Page 1
1
Pathology week 1 – Cellular adaptation, injury and death
Cellular responses to injury
Cellular Responses to Injury
Nature and Severity of Injurious Stimulus Cellular Response
Altered physiologic stimuli: Cellular adaptations:
• ↑demand, ↑ trophic stimulation (e.g. growth factors, hormones) • Hyperplasia, hypertrophy
• ↓ nutrients, stimulation • Atrophy
• Chronic irritation (chemical or physical) • Metaplasia
Reduced oxygen supply; chemical injury; microbial infection Cell injury:
• Acute and self-limited • Acute reversible injury
• Progessive and severe (including DNA damage) • Irreversible injury → cell death
Necrosis
Apoptosis
• Mild chronic injury • Subcellular alterations in organelles
Metabolic alterations, genetic or acquired Intracell accumulations; calcifications
Prolonged life span with cumulative sublethal injury Cellular aging
Hyperplasia
- response to increased demand and external stimulation
- ↑ number cells - ↑ volume of organ
- often occurs with hypertrophy
- occurs if cells able to synthesize DNA – mitotic division
- physiologic or pathologic
Physiological hyperplasia
A) hormonal – ↑ functional capacity tissue when needed (breast in puberty, uterus in pregnancy)
B) compensatory - ↑ tissue mass after damage/resection (post-nephrectomy)
Mechanisms:
- ↑ local production growth factors or activation intracellular signaling pathways
o both → production transcription factors that turn on cellular genes incl those encoding growth factors,
receptors for GFs, cell cycle regulators →→ cellular proliferation
- in hormonal hyperplasia hormones may act as growth factors
- ↑ tissue mass secondary to proliferation and development of new cells from stem cells
Pathological hyperplasia
- mostly due to xs hormonal stimulation or GFs
o endometrial hyperplasia (xs oestrogen), BPH
- abnormal but controlled because if hormonal stimulation removed then hyperplasia regresses
- but can turn into cancerous proliferation (endometrial cancer)
- in wound healing proliferation of fibroblasts and blood vessels aids repair
- certain viruses can cause hyperplasia → HPV – warts
Hypertrophy
- response to ↑ functional demand or hormonal stimulation
- ↑ size of cells → ↑ size of organ (no new cells), due to ↑ synthesis structural components
- occurs in nondividing cells (eg myocardium)
- physiologic or pathologic
- usually due to ↑ workload (striated muscle)
- in heart due to chronic haemodynamic overload (HTN or valve disease)
- ↑ number myofilaments per cell
- hormonally causes hypertrophy AND hyperplasia in pregnant uterus
Page 2
2
Mechanisms:
- heart: signal transduction pathways – stimulate synthesis of cellular proteins
o genes induced incl. encoding transcription factors, growth factors (IGF-1, TGF-1), and vasoactive agents
(alpha-adrenergic agonists, endothelin-1, angiotensin II)
o may switch from adult to fetal or neonatal forms
� some genes only found in early development reappear in hypertrophy eg ANF
- triggers: mechanical or tropic (eg IGF1, angiotensin II)
- cardiac hypertrophy reaches point where unable to compensate for ↑ burden → heart failure
- limiting factors for hypertrophy: vascular supply, ↓ oxidative capacities of mitochondria, ↓ protein synthesis,
cytoskeletal alterations
Atrophy
- shrinkage in size of cells by loss of structural components of cell
- adaptive response, may lead to cell death
- apoptosis may be induced by same signals causing atrophy
- physiologic atrophy: common in early development (thyroglossal duct)
o uterus post delivery
- pathologic atrophy: local or generalised
� ↓ workload (atrophy of disuse) – POP: ↓ cell size, then ↓ cell no’s; with bone resorption –
osteoporosis
o loss of innervation (denervation atrophy) – carpal tunnel
o ↓ blood supply – arterial occlusive disease/ischaemia: brain
o inadequate nutrition (protein/calorie malnutrition – muscle breakdown – cachexia
� chronic inflammation overproduction TNF – appetite suppression and atrophy
o loss of endocrine stimulation – endocrine/reproductive glands, breast – loss oestrogen
o aging (senile atrophy) – cell loss in permanent cells: brain, heart
o pressure (tissue compression) – tumours: ischaemia
Mechanisms:
- affects balance between protein synthesis and degradation
- lysosomes contain acid hydrolases and other enzymes that degrade endocytosed proteins
- ubiquitin and proteasome pathway responsible for degradation of many cytosolic and nuclear proteins (proteins
conjugated to ubiquitin then degraded with the proteasome – this pathway responsible for accelerated proteolysis
in cachexia
- glucocorticoids and thyroid hormone stimulate proteasome-mediated protein degradation, insulin opposes these
actions
- cytokines eg TNF can ↑ muscle proteolysis this way
- atrophy often accomp by ↑↑ in no. of autophagic vacuoles (membrane-bound vacuoles in cell containing fragments
of cell components into which lysosomes discharge their hydrolytic contents – digested
- some of cell debris resists digestion – persist in membrane-bound bodies in the cytoplasm eg lipofuscin
granules – brown colour in tissue (brown atrophy)
Metaplasia
- reversible change where one adult cell type is replaced by another type
- may be adaptive to withstand chronic stress such as chemical or physical irritants
- most common epithelial metaplasia: columnar to squamous
� resp tract due to chronic irritation (smoking)
� resp tract due to Vit A deficiency
� salivary, pancreatic, biliary stones
o squamous epithelium withstands more stress but resp tract protective mucus lost
o may go on to malignant transformation
- metaplasia from squamous to columnar
� Barretts, due to increase acid (go on to adenoCa)
- connective tissue metaplasia – formation cartilage, bone or adipose in tissues not normally containing these
o myositis ossificans post #
Mechanisms:
- reprogramming of stem cells that exist in normal tissues or of undifferentiated matrix components present in
connective tissue
- differentiation of stem cells due to signals by cytokines, GFs and ECM components
Page 3
3
- certain cytostatic drugs cause disruption of DNA methylation patterns and can transform mesenchymal cells from
one type (fibroblast) to another (muscle, cartilage)
Cell Injury and Cell Death
- reversible injury: initially functional and morphological changes that are reversible if damaging stimulus removed
o ↓ oxidative phosphorylation
o ATP depletion
o cellular swelling (change in ion conc’s and water influx)
- irreversible injury and death: with continuing damage
o apoptosis (not always assoc with cell injury) and necrosis (always pathological)
- causes of cell injury:
o hypoxia → ↓aerobic oxidative respiration (incl CO, anaemia)
� cf ischaemia – loss of blood supply with ↓ O2 and metabolic substances ie glucose → more
rapid/severe cell injury)
o physical agents – mechanical trauma, burns, cold, pressure, radiation, electric shock
o chemical agents (gluc/salt if hypertonic, ↑↑ O2, poisons, pollutants, insecticides, CO, asbestos, drugs,
EtOH)
o infectious agents
o immunological reactions (anaphylaxis)
o genetic derangements (Downs, Sickle cell, enzymes)
o nutritional imbalances (starvation, anorexia, obesity)
- morphology:
o reversible: swelling ER and mitochondria, clumping chromatin
o irreversible: swelling ER, loss ribosomes, lysosome rupture, membrane blebs, myelin figures, swollen
mitochondria, nuclear condensation (pyknosis)
o necrosis: fragmentation cell membrane and nucleus (karyorrhexis) then dissolution nucleus (karyolysis)
Features of Necrosis and Apoptosis
Features of Necrosis and Apoptosis
Feature Necrosis Apoptosis
Cell size Enlarged (swelling) Reduced (shrinkage)
Nucleus Pyknosis → karyorrhexis →
karyolysis
Fragmentation into nucleosome size fragments
Plasma membrane Disrupted Intact; altered structure, especially orientation of lipids
Cellular contents Enzymatic digestion; may leak out
of cell
Intact; may be released in apoptotic bodies
Adjacent
inflammation
Frequent No
Physiologic or
pathologic
Invariably pathologic (culmination
of irreversible cell injury)
Often physiologic, means of eliminating unwanted cells; may
be pathologic after some forms cell injury, esp DNA damage
Cellular and biochemical sites of damage in cell injury
Page 4
4
Mechanisms of Cell Injury
- cellular response to injurious stimuli depends on type of injury, duration and severity
- consequences of cell injury depend on type, state and adaptability of injured cell – cell nutritional status and
metabolic needs important in response to injury
o ?genetic polymorphisms affect response
- biochemical mechanisms:
o cell injury results in abnormalities of 1+ of 5 cellular components:
� aerobic respiration (mitochondrial oxidative phosphorylation/ATP)
� maintenance of cell membrane integrity
� protein synthesis
� intracellular cytoskeleton
� integrity of genetic apparatus
Intracellular mechanisms:
5 pathways
1) ATP depletion
- ↓ synthesis/ATP depletion due to ischaemic and
toxic injury
o ATP made by glycolysis (anaerobic,
inefficient) + oxidation
phosphorylation in mitochondria
(aerobic, efficient)
o hypoxia → glycolysis/glycogen
depletion, ↑ lactate, acidosis
o ATP also for membrane transport,
maintain ion gradients, protein synth
2) mitochondrial damage
- directly (hypoxia/toxins) or due to ↑ cytosolic
Ca, oxidative stress, phospholipid breakdown
- damage causes formation high-conductance
channel (mitochondrial permeability transition
(MPT), leaks protons, loss of ion potential
driving oxidative phosphorylation
- leaks cytochrome c, triggers apoptosis
3) influx Ca 2+
- usually CaMgATPase keeps intracell Ca low,
but ischaemia/toxins cause influx Ca, release Ca
from mitochondria/ER
- ↑ Ca activates phospholipases degrading
membrane phospholipids, proteases breakdown
membranes/cytoskeletal proteins, ATPases →
ATP depletion, endonucleases → chromatin
fragmentation
4) accumulation O2-derived free radicals
- partially reduced, highly reactive, unstable O2
molecules
- cause free radical formation in autocatalytic
chain reaction (propagation)
- damage lipids, proteins, nucleic acid (oxidative
stress)
- generation occurs by:
o ionizing radiation (XR, UV) hydrolyze
water to hydroxyl and H free radicals
o enzymatic metabolism chemicals/drugs
eg carbon tetrachloride
o redox reactions as part of normal
metabolism (respiration)
o transition metals (iron, copper)
catalyze free radical formation
o NO acts as free radical
- free radicals unstable – decay spontaneously
o antioxidants block free radical
formation or scavenge them (Vit E, A,
C, glutathione)
o reactive forms metals minimized by
transport/storage proteins (ferritin)
o scavenging enzymes catabolize H2O2
(catalase, glutathione peroxidase,
superoxide dismutase)
5) defects in membrane permeability
- damaged directly by toxins, physical/chemical
agents, lytic complement components, perforins
- damaged indirectly by free radicals
- ↑ permeability → change intracellular
osmolarity and enzyme activity
Page 5
5
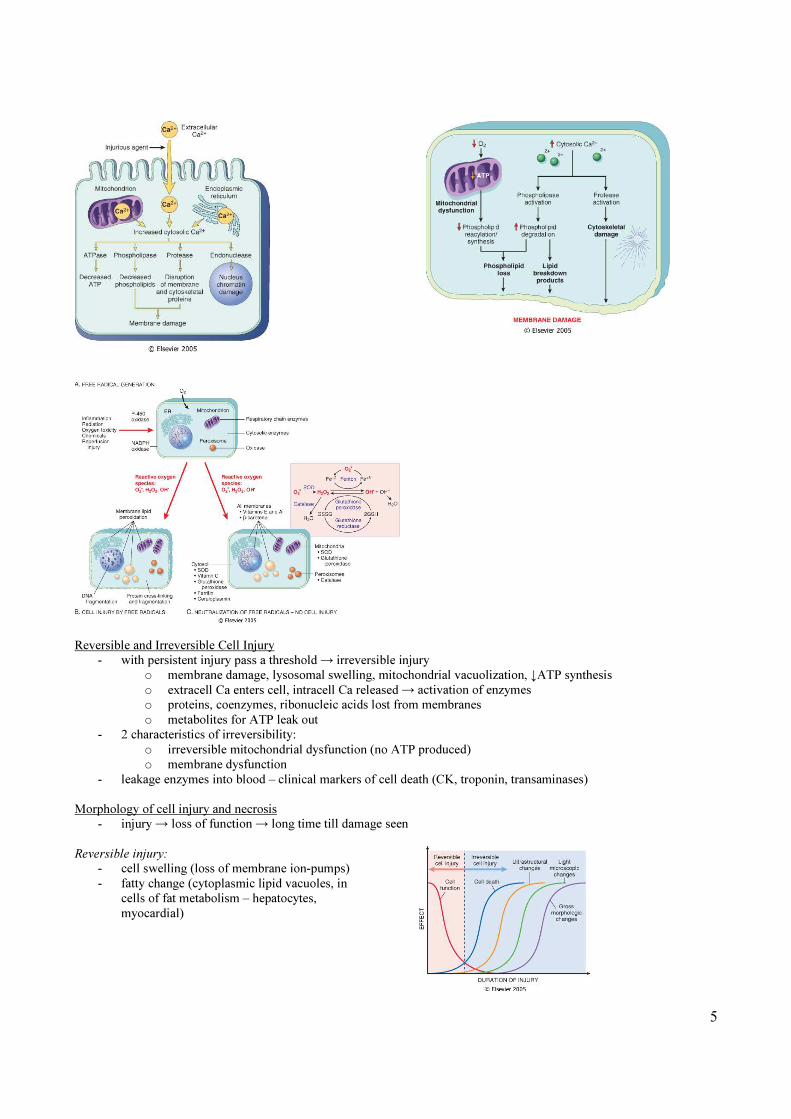
Reversible and Irreversible Cell Injury
- with persistent injury pass a threshold → irreversible injury
o membrane damage, lysosomal swelling, mitochondrial vacuolization, ↓ATP synthesis
o extracell Ca enters cell, intracell Ca released → activation of enzymes
o proteins, coenzymes, ribonucleic acids lost from membranes
o metabolites for ATP leak out
- 2 characteristics of irreversibility:
o irreversible mitochondrial dysfunction (no ATP produced)
o membrane dysfunction
- leakage enzymes into blood – clinical markers of cell death (CK, troponin, transaminases)
Morphology of cell injury and necrosis
- injury → loss of function → long time till damage seen
Reversible injury:
- cell swelling (loss of membrane ion-pumps)
- fatty change (cytoplasmic lipid vacuoles, in
cells of fat metabolism – hepatocytes,
myocardial)
Page 6
6
Necrosis:
- 2 processes underlie morphologic changes:
o protein denaturation
o enzyme digestion of organelles and cytosolic components
- eosinophilic (pink) on H+E stains
- ‘glassy’ (glycogen loss and vacuolated)
- cell membranes fragmented
- attract Ca salts → fatty soaps
- pyknosis (small, dense nucleus)
- karyolysis (faint, dissolved nucleus)
- karyorrhexis (fragmented nucleus)
- tissue patterns of necrosis:
o coagulative (most common):
� protein denaturation, preservation cell framework
� in hypoxic death of tissues except brain
� undergoes heterolysis (digestion by autosomal enzymes of invading leukocytes) or autolysis
(digestion by own lysosomal enzymes)
o liquefactive necrosis:
� autolysis and heterolysis predominates over protein denaturation
� soft and filled with fluid
� common in abscesses and brain
o caseous necrosis:
� in TB – grossly soft, friable, ‘cheesey’ material
� microscopically amorphous eosinophilic with cell debris
o fat necrosis:
� in adipose
� lipase activation (from pancreas or macrophages) release fatty acids from triglycerides, then
complex with Ca to create soaps
� grossly white, chalky (fat saponification)
� histo: vague cell outlines, Ca deposition
Examples of cell injury and necrosis
Ischaemic and hypoxic injury
o up to a point can recover if blood flow restored
o after this can cause reperfusion injury if flow restored eg MI, CVA
- reversible injury:
o hypoxia
� → loss of ATP generation by mitochondria
� → failure of Na/K ATPase membrane transport → Na enters cell and K exits, Ca influx and
intracell Ca release → net gain of solute and isosmotic gain of water, cell swelling, ER dilation
� also accumulation metabolic breakdown products
o cellular energy metabolism altered: anaerobic glycolysis → glycogen depleted, lactate accumulation, ↓pH
o detachment ribosomes from RER → ↓ protein synthesis
- irreversible injury:
o if ischaemia persists, all changes above + due to ATP depletion and membrane dysfxn
� ATP depletion induce MPT (membrane permeability transition - ↑ perm of membrane) in
mitochondria; pores form → ↓memb potential/diffusion solutes
� ATP depletion → releases cytochrome c (component of electron transport chain, key regulator in
driving apoptosis)
� ↑ cytosolic Ca activates membrane phospholipases → loss phospholipids
� ↑ Ca activates intracell proteases → degradation cytoskeletal elements → cell membrane
susceptible to stretching and rupture
� FFAs and lysophospholipids accumulate, toxic to membrane
Page 7
7
Ischaemia-reperfusion injury
o additional cells may die after blood flow restored, due to necrosis or apoptosis
o neutrophilic infiltrates
o important in MI, CVA, ARF
o mechanisms:
� ↑ generation O2-derived free radicals
� reactive O2 species protome MPT, precluding mitochondrial recovery
� inflammation by recruitment PMNs → ↑ cytokine + adhesion molecule expression, ↑ blood flow
→ ↑ inflam cell infiltration
� activation complement (IgM Abs deposit in ischaemic tissue, when blood flow resumes
complement binds to Abs, activated, cause injury and inflammation
Chemical Injury
2 mechanisms:
1) directly - bind to critical molecular components
- cyanide poisons mitochondrial cytochrome
oxidase and blocks oxidative phosphorylation
- mercury binds to cell membrane protein
sulfhydryl groups, inhibits ATPase dependent
transport)
2) indirectly – conversion to toxic metabolites
- paracetamol
- carbon tetrachloride CCl4
Apoptosis
- programmed cell death, induced by tightly regulated intracellular programmes
- eliminates unwanted cells selectively, minimal disturbance to surrounding cells and host
- cells activate enzymes that degrade cells own DNA and cytoplasmic proteins
- plasma membrane remains intact, structure altered so cell avid target for phagocytosis
- dead cells cleared, contents not leaked, no inflammatory reaction, cell shrinks
o cf necrosis – loss membrane integrity, enzyme digestion, host reaction
- necrosis and apoptosis may coexist, may share common features
Page 8
8
Causes of apoptosis:
- physiologic:
o programmed destruction cells in embryogenesis
o hormone-dependent involution of tissues (endometrium, prostate)
o cell deletion proliferating cell populations (get cell epithelium) to maintain constant no.
o cells already served purpose (neutrophils after inflam response)
o deletion potentially harmful self-reactive lymphocytes
o cell death induced by cytotoxic T cells, to eliminate viral-infected or neoplastic cells
- pathologic:
o cell death produced by injury (radiation, cytotoxic drugs – risk DNA mutations)
� mild injury (heat) → apoptosis, severe injury → necrosis
� some viruses (hepatitis)
� pathologic atrophy in organs with duct obstruction (pancreas)
� cell death in tumours
Morphologic features:
- cell shrinkage, chromatin condensation and fragmentation, cellular blebbing and fragmentation into apoptotic
bodies, phagocytosis of apoptotic bodies
- no inflam response so hard to detect histologically
Biochemical features:
- protein cleavage by family of proteases called caspases; also DNAases, breakdown DNA
- internucleosomal cleavage of DNA into fragments
- plasma membrane alters (protein flips inner → outer side) to allow recognition by phagocytes
Mechanisms of apoptosis:
- apoptosis induce by cascade of events
- divided into initiation phase (caspases activated), execution phase (enzymes cause cell death)
- initiation of apoptosis through 2 distinct but convergent pathways:
o extrinsic (receptor-initiated) or intrinsic (mitochondrial)
- initiation phase:
o extrinsic (death receptor) pathway:
� death receptors members of TNF receptor family → bring multiple inactive caspases close
together → activates them, rapid cascade of caspase activation
o intrinsic (mitochondrial) pathway:
� ↑ mitochondrial permeability → pro-apoptotic molecules release into cytoplasm
� Bcl-2 family of proteins regulate apoptosis
� balance between pro-apoptotic and protective molecules (Bcl-2) regulate mitochondrial
permeability
- execution pathway:
o mediated by proteolytic caspases
Page 9
9
o divided into 2 groups: initiator (8,9) and executioner (3,6)
o exist as inactive proenzymes → cleaved to activate
o either hydrolyzed by other caspases or activated autocatalytically
o executioner caspases act on many cell components, cleave cytoskeleton and nuclear matrix proteins →
nuclear breakdown
o in nucleus caspases cleave proteins involved in transcription, DNA replication/repair
Examples of apoptosis:
- growth factor deprivation
o affects hormone-sensitive cells, lymphocytes not stimulated by antigens or cytokines, neurons deprived of
nerve growth factor
� apoptosis triggered by intrinsic (mitochondrial) pathway due to relative excess of pro-apoptotic
proteins
o DNA damage
� radiation/chemotherapy induce apoptosis via mechanism due to DNA damage
� DNA damage → p53 (tumour suppressor) accumulates → cell cycle arrest at G1 phase to allow
repair → if repair can’t take place then p53 triggers apoptosis
� if p53 ↓/absent → apoptosis doesn’t occur (some cancers)
o TNF family receptors
� cell receptor Fas induces apoptosis when cross-linked by Fas ligand (protein produced by
immune system)
� important for eliminating lymphocytes that recognize self-antigens (mutations in Fas can cause
autoimmune disease)
� TNF can induce apoptosis or promote cell survival depending on which adapter protein attaches
to it
o cytotoxic T lymphocytes
� recognize foreign antigen on infected host cells and secrete porforin (transmembrane pore-
forming molecule allowing entry of protease granzyme B)
• granzyme B activates multiple caspases → apoptosis
o dysregulated apoptosis
� too much or too little, underlies many disease
� disorders with defective apoptosis/↑ cell survival
• cancers
• autoimmune disorders
� disorders with ↑ apoptosis and excess cell death
• neurodegenerative diseases (SMA)
• ischaemic injury (MI, CVA)
• death of virus-infected cells
Subcellular responses to injury
Lysosomal Catabolism
- primary lysosomes: membrane-bound organelles containing hydrolytic enzymes
o fuse w membrane-bound vacuoles containing ingested material→phagolysosomes (secondary lysosomes)
- 2 mechanisms:
o Heterophagy: uptake/degrad material from external environ by phagocyt eg ingestion bacteria by leukos
o Autophagy: degradation intracellular organelles, esp in cells undergoing atrophy
- if substances not fully digested persist in cells as residual bodies eg lipofuscin pigment, carbon
- lysosomal storage disorders – accum in lysosomes – esp problem in CNS
Page 10
10
Induction (hypertrophy) of smooth ER
- SER site for metabolizing exogenous agents, usually by P450 pathway
- prolonged exposure induces SER → adaptive hypertrophy (CBZ, phenytoin)
Mitochondrial alterations
- cell hypertrophy/atrophy → ↑/↓ in mitochondria
- megamitochondria (large, abnormal shape) in hepatocytes in EtOH liver disease
- mitochondrial myopathies → ↑ number morphologically abnormal mitochondria
Cytoskeletal abnormalities
- can cause defects in cell function or cell injury can cause cytoskeletal abnormalities
o eg EtOH liver disease → accum intracellular intermediate filaments (Mallory bodies)
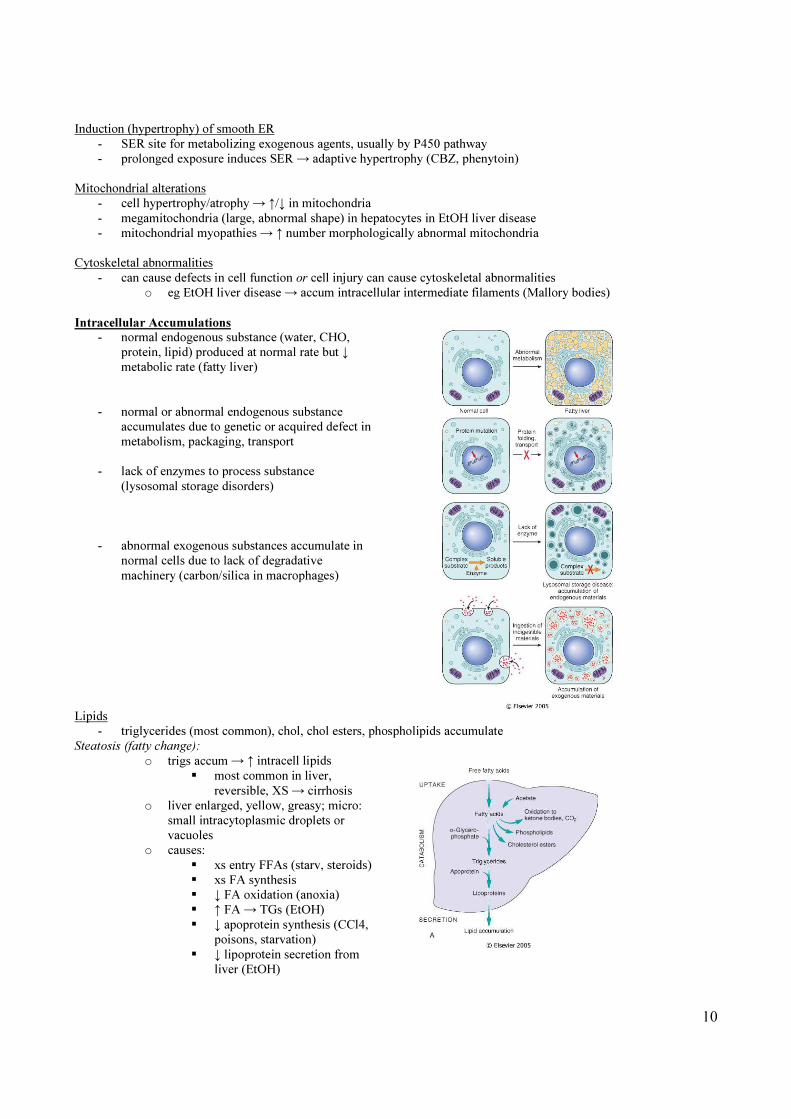
Intracellular Accumulations
- normal endogenous substance (water, CHO,
protein, lipid) produced at normal rate but ↓
metabolic rate (fatty liver)
- normal or abnormal endogenous substance
accumulates due to genetic or acquired defect in
metabolism, packaging, transport
- lack of enzymes to process substance
(lysosomal storage disorders)
- abnormal exogenous substances accumulate in
normal cells due to lack of degradative
machinery (carbon/silica in macrophages)
Lipids
- triglycerides (most common), chol, chol esters, phospholipids accumulate
Steatosis (fatty change):
o trigs accum → ↑ intracell lipids
� most common in liver,
reversible, XS → cirrhosis
o liver enlarged, yellow, greasy; micro:
small intracytoplasmic droplets or
vacuoles
o causes:
� xs entry FFAs (starv, steroids)
� xs FA synthesis
� ↓ FA oxidation (anoxia)
� ↑ FA → TGs (EtOH)
� ↓ apoprotein synthesis (CCl4,
poisons, starvation)
� ↓ lipoprotein secretion from
liver (EtOH)
Page 11
11
Cholesterol and cholesterol esters:
- cholesterol needed for cell membrane/hormone synthesis
- accumulation (intracellular cytoplasmic vacuoles) can cause:
o atherosclerosis: chol and chol esters accum in arterial wall SMCs/macrophages
o xanthomas: in acquired and hereditary hyperlipidaemias, accum in clusters of “foamy” macrophages and
mesenchymal cells (under skin, in tendons)
o inflam and necrosis: lipid-laden (foamy) macrophages result from phagocytosis of membrane lipids
derived from injured cells
o cholesterolosis: focal accum chol-laden macrophages in lamina propria of gallbladder
o Neimann-Pick disease, type C: lysosomal storage dis - mutation enzyme chol catab
Proteins:
- intracellular protein accum secondary to xs synthesis, absorption or defect in transport
- intracellular protein deposition in:
o PCT kidney epithelium due chronic reabsorption in proteinuria
o Ig distending ER of plasma cells due to ↑ synthesis (form Russell bodies)
o extracellular deposition in amyloidosis
- defects in protein folding sometimes causes protein depositions
o after synthesis partially folded proteins form intracell aggregates that entangle proteins
o this is prevented by stabilization by molecular chaperones
� chaperones also involved in transporting proteins into organellar destinations
o chaperones repair misfolded proteins; if ineffective, proteins targeted for degradation by proteasome; if
misfolded proteins accumulate they trigger apoptosis
- altered protein folding causes disease by:
o defects in intracellular transport or secretion (eg α1AT def – accum in ER of hepatocytes and lung; in CF
mutation delays dissociation of Cl channel protein from chaperone → abnormal folding → degradation)
o toxicity of abnormally folded proteins: in neurodegenerative disease (AD), amyloidosis
Hyaline changes:
- any alteration to tissues that causes a homogenous, glassy pink appearance in H+E stain
- intracellular hyaline change eg Russell bodies, viral inclusions, Mallory bodies
- extracellular hyaline change eg damaged arterioles in HTN/DM, due to extravasation proteins
Glycogen:
- xs intracellular deposits (clear vacuoles) seen in glycogen storage diseases → accum in cell → cell injury/death;
and in altered glucose metabolism (DM)
Pigments:
- accumulated materials
- exogenous:
o carbon/coal dust deposits called anthracosis when accum in pulm macrophages and LNs
o pigments from tattoos live in macrophages for life of cell
- endogenous:
o lipofuscin (‘wear and tear’ pigment): assoc with cellular atrophy
� seen as fine yellow-brown intracytoplasmic granules
� made of lipids, phospholipids, protein, derived from cell membrane lipid peroxidation
o haemosiderin: Hb-derived, yellow-brown granular intracellular pigment made of ferritin
� localized (macrophage-mediated metabolism of focal haemorrhage – bruise)
� systemic (↑ dietary iron absorption – haemochromatosis, ↓ utilization – thalassemia, or
haemolysis or chronic transfusions)
o melanin: endogenous, non-Hb derived, brown-black pigment, from oxidation of tyrosine to
dihydroxyphenylalanine in melanocytes
Pathological Calcification
- abnormal tissue deposition of calcium salts
- 2 forms:
o dystrophic: in nonviable/dying tissues with normal serum Ca
� atherosclerosis, calcific AS, tuberculous node
Page 12
12
o metastatic: in viable tissues with hypercalcaemia
� nephrocalcinosis, pulmonary calcinosis, gastric mucosal
Dystrophic calcification:
- in arteries in atherosclerosis and damaged heart valves and in areas of necrosis
- intracellular and extracellular
- precipitation of crystalline CaPO4 similar to bone hydroxyapatite
o initiation (nucleation)
� extracellular on membrane-bound vesicles from dead/dying cells that concentrate Ca
• membrane-bound phosphatases then generate phosphate → binds to Ca
� intracellular occurs in mitochondria of dead/dying cells
o propagation: depends on conc Ca/phos, presence inhibitors/structural components ECM
- psammoma bodies in thyroid Ca (lamellated)
Metastatic calcification:
- amorphous basophilic densities throughout body → no consequence unless massive deposition → renal and lung
problems
- 4 main causes:
o ↑ PTH (parathyroid tumours, ectopic PTHrP)
o destruction bone tissue (marrow malignancy – myeloma, mets, ↑bone turnover – pagets)
o Vit D toxicity, sarcoidosis
o CRF → secondary hyperparathyroidism due to phosphate retention
Cellular Ageing
- reflects accumulative effects of sublethal cellular/molecular damage
↓ Metabolic functions of ageing cells:
- ↓ mitochondrial ATP generation
- ↓ synthesis structural, enzymatic, regulatory proteins
- ↓ capacity nutrient uptake
- ↑ DNA damage/↓ repair
- accum oxidative damage (eg lipofuscin)
- accum advanced glycation end products → protein cross-linking
Morphological alterations:
- irregular and abnormally lobed nuclei
- pleomorphic and vacuolated mitochondria
- ↓ ER
- distorted golgi apparatus
Mechanisms of cellular ageing:
3 processes:
- replicative senescence (cells have limited capacity for replication)
o finite life span, population doublings age dependent (ie have clocks – Hayflick theory)
o telomere shortening (incomplete replication of chromosome ends) – as cells repeatedly divide telomeres
progressively shorten
� in some cancers telomerase reactivated – important in cell immortality
Page 13
13
- genetic factors that influence ageing process
o individual genes can affect longevity → ↓ signaling thru IGF1 can prolong lifespan
- environmental factors that cause progressive accumulation metabolic and genetic damage
o aging a balance between metabolic damage and repair mechanisms
� reactive O2 metabolites → oxidative damage which ↑ with age
� ↑ production due to high calorie diet, exposure to radiation → ↓ life span
o counterbalancing protective responses:
� antioxidants
� recognition and repair of damaged DNA
• Werner syndrome – premature aging – defect in DNA helicase, rapid accumulation
chromosomal damage