26
Paediatric Transplantation in Metabolic Disease Jo Page November 2011
| Date post: | 18-Dec-2015 |
| Category: |
Documents |
| Upload: | asher-poole |
| View: | 213 times |
| Download: | 0 times |

Paediatric Transplantation in Metabolic Disease
Jo Page
November 2011

Introduction
Biology of disease Severity of disease Pre transplant care Post transplant care Manchester Data Summary
Genetic-Inherited Disease-MPS I
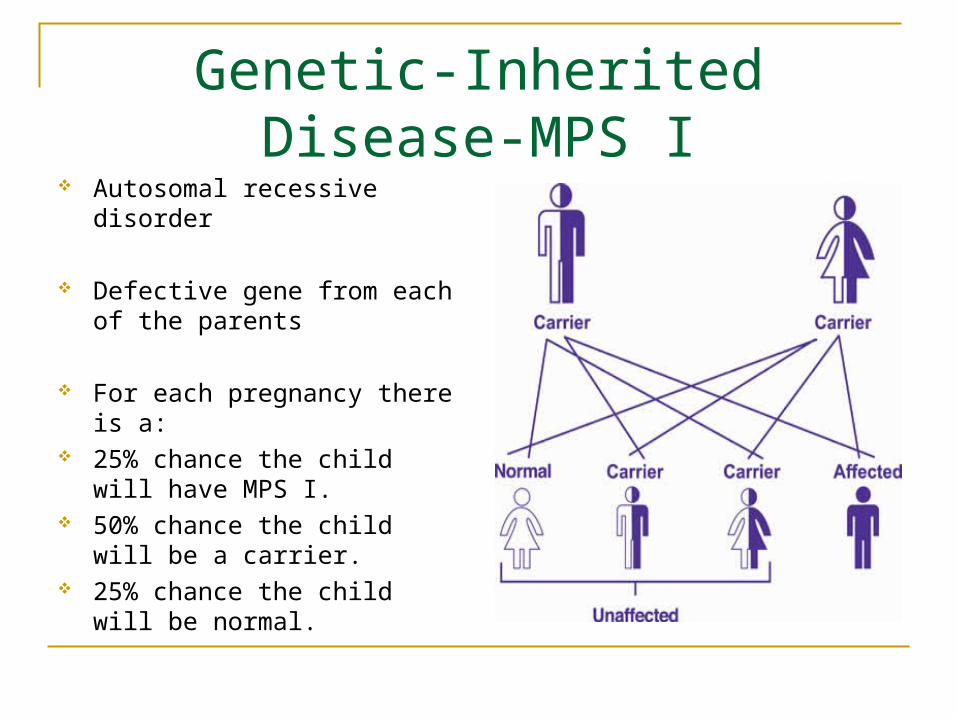
Autosomal recessive disorder
Defective gene from each of the parents
For each pregnancy there is a: 25% chance the child will have
MPS I. 50% chance the child will be a
carrier. 25% chance the child will be
normal.

Metabolic Disease-MPS I
MPS I (Hurlers Disease) is a Lysosomal Storage Disorder. Incidence 1:100 000
Proteins, carbohydrates and fats are broken down in the lysosomes within all cells by certain enzymes
The enzyme α-L-iduronidase breaks down complex proteins into simple proteins
In MPS I, the missing enzyme is α-L-iduronidase and it is found in every cell in the body
Without this enzyme the cell accumulates protein causing an alteration of cell, tissue and organ function

A LYSOSOMAL STORAGE DISEASE
NucleusAbnormal cell
Nucleus
Lysosomes with accumulated
substrate
Normal cell lysosomes

Severity of Disease
Severe Less Severe
a-L-iduronidase deficiency
Hurler
Hurler Scheie
Scheie
50-80 % patients have MPS I H

Spectrum of Phenotypes
MPS I H MPS I H S MPS I S
Mean age at diagnosis 1
Before 1 y
[0,2 y – 7 y]
4 y
[0,2 y – 36 y]
9 y
[2 y – 54 y]
Cognition Pronounced mental delay with loss of
acquired skills
No or mild mental delay
Learning disabilities
No impairment
Life expectancy
Mean age of survival 2
7y
Estimated20 years
Estimated
Adulthood
Each patient is unique and may exhibit a distinct clinical courseDiagnostic delay is common, and partly due to a lack of disease
awareness

How MPS I affects the body
Progressive multi-organ involvement
Brain Eyes Ears, nose and throat Lungs Heart Liver Spleen Joint and bones
Onset of disease in first 2 years of life
Neurological regression Corneal clouding Enlarged tonsils/adenoids Respiratory/ENT Infections Valvular heart
disease/Cardiomyopathy Hepatomegaly/Splenomegaly Multiple skeletal
deformities/joint stiffness Hernias

Corneal clouding
Severe Hip Dysplasia
Severe vertebral abnormalities

Severe MPS I

Pre Transplant
Enzyme Replacement Therapy given weekly in an attempt to improve/stabilise the child’s medical condition prior to BMT. ERT is given currently for approx 12 weeks prior and 2-4 weeks post HSCT:
Donor search Information for families Genetic counselling Recipient baseline investigations: (ENT, Opthalmology,
Cardiology, Physiotherapy, Radiology, Dental, Child Psychology, Sleep Study, Virology)
Sibling donor Investigations: (HTA, IMA, Virology) Additional factors (geography, family needs, complex
families)

Why HSCT?
• In HSCT donor cells repopulate the blood system and release enzyme which cross-corrects affected cells
• Blood cells cross the brain barrier and secrete enzyme cross-correcting neuronal cells
• Ideally HSCT should be undertaken in children under the age of 2 years before they have significant neuro-developmental involvement. Children tend to reach a developmental plateau before beginning to decline
• HSCT can reverse many of the somatic manifestations of the disease including:
Obstructive sleep apnoea, Hepatosplenomegaly, and Cardiomyopathy (but not valve disease) Reduce coarse facial features Importantly appears to stabilise the neurological decline.

Planning
Individual Assessment: Age at diagnosis, Symptoms at diagnosis, Mutational analysis, Family history.
Donor availability
Risk of therapy• short term mortality risk• long term morbidity risk
Risk of no therapy i.e. risk of underlying disease
Multiple therapies (ERT + HSCT). ERT alone does not cross brain barrier and can develop antibodies

Transplantation
Donor Hierachy: Sibling (unaffected) Mud cord rather than Mud adult if equivalent match 6/6 cord> 10/10 Mud>5/6 cord>9/10 Mud>4/6 cord
Busulfan: IV with PK monitoring, 16 doses over 4 days
Cyclophosphamide: 4 doses
ERT: given longer if specific pre transplant co-morbidities eg cardiac
Serotheraphy: ATG if cord, Alemtuzemab if MUD, None if sibling
GCSF prophylaxis: Ciclosporin/Prednisolone if cord, Ciclosporin / MMF if MUD PBSC or Ciclosporin alone if MUD marrow, Ciclosporin/short Methotrxate if sibling

EFS after UCBT by number of HLA disparities and CD34+ at collection (n=116)
6050403020100
1,0
,9
,8
,7
,6
,5
,4
,3
,2
,1
0,0
HLA 6/6 n= 22 81±8 %
HLA 5/6 n= 66 68±6 %
HLA 4 or 3/6 n= 28 57±9 %
P=0.166050403020100
1,0
,9
,8
,7
,6
,5
,4
,3
,2
,1
0,0
<2.8 x105/kg 60±7 %
>2.8 x105/kg 77±6 %
P=0.046

Post Transplant
Monitor enzyme levels
Monitor chimerism
Monitor urine Gags (Substrate of iduronidase Glycosaminoglycans stored in the urine)
Annual assessment


FOLLOW UP CLINIC FOR POST HSCT CHILDREN
MPS SOCIETY
CLINICAL NURSE SPECIALIST
METABOLICCONSULTANT
ENDOCRINOLOGIST(Over 6yr old)
BMTCONSULTANT (under 6yr old)
PHYSIOTHERAPIST
SPINAL CONSULTANT
POST BMT PATIENT

Post Transplant

Manchester
2 main centres in UK From September 2004: 33 patients receiving 35
grafts Median age at ERT 9 months (3-19 months) Median age at HSCT 14 months (2-22 months) Oral Busulfan N=7, IV Busulfan N=26 Family donors N=10 (5 MSD, 4 PBSC, 1 9/10) Unrelated donors N=23 (18 cord, one received 2, 3
MUD)

Transplants for MPS I
0
1
2
3
4
5
6
7
8
2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010
Year
No
of
pat
ien
ts
no of tx

Outcome
2 primary graft failure 1 Rescued with immediate second cord after RIC (RIP 12 months Adeno) 1 later successful MUD after “autologous” back up given on ICU
All other patients survived
Single limited GvHD (skin)
All cord fully engrafted and maintain full donor chimerism, others acceptable with adequate enzyme level

Manchester’s Survival

Summary
MPS I is a complex and rare disorder that is treatable (but not curable) by HSCT-the disease is stabilised at time of transplant (higher level of donor enzyme is thought to be more effective in improving organ function)
The treatment choice is dependent upon factors at diagnosis.
Clinical data from MPSI HSCT in Manchester (transplant outcome) show improved results that are also in accordance with European data from EBMT Registry
Aim of both HSCT and ERT is to prevent further damage caused by the disease (limitation in bone/joint abnormalities)
Enzyme Delivery is the future of HSCT by raising awareness of disease and newborn screening
All patients will have long term follow up and assessments to achieve the best outcome and quality of life

Publications
“Outcomes of HSCT for Hurler Syndrome in Europe: a risk factor analysis for graft failure” BMT, 2007, 40, 225-33
Reduced conditioning, T cell depletion and no Bu pK monitoring leads to increased risk of graft failure
“Risk Factor analysis of outcomes after unrelated cord transplantation in patients with Hurler Syndrome”. BBMT, 2009, 15, 618-25
Early transplantGood cord matchUse of Bu Cy

Thank You !

















