ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2017 Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy 233 Palladium(II)-Catalyzed Addition Reactions Synthesis of Aryl Amidines and Aryl Ketones JONAS RYDFJORD ISSN 1651-6192 ISBN 978-91-513-0012-2 urn:nbn:se:uu:diva-326816
Transcript
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Pharmacy 233
Dissertation presented at Uppsala University to be publicly examined in B22, BMC,Husargatan 3, Uppsala, Friday, 15 September 2017 at 09:15 for the degree of Doctor ofPhilosophy (Faculty of Pharmacy). The examination will be conducted in English. Facultyexaminer: Professor Adriaan Minnaard (University of Groningen).
AbstractRydfjord, J. 2017. Palladium(II)-Catalyzed Addition Reactions. Synthesis of ArylAmidines and Aryl Ketones. Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Pharmacy 233. 97 pp. Uppsala: Acta Universitatis Upsaliensis.ISBN 978-91-513-0012-2.
Palladium-catalyzed reactions have become one of the most important tools in modern organicchemistry due to its ability to catalyze the formation of new carbon-carbon bonds.
The aim of the work presented in this thesis was to develop new palladium(II)-catalyzedaddition reactions. In this work, cyanamides were investigated as a new substrate to give arylamidines as products. The first protocol developed employed aryltrifluoroborates as the arylpartner, and the insertion of the aryl group into un-, mono-, and di-substituted cyanamideswas successful for a wide variety of aryltrifluoroborates. An alternative method of generatingthe necessary intermediate for insertion into the cyanamide is the decarboxylative formationof aryl-palladium from aryl carboxylic acids. A protocol was developed for this reaction, butwas unfortunately limited to a small number of ortho-substituted electron-rich aryl carboxylicacids. The mechanism was investigated by the means of DFT calculations and ESI-MS studies,and the rate-determining step was suggested to be the 1,2-carbopalladation based upon thoseresults. A translation of the batch protocol to continuous-flow conditions was also demonstrated.The ideal method of generating the aryl-palladium species is by C-H bond activation, and thisapproach was demonstrated with indoles, giving a variety of 3-amidinoindoles as products. Themechanism was investigated by DFT calculations and a plausible catalytic cycle was proposed.
A continuous-flow application of a desulfitative palladium(II)-catalyzed addition to nitrilesto give ketones was developed. In addition, different reactor materials were evaluated in themicrowave heated reactor cavity. Thus the reaction was shown to proceed with microwaveheating in a borosilicate glass and an aluminum oxide reactor, and also in conditions mimickingconventional heating in a silicon carbide reactor.
Finally, a protocol was developed for the convenient synthesis of sodium aryl sulfinates fromGrignard and lithium reagents using a solid sulfur dioxide source as a safe alternative to the gas.The products of this protocol can be used as aryl-palladium precursors by a desulfitative process.
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Sävmarker, J., Rydfjord, J., Gising, J., Odell, L. R., Larhed, M.
(2012) Direct Palladium(II)-Catalyzed Synthesis of Arylamidines from Aryltrifluoroborates. Organic Letters, 14(9):2394-2397
II Rydfjord, J., Svensson, F., Trejos, A., Sjöberg, P.J.R., Sköld, C., Sävmarker, J., Odell, L.R., Larhed, M. (2013) Decarboxylative Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Aryl Carboxylic Acids: Development and Mechanistic Investigation. Chemistry – A European Journal, 19(41):13803-13810
III Rydfjord, J., Skillinghaug, B., Brandt, P., Odell, L.R., Larhed, M. Route to 3-Amidino Indoles via Pd(II)-Catalyzed C-H Bond Activation. Organic Letters, 19(15):4066-4069
IV Skillinghaug, B., Rydfjord, J., Sävmarker, J., Larhed, M. (2016) Microwave Heated Continuous Flow Palladium(II)-Catalyzed Desulfitative Synthesis of Aryl Ketones. Organic Process Research & Development, 20(11):2005-2011
V Skillinghaug, B., Rydfjord, J., Odell, L.R. (2016) Synthesis of sodium aryl sulfinates from aryl bromides employing 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct (DABSO) as a bench-stable, gas-free alternative to SO2. Tetrahedron Letters, 57 (5):533-536
Reprints were made with permission from the respective publishers.
Papers Not Included in This Thesis
2014 Konda, V., Rydfjord, J., Sävmarker, J., Larhed, M. Safe Palladium-Catalyzed Cross-Couplings with Microwave Heating Using Continuous-Flow Silicon Carbide Reactors. Organic Process Research & Development, 18(11):1413-1418
2014 Skillinghaug, B., Sköld, C., Rydfjord, J., Svensson, F., Behrends, M., Sävmarker, J., Sjöberg, P.J.R., Larhed, M. Palladium(II)-Catalyzed Desulfitative Synthesis of Aryl Ketones from Sodium Arylsulfinates and Nitriles: Scope, Limitations, and Mechanistic Studies. The Journal of Organic Chemistry, 79(24):12018-12032
2013 Rydfjord, J., Svensson, F., Fagrell, M., Sävmarker, J., Thulin, M., Larhed, M. Temperature measurements with two different IR sensors in a continuous-flow microwave heated system. Beilstein Journal of Organic Chemistry, 9(1):2079-2087
1.3 Mechanistic Investigations ................................................................. 24 1.3.1 Density Functional Theory Calculations .................................... 24 1.3.2 Electrospray Ionization Mass Spectrometry for Investigation of Live Reaction Mixtures ................................................................... 25
1.4 Amidines ............................................................................................ 27 1.4.1 Synthesis of amidines ................................................................. 29
1.5 Continuous-flow Chemistry ............................................................... 30 1.5.1 Heating by a Non-Resonant Microwave Applicator ................... 31
2. Aims of the Present Study ......................................................................... 35
3. Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Cyanamides and Aryltrifluoroborates (Paper I)................................................................. 36
3.1 Background ........................................................................................ 36 3.2 Optimization of Reaction Conditions ................................................. 36 3.3 Investigation of the Scope of the Reaction ......................................... 38 3.4 Suggested Mechanism ........................................................................ 41
4. Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Cyanamides and Aryl Carboxylic Acids (Paper II) ........................................................... 42
4.1 Background ........................................................................................ 42 4.2 Optimization of Reaction Conditions ................................................. 43 4.3 Investigation of the Scope of the Reaction ......................................... 45 4.4 Continuous-Flow Scale-Out ............................................................... 48 4.5 Mechanistic Investigation................................................................... 49
4.5.1 DFT Calculations ........................................................................ 50 4.5.2 Live Reaction Studies Using ESI-MS ........................................ 52
5. Palladium(II)-Catalyzed Synthesis of 3-Amidino Indoles from Cyanamides and Indoles (Paper III) .............................................................. 54
5.1 Background ........................................................................................ 54 5.2 Optimization of Reaction Conditions ................................................. 55 5.3 Investigation of the Scope of the Reaction ......................................... 56 5.4 Mechanistic Investigation................................................................... 59
6.1 Background ........................................................................................ 64 6.2 Reactor Properties .............................................................................. 65 6.3 Optimization of Reaction Conditions ................................................. 66 6.4 Investigation of the Scope of the Reaction ......................................... 69
7. Synthesis of Sulfinates Using a Solid SO2 Source (Paper V).................... 71 7.1 Background ........................................................................................ 71 7.2 Optimization of Reaction Conditions ................................................. 72 7.3 Investigation of the Scope of the Reaction ......................................... 73 7.4 Application in Palladium(II)-Catalysis .............................................. 75
Ar bpy CMD CSA DABSO DAF DCM DFT DMA DMF dmphen dppp equiv FDA IR 6-Mebpy MSA MW NMA NMF NMP NMR OAc PES pTSA TFA TFMSA TS
Aryl 2,2'-bipyridine Concerted metalation-deprotonation Camphorsulphonic acid 1,4-diazabicyclo[2.2.2]octane bis(SO2) adduct 4,5-diazafluoren-9-one Dichloromethane Differential functional theory N,N-Dimethylacetamide N,N-Dimethylformamide 2,9-dimethyl-1,10-phenanthroline 1,3-bis(diphenylphosphino)propane Equivalent U.S. Food and Drug Administration Infrared 6-Methyl-2,2'-bipyridyl Methylsulphonic acid Microwave N-Methylacetamide N-Methylformamide N-Methylpyrrolidone Nuclear Magnetic Resonance Acetate Potential energy surface p-Tolylsulphonic acid Trifluoroacetic acid Trifluoromethylsulphonic acid Transition state
11
1. Introduction
1.1 Catalysis The term catalysis was first coined by Jöns Jacob Berzelius in 1835.1,2 In 1894, Wilhelm Ostwald arrived at the notion that catalysis is an acceleration of a reaction by the presence of a foreign substance.3 Today the IUPAC Gold Book defines a catalyst as a “substance that increases the rate of a reaction without modifying the overall standard Gibbs energy change in the reaction”, and ca-talysis as “the action of a catalyst”.4,5 In Figure 1, this is illustrated in a simplified energy diagram. The energy barrier (or free energy of activation) of the reaction is lowered in the presence of the catalyst even though the en-ergies of the reactants or products are not changed. The catalyst should not be consumed during the reaction.
Figure 1. Schematic representation of a catalyzed reaction. The energy of the reac-tants and products are the same for the catalyzed and non-catalyzed reaction, however, the energy barrier of the reaction is lowered with the catalyst present.
12
Catalysis can be subdivided into heterogeneous and homogeneous catalysis (Figure 2). In heterogeneous catalysis, the catalyst and the reactants are in dif-ferent phases, most often with the catalyst as a solid and the reaction occurring on its surface. Some prominent examples of heterogeneous catalysis include the Haber-Bosch process for synthesis of ammonia from nitrogen and hydro-gen gas,6 palladium on charcoal7 or Raney nickel7–9 catalyzed hydrogenations, and the Ostwald process for producing nitric acid.10,11 In homogeneous catal-ysis the catalyst and the reactants are in the same phase, most often a liquid. Prominent examples of homogeneous catalysis include the Wacker oxida-tion,12–14 the Monsanto process15 and the Shell higher olefins process.16 Heterogeneous catalysis have found more use in industrial applications due to (generally) easier separation of the catalyst from the product while homoge-neous catalysis generally excels in terms of selectivity.17
Figure 2. Types of catalysis (main classification based on aggregation state)
Homogeneous catalysis is usually divided into four categories depending on the nature of the catalyst. Acid/base catalysis is one, exemplified by acid cat-alyzed esterification reactions. Biocatalysis is another, and this field mostly deals with enzymatic catalysis. In transition metal catalysis, the catalyst is a transition metal such as palladium, platina or nickel. Organocatalysis is a ra-ther young field, with little activity until the late 1990s, and the catalysts are, in these cases, small organic molecules such as proline derivatives.18
This thesis will deal with transition metal catalysis, in particular palladium(II)-catalyzed reactions.
13
1.1.1 Palladium Catalysis
The transition metals are characterized by their incompletely filled d orbitals, which also give them properties suitable for catalyzing reactions. The acces-sible d orbitals allow the metal to form π- and σ-bonds to other molecules. The type of reactions a single transition metal catalyzes is mainly dependent on the number of d electrons in the outer shell and other properties such as its size.19
Palladium has some interesting properties (see Table 1) making it espe-cially suitable for catalytic reactions, and especially the formation of new C-C bonds. It has easily accessible HOMO and LUMO orbitals, enabling a broad reactivity. Compared to other transition metals it has a relatively high electronegativity, similar to carbon (2.20 for palladium compared to 2.55 for carbon on the Pauling scale). This makes the carbon-palladium bond relatively non-polar. Palladium favors the oxidation states of 0 (d10) and +2 (d8), and thus undergoes processes like oxidative addition and reductive elimination.
Palladium prefers to bind four ligands to form stable 16- and 18-electron for palladium(II) and palladium(0) complexes, respectively, as each ligand contributes with two electrons. A ligand could be the solvent, a reactant, an additive, or any Lewis base capable of binding to the palladium center. Gen-erally though, the term ligand is used for dedicated molecules that should not react, rather they should modulate and fine-tune the electronic and steric en-vironment of the palladium center. These ligands can be mono- or polydentate and the most common types are phosphine and nitrogen based ligands.
Table 1. Properties of palladium
14
Palladium catalysis is usually divided into palladium(0)-catalyzed and palla-dium(II)-catalyzed reactions, depending on the oxidation state of the metal when it enters the catalytic cycle. Palladium(0)-catalyzed reactions start with the oxidative addition of an electrophile, such as an aryl halide or pseudohal-ide, to generate an organopalladium species that can undergo further reactions. In palladium(II)-catalysis the organopalladium species is formed by a process such as transmetalation, rather than a redox process.
Palladium(0)-catalysis have found widespread use with transformations such as the Mizoroki-Heck,20,21 Suzuki-Miyaura,22,23 Negishi,24 and So-nogashira reaction (Figure 3),25 and is widely applied in medicinal chemistry26 and in the production of fine chemicals.26 Palladium(II)-catalysis on the other hand has long been an overlooked area of chemistry, even though the Wacker process12–14 constitutes an early example of an extremely relevant industrial application.
1.2 Palladium(II)-Catalysis In the late 1950s-1970s the Wacker process,12–14 vinyl acetate synthesis from ethylene and acetic acid,27 the Fujiwara-Moritani reaction,28–32 and the oxida-tive Heck reaction33,34 were discovered, all of which are examples of well-known and useful palladium(II)-catalyzed reactions.
Figure 4. Palladium(II)-catalyzed reactions discovered in the 1960’s to 1970’s. The Wacker process and the vinyl acetate synthesis also represent reactions of great im-portance in industrial applications
After these reports on palladium(II)-catalyzed reactions there was little activ-ity in this research field until the 1990s, when more reports surfaced on a manifold of palladium(II)-catalyzed reactions.
It has been previously mentioned that the difference between palladium(0)- and palladium(II)-catalysis lies in the generation of the initial organopalladium species. The organopalladium derivative is generated in pal-ladium(II)-catalysis by an organyl transmetalation reaction,33–38 C-H bond activation,39,40 a decarboxylation,41,42 or a desulfination.43–46 Following this, a number of processes can occur, such as coordination of and addition to a π-bond, transmetalation or reductive elimination. The regeneration of the cata-lyst often requires stoichiometric amounts of a reoxidant, as many palladium(II)-catalyzed reactions generate palladium hydride or metallic pal-ladium.
16
1.2.1 Aryl-Palladium Precursors The generation of the organopalladium species, often an aryl-palladium spe-cies, is a crucial step in palladium(II)-catalysis as mentioned before. Here will follow an overview of some of the more common aryl-palladium precursors, and in particular those used in this work.
Organoboronic Acids
Figure 5. Boronic acids as aryl-palladium precursors
Boronic acids are arguably the most frequently used organopalladium precur-sor and should thus be mentioned even though only one example of their use is included in this work.
Boronic acids are today attractive coupling partners due to the very good commercial availability, something that was fueled by the success of the Suzuki-Miyaura reaction and other coupling reactions. These substrates are most often air-stable and not very moisture sensitive, which contributes to their popularity and widespread use.47 In addition, boronic acids are relatively benign from both an environmental as well as a toxicological perspective.47
The organopalladium intermediate is generated from boronic acids by transmetalation. Even though this process is thoroughly investigated for palladium(0)-catalyzed reactions,48,49 it is not studied in such great detail for palladium(II)-catalyzed reactions under acidic conditions. Although boronic acids are very useful organopalladium precursors, their use is associated with some drawbacks. Boronic acids can be difficult to purify due to their non-monomeric nature.47,50 Many boronic acids, especially electron-deficient heteroaryl boronic acids, easily undergo protodeboronation, resulting in short shelf-life and requiring storage at lower temperatures. This reaction is also seen in protic solvents, influencing the outcome of reactions carried out in these solvents.50,51
17
Organotrifluoroborates
Figure 6. Trifluoroborates as aryl-palladium precursors
Trifluoroborates pose an interesting alternative to the use of boronic acids in palladium-catalyzed reactions. These compounds are solids with superior shelf-life compared to the corresponding boronic acids. The trifluoroborates hydrolyze to more active transmetalating boron species, which can be used as a strategy for keeping the concentration of the transmetalating species low by controlling the rate of this reaction.52 This can combat side reactions such as protodeboronation and homocoupling observed with the use of certain boronic acids. The trifluoroborates can easily be generated from the corresponding boronic acid by treatment with KHF2.53
Trifluoroborates are now also widely available commercially, much owed to the pioneering work by the groups of Genet,54 Batey,55 and Molander.56
Figure 7. Aryl carboxylic acids as aryl-palladium precursors
Carboxylic acids can decarboxylate spontaneously at moderate temperatures in case of highly activated substrates such as β-keto acids. The use of a metal can facilitate this decarboxylative process, and enable the decarboxylation of less activated carboxylic acids. Pesci showed that mercury could effect the formation of an organomercury species by decarboxylation57 while Shepard58 reported the use of catalytic copper or nickel for protodecarboxylation of electron-deficient furoic acids in 1930. A protocol by Nilsson et al. in 1966 describing a crossed Ullman type coupling reaction between aryl-copper rea-gents and aryl iodides was the first example of a cross coupling using aryl-metal species derived from carboxylic acids.59 Palladium can also effect decarboxylations of aryl carboxylic acids. An intramolecular example using one equivalent of palladium acetate was reported by Peschko et al. in 200041 and an intermolecular reaction was reported by Myers in an oxidative Heck reaction in 200242 using catalytic amounts of palladium.
Palladium-catalyzed decarboxylative reactions are unfortunately limited by the requirement of ortho substituted electron-rich aryl carboxylic acids as sub-strates and can thus not be applied generally as organopalladium
18
precursors.60,61 This ortho substitution requirement can be explained by the mechanism as it has been suggested that the carboxylic acid functionality must rotate out of the plane so that it is orthogonal to the aromatic system for de-carboxylation to occur.62,63 This process is facilitated by ortho substituents as they help to force the carboxylic acid functionality out of the plane.
Meanwhile the applicability of mercury-mediated reactions is limited by the toxicity of mercury and organomercury compounds. Copper and silver are better at facilitating the decarboxylation process and do not show the same requirement of electron-rich ortho-substituted substrates as the corresponding palladium-catalyzed reactions. Thus bimetallic catalytic systems have been developed to combine the more facile decarboxylation process of copper and silver with palladium-catalysis.64,65
Aryl Sulfinic Acid Derivatives
Figure 8. Aryl sulfinic acids as aryl-palladium precursors
A report by Garves43 in 1970 detailing an example of a palladium-catalyzed desulfitative reaction was the first clue to the potential to use aryl sulfinic acids or sulfinates as aryl-palladium precursors. The desulfitative reaction does not require ortho-substituents to proceed and thus represents an alternative to the decarboxylative process. In 2011 a number of papers appeared almost simul-taneously using this approach,44–46 marking new-found interest in this research topic.
Skillinghaug et al.66 reported on a DFT study of the energy requirement for rotation of the acidic functionality of benzenesulfinic acid, benzoic acid, and 2,6-dimethoxybenzoic acid. Their results indicate that the sulfinic acid does in fact have a lower energy requirement of rotating the acid functionality so that it is orthogonal to the plane of the aromatic ring by 13 kJ mol-1. The di-ortho-substituted 2,6-dimethoxybenzoic acid on the other hand has its min-imum energy with the acid functionality orthogonal to the plane of the aromatic ring, representing a 30 kJ mol-1 lower energy than benzoic acid.
The commercial availability of sulfinic acids and sulfinates was limited in 2011, but with an increasing interest in desulfitative reactions the availability of sulfinic acid derivatives is greatly improved at the time of writing this the-sis.
19
C-H bond Activation Substrates
Figure 9. C-H bond activation to generate the aryl-palladium species
To be able to pick any C-H bond in a molecule and functionalize it can be considered somewhat of a holy grail in chemistry. The previous approaches described to generate an aryl-palladium complex all require a functional group as a handle that determines the structure of the aryl-palladium species. Since the installation of a functional group is often cumbersome, this approach may not be ideal, especially not from an atom-economical perspective.
C-H bond activation facilitated by metal catalysts often make use of a di-recting group to bring the catalyst in the vicinity to the C-H bond of interest and/or to modify the reactivity of the catalyst to allow it to react with that carbon.39 Another approach, relevant to this work, is to generate an electro-philic catalyst which reacts preferentially with electron-rich positions of arenes.40
Selective functionalization of electronically and sterically unbiased sub-strates lacking directing groups is inherently difficult and thus limits the applicability of this strategy, although significant progress has been made over the past decade.
1.2.2 Palladium(II)-Catalyzed Addition Reactions The term addition reaction is a broad one that generally describes the addition of two atoms over a π-bond. Commonly encountered addition reactions are electrophilic additions to alkenes and nucleophilic additions to carbonyl com-pounds. In the case of palladium(II)-catalyzed addition reactions the palladium metal helps or enables the reaction by modulating reactivity of the unsaturated substrate and/or the reagents adding to the π-bond. In the case of an organopalladium species adding to a π-bond, the carbopalladation of the π-bond can occur in different manners, either in a 1,1 fashion where palladium and the organyl group adds to the same atom or in a 1,2 fashion, adding to both atoms in the unsaturated substrate (Figure 10). The terminology insertion or migratory insertion is also used, and reflects a change in wording relating to which of the reactants is considered to perform an action. An insertion re-action would involve an intermolecular attack by a nucleophile or an electrophile on the π-bond, while a migratory insertion happens intramolecu-larly with both partners coordinated to palladium. To exemplify in the case of an organopalladium species reacting with a carbon monoxide molecule, this is a 1,1-addition to carbon monoxide by the organopalladium species, by a
20
carbopalladation of the carbon monoxide, or it can be described as the migra-tory insertion of carbon monoxide into the organopalladium bond.
Figure 10. Addition of organopalladium to a π-bond, general scheme. The curved arrows show 1,2-carbopalladation
Examples of palladium(II)-catalyzed addition reactions include additions to alkenes28,29,32,33,67–69 and alkynes70,71 by oxypalladation, aminopalladation, and carbopalladation reactions. Addition to polar unsaturated functional groups such as aldehydes,72 α,β-unsaturated ketones,73 isocyanides,74 and nitriles have also been reported.
1.2.3 Palladium(II)-Catalyzed Addition to Nitriles This thesis will in particular deal with reactions related to the palladium(II)-catalyzed addition to nitriles. In general, this reaction proceeds by the addition of aryl-palladium to a nitrile bond, generating a ketimine upon release from the palladium catalyst. This ketimine is then most often hydrolyzed to the cor-responding ketone in situ. This addition was first reported by Garves in 1970 using aryl sulfinic acids as the aryl-palladium precursor, although using stoi-chiometric amounts of palladium.43 A number of palladium(0)-catalyzed reactions were also reported75–79, these reactions start by the oxidative addition of an aryl-halide and is followed by 1,2-carbopalladation of a nitrile. Then a palladium(II)-catalyzed addition to nitriles, a cyclization, was published by Zhao and Lu in 2002.70 Thereafter came two reports by Zhou and Larock de-tailing the intermolecular reaction between arylboronic acids or arenes with nitriles.80,81 The scope of the intermolecular reaction was later expanded by Zhao and Lu.82,83 After that followed numerous reports on this transformation, employing vari-ous other aryl-palladium precursors.
21
Figure 11. Reported aryl-palladium precursors for the palladium(II)-catalyzed addi-tion to nitriles, giving ketones after hydrolysis of the intermediate ketimine.
Ketones and Ketimines from Arylboronic Acids In the first report by Zhou and Larock detailing the intermolecular reaction between arylboronic acids and nitriles, the scope demonstrated was limited to three arylboronic acids combined with benzonitrile.80 The authors demon-strated the isolation of the corresponding ketones for phenylboronic acid and 4-methylphenylboronic acid, while 2,4,6-trimethylphenylboronic acid af-forded the corresponding ketimine.
Later, the scope of the reaction was further investigated by Zhou and Larock, showing good yields for electron-rich arylboronic acids and lower yield for the more electron-deficient 4-nitrophenylboronic acid.81
Zhao and Lu, employed another catalytic system with bpy as the ligand.82 In this protocol, aliphatic nitriles were shown to work, albeit with low yield, and in general it could be observed that electron-deficient nitriles worked bet-ter than electron-rich nitriles. In another protocol by the same authors, a new catalyst system employing a cationic palladium complex with bpy as the lig-and was reported.83 This generally shows higher yields than the previous protocol by the same authors and the yields of alkyl nitriles are good employ-ing this catalytic system.
Yu et al. reported on a protocol employing a water-soluble palladacycle as a catalyst, thus being able to use water as the sole solvent.84 The trends in terms of reactivity parallel those of the protocols already disclosed at that time.
Two later protocols by Wang et al.85 and Das et al.86 report similar reactiv-ity, with the later protocol delivering impressive yields for a range of nitrile derivatives.
Ketones from Aryltrifluoroborates Aryltrifluoroborates represent a more air- and moisture-stable alternative to boronic acids. The synthesis of aryl ketones from aryltrifluoroborates by palladium(II)-catalysis was first reported by Zhao and Lu in their investigation of possible arylboronic acid derivatives in 2006.82 Several years later, in 2013,
22
a protocol developed especially for the use of aryltrifluoroborates as the aryl-palladium precursor was reported.87 This protocol showed a wide scope with regards to alkyl nitriles, even though it suffers from the same limitation in terms of low yields for electron-deficient aryl-palladium precursors.
Ketones from Aryl Carboxylic Acids A protocol for the decarboxylative synthesis of aryl ketones from aryl carbox-ylic acids was developed by Lindh et al. in 2010.88 The protocol showed a substrate scope consistent with the known limitations of palladium-catalyzed decarboxylations, namely the need for electron-rich ortho-substituted aryl car-boxylic acids. The authors demonstrated the use of several heterocyclic carboxylic acids as well, including derivatives of thiophene, benzothiophene, benzofuran, and pyridine. Interestingly these heteroaryl derivatives are not re-ported starting from arylboronic acids. The scope with regards to different nitriles was limited to liquid nitriles, which were used in large excess as sol-vents.
This decarboxylative reaction was further developed by Axelsson et al. to enable the use of solid nitriles and the nitrile scope was thus expanded to in-clude aromatic, heteroaromatic and benzylic nitriles.89 Additionally the authors demonstrated the use of a pyrrole derivative as the carboxylic acid partner. A computational study suggest that 1,2-carbopalladation is the rate-determining step of the reaction.63 This study also highlighted the importance of a polar solvent to stabilize the positively charged transition states.
Ketones from Aryl Sulfinates The report by Garves43 detailing an example of a desulfitative reaction to give an aryl ketone upon addition to a nitrile, was long overlooked until three pa-pers describing this reaction with catalytic amounts of palladium were published in 2011.44–46
The scope of the reactions described in these three papers with regards to the aryl sulfinic acid or sulfinate does show that the reaction proceeds readily for electron-rich and neutral substrates while the yield drops for electron-de-ficient aryl sulfinates.
In an extended study by Skillinghaug et al.,66 the scope was further studied, demonstrating orthogonality between the decarboxylative approach and the desulfitative, as the use of sodium 2,4,6-trimethylbenzenesulfinate only af-forded 5% of the corresponding product with acetonitrile, compared to 85% reported88 for 2,4,6-trimethylbenzoic acid. The extended study also demon-strated the use of various heterocyclic nitriles and nitriles with other functional groups such as aldehydes and esters present.
Ketones by C-H Bond Activation of Arenes The reports by Zhou and Larock describing boronic acids as aryl-palladium precursors mainly detailed the use of arenes as aryl-palladium precursors by
23
C-H bond activation.80,81 The reaction employs rather harsh conditions, using TFA as the solvent and yields the corresponding product from electron-rich and neutral arenes with the carbon-carbon bond forming at the most electron-rich position of the arene, even though mixtures of different regioisomers were obtained for some substrates.
Ketones by C-H Bond Activation of Indoles Zhou and Larock also tested 1-methylindole as a C-H bond activation sub-strate, with no product formed, and speculated that the substrate was not stable in the reaction conditions employed.81
Using milder conditions, there were two reports in 2013 followed by one in 2014 describing the palladium(II)-catalyzed C-H bond activation of indoles to give 3-acylindoles.86,90,91 Ma et al. demonstrated the use of a number of N-protected and free (N-H) indoles which give the corresponding 3-acylindoles in good yield with various nitriles.90 The papers by Jiang and Wang,91 and Das et al.86 focused on free (N-H) indoles, reporting a scope similar to that of Ma et al. The scope reported in these studies is limited to indoles that are moder-ately electron-deficient to electron-rich.
Ketones by C-H Bond Activation of Thiophenes Jiang and Wang also published a paper on the synthesis of 2-acylthiophenes using palladium(II)-catalyzed C-H bond activation of thiophenes.92 The reac-tion proceeds well with electron-rich and neutral thiophenes as well as various aromatic and aliphatic nitriles.
Hydration of Nitriles to give Amides The use of palladium(II) salts to catalyze the conversion of nitriles into amides was disclosed in a patent in 1972.93 This was followed by several academic publications, examining the reaction in more detail.94–101 The reactions pro-ceed either by addition of water93–99 or by water transfer from an amide100 or acetaldoxime101. This is an interesting transformation, as the amide can be iso-lated, rather than for the acid- or base-catalyzed process where the hydration of the resulting amide is usually faster, resulting in conversion of the nitrile to a carboxylic acid.
24
1.3 Mechanistic Investigations There are many tools available to the chemist when it comes to elucidating the finer details of the mechanism of a reaction. In this thesis, two such techniques have been used to provide a better mechanistic understanding of the reactions investigated.
1.3.1 Density Functional Theory Calculations A computational investigation of a reaction mechanism can be seen as an at-tempt at probing a potential energy surface (PES) where a point on the surface represents a given geometry of a molecule or a system. By making an educated starting assumption, one can optimize the geometry to find a local energy min-imum on the PES followed by an investigation of conformations and isomeric forms to find the lowest energy minimum. Then one can try to find a transition state (TS), which is a saddle point on the PES – a local minimum in all other dimensions except for the reaction coordinate that connects the reactant(s) and the product(s).
Figure 12. A representation of a general reaction profile generated by DFT calcula-tions.
One method of calculating the energy of a given point on the PES is by using density functional theory (DFT) calculations. DFT is a computational method that relies on solving an electron density functional as an approximate solution to the Schrödinger equation. DFT relies on the use of basis sets that are a set of mathematical functions that typically are designed to describe the atomic orbitals. Linear combination of the basic functions then gives a model for the molecular orbitals. DFT is sometimes merged with the Hartree-Fock method, giving hybrid functionals.102 This approach, along with corrections to account for dispersion forces,103 has been used in the DFT calculations presented within this thesis.
DFT calculations provide an excellent tool to investigate a hypothesized mechanism and can, in conjunction with other techniques, provide valuable insight into the mechanism.
25
1.3.2 Electrospray Ionization Mass Spectrometry for Investigation of Live Reaction Mixtures One technique that is suitable for studying sensitive organometallic species is electrospray ionization mass spectrometry (ESI-MS). This is a technique pop-ularized due to its use for identification and structure analysis of biologically relevant macromolecules such as peptides and proteins.104 In 2002, Fenn and Tanaka shared the Nobel Prize in chemistry for their development of this tech-nique, highlighting its importance.
In brief, the technique works by the application of an electric field over a capillary to enrich ions which assist the formation of smaller and smaller drop-lets, eventually leaving the charged analyte after evaporation of the solvent. The technique is thus especially suited for the study of charged species in so-lution. The generated ions are then separated in a mass analyzer before detection. A triple-quadrupole was used as the analyzer in the work described in this thesis. Thus MS/MS techniques such as product ion, precursor ion and neutral loss scan could be used.105
The use of ESI-MS for the study of reaction mechanisms is thoroughly de-scribed in a microreview by Santos,106 and several articles also detail the use of ESI-MS as a tool for studying palladium-catalyzed reactions.45,66,88,107–111 The decarboxylative and desulfitative synthesis of aryl ketones from nitriles by palladium(II)-catalysis has been studied with ESI-MS by our group, reveal-ing several species that could be catalytically relevant for this addition reaction.45,66,88
In terms of workflow, the investigation of a palladium-catalyzed reaction can be performed by 1) identification of peaks containing palladium by the characteristic isotopic pattern of palladium, 2) product ion scan of the identi-fied peaks. To determine the identity of the ionized species, neutral loss scans for masses equaling that of possible ligands can be performed along with pre-cursor ion scans.
Figure 13. Techniques used in the ESI-MS studies of live reactions
26
By performing several experiments in which, for instance, the substrates and the ligand are changed one at a time, additional information is gained which helps verify the identity of the complexes observed as well as help determine which species are more likely to be catalytically relevant. To exemplify, add-ing a methyl group to one of the substrates would increase the mass of any palladium complex, which is derived from this substrate.
Even though this technique allows for easy identification of palladium complexes, one should be aware that limitations to this approach are posed by the fact that only ionic species are detected. The detected species may also not be catalytically relevant and rather represent stable complexes which are not relevant for the progression of the reaction. Thus, ESI-MS studies should be used in conjunction with other techniques to advance the mechanistic under-standing of a reaction.
27
1.4 Amidines Carboxamidines or carboximidamides, henceforth termed amidines, are com-pounds with the general structure RC(=NR)NR2 as depicted in Figure 14 below.
Figure 14. General structure of an amidine, as well as commonly encountered cyclic amidine, 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU).
Most organic chemists recognize amidines as strong bases (pKa values of around 10-12 is common), the commonly used 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), being a prime example.
Amidines have also found use in the emerging field of organocatalysis as nucleophilic catalysts, catalyzing acyl transfer reactions, amidation-, and es-terification reactions to name some examples.112,113
Amidines also serve as useful precursors for various heterocycles (Figure 15) such as imidazoles,114 1,2,4-triazoles,115,116 tetrazoles,117 pyrimidines,118 benzimidazoles,119 quinazolines,120,121 and purines.122
Figure 15. Various hetereocycles that can be prepared from amidines.
The amidine functionality is relevant in the field of medicinal chemistry. Ar-omatic diamidines are interesting molecules that are active against intracellular parasites responsible for diseases such as sleeping sickness, Cha-gas disease, toxoplasmosis, leishmaniasis and malaria.123 One such compound, pentamidine, has been used since 1937 and is placed on the World Health Organization’s List of Essential Medicines as a treatment for African trypanosomiasis.124 It is also used in the treatment of leishmaniasis, babesiosis and to prevent pneumocystis pneumonia in immunocompromised patients. Another aromatic diamidine, phenamidine, is used for babesiosis in veterinary medicine, along with other diamidine analogues such as diminazene.125
28
Brunfelsamidine is a pharmacologically active substance found in plants of the nightshade family which has convulsant and neurotoxic effects.126 Root or bark extracts containing this substance have been used among natives in the Amazon as a remedy against arthritis. Thrombin inhibitor dabigatran, an oral anticoagulant (as the prodrug dabigatran etexilate), contain the amidine mo-tif.127 Taribavirin was being developed as a prodrug for ribavirin in which the amidine motif is converted in vivo to the corresponding amide.128 The struc-ture of the pharmacologically active amidines which has been exemplified is given in Figure 16.
Among other examples from drug discovery are the use of amidines as m1 muscarinic receptor agonists129 and as inhibitors of serine proteases,130,131 acid-sensing ion channels,132 and BACE1.133
Figure 16. Selected pharmacologically active amidines.
29
1.4.1 Synthesis of amidines Amidines are traditionally prepared by the Pinner reaction134–136 where a ni-trile is treated with HCl and an alcohol to give a Pinner salt which can be reacted with an amine to give the hydrochloride salt of the corresponding am-idine. Ortho-substituted benzamidines are difficult to make by this strategy.137 Another strategy is to use base catalysis to generate an imidate from a nitrile and an alcohol.138,139 The imidates can then react with ammonium salts to give primary amidines. A modified Pinner reaction (Thio-Pinner reaction) that does not require the use of acid relies on the use of thiols to form a thioimidic ester that can react with amines to form amidines.140,141 Garigipati reported on the addition of alkylchloroaluminum amides to nitriles, affording un-, mono-, or di-substituted amidines.142 The alkylchloroaluminum amide is readily made from trimethylaluminum and ammonium chlorides or amine hydrochlorides. Another method for the synthesis of amidines is the addition of a Grignard or lithium reagent to cyanamides followed by acidic workup to give the amidine salts.143,144
An alternative starting material is amides, and imidoyl chlorides or imidate tetrafluoroborates can be generated by reacting the amide with a chlorinating agent or triethyloxonium tetrafluoroborate, respectively.137 Subsequent reac-tion with amines give the corresponding amidines. This method is useful for the generation of tri-substituted amidines.
There are also several palladium(0)-catalyzed methods describing the syn-thesis of amidines from aryl halides or alkenyl halides.145–149 An Ytterbium catalyzed addition of amines has also been reported.150
In Table 2 an overview of some of the important approaches to amidines described is presented. For a more thorough overview of synthetic methods for amidine synthesis, the reader is referred to selected reviews and book chap-ters covering this topic.151–154
Table 2. Common methods for the synthesis of amidines
30
1.5 Continuous-flow Chemistry Most people associate organic chemistry with the mixing of reagents in a sol-vent under stirring in a glass vessel such as a round-bottom flask or an Erlenmeyer flask. This is the standard way of setting up reactions, usually re-ferred to as batch chemistry. In a typical batch chemistry setup, reagents are mixed for a fixed time at a certain temperature before quenching of the reac-tion and isolation of the product from the resulting product mixture. If one would need to produce more of the product one could either run multiple iter-ations of the experiment or run a reaction at a larger scale. Typical problems encountered is batch-to-batch variability, and problematic scale-up due to rea-sons such as heat transfer limitations155 or dangerous intermediates.156,157
Another way of running a chemical reaction is to run it as a continuous operation. One approach to this, when the reaction mixture is liquid, is continuous-flow chemistry (see Figure 17 for an illustration). By using a pump, reagents can be transferred into a reactor where the conditions for the transformation into product is applied, and then further transported to a col-lection vessel for subsequent processing.
Figure 17. General principle of batch and continuous-flow chemistry.
In comparison to batch chemistry, the volume of the reaction mixture which is processed at a given time can be kept low as the scale of the reaction effec-tively will be determined by the time the system is operational.155 This is also reflected in the difference in terminology; in batch chemistry one scales up the reaction, while in continuous-flow chemistry one scales out the reaction. The batch-to-batch variability encountered in batch chemistry can also be avoided in continuous-flow chemistry, since the process, once it reaches equilibrium, should give the same output per time unit. The process can also be monitored by inline or online analysis of the reaction mixture.158–160
In terms of safety, the large surface-to-volume ratio of the continuous-flow reactor allows for fast temperature changes, allowing better process control. This is useful for highly exothermic processes which can be difficult to scale up using batch chemistry.161 The small volume which is in the reactor at any
31
given moment is also advantageous when processing highly toxic or explosive reagents, or when intermediates formed pose such a risk.156,161
There are of course disadvantages with continuous-flow methodology as well. The pumps used are usually in the laboratory setting either HPLC or syringe pumps, neither are well suited for handling particles. The reaction mixture, reagent mixtures as well as the product mixture should thus ideally be homogenous. The commercially available continuous-flow chemistry in-strumentation are also typically costly and can be experienced as complicated to setup and maintain.
An overview of the advantages and disadvantages with continuous-flow chemistry can be seen in Table 3 below, compared with batch chemistry.
Table 3. Advantages and disadvantages of continuous-flow processing
Advantages Scale-out rather than scale-up,
operate for a longer time Batch-to-batch variability is not a
problem Fast heat transfer – better process
control Safer operation with highly exo-
thermic reactions Safer processing of explosive/
toxic reagents/intermediates
Disadvantages Homogenous reagent/reaction/prod-
uct mixtures highly preferred Initial investment cost Can be complicated to setup and
maintain
1.5.1 Heating by a Non-Resonant Microwave Applicator In this work a setup for continuous-flow chemistry has been used which utilize a non-resonant microwave (MW) applicator for heating. The principle is de-scribed after an introduction to the concept of MW heating.
Microwave-Assisted Organic Synthesis The prevalent heating method in organic synthesis has evolved from open flames, through the Bunsen burner, to today’s use of safer alternatives such as hot plates combined with oil baths or metal heating blocks, and heating man-tels. Despite this, the fundamental principle of how the heat is transferred to the reaction mixture has not changed and rely on the wall of the reaction vessel heating the content by conduction. This process can be quite slow, depending on the surface-to-volume ratio of the vessel and the conductive properties of the involved materials. This heating process also typically results in a higher temperature of the walls of the reaction vessel than the temperature acquired by the reaction mixture. Under reflux conditions this is even more apparent, as the temperature of the heater is usually kept tens of degrees Celsius above the boiling point of the solvent.
32
The use of MW radiation to heat things, in particular foodstuff, should not have passed unnoticed, and MW ovens are now found in most households. MW radiation, an electromagnetic radiation, heat by transferring energy to molecules by dipolar polarization or ionic conduction. In both cases, heat is generated when a dipole or an ion moves in the direction of the oscillating electromagnetic field. Ions will oscillate while dipoles rotate. These move-ments correspond to kinetic energy which will in turn generate volumetric heating by induced losses.
The ability of a substance to be heated by MWs can be related to the loss tangent; tan δ = ε’’/ε’ (in which ε’’ is the dielectric loss constant and ε’ is the dielectric constant), with a higher value meaning that the substance will gen-erate more heat when exposed to MW radiation than a substance with a low tan δ value. These values are tabulated for many solvents used in organic chemistry, and are related to the dipole moment of the solvents, with a higher tan δ for polar solvents.162
Figure 18. Water molecule affected by the electric field of the electromagnetic wave that is MWs
MWs can be transmitted, reflected or absorbed depending on the properties of the irradiated material. The standard material used in the glass vessels that chemists use, borosilicate glass, has a very low tan δ value, and is thus rela-tively MW transparent. This means that a reaction conducted in a glass vessel will be directly heated by the MW radiation rather than by the wall, given that polar substances (with higher tan δ values) are present in the reaction mixture.
This direct way of heating has become increasingly popular in organic chemistry, especially due to the many reports on enhanced reaction rates and selectivity when compared to reactions run with conventional heating. The majority of these observed differences are due to different temperature profiles during the heating and cool-off period, as well as differences in the actual re-action temperature. By using tools such as fiber optic temperature probes that can accurately measure the temperature of the reaction solution, the group of
33
Kappe, among others, has repeatedly been able to show that the results using MW processing can be mimicked by conventional heating, given that the same heating profile can be achieved.163–168
Regardless of any benefits from the actual method of applying the heating, the dedicated MW instruments developed for synthetic chemistry applications has enabled safe processing at elevated pressures and temperatures. Reactions can thus be performed at temperatures well above the boiling point of the sol-vent used.
The dominating designs of MW apparatus are called single mode and multi-mode, which reflect how the MWs are distributed in the cavity. A single mode device would create a standing wave pattern in a resonating cavity, re-sulting in nodes with zero intensity, and antinodes which would have the highest intensity. The single mode design is common in dedicated MW instru-ments for small scale synthesis, where the reaction vessel can be placed in the antinode. A multi-mode apparatus has a cavity where the MWs are dispersed randomly so that a single standing wave is not created. This allows for heating to be applied over a larger volume, even if the average effect at a given point will be lower than in the antinode of a single mode instrument. This is the standard design of the domestic MW oven. For synthetic chemistry purposes, the multimode cavities are mostly used when larger volumes, usually above 50 ml, are heated. The disadvantage with this design is that the MW field cre-ated is non-homogenous which can produce local differences in heating of a sample.169,170
A Non-Resonant Microwave Applicator for Continuous-Flow Chemistry A modified batch MW system allowed the first continuous-flow application of MW heating described by Cablewski et al. in 1994.171 Other examples fol-lowed, using both continuous-flow or stop/flow approaches.172–178 The benefits making up the rationale for combining MW heating and continuous-flow methodology is manifold and includes rapid changes of the temperature of the reaction mixture, convenient access to high temperatures, and heating that can instantaneously be switched off for the reaction mixture (given that it is directly heated).
In 2012 our group described the evaluation of a new type of MW applicator, designed for continuous-flow chemistry.179 This applicator, designed by Wavecraft AB, applies MWs to a straight borosilicate glass tubular reactor by a helical antenna surrounding the reactor. This design has the advantage that it is non-resonant, avoiding hot and cold spots in the irradiated volume. This contrasts with the previously described approaches to distributing the MWs and is suitable for an evenly distributed axial field across the reactor.
The lessons learnt during the initial evaluation as well as during continued use of the instrument180–183 has resulted in some changes from the initial de-sign. For instance, the system originally featured a setup of five infrared (IR) sensors that measured the temperature of the outside wall of the borosilicate
34
glass reactor. This has been reduced to one sensor measuring the temperature at a representative position on the reactor. Additionally, the temperature meas-ured by the IR sensor has been correlated to the measurements of a fiber optic probe in the reactor to provide a model for estimating the true reaction tem-perature based on the IR sensor reading.184 The use of a fiber optic probe, instead of the standard thermocouple approach to temperature measurement was necessary as a thermocouple cannot be used in the MW field. An IR sen-sor that can measure the temperature through the borosilicate glass wall was also evaluated with good results. A new reactor type, a straight tubular silicon carbide (SiC) reactor has also been introduced.181 The silicon carbide material absorbs MWs to a large extent, and thus heats the reaction mixture by conven-tional heating, even though the reactor itself is heated by MWs. This effectively means that, for reactions that can cause metal-precipitation on the glass surface and subsequent rapid heating of the metal followed by reactor failure, a SiC reactor can be used in place of the borosilicate glass reactor for certain applications.
35
2. Aims of the Present Study
The overall aim of the work presented in this thesis has been to develop new palladium(II)-catalyzed addition reactions. More specifically, the aims of this thesis were related to addition of aryl-palladium to nitrile derivatives:
to develop a synthetic method for the reaction between an aryl-palladium
species and cyanamides to give aryl amidines,
to investigate the reaction between an aryl-palladium species and cyanamides with respect to the aryl-palladium precursor,
to use continuous-flow methodology to develop protocols, for palladium(II)-catalyzed reactions addition reactions.
During the course of this work, a need for more convenient, general, and safe methodology for the synthesis of the aryl-palladium precursor aryl sulfinates was found. Thus the aims were expanded as follow: to develop a synthetic method for the synthesis of sulfinic acids or sodium
sulfinates utilizing a safe source of SO2.
36
3. Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Cyanamides and Aryltrifluoroborates (Paper I)
3.1 Background During the work on new aryl sources for the palladium(II)-catalyzed 1,2-addition to nitriles within our group, we also considered the use of other nitrile derivatives which would generate a more stable imine species. One such class of nitrile derivatives was identified as cyanamides, which upon 1,2-addition of aryl-palladium would give aryl amidines as products (see Fig-ure 19 below).
Figure 19. Palladium(II)-catalyzed 1,2-addition to nitriles vs cyanamides
The amidine functionality is a versatile precursor for the generation of a large variety of heterocycles and can be found in many pharmacologically active molecules including FDA approved drugs. For more details about the use of and synthesis of amidines, the reader is referred to the introductory chapter.
3.2 Optimization of Reaction Conditions We started our investigation by an explorative screening of aryl sources where aryltrifluoroborates were identified as the most promising substrates. A good starting point was conditions identified as productive in an earlier developed protocol by our group for the synthesis of aryl ketones from nitriles and so-dium sulfinates. The conditions were slightly modified, switching the solvent to methanol, which had been successfully employed in an oxidative Heck pro-tocol, involving aryltrifluoroborates as aryl-palladium precursors, by our group.185 Thus the starting conditions were as follow: a mixture of 4% Pd(O2CCF3)2, 6% 6-methyl-2,2’-bipyridyl (6-Mebpy) as the catalytic system, potassium 4-methylphenyltrifluoroborate, 2 equiv cyanamide and 5 equiv
37
TFA in methanol were mixed and MW heated for 20 minutes at 120 °C. These conditions gave full conversion of the yield determining trifluoroborate, while formation of the corresponding aryl amidine product could be observed by 1H NMR analysis of the reaction mixture. We were able to reduce the amount of acid to 2 equiv of TFA without affecting the productivity of the reaction. It was also possible to reverse the stoichiometry of the cyanamide and aryltri-fluoroborate substrate, while reducing the excess of aryltrifluoroborate to 1.1 equiv and changing the cyanamide substrate from cyanamide to 1-piperidine-carbonitrile to facilitate 1H NMR analysis of the reaction outcome.
The change in stoichiometry from having the aryltrifluoroborate as the yield determining substrate to using it in excess was made to simplify the pu-rification procedure, a liquid-liquid extractive workup of the crude mixture. A dilution of the reaction mixture with DCM followed by extraction with satu-rated NaHCO3(aq) leaves the 6-Mebpy ligand in the organic phase. The protonated aryl amidine product can then be extracted from the aqueous phase upon basification by NaOH addition followed by extraction with DCM to give the aryl amidine upon concentration of the organic phase.
A final ligand screen was next performed based upon these optimized con-ditions (see Table 4).
Table 4. Optimization of reaction conditions, ligand screen
Isolated yields, >95% pure by 1H NMR. Reaction conditions: Pd(O2CCF3)2 (0.04 mmol), ligand (0.06 mmol), TFA (2 mmol), 4-methylphenyltrifluoroborate (1a) (1.1 mmol), 1-piperidinecarboni-trile 2a (1 mmol), and MeOH (3 mL) is heated in a sealed vial at 120 °C for 20 min by MW. aNo TFA added. bNo Pd(TFA)2 added. n.d.: Product could not be detected by LC-MS.
The two bipyridyl ligands 6-Mebpy and bpy gave similar yields, 88% and 86%, respectively (Table 4, entries 1-2). 1,10-Phenanthroline, which is more rigid than the bipyridyl scaffold, furnished a 69% yield of the corresponding amidine (entry 3). Dmphen, surprisingly, only gave trace amounts of product while the phosphine based ligand dppp did not show any formation of product (entries 4-5).
38
To verify the importance of each component, we also performed reactions omitting, one by one, ligand, TFA, and Pd(O2CCF3)2. Product formation could not be detected, or was only detected in trace amounts for these reactions (en-tries 6-8).
3.3 Investigation of the Scope of the Reaction The reaction conditions identified in entry 1, Table 4 were chosen for further investigation of the scope of the reaction, starting with the scope with regards to the aryltrifluoroborates (Table 5).
Table 5. Aryltrifluoroborate scope
Isolated yields, >95% pure by 1H NMR. Reaction conditions: Pd(O2CCF3)2 (0.04 mmol), 6-Mebpy (0.06 mmol), TFA (2 mmol), po-tassium aryltrifluoroborates 1a-j (1.1 mmol), 1-piperidinecarbonitrile 2a (1 mmol), and MeOH (3 mL) is heated in a sealed vial at 120 °C for 20 min by MW. aNo TFA added. bNo Pd(TFA)2 added. n.d.: Product could not be de-tected by LC-MS.
High yields were isolated with electron-rich 4-methoxy and 4-tert-butyl sub-stituted trifluoroborate substrates giving 3b and 3c in 86% and 74% yield, respectively. Phenyl trifluoroborate gave benzamidine, 3d, in good yield (73%). Ortho substitution in the aryltrifluoroborate was tolerated, giving the corresponding amidine 3e in 66% yield. Chemoselectivity was demonstrated by a 4-bromo substituted aryltrifluoroborate, resulting in an isolated yield of
39
63% of aryl amidine, 3f. Heteroaryl amidine 3g could be isolated in 66% yield while 2-naphtyltrifluoroborate gave 40% yield of 3h. Electron-deficient tri-fluoroborates only furnished 37% of 3i and trace amounts of 3j, respectively.
One boronic acid, 4a, and one boronic acid pinacol ester, 4b, were also evaluated as aryl-palladium precursors under these conditions (Table 6), providing yields of 3a which were lower than the corresponding aryltri-fluoroborate, 1a (33% and 45%, respectively, compared to 88%).
Table 6. Other boronic acid derivatives tested
Isolated yields, >95% pure by 1H NMR. Reaction condi-tions: Pd(O2CCF3)2 (0.04 mmol), 6-Mebpy (0.06 mmol), TFA (2 mmol), 4 or 5 (1.1 mmol), 1-piperidinecarbonitrile 2a (1 mmol), and MeOH (3 mL) is heated in a sealed vial at 120 °C for 20 min by MW.
The scope of the cyanamide was also extended to include cyanamide (2b), dimethylcyanamide (2c), diisopropylcyanamide (2d), tert-butylcyanamide (2e), and 4-morpholinecarbonitrile (2f). Cyanamide substrates 2b and 2c were productive with 1a as the aryltrifluoroborate coupling partner, furnishing 73% and 82% yield of 3k and 3l, respectively (see Table 7). The bulkier 2d afforded 24% yield of 3m while monosubstituted 2e gave 64% yield of the correspond-ing amidine 3n. Cyclic 2f was also productive, affording 58% yield of 3o. The more electron-rich aryltrifluoroborate 1b was also reacted with cyanamides 2b, 2d, and 2f, generally giving as good or better yield when compared to the corresponding cyanamide with 1a. The electron-deficient 1i gave traces of product 3s when combined with cyanamide 2b and only 33% yield of 3t when combined with cyanamide 2f.
40
Table 7. Cyanamide scope
Isolated yields, >95% pure by 1H NMR. Reaction conditions: Pd(O2CCF3)2 (0.04 mmol), 6-Mebpy (0.06 mmol), TFA (2 mmol), potassium aryltrifluoroborates 1a-b or 1i (1.1 mmol), cyan-amides 2b-f (1 mmol), and MeOH (3 mL) is heated in a sealed vial at 120 °C for 20 min by MW. R is an un-, mono-, or di-substituted amino group.
41
3.4 Suggested Mechanism Based on previous live reaction studies using HRMS on the 1,2-addition reac-tions with nitriles using carboxylic acids88 or sodium aryl sulfinates,45 a catalytic cycle was proposed. The Pd(II) ligand-coordinated complex A un-dergoes transmetallation generating the aryl-palladium intermediate B which, upon ligand exchange, affords complex C with cyanamide coordinated. A 1,2-carbopalladation then affords D which, upon protonation of the imid-amidate, regenerates complex A.
Figure 20. Proposed reaction mechanism for the palladium(II)-catalyzed addition of aryltrifluoroborates to cyanamides.
42
4. Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Cyanamides and Aryl Carboxylic Acids (Paper II)
4.1 Background Following the successful development of a protocol for the palladium(II)-catalyzed 1,2-addition to cyanamides described in Paper I,186 there was reason to believe that other aryl-palladium sources than aryltrifluoroborates could be employed in a similar manner for the synthesis of aryl amidines. In Paper I, the results using the related aryl-palladium precursors boronic acid and bo-ronic acid pinacol ester were reported (Table 6), but with lower isolated yield than the corresponding aryltrifluoroborate.186 Efforts were made to optimize the reaction for these substrates, however with limited success. It was not in-vestigated in detail, and the nature of the challenges with these substrates are therefore not known.
As an interesting alternative to the transmetalation with boronic acid deriv-atives there is the possibility to make use of a decarboxylative process for aryl-palladium generation, and thus use carboxylic acids as a precursor.
The merits of aryl carboxylic acids as aryl-palladium precursors have been discussed in more detail in the introduction, the main points being the atom economy and environmental aspects related to the fact that CO2 would be the byproduct of the aryl-palladium generation.187 There is also a potential orthog-onality between boronic acid derivatives and carboxylic acids. Sterically congested aryl carboxylic acids are often preferred substrates as the occur-rence of ortho-substituents is known to be beneficial for the aryl-palladium generation.188 This is not necessarily the case for boronic acid derivatives, and thus carboxylic acids could add to the scope of palladium(II)-catalyzed 1,2-addition to cyanamides, making new aryl amidines available by this reaction class.
The corresponding reaction with nitriles as 1,2-addition substrates and aryl carboxylic acids as aryl-palladium precursors was developed by our group in 2010,88 and this in conjunction with the work described in Paper I186 lays the foundation for the development of a new amidine synthesis from aryl carbox-ylic acids and cyanamides.
43
4.2 Optimization of Reaction Conditions The investigation was started with a solvent screen, making use of the previ-ously developed catalytic system for aryl amidine synthesis from aryltrifluoroborates. Pd(O2CCF3)2 and 6-Mebpy was thus used as palladium salt and ligand, the loading set at 2% and 3% respectively for the palladium salt and ligand. TFA was used as the proton donor, adding 1 equivalents, an adjustment from the 2 equivalents used in the protocol developed in Paper I, as the aryl carboxylic acid will also serve as a proton donor. The benzoic acid was used in slight excess in the solvent screen and the reaction was heated in a sealed vessel in MW for 30 minutes at 120 °C. The product was isolated using the liquid-liquid extractive workup described in Paper I.
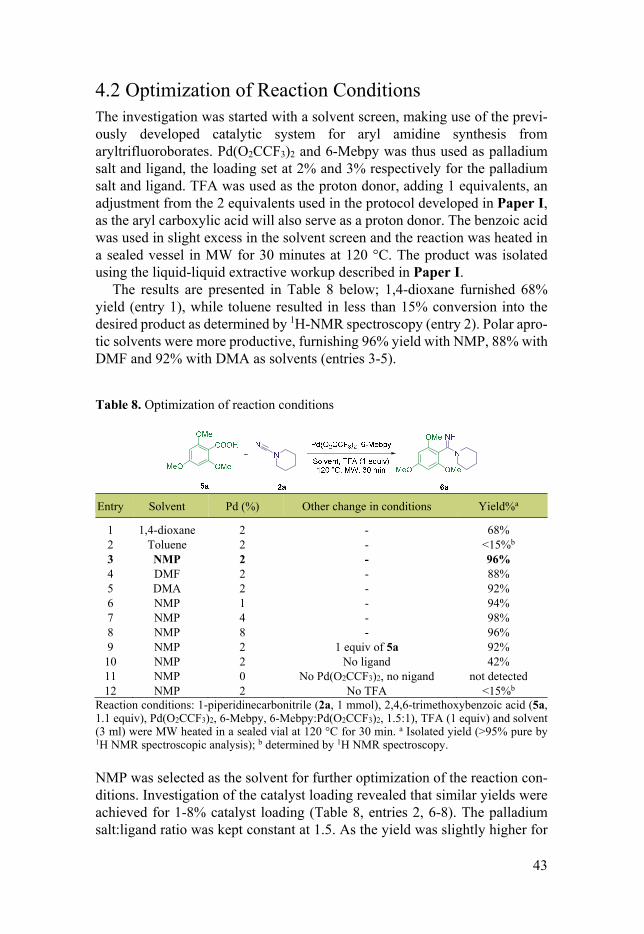
The results are presented in Table 8 below; 1,4-dioxane furnished 68% yield (entry 1), while toluene resulted in less than 15% conversion into the desired product as determined by 1H-NMR spectroscopy (entry 2). Polar apro-tic solvents were more productive, furnishing 96% yield with NMP, 88% with DMF and 92% with DMA as solvents (entries 3-5).
Table 8. Optimization of reaction conditions
Entry Solvent Pd (%) Other change in conditions Yield%a
Reaction conditions: 1-piperidinecarbonitrile (2a, 1 mmol), 2,4,6-trimethoxybenzoic acid (5a, 1.1 equiv), Pd(O2CCF3)2, 6-Mebpy, 6-Mebpy:Pd(O2CCF3)2, 1.5:1), TFA (1 equiv) and solvent (3 ml) were MW heated in a sealed vial at 120 °C for 30 min. a Isolated yield (>95% pure by 1H NMR spectroscopic analysis); b determined by 1H NMR spectroscopy.
NMP was selected as the solvent for further optimization of the reaction con-ditions. Investigation of the catalyst loading revealed that similar yields were achieved for 1-8% catalyst loading (Table 8, entries 2, 6-8). The palladium salt:ligand ratio was kept constant at 1.5. As the yield was slightly higher for
44
2, 4 and 8% as compared to 1%, 2% catalyst loading was chosen for continued optimization. Finally, a change of stoichiometry to use 1 equiv of 5a instead of 1.1 equiv resulted in a slightly lower yield. Control experiments revealed that although removal of the ligand from the reaction mixture resulted in a less productive reaction, the desired product could still be isolated in 42% yield (Table 8, entry 10). Removal of the palladium salt and ligand results in a non-productive reaction, no product formation could be detected (entry 11). Omis-sion of TFA resulted in low conversion into the desired product as determined by 1H-NMR spectroscopy (entry 12). Based on these results the conditions presented in Table 8, entry 3, were chosen for further investigation of the scope of the reaction.
45
4.3 Investigation of the Scope of the Reaction First the scope of the reaction was investigated with regards to the cyanamide partner, using an expanded set of substrates as compared to what was investi-gated in Paper I.186 The results are presented below in Table 9.
Table 9. Cyanamide scope
Reaction conditions: cyanamide (2a-j, 1 mmol), 2,4,6-trimethoxyben-zoic acid (5a, 1.1 equiv), Pd(O2CCF3)2 (2%), 6-Mebpy (3%), TFA (1 equiv) and NMP (3 ml) were MW heated in a sealed vial at 120 °C for 30 min. Isolated yield (>95% pure by 1H NMR spectroscopic analysis). R is an un-, mono-, or di-substituted amino group.
In general, good to excellent yields could be isolated. Cyanamide 2b furnished primary amidine 6b in 76% yield. Di-alkyl substituted cyanamides gave the corresponding products 6c, 6d, and 6e in 93%, 98% and 64% yield, respec-tively. The dibenzyl derivative 6f was isolated in 68% yield while the monosubstituted cyanamide 2e furnished 6g in 90% yield. Cyclic cyanamides provided 6h and 6i in 93% and 74% yield, in addition to 6a, which was iso-lated in 96% yield during the optimization of the reaction.
The continued investigation of the scope of the reaction focused on the car-boxylic acid partner, and it was soon realized that decreased reactivity required longer reaction times as well as a higher temperature, 60 minutes
46
compared to 30 minutes and 140 °C compared to 120 °C. Using these condi-tions 6j, 6k, and 6l could be isolated in 65%, 9%, and 8% yield, respectively, from the corresponding aryl carboxylic acids 5b, 5c, and 5d (see Table 10).
Table 10. Carboxylic acid scope
+5b-d
6j-l
Ar N
NH
N
NH
MeO
N
NH
6j
2a
6l
OMe
OMe OMe
N
NH
6k
OMe
OMe
65% 9%[a] 8%a
Ar COOH
OMe
MeO
NMP, TFA (1 equiv)140 °C, MW, 60 min
Pd(O2CCF3)2, 6-Mebpy
Reaction conditions: 2a (1 mmol), aryl carboxylic acid 5 (1.1 equiv), Pd(O2CCF3)2 (2%), 6-Mebpy (3%), TFA (1 equiv) and NMP (3 ml) were MW heated in a sealed vial at 140 °C for 60 min. Isolated yield (>95% pure by 1H NMR spectroscopic anal-ysis). aPurified using preparative HPLC.
A number of other aryl carboxylic acids were also tested in the protocol (2,4,6-trimethylbenzoic acid, 3-methylthiophene-2-carboxylic acid, 2,4-dimethox-ybenzoic acid, benzoic acid, 2,6-dimethoxypyridine-3-carboxylic acid, ortho-anisic acid, 2,6-dimethoxy-3-nitrobenzoic acid, 3-bromo-2,6-dimethoxyben-zoic acid and 2,6-difluoro-4-methoxybenzoic acid), however with poor result, only showing trace amounts by HPLC-MS analysis.
In an attempt to broaden the scope with regards to the aryl carboxylic acid substrate, the addition of 2 equiv of silver trifluoroacetate or copper(I)oxide to a reaction with 2,4,6-trimethylbenzoic as the substrate was tested. Even though these salts are known to facilitate the decarboxylation process,189 the addition did not improve the reaction outcome.
Other efforts made to improve the scope include various changes in tem-perature, time, and stoichiometry, however without any improvement in the reaction outcome for non-productive substrates.
Instead, attention was turned to the other productive carboxylic acid 5b which gave a 65% yield of the corresponding amidine 6j, and an investigation of the cyanamide scope for the reaction using this carboxylic acid was under-taken (see Table 11). The yields obtained were in general lower for the reaction of 5b with a given cyanamide substrate as compared to 5a, affording 36-65% yield of the corresponding amidines 6m-o and 6r-t. The exception was diisopropylcyanamide and dibenzylcyanamide, which both furnished 74% yield of the corresponding products 6p and 6q.
47
Table 11. Cyanamide scope with 5b as the aryl carboxylic acid substrate
Reaction conditions: 2a-i (1 mmol), aryl carboxylic acid 5b (1.1 equiv), Pd(O2CCF3)2 (2%), 6-Mebpy (3%), TFA (1 equiv) and NMP (3 ml) were MW heated in a sealed vial at 140 °C for 60 min. Isolated yield (>95% pure by 1H NMR spectroscopic anal-ysis). a Purified using preparative HPLC. R is an un-, mono-, or di-substituted amino group.
48
4.4 Continuous-Flow Scale-Out An adaption of the protocol to continuous-flow conditions was performed. The reaction mixture was diluted somewhat compared to the batch conditions, to make up a stock solution with 0.2M of 5a, and stoichiometry otherwise as in the batch protocol. The dilution was made to ensure that the reaction mix-ture was homogenous, to work with the HPLC pumps used in the continuous-flow setup. The setup utilized a non-resonant MW reactor, which has been described in the Introduction, for heating. See Figure 21 for a schematic illus-tration of the setup.
Figure 21. Schematic illustration of the continuous-flow setup used
The use of continuous-flow allows for quick variation of various reaction pa-rameters, in this setup mainly time (in heated zone) and temperature, and thus the flow rate and temperature could be set to 1 ml/min (1 min in the heated zone) and 120 °C. Using these optimized conditions, the yield was 92% based on the purification of a 5 ml aliquot of the collected crude product mixture. This corresponds to a theoretical throughput of 11 mmol/h or 3 g/h.
49
4.5 Mechanistic Investigation To better understand the limited scope with regards to the aryl carboxylic acid substrate, a mechanistic investigation was initiated.
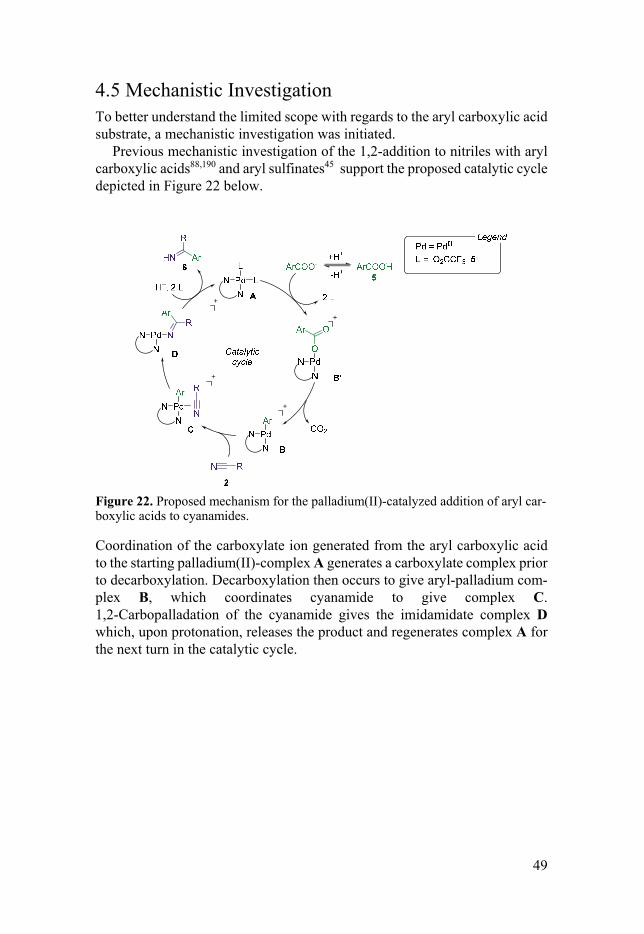
Previous mechanistic investigation of the 1,2-addition to nitriles with aryl carboxylic acids88,190 and aryl sulfinates45 support the proposed catalytic cycle depicted in Figure 22 below.
Figure 22. Proposed mechanism for the palladium(II)-catalyzed addition of aryl car-boxylic acids to cyanamides.
Coordination of the carboxylate ion generated from the aryl carboxylic acid to the starting palladium(II)-complex A generates a carboxylate complex prior to decarboxylation. Decarboxylation then occurs to give aryl-palladium com-plex B, which coordinates cyanamide to give complex C. 1,2-Carbopalladation of the cyanamide gives the imidamidate complex D which, upon protonation, releases the product and regenerates complex A for the next turn in the catalytic cycle.
50
4.5.1 DFT Calculations Based on the proposed catalytic cycle depicted in Figure 22 we performed DFT calculations to further investigate the mechanism. Aryl carboxylic acids 5a-c were included in the study, representing two productive substrates (5a and 5b) and one less productive (5c). Cyanamide 2b was chosen as the cyan-amide substrate and 6-Mebpy was used as the ligand. To simplify the calculations Pd(OAc)2 was used as the palladium salt in place of Pd(O2CCF3)2. Although the use of Pd(OAc)2 resulted in a slight reduction of yield of 6a from 96% (Table 8, entry 3) to 91%, it still represents a highly productive catalytic system and thus a good model for the optimized catalytic system.
Figure 23. Free energy profile for decarboxylation of 5a, 5b, and 5c and following 1,2-carbopalladation of cyanamide 2b. The optimized geometry of TS-II is shown to the right.
The resulting energy pathway is depicted in Figure 23 and starts with I, a di-acetate chelated species with a ligand associated in a bidentate manner. From I, through exchange of one (for 5b) or two (for 5a and 5c) of the acetates with the corresponding carboxylate ion or ions of 5a-c, the lowest energy complex II prior to decarboxylation is formed. The dissociation of one anionic ligand from II generates cationic species III, which, upon a change in binding mode gives IV. Aryl-palladium complex V is then formed over transition state TS-I, releasing CO as complex VI is formed. Then, coordination of a carboxylate ion gives the lowest energy intermediate prior to 1,2-carbopalladation, the neutral complex VII. Association of the cyanamide gives VIII which upon 1,2-carbopalladation over TS-II (see Figure 23 for the optimized geometry of
51
the complex), generates the imidamidate coordinated complex IX. Associa-tion of the carboxylate ion of 5 generates complex X which, upon subsequent product release by protonation and replacement with carboxylate ion of 5 (or in the case of 5b an acetate ion) regenerates complex II, resulting in an overall exergonic reaction.
Table 12. Free energy requirement of decarboxylation and 1,2-carbopalladation for the reaction using 5a, 5b, or 5c as the aryl carboxylic acid substrate
For all three substrates the calculated energy required for 1,2-carbopalladation is higher than for decarboxylation, although only slightly (0.5 kJ/mol) for 5a (Table 12). The 1,2-carbopalladation is therefore expected to be rate-deter-mining, in particular for 5b and 5c. The free energy requirements for the rate-determining 1,2-carbopalladation are significantly lower for 5a and 5b com-pared to 5c.
To summarize, the calculations and the experimental findings both support the need for electron donating substituents on the benzoic acid.
52
4.5.2 Live Reaction Studies Using ESI-MS The suggested mechanism (Figure 22) was further investigated by studying live reaction mixtures by ESI-MS(+). This technique allows for the study of sensitive complexes,106,110 and is suitable for the observation of the cationic palladium complexes suggested to be present in this reaction.
Palladium-containing complexes are found by the characteristic isotopic pattern produced by palladium (106Pd and 108Pd have almost the same natural abundance, see Table 1), and their identity deduced by comparison of multiple productive catalytic reactions. In these reactions, one component was changed at a time according to Table 13 below.
Even though several palladium-containing complexes could be identified, only four were present in all of the reactions. These complexes were further examined by MS/MS(+) studies as well as neutral loss experiments. In Figure 24 below, the suggested cationic palladium complexes are depicted along with the spectrum from reaction 1 in Table 13.
Figure 24. ESI-MS(+) scan of reaction mixture at 30–70 % conversion. Spectrum from a reaction between 5a and 2a using standard reaction conditions. Below the proposed PdII intermediates are shown. The drawing of cis and trans geometries are based on the corresponding DFT calculated configurations with Pd(OAc)2.
53
Several of the identified complexes are relevant for the suggested mechanism. The complex prior to decarboxylation, B’, was identified (please refer back to Figure 22 for the denotations of complexes in the catalytic cycle). In addition, this palladium-containing species also produces a neutral loss corresponding to carbon monoxide (44 Da). This could be due to decarboxylation occurring in the mass spectrometer, or, alternatively this could be due to a complex in which decarboxylation has already occurred, with carbon monoxide still bound to the palladium complex (see complex V in Figure 23). The aryl-palladium complex which is formed upon decarboxylation, B, could also be identified. C, the σ-complex formed when the cyanamide coordinates to B and the complex formed upon 1,2-carbopalladation, D, would have the same mass. Complex C could be identified by neutral loss experiments, while com-plex D could not be conclusively identified.
The ESI-MS study thus supports the suggested catalytic cycle, by three identified complexes B’, B, and C, which should be mechanistically relevant.
54
5. Palladium(II)-Catalyzed Synthesis of 3-Amidino Indoles from Cyanamides and Indoles (Paper III)
5.1 Background In Papers I-II186,191 the palladium(II)-catalyzed 1,2-addition to cyanamides using aryltrifluoroborates and aryl carboxylic acids as aryl-palladium precur-sors was described. These aryl-palladium precursors work by a transmetalation or decarboxylation process, respectively, to generate the aryl-palladium species. Another process for generating a (hetero)aryl-palladium species is by C-H bond activation. This is highly sought after, as it represents ideal atom economy and does not require installation of a functional group as a handle for the reaction. However, limitations are posed by the fact that most C-H bonds are relatively inert and that selectivity can be hard to achieve as C-H bonds are usually abundant.192 The selectivity issue can be overcome by either a careful choice of substrate class, or use of directing groups in the sub-strate. One substrate class which displays good selectivity and reactivity in metal-catalyzed C-H functionalization reactions is indoles. Indoles display high reactivity in the C-3 position related to the higher electron-density of this position.
3-Acylindoles have been successfully synthesized by palladium(II)-catalyzed C-H activation of indoles followed by 1,2-addition to nitriles,86,90,91 and other C-H functionalization substrates have also been used for the same type of transformation.80,81,193 The papers on the synthesis on 3-acylindoles were all published after our work on Paper II, and this inspired us to consider indoles as a C-H activation substrate for the synthesis of 3-amidino indoles from cyanamides.
55
5.2 Optimization of Reaction Conditions An initial investigation of the reaction conditions was carried out using HPLC-UV to determine the ratio between product and starting indole. This method gives a crude measure of reaction outcome, and for some reactions this was combined with an extractive purification procedure similar to what has been described in papers I-II.186,191
This initial investigation started from conditions inspired by the published protocols for synthesis of 3-acylindoles86,90,91 and papers I-II.186,191 First the reaction solvent was determined by screening a selection of solvents (DMF, DMA, NMP, NMF and NMA) for a reaction using Pd(OAc)2 as the palladium salt (5%), bpy as the ligand (6%), CSA (1.5 equiv) as the acid additive, N-methylindole as the yield-determining reagent and dimethylcyanamide (1.5 equiv). 1 ml of solvent was used and the reaction mixture was heated for 2 h at 100 °C by MW irradiation. DMA was chosen as the solvent based on the solvent screen, and as conventional heating at 100 °C for 18 hours in a heating block gave comparable yield of the desired product, the heating method was changed to allow for easier parallelization of reactions.
A ligand screen was then undertaken (with Pd(O2CCF3)2 as the palladium salt), using 1H qNMR to estimate the yield of the reaction for a number of bidentate nitrogen ligands. The method chosen was to use an electronic refer-ence to estimate the concentration of the product in a sample of the crude mixture after reaction.194 The results of the screen are presented in Table 14 below.
Table 14. Optimization of reaction conditions, ligand screen
a By 1H-qNMR analysis of crude reaction. Reaction conditions: Pd(O2CCF3)2 (0.02 mmol), ligand (0.024 mmol), 7a (0.4 mmol), 2c (0.6 mmol), CSA (0.6 mmol), and DMA (1 mL) at 100 °C for 18 h.
As can be seen from the table, one ligand stands out, namely 4,5-diazafluoren-9-one, henceforth referred to as DAF.
56
Different acid additives were also investigated by exchanging CSA for pTSA, MSA, TFMSA, TFA and H2SO4. Good product:starting material ratios, as well as similar extracted crude yields of 87-89% were achieved for the sul-fonic acids CSA, pTSA, MSA, and TFMSA. Given that pTSA is an easy-to-handle solid which has a lower molecular weight than CSA, this acid was cho-sen for further development. Reversing the stoichiometry did not prove beneficial, thus the cyanamide was kept in excess. Finally, variations of the palladium salt (Pd(OAc)2 or Pd(O2CCF3)2), acid additive (HOAc, TFA, MSA or pTSA), and base addition (NaOAc or NaO2CCF3) were tested, resulting in the choice of Pd(O2CCF3)2 as the palladium salt and pTSA as the acid.
This optimized protocol, using Pd(O2CCF3)2 (5%), DAF (6%), pTSA (1.5 equiv), indole 7 as the yield-determining reagent (0.4 mmol scale) and cyana-mide 2 (1.5 equiv) in 1 ml of DMA at 100 °C for 18 hours, was used for further investigation of the scope of the reaction.
5.3 Investigation of the Scope of the Reaction A protocol for the isolation of the 3-amidinoindoles produced in this reaction had to be established, and unfortunately the previously used strategy in Papers I-II186,191 of an extractive workup to get the pure product was not viable. Nor-mal-phase chromatography on silica or aluminum oxide did not afford pure product. Fortunately, the use of a reverse-phase C18 column on an automated flash chromatography system proved to be a working strategy, affording the pure amidine after basification and extraction of the combined fractions.
With a working protocol for the isolation of the product of the reaction, the scope was investigated by varying the cyanamide partner (Table 15).
57
Table 15. Cyanamide scope
Isolated yield (>95% pure by 1H NMR). Reaction conditions: Pd(O2CCF3)2 (0.02 mmol), DAF (0.024 mmol), 7a (0.4 mmol), cy-anamide substrate 2a-i (0.6 mmol), pTSA (0.6 mmol), and DMA (1 ml) at 100 °C for 18 h. aIsolated yield (>90% pure by 1H NMR). bTFA in place of pTSA. R is an un-, mono-, or di-substituted amino group.
Cyanamide (2b) gave 55% yield of the corresponding primary amidine 8b, while the di-alkyl substituted 8a, 8c, and 8d were isolated in 73%, 78% and 52%, respectively. The dibenzyl derivative 8e and mono-substituted 8f was isolated in 48% and 42% yield, respectively. The six-membered cyclic cyan-amide substrates 1-cyanopiperidine and 1-cyanomorpholine both afforded the corresponding amidines 8g and 8h in good yields (70% and 76%, respec-tively). Finally 1-cyanopyrrolidine furnished 8i in 29% yield.
The scope of the reaction with regards to the indole partner was also inves-tigated, see Table 16.
58
Table 16. Indole scope
Isolated yield (>95% pure by 1H NMR). Reaction conditions: Pd(O2CCF3)2 (0.02 mmol), DAF (0.024 mmol), indole substrate 7b-7h (0.4 mmol), cyanamide substrate 2 (0.6 mmol), pTSA (0.6 mmol), and DMA (1 ml) at 100 °C for 18 h. aIsolated yield (>90% pure by 1H NMR). R is a dimethylamino or diethylamino group.
Unprotected indole (7b) afforded 8j and 8k in 59% and 66% yield, respec-tively, from the reaction with dimethyl- or diethylcyanamide. 2-methylindole afforded the corresponding amidine 8l in 74% yield, while 5-methoxy-2-me-thylindole gave 8m in 77% yield. The bulkier 2-phenylindole and 1-methyl-2-phenylindole furnished 8n and 8o in 42% and 30% yield, respectively. 5-methylindole gave 8p in 70% yield while the more electron-deficient sub-strates 5-bromoindole and 5-cyanoindole afforded a 35% and 24% yield of 8q and 8r, respectively.
59
5.4 Mechanistic Investigation In order to better understand the reaction mechanism, and especially the role of the ligand and acid additive, a mechanistic investigation was initiated. A generic catalytic cycle is depicted in Figure 25 below, based upon the accu-mulated experience from paper I and paper II.186,191
Figure 25. Proposed mechanism for the palladium(II)-catalyzed addition of indoles to cyanamides, exemplified with substrates 7a and 2c.
The reaction begin with the generation of aryl-palladium complex B from the indole. Next coordination of the cyanamide gives complex C. 1,2-Carbopal-ladation of the coordinated cyanamide then gives the corresponding imidamidate complex D, which releases the product upon protonation and re-generates the catalyst A.
60
5.4.1 DFT Calculations Based on the proposed catalytic cycle depicted in Figure 25 we performed DFT calculations to further investigate the mechanism. The study is based upon the use of substrates 7a and 2c. As a simplification, in place of pTSA, TFA was used as the acid additive. TFA also gives a productive reaction, how-ever with a decrease in yield, see Table 15, product 8c.
In order to investigate the reasons for the particular productivity of the cat-alytic system, both DAF and the less productive ligand bpy (see Table 14), were included in the computational study.
The C-H bond activation was postulated to proceed by a concerted meta-lation-deprotonation (CMD) mechanism, and the DFT calculations indeed indicated that the CMD step should proceed readily for both ligands (Figure 26). One observation from this comparison is that the DAF ligand strongly disfavors κ2-coordination of TFA. The CMD step is according to the compu-tational study completely reversible for both catalysts, this agrees with deuterium incorporation studies for a similar study which has been reported by Campbell et al.195 The difference in free energy of activation between DAF and bpy for the CMD step is 6.3 kJ mol-1 in favor of DAF.
The next step, 1,2-carbopalladation of the cyanamide, also proceeds readily for both ligands, with a small advantage for DAF of 12.6 kJ mol-1. Complex XVI, after 1,2-carbopalladation of the cyanamide, has the product coordinated in a bidentate fashion. Regeneration of the catalyst then requires displacement of the product by an indole moiety and a trifluoroacetate ion, presumably ini-tiated by protonation of the imidamidate XVI. This step is associated with a significant difference in free energy of activation for the DAF and bpy ligands. For DAF, the barrier for formation of the transition state TS-V is 51.9 kJ mol-1 (relative to XVI) while the bpy has a higher barrier of 68.2 kJ mol-1. The pathway is depicted Figure 26, and involves a change in binding mode of the ligand and a change in the square planar coordination of the pal-ladium complex.
61
0
100
C-H bond activation 1,2-carbopalladation
XI
XII
XIII
TS-III XIV
XV
TS-IV
XVI0
-100
-50
50
Protonation of XVI
PdNN
O O
PdNN
O
OF3C
PdNN
O O
CF3
N
H
PdNN
OOF3C
N
H
PdNN
OOF3C
N
H
O
F3C CF3
OC-H bond activation
XI XII XIII TS-III XIV
PdNN
N
N
N PdNN
N
N
N PdNN
N
N
N1,2-carbopalladation
XV TS-IV XVI
Protonation of XVI PdNN
N
N
N
OF3C
OH
PdNN
N
N
N
OF3C
OH
PdN
N
NN
O
F3C
O
H NPd
N
N
NN
O
F3C
O
HN
PdN
N
NN
O
F3C
O
HN
PdN
N
NN
O
CF3O
H
N
HH
PdN
N
N
N
O
CF3OH
N
XVII TS-V XVIII TS-VI
XIX TS-VII XX
XVII
TS-VXVIII
TS-VI
XIX
XX
TS-VII
DAF
bpy
Figure 26. CMD, 1,2-carbopalladation and protonation of XVI for catalysts with DAF or bpy as ligands. Free energies of the complexes are given in kJ mol-1 in the reaction profile.
The free energies of activation for the CMD, 1,2-carbopalladation and proto-nation of the imidamidate are all relatively low. XX is therefore the likely resting state of the reaction. To regenerate the catalyst, one alternative to con-sider is a protonation of the amidine complex XX. A concerted mechanism could thus be identified leading to complex XII (Figure 27).
62
Figure 27. Catalyst regeneration. Free energies of the complexes are given in kJ mol-1 in the reaction profile relative XIII.
The barrier for this step is quite high (172.8 and 154.4 kJ mol-1 relative XX for DAF and bpy, respectively), and an alternative mechanism was thus con-sidered. A concerted ligand exchange of the amidine for an indole moiety would regenerate catalytic complex XIII, see Figure 27.
63
This alternative mechanism has a lower free energy of activation (135.6 and 146.4 kJ mol-1 relative to XX, for DAF and bpy, respectively), compared to the mechanism involving a concerted protonation and ligand exchange. Prod-uct release in neutral form is an endergonic process. However, taking into account the acidic environment and protonating the product does give an ex-ergonic process, thus accounting for the thermodynamic driving force of the reaction.
Overall the DFT calculations support the proposed catalytic cycle. The cal-culations also show that DAF is slightly faster compared to bpy for all key steps in the reaction. Although it may not fully explain the observed differ-ences in the experimental outcome, it still supports that DAF should perform better in this reaction. The DFT calculations also provide an explanation for the acid dependency of the reaction, as the catalyst regeneration relies heavily on this.
64
6. Continuous-Flow Palladium(II)-Catalyzed Desulfitative Synthesis of Aryl Ketones (Paper IV)
6.1 Background One specific application of the 1,2-addition to nitriles is the use of sodium sulfinates or sulfinic acids as aryl-palladium precursors.44–46,66 These reagents have recently been recognized as useful alternatives to aryl carboxylic acids as aryl-palladium precursors. The limitations of the aryl carboxylic acids in terms of the need for electron-rich substrates with substitutents ortho to the carboxylic acid moiety are not present for the aryl sulfinic acid derivatives. The reaction conditions used in previous investigations by our group45,66 con-stitute an interesting system to develop further for application in continuous-flow conditions. Continuous-flow allows for a convenient and safe way to scale up, or more accurately, scale out, a reaction. The main challenge in trans-lating from batch to continuous-flow operation is the solubility of the starting materials, as most pumps are not suitable for pumping mixtures with particles.
The use of a (non-resonant) MW applicator for the heating of the reaction mixture in the system opens up for some exciting possibilities in terms of the use of different reactor materials to vary the properties of the system.
The standard reactor material is borosilicate glass which is MW transpar-ent. This means that the reaction mixture is directly heated by the MWs. Combined with an IR sensor that can measure the temperature through the reactor wall184 this constitutes a system with excellent control of the process. However, with borosilicate glass, any MW induced superheating of potential metal precipitate could lead to reactor failure. As the system is biphasic for some nitriles the use of a borosilicate glass reactor with the IR sensor that can measure the temperature of the actual reaction mixture also allows for direct compensation in terms of applied MW power depending on which phase is currently passing the IR sensor.
An alternative reactor material is the ceramic material silicon carbide. This material readily absorbs MWs and can withstand high temperatures (thermally and chemically resistant up to 1500 °C with a melting point of ~2700 °C).196 It has previously been suggested both as a passive heating element to enable indirect heating by MWs of reaction mixtures with low tan δ values,163,196 as a
65
MW reactor/vial material,164,197,198 and recently it has been applied as contin-uous-flow reactors for use with MW heating.178,181 The use of a silicon carbide reactor would effectively mimic conventional heating of the reaction mixture.
A third alternative, in part combining the properties of the borosilicate glass and silicon carbide reactors, is the use of aluminum oxide as a reactor material. This material is ceramic and very thermostable – and MW transparent. It has been extensively used in standard continuous-flow chemistry setups,199–202 alt-hough not, to the author’s knowledge, as a reactor material for MW heating. Aluminum oxide reactors could thus enable direct MW heating with the added benefit of being a more thermostable material than borosilicate glass.
6.2 Reactor Properties A control experiment to verify the properties of the reactor materials in the MW field was performed (Figure 28).
Figure 28. Control experiment for the reactor materials
In the control experiment the heating profiles of the different reactors were compared for two different solvents, namely benzene and a 1:1 THF/H2O mix-ture. These solvents have a large difference in their MW absorbing capabilities (tan δ for benzene = 0.0014,203 THF = 0.047,162 and H2O = 0.122162) , and the heating profile should thus differ between solvents for a given reactor, if the solvent is heated rather than the reactor itself. The temperature is measured with an Optris CSmicro 3M IR sensor for the borosilicate glass reactor which enables direct measurement of the temperature of the reaction mixture at the glass-liquid interface.184 For the silicon carbide and the aluminum oxide reac-tor an Optris CT IR sensor with a LT22 sensing head was used. This sensor measures the temperature on the outer wall of the reactor.
The expected behavior of the different reactor materials with these solvents was indeed observed. For the silicon carbide reactor which is MW absorbing
0
20
40
60
80
100
0 60 120 180
Tem
pera
ture
(C
)
Time (s)
Benzene
0 60 120 180Time (s)
1:1 THF/H2O
Borosilicate glass
Aluminium oxide
Silicon carbide
66
the heating profile is very similar for benzene and THF/H2O as the reactor itself is heated. For the borosilicate glass and the aluminum oxide reactor the heating of the THF/H2O mixture is clearly faster than for benzene, as expected given that the MW absorbing capabilities of the solvents should be a signifi-cant factor. The slower rise in temperature for the aluminum oxide reactor compared to the borosilicate glass reactor can be explained by the difference between the IR sensors (surface of the reactor or glass-liquid interface).
6.3 Optimization of Reaction Conditions The catalyst system, acid additive and stoichiometry were taken from the ear-lier published protocols,45,66 and adapted slightly to better suit continuous-flow processing by diluting the reaction mixture. Initial attempts using a model re-action between sodium 4-methylbenzenesulfinate and benzyl cyanide produced a biphasic reaction mixture, which upon vigorous stirring gave an emulsion suitable for processing. One stock solution with all reagents in one flask was used rather than a setup with reagents separated into two or more stock solutions. As the biphasic solution was pumped, the emulsion separates into two phases, resulting in a segmented flow, see Figure 29.
Figure 29. Reaction mixture in reactor with applied MW heating. The background has been removed digitally for increased clarity.
67
The starting point of the optimization was thus a stock solution with 75 mM of 9a (1.0 equiv), Pd(O2CCF3)2 (8%), 6-Mebpy (12%), 2a (5.0 equiv) and TFA (10 equiv) in a 1:1 H2O/THF mixture. This solution is pumped through a straight tube reactor. The reactor is 200 mm long, has an inner diameter of 3 mm and the MW field covers 120 mm of the reactor.
First a suitable temperature and flow rate was determined using a borosili-cate glass reactor by screening different temperatures and flow rates (Table 17).
Table 17. Optimization of reaction conditions for the borosilicate glass reactor
a Measured using an Optris CSmicro 3M IR sensor. b Time in the MW irradiated zone of the reactor. c Isolated yield at 0.6 mmol scale (>95% pure by 1H NMR spectroscopic analysis).
Purification of an aliquot by silica gel chromatography, corresponding to a 0.6 mmol scale of the reaction, furnished ketone 11a in 79% yield using the opti-mal conditions found during the screening shown in Table 17 (120 °C and a flow rate of 1 ml/min, entry 4). This corresponds to an extrapolated production rate of 4.3 mmol/h. The lower yield at a lower flow rate (entry 3) is likely due to problems experienced controlling the temperature at lower flow rates, which could arise from the different MW absorbing characteristics of the two phases.
Next the three different reactors were compared by running the same reac-tion. The actual temperature of the reaction mixture differs somewhat depending on the reactor and the sensor used (Optris CSmicro 3M IR sensor for the borosilicate glass reactor and an Optris CT IR sensor with LT22 sens-ing head for the aluminum oxide and silicon carbide reactors).184 To account for this three different temperatures (100, 120, and 140 °C) were tested for each reactor and the condition that gave the best outcome by GC-MS analysis was chosen for purification (Table 18).
68
Table 18. Reactor comparison
a Measured using an Optris CSmicro 3M IR sensor for the borosilicate glass re-actor or an Optris CT IR sensor with a LT22 sensing head for the aluminum oxide and silicon carbide reactor. b The heated zone is considered to be the zone covered by the MW field for the borosilicate glass and aluminum oxide, or the full length of the reactor for the silicon carbide reactor. c Time in the heated zone of the re-actor. d Isolated yield at 0.6 mmol scale (>95% pure by 1H NMR spectroscopic analysis).
The use of the silicon carbide reactor mimic conventional heating and the re-sult (74%) is on par with the borosilicate glass and aluminum oxide reactors that both allow MW heating of the reaction mixture (79% and 78%, respec-tively). The latter two were chosen for further investigation of the scope of the reaction.
69
6.4 Investigation of the Scope of the Reaction The aryl sulfinate scope of the continuous-flow protocol was first investigated with acetonitrile (10b) as the nitrile substrate, see Table 19.
Table 19. Aryl sulfinate scope
Three temperatures (100, 120, and 140 °C) were carried out and checked by GC-MS. The optimal temperature was 120 °C for all reactions except for the one giving product 11c. a Temperature controlled with the help of an Optris CSmicro 3M IR sensor for the boro-silicate glass reactor. Isolated yield at 0.6 mmol scale (>95% pure by 1H NMR spectroscopic analysis). b Temperature controlled with the help of an Optris CT IR sensor with a LT22 sensing head for the aluminum oxide reactor. Isolated yield at 0.6 mmol scale (>95% pure by 1H NMR spectroscopic analysis). c Reaction run at 140 °C. d Deter-mined by GC-MS analysis.
The borosilicate glass reactor gave slightly higher yields than the aluminum oxide reactor. The yields are lower than what has been reported in batch using similar conditions.66 Ketone 11e was reported as a somewhat problematic sub-strate, requiring lowering of the water content in batch.66 In this protocol, the product could be isolated in good yield without any modifications to the pro-tocol to avoid hydrolysis of the amide functionality. This might be attributed to the very short time the reaction mixture spends in the heated zone (50 s).
70
Table 20. Nitrile scope
Three temperatures (100, 120, and 140 °C) were carried out and checked by GC-MS. The optimal temperature was 120 °C for all reactions. a Temperature controlled with the help of an Optris CSmicro 3M IR sensor for the borosilicate glass reactor. Isolated yield at 0.6 mmol scale (>95% pure by 1H NMR spectroscopic analysis). b Temperature controlled with the help of an Optris CT IR sensor with a LT22 sensing head for the aluminum oxide reac-tor. Isolated yield at 0.6 mmol scale (>95% pure by 1H NMR spectroscopic analysis).
The nitrile scope was then investigated with sodium benzenesulfinate (9b) as the aryl sulfinate substrate (Table 20). The same trends were observed as for the aryl sulfinate scope study, higher yields in general for the borosilicate glass reactor with the exception of 11k, and lower yields in general than the batch protocol66. Running three control reactions in batch mode (11b, 11g, and 11k) with the same reaction conditions (10x dilution compared to the published batch protocol)66 furnished similar yields to the continuous-flow operation (53%, 55%, and 46% respectively).
It should further be noted that no reactor failure was observed for either of the reactors tested.
71
7. Synthesis of Sulfinates Using a Solid SO2
Source (Paper V)
7.1 Background Sodium sulfinates have recently been recognized as versatile aryl-palladium precursors. In the last decade several protocols employing these substrates have been reported. Examples include Heck204–207 and Sonogashira208 type re-actions, conjugate 1,4-addition,209,210 phosphonation211,212 and C-H arylation reactions213–218. In addition, several reports on the use of sodium aryl sulfinates for the 1,2-addition to nitriles have been published, including three by our group.44–46,66
As sodium aryl sulfinates are not widely available commercially, many of the substrates used in these protocols need to be synthesized. Methods of prep-aration include reduction of sulfonyl chlorides by sodium sulfite219 or zinc,220,221 reaction of SO2 with organometal reagents such as Grignard222,223 or organolithium,224 Friedel-Crafts sulfination with sulfonyl chlorides,225 and palladium-catalyzed coupling of diazonium tetrafluoroborates, hydrogen and SO2.226 The protocol employing sodium sulfite as a reductant for the synthesis of sulfinates is performed in water and is troublesome for synthesis of certain more hydrophobic sulfinates. Many of the other strategies are hampered by the toxic and corrosive nature of the SO2 gas. Fortunately a useful solid SO2 surrogate has been developed, 1,4-diazabicyclo[2.2.2]octane bis(sulfur diox-ide) adduct (DABSO). 227,228 DABSO has indeed been employed with organometallic reagents, generating the corresponding sulfinates.227,229–242 These protocols used the sulfinates as intermediates to give aryl sulfones,230–
232,235 arylsulfonyl alkynes233, sulfoxides,237 sulfonyl fluorides,240 or sulfona-mides227,229,234,236,238,241,242 in one-pot fashion.
A similar one-pot, two step approach to the synthesis of aryl ketones, start-ing from aryl bromides was attempted, however only trace amounts of the ketone was observed along with significant disproportionation of the sulfinate. Thus, development of a protocol for the synthesis and isolation of sodium sul-finates, based on the use of DABSO as a SO2 surrogate was initiated.
72
7.2 Optimization of Reaction Conditions A model reaction using commercially available Grignard reagent phenyl mag-nesium chloride in THF was conducted to verify the viability of the approach and to work out a suitable purification procedure.
This model reaction gave near quantitative conversion of the starting Gri-gnard reagent to the corresponding magnesium sulfinate. The purification was not straight-forward however; extraction using 2M HCl gave a mixture of the corresponding sulfinic and sulfonic acid. This is explained by a known reac-tion, disproportionation of sulfinic acids gives the corresponding sulfonic acid.243,244 Switching to a literature protocol employing sulfuric acid instead of HCl226 mitigated the problem with disproportionation, but subsequent puri-fication by silica gel, aluminum oxide or reverse-phase chromatography gave almost exclusively the corresponding sulfonic acid. Thus the product was iso-lated as the sodium salt by acidification with sulfuric acid followed by extraction into diethyl ether, subsequent extraction of the organic phases with a sodium carbonate solution followed by evaporation of the aqueous phase. Finally, a solid-liquid extraction using ethanol gave the pure sodium sulfinate (90% yield from commercial phenyl magnesium chloride in THF).
To access a wider range of sulfinates than what is available using commer-cially available Grignard reagents, the generation of the Grignard reagents was optimized starting from a MW heated protocol.245
A protocol for the corresponding lithium reagents was also developed, forming the lithium reagent at -78 °C in THF with n-butyl lithium. The addi-tion of the lithium reagents to the DABSO suspension was performed at -78 °C except in cases where poor solubility of the lithium reagent at this temper-ature prohibited transfer. In these cases, the lithium reagent was allowed to warm until transfer was possible.
73
7.3 Investigation of the Scope of the Reaction The scope of the reaction was first investigated with regards to synthesizing the aryl sulfinates from the corresponding Grignard reagents generated from aryl bromides. The results are presented in Table 21.
Table 21. Sodium sulfinates from Grignard reagents
Isolated yield (>95% pure by 1H NMR). Reaction conditions: 1) aryl bromide (12, 2.0 mmol), Mg (4 equiv), and one I2 crystal in 2.5 ml dry THF under N2 atmosphere was heated by MW to 100 °C for 1 h. 2) the preformed Grignard reagent is added to DABSO (0.5 mmol) at 0-4 °C and stirred for 3 h. 3) reaction mixture allowed to reach room temperature, then extracted with a Na2CO3 so-lution (10% in water). a Starting from chlorobenzene. b Starting from commercially available phenyl magnesium chloride. cStarting from iodoben-zene.
Most aryl bromides gave the corresponding aryl sulfinate in good yield. Elec-tron-poor substrates generally performed better, exemplified with 13b with a p-methoxy substituent which was isolated in 48% yield while m-methoxy sub-stituted 13c could be isolated in 85% yield. 4-bromobiphenyl gave 13j in 87% yield, this sodium sulfinate was not accessible by reduction of the correspond-ing sulfonyl chloride. Using chlorobenzene for the generation of 13a gave 64% yield, while iodobenzene furnished 13a in 25% yield. For the iodoben-zene, biaryl formation was observed from a competing Wurtz-Fittig type reaction.246
Next the scope of the reaction using lithium reagents generated by treat-ment with n-butyllithium was investigated, see Table 22.
74
Table 22. Sodium sulfinates from lithium reagents
Isolated yield (>95% pure by 1H NMR). Reaction conditions: 1) aryl bromide (12, 2.0 mmol) in 2.5 ml dry THF under N2 atmosphere was cooled to -78 °C and n-BuLi (1 equiv, 2.5 M solution in hexanes) was added slowly and then stirred for 1 h. 2) the preformed lithium reagent is added to DABSO (0.5 mmol) at -78 °C and stirred for 3 h. 3) reaction mixture is allowed to reach room tem-perature, then extracted with a Na2CO3 solution (10% in water). a Using t-BuLi (1 equiv, 1.7 M solution in hexanes) instead of n-BuLi.
The yields are similar for the lithium reagents when compared to the corre-sponding Grignard reagents. Electron-poor substrates seem to be favored for the lithium reagents as for the Grignard reagents. The trend seen for the Gri-gnard reagents where 4-bromobenzotrifluoride performed better than 2-bromobenzotrifluoride (91% and 75% of 13d and 13e, respectively, Table 21) was reversed for the lithium reagents (48% and 99% of 13d and 13e, respec-tively, Table 22). The limited solubility of the aryl lithium reagent partly explains the relatively low yield of 13j.
Generation of the corresponding lithium reagents from thiophene, furan, and benzofuran was performed using t-butyllithium after initial attempts using thiophene and n-butyl lithium gave a mixture of sodium thiophenesulfinate and sodium n-butylsulfinate. The yields of the small and relatively polar thio-phene and furan derivatives 13l and 13m was low (15% and 21%,
75
respectively) due to problems with the workup. The more hydrophobic 13n was isolated in 50% yield.
2-Bromo-thiophene treated with n-butyllithium gave sodium n-butyl-thio-phenesulfinate (13o) in 68% yield by nucleophilic attack on the n-butylbromide formed upon lithiation.
7.4 Application in Palladium(II)-Catalysis Finally, the previously unattainable (by sulfonyl chloride reduction) sulfinate 13j was tested in the palladium(II)-catalyzed 1,2-addition to acetonitrile to at-tain 4-acetylbiphenyl in 48% yield (Figure 30).
Figure 30. Palladium(II)-catalyzed 1,2-addition of 13j to acetonitrile. Isolated yield (>95% pure by 1H NMR). Reaction conditions: Pd(O2CCF3)2 (8%), 6-Mebpy (12%), were stirred in THF/H2O (1:1, 1.4 ml) for 1 min before addition of 13j (0.5 mmol), acetonitrile (5 equiv) and TFA (10 equiv). The reaction was heated by MW to 100 °C for 1h.
76
8. Concluding Remarks
The work presented in this thesis describes the development of palladium(II)-catalyzed additions to nitriles and cyanamides; giving ketones and amidines. In conjunction with these new methods, mechanistic investigations and new continuous-flow methodology is presented. In addition, the development of a synthetic route to sodium aryl sulfinates, which can serve as aryl-palladium precursors, is described. The specific results and conclusions derived from these studies are summarized below.
In paper I the development of a palladium(II)-catalyzed addition reaction
using cyanamides to generate aryl amidines is described. Aryltrifluorob-orates were used to generate the aryl-palladium species and in combination with primary, secondary, and tertiary cyanamides this proto-col represents a new and mild synthetic route to aryl amidines. An arylboronic acid and a boronic acid pinacol ester were also productive us-ing the same conditions.
With these positive results in hand a new protocol, described in paper II, was developed that used aryl carboxylic acids as the aryl-palladium pre-cursor. These substrates are challenging and the reaction required very electron-rich substrates with at least one ortho substituent. Primary, sec-ondary, and tertiary cyanamides were all productive, and the sterically congested aryl amidines produced from this reaction are interesting prod-ucts, not easily derived by other methods. A continuous-flow method was also developed, producing the desired aryl amidine in excellent yield, cor-responding to an output of 11 mmol/h. The mechanism of the reaction was also investigated by the means of DFT calculations in combination with ESI-MS studies of the reaction mixture. This resulted in a plausible mechanism being suggested, supported by these data.
Another type of aryl-palladium precursor, namely indole derivatives, was investigated in paper III as a substrate in the palladium(II)-catalyzed ad-dition to cyanamides. Relying on C-H activation to generate the aryl-palladium species this class of substrates provides an atom-efficient strat-egy for the synthesis of 3-amidinoindoles. A variety of indoles were productive with primary, secondary, and tertiary cyanamides. Mechanistic investigation by DFT calculations provided a plausible mechanistic path-way.
77
Some trends can be discerned regarding the palladium(II)-catalyzed addi-tion to cyanamides by examining papers I-III together. Primary, secondary, and tertiary cyanamides all provide good results combined with the different aryl-palladium precursors. Similar reactivity of the dif-ferent cyanamide substrates is also observed through papers I-III. Electron-rich and neutral aryl-palladium precursors work good in general, while electron-deficient substrates generally gives low or no conversion to the desired product.
In paper IV a continuous-flow method was developed for the synthesis of aryl ketones by a palladium(II)-catalyzed desulfitative addition of so-dium aryl sulfinates to nitriles. The developed protocol provided the ketones in fair to good yields, showing the same disfavor of electron-poor aryl-palladium precursors as was observed in papers I-III. An additional point made in paper IV is that the use of an aluminum oxide reactor was demonstrated, constituting a safer alternative to the standard borosilicate glass reactor used for direct heating of the reaction mixture by MW heating.
The use of the SO2 surrogate DABSO for the synthesis of sodium aryl-sulfinates was demonstrated in paper V. These substrates are of interest in palladium(II)-catalysis as aryl-palladium precursors through a desul-fitative process. Grignard and organolithium reagents are used as the nucleophiles and the scope of the reaction is wide. The protocol constitute a safe and convenient method for the synthesis of sodium arylsulfinates.
78
Acknowledgements
There are many people to thank that have helped making this thesis possible. I would like to thank the following people in particular: My supervisor professor Mats Larhed, for taking me on as a PhD student and your patience, allowing me to explore new ideas. I’m also grateful for the chance to do some medicinal chemistry on the side, as well as the opportunity to work with the continuous-flow system. My co-supervisors; Dr. Jonas Sävmarker, I am very thankful for all the support and mentoring. All the discussions on chemistry and what-not, as well as your positive atti-tude, has been extra appreciated. Dr. Luke Odell, for always being available to discuss the theoretical and prac-tical aspects of chemistry, you’re really the go-to-guy in these cases. Dr. Christian Sköld, for discussions and practical help when it comes to the mechanistic investigations presented in this thesis. Professor Anders Hallberg, for inspiring guidance in the medicinal chemistry project. Dr. Per Sjöberg, for invaluable help with the ESI-MS investigations, you re-ally can make (almost) any complex appear (given that it is charged…). Magnus Fagrell. I appreciate the chance to work with you on the flow equip-ment, it is inspiring to see such a formidable attitude to problem solving and product improvement. Dr. Peter Brandt, your contribution to the last paper included in the thesis was invaluable, I appreciate the swift work on the DFT calculations towards all kinds of deadlines and all the mechanistic discussions. Dr. Bobo Skillinghaug, Thank you for all the collaborative efforts, you’re an excellent colleague as well as a friend! Your only flaw must be your inability to designate the roles of hero and sidekick ;) …
79
Co-authors; Dr. Måns Thulin – for statistically significant contribution to the temperature measurement paper. Dr. Fredrik Svensson for all the nice work on DFT calculations, statistics, coffee table discussions, and (pub)paper writ-ing sessions. Dr. Alejandro Trejos – for help with the second paper and for being so welcoming during my first time at the department. Dr. Vivek Konda, excellent, humble chemist and good guy – I really appreciate the work we did together on the SiC paper. Co-authors to be, in the near future; Sara Roslin, Dr. Patrik Nordeman, Dr. Prajakta Naik, and Dr. Tamal Roy – you also de-serve mention for the great work on projects soon to be published. Luke, Jonas, Christian, and Bobo for critical examination of the thesis. Apotekarsocieteten and Smålands Nation for financial support in the form of travel grants, allowing for parts of this work to be presented at conferences. Dr. Ulf Bremberg, for inspiring discussions on things of all nature, as well as for giving me the opportunity to do the internship at Beactica. Dr. Ashkan Fardost, TV-personality, as well as excellent office-mate for the first 4 years of my PhD period, for great discussions on things chemistry- and non-chemistry related. Tamal, I’ve been lucky in the office-mate lottery, it’s been a pleasure to be able to work on some projects with you as well! Karin Engen, Dr. Johan Gising, Dr. Marc Stevens, Dr. Johanna Larsson, and Sävmarker – current and past NMR team members, for all the enjoyable time spent in the lower levels of BMC! All the people that have made travelling to conferences of all sorts extra en-joyable; I especially enjoyed the trips to California, Boston, and London. Shiao, Natalia, and Mariama for being good colleagues and for fun get-togeth-ers. Project workers, Alaa Abbas for valuable contribution to one of the ”still to come” papers. Mostafa Haitham for valuable contribution to the initial phase of the work on the indoles. Gunilla Eriksson, thank you for your invaluable contribution to making the division such a great workplace! Sorin Srbu, sys-admin, for making the com-puters run smoothly, (admin rights…,) and for helping out whenever it was needed. All other administrative personnel, none mentioned, none forgotten – you all make the essential things run smoothly at the department. My high school teacher Folke, you deserve thanks for conveying your interest in chemistry, and thus getting me started on this path.
80
Vänner och ”bonus-släkt” som ställt upp och stöttat extra inför alla möjliga deadlines nu mot slutet. Alla ni som förgyller min fritid med allt möjligt skoj så som spelkvällar, fot-boll, pumpakarvning osv… Mina föräldrar och familj för en fantastisk stöttning, under hela resan fram till denna avhandling! (Almost) Last but certainly not least, Martina! Du har varit fantastisk under denna (utdragna) slutspurt. Faktiska bidrag till avhandlingen (motiverat mig att börja skriva och läsning) tillsammans med stöttning på hemmaplan har gjort detta möjligt. Tack för allt jobb du lagt ner, och för att du samtidigt är en helt fantastisk mamma till Linn. ♥ And finally, I must express my gratitude to all the (present and former) col-leagues at the division of organic pharmaceutical chemistry for creating such a good work environment, I have really enjoyed this time!
81
References
(1) Berzelius, J. J. Årsberättelsen om Fram. i Fys. och kemi, R. Swedish Acad. Sci. 1835.
(2) Lindström, B.; Pettersson, L. J. A Brief History of Catalysis. CATTECH 2003, 7 (4), 130–138.
(3) Ostwald, W. Abstracts. Zeitschrift für Phys. Chemie 1894, 15, 705–706. (4) Laidler, K. J. A Glossary of Terms Used in Chemical Kinetics, Including
Reaction Dynamics. Pure Appl. Chem. 1996, 68 (1), 149–192. (5) IUPAC Compendium of Chemical Terminology, Electronic version
http://goldbook.iupac.org/C00876.html (accessed Jul 2, 2017). (6) Bosch, C. Process of Producing Ammonia. U.S. Patent 990,191 (A), 1911. (7) Blaser, H.-U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M.
Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345 (12), 103–151.
(8) Raney, M. Method of Preparing Catalytic Material. U.S. Patent 1,563,587, 1925.
(9) Raney, M. Method of Producing Finely-Divided Nickel. U.S. Patent 1,628,190, 1927.
(10) Ostwald, W. Improvements in the Manufacture of Nitric Acid and Nitrogen Oxides. U.K. Patent GB 190200698 (A), 1902.
(11) Ostwald, W. Improvements in and Relating to the Manufacture of Nitric Acid and Oxides of Nitrogen. U.K. Patent GB 190208300 (A), 1903.
(12) Smidt, J.; Hafner, W.; Jira, R.; Sedlmeier, J.; Sieber, R.; Rüttinger, R.; Kojer, H. Katalytische Umsetzungen von Olefinen an Platinmetall-Verbindungen Das Consortium-Verfahren Zur Herstellung von Acetaldehyd. Angew. Chemie 1959, 71 (5), 176–182.
(13) Smidt, J.; Hafner, W.; Jira, R.; Sieber, R.; Sedlmeier, J.; Sabel, A. The Oxidation of Olefins with Palladium Chloride Catalysts. Angew. Chemie Int. Ed. English 1962, 1 (2), 80–88.
(14) Jira, R. Acetaldehyde from Ethylene-A Retrospective on the Discovery of the Wacker Process. Angew. Chemie Int. Ed. 2009, 48 (48), 9034–9037.
(15) Hershman, A.; Knox, W.; Paulik, F.; Roth, J. Production of Carboxylic Acids and Esters. U.S. Patent 3,769,329 (A), 1973.
(16) Keim, W. Oligomerization of Ethylene to α-Olefins: Discovery and Development of the Shell Higher Olefin Process (SHOP). Angew. Chemie Int. Ed. 2013, 52 (48), 12492–12496.
(18) MacMillan, D. W. C. The Advent and Development of Organocatalysis. Nature 2008, 455 (7211), 304–308.
82
(19) Crabtree, R. H. General Properties of Organometallic Complexes. In The Organometallic Chemistry of the Transition Metals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; pp 29–52.
(20) Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of Olefin with Aryl Iodide Catalyzed by Palladium. Bull. Chem. Soc. Jpn. 1971, 44 (2), 581–581.
(21) Nolley, J. P.; Heck, R. F. Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem. 1972, 37 (14), 2320–2322.
(22) Miyaura, N.; Suzuki, A. Stereoselective Synthesis of Arylated (E)-Alkenes by the Reaction of Alk-1-Enylboranes with Aryl Halides in the Presence of Palladium Catalyst. J. Chem. Soc. Chem. Commun. 1979, No. 19, 866–867.
(23) Miyaura, N.; Yamada, K.; Suzuki, A. A New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides. Tetrahedron Lett. 1979, 20 (36), 3437–3440.
(24) King, A. O.; Okukado, N.; Negishi, E. Highly General Stereo-, Regio-, and Chemo-Selective Synthesis of Terminal and Internal Conjugated Enynes by the Pd-Catalysed Reaction of Alkynylzinc Reagents with Alkenyl Halides. J. Chem. Soc., Chem. Commun., 1977, No. 19, 683–684.
(25) Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16 (50), 4467–4470.
(26) Torborg, C.; Beller, M. Recent Applications of Palladium-Catalyzed Coupling Reactions in the Pharmaceutical, Agrochemical, and Fine Chemical Industries. Adv. Synth. Catal. 2009, 351 (18), 3027–3043.
(27) Nakamura, S.; Yasui, T. The Mechanism of the Palladium-Catalyzed Synthesis of Vinyl Acetate from Ethylene in a Heterogeneous Gas Reaction. J. Catal. 1970, 17 (3), 366–374.
(28) Moritani, I.; Fujiwara, Y. Aromatic Substitution of Styrene-Palladium Chloride Complex. Tetrahedron Lett. 1967, 8 (12), 1119–1122.
(29) Fujiwara, Y.; Moritani, I.; Matsuda, M.; Teranishi, S. Aromatic Substitution of Styrene-Palladium Chloride Complex. II Effect of Metal Acetate. Tetrahedron Lett. 1968, 9 (5), 633–636.
(30) Danno, S.; Moritani, I.; Fujiwara, Y. Aromatic Substitution of olefin—IX. Tetrahedron 1969, 25 (19), 4819–4823.
(31) Fujiwara, Y.; Moritani, I.; Matsuda, M.; Teranishi, S. Aromatic Substitution of Olefin. IV Reaction with Palladium Metal and Silver Acetate. Tetrahedron Lett. 1968, 9 (35), 3863–3865.
(32) Fujiwara, Y.; Moritani, I.; Danno, S.; Asano, R.; Teranishi, S. Aromatic Substitution of Olefins. VI. Arylation of Olefins with palladium(II) Acetate. J. Am. Chem. Soc. 1969, 91 (25), 7166–7169.
(33) Heck, R. F. Allylation of Aromatic Compounds with Organopalladium Salts. J. Am. Chem. Soc. 1968, 90 (20), 5531–5534.
(34) Dieck, H. A.; Heck, R. F. Palladium-Catalyzed Conjugated Diene Synthesis from Vinylic Halides and Olefinic Compounds. J. Org. Chem. 1975, 40 (8), 1083–1090.
83
(35) Oda, H.; Morishita, M.; Fugami, K.; Sano, H.; Kosugi, M. A Novel Diarylation Reaction of Alkynes by Using Aryltributylstannane in the Presence of Palladium Catalyst. Chem. Lett. 1996, 25 (9), 811–812.
(36) Hirabayashi, K.; Nishihara, Y.; Mori, A.; Hiyama, T. A Novel C-C Bond Forming Reaction of Aryl- and Alkenylsilanols. A Halogen-Free Mizoroki-Heck Type Reaction. Tetrahedron Lett. 1998, 39 (43), 7893–7896.
(37) Matoba, K.; Motofusa, S.; Sik Cho, C.; Ohe, K.; Uemura, S. Palladium(II)-Catalyzed Phenylation of Unsaturated Compounds Using Phenylantimony Chlorides under Air. J. Organomet. Chem. 1999, 574 (1), 3–10.
(38) Inoue, A.; Shinokubo, H.; Oshima, K. Oxidative Heck-Type Reaction Involving Cleavage of a Carbon-Phosphorus Bond of Arylphosphonic Acids. J. Am. Chem. Soc. 2003, 125 (6), 1484–1485.
(39) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chemie Int. Ed. 2009, 48 (28), 5094–5115.
(40) Kuhl, N.; Hopkinson, M. N.; Wencel-Delord, J.; Glorius, F. Beyond Directing Groups: Transition-Metal-Catalyzed C-H Activation of Simple Arenes. Angew. Chemie Int. Ed. 2012, 51 (41), 10236–10254.
(41) Peschko, C.; Winklhofer, C.; Steglich, W. Biomimetic Total Synthesis of Lamellarin L by Coupling of Two Different Arylpyruvic Acid Units. Chem. - A Eur. J. 2000, 6 (7), 1147–1152.
(42) Myers, A. G.; Tanaka, D.; Mannion, M. R. Development of a Decarboxylative Palladation Reaction and Its Use in a Heck-Type Olefination of Arene Carboxylates. J. Am. Chem. Soc. 2002, 124 (38), 11250–11251.
(43) Garves, K. Coupling, Carbonylation, and Vinylation Reactions of Aromatic Sulfinic Acids via Organopalladium Intermediates. J. Org. Chem. 1970, 35 (10), 3273–3275.
(44) Liu, J.; Zhou, X.; Rao, H.; Xiao, F.; Li, C.-J.; Deng, G.-J. Direct Synthesis of Aryl Ketones by Palladium-Catalyzed Desulfinative Addition of Sodium Sulfinates to Nitriles. Chem. - A Eur. J. 2011, 17 (29), 7996–7999.
(45) Behrends, M.; Sävmarker, J.; Sjöberg, P. J. R.; Larhed, M. Microwave-Assisted Palladium(II)-Catalyzed Synthesis of Aryl Ketones from Aryl Sulfinates and Direct ESI-MS Studies Thereof. ACS Catal. 2011, 1 (11), 1455–1459.
(46) Miao, T.; Wang, G.-W. Synthesis of Ketones by Palladium-Catalysed Desulfitative Reaction of Arylsulfinic Acids with Nitriles. Chem. Commun. 2011, 47 (33), 9501–9503.
(48) Carrow, B. P.; Hartwig, J. F. Distinguishing between Pathways for Transmetalation in Suzuki-Miyaura Reactions. J. Am. Chem. Soc. 2011, 133 (7), 2116–2119.
(49) Amatore, C.; Le Duc, G.; Jutand, A. Mechanism of Palladium-Catalyzed Suzuki-Miyaura Reactions: Multiple and Antagonistic Roles of Anionic “Bases” and Their Countercations. Chem. - A Eur. J. 2013, 19 (31), 10082–10093.
84
(50) Molander, G. A.; Canturk, B.; Kennedy, L. E. Scope of the Suzuki-Miyaura Cross-Coupling Reactions of Potassium Heteroaryltrifluoroborates. J. Org. Chem. 2009, 74 (3), 973–980.
(51) Fleckenstein, C. A.; Plenio, H. Efficient Suzuki-Miyaura Coupling of (Hetero)aryl Chlorides with Thiophene- and Furanboronic Acids in Aqueous N-Butanol. J. Org. Chem. 2008, 73 (8), 3236–3244.
(52) Lennox, A. J. J.; Lloyd-Jones, G. C. Organotrifluoroborate Hydrolysis: Boronic Acid Release Mechanism and an Acid-Base Paradox in Cross-Coupling. J. Am. Chem. Soc. 2012, 134 (17), 7431–7441.
(53) Vedejs, E.; Chapman, R. W.; Fields, S. C.; Lin, S.; Schrimpf, M. R. Conversion of Arylboronic Acids into Potassium Aryltrifluoroborates: Convenient Precursors of Arylboron Difluoride Lewis Acids. J. Org. Chem. 1995, 60 (10), 3020–3027.
(54) Darses, S.; Genet, J.-P. Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis. Chem. Rev. 2008, 108 (1), 288–325.
(55) Batey, R. A.; Quach, T. D. Synthesis and Cross-Coupling Reactions of Tetraalkylammonium Organotrifluoroborate Salts. Tetrahedron Lett. 2001, 42 (52), 9099–9103.
(56) Molander, G. A.; Ellis, N. Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res. 2007, 40 (4), 275–286.
(57) O’Hair, R. A. J.; Rijs, N. J. Gas Phase Studies of the Pesci Decarboxylation Reaction: Synthesis, Structure, and Unimolecular and Bimolecular Reactivity of Organometallic Ions. Acc. Chem. Res. 2015, 48 (2), 329–340.
(58) Shepard, A. F.; Winslow, N. R.; Johnson, J. R. The Simple Halogen Derivatives of Furan. J. Am. Chem. Soc. 1930, 52 (5), 2083–2090.
(59) Nilsson, M.; Kulonen, E.; Sunner, S.; Frank, V.; Brunvoll, J.; Bunnenberg, E.; Djerassi, C.; Records, R. A New Biaryl Synthesis Illustrating a Connection between the Ullmann Biaryl Synthesis and Copper-Catalysed Decarboxylation. Acta Chem. Scand. 1966, 20, 423–426.
(60) Tanaka, D.; Romeril, S. P.; Myers, A. G. On the Mechanism of the palladium(II)-Catalyzed Decarboxylative Olefination of Arene Carboxylic Acids. Crystallographic Characterization of Non-Phosphine palladium(II) Intermediates and Observation of Their Stepwise Transformation in Heck-like Processes. J. Am. Chem. Soc. 2005, 127 (29), 10323–10333.
(61) Dickstein, J. S.; Mulrooney, C. A.; O’Brien, E. M.; Morgan, B. J.; Kozlowski, M. C. Development of a Catalytic Aromatic Decarboxylation Reaction. Org. Lett. 2007, 9 (13), 2441–2444.
(62) Xue, L.; Su, W.; Lin, Z. A DFT Study on the Pd-Mediated Decarboxylation Process of Aryl Carboxylic Acids. Dalt. Trans. 2010, 39 (41), 9815–9822.
(63) Svensson, F.; Mane, R. S.; Sävmarker, J.; Larhed, M.; Sköld, C. Theoretical and Experimental Investigation of palladium(II)-Catalyzed Decarboxylative Addition of Arenecarboxylic Acid to Nitrile. Organometallics 2013, 32 (2), 490–497.
(64) Goossen, L. J. Synthesis of Biaryls via Catalytic Decarboxylative Coupling. Science 2006, 313 (5787), 662–664.
85
(65) Gooßen, L. J.; Lange, P. P.; Rodríguez, N.; Linder, C. Low-Temperature Ag/Pd-Catalyzed Decarboxylative Cross-Coupling of Aryl Triflates with Aromatic Carboxylate Salts. Chem. - A Eur. J. 2010, 16 (13), 3906–3909.
(66) Skillinghaug, B.; Sköld, C.; Rydfjord, J.; Svensson, F.; Behrends, M.; Sävmarker, J.; Sjöberg, P. J. R.; Larhed, M. Palladium(II)-Catalyzed Desulfitative Synthesis of Aryl Ketones from Sodium Arylsulfinates and Nitriles: Scope, Limitations, and Mechanistic Studies. J. Org. Chem. 2014, 79 (24), 12018–12032.
(67) Cho, C. S.; Uemura, S. Palladium-Catalyzed Cross-Coupling of Aryl and Alkenyl Boronic Acids with Alkenes via Oxidative Addition of a Carbonboron Bond to palladium(O). J. Organomet. Chem. 1994, 465 (1–2), 85–92.
(68) Jung, Y. C.; Mishra, R. K.; Yoon, C. H.; Jung, K. W. Oxygen-Promoted Pd(II) Catalysis for the Coupling of Organoboron Compounds and Olefins. Org. Lett. 2003, 5 (13), 2231–2234.
(69) Andappan, M. M. S.; Nilsson, P.; Larhed, M. The First Ligand-Modulated Oxidative Heck Vinylation. Efficient Catalysis with Molecular Oxygen as palladium(0) Oxidant. Chem. Commun. 2004, 2 (2), 218–219.
(70) Zhao, L.; Lu, X. PdII-Catalyzed Cyclization of Alkynes Containing Aldehyde, Ketone, or Nitrile Groups Initiated by the Acetoxypalladation of Alkynes. Angew. Chemie Int. Ed. 2002, 41 (22), 4343–4345.
(71) Tsukamoto, H.; Kondo, Y. Palladium(II)-Catalyzed Annulation of Alkynes with Ortho-Ester-Containing Phenylboronic Acids. Org. Lett. 2007, 9 (21), 4227–4230.
(72) Jia, X.; Zhang, S.; Wang, W.; Luo, F.; Cheng, J. Palladium-Catalyzed Acylation of Sp 2 C−H Bond: Direct Access to Ketones from Aldehydes. Org. Lett. 2009, 11 (14), 3120–3123.
(73) Lu, X.; Lin, S. Pd(II)-Bipyridine Catalyzed Conjugate Addition of Arylboronic Acid to α,β-Unsaturated Carbonyl Compounds. J. Org. Chem. 2005, 70 (23), 9651–9653.
(74) Wang, Y.; Zhu, Q. Palladium(II)-Catalyzed Cycloamidination via C(sp2)-H Activation and Isocyanide Insertion. Adv. Synth. Catal. 2012, 354 (10), 1902–1908.
(75) Yang, C.-C.; Tai, H.-M.; Sun, P.-J. Palladium-Catalyzed Chemoselective Intramolecular Cyclization of Bromoanilinoalkenenitriles. J. Chem. Soc. Perkin Trans. 1 1997, No. 19, 2843–2850.
(76) Luo, F.-H.; Chu, C.-I.; Cheng, C.-H. Nitrile-Group Transfer from Solvents to Aryl Halides. Novel Carbon−Carbon Bond Formation and Cleavage Mediated by Palladium and Zinc Species. Organometallics 1998, 17 (6), 1025–1030.
(77) Larock, R. C.; Tian, Q.; Pletnev, A. A. Carbocycle Synthesis via Carbopalladation of Nitriles. J. Am. Chem. Soc. 1999, 121 (13), 3238–3239.
(78) Pletnev, A. A.; Tian, Q.; Larock, R. C. Carbopalladation of Nitriles: Synthesis of 2,3-Diarylindenones and Polycyclic Aromatic Ketones by the Pd-Catalyzed Annulation of Alkynes and Bicyclic Alkenes by 2-Iodoarenenitriles. J. Org. Chem. 2002, 67 (26), 9276–9287.
86
(79) Tian, Q.; Pletnev, A. a; Larock, R. C. Carbopalladation of Nitriles : Synthesis of 3,4-Disubstituted 2-Aminonaphthalenes and 1,3-Benzoxazine Derivatives by the Palladium-Catalyzed Annulation of Alkynes by ( 2-Iodophenyl ) Acetonitrile. J. Org. Chem. 2003, 2 (68), 339–347.
(80) Zhou, C.; Larock, R. C. Synthesis of Aryl Ketones by the Pd-Catalyzed C-H Activation of Arenes and Intermolecular Carbopalladation of Nitriles. J. Am. Chem. Soc. 2004, 126 (8), 2302–2303.
(81) Zhou, C.; Larock, R. C. Synthesis of Aryl Ketones or Ketimines by Palladium-Catalyzed Arene C-H Addition to Nitriles. J. Org. Chem. 2006, 71 (9), 3551–3558.
(82) Zhao, B.; Lu, X. Palladium(II)-Catalyzed Addition of Arylboronic Acid to Nitriles. Tetrahedron Lett. 2006, 47 (38), 6765–6768.
(83) Zhao, B.; Lu, X. Cationic Palladium(II)-Catalyzed Addition of Arylboronic Acids to Nitriles. One-Step Synthesis of Benzofurans from Phenoxyacetonitriles. Org. Lett. 2006, 8 (26), 5987–5990.
(84) Yu, A.; Li, J.; Cui, M.; Wu, Y. Addition of Arylboronic Acids to Nitriles in Aqueous Media Catalyzed by a 2,2’-bipyridine-Cyclopalladated Ferrocenylimine Complex. Synlett 2007, 2007 (19), 3063–3067.
(85) Wang, X.; Wang, X.; Liu, M.; Ding, J.; Chen, J.; Wu, H. Palladium-Catalyzed Reaction of Arylboronic Acids with Aliphatic Nitriles: Synthesis of Alkyl Aryl Ketones and 2-Arylbenzofurans. Synth. 2013, 45 (16), 2241–2244.
(86) Das, T.; Chakraborty, A.; Sarkar, A. Palladium Catalyzed Addition of Arylboronic Acid or Indole to Nitriles: Synthesis of Aryl Ketones. Tetrahedron Lett. 2014, 55 (52), 7198–7202.
(87) Wang, X.; Liu, M.; Xu, L.; Wang, Q.; Chen, J.; Ding, J.; Wu, H. Palladium-Catalyzed Addition of Potassium Aryltrifluoroborates to Aliphatic Nitriles: Synthesis of Alkyl Aryl Ketones, Diketone Compounds, and 2-Arylbenzo[b]furans. J. Org. Chem. 2013, 78 (11), 5273–5281.
(88) Lindh, J.; Sjöberg, P. J. R.; Larhed, M. Synthesis of Aryl Ketones by Palladium(II)-Catalyzed Decarboxylative Addition of Benzoic Acids to Nitriles. Angew. Chemie Int. Ed. 2010, 49 (42), 7733–7737.
(89) Axelsson, L.; Veron, J. B.; Sävmarker, J.; Lindh, J.; Odell, L. R.; Larhed, M. An Improved palladium(II)-Catalyzed Method for the Synthesis of Aryl Ketones from Aryl Carboxylic Acids and Organonitriles. Tetrahedron Lett. 2014, 55 (15), 2376–2380.
(90) Ma, Y.; You, J.; Song, F. Facile Access to 3-Acylindoles through Palladium-Catalyzed Addition of Indoles to Nitriles: The One-Pot Synthesis of Indenoindolones. Chem. Eur. J. 2013, 19 (4), 1189–1193.
(91) Jiang, T.-S.; Wang, G.-W. Synthesis of 3-Acylindoles by Palladium-Catalyzed Acylation of Free (N–H) Indoles with Nitriles. Org. Lett. 2013, 15 (4), 788–791.
(92) Jiang, T.-S.; Wang, G.-W. Synthesis of 2-Acylthiophenes by Palladium-Catalyzed Addition of Thiophenes to Nitriles. Adv. Synth. Catal. 2014, 356 (2–3), 369–373.
(93) Goetz, R. W.; Mador, I. L. Catalyzed Hydrolysis of Nitriles to Amides. U.S. Patent 3,670,021 A, 1972.
87
(94) Paraskewas, S. Synthese von Carbonsäureamiden Durch Partielle Hydrolyse von PdCl 2 -Nitril-Komplexen. Synthesis (Stuttg). 1974, No. 8, 574–575.
(95) Villain, G.; Constant, G.; Gaset, A.; Kalck, P. Selective Catalysis of the Hydration of Nitriles to Amides: Evidence for an Efficient Homogeneous Precursor Containing a PdOH Bond. J. Mol. Catal. 1980, 7 (3), 355–364.
(96) Villain, G.; Kalck, P.; Gaset, A. Regioselective Hydration of Acrylonitrile to Give Acrylamide Catalyzed by (Pd(OH)2(bipyridine)(H2O)). Tetrahedron Lett. 1980, 21 (30), 2901–2904.
(97) Villain, G.; Gaset, A.; Kalck, P. Selective Catalysis of the Hydration of Nitriles into Amides. Part II[1]. Kinetic Investigation of the Reaction Catalyzed by [PdCl(OH)(bipy)(H2O)]. Generalization of the Reaction. J. Mol. Catal. 1981, 12 (1), 103–111.
(98) McKenzie, C. J.; Robson, R. High Turnover Catalysis at Bimetallic Sites of the Hydration of Nitriles to Carboxamides Co-Catalysed by Acid. Highly Specific Hydration of Acrylonitrile to Acrylamide. J. Chem. Soc. Chem. Commun. 1988, No. 2, 112–114.
(99) Kaminskaia, N. V.; Kostic, N. M. Nitrile Hydration Catalysed by palladium(II) Complexes. J. Chem. Soc. Dalt. Trans. 1996, No. 18, 3677–3686.
(100) Maffioli, S. I.; Marzorati, E.; Marazzi, A. Mild and Reversible Dehydration of Primary Amides with PdCl 2 in Aqueous Acetonitrile. Org. Lett. 2005, 7 (23), 5237–5239.
(101) Kim, E. S.; Kim, H. S.; Kim, J. N. An Efficient Pd-Catalyzed Hydration of Nitrile with Acetaldoxime. Tetrahedron Lett. 2009, 50 (24), 2973–2975.
(102) Becke, A. D. A New Mixing of Hartree–Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98 (2), 1372–1377.
(103) Grimme, S. Density Functional Theory with London Dispersion Corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1 (2), 211–228.
(104) Fenn, J.; Mann, M.; Meng, C.; Wong, S.; Whitehouse, C. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246 (4926), 64–71.
(105) Mass Spectrometry; Ekman, R., Silberring, J., Westman-Brinkmalm, A., Kraj, A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009.
(106) Santos, L. S. Online Mechanistic Investigations of Catalyzed Reactions by Electrospray Ionization Mass Spectrometry: A Tool to Intercept Transient Species in Solution. European J. Org. Chem. 2008, 2008 (2), 235–253.
(107) Aliprantis, A. O.; Canary, J. W. Observation of Catalytic Intermediates in the Suzuki Reaction by Electrospray Mass Spectrometry. J. Am. Chem. Soc. 1994, 116 (15), 6985–6986.
(108) Brown, J. M.; Hii, K. K. Characterization of Reactive Intermediates in Palladium-Catalyzed Arylation of Methyl Acrylate(Heck Reaction). Angew. Chemie Int. Ed. English 1996, 35 (6), 657–659.
(109) Sabino, A. A.; Machado, A. H. L.; Correia, C. R. D.; Eberlin, M. N. Probing the Mechanism of the Heck Reaction with Arene Diazonium Salts by Electrospray Mass and Tandem Mass Spectrometry. Angew. Chemie Int. Ed. 2004, 43 (19), 2514–2518.
88
(110) Enquist, P. A.; Nilsson, P.; Sjöberg, P.; Larhed, M. ESI-MS Detection of Proposed Reaction Intermediates in the Air-Promoted and Ligand-Modulated Oxidative Heck Reaction. J. Org. Chem. 2006, 71 (23), 8779–8786.
(111) Andaloussi, M.; Lindh, J.; Sävmarker, J.; J. R. Sjöberg, P.; Larhed, M. Microwave-Promoted Palladium(II)-Catalyzed C-P Bond Formation by Using Arylboronic Acids or Aryltrifluoroborates. Chem. Eur. J. 2009, 15 (47), 13069–13074.
(112) Superbases for Organic Synthesis; Ishikawa, T., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2009.
(113) Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Amidines, Isothioureas, and Guanidines as Nucleophilic Catalysts. Chem. Soc. Rev. 2012, 41 (6), 2109–2121.
(114) Li, J.; Neuville, L. Copper-Catalyzed Oxidative Diamination of Terminal Alkynes by Amidines: Synthesis of 1,2,4-Trisubstituted Imidazoles. Org. Lett. 2013, 15 (7), 1752–1755.
(115) Castanedo, G. M.; Seng, P. S.; Blaquiere, N.; Trapp, S.; Staben, S. T. Rapid Synthesis of 1,3,5-Substituted 1,2,4-Triazoles from Carboxylic Acids, Amidines, and Hydrazines. J. Org. Chem. 2011, 76 (4), 1177–1179.
(116) Huang, H.; Guo, W.; Wu, W.; Li, C. J.; Jiang, H. Copper-Catalyzed Oxidative C(sp3)-H Functionalization for Facile Synthesis of 1,2,4-Triazoles and 1,3,5-Triazines from Amidines. Org. Lett. 2015, 17 (12), 2894–2897.
(117) Ramanathan, M.; Wang, Y. H.; Liu, S. T. One-Pot Reactions for Synthesis of 2,5-Substituted Tetrazoles from Aryldiazonium Salts and Amidines. Org. Lett. 2015, 17 (23), 5886–5889.
(118) Guo, W.; Li, C.; Liao, J.; Ji, F.; Liu, D.; Wu, W.; Jiang, H. Transition Metal Free Intermolecular Direct Oxidative C-N Bond Formation to Polysubstituted Pyrimidines Using Molecular Oxygen as the Sole Oxidant. J. Org. Chem. 2016, 81 (13), 5538–5546.
(119) Peng, J.; Ye, M.; Zong, C.; Hu, F.; Feng, L.; Wang, X.; Wang, Y.; Chen, C. Copper-Catalyzed Intramolecular C-N Bond Formation: A Straightforward Synthesis of Benzimidazole Derivatives in Water. J. Org. Chem. 2011, 76 (2), 716–719.
(120) McGowan, M. A.; McAvoy, C. Z.; Buchwald, S. L. Palladium-Catalyzed N-Monoarylation of Amidines and a One-Pot Synthesis of Quinazoline Derivatives. Org. Lett. 2012, 14 (14), 3800–3803.
(121) Liu, Q.; Zhao, Y.; Fu, H.; Cheng, C. Copper-Catalyzed Sequential N-Arylation and Aerobic Oxidation: Synthesis of Quinazoline Derivatives. Synlett 2013, 24 (16), 2089–2094.
(122) Szczepankiewicz, B. G.; Rohde, J. J.; Kurukulasuriya, R. Synthesis of Purines and Other Fused Imidazoles from Acyclic Amidines and Guanidines. Org. Lett. 2005, 7 (9), 1833–1835.
(123) Soeiro, M. N. C.; Werbovetz, K.; Boykin, D. W.; Wilson, W. D.; Wang, M. Z.; Hemphill, A. Novel Amidines and Analogues as Promising Agents against Intracellular Parasites: A Systematic Review. Parasitology 2013, 140 (8), 1–23.
89
(124) World Health Organization. WHO List of Essential Medicines (EML) 20th Edition. 2017, No. 20th Edition, 62.
(125) Peregrine, A. S.; Mamman, M. Pharmacology of Diminazene: A Review. Acta Trop. 1993, 54 (3–4), 185–203.
(126) Lloyd, H. A.; Fales, H. M.; Goldman, M. E.; Jerina, D. M.; Plowman, T.; Schultes, R. E. Brunfelsamidine: A Novel Convulsant from the Medicinal Plant Brunfelsia Grandiflora. Tetrahedron Lett. 1985, 26 (22), 2623–2624.
(127) Blommel, M. L.; Blommel, A. L. Dabigatran Etexilate: A Novel Oral Direct Thrombin Inhibitor. Am. J. Heal. Pharm. 2011, 68 (16), 1506–1519.
(128) Deming, P.; Arora, S. Taribavirin in the Treatment of Hepatitis C. Expert Opin. Investig. Drugs 2011, 20 (10), 1435–1443.
(129) Ojo, B.; Dunbar, P. G.; Durant, G. J.; Nagy, P. I.; Huzl, J. J.; Periyasamy, S.; Ngur, D. O.; El-Assadi, A. A.; Hoss, W. P.; Messer, W. S. Synthesis and Biochemical Activity of Novel Amidine Derivatives as m1 Muscarinic Receptor Agonists. Bioorg. Med. Chem. 1996, 4 (10), 1605–1615.
(130) Kotthaus, J.; Steinmetzer, T.; van de Locht, A.; Clement, B. Analysis of Highly Potent Amidine Containing Inhibitors of Serine Proteases and Their N-Hydroxylated Prodrugs (Amidoximes). J. Enzyme Inhib. Med. Chem. 2011, 26 (1), 115–122.
(131) Goswami, R.; Wohlfahrt, G.; Törmäkangas, O.; Moilanen, A.; Lakshminarasimhan, A.; Nagaraj, J.; Arumugam, K. N.; Mukherjee, S.; Chacko, A. R.; Krishnamurthy, N. R.; Jaleel, M.; Palakurthy, R. K.; Samiulla, D. S.; Ramachandra, M. Structure-Guided Discovery of 2-Aryl/pyridin-2-Yl-1H-Indole Derivatives as Potent and Selective Hepsin Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25 (22), 5309–5314.
(132) Chen, X.; Orser, B. A.; MacDonald, J. F. Design and Screening of ASIC Inhibitors Based on Aromatic Diamidines for Combating Neurological Disorders. Eur. J. Pharmacol. 2010, 648 (1–3), 15–23.
(133) Oehlrich, D.; Prokopcova, H.; Gijsen, H. J. M. The Evolution of Amidine-Based Brain Penetrant BACE1 Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24 (9), 2033–2045.
(134) Pinner, A.; Klein, F. Umwandlung Der Nitrile in Imide. Berichte der Dtsch. Chem. Gesellschaft 1877, 10 (2), 1889–1897.
(135) Pinner, A.; Klein, F. Umwandlung Der Nitrile in Imide. Berichte der Dtsch. Chem. Gesellschaft 1878, 11 (2), 1475–1487.
(136) Pinner, A. Ueber Die Umwandlung Der Nitrile in Imide. Berichte der Dtsch. Chem. Gesellschaft 1883, 16 (2), 1643–1655.
(137) Weintraub, L.; Oles, S. R.; Kalish, N. Convenient General Synthesis of Amidines. J. Org. Chem. 1968, 33 (4), 1679–1681.
(138) Schaefer, F. C.; Peters, G. A. Base-Catalyzed Reaction of Nitriles with Alcohols. A Convenient Route to Imidates and Amidine Salts. J. Org. Chem. 1961, 26 (2), 412–418.
(139) Schaefer, F.; Krapcho, A. Preparation of Amidine Salts by Reaction of Nitriles with Ammonium Salts in the Presence of Ammonia. J. Org. Chem. 1962, 27 (4), 1255–1258.
90
(140) Barker, P. L.; Gendler, P. L.; Rapoport, H. Acylation of Dibasic Compounds Containing Amino Amidine and Aminoguanidine Functions. J. Org. Chem. 1981, 46 (12), 2455–2465.
(141) Lange, U. E. W.; Schäfer, B.; Baucke, D.; Buschmann, E.; Mack, H. A New Mild Method for the Synthesis of Amidines. Tetrahedron Lett. 1999, 40 (39), 7067–7070.
(142) Garigipati, R. S. An Efficient Conversion of Nitriles to Amidines. Tetrahedron Lett. 1990, 31 (14), 1969–1972.
(143) Adams, R.; Beebe, C. H. Action of the Grignard Reagent on CN Compounds. Synthesis of Amidines From Cyanamides. J. Am. Chem. Soc. 1916, 38 (12), 2768–2772.
(144) Anderson, H. J.; Wang, N.-C.; Jwili, E. T. P. Reaction of Phenyllithium with Some N,N -Disubstituted Cyanamides. Can. J. Chem. 1971, 49 (13), 2315–2320.
(145) Saluste, C. G.; Whitby, R. J.; Furber, M. A Palladium-Catalyzed Synthesis of Amidines from Aryl Halides. Angew. Chemie 2000, 39 (22), 4156–4158.
(146) Saluste, C. G.; Whitby, R. J.; Furber, M. Palladium-Catalysed Synthesis of Imidates, Thioimidates and Amidines from Aryl Halides. Tetrahedron Lett. 2001, 42 (35), 6191–6194.
(147) Kishore, K.; Tetala, R.; Whitby, R. J.; Light, M. E.; Hursthouse, M. B. Corrigendum to “Palladium-Catalysed Three Component Synthesis of Α,β-Unsaturated Amidines and Imidates” [Tetrahedron Lett. 45 (2004) 6991]. Tetrahedron Lett. 2004, 45 (45), 8427.
(148) Tetala, K. K. R.; Whitby, R. J.; Light, M. E.; Hurtshouse, M. B. Palladium-Catalysed Three Component Synthesis of Α,β-Unsaturated Amidines and Imidates. Tetrahedron Lett. 2004, 45 (38), 6991–6994.
(149) Saluste, C. G.; Crumpler, S.; Furber, M.; Whitby, R. J. Palladium Catalysed Synthesis of Cyclic Amidines and Imidates. Tetrahedron Lett. 2004, 45 (38), 6995–6996.
(150) Wang, J.; Xu, F.; Cai, T.; Shen, Q. Addition of Amines to Nitriles Catalyzed by Ytterbium Amides: An Efficient One-Step Synthesis of Monosubstituted N-Arylamidines. Org. Lett. 2008, 10 (3), 445–448.
(151) Shriner, R. L.; Neumann, F. W. The Chemistry of the Amidines. Chem. Rev. 1944, 35 (3), 351–425.
(152) Dunn, P. J. 5.19 – Amidines and N-Substituted Amidines. In Comprehensive Organic Functional Group Transformations; Katritzky, A. R., Taylor, R. J. K., Eds.; Elsevier: Oxford, 2005; pp 655–699.
(153) Aly, A. A.; Nour-El-Din, A. M. Functionality of Amidines and Amidrazones. Arkivoc 2008, 2008 (1), 153.
(154) Ishikawa, T.; Kumamoto, T. Amidines in Organic Synthesis. In Superbases for Organic Synthesis; John Wiley & Sons, Ltd: Chichester, UK, 2009; pp 49–91.
(155) Anderson, N. G. Practical Use of Continuous Processing in Developing and Scaling up Laboratory Processes. Org. Process Res. Dev. 2001, 5 (6), 613–621.
(156) Malet-Sanz, L.; Susanne, F. Continuous Flow Synthesis. A Pharma Perspective. J. Med. Chem. 2012, 55 (9), 4062–4098.
91
(157) Watts, P.; Haswell, S. J. Continuous Flow Reactors for Drug Discovery. Drug Discov. Today 2003, 8 (13), 586–593.
(158) Welch, C. J.; Gong, X.; Cuff, J.; Dolman, S.; Nyrop, J.; Lin, F.; Rogers, H. Online Analysis of Flowing Streams Using Microflow HPLC. Org. Process Res. Dev. 2009, 13 (5), 1022–1025.
(159) Browne, D. L.; Wright, S.; Deadman, B. J.; Dunnage, S.; Baxendale, I. R.; Turner, R. M.; Ley, S. V. Continuous Flow Reaction Monitoring Using an on-Line Miniature Mass Spectrometer. Rapid Commun. Mass Spectrom. 2012, 26 (17), 1999–2010.
(160) Sadler, S.; Moeller, A. R.; Jones, G. B. Microwave and Continuous Flow Technologies in Drug Discovery. Expert Opin. Drug Discov. 2012, 7 (12), 1107–1128.
(161) Wiles, C.; Watts, P. Continuous Flow Reactors: A Perspective. Green Chem. 2012, 14 (1), 38–54.
(162) Gabriel, C.; Gabriel, S.; H. Grant, E.; H. Grant, E.; S. J. Halstead, B.; Michael P. Mingos, D. Dielectric Parameters Relevant to Microwave Dielectric Heating. Chem. Soc. Rev. 1998, 27 (3), 213–223.
(163) Razzaq, T.; Kremsner, J. M.; Kappe, C. O. Investigating the Existence of Nonthermal/specific Microwave Effects Using Silicon Carbide Heating Elements as Power Modulators. J. Org. Chem. 2008, 73 (16), 6321–6329.
(164) Obermayer, D.; Gutmann, B.; Kappe, C. O. Microwave Chemistry in Silicon Carbide Reaction Vials: Separating Thermal from Nonthermal Effects. Angew. Chemie 2009, 121 (44), 8471–8474.
(165) Kappe, C. O. How to Measure Reaction Temperature in Microwave-Heated Transformations. Chem. Soc. Rev. 2013, 42 (12), 4977–4990.
(166) Kappe, C. O. Unraveling the Mysteries of Microwave Chemistry Using Silicon Carbide Reactor Technology. Acc. Chem. Res. 2013, 46 (7), 1579–1587.
(167) Kappe, C. O.; Pieber, B.; Dallinger, D. Microwave Effects in Organic Synthesis: Myth or Reality? Angew. Chemie Int. Ed. 2013, 52 (4), 1088–1094.
(168) Yeboah, K. A.; Boyd, J. D.; Kyeremateng, K. A.; Shepherd, C. C.; Ingersoll, I. M.; Jackson, D. L.; Holland, A. W. Large Accelerations from Small Thermal Differences: Case Studies and Conventional Reproduction of Microwave Effects on Palladium Couplings. React. Kinet. Mech. Catal. 2014, 112 (2), 295–304.
(169) Larhed, M.; Moberg, C.; Hallberg, A. Microwave-Accelerated Homogeneous Catalysis in Organic Chemistry. Acc. Chem. Res. 2002, 35 (9), 717–727.
(170) Kappe, C. O. O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chemie Int. Ed. 2004, 43 (46), 6250–6284.
(171) Cablewski, T.; Faux, a F.; Strauss, C. R. Development and Application of a Continuous Microwave Reactor for Organic-Synthesis. J. Org. Chem. 1994, 59 (12), 3408–3412.
(172) Bagley, M. C.; Jenkins, R. L.; Lubinu, M. C.; Mason, C.; Wood, R. A Simple Continuous Flow Microwave Reactor. J. Org. Chem. 2005, 70 (17), 7003–7006.
92
(173) Comer, E.; Organ, M. G. A Microreactor for Microwave-Assisted Capillary (Continuous Flow) Organic Synthesis. J. Am. Chem. Soc. 2005, 127 (22), 8160–8167.
(174) Baxendale, I.; Hayward, J.; Ley, S. Microwave Reactions Under Continuous Flow Conditions. Comb. Chem. High Throughput Screen. 2007, 10 (10), 802–836.
(175) Glasnov, T. N.; Kappe, C. O. Microwave-Assisted Synthesis under Continuous-Flow Conditions. Macromol. Rapid Commun. 2007, 28 (4), 395–410.
(176) Gjuraj, E.; Kongoli, R.; Shore, G. Combination of Flow Reactors with Microwave-Assisted Synthesis : Smart Engineering Concept for Steering Synthetic Chemistry on the “ Fast Lane .” Chem. Biochem. Eng. Q. 2012, 26 (3), 285–307.
(177) Morschhäuser, R.; Krull, M.; Kayser, C.; Boberski, C.; Bierbaum, R.; Püschner, P. A.; Glasnov, T. N.; Kappe, C. O. Microwave-Assisted Continuous Flow Synthesis on Industrial Scale. Green Process. Synth. 2012, 1 (3), 281–290.
(178) Sauks, J. M.; Mallik, D.; Lawryshyn, Y.; Bender, T.; Organ, M. A Continuous-Flow Microwave Reactor for Conducting High-Temperature and High-Pressure Chemical Reactions. Org. Process Res. Dev. 2014, 18 (11), 1310–1314.
(179) Öhrngren, P.; Fardost, A.; Russo, F.; Schanche, J. S.; Fagrell, M.; Larhed, M. Evaluation of a Nonresonant Microwave Applicator for Continuous-Flow Chemistry Applications. Org. Process Res. Dev. 2012, 16 (5), 1053–1063.
(180) Fardost, A.; Russo, F.; Larhed, M. A Non-Resonant Microwave Applicator Fully Dedicated to Continuous Flow Chemistry. Chim. Oggi 2012, 30 (4), 14–16.
(181) Konda, V.; Rydfjord, J.; Sävmarker, J.; Larhed, M. Safe Palladium-Catalyzed Cross-Couplings with Microwave Heating Using Continuous-Flow Silicon Carbide Reactors. Org. Process Res. Dev. 2014, 18 (11), 1413–1418.
(182) Engen, K.; Sävmarker, J.; Rosenström, U.; Wannberg, J.; Lundbäck, T.; Jenmalm-Jensen, A.; Larhed, M. Microwave Heated Flow Synthesis of Spiro-Oxindole Dihydroquinazolinone Based IRAP Inhibitors. Org. Process Res. Dev. 2014, 18 (11), 1582–1588.
(183) Kumpina, I.; Isaksson, R.; Sävmarker, J.; Wannberg, J.; Larhed, M. Microwave Promoted Transcarbamylation Reaction of Sulfonylcarbamates under Continuous-Flow Conditions. Org. Process Res. Dev. 2016, 20 (2), 440–445.
(184) Rydfjord, J.; Svensson, F.; Fagrell, M.; Sävmarker, J.; Thulin, M.; Larhed, M. Temperature Measurements with Two Different IR Sensors in a Continuous-Flow Microwave Heated System. Beilstein J. Org. Chem. 2013, 9, 2079–2087.
(185) Sävmarker, J.; Lindh, J.; Nilsson, P.; Sjöberg, P. J. R.; Larhed, M. Oxidative Heck Reactions Using Aryltrifluoroborates and Aryl N -Methyliminodiacetic Acid (MIDA) Boronates. ChemistryOpen 2012, 1 (3), 140–146.
93
(186) Sävmarker, J.; Rydfjord, J.; Gising, J.; Odell, L. R.; Larhed, M. Direct palladium(II)-Catalyzed Synthesis of Arylamidines from Aryltrifluoroborates. Org. Lett. 2012, 14 (9), 2394–2397.
(187) Gooßen, L. J.; Rodríguez, N.; Gooßen, K. Carboxylic Acids as Substrates in Homogeneous Catalysis. Angew. Chemie Int. Ed. 2008, 47 (17), 3100–3120.
(188) Goossen, L. J.; Rodríguez, N.; Melzer, B.; Linder, C.; Deng, G.; Levy, L. M. Biaryl Synthesis via Pd-Catalyzed Decarboxylative Coupling of Aromatic Carboxylates with Aryl Halides. J. Am. Chem. Soc. 2007, 129 (15), 4824–4833.
(189) Rodríguez, N.; Goossen, L. J. Decarboxylative Coupling Reactions: A Modern Strategy for C-C-Bond Formation. Chem. Soc. Rev. 2011, 40 (10), 5030–5048.
(190) Svensson, F.; Mane, R. S.; Sävmarker, J.; Larhed, M.; Sköld, C. Theoretical and Experimental Investigation of palladium(II)-Catalyzed Decarboxylative Addition of Arenecarboxylic Acid to Nitrile. Organometallics 2013, 32 (2), 490–497.
(191) Rydfjord, J.; Svensson, F.; Trejos, A.; Sjöberg, P. J. R.; Sköld, C.; Sävmarker, J.; Odell, L. R.; Larhed, M. Decarboxylative Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Aryl Carboxylic Acids: Development and Mechanistic Investigation. Chem. - A Eur. J. 2013, 19 (41), 13803–13810.
(192) Lyons, T. W.; Sanford, M. S. Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110 (2), 1147–1169.
(193) Miao, T.; Wang, L. Palladium-Catalyzed Desulfitative Direct C-H Arylation of Electron-Deficient Polyfluoroarenes with Sodium Arenesulfinates. Adv. Synth. Catal. 2014, 356 (2–3), 429–436.
(194) Watanabe, R.; Sugai, C.; Yamazaki, T.; Matsushima, R.; Uchida, H.; Matsumiya, M.; Takatsu, A.; Suzuki, T. Quantitative Nuclear Magnetic Resonance Spectroscopy Based on PULCON Methodology: Application to Quantification of Invaluable Marine Toxin, Okadaic Acid. Toxins (Basel). 2016, 8 (10), 294–302.
(195) Campbell, A. N.; Meyer, E. B.; Stahl, S. S. Regiocontrolled Aerobic Oxidative Coupling of Indoles and Benzene Using Pd Catalysts with 4,5-Diazafluorene Ligands. Chem. Commun. 2011, 47 (37), 10257–10259.
(196) Kremsner, J. M.; Kappe, C. O. Silicon Carbide Passive Heating Elements in Microwave-Assisted Organic Synthesis. J. Org. Chem. 2006, 71 (12), 4651–4658.
(197) Damm, M.; Kappe, C. O. Parallel Microwave Chemistry in Silicon Carbide Reactor Platforms: An in-Depth Investigation into Heating Characteristics. Mol. Divers. 2009, 13 (4), 529–543.
(198) Gutmann, B.; Obermayer, D.; Reichart, B.; Prekodravac, B.; Irfan, M.; Kremsner, J. M.; Kappe, C. O. Sintered Silicon Carbide : A New Ceramic Vessel Material for Microwave Chemistry in Single-Mode Reactors. 2010, 12182–12194.
(199) Cho, S.-H.; Walther, N. D.; Nguyen, S. T.; Hupp, J. T. Anodic Aluminium Oxide Catalytic Membranes for Asymmetric Epoxidation. Chem. Commun. 2005, No. 42, 5331–5333.
94
(200) Duque, R.; Pogorzelec, P. J.; Cole-Hamilton, D. J. A Single Enantiomer (99 %) Directly from Continuous-Flow Asymmetric Hydrogenation. Angew. Chemie Int. Ed. 2013, 52 (37), 9805–9807.
(201) Aellig, C.; Jenny, F.; Scholz, D.; Wolf, P.; Giovinazzo, I.; Kollhoff, F.; Hermans, I. Combined 1,4-Butanediol Lactonization and Transfer Hydrogenation/hydrogenolysis of Furfural-Derivatives under Continuous Flow Conditions. Catal. Sci. Technol. 2014, 4 (8), 2326–2331.
(202) Amara, Z.; Poliakoff, M.; Duque, R.; Geier, D.; Franciò, G.; Gordon, C. M.; Meadows, R. E.; Woodward, R.; Leitner, W. Enabling the Scale-Up of a Key Asymmetric Hydrogenation Step in the Synthesis of an API Using Continuous Flow Solid-Supported Catalysis. Org. Process Res. Dev. 2016, 20 (7), 1321–1327.
(203) Jansen, K. Microwave Technology in Zeolite Synthesis. In Verified Syntheses of Zeolitic Materials; Robson, H. E., Ed.; Elsevier: Amsterdam, The Netherlands, 2001; pp 39–42.
(204) Zhou, X.; Luo, J.; Liu, J.; Peng, S.; Deng, G.-J. Pd-Catalyzed Desulfitative Heck Coupling with Dioxygen as the Terminal Oxidant. Org. Lett. 2011, 13 (6), 1432–1435.
(205) Hu, S.; Xia, P.; Cheng, K.; Qi, C. Pd-Catalyzed Ligand-Free Desulfitative Heck Reaction with Arenesulfinic Acid Salts under Air. Appl. Organomet. Chem. 2013, 27 (3), 188–190.
(206) Bal Raju, K.; Mari, V.; Nagaiah, K. Regioselective Palladium(II)-Catalyzed Desulfitative Heck-Type Reaction: Access to α-Benzyl-β-Keto Esters from Baylis-Hillman Adducts and Sodium Sulfinates. Synthesis (Stuttg). 2013, 45 (20), 2867–2874.
(207) Wang, G.-W.; Miao, T. Palladium-Catalyzed Desulfitative Heck-Type Reaction of Aryl Sulfinic Acids with Alkenes. Chem. - A Eur. J. 2011, 17 (21), 5787–5790.
(208) Xu, Y.; Zhao, J.; Tang, X.; Wu, W.; Jiang, H. Chemoselective Synthesis of Unsymmetrical Internal Alkynes or Vinyl Sulfones via Palladium-Catalyzed Cross-Coupling Reaction of Sodium Sulfinates with Alkynes. Adv. Synth. Catal. 2014, 356 (9), 2029–2039.
(209) Chen, W.; Zhou, X.; Xiao, F.; Luo, J.; Deng, G. J. Palladium-Catalyzed Desulfitative Addition of Sodium Sulfinates with Α,β-Unsaturated Carbonyl Compounds. Tetrahedron Lett. 2012, 53 (33), 4347–4350.
(210) Wang, H.; Li, Y.; Zhang, R.; Jin, K.; Zhao, D.; Duan, C. Palladium-Catalyzed Desulfitative Conjugate Addition of Aryl Sulfinic Acids and Direct ESI-MS for Mechanistic Studies. J. Org. Chem. 2012, 77 (10), 4849–4853.
(211) Miao, T.; Wang, L. Palladium-Catalyzed Desulfitative Cross-Coupling Reaction of Sodium Arylsulfinates with H-Phosphonate Diesters. Adv. Synth. Catal. 2014, 356 (5), 967–971.
(212) Li, J.; Bi, X.; Wang, H.; Xiao, J. Palladium-Catalyzed Desulfitative C–P Coupling of Arylsulfinate Metal Salts and H-Phosphonates. RSC Adv. 2014, 4 (37), 19214–19217.
95
(213) Wang, S.; Liu, W.; Lin, J.; Jiang, Y.; Zhang, Q.; Zhong, Y. Palladium-Catalyzed Direct and Regioselective C-5 Desulfitative Arylation of Thiazolo[3,2-B]-1,2,4-Triazoles with Sodium Sulfinates. Synlett 2013, 25 (4), 586–590.
(214) Miao, T.; Li, P.; Wang, G.-W.; Wang, L. Microwave-Accelerated Pd-Catalyzed Desulfitative Direct C2-Arylation of Free (NH)-Indoles with Arylsulfinic Acids. Chem. - An Asian J. 2013, 8 (12), 3185–3190.
(215) Wu, M.; Luo, J.; Xiao, F.; Zhang, S.; Deng, G.-J.; Luo, H.-A. Palladium-Catalyzed Direct and Site-Selective Desulfitative Arylation of Indoles with Sodium Sulfinates. Adv. Synth. Catal. 2012, 354 (2–3), 335–340.
(216) Jafarpour, F.; Olia, M. B. A.; Hazrati, H. Highly Regioselective α-Arylation of Coumarins via Palladium-Catalyzed C-H Activation/Desulfitative Coupling. Adv. Synth. Catal. 2013, 355 (17), 3407–3412.
(217) Wang, M.; Li, D.; Zhou, W.; Wang, L. A Highly Efficient Palladium-Catalyzed Desulfitative Arylation of Azoles with Sodium Arylsulfinates. Tetrahedron 2012, 68 (7), 1926–1930.
(218) Liu, B.; Guo, Q.; Cheng, Y.; Lan, J.; You, J. Palladium-Catalyzed Desulfitative C-H Arylation of Heteroarenes with Sodium Sulfinates. Chem. - A Eur. J. 2011, 17 (48), 13415–13419.
(219) Liu, L.; Chi, Y.; Jen, K. Copper-Catalyzed Additions of Sulfonyl Iodides to Simple and Cyclic Alkenes. J. Org. Chem. 1980, 45 (3), 406–410.
(220) Fujiwara, Y.; Dixon, J. A.; O’Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. a; Baxter, R. D.; Herlé, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Practical and Innate Carbon–hydrogen Functionalization of Heterocycles. Nature 2012, 492 (7427), 95–99.
(221) O’Hara, F.; Baxter, R. D.; O’Brien, a G.; Collins, M. R.; Dixon, J. a; Fujiwara, Y.; Ishihara, Y.; Baran, P. S. Preparation and Purification of Zinc Sulfinate Reagents for Drug Discovery. Nat. Protoc. 2013, 8 (6), 1042–1047.
(222) Allen, P. Aliphatic Sulfinic Acids. I. Analysis and Identification. J. Org. Chem. 1942, 7 (1), 23–30.
(223) Allen, Jr., P.; Rehl, W. R.; Fuchs, P. E. Sulfone Formation During Sulfination of the Alkyl Grignard Reagent. J. Org. Chem. 1955, 20 (9), 1237–1239.
(224) Umierski, N.; Manolikakes, G. Arylation of Lithium Sulfinates with Diaryliodonium Salts: A Direct and Versatile Access to Arylsulfones. Org. Lett. 2013, 15 (19), 4972–4975.
(226) Pelzer, G.; Keim, W. Palladium-Catalyzed Synthesis of Sulfinic Acids from Aryldiazonium Tetrafluoroborates, Sulfur Dioxide and Hydrogen. J. Mol. Catal. A Chem. 1999, 139 (2–3), 235–238.
(227) Woolven, H.; González-Rodríguez, C.; Marco, I.; Thompson, A. L.; Willis, M. C. DABCO- Bis (Sulfur Dioxide), DABSO, as a Convenient Source of Sulfur Dioxide for Organic Synthesis: Utility in Sulfonamide and Sulfamide Preparation. Org. Lett. 2011, 13 (18), 4876–4878.
(228) Emmett, E. J.; Willis, M. C. The Development and Application of Sulfur Dioxide Surrogates in Synthetic Organic Chemistry. Asian J. Org. Chem. 2015, 4 (7), 602–611.
96
(229) Waldmann, C.; Schober, O.; Haufe, G.; Kopka, K. A Closer Look at the Bromine–Lithium Exchange with Tert -Butyllithium in an Aryl Sulfonamide Synthesis. Org. Lett. 2013, 15 (12), 2954–2957.
(230) Rocke, B. N.; Bahnck, K. B.; Herr, M.; Lavergne, S.; Mascitti, V.; Perreault, C.; Polivkova, J.; Shavnya, A. Synthesis of Sulfones from Organozinc Reagents, DABSO, and Alkyl Halides. Org. Lett. 2014, 16 (1), 154–157.
(231) Richards-Taylor, C. S.; Blakemore, D. C.; Willis, M. C. One-Pot Three-Component Sulfone Synthesis Exploiting Palladium-Catalysed Aryl Halide Aminosulfonylation. Chem. Sci. 2014, 5 (1), 222–228.
(232) Deeming, A. S.; Russell, C. J.; Hennessy, A. J.; Willis, M. C. DABSO-Based, Three-Component, One-Pot Sulfone Synthesis. Org. Lett. 2014, 16 (1), 150–153.
(233) Chen, C. C.; Waser, J. One-Pot, Three-Component Arylalkynyl Sulfone Synthesis. Org. Lett. 2015, 17 (3), 736–739.
(234) Deeming, A. S.; Russell, C. J.; Willis, M. C. Combining Organometallic Reagents, the Sulfur Dioxide Surrogate DABSO, and Amines: A One-Pot Preparation of Sulfonamides, Amenable to Array Synthesis. Angew. Chemie Int. Ed. 2015, 54 (4), 1168–1171.
(235) Fan, W.; Su, J.; Shi, D.; Feng, B. An Efficient One-Pot, Three-Component Synthesis of Vinyl Sulfones via Iodide-Catalyzed Radical Alkenylation. Tetrahedron 2015, 71 (38), 6740–6743.
(236) Flegeau, E. F.; Harrison, J. M.; Willis, M. C. One-Pot Sulfonamide Synthesis Exploiting the Palladium-Catalyzed Sulfination of Aryl Iodides. Synlett 2016, 27 (1), 101–105.
(237) Lenstra, D. C.; Vedovato, V.; Ferrer Flegeau, E.; Maydom, J.; Willis, M. C. One-Pot Sulfoxide Synthesis Exploiting a Sulfinyl-Dication Equivalent Generated from a DABSO/Trimethylsilyl Chloride Sequence. Org. Lett. 2016, 18 (9), 2086–2089.
(238) Tsai, A. S.; Curto, J. M.; Rocke, B. N.; Dechert-Schmitt, A. M. R.; Ingle, G. K.; Mascitti, V. One-Step Synthesis of Sulfonamides from N-Tosylhydrazones. Org. Lett. 2016, 18 (3), 508–511.
(239) Deeming, A. S.; Russell, C. J.; Willis, M. C. Palladium(II)-Catalyzed Synthesis of Sulfinates from Boronic Acids and DABSO: A Redox-Neutral, Phosphine-Free Transformation. Angew. Chemie Int. Ed. 2016, 55 (2), 747–750.
(240) Davies, A. T.; Curto, J. M.; Bagley, S. W.; Willis, M. C. One-Pot Palladium-Catalyzed Synthesis of Sulfonyl Fluorides from Aryl Bromides. Chem. Sci. 2017, 8 (2), 1233–1237.
(241) Du, B.; Wang, Y.; Sha, W.; Qian, P.; Mei, H.; Han, J.; Pan, Y. Copper-Catalyzed Selective Aerobic Oxidative Cascade Reaction of Hydrazines, DABSO, and Amines for the Direct Synthesis of Sulfonamides. Asian J. Org. Chem. 2017, 6 (2), 153–156.
(242) Zhu, H.; Shen, Y.; Deng, Q.; Huang, C.; Tu, T. One-Pot Bimetallic Pd/Cu-Catalyzed Synthesis of Sulfonamides from Boronic Acids, DABSO and O -Benzoyl Hydroxylamines. Chem. - An Asian J. 2017, 12 (6), 706–712.
(243) Kice, J. L.; Bowers, K. W. The Mechanism of the Disproportionation of Sulfinic Acids. J. Am. Chem. Soc. 1962, 84 (4), 605–610.
97
(244) Kice, J. L.; Guaraldi, G.; Venier, C. G. The Mechanism of the Disproportionation of Sulfinic Acids. Rate and Equilibrium Constants for the Sulfinic Acid-Sulfinyl Sulfone (Sulfinic Anhydride) Equilibrium 1. J. Org. Chem. 1966, 31 (11), 3561–3567.
(245) Gold, H.; Larhed, M.; Nilsson, P. Microwave Irradiation as a High-Speed Tool for Activation of Sluggish Aryl Chlorides in Grignard Reactions. Synlett 2005, 2005 (10), 1596–1600.
(246) Kadam, A.; Nguyen, M.; Kopach, M.; Richardson, P.; Gallou, F.; Wan, Z.-K.; Zhang, W. Comparative Performance Evaluation and Systematic Screening of Solvents in a Range of Grignard Reactions. Green Chem. 2013, 15 (7), 1880–1888.
Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Pharmacy 233
Editor: The Dean of the Faculty of Pharmacy
A doctoral dissertation from the Faculty of Pharmacy, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofPharmacy. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Pharmacy”.)