decrease the H2O2-dependent activation of NF-κB. Based on these molecular events, a
new mechanism of NO-mediated sensitization to apoptosis is revealed.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 21
21
Acknowledgments
This work was supported in part by the Boiron Research Foundation, the UCLA Gene Therapy
Program, the National Council of Science and Technology of Venezuela (CONICIT-Venezuela)
and the Vollmer Foundation [H.G.]. We thank Dr. Diana Márquez and Dr. Jon Fukuto for their
thorough discussion of this study and their expert assistance with the technical aspects of the
regulation of gene expression and NO-mediated effects, respectively. We are grateful to Chuen-
Pei Ng for his assistance with the fluorescence cytometry analysis. We also thank Ms. Stephanie
Louie for secretarial assistance in the preparation of the manuscript.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 22
22
References
1. Dive, C., and Hickman, J. A. (1991) Br J Cancer 64(1), 192-6
2. Eastman, A. (1990) Cancer Cells 2(8-9), 275-80
3. Wang, C. Y., Mayo, M. W., and Baldwin, A. S., Jr. (1996) Science 274(5288), 784-7
4. Wang, C. Y., Cusack, J. C., Jr., Liu, R., and Baldwin, A. S., Jr. (1999) Nat Med 5(4),
412-7
5. Beg, A. A., and Baltimore, D. (1996) Science 274(1 November), 782-784
6. Tartaglia, L. A., and Goeddel, D. V. (1992) Immunol Today 13(5), 151-3
7. Hsu, H., Shu, H. B., Pan, M. G., and Goeddel, D. V. (1996) Cell 84(2), 299-308
8. Wang, C. Y., Mayo, M. W., Korneluk, R. G., Goeddel, D. V., and Baldwin, A. S., Jr.
(1998) Science 281(5383), 1680-3
9. Garcia-Ruiz, C., Colell, A., Mari, M., Morales, A., and Fernandez-Checa, J. C. (1997) J
Biol Chem 272(17), 11369-77
10. Hennet, T., Richter, C., and Peterhans, E. (1993) Biochem J 289(Pt 2), 587-92
11. Suzuki, Y. J., Forman, H. J., and Sevanian, A. (1997) Free Radic Biol Med 22(1-2), 269-
85
12. Huie, R. E., and Padmaja, S. (1993) Free Radic Res Commun 18(4), 195-9
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 23
23
13. Szabo, C., and Ohshima, H. (1997) Nitric Oxide 1(5), 373-85
14. Fukuto, J. M. (1995) Adv Pharmacol 34, 1-15
15. Han, S. Y., Choung, S. Y., Paik, I. S., Kang, H. J., Choi, Y. H., Kim, S. J., and Lee, M. O.
(2000) Biol Pharm Bull 23(4), 420-6
16. Mizutani, Y., Bonavida, B., Nio, Y., and Yoshida, O. (1994) J Urol 151(6), 1697-702
17. Safrit, J. T., Berek, J. S., and Bonavida, B. (1993) Gynecol Oncol 48(2), 214-20
18. Safrit, J. T., Belldegrun, A., and Bonavida, B. (1993) J Urol 149(5), 1202-8
19. Morimoto, H., Yonehara, S., and Bonavida, B. (1993) Cancer Res 53(11), 2591-6
20. Frost, P. J., Belldegrun, A., and Bonavida, B. (1999) Prostate 41(1), 20-30
21. Garban, H. J., and Bonavida, B. (1999) Gynecol Oncol 73(2), 257-64
22. Stamler, J. S. (1995) Curr Top Microbiol Immunol 196, 19-36
23. Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990)
Proc Natl Acad Sci U S A 87(4), 1620-4
24. Beckman, J. S. (1991) J Dev Physiol 15(1), 53-9
25. Decker, T., and Lohmann-Matthes, M. L. (1988) J Immunol Methods 115(1), 61-9
26. Bradford, M. M. (1976) Anal Biochem 72, 248-54
27. Lenardo, M. J., and Baltimore, D. (1989) Cell 58(2), 227-9
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 24
24
28. Liu, H., Sidiropoulos, P., Song, G., Pagliari, L. J., Birrer, M. J., Stein, B., Anrather, J.,
and Pope, R. M. (2000) J Immunol 164(8), 4277-85
29. Ziegler-Heitbrock, H. W., Sternsdorf, T., Liese, J., Belohradsky, B., Weber, C., Wedel,
A., Schreck, R., Bauerle, P., and Strobel, M. (1993) J Immunol 151(12), 6986-93
30. Park, S. K., Lin, H. L., and Murphy, S. (1997) Biochem J 322(Pt 2), 609-13
31. Hong, Y. H., Peng, H. B., La Fata, V., and Liao, J. K. (1997) J Immunol 159(5), 2418-23
32. Schreck, R., Albermann, K., and Baeuerle, P. A. (1992) Free Radic Res Commun 17(4),
221-37
33. Schreck, R., Rieber, P., and Baeuerle, P. A. (1991) Embo J 10(8), 2247-58
34. Moncada, S., Palmer, R. M., and Higgs, E. A. (1991) Pharmacol Rev 43(2), 109-42
35. Jenkins, D. C., Charles, I. G., Thomsen, L. L., Moss, D. W., Holmes, L. S., Baylis, S. A.,
Rhodes, P., Westmore, K., Emson, P. C., and Moncada, S. (1995) Proc Natl Acad Sci U S
A 92(10), 4392-6
36. Liu, L., and Stamler, J. S. (1999) Cell Death Differ 6(10), 937-42
37. Li, J., and Billiar, T. R. (2000) Semin Perinatol 24(1), 46-50
38. Shi, Q., Xiong, Q., Wang, B., Le, X., Khan, N. A., and Xie, K. (2000) Cancer Res
60(10), 2579-83
39. Taylor, B. S., Alarcon, L. H., and Billiar, T. R. (1998) Biochemistry (Mosc) 63(7), 766-81
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 25
25
40. Khwaja, A., and Tatton, L. (1999) J Biol Chem 274(51), 36817-23
41. Sawyer, S. T., and Jacobs-Helber, S. M. (2000) J Hematother Stem Cell Res 9(1), 21-9
42. Selinsky, C. L., and Howell, M. D. (2000) Cell Immunol 200(2), 81-7
43. Nemeth, Z. H., Hasko, G., and Vizi, E. S. (1998) Shock 10(1), 49-53
44. Schulze-Osthoff, K., Bakker, A. C., Vanhaesebroeck, B., Beyaert, R., Jacob, W. A., and
Fiers, W. (1992) J Biol Chem 267(8), 5317-23
45. Schulze-Osthoff, K., Beyaert, R., Vandevoorde, V., Haegeman, G., and Fiers, W. (1993)
Embo J 12(8), 3095-104
46. Moreno-Manzano, V., Ishikawa, Y., Lucio-Cazana, J., and Kitamura, M. (2000) J Biol
Chem 275(17), 12684-91
47. Baeuerle, P. A., and Henkel, T. (1994) Annu Rev Immunol 12, 141-79
48. Clancy, R. M., Leszczynska-Piziak, J., and Abramson, S. B. (1992) J Clin Invest 90(3),
1116-21
49. Fujii, H., Ichimori, K., Hoshiai, K., and Nakazawa, H. (1997) J Biol Chem 272(52),
32773-8
50. Peng, H. B., Libby, P., and Liao, J. K. (1995) J Biol Chem 270(23), 14214-9
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 26
26
Footnotes
1The abbreviations used are: IFN-γ, interferon gamma; iNOS, inducible nitric oxide synthase;
NO, nitric oxide; IL-1β, interleukin 1-beta; LPS, lipopolysaccharide; SNAP, S-Nitroso-N-
acetylpenicillamine; L-NMA, NG-Monomethyl-L-arginine; TNF, tumor necrosis factor; H2O2,
Hydrogen Peroxide; NF-κ B, Nuclear Factor kappa B; NOC18: DETA NONOeate; ODQ, 1H-
[1,2,4]oxadiazolo- [4,3-a]quinoxalin-1-one; SOD, Superoxide dismutase.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 27
27
Figure Legends
Fig. 1. Effect of nitric oxide on the sensitization of AD10 cells to TNF- -induced
cytotoxicity. The cytotoxic effect of increasing concentrations of TNF-α (0, 0.01, 0.1 and 1
ng/mL) in a 24 hour incubation assay was assessed by the LDH release into the culture medium.
A, AD10 cells were pretreated with 100 U/mL of IFN-γ for 18 hours in the presence or absence
of 1 mM of the NOS inhibitor L-NMA. B, AD10 cells were treated with the NO donor SNAP (0,
10 and 100 µM) 18 hours prior to exposure to TNF-α. **, P<0.005; ***, P<0.001.
Fig. 2. Role of NO and PDTC on endogenous TNF- gene expression. The relative expression
of TNF-α mRNA was assessed by RT-PCR. A, AD10 cells were pretreated in the presence or
absence of increasing concentrations of the NO donor SNAP (1, 10, 100 and 500 µM) for 18
hours and then stimulated with 100 U/mL of TNF-α for 4 hours. B, AD10 cells were pretreated
in the presence or absence of increasing concentrations of PDTC (1, 10, 100 and 500 µM) for 18
hours and then stimulated with 100 U/mL of TNF-α for 4 hours. Amplification of G-3-PDH
mRNA was used as internal standard control of gene expression for relative comparison.
Fig. 3. Effect of NO on the TNF- -mediated NF- B nuclear translocation. Nuclear extracts
from AD10 cells pretreated with TNF-α in the presence or absence of the NO donor SNAP (500
µM) were analyzed for their NF-κB DNA-binding ability using EMSA. Exogenous H2O2 was
used as a positive NF-κB activator that bypasses the generation of O•2-. ***, P<0.001.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 28
28
Fig. 4. Effect of NO on TNF- -dependent generation of H2O2. Changes in the intracellular
H2O2 levels of AD10 cells treated with TNF-α in the presence or absence of the NO donor
SNAP (500 µM) were assessed by fluorescence flow cytometry. ***, P<0.001.
Fig.5. Effect of NO on the TNF- -dependent activation of NF- B. Transiently transfected
AD10 cells with the NF-κB/2EGFP reporter vector were stimulated with TNF-α (10 and 100
U/mL) for two to three hours in the presence or absence of the NO donor SNAP (500 µM).
Exogenous H2O2 was used as a positive NF-κB activator that bypasses the generation of O•2-.
Total mean intensity was determined by fluorescence flow cytometry. Statistical paired
comparisons were established between columns 6-7 against 2-3 and 8-9 against 6-7. ***,
P<0.001.
Fig. 6. Schematic representation of possible site of action of NO in the disruption of the
TNF- -dependent generation of H2O2 and subsequent inhibition of the activation of NF-
B. The autocrine-paracrine loop involving the NF-κB-dependent expression of TNF-α
maintains cell’s resistance to apoptotic stimuli and survival. This loop could be interrupted by
the inhibitory activity that nitric oxide exerts on the TNF-α-induced activation of NF-κB. In an
in vivo situation, the exposure of tumor cells to pro-inflammatory cytokines, such as IFN-γ, will
promote the induction of iNOS by the tumor cells or neighboring lymphocytes and resulting in
the generation of nitric oxide. Hence, the endogenously generated or the exogenously provided
NO would scavenge the TNF-α-generated O•2- and decrease the H2O2-dependent activation of
NF-κB. This results in inhibition of NF-κB-dependent gene expression of anti- apoptotic factors
and sensitization of cells to apoptosis.
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 29
TNF- [ng/ml]
% C
ytot
oxic
ity
0
10
30
50
Control
IFN-γ [100 U/mL]
L-NMA [1 mM] +IFN-γ [100 U/mL]
0.01 0.1 10
******
***
20
40
% C
ytot
oxic
ity
TNF- [ng/ml]
Control
SNAP[10µM]
SNAP[100µM]
0
20
40
60
0 0.01 0.1 1
***
***
****
A
B
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 30
TNF-α [100 U/mL]
+1
+2
+3
+4
+5
-6
SNAP
TNF-α
G-3-PDH
PDTC
TNF-α
G-3-PDH
A
B
TNF-α [100 U/mL]
+1
+2
+3
+4
+5
-6
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 31
NF-κ B
p50-p50
p65-p50
Den
sito
met
ric U
nits
TNFα-10 U/mLTNFα-100 U/mL
H2O2 200 µMH2O2 400 µMSNAP 500µM
-----
+----
-+---
--+--
---+-
+---+
-+--+
-++-+
+-+-+
----+
0
2040
6080
100
120140
160
***
***
******
1 2 3 4 5 6 7 8 9 10
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 32
0
100
200
300
400
500
600
700
Tot
al M
ean
Inte
nsit
y
TNFα-10 U/mLTNFα-100 U/mL
SNAP 500µM
---
+--
-+-
+-+
-++
******
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 33
NF-k B/2EGFPTNFα-10 U/mL
TNFα-100 U/mLH2O2 200 µMH2O2 400 µMSNAP 500µM
+-----
++----
+-+---
+--+--
+---+-
++---+
+-+--+
+-++-+
++-+-+
+----+
******
******
Tot
al M
ean
Inte
nsit
y
4000
5000
6000
7000
8000
9000
10000
11000
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 34
TNFR1
Superoxide
NF- B
Anti ApoptosisGene Expression
Hydrogen Peroxide
TNF-
iNOS
NO
SOD
IFN- γ
NO-donors
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Page 35
Hermes J. Garbán and Benjamin Bonavida-induced cytotoxicityαhuman tumor cells to TNF-
B: Role in sensitization ofκNitric oxide disrupts H2O2-dependent activation of NF-
published online December 15, 2000J. Biol. Chem.
10.1074/jbc.M008471200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from