35
Simulating biomolecular function from motions across multiple scales (I) Peter J. Bond (BII) [email protected]

Simulating biomolecular function from motions
across multiple scales (I)
Peter J. Bond (BII) [email protected]

125,000
2017
1972no.ofstructures0
StructuralBiology:WhytheNeedforSimulation?year
• Explosioninnumberof
structuresdepositedtoPDB
overpast~15years…dueto:
- Post-genomicsera:accessibility
tonumerousgenomes,more
stableproteomesetc.
- Automationincrystallization
protocols,robotics.
- Structuralbiologyconsortia(and
money!)
• AlsoimprovementsinNMR,
cryoEM,&biophysicalmethods.
• Sowithallthisstructuraldata,
whytheneedforsimulation? 2
RCSBPDB:RCSBProteinDataBankhttps://www.rcsb.org/

TheImportanceofDynamicsand“Landscape”…
single “snapshot”
3
ligand binding
10-15
10-12
10-9
10-6
10-3
100
10-10 10-9 10-8 10-7 10-6 10-5 10-4(nm) (µm)
(fs)
(ps)
(ns)
(µs)
(ms)
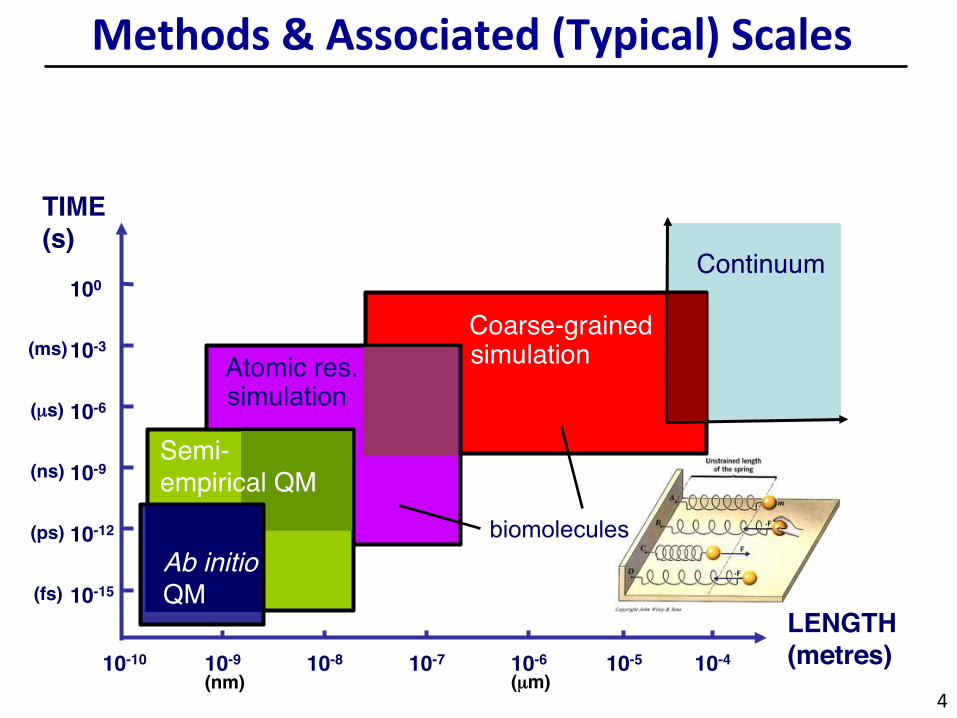
LENGTH (metres)
TIME (s)
Coarse-grained
Semi-empirical QM
Ab initio QM
Continuum
simulation Atomic res.
biomolecules
4
Methods&Associated(Typical)Scales
simulation

BiomolecularSimulations:FromStructuretoDynamics
o Staticstructure– invitroconditions.o Simulation:~300K,biologicalmodel...o 103–105atoms…o ~106pair-wiseinteractions:“forcefield”o NumericalintegrationofF=ma.o Coordinatescalculatedevery
0.000000000000001sec,~1CPUsec…
FFusedtocalculateresultantforcesFi(&accelerationa
i
viaNewton’s2ndlaw)onparticleiwithmassmi
Fi = −∇iEsystem =miai
−δEsystemδri
=miδviδt
=miδ 2riδt2
thuswecanrelategradientofPEtochangesinpositions/velocitiesasafunctionoftime:
5

BiomolecularSimulations:FromStructuretoDynamics
real… explicit
COMPUTATIONAL COST...
implicit (e.g. ε, ±ξ)
o Staticstructure– invitroconditions.o Simulation:~300K,biologicalmodel...o 103–105atoms…o ~106pair-wiseinteractions:“forcefield”o NumericalintegrationofF=mao Coordinatescalculatedevery
0.000000000000001sec,~1CPUsec…
Periodicity mimics infinite system (e.g. cube). Minimum image convention. Good rule of thumb: ≥2 nm between “images”.
6

A
B i ii
21 Å
35 Å
MolecularSimulation–“ComputationalMicroscope”
• Computationalmodelling–nowanindispensibletoolforcomplementingtraditionalexperiments.
• ArielWarshel:“…thebesttoolwehavetoseehowmoleculesareworking.”(awardedNobelPrizeinChemistry,2013withLevitt&Karplus).• KlausSchultencoinedtheterm“computationalmicroscope”.• Notsimplyaninsilico“imaging”technique– notjustformovies…- dynamics,interactions,conformationalchanges,mechanisms!- nolimitationsonspatio-temporal“zoom”!- abilitytocarryout“alchemistry”!- abilitytodo“thoughtexperiments”!- powerfultool:integratemodel&experiment.
But...PotentialLimitations:
• Accuracyofstartingmodel/availableexperimentaldata…
• Accuracyoftheunderlyingforcefield…
• Limitedsamplingintime/space… 7

8
Simulating(andwaitingfor)Motions…
Zwier&Chong.CurrentOpinioninPharmacology.2010.10:745-752.
energy
conformation

supe
rcom
putin
gpo
wer
Theincreasingpowerofbiomolecularsimulation
life cycle of E. coli
• <decade:~103↑simulationperformance…-thankstoalgorithms,architectures,cost…-alsoimprovesFFaccuracy.
Schlicketal.Biomolecularmodelingandsimulation:afieldcomingofage.QRevBiophys.2011.44:191-228.
9
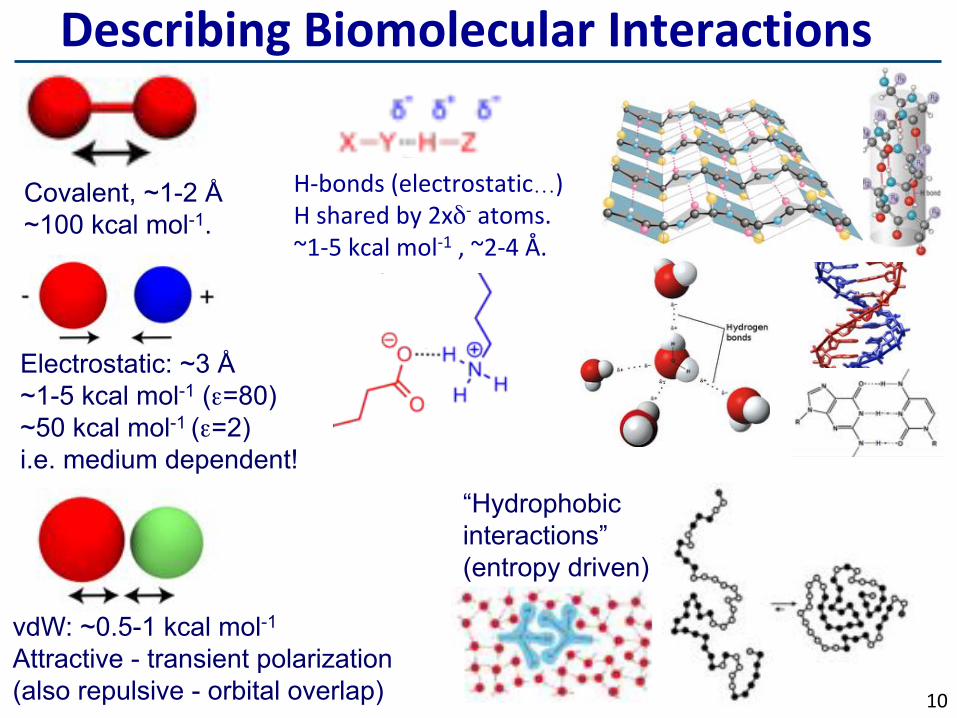
Electrostatic: ~3 Å ~1-5 kcal mol-1 (ε=80) ~50 kcal mol-1 (ε=2)
i.e. medium dependent!
Covalent, ~1-2 Å ~100 kcal mol-1.
DescribingBiomolecularInteractions
H-bonds(electrostatic…)Hsharedby2xδ-atoms.~1-5kcalmol-1,~2-4Å.
vdW: ~0.5-1 kcal mol-1 Attractive - transient polarization (also repulsive - orbital overlap)
“Hydrophobic interactions” (entropy driven)
10
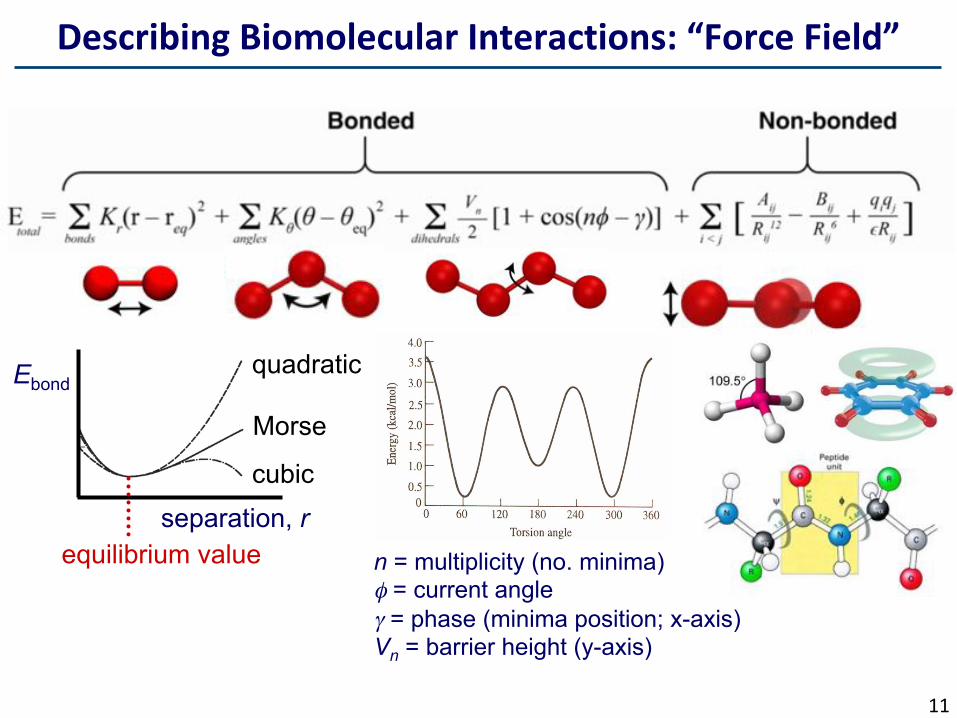
Ebond
separation, r cubic
Morse
quadratic
equilibrium value n = multiplicity (no. minima) φ = current angle γ = phase (minima position; x-axis) Vn = barrier height (y-axis)
DescribingBiomolecularInteractions:“ForceField”
11

Evdw = 4ε{(σ/R)12 - (σ/R)6}
σ
E
R
Lennard-Jones (“6-12”) potential:
DescribingBiomolecularInteractions:“ForceField”
Pair-wise sum of all possible interacting non bonded atoms i and j… O(n2)
Electrostatics – decays slowly (i.e. 1/R)… many methods to treat this.. *** Stick with FF recommendation! ***

Energies&ForceFields(FFs)…Describe total energy of the system such that there are penalties for deviations from reference values.
§ Energiesarecalculatedusinganempiricallyderivedforcefield(FF).
§ “Balls&springs”:Bonded(+fc/E
o),
non-bondedinteractions(LJ),particlemass,size,partialcharge.
§ Parametersfromwhere?§ Fragmentgeometries–X-raystudies.
Biomolecules-highlyspecificrefinementsovertheyears(butcf.over-fitting,e.g.IDPs…)
§ Rotationalbarriers/vibrationalfrequenciesfromspectroscopy.
§ Chargesfrome.g.QMcalculations.§ vanderWaal’s–trialanderror
e.g.tomatchexperimentaldensities.§ Thermodynamicproperties…§ ManyaccurateFFsarenowavailable!
ETOTAL = EBONDED + ENON-BONDED
13

RealSimulationCodes&ForceFieldsCHARMM (Chemistry at Harvard Molecular Mechanics) www.charmm.org
♦ Interface through fortran like scripting language - tough! ♦ Very powerful, many different features. Slow. ♦ $600 (academic) but also free reduced-functionality version.
AMBER (Assisted Model Building with Energy Refinement) www.ambermd.org
♦ Suite of about 60 programs based around a few central ones ♦ Slow on standard CPUs; fast with GPU-optimization ♦ $500 (academic) $15-20,000 (industry).
GROMACS (Groningen Machine for Chemistry Simulation) www.gromacs.org
♦ Simple interface (not scripting based) ♦ The fastest codes on 100’s cores (CPU/GPU) ♦ GNU licensed (i.e. free!)
NAMD (Not just Another Molecular Dynamics program) www.ks.uiuc.edu/Research/namd
♦ Optimized for many 1000’s of cores ♦ Written in C++ with a TCL-based scripting interface. ♦ Also free of charge.
14

http://bio.demokritos.gr/gromita/-GraphicalUserInterfaceforGROMACSv4+
http://haddock.science.uu.nl/enmr/services/GROMACS/main.php-Web-basedportalforautomatedGROMACSsimulations,distributedEuropeanGridnetwork(10nssims).http://py-enmr.cerm.unifi.it-similarforAMBER-basedNMRrefinement.http://mmb.irbbarcelona.org/MDWeb/-Settingup/running/analysisofsimulationsinAmber,NAMD,GROMACSandrelated…
https://www.charmming.org-CHARMMinginterface–preparation/submission/analysis.
15
AutomatedSimulations…butbewary…
http://www.bevanlab.biochem.vt.edu/

Obtain structure – X-ray / NMR / model
Add H’s, consider pkA, prepare topology
Solvate + add ions
Minimize
Analyze
Ene
rgy
Geometry Production
Equilibration
♦ missing atoms / residues / loops & mutations (Pymol, Modeller, Swiss-model etc.)
♦ oligomer state ♦ disulfides (assess via distance only?) ♦ ligands (CGenFF, PRODRG, SwissParam, VMD QMTool – Gaussian.)
VF ii −∇=
e.g.Steepestdescents– followgradient“downhill”untilthreshold(ΔEorFmax)
Bulk/structural/crystalwater/ions
Aimto“relax”system,e.g.:solvent/iondistribution,temperature,boxsize/density…Cf.ensemble(e.g.NPT)
Erestr = k (r - r0)2
SimulationWorkflow
16
Early Steps: Know your system! (PDB “headers” & papers are your friend!)

Cα
RM
SD
(Å)
time (ns)
1
2
10
3
Take frames from here
AssessingErrors&Convergence...
• Checkdistributionofpropertiesagainstaverage–evendistribution?
• Calculateblockaveragesforasingletrajectory.
• Calculatemultiplesimulationreplicasandcompare…(Ergodic…)
Simple - look at it! Sampling & Convergence
each τblock should > τrelax x no. steps
0
Care… this is a very limited indicator alone…
Comparison to Experiment
Protein structural deviation
e.g. RMSF vs B-factors
… remember experimental error!
22
38 RMSFBiπ
=
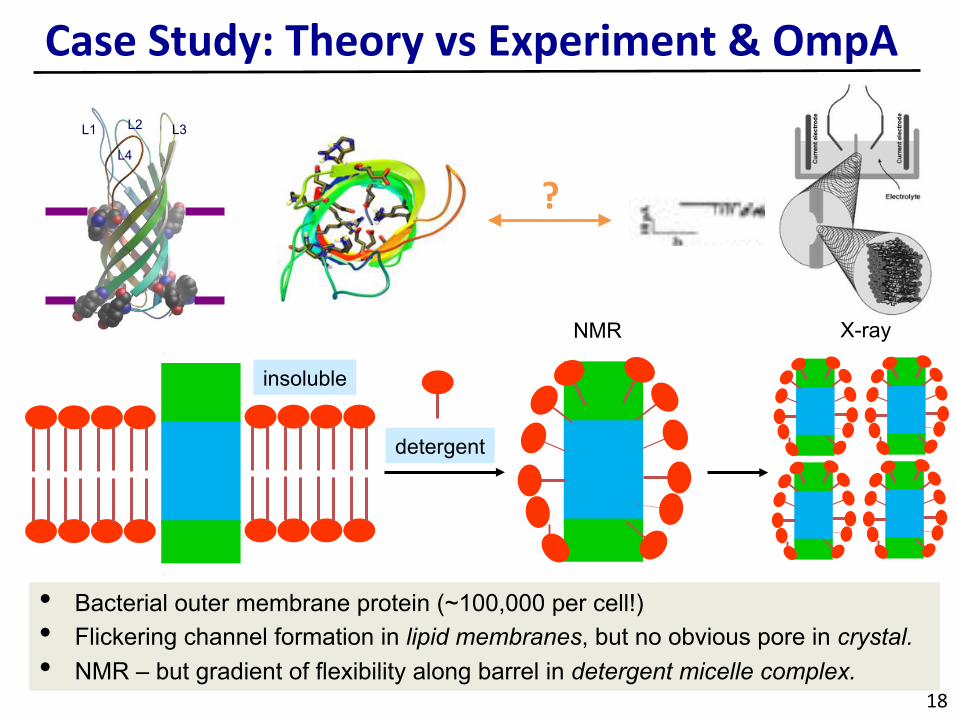
L1 L3
L4
L2
• Bacterial outer membrane protein (~100,000 per cell!) • Flickering channel formation in lipid membranes, but no obvious pore in crystal. • NMR – but gradient of flexibility along barrel in detergent micelle complex.
?
insoluble
detergent
NMR X-ray
18
CaseStudy:TheoryvsExperiment&OmpA
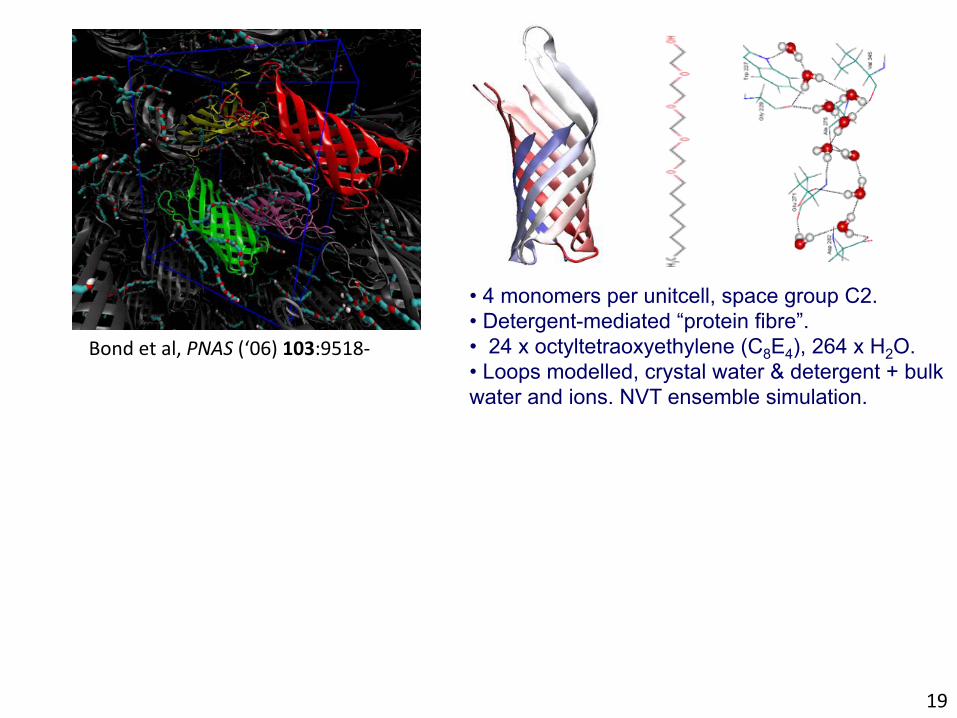
Bondetal,PNAS(‘06)103:9518-
19
• 4 monomers per unitcell, space group C2. • Detergent-mediated “protein fibre”. • 24 x octyltetraoxyethylene (C8E4), 264 x H2O. • Loops modelled, crystal water & detergent + bulk water and ions. NVT ensemble simulation.

Bondetal,PNAS(‘06)103:9518-
20
RMSD
(Å)
0
2
4
6
0 10 20 30 40 50time(ns)
crystal simulation
L4 L1 L3 L2
T1 T2 T3
Bi =
[8π2
/3].R
MS
F i2 (Å
2 )
• Detergent molecules dynamically cover protein fibre – membrane-like environment. • β-barrel RMSD low. Higher for loops – low crystal density & inherent high mobility. • B-factor correlation... Missing density - vibrations, fluctuations, and lattice disorder…

OmpA: Dynamics vs. Environment
Bond & Sansom, J Mol Biol (‘03) 329:1035-
21
Membrane Insertion Protocols • Simplified lipid membrane – in vitro system. (Now bacterial membranes possible). • g_membed, GROMACS (also mdrun_hole): protein “contracted” in xy-plane, overlapping
lipids deleted, then protein grown back during EM/MD to push remaining lipids away. • CHARMM GUI Membrane Builder – NAMD, GROMACS, AMBER, CHARMM: random
lipids from a membrane library packed against protein surface. • Or nowadays: just “insert, delete, and equilibrate”… Micelle Insertion Protocols • ~60 DPC detergent molecules based on DLS measurements. Concentration > CMC. • “Spoke-like” DPC placement + equilibration. (Also CHARMM Micelle builder). • Simulations match protein-detergent NOEs detected from NMR.
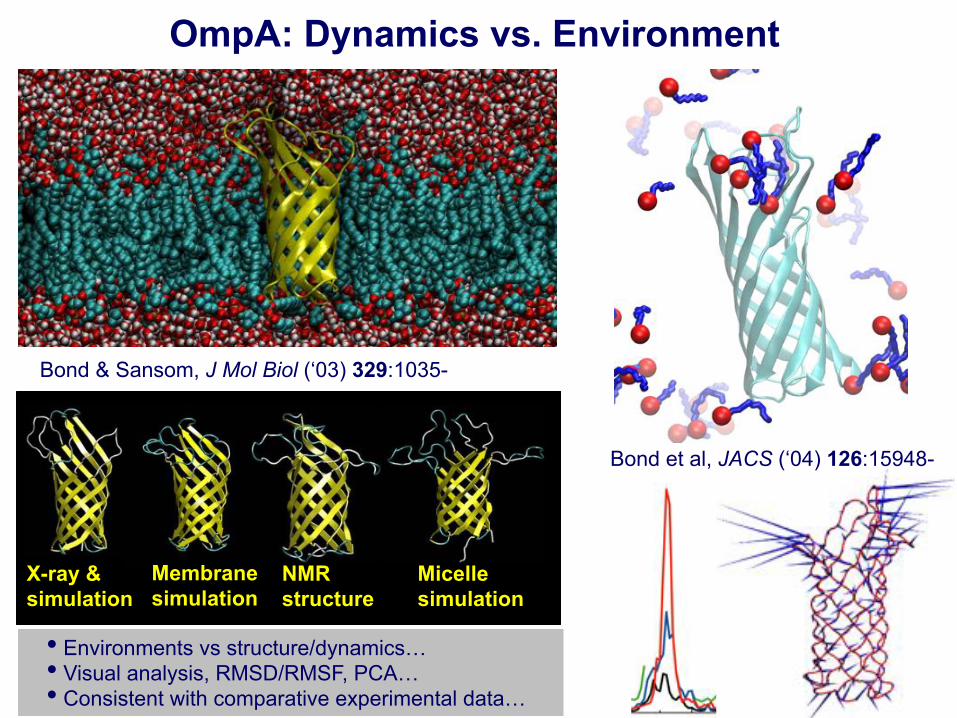
OmpA: Dynamics vs. Environment
Bond & Sansom, J Mol Biol (‘03) 329:1035-
22
• Environments vs structure/dynamics… • Visual analysis, RMSD/RMSF, PCA… • Consistent with comparative experimental data…
X-ray & simulation
Membrane simulation
NMR structure
Micelle simulation
Bond et al, JACS (‘04) 126:15948-

z (n
m)
time (ps)
• Water trajectories: difference in permeation properties in different environments. • Single “gate” region with alternating electrostatic switch proposed. • Bond et al., Biophys. J. (‘02) 83:763-. • Open state conductance estimated as ~60 pS at 0.1 V in 1M KCl... = expt! • Double-mutant cycles & conformational exchange experiments confirm the hypothesis! Hong et al., Nat. Chem. Biol. (‘06) 2:627-.
23

A
B i ii
21 Å
35 Å
TheComputationalMicroscope:Fast-Forward
• Needfor“enhancedsampling”…e.g.:- Heating–proteinfolding,integrationofexperimentaldata.- Biasingpotentials–molecularbinding&energies.- Coarse-graining– simplifyingthelandscape.
-30
-3
-6-9
-12
-15time(logseconds)
fs
ps
ns µs
ms
bondvibrations
sidechainrotation
loopmotions
conformational
changes,
ligandbinding
proteinfolding,
macromolecular
assembly
24

Sampling,Constraining,&Heating!
• ReplicaexchangeMD(“paralleltempering).• RunNcopiesofsystematdifferenttemperatures;
Metropoliscriteriontoexchangeconfigurations;acceptancebasedonBoltzmann-weightedΔE…
(MoredynamicthanX-ray:spectrofluorometry&CD)MarzinekJKetal.CharacterizingtheConformationalLandscapeofFlavivirusFusionPeptidesviaSimulationandExperiment.2016,ScientificReports.5,19160.
X-raystructures
25
Energy
conformation
• Simulatedannealing–“heat&cool”.• Usefulforinterpretingexperimentaldata–
integrateasrestraints.• E=EBONDED+ENON-BONDED+w.ERESTRAINTS• ERESTRAINTS=EX-RAYorENMR(e.g.NOEdistances)
time
folding
ΔE≥0ΔE<0
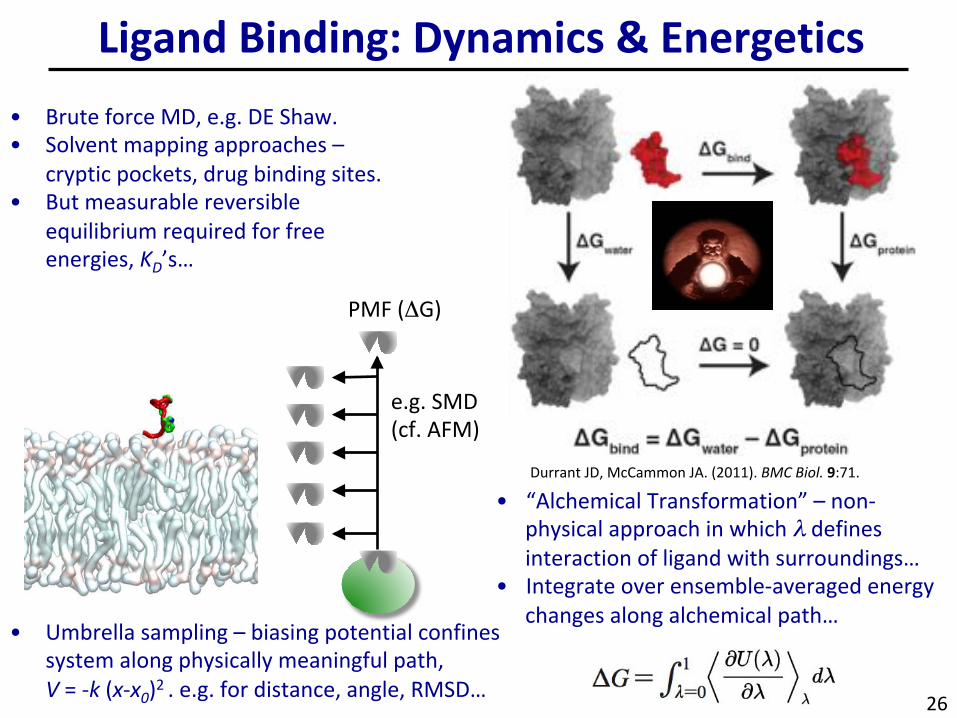
• BruteforceMD,e.g.DEShaw.• Solventmappingapproaches–
crypticpockets,drugbindingsites.• Butmeasurablereversible
equilibriumrequiredforfreeenergies,K
D’s…
LigandBinding:Dynamics&Energetics
26
• “AlchemicalTransformation”–non-physicalapproachinwhichλdefinesinteractionofligandwithsurroundings…
• Integrateoverensemble-averagedenergychangesalongalchemicalpath…
• Umbrellasampling–biasingpotentialconfinessystemalongphysicallymeaningfulpath,V=-k(x-x
0)2.e.g.fordistance,angle,RMSD…
PMF(ΔG)
e.g.SMD(cf.AFM)
DurrantJD,McCammonJA.(2011).BMCBiol.9:71.

• biological membrane: lipid bilayer + proteins (α-helical or β-barrel).
• membrane proteins: ~25% of genes. • drug targets: ion channels & receptors.
cells membranes
proteins
~10Å
~10nm
~100nm
~1µm
ComputationalMicroscope:TuningtheResolution
• Biasedsamplingapproachesusefulforspeedingupspecificsystems.• Butwhataboutgeneralimprovementoftime/length-scalesinbiological
systems,whichspanseveralregimes…• e.g.:crowdedcytoplasmicenvironment,extendedlipidmembranes.
27
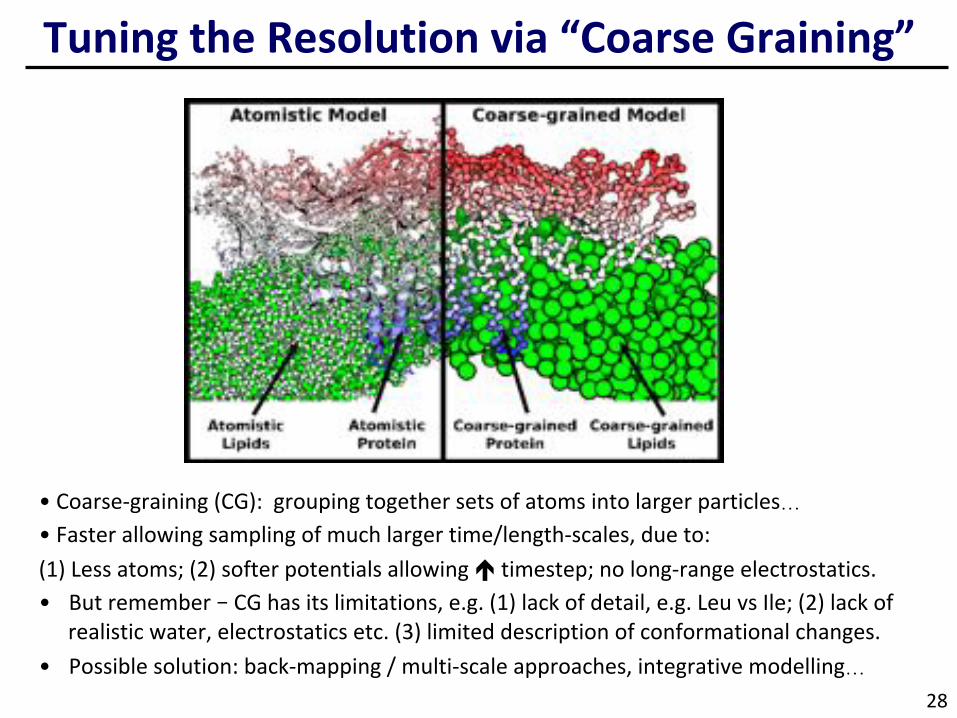
TuningtheResolutionvia“CoarseGraining”
• Coarse-graining(CG):groupingtogethersetsofatomsintolargerparticles… • Fasterallowingsamplingofmuchlargertime/length-scales,dueto:(1)Lessatoms;(2)softerpotentialsallowingétimestep;nolong-rangeelectrostatics.• Butremember–CGhasitslimitations,e.g.(1)lackofdetail,e.g.LeuvsIle;(2)lackof
realisticwater,electrostaticsetc.(3)limiteddescriptionofconformationalchanges.• Possiblesolution:back-mapping/multi-scaleapproaches,integrativemodelling…
28

MartiniCoarse-GrainedForceField&Variants
water
+ve ion
-ve ion
lipid • ~1 particle per 4 heavy atoms. • Bond/angle potentials with weak fc’s. • Limited number of particle types with
different levels of LJ interaction, from strong polar interactions in bulk solvent to repulsion between polar & nonpolar phases.
• Typically short-range electrostatics, fully charged ions/groups…
http://cgmartini.nl/– martinize.py,insane.py,backward.py,etc.
29
Marrinkandco-workers.1stlipids,morerecentlyotherbiomolecules.J.Phys.Chem.B(2004)108:750-;J.Phys.Chem.B(2007)111:7812-;JCTC(2008)4:819-;JCTC(2009)5:2531-

• Example extension to proteins - 1-3 particles/AA, H-bonding. 2o structure restraints based on analysis of native state. Bond & Sansom (2006) JACS 128:2697. Bond et al (2007) J. Struct. Biol. 157:593. Parameterization: Amino acids transfer free energies. Validation: membrane PMFs & compare with spectroscopic data.
• Martini: 2o structure maintained via weak dihedrals (but structure more flexible).
WALPLS-helix fd-coat
Biophys J. (2008) 94:3393-
Coarse-GrainedSimulationsofPeptides
30

• LacY test-case – CG-ENM vs. atomistic (Rc = 0.7 nm). • All-Atom, AA (docked) vs. CG (assembled): similar lipid-protein interactions. • OmpA: Tuning of ENM cutoffs & force-constants. Similar dynamics in AA vs. CG.
CGProteins:ElasticNetworkModels
residue
RM
SF
(nm
)
atomistic
0
0.5
1.0
1.5
0 40 80 120 160
CG
31

• Spontaneous assembly of membrane proteins into lipid / detergent. • Similar approaches for e.g. DNA, bio/nano systems (in preparation). • ~102-103 x speedup vs. all-atom simulations; can be back-mapped...
UnbiasedLipid/ProteinAssemblyUsingCGSimulations
32
Ω ∼ �40º
TG XXXG
JACS(‘06)128:2697-.BiophysJ.(‘08)95:3790-.JRSocInterface(2008)5:S241-

◆ Maculatin 1.1: cell lysis. Flurophore leakage but lipid maintained? (confocal microscopy). ◆ Self-assembly to induce membrane disruption and cell lysis at high concentration. ◆ 100 peptides, 900 POPC lipids, ~60,000 water beads (equivalent to ~500k atoms). ◆ Surface binding → peptide aggregation → membrane stretching & vesicle deformation. ◆ Disordered aggregates - contrast with e.g. ordered WALP peptide insertion.
750 ns
BIGSYSTEMS!–e.g.AntimicrobialPeptideAttack
Ambroggioetal(2005)Biophys.J.89:1874-1881Chiaetal(2000)Eur.J.Biochem.267:1894.
◆ Bond et al (2008) Biophys. J. 95:3802
33

• MolecularSimulations–WhatandWhy?• AccessibleTimes&LengthScales• PotentialLimitations• Interactions,Energies,andForceFields• Long-RangeInteractions&Boundaries• TheSimulationWorkflow
• WhatCanaSimulationTellUs?• TestCase:MembraneProteinDynamics• StateoftheArt:EnhancedSampling&Coarse-
Grained/MultiscaleApproaches
Introduction to Simulation
Practicalities of Simulation
Uses, Now & the Future
34
Biomolecular Simulations: Summary
Next:SimulationsinAction

ComputerSimulationofLiquids:Allen&Tildesley
MolecularModelling:PrinciplesandApplications:Leach
UnderstandingMolecularSimulation:FromAlgorithmstoApplications:Frenkel&Smit
GROMACSmanual–www.gromacs.org/
ReferenceTexts,Manuals,Reviews
• HospitalA,GoñiJR,OrozcoM,GelpíJL.(2015).Moleculardynamicssimulations:advancesandapplications.AdvApplBioinformChem.8:37-47.
• DrorRO,DirksRM,GrossmanJP,XuH,ShawDE.(2012).Biomolecularsimulation:acomputationalmicroscopeformolecularbiology.AnnuRevBiophys.41:429-52.
• DurrantJD,McCammonJA.(2011).Moleculardynamicssimulationsanddrugdiscovery.BMCBiol.9:71.• KarplusM,McCammonJA.(2002).MolecularDynamicsSimulationsofBiomolecules.NatStructBiol.9:646-52.• LeeEH,HsinJ,SotomayorM,ComellasG,SchultenK.(2009).Discoverythroughthecomputationalmicroscope.
Structure.17:1295-306.• BigginPC,BondPJ.(2015).Moleculardynamicssimulationsofmembraneproteins.MethodsMolBiol.
1215:91-108.• KhalidS,BondPJ.(2013).Multiscalemoleculardynamicssimulationsofmembraneproteins.MethodsMol.
Biol.924:635-57.

















