Nuclear Magnetic Resonance Spectroscopy Notes adapted by Audrey Dell Hammerich, October 3, 2013 Nuclear magnetic resonance (NMR), as all spectroscopic methods, relies upon the interaction of the sample being examined with electromagnetic radiation, here in the low energy range of radio frequencies (1-1000 MHz). To absorb a photon of electromagnetic radiation the sample must exhibit periodic motion whose frequency matches that of the absorbed radiation Δ E sample = E photon = ħ ω = h ν = hc / λ (1) The experiments will sample from the broad range of information accessible from NMR interrogation of a sample, in particular how NMR frequencies can be used to obtain the pK a of as well as structural information on a sample. Angular Momentum and Spin Classically a rotating particle possesses angular momentum. The nucleus of an atom can be visualized as “rotating” and, consequently, has a spin angular momentum, I. This angular momentum is intrinsic to the nucleus, rather than the angular momentum arising from the nucleus physically rotating or spinning in space, and properly requires a quan- tum mechanical treatment. Nevertheless, employing an analogy of spin with that of a rotating classical particle is instructive. The magnitude of the spin angular momentum is given in quantum mechanics by | I | = [ I ( I +1)] 1/2 ħ I = 0, 1 / 2 , 1, 3 / 2 , … (2) where ħ is Planck’s constant h divided by 2π and I is the spin angular momentum quan- tum number or the “spin” of the nucleus. I has a quantized z-component I z = m ħ m = −I, …, 0, …, I (3) where m is the magnetic quantum number with 2I +1 values. (The z-component is im- portant as the direction of the static magnetic field is chosen as the z axis and the compo- nent of the magnetic moment which will interact with this magnetic field - generated by the NMR spectrometer - lies along this axis.) Note the parallel between the orbital angu- lar momentum quantum number l and magnetic quantum number m l for the electron in a hydrogen atom and the I and m quantum numbers here. Nuclei of all elements are composed of protons (p) and neutrons (n), both of which have spin I = 1 / 2 . Thus the total nuclear spin is the resultant of the spin and orbital angu- lar momenta of all the nucleons. The quantum treatment indicates that protons and neu- trons pair up separately and that even numbers of either have zero spin angular momen- tum. The model leads to the three cases summarized in Table I.
Transcript
Nuclear Magnetic Resonance Spectroscopy Notes adapted by Audrey Dell Hammerich, October 3, 2013
Nuclear magnetic resonance (NMR), as all spectroscopic methods, relies upon the interaction of the sample being examined with electromagnetic radiation, here in the low energy range of radio frequencies (1-1000 MHz). To absorb a photon of electromagnetic radiation the sample must exhibit periodic motion whose frequency matches that of the absorbed radiation
ΔE sample = Ephoton = ħω = hν = hc /λ (1)
The experiments will sample from the broad range of information accessible from NMR interrogation of a sample, in particular how NMR frequencies can be used to obtain the pKa of as well as structural information on a sample.
Angular Momentum and Spin
Classically a rotating particle possesses angular momentum. The nucleus of an atom can be visualized as “rotating” and, consequently, has a spin angular momentum, I. This angular momentum is intrinsic to the nucleus, rather than the angular momentum arising from the nucleus physically rotating or spinning in space, and properly requires a quan-tum mechanical treatment. Nevertheless, employing an analogy of spin with that of a rotating classical particle is instructive. The magnitude of the spin angular momentum is given in quantum mechanics by
|I | = [I(I +1)]1 /2 ħ I = 0, 1/2 , 1, 3/2 , … (2) where ħ is Planck’s constant h divided by 2π and I is the spin angular momentum quan-tum number or the “spin” of the nucleus. I has a quantized z-component
Iz = mħ m = −I, …, 0, …, I (3) where m is the magnetic quantum number with 2I +1 values. (The z-component is im-portant as the direction of the static magnetic field is chosen as the z axis and the compo-nent of the magnetic moment which will interact with this magnetic field - generated by the NMR spectrometer - lies along this axis.) Note the parallel between the orbital angu-lar momentum quantum number l and magnetic quantum number ml for the electron in a hydrogen atom and the I and m quantum numbers here.
Nuclei of all elements are composed of protons (p) and neutrons (n), both of which have spin I = 1/2 . Thus the total nuclear spin is the resultant of the spin and orbital angu-lar momenta of all the nucleons. The quantum treatment indicates that protons and neu-trons pair up separately and that even numbers of either have zero spin angular momen-tum. The model leads to the three cases summarized in Table I.
Table I – Distinct Ways to Combine Spin of the Nucleons in an Atom
Spin Nucleon Description Examples I = 0 even numbers of both p and n 12C: 6p, 6n
16O: 8p, 8n I = n (integer) odd numbers of both p and n 2H: 1p, 1n, I = 1
10B: 5p, 5n, I = 3 I = n/2 (half integer) even p (n) and odd n (p) 13C: 6p, 7n, I = 1/2
23Na: 11p, 12n, I = 3/2 Nuclear Magnetic Moment
Classically if a rotating particle is charged it generates a magnetic dipole which cre-ates a magnetic field. The dipole has a magnetic moment. Nuclear spin angular mo-mentum has an associated nuclear magnetic dipole moment μ which can interact with a magnetic field
μ = γI (4) where γ is the magnetogyric ratio, a constant characteristic of each nuclide. The mag-netic moment also has a quantized z-component.
μz = γIz = mħγ (5) Protons and neutrons are actually not true elementary particles but consist of charged fundamental particles known as quarks. Both nucleons (protons and neutrons) contribute to the spin and also can contribute to the nuclear magnetic moment, μ . However I = 0 spins have no spin and, thus, no magnetic moment. Table II summarizes properties of some nuclides. Table II. Properties of Selected Nuclides
Nuclide I γ [× 107 rad (Ts)-1] % Natural Abundance
23Na 3/2 7.0801 100 26.466 1relative to a proton frequency of 100 MHz Nuclear Spin in an External Magnetic Field (Zeeman Effect)
There is no preferred orientation for a magnetic moment in the absence of external fields. In the absence of Bo the magnetic moments of individual nuclei are randomly ori-
2
ented and all have essentially the same energy. Application of an external magnetic field removes the randomness, forcing the nuclei to align with or against the direction of Bo. This change from a random state to an ordered state is known as polarization. Such po-larization means there is a difference in the population of the various spin states. Fur-thermore the different spin states are no longer degenerate (the same) in energy.
Classically, in the presence of an external magnetic field Bo the energy of a magnetic
moment μ depends on its orientation relative to the field
E = − μ . Bo (6) being a minimum when the magnetic moment is aligned parallel to the magnetic field and a maximum when it is anti-parallel. From the quantum mechanical prospective, when a nucleus is introduced into a magnetic field its magnetic moment will align itself in 2I +1 orientations (number of values of the m quantum number) about the z direction of Bo where the energy is given by
Em = − μz Bo = − mħγB o (7)
For an I = 1/2 nucleus there are only two orientations for the magnetic moment μ: 1) a lower energy orientation parallel to Bo with a magnetic quantum number m = 1/2 often referred to as the α spin state and 2) a higher energy anti-parallel orientation with m = − 1/2 referred to as the β spin state.
β
anti-parallel to Bo Eβ = 1/2 ħγB o α parallel to Bo
Eα = − 1/2 ħγB o
Energy levels for a nucleus with spin quantum number I = 1/2
For an I > 1/2 nucleus there are more than two orientations. The physical description
can be portrayed in a vector diagram where the length of the angular momentum vector is a constant whose magnitude |I | = [I(I +1)]1 /2 ħ and whose projection on the z-axis is Iz = mħ . The number of possible orientations of this vector is given by the number of values of the magnetic quantum number m. Note that even with the magnitude of the vector and m known the uncertainty principle does not allow one to specify the vector I. It can lie anywhere on the base of the cones about the z-axis as shown on the next page.
3
For clarity the vector is only shown for the highest and lowest values for m though all of the cone bases are given.
Orientations of the spin angular momentum vector for I = 1/2 , 1, and 2 nuclei
Transition Frequencies NMR spectroscopy induces transitions between adjacent nuclear spin energy states
(the selection rule is Δm = ±1). The energy change for a nucleus undergoing an NMR transition from the spin state characterized by the magnetic quantum number m to the state with quantum number m − 1 is
Equation (8) follows from the previous discussions. The difference between the z-component of the angular momentum of adjacent m states is ħ [Eq. (2)]. This differ-ence is multiplied by γ to obtain the difference in the magnetic moment z-component [Eq. (5)]. This result is then multiplied by the magnetic field strength to obtain the energy dif-ference between adjacent m states in a magnetic field [(Eq.7)].
The frequency ν of the electromagnetic radiation used to induce an NMR transition between adjacent m levels in an external magnetic field Bo is found from Eq. (1)
ν = ΔE /h = ħγBo/h = γBo/2π (9)
The units of frequency (ν) are cycles/second, hertz (Hz) in SI units. In NMR spectros-copy, it is often more convenient to use angular frequency (ω) with units of radi-ans/second. Since one cycle equals 2π radians,
4
ω ≡ 2πν (10) As cycles and radians are not SI units, both ν and ω have the same SI units (s−1). The an-gular frequency of an NMR transition is more commonly written as
ωo = γBo (11) which is the Larmor equation. Note that the use of ω eliminates the occurrence of 2π in the Larmor equation.
The Larmor frequency has two important physical interpretations. It is the frequency of the electromagnetic radiation that induces a transition between nuclear spin quantum states in the magnetic field. It is also the precessional (or rotational) frequency of the nu-clear magnetic moment about the magnetic field. While the actual origin is quantum me-chanical and involves the Heisenberg uncertainty princi-ple, this precession has a classical analogy. The interac-tion between a magnetic field and a magnetic dipole moment produces a torque on the dipole that makes it revolve around the direction of the magnetic field at a fixed angle, sweeping out a conical surface. This precession is illustrated on the right for an I = 1/2 nucleus and shows the two allowed orientations of the spin vector I (and, by Eq. (4), its associated magnetic moment μ). The length of the vector is ħ√3/2, Eq. (2), and its projection on the z-axis is either 1/2 ħ or – 1/2 ħ , Eq. (3). Boltzmann Statistics
In the presence of an external magnetic field different nuclear spin states (with dif-
ferent values of m) have different energies. The energy difference is proportional to Bo . At thermal equilibrium, these states will also have different populations, their ratio given by the Boltzmann equation
kT/E
NN Δ−= e
low
high (12)
with Nhigh and N low the respective populations of the upper and lower spin states (such as β and α for an Ι = 1/2 nucleus), ΔE = Ehigh − E low the energy difference between the two states, k the Boltzmann constant, and T the absolute temperature. In currently achievable magnetic fields, the difference between nuclear spin energy levels ΔE is much smaller than kT, implying that N low is only very slightly in excess of Nhigh. For 1H in a 9.4 tesla field (400 MHz) and 300 K one obtains a population ratio N(α)/N(β) of 1.000064, i.e., for one million spins in the upper β state there are one million and sixty-four in the lower
5
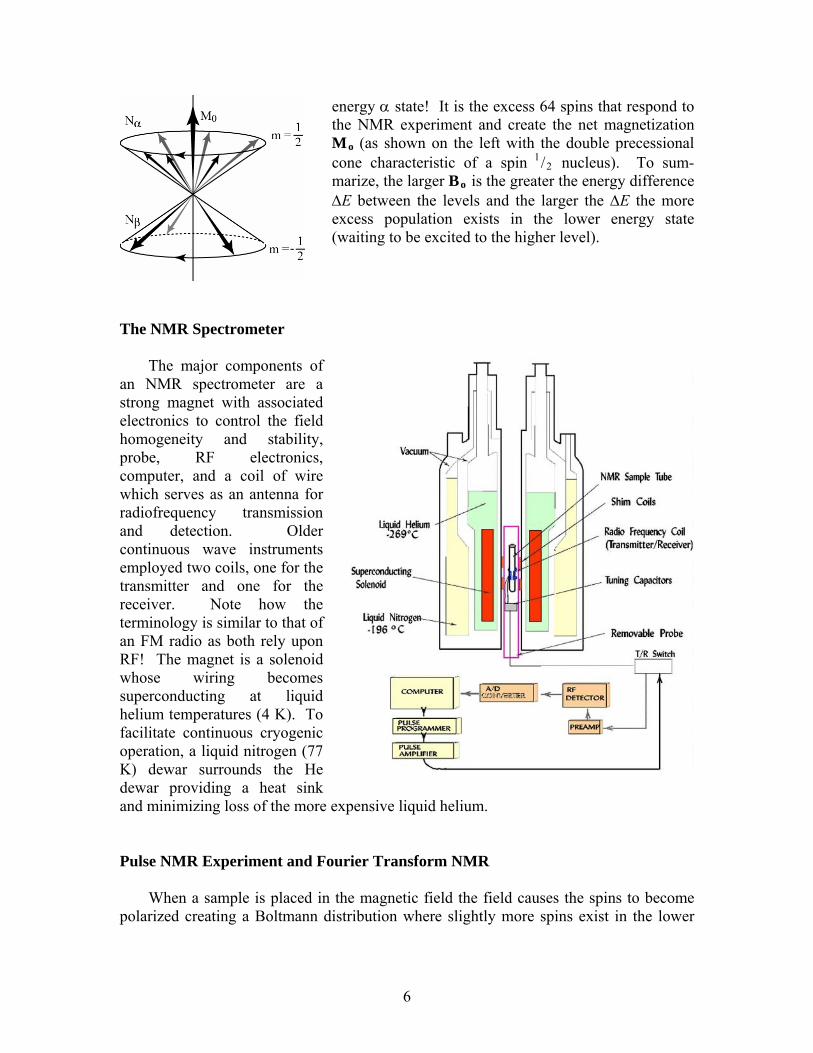
energy α state! It is the excess 64 spins that respond to the NMR experiment and create the net magnetization Mo (as shown on the left with the double precessional cone characteristic of a spin 1/2 nucleus). To sum-marize, the larger Bo is the greater the energy difference ΔE between the levels and the larger the ΔE the more excess population exists in the lower energy state (waiting to be excited to the higher level).
The NMR Spectrometer
The major components of an NMR spectrometer are a strong magnet with associated electronics to control the field homogeneity and stability, probe, RF electronics, computer, and a coil of wire which serves as an antenna for radiofrequency transmission and detection. Older continuous wave instruments employed two coils, one for the transmitter and one for the receiver. Note how the terminology is similar to that of an FM radio as both rely upon RF! The magnet is a solenoid whose wiring becomes superconducting at liquid helium temperatures (4 K). To facilitate continuous cryogenic operation, a liquid nitrogen (77 K) dewar surrounds the He dewar providing a heat sink and minimizing loss of the more expensive liquid helium.
Pulse NMR Experiment and Fourier Transform NMR When a sample is placed in the magnetic field the field causes the spins to become
polarized creating a Boltmann distribution where slightly more spins exist in the lower
6
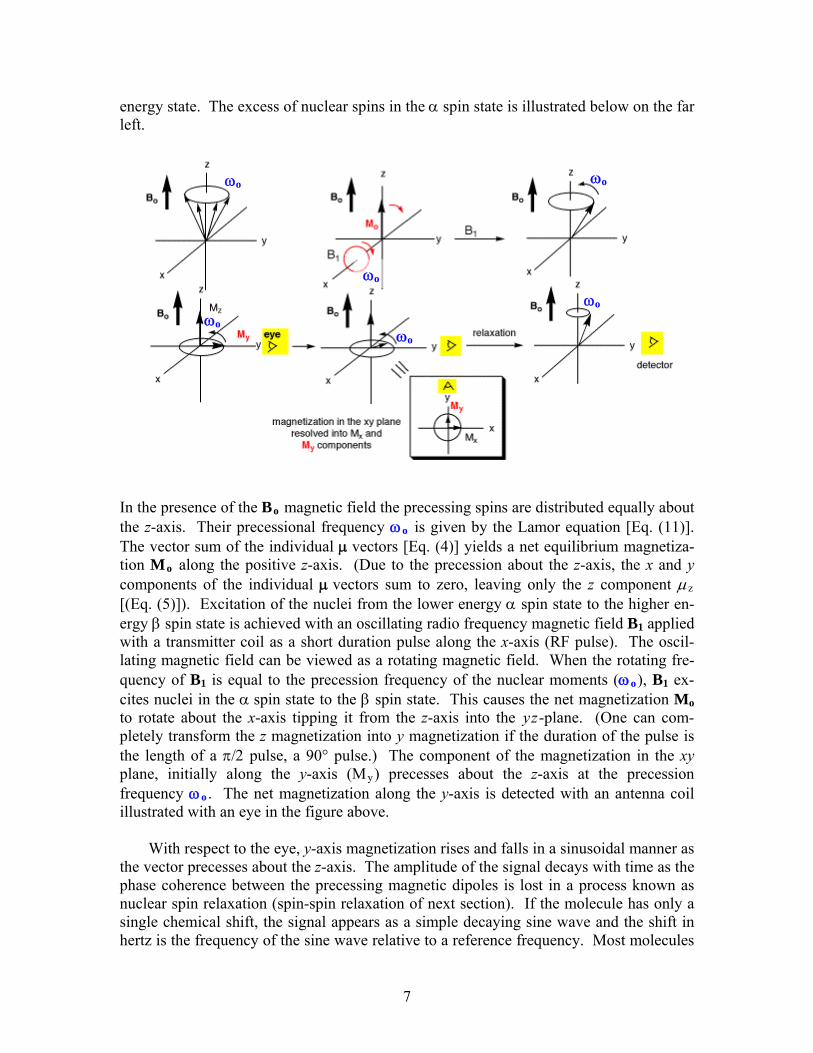
energy state. The excess of nuclear spins in the α spin state is illustrated below on the far left.
ωo ωo
ωo
ωo ωo
ωo
In the presence of the Bo magnetic field the precessing spins are distributed equally about the z-axis. Their precessional frequency ωo is given by the Lamor equation [Eq. (11)]. The vector sum of the individual μ vectors [Eq. (4)] yields a net equilibrium magnetiza-tion Mo along the positive z-axis. (Due to the precession about the z-axis, the x and y components of the individual μ vectors sum to zero, leaving only the z component μz [(Eq. (5)]). Excitation of the nuclei from the lower energy α spin state to the higher en-ergy β spin state is achieved with an oscillating radio frequency magnetic field B1 applied with a transmitter coil as a short duration pulse along the x-axis (RF pulse). The oscil-lating magnetic field can be viewed as a rotating magnetic field. When the rotating fre-quency of B1 is equal to the precession frequency of the nuclear moments (ωo), B1 ex-cites nuclei in the α spin state to the β spin state. This causes the net magnetization Mo to rotate about the x-axis tipping it from the z-axis into the yz-plane. (One can com-pletely transform the z magnetization into y magnetization if the duration of the pulse is the length of a π/2 pulse, a 90° pulse.) The component of the magnetization in the xy plane, initially along the y-axis (My) precesses about the z-axis at the precession frequency ωo . The net magnetization along the y-axis is detected with an antenna coil illustrated with an eye in the figure above.
With respect to the eye, y-axis magnetization rises and falls in a sinusoidal manner as the vector precesses about the z-axis. The amplitude of the signal decays with time as the phase coherence between the precessing magnetic dipoles is lost in a process known as nuclear spin relaxation (spin-spin relaxation of next section). If the molecule has only a single chemical shift, the signal appears as a simple decaying sine wave and the shift in hertz is the frequency of the sine wave relative to a reference frequency. Most molecules
7
have many nuclei with many different chemical shifts and correspondingly many different precession frequencies. The B1 field actually contains a broad band width of frequencies that excite all nuclei in a molecule at the same time. Since the RF pulse is on
the order of microseconds, the time-energy uncertainty principle (ΔEΔt = ħΔωΔ t ≥ ħ) shows that the pulse will consist of a range of frequencies Δω able to simultaneously excite all the spins of a given nuclide. The net magnetization along the y-axis is then the sum of the magnetization of each set of equivalent nuclei, all precessing at different frequencies. The resulting waveform is called the free induction decay (FID) or the time domain spectrum. It is a measure of y-axis magnetization My as a function of time after the B1 pulse. A Fourier transformation (FT) of the signal yields the individual precession frequencies or chemical shifts, the frequency domain spectrum or simply the NMR spectrum:
The precession of spins in the xy-plane does not last forever. It decays due to three distinct effects:
1. The magnetic field is not perfectly uniform. Nuclei in different parts of the sam-
ple precess at slightly different frequencies and get out of phase with one another, thereby gradually decreasing the net magnetization of the sample.
2. Spin-Lattice (or Longitudinal) Relaxation, T1 (mechanism which involves a
net transfer of energy from spin system to surroundings to reestablish Boltzmann distribution – an enthalpy effect). The applied RF pulse and consequent rotation of the net magnetization Mo from the z-axis is a disruption of the thermal equilibrium of the spins. The system responds to reestablish equilibrium by transferring energy from the spin system to the environment (lattice) until the populations of the energy states regain the Boltzmann distribution given in Eq. (12). The process is attributable to electromag-netic interactions between the nuclei and the surrounding particles which cause transi-tions between the spin states (α and β when I = 1/2). As it is the coherent combination of these spin states that contribute to the magnetization rotating in the xy-plane, the result is a gradual decay of these coherent combinations and a return to the state of equilibrium in which the magnetization is in the z-direction and no longer capable of inducing a signal
8
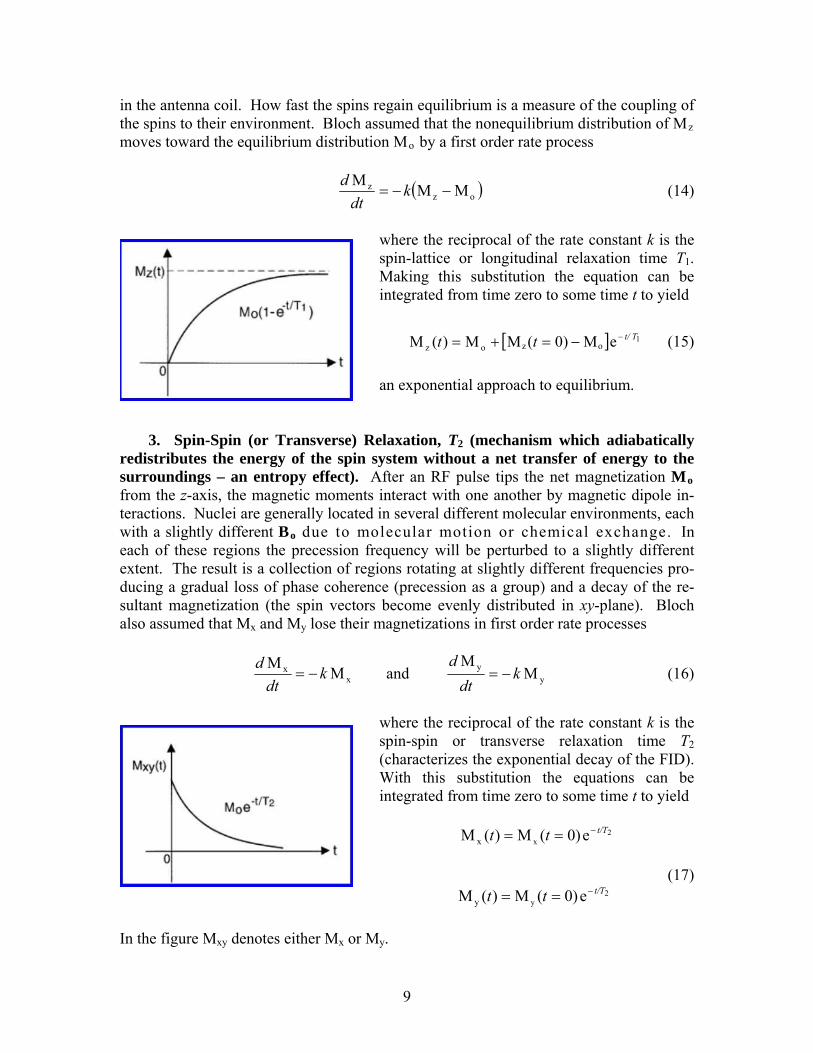
in the antenna coil. How fast the spins regain equilibrium is a measure of the coupling of the spins to their environment. Bloch assumed that the nonequilibrium distribution of Mz moves toward the equilibrium distribution Mo by a first order rate process
( ozz MMM
−−= kdt
d )
ld
(14)
where the reciprocal of the rate constant k is the spin-lattice or longitudinal relaxation time T1. Making this substitution the equation can be integrated from time zero to some time t to yie
[ ] 1eM)0(MM)(M ozoz
Tt/tt −−=+= (15)
an exponential approach to equilibrium.
3. Spin-Spin (or Transverse) Relaxation, T2 (mechanism which adiabatically
redistributes the energy of the spin system without a net transfer of energy to the surroundings – an entropy effect). After an RF pulse tips the net magnetization Mo from the z-axis, the magnetic moments interact with one another by magnetic dipole in-teractions. Nuclei are generally located in several different molecular environments, each with a slightly different Bo due to molecular motion or chemical exchange. In each of these regions the precession frequency will be perturbed to a slightly different extent. The result is a collection of regions rotating at slightly different frequencies pro-ducing a gradual loss of phase coherence (precession as a group) and a decay of the re-sultant magnetization (the spin vectors become evenly distributed in xy-plane). Bloch also assumed that Mx and My lose their magnetizations in first order rate processes
xx MM k
dtd
−= and yy M
Mk
dtd
−= (16)
where the reciprocal of the rate constant k is the spin-spin or transverse relaxation time T2 (characterizes the exponential decay of the FID). With this substitution the equations can be integrated from time zero to some time t to yield
x )0(M)(M x tt ==
2e t/T−
(17) 2
y e)0(M)(M yt/Ttt −==
In the figure Mxy denotes either Mx or My.
9
The spin-spin relaxation time does not exceed the spin-lattice relaxation time, T2 ≤ T1. Since the relaxation times are reciprocals of rate constants this implies that spin-lat-tice relaxation processes are not faster than spin-spin relaxation processes. In many cases, the same physical relaxation mechanisms determine T1 and T2 so that they are then equal. In spectroscopies involving higher energy excitation such as in the ultraviolet or visible region of the electromagnetic spectrum, the return to the ground state of an ex-cited molecule is very rapid. The situation is quite different in NMR where the small en-ergy difference between nuclear spin states means that spontaneous emission is very slow. (The lifetime of an unperturbed excited nucleus is in the range of years!) Conse-quently the excited nucleus must be induced to flip its spin and return to the ground state by some external means. An analysis of the interaction of electromagnetic radiation with matter shows that a spin subjected to a fluctuating magnetic field will be induced to un-dergo transitions between all available energy levels at a rate that is proportional to the intensity of the field. The principal sources for producing fluctuating magnetic fields are the movement of spins in space due to molecular motion or due to molecular rotation. The principal mechanisms by which these fields are produced are
• dipole-dipole interactions with other nuclei – generally dominant mechanism in
solution; most important for I = 1/2 nuclei with nearby protons • chemical shift (or shielding) anisotropy – molecular tumbling of a nonspherical
distribution of electron density causes the local magnetic field acting on a nucleus to change the shielding of the Bo field
• scalar interactions – indirect spin-spin coupling of nuclear spins through elec-trons; the nuclei are either involved in chemical exchange or one of the magnetic moments is that of an electron (and thus requires a quadrupolar nucleus)
• spin-rotation interactions – molecular collisions interrupt the coupling between rotational angular momentum of the electrons and nuclear spin
• quadrupolar interactions – nuclei with I > 1/2 have a nonspherical nuclear charge distribution and possess an electric quadrupole moment that can interact with electric fields
Attributes of 1H NMR Spectroscopy
NMR Chemical Shifts
610Hzin frequency er spectromet
Hzin reference) offrequency signal of (frequencyδppm shift, chemical ×
− =
The chemical shift is the position on the δ scale (in ppm) where the peak occurs. In proton and 13C NMR the reference at 0 ppm is the chemical shift of tetramethylsilane, (CH3)4Si (i.e., TMS).
10
Table III – Proton Chemical Shifts
There are two major factors that influence chemical shifts:
• deshielding due to reduced electron density (e.g., due to electronegative atoms) • anisotropy due to magnetic fields (e.g., those generated by π bonds)
Shielding in NMR
Nuclei are shielded by valence electrons surrounding them which circulate in an ap-plied magnetic field producing a local diamagnetic current in the opposite direction. This diamagnetic shielding will affect the frequency of radiation necessary to cause a nucleus to spin flip (the resonance frequency). Therefore nuclei will absorb radiation of slightly different frequency depending upon their local magnetic environment which is deter-mined by the structure of the compound. Hence magnetically different types of nuclei will occur at different chemical shifts. This is what makes NMR so useful for structure determination; otherwise all nuclei would have the same chemical shift. Some important factors include:
• inductive effects by electronegative groups • magnetic anisotropy
Electronegativity
Electrons around the nucleus create a magnetic field that opposes the applied field. This reduces the field experienced at the nucleus. Since the induced field opposes the applied field the electrons are said to be diamagnetic and the effect on the nucleus is re-
11
ferred to as diamagnetic shielding. Since the field experienced by the nucleus defines the energy difference between the different spin states, the frequency and hence the chemical shift δ will change depending on the electron density around the nucleus. Electronegative groups decrease the electron density around the nucleus, and there is less shielding (i.e. deshielding) so the chemical shift increases.
Magnetic Anisotropy Magnetic anisotropy means that there is a non-uniform magnetic field. Electrons in
π systems (e.g. aromatics, alkenes, alkynes, carbonyls, etc.) interact with the applied field which induces a magnetic field that causes the anisotropy. As a result, the nearby nuclei will experience three fields: the applied field, the shielding field of the valence electrons, and the field due to the π system. Depending on the position of the nucleus in this third field, it can be either shielded (smaller δ) or deshielded (larger δ), which implies that the energy required for and the frequency of the absorption will change
Magnetic Anisotropy
Different ways to express the relative chemical shifts are summarized in Table IV.
12
Table IV. Nomenclature to Express Relative Peak Positions in NMR
low field high field down field up field deshielded shielded less electron density more electron density high frequency low frequency large δ (ppm) small δ (ppm)
Chemical Shifts in 1H NMR Magnetically different types of nuclei will occur at different chemical shifts resulting
in an NMR spectrum which contains peaks for each of these different types of nuclei.
Coupling in 1H NMR
Spectra generally have peaks that appear in clusters due to coupling (referred to as scalar, spin-spin, or J-coupling) with neighboring protons The coupling constant J (measured in frequency units, Hz) is a measure of the interaction between a pair of pro-tons and is independent of the magnetic field strength. The interaction is through chemi-cal bonds via coupling of the nuclear spins with the spin of the electrons and rapidly de-creases with the number of bonds.
13
Before addressing the coupling, examine the peak assignments in the above spectra:
• δ = 5.9 ppm, integration = 1H; deshielded: agrees with the −CHCl2 unit • δ = 2.1 ppm, integration = 3H; agrees with −CH3 unit.
What about the coupling patterns? Coupling arises because the magnetic field of
adjacent protons influences the field that the proton experiences. To understand the im-plications of this, first consider the effect the −CH group has on the adjacent −CH3. The methine −CH can adopt two alignments with respect to the applied magnetic field, one which deshields neighboring protons and the other which shields them. As a result the
methyl −CH3 is split into a doublet, two lines of equal intensity due to the equal prob-ability of the methine proton being aligned either parallel or anti-parallel to the applied field. Remember that the excess of spins in the lower energy state
is only very, very slightly larger than the number in the higher energy state. When con-sidering coupling patterns, for practical purposes, they can be considered to be equal.
Now consider the effect the −CH3 group has on the adjacent −CH. The methyl -CH3 protons have 8 possi-ble combinations with respect to the applied field, only four of which are magneti-cally distinct. The re-sulting signal for the adjacent methine −CH is a quartet, four
lines with the intensity ratio 1:3:3:1.
14
n + 1 Rule
As protons on a carbon atom experience the magnetic field of protons on adjacent carbon atoms the signal for a particular proton will be split by these protons into n + 1 peaks where n is the number of adjacent protons. This rule can be extended to any spin 1/2 nucleus. Pascal’s Triangle
The relative intensities of the lines in a coupling pattern are given by a binomial expansion or more conveniently by Pascal's triangle. Individual resonances are split due to coupling with n adjacent protons. The number of lines in a coupling pattern is given, in general, by 2nI + 1 for coupling with n spin I nuclei. Interpreting 1H NMR Spectra What can be obtained from a 1H NMR spectrum:
1. number of equivalent types of H – number of groups of signals in the proton NMR spectrum
2. types of H – chemical shift of each group; protons found in chemically identical
environments are chemically (and usually also magnetically) equivalent; chemi-cally equivalent protons will have the same chemical shift.
3. number of H of each type – NMR spectrometer can integrate all peaks (deter-
mine the area under each peak) to determine the relative numbers of protons re-sponsible for all peaks.
4. connectivity – spin-spin splitting (J coupling with the n + 1 rule); coupling pat-
tern gives what is adjacent to each group of protons
15
Chemical shift
• position on the δ scale (in ppm) where the peak occurs • major factors influencing shifts: 1) deshielding due to reduced electron density
(electronegative atoms) and 2) anisotropy (magnetic fields generated by π bonds).
Integration
• area of a peak is proportional to the number of H that the peak represents • integral measures the area of the peak • integral gives the relative ratio of the number of H for each peak
Coupling
• proximity of other n H atoms on neighboring carbon atoms, causes the signals to be split into n +1 lines (to first order).
• this is also known as the multiplicity or splitting of each signal. Table V. Magnitude of Some Typical Coupling Constants1
1Magnitude of the coupling constant is independent of the strength of the applied field.
It is useful to compare and contrast 1H NMR and C NMR:
• C isotope does not exhibit NMR behavior (nuclear spin I = 0)
r than γ for 1H in
• seldom observed due to the low natural abundance of C )
ing J coupling be-
no meaningful peak
he ge of these points are that 13C take longer to acquire, though they
1
able VI. Carbon Chemical Shifts
13C chemical shifts in the order: sp2 < sp < sp3. In the following are three ex mples of the simplicity of 13C spectra over 1H spectra which also illustrate the large range of the carbon chemical shifts.
13
12
• 13C isotope has a natural abundance of 1.108% (of all C atoms) • Magnetogyric ratio γ for 13C is approximately four times smalle• As a result, a 13C nucleus is about 400 times less sensitive than an 1H nucleus
NMR spectroscopy 13C - 13C coupling is 13
• Chemical shifts measured with respect to tetramethylsilane, (CH3)4Si (i.e., TMS• Chemical shift range is normally 0 to 220 ppm
13 as in 1H NMR • Similar factors affect the chemical shifts in C 13• C spectra are normally broadband proton decoupled, remov
13 1tween C and H, so peaks appear as single lines • Number of peaks indicates the number of distinct types of C • Long T1 relaxation times (excited state to ground state) mean
area integrations
Tte
neral implicationsnd to look simpler. Overlap of peaks is much less common than for H NMR which
makes it easier to determine how many distinct types of C are present. T
Note the importance of hybridization in the shielding of
a
17
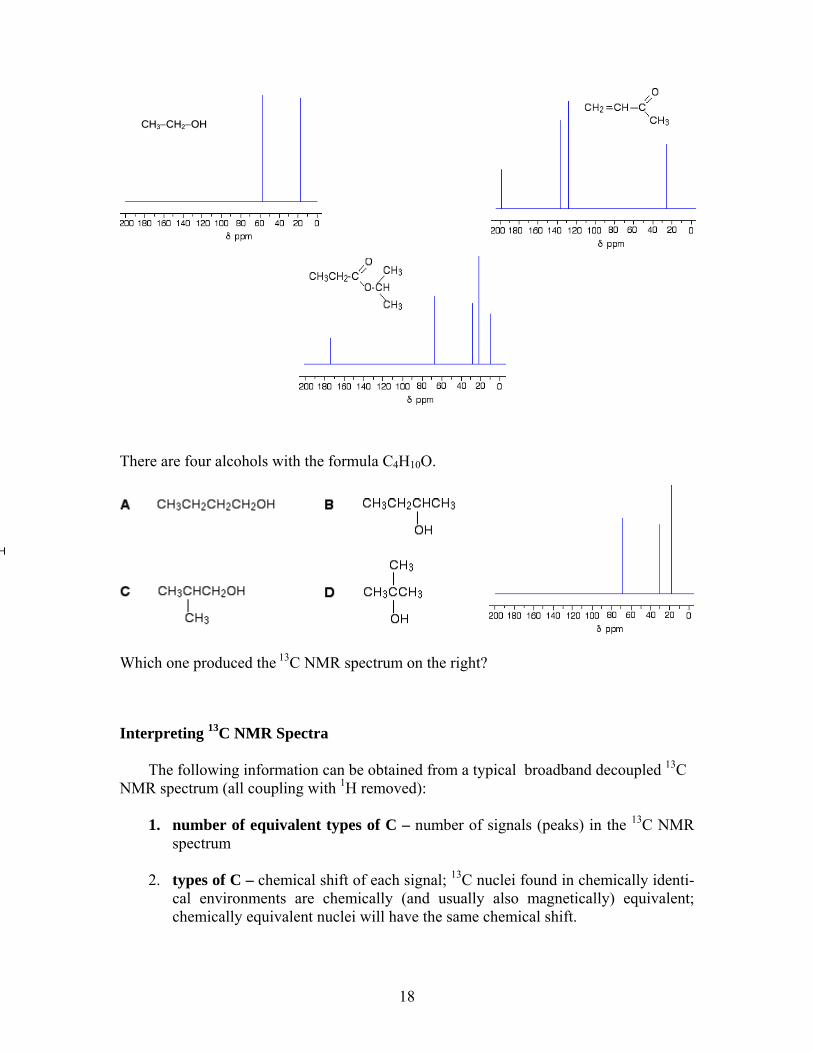
There are four alcohols with the formula C4H10O.
Which one produced the 13C NMR spectrum on the right?
The following information can be obtained from a typical broadband decoupled 13C MR spectrum (all coupling with 1H removed):
1. number of equivalent types of C – number of signals (peaks) in the 13C NMR
. types of C – chemical shift of each signal; 13C nuclei found in chemically identi-
equivalent nuclei will have the same chemical shift.
Interpreting 13C NMR Spectra
N
spectrum 2
cal environments are chemically (and usually also magnetically) equivalent; chemically
H
CCHH33−−CCHH22−−OOHH
18
NMR Experiments to Aid Spectral Interpretation
h dimension of an NMR experiment represents a different obs1 13
Eac ervable nucleus.
ormal H and C NMR examine a single type of nucleus at one time by plotting inten-sity versus frequen using the Fourier
ansform (FT) technique coupled with a computer capable of directing RF pulses on both nucl
o carbon (via the process of polarization transfer). The gain arises due to the larger population differences associated with
90 decoupler pulse - carbon spectrum contains only carbons with a single at-
• 135 decoupler pulse - carbon spectrum with methyl (CH3) and methine (CH) car-
B. HETCOR (HMQC): 2D experiment used to identify couplings between
hetewith th e 2D spectrum. It
lies on scalar coupling (spin-spin or J coupling) between the different nuclei. The HET
enerally used to analyze coupling relationships between protons but may e used to correlate any high-abundance homonuclear spins. The COSY spectra in our
Ncy. One can examine multiple nuclei simultaneously
trei during the same time period. In such experiments intensity is plotted as a function
of two frequencies generally in the form of a contour plot. This part of the NMR lab will examine three one-dimensional techniques, normal 1H and 13C NMR and DEPT, and two two-dimensional NMR techniques, HETCOR and COSY.
A. DEPT: 1D experiment used for enhancing the sensitivity of carbon signals and
for editing 13C spectra. The sensitivity gain comes from starting the experiment with proton excitation and transferring the magnetization ont
1H, which are four times those of 13C (γ is four times larger). The editing feature alters the amplitude and sign of the carbon resonances according to the number of directly bonded protons:
• 45o decoupler pulse - carbon spectrum contains only carbons with protons at-
tached (quaternary carbons are not observed).
o•tached proton, methine CH
o
bon peaks up, methylene (CH2) carbon peaks down (negative).
ronuclear spins separated by one bond. Most often employed to correlate carbons eir directly bonded protons by the presence of cross-peaks in th
reCOR spectra in our experiments plot proton versus carbon with the 1D spectra
displayed along the appropriate axes. The 2D peaks show which protons are coupled to which carbons. C. COSY: 2D experiment used to identify nuclei that share a scalar (J) coupling. The presence of off-diagonal peaks (cross-peaks) in the spectrum directly correlates the cou-pled partners. Gbexperiments plot the proton spectrum versus itself. The 2D peaks show which 1H are coupled over three bonds.
19
A. DEPT: Distortionless Enhancement by Polarization Transfer
A 1D experiment that utilizes polarization transfer from a nucleus with a relatively rger magnetogyric ratio γ with a smaller γ to incre e signal from the latter
pulse from 45 to 0 to 135o the multiplicity of the carbon nucleus can be determined.
at when 1) θ = 45o
signals from all CH, CH , and CH
at 45, 90, and 180 for the compound on the
tify which of A-E below is
la to one ase thnucleus, here from 1H to 13C. By changing the length of the last proton9
Pulse sequence: Observed 13C signals are mod- ulated by the 13C−1H coupling con- constant so th
2 3carbons are observed (no qua-ternary C or C attached to D, as in a deuterated solvent), 2) θ = 90o sig-nals seen from only CH carbons, and 3) θ = 135o signals from all CH, CH2, and CH3 carbons but the CH2 signals are negative.
On the right are the proton decoupled 13C spec-
trum and DEPT spectra o
left. DEPT 45 only shows C with a directly bonded H, DEPT 90 CH, and DEPT 135 shows carbon with a di-rectly bonded H but peaks for CH2 carbon atoms are negative.
The DEPT 90 and 135 spectra on the left are sufficient to iden
the structure of the compound. On top is the normal 1H de-coupled 13C spectrum.
20
B. HETCOR: HETeronuclear CORrelation (also 13C/1H COSY or 13C/1H HMQC)
A 2D heteronuclear correlation experiment where cross peaks yield information y of
nuclei. The experiment takes advantage of the large one-bond heteronucler J coupling for polarization transfer between the H and C nuclei. The experiment can be modified to give coupling information ove
polarization transfer. It encodes the proton
the
1D
13
served at inter-
about the connectivit two different spin coupled spin 1/2 nuclei, here protons with 13C
1 13
r more than one bond.
Pulse sequence: The HETCOR ex- periment involves 13C−1H correlation by
chemical shift informa-tion into observed 13C signals and yields cross signals for all 1H and 13C nuclei that are connected by 13C−1H coupling over one bond.
1H NMR plotted vs. 1D
C NMR; cross peaks ob-
section of the x and y values denoting the CH interac-tions. Peak A shows that the H at ~ 4 is bonded to the C at ~ 60 ppm. Peak B shows that H at ~ 1.8 ppm is bonded to C at ~ 18 ppm. The qua-ternary C is identifiable as no cross-peak appears (*).
1H Spe
NMR ctrum
13C NMR Spectrum
*
21
C. COSY: COrrelation SpectroscopY
A 2D homonuclear correlation experiment where cross peaks yield information on the
Pulse sequence: COSY experiment involves
1D 1H NMR plotted vs.
protons which are spin-spin coupled to each other. The experiment uses polarization transfer between the coupled spins. The technique can be modified to yield COSY spec-tra for four-, five-, and occasionally six-bond couplings.
1H−1H correlation by polari- zation transfer. It encodes the proton coupling infor-mation into the observed 1H signals and yields cross signals for all 1H that are coupled over three bonds.
1D 1H NMR generat-ing a 2D xy plot. If a signal on the x-axis has an interaction with a signal on the y, a cross- peak is observed at the intersection of the x and y values, denoting the interaction. The peaks on the diagonal represent the 1H spec-trum and the COSY is symmetric with respect to the diagonal. Peak A indicates that the peak at ~ 6.9 ppm is proton coupled to the peak at ~ 1.8. Peak B indicates that the peak at ~ 4.2 ppm is coupled to the 1H at ~ 1.3.
1H NMR Spectrum
1H NMR Spectrum
22
Web References
. Jim Clark emguide.co.uk/analysis/nmrmenu.html#top
1http://www.ch 2. Joseph P. Hornak, Rochester Institute of Technology http://www.cis.rit.edu/htbooks/nmr/bnmr.htm 3. Ian Hunt, University of Calgary
s/351/Carey5th/Ch13/ch13-2dnmr-1.html#cosyhttp://www.chem.ucalgary.ca/courseOn-Line Learning Center for "Organic Chemistry" (Francis A. Carey), University of Cal-gary, http://www.chem.ucalgary.ca/courses/351/Carey/Ch13/ch13-nmr-1.html 4. Tad Koch, University of Colorado
port/nmrtheory/main.htmlhttp://orgchem.colorado.edu/hndbksup 5. Brent P. Krueger, Hope College
hem348_2002/NMR/Principles_of_NMR_Spectroschttp://www.chem.hope.edu/~krieg/Copy.html 6. Arvin Moser, Advanced Chemistry Development, Inc (ACD Labs) http://acdlabs.typepad.com/elucidation/hsqchmqc 7. Tom Newton, University of Southern Maine
3/Lectures/DEPT/DEPT.htmlhttp://www.usm.maine.edu/~newton/Chy251_25 8. Hans J. Reich, University of Wisconsin
605/index.htmhttp://www.chem.wisc.edu/areas/reich/chem 9. William Reusch, Michigan State University: