324


Pharmaceutical Biotechnology
Edited byO. Kayser and R.H. M
..uller
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

ii
Related Titles
H.-J. Rehm, G. Reed, A. P..uhler,
P. Stadler, G. Stephanopoulos
Biotechnology, Second,Completely Revised Edition,Volume 3/Bioprocessing
1993, ISBN 3-527-28313-7
H. Klefenz
Industrial PharmaceuticalBiotechnology
2002, ISBN 3-527-29995-5
G. Walsh
Proteins/Biochemistry andBiotechnology
2001, ISBN 0-471-89906-2
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

Oliver Kayser, Rainer H. M..uller
Pharmaceutical Biotechnology
Drug Discovery and Clinical Applications
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

Edited by
Dr. Oliver KayserFree University BerlinInstitute of PharmacyPharmaceutical TechnologyBiopharmacy & BiotechnologyKelchstr. 3112169 BerlinGermany
Prof. Dr. Rainer H. M..uller
Free University BerlinInstitute of PharmacyPharmaceutical TechnologyBiopharmacy & BiotechnologyKelchstr. 3112169 BerlinGermany
� This book was carefully producednevertheless, authors, editors, and publisherdo not warrant the information containedtherein to be free of errors. Readers areadvised to keep in mind that statements, dataillustrations, procedural details or other itemsmay inadvertently be inaccurate.
Library of Congress Card No.: applied for
British Library Cataloguing-in-PublicationData. A catalogue record for this book isavailable from the British Library.
Bibliographic information published by DieDeutsche Bibliothek Die Deutsche Bibliotheklists this publication in the DeutscheNationalbibliografie; detailed bibliographicdata is available in the Internet athttp://dnb.ddb.de.
2004 WILEY-VCH Verlag GmbH & Co.KGaA, WeinheimAll rights reserved (including those oftranslation into other languages). No part ofthis book may be reproduced in anyform – nor transmitted or translated into amachine language without writtenpermission from the publishers. Registerednames, trademarks, etc. used in this book,even when not specifically marked as such,are not to be considered unprotected by law.
Printed in the Federal Republic of GermanyPrinted on acid-free paper.
Composition: Laserwords Private Ltd,Chennai, IndiaPrinting: betz-druck GmbH, DarmstadtBookbinding: Litges & Dopf BuchbindereiGmbH, HeppenheimISBN 3-527-30554-8

v
Preface
Pharmaceutical biotechnology has a long tradition and is rooted in the last century,first exemplified by penicillin and streptomycin as low molecular weight biosyntheticcompounds. Today, pharmaceutical biotechnology still has its fundamentals infermentation and bioprocessing, but the paradigmatic change affected by biotechnologyand pharmaceutical sciences has led to an updated definition. Upon a suggestionby the European Association of Pharma Biotechnology (EAPB), pharmaceuticalbiotechnology is defined as a science covering all technologies required for theproduction, manufacturing, and registration of biotechnological drugs.
The biopharmaceutical industry has changed dramatically since the first recombinantprotein (Humulin) was approved for marketing in 1982. The range of resourcesrequired for the pharmaceutical industry has expanded from its traditional fields.Advances in the field of recombinant genetics allows scientists to routinely clone genesand create genetically modified organisms that can be used in industrial productionprocesses. Also, specific therapeutic proteins can be synthesized in nonbiological ways,and recombinant proteins can be isolated from complex mixtures in commercially viableprocesses. In contrast to academic research, industrial development and manufacturingis guided by cost and time effectiveness, patent protection, exclusivity periods, andregulatory compliance. There are many critical industry issues that companies have toface; hence there is a need for new pharmaceutical biotechnology textbooks focussingon industrial needs.
Therapeutic proteins and the recently approved antisense oligonucleotideFomivirsen represent new and innovative biotech drugs that are different fromclassical drugs in the development and production process. In this area, pharmaceuticalcompanies are confronted with new challenges to develop new products and to applynew technologies. Industrial needs are particularly different and are either not discussedor are only marginally discussed in existing textbooks, which is why we feel that thereis a need for a new pharmaceutical biotechnology textbook.
We asked experts from the pharmaceutical biotech area to present their integrated viewto answer questions focussing on industrial needs in the discovery and manufacture ofrecombinant drugs and new therapies. We are glad that a majority of contributors,active in the pharmaceutical industry, have participated and shared their viewson new developments in protein production, production organisms, DNA vaccines,bioinformatics, and legal aspects. Distinct problems related to recombinant proteins that
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

vi Preface
have arisen in recent years, such as drug stability, pharmacokinetics, and metabolization,are discussed in detail. It should be mentioned that for the first time the topic of genericrecombinant drugs is presented in this textbook.
Biotechnology is a fast-moving area and crucial topics for future technologies can berecognized today. We wanted to give an insight into these future enterprise technologiesand had asked for contributions to highlight new developments in gene therapy, tissueengineering, personalized medicine, and xenotransplantation having a realistic chanceof being used in industrial applications.
In this textbook, you will find updated facts and figures about the biotech industry,product approvals, and discussions of how biotechnology is applied in human andanimal health care, and in industrial and environmental processes. We address howbiotech is being employed in national security efforts as well as the ethical issues thatare frequently debated when people discuss the use of biotechnology in health sciences.
We would like to thank all contributors for their contributions, because we knowthat time was short and most of the papers were written alongside their regular duties.Special thanks to Dr. Andrea Pillmann, Wiley VCH, for her support in the layout,proofreading, and production of this textbook.
We are convinced that this textbook is filling a niche and covering industrial needsand interests in the pharmaceutical biotech area. Our point of view is that this textbookwill cater to scientists and decision makers in pharmaceutical and biotechnologicalcompanies, venture capitals/finance, and politics.
Berlin, December 2003O. KayserR.H. Mullers

vii
Foreword
Pharmaceutical Biotechnology is a multidisciplinary scientific field undergoing anexplosive development. Advances in the understanding of molecular principles andthe existence of many regulatory proteins have established biotechnological ortherapeutic proteins as promising drugs in medicine and pharmacy. More recentdevelopments in biomedical research highlight the potential of nucleic acids ingene therapy and antisense RNAi technology that may become a medical reality inthe future.
The book attempts to provide a balanced view of the biotechnological industry,and the number of experts from the industry sharing their knowledge andexperience with the readers gives the book an outstanding value. All contributorsprovide with each chapter an up-to-date review on key topics in pharmaceuticalbiotechnology. Section 1 serves as an introduction to basics in protein productionand manufacturing. Particular emphasis not only on production organisms likemicroorganisms and plants but also on industrial bioprocessing will be appreciatedby the reader.
The advent and development of recombinant proteins and vaccines is describedin detail in Part 2. Biotech drugs have created a number of unique problemsbecause of their mostly protein nature. The production, downstream processing,and characterization is in many aspects different from conventional low molecularweight drugs and is highlighted by selected experts still in touch with the labbench. Bringing the therapeutic protein to the patient is a major challenge. Proteinformulation, biopharmaceutical aspects, and drug regulation are fields that are fastdeveloping and well recognized by their new and innovative techniques. Drugregulation has a major impact on the whole drug manufacturing process, which iswhy special chapters on the drug approval process in Europe and the United States,and biogenerics are of high interest. Finally, in Part 4, experts provide an outlookon potential drugs and therapeutic strategies like xenotransplantation that are underinvestigation. Hopefully, some of these concepts will find clinical application in thefollowing years.
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

viii Foreword
I believe that there is a distinct need for a pharmaceutical biotech book focusing onthe industrial needs of recombinant drugs and providing detailed insight into industrialprocesses and clinical use. Therefore, this work is not only a valuable tool for theindustrial expert but also for all pharmacists and scientists from related areas who wishto work with biotech drugs. In life-learning courses and the professional environment,this compact book is the basis for a solid understanding for those who wish to gain abetter overview of the industry they are working in.
Robert LangerMIT Boston, November 2003

ix
Contents
List of Contributors ix
Color Plates xv
Part I. Introduction to Concepts and Technologies in PharmaceuticalBiotechnology 1
1 A Primer on Pharmaceutical Biotechnology and Industrial Applications 3Oliver Kayser, Rainer H. M
..uller
2 Procaryotic and Eucaryotic Cells in Biotech Production 9Stefan Pelzer, Dirk Hoffmeister, Irmgard Merfort, Andreas Bechthold
3 Biopharmaceuticals Expressed in Plants 35J..org Kn
..ablein
Part II. Industrial Development and Production Process 57
4 Scientific, Technical and Economic Aspects of Vaccine Research andDevelopment 59Jens-Peter Gregersen
5 DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 79Jeffrey Ulmer, John Donnelly, Jens-Peter Gregersen
6 Characterization and Bioanalytical Aspects of Recombinant Proteins asPharmaceutical Drugs 103Jutta Haunschild, Titus Kretzschmar
7 Biogeneric Drugs 119Walter Hinderer
Part III. Therapeutic Proteins – Special Pharmaceutical Aspects 145
8 Pharmacokinetics and Pharmacodynamics of Biotech Drugs 147Bernd Meibohm, Hartmut Derendorf
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

x Contents
9 Formulation of Biotech Products 173Ralph Lipp, Erno Pungor
10 Patents in the Pharmaceutical Biotechnology Industry: Legal and EthicalIssues 187David B. Resnik
11 Drug Approval in the European Union and the United States 201Gary Walsh
Part IV. Biotech 21 – Into the Next Decade 211
12 Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer213Antonio J. Grillo-Lopez
13 Somatic Gene Therapy – Advanced Biotechnology Products in ClinicalDevelopment 231Matthias Schweizer, Egbert Flory, Carsten Muenk, Klaus Cichutek,Uwe Gottschalk
14 Nonviral Gene Transfer Systems in Somatic Gene Therapy 249Oliver Kayser, Albrecht F. Kiderlen
15 Xenotransplanation in Pharmaceutical Biotechnology 265Gregory J. Brunn, Jeffrey L. Platt
16 Sculpturing the Architecture of Mineralized Tissues: Tissue Engineering ofBone from Soluble Signals to Smart Biomimetic Matrices 281Ugo Ripamonti, Lentsha Nathaniel Ramoshebi, Janet Patton, June Teare, ThatoMatsaba, Louise Renton
Index 299

xi
List of Contributors
Dr. Albrecht F. KiderlenRobert Koch-InstitutNordufer 2013353 BerlinGermany
Prof. Dr. Andreas BechtholdAlbert-Ludwigs-Universit
..at Freiburg
Pharmazeutische BiologieStefan-Meier-Straße 1979104 FreiburgGermany
Dr. Antonio J. Grillo-LopezNeoplastic and Autoimmune DiseasesResearch InstituteP. O. Box 3797Rancho Santa Fe, CA 92067USA
Prof. Dr. Bernd MeibohmDepartment of Pharmaceutical SciencesCollege of Pharmacy, University ofTennessee, Health Science CenterMemphis, TN 38163USA
Prof. Dr. David B. ResnikThe Brody School of MedicineEast Carolina UniversityGreenville, NC 27858USA
Prof. Dr. Dirk HoffmeisterThe University of WisconsinSchool of Pharmacy777 Highland AvenueMadison, WI 53705USA
Dr. Erno PungorBerlex Biosciences2600 Hilltop DriveRichmond, CA 94804USA
Dr. Gary WalshIndustrial Biochemistry ProgramUniversity of LimerickLimerick CityIreland
Prof. Dr. Gregory J. BrunnTransplantation Biology and the Depart-ments of Pharmacology and ExperimentalTherapeuticsMayo ClinicRochester, MI 55905USA
Prof. Dr. Hartmut DerendorfDepartment of Pharmaceutics, College ofPharmacyUniversity of FloridaGainesville, FL 32610USA
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

xii List of Contributors
Prof. Dr. Irmgard MerfortAlbert-Ludwigs-Universit
..at Freiburg
Pharmazeutische BiologieStefan-Meier-Straße 1979104 FreiburgGermany
Dr. Janet PattonBone Research UnitMedical Research Council/University of the Witwatersrand7 York RoadParktown 2193 JohannesburgSouth Africa
Prof. Dr. Jeffrey L. PlattTransplantation Biology and the Depart-ments of Pharmacology and ExperimentalSurgery, Immunology and PediatricsMayo ClinicRochester, MI 55905USA
Dr. Jeffrey UlmerChiron Corporation4560 Horton StreetEmeryville, CA 94608-2916USA
Dr. Jens-Peter GregersenChiron-Behring GmbHPostfach 163035006 MarburgGermany
Dr. John DonnellyChiron Corporation4560 Horton StreetEmeryville, CA 94608-2916USA
Dr. J..org Kn
..ablein
Schering AGAnalytical Development BiologicalsM
..ullerstraße 178
13342 BerlinGermany
June TeareBone Research UnitMedical Research Council/University of the Witwatersrand7 York RoadParktown 2193 JohannesburgSouth Africa
Dr. Jutta HaunschildMorphoSys AGLena-Christ-Strasse 4882152 MartinsriedGermany
Prof. Dr. Klaus CichutekPaul-Ehrlich-InstitutPaul-Ehrlich-Straße 51–5963225 LangenGermany
Dr. Lentsha Nathaniel RamoshebiBone Research UnitMedical Research Council/University of the Witwatersrand7 York RoadParktown 2193 JohannesburgSouth Africa
Louise RentonBone Research UnitMedical Research Council/University of the Witwatersrand7 York RoadParktown 2193 JohannesburgSouth Africa

List of Contributors xiii
Priv. Doz. Dr. Oliver KayserFreie Universit
..at Berlin
Institut f..ur Pharmazie
Pharmazeutische TechnologieBiopharmazie & BiotechnologieKelchstraße 3112169 BerlinGermany
Prof. Dr. Rainer H. M..uller
Freie Universit..at Berlin
Institut f..ur Pharmazie
Pharmazeutische TechnologieBiopharmazie & BiotechnologieKelchstraße 3112169 BerlinGermany
Priv. Doz. Dr. Ralf LippSchering AGM
..ullerstraße 178
13342 BerlinGermany
Dr. Stefan PelzerCombinature Biopharm AGRobert-R
..ossle-Straße 10
13125 BerlinGermany
Thato MatsabaBone Research UnitMedical Research Council/University of the Witwatersrand7 York RoadParktown 2193 JohannesburgSouth Africa
Dr. Titus KretzschmarMorphoSys AGLena-Christ-Strasse 4882152 MartinsriedGermany
Dr. Udo GottschalkBayer AGGB Pharma-BiotechnologieFriedrich-Ebert-Straße 21742096 WuppertalGermany
Dr. Ugo RipamontiBone Research UnitMedical Research Council/University of the Witwatersrand7 York RoadParktown 2193 JohannesburgSouth Africa
Dr. Walter HindererBioGeneriX AGJanderstraße 368199 MannheimGermany

Part IIntroduction to Concepts andTechnologies in PharmaceuticalBiotechnology
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

3
1A Primer on PharmaceuticalBiotechnology and IndustrialApplications
Oliver Kayser and Raimer H. M..uller
Freie Universit..at Berlin, Berlin, Germany
1.1Introduction
Today we can mark historic milestonesand achievements in the pharmaceuticalindustry (Table 1). The year 2003 repre-sents the 50th anniversary of the discoveryof the double helix structure of DNA andit also marks the 30th anniversary of thediscovery of the technique for creating re-combinant DNA by Stanley Cohen andHerbert Boyer. This technique still influ-ences modern medicine and the develop-ment of new recombinant and therapeuticproteins today. Also, 50 years after Watsonand Crick’s discovery, the completion ofsequencing of the human genome is an-other milestone in biotechnology leadingto new genomic-based drugs [1].
In fact, pharmaceutical biotechnology isone of the key industries today. Recombi-nant DNA technologies have entered drugdiscovery and all fields in the developmentand manufacture of therapeutic proteinsand nucleotides. Biotechnology has a ma-jor impact on pharmaceutical industrybecause recent advances in recombinantprotein chemistry, vaccine production, anddiagnostics have and will revolutionize the
treatment paradigms for many seriousand unmet diseases. Currently, approxi-mately 150 approved therapeutic proteinsand vaccines are available. Recently, thefirst oligonucleotide for the treatmentof cytomegalovirus (CMV) infection ofthe eyes was approved by the Food andDrug Adiministration (FDA). The drugFomivirsen (Vitravene) is an antisenseoligonucleotide and represents a newbiotechnological group of compounds withnew, promising therapeutic purposes [2].
1.2Actual Status of Biotechnology and itsApplications in Pharmaceutical Industry
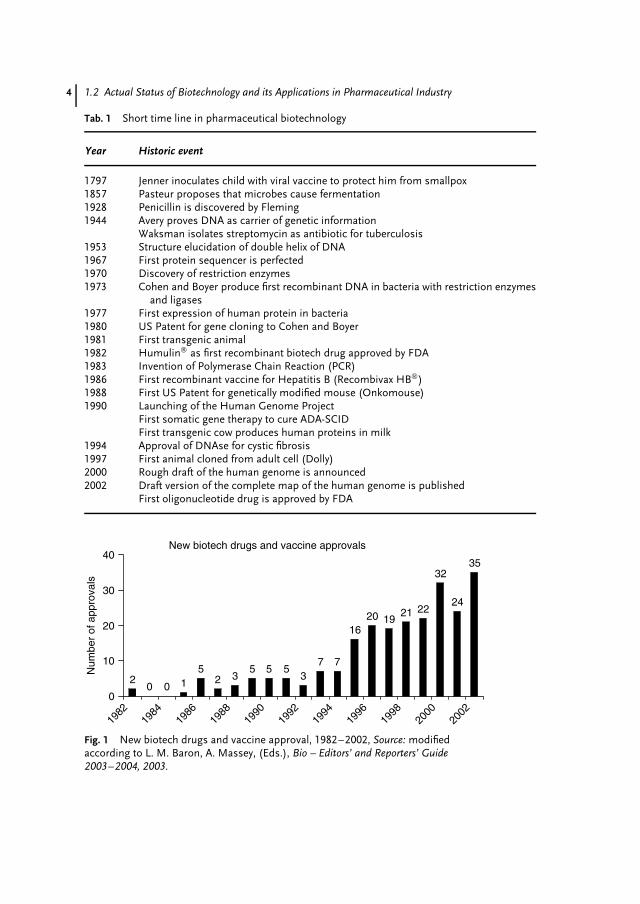
As mentioned earlier, more than 150approved biotech drugs or vaccines areon the market and 70% were approvedin the last six years (Fig. 1). A recentsurvey by the Pharmaceutical Researchand Manufacturers of America (PhRMA)found 369 drugs in the pipeline meet-ing the criteria as biotechnological drugsand medicines. These drugs target 200potential diseases [3] and provide new ther-apies for autoimmune diseases, asthma,Alzheimer, multiple sclerosis, and cancer,
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8
4 1.2 Actual Status of Biotechnology and its Applications in Pharmaceutical Industry
Tab. 1 Short time line in pharmaceutical biotechnology
Year Historic event
1797 Jenner inoculates child with viral vaccine to protect him from smallpox1857 Pasteur proposes that microbes cause fermentation1928 Penicillin is discovered by Fleming1944 Avery proves DNA as carrier of genetic information
Waksman isolates streptomycin as antibiotic for tuberculosis1953 Structure elucidation of double helix of DNA1967 First protein sequencer is perfected1970 Discovery of restriction enzymes1973 Cohen and Boyer produce first recombinant DNA in bacteria with restriction enzymes
and ligases1977 First expression of human protein in bacteria1980 US Patent for gene cloning to Cohen and Boyer1981 First transgenic animal1982 Humulin as first recombinant biotech drug approved by FDA1983 Invention of Polymerase Chain Reaction (PCR)1986 First recombinant vaccine for Hepatitis B (Recombivax HB)1988 First US Patent for genetically modified mouse (Onkomouse)1990 Launching of the Human Genome Project
First somatic gene therapy to cure ADA-SCIDFirst transgenic cow produces human proteins in milk
1994 Approval of DNAse for cystic fibrosis1997 First animal cloned from adult cell (Dolly)2000 Rough draft of the human genome is announced2002 Draft version of the complete map of the human genome is published
First oligonucleotide drug is approved by FDA
New biotech drugs and vaccine approvals
20 0 1
52 3
5 5 53
7 7
1620 19
21 22
32
24
35
0
10
20
30
40
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
2002
Num
ber
of a
ppro
vals
Fig. 1 New biotech drugs and vaccine approval, 1982–2002, Source: modifiedaccording to L. M. Baron, A. Massey, (Eds.), Bio – Editors’ and Reporters’ Guide2003–2004, 2003.

A Primer on Pharmaceutical Biotechnology and Industrial Applications 5
including immunization and different in-fectious diseases (AIDS, Malaria) [3, 4].Biotechnology-produced pharmaceuticalscurrently account for 5% of the worldwidepharmaceutical market and are expected toreach approximately 15% by the year 2050.At the same time, the explosive growthof genetic diagnostic techniques will al-low personalized genetic profiling of eachindividual in one hour and for less thanUS$100.
Not only drugs but also new medi-cal diagnostic tests will be produced anddistributed by pharmaceutical biotech in-dustry. Hundreds of tests will be availableto increase the safety of blood products.Also, costs for clinical analysis will bereduced. One example is the testing ofLow Density Lipoproteins (LDL), choles-terol, and other parameters in one testdesign. In comparison to conventionaltests, cholesterol, total triglycerides, andLDL were determined separately at highcosts. In the future, biotechnology-derivedtests will be more accurate and quickerthan previous tests and will allow ear-lier diagnosis of the disease. Proteomicsmay increase sensitivity and may discovertoday unknown molecular markers that in-dicate incipient diseases before symptomsappear, helping to prevent diseases andconduct therapies much earlier [5–7].
Xenotransplantation from transgenicanimals is a future field in pharmaceuticalindustry. In general, organ transplantationis an effective and cost-efficient treatmentfor severe and life-threatening diseasesof organs, mostly heart, liver, and kid-ney. In Europe, there are 35 000, andin the United States, there are 60 000people on organ-recipient lists. Organtransplantation costs vary from ¤60 to120 000 and require a lifelong drug therapywith immunosuppressive drugs to avoidtransplant rejection. Genetically modified
organs and cells from other organisms likepigs – called as xenotransplantation – arepromising sources of donor organs thatcan be used to overcome the lack of a suf-ficient number of human organs. But, thespread of infectious pathogens by trans-plantation of nonhuman organs and theinduction of oncogenes is a potential riskand needs close attention [8, 9].
Tissue engineering, in relation to xeno-transplantation, is another attractive fieldin pharmaceutical biotechnology. Tissueengineering combines advances in cell bi-ology and biomaterial science. Tissues con-sist of scaffolding material (e.g. collagen,biodegradable polymers), which eventuallydegrades after forming organs or cell im-plants. Skin tissues and cartilages werethe first tissues successfully engineeredand tested in vivo; recently, biohybrid sys-tems to maintain patients’ liver or kidneyfunction were also successfully tested [10].
Stem cells are considered today as a newavenue in therapy to cure most deadlyand debilitating diseases such as Parkin-son, Alzheimer, leukemia, and geneticdisorders like adenosine deaminase (ADA)deficiency and cystic fibrosis (CF). The po-tential of embryonic and adult stem cellsare intensively discussed, but no majorbreakthrough can be expected in the next10 years to turn these cells and techniquesinto industrial applications. It should alsobe clear that therapeutic cloning, whichis related to stem cell research, will bringethical questions [11]. Discussions of eth-ical and social implications are importanttoday to convince the public of potentialbenefits and to explain the future risksof applied techniques. Significant imped-iments of diagnostics and therapeuticsexist, and deep concerns must be respectedbefore any genetic therapy like somaticgene therapy, stem cell, or cloning willever be accepted.

6 1.3 What is the Impact of Biotechnology and Genomics on the Drug Development Process?
1.3What is the Impact of Biotechnology andGenomics on the Drug DevelopmentProcess?
The most frequent trends for the phar-maceutical industry in biotechnology aresurely new technologies and innovations,especially genomics, and also influencesof government regulations, health carelegislation affecting own product pricing,changes in demographics, and an ag-ing population [4]. With special emphasis,more topics with minor influence relateto patent protection, e-business, multina-tional scope of industry, requirements fornew drugs, and changes in informationtechnology to address some of them [3]. Inthe first decade of this century, the biotechindustry is likely to show even more ex-plosive growth as progress in computermodeling, automated lab techniques, andknowledge of human genes and proteinscontinues. As a result, more life-savingtherapies will reach the people who mostneed them. Today, pharma industry seemsto be in good shape to work on thesechallenges, as indicated by some impres-sive statistics that emphasize the industry’sgrowth [12]:
• During the 1980s, the biotech industryturned out 18 new drugs and vaccines.By comparison, 33 biotech medicineswere approved in both 1998 and 1999,and 25 more were approved in the firsthalf of 2000.
• Most of the 1998–1999 approvals werefor new products, though a few were forexpanding the application of drugs orvaccines to more diseases.
• The number of patents granted tobiotech companies has tripled fromnearly 3000 per year in the early 1990sto more than 9000 in 1998.
• After a decade of slow, steady growth,biotechnology patent awards began asteep ascent in 1995, when nearly 4000patents were granted. Since then, thenumber of patents has skyrocketed at arate of 25% or more each year.
• Pharmaceutical companies, which tra-ditionally have focused on chemicalapproaches to treating disease, have be-come increasingly supportive of biotechR&D – in their own labs, in partnershipswith biotech firms, and through acqui-sitions of biotech firms. Alliances in thebiotech industry doubled to nearly 250between 1998 and 2000.
• Between 1998 and 1999, industry-widesales and revenues increased by 13%to $16.1 billion and $22.3 billion respec-tively.
For the future, the biotechnology andgenomic way is technology-driven andformed by the integration of high-throughput technologies, genomics, andbioinformatics. Even the genetics waveis data-driven and is an applied new lifescience field to identify genes that makeindividuals as their carriers susceptible toparticular diseases and allows personal-ized medicines based on pharmacogeneticfacts. So, what is the impact of pharmaceu-tical biotechnology and genomics on theeconomics of R&D?
1.3.1Reducing Costs in R&D
Before biotechnology had been intensivelyintroduced to industrial research, develop-ing costs of each drug had cost companieson average US$880 million and had taken15 years from start to market authoriza-tion. About 75% of these costs werespent on failures. Using genomic tech-nologies, there is a realistic chance of

A Primer on Pharmaceutical Biotechnology and Industrial Applications 7
reducing companies’ costs to US$500 mil-lion, largely as a result of efficacy gains.Significant savings not only of money butalso of time by 15% are possible [12, 13].
1.3.2Increase in Productivity
From trial-and-error approaches and com-plex biochemical in vitro assays, biotech-nology allows industrialized target de-tection and validation. By the use ofmicro array technologies and bioinformat-ics, thousands of genes in diseased andhealthy tissues will be analyzed by a singleDNA chip. By the use of bioinformatics,results from different assays can be ana-lyzed and linked to an integrated follow-upof information in databases. In total, thepotential savings per drug by intelligentinformation retrieval systems and geneticanalytics are estimated at about US$140million per drug and less than one yearof time to market. The Boston ConsultingGroup (BCG) calculated a sixfold increasein productivity at the same level of invest-ment [12].
1.3.3Accelerating the Drug DevelopmentProcess
There is not only an effect on the preclini-cal development of a drug by biotechnologyand genomics but pharmaceutical biotech-nology will also help predict drug prop-erties and pharmacokinetic parameters(ADMA/tox) to accelerate the industrialdrug development process. Companieswill be in the position to pull certainpreclinical activities into the chemistryand drug validation part of the valuechain. Potential savings are in the or-der of US$20 million and 0.3 years perdrug [12].
1.3.4Maintaining High Standards in QualityAssurance
Biotechnological drugs have the same highstandard in quality and safety as con-ventional drugs. Of high interest is thequestion of costs of quality control forrecombinant drugs. Boston ConsultingGroup expects an increase of US$200 mil-lion and more than one year per drug [12].The main reason for this is explainedby the extra time needed for unknownchemical and physical properties of re-combinant proteins and oligonucleotides.Another time- and cost-consuming aspectis the importance of developing new drug-specific appropriate test assays for drugvalidation, standardization, activity deter-mination (e.g. biological units), toxicity,and bioanalytical methods.
1.4Future Outlook
Integrating biotechnology and genomicsin the whole drug development processgives companies the opportunity to saveup to US$300 million per drug – aboutone-third of the costs today – and theprospect of bringing the drug two yearsearlier on the market [3]. Each day lostbefore market entry will lead to a lossof US$1.5 million per day, indicatingthe value of recombinant drugs and theneed for making manufacturing processesoperational and effective.
Any predictions for the near futureare challenging. Future reports estimatea significant increase of recombinantdrugs replacing up to 30% of commer-cially used low-molecular drugs up to2015. For the production of recombi-nant biotech drugs, bioprocessing in all

8 1.4 Future Outlook
reactor sizes will be routinely used [14].From 2010, genetically modified plantsand animals – transgenic organisms – willalso be routinely used to produce recom-binant drugs (Gene Pharming). Somaticgene therapy and the introduction ofnanorobotic devices may be expected inthe time period between 2010 and 2018to end up with individual genome pro-filing for ¤100 in 2050. Personalizedmedicine and diagnostics on a biochipmay also find industrial interest in thenext 10 years. Interestingly, creation ofartificial life or complex biochemical net-works is expected to be unrealistic in thenext 25 years.
References
1. J. A. Miller, V. Nagarajan, Trend Biotechnol.2000, 18, 109–191.
2. J. Kurrek, Eur. J. Biochem. 2003, 270,1628–1644.
3. A. M. Baron, A. Massey Editors’ andReporters’ Guide to Biotechnology, URL: http://www.bio.org (24.07.2003), 2001.
4. J. M. Reichert, New biopharmaceuticals in theUSA: trends in development and marketingapprovals 1995–1999, Trends in Biotechnology2000, 18, 364–369.
5. K. K. Jain, Nanodiagnostics: application ofnanotechnology in molecular diagnostics ExpertRev Mol Diagn 2003, 3, 153–161.
6. K. K. Jain, Proteomics: delivering new routesto drug discovery – Part 1. Drug Discov. Today2001 Aug 1; 6(15): 772–774.
7. K. K. Jain, Proteomics: delivering new routesto drug discovery – Part 2. Drug Discov. Today2001 Aug 15; 6(16): 829–832.
8. D. K. Langat, J. M. Mwenda, Acta Tropica2000, 76, 147–158.
9. F. H. Bach, Annu. Rev. Med. 1998, 49,301–310.
10. C. K. Colton, Cell Transplantation 1995, 4,415–436.
11. R. Eisenberg, Nat. Genet. 2000, 1, 70–74.12. P. Tollman, P. Guy, J. Altshuler, N. Vrettos,
C. Wheeler, A Revolution in R&D – TheImpact of Genomics, The Boston ConsultingGroup, Boston, 2001.
13. Anonymous, Convergence – The BiotechnologyIndustry Report, Ernst & Young, Score RetrievalFiles, URL: www.ey.com/industry/health(24.07.2003), 2000.
14. Anonymous, Endurance – The European Bio-technology Report 2003, URL: www.ey.com/industry/health (24.07.2003), 2000.

9
2Procaryotic and Eucaryotic Cellsin Biotech Production
Stefan PelzerCombinature Biopharm AG, Berlin, Germany
Dirk HoffmeisterThe University of Wisconsin, Madison, WI, USA
Irmgard Merfort and Andreas BechtholdAlbert-Ludwigs-Universit
..at Freiburg, Freiburg, Germany
2.1Introduction
The production of compounds used inthe food and pharmaceutical industriesby biotech processes is both an old anda very young business. Over the past 70years, fermentation of microorganismsor the use of yeast and plants in theproduction of important pharmaceuticalshas been well established. The promisesof genomics in drug discovery and drugproduction, which were eagerly embracedin the mid-1990s, have now been fulfilledin many areas. A systematic integrationof technologies results in a superioroutput of data and information, andthereby enhances our understanding ofbiological function – drug discovery anddevelopment is hence facing a new age.
Bacterial strains, especially Actino-mycetes have been used in biotechproduction and drug discovery for years.
Genetic methods now open the fieldof combinatorial biosynthesis that hasimproved impressingly in the past cou-ple of years. Also, the productivity of yeastand other fungi in a variety of differentprocesses has improved significantly sincegenetic methods have been introduced.In addition, a number of recent worksconsiderably widens the potential of plantbiotechnology. This review covers exam-ples describing the use of procaryotic cellsand plant cells in biotech production. Theuse of other eucaryotic cells, especially ofanimal origin, is reviewed in other chap-ters of this book.
2.2Actinomycetes in Biotech Production
Soil bacteria of the order Actinomycetesare the most important producers of phar-maceutically relevant bioactive metabolites
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

10 2.2 Actinomycetes in Biotech Production
including antibiotics, antitumor agents,immunosuppressants, antiparasitic ag-ents, herbicides, and enzyme-inhibitingagents. The success story of these bac-teria began about 60 years ago with thegroundbreaking work of Waksman, whodiscovered and described streptomycinas the first antibiotic synthesized by anActinomycete [1]. Ever since, systematiclarge-scale screens performed by the phar-maceutical industry have revealed numer-ous therapeutically relevant drugs. Morethan two-thirds of all naturally derived an-tibiotics currently used are produced byActinomycetes strains, underlining theirimportance to medicine [2].
2.2.1Actinomycetes: Producer of CommerciallyImportant Drugs
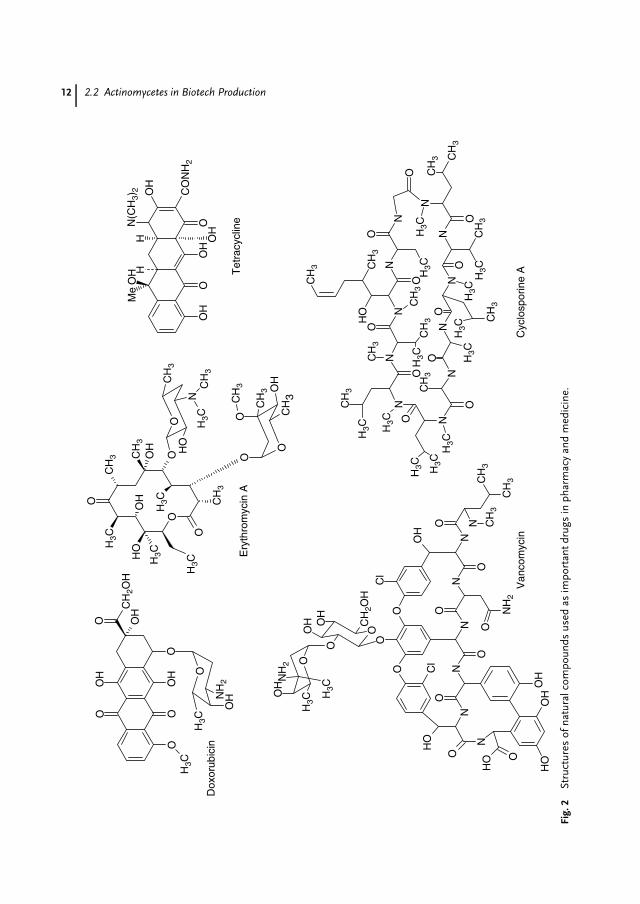
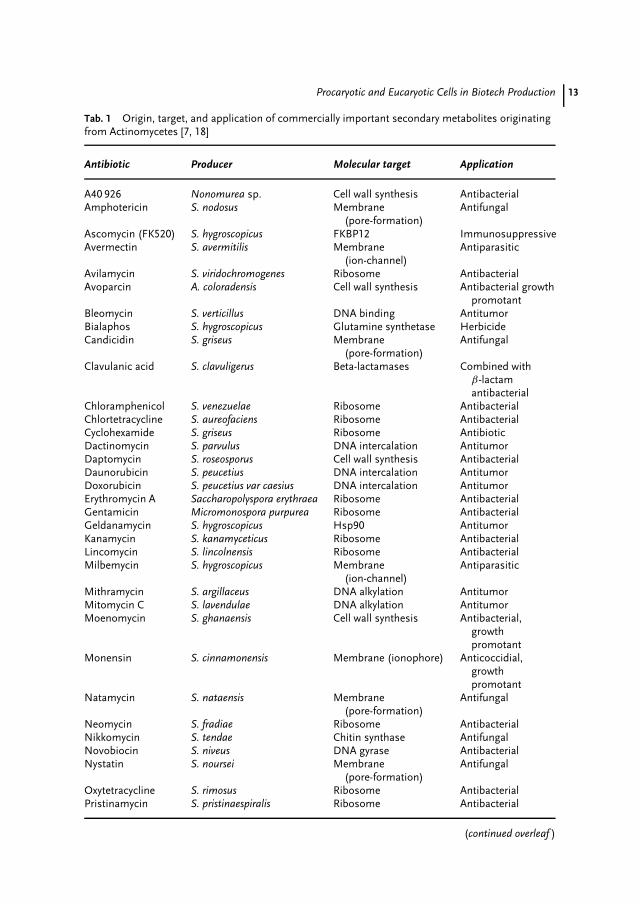
Natural products (‘‘secondary metabo-lites’’) have been the largest contributorsto drugs in the history of medicine. Beforeantibiotics were introduced in the 1940sand 1950s (see above), patients with bac-teraemia faced low survival chances [3],and the mortality from tuberculosis was50% [4]. It has been stated that the dou-bling of our life span in the twentiethcentury is mainly due to the use ofplant and microbial secondary metabo-lites [5]. Of the 520 new drugs approvedbetween 1983 and 1994, 39% were naturalproducts or those derived from naturalproducts and 60 to 80% of antibacte-rial and anticancer drugs were derivedfrom natural products [6]. Almost half ofthe best-selling pharmaceuticals are nat-ural or related to them [7,8]. In 2001,over 100 natural product–derived com-pounds were in clinical development [9].Natural products and their derivativesaccount for annual revenues of aboutUS$30 billion in the antiinfectives market,
US$20 billion in the anticancer market,and US$14 billion in the lipid-loweringmarket [10]. Actinomycetes and, particu-larly, Streptomycetes (Fig. 1) are the largestantibiotic-producing genus in the mi-crobial world discovered so far. Of the12 000 or so antibiotics known in 1995,55% were produced by Streptomycetesand an additional 11% by other Acti-nomycetes [11]. A compilation of numer-ous bioactive and commercially importantmetabolites, which are all synthesizedby Actinomycetes strains, is shown inTable 1. This list includes not only veryimportant drugs such as the macrolideerythromycin A synthesized by Saccha-ropolyspora (Sac.) erythraea (in 2000, theannual sales of semisynthetic derivativesreached US$2.6 billion [12]), the glycopep-tide vancomycin synthesized by Amy-colatopsis (A.) orientalis (in 2000, theannual sales of glycopeptides reachedUS$424 million, [12]) and tetracycline syn-thesized by Streptomyces (S.) aureofa-ciens (in 2000, the annual sales reachedUS$217 million [12]) but also anticanceragents like doxorubicin synthesized by S.peucetius (Fig. 2). Many compounds pro-duced by Actinomycetes belong to thelarge family of polyketides. Polyketides arestructurally diverse (Fig. 2) and exhibit awide scope of bioactivities. More than 500aromatic polyketides have been character-ized from Actinomycetes [13]. Polyketidesare particularly important for drug discov-ery, since statistics show that 1 out of 100polyketides will make its way to commer-cialization. With an average of as low as 1out of 5000 compounds, other substancesare far less likely to hit the market [14].Sales of drugs based on polyketides ex-ceed US$15 billion a year [14]. In general,for industrial production, overproducingstrains have to be developed. Today, mod-ern processes allow the production of
Procaryotic and Eucaryotic Cells in Biotech Production 11
Fig. 1 Photography of a sporulated Streptomyces strain growing on solid medium.The blue drops indicate the production of an antibiotic (aromatic polyketide). (SeeColor Plate p. xv).
compounds at concentrations even higherthan 10 g L−1 [15–17].
2.2.2Actinomycetes Genetics: The Basis forUnderstanding Antibiotic Biosynthesis
Streptomyces coelicolor A3(2) is the ge-netically best characterized strain amongthe filamentous Actinomycetes [19]. Acti-nomycetes genetics has been the sub-ject of research since 1958 when Prof.Dr. Sir D. Hopwood published the firstlinkage map of S. coelicolor, performing thefirst genetic recombination experimentsconsidering six marker genes [20]. After-wards, Actinomycetes genetics developedcontinuously, by the identification of mu-tants interrupted in the biosynthesis ofactinorhodin [21]. After identification and
isolation of easily selectable antibiotic re-sistance genes [22], the first gene cloningin Streptomyces was described in 1980 [23].In 1984, Malpartida and Hopwood demon-strated for the first time that antibioticbiosynthesis genes are usually organizedas a gene cluster of structural, regula-tory, export, and self-resistant genes [24].Hence, once a single gene within a clus-ter has been located, the others may beidentified quickly by chromosomal walk-ing. In the course of the last two decades,many molecular tools including vector sys-tems (phage and plasmid-based) have beendeveloped along with DNA transfer andgene inactivation techniques, which wereall necessary for targeted manipulation ofActinomycetes [2, 25]. The excellent man-ual published by the John Innes Institutesummarizes all the necessary information
12 2.2 Actinomycetes in Biotech Production
OC
H3
NH
OO
O
CH
3
O
H3C
HO
H3C
H3C
H3C
O
CH
3
CH
3
CH
3
CH
3
H3C
OH
CH
3O
H
OC
H3
OH
OO
OH
OO
HO
HO
CO
NH
2
OH
N(C
H3)
2H
Me
OH H
OOH O
H
CH
2OH
O
OH
O
NH
2
N
OO
Cl
Cl
O
NN
NN
N
O
O
OO N C
H3
CH
3CH
3O
NH
2
O
HO
HO O
HO
OH
OH
O
OH
NN
NN
N
N
NN
N
CH
3
CH
3
CH
3C
H3
CH
3
CH
3
O
O
CH
3
OH
O
O
CH
3
CH
3
O
NN
CH
3
O
CH
3
OO
O
OO
O
O
O O
OH
OH
OH
OH
3C
O
CH
2OH
OH
H3C
H3C H
3C
H3C H
3C
H3C
H3C
H3C
H3C
H3C
H3C
H3C
H3C H3C
H3C
NH
2
Dox
orub
icin
Ery
thro
myc
in A
Tet
racy
clin
e
Van
com
ycin
Cyc
losp
orin
e A
Fig.
2St
ruct
ures
ofna
tura
lcom
poun
dsus
edas
impo
rtan
tdru
gsin
phar
mac
yan
dm
edic
ine.
Procaryotic and Eucaryotic Cells in Biotech Production 13
Tab. 1 Origin, target, and application of commercially important secondary metabolites originatingfrom Actinomycetes [7, 18]
Antibiotic Producer Molecular target Application
A40 926 Nonomurea sp. Cell wall synthesis AntibacterialAmphotericin S. nodosus Membrane
(pore-formation)Antifungal
Ascomycin (FK520) S. hygroscopicus FKBP12 ImmunosuppressiveAvermectin S. avermitilis Membrane
(ion-channel)Antiparasitic
Avilamycin S. viridochromogenes Ribosome AntibacterialAvoparcin A. coloradensis Cell wall synthesis Antibacterial growth
promotantBleomycin S. verticillus DNA binding AntitumorBialaphos S. hygroscopicus Glutamine synthetase HerbicideCandicidin S. griseus Membrane
(pore-formation)Antifungal
Clavulanic acid S. clavuligerus Beta-lactamases Combined withβ-lactamantibacterial
Chloramphenicol S. venezuelae Ribosome AntibacterialChlortetracycline S. aureofaciens Ribosome AntibacterialCyclohexamide S. griseus Ribosome AntibioticDactinomycin S. parvulus DNA intercalation AntitumorDaptomycin S. roseosporus Cell wall synthesis AntibacterialDaunorubicin S. peucetius DNA intercalation AntitumorDoxorubicin S. peucetius var caesius DNA intercalation AntitumorErythromycin A Saccharopolyspora erythraea Ribosome AntibacterialGentamicin Micromonospora purpurea Ribosome AntibacterialGeldanamycin S. hygroscopicus Hsp90 AntitumorKanamycin S. kanamyceticus Ribosome AntibacterialLincomycin S. lincolnensis Ribosome AntibacterialMilbemycin S. hygroscopicus Membrane
(ion-channel)Antiparasitic
Mithramycin S. argillaceus DNA alkylation AntitumorMitomycin C S. lavendulae DNA alkylation AntitumorMoenomycin S. ghanaensis Cell wall synthesis Antibacterial,
growthpromotant
Monensin S. cinnamonensis Membrane (ionophore) Anticoccidial,growthpromotant
Natamycin S. nataensis Membrane(pore-formation)
Antifungal
Neomycin S. fradiae Ribosome AntibacterialNikkomycin S. tendae Chitin synthase AntifungalNovobiocin S. niveus DNA gyrase AntibacterialNystatin S. noursei Membrane
(pore-formation)Antifungal
Oxytetracycline S. rimosus Ribosome AntibacterialPristinamycin S. pristinaespiralis Ribosome Antibacterial
(continued overleaf )

14 2.2 Actinomycetes in Biotech Production
Tab. 1 (continued)
Antibiotic Producer Molecular target Application
Ramoplanin Actinoplanes spec. Cell wall synthesis AntibacterialRapamycin S. hygroscopicus FKBP ImmunosuppressiveRifamycin A. mediterranei RNA polymerase AntibacterialSalinomycin S. albus Membrane (ionophore) Anticoccidial,
growthpromotant
Spinosyn Sac. spinosa unknown InsecticidalSpiramycin S. ambofaciens Ribosome AntibacterialStaurosporin S. staurosporeus Protein kinase C AntibacterialStreptomycin S. griseus Ribosome AntibacterialTacrolimus (FK506) Streptomyces spec. FKBP ImmunosuppressiveTeicoplanin A. teicomyceticus Cell wall synthesis AntibacterialTetracycline S. aureofaciens Ribosome AntibacterialThienamycin S. cattleya Cell wall synthesis AntibacterialTylosin S. fradiae Ribosome Growth promotantVancomycin A. orientalis Cell wall synthesis AntibacterialVirginiamycin S. virginiae Ribosome Growth promotant
O
OO
O
OOO
OO
O
CH3CH3
OO
OO
OH
CH3
OHCH3
CH3
OH OH
OH
Cl
OHCl
CH3
OCH3
OO
CH3CH3
O
O
OCH3
OH
OO O
CH3
OH
H
Gavibamycin H1
aviG5
aviG2
aviG4
HO–CH2
Fig. 3 Production ofgavibamycin H1 by a mutantwith deletions in threemethyltransferase genes.
related to handling and molecular genetictools essential for working with Actino-mycetes [18]. Several hundred biosyntheticgene clusters have so far been identifiedand genes encoding about 80 pathways for
secondary metabolites have been cloned, atleast partially sequenced and made avail-able to the public [26–28]. The average sizeof an antibiotic biosynthetic gene clusterranges from about 20 kb for a simple

Procaryotic and Eucaryotic Cells in Biotech Production 15
aromatic polyketide like actinorhodin to120 kb for complex polyketides like theantifungal antibiotic nystatin [29].
A highlight in Actinomycetes geneticswas the completion of the first genomesequence of the model Actinomycete Strep-tomyces coelicolor A3(2) in July 2001 [30].Recently, the genome sequence of a sec-ond Streptomyces strain, the avermectinproducer S. avermitilis, has been pub-lished [31,32], opening up new perspec-tives for comparative genomics with Acti-nomycetes. A characteristic feature ofActinomycete chromosomes is their lin-ear structure [33, 34]. The genome of S.coelicolor comprises 8 667 507 bp (G/C con-tent of 72.1%), whereas the S. avermitilisgenome contains 9 025 608 bp (G/C con-tent of 70.7%). Both genomes are denselypacked and harbor 7574 ORFs in S. aver-mitlis and 7825 ORFs in S. coelicolor,respectively [30]. Comparative analysis ofthe S. coelicolor and S. avermitilis chro-mosomes revealed that both the genomeshad an unusual biphasic structure witha core region of 5 Mb and 6.5 Mb, re-spectively [30, 31]. The most interestingfeature in both the completed Streptomycesgenomes that will impact biotechnologyis the abundance of secondary metabolitegene clusters. Before the genome of S.coelicolor was sequenced, three antibioticsand a spore pigment were known to besynthesized from this strain. The genomesequence revealed that 23 gene clusters(about 5% of the total genome) are di-rectly dedicated to secondary metabolismincluding clusters for further putativeantibiotics, pigments, complex lipids, sig-naling molecules and iron-scavengingsiderophores [35]. In S. avermitilis, 30 geneclusters related to secondary metaboliteswere identified, corresponding to 6.6%of the genome. From these clusters, 5
out of 30 are putatively involved in pig-ments and siderophores, 5 in terpenes, 8in nonribosomal peptides and 12 in polyke-tide biosynthesis [32]. With avermectin,oligomycin, and a polyene antibiotic, onlythree complex polyketide clusters havebeen characterized from this strain before.The completed genome-sequence data canalso be used to study the regulatory net-work of primary metabolism pathways andthe cross-talk between primary and sec-ondary metabolism (i.e. the carbon flux).The knowledge gained by these analy-ses will be useful for the constructionof improved strains produced in a ratio-nal approach by deleting undesired path-ways or adding advantageous pathways,generating precursors and essential co-factors. Moreover, targeted modificationswill improve cell growth and fermentationproperties (metabolic engineering) [16,17].Successful metabolic engineering of astrain producing doramectin, a commer-cial antiparasitic avermectin analog, is anexcellent example for the importance ofthis technology [36].
2.2.3Urgent Needs for the Development of NewAntimicrobial Drugs
Stimulated by the discovery of numerousnovel antibacterial agents, which reacheda peak in the 1970s [37], US SurgeonGeneral William Stewart declared in1969 in the US congress that it was‘‘time to close the book on infectiondiseases’’ [38]. Today, unfortunately, weknow that antibiotics have not won thefight against infectious microorganismsand therefore there is a permanent needfor new antibiotics.
One main reason for this development isthe problem of emerging resistant formsof pathogens. As an example, according

16 2.2 Actinomycetes in Biotech Production
to the WHO more than 95% of S.aureus strains worldwide are resistantto penicillin G, and up to 60% areresistant to its derivative methicillin [39].Several reasons (e.g. use of antibioticsas growth promoters, changes in thespectrum of pathogens) are responsiblefor this development [40–43].
The past decades witnessed a major de-crease in the number of newly discoveredcompounds. In an almost 40-year period(1962–2000), no new class of antibioticwas introduced to the market (nalidixicacid in 1962, the oxazolidinone antibioticlinezolid in 2000) [44, 45]. The major rea-son for the decrease in the number ofnewly discovered compounds might bea decline in screening efforts [37]. Ironi-cally, some of the leading pharmaceuticalcompanies are currently cutting back theirantiinfective programs, especially for nat-ural products [46]. They rather focus theiractivities on the semisynthetic modifica-tion of existing antibiotics to producesecond- and third-generation antibioticswith improved properties.
Nevertheless, there is no need to resign.According to biomathematical modeling,only 3% of all antibacterial agents synthe-sized in Streptomyces have been reportedso far [37]. Additionally, less than 10% ofthe world’s biodiversity has been testedfor biological activity, and many moreuseful natural lead compounds are yet tobe discovered [47].
2.2.4Strategies for the Identification andDevelopment of New Antimicrobial Drugs
2.2.4.1 Approaches to Explore Nature’sChemical DiversityVicuron Pharmaceuticals Inc., formerlyBiosearch Italia, a company screening fornew antibiotics, focuses its activities on
a proprietary strain collection of 50 000microorganisms, including unusual fila-mentous Actinomycetes and filamentousfungi or strains that are difficult to isolate.The rationale behind this campaign is thatthese organisms have not been intensivelyscreened in the past and that they may beproducers of novel compound classes [48].
Another strategy to reveal the chemi-cal diversity of a single strain is the Onestrain – many Compounds (OSMAC) ap-proach described by Bode et al. [49]. Bysystematic alteration of cultivation para-meters, the number of secondary metabo-lites increased tremendously in a singlestrain. When this method was applied,up to 20 different metabolites with, insome cases, high production titers weredetected. Since recent estimates suggestthat only 0.1 to 1% of the microbialflora in the environment can be keptin culture [50], the ‘‘metagenome’’ of theunculturable microorganisms should alsohave a potential to generate novel sec-ondary metabolites. Indeed, several re-ports demonstrated that it is possible toconstruct DNA libraries from ‘‘soil-DNA’’and to use them for the production of novelmetabolites in a heterologous Streptomyceshost [51, 52].
2.2.4.2 Exploiting the EnormousGenotypic Potential of Actinomycetes by‘‘Genome Mining’’The completion of the sequence of the twoStreptomyces genomes demonstrated thatbetween 5 and 6.6% of the whole genomeare directly involved in the biosynthe-sis of predominantly unknown secondarymetabolites (see above). Prior to genomesequencing, a number of reports were pub-lished in which cryptic or silent secondarymetabolite pathways were identified dur-ing the search for gene clusters for knownmetabolites. Hence, the occurrence of

Procaryotic and Eucaryotic Cells in Biotech Production 17
multiple ‘‘orphan’’ gene clusters has beenreported for various compound classes likenonribosomal peptides [53, 54], PKSI [55,56] and PKSII [57, 58]. Combinature Bio-pharm AG is a Berlin-based companyusing modern high throughput genomicsfor the systematic genetic screening ofseveral Actinomycetes genomes to iden-tify known and ‘‘orphan’’ clusters [27].Recently, Zazopoulos et al. [59] describedhow a genomics-guided approach can berewarding for the discovery and expressionof cryptic metabolic pathways (genomemining). The genetic information of thesebiosynthesis clusters is used for the tar-geted generation and modification of novelcompounds in an approach termed ‘‘com-binatorial biosynthesis.’’
2.2.4.3 Generation of Novel Antibiotics byTargeted Manipulation of the Biosynthesis(Combinatorial Biosynthesis)Researchers have started using biosyn-thetic genes to alter the structure of naturalcompounds by genetic engineering or tocombine genes from different biosyntheticpathways. This new technology named‘‘combinatorial biosynthesis’’ results inthe formation of novel natural products.
New Drugs by Targeted Gene DisruptionInactivation of specific selected genes isa very common methodology for the gen-eration of novel structural variations ofknown natural products. Erythromycin is amacrolide antibiotic that is clinically usefulin the treatment of infections by Gram-positive bacteria. A hydroxyl group at C6 ofthe erythronolide macrolactone is respon-sible for acidic inactivation in the stomachby conversion into anhydroerythromycin.Erythromycin derivatives lacking this hy-droxyl group are therefore interestingfrom the therapeutic and pharmacological
point of view. The gene eryF that en-codes a cytochrome P450 monooxygenaseresponsible for the introduction of this hy-droxyl group into the macrolactone wasinactivated and the mutant produced 6-deoxyerythromycin A. This is a muchmore acid-stable antibiotic and as effi-cient as erythromycin because of its higherstability [60].
The orthosomycins are a prominentclass of antibiotics produced by variousActinomycetes. Members of this classare active against a broad range ofGram-positive pathogenic bacteria. Promi-nent examples of orthosomycins arethe avilamycins and the everninomicinsproduced by S. viridochromogenes Tu57and Micromonospora carbonacea, respec-tively. Avilamycins and everninomicins arepoorly soluble in water, which poses a ma-jor obstacle for their use as therapeutics.The avilamycin biosynthetic gene clusterhas been cloned and sequenced [61]. Sev-eral putative methyltransferase genes havebeen found in the cluster. Double andtriple mutants have been generated bydeleting two or three methyltransferasegenes in the chromosome of the producerstrain (Fig. 3). All mutants produced novelavilamycin derivatives with improved wa-ter solubility.
Improved Yield by Expression of GenesPristinamycin, produced by S. pristinaes-piralis, is a mixture of two types of macro-cyclic lactone peptolides, pristinamycins I(PI), a branched cyclic hexadepsipeptideof the streptogramin B group, and pristi-namycins II (PII), a polyunsaturated cyclicpeptolide of the streptogramin A group.Both the compounds inhibit the growthof bacteria. In combination, they displaya synergistic bactericidal activity. The PIIcomponent of pristinamycin is producedmainly in two forms, called PIIA (80%)

18 2.2 Actinomycetes in Biotech Production
and PIIB (20%). A water-soluble derivateof pristinamycin, now being marketed un-der the trade name Synercid, was obtainedby the chemical modification of PIIA. Togenerate a PIIA-specific producer strain,two genes, snaA and snaB, were isolatedfrom the biosynthetic gene cluster. The en-zymes encoded by snaA and snaB catalyzethe conversion of PIIB to PIIA. Both geneswere placed under the transcription con-trol of a strong promoter and were clonedinto an integrative vector. The integrationof this vector into the chromosome of theproducer strain resulted in the productionof 100% PIIA and this was achieved inhigh concentrations [62].
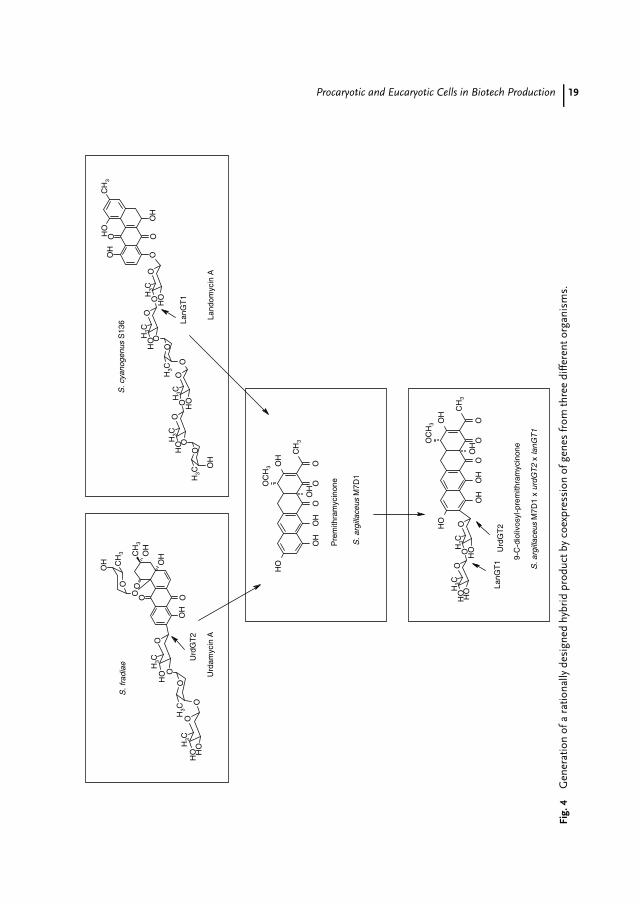
New Drugs by Expression of GenesMithramycin is an aromatic polyketide,which is clinically used as an anti-cancer agent. It possesses a tricyclicchromophore and is glycosylated at twodifferent positions [63]. Urdamycin A isan angucycline polyketide produced byS. fradiae Tu2717, which also shows an-titumor activity. It consists of the agly-con aquayamycin, which contains a C-glycosidically linked D-olivose, and threeadditional O-glycosidically linked deoxy-hexoses [64, 65]. The UrdGT2 glycosyl-transferase catalyzes the C-glycosyl trans-fer of activated D-olivose as the firstglycosylation step during the urdamycinbiosynthesis. Landomycins are producedby S. cyanogenus S136 and contain an un-usual hexasaccharide consisting of fourD-olivose and two L-rhodinose units. Thesepolyketides also show antitumor activi-ties, in particular, against prostata cancercell lines [66]. To generate novel com-pounds, genes out of the urdamycin andlandomycin clusters were expressed inmutants of S. argillaceus: coexpression ofurdGT2 (urdamycin biosynthesis) togetherwith lanGT1, (landomycin biosynthesis) in
a mutant of the mithramycin producerled to the hybrid molecule 9-C-diolivosyl-premithramycinone [67]. This examplewas listed as a highlight in the field ofcombinatorial biosynthesis as genes fromthree different organisms yielded a ratio-nally designed product (Fig. 4) [68].
Recently, a plasmid-based strategy hasbeen described that allows the use of de-oxysugar biosynthetic genes to producea variety of deoxysugars in a cell, whichcan then be attached to an aglycon bythe use of different glycosyltransferases.As an example, a plasmid was generatedharboring all the genes necessary for thebiosynthesis dTDP-D-olivose. This plasmidwas coexpressed with the highly substrate-flexible glycosyltransferase gene elmG inS. albus. When 8-demethyl-tetracenomycinC was fed to this strain, D-olivosyl-tetracenomycin was produced. In asimilar way, L-rhamnosyl-tetracenomycinC, L-olivosyl-tetracenomycin C, and L-rhodinosyl-tetracenomycin C were gener-ated depending on the deoxysugar biosyn-thetic genes used in each case [69].
Polyketides are synthesized by the actionof polyketide synthases (PKSs), which havebeen classified into two types, type I (mod-ular PKSs) and type II (iterative PKSs).
Modular PKSs are large multifunc-tional enzymes. Active sites (domains)within these enzymes ketosynthases (KS),acyltransferases (AT), dehydratases (DH),enoyl reductases (ER), ketoreductases(KR), acyl carrier proteins (ACP) andthioesterases (TE) are organized into mod-ules such that each module catalyzes thestereospecific addition of a new monomeronto a growing polyketide chain and alsosets the reduction level of the carbonatoms of the resulting intermediate [70].In 1994, the heterologous expression ofthe complete erythromycin polyketide syn-thase was accomplished. The recombinant
Procaryotic and Eucaryotic Cells in Biotech Production 19
OC
H3
OO
OH
CH
3
OH
OO
CH
3
OH
O
OC
H3
OO
OH
CH
3
OH
OO
CH
3O
O
CH
3O
OH
OH
OH
OH
OO
HC
H3
OH
OO
CH
3
OH
CH
3
OO
OO
HO
H
OC
H3
HO
OH
OO
HOH
CH
3
O
OO
CH
3O
OH
OO
HC
H3
OC
H3
OH
OH
O
OH
O
O
CH
3
OH
CH
3
OO
OO
HO
H
OC
H3
HO
OH
Urd
GT
2
Pre
mith
ram
ycin
one
S. a
rgill
aceu
s M
7D1
9-C
-dio
livos
yl-p
rem
ithra
myc
inon
e
Land
omyc
in A
S. c
yano
genu
s S
136 La
nGT
1
Urd
amyc
in A
S. f
radi
ae
Urd
GT
2La
nGT
1
S. a
rgill
aceu
s M
7D1
x ur
dGT
2 x
lanG
T1
Fig.
4G
ener
atio
nof
ara
tiona
llyde
sign
edhy
brid
prod
uctb
yco
expr
essi
onof
gene
sfr
omth
ree
diffe
rent
orga
nism
s.

20 2.2 Actinomycetes in Biotech Production
strain produced 6-deoxyerythronolide B.This polyketide synthase was then usedfor a variety of experiments in whichmodules/domains of the polyketide syn-thase were exchanged. As an example,compounds produced by the substitutionof KR domains are shown in Fig. 5. Inthese examples, KR domains from the ery-thromycin PKS have been replaced by do-mains from the rapamycin producer [71].
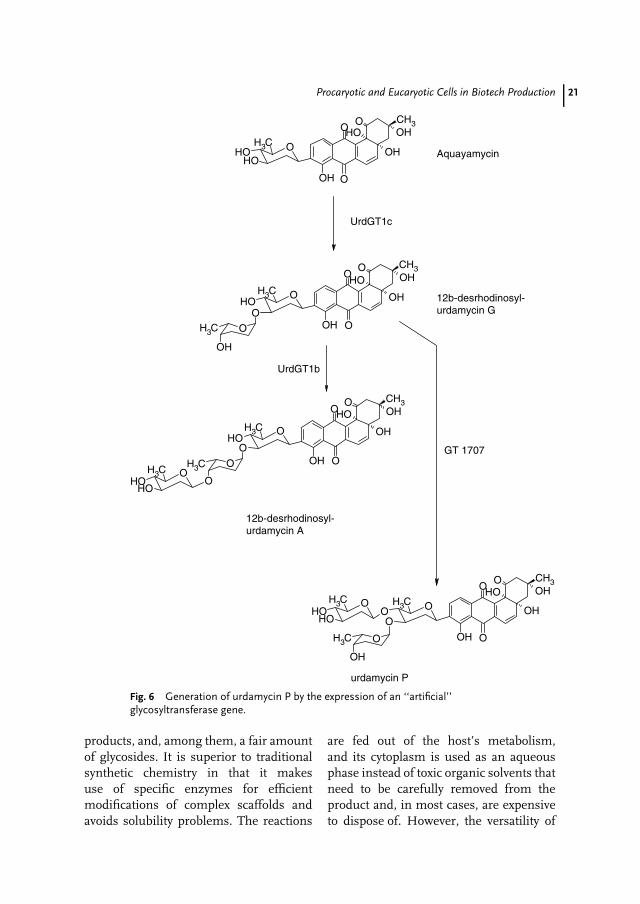
New Drugs by Expression of ‘‘Artificial’’Genes UrdGT1b and UrdGT1c, involvedin urdamycin biosynthesis, share 91%identical amino acids. However, the twoGTs show different specificities for bothnucleotide sugar and acceptor substrate.Targeted amino acid exchanges reducedthe number of amino acids, potentially
dictating the substrate specificity to 10 ineither enzyme. Subsequently, a gene li-brary was created such that only codonsof these 10 amino acids from bothparental genes were independently com-bined. Screening of almost 600 librarymembers in vivo revealed 40 active mem-bers, acting either like the parental en-zymes UrdGT1b and UrdGT1c or display-ing a novel specificity. The novel enzymaticspecificity is responsible for the biosynthe-sis of urdamycin P carrying a branchedsaccharide side chain, hitherto unknownfor urdamycins (Fig. 6) [72, 73].
2.2.4.4 Novel Natural Compounds byGlycorandomizationCombinatorial biosynthesis represents anin vivo methodology to diversify natural
OHO
CH3
O
CH3
CH3
CH3 O
CH3
CH3
CH3OH
OH
OHO
CH3
O
CH3
CH3
CH3 O
CH3
CH3
CH3OH
O
CH3
O
CH3
CH3
CH3 O
CH3
CH3
CH3OH
OH
OHO
CH3
O
CH3
CH3
CH3 O
CH3
CH3
CH3
OH
6-deoxyerythronolide B (6dEB) 2,3-anhydro-6dEBKR6 was substituted by rapDH/KR1
5-deoxy-6dEBKR5 was substituted by rapDH/ER/KR1
10,11-anhydro-6dEBKR2 was substituted by rapDH/KR4
Fig. 5 Production of novel natural compounds by exchanging of modules ofpolyketide synthases.
Procaryotic and Eucaryotic Cells in Biotech Production 21
OOH
OOH
OHCH3
O CH3OH
OH
O OH
O
OHOH
CH3O
O
O
CH3
OH
OOH
OOH
CH3
O CH3OH
OH
O
O
OH
OCH3
OH
OOH
OOH
CH3
O CH3OH
OH
O
OH
O
O
CH3
OH
OOH
OCH3
O CH3OH
OH
O
O
OHOH
CH3O
UrdGT1c
UrdGT1b
Aquayamycin
12b-desrhodinosyl-urdamycin G
12b-desrhodinosyl-urdamycin A
urdamycin P
GT 1707
Fig. 6 Generation of urdamycin P by the expression of an ‘‘artificial’’glycosyltransferase gene.
products, and, among them, a fair amountof glycosides. It is superior to traditionalsynthetic chemistry in that it makesuse of specific enzymes for efficientmodifications of complex scaffolds andavoids solubility problems. The reactions
are fed out of the host’s metabolism,and its cytoplasm is used as an aqueousphase instead of toxic organic solvents thatneed to be carefully removed from theproduct and, in most cases, are expensiveto dispose of. However, the versatility of
22 2.2 Actinomycetes in Biotech Production
combinatorial biosynthesis is somewhatlimited. A still unsolved issue is how anovel active compound is dealt with bythe host strain. Highly antimicrobial orcytotoxic agents that are not immediatelyand effectively detoxified by the host’sintrinsic mechanisms will kill the hoststrain long before the compound isdetected in a screen. Therefore, especiallyin case of antimicrobials, scientists run therisk of selecting for structures innocuousto pathogens. In the case of glycosidicstructures, true combinatorial approachessuffer from a limited sugar diversity asa second drawback. Only those sugarsare available for drug-lead diversificationwhose biosynthetic routes are understoodand the genes involved cloned.
To make use of the possibilitiesmicrobial enzymes, particularly natural
OPO3
R6 R2
R3
HR5
R8
O
R4R1
R7
ONDP
R6 R2
R3
HR5
R8
O
R4R1
R7
H
R6 R2
R3
O-aglyconR5
R8
O
R4R1
R7
2−
Chemical synthesis
Sugar phosphatelibrary
Nucleotidylyl-transferase
Sugar nucleotidelibrary
Glycosyltransferase
Library ofglycosides
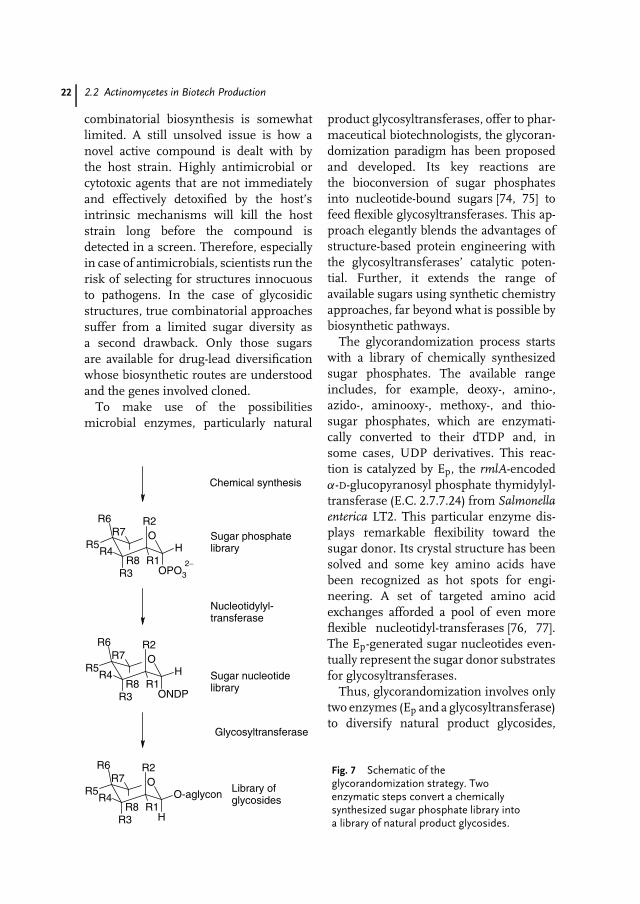
Fig. 7 Schematic of theglycorandomization strategy. Twoenzymatic steps convert a chemicallysynthesized sugar phosphate library intoa library of natural product glycosides.
product glycosyltransferases, offer to phar-maceutical biotechnologists, the glycoran-domization paradigm has been proposedand developed. Its key reactions arethe bioconversion of sugar phosphatesinto nucleotide-bound sugars [74, 75] tofeed flexible glycosyltransferases. This ap-proach elegantly blends the advantages ofstructure-based protein engineering withthe glycosyltransferases’ catalytic poten-tial. Further, it extends the range ofavailable sugars using synthetic chemistryapproaches, far beyond what is possible bybiosynthetic pathways.
The glycorandomization process startswith a library of chemically synthesizedsugar phosphates. The available rangeincludes, for example, deoxy-, amino-,azido-, aminooxy-, methoxy-, and thio-sugar phosphates, which are enzymati-cally converted to their dTDP and, insome cases, UDP derivatives. This reac-tion is catalyzed by Ep, the rmlA-encodedα-D-glucopyranosyl phosphate thymidylyl-transferase (E.C. 2.7.7.24) from Salmonellaenterica LT2. This particular enzyme dis-plays remarkable flexibility toward thesugar donor. Its crystal structure has beensolved and some key amino acids havebeen recognized as hot spots for engi-neering. A set of targeted amino acidexchanges afforded a pool of even moreflexible nucleotidyl-transferases [76, 77].The Ep-generated sugar nucleotides even-tually represent the sugar donor substratesfor glycosyltransferases.
Thus, glycorandomization involves onlytwo enzymes (Ep and a glycosyltransferase)to diversify natural product glycosides,

Procaryotic and Eucaryotic Cells in Biotech Production 23
thereby eliminating the need for large setsof biosynthetic genes (Fig. 7).
Although being a very recent tech-nique, glycorandomization has alreadydemonstrated its versatility in its firstapplication toward diversifying the Actino-mycete natural products vancomycin andteicoplanin [78], nonribosomally gener-ated sugar-decorated heptapeptides, whichare in clinical use as antimicrobial drugsof last resort.
Like conventional combinatorial biosyn-thesis, glycorandomization requires flex-ible glycosyltransferases. As recentlypointed out in the case of novobiocin [79],a highly specific glycosyltransferase limitsthe library size. Despite such issues thatneed to be addressed in future work, gly-corandomization is a promising approachto make use of the metabolic potential ofprocaryotic cells and should promote drugdevelopment in the future.
2.2.4.5 Novel Natural Compounds byMutasynthesisThe substrate flexibility of enzymes isalso the basis for the ‘‘mutasynthesis’’approach. During ‘‘mutasynthesis,’’ a mi-croorganism containing a defined muta-tion in an important precursor biosynthe-sis gene of an interesting metabolite can befed with alternative or even synthetic pre-cursors. Consequently, derivatives of com-plex natural products are generated, whichmay not have been obtained by syntheticmethods [80]. This technology was suc-cessfully applied to generate the first fluo-rinated vancomycin-type antibiotics [81].
2.3Saccharomyces cerevisiae and Other Fungiin Biotech Production
Saccharomyces (Sa.) cerevisiae might beviewed to be one of the most important
fungal organisms used in biotechnology.It has been used in the ‘‘old biotechnology’’for baking and brewing since prehistory.Yeast genetics, yeast biochemistry, and,finally, yeast molecular biology have sub-stantially contributed to the importance ofSa. cerevisiae also in the ‘‘new biotechnol-ogy’’ area.
Yeast is a unicellular organism, which,unlike more complex eukaryotes, isamenable to mass production. It canbe grown on defined media, giving theinvestigator a complete control over en-vironmental parameters. The availabilityof the complete genome sequence ofSa. cerevisiae opened the age of ‘‘newbiotechnology’’ [82]. In this chapter, wefirst review how genetic engineering ofSa. cerevisiae resulted in improved pro-ductivity and yield of important biotechproducts. Later, three examples of fungalnatural products (or their derivatives) aredescribed that have found their way intoclinical use.
2.3.1Generation of Engineered Strains ofSaccharomyces cerevisiae for theProduction of Alcoholic Beverages
Alcohol fermentation is one of the mostimportant processes of biotechnology.Generally, it is initiated by adding yeastto a carbon source and discontinues ata given alcohol concentration. The exten-sion of the substrate range of Sa. cerevisiaeis of major importance for the large-scale production of several metabolites.Sa. cerevisiae is not able to degrade starchand dextrin, since it does not producestarch-decomposing enzymes. Therefore,it is necessary to add starch-decomposingenzymes before fermentation. Attemptshave been undertaken to use recombinantstrains that contain the decomposing

24 2.3 Saccharomyces cerevisiae and Other Fungi in Biotech Production
enzymes-encoding genes in order to avoidthe preincubation process. The completeassimilation of starch (>98%) was accom-plished by coexpression of the sta2 geneof Sa. diastaticus encoding a glucoamylase,the amy1 gene of Bacillus amyloliquefaciensencoding an α-amylase, and the pulA geneof Klebsiella pneumoniae encoding a pullu-lanase [83].
Further, genetically engineered strainshave been developed, which are able to uti-lize lactose, melobiose, xylose, and othermaterials. A thermostable β-galactosidaseencoded by lacA from Aspergillus nigerwas expressed in Sa. cerevisiae, en-abling the strain to use lactose as car-bon source [84]. A melibiase-producingyeast was constructed by overexpress-ing the melI gene from another Sa.strain [85]. Moreover, after the coexpres-sion of xyl1 and xyl2, encoding a xy-lose reductase and xylitol dehydroge-nase from Pichia stipitis along with theoverexpressed xylulolkinase XKS1 fromSa. cerevisiae, xylose was converted toethanol [86].
Especially in the large-scale produc-tion of beer, of highest significance isnot ethanol production but a balancedflavor to obtain the desired taste. Oneunpleasant off-flavor compound is di-acetyl, which is a nonenzymatically de-graded product of α-acetolactate. Diacetylis then enzymatically converted to ace-toin and subsequently to 2,3-butanediol.The nonenzymatic-degradation step is veryslow and requires long lager periods.
One way to avoid the off-flavor is tointroduce an alternative route of degrada-tion of α-acetolactate directly to acetoin.α-Acetolactate decarboxylases from dif-ferent organisms were successfully over-expressed in the beer-producer strains,accelerating the brewing process by dimin-ishing the time of lagering by weeks [87].
Another interesting example is the ex-pression of a β-glucanase of Bacillussubtilis in yeast. β-Glucans, the highlyviscous side products during fermenta-tion, impede beer filtration, which isstill an important separation techniquein the brewing industry. The pres-ence of β-glucanase during fermenta-tion did not affect the beer quality andtaste but improved the filtration pro-cess [88].
2.3.2Generation of Engineered Strains ofSaccharomyces cerevisiae for Lactic acid,Xylitol, and Strictosidine Production
Several lactate dehydrogenases (LDHs)were expressed in Sa. cerevisiae in orderto produce lactic acid. Most successfulwas the expression of a fungal (Rhizo-pus oryzae) lactate dehydrogenase (LDH).A recombinant strain accumulated ap-proximately 40% more lactic acid witha final concentration of 38 g L−1 lacticacid and a yield of 0.44 g of lactic acidper gram of glucose [89]. Xylitol is anattractive sweetener used in the food in-dustry. Xylitol production in yeast wasperformed by the expression of xyl1of P. stipitis, encoding a xylitose reduc-tase [90].
A transgenic Sa. cerevisiae was con-structed harboring the cDNAs that en-codes strictosidine synthase (STR) andstrictosidine beta-glucosidase (SGD) fromthe medicinal plant Catharanthus roseus.Both enzymes are involved in the biosyn-thesis of terpenoid indole alkaloids. Theyeast culture was found to express highlevels of both enzymes. Upon feeding oftryptamine and secologanin, this trans-genic yeast culture produced high levelsof strictosidine [91].

Procaryotic and Eucaryotic Cells in Biotech Production 25
2.3.3The Use of Fungi in the Production ofStatins, Cyclosporin, and ß-LactamAntibiotics
2.3.3.1 StatinsStatins are the secondary metabolites ofa number of different filamentous fungi.Their medical importance and commer-cial value stem from their ability to inhibitthe enzyme (3S)-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase. Since thisenzyme catalyzes a key step in the endoge-nous cholesterol biosynthetic pathway,statins have become the widely used an-tihypercholesterolemic drugs. Along withsome synthetic statins, the most promi-nent examples are lovastatin, mainly fromAspergillus terreus, and mevastatin pro-duced by Penicillium citrinum, which wasthe first statin to be discovered [92, 93].
Chemical modification, for example hy-droxylation, turned out to be rather un-productive during derivatization efforts.Thus, biotransformation and biotechno-logical approaches have become the strat-egy of choice relatively early. For example, atwo-step fermentation/biotransformationprocess has been established for the clin-ically important pravastatin: Its directprecursor mevastatin is obtained out ofa P. citrinum culture in the first stepand is then subjected to biotransforma-tion, for example, by S. carbophilus tocomplete pravastatin biosynthesis by in-troducing a hydroxyl group at C6 [94].Later, improved Aspergillus and Monascusstrains for direct pravastatin productionwere described [95]. Pioneering work onthe genetics and enzymology underly-ing lovastatin biosynthesis was publishedin 1999 [96, 97], paving the way for thegeneration of novel derivatives. Recently,an approach termed ‘‘association analy-sis’’ [98] was developed to further improve
statin producers. The interrelationship be-tween secondary metabolite productionlevels and genome-wide gene expressionwas profiled for a minilibrary of A. terreusstrains engineered to express either wildtype or engineered genes that are part ofthe lovastatin cluster itself or implicated insecondary metabolite regulation. The au-thors found that multiple genes/proteinsattributed to cellular processes as di-verse as primary metabolism, secondarymetabolism, carbohydrate utilization, sul-fur assimilation, transport, proteolysis,and many more correlate with increased(or decreased) lovastatin production lev-els. This approach revealed multiple pointsfrom which to start engineering and mayhelp manipulate statin-producing filamen-tous fungi for industrial purposes.
2.3.3.2 CyclosporinCyclosporin A (INN: ciclosporin) is acyclic, nonribosomally synthesized un-decapeptide from Tolypocladium inflatum(Fig. 2). Apart from its antifungal prop-erties, it represents a potent immuno-suppressive drug as it interferes withlymphokine production [99]. CyclosporinA has been introduced into clinical useto prevent allograft rejection after organtransplants.
The biosynthesis of cyclosporin A hasbeen extensively investigated. A huge 45.8-kb open reading frame was identifiedas a putative gene coding for the cy-closporine synthetase, a multifunctionalpeptide synthetase [100]. With a molecu-lar mass of 1689 kDa, it represents one ofnature’s largest enzymes. Definitive func-tional evidence arose from targeted geneinactivation, which abolished cyclosporinA biosynthesis [101]. Fungal peptide syn-thetases are somewhat different fromtheir prokaryotic counterparts in that they

26 2.3 Saccharomyces cerevisiae and Other Fungi in Biotech Production
usually consist of one huge single en-zyme, as is the case for cyclosporin,whereas bacteria generally use multiunitsynthetases. Further, fungal peptides quiteoften include D-configured amino acids.However, these peptide synthetases donot harbor an epimerization domain [102].In the case of cyclosporin, an externalalanin racemase responsible for supply-ing the synthetase with D-alanine hasbeen purified and characterized [103]. Bothcyclosporin synthetase and D-alanine race-mase were localized as vacuolar mem-brane–associated enzymes [104]. Even be-fore the cyclosporin synthetase was char-acterized, it had become obvious that thebiosynthetic pathway tolerates a numberof substrate analogs. For example, D-alanine can be replaced by D-serine aswas demonstrated by precursor-directedbiosynthesis [105], thereby leading to novelcyclosporin derivatives.
For enhanced scaled-up cyclosporin pro-duction, a modified sporulation/immobili-zation method has been proposed, increas-ing the cyclosporin yield up to tenfold,compared to suspended culture tech-niques [106]. The immobilization of cellson celite carrier beads decreases the cul-ture’s viscosity and therefore increases themass transfer. It is possible to run thefermentation continuously since the fun-gal spores can be trapped in the fermentervessel to populate and germinate on freshlyadded beads.
2.3.3.3 β-LactamsEver since penicillin was discovered andfurther developed into a drug for use inhumans, there has hardly been a natu-ral product that parallels its impact onmedicine and pharmacy. Yet, penicillin isonly one example of the β-lactam antibi-otics class, along with other fungal (andstreptomycete) secondary metabolites, for
example, cephalosporin C from Acremo-nium chrysogenum. The commercial im-portance of penicillins and cephalosporinsis evident from the worldwide annualsales for these compounds (including theirsemisynthetic derivatives), which were es-timated to reach US$15 billion, and ofthat, US$4.8 billion derived from salesin the United States [107, 108]. β-lactambiosynthesis has been thoroughly inves-tigated, and been reviewed in a numberof compilations [109]. In brief, a nonribo-somal peptide synthetase assembles thebuilding blocks, L-α-aminoadipic acid, L-valine (which is epimerized to D-valin),and L-cysteine, into a linear tripeptide,which is then cyclized to isopenicillinN (IPN) by the enzyme IPN synthase.The latter enzyme has been crystallizedand, by elegant X-ray diffraction investi-gations, been used to support the notionthat the bicyclic enzymatic product IPN issynthesized in a two-step process, with β-lactam formation preceding the closureof the thiazolidine ring [110, 111]. IPNrepresents the last common intermediatealong the pathways toward penicillin andcephalosporins. While penicillin biosyn-thesis requires only side-chain modifica-tions, the five-membered penicillin thiazo-lidine ring is expanded to a dihydrothiazinesystem on the route to cephalosporins.Gene clusters coding for the enzymaticmachinery of β-lactam biosynthesis havebeen cloned from Penicillium chrysogenum,Aspergillus nidulans (Penicillins), and Acre-monium chrysogenum (Cephalosporin C).In the latter case, the cluster is splitup and located on two different chromo-somes. Strain development, fermentation,recovery, and purification conditions forβ-lactam producers have been subjectsof optimization ever since commercialproduction started. For example, modernfed-batch fermentations yield penicillin

Procaryotic and Eucaryotic Cells in Biotech Production 27
titers exceeding 40 g L−1 (compared to lessthan 1 g L−1 in 1950). Penicillin G and Vare produced in highly automated fermen-tation vessels with a capacity in the 100to 400 000 liter range. Although costs forenergy and labor have increased, the ad-vances in penicillin production techniqueshave led to a drastic decrease in bulk pricesfrom ∼US$300 per kilogram in the early1950s to ∼US$15 to 20 per kilogram to-day [108].
2.4Plants in Biotech Production
From the very roots of humanity, plantshave made a crucial contribution to ourwell-being. Plant products have been usedas food and as medicine. Even today, plantsare an important source for the discoveryof novel pharmacologically active com-pounds, though the recent competitionfrom combinatorial chemistry and com-putational drug design has declined theinterest. In recent years, gene technol-ogy has opened up exciting perspectivesand offers tools not only to improvethe existing properties of plants, such asthe amount of bioactive compounds, but
also to create transgenic plants with newproperties. Facile transformation and culti-vation not only make plants suitable for theproduction of secondary metabolites butalso for recombinant proteins. Plants arecapable of carrying out acetylation, phos-phorylation, and glycosylation as well asother posttranslational protein modifica-tions required for the biological activityof many eukaryotic proteins. This chap-ter gives some examples describing theuse of plant cells in biotech productionby which pharmaceuticals as well as func-tional and medicinal food are obtained.In this Chapter, we restrict ourselves toa secondary metabolite plant in the low-molecular-weight range.
2.4.1Transgenic Plants as Functional Foods orNeutraceuticals
A few years ago, industry started the ageof engineered functional food. Numer-ous examples such as the generation ofgolden rice, the production of healthy plantoils, and engineered plants with increasedlevels of essential vitamins and nutri-ents [112] have been published (Table 2).Golden rice was engineered with two
Tab. 2 Examples of new properties in transgenic plants used asneutraceuticals
Property Plant References
Resveratrol Peanut (114)Increased amount of iron and its
bioavailability by the reduction ofphytic acid
Maize (115)
Lactoferrin Rice (116, 117)Enriched ferritin leading to the binding
of iron and consequently to itsaccumulation
Lettuce (118)
Removal of bitter-tasting compounds(glycoalkaloids)
Potatoes (119)

28 2.4 Plants in Biotech Production
plant genes from Narcissus pseudonarcis-sus encoding a phytoene synthase and alycopene ß-cyclase along with one bacte-rial gene from Erwinia uredovora encodinga phytoene desaturase to synthesize ß-carotene, a precursor of vitamin A [113].This was possible because the transfor-mation of rice was well established andall carotinoid biosynthetic pathway geneshad been identified. Despite these promis-ing results, golden rice is not yet on themarket. Further work aims to increase theprovitamin A amount and to unify high-iron rice lines with provitamin A lines, asit is known that provitamin A potentiallyincreases iron bioavailability.
Wheat is a further target in the area of en-gineered functional food. A gene encodinga stilbene synthase was expressed in ricethat enabled wheat to produce resveratrol.This natural antioxidant possesses positiveeffects against the development of throm-bosis and arteriosclerosis. Moreover, in itsglycosidic form it enhances resistance tofungal pathogenes, which also occurs intransgenic plants [120].
The current health-related objective ofplant seed engineering is to increase thecontent of ‘‘healthy fatty acids’’ and re-duce ‘‘unhealthy fatty acids’’ in oilseedcrops, such as soybean, oil palm, rape-seed, and sunflower. Genetic engineeringwas successful in reducing the levels oftrans-unsaturated fatty acids and in re-ducing the ratio between omega-6 andomega-3 unsaturated fatty acids in somevegetable oils [121, 122]. Metabolic engi-neering also succeeded in increasing thevitamin E, vitamin C, and the lycopenecontent in plants [112, 123], as well asthe content of bioflavonoids, known fortheir antioxidant, anticancer, and estro-genic properties [124]. In addition, humanmilk proteins like lactoferrin can now beexpressed in plants [116]. These proteins
are believed to have a multitude of bio-logical activities that benefit the newborninfant. Functional food selected and adver-tised for its high content of therapeuticallyactive molecules is already offered in theshelves of supermarkets, leaving the deter-mination of their true medical benefit tothe consumer.
A further research area is the eliminationof natural compounds from a plant toavoid severe side effects. As an example,peanut causes allergies due to severalproteins. Researchers are now workingon the generation of plants with reducedlevels of these proteins.
2.4.2Transgenic Plants and Plant Cell Culture asBioreactors of Secondary Metabolites
Biotech methods are also used to in-crease the amount of pharmaceuticallyinteresting compounds in plants. Leavesof Atropa belladonna contain high amountsof L-hyoscyamin, but negligible contentsof L-scopolamin due to the low activ-ity of hyoscyamin-6ß-hydroxylase (H6H)in roots. H6H-cDNA was isolated fromHyoscyamus niger, cloned, and introducedin Atropa belladonna using Agrobacteriumtumefaciens. Transgenic plants as wellas the sexual descendents contain thetransgene and accumulate up to 1%of L-scopolamin, but only traces of L-hyoscyamin (Table 3) [125].
Another approach to produce biologi-cally active secondary metabolites is theuse of plant cell cultures. Plant cell cul-tures are advantageous in that they arenot limited by environmental, ecological,or climatic conditions. Further, cells canproliferate at higher growth rates thanwhole plants in cultivation. As shown inTable 4, some metabolites in plant cell cul-tures have been reported to accumulate
Procaryotic and Eucaryotic Cells in Biotech Production 29
Tab. 3 Transgenic plants with selected improved production of secondary metabolites [125]
Compound Target protein Gene donor Gene recipient
Cadaverin Lysin-decarboxylase Hafnia alvei Nicotiana tabacumSterols HMG-CoA-reductase Hevea brasiliensis Nicotiana tabacumNicotin Ornithin-decarboxylase Saccharomyces cerevisiae Nicotiana tabacumResveratrol Stilbene-synthase Vitis vinifera Nicotiana tabacumScopolamin Hyoscyamin-6ß-hydoxylase Hyoscyamus niger Atropa belladonna
Tab. 4 Comparison of product yield of secondary metabolites in cell cultureand parent plants [126]
Product Plant Yield (% DWa)
Culture Plant
Anthocyanin Vitis sp. 16 10Euphorbia milli 4 0.3Perilla frutescens 24 1.5
Anthraquinone Morinda citrifolia 18 2.2Berberine Coptis japonica 13 4
Thalictrum minor 10 0.01Rosmarinic acid Coleus blumei 27 3.0Shikonin Lithospermum erythrorhizon 14 1.5
aDry weight.
with a higher titer compared to thosein the parent plants. Some industrialprocesses harness this potential, for exam-ple, for shikonin, ginseng, and paclitaxelproduction [126]. Especially Taxus cell cul-tures are an interesting alternative to theisolation of paclitaxel from plantation-grown plants as the slow growth of Taxusspecies, the significant variation in tax-oid content, and the costly purification of10-deacetylbaccatin III from co-occurringtaxoids are significantly limiting parame-ters [127–129]. Using cell cultures, taxolproduction rates up to 23.4 mg L day−1
with paclitaxel comprising 13 to 20%of the total taxoid fraction can now beachieved [130].
However, the use of plant cell culturesfor the production of interesting molecules
has not gained acceptance in industry asyet. Usually, low productivity and highcosts are the most important negative pa-rameters [131]. Nevertheless, research isstill going on and is well described inthe next chapter. Further application ofplant-cell-suspension cultures are aimed atthe production of ajmalicine, vinblastine,vincristine, podophyllotoxins, and camp-tothecin [132–134].
2.4.3Transgenic Plants as Bioreactors ofRecombinant Proteins
Nowadays, plants such as tobacco, potato,tomato, banana, legumes, and cerealsas well as alfalfa, are used in molec-ular farming and have emerged as

30 2.4 Plants in Biotech Production
Tab. 5 Examples of therapeutic antibodies produced in plants [135]
Antibody Antigen Cellular Transgenic Max. expression
format location plant level
dAb Substance P Apoplast Nicotianabenthamiana
1% TSP leaves
IgG1, Fab Human creatinekinase
Apoplast N. tabacumArabidopsis thaliana
0.044% TSP leaves1.3% TSP leaves
IgG1 Streptococcalsurface antigen(I/II)
Plasmamembrane
N. tabacum 1.1% TSP leaves
IgG1 Human IgG Apoplast Medicago sativa 1% TSP
IgG1 Herplex simplexvirus 2
Apoplast Glycine max Not reported
SigA Streptococcalsurface antigen(I/II)
Apoplast N. tabacum 0.5 mg g−1 FWleaves
scFv Carcinoembryonicantigen
ER Pisum sativum 0.009 mg g−1 seed
Note: dAb: single domain antibody; FW: fresh weight; TSP: total soluble protein.
promising biopharming systems for pro-duction of pharmaceutical proteins, suchas antibodies, vaccines, regulatory proteinsand enzymes, [112, 135–138] (Table 5).The advantages offered by plants includelow cost of cultivation, high biomass pro-duction, relatively fast protein synthesis,low operating costs, excellent scalability,eucaryotic posttranslational modifications,low risk of pathogenicity toward humansand endotoxins, and a relatively highprotein expression level [112, 139]. Usingtransgenic plants as a host is highly attrac-tive in that proteins can be administeredin fruits and vegetables as a source ofantigens for oral vaccination [135]. Thus,potatoes expressing a synthetic LT-B gene,a labile toxin from Escherichia coli were suc-cessfully used in a clinical study to examinean edible plant vaccine [137]. Further, in-teresting vaccines that have been testedclinically are directed against viral diarrhea
and hepatitis B. No plant-derived proteinhas still been developed to be used as adrug, but molecular farming has gainedattention as plants can be turned intomolecular medicine factories.
Recently, plant cells have also been con-sidered to be an alternative host for theproduction of recombinant proteins sincethey are able to glycosylate proteins [133,139]. Of the various systems used for cul-tivation, such as hairy roots, immobilizedcells, and free cell suspensions, the lat-ter is regarded to be most suitable forlarge-scale applications. Full-size antibod-ies, antibody fragments, and fusion pro-teins can be expressed in transgenic-plant-cell systems, such as Nicotiana tabacum,pea, wheat, and rice using shake-flaskor fermentation cultures [136]. Yet, thesesystems are still of low commercial im-portance due to their unadvantageousproductivity.

Procaryotic and Eucaryotic Cells in Biotech Production 31
References
1. A. Schatz, E. Bugie, S. Waksman, Proc. Soc.Exp. Biol. Med. 1944, 55, 66–69.
2. R. H. Baltz, Trends Microbiol. 1998, 6,76–83.
3. R. Austrian, J. Gold, Ann. Intern. Med. 1964,60, 759–776.
4. P. Dineen, W. P. Homan, W. R. Grafe,Ann. Surg. 1976, 184, 717–722.
5. G. L. Verdine, Nature 1996, 384, 11–13.6. G. M. Cragg, D. J. Newman, K. M. Snader,
J. Nat. Prod. 1997, 60, 52–60.7. A. L. Demain, Appl. Microbiol. Biotechnol.
1999, 52, 455–463.8. T. Henkel, R. M. Brunne, H. Muller, Angew.
Chem., Int. Ed. Engl. 1999, 38, 643–647.9. A. Harvey, Scrip Reports, PJB Publications
Ltd, Richmond, Surrey, UK, 2001.10. H. H. Cowen, Pharm. Therap. Categories
Outlook 2001, 10.11. J. Berdy, Proceedings of the 9th International
Symposium on The Biology of Actinomycetes,Part 1, Allerton Press, New York, 1995,pp. 3–23.
12. L. Gray, Business Communication CompanyInc, Norwalk, CT, 2002, p. 114.
13. B. S. Moore, J. Piel, Antonie Van Leeuwen-hoek 2000, 78, 391–398.
14. J. K. Borchardt, Modern Drug Discov. 1999,2, 22–29.
15. S. Donadio, M. Sosio, G. Lancini, Appl.Microbiol. Biotechnol. 2002, 60, 377–380.
16. W. R. Strohl, Metab. Eng. 2001, 3, 4–14.17. H. Liu, K. A. Reynolds, Metab. Eng. 2001, 3,
40–48.18. T. Kieser, M. J. Bibb, M. J. Buttner et al.,
Practical Streptomyces Genetics, The JohnInnes Foundation, Norwich, UK, 2000.
19. D. A. Hopwood, Microbiology 1999, 145,2183–2202.
20. D. A. Hopwood, Genetic Recombination inStreptomyces coelicolor, Ph. D. Thesis,University of Cambridge, Cambridge, 1958.
21. B. A. M. Rudd, D. A. Hopwood, J. Gen.Microbiol. 1979, 114, 35–43.
22. C. J. Thompson, J. M. Ward, D. A.Hopwood, Nature 1980, 286, 525–527.
23. M. J. Bibb, J. L. Schottel, S. N. Cohen,Nature 1980, 284, 526–531.
24. F. Malpartida, D. A. Hopwood, Nature 1984,309, 462–464.
25. G. Muth, D. F. Brolle, W. Wohllebenin Manual of Industrial Microbiologyand Biotechnology (Eds.: A. L. Demain,J. E. Davis, R. M. Atlas), ASM Press,Washington, DC, 1999, pp. 353–367.
26. A. Paradkar, A. Trefzer, R. Chakraburttyet al., Crit. Rev. Biotechnol. 2003, 23, 1–27.
27. T. Weber, K. Welzel, S. Pelzer et al., J.Biotechnol. 2003, in press.
28. P. R. August, T.-W., Yu, H. G. Floss in DrugDiscovery from Nature (Eds.: S. Grabley,R. Thiericke), Springer, Berlin-Heidelberg,2000.
29. T. Brautaset, O. N. Sekurova, H. Slettaet al., Chem. Biol. 2000, 7, 395–403.
30. S. D. Bentley, K. F. Chater, A. M. Cerdeno-Tarraga et al., Nature 2002, 417, 141–147.
31. S. Omura, H. Ikeda, J. Ishikawa et al., Proc.Natl. Acad. Sci. U. S. A. 2001, 98,12215–12220.
32. H. Ikeda, J. Ishikawa, A. Hanamoto et al.,Nat. Biotechnol. 2003, 21, 526–531.
33. Y. S. Lin, H. M. Kieser, D. A. Hopwoodet al., Mol. Microbiol. 1993, 10, 923–933.
34. M. Redenbach, J. Scheel, U. Schmidt,Antonie Van Leeuwenhoek 2000, 78,227–235.
35. D. A. Hopwood, Nat. Biotechnol. 2003, 21,505–506.
36. T. A. Cropp, D. J. Wilson, K. A. Reynolds,Nat. Biotechnol. 2000, 18, 980–983.
37. M. G. Watve, R. Tickoo, M. M. Jog et al.,Arch. Microbiol. 2001, 176, 386–390.
38. N. Johnston, Modern Drug Discov. 2002, 6,28–32.
39. H. Breithaupt, Nat. Biotechnol. 1999, 17,1165–1169.
40. A. Coates, Y. Hu, R. Bax et al., Nat. Rev.Drug. Discov. 2002, 11, 895–910.
41. H. Zahner, H.-P. Fiedler in Past Perspectivesand Future Friends (Eds.: P. A. Hunter,G. K. Darby, N. J. Russel), CambridgeUniversity Press, Cambridge, 1995,pp. 67–84.
42. W. R. Strohl in Biotechnology of Antibiotics(Eds.: W. R. Strohl), 2nd ed., MarcelDekker, New York, 1997, pp. 1–47.
43. J. Rosamond, A. Allsop, Science 2000, 287,1973–1976.
44. R. P. Bax, R. Anderson, J. Crew et al., Nat.Med. 1998, 4, 545–546.
45. S. Tsiodras, H. S. Gold, G. Sakoulas et al.,Lancet 2001, 358, 207–208.
46. A. L. Demain, Nat. Biotechnol. 2002, 20, 331.

32 2.4 Plants in Biotech Production
47. A. Harvey, Drug Discov. Today 2000, 5,294–300.
48. S. Donadio, P. Monciardini, R. Alduinaet al., J. Biotechnol. 2002, 99, 187–198.
49. H. B. Bode, B. Bethe, R. Hofs et al.,ChemBiochem 2002, 3, 619–627.
50. T. V. J. Goksoyr, F. L. Daae, Appl. Environ.Micobiol. 1990, 56, 782–787.
51. G. Y. Wang, E. Graziani, B. Waters et al.,Org. Lett. 2000, 2, 2401–2404.
52. I. A. MacNeil, C. L. Tiong, C. Minor et al., J.Mol. Microbiol. Biotechnol. 2001, 3, 301–308.
53. S. Pelzer, W. Reichert, M. Huppert et al., J.Biotechnol. 1997, 56, 115–128.
54. M. Sosio, E. Bossi, A. Bianchi et al., Mol.Gen. Genet. 2000, 264, 213–221.
55. X. Ruan, D. Stassi, S. A. Lax et al., Gene1997, 203, 1–9.
56. B. Shen, L. Du, C. Sanchez et al., Bioorg.Chem. 1999, 27, 155–171.
57. C. Mendez, E. Kunzel, F. Lipata et al., J.Nat. Prod. 2002, 65, 779–782.
58. M. Metsa-Ketela, K. Palmu, T. Kunnariet al., Antimicrob. Agents Chemother. 2003,47, 1291–1296.
59. E. Zazopoulos, K. Huang, A. Staffa et al.,Nat. Biotechnol. 2003, 21, 187–190.
60. J. Weber, J. Leung, S. Swanson et al.,Science 1991, 252, 114–117.
61. G. Weitnauer, A. Muhlenweg, A. Trefzeret al., Chem.Biol. 2001, 8, 569–581.
62. G. Sezonov, V. Blanc, N. Bamas-Jacqueset al., Nat. Biotechnol. 1997, 15, 349–353.
63. J. Skarbek, M. Speedie in AntitumorCompounds Of Natural Origin (Ed.:A. Aszalos), CRC Press, Boca Raton, FL,1981, pp. 191–235, Vol. 1.
64. H. Drautz, H. Zahner, J. Rohr et al., J.Antibiot. 1986, 39, 1657–1669.
65. A. Trefzer, D. Hoffmeister, E. Kunzel et al.,Chem. Biol. 2000, 7, 133–142.
66. R. Crow, B. Rosenbaum, R. Smith et al.,Bioorg. Med. Chem. Lett. 1999, 9, 1663–1666.
67. A. Trefzer, G. Blanco, L. Remsing et al., J.Am. Chem. Soc. 2002, 124, 6056–6062.
68. U. Rix, C. Fischer, L. Remsing et al., Nat.Prod. Rep. 2002, 19, 542–580.
69. L. Rodriguez, I. Aguirrezabalaga, N. Allendeet al., Chem. Biol. 2002, 9, 721–729.
70. D. Hopwood, Chem. Rev. 1997, 10,2465–2498.
71. R. McDaniel, A. Thamchaipenet, C.Gustafsson et al., Proc. Natl. Acad. Sci. U. S.A. 1999, 96, 1846–1851.
72. D. Hoffmeister, K. Ichinose, A. Bechthold,Chem. Biol. 2001, 8, 557–567.
73. D. Hoffmeister, B. Wilkinson, G. Fosteret al., Chem. Biol. 2002, 9, 287–295.
74. J. Jiang, J. B. Biggins, J. S. Thorson, J. Am.Chem. Soc. 2000, 122, 6803–6804.
75. J. Jiang, J. B. Biggins, J. S. Thorson, Angew.Chem., Int. Ed. 2001, 40, 1502–1505.
76. W. A. Barton, J. Lesniak, J. B. Biggins et al.,Nat. Struct. Biol. 2001, 8, 545–551.
77. W. A. Barton, J. B. Biggins, J. Jiang et al.,Proc. Natl. Acad. Sci. 2002, 99, 13397–13402.
78. H. C. Losey, J. Jiang, J. B. Biggins et al.,Chem. Biol. 2002, 9, 1305–1314.
79. C. Albermann, A. Soriano, J. Jiang et al.,Org. Lett. 2003, 5, 933–936.
80. W. Wohlleben, S. Pelzer, Chem. Biol. 2002,9, 1163–1164.
81. S. Weist, B. Bister, O. Puk et al., Angew.Chem., Int. Ed. Engl. 2002, 41, 3383–3385.
82. S. Ostergaard, L. Olsson, J. Nielsen, Mico-biol. Mol. Biol. Rev. 2000, 64, 34–50.
83. B. Janse, I. Pretorius, Appl. Microbiol.Biotechnol. 1995, 42, 873–883.
84. V. Kumar, S. Ramakrishnan, T. Teeri et al.,Biotechnology 1992, 10, 82–85.
85. S. Vincent, P. Bell, P. Bissinger et al., Lett.Appl. Microbiol. 1999, 28, 148–152.
86. N. Ho, Z. Chen, A. Brainard et al., Adv.Biochem. Eng. Biotechnol. 1999, 65, 163–192.
87. S. Yamano, K. Kondo, J. Tanaka et al., J.Biotechnol. 1994, 14, 173–178.
88. B. Cantwell, G. Brazil, N. Murphy et al.,Curr. Genet. 1986, 11, 65–70.
89. C. Skory, J. Ind. Microbiol. Biotechnol. 2003,30, 22–27.
90. R. Govinden, B. Pillay, W. Van Zyl et al.,Appl. Microbiol. Biotechnol. 2001, 55, 76–80.
91. A. Geerlings, F. Redondo, A. Contin et al.,Appl. Microbiol. Biotechnol. 2001, 56,420–424.
92. A. Endo, M. Kuroda, K. Tanzawa, FEBSLett. 1976, 72, 323–326.
93. M. Manzoni, M. Rollini, Appl. Microbiol.Biotechnol. 2002, 58, 555–564.
94. T. Matsuoka, S. Miyakoshi, K. Tanzawaet al., Eur. J. Biochem. 1989, 184, 707–713.
95. M. Manzoni, S. Bergomi, M. Rollini et al.,Biotechnol. Lett. 1999, 21, 253–257.
96. J. Kennedy, K. Auclair, S. G. Kendrew et al.,Science 1999, 284, 1368–1372.
97. L. Hendrickson, C. R. Davis, C. Roachet al., Chem. Biol. 1999, 6, 429–439.

Procaryotic and Eucaryotic Cells in Biotech Production 33
98. M. Askenazi, E. M. Driggers, D. A.Holtzman et al., Nat. Biotechnol. 2003, 21,150–156.
99. C. Randak, T. Brabletz, M. Hergenrotheret al., EMBO J. 1990, 9, 2529–2536.
100. G. Weber, K. Schorgendorfer, E. Schneider-Scherzer et al., Curr. Genet. 1994, 26,120–125.
101. G. Weber, E. Leitner, Curr. Genet. 1994, 26,461–467.
102. S. Doekel, M. A. Marahiel, Metab. Eng.2001, 3, 64–77.
103. K. Hoffmann, E. Schneider-Scherzer,H. Kleinkauf et al., J. Biol. Chem. 1994, 269,12710–12714.
104. M. Hoppert, C. Gentzsch, K. Schorgen-dorfer, Arch. Microbiol. 2001, 176, 285–293.
105. R. Traber, H. Hofmann, H. Kobel, J.Antibiot. 1989, 42, 591–597.
106. T. H. Lee, G.-T. Chun, Y. K. Chang, Biotech-nol. Prog. 1997, 13, 546–550.
107. W. R. Strohl in Encyclopedia of BioprocessTechnology: Fermentation, Biocatalysis andBioseparations (Eds.: M. C. Flickinger,S. W. Drew), Wiley, New York, 1999,pp. 2348–2365, Vol. 5.
108. R. P. Elander, Appl. Microbiol. Biotechnol.2003, 61, 385–392.
109. J. F. Martin, Appl. Microbiol. Biotechnol.1998, 50, 1–15.
110. P. L. Roach, I. J. Clifton, C. M. H. Hensgenset al., Nature 1997, 387, 827–830.
111. N. I. Burzlaff, P. J. Rutledge, I. J. Cliftonet al., Nature 1999, 401, 721–724.
112. I. D. Raskin, D. M. Ribnicky, S. Komar-nytsky et al., Trends Biotechnol. 2002, 20,522–531.
113. P. Beyer, S. Al-Babili, X. Ye et al., Am. Soc.Nutr. Sci. 2002, 132, 506–510.
114. I. M. Chung, M. R. Park, S. Rehmann et al.,Mol. Cells 2001, 12, 353–359.
115. C. Mendoza, F. E. Viteri, B. Lonnerdalet al., Am. J. Clin. Nutr. 2001, 73, 80–85.
116. B. Lonnerdal, J. Am. Coll. Nutr. 2002,21(Suppl. 3), 218S–221S.
117. Y. A. Suzuki, S. L. Kelleher, D. Yalda et al.,J. Pediatr. Gastroenterol. Nutr. 2003, 36,190–199.
118. F. Goto, T. Yoshihara, H. Saiki, Theor. Appl.Gen. 2000, 100, 658–664.
119. L. Arnqvist, P. C. Dutta, L. Jonsson et al.,Plant Physiol. 2003, 131, 1792–1799.
120. S. Fettig, D. Heß, Transgenic Res. 1999, 8,179–189.
121. J. Thelen, J. B. Ohlrogge, Metab. Eng. 2002,4, 12–21.
122. K. Liu, Proceedings of the WorldConference on Oilseed ProcessingUtilization, Cancun, Mexiko, 2000pp. 84–89.
123. D. Shintani, D. Della Penna, Science 1998,282, 2098–2100.
124. G. Forkman, S. Martens, Curr. Opin.Biotechnol. 2001, 12, 155–160.
125. W. Kreis, D. Baron, G. Stoll, Biotechnologieder Arzneistoffe, Dtsch. Apoth. Verlag,Stuttgart, 2001, pp. 250–260.
126. J. Zhong, Adv. Biochem. Eng. Biotechnol.2001, 72, 1–26.
127. S. Jennewein, R. Croteau, Appl. Microbiol.Biotechnol. 2001, 57, 13–19.
128. Y. Yukimune, H. Tabata, Y. Higashi et al.,Nat. Biotechnol. 1996, 14, 1129–1132.
129. R. Cusido, J. Palazon, M. Bonfill et al.,Biotechnol. Prog. 2002, 18, 418–423.
130. R. Ketchum, D. Gibson, R. Croteau et al.,Biotechnol. Bioeng. 1999, 62, 97–105.
131. J. Choi, G. Cho, S. Byun et al., Adv.Biochem. Eng. Biotechnol. 2001, 72, 63–102.
132. R. M. Moraes, E. Bedir, H. Barrett et al.,Planta Med. 2002, 68, 341–344.
133. R. Fischer, C. Vaquero-Martin, M. Sacket al., Biotechnol. Appl. Biochem. 1999, 30,113–116.
134. S. Hiroshi, T. Yamakawa, M. Yamazakiet al., Biotechnol. Lett. 2002, 24, 359–363.
135. S. Schillberg, R. Fischer, N. Emans, Natur-wissenschaften 2003, 90, 115–155.
136. R. Fischer, J. Drossard, U. Commandeuret al., Biotechnol. Appl. Biochem. 1999, 30,101–108.
137. H. S. Mason, H. Warzecha, T. Mor et al.,Trends Mol. Med. 2002, 8, 324–329.
138. A. M. Walmsley, C. J. Arntzen, Curr. Opin.Biotechnol. 2000, 11, 126–129.
139. J. Miele, Trends Biotechnol. 1997, 15, 45–50.

35
3Biopharmaceuticals Expressed inPlants
J..org Kn
..ablein
Analytical Development Biologics Schering AG, 13342 Berlin, Germany
3.1Introduction
Biopharmaceuticals, which are large mole-cules produced by living cells, are currentlythe mainstay products of the biotechnologyindustry. Indeed, biologics such as Genen-tech’s (Vacaville, CA, USA) human growthfactor somatropin or Amgen’s (ThousandOaks, CA, USA) recombinant erythropoi-etin have shown that biopharmaceuticalscan benefit a huge number of patients andalso generate big profits for these com-panies at the same time. But it has alsobecome obvious over the last couple ofyears that current fermentation capacitieswill not be sufficient to manufacture allbiopharmaceuticals (in the market alreadyor in development), because the marketand demand for biologics is continuouslyand very rapidly growing; for antibodiesalone (with at least 10 monoclonal an-tibodies approved and being marketed),the revenues are predicted to expand toUS$3 billion in 2002 [1] and US$8 billionin 2008 [2]. The 10 monoclonal antibodieson the market consume more than 75%of the industry’s manufacturing capabil-ity. And there are up to 60 more that are
expected to reach the market in the nextsix or seven years [3]. Altogether, there areabout 1200 protein-based products in thepipeline with a 20% growth rate and themarket for current and late stage (PhaseIII) is estimated to be US$42 billion in2005, and even US$100 billion in 2010 [4].But, there are obvious limitations oflarge-scale manufacturing resources andproduction capacities – and pharmaceuti-cal companies are competing [5].
To circumvent this capacity crunch, itis necessary to look into other technolo-gies rather than the established ones, like,for example, Escherichia coli or CHO (Chi-nese hamster ovary) cell expression. Onesolution to avoid these limitations couldbe the use of transgenic plants to ex-press recombinant proteins at low cost,in GMP (good manufacturing practice)quality greenhouses (with purification andfill finish in conventional facilities). Plantstherefore provide an economically soundsource of recombinant proteins [6], suchas industrial enzymes [7], and biophar-maceuticals [8, 9]. Furthermore, using theexisting infrastructure for crop cultivation,processing, and storage will reduce theamount of capital investment required for
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

36 3.2 Alternative Expression Systems
commercial production. For example, itwas estimated that the production costsof recombinant proteins in plants couldbe between 10 and 50 times lower thanthose for producing the same protein in E.coli [10] and Alan Dove describes a factorof thousand for cost of protein (US dol-lar per gram of raw material) expressedin, for example, CHO cells compared totransgenic plants [11]. So, at the dawnof this new millennium, a solution isrising to circumvent expression capacitycrunches and to supply mankind withthe medicines we need. Providing theright amounts of biopharmaceuticals cannow be achieved by applying our knowl-edge of modern life sciences to systemsthat were on this planet long time beforeus – plants.
3.2Alternative Expression Systems
Currently, CHO cells are the most widelyused technology in biomanufacturing be-cause they are capable of expressingeukaryotic proteins (processing, folding,and post-translational modifications) thatcannot be provided by E. coli. A long
track record exists for CHO cells, butunfortunately they bring some problemsalong when it comes to scale-up for pro-duction. Transport of oxygen (and othergases) and nutrients is critical for thefermentation process, as well as heatmust diffuse evenly to all cultured cells.According to the Michaelis–Menten equa-tion, the growth rate depends on theoxygen/nutrient supply, therefore goodmixing and aeration are a prerequisitefor the biomanufacturing process and areusually achieved by different fermentationmodes (see Fig. 1). But the laws of physicsset strict limits on the size of bioreac-tors. For example, an agitator achievesgood heat flow and aeration, but with in-creased fermenter size, shear forces alsoincrease and disrupt the cells – and build-ing parallel lines of bioreactors multipliesthe costs linearly. A 10 000-L bioreac-tor costs between US$250 000 to 500 000and takes five years to build (concep-tual planning, engineering, construction,validation, etc.). An error in estimatingdemand for, or inaccurately predictingthe approval of, a new drug can beincredibly costly. To compound the prob-lem, regulators in the United States andEurope demand that drugs have to be
(a)
Mechanic: agitator Pneumatic: gassing Hydrodynamic: pumps Airlift reactor
(b) (c) (d)
Fig. 1 Different fermentation modes for bioreactors. In order to achievebest aeration and mixing and to avoid high shear forces, differentfermentation modes are applied. (a) Mechanical, (b) pneumatical,(c) hydrodynamic pumps, and (d) airlift reactor [12].

Biopharmaceuticals Expressed in Plants 37
produced for the market in the samesystem used to produce them for thefinal round of clinical trials, in orderto guarantee bioequivalence (e.g. toxicity,bioavailability, pharmacokinetics, pharma-codynamics) of the molecule. So, compa-nies have to choose between launching aproduct manufactured at a smaller devel-opment facility (and struggling to meetmarket demands) or building larger, dedi-cated facilities for a drug that might neverbe approved!
Therefore, alternative technologies areused for the expression of biopharmaceu-ticals, some of them also at lower costsinvolved (see Fig. 2). One such alterna-tive is the creation of transgenic animals(‘‘pharming’’), but this suffers from thedisadvantage that it requires a long timeto establish such animals (approximately2 years). In addition to that, some of thehuman biopharmaceuticals could be detri-mental to the mammal’s health, whenexpressed in the mammary glands. This iswhy ethical debates sometimes arise fromthe use of transgenic mammals for pro-duction of biopharmaceuticals. Althoughthere are no ethical concerns involvedwith plants, there are societal ones thatwill be addressed later. Another expres-sion system (see Fig. 2) utilizes transgenicchicken. The eggs, from which the proteinsare harvested, are natural protein produc-tion systems. But production of transgenicbirds is still several years behind trans-genic mammal technology. Intensive ani-mal housing constraints also make themmore susceptible to disease (e.g. Asia 1997or Europe 2003: killing of huge flocks withthousands of chicken suffering from fowlpest). In the light of development time,experience, costs and ethical issues, plantsare therefore the favored technology, sincesuch systems usually have short gene-to-protein times (weeks), some are already
well established, and as mentioned before,the involved costs are comparatively low.This low cost of goods sold (COGS) forplant-derived proteins is mainly due to lowcapital costs: greenhouse costs are onlyUS$10/m2 versus US$1000/m2 for mam-malian cells.
3.3History of Plant Expression
Plants have been a source of medicinalproducts throughout human evolution.These active pharmaceutical compoundshave been primarily small molecules, how-ever. One of the most popular examples isaspirin (acetylsalicylic acid) to relieve painand reduce fever. A French pharmacistfirst isolated natural salicin (a chemicalrelative of the compound used to makeaspirin) from white willow bark in 1829.Advances in genetic engineering are nowallowing for the production of therapeuticproteins (as opposed to small molecules) inplant tissues. Expression of recombinantproteins in plants has been well docu-mented since the 1970s and has slowlygained credibility in the biotechnology in-dustry and regulatory agencies. The firstproof of concept has been the incorpo-ration of insect and pest resistance intograins. For example, ‘‘Bt corn’’ containsgenes from Bacillus thuringensis and is cur-rently being grown commercially. Geneticengineering techniques are now availablefor the manipulation of almost all commer-cially valuable plants. Easy transformationand cultivation make plants suitable forproduction of virtually any recombinantprotein.
Plants have a number of advantagesover microbial expression systems, butone of them is of outmost importance:they can produce eukaryotic proteins in
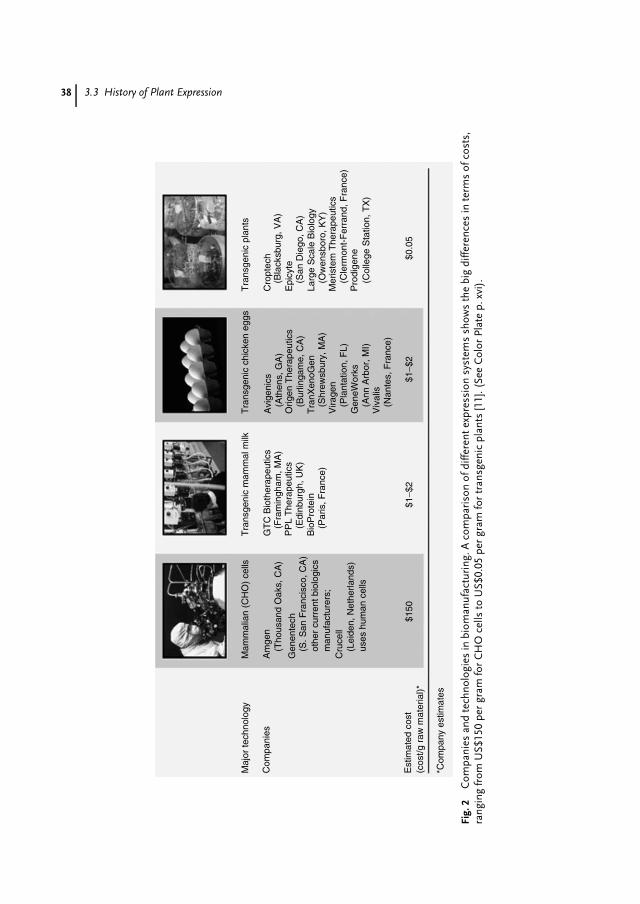
38 3.3 History of Plant Expression
Maj
or te
chno
logy
Mam
mal
ian
(CH
O)
cells
Tra
nsge
nic
mam
mal
milk
Tra
nsge
nic
chic
ken
eggs
Tra
nsge
nic
plan
ts
Cro
ptec
h(B
lack
sbur
g, V
A)
(San
Die
go, C
A)
(Ow
ensb
oro,
KY
)
(Cle
rmon
t-F
erra
nd, F
ranc
e)
(Col
lege
Sta
tion,
TX
)
Epi
cyte
Larg
e S
cale
Bio
logy
Mer
iste
m T
hera
peut
ics
Pro
dige
ne
Avi
geni
cs(A
then
s, G
A)
(Bur
linga
me,
CA
)
(Shr
ewsb
ury,
MA
)
(Pla
ntat
ion,
FL)
(Ann
Arb
or, M
I)
(Nan
tes,
Fra
nce)
Orig
en T
hera
peut
ics
Tra
nXen
oGen
Vira
gen
Gen
eWor
ks
Viv
alis
GT
C B
ioth
erap
eutic
s
PP
L T
hera
peut
ics
Bio
Pro
tein
(Par
is, F
ranc
e)
(Edi
nbur
gh, U
K)
(Fra
min
gham
, MA
)(T
hous
and
Oak
s, C
A)
(Lei
den,
Net
herla
nds)
uses
hum
an c
ells
(S. S
an F
ranc
isco
, CA
)ot
her
curr
ent b
iolo
gics
man
ufac
ture
rs;
Am
gen
Gen
ente
ch
Cru
cell
$150
*Com
pany
est
imat
es
$1−$
2$1
−$2
$0.0
5
Com
pani
es
Est
imat
ed c
ost
(cos
t/g r
aw m
ater
ial)*
Fig.
2C
ompa
nies
and
tech
nolo
gies
inbi
oman
ufac
turi
ng.A
com
pari
son
ofdi
ffere
ntex
pres
sion
syst
ems
show
sth
ebi
gdi
ffere
nces
inte
rms
ofco
sts,
rang
ing
from
US$
150
per
gram
for
CH
Oce
llsto
US$
0.05
per
gram
for
tran
sgen
icpl
ants
[11]
.(Se
eC
olor
Plat
ep.
xvi)
.

Biopharmaceuticals Expressed in Plants 39
their native form, as they are capable ofcarrying out post-translational modifica-tions required for the biological activity ofmany such proteins. These modificationscan be acetylation, phosphorylation, andglycosylation as well as others. Per se,there is no restriction to the kind ofproteins that can be expressed in plants:vaccines (e.g. pertussis or tetanus toxins),serum proteins (e.g. albumin), growth fac-tors (e.g. vascular endothelial growth factor(VEGF), erythropoietin), or enzymes (e.g.urokinase, glucose oxidase, or glucocere-brosidase). However, enzymes sometimeshave very complex cofactors, which are es-sential for their catalytic mode of action,but cannot be supplied by most expressionsystems. This is why, for the expression ofsome enzymes, expression systems withspecial features and characteristics need tobe developed [13]. Another very importantclass of proteins are the antibodies (e.g.scFv, Fab, IgG, or IgA). More than 100antibodies are currently used in clinicaltrials as therapeutics, drug delivery vehi-cles, in diagnostics and imaging, and indrug discovery research for both screeningand validation of targets [14]. Again, plantsare considered as the system of choicefor the production of antibodies (‘‘plan-tibodies’’) in bulk amounts at low costs.Since the initial demonstration that trans-genic tobacco (Nicotiana tabacum) is able toproduce functional IgG1 from mouse [15],full-length antibodies, hybrid antibodies,antibody fragments (Fab), and single-chainvariable fragments (scFv) have been ex-pressed in higher plants for a number ofpurposes. These antibodies can serve inhealth care and medicinal applications, ei-ther directly by using the plant as a foodingredient or as a pharmaceutical or di-agnostic reagent after purification fromthe plant material. In addition, antibodies
may improve plant performance, for ex-ample, by controlling plant disease or bymodifying regulatory and metabolic path-ways [16–19].
3.4SWOT Analysis Reveals a Ripe Market forPlant Expression Systems
When I analyzed the different expres-sion systems regarding their strengths,weaknesses, opportunities, and threats(SWOT), the advantages of plants and theirpotential to circumvent the worldwide ca-pacity limitations for protein productionbecame quite obvious (see Fig. 3). Compar-ison of transgenic animals, mammaliancell culture, plant expression systems,yeast, and bacteria shows certain advan-tages for each of the systems. In theorder in which the systems were just men-tioned, we can compare them in termsof their development time (speed). Trans-genic animals have the longest cycle time(18 months to develop a goat), followedby mammalian cell culture, plants, yeast,and bacteria (one day to transform E.coli). If one looks at operating and cap-ital costs, safety and scalability, the datashow that plants are beneficial: therefore,in the comparison (see Fig. 3) they areshown on the right-hand side already.But even for glycosylation, multimeric as-sembly and folding (where plants are notshown on the right-hand side, meaningother systems are advantageous), someplant expression systems are moving inthat direction. An example for this is themoss system from the company greenova-tion Biotech GmbH (Freiburg, Germany),which will be discussed in detail in theexample section. This system performsproper folding and assembly of even suchcomplex proteins like the homodimeric

40 3.4 SWOT Analysis Reveals a Ripe Market for Plant Expression Systems
Strengths• Access new manufacturing facilities• High production rates/high protein yield• Relatively fast ‘gene-to-protein’ time• Safety benefits; no hum. pathogens/no TSE• Stable cell lines/high genetic stability• Simple medium (water, minerals & light)• Easy purification (ion exchange vs. prot A)
TRENDS in Biotechnology Vol.20 No.12, 2002
Minus Plus
Opportunities • Reduce projected COGS • Escape capacity limitations • Achieve human-like glycosylation
Weaknesses• No approved products yet (but Phase III)• No final guidelines yet (but drafts available)
Threats• Food chain contamination• Segregation risk
Fig. 3 SWOT analysis of plant expressionsystems. Plant expression systems have a lot ofadvantages (plus) over other systems and aretherefore mostly shown on the right-hand side ofthe picture (Raskin I et al., Plants and humanhealth in the twenty-first century. Trends inBiotechnol. 2002 20, 522–531.). Herein differentsystems (transgenic animals, mammalian cellculture, plants, yeast, and bacteria) arecompared in terms of speed (how quickly theycan be developed), operating and capital costs
and so on, and plants are obviouslyadvantageous. Even for glycosylation, assemblyand folding, where plants are not shown on theright-hand side (meaning other systems areadvantageous), some plant expression systemsare moving in that direction (as will be shownexemplarily in the section for moss). Also, theweaknesses and threats can be dealt with, usingthe appropriate plant expression system [20].(See Color Plate p. xvii).
VEGF. Even the sugar pattern could suc-cessfully be reengineered from plant tohumanlike glycosylation.
In addition to the potential of perform-ing human glycosylation, plants also enjoythe distinct advantage of not harboringany pathogens, which are known to harmanimal cells (as opposed to animal cell cul-tures and products), nor do the productscontain any microbial toxins, TSE (Trans-missible Spongiform Encephalopathies),prions, or oncogenic sequences [21]. Infact, humans are exposed to a large, con-stant dose of living plant viruses in thediet without any known effects/illnesses.Plant production of protein therapeutics
also has advantages with regard to theirscale and speed of production. Plantscan be grown in ton quantities (usingexisting plant/crop technology, like com-mercial greenhouses), be extracted withindustrial-scale equipment, and producekilogram-size yields from a single plot ofcultivation. These economies of scale areexpected to reduce the cost of productionof pure pharmaceutical-grade therapeuticsby more than two orders of magnitudeversus current bacterial fermentation orcell culture reactor systems (plus rawmaterial COGS are estimated to be aslow as 10% of conventional cell cultureexpenses).

Biopharmaceuticals Expressed in Plants 41
Although a growing list of heterologousproteins were successfully produced in anumber of plant expression systems withtheir manifold advantages, there are alsoobvious downsides. One weakness is thatno product has been approved for themarket yet (but will be soon, since someare in Phase III clinical trials already, seeTable 1). The other weakness is that nofinal regulatory guidelines exist. But asmentioned before, regulatory authorities(Food and Drug Administration (FDA),European Medicine Evaluation Agency(EMEA), and Biotechnology RegulatoryService (BRS)) and the Biotechnology In-dustry Organization (BIO) have draftedguidelines on plant-derived biopharma-ceuticals (see Table 2) and have askedthe community for comments. The FDAhas also issued several PTC (Points ToConsider) guidelines about plant-based bi-ologics, and review of the July 2002 PTCconfirms that the FDA supports this fieldand highlights the benefits of plant ex-pression systems – including the absenceof any pathogens to man from plant ex-tracts. The main concerns of using plantexpression systems are societal ones about
environmental impacts, segregation risk,and contamination of the food chain.But these threats can be dealt with, us-ing non-edible plants (non-food, non-feed),applying advanced containment technolo-gies (GMP greenhouses, bioreactors) andavoiding open-field production.
Owing to the obvious strengths ofplant expression systems, there has beenexplosive growth in the number of start-upcompanies. Since the 1990s, a numberof promising plant expression systemshave been developed, and in response tothis ‘‘blooming field’’ big pharmaceuticalcompanies have become more interested.Now, the plant expression field is ‘‘ripe’’for strategic alliances, and, in fact, thelast year has seen several major biotechcompanies begin partnerships with suchplant companies. The selection of severalsuch partnerships shown in Table 1 clearlydemonstrates that, in general, there hasbeen sufficient experimentation with var-ious crops to provide the overall proof ofconcept that transgenic plants can producebiopharmaceuticals. However, and thiscan be seen in the table as well, the com-mercial production of biopharmaceuticals
Tab. 1 Plant-derived biopharmaceuticals in clinical trials
Company Partner Protein /indication Host Stage
Monsanto Guy’s Hospital London Anticaries antibody Corn Phase IIILarge Scale Biology Own product scFv (non-Hodgkin) Tobacco Phase IIIMeristem Therapeutics Solvay Pharmaceuticals Gastric lipase Corn Phase IILarge Scale Biology ProdiGene, Plant
BioscienceAnti-idiotype antibody Tobacco Phase I
Monsanto NeoRx Antitumor antibody Corn Phase IProdiGene Own product TGEV vaccine Corn Phase IEpicyte Pharmaceutical Dow, Centocor Anti-HSV antibody Corn Phase ICropTech Immunex Enbrel (arthritis) Tobacco PreclinicalCropTech Amgen Therapeutic antibodies Tobacco PreclinicalAltaGen Bioscience Inc. US Army + 3 biotechs Antibodies Potato PreclinicalMeristem Therapeutics CNRS Human lactoferrin Corn PreclinicalMPB Cologne GmbH Aventis CropScience Confidential Potato Preclinical

42 3.5 Risk Assessment and Contingency Measures
Tab. 2 Drafted guidelines on plant-derived biopharmaceuticals
Agency Guideline Status
BRS (BiotechnologyRegulatory Services)
‘‘Case study on plant-derived biologics’’for Office of Science and TechnologyPolicy/Council on EnvironmentalQuality
Released: Mar 5,2001
BIO (BiotechnologyIndustry Organization)
‘‘Reference Document for Confinementand Development of Plant-MadePharmaceuticals in the United States’’
Released: May17, 2002
EMEA (European MedicineEvaluation Agency)
‘‘Concept Paper on the Development of aCommittee for Proprietary MedicinalProducts (CPMP) Points to Consider onthe Use of Transgenic Plants in theManufacture of Biological MedicinalProducts for Human Use’’
Released: Mar01, 2001
FDA (Food and DrugAdministration)
‘‘Drugs, Biologics, and Medical DevicesDerived from Bioengineered Plants forUse in Humans and Animals’’
Issued: Sep 6,2002
EMEA (European MedicineEvaluation Agency)
‘‘Points To Consider Quality Aspects ofMedicinal Products containing activesubstances produced by stabletransgene expression in higher plants’’
Issued: Mar 13,2002
in transgenic plants is still in the earlystages of development and yet the mostadvanced products are in Phase III clinicaldevelopment.
3.5Risk Assessment and ContingencyMeasures
For a number of reasons, including theknowledge base developed on geneticallymodifying its genome, industrial pro-cesses for extracting fractionated productsand the potential for large-scale produc-tion, the preferred plant expression systemhas been corn. However, the use ofcorn touches on a potential risk: someenvironmental activist groups and tradeassociations are concerned about the effecton the environment and possible contam-ination of the food supply. These issues
are reflected in the regulatory guidelinesand have been the driving force to inves-tigate other plants as well. While manymature and larger companies have beenworking in this area for many years, thereare a number of newcomers that are de-veloping expertise as well. These smallercompanies are reacting to the concernsby looking at the use of non-edible plantsthat can be readily raised in greenhouses.All potential risks have to be assessedand contingency measures need to be es-tablished. Understanding the underlyingissues is mandatory to make sophisticateddecisions about the science and subse-quently on the development of appropriateplant expression systems for production ofbiopharmaceuticals.
Ongoing public fears from the foodindustry and the public, particularlyin Europe (‘‘Franken Food’’) couldhave spillover effects on plant-derived

Biopharmaceuticals Expressed in Plants 43
pharmaceuticals. Mistakes and misunder-standings have already cost the geneticallyenhanced grain industry hundreds of mil-lions of dollars [21]. The only way toprevent plant expression systems fromsuffering the same dilemma is to providethe public with appropriate information onemerging discoveries and newly developedproduction systems for biopharmaceuti-cals. Real and theoretical risks involvethe spread of engineered genes into wildplants, animals, and bacteria (horizontaltransmission). For example, if herbicide-resistance was transmitted to weeds, orantibiotic resistance was to be transmittedto bacteria, superpathogens could result. Ifthese genetic alterations were transmittedto their progeny (vertical transmission), anexplosion of the pathogens could cause ex-tensive harm. An example of this occurredseveral years ago, when it was feared thatpest-resistant genes had been transmittedfrom Bt corn to milkweed – leading to thewidespread death of Monarch butterflies.Although this was eventually not foundto be the case, the public outcry over theincident was a wake-up call to the possi-ble dangers of transgenic food technology.To avoid the same bad perception for bio-pharmaceuticals expressed in plants, thereis the need for thorough risk assessmentand contingency planning. One methodis the employment of all feasible safetystrategies to prevent spreading of engi-neered DNA (genetic drift), like a basiccontainment in a greenhouse environ-ment. Although no practical shelter cantotally eradicate insect and rodent intru-sion, this type of isolation is very effectivefor self-pollinators and those plants withsmall pollen dispersal patterns. The useof species-specific, fragile, or poorly trans-missible viral vectors is another strategy.Tobacco mosaic virus (TMV), for exam-ple, usually only infects a tobacco host.
It requires an injury of the plant to gainentry and cause infection. Destruction ofa field of TMV-transformed tobacco re-quires only plowing under or applicationof an herbicide. These factors prevent bothhorizontal and vertical transmission. Inaddition, there is no known incidence ofplant viruses infecting animal or bacterialcells. Another approach is to avoid stabletransgenic germ lines and therefore mostuses of transforming viruses do not involvethe incorporation of genes into the plantcell nucleus. By definition, it is almost im-possible for these genes to be transmittedvertically through pollen or seed. The en-gineered protein product is produced onlyby the infected generation of plants. An-other effective way to reduce the risk ofgenetic drift is the use of plants that donot reproduce without human aid. Themodern corn plant cannot reproduce with-out cultivation and the purposeful plantingof its seeds. If a plant may sprout fromgrain, it still needs to survive the wintering-over process and gain access to the properplanting depth. This extinction process isso rapid, however, that the errant loss ofan ear of corn is very unlikely to grow anew plant. Another very well-known ex-ample of self-limited reproduction is themodern banana. It propagates almost ex-clusively through vegetative cloning (i.e.via cuttings).
Pollination is the natural way for mostplants to spread their genetic information,make up new plants, and to deliver theiroffspring in other locations. The use ofplants with limited range of pollen dis-persal and limited contact with compatiblewild hosts therefore is also very effectiveto prevent genetic drift. Corn, for exam-ple, has pollen, which survives for only10 to 30 min and, hence, has an effectivefertilizing radius of less than 500 m. InNorth America, it has no wild-type relatives

44 3.5 Risk Assessment and Contingency Measures
with which it could cross-pollinate. Inaddition to being spatially isolated fromnearby cornfields, transgenic corn can be‘‘temporally isolated’’ by being plantedat least 21 days earlier or 21 days laterthan the surrounding corn, to ensure thatthe fields are not producing flowers atthe same time [11]. Under recent USDA(U.S. Department of Agriculture) regula-tions, the field must also be planted withequipment dedicated to the geneticallymodified crop. For soybeans, the situa-tion is different, since they are virtually100% self-fertilizers and can be planted invery close proximity to other plants withoutfear of horizontal spread. Another optionis the design of transgenic plants that haveonly sterile pollen or – more or less onlyapplicable for greenhouses – completelyprevent cross-pollination by covering theindividual plants. One public fear re-gards spreading antibiotic resistance fromone (transgenic donor) plant to otherwild-type plants or bacteria in the environ-ment. Although prokaryotic promoters forantibiotic-resistance are sometimes usedin the fabrication and selection of trans-genic constructs, once a transgene hasbeen stably incorporated into the plantgenome, it is under the control of plant(eukaryotic) promoter elements. Hence,antibiotic-resistance genes are unable topass from genetically altered plants intobacteria and remain functional. As statedearlier, another common fear is the cre-ation of a ‘‘super bug.’’ The chance ofcreating a supervirulent virus or bacteriumfrom genetic engineering is unlikely, be-cause the construction of expression cas-settes from viral or bacterial genomesinvolves the removal of the majority ofgenes responsible for the normal functionof these organisms. Even if a resultant or-ganism is somewhat functional, it cannot
compete for long in nature with normal,wild-type bacteria of the same species.
As one can see from the aforementionedsafety strategies, considerable effort isput into the reduction of any potentialrisk from the transgenic plant for theenvironment. In general, the scientificrisk can be kept at a minimum, ifcommon sense is applied – in accordancewith Thomas Huxley (1825–1895) that‘‘Science is simply common sense at itsbest.’’ For example, protein toxins (forvaccine production) should never be grownin food plants.
Additionally, the following can be em-ployed as a kind of risk management toprevent the inappropriate or unsafe use ofgenetically engineered plants [21]:
• An easily recognized phenotypic char-acteristic can be coexpressed in anengineered product (e.g. tomatoes thatcontain a therapeutic protein can beselected to grow in a colorless varietyof fruit).
• Protein expression can be induced onlyafter harvesting or fruit ripening. Forexample, CropTech’s (Blacksburg, VA,USA) inducible expression system intobacco, MeGA-PharM, leads to veryefficient induction upon leaf injury (har-vest) and needs no chemical inducers.This system possesses a fast inductionresponse and protein synthesis rate,thus leads to high expression levels withno aged product in the field (no environ-mental damage accumulation).
• Potentially antigenic or immunomodu-latory products can be induced to growin, or not to grow in, a certain plant tis-sue (e.g. root, leaf/stem, seed, or pollen).In this way, for example, farmers can beprotected from harmful airborne pollenor seed dusts.

Biopharmaceuticals Expressed in Plants 45
• Although no absolute system can pre-vent vandalism or theft of the transgenicplants, a very effective, cheap solutionhas been used quietly for many yearsnow in the United States. Plots of thesemodified plants are being grown withabsolutely no indication that they aredifferent from a routine crop. In theMidwest, for example, finding a trans-genic corn plot among the millions ofacres of concurrently growing grain isvirtually impossible. The only questionhere is, if this approach really helps fa-cilitating a fair and an open discussionwith the public. Asking the same ques-tion for the EU is not relevant: owingto labeling requirements, this approachwould not be feasible, as, in general, itis much more difficult to perform open-field studies with transgenic plants.
3.6Moving Plants to Humanlike Glycosylation
As discussed earlier, plant production oftherapeutic proteins has many advantagesover bacterial systems. One very impor-tant feature of plant cells is their capabilityof carrying out post-translational modifi-cations [22]. Since they are eukaryotes (i.e.have a nucleus), plants produce proteinsthrough an ER (Endoplasmatic Reticulum)pathway, adding sugar residues also tothe protein – a process called glycosylation.These carbohydrates help determine thethree-dimensional structures of proteins,which are inherently linked to their func-tion and their efficacy as therapeutics. Thisglycosylation also affects protein bioavail-ability and breakdown of the biophar-maceutical; for example, proteins lackingterminal sialic acid residues on their sugargroups are often targeted by the immunesystem and are rapidly degraded [23]. The
glycosylation process begins by targetingthe protein to the ER. During translationof mRNA (messenger RNA) into protein,the ribosome is attached to the ER, andthe nascent protein fed into the lumenof the ER as translation proceeds. Here,one set of glycosylation enzymes attachescarbohydrates to specific amino acids ofthe protein. Other glycosylation enzymeseither delete or add more sugars to thecore structures. This glycosylation processcontinues into the Golgi apparatus, whichsorts the new proteins, and distributesthem to their final destinations in the cell(see Fig. 4). Bacteria lack this ability andtherefore cannot be used to synthesize pro-teins that require glycosylation for activity.Although plants have a somewhat differentsystem of protein glycosylation from mam-malian cells, the differences are usually notproving to be a problem. Some proteins,however, require humanlike glycosylation(see Fig. 5) – they must have specific sugarstructures attached to the correct sites onthe molecule to be maximally effective [23].Therefore, some efforts are being madein modifying host plants in such a waythat they provide the protein with hu-man glycosylation patterns. One exampleof modifying a plant expression system inthis way is the transgenic moss, which willbe discussed in the next section.
3.7Three Promising Examples: Tobacco(Rhizosecretion, Transfection) and Moss(Glycosylation)
To further elaborate on improving gly-cosylation and downstream processing,three interesting plant expression systemswill be discussed. All systems share theadvantage of utilizing non-edible plants(non-food and non-feed) and can be kept ineither a greenhouse or a fermenter to avoid

46 3.7 Three Promising Examples: Tobacco (Rhizosecretion, Transfection) and Moss (Glycosylation)
Lumen
Golgi
Cytosol
Synthesis oflipid-linkedprecursor
Glycantransfer
Trimmingand
processing
Furthertrimming
Terminalglycosylation
ER
Fig. 4 The glycosylation pathway via ER andGolgi apparatus. In the cytosol carbohydrates areattached to a lipid precursor, which is thentransported into the lumen of the ER to finishcore glycosylation. This glycan is now attached tothe nascent, folding polypeptide chain (which issynthesized by ribosomes attached to the
cytosolic side of the ER from where ittranslocates into the lumen) and subsequentlytrimmed and processed before it is folded andmoved to the Golgi apparatus. Capping of theoligosaccharide branches with sialic acid andfucose is the final step on the way to a matureglycoprotein [23]. (See Color Plate p. xviii).
Bacteria Yeast Transgenicplants
Transgenicanimals
Nativeglycoproteins
N-glycolylneuraminic acid
N-acetylneuraminic acid
Mannose
Fucose
Galactose
Xylose
Peptide
N-acetylglucosamine
Fig. 5 Engineering plants to humanlike glycosylation. The first step to achieve humanlikeglycosylation in plants is to eliminate the plant glycosylation pattern, that is, the attachment ofβ-1-2-linked xylosyl and α-3-linked fucosyl sugars to the protein. Because these two residueshave allergenic potential, the corresponding enzymes xylosyl and fucosyl transferase areknocked out. In case galactose is relevant for the final product, galactosyl transferase is insertedinto the host genome. Galactose is available in the organism so that this single-gene insertionis sufficient to ensure galactosylation [24]. (See Color Plate p. xviii).

Biopharmaceuticals Expressed in Plants 47
any segregation risk. Another obvious ad-vantage is secretion of the protein into themedium so that no grinding or extractionis required. This is very important in lightof downstream processing: protein purifi-cation is often as expensive as the bioman-ufacturing and should never be underesti-mated in the total COGS equation [22].
3.8Harnessing Tobacco Roots to SecreteProteins
Phytomedics (Dayton, NJ, USA) uses to-bacco plants as an expression systemfor biopharmaceuticals. Besides the ad-vantage of being well characterized andused in agriculture for some time, to-bacco has a stable genetic system, pro-vides high-density tissue (high proteinproduction), needs only simple medium,
and can be kept in a greenhouse (seeFig. 6). Optimized antibody expression canbe rapidly verified using transient ex-pression assays (short development time)in the plants before creation of trans-genic suspension cells or stable plantlines (longer development time). Differ-ent vector systems, harboring targetingsignals for subcellular compartments, areconstructed in parallel and used for tran-sient expression. Applying this screeningapproach, high expressing cell lines canrapidly be identified. For example, trans-genic tobacco plants, transformed withan expression cassette containing theGFP (Green Fluorescent Protein) genefused to an aps (amplification-promotingsequence), had greater levels of corre-sponding mRNAs and expressed pro-teins compared to transformants lackingaps [25]. Usually, downstream processing(isolation/extraction and purification of
• Root secretion, easy recovery• Greenhouse-contained tanks• High-density tissue• Salts and water only• Tobacco is well characterized• Stable genetic system
Phytomedics (tobacco):
Fig. 6 Secretion of the biopharmaceuticals via tobacco roots. The tobacco plants aregenetically modified in such a way, that the protein is secreted via the roots into themedium (‘‘rhizosecretion’’). In this example, the tobacco plant takes up nutrients andwater from the medium and releases GFP (green fluorescent protein). Examination ofroot-cultivation medium by its exposure to near-ultraviolet illumination reveals the brightgreen-blue fluorescence characteristics of GFP in the hydroponic medium (left flask inpanel lower left edge). The picture also shows a schematic drawing of the hydroponictank, as well as tobacco plants at different growth stages, for example, callus,–fullygrown and greenhouse plantation [24]. (See Color Plate p. xix).

48 3.9 High Protein Yields Utilizing Viral Transfection
the target protein) is limiting for sucha system, for example, if the proteinhas to be isolated from biochemicallycomplex plant tissues (e.g. leaves), thiscan be a laborious and expensive pro-cess and a major obstacle to large-scaleprotein manufacturing. To overcome thisproblem, secretion-based systems utiliz-ing transgenic plant cells or plant organsaseptically cultivated in vitro would beone solution. However, in vitro systemscan be expensive, slow growing, unstable,and relatively low yielding. This is whyanother interesting route was followed.Secretion of molecules is a basic func-tion of plant cells and organs in plants,and is especially developed in plant roots.In order to take up nutrients from thesoil, interact with other soil organisms,and defend themselves against numerouspathogens, plant roots have evolved sophis-ticated mechanisms based on the secre-tion of different biochemicals (includingproteins like toxins) into their neighbour-hood (rhizosphere). In fact, Borisjuk andcoworkers [26] could demonstrate that rootsecretion can be successfully exploited forthe continuous production of recombinantproteins in a process termed ‘‘rhizosecre-tion.’’ Here, an endoplasmic reticulumsignal peptide is fused to the recombi-nant protein, which is then continuouslysecreted from the roots into a simple hy-droponic medium (based on the naturalsecretion from roots of the intact plants).The roots of the tobacco plant are sit-ting in a hydroponic tank (see Fig. 6),taking up water and nutrients and con-tinuously releasing the biopharmaceutical.By this elegant set up, downstream pro-cessing becomes easy and cost-effective,and also offers the advantage of continu-ous protein production that integrates thebiosynthetic potential of a plant over itslifetime and might lead to higher protein
yields than single-harvest and extractionmethods. Rhizosecretion is demonstratedin Fig. 6, showing a transgenic tobaccoplant expressing GFP and releasing it intothe medium.
3.9High Protein Yields Utilizing ViralTransfection
ICON Genetics (Halle, Germany) has de-veloped a protein-production system thatrelies on rapid multiplication of viral vec-tors in an infected tobacco plant (seeFig. 7). Viral transfection systems offera number of advantages, such as veryrapid (1 to 2 week) expression time,possibility of generating initial milligramquantities within weeks, high expressionlevels, and so on. However, the existingviral vectors, such as TMV-based vectorsused by, for example, Large Scale BiologyCorp. (Vacaville, CA, USA) for productionof single-chain antibodies for treatmentof non-Hodgkin lymphoma (currently inPhase III clinical trials, see Table 1), hadnumerous shortcomings, such as inabilityto express genes larger than 1 kb, inabil-ity to coexpress two or more proteins(a prerequisite for production of mon-oclonal antibodies, because they consistof the light and heavy chains, which areexpressed independently and are subse-quently assembled), low expression levelin systemically infected leaves, and so on.ICON has solved many of these problemsby designing a process that starts withan assembly of one or more viral vec-tors inside of a plant after treating theleaves with agrobacteria, which deliverthe necessary viral vector components.ICON’s proviral vectors provide advan-tages of fast and high-yield amplification

Biopharmaceuticals Expressed in Plants 49
• Viral transfection• Fast development• High-protein yields• Coexpression of genes
ICON Genetics (tobacco):
RbcSCP
GFP
Expression in plant tissue
Coomassie gel
I
IVIII
II
RbcL
Fig. 7 Viral transfection of tobacco plants. This new generation platform for fast (1 to 2 weeks),high-yield (up to 5 g per kilogram of fresh leaf weight) production of biopharmaceuticals isbased on proviral gene amplification in a non-food host. Antibodies, antigens, interferons,hormones, and enzymes could successfully be expressed with this system. The picture showsdevelopment of initial symptoms on a tobacco following the agrobacterium-mediated infectionwith viral vector components that contain a GFP gene (I); this development eventually leads toa systemic spread of the virus, literally converting the plant into a sack full of protein of interestwithin two weeks (II). The system allows to coexpress two proteins in the same cell, a featurethat allows expression of complex proteins such as full-length monoclonal antibodies. Panel IIIand IV show the same microscope section with the same cells, expressing green fluorescentprotein (III) and red fluorescent protein (IV) at the same time. The yield and total proteinconcentration achievable are illustrated by a Coomassie gel with proteins in the system: GFP(protein of interest), CP (coat protein from wild-type virus), RbcS and RbcL (small and largesubunit of ribulose-1,5-bisphosphate carboxylase) [24]. (See Color Plate p. xx).
processes in a plant cell, simple and inex-pensive assembly of expression cassettesin planta, and full control of the process.The robustness of highly standardized pro-tocols allow to use inherently the same safeprotocols for both laboratory-scale as wellas industrial production processes. In thissystem, the plant is modified transientlyrather than genetically and reaches thespeed and yield of microbial systems whileenjoying post-translational capabilities ofplant cells. De- and reconstructing of thevirus adds some safety features and alsoincreases efficiency. There is no ‘‘physiol-ogy conflict,’’ because the ‘‘growth phase’’is separated from the ‘‘production phase,’’so that no competition occurs for nutrients
and other components required for growthand also for expression of the biopharma-ceutical at the same time.
This transfection-based platform allowsto produce proteins in a plant host at acost of US$1 to 10 per gram of crudeprotein. The platform is essentially freefrom limitations (gene insert size limit,inability to express more than one gene) ofcurrent viral vector-based platforms. Theexpression levels reach 5 g per kilogram offresh leaf tissue (or some 50% of totalcellular protein!) in 5 to 14 days afterinoculation. Since the virus process (inaddition to superhigh production of itsown proteins, including the protein ofinterest) leads to the shut-off of the other

50 3.10 Simple Moss Performs Complex Glycosylation
cellular protein synthesis, the amount ofprotein of interest in the initial extract isextremely high (Fig. 7). It thus results inreduced costs of downstream processing.Milligram quantities can be producedwithin two weeks, gram quantities in 4to 6 months, and the production systemis inherently scalable. A number ofhigh-value proteins have been successfullyexpressed, including antibodies, antigens,interferons, hormones, and enzymes.
3.10Simple Moss Performs ComplexGlycosylation
Greenovation Biotech GmbH (Freiburg,Germany) has established an innovativeproduction system for human proteins.The system produces pharmacologicallyactive proteins in a bioreactor, utilizinga moss (Physcomitrella patens) cell culturesystem with unique properties (see Fig. 8).It was stated before that post-translationalmodifications for some proteins are crucial
to gain complete pharmacological activ-ity. Since moss is the only known plantsystem that shows a high frequency of ho-mologous recombination, this is a highlyattractive tool for production strain de-sign. By establishing stable integrationof foreign genes (gene knockout andnew transgene insertion) into the plantgenome, it can be programmed to pro-duce proteins with modified glycosyla-tion patterns that are identical to animalcells. The moss is photoautotrophic andtherefore only requires simple media forgrowth, which consist essentially of wa-ter and minerals. This reduces costs andalso accounts for significantly lower in-fectious and contamination risks, but inaddition to that, the system has somemore advantages:
• The transient system allows productionof quantities for a feasibility studywithin weeks – production of a stableexpression strain takes 4 to 6 months.
• On the basis of transient expressiondata, the yield of stable production lines
• Simple medium (photoautotrophic plant needs only water and minerals)• Robust expression system (good expression levels from 15 to 25°C)
• Secretion into medium via human leader sequence (broad pH range: 4−8)• Easy purification from low-salt medium via ion exchange
• Easy genetic modifications to cell lines• Stable cell lines/high genetic stability
• Codon usage like human (no changes required)• Inexpensive bioreactors from the shelf
• Nonfood plant (no segregation risk)• Good progress on genetic modification of glycosylation
pathways (plant to human)
Greenovation (moss system):
Fig. 8 Greenovation use a fully contained moss bioreactor. This company hasestablished an innovative production system for human proteins. The systemproduces pharmacologically active proteins in a bioreactor, utilizing a moss(Physcomitrella patens) cell culture system with unique properties [24]. (See ColorPlate p. xxi).
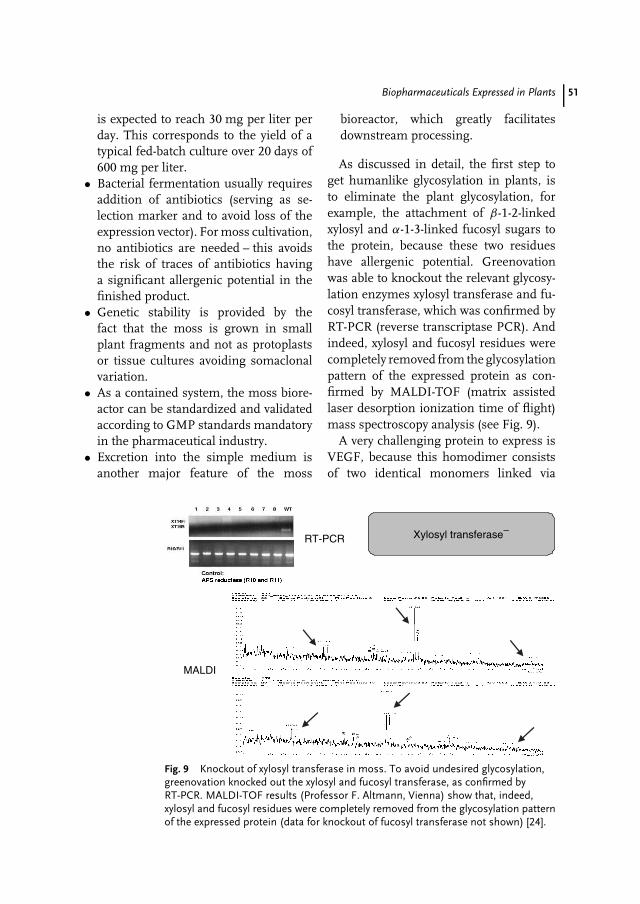
Biopharmaceuticals Expressed in Plants 51
is expected to reach 30 mg per liter perday. This corresponds to the yield of atypical fed-batch culture over 20 days of600 mg per liter.
• Bacterial fermentation usually requiresaddition of antibiotics (serving as se-lection marker and to avoid loss of theexpression vector). For moss cultivation,no antibiotics are needed – this avoidsthe risk of traces of antibiotics havinga significant allergenic potential in thefinished product.
• Genetic stability is provided by thefact that the moss is grown in smallplant fragments and not as protoplastsor tissue cultures avoiding somaclonalvariation.
• As a contained system, the moss biore-actor can be standardized and validatedaccording to GMP standards mandatoryin the pharmaceutical industry.
• Excretion into the simple medium isanother major feature of the moss
bioreactor, which greatly facilitatesdownstream processing.
As discussed in detail, the first step toget humanlike glycosylation in plants, isto eliminate the plant glycosylation, forexample, the attachment of β-1-2-linkedxylosyl and α-1-3-linked fucosyl sugars tothe protein, because these two residueshave allergenic potential. Greenovationwas able to knockout the relevant glycosy-lation enzymes xylosyl transferase and fu-cosyl transferase, which was confirmed byRT-PCR (reverse transcriptase PCR). Andindeed, xylosyl and fucosyl residues werecompletely removed from the glycosylationpattern of the expressed protein as con-firmed by MALDI-TOF (matrix assistedlaser desorption ionization time of flight)mass spectroscopy analysis (see Fig. 9).
A very challenging protein to express isVEGF, because this homodimer consistsof two identical monomers linked via
MALDI
Xylosyl transferase−RT-PCR
Fig. 9 Knockout of xylosyl transferase in moss. To avoid undesired glycosylation,greenovation knocked out the xylosyl and fucosyl transferase, as confirmed byRT-PCR. MALDI-TOF results (Professor F. Altmann, Vienna) show that, indeed,xylosyl and fucosyl residues were completely removed from the glycosylation patternof the expressed protein (data for knockout of fucosyl transferase not shown) [24].
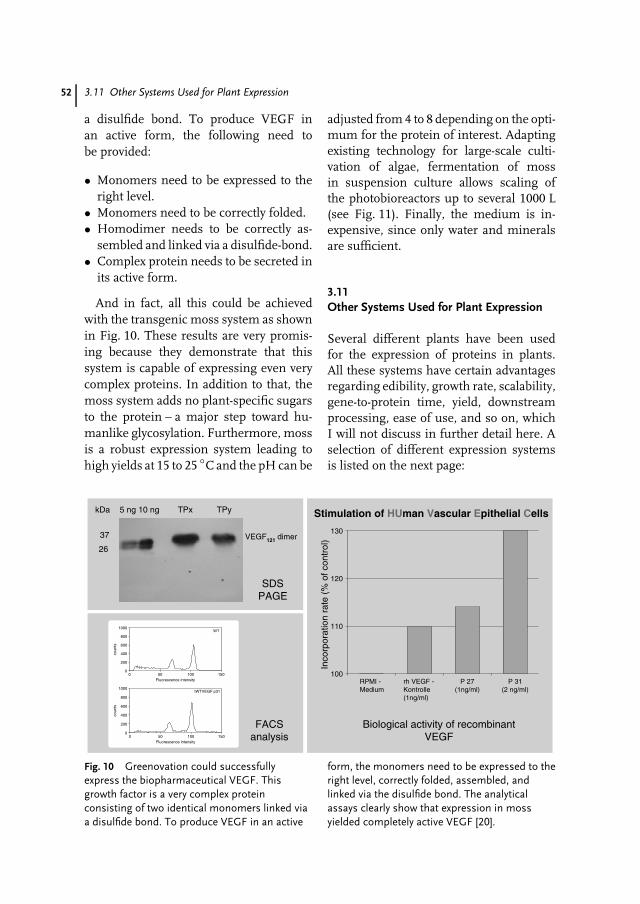
52 3.11 Other Systems Used for Plant Expression
a disulfide bond. To produce VEGF inan active form, the following need tobe provided:
• Monomers need to be expressed to theright level.
• Monomers need to be correctly folded.• Homodimer needs to be correctly as-
sembled and linked via a disulfide-bond.• Complex protein needs to be secreted in
its active form.
And in fact, all this could be achievedwith the transgenic moss system as shownin Fig. 10. These results are very promis-ing because they demonstrate that thissystem is capable of expressing even verycomplex proteins. In addition to that, themoss system adds no plant-specific sugarsto the protein – a major step toward hu-manlike glycosylation. Furthermore, mossis a robust expression system leading tohigh yields at 15 to 25 ◦C and the pH can be
adjusted from 4 to 8 depending on the opti-mum for the protein of interest. Adaptingexisting technology for large-scale culti-vation of algae, fermentation of mossin suspension culture allows scaling ofthe photobioreactors up to several 1000 L(see Fig. 11). Finally, the medium is in-expensive, since only water and mineralsare sufficient.
3.11Other Systems Used for Plant Expression
Several different plants have been usedfor the expression of proteins in plants.All these systems have certain advantagesregarding edibility, growth rate, scalability,gene-to-protein time, yield, downstreamprocessing, ease of use, and so on, whichI will not discuss in further detail here. Aselection of different expression systemsis listed on the next page:
WT
0
200
400
600
800
1000
0 50 100 150Fluorescence intensity
coun
ts
tWTVEGF p31
0
200
400
600
800
1000
0 50 100 150Fluorescence intensity
coun
ts
FACSanalysis
kDa 5 ng 10 ng TPx TPy
37
26
VEGF121 dimer
SDSPAGE
Stimulation of HUman Vascular Epithelial Cells
RPMI - Medium
rh VEGF -Kontrolle(1ng/ml)
P 27(1ng/ml)
P 31(2 ng/ml)
100
110
120
130
Inco
rpor
atio
n ra
te (
% o
f con
trol
)
Biological activity of recombinantVEGF
Fig. 10 Greenovation could successfullyexpress the biopharmaceutical VEGF. Thisgrowth factor is a very complex proteinconsisting of two identical monomers linked viaa disulfide bond. To produce VEGF in an active
form, the monomers need to be expressed to theright level, correctly folded, assembled, andlinked via the disulfide bond. The analyticalassays clearly show that expression in mossyielded completely active VEGF [20].

Biopharmaceuticals Expressed in Plants 53
30 L pilot reactor for moss Two weeks after incubation
Fig. 11 Scaling of photobioreactors up to several 1000 L. The moss bioreactor isbased on the cultivation of Physcomitrella patens in a fermenter. The mossprotonema is grown under photoautotrophic conditions in a medium that consistsessentially of water and minerals. Light and carbon dioxide serve as the only energyand carbon sources. Cultivation in suspension allows scaling of thephotobioreactors up to several 1000 L. Adaptation of existing technology forlarge-scale cultivation of algae is done in cooperation with the Technical Universityof Karlsruhe. Courtesy of greenovation Biotech GmbH (Freiburg, Germany) andProfessor C. Posten, Technical University (Karlsruhe, Germany). (See Color Platep. xxi).
Alfalfa Ethiopianmustard
Potatoes
Arabidopsis Lemna RiceBanana Maize SoyabeanCauliflower Moss TomatoesCorn Oilseeds Wheat
Some of these systems have been usedfor research on the basis of their ease oftransformation, well-known characteriza-tion, and ease to work with. However, theyare not necessarily appropriate for commer-cial production. Which crop is ultimatelyused for full-scale commercial productionwill depend on a number of factors [21]including
• time to develop an appropriate system(gene-to-protein);
• section of the plant expressing theproduct/possible secretion;
• cost and potential waste products fromextraction;
• ‘‘aged’’ product/ease of storage;• long-term stability of the storage tissue;• quantities of protein needed (scale of
production).
Depending on the genetic complexityand ease of manipulation, the develop-ment time to produce an appropriatetransgenic plant for milligram productionof the desired protein can vary from 10to 12 months in corn as compared to onlyweeks in moss. Estimates for full GMPproduction in corn are 30 to 36 monthsand approximately 12 months for moss.Expression of the protein in various tissuesof the plant can result in a great variation inyield. Expression in the seed can often leadto higher yields than in the leafy portion ofthe plant. This is another explanation forthe high interest in using corn, which has arelatively high seed-to-leaf ratio. Extraction

54 3.13 Conclusion
from leaf can be costly as it contains ahigh percentage of water, which could re-sult in unavoidable proteolysis during theprocess. Proteins stored in seeds can bedesiccated and remain intact for long peri-ods of time. The purification and extractionof the protein is likely to be done by adapta-tions of current processes for the extractionand/or fractionation. For these reasons, itis anticipated that large-scale commercialproduction of recombinant proteins willinvolve grain and oilseed crops such asmaize, rice, wheat, and soybeans. On thebasis of permits for open-air test plotsissued by the USDA for pharmaceuticalproteins and industrial biochemicals, cornis the crop of choice for production with73% of the permits issued. The other majorcrops are soybeans (12%), tobacco (10%)and rice (5%).
In general, the use of smaller plants thatcan be grown in greenhouses is an effectiveway of producing the biopharmaceuticalsand alleviating concerns from environ-mental activist groups that the transgenicplant might be harmful to the environ-ment (food chain, segregation risk, geneticdrift, etc.).
3.12Analytical Characterization
Validated bioanalytical assays are essentialand have to be developed to characterizethe biopharmaceuticals during the produc-tion process (e.g. in-process control) andto release the final product for use as adrug in humans. These assays are appliedto determine characteristics such as pu-rity/impurities, identity, quantity, stability,specificity, and potency of the recombi-nant protein during drug development.Since the very diverse functions of dif-ferent proteins heavily depend on their
structure [27], one very valuable parameterin protein characterization is the elucida-tion of their three-dimensional structure.Although over the last couple of years a lotof efforts were put into method improve-ment for the elucidation of protein struc-tures (during my PhD thesis I was alsoworking in this fascinating field togetherwith my boss Professor Robert Huber [25],Nobel Prize Laureate in 1988 ‘‘for thedetermination of the three-dimensionalstructure of a photosynthetic reaction cen-tre’’) it is still very time-consuming to solvethe 3D structure of larger proteins. This iswhy despite the high degree of informationthat can be obtained from the protein struc-ture, this approach cannot be applied ona routine basis. Therefore, tremendous ef-forts are put into the development of otherassays to guarantee that a potent biophar-maceutical drug is indeed ready for usein humans. A comprehensive overview isgiven in this textbook in Chapter 6 (JuttaHaunschild and Titus Kretzschmar ‘‘Char-acterization and bioanalytical aspects ofrecombinant proteins as pharmaceuticaldrugs’’).
3.13Conclusion
The production of protein therapeuticsfrom transgenic plants is becoming areality. The numerous benefits offeredby plants (low cost of cultivation, highbiomass production, relatively fast gene-to-protein time, low capital and operatingcosts, excellent scalability, eukaryotic post-translational modifications, low risk ofhuman pathogens, lack of endotoxins,as well as high protein yields) virtu-ally guarantee that plant-derived proteinswill become more and more commonfor therapeutic uses. Taking advantage

Biopharmaceuticals Expressed in Plants 55
of plant expression systems, the avail-ability of cheap protein-based vaccines inunderdeveloped countries of the world isin the near future. The cost of very expen-sive hormone therapies (erythropoietin,human growth hormone, etc.) could falldramatically within the next decade dueto the use of, for example, plant expres-sion systems. Fears about the risks of theplant expression technology are real andwell founded, but with a detailed under-standing of the technology, it is possibleto proactively address these safety issuesand create a plant expression industry al-most free of mishaps. For this purpose,the entire set up, consisting of the specificplant expression system and the proteinbeing produced, needs to be analyzed andits potential risks assessed on a case-by-case basis. As plant-derived therapeuticsbegin to demonstrate widespread, tangi-ble benefits to the population, and asthe plant expression industry develops alonger safety track record, public accep-tance of the technology is likely to improvecontinuously. Plants are by far the mostabundant and cost-effective renewable re-source uniquely adapted to complex bio-chemical synthesis. The increasing cost ofenergy and chemical raw materials, com-bined with the environmental concernsassociated with conventional pharmaceu-tical manufacturing, will make plants evenmore compatible in the future. With thewords of Max Planck (1858–1947) ‘‘Howfar advanced Man’s scientific knowledgemay be, when confronted with Nature’simmeasurable richness and capacity forconstant renewal, he will be like a mar-veling child and must always be preparedfor new surprises,’’ we will definitely dis-cover more fascinating features of plantexpression systems. But there is no needto wait: combining the advantages of sometechnologies that we have in hand by now
could already lead to the ultimate plantexpression system. This is what we shouldfocus on, because, then, at the dawn ofthis new millennium, this would for thefirst time yield large-enough amounts ofbiopharmaceuticals to treat everybody onour planet!
Acknowledgments
I would like to thank the companies green-ovation Biotech GmbH (Freiburg, Ger-many), ICON Genetics (Halle, Germany),and Phytomedics (Dayton, NJ, USA) forproviding some data and figures to preparethis manuscript.
References
1. K. J. Morrow, Genet. Eng. News 2002, 22,34–39.
2. The Context Network, BiopharmaceuticalProduction in Plants, Biopharma Prospectus,West Des Moines, IA, USA, Copyright2002.
3. L. Davies, J. Plieth, Scr. Mag. 2001, 10,25–29.
4. Arthur D. Little, Inc. (ADL), Cambridge, MA,USA, AgIndustries Research, Copyright2002.
5. K. Garber, Nat. Biotechnol. 2001, 19,184–185.
6. R. L. Evangelista, A. R. Kusnadi, J. A. How-ard et al., Biotechnol. Prog. 1988, 14, 607–614.
7. A. S. Ponstein, T. C. Verwoerd, J. Pen,Production of enzymes for industrialuse in Engineering Plants for CommercialProducts and Applications (Eds.: G. B. Collins,R. J. Sheperd), New York Academy ofSciences, New York, 1996, pp. 91–98,Vol. 792.
8. G. Giddings, G. Allison, D. Brooks et al.,Nat. Biotechnol. 2000, 18, 1151–1155.
9. R. Fischer, N. Emans, Transgenic Res. 2000,9, 279–299.
10. A. Kusnadi, Z. L. Nikolov, J. A. Howard, Bio-technol. Bioeng. 1997, 56, 473–484.

56 3.13 Conclusion
11. A. Dove, Nat. Biotechnol. 2002, 20, 777–779.12. J. Knablein, Transport Processes in Bioreactors
and Modern Fermentation Technologies,Lecture at University of Applied Sciences,Emden, Germany, 2002.
13. J. Knablein, Isolation, Cloning And Sequencingof the Respiratory Operon of Rhodobactercapsulatus and the Development of aGeneral Applicable System for the HomologueExpression of Difficult-to-Express Proteins,ISBN 3-933083-23-0, Hieronymus Verlag,Munchen, 1997.
14. Drug & Market Development Publications,Antibody Engineering: Technologies, Applica-tions and Business Opportunities, Westbor-ough, MA, USA, Copyright 2003.
15. A. Hiatt, R. Cafferkey, K. Bowdish, Nature1989, 342, 76–78.
16. U. Conrad, U. Fiedler, Plant Mol. Biol. 1994,26, 1023–1030.
17. J. K. C. Ma, M. B. Hein, Plant Physiol. 1995,109, 341–346.
18. M. D. Smith, Biotechnol. Adv. 1996, 14,267–281.
19. G. C. Whitelam, W. Cockburn, Trends PlantSci. 1996, 1, 268–272.
20. J. Knablein, Biotech: A New Era in theNew Millennium – From Plant Fermentationto Plant Expression of Biopharmaceuticals,PDA International Congress, Prague, CzechRepublic, 2003.
21. Technology Catalysts International Corpo-ration, Biopharmaceutical Farming, FallsChurch, VA, USA, Copyright 2002.
22. I. Raskin, B. Fridlender et al., Trends Bio-technol. 2002, 20, 522–531.
23. A. Dove, Nat. Biotechnol. 2001, 19, 913–917.24. J. Knablein, Biotech: A New Era in the New
Millennium – Biopharmaceutic Drugs Manu-factured in Novel Expression Systems, 21.DECHEMA-Jahrestagung der Biotechnolo-gen, Munich, Germany, 2003.
25. N. V. Borisjuk, I. Raskin et al., Nat.Biotechnol. 2000, 18, 1303–1306.
26. N. V. Borisjuk, I. Raskin et al., Nat.Biotechnol. 1999, 17, 466–469.
27. M. J. Romao, J. Knablein, R. Huber et al.,Prog. Biophys. Mol. Biol. 1998, 68, 121–144.
28. J. Knablein, R. Huber et al., J. Mol. Biol. 1997,270, 1–7.

Part IIIndustrial Development andProduction Process
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

59
4Scientific, Technical andEconomic Aspects of VaccineResearch and Development
Jens-Peter GregersenChiron-Behring GmbH, Marburg, Germany
4.1Introduction
Vaccine research and vaccine developmentare commonly combined by the term R&Dbecause, in practice, these two differentdisciplines cannot be easily separated. Vac-cine research and development have muchin common: they both use the same tech-nical language, apply very similar methodsand tools, and have the same ultimate goal,but there is also a fundamental difference,as the underlying motivating factors, work-ing habits, and the final output and resultsare entirely different. Research is mainlymotivated by, and aiming at, scientific pub-lications, which are best achieved by newmethods, inventions, and discoveries. Assoon as these have been published, a re-searcher’s attention must turn to anotherand new subject. Developers normally starttheir work when new discoveries or inven-tions have been made and may well workon one and the same objective for an entiredecade without publishing anything. Theyare not aiming at inventions; their inten-tion must be to arrive at innovations, thatis, products that will have an impact onour daily life. For the researcher, a vaccine
could be an antigen or a preparation thathas the potential of eliminating or inhibit-ing microorganisms. In order to convertthis into a useful vaccine, developers mustthen add several other dimensions to theresearch product, namely quality, safety, aspecifically defined clinical efficacy, andpractical utility. Building practical util-ity into a product is probably the mostdemanding or far-reaching one of thosefour categories. It encompasses and com-bines almost any aspect of the product,including its local reactivity, acceptableapplication schemes with only few vaccina-tions, a proven and perceived effectiveness,comfortable presentation forms, formu-lations that guarantee good stability andshelf-lives, and, of course, adequate prod-uct prices.
4.2From the Research Concept to aDevelopment Candidate
Concepts for new vaccines arise fromresearch and are based on combined sci-entific findings collected over many yearsand by various scientific institutions and
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

60 4.2 From the Research Concept to a Development Candidate
disciplines. New vaccine concepts are regu-larly presented and proposed in large num-bers by scientific publications or patentapplications, but these concepts rarely re-sult in new vaccines. After being tested inmice, most concepts slowly fade, since theoriginal results cannot be reproduced un-der more practical conditions or turn outto be insufficiently effective to justify addi-tional work. On the other hand, there arealso organizational and financial aspectsthat represent serious hurdles. Most aca-demic institutions and scientists simplydo not have adequate resources to performvaccine studies in specific models or evenin monkeys or primates. Whereas vac-cine antigen candidates can be designedand made by only one or a few indi-viduals, studying these more intensivelywould normally require other specialists,specific facilities, and, of course, muchmore money. The initial research projectnow competes for scarce resources andneeds very convincing data to make it tothe next stage.
Scientific collaborations across institu-tional walls are an almost absolute pre-requisite for continuing projects beyondtesting in small laboratory animals andin order to proceed into a more intensiveand application-oriented research phase.During this secondary research phase,promising concepts are taken up, repro-duced, and improved until finally – andin only very few cases – a viable productand development concept can be put to-gether. Almost invariably the efficacy of thecandidate vaccine needs to be improvedand made more reliable. For many newindications, even the tools and modelsmust be established first, by which im-munological effects or protection can beadequately measured.
For those few candidates that remainattractive after being studied in a more
reliable way or in better models, it willthen be important to assess the technicaland economic aspects of the vaccinecandidate very carefully. As these stronglydepend on available facilities, generalexpertise, and specific experience withcertain techniques needed, these aspectsare normally evaluated by the developingorganization during a project evaluationor ‘‘predevelopment’’ phase. At the end ofthis phase, a development concept shouldbe available, which at least fulfills thefollowing three criteria:
1. There should be sufficient evidencethat the vaccine candidate is effectiveand protective in humans or in thetarget animal species. This normallypresupposes that meaningful animalmodels have been established and thatthe vaccine has been tested successfully.
2. There should be a defined technicalbase or verified options by which thevaccine can be reliably and safely pro-duced on a large scale. This includes,for example, cell culture or expressionsystems, purification schemes, and for-mulations that are qualified for theproduction of pharmaceutical productsand do not contain hazardous compo-nents that cannot be removed duringlater process steps.
3. The expected product cost and theresulting sales prices must be in balancewith the envisaged benefit of using thevaccine and expected revenues shouldbe able to recoup the development costin a reasonable period of time.
Thus, there should be rather clear ideas asto how the vaccine is to be manufacturedand how it is characterized in its mainqualities. If this base is not yet known oris based only on assumptions, a targetedproduct development in its strict sense is

Scientific, Technical and Economic Aspects of Vaccine Research and Development 61
not possible, as neither the way to go northe target or end result are known. In thiscase, the project should still be consideredto be a research project. But particularly inthe case of vaccines, development projectsare frequently started with many uncer-tainties, assumptions, and compromises,as vaccines are highly complex compo-sitions, which cannot be characterizedentirely and completely by analytical meth-ods. Vaccines are products that are definedto a great deal by the process by which theyare made, by the analytical tools by whichthey are tested, and even by the facili-ties in which they are manufactured. Asa consequence, most vaccine developmentprojects have no clear starting point and re-search and process development activitiesrun in parallel. Although partly impossible,this should be avoided as far as possi-ble, as development activities need manymore people and are considerably moreexpensive than research. No developing or-ganization has sufficient resources to runnumerous complex development projectsin parallel or to change the direction of adevelopment again and again. By definingadequate criteria and by a proper projectorganization, critical aspects of a develop-ment project can be identified early, so that
these are evaluated during the applied re-search phase prior to the onset of productdevelopment.
4.3Vaccine Research Projects
An excellent overview of ongoing researchactivities for vaccines is provided by theJordan Reports issued by the US NationalInstitutes of Health [1]. According to thelatest issue of these reports, the number ofvaccine R&D projects in the United Statesin the year 2000 amounted to more than500 projects. Almost one-third of thesewere various efforts to develop vaccinesagainst AIDS. A list of the main targetindications pursued by recent vaccineresearch and development efforts is givenin Table 1.
The top positions of the vaccine research‘‘hit list’’ have not changed very much overthe past few decades. Well-known viral andbacterial infections continue to occupy themost prominent positions. However, thenumber of individual projects for manyof these vaccine indications has increasedconsiderably. The simple reason for thisis that formerly complete microorganisms
Tab. 1 Main infectious agents or targets for new vaccines in advanced R&D
Viruses Bacteria Parasites Tumours
HIV/AIDS Streptococcus Malaria B-cell lymphomasHepatitis C virus Helicobacter pylori Leishmania MelanomasHerpes simplex viruses Borrelia/Lyme disease Schistosoma Prostata carcinomaCytomegalovirus Salmonella Toxoplasma CEA-tumorsHRSV Enterotoxigenic E. coli TrypanosomaParainfluenza ShigellaRotavirus
Note: HIV/AIDS: Human immunodeficiency virus/acquired immunodeficiency syndrome;HRSV: Human respiratory syncytialvirus; CEA: Carcino-embryonal antigen: an antigen that isfrequently found on colorectal, bronchial, and breast cancer cells.

62 4.4 Scientific Challenges of Vaccine R&D
or subfractions thereof, but rarely purifiedsingle antigens, had to be used asvaccine candidates. Modern molecularbiology and recombinant techniques resultin individual antigens or even singleepitope peptides, which may be variedor combined by almost endless options.Of course, this increases the number ofcandidates significantly and offers manynew chances and possibilities, but itdoes not necessarily increase the chancesof success for each individual approach.Molecular biology has not only openedup various new possibilities to approachantiparasite vaccines and tumor vaccinesbut also, in these particularly complexfields, the number of projects dealingwith conventional ‘‘whole’’ organismsor cells is quite remarkable. Antitumorvaccine projects indicate that vaccinesshould no longer be regarded only asinfection prophylaxis. Immunizations canand will in future also be used astherapeutic measures. Vaccine researcheven covers approaches that attempt toinduce temporal infertility by the inductionof antihormonal antibodies.
In comparison to current vaccine R&Dprojects, the number of newly licensedvaccines is extremely small. Most newlylicensed products are improvements orcombinations of existing vaccines; realvaccine novelties are very rare. Thus, thechances that a vaccine project in advancedresearch finally ends up as a vaccineproduct is minimal and is certainly farbelow 1%. These low success rates inresearch inevitably lead to long researchphases. Short time intervals of aroundfive years between the first publication orpatent application of a new vaccine conceptand the start of full development are anextremely short, applied research phase forvaccines. These may be applicable to someveterinary vaccines, for which vaccine
protection of a candidate vaccine can bemeasured directly in the target species.For vaccines against human diseases, 10or more years appear to be a morerealistic average estimate for this phase.If one adds those further 10 to 12 yearsthat it takes on average to develop avaccine product, one must assume thatafter the basic concept has been publishedor patented for the first time, about20 years are needed to successfully developa new vaccine product. Those who considerthese figures as unrealistic estimates arereminded that the average time intervalbetween concept and first appearanceon the market for various innovativetechnical products developed during thepast 100 years (including e.g. not onlycomplex products, such as antibiotics, thepace-maker, and radar but also presumablysimple products such as the zipper, drysoup mixes, powdered coffee, ball-pointpens, and liquid shampoo) was also20 years [2, 3]. At that time, all innovationshad to overcome existing hurdles, such asscientific challenges, technical difficulties,and usually financial limitations also.
4.4Scientific Challenges of Vaccine R&D
Science and technologies are the drivingforces that enable us to develop new vac-cines. Regarding the basic technologies,there are few discoveries to be named thathad a significant positive influence and re-sulted in new vaccines. Cultivation of purebacterial cultures is still the fundamentalbase for most bacterial vaccines. A remark-able breakthrough came with the inventionand development of cell culture techniquesin the 1950s, which led to several new orsignificantly improved antiviral vaccines,including the currently still exclusively

Scientific, Technical and Economic Aspects of Vaccine Research and Development 63
used ‘‘state-of-the-art’’ vaccines against po-liomyelitis, mumps, measles, rubella, andcell culture rabies vaccines. Compared tothese technologies, molecular biology andrecombinant techniques up to now had arather limited success with essentially onlyone recombinant human vaccine for hep-atitis B. DNA vaccines may be regardedas yet another new and basic technologyfor new vaccines, but only a decade af-ter their discovery they certainly did notyet have enough time to mature to prac-tical applications. Monoclonal antibodiesor anti-idiotype antibodies, however, didnot lead to new vaccines as expected, al-though these basic techniques were oftenquoted as a major breakthrough in vac-cine research.
Apart from a few essential technologies,continuous research in virology, micro-biology, parasitology, and immunologyare the foundations for vaccine research.However, even the most detailed knowl-edge about cytokines and their regulationof immune responses, or of fundamen-tal genetic mechanisms controlling thegrowth and replication of microorganismscannot be expected to bring any director immediate success. For the past andfor the foreseeable future, it seems thatit is more the pragmatic, application-oriented research that primarily fostersvaccine development. Complex immuno-logical hurdles must be overcome in orderto arrive at a new vaccine target, and thatis mainly done by establishing suitableanimal models and by testing all sorts ofvaccine candidate antigens in these modelsin a very pragmatic way for their protec-tive effects.
Current efforts to develop a vaccineagainst AIDS serve as a good exampleof illustrating the importance of suitablemodels for vaccine development. TheJordan Report 2000 [1] lists 135 different
AIDS vaccine projects. Only 10% of thesewere considered to be basic researchand development (R&D) projects, that is,they are mainly in a phase of selecting,constructing, and making the desiredantigen. The rest of all these projects wereallocated to preclinical testing phases inanimals or to clinical testing in humans(compare Fig. 1). Less than one-third ofthese projects seemed to have passed smallanimal testing successfully and appearedto be worth testing in monkeys. Only4.4% of the antigen candidates proceededto trials in chimpanzees. A substantialproportion of 44% of vaccine candidateswas tested in humans for safety andefficacy, however, only 1.5% were alreadyin Phase III clinical trials, indicating thatthese two different vaccine candidatesappear interesting enough to go intowidespread field-testing for efficacy. Thelow number of projects in basic R&Dshows that after two decades of AIDSresearch, there are not too many newantigens or entirely new approaches tobe discovered. In the absence of reliableanimal models, the relatively high numberof projects in early human clinical trialsand the low number in later stage clinicaltrials very clearly demonstrate that inthis case research is essentially performedin human clinical trials – with all theinherent limitations. Consequently, thechances of success are low, while at thesame time the cost of such research isextremely high.
What are the scientific challenges anddifficulties to be overcome on the waytoward an effective AIDS vaccine? As sum-marized in Table 2, infectious microor-ganisms and parasites have developedvarious mechanisms by which they ef-fectively prevent their elimination by thehost’s immune response. All of these neg-ative attributes have been found to be
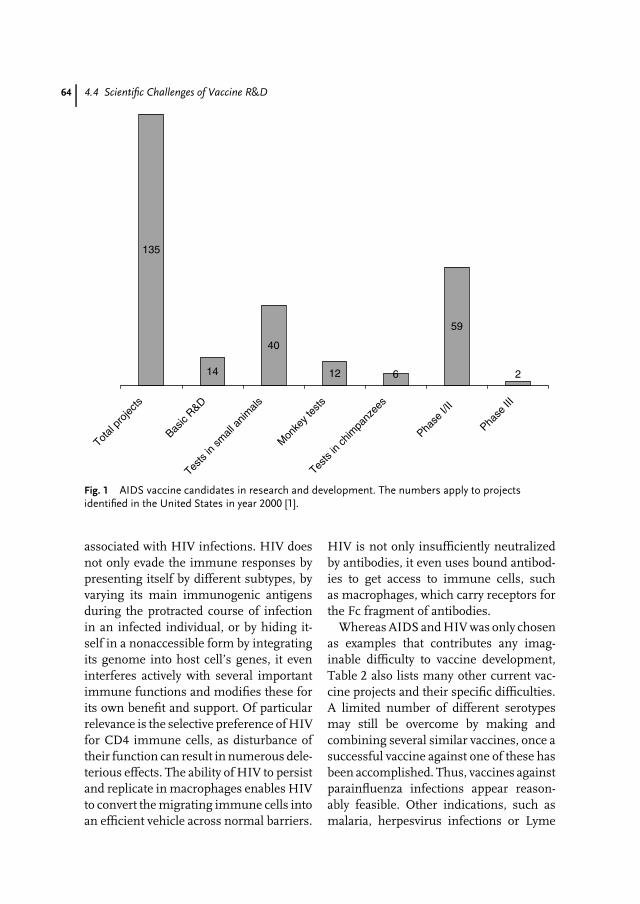
64 4.4 Scientific Challenges of Vaccine R&D
135
14
40
12 6
59
2
Total
proje
cts
Basic
R&D
Tests
in sm
all a
nimals
Mon
key t
ests
Tests
in ch
impa
nzee
s
Phase
I/II
Phase
III
Fig. 1 AIDS vaccine candidates in research and development. The numbers apply to projectsidentified in the United States in year 2000 [1].
associated with HIV infections. HIV doesnot only evade the immune responses bypresenting itself by different subtypes, byvarying its main immunogenic antigensduring the protracted course of infectionin an infected individual, or by hiding it-self in a nonaccessible form by integratingits genome into host cell’s genes, it eveninterferes actively with several importantimmune functions and modifies these forits own benefit and support. Of particularrelevance is the selective preference of HIVfor CD4 immune cells, as disturbance oftheir function can result in numerous dele-terious effects. The ability of HIV to persistand replicate in macrophages enables HIVto convert the migrating immune cells intoan efficient vehicle across normal barriers.
HIV is not only insufficiently neutralizedby antibodies, it even uses bound antibod-ies to get access to immune cells, suchas macrophages, which carry receptors forthe Fc fragment of antibodies.
Whereas AIDS and HIV was only chosenas examples that contributes any imag-inable difficulty to vaccine development,Table 2 also lists many other current vac-cine projects and their specific difficulties.A limited number of different serotypesmay still be overcome by making andcombining several similar vaccines, once asuccessful vaccine against one of these hasbeen accomplished. Thus, vaccines againstparainfluenza infections appear reason-ably feasible. Other indications, such asmalaria, herpesvirus infections or Lyme
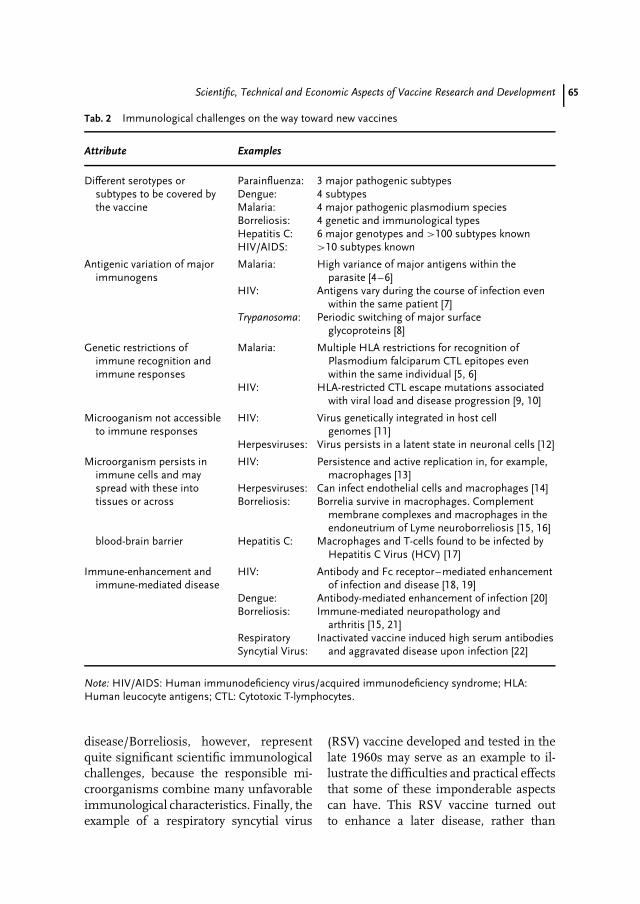
Scientific, Technical and Economic Aspects of Vaccine Research and Development 65
Tab. 2 Immunological challenges on the way toward new vaccines
Attribute Examples
Different serotypes or Parainfluenza: 3 major pathogenic subtypessubtypes to be covered by Dengue: 4 subtypesthe vaccine Malaria: 4 major pathogenic plasmodium species
Borreliosis: 4 genetic and immunological typesHepatitis C: 6 major genotypes and >100 subtypes knownHIV/AIDS: >10 subtypes known
Antigenic variation of majorimmunogens
Malaria: High variance of major antigens within theparasite [4–6]
HIV: Antigens vary during the course of infection evenwithin the same patient [7]
Trypanosoma: Periodic switching of major surfaceglycoproteins [8]
Genetic restrictions ofimmune recognition andimmune responses
Malaria: Multiple HLA restrictions for recognition ofPlasmodium falciparum CTL epitopes evenwithin the same individual [5, 6]
HIV: HLA-restricted CTL escape mutations associatedwith viral load and disease progression [9, 10]
Microoganism not accessibleto immune responses
HIV: Virus genetically integrated in host cellgenomes [11]
Herpesviruses: Virus persists in a latent state in neuronal cells [12]
Microorganism persists inimmune cells and may
HIV: Persistence and active replication in, for example,macrophages [13]
spread with these into Herpesviruses: Can infect endothelial cells and macrophages [14]tissues or across Borreliosis: Borrelia survive in macrophages. Complement
membrane complexes and macrophages in theendoneutrium of Lyme neuroborreliosis [15, 16]
blood-brain barrier Hepatitis C: Macrophages and T-cells found to be infected byHepatitis C Virus (HCV) [17]
Immune-enhancement andimmune-mediated disease
HIV: Antibody and Fc receptor–mediated enhancementof infection and disease [18, 19]
Dengue: Antibody-mediated enhancement of infection [20]Borreliosis: Immune-mediated neuropathology and
arthritis [15, 21]RespiratorySyncytial Virus:
Inactivated vaccine induced high serum antibodiesand aggravated disease upon infection [22]
Note: HIV/AIDS: Human immunodeficiency virus/acquired immunodeficiency syndrome; HLA:Human leucocyte antigens; CTL: Cytotoxic T-lymphocytes.
disease/Borreliosis, however, representquite significant scientific immunologicalchallenges, because the responsible mi-croorganisms combine many unfavorableimmunological characteristics. Finally, theexample of a respiratory syncytial virus
(RSV) vaccine developed and tested in thelate 1960s may serve as an example to il-lustrate the difficulties and practical effectsthat some of these imponderable aspectscan have. This RSV vaccine turned outto enhance a later disease, rather than

66 4.5 Technical Aspects of Vaccine Development
preventing it [22]. More than 30 years af-ter those results were published, there isstill no real explanation for the underlyingmechanism and almost all further effortsto develop a new vaccine were stuck in apreclinical phase.
Another important aspect, which seemsto be underrated in many current vaccineresearch projects, is the fact that most vac-cines are not sufficiently effective if theseare based on only single antigens. Con-trolled vaccine studies performed underideal conditions in genetically homoge-nous or inbred animals quite often leadto the false assumption that a fully protec-tive vaccine antigen has been identified.But, when the same vaccine is then stud-ied under more practical conditions byfewer numbers of immunizations, in thepresence of acceptable and better-toleratedadjuvants, it becomes evident that the se-lected antigen candidate alone is simplynot effective enough. Table 3 summarizesthe experiences made with different foot-and-mouth-disease (FMD) experimentalvaccines. Results from model studies withthis type of vaccine can be correlatedreasonably well with protective responsein the target species. The FMD virusconsists only of three structural proteinsand the most relevant virus-neutralizingantigenic epitopes are known to be located
on virus protein 1. Thus, FMD vaccinesappeared as an excellent target for new vac-cines based on recombinant technologies.The standard vaccine, made of inactivatedwhole virus particles, required a relativelylow amount of antigen and only oneimmunization in order to confer protec-tion. Efforts to make smaller subunit orsingle-protein vaccines resulted only ina similar protection if several immuniza-tions and/or massively increased antigendoses were given. These results had beenestablished rather early in the molecularbiology vaccine era, but despite intensivefurther research, a commercially viable re-combinant FMD vaccine has never beenachieved. Meanwhile, conventional wholevirus vaccines were successful enough toallow for measures to eradicate the diseasecompletely in those countries where thevaccine has been used intensively.
4.5Technical Aspects of Vaccine Development
In an ideal situation, vaccine develop-ment commences with a proven, protectiveand well-defined antigenic composition.Successful vaccine development then nor-mally takes 10 or more years, but onlya small proportion of all development
Tab. 3 Protectivity of different forms of vaccine antigens
Type of antigen Amount ofantigen [µg]
Vaccinations
Purified whole virus particles 1 1Virion subunits (12 S) 10 2Virus protein 1 200 3Oligopeptide (N-142-160-C) 200 1
Note: Type of antigen, amount of antigen, and frequency ofimmunization required to achieve protection againstfoot-and-mouth-disease virus infection in the guinea pig model [23].

Scientific, Technical and Economic Aspects of Vaccine Research and Development 67
candidates finally end up as a licensedproduct; the vast majority remains stuckin early development phases or is aban-doned [24, 25]. Figure 2 summarizes theessential tasks of a vaccine developmentproject and may give a rough impressionof what is to be expected. For the sakeof clarity, several time dependencies andoverlaps during the preclinical phase havebeen neglected in this graphic overview.
An extensive range of national andinternational rules and guidelines exist,covering almost any aspect of pharma-ceutical and vaccine development andregistration [26–28]. These guidelines de-scribe standards that are not binding ina legal sense, but adherence to these isstrongly recommended, as during laterregistration and licensing, the productwill be judged by the same rules. Devi-ations from guideline recommendationsmay be inevitable for certain aspectsand particularly for vaccines, but theseshould only be considered if convinc-ing reasons for doing so can be pre-sented. A summary of relevant guide-line’s requirement along with specificinterpretations and applications for bio-pharmaceuticals and vaccines is givenin [29] and may be helpful for prospec-tive developers in order to get a rea-sonable understanding of the guidingprinciples.
4.5.1Preclinical Development
Preclinical development comprises thetechnical and scientific elaboration ofa process to manufacture the desiredproduct on a large scale. Firstly, cellcultures and microorganisms to be usedmust be established as Master Cell Banksand Working Cell Banks, or as Master andWorking Seeds, respectively. These ensure
a constant supply of well-characterized,uniform, biological starting materials.Numerous tests in vitro and in vivoare required to guarantee the absenceof undesired adventitious agents and toconfirm the identity of these cell banksand microbial seeds.
Starting with a single aliquot of theWorking Cell Bank and/or the WorkingSeed material, a process is then estab-lished and brought up to a final scale. Theterm ‘‘upstream process’’ typically meansa cell culture or fermentation process up tosome 100 L, but for very common vaccineslarger scales may be chosen. Downstreamprocessing summarizes activities duringthe purification process and typically in-cludes recovery and concentration steps,followed by a secondary purification or‘‘polishing’’ to remove specific impuritiesand process-related impurities introducedduring earlier steps. Inactivation of bacte-ria or viruses or detoxifying steps for toxoidvaccines is usually included after an initialconcentration step.
Formulation development includes thedesign of adequately buffered and well-tolerated, stable formulations, adjuvanta-tion, the development of specific applica-tion forms, combination of vaccines intocompatible vaccines, and, particularly forlive attenuated vaccines, the developmentof a lyophilization process. Formulationdevelopment also extends to the selectionor design of final syringes or other pre-sentation forms and to filling and packingprocesses. Stability monitoring programsfor intermediate and final products areof adamant importance for any develop-ment work and should be started as earlyas possible to avoid difficulties at a latedevelopment stage.
Analytical development encompasses allactivities to design and use adequatemethods to control and specify all parts of
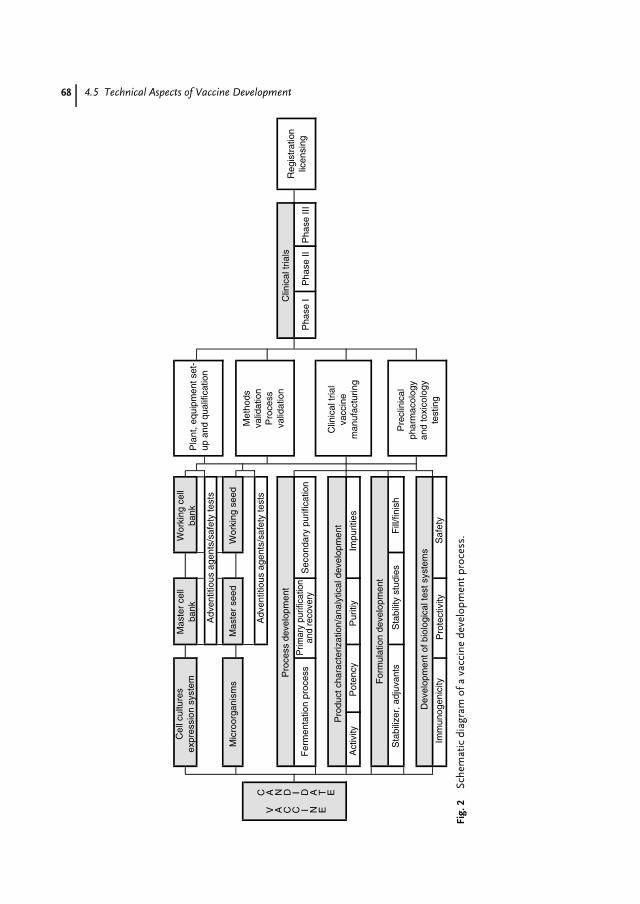
68 4.5 Technical Aspects of Vaccine Development
Cel
l cul
ture
sex
pres
sion
sys
tem
Mas
ter
cell
bank
Wor
king
cel
lba
nk
Adv
entit
ious
age
nts/
safe
ty te
sts
Mic
roor
gani
sms
Mas
ter
seed
Wor
king
see
d
Adv
entit
ious
age
nts/
safe
ty te
sts
Pro
cess
dev
elop
men
t
Fer
men
tatio
n pr
oces
sP
rimar
y pu
rific
atio
nan
d re
cove
ryS
econ
dary
pur
ifica
tion
Pha
se I
Pha
se II
Pha
se II
I
Pro
duct
cha
ract
eriz
atio
n/an
alyt
ical
dev
elop
men
t
Act
ivity
Pot
ency
Pur
itiy
Impu
ritie
s
For
mul
atio
n de
velo
pmen
t
Sta
biliz
er, a
djuv
ants
Sta
bilit
y st
udie
sF
ill/fi
nish
Dev
elop
men
t of b
iolo
gica
l tes
t sys
tem
s
Imm
unog
enic
ityP
rote
ctiv
ityS
afet
y
Pla
nt, e
quip
men
t set
-up
and
qua
lific
atio
n
Met
hods
valid
atio
nP
roce
ssva
lidat
ion
Clin
ical
tria
lva
ccin
em
anuf
actu
ring
Pre
clin
ical
phar
mac
olog
yan
d to
xico
logy
test
ing
Clin
ical
tria
lsR
egis
trat
ion
licen
sing
V A C C I N E
C A N D I D A T E
Fig.
2Sc
hem
atic
diag
ram
ofa
vacc
ine
deve
lopm
entp
roce
ss.

Scientific, Technical and Economic Aspects of Vaccine Research and Development 69
the process and the product. This includestesting of starting materials, intermediateproducts, and the final product, forexample, for identity, specific activity,conformation, purity, and impurities. Fora vaccine developed according to today’sstandards, a range of about 100 differenttests and methods will be required. Mostof these tests need to be validated for theirspecific purpose in order to assess themethods specificity, sensitivity, statisticalexactness, and its limitations.
In parallel with process development,biological tests and model systems mustbe available to monitor the vaccine’s po-tency, immunogenicity, or protectivity atany stage, as vaccines are particularly la-bile products and minor modifications ofthe process can have a significant – mainlynegative – influence on the vaccine anti-gen. Likewise, biological models mustbe at hand to study the vaccine’s basicpharmacological, immunological, toxico-logical, and potential immunotoxicologicalcharacteristics. As far as this can beadequately studied, these include doseresponses, characterization of induced hu-moral and cellular immune responsesor of their major contributing protectivemechanisms, longevity of immune re-sponses, and potential immunological sideeffects. Although vaccines rarely presentsevere tolerability or toxicological risks,abbreviated classic toxicological testing ismandatory before the onset of studiesin humans. Most vaccines need to betested only in local and systemic toler-ance studies and in repeated dose studiesin standard toxicology models, but for newadjuvants and certain new excipients, rep-resenting a significant part of the vaccinecomposition, even a complete toxicologyprogram, including two years of carcino-genicity studies, may be needed. Furthertoxicology and safety studies addressing
specific risks, such as embryonal, fetal,or peri- and neonatal toxicity may berequired for certain vaccines and appli-cations or if risks are expected or known.The recent withdrawal of a newly licensedRotavirus vaccine that was suspected tocause intussusceptions and fatal bowl ob-structions in vaccinated children may serveas an example that such studies maybe required not on entirely hypotheticalgrounds alone. In this particular case,however, the true reason for the fatal-ities could also be a mere coincidenceand the higher medicinal attention andreporting of fatality cases in vaccinatedindividuals.
Owing to the biological origin ofmany starting materials, risks associatedwith prions and potentially contaminatingviruses must be addressed. Organizationalmeasures are to be put in place to avoid riskmaterials in addition to testing for adven-titious agents. Potential risks by startingmaterials or process contaminants can fur-ther be evaluated and assessed by modelstudies with various viral and microbialagents. If specific risks are identified and ifsafety margins appear low, specific coun-termeasures are to be included into theprocess. As far as possible, within the tech-nical limitations of these safety studies, aresidual risk of less than 1 in 1 000 000cases should be aimed for. In practicalterms this means, for example, that anunnoticed contaminating virus is inac-tivated or eliminated by the process toa degree that no active virus would befound in a vaccine volume equivalent to1 000 000 doses. For live attenuated vac-cines, viral safety must also be assuredby assessing the genetic and phenotypicstability of the vaccine virus and by eval-uating the chances and consequences oftransmissions of the virus to unvaccinatedindividuals.

70 4.5 Technical Aspects of Vaccine Development
4.5.2Production Facilities
Facilities and equipment for the manu-facturing of a vaccine are an immanentpart of the registration dossier for theproduct. Any major change would haveto be approved by the regulating author-ities. Thus, at least for the later clinicalphases, the product should be made in aspecific plant and with dedicated equip-ment. For a development project, thismeans that after the process has been de-fined, large investments into buildings,facilities, and equipment are to be ex-pected. Owing to the inherent risks ofthese investments, pilot plants should beavailable to produce initial clinical trialvaccine lots on an intermediate scale. Adeveloping organization may even chooseto go into Phase III clinical trials witha vaccine that has been produced in apilot plant and to seek registration forthis ‘‘preliminary’’ product. This approachdelays the investment decision to a laterpoint of time when all development riskshave been abolished, but inevitably re-quires new clinical trials for the vaccinethat is later on made in the final plantand extends the time to the market byseveral years. The sum to be investedgreatly depends upon the scale of oper-ation and dosage volume of the vaccine.For a complete vaccine plant including allauxiliary functions, the total investmentmay well accumulate to far above ¤ orUS$100 million. Vaccine producers whocan use their existing infrastructure, suchas buildings, filling and packaging facili-ties, raw material and media productionareas, quality control laboratories, and soon, would have to invest significantly less.For small or start-up companies, outsourc-ing and outlicensing may be chosen toreduce risky capital investments, as only
vaccines with high market expectationsjustify establishing a complete, own man-ufacturing operation.
4.5.3Clinical Development
The clinical development of a new vac-cine is done in three phases and normallylasts three to seven years. The durationmainly depends not only on the novelty andcomplexity of the vaccine indication to beexplored but also on the availability of mea-surable immunological surrogate markersof protection. If the vaccine’s efficacy mustbe evaluated by comparing randomly oc-curring cases of the disease in test groupsand in alternatively treated control groups,clinical studies can be extremely long last-ing, demanding, and risky.
Prerequisites of all clinical trials areadequate preclinical pharmacological andtoxicological safety assessments, includ-ing animal studies, to justify tests inhumans. On the basis of the availablesafety data and documentation, approvalfor clinical trials must be obtained by therelevant ethics committees and health au-thorities. Trials will only be admitted ifthese are conducted according to preestab-lished, systematic, and written proceduresfor the organization and conduct of the tri-als for data collection, documentation, andstatistical verification of the trial results.The ‘‘informed consent’’ of all participat-ing trial subjects and medical personnelis essential. For trials involving childrenor mentally handicapped persons, the in-formed consent must be given by parentsor by the person responsible. Clinical trialsare to be planned and conducted accord-ing to ‘‘good clinical practice’’ standardsthat require controlled and randomizedtrials where possible. Control groups areto be treated by established products

Scientific, Technical and Economic Aspects of Vaccine Research and Development 71
or treatments. Placebo treatments areonly admitted where no alternative treat-ment exists.
During the initial Phase I, the basicsafety features of the vaccine candidate areintensively studied in a limited number(<100) of patients or healthy volunteers.The main purpose of these studies is toconfirm the vaccine’s local and generaltolerance before it is applied in furtherclinical trial subjects, but Phase I vaccinestudies can partly be used for a first dose-finding, and immunological evaluationsfor adequate immune responses. DuringPhase I trials, vaccines rarely fail due tosafety concerns, but quite frequently dueto insufficient or inconsistent immuneresponses below expected levels.
Phase II clinical studies usually com-prise no more than several hundredsubjects and are normally done as con-trolled studies comparing the test vaccinealong with an alternative prophylactic ortherapeutic treatment. Clinical evaluationsare mainly addressing the vaccine’s ef-fectiveness and safety, doses, applicationschemes, and possibly also different tar-get groups selected by age, specific risks,countries, or by epidemiological criteria.
Phase III clinical studies are expanded,controlled, or uncontrolled trials on effi-cacy and safety in various clinical settingsand under practical conditions. Altogetherseveral hundred to several thousand trialsubjects are enrolled at various trialsites, which are often distributed overseveral countries in order to study dif-ferent epidemiological situations, ethnicpopulations, and deviating local medicalpractices. Phase III studies can also beevaluated for risk–benefit relationshipsand address practicability aspects as wellas interactions by other products or con-comitantly applied medical treatments.Postmarketing clinical trials of the licensed
product, often referred to as Phase IV clin-ical trials, are nowadays, rather often, alsorequested as part of a conditioned licens-ing of pharmaceutical products, mainly inorder to specifically investigate those as-pects that can only be assessed by largestatistical cohorts.
For live attenuated vaccines, specificsafety aspects must also be studied clini-cally. As live viruses or bacteria replicate inthe vaccine and may be shed into the en-vironment, the potential transmission ofvaccine microorganisms to unvaccinatedsubjects must be studied. If transmissionis possible or likely, the vaccine’s genotypicand phenotypic stability must be carefullystudied and confirmed.
4.5.4Licensing and Registration of VaccineProducts
The formal aspects of pharmaceuticalproduct licensing will be dealt with in an-other chapter of this book [see Chapter 10in this volume]. On the basis of previousexperience and evaluations, the process ofgetting a vaccine through the evaluationat different national licensing authoritieson average takes about two years, whichincludes time periods for working off andanswering questions not adequately cov-ered by the registration dossier.
As vaccines and other biological phar-maceuticals are particularly complex com-positions that cannot be adequately charac-terized by specific quality control methods,the entire process, manufacturing facili-ties, analytical methods used to specifythe product and its starting materials,and all ingredients are considered as be-ing an inherent characterizing part of theproduct. Any change to these affects theproduct’s license and requires approval bythe licensing authority. Changing essential

72 4.6 Economic Aspects of Vaccine Development
elements, such as production cell sub-strates or microbial strains, critical testmethods such as potency assays, purifi-cation methods, or formulations wouldalmost inevitably be seen as a change tothe product that needs to be verified bynew clinical trials. Furthermore, each in-dividual batch produced must be approvedand released by the authorities.
4.6Economic Aspects of Vaccine Development
Without any doubt, the development ofvaccines is a very costly and long-lastingprocess that bears a significant risk offailure. The following paragraphs intendto provide some deeper insights intothe specific risks and chances, cost, andtime requirements to develop a newvaccine, as the knowledge of those basicsdrawn from experience may by helpfulin decision making. After all, successfulvaccine development depends not onlyon good science and technical methodsbut also to a great extent on adequatemanagement decisions.
4.6.1Vaccine Development Cost
The number of successful vaccine projectsis fairly low and retrospective evaluationsof the specific cost incurred by these de-velopment projects over a time period of10 or more years are difficult. However,cost evaluations covering developmentsfrom the late 1960s to the early 1990sexist, which summarize the developmentcost of various pharmaceutical develop-ments [30–33]. Although chemical drugproducts dominated these figures, sev-eral vaccine projects were also assessed.With all the inherent variability, we can
reasonably assume that these figures alsogive adequate estimates for vaccine prod-ucts. These evaluations show that phar-maceutical development cost during thoseyears tended to increase by a factor of about10 within a decade. As demonstrated by asimple graph (Fig. 3), the rising cost is onlyin part due to the normal inflation rate, butclearly correlates with the increasing reg-ulatory demands, as exemplified by thenumber of applicable guidelines.
The latest figure of US$231 million, pub-lished in 1991, was based on evaluationsof 93 successful product developments.This sum has since been quoted on manyoccasions and has been willfully and gener-ously projected to later dates. Thus, quotedsums of US$500 to 600 million may be en-countered to describe the cost and risksof pharmaceutical development projects.However, these figures are misleading ifseveral important details about the origi-nal calculations are not mentioned: those231 millions include to a great extent, op-portunity cost (calculated by an interestrate of 9% of the invested capital overa period of 12 years) and the cost ofmany unsuccessful or abandoned projects(assuming a success rate of 23%), fur-thermore, tax credits were not accountedfor. All in all, the underlying ex-pocket ex-penses must be assumed to be only aboutone-fourth of the total sum and accordingto today’s standards, direct cost between ¤or US$60 and 100 million may be assumedas a realistic estimate for the developmentof an new vaccine. If however there is nosuitable infrastructure and if investmentsinto completely new production facilitiesare to be made, this could easily doublethe cost.
Apart from capital investments, person-nel is the most relevant cost factor to beconsidered. Owing to the high number of
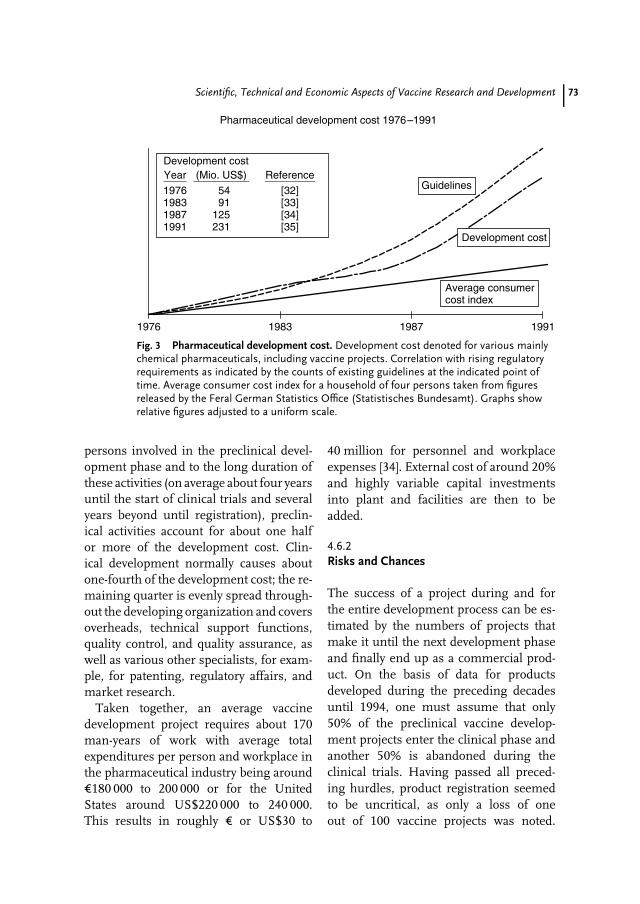
Scientific, Technical and Economic Aspects of Vaccine Research and Development 73
Pharmaceutical development cost 1976–1991
1976 1983 1987 1991
Guidelines
Development cost
Average consumercost index
Development costYear (Mio. US$) Reference
1976198319871991
5491
125231
[32][33][34][35]
Fig. 3 Pharmaceutical development cost. Development cost denoted for various mainlychemical pharmaceuticals, including vaccine projects. Correlation with rising regulatoryrequirements as indicated by the counts of existing guidelines at the indicated point oftime. Average consumer cost index for a household of four persons taken from figuresreleased by the Feral German Statistics Office (Statistisches Bundesamt). Graphs showrelative figures adjusted to a uniform scale.
persons involved in the preclinical devel-opment phase and to the long duration ofthese activities (on average about four yearsuntil the start of clinical trials and severalyears beyond until registration), preclin-ical activities account for about one halfor more of the development cost. Clin-ical development normally causes aboutone-fourth of the development cost; the re-maining quarter is evenly spread through-out the developing organization and coversoverheads, technical support functions,quality control, and quality assurance, aswell as various other specialists, for exam-ple, for patenting, regulatory affairs, andmarket research.
Taken together, an average vaccinedevelopment project requires about 170man-years of work with average totalexpenditures per person and workplace inthe pharmaceutical industry being around¤180 000 to 200 000 or for the UnitedStates around US$220 000 to 240 000.This results in roughly ¤ or US$30 to
40 million for personnel and workplaceexpenses [34]. External cost of around 20%and highly variable capital investmentsinto plant and facilities are then to beadded.
4.6.2Risks and Chances
The success of a project during and forthe entire development process can be es-timated by the numbers of projects thatmake it until the next development phaseand finally end up as a commercial prod-uct. On the basis of data for productsdeveloped during the preceding decadesuntil 1994, one must assume that only50% of the preclinical vaccine develop-ment projects enter the clinical phase andanother 50% is abandoned during theclinical trials. Having passed all preced-ing hurdles, product registration seemedto be uncritical, as only a loss of oneout of 100 vaccine projects was noted.
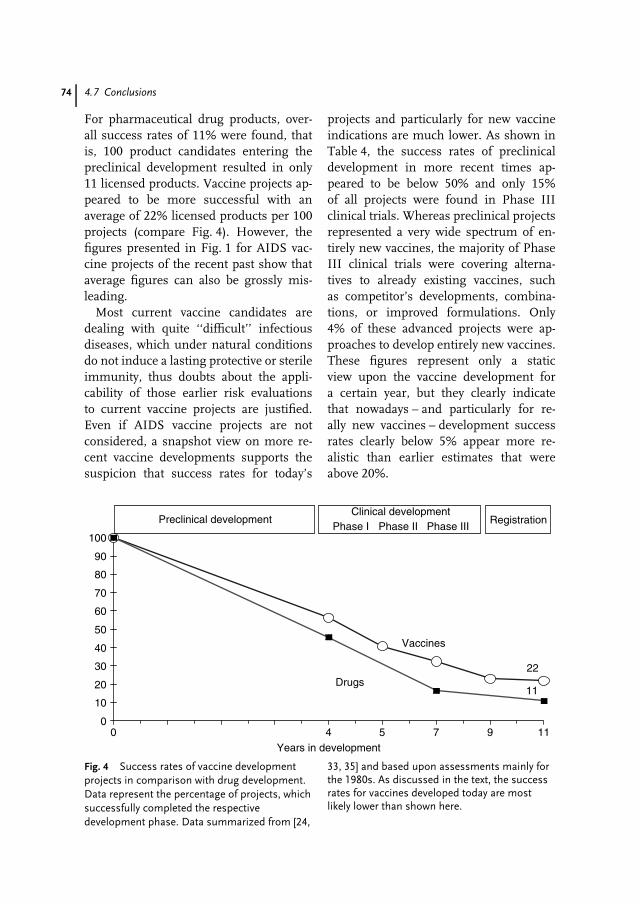
74 4.7 Conclusions
For pharmaceutical drug products, over-all success rates of 11% were found, thatis, 100 product candidates entering thepreclinical development resulted in only11 licensed products. Vaccine projects ap-peared to be more successful with anaverage of 22% licensed products per 100projects (compare Fig. 4). However, thefigures presented in Fig. 1 for AIDS vac-cine projects of the recent past show thataverage figures can also be grossly mis-leading.
Most current vaccine candidates aredealing with quite ‘‘difficult’’ infectiousdiseases, which under natural conditionsdo not induce a lasting protective or sterileimmunity, thus doubts about the appli-cability of those earlier risk evaluationsto current vaccine projects are justified.Even if AIDS vaccine projects are notconsidered, a snapshot view on more re-cent vaccine developments supports thesuspicion that success rates for today’s
projects and particularly for new vaccineindications are much lower. As shown inTable 4, the success rates of preclinicaldevelopment in more recent times ap-peared to be below 50% and only 15%of all projects were found in Phase IIIclinical trials. Whereas preclinical projectsrepresented a very wide spectrum of en-tirely new vaccines, the majority of PhaseIII clinical trials were covering alterna-tives to already existing vaccines, suchas competitor’s developments, combina-tions, or improved formulations. Only4% of these advanced projects were ap-proaches to develop entirely new vaccines.These figures represent only a staticview upon the vaccine development fora certain year, but they clearly indicatethat nowadays – and particularly for re-ally new vaccines – development successrates clearly below 5% appear more re-alistic than earlier estimates that wereabove 20%.
22
11
0
10
20
30
40
50
60
70
80
90
100
0 4 5 7 9 11Years in development
Preclinical developmentClinical development
Phase I Phase II Phase IIIRegistration
Vaccines
Drugs
Fig. 4 Success rates of vaccine developmentprojects in comparison with drug development.Data represent the percentage of projects, whichsuccessfully completed the respectivedevelopment phase. Data summarized from [24,
33, 35] and based upon assessments mainly forthe 1980s. As discussed in the text, the successrates for vaccines developed today are mostlikely lower than shown here.

Scientific, Technical and Economic Aspects of Vaccine Research and Development 75
Tab. 4 Human vaccine development projects in the year 2000
Project phase No. of vaccine projects
Preclinical development 349 100%Phase I clinical development 158 45%Phase II clinical development 102 29%Phase III clinical development 51 15%
Thereof alternatives to existing vaccines in Phase III 37 11%Thereof new vaccine indications in Phase III 14 4%
Note: Data extracted from listed vaccine projects in the year 2000 [1] withoutconsideration of AIDS vaccine projects. Vaccine projects in Phase III clinical trialsfor new indications include vaccines against Coccidioides immitis, group Bstreptococcus, Streptococcus pneumoniae, Plasmodium falciparum, Trypanosomsacruzi, Leishmania major, Mycobacterium leprae, Meningococcus B and C, Rotavirus,and Vibrio cholerae.
4.7Conclusions
Judged by the number of scientific publica-tions in microbiological and biotechnologyjournals, vaccine R&D appears to havea great attraction for scientists from allpertinent scientific disciplines. Whenevernew methods and technologies becameavailable, these have always and imme-diately triggered a huge number of newvaccine research projects and stimulatedresearch into formerly hopeless vaccines.Along with the good reputation that vac-cines enjoy, this scientific enthusiasm isan excellent base for new vaccines, and agood base to attract the required capital aswell. But considering the high risks andthe long duration of vaccine R&D, theremust also be other reasons why investorsand pharmaceutical companies invest inthis field.
Vaccines represent only a small pro-portion of the pharmaceutical market,but nevertheless vaccines are extremelysuccessful products. Firstly, vaccines ef-fectively prevent diseases, rather than onlycuring these. Owing to these advantages,vaccines have often created their own
markets and have even defended theirmarket shares against competition by veryeffective therapeutics or antibiotics. Sec-ondly, most vaccines are recommendedby public health authorities and thus en-joy a rather safe position on the market.Furthermore, there are usually only ratherlimited numbers of competitive productsbecause vaccines are far too complex tobecome an easy target for producers ofgeneric imitations. And finally, vaccinesusually have a very long life span. Aslong as vaccine products are not neglectedand become outdated, but are constantlyadapted to a better state of the art, vac-cines do not lose their market position,unless they are too successful and by andby eliminate the need to use the vaccine.
Thus, vaccine R&D can be very re-warding for both scientists and investors.Regarding the risks, however, the investorhas a quite different perspective than thescientist. The investor may contain risks byputting capital into many different projectsand enterprises, thus participating in thestatistically very successful ‘‘average vac-cine’’. To a limited extent, large companieswho develop vaccines can also apply thesame strategy. But small enterprises and

76 4.7 Conclusions
individual scientists working for only oneor a few R&D projects have only few op-tions to manage and reduce risks. Theyoften choose a high-risk approach by aim-ing only for ‘‘block buster’’ products. Inthis case, they must be aware that compe-tition in this field will be also very strong,which increases the risks even more. Butwithin the given financial limitations, riskscould also be spread over a certain numberof projects in early R&D phases, prefer-ably by approaching different indicationsand concentrating on an attractive new orimproved technology.
In any case, vaccine R&D is certainly nota playing ground for those who expect fastsuccess and revenues. Any organizationthat intends to invest into vaccine R&Dshould be prepared – both mentally andfinancially – to endure for at least 10to 20 years.
References
1. The Jordan Report 2000, Accelerated De-velopment of Vaccines, Division of Mi-crobiology and Infections Diseases, Na-tional Institutes of Health. Washington DC,http://www.niaid.nih.gov/publications/pdf/jordan.pdf.
2. S. Rosen, Future Facts, Simon & Schuster,New York, 1976, (see also: D. J. Ellis, P.P. Pekar, Planning Basics for Managers,Amacon, New York, 1983).
3. Batelle Memorial Laboratories, Science,Technology and Innovation, Report to theNational Science Foundation, USA, 1973.
4. J. D. Smith, C. E. Chitnis, A. G. Craig et al.,Cell 1995, 82, 101–110.
5. R. Wang, D. L. Doolan, T. P. Le et al., Science1998, 282, 476–480.
6. T. P. Le, K. M. Coonan, R. C. Hedstrom et al.,Vaccine 2000, 18, 1893–1901.
7. H. C. Lance, J. M. Depper, W. C. Green et al.,N. Engl. J. Med. 1985, 313, 79–84.
8. J. E. Donelson, K. L. Hill, N. N. El-Sayed,Mol. Biochem. Parasitol. 1998, 91, 51–66.
9. P. J. Goulder, C. Brander, Y. Tang et al.,Nature 2001, 412, 334–338.
10. C. B. Moore, M. John, I. R. James et al.,Science 2002, 296, 1439–1443.
11. P. Luciw, Human immunodeficiency virusesand their replication Chapter 60 in FieldsVirology (Eds.: B. N. Fields, D. N. Knipe,P. M. Howley), 3rd ed., Lippincott-Raven,1996, pp. 1881–1952, Vol. 2.
12. B. Roizman, A. E. Sears, Herpes simplexviruses and their replication Chapter 72 inFields Virology (Eds.: B. N. Fields, D. N.Knipe, P. M. Howley), 3rd ed., Lippincott-Raven, 1996, pp. 2231–2296, Vol. 2.
13. A. T. Haase, Nature 1986, 322, 130–136.14. J. Odeberg, C. Cerboni, H. Browne et al.,
Scand. J. Immunol. 2002, 55, 149–161.15. D. Maimone, M. Villanova, G. Stanta et al.,
Muscle Nerve 1997, 20, 969–975.16. R. R. Montgomery, M. H. Nathanson, S. E.
Malawista, J. Immunol. 1993, 150, 909–915.17. A. L. Zignego, M. Decarli, M. Monti et al., J.
Med. Virol. 1995, 47, 58–64.18. A. Takeda, C. Tuazon, F. A. Ennis et al.,
Science 1988, 242, 550–583.19. J. Homsy, M. Meyer, J. A. Levy, J. Virol. 1990,
64, 1437–1440.20. S. B. Halstead, J. Infect. Dis. 1979, 140,
527–533.21. B. K. Du Chateau, E. L. Munson, D. M. Eng-
land et al., J. Leukoc. Biol. 1999, 65, 162–170.22. H. W. Kim, J. G. Canchola, C. D. Brandt
et al., Am. J. Epidemiol. 1969, 89, 422–434.23. M. C. Horzinek, Kompendium der allge-
meinen Virologie, Enke Verlag, Stuttgart,1984.
24. M. M. Struck, Biotechnology 1994, 12,674–677.
25. M. M. Struck, Nat. Biotechnol. 1996, 14,591–593.
26. The Rules Governing Medicinal Productsin the European Community, Volume I–IV(1989), Vol. III plus Addendum 1, 2, 3(1990–1994), as well as numerous newerguidelines, which are not contained inthe above mentioned volumes can beobtained from: Office des PublicationsOfficielles des Communantes Europeenes,Luxemburg. These guidelines are retrievablevia http://www.emea.eu.int/.
27. US-FDA guidelines and Points to Consider(PCT) documents, as well as lists of allavailable documents about human biologicalmedicines are available via: Congressional,

Scientific, Technical and Economic Aspects of Vaccine Research and Development 77
Consumer and International Affairs Staff,Metro Park North, Building 3, 5600 FishersLane, Rockville, MD 20857 and via internetunder http://www.fda.gov/cber/guidelines.htm.
28. International regulatory guidelines, har-monized between the EU, USA, andJapan, and covering various aspects areavailable as ICH (International Confer-ence on Harmonization) documents under:http://www.ich.org/ich5.html.
29. J. P. Gregersen, Research and Developmentof Vaccines and Pharmaceuticals fromBiotechnology. A Guide to EffectiveProject Management, Patenting and ProductRegistration, VCH, Weinheim, New York,Basel, Cambridge, Tokyo, 1994.
30. R. W. Hansen, The pharmaceutical develop-ment process: Estimates of development andthe effects of proposed regulatory changes
in Issues in Pharmaceutical Economics (Ed.:R. J. Chien), Lexington Books, Cambridge,MA, 1979, pp. 151–186.
31. L. Langle, R. Occelli, J. Econ. Med. 1983, 1,77–106.
32. S. N. Wiggins, The Cost of Developing aNew Drug, Pharmaceutical Manufacturers’Association, Washington, DC, 1987.
33. J. A. Di Masi, R. W. Hansen, H. G.Grabowski et al., J. Health Econ. 1991, 10,107–142.
34. J. P. Gregersen, Vaccine development:the long road from initial idea toproduct licensure Chapter 71 in NewGeneration Vaccines (Eds.: M. M. Levine,G. W. Woodrow, J. B. Kaper et al.), 2nd ed.,Marcel Dekker, New York, Basel, HongKong, 1997, pp. 1165–1177.
35. B. Bienz-Tadmore, P. A. Dicerbo, G. Tad-more et al., Biotechnology 1992, 10, 521–525.

79
5DNA Vaccines: from ResearchTools in Mice to Vaccines forHumans
Jeffrey Ulmer and John DonnellyChiron Corporation, Emeryville, CA, USA
Jens-Peter GregersenChiron-Behring GmbH, Marburg, Germany
5.1Introduction
Expression of foreign genes in animalscan be achieved through the simple ad-ministration of recombinant DNA, as wasfirst demonstrated more than 20 yearsago [1, 2], although the impetus for therecent application to vaccines is typicallytraced to the work of Wolff et al. in 1990 [3].Shortly thereafter, the induction of anti-body responses [4], cytotoxic T-lymphocyte(CTL) responses [5], and protective immu-nity by DNA vaccines in a lethal animalchallenge model [5, 6] were reported. Sincethen, the field of DNA vaccines (alsotermed genetic vaccines) has been veryactive. Over the past decade, the generalutility of this approach for prophylaxis andtherapy of infectious and noninfectiousdiseases has been well established (for re-views see [7, 8]), culminating in the humanclinical testing of many different DNA vac-cines. During this time, an understandingof the mode of action of DNA vaccineshas been gained, as well as insights into
their limitations. As a consequence, severalsecond-generation DNA vaccine technolo-gies have been developed and some ofthese are now entering clinical evaluation.This review will address the technologi-cal developments that have been achieved,with a look into the issues that will need tobe considered if a DNA vaccine approachesregistration.
5.2DNA Vaccine Construction andImmunology
Effective vaccines have three key com-ponents: (1) an antigen against whichadaptive immune responses are generated,(2) an immune stimulus (or adjuvant) tosignal the innate immune system to po-tentiate the antigen-specific response, and(3) a delivery system to ensure that the anti-gen and adjuvant are delivered togetherat the right time and location. For DNAvaccines, the antigen is produced in situ,
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

80 5.2 DNA Vaccine Construction and Immunology
albeit at very low levels. Thus, the potencyof DNA vaccines depends, in part, on ef-fective expression plasmids. With regardto immune stimulation, DNA vaccines ap-pear to contain a built-in adjuvant in theform of immunostimulatory CpG motifs.However, even simple addition of conven-tional adjuvants can increase DNA vaccinepotency, suggesting that there is room forstronger innate immune signaling by DNAvaccines. Finally, DNA vaccines on theirown do not have an inherent ability to effi-ciently enter cells in a functional way (i.e. totransfect them) and thus require means ofdelivery. Hence, for DNA vaccines to be op-timally effective, enabling technologies inall of these three areas must be developed.
5.2.1DNA Vaccine Expression Plasmids
Most DNA vaccines tested over the pastdecade have consisted of conventionalplasmids with a eukaryotic expression cas-sette. The important elements of suchplasmids are the promoter, the gene insert,the polyadenylation termination sequence,a bacterial origin of replication for produc-tion in Escherichia coli, and an antibioticresistance gene for selection (see Fig. 1).Typically, strong viral promoters, suchas the intermediate early promoter ofcytomegalovirus with the intron A, areused. This ensures constitutive, high lev-els of antigen production in many tissue
pCMV
Intron A
Conventional DNAvaccine Antigen gene
a replicase genes Antigen geneSG
3′5′
5′ 3′
Alphavirus-basedDNA vaccine
BGHpA
pCMVIE
ori
Kanr
Fig. 1 Schematic representation of a typical DNA vaccineplasmid. Shown are the promoter (pCMVIE), transcriptionterminator (BGHpa), bacterial origin of replication (ori), andantibiotic resistance gene (Kanr). Several different types of insertsmay be included in a DNA vaccine. Shown are ones containing adiscrete open reading frame and those containing an alphavirusRNA replicon. SG = subgenomic promoter.

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 81
types and thus increases the likelihoodof inducing an immune response. Cer-tain other types of promoters, includingthose whose expression may be limitedto specific types of tissues, have alsobeen used with success. These includethe muscle creatine kinase [9, 10], majorhistocompatibility complex (MHC) classI [11], desmin [12], and elongation factor1–α [13] promoters. A potential advantageof tissue-specific promoters over viral pro-moters is the additional measure of safetythey may provide because of a more limiteddistribution of antigen production aftervaccination. Also, certain viral promoterscan be downregulated by cytokines [14],which are produced in situ after DNA vac-cination.
Another type of plasmid DNA vac-cine introduced more recently encodesan alphavirus RNA replicon. Alphavirusplasmid replicons based on Sindbis virusincorporate the nonstructural ‘‘replicase’’protein genes and cis replication signals,such that primary transcription from anRNA polymerase II promoter (e.g. CMV)gives rise to an RNA vector replicon capa-ble of directing its own cytoplasmic am-plification and expressing an encoded het-erologous gene [15, 16]. Similar plasmidsbased on Semliki Forest virus repliconshave also been generated [17–19]. Theseplasmids have been shown to be more po-tent than conventional CMV-based DNAvaccines, particularly at low DNA doses.Possible explanations for this increasedeffectiveness include: (1) amplification ofmRNA in the cytoplasm by the RNAreplicon that may enhance expressionlevels, (2) the presence of a dsRNA in-termediate that may act to stimulate theinnate immune system, (3) expression ofother alphavirus nonstructural proteinsthat may provide additional helper T-cellepitopes, and (4) induction of apoptotic
cell death in cells transfected with pSIN,which may facilitate cross-priming of T-cell responses.
A third type of DNA vaccine consists oflinear DNA sequences containing simplythe promoter, gene, and polyadenylationsite. These DNA vaccines can be in theform of a contiguous DNA sequence con-taining all of the above elements [20] or agene hybridized to the promoter and ter-mination sequence [21]. The latter versionprovides the opportunity to rapidly gen-erate DNA vaccines by PCR amplificationwithout the need for bacterial transforma-tion, thereby facilitating the screening oflarge numbers of vaccine candidates (seeChapter 4).
Irrespective of the type of DNA vaccinevector, antigens expressed from it canbe in one of several possible forms,ranging from short peptides of definedT-cell epitopes (as small as 8 aminoacids in length) to large polyproteins(>1000 amino acids in length). Severalreports have demonstrated that singleT-cell epitopes expressed in minigeneDNA vaccines can induce potent T-cellresponses [22]. In some cases, potency wasincreased by the addition of an N-terminalsignal sequence to facilitate targeting of theepitope to the endoplasmic reticulum [23].Multiple epitopes expressed end to end ina ‘‘string-of-beads’’ fashion have the abilityto elicit multiple T-cell responses [24], andthis approach is currently being testedin human clinical trials for malaria andHIV [25] (A. Hill, unpublished). Thisapproach can focus the response ondefined dominant epitopes, but requiresprior knowledge of these epitopes and maynot be sufficient for complete coverageof the diversity of human leukocyteantigen (HLA) haplotypes.
A means around this issue is to ex-press whole antigens to allow determinant

82 5.2 DNA Vaccine Construction and Immunology
selection of epitopes by the host. Expres-sion of whole antigens in their nativestate is also important for the inductionof neutralizing antibodies. Most DNA vac-cines reported so far have encoded wholeantigens. For antigens whose expressionlevels are limited by the use of subop-timal codons, synthetic genes containingappropriate codons commonly utilized ineukaryotic cells are often very effective [26,27]. The nature of the antigen can alsoaffect the quantity and quality of immuneresponses. Secreted antigens are most ef-fective for induction of antibodies andoften for T-cell responses, as well [28, 29].Targeting antigens for processing and pre-sentation by MHC class I and II moleculescan be achieved by the use of ubiqui-tin [30, 31] and lysosomal [32, 33] targetingsignals, respectively. Alternatively, extra-cellular delivery of expressed antigens toantigen-presenting cells (APCs) throughthe use of fusion proteins containing lig-ands for receptors on these cells has beenused effectively [34–37].
For many vaccine targets, a singleantigen is not sufficient to provide opti-mal protection. This is particularly truefor viruses with variant surface glyco-proteins, such as HIV, where multipleclades must also be represented in avaccine. Thus, the vaccine may requireseveral components. In theory, this canbe accomplished by simple mixtures ofplasmids, as DNA vaccines are amenableto combinations. However, this adds tothe complexity and cost of the vaccine.A potential means to minimize theseissues is to express several antigens si-multaneously from a minimum numberof plasmids, through the use of multi-cistronic vectors [38, 39], polyprotein genecassettes [40], or partial genomic con-structs [41, 42].
5.2.2Antigen Presentation and Stimulation ofImmune Responses
After DNA vaccination or during viral in-fection, antigens may be produced directlywithin APCs, such as Langerhans cells(LC) and dendritic cells (DC), or theymay be acquired by APCs in a processtermed ‘‘cross-presentation,’’ where anti-gens are produced by one cell type and thentransferred to APCs (see Fig. 2). Eitherway, this is sufficient for the priming ofantigen-specific lymphocytes. In practice,DNA vaccination results primarily in thetransfection of non-APCs, either myocytesafter im injection [3, 43] or fibroblasts andkeratinocytes after gene-gun administra-tion [44]. For B-cell responses, non-APCscan be considered simply as factories forthe production of antigens, which can thenbe presented to B-lymphocytes for prim-ing of antibody responses. In this regard,antigens that are secreted or those thatspontaneously form higher-order struc-tures, such as virus-like particles, areparticularly immunogenic.
Both the gene-gun and im injectionmethods of DNA vaccination effectivelyprime T-cell responses, including CD8+CTL. Because the gene gun can propelDNA-coated gold beads directly into LCresident in the skin, the primary meansof CTL induction appears to be by directpriming of LC [45]. In contrast, DNA in-jected into the muscle has no inherentability to efficiently enter and transfect DC,thus the predominant mode of CTL prim-ing is by cross-priming [46]. In support ofdirect priming are the following observa-tions: (1) plasmid DNA, antigen-encodingmRNA, and expressed antigen have beenshown to be present in DC and LC afterDNA immunization by im injection andgene gun, respectively [45, 47, 48], (2) DC

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 83
Cross-priming
Direct priming
Myocyte
Myocyte
Ag Ag
(a) (b)
(c)
Ag
Ag
APC
APC
Ag
MHC I
MHC I
MHC I
DNA vaccine
T-cell
T-cell
T-cell
DNA vaccine DNA vaccine
Fig. 2 Possible means of inducing a CTLresponse by a DNA vaccine. Upon injection of aDNA vaccine, there are three possible modes ofCTL induction: (1) direct activation of na
..ı ve
T-cells by transfected muscle cells (a), (2) directactivation of T-cells by transfected
antigen-presenting cells (b), and (3) indirectactivation of T-cells involving transfer of antigenproduced by muscle cells to a professionalantigen-presenting cell (c). A current workinghypothesis is that CTLs are induced by DNAvaccines utilizing modes (b) and (c), but not (a).
isolated from DNA vaccine–injected tissuecan present antigen to T-cells in vitro [49,50], and (3) passive transfer of DC trans-fected in vitro with DNA vaccines inducesstrong T-cell responses in naıve recip-ients [51]. In support of cross-primingare the following data: (1) transplantationof DNA vaccine-transfected myocytes ortumor cells to naıve or bone marrowchimeric mice induced strong T-cell re-sponses [52–54], (2) DNA encoding FAS(CD95) [55] or mutant caspases [56] tofacilitate apoptotic cell death increasedinduction of T-cell responses by DNAvaccines, (3) in vitro studies suggestedthat a heat-shock protein chaperone isinvolved in cross-priming induced byDNA vaccination [57], (4) T-cell responsesto DNA vaccines can be increasedby targeting the expressed antigen toAPCs by utilizing fusion proteins en-coding CTLA4 [37] or chemokines [35], or
extracellular transfer by HIV tat [58] orherpes simplex vims (HSV) VP22 [59], and(5) in vivo electroporation, which facilitatesuptake of DNA vaccines by myocytes,enhanced CTL responses [60, 61]. Thus,taken collectively, the data indicate thatboth means of priming are sufficient forthe induction of CTL.
DNA vaccines appear to have a built-inadjuvant for signaling the innate immunesystem, in the form of immunostimula-tory CpG motifs. It has been well es-tablished that oligonucleotides containingunmethylated CpG motifs signal throughTLR9 present on plasmacytoid DC and B-lymphocytes, resulting in the activation ofDC and the production of cytokines [62].Oligonucleotides derived from DNA vac-cines presumably can be generated invivo by nuclease digestion and therebyprovide an immune stimulus or adju-vant effect for antigens expressed by DNA
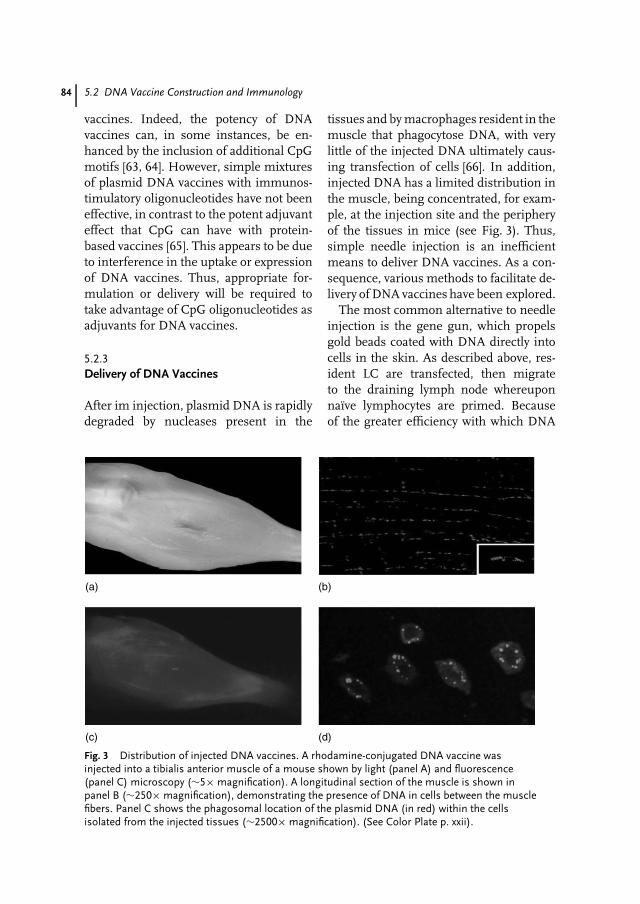
84 5.2 DNA Vaccine Construction and Immunology
vaccines. Indeed, the potency of DNAvaccines can, in some instances, be en-hanced by the inclusion of additional CpGmotifs [63, 64]. However, simple mixturesof plasmid DNA vaccines with immunos-timulatory oligonucleotides have not beeneffective, in contrast to the potent adjuvanteffect that CpG can have with protein-based vaccines [65]. This appears to be dueto interference in the uptake or expressionof DNA vaccines. Thus, appropriate for-mulation or delivery will be required totake advantage of CpG oligonucleotides asadjuvants for DNA vaccines.
5.2.3Delivery of DNA Vaccines
After im injection, plasmid DNA is rapidlydegraded by nucleases present in the
tissues and by macrophages resident in themuscle that phagocytose DNA, with verylittle of the injected DNA ultimately caus-ing transfection of cells [66]. In addition,injected DNA has a limited distribution inthe muscle, being concentrated, for exam-ple, at the injection site and the peripheryof the tissues in mice (see Fig. 3). Thus,simple needle injection is an inefficientmeans to deliver DNA vaccines. As a con-sequence, various methods to facilitate de-livery of DNA vaccines have been explored.
The most common alternative to needleinjection is the gene gun, which propelsgold beads coated with DNA directly intocells in the skin. As described above, res-ident LC are transfected, then migrateto the draining lymph node whereuponnaıve lymphocytes are primed. Becauseof the greater efficiency with which DNA
(a) (b)
(c) (d)
Fig. 3 Distribution of injected DNA vaccines. A rhodamine-conjugated DNA vaccine wasinjected into a tibialis anterior muscle of a mouse shown by light (panel A) and fluorescence(panel C) microscopy (∼5× magnification). A longitudinal section of the muscle is shown inpanel B (∼250× magnification), demonstrating the presence of DNA in cells between the musclefibers. Panel C shows the phagosomal location of the plasmid DNA (in red) within the cellsisolated from the injected tissues (∼2500× magnification). (See Color Plate p. xxii).

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 85
is delivered into cells by this technique,less DNA is required for induction of im-mune responses than by needle injection.However, as this technology is currentlypracticed, a significant limitation is im-posed on the quantity of DNA that canbe delivered at one time. Nevertheless,this technology has been shown to in-duce both humoral and cellular immuneresponses in human clinical trials [67].Various noninvasive routes of DNA deliv-ery have also been evaluated. These includeintranasal, oral, intravaginal, and topicaladministration onto the skin. In manycases, particularly by the oral route, nakedDNA was not effective due to rapid degra-dation by hydrolytic enzymes. Thus, for-mulations designed to protect DNA fromdigestion are required, such as encapsula-tion into chitosan particles [68], polylactidecoglycolide (PLG) microspheres [69], or li-posomes [70].
For parenteral injection of DNA vac-cines, naked DNA has been effective insmall animal models, but, as mentionedabove, there is much room for improve-ment in the efficiency of DNA delivery.To this end, two basic approaches havebeen taken: (1) to increase the efficiencyof uptake of DNA by cells in the in-jected tissue (e.g. myocytes) to facilitatecross-priming of immune responses and(2) to target DNA to APCs to facilitatedirect priming of immune responses.First, to increase DNA distribution anduptake in the injected tissues, physicaltechniques generally have been most ef-fective. These include the aforementionedgene-gun approach, needle-free devices(such as the Biojector) designed to pro-duce better distribution of vaccine [71],in vivo electroporation [60] or sonopora-tion [72] to induce transient discontinu-ities in the plasma membranes of cells,and the use of large-volume inoculation to
induce high hydrostatic pressure locally inthe tissues [73]. These techniques requiredevices and some are cumbersome, in-volving invasive procedures that may notbe appropriate or practical for widespreaduse with prophylactic vaccines. Second,to target APCs for DNA vaccine uptake,formulations have generally been used.Liposomes [74] and microparticles basedon PLG [75] and chitosan [76] have beena particularly effective strategy, in theorybecause of their similar size to pathogens.Work with DNA vaccines adsorbed ontothe surface of PLG microparticles hasshown efficient delivery into DC in vitro,enhancement of transfection of cells in thedraining lymph nodes of injected mice,and marked increases in DNA vaccine po-tency, with a dependence on the size of themicroparticles [77]. These observations areconsistent with the hypothesis that the mi-croparticles target DNA vaccines to APCsin vivo. These techniques for increasingDNA uptake by cells address the first stepin facilitating transfection. Once in the en-dosome, though, the DNA plasmids mustfind their way into the cytoplasm and thenthe nucleus. Thus, further improvementsin DNA delivery may be achieved throughthe inclusion of components that desta-bilize the endosomal membrane [78] andtarget DNA to the nucleus [79]. The lattermay be particularly important for transfec-tion of muscle cells, which are terminallydifferentiated; thus the nuclear membraneremains intact.
5.3Screening for Protective Antigen Candidates
Various efforts have been made to har-ness the ease of preparation of DNAvaccines for mass screening of candi-date antigens for inclusion in vaccinesagainst infectious diseases. The ability of

86 5.3 Screening for Protective Antigen Candidates
gene-gun immunization to evoke immuneresponses with small quantities of DNAled to the hypothesis that a small amountof DNA encoding a protective immunogencould still provide protection when mixedwith many other DNAs that encoded weakimmunogens or no antigen at all. Thishypothesis was tested initially using my-coplasma pulmonis [80]. Immunization ofmice by gene gun with a random li-brary of M. pulmonis DNA fragmentsdid confer protection from challenge. Acomparable library approach to immuniza-tion has also been used against murinemalaria [81] and murine cysticercosis [82].Immunization with random libraries hasalso been used in studies of HIV-2 in-fection in Hamadryas baboons [83] andsimian immunodeficiency vims (SIV) inrhesus macaques [84]. Refinements of thistechnique include the use of cDNA li-braries to ensure that each DNA plasmidencodes a gene [85, 86] or simply to useunligated PCR products that have beenhybridized with a synthetic promoter andterminator region [21]. The latter methodcan also produce sufficient gene expres-sion to elicit immune responses in micewithout the need for constructing sets ofplasmid vectors. This approach also al-lows different fragments of genes to beused by the use of random primers orprimers selective for particular fragmentsin the PCR reaction used to generate thecoding regions.
The next step after demonstrating pro-tection in a challenge model is to deconvo-lute the pool and identify individual anti-gens that are responsible for protection.In theory, this can be done by immu-nizing with multiple distinct pools in amatrix array, where the intersection of eachrow and column contains a unique set ofgenes. However, mixtures of antigens maysynergize to provide a protective immune
response. Thus, when the mixtures arebroken up into smaller sets, protective effi-cacy may be lost. Alternatively, some geneproducts may interfere with protection andcould cancel out an active component ina pool. The concept of beginning withrandom expression libraries and carryingthrough to identification of single antigenshas not yet been reported. This may be anindication of the complexity of the biologi-cal phenomena that underlie immunity tocomplex microorganisms.
A more conservative approach to thescreening of antigens in complex patho-gens has been attempted using individualgenes cloned from expression libraries.Here, the limitations are experienced notat the macrolevel, as in complex mixturesof genes, but at the microlevel, as in theexpression and trafficking of the singleantigens themselves. For example, expres-sion of individual genes may be limitedby nucleic acid content (A-T-rich genesmay form less stable mRNA), or may re-quire cofactors for transport of messageout of the nucleus. Amino acid content,for example, high hydrophobicity, may re-sult in insolubility or in a requirement fora cofactor provided by the pathogen foroptimal assembly, resulting in inefficientprocessing and presentation. Sequence el-ements that target proteins for degradationor add bulky posttranslational modifica-tions may bias T-cell recognition or preventprocessing altogether. Hepatitis C virusprovides examples of many of these limi-tations: insoluble proteins, cotranslationalassembly of subunits, and glycosylationsequences that are not used in the nativevirion but are glycosylated when the sameproteins exit the cell through normal se-cretory pathways [87]. The end result insuch a situation is that a quick experimentmay determine whether an antigen per-forms well or poorly as a DNA vaccine,

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 87
but detailed study and a comprehensiveunderstanding of the structural biology ofthe pathogen may be required to create afunctional DNA vaccine for an antigen ofinterest. Thus, the level of understandingof the antigen that is required to cre-ate a good DNA vaccine has served as alimitation to the use of this technologyfor screening approaches to vaccination.Nevertheless, it has been possible usingdirected screening of a cDNA expressionlibrary from the tick Ixodes scapularis toidentify individual protective antigens inan animal challenge model [86].
5.4The Development Path of a DNA VaccineCandidate
An overview of a vaccine development pro-cess along with basic scientific, technical,and economic aspects of vaccine researchand development is given in Chapter 4.Most of the contents of that chapter alsoapply to DNA vaccines and will not berepeated here. Rather, in the followingsection, we will concentrate more on regu-latory quality and safety requirements forpotential DNA vaccines.
Anticipating the need for an early reg-ulatory guidance, specific points to con-sider have been issued by the Food andDrug Administration (FDA) in the UnitedStates and by the World Health Orga-nization (WHO) [88, 89]. These specificguidelines should be viewed in the con-text of the entire regulatory framework.Therefore, a limited list of other relevantguidelines is also given in the refer-ence section [90]. Partly, these guidelinesare not directly applicable to vaccinesin general (including DNA vaccines) oreven expressively exclude these from theirscope. Nevertheless, they describe the ba-sic rationale and define a technical state
of the art that must be met. An overviewon how to apply regulatory requirementspragmatically to biologicals and vaccinesis given in Ref. [91].
Several DNA vaccines have been testedin clinical trials. As such, DNA vaccineshave progressed part of the way throughthe development process. However, theseclinical trials were done at an early stageand were more or less extended studies ofresearch candidates. Such early stage clini-cal trials differ greatly from those requiredfor licensable products, as usually only anabbreviated preclinical development hasbeen performed in order to make clini-cal trial products available at the earliestpossible time. Many more detailed qual-ity analyses and very long, complex, andexpensive safety studies will have to beperformed to arrive at a marketable DNAvaccine that is sufficiently safe and effica-cious to justify its widespread applicationin healthy individuals.
5.4.1Quality of Starting Materials
Any pharmaceutical product needs to bethoroughly and completely defined andcharacterized by adequate analytical meth-ods. This includes all starting materials,the production process, purified bulk ma-terials, the formulated vaccine, and anyexcipient, adjuvant, or other constituent ofthe vaccine and may well mean that in to-tal a set of 100 or more analytical methodsmust be applied. Table 1 explains how es-sential starting materials of a DNA vaccineneed to be tested and characterized, mainlyin order to provide sufficient informationfor appropriate risk and safety evaluationsand also for a proper and reproduciblespecification or definition of the product.For safety reasons, any unintended by-product that could be expressed by the

88 5.4 The Development Path of a DNA Vaccine Candidate
Tab. 1 Characterization and quality aspects of starting materials of a plasmid DNAvaccine
PlasmidConstruction of the entire plasmidDetailed functional mapSource and function of plasmid componentsRegions of eukaryotic origin
Antigen-coding sequenceIdentificationOriginMeans and ways of isolationSequence
Bacterial cells to produce plasmidsEstablished Master Cell Bank and Working Cell BankConfirmed identityAbsence of microbial contaminants, bacteriophagesStability upon passagingDefined maximum passage number
Use only if absolutely unavoidable:• Retroviral-like long terminal repeats• Oncogene sequences• Extended sequences with homologies to the human genome• Sequences encoding for cell growth-regulating functions• Alternative, unintended reading frames
plasmid should be avoided. This appliesto oncogenes, alternative open readingframes, gene-regulating (long terminal re-peat) sequences, and to resistance markersbased on allergy-prone antibiotics, suchas penicillins. If antibiotic-selection mark-ers are used, one should preferably usekanamycin or neomycin, as these are lessoften used for critical clinical indications.The minimum levels of these antibioticsin the final vaccine must be specified andshould be below levels that could causeunintentional effects. The same degree ofcharacterization applies to any formulationcomponent and to the bacterial cells usedto produce the plasmids. The cells mustcome from established and pretested cellbanks, and these are to be used withina defined number of passages, duringwhich time the bacteria and plasmidsremain stable.
5.4.2Process Development and In-processControls
Purification of plasmids at a small scalenormally follows well-established meth-ods, which can be applied to produceclinical trial product of adequate qualityand are adaptable to a larger scale. Butbefore a plasmid can be made at a finalproduct scale, significant investments intofacilities and equipment must be made tocomply with current Good ManufacturingPractices (cGMP). Furthermore, there arenormally various technical difficulties tobe solved, as it is never trivial to scaleup even a seemingly simple process. Forexample, the control of microbial contami-nations becomes a much more demandingtask during large-scale operations andthe inevitable, longer holding times often

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 89
adversely affect the product quality andyields. If complex formulations are envis-aged, this will certainly compound thesescale-related difficulties.
The purification process must be ade-quately controlled by a range of methodsfor monitoring and quantifying impuri-ties, such as RNA and genomic DNA,endotoxins, bacterial proteins, carbohy-drates, and impurities derived from start-ing materials or from substances intro-duced during the process, for example, bychromatography column matrix bleeding.Degraded plasmids (linear and relaxed,open circular forms, dimers), modified orinadequately complexed/formulated plas-mids could be less efficient and shouldtherefore be monitored and limited by suit-able control methods (compare Table 2).Much experience has already been accu-mulated on how to produce and analyzeDNA vaccine and gene therapy plas-mids [92].
A stable manufacturing process shouldideally yield a consistent quality at allintermediate steps, so that upper lim-its for impurities and lower limits forthe product’s quality can be adequately
controlled by keeping the process constant.In these cases, it may be sufficient to proveand validate the consistency of the process.But for critical components and in thosecases where limits are narrow, analyseswill be necessary and advisable to monitorthe quality.
But what are acceptable limits forimpurities? The more formal answer tothis question is that preclinical and clinicaldata should justify the acceptability ofthe chosen limits, that is, neither theefficacy nor the safety of the vaccineis adversely affected by the remainingimpurities. Since this is not really ameasure that can be applied duringprocess development, the basic rationaleshould be that whatever is avoidable bycurrent standards must be avoided. Inpractical terms, this means that state-of-the-art technologies should be appliedand reasonable efforts should be madeto achieve the highest levels of purity. Ifadditional measures would adversely affectvaccine quality (stability, specific activity)or significantly reduce the yield withoutgiving any benefit, these would appearinappropriate.
Tab. 2 Characterization and quality testing of a DNA vaccine plasmidpurification process
Elimination of impuritiesBacterial RNA and genomic DNAEndotoxinsBacterial proteins, carbohydrates, and other impuritiesMedia components and substances used or added during purification
Product-related impuritiesLinear plasmid DNAOpen circular plasmidsDimeric or oligomeric plasmid componentsComplexed (e.g. formulated, encapsulated, etc.) plasmids versusnoncomplexed proportions
• Acceptable amounts and upper/lower limits to be defined by processvalidation or alternatively by analyses of each batch
• Specifications to be justified by preclinical and clinical safety data

90 5.5 Preclinical Safety and Efficacy
5.4.3Quality of the Final Product
As listed in Table 3, quality of the vaccinemust again be confirmed for bulk plas-mids, for the formulated, and for the filledfinal product. Apart from routine tests forplasmid identity, purity, and sterility, con-sistency of the product and process mustbe demonstrated in at least three consecu-tive runs of the entire process. These runsmust result in a product that meets all pre-defined specifications. Stability of DNAvaccines must be evaluated by long-termstudies to demonstrate that the definedspecifications are met until the end ofthe envisaged shelf live. For naked DNA,it is not anticipated that stability will bean issue.
The absolute requirement to measurethe potency of each DNA vaccine lotrepresents particular challenges, as a po-tency assay should quantitatively measurethe active ingredient for its ability toraise adequate immune responses. Most
likely, this will require titrations of thevaccine in an animal model. If certainlevels of antibodies can be correlated withprotective effects, measurements of theinduced immune responses may be suf-ficient. However, for most new vaccinessuch correlations do not exist, hence chal-lenge infection studies (if available) withdeterminations of protective doses may berequired. For naked DNA, it could be ar-gued that potency assays based upon aquantitative measurement of expressionafter transfection of suitable cell linescould be sufficient, since this could cor-relate with antigen expression in vivo.However, formulated DNA vaccines maynot behave similarly in vitro and in vivo.
5.5Preclinical Safety and Efficacy
Before a new vaccine is tested in clinicaltrials, a reasonable set of data shouldbe accumulated to provide evidence that
Tab. 3 Characterization and quality testing of bulk plasmids and formulated vaccines
Plasmid identity Partial or complete sequence verification
Plasmid purity Absence or degree of denaturation or degradationSpecified minimum level of supercoiled DNA
Residual impurities Unavoidable impurities within specified limitsSterility (absence of microbial contaminants)
Stability Real-time studies in final containers (mainly for supercoiled plasmidDNA)• Specifications must be met for entire shelf life
Accelerated stability studies at elevated temperature and otherstrenuous conditions useful and recommended
Consistency 3 (−5) consecutive batches in final facilities and with final equipment• Intermediates and products must meet specifications.
Potency assay Quantitative measurement of the active component of the vaccine by,for example,– Titration in a suitable animal model– Quantitative measurement of expression after transfection of cell
lines• Comparison with an internal reference standard

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 91
the vaccine can be safely administered tohumans and is expected to be effective.For early clinical trials, a complete safetyevaluation is normally not yet available. Ata minimum, local reactions and toxicityupon single and repeated applicationsshould be studied. It may be difficult toprovide preclinical efficacy data for a newvaccine indication, if reliable models arenot yet available. But even if permissionto perform clinical testing were grantedwithout supporting efficacy data frommodel studies, what would be an adequatedose range to be studied and how arethe clinical studies to be evaluated forefficacy? Preclinical model studies anda good understanding of the protectivefunction of the vaccine beyond its capacityto induce immune responses are criticalbefore clinical trials are commenced.Studies in larger animal species can, inmany cases, yield similar or even betterinformation at lower risk and less costthan a premature Phase I clinical trial withits inherently limited possibilities to assessefficacy. There may be exceptions, wherethe vaccine’s mode of action requirestests in humans, because the vaccine istargeted very specifically at certain celltypes or is dependent on highly specificimmune responses.
5.5.1Pharmacodynamic and PharmacokineticProperties of the Vaccine
Pharmacodynamic and pharmacokineticstudies are normally applied only to chem-ical drugs; in the field of vaccines, theseterms are uncommon. Pharmacodynamiceffects of a medicinal product describe itsprecise mode of action, dose-effect rela-tionships, and side effects that are relatedto the mode of action. Pharmacokineticproperties of a medicinal (drug) product
include its in vivo adsorption, distribu-tion, metabolism, and excretion. Whereasfor drugs pharmacodynamics and phar-macokinetics are mandatory and provideessential information about basic safetyand efficacy characteristics, these are notnormally studied in depth for conven-tional vaccines, as there are technical andscientific limitations. However, for DNAvaccines, an in-depth knowledge of theirmode of action, distribution, persistence,or elimination in vivo will be importantto foresee and analyze potential risks andside effects.
Primary pharmacodynamic studies ofa vaccine should provide an under-standing about the immunological andprotective mode of action along withdose-response relationships. (Secondarypharmacodynamics mainly reveal unin-tentional side effects and will be coveredlater in the context of safety studies). Re-garding the humoral immune responses,the duration and titers of antibodies shouldbe measured and specific functional re-sponses should be studied, including, forexample, virus-neutralizing, bactericidal,and complement-binding antibodies, orantibody-dependent cytotoxicity (ADCC).Classes and subclasses of antibodies canbe evaluated to define the types of immuneresponses induced. For example, humoralimmune responses predominantly induceIgG1, whereas cellular responses are moreassociated with IgG2a antibodies.
Since DNA vaccines have the potentialto induce cytotoxic T-cells (CTL) moreefficiently than conventional, inactivatedvaccines, it may be desirable or necessaryto study and characterize the specificityand functional effects of CTL responses.However, this represents a technical andpractical challenge, because the assays tomeasure CTL are not strictly quantitativeand immune cells from each vaccinated

92 5.5 Preclinical Safety and Efficacy
individual must be cultured and studied.Alternatively, cytokine profiles could beanalyzed to identify the predominantimmune regulatory pathway. In man,cellular (T-helper type 1) responses leadingto CTL activation are mainly controlledand stimulated by IL-12 and Interferon-γ , whereas humoral responses are moreassociated with elevated levels of IL-4,5, 10, and 13. If vaccine plasmids arecoexpressing cytokines or costimulatorymolecules of antigen-presenting cells, thefunction of these cytokines needs to becarefully assessed with particular attentionto potential interactive dysfunctions. Otherpharmacodynamic effects to be consideredare immune complex formations andinteractions with other vaccinations ortherapies, which could be administeredconcurrently with the new vaccine.
Pharmacokinetic studies will be neededto understand how the vaccine plasmidsare distributed in the body and internalizedby certain cell types, whether they per-sist and express the encoded antigens orother sequences, and whether the plasmidDNA can be integrated into the cellulargenome. Pharmacokinetic biodistributionstudies in animal models should be eval-uated by high-sensitivity PCR (polymerasechain reaction) methods using primersderived from the vaccine plasmid. Whendifferent tissues are analyzed, separationof genomic DNA from plasmid DNA willbe needed for a distinction between inte-grated or nonintegrated plasmid DNA.
From studies in mice, it is to be expectedthat pure or encapsulated plasmids willmainly be distributed at the site of injec-tion and in the lymphoid organs and willpersist for at least a few months [93]. Afterintravenous injection, plasmids were dis-seminated to all examined tissues except tothe brain and to the gonads [93, 94]. In con-trast, after im injection, DNA was detected
in most tissues within a few weeks, butthereafter only in injected muscle. A smallnumber of plasmids may persist for longperiods of time, as intramuscularly in-jected plasmid DNA was still found aftermore than a year and expressed a reportermolecule during the entire observation pe-riod of 19 months [95], which is almost theentire life span of a mouse. Plasmids de-livered to the skin or propelled into uppercellular layers of the skin by the gene gunare mainly taken up by terminally differen-tiated keratinocytes, which will be lost afterseveral days or weeks [96]. But, migratingdendritic or Langerhans’ cells in the skincan also take up the plasmids and couldpossibly carry these to the draining lymphnodes [97, 98].
If a particular construct exhibits inte-gration activity, that is, if plasmid se-quences are found to be inserted into ahost cell genome, one will need to as-sess risk benefits for the disease to betargeted. In other words, ‘‘. . . manufactur-ers should carefully evaluate whether toproceed with product development’’. Thisjudgment reflects the views as expressedby the U.S. Food and Drug Adminis-tration in 1996 [88, see part V, F] andrelates to the risks of insertional mu-tagenesis. Animal model studies led tothe conclusion that the risks are low, asthe probability of an integration eventwas assessed as being several orders ofmagnitude below the spontaneous mu-tation rate [99, 100]. However, evidenceof genomic insertion must be consideredonly as a first step toward uncontrolledcritical events, such as an activation ofoncogenic sequences, deactivation of sup-pressor genes, or rearrangements. If in-tegration of DNA vaccines is observed,very long and costly tumorigenicity stud-ies may be needed. However, it must bekept in mind that such studies are meant

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 93
to identify risks, but are rarely able toprove absence of a specific risk beyondany doubt.
5.5.2Preclinical Safety and Toxicology Testing
Beyond standard toxicology and histopath-ology, the safety package of a DNAvaccine must be assembled by a vari-ety of complementary methodological ap-proaches, including biological, molecularbiological, biochemical, immunological,and immune-histological methods. Notonly the obvious risks but also all potentialrisks and unexpected or undesirable sideeffects of the vaccine must be assessed.Worst-case assumptions should be madewherever data is limited or inconclusive,for example, regarding the distribution invivo or the longevity of persistence of plas-mids and antigen expression.
Table 4 summarizes the main preclin-ical safety aspects to be considered andevaluated. Conventional toxicology studiesfollowed by histopathological examinationof all affected organs should be done inorder to assess basic safety issues. Thesetrials are normally conducted in two dif-ferent species, for example, a rodent anda nonrodent species. Mice or rats anddogs are often chosen, but with regard tocertain immunotoxicological risks, mon-keys, primates, or even transgenic animalsmay be more appropriate to study DNAvaccines. Choosing adequate safety trialsand models for vaccines (and in par-ticular for DNA vaccines) may be verydifficult, hence regulatory guidelines arealso not very specific. Therefore, guidelinesencourage developers to contact the regula-tory authorities responsible for advice andconsultation on the intended toxicologyprogram.
Tab. 4 Preclinical safety testing of DNA vaccines
An evaluation of potential unexpected and undesirable effects . . .
must consider the vaccine’s . . . in order to assess the• mode of application; • toxicity of single doses/overdoses;• intended application scheme; • local tolerance• formulation; • toxicity upon repeated application with particular attention
to potential risk factors, such as• dose range; – influences on specific organ systems (secondary
pharmacodynamics)• distribution in vivo; – reproductive functions and fertility• longevity of persistence and
antigen expression;– immunotoxicology
• acute and chronic effects ofantigen expression andimmune responses;
If risk factors exist or have been identified, additional studies arerequired to address
• incorporation of other activegenes, such as costimulatorycytokines;
• embryonal, fetal, and perinatal toxicity;
• reactions of particularlysensitive individuals; and
• mutagenicity and tumorigenic potential
• potential ofimmunopathological reactions

94 5.5 Preclinical Safety and Efficacy
Local tolerability testing is an absoluterequirement for any vaccine. One mayreasonably expect that pure plasmids willbe well tolerated, but technologies toimprove the uptake of plasmids intocells or to render DNA vaccines moreimmunogenic may affect local tolerance.For example, coadministrations of hypo-osmotic solutions, local anesthetics, orcardiotoxin have been used in research, butinduce cell necrosis at the injection site.
Animal toxicity studies using singledoses of the vaccine and overdoses (e.g. 10-fold overdoses) will most likely not revealsystemic intolerances of a DNA vaccine.Trials applying repeated doses will be moreinformative and should thus be plannedvery carefully. The number of doses andthe potency studied should correspond tothe intended application scheme, whichnormally consists of a primer dose fol-lowed by one or two booster vaccinations.Safety margins could be extended by ad-ministering extra vaccinations with shortertime intervals and by increasing the dose.Histopathology must be done with all or-gans and tissues that show changes orare expected to contain vaccine plasmids.Particular attention must be paid to poten-tial immunotoxicological side reactions:pathologic immune complex formationcould cause obstructing protein depositsin the kidney tubuli and autoimmunereactions can be identified either histo-logically as inflammatory cell invasions inaffected tissues or by elevated cell enzymelevels released as a consequence of a celldestruction.
Vaccines that are expected to affectphysiological processes related to fertil-ity, pregnancy, or fetal development wouldrequire various further studies regardingthe vaccine’s reproductive toxicology. Thesame applies if alterations to male or fe-male germ-line cells would be detected
during other toxicology studies. Suitabletest animal species for these studies mustbe chosen on the basis of immunologicaland functional properties of the vaccine.Mice, rats, or hamsters are normally used,but may not always be the ideal choice fora DNA vaccine. Reproductive toxicologyconsists of three different elements, whichcan partly be combined in a common studyprotocol. Fertility studies would evaluateadverse effects on spermatogenesis, for-mation of ovarian follicles, conception,implantation, and organogenesis. Teratol-ogy studies would cover the organogenesisperiod and usually end near or at the termof delivery. Peri- and postnatal toxicity test-ing normally commences before mating orin early pregnancy, covers the entire preg-nancy period, and extends over the entirelactation period until weaning. Recently,fertility studies have been requested moreoften in order to assess unexpected risksin female recipients of medicinal prod-ucts. Thus, fertility studies may be neededfor any new DNA vaccine that is intendedalso for women of childbearing ages. Afull reproductive toxicology program, ex-tending over two to three generations andcombined with teratology testing may berequired if vaccine plasmids have beenfound in gonadal tissues and could inducegerm-line alterations. This will also raisethe issue of whether the plasmids can bepassed on to the next generation. There-fore, the theoretical and unforeseeable riskof plasmids persisting in the gonads wouldsignificantly complicate the safety assess-ment of a DNA vaccine.
Tumorigenicity studies may only beappropriate if a DNA vaccine shows abroad tissue distribution, persists for longperiods of time, or is intended for frequentand chronic use. They would also be rel-evant if it is decided to continue thedevelopment of a DNA vaccine shown

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 95
to induce genomic integration or knownto contain oncogenic sequences or exten-sive sequence homologies to the humangenome. Such long-term studies may,however, be dispensable in those rarecases in which the vaccine is targeted to aseverely life-threatening, acute disease andis not intended for a prophylactic use.
DNA vaccines that coexpress cytokinesor contain toxins or toxin conjugates mayof course trigger a variety of unexpectedimmunological or even direct adverse ef-fects. These will mainly be assessed duringsingle and repeated dose toxicity trials, butfurther studies addressing specific ques-tions may be needed. For vector andcarrier microorganisms (e.g. salmonella orshigella species), specific studies to evalu-ate the likelihood and consequences ofa distribution to unvaccinated individualsare mandatory, as these are also standardrequirements for conventional live attenu-ated vaccines.
5.5.3Immunotoxicology Aspects
DNA vaccines are derived from bacte-rial DNA plasmid and, thus, are able tostimulate anti-DNA antibodies. If theseantibodies are cross-reactive with hostchromosomal DNA, they could act likeautoantibodies and induce autoimmunediseases (such as systemic lupus erythe-matosus) characterized by the accumu-lation of DNA, antinuclear antibodies,and complement in various organs alongwith local inflammatory responses. Spe-cific ‘‘lupus-prone’’ mouse strains existthat develop a similar disease. Repeatedapplication of DNA vaccines to these micedid not alter the onset or the course ofthe disease. In normal mice, anti-DNA an-tibodies were induced by DNA vaccines,but these remained far below those levels
found in lupus-prone mice [101]. Duringclinical trials in humans, significant risesof anti-DNA antibodies have not beenobserved [102]. Thus, the risk of anti-DNAautoimmune responses seems low, butshould be monitored during clinical trials.
Cells harboring vaccine plasmids andexpressing foreign antigens normallypresent these antigens on their cell sur-face. They can not only stimulate animmune response but can as well be-come the target of an immune attack,resulting in inflammatory reactions andin cell-mediated cytolysis in tissues withhigh expression rates. These effects maybe more prominent after a second or re-peated application of the vaccine. Acuteinflammation of muscle tissues associatedwith the destruction of myocytes has infact been observed during some animalstudies [103, 104], whereas other studiesdetected no anti-myocyte antibodies ormuscle tissue reactions [101]. Therefore,monitoring of muscle cell enzyme levelsduring early clinical trials should be con-sidered to assess the risks associated withtissue destruction and autoantibodies. Sofar, there have been no clinical reportsabout such problems. However, a higherefficiency of plasmid uptake and expres-sion or plasmids targeted to specific organsmay result in quite different findings. Inprinciple, animal toxicology studies andhistological evaluations should be able todetect such autoimmune effects, but dueto animal species and human genotypedifferences, the matter needs to be studiedmainly during clinical trials.
A classical way of actively inducingimmune tolerance, for example, against al-lergens, would be to repeatedly administerlow doses of the same antigen over a longperiod of time. In principle, this couldalso occur by a prolonged antigen expres-sion after DNA vaccination. Neonates with

96 5.6 Clinical Safety and Efficacy Trials
their immature immune systems or indi-viduals with reduced immune responsescould be particularly prone to this kindof tolerance. In one study, tolerance wasobserved after DNA vaccination of neona-tal mice. These mice did not respond to asecond vaccination given several monthslater, even if the same antigen was in-jected directly as a conventional vaccine.If immunized at an age of two monthsand revaccinated four months later, miceresponded well to the same DNA vaccine.Aged mice also showed reduced immuneresponses and reduced protection frominfection [105, 106]. In contrast, severalother studies did not reveal similar ef-fects, thus immune tolerance appears tobe associated only with certain antigens.As long as the underlying mechanisms re-main unknown, preclinical studies couldbe useful to predict clinical results, partic-ularly in those cases in which the vaccineis to be used in neonates, at advanced age,or in other immunocompromised individ-uals. As for any other vaccine intendedfor those target groups, separate clinicaltrials should also be done to assess safetyand efficacy.
5.6Clinical Safety and Efficacy Trials
A DNA vaccine candidate that has success-fully passed all preclinical hurdles needsto be tested clinically by a three-phasedscheme as outlined in Chapter 4. As de-fined by a European Council directive, aclinical trial means ‘‘. . .any investigationin human subjects intended to discoveror verify the clinical, pharmacologicaland/or other pharmacodynamic effects of. . . investigational medicinal product(s),and/or to identify any adverse reactions. . .and/or to study absorption, distribution,
metabolism, and excretion . . . with theobject of ascertaining . . .safety and/or ef-ficacy.’’ [107]. This definition explains thatthe expectations for the conduct of clinicaltrials go far beyond efficacy and a basicsafety assessment, but include pharmaco-dynamic and pharmacokinetic elements,which could be of particular relevance forDNA vaccines and even more so for vectororganisms applied to DNA vaccines. Thus,clinical trials within that scope would beimpossible without having established arelevant database from preclinical studies.Certain pharmacological and pharmacoki-netic aspects cannot be studied directly inhumans, so compromises must be madeand the gaps must be filled by appropriateanimal studies.
On the basis of the available preclinicaldata and on predefined clinical trialplans, ethics committees will evaluatewhether the anticipated benefits andrisks would justify clinical testing. Inaddition, local regulatory authorities mustbe informed and may deny consent.The clinical trial product must be madein compliance with Good ManufacturingPractices with increasing demands (e.g.regarding the qualification of equipmentand facilities, product specifications, andvalidation of methods) as the trials proceedto later phases.
Phase I clinical trials mainly evaluatethe vaccine’s basic safety and also measurethe immune responses induced. Safetyaspects to be studied include, for example,general local and systemic reactions,potential immunological complications,and specific side effects on certain organsand tissues that might occur because oftheoretical considerations or have beenidentified during toxicology studies. Forlive vector organisms, additional safetyfeatures, such as shedding and distributionof the organisms, will have to be studied

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 97
in order to confirm preclinical safetyevaluations and to rule out risks associatedwith the practical use of the vaccine.
Experience from published clinicalPhase I studies will most likely lead to adose-escalation study design, starting withdoses that were shown to be of adequateimmunogenicity during preclinical test-ing. According to existing – still rather lim-ited – experience, increased DNA dosesmay then be needed to achieve immuneresponses in humans, similar to thoseseen in animal models. For example, an-tibody responses against a Plasmodiumfalciparum circumsporoite antigen wereinduced in mice at doses of a few micro-grams of DNA per vaccination. The firsttrials of this vaccine in humans initiallyused doses from 20 µg to 100, 500, andfinally up to 2500 µg, but even the high-est doses did not stimulate any detectableantibodies. However, CTL responses in upto four out of five trial individuals in thehighest dose group were noted [108, 109].A similar experience has been seen withgene-gun delivery of plasmids, for exam-ple, for a DNA vaccine encoding hepatitisB surface antigen, where low antibodyresponses were observed in human tri-als [110]. Owing to the capacity of the goldparticles to bind DNA, there have beenlimitations in the amount of DNA thatcan be delivered by the gene gun. So,repeated boosting and simultaneous ap-plications at multiple sites were neededto deliver higher doses of DNA plasmids.In this case, the DNA vaccine finally in-duced antibody levels, which would havebeen considered as being protective, if in-duced by a conventional vaccine. The firstexample of a malaria DNA vaccine illus-trate a dilemma, which is only, in part,specific to DNA vaccines. The vaccine in-duced CTL but no antibody response. Thequestion now is whether CTL responses
alone can be taken as a marker of pro-tection, and if so, which level would berequired? This question cannot be an-swered until the vaccine has been testedclinically under conditions that expose trialsubjects to the parasites and monitor thenumber and severity of cases occurringin comparison to a control group. Un-til then, using CTL as an indicator ofpotency raises logistic and technical chal-lenges, since measuring CTL responsesrequires viable peripheral blood mononu-clear cells from each trial individual andhighly specialized laboratories to performthe tests.
A Hepatitis B DNA vaccine may bea much more suitable candidate to es-tablish a first DNA vaccine product andto successfully pass all clinical hurdles.There is a well-known, single antigen thatconfers protection, whose serological re-sponses are easily measured and can becorrelated with protective effects. OtherDNA vaccine candidates include antitu-mor vaccines [111–113], which may passclinical phases more rapidly than prophy-lactic vaccines, as their efficacy could bea directly measurable clinical effect, suchas tumor regression or reduced or absentmetastases. Furthermore, their use couldbe justified even with a partial effect andsome unresolved safety concerns, whereasfor a prophylactic vaccine for healthy peo-ple, higher standards must be met.
5.7Registration and Licensing of DNA Vaccines
The formal process of licensing a newvaccine in different countries is describedin Chapter 10 of this volume. In thischapter, only a few general aspects oflicensing of an entirely new product andactive principle will be briefly discussed.

98 5.8 Conclusions
Potential developers of a DNA vaccineproduct should not only consult specificguidelines and recommendations for vac-cines and DNA vaccines but should alsobe advised to carefully monitor the regu-lations that evolve around gene therapy.There are some parallels and similaritiesbetween DNA vaccines and gene therapyapproaches, thus insight may be gainedabout potential quality and safety aspects.For safety and toxicology studies andduring the preparation of clinical trials,contact should be sought with regulatoryauthorities in the respective countries andpreferably with the leading authorities,such as the European Medicines Evalu-ation Agency (EMEA) or the US FDA,who welcome such contacts and have im-plemented official channels to obtain amore binding opinion for specific ques-tions. As the EMEA relies on rapporteursfrom the individual country’s authorities,certain national authorities (particularlythose who are actively pursuing researchin this field) may also be open to discussdevelopment proposals on a more infor-mal level. Through these consultations,developers will not only obtain useful ad-vice on specific questions but may alsodetermine where a specific applicationshould be filed. There may occasionallybe doubts regarding the regulatory path-ways in the United States and whethera certain product (e.g. a DNA vaccineintended to induce antihormonal antibod-ies) will be considered a vaccine/biologicalproduct or a gene therapy/drug. Thesetwo categories are regulated quite differ-ently and by different departments, butthe definitions for these products partlyoverlap and are evaluated on a case-by-casebasis.
DNA vaccines raise certain safety-relatedissues that may remain unsolved even afterintensive studies. Hence, a scientifically
grounded, quantitative risk evaluationshould be made. On the basis of ex-isting data from the developer’s ownstudies, combined with published infor-mation, and complemented by reasonedassumptions where specific informationis missing, such a risk assessment can bea valuable tool to aid the decision process.In the absence of quantitative data, worst-case assumptions should be made, butrealistic case scenarios may be included toindicate a possible range. An example ofhow this done in a quantitative mannerfor the risk of insertional mutagenesis isgiven by Kurth [99].
As with other new technologies, it ispossible that the first licensed DNA vac-cines will receive conditional approval. Inthis case, postlicensing ‘‘Phase IV’’ clini-cal trials may be requested to address andmonitor remaining unresolved issues byspecific studies or on a larger scale. Fur-thermore, as for any medicinal product,effective and intensive pharmacovigilanceprocedures will be used to closely mon-itor the appearance of any side effects,while the vaccine is applied widely androutinely.
5.8Conclusions
Studies of DNA vaccines in animal mod-els have borne out many of the theoreticaladvantages of this approach over conven-tional methods of immunization. Fast andrelatively simple to construct using stan-dard molecular biologic techniques, theyalso have become easier to produce, ow-ing to the development of various kits forproducing plasmid in quantities neededfor clinical trials. The antigens that areexpressed from the plasmid DNA arein their native conformations and are

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 99
processed for presentation to T-cells com-parable to that of the native protein inviruses or intracellular bacteria. In labo-ratory animal studies, it is clear that thisapproach can provide protective immunityfrom infectious challenges in a variety ofmodel systems. However, some importantlimitations remain. A good understand-ing of the structure of the antigen andits intracellular processing is required.Attempts to generate DNA vaccines ofhigh quality without this understandingare often frustrating because of poor ex-pression, antigenicity, or processing. Theuptake of the DNA and the efficiency ofits delivery can vary significantly amongspecies. This is not only related to bodymass but also to muscle size, volume ofinoculum, and structure of muscle compo-nents. By far, the most drastic limitationis the lack of immunogenicity of thesevaccines in humans. The basis for thisdifference between humans and animalmodels is still not well understood. Variousmethod of targeting, particulate delivery,and formulation offer promise, but haveyet to be fully tested in humans. Theinability to compare an ineffective DNAvaccine with an effective one (since noneyet exists) is a major handicap to un-derstanding how to make DNA vaccinesimmunogenic in humans. A further con-sequence of the lack of effectiveness inearly human trials is that the regulatoryenvironment for DNA vaccines is notyet mature. Many Phase I studies havebeen done and regulatory agencies haveformed a comprehensive set of expecta-tions for which safety parameters are tobe measured and how to measure them.However, since no DNA vaccine has ad-vanced beyond Phase II, regulators havenot yet confronted the need to createregulatory or safety requirements for regis-tration studies. The future of DNA vaccines
depends on one or more technologies,possibly not yet invented, that will giveconsistent high immune responses. Onlywhen that problem has been solved willthe manufacturing, clinical, and regula-tory pieces of the development puzzle fallinto place.
References
1. T. W. Dubensky, B. A. Campbell, L. P. Vill-arreal, Proc. Natl. Acad. Sci. U. S. A. 1984,81, 7529–7533.
2. H. Will, R. Cattaneo, H. G. Koch et al.,Nature 1982, 299, 740–742.
3. J. A. Wolff, R. W. Malone, P. Williamset al., Science 1990, 247, 1465–1468.
4. D. C. Tang, M. De Vit, S. A. Johnston,Nature 1992, 356, 152–154.
5. J. B. Ulmer, J. J. Donnelly, S. E. Parkeret al., Science 1993, 259, 1745–1749.
6. E. F. Fynan, R. G. Webster, D. H. Fulleret al., Proc. Natl. Acad. Sci. U. S. A. 1993,90, 11478–11482.
7. J. J. Donnelly, J. B. Ulmer, J. W. Shiveret al., Annu. Rev. Immunol. 1997, 15,617–648.
8. S. Gurunathan, D. M. Klinman, R. A. Seder,Annu. Rev. Immunol. 2000, 18, 927–974.
9. J. R. Gebhard, J. Zhu, X. Cao et al., Vaccine2000, 18, 1837–1846.
10. A. Bojak, D. Hammer, H. Wolf et al.,Vaccine 2002, 20, 1975–1979.
11. E. Kurar, G. A. Splitter, Vaccine 1997, 15,1851–1857.
12. M. Kwissa, V. K. Von Kampen, R. Zur-briggen et al., Vaccine 2000, 18, 2337–2344.
13. A. Kamei, S. Tamaki, H. Taniyama et al.,Virology 2000, 273, 120–126.
14. Z. Q. Xiang, Z. He, Y. Wang et al., Vaccine1997, 15, 896–898.
15. M. J. Hariharan, D. A. Driver, K. Townsendet al., J. Virol. 1998, 72, 950–958.
16. W. W. Leitner, H. Ying, D. A. Driver et al.,Cancer Res. 2000, 60, 51–55.
17. O. Vidalin, A. Fournillier, N. Renard et al.,Virology 2000, 276, 259–270.
18. M. M. Morris-Downes, K. V. Phenix, J.Smyth et al., Vaccine 2001, 19, 1978–1988.
19. C. Smerdou, P. Liljestrom, Curr. Opin. Mol.Ther. 1999, 1, 244–251.

100 5.8 Conclusions
20. C. M. Leutenegger, F. S. Boretti, C. N.Mislin et al., J. Virol. 2000, 74,10447–10457.
21. K. F. Sykes, S. A. Johnston, Nat. Biotechnol.1999, 17, 355–359.
22. I. F. Ciernik, J. A. Berzofsky, D. P. Carbone,J. Immunol. 1996, 156, 2369–2375.
23. A. Iwasaki, C. S. Dela Cruz, A. R. Younget al., Vaccine 1999, 17, 2081–2088.
24. G. Y. Ishioka, J. Fikes, G. Hermanson et al.,J. Immunol. 1999, 162, 3915–3925.
25. T. Hanke, J. Schneider, S. C. Gilbert et al.,Vaccine 1998, 16, 426–435.
26. J. Haas, E. C. Park, B. Seed, Curr. Biol.1996, 6, 315–324.
27. J. Zur Megede, M. C. Chen, B. Doe et al., J.Virol. 2000, 74, 2628–2635.
28. S. L. Baldwin, C. D. D’souza, I. M. Ormeet al., Tuber. Lung Dis. 1999, 79, 251–259.
29. C. Rush, T. G. Mitchell, P. Arside, J.Immunol. 2002, 169, 4951–4960.
30. M. P. Velders, S. Weijzen, G. L. Eiben et al.,J. Immunol. 2001, 166, 5366–5373.
31. F. Rodriguez, J. Zhang, J. L. Whitton, J.Virol. 1997, 71, 8497–8503.
32. F. Rodriguez, S. Harkins, J. M. Redwineet al., J. Virol. 2001, 75, 10421–10430.
33. H. Ji, T. L. Wang, C. H. Chen et al., Hum.Gene Ther. 1999, 10, 2727–2740.
34. G. Deliyannis, J. S. Boyle, J. L. Brady et al.,Proc. Natl. Acad. Sci. U. S. A. 2000, 97,6676–6680.
35. A. Biragyn, K. Tani, M. C. Grimm et al.,Nat. Biotechnol. 1999, 17, 253–258.
36. T. M. Ross, Y. Xu, R. A. Bright et al., Nat.Immunol. 2000, 1, 127–131.
37. J. S. Boyle, J. L. Brady, A. M. Lew, Nature1998, 392, 408–411.
38. J. Wild, B. Gruner, K. Metzger et al.,Vaccine 1998, 16, 353–360.
39. Y. H. Chow, W. L. Huang, W. K. Chi et al.,J. Virol. 1997, 71, 169–178.
40. Y. Huang, W. P. Kong, G. J. Nabel, J. Virol.2001, 75, 4947–4951.
41. R. R. Amara, F. Villinger, J. D. Altmanet al., Science 2001, 292, 69–74.
42. E. J. Gordon, R. Bhat, Q. Liu et al., J. Infect.Dis. 2000, 181, 42–50.
43. J. A. Wolff, J. J. Ludtke, G. Acsadi et al.,Hum. Mol. Genet. 1992, 1, 363–369.
44. T. Tuting, W. J. Storkus, L. D. Falo, Jr J.Invest. Dermatol. 1998, 111, 183–188.
45. A. Porgador, K. R. Irvine, A. Iwasaki et al.,J. Exp. Med. 1998, 188, 1075–1082.
46. M. Corr, A. Von Damm, D. J. Lee et al., J.Immunol. 1999, 163, 4721–4727.
47. O. Akbari, N. Panjwani, S. Garcia et al., J.Exp. Med. 1999, 189, 169–178.
48. M. A. Chattergoon, T. M. Robinson, J. D.Boyer et al., J. Immunol. 1998, 160,5707–5718.
49. S. Casares, K. Inaba, T. D. Brumeanu et al.,J. Exp. Med. 1997, 186, 1481–1486.
50. A. Bot, A. C. Stan, K. Inaba et al., Int.Immunol. 2000, 12, 825–832.
51. E. Manickan, S. Kanangat, R. J. Rouseet al., J. Leukoc. Biol. 1997, 61, 125–132.
52. J. B. Ulmer, R. R. Deck, C. M. Dewitt et al.,Immunology 1996, 89, 59–67.
53. T. M. Fu, J. B. Ulmer, M. J. Caulfield et al.,Mol. Med. 1997, 3, 362–371.
54. A. Y. Huang, P. Golumbek, M. Ahmad-zadeh et al., Science 1994, 264, 961–965.
55. M. A. Chattergoon, J. J. Kim, J. S. Yanget al., Nat. Biotechnol. 2000, 18, 974–979.
56. S. Sasaki, R. R. Amara, A. E. Oran et al.,Nat. Biotechnol. 2001, 19, 543–547.
57. U. Kumaraguru, R. J. Rouse, S. K. Nairet al., J. Immunol. 2000, 165, 750–759.
58. J. A. Leifert, S. Harkins, J. L. Whitton, GeneTher. 2002, 9, 1422–1428.
59. S. C. Oliveira, J. S. Harms, R. R. Afonsoet al., Hum. Gene Ther. 2001, 12,1353–1359.
60. G. Widera, M. Austin, D. Rabussay et al., J.Immunol. 2000, 164, 4635–4640.
61. S. Zucchelli, S. Capone, E. Fattori et al., J.Virol. 2000, 74, 11598–11607.
62. A. M. Krieg, Annu. Rev. Immunol. 2002, 20,709–760.
63. Y. Sato, M. Roman, H. Tighe et al., Science1996, 273, 352–354.
64. R. A. Pontarollo, L. A. Babiuk, R. Heckeret al., J. Gen. Virol. 2002, 83, 2973–2981.
65. R. Weeratna, C. L. Brazolot Millan, H. L.Krieg Amdavis, Antisense Nucleic Acid DrugDev. 1998, 8, 351–356.
66. M. Dupuis, K. Denis-Mize, C. Woo et al., J.Immunol. 2000, 165, 2850–2858.
67. M. J. Roy, M. S. Wu, L. J. Barr et al., Vaccine2000, 19, 764–778.
68. M. Kumar, A. K. Behera, R. F. Lockey et al.,Hum. Gene Ther. 2002, 13, 1415–1425.
69. D. H. Jones, C. D. Partidos, M. W. Stewardet al., Behring Inst. Mitt. 1997, 220–228.
70. M. G. Cusi, R. Zurbriggen, M. Valassinaet al., Virology 2000, 277, 111–118.

DNA Vaccines: from Research Tools in Mice to Vaccines for Humans 101
71. W. O. Rogers, J. K. Baird, A. Kumar et al.,Infect. Immun. 2001, 69, 5565–5572.
72. M. Fernandez-Alonso, A. Rocha, J. M. Coll,Vaccine 2001, 19, 3067–3075.
73. G. Zhang, V. Budker, P. Williams et al.,Hum. Gene Ther. 2001, 12, 427–438.
74. G. Gregoriadis, Curr. Opin. Mol. Ther. 1999,1, 39–42.
75. D. O’hagan, M. Singh, M. Ugozzoli et al., J.Virol. 2001, 75, 9037–9043.
76. K. Roy, H. Q. Mao, S. K. Huang et al., Nat.Med. 1999, 5, 387–391.
77. K. S. Denis-Mize, M. Dupuis, M. L. Macki-chan et al., Gene Ther. 2000, 7, 2105–2112.
78. H. Lee, J. H. Jeong, T. G. Park, J. ControlRelease 2001, 76, 183–192.
79. R. Schirmbeck, S. A. Konig-Merediz,P. Riedl et al., J. Mol. Med. 2001, 79,343–350.
80. M. A. Barry, W. C. Lai, S. A. Johnston,Nature 1995, 377, 632–635.
81. P. M. Smooker, Y. Y. Setiady, T. W. Rain-czuk Aspithill, Vaccine 2000, 18, 2533–2540.
82. K. Manoutcharian, L. I. Terrazas, G. Gevo-rkian et al., Immunol. Lett. 1998, 62,131–136.
83. C. P. Locher, K. F. Sykes, D. J. Blackbournet al., J. Med. Primatol. 2002, 31, 323–329.
84. K. F. Sykes, M. G. Lewis, B. Squires et al.,Vaccine 2002, 20, 2382–2395.
85. P. C. Melby, G. B. Ogden, H. A. Floreset al., Infect. Immun. 2000, 68, 5595–5602.
86. C. Almazan, K. M. Kocan, D. K. Bergmanet al., Vaccine 2003, 21, 1492–1501.
87. M. Houghton, Curr. Top. Microbiol.Immunol. 2000, 242, 327–39.
88. Food and Drug Administration, Centerfor Biologics Evaluation and Research:Points to consider on plasmid DNA vac-cines from preventive infectious diseaseindications, Docket No. 96 N-0400, 1996,http://www.fda.gov/CBER/gdlns/plasmid.pdf
89. WHO Guidelines for assuring the quality ofDNA vaccines. Annex 3 in: WHO TechnicalReport Series, No. 876, 1998.
90. European (CPMP) and internationallyharmonized (ICH) guidance documentswith basic applicability to DNA vaccines:Note for guidance on specifications:Test procedures and acceptance criteriafor biotechnological/biological products.CPMP/ICH/356/96 Note for guidance onpreclinical pharmacology and toxicology
testing of vaccines. CPMP/SWP/465/95 forguidance on preclinical safety evaluationof biotechnology-derived Note pharmaceuti-cals. CPMP/ICH/302/95 Note for Guidanceon the quality, preclinical and clinical as-pects of gene transfer medicinal products.CPMP/BWP/3088/99 Safety studies forgene therapy products. CPMP/SWP/112/98Retrievable via http://www.emea.eu.int/ICH guidelines are also available underhttp://www.ich.org/ich5.html.
91. J. P. Gregersen, Requirements for thepreclinical pharmacology and safetyassessment and: Outline of majorregistration requirements (Annex A inResearch and Development of Vaccinesand Pharmaceuticals from Biotechnology.A Guide to Effective Project Management,Patenting and Product Registration (Ed.:J. P. Gregersen), VCH, Weinheim, NewYork, Basel, Cambridge, Tokyo, 1994, pp.119–147.
92. M. Schleef, Plasmids for gene therapyand vaccination, Wiley-VCH, WeinheimGermany, 2001.
93. S. E. Parker, F. Borellini, M. L. Wenk et al.,Hum. Gene Ther. 1999, 10, 741–758.
94. L. Lunsford, U. Mckeever, V. Eckstein et al.,J. Drug. Target 2000, 8, 39–50.
95. J. A. Wolff, J. J. Ludtke, G. Acsadi et al.,Hum. Mol. Genet. 1992, 1, 363–369.
96. E. Raz, D. A. Carson, S. E. Parker et al.,Proc. Natl. Acad. Sci. U. S. A. 1994, 91,9519–9523.
97. C. Condon, S. C. Watkins, C. M. Celluzziet al., Nat. Med. 1996, 2, 1122–1128.
98. C. A. Torres, A. Iwasaki, B. H. Barber et al.,J. Immunol. 1997, 158, 4529–4522.
99. R. Kurth, Ann. N. Y. Acad. Sci. 1995, 772,140–151.
100. T. Martin, S. E. Parker, R. Hedstrom et al.,Hum. Gene Ther. 1999, 10, 759–768.
101. G. Mor, M. Singla, A. D. Steinberg et al.,Hum. Gene Ther. 1997, 10, 293–300.
102. R. R. Macgregor, J. D. Boyer, K. E. Ugenet al., J. Infect. Dis. 1998, 178, 92–100.
103. H. L. Davis, C. L. Brazolot Millan, S. C.Watkins, Gene Ther. 1997, 4, 181–188.
104. J. L. Whitton, F. Rodriguez, J. Zhang et al.,Vaccine 1999, 7, 1612–1619.
105. G. Mor, M. Yamshchikov, M. Sedegahet al., J. Clin. Invest. 1996, 98, 2700–2705.

102 5.8 Conclusions
106. D. M. Klinman, M. Takeno, M. Ichinoet al., Springer Semin. Immunopathol. 1997,19, 245–256.
107. European Parliament and Council Directive2001/20/EC. Official J. Eur. Commun. L121/34, (1.5.2001).
108. R. Wang, D. L. Doolan, T. P. Le et al.,Science 1998, 282, 476–480.
109. T. P. Le, K. M. Coonan, R. C. Hedstromet al., Vaccine 2000, 18, 1893–1901.
110. M. J. Roy, M. S. Wu, L. J. Barr et al., Vaccine2001, 19, 764–778.
111. F. K. Stevenson, D. Zhu, M. B. Spellerberget al., Rev. Clin. Exp. Hematol. 1999, 9,2–21.
112. M. Mincheff, S. Tchakarov, S. Zoubaket al., Eur. Urol. 2000, 38, 208–217.
113. P. Walsh, R. Gonzales, S. Dow et al., Hum.Gene Ther. 2000, 11, 1355–1368.

103
6Characterization andBioanalytical Aspects ofRecombinant Proteins asPharmaceutical Drugs
Jutta Haunschild and Titus KretzschmarMorphoSys AG Lena-Christ-Strasse 48 D-82152 Martinsried Germany
6.1Introduction
The development of recombinant proteinsas pharmaceutical drugs demands robust,sensitive, and specific analytical assays tocharacterize the purified drug with respectto its physicochemical as well as biologicalfeatures, and bioanalytical assays to quan-tify proteins or their activities in biolog-ical matrices. Well-established analyticalassays are applied to determine character-istics such as purity/impurities, identity,quantity, stability, specificity, and potencyof the purified recombinant protein dur-ing drug development. The determinationof the purity and identity of a proteindrug is a particularly challenging tasksince recombinant proteins are producedfrom living systems that inherently leadto protein variants (e.g. posttranslation-ally modified and/or fragmented proteins)with altered characteristics, which may behard to separate from the original proteindrug. In stability studies, those proteinfeatures are evaluated that might be sub-ject to change during storage/handling ofthe drug. Specificity measurements leadto a closer understanding of drug-target
interaction(s), which might result in earlyhints about possible side effects in clinicaltrials. Finally, potency determinations areused to quantify the biological activity ofthe therapeutic protein.
On the other hand, bioanalytical assaysare necessary to determine and quan-tify the protein drug in biological fluids.For example, validated bioanalytical as-says are the key in the quantitation ofthe protein drug in the course of phar-macokinetic studies (see Chapter 8). Espe-cially in the case of humanized/humanmonoclonal antibodies, bioanalytical as-say development in human serum/plasmais challenging because the therapeuticconcentration of antibodies can be verylow (0.1 to 10 µg mL−1 or even lower), andbecause these antibodies are so similar tothe native human antibodies that circulatein the blood in very high concentrations of10 000 µg mL−1.
Another major topic of scientific and reg-ulatory consideration in the developmentof therapeutic proteins is the assessment ofundesired immune responses to the drugthat may lead to a reduction in efficacyand to adverse reactions. This assessmentalso requires validated bioanalytical assays,
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

104 6.2 Characterization of Purified Recombinant Proteins
which allow to precisely measure the im-mune response.
In future, there will be an even greateremphasis on the (bio)analytical descriptionof biological substances because of anincrease in numbers of biologicals inclinical development, and of the adventof generics of biological drugs and the taskof evaluating these compounds for clinicaluse. Developing methods and protocolsfor assessing bioequivalence of an originaldrug and its generics is a high priorityfor the FDA according to the current FDACommissioner [1].
The scope of this article is to summarizethe analytical methods used to characterizethe purified recombinant protein in the dif-ferent stages of drug development, and tosummarize bioanalytical methods with fo-cus on their validation and standardizationas well as in the determination of immuno-genicity of the therapeutic protein.
6.2Characterization of Purified RecombinantProteins
6.2.1Purity and Impurities
The absolute purity of a biological sub-stance is hard – if at all possible – to deter-mine. Regular and sometimes only subtleprotein modifications such as glycosyla-tion, alternative disulphide bond forma-tion, deamidation, oxidation, phosphoryla-tion, acetylation, sulfation, sulfoxidation,γ -carboxylation, and pyroglutamate for-mation lead to protein variants that mayhave more or less different characteris-tics. Also, truncated protein variants mightbe generated by the presence of crypticor alternative start sites of transcription,by premature stop of the peptide chain
elongation process, or by the action ofhost cell peptidases. Peptide mapping andmass spectrometry (MS) usually achievedetection of most of such protein vari-ants. Aggregation is another modificationof a protein, which can be the resultof, for example, underglycosylation, oxi-dation, and/or deamidation, and can bedetected by size-exclusion chromatogra-phy. The amount of aggregated proteinusually should stay below 5%. It is highlyrecommended to investigate the natureand potential toxicity of such alterations.To analyze these variants is an essential yetchallenging task, as their physicochemicalfeatures might not be very different fromeach other. Owing to the possible pres-ence of highly related protein variants inthe preparation, it is recommended to de-termine the purity of a protein drug byat least two independent methods, that is,methods that use different physicochemi-cal principles such as SDS-polyacrylamidegel electrophoresis and reverse-phase highpressure liquid chromatography (HPLC).
Besides these protein variants, so-calledprocess-related impurities have to be con-sidered. Of major concern are residual an-tibiotics from fermentation, enzymes andantibodies from chromatography columnsand other column leachates, endotoxinfrom bacterial hosts [2], (retro-) viruses [3],bacteria, fungi, mycoplasma, prions, var-ious other media components such assolvents, antifoam agents, heavy metalions, as well as preservatives and ex-pression host components. As for DNAcontaminations, less than 10 to 100 pgper dose are allowed in the final drugproduct [4]. To check for the presence ofantigenic expression host-related impuri-ties, a polyclonal antibody serum to the‘‘empty’’ host, that is, host cells that are notharboring the product-encoding gene, isvery helpful. Moreover, whenever possible,

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 105
specific impurity standards should be usedfor impurity quantification, and the limitof detection/-quantification (LOD/-Q) forimpurity assays should be indicated. Theacceptance limits should not be set higherthan safety data justify, and it should notbe lower than what is historically achiev-able by the manufacturing process and byreasonable analytical efforts.
In some instances, the protein drugis conjugated to effector functions suchas radioisotopes, toxins, or other proteinssuch as cytokines that mediate a biologicaleffect. Besides considering all aspectsmentioned above and below for theindividual components of the conjugate,special care has to be taken to determinethe average coupling ratio as well as theamount of free components, if any, in thepreparations.
6.2.2Identity
Identity assays aim at confirming themolecular composition and, if technicallypossible and commercially reasonable,structure of the drug substance, and thusshould be suited to allow the detection ofeven minor alterations in the molecularcomposition of the drug.
Amino acid analysis and peptide map-ping are standard methods in the courseof protein identification processes [5, 6].Molecular mass determination of wholemolecules as well as peptide frag-ments with accuracies of about 0.01%by either matrix-assisted laser desorp-tion/ionization (MALDI)-MS for surfaceimmobilized samples, or electrospray ion-ization (ESI)-MS for liquid samples, isanother highly efficient protein iden-tification method. These methods ad-ditionally support the identification ofposttranslational modifications such as
glycosylation, glycation, phosphorylation,sulfation, etc. [7–11].
Besides amino acid analysis and elabo-rated mass spectroscopy techniques, manymore analytical methods are applied tosupport the identity examinations of theprotein drug, such as determination of theextinction coefficient, isoelectric point, andcrystal structure, as well as recording thenuclear magnetic resonance (NMR) andcircular dichroism (CD) spectra and deter-mining the chromatographic profiles fromHPLC-runs as well as from capillary andpolyacrylamide gel electrophoresis (CEand PAGE, respectively).
Automated systems as well as microflu-idic devices (‘‘lab-on-the-chip’’) for chro-matographic separation of proteins andtheir subsequent analysis have a huge po-tential to dominate the analytics field inthe future [12].
6.2.3Quantification of the Protein
Many physicochemical assays are estab-lished to quantify the protein mass. It isdetermined by exploiting the extinction co-efficient in optical density measurementsor by colorimetric assays such as theBradford, Lowry, bicinchoninic (BCA), andbiuret assay [13, 14]. Albeit easy to per-form, these colorimetric assays suffer frominaccuracies that are due to the use of inap-propriate standards like bovine serum al-bumin. If relevant standards are not avail-able, quantitative amino acid analysis [6],the (micro-)Kjeldahl nitrogen method [14,15] or gravimetry as very accurate but time-consuming alternatives can be applied.
Bioassays and immunoassays are alsoexploited to quantify the protein amount,which have to be validated for accuratemeasurements and definitively require areference standard (see Sect. 6.2.6.1).

106 6.2 Characterization of Purified Recombinant Proteins
6.2.4Stability, Storage, and Sterility
Stability studies include the evaluation ofthose protein features that are susceptibleto change during storage and mightinfluence the quality, safety, and efficacy.The testing should cover physicochemical,biological, and microbiological aspects, aswell as the preservative content such asantioxidants or antimicrobials.
Stability-indicating assays are validatedquantitative analytical procedures that candetect drug alterations over time. Theyshould include tests for integrity of thedrug, potency, sterility, and, if applicable,moisture, pH, and preservative stabilitymeasured at regular intervals throughoutthe dating period [16–19].
One key parameter for stability testingis temperature. Real-time stability studiesmay be confined to the proposed storagetemperature. Accelerated stability tests canbe conducted at elevated temperaturesexceeding standard storage temperatures.Data from accelerated stability studiesare supportive but do not substitutefor real-time data. They may help tovalidate the respective analytical assays,to elucidate the degradation profile ofthe drug, and to assess the drug stabilityunder storage conditions other than thoseproposed.
Evaluation of the effect of humidityon drug stability may be omitted if thedrug container gives appropriate protec-tion against variations in humidity. Otherparameters that, if indicated, have to beconsidered for stability testing are the ef-fect of light exposure, container/closuredrug interactions, and stability after recon-stitution of a freeze-dried product. Sterilitytesting should be performed initially andat the end of the proposed shelf life (fordetails see [20, 21]).
Besides stability and sterility, othercharacteristics of the sample such as visualappearance (color, opacity, particulates),dissolution time, and osmolality also haveto be described for the drug product in itsfinal container.
Although the biological drug may besubject to substantial losses of activity overstorage time, the regulatory authoritieshave provided little guidance concerningrelease and end of shelf-life specifications.It thus remains a case-by-case decisionwhether the loss of activity is consideredacceptable.
6.2.5Specificity and Cross-reactivity
Assays have to be designed that allowthe evaluation of the specificity of agiven drug-target interaction, especiallywhen considering antibodies as a veryimportant class of protein drugs. Onemay distinguish three types of assays forspecificity assessment:
1. Binding assays (see also Sect. 6.2.6.1),which include appropriate positive andnegative controls as well as targetmolecule controls that ideally consistof the closest target-related variant(s).Preferably, quantitative inhibition as-says are performed with soluble targetpreparations, which distinctly enhancethe confidence in the specificity of thedrug-target interaction.
2. Determination of the molecular natureof the drug binding site by epitopemapping or by measuring the impactof carbohydrates on the binding siteand of other modifications that mightmodulate the binding event.
3. Immunohistochemistry (IHC) to scanfor cross-reactivity with human (and an-imal) tissues [22]. Quick-frozen surgical

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 107
samples are preferred over post-mortem tissue samples. Tissues fromat least three unrelated human donorsshould be evaluated.
If with these assays cross-reactivity invitro to nontarget molecules or tissues isdetected, then testing in vivo for cross-reactivity in animal models, if available, isindicated.
6.2.6Potency Determination
In the first part of this chapter, generalaspects of potency determinations aredescribed, whereas in the second part,special features of potency assessment inbiological matrices are summarized.
6.2.6.1 General Aspects and AssaysProtein therapeutics often exert an exqui-site biological function at very low concen-trations, that is, in the pico- to nanomolarrange. Sensitive methods for the deter-mination of the biological activity haveto be established that reliably allow thequantification of this prominent featureof protein drugs. It is of utmost impor-tance to define a reference standard inquantitative terms, which is a difficult un-dertaking for protein drugs [23]. Owing tomicroheterogeneities, denatured inactivematerial, various glycosylation patterns,and quite different set-ups of potency as-says, it remains an elusive challenge todefine appropriate standards. Applying themildest purification schemes starting fromthe natural protein source (and not fromrecombinant expression systems), usingappropriate storage conditions to keepthe protein in its active conformation, aswell as robust and sensitive potency assayconditions are the essentials for prepara-tion of a reference standard. With such
reference in hand, the potency is routinelyexpressed in activity units per milligram ofpure protein. The WHO is providing cali-brated potency standards that are availablefor commercial and academic organiza-tions [24]. If there is no official source, areference standard should be of the high-est possible purity that can be obtainedwith reasonable efforts, and should be fullycharacterized as described above.
As for antibodies, the prominent bio-logical function is binding to the antigen.Besides the manifold antibody binding as-says, isothermal titration calorimetry (ITC)could become an elegant means for refer-ence standard evaluation. The fraction ofbinding antibody in the sample is deter-mined by measuring the stoichiometry ofthe antigen–antibody interaction [25, 26].For example, with a monovalent Fab an-tibody fragment, a 1 : 1 stoichiometry forbinding to a monomeric antigen is ex-pected, if all Fab molecules are in theiractive binding conformation. For correctinterpretation of data, antibody-ITC relieson the presence of homogenous, epitope-presenting antigen preparations, and onnot too high affinities of the interactions.
In the case that a protein exerts morethan one biological activity, the assaythat most closely reflects the clinicalsituation should be chosen. Assays formeasuring further biological function(s)of the drug should also be established toassess possible side effects in vivo.
However, two protein preparations withidentical specific activity units or function-ality in vitro may behave very differently invivo. For example, different glycosylationpatterns may lead to very similar or evenidentical molecular masses and potency invitro, but the serum half-life and immuno-genicity of both variants may significantlyvary from each other, and thus may lead todistinctly different effects in vivo.

108 6.2 Characterization of Purified Recombinant Proteins
In general, potency determination meth-ods can be grouped into (1) biochemicalassays that use defined reagents in vitro,and into (2) cell-based as well as (3) animal-based assays that rely on living systems.
Biochemical Assays In most biochemicalassays, the specific binding of the proteindrug to its target molecule(s) is deter-mined. The target can be either the nativeor recombinant protein or cells express-ing the target molecules. Typically, the lawof mass action governs these assays. To-day’s most often exploited binding assayis the immunoassay that relies on anti-bodies as detection reagent. Historically,the radioimmunoassay (RIA), which usesradioisotope labeling to track the bind-ing event, paved the way to most of thenonradioactive immunoassays such as theoutstanding enzyme-linked immunosor-bent assay (ELISA) and its many variationsas, for example, sandwich ELISA, captureELISA, inverted ELISA, and competitiveELISA (for review, see [27–29]). Besidesimmunoassays, receptor-binding assaysand substrate-binding assays are also com-monly applied.
The quality of any immunoassay heavilydepends on the specificity, affinity, andstability of the used antibody reagents.Binding of the antibody reagent to theprotein drug yields a signal via someenzyme, fluorescence, luminescence, orradioisotope label on the detector antibody.This signal is then transformed intobinding constants (e.g. IC50 values) and/orconcentrations.
Another very elegant binding assay isbased on surface plasmon resonance (SPR)measurements. It is a label-free method inwhich one reaction partner is immobilizedon a chip, while the other reactant insolution is flowing over the chip. Uponbinding, an optical signal is generated
and recorded as a function of time [30,31]. With this fast method, high- andlow-affinity interactions are detected withonly minute amounts of sample. Hence,surface plasmon resonance determinationhas become a very attractive technique notonly for comprehensive evaluation of thebinding event but also for the detectionof anti-drug antibodies in the sera ofpatients [32–34] (see also Sect. 6.4).
In general, biochemical assays are cheapand easy to perform, and allow high-throughput binding measurements at highaccuracy and precision. On the other hand,the biological activity of the protein drugcannot be appropriately assessed by suchassays since the biologically inactive frac-tion of the drug is often detected as well. Asfor antibodies, biochemical assays are keyfor evaluation of their binding character-istics, but antibody potency assays shouldalso consider the anticipated mode of ac-tion of the therapeutic antibody, beyondthe mere antigen-binding event for ex-ample, induction of signal cascades thattrigger cell proliferation or cell death.
Cell-based Assays The basis for settingup cell-based assays for potency deter-mination is the availability of responsivecells that are either immortalized, freshlyisolated from tissues, or generated byengineering to obtain heterologous ex-pression of the target molecule of choice.The typical read outs are cell prolifera-tion, differentiation, apoptosis, cytotoxicity(e.g. for antibodies: ADCC (antibody-dependent cell-mediated cytotoxicity) andCDC (complement-dependent cytotoxic-ity)), chemotaxis, signal transduction, se-cretion of biologically active substances,or reporter gene approaches relying ongreen fluorescent protein or luciferaseconstructs. Owing to the complex re-active responses of cells, the thorough

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 109
validation of cell-based assays is highlyrecommended for a meaningful inter-pretation of data [23]. For example, theaddition of drug-neutralizing compoundsto the assay is recommended as a checkfor specificity.
Critical aspects of cell-based assaysinclude the cell immortalization method,cell culture history, passage number,stability of cell line, mycoplasma infection,surface marker pattern, and the effect offetal calf serum in the medium.
Animal-based Assays Animal-based as-says are briefly described here as potencyassays, and not as in vivo proof-of-principlestudies in preclinical development.
Animals, in particular non-human pri-mates, as well as tissues and organsfrom animals provide a metabolizing en-vironment and thus offer the advantageof being closer to the ‘‘real-life’’ situa-tion in patients. Especially, bioavailabilityand toxicity aspects are thus integratedin animal-based assays. However, the ex-ploitation of animal-based potency studiesis significantly hampered by the fact thatthey only provide a low throughput athigh costs, take long time while yieldinghigh variability in the results, and finallyraise serious ethical questions. Hence,animal-based assays are only rarely usedfor potency determinations (see, for exam-ple, [35]).
6.2.6.2 Potency Determination inBiological MatricesBesides potency assessment of the purifieddrug, potency assays are also applied forquantification of the protein activity in bi-ological matrices from animals/humans.Since the concentration of the therapeuticprotein in the biological sample is gen-erally low (pico- to nanomolar), standard
chromatographic methods to enrich theprotein prior to analysis are often notapplicable. Hence, the functionality ofthe protein has to be determined in acomplex matrix of other accompanyingunrelated (or sometimes even related) pro-teins, non-protein macromolecules, andlow molecular weight compounds suchas salts and colored ingredients. Dur-ing the past decade, many assays havebeen devised for measuring the potencyof proteins under these conditions [36]. Byspiking in the purified drug as well as thereference standard into the matrix and bytesting series of dilutions with the matrixas diluent, the matrix effect can be assessedand adequately considered in the calcu-lations. Special focus has to be directedtowards the interference with endogenousnative, drug-related protein(s) in the ma-trix that have to be subtracted from thesignal. In general, the storage of biologicalsamples at −80 ◦C in aliquots is indicatedin order to avoid degradation events.
Consequently, the thorough validationof potency assays, as well as their refine-ment from the very beginning of drugdevelopment on, is of key importance.
6.3Validation of Bioanalytical Assays
This section focuses on the bioanalyt-ical methods of validation applied topreclinical studies (nonhuman pharma-cology/toxicology) and human clinicalpharmacological studies such as bioavail-ability and bioequivalence studies re-quiring pharmacokinetics (PK) evalua-tion [37, 38]. Inherently, differences ex-ist between bioanalytical methods ap-plied to preclinical and clinical develop-ment of small molecules versus recombi-nant proteins (macromolecules). Whereas

110 6.3 Validation of Bioanalytical Assays
small molecules are commonly analyzedby chromatography and mass spectrome-try (e.g. HPLC and LC/MS/MS), recom-binant proteins are mainly characterizedby biochemical (e.g. immunoassays suchas ELISA) and cell-based assays. As thecurrently existing FDA guideline ‘‘Bio-analytical method validation’’ mainly fo-cuses on small molecules [38], workshopson ‘‘Bioanalytical methods validation formacromolecules’’ were and are beingheld, which will soon result in a newguideline for the method validation ofmacromolecules [39]. For this reason, onlyguiding principles can be given in thissection.
Several different assay formats, as al-ready mentioned earlier, can generally beused such as ELISA, fluoroimmunoassay(FIA), dissociation-enhanced lanthanidefluoroimmunoassay (DELFIA), RIA, andSPR. For the quantification of proteins inbiological matrices such as blood, plasma,serum or urine, most often ELISAs arevalidated.
In the following part, an overview ondifferent validation aspects mainly con-sidering ELISAs is presented. The studyvalidation comprises the method establish-ment, pre-study and in-study validation.
6.3.1Method Establishment
For setting up a bioanalytical method,calibration standards and quality controls(QC) in which the reference standard isspiked into blank samples are needed.The quality of the reference standardis pivotal to the success of the assayfor deriving accurate measurements. Thedocumentation of the reference standardideally includes the lot number as well ascertificates of analysis, stability, identity,and purity.
6.3.2Pre-study Validation
Pre-study validation defines that themethod produces reliable results. Duringpre-study validation, fundamental param-eters such as selectivity, assay calibration,accuracy, precision, linearity, and stabilityare evaluated.
6.3.2.1 SelectivityThe selectivity is the ability of an analyt-ical method to differentiate and quantifythe analyte (protein) in the presence ofother components in the sample. Se-lectivity investigations focus on reliablequantitation of the analyte against a back-ground of interferences from endogenousmatrix components. Measurements are as-sessed by spiking the analyte into thebiological matrix (e.g. serum) from a re-presentative number of individual subjects(at least six) at concentrations near thelower limit of quantitation (LLOQ, see be-low), [37].
6.3.2.2 Assay CalibrationA calibration (standard) curve describesthe concentration-response curve typicallyincluding more than eight calibrators andadditional ones serving as anchor pointsthus facilitating curve fitting. All calibra-tors are prepared in duplicates in the ma-trix analyzed. The concentration-responserelationship is most often fitted with afour- to five-parameter logistic model.
6.3.2.3 Accuracy and PrecisionQC samples are spiked samples usedto evaluate accuracy and precision. Ac-curacy describes the mean deviation ofthe QCs from the target (nominal, true)concentration and is mainly provided inpercent deviation. Precision describes the

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 111
closeness of QCs and is mainly expressedas coefficient of variation (CV) in percent.
At least three sets of QCs representingthe entire range of the calibration (stan-dard) curve are included: low, medium,and high QC. The low QC sample oftenserves also as the LLOQ, and the high QCas the upper limit of quantitation (ULOQ)both of which can be measured with ac-ceptable accuracy and precision.
Accuracy and precision are determinedusing a minimum of five to six determina-tions per concentration and are assessedfor ‘‘intra-assay’’ (intra-batch or intra-run)and ‘‘inter-assay’’ (inter-batch or inter-run)conditions. Accuracy should be within ±20(30)% of the target concentration and pre-cision should not exceed ±20 (30)% ofCV [37, 38].
6.3.2.4 LinearityAs samples in immunoassays are generallydiluted, the linearity has to be determinedby keeping the matrix component constantand diluting the protein over the expecteddilution range.
6.3.2.5 StabilityThe stability of the protein in the biologicalmatrix at intended storage temperatures(e.g., −20 ◦C or −80 ◦C) is assessedby determining freeze-thaw stability ata minimum of three cycles, short-termroom temperature stability, and long-termstorage stability for the time period oftypical storage times (e.g. six months).
6.3.3In-study Validation
Currently, the following in-study valida-tion acceptance criteria are recommended:at least four of every six QC samples shouldbe within about 20 (30)% of their respective
nominal value. Two of the six QC samplesmay be outside the 20 (30)% of their re-spective nominal value, but not both atthe same concentration. Thus, QC resultscannot be reported from a truncated stan-dard curve. For example, a typical 96wellELISA-plate should contain the followingsamples each in duplicates: 8 to 10 calibra-tion samples, 1 blank, 3 QCs and 34 to 36unknowns.
If the bioanalytical method is performedaccording to good laboratory practice(GLP), the method is described in a stan-dard operating procedure (SOP) and thevalidation method is reported accordingly.In general, validated methods are used inpreclinical development for toxicokineticstudies and in clinical development forall studies in which pharmacokinetics isevaluated.
6.4Immunogenicity of Recombinant Proteins
Nowadays, many biotherapeutics in clin-ical trials are of human or humanizedcomposition. In patients, one would ex-pect these drugs not to be recognizedand attacked by the immune system. Butapparently all therapeutic proteins elicitanti-drug antibodies to a varying extent assummarized in recent overviews [40–42].
Hence, immunogenicity of recombinantproteins is a high-profile concern forindustry and regulatory authorities.
6.4.1Examples
The following section describes (1) exam-ples for the incidence of anti-protein drugantibodies, (2) the potential impact of suchantibodies, and (3) two cases of clinicalconsequences.

112 6.4 Immunogenicity of Recombinant Proteins
Incidences The following short list in-cludes murine, chimeric, and humanizedmonoclonal antibodies as well as inter-ferons and interleukins against whichanti-protein antibodies have been raisedin patients:
(a) Murine antibodies: OKT3 (anti-CD3):∼80% immune responses [42].
(b) Chimeric antibodies: Remicade (anti-CD20): ∼10–57%; Simulect (anti-IL2receptor): ≤2%; ReoPro (Fab, anti-GPIIb/IIIa): 7–19% [42].
(c) Humanized antibodies: Herceptin (anti-HER2): ≤0.1%; Zenapax (anti-IL2 recep-tor): 8% [42].
(d) Interferons/interleukins: Roferon (int-erferon-α2a): 20–50% [43]; Intron (inter-feron-α2b): 0–24% [43]; Betaferon (int-erferon-ß1b): ∼44% [44]; Proleukin (inter-leukin-2): 47–74% [42].
Impact The induction of allergic anti-drug reactions such as the anaphylactic ordelayed type is a relatively rare event. Butanti-drug antibodies bind to the drug andmight neutralize or modulate its bioactivityin in vitro assays as well as in vivo. Anti-protein drug antibodies might (1) have noimpact at all in clinical settings (indeed,in many cases the presence of anti-drugantibodies yielded no detectable side ef-fects or influence on the drug safetyand efficacy), (2) affect the kidney clear-ance parameters and serum half-life dueto antibody–drug complex formation, (3)neutralize and thus reduce the efficacy ofthe protein drug, or (4) in the worst casereact with the endogenous, drug-relatedprotein to deplete for a long durationor even forever the naturally occurringprotein from the patient as seen with ery-thropoietin (EPO).
Cases Recently, serious red-cell aplasiareactions have been notified in France thatare linked to anti-EPO antibodies [45]. Theexact cause for the appearance of suchantibodies is still unclear but changesin the production process might haveplayed a role. Another example is themegakaryocyte growth and differentiationfactor (MGDF) against which neutraliz-ing anti-MGDF antibodies were raised inpatients leading to severe thrombocytope-nia [46, 47].
6.4.2Factors Leading to Immune Responses
There is a plethora of reasons whytherapeutic recombinant protein productscan lead to an immune response [48]:
1. Obviously, the more the drug’s primarysequence deviates from natural humansequences, the higher the likelihoodof developing an immune response.Even a single amino acid deviationmay elicit an anti-drug response [49].As for antibodies, the variability withinthe sequences of the complementaritydetermining regions (CDR), which aremainly responsible for binding to theantigen, might lead to the induction ofso-called anti-idiotypic antibodies. An-tibodies are an excellent example ofthe steadily ongoing progress in thedevelopment of biotherapeutics. Thefirst therapeutically relevant antibod-ies were of mouse origin provokingmassive human anti-mouse antibody(HAMA) reactions in patients. Duringthe past 10 to 15 years, more and morechimeric (human antibody Fc-part withgrafted mouse variable region) andhumanized (human antibody frame-work sequences with grafted mouseCDRs) antibodies have been developed

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 113
to minimize immune reactions. Thefirst fully human antibody got marketapproval in 2003 (Humira). With re-gard to antibodies one may now ask,how human is human? Antibodiesoften have acquired in vivo point muta-tions by somatic maturation processesto give a better fit to the antigen. Hence,there is nothing like one defined anti-body species binding to a given antigen,but a plethora of antibodies, of whichthe sequences differ from individual toindividual. Latest antibody libraries assource of therapeutic human antibodiessuch as HuCAL (MorphoSys AG; Ger-many) thus rely on antibody sequencesthat are very close or even identicalto the human germline sequences tofurther minimize the risk of elicitingimmune reactions in a patient popula-tion [50–52]. The rationale behind thisapproach is that germline-based anti-body sequences are more conservedin humans than somatically maturedones.
2. The glycosylation pattern of the re-combinant protein drug might varyfrom that of the natural human proteinowing to very different glycosylationcapabilities of the expression host sys-tem used for production. For example,Gribben et al. observed the develop-ment of anti-recombinant human gran-ulocyte macrophage-colony stimulatingfactor (rhGM-CSF) antibodies directedagainst the protein backbone, which isnormally protected in the native proteinby O-linked glycosylation but becomesexposed upon expression from yeastand Escherichia coli [53].
3. Impurities such as protein drug vari-ants might lead to anti-impurity anti-bodies, although minor modificationssuch as protein oxidation or deamida-tion are assumed to be tolerated by
the human immune system [54]. Onthe other hand, oxidation and deami-dation often result in protein aggrega-tion, which is known to substantiallypromote immunogenicity. Aggregationcan also be promoted by the formula-tion and storage conditions as well asthe production process per se [55–57].
4. The route of application has a sub-stantial impact on eliciting anti-proteinantibodies. Intramuscular and subcu-taneous applications seem to be moreprone to raise anti-protein antibodiesas compared to intravenous applica-tion [58, 59].
5. The repeated administration of proteindrugs as in chronic diseases resultsin a higher likelihood of developinganti-protein antibodies as compared toapplication in acute indications. In par-ticular, nonphysiologically high dosesof the drug may break the natural tol-erance due to activation of otherwiseclonally silenced B-cells. On the otherhand, ultrahigh drug doses may havethe opposite effect and induce toleranceby eradication of drug-reactive immunecells [60, 61]. Obviously, immunosup-pressed cancer or transplantation pa-tients most frequently have reducedlevels of or even do not develop anti-drug antibodies [62].
Despite the progress in explaining theimmunogenicity of protein drugs, un-fortunately rather often immunogenicitycannot be assigned to the above listedfactors, and the underlying mechanism(s)remain unknown.
In summary, it is of utmost interestto determine the level of anti-proteinantibodies and their impact on the invivo situation in order to allow theimplementation of countermeasures tominimize immune reactions. One major

114 6.4 Immunogenicity of Recombinant Proteins
challenge is to set up validated in vitroassays with appropriate sensitivity thatinclude relevant positive controls forreliable detection of anti-drug antibodies(see below and [63]).
6.4.3Methods to Determine Anti-proteinAntibodies
Assays used for the detection of anti-protein antibodies generally fall into twoclasses: first, assays detecting antibod-ies that bind to a drug, and second,bioassays measuring antibodies that neu-tralize/modulate the biological effect of adrug.
In a first assay, the level of antibodiesthat simply bind to the protein is deter-mined. As already described above, thereare several different assay formats avail-able (ELISA, FIA, DELFI, RIA, SPR), eachhaving its advantages and disadvantagesdepending on the nature of the product.Calibration of anti-drug antibody assays isan area of considerable debate. As reg-ulators have expressed concerns aboutbioanalytical data that are expressed in ar-bitrary units such as titers, it is emphasizedthat antibody reference standards (i.e.anti-drug immunoglobulins purified fromantiserum) should be used for calibrationso that results can be expressed in massunits (e.g. ng mL−1 immunoglobulin). Ac-cording to Mire–Sluis (FDA), more thanone positive control should be produced innonhuman primates (or in immunoglobu-lin humanized mice), and the specific an-tibodies purified resulting in a polyclonalpreparation [64]. A useful process may beaffinity purification with three differentstringencies providing high-, medium-,and low-affinity antibodies, which can beapplied to qualify the assay. Sometimesalso monoclonal anti-idiotypic antibodies
are used for calibration purposes. The limitof quantitation in mass units should bemeasured by using, for example, a dilu-tion series of the anti-drug antibody spikedinto serum and by assessing the precisionat higher dilutions. The nonspecific back-ground (NSB) of the assay can best bedetermined by applying a significant num-ber of negative control samples in order toprovide a mean level of NSB. Mean NSBplus 2 or 3 standard deviations appears tobe the most general practice for determin-ing the nonspecific background value.
Binding anti-drug antibodies are ideallyfurther examined for their capacity to neu-tralize/modulate a functionally relevantresponse of the drug. This often involvescell-based assays, similar in design to theones used in potency assays.
Alternatively to direct measurement ofanti-protein antibodies, there seems to bean increased interest in the use of T-cellresponses as a marker of immunogenic-ity [64] (see also below).
In general, the selection of samplingtimes is crucial to appropriate anti-proteinantibody detection. A predose sample isnecessary to determine potentially pre-existing antibodies. The timing of post-dose sampling depends on the frequencyof the dosing regimen, but is best per-formed at time points when drug proteinconcentration in the systemic circulationis minimal in order to avoid interferences.
6.4.4Prediction of Immunogenicity ofTherapeutic Proteins in Humans
Besides the various causes for eliciting animmune response in patients, the proteindrug sequence as such is subject to de-tailed investigations with the final goal ofengineering out potentially immunogenicsequence stretches. Several approaches for

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 115
the investigation of potential immuno-genicity in humans are currently applied.
Animal Models Early experimental hintsfor protein immunogenicity may arisefrom animal models, especially from ex-periments with transgenic animals thatare tolerant to the human protein [65], butmight develop anti-protein variant anti-bodies. On the other hand, if the drug failsto stimulate an immune response in nor-mal (nonengineered) animals, preferablyin non-human primates, even when ad-ministered with some adjuvant, then thereis a good probability of low immunogenic-ity in humans [66, 67].
Hydrophilicity There has been significantprogress in the computational approach topredict immunogenic epitopes in proteindrugs. Early experiments demonstratedthat regions at the surface of proteinsare more immunogenic than core regions.Surface sites such as N- and C-termini aswell as interdomain loops contain ratherhydrophilic amino acids that are accessibleto the environment. The immune systemtends to select epitopes from such sitesto develop anti-protein antibodies [68, 69].Thus, algorithms for hydrophilicity assess-ment have been applied to identify poten-tially immunogenic epitopes [70, 71].
Peptide-MHC Interaction Analysis The de-velopment of an anti-protein antibodyresponse frequently requires activation ofT-helper cells that induce B-cells to secretespecific antibodies. The key unit for reg-ulation of this process is a trimolecularcomplex consisting of the T-cell receptoron T-cells, and the class II major histocom-patibility complex (MHC) harboring anMHC-ligand on antigen presenting cells.The MHC-ligands are peptides derived
from the degradation of either self orforeign proteins. During the past years,there has been substantial progress inthe understanding of the interaction be-tween the MHC and its peptide ligands.MHC molecules are peptide receptorswith a certain degree of plasticity, be-ing able to accommodate a great varietyof different peptides provided they sharesome common features. For example, pep-tides naturally presented by the MHCare uniform in length and have a spe-cific sequence motif, both defined by therespective MHC allele [72]. These allele-specific motifs allow prediction of T-cellepitopes as demonstrated by Rotzschkeet al., and rendered possible algorithmsto scan protein sequences for potentialMHC-binding motifs [73, 74]. These algo-rithms were further developed and refinedover the years to include latest struc-tural data of the MHC as well as resultsfrom studies of antigen processing [75,76]. With thrombopoietin, an example hasbeen published that illustrates the powerof these in silico analyses for antigenicepitopes [41]. Ideally, the results of an insilico analysis have to be experimentallyconfirmed by, for example, MHC-peptidebinding assays or T-cell recognition as-says. Alternatively, responses of specific,primed T-cells such as chromium-release,cytokine generation, or thymidine incorpo-ration can be determined. Recently, an invitro assay relying on naıve T-cells has beendescribed for the assessment of immuno-genicity [77]. The potential of the predic-tion of MHC-peptide interactions is nicelydemonstrated with the J591 antibody thatoriginated as a mouse monoclonal anti-body against prostate-specific membraneantigen. This antibody has undergone aso-called DeImmunisation process (Bio-vation Ltd., UK), and Phase I clinicalstudies have been undertaken. To date,

116 6.4 Immunogenicity of Recombinant Proteins
over 75 patients have received the modifiedJ591 antibody without immune responsesas verified by either standard ELISA orSPR assays (www.biovation.co.uk). Oneissue that is still open to prediction of im-munogenicity by peptide-MHC interactionanalysis is its restriction to T-cell epitopes.Ideally, algorithms have to be developed tocover B-cell epitopes as well.
Besides identification of antigenic sitesof protein drugs by elaborated predictiontools and their removal by engineering ap-proaches, a completely different approachis available to address immunogenicityproblems. The modification of the proteindrug by conjugating with polyethylenegly-col to mask antigenic sites is a major andsuccessful alternative to ‘‘de-immunizing’’protocols [78–80].
Despite all these efforts to predict proteindrug immunogenicity in humans, the finalproof for the presence of immunogenicepitopes are clinical trials in combinationwith sensitive and validated assays toreliably determine the level of anti-proteindrug antibodies and their impact on drugsafety and efficacy.
Since the number of recombinant pro-tein drugs will substantially increase overthe next decade, analytical as well as bioan-alytical methods for the characterization ofsuch macromolecules will definitively gainin importance. Especially the validationof bioanalytical methods is of key impor-tance to allow an accurate description ofprotein drugs. This requires harmonizedguidelines, which are not yet existing butseem to be on the way, to accelerate drugdevelopment.
Acknowledgment
We would like to thank Sabine Brettreichfor preparing the manuscript.
References
1. M. Schuppenhauer, BioCentury Feb 3, 2003,A5.
2. CDER/CBER/CDRH/CVR-FDA, Guidelineson validation of the limulus amebocytelysate test as an end-product endotoxin testfor human and animal parenteral drugs,biological products, and medical devices,December 1987.
3. ICH, Q5A Guidance for industry: viral safetyevaluation of biotechnology products derivedfrom cell lines of human or animal origin,September 1998.
4. W. Werz, R. G. Werner, J. Biotechnol. 1998,61, 157–161.
5. C. Bayard, F. Lottspeich, J. Chromatogr., B:Biomed. Sci. Appl. 2001, 25, 113–122.
6. M. Fountoulakis, H. W. Lahm, J. Chro-matogr., A 1998, 826, 109–134.
7. A. Dell, H. R. Morris, Science 2001, 291,2351–2356.
8. J. J. Dalluge, Curr. Protein Pept. Sci. 2002, 3,181–190.
9. J. Leushner, Expert Rev. Mol. Diagn. 2001, 1,11–18.
10. F. K. Yeboah, V. A. Yaylayan, Nahrung 2001,45, 164–171.
11. E. Mirgorodskaya, T. N. Krogh, P. Roep-storff, Methods Mol. Biol. 2000, 146, 273–292.
12. J. Khandurina, A. Guttman, J. Chromatogr.,A 2002, 943, 159–183.
13. C. V. Sapan, R. L. Lundblad, N. C. Price,Biotechnol. Appl. Biochem. 1999, 29, 99–108.
14. R. P. Keller, M. C. Neville, Clin. Chem. 1986,32, 120–123.
15. M. Thompson, L. Owen, K. Wilkinson et al.,Analyst 2002, 127, 1666–1668.
16. FDA, Guideline for submitting documenta-tion for the stability of human drugs andbiologics, February 1987.
17. ICH, Q5C Guideline for industry: qualityof biotechnological products: stability testingof biotechnological/biological products, July1996.
18. ICH, Q1A (R1) Stability testing of new drugsubstances and products, August 2001.
19. ICH, Q1E Evaluation of stability data,February 2002.
20. U.S. Pharmacopeia, USP 26-NF 21, 2003.21. European Pharmacopoeia, 4th ed., 2002,
Sections 2.6.1, 5.1, European Directorate

Characterization and Bioanalytical Aspects of Recombinant Proteins as Pharmaceutical Drugs 117
for the Quality of Medicines (EDQM),Strasbourg.
22. FDA (CBER), Points to consider in themanufacture and testing of monoclonalantibody products for human use, February1997.
23. A. R. Mire-Sluis, Dev. Biol. Stand. 1999, 100,83–93.
24. International Biological Standards, http://www.nibsc.ac.uk/catalog/standards/preps/sub cyto.html.
25. S. Leavitt, E. Freire, Curr. Opin. Struct. Biol.2001, 11, 560–566.
26. W. H. Ward, G. A. Holdgate, Prog. Med.Chem. 2001, 38, 309–376.
27. S. Sivieri, A. M. Ferrarini, P. Gallo, Mult.Scler. 1998, 4, 7–11.
28. J. E. Butler, J. Immunoassay 2000, 21,165–209.
29. R. S. Schrijver, J. A. Kramps, Rev. Sci. Tech.1998, 17, 550–561.
30. M. A. Cooper, Nat. Rev. Drug Discov. 2002, 1,515–528.
31. J. M. McDonnell, Curr. Opin. Chem. Biol.2001, 5, 572–577.
32. S. J. Swanson, D. Mytych, J. Ferbas, Dev.Biol. (Basel) 2002, 109, 71–78.
33. N. R. Gonzales, P. Schuck, J. Schlom et al.,J. Immunol. Methods 2002, 268, 197–210.
34. M. A. Takacs, S. J. Jacobs, R. M. Bordenset al., J. Interferon Cytokine Res. 1999, 19,781–789.
35. J. Ashby, P. A. Lefevre, J. Appl. Toxicol. 2000,20, 35–47.
36. A. R. Mire-Sluis, Pharm. Res. 2001, 18,1239–1246.
37. K. J. Miller, R. R. Bowsher, A. Celniker et al.,Pharm. Res. 2001, 18, 1373–1383.
38. FDA, Guidance for industry: bioanalyticalmethod validation, May 2001.
39. AAPS, Workshop on bioanalytical methodvalidation for macromolecules in sup-port of pharmacokinetic studies, May2003.
40. P. Chamberlain, A. R. Mire-Sluis, Dev. Biol.(Basel) 2003, 112, 3–11.
41. E. Koren, Dev. Biol. (Basel) 2002, 109, 87–95.42. E. Koren, L. A. Zuckerman, A. R. Mire-
Sluis, Curr. Pharm. Biotechnol. 2002, 3,349–360.
43. P. Kontsek, H. Liptakova, E. Kotsekova, ActaVirol. 1999, 43, 63–70.
44. P. Kivisakk, G. V. Alm, S. Fredrikson et al.,Eur. J. Neurol. 2000, 7, 27–34.
45. N. Casadevall, J. Nataf, B. Viron et al., N.Engl. J. Med. 2002, 346, 469–475.
46. J. Li, C. Yang, Y. Xia et al., Blood 2001,, 98,3241–3248.
47. I. Zipkin, BioCentury Sep 14, 1998, 6(74),A8.
48. H. Schellekens, Nat. Rev. Drug Discov. 2002,1, 457–462.
49. R. M. Maizels, J. A. Clarke, M. A. Harveyet al., Eur. J. Immunol. 1980, 10, 509–515.
50. A. Knappik, L. Ge, A. Honegger et al., J. Mol.Biol. 2000, 296, 57–86.
51. B. Krebs, R. Rauchenberger, S. Reiffert et al.,J. Immunol. Methods 2001, 254, 67–84.
52. T. Kretzschmar, T. von Ruden, Curr. Opin.Biotechnol. 2002, 13, 598–602.
53. J. G. Gribben, S. Devereux, N. S. Thomaset al., Lancet 1990, 335, 434–437.
54. J. L. Cleland, M. F. Powell, S. J. Shire, Crit.Rev. Ther. Drug Carrier Syst. 1993, 10,307–377.
55. E. Hochuli, J. Interferon Cytokine Res. 1997,17, SS15–SS21.
56. J. C. Ryff, J. Interferon Cytokine Res. 1997, 17,SS29–SS33.
57. W. V. Moore, P. Leppert, J. Clin. Endocrinol.Metab. 1980, 51, 691–697.
58. W. E. Paul, (Ed.), Fundamental Immunology,4th ed., Lippincott Williams & WilkinsPublishers, Philadelphia, 1999.
59. A. Braun, L. Kwee, M. A. Labow et al.,Pharm. Res. 1997, 14, 1472–1478.
60. C. C. Goodnow, R. Brink, E. Adams, Nature1991, 352, 532–536.
61. K. M. Shokat, C. C. Goodnow, Nature 1995,375, 334–338.
62. F. Baert, M. Noman, S. Vermeire et al., N.Engl. J. Med. 2003, 348, 601–608.
63. A. R. Mire-Sluis, Dev. Biol. (Basel) 2002, 109,59–69.
64. A. R. Mire-Sluis, Immunogenicity testing.Immunogenicity of therapeutic biologicalproducts, IABs international meeting,October 2001.
65. A. V. Palleroni, A. Aglione, M. Labow et al.,J. Interferon Cytokine Res. 1997, 17 (Suppl. 1),23–27.
66. C. M. Zwickl, B. L. Hughes, K. S. Pirooziet al., Fundam. Appl. Toxicol. 1996, 30,243–254.
67. D. Wierda, H. W. Smith, C. M. Zwickl,Toxicology 2001, 158, 71–74.

118 6.4 Immunogenicity of Recombinant Proteins
68. E. H. Kemp, E. A. Waterman, R. A. Ajjanet al., Clin. Exp. Immunol. 2001, 124,377–385.
69. D. C. Benjamin, J. A. Berzofsky, I. J. Eastet al., Annu. Rev. Immunol. 1984, 2, 67–101.
70. J. Janin, Nature 1979, 277, 491–492.71. M. Z. Atassi, Eur. J. Biochem. 1984, 145,
1–20.72. K. Falk, O. Rotzschke, S. Stevanovic et al.,
Nature 1991, 351, 290–296.73. O. Rotzschke, K. Falk, S. Stevanovic et al.,
Eur. J. Immunol. 1991, 21, 2891–2894.74. K. C. Parker, M. A. Bednarek, J. E. Coligan,
J. Immunol. 1994, 152, 163–175.
75. T. Sturniolo, E. Bono, J. Ding et al., Nat.Biotechnol. 1999, 17, 555–561.
76. M. Schirle, T. Weinschenk, S. Stevanovic, J.Immunol. Methods 2001, 257, 1–16.
77. M. M. Stickler, D. A. Estell, F. A. Harding, J.Immunother. 2000, 23, 654–660.
78. A. M. Chen, M. D. Scott, BioDrugs 2001, 15,833–847.
79. N. Trakas, S. J. Tzartos, J. Neuroimmunol.2001, 120, 42–49.
80. A. P. Chapman, Adv. Drug Deliv. Rev. 2002,54, 531–545.

119
7Biogeneric Drugs
Walter HindererBioGeneriX AG, Mannheim, Germany
7.1Introduction
Biopharmaceuticals, and especially thera-peutic proteins, represent an exceptionallyfast growing segment within the pharma-ceutical market. In 2003, approximately 50different recombinant proteins, which areapplied as active ingredients in pharma-ceuticals, are registered in Europe. Theseprotein drugs gain more than 10% of thetotal market of pharmaceuticals, whichwas around US$400 billion in the year2002. Moreover, considering new pharma-ceuticals coming for approval, the percent-age of recombinant proteins is expectedto rise over 50% [1]. Among these bio-pharmaceuticals, many are blockbustersand most of them are high priced. Inaddition, the first wave of the therapeu-tic proteins will run off patent protectionwithin the next five years and in principlethis market segment will open up to com-petitors like the generic drug suppliers.Taking all these facts together, biopharma-ceuticals might be highly attractive for thegeneric industry. Many financial analystsand market observers promise golden op-portunities that lie ahead for biogenerics.
It is believed that this cluster of productsoffers a multibillion Euro marketplace inthe near future.
Nowadays, the field of biogenerics isfrequently reviewed and controversiallydiscussed. This chapter necessarily givesan incomplete overview of a very complexsituation, for which neither a universalstrategy nor a clear regulatory pathwayexists today. It is written from the viewof a German biogeneric company, Bio-GeneriX, a subsidiary of a big generic man-ufacturer, ratiopharm, and unavoidablymixed up with personal interpretations ofthe author. The decision of ratiopharm tospin-off a specialized company responsiblefor biogeneric pharmaceuticals was basedon thorough analyses of opportunities,risks, and strategic possibilities for thiskind of business. This article will narrowthe subject to practical aspects, emergingfrom four years of experience with the bio-generic drug development. Furthermore, Iwill mainly focus on the first wave of bio-generic drugs, which are products that runoff patent in the next five years. Finally, itmust be emphasized that this chapter isdirected exclusively to the European situa-tion. Considering that patents will expire in
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

120 7.2 Recombinant Therapeutic Proteins
most cases substantially earlier in Europethan in the United States, the first wave ofbiogeneric drugs is awaited in Europe first.The differences in patent expiry betweenEurope and the United States is readilyexplained by the different patent laws.
7.2Recombinant Therapeutic Proteins
The term ‘‘biopharmaceuticals,’’ althoughhaving a wider definition, is often usedequivalently to ‘‘therapeutic proteins.’’ Thefirst generation of these potent drug sub-stances were exclusively derived fromhuman or animal sources and appliedin a replacement therapy of hormoneor blood-clotting factor deficiencies inchronic diseases. Examples for these firstgeneration of proteins used as pharmaceu-tical drugs are human growth hormone(hGH), isolated from cadaveric humanpituitary glands, insulin, extracted frompig or bovine pancreas, and several hu-man blood proteins, derived out of pooledplasma fractions, for example, albumin,immunoglobulins, or coagulation factors.These natural sources, however, are linkedto many safety problems in the past, andthe resulting protein products have causedthe transmission of infective agents, lead-ing to Creutzfeld–Jacob disease in thecase of hGH [2], and chronic hepatitisB and C, and AIDS in the case of co-agulation factor VIII and IX [3]. Furtherproblems emerged from the immuno-genicity of nonhuman proteins, for in-stance, in the case of bovine or porcineinsulin [4], which can lead to allergic orother immune reactions like neutraliza-tion of the drug activity by antibodies insome cases.
Along with the emergence of methodsof genetic engineering in the 1970s
and 1980s, such as recombinant DNAtechnology and hybridoma technology,these natural proteins were more and morereplaced by recombinant versions. Onlyfew exceptions exist, for example insulinsand factor VIII and IX, where naturaltherapeutic proteins keep a significant rolein the market. The first human healthcare product derived from recombinantDNA technology was Eli Lilly’s insulin(Humulin) approved in the United Statesand Europe more than two decades ago [5].This was a milestone for the biotechnologyindustry. The continuous improvement ofthese new technologies built the basisfor the successful development of theyoung biotechnology industry. Some ofthe originally small biotech start-ups, suchas Genentech, Amgen, or Biogen, grew upto become big pharmaceutical companiesduring the last two decades and todaythey represent a significant portion of thepharmaceutical industry.
Besides the safety aspect, the recom-binant DNA technology provides furtheradvantages. Most important are (1) thelarge-scale production of high amounts ofprotein with defined and homogeneousquality to lower costs; (2) the develop-ment of novel drugs, directed to newtargets, which could not be isolated in suf-ficient amounts and qualities from naturalsources; and (3) creating protein variants,muteins, having even improved propertiesover the natural polypeptides. Today, itwould be a very difficult approach to reg-ister a natural protein, derived from livingorganisms or organs, in view of existing re-combinant versions, the biological sourcesof which are regarded as less inferior bythe regulatory authorities.
Considering all these aspects, one caneasily conclude that a product port-folio of a pure biogeneric companywill exclusively consist of recombinant

Biogeneric Drugs 121
proteins. Therefore, this overview isrestricted to recombinant proteins, includ-ing glycoproteins.
7.3Definition of Biogenerics
The commonly used term ‘‘biogener-ics’’ is not appropriate and not accuratein the eyes of regulatory authorities.At the moment, the industry faces arather unweighable regulatory situationthat makes the definition of biogenerics,better named ‘‘comparable biotech prod-ucts’’ or ‘‘biosimilar products’’ accordingto recent guidelines, a difficult task. How-ever, it is essential for the following consid-erations to find a common understandingof what biogenerics really is. At first, we
have to look at chemically synthesizedgenerics and ask, ‘‘how are they defined?’’By no means will this lead to a straightfor-ward, uniform answer. Depending on thepoint of view, such as trademark, patent, orregulation, or even on the country wherethe question is asked, the answer can varyconsiderably. The knowledge about, andthe occurrence of, generics depends on thehealth policy, which is different among theEuropean countries.
At least three aspects seem uniformand helpful for a general definition ofgenerics: Generics are pharmaceuticalsthat (1) appear after patent expiry, (2) aresold with a price reduction compared tothe original product, and (3) have an ac-tive substance identical to the originator’sone. In Table 1, some other items con-tributing to a full definition of generics
Tab. 1 Definition criteria for generic pharmaceuticals and their transferability to biogenerics
Definitions Chemical generic(‘‘generic’’)
Biotech generic(‘‘biogeneric’’)
Pharmaceutical, launched afterpatent expiry of the activesubstance
Yes Yes
Will be offered with a low price Yes (significant pricereduction)
Yes (moderate pricereduction)
Will be sold under a generic name(INN)
Yes (exceptions: brandedgenerics)
Yes/No (brands are morelikely)
Will be distributed without oralmost little product-specificpromotional efforts
Yes/No No
The active ingredient isqualitatively and quantitativelyessentially similar to an originalproduct
Yes (proven byphysical/chemicalanalysis andbioequivalence)
Yes/No (‘‘biosimilarity’’ tobe confirmed by clinicalstudies)
Approved without the proof ofefficacy and safety in patients.Only a bioequivalence studywas required.
Yes No (efficacy and safetyproven by phase I andII/III studies)
The kind of European approval isfacultative
Yes (mutual recognition orcentralized procedure)
No (centralized procedureis mandatory)
A simplified dossier is sufficientfor filing a MAA
Yes (reference to theoriginator’s dossier)
No (full dossier required,no reference possible)

122 7.4 Regulatory Situation
are mentioned. Since chemical genericshave a well established and a simpli-fied route of approval, this regulatoryfeature contributes substantially to theirdefinition. Classical generics are typicallyapproved on the basis of an abridged filingdossier, which makes use of the possi-bility to refer to the originator’s dossierof approval. The applicant proves essen-tial similarity utilizing analytical data andshows bioequivalence in a small pharma-cokinetic study, usually performed withhealthy volunteers. It is also commonthat generics are sold under the interna-tional nonproprietary name (INN), oftenwithout any product-specific promotionalactivities. Branded generics, however, areexceptions and they still have a significantmarket share, especially in Europe.
Looking at the aforementioned char-acteristics, how can a definition of bio-generics be achieved? What is fulfilled forbiotechnology-derived generics? Table 1gives the answer. The aforementionedgeneral three items, namely, patent ex-piry, lower price, and essential similarity,are fulfilled except for the latter, althoughwe expect price differences to the propri-etor product to be more moderate andmore stable compared to chemical gener-ics, where substantial price erosion canoccur within a few months after the end ofpatent protection. The ‘‘essential similar-ity,’’ ‘‘comparability,’’ or ‘‘biosimilarity’’ isan ongoing dispute between regulatory au-thorities, European commissions, and thedifferent lobbies competing for politicalinfluence. For the time being, generic ap-plicants need to file a more or less completedossier, including preclinical and clinicalstudies showing efficacy and safety. Asthe approval of biopharmaceuticals is notonly linked to the product but also to theprocess and the site of production, thestandard route for registration of generics
via reference to the originator’s file is notpossible. This will be elucidated in moredetail in the following section. For thesekinds of products, own brands and spe-cific promotions are more likely than forthe classical generics.
In summary, biogenerics are best de-fined as copies of therapeutic proteins,launched after patent expiry of the activepharmaceutical ingredient, and sold with amoderate price reduction. They have to beapproved via the route of the centralizedprocedure in Europe and currently requirea complete stand-alone dossier includingclinical studies proving efficacy and safety.
7.4Regulatory Situation
At present, there is neither in Europe norin the United States a regulatory pathwayfor obtaining a generic type of approvalfor a biotech drug. To understand the po-sition of the regulatory authorities, it isnecessary to consider the specific differ-ences between low-molecular weight sub-stances and complex macromolecules likepolypeptides. In contrast to synthetic smallmolecules, large proteins and especiallyglycoproteins have a complex tertiary struc-ture that is sensitive to modifications in so-lution. Slight conformational changes mayreduce efficacy and/or lead to an increasein immunogenicity. Degradation or oxida-tion of amino acid residues are examplesfor such undesired alterations, which canoccur during the process or later on duringthe shelf life of the product. Aggregation,often favored by oxidation, can be a criticalparameter during the production processand for the storage of bulk substanceor final product. Sophisticated formula-tion is necessary to keep the protein as amonomer, stable in solution. Aggregation

Biogeneric Drugs 123
is correlated with an increase in immuno-genicity. It is known that immunogenicitycan cause severe clinical consequencesat worst leading to life-threatening com-plications. Unfortunately, the analyticalmethods established today cannot fullypredict the biological and clinical prop-erties of a protein and cannot establishwhether the structures of two biophar-maceuticals are completely identical [6].According to these scientific arguments, asubstantially abbreviated clinical programfor biogenerics is currently not realistic.Preclinical and clinical trials demonstrat-ing efficacy and safety is mandatory.
Another panel of arguments against anessential similarity or a pure compara-bility approach results from the specificimpurity profile related to process and bi-ological sources. This is the reason for thelinking of the market authorization appli-cation (MAA) to product, process, and siteof production, which build an inseparablepackage for the approval. Changing theprocess or transfer to another productionsite would require a new registration. Withthe help of batch record data, a compara-bility approach is possible in such casesand the 2001 guideline on comparabilityof the Committee for Proprietary Medic-inal Products (CPMP) is appropriate [7].Undoubtedly, biogeneric players have toapply new manufacturing processes to ex-isting products, without having access tothe methods of the innovators or to thematerial from intermediate steps. Thus,the claim for a comparability is difficult.Consequently, the authorities will regardthese biogenerics case by case. The actualterm used for ‘‘biogeneric drug’’ by theEuropean Medicines Evaluation Agency(EMEA) is ‘‘biosimilar product’’; a clearpolicy or a decision, however, is still notreleased. Several of the biogeneric compa-nies are aware of this situation and will
follow a conservative approach that en-tails running significant trials and seekingregulatory approval as stand-alone prod-ucts [8].
What changes can be expected in thenear future? The political influence of thedifferent lobbies will put pressure on theemergence of a clear regulatory outlinefor follow-on biotech drugs. Unlike in theUnited States, where a fundamental le-gal differentiation is made between drugsand biologics, in the European Union thelegislation is based on a single definitionframework, and so the introduction of alegal provision and regulatory pathway forcomparable biological medicinal productswould be easier [9]. A new perspective isprovided by draft Annex 1 to the Euro-pean Directive 2001/83 [10], which setsout the legal basis for biogenerics to refer-ence the originator product. This guidelinestill requires transposition into nationallegislation. On the basis of this policy,a comparability approach cannot be ex-cluded for the future; however, this mustnot be mistaken for a classical genericpathway. Substantial, case-dependent pre-clinical and clinical work still would benecessary to convince authorities aboutcomparable efficacy and noninferiority tothe reference product. Their major con-cern seems to be immunogenicity (seealso Sect. 7.6.1).
On the other hand, the Food and DrugAdministration (FDA), though already al-lowing a slim approval pathway for follow-on versions of insulin and hGH, has nolegal basis for an ‘‘Abbreviated New DrugApplication’’ (ANDA) of a biotech product.If the FDA will change its view on com-parability aspects is a matter of politicaldiscussions too. Whether the recent mas-sive drug review reorganization within theFDA will go further in this direction is aspeculation. Most categories of therapeutic

124 7.5 Patent Situation
proteins, including cytokines, growth fac-tors, interferons (IFN), and enzymes aretransferred from the Center for Biolog-ics Evaluation and Research (CBER) tothe Center for Drug Evaluation and Re-search (CDER). Insulin and hGH alreadybelonged to CDER. It remains to be seen,if this will lead to benefits for the regula-tion of biogenerics in the United States.For more information on the US regula-tory situation and the changes in the FDA,refer to recent reviews [11–13].
Defining categories according to differ-ent risk profiles may be a solution forfuture regulation. It should be possibleto use scientific data to create categoriesof biologics that conceivably could be ap-proved on the basis of relatively less clinicaldata and those that would require muchmore extensive human testing prior to ap-proval [11].
In conclusion, biogeneric players arecurrently faced with a complex and unsat-isfactory regulatory situation. A regulatorybasis for an EMEA approval based on sub-stantially abridged clinical trials is notpresent today. The first wave of bio-generic drugs now enters clinical phases.The companies have to decide whetherto follow a classical stand-alone approachwith extended preclinical and clinical de-velopment, which is a low-risk, high-coststrategy, or to speculate for simplificationsand follow a slim clinical program. Thelatter is certainly a high-risk strategy.
7.5Patent Situation
Patents are the most relevant and themost effective means of intellectual prop-erty protection. Obviously, the knowledgeabout relevant patents is of crucial impor-tance for the generic business. The leading
companies in this field have establishedstrategies to launch their products imme-diately after patent expiry. Thus, they haveto be sure, country by country, when cor-responding patents will expire. The searchof relevant patents, the understanding ofthe scope, the analysis of the patent fam-ily, and the examination of the legal status,including supplementary protection cer-tificates (SPCs), have to be performedthoroughly. For big generic companies likeratiopharm, which started with this kindof business in 1974, this is mainly routine.
Unfortunately, the patent situation isconsiderably more complicated in the fieldof biotechnology. Some of the specificproblems should be mentioned here. Thetotal number of patents dealing with aspecific protein can be incredibly high.For instance, our in-house patent searchesfor potential biogeneric target products re-vealed between 1000 and 5000 hits fora given therapeutic protein. These hugenumbers of documents have to be re-stricted to those few that are really relevantand which cannot be circumvented. Thisrequires specific biotech knowledge alongwith patent know-how. Patent analysis fora generic development is mainly a sur-vey of the past, a historical work-up. If apatent expires in Europe today, it musthave been filed 20 years ago. Although themethods used then are free for use today,they are in most cases old fashioned andnot suitable for a state-of-the-art produc-tion process. Moreover, since patents arerapidly filed in the earliest stage of develop-ment, the methods described, in general,are far away from the final biopharmaceu-tical manufacturing process. The pressureto be the first in filing a patent in order tosecure the intellectual property status washigh and of strategic importance for thepreviously small biotech companies. Addi-tionally, at that time when gene technology

Biogeneric Drugs 125
was just emerging, attorneys and examin-ers had only little experience, if any, withthis subject, and the corresponding patentclaims are often not clear and are difficultto interpret.
In addition, the majority of the relevantbiotechnology patents are related to spe-cific methods for production, sometimeseven restricted to a specific expression sys-tem, rather to appear as broad substancepatents. This is related to the fact thatrecombinant versions of already knownnatural proteins, in principle, lack novelty.The recombinant protein itself was not re-garded as new if the natural protein wasstate of the art. Therefore, patents have toclaim a recombinant protein in combina-tion with modes of production, includingvector constructs, expression systems, andpurification methods, or more impor-tantly, together with an application. It isobvious that for nearly all examples ofproducts, which are mentioned in Table 2,one will not find a monopolistic marketsituation. Typically, parallel developmentsusing distinct solutions lead to two or morecompeting products.
Besides ‘‘primary patents’’ that are ba-sic patents that cannot be circumventedand whose expiry has to be waited for,there are other categories of patents thatmay appear as severe hurdles. One exam-ple is the pharmaceutical formulation oftherapeutic proteins, a dangerous mine-field also for classical generics. Numerouspatents claim distinct product-specific for-mulations. These inventions are relatedto a more advanced development stage,and they usually expire several yearslater. I believe that some of the bio-generic players are not fully aware ofthis difficult situation. Again, it requiresspecific experience to find a gap withina dense net of formulation patents. Thebiogeneric products will probably appear
with modified formulations on the market,which differ from the original products.Otherwise, the generic competitors haveto wait some more years for launch orthey would undergo the risk of beingsued for patent infringement. An alteredformulation, however, is a step furtherfrom a regulatory comparability approach(see above).
Another category of such ‘‘secondarypatents’’ is applications claiming thetherapeutic use of the drug for a newmedical indication. These kinds of patentshave to follow special rules for wordingthe claims. Nevertheless, they can hindergeneric developments for a long time. Anexample is given by interferon alfa (IFN-α), which was claimed in recent yearsfor its therapeutic application in hepatitisC–infected patients in combination withribavirin, a classical antiviral compound(see also Sect. 7.6.7). Although both thedrugs run off patent, their combined usein the major indication seems to be blockedat least until 2018.
Once a disturbing patent or patentapplication has been identified, there arein principle four strategic possibilitiesto overcome infringement: (1) waitingfor its expiration; (2) taking a license;(3) working out a circumventing strategy,if possible; or (4) performing legal actions,such as filing an opposition or an appeal,or, during an advanced step, filing anullity suit. It is assumed that thebiogeneric companies will presumablyadopt strategies (1) and (3).
An indispensable legal aspect forthe generic industry is the so-called‘‘Roche–Bolar’’-type exemptions or ‘‘Bo-lar’’ provisions. So far, I have only consid-ered infringement of patents by the sub-stance itself or by methods used. However,even if a substance or method infringes theclaims of a patent, there are exceptions that
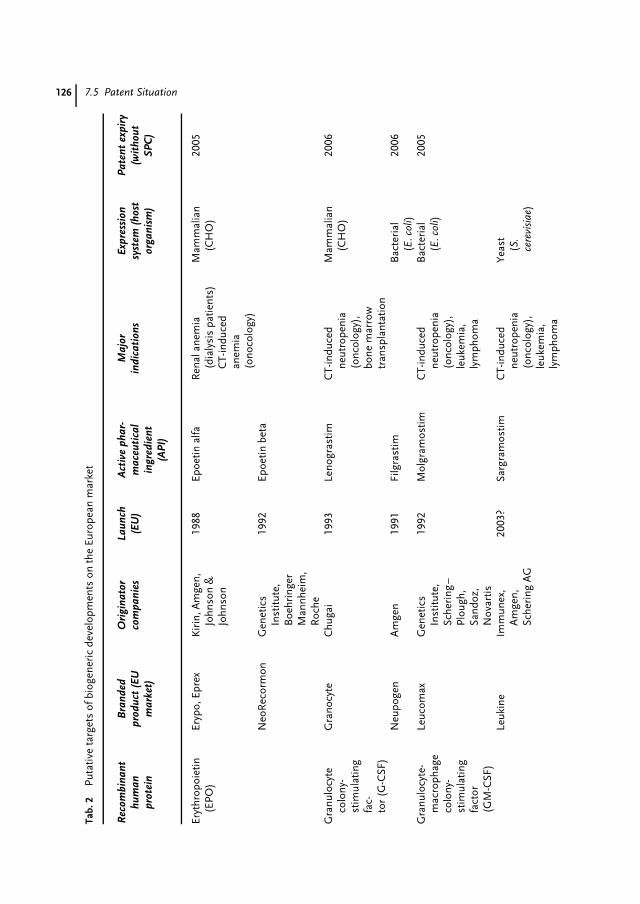
126 7.5 Patent SituationTa
b.2
Puta
tive
targ
ets
ofbi
ogen
eric
deve
lopm
ents
onth
eEu
rope
anm
arke
t
Rec
ombi
nant
hum
anpr
otei
n
Bra
nded
prod
uct
(EU
mar
ket)
Ori
gina
tor
com
pani
esLa
unch
(EU
)A
ctiv
eph
ar-
mac
euti
cal
ingr
edie
nt(A
PI)
Maj
orin
dica
tion
sEx
pres
sion
syst
em(h
ost
orga
nism
)
Pate
ntex
piry
(wit
hout
SPC
)
Eryt
hrop
oiet
in(E
PO)
Eryp
o,Ep
rex
Kir
in,A
mge
n,Jo
hnso
n&
John
son
1988
Epoe
tinal
faR
enal
anem
ia(d
ialy
sis
patie
nts)
CT-
indu
ced
anem
ia(o
noco
logy
)
Mam
mal
ian
(CH
O)
2005
Neo
Rec
orm
onG
enet
ics
Inst
itute
,B
oehr
inge
rM
annh
eim
,R
oche
1992
Epoe
tinbe
ta
Gra
nulo
cyte
colo
ny-
stim
ulat
ing
fac-
tor
(G-C
SF)
Gra
nocy
teC
huga
i19
93Le
nogr
astim
CT-
indu
ced
neut
rope
nia
(onc
olog
y),
bone
mar
row
tran
spla
ntat
ion
Mam
mal
ian
(CH
O)
2006
Neu
poge
nA
mge
n19
91Fi
lgra
stim
Bac
teri
al(E
.col
i)20
06
Gra
nulo
cyte
-m
acro
phag
eco
lony
-st
imul
atin
gfa
ctor
(GM
-CSF
)
Leuc
omax
Gen
etic
sIn
stitu
te,
Sche
ring
–Pl
ough
,Sa
ndoz
,N
ovar
tis
1992
Mol
gram
ostim
CT-
indu
ced
neut
rope
nia
(onc
olog
y),
leuk
emia
,ly
mph
oma
Bac
teri
al(E
.col
i)20
05
Leuk
ine
Imm
unex
,A
mge
n,Sc
heri
ngA
G
2003
?Sa
rgra
mos
timC
T-in
duce
dne
utro
peni
a(o
ncol
ogy)
,le
ukem
ia,
lym
phom
a
Yeas
t(S
.ce
revi
siae
)
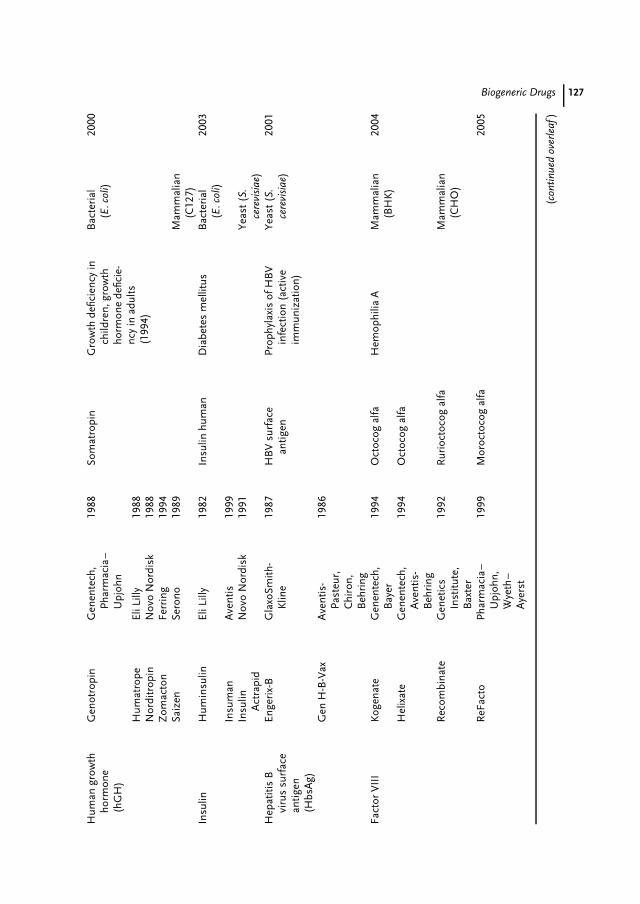
Biogeneric Drugs 127H
uman
grow
thho
rmon
e(h
GH
)
Gen
otro
pin
Gen
ente
ch,
Phar
mac
ia–
Upj
ohn
1988
Som
atro
pin
Gro
wth
defic
ienc
yin
child
ren,
grow
thho
rmon
ede
ficie
-nc
yin
adul
ts(1
994)
Bac
teri
al(E
.col
i)20
00
Hum
atro
peEl
iLill
y19
88N
ordi
trop
inN
ovo
Nor
disk
1988
Zom
acto
nFe
rrin
g19
94Sa
izen
Sero
no19
89M
amm
alia
n(C
127)
Insu
linH
umin
sulin
EliL
illy
1982
Insu
linhu
man
Dia
bete
sm
ellit
usB
acte
rial
(E.c
oli)
2003
Insu
man
Ave
ntis
1999
Insu
linA
ctra
pid
Nov
oN
ordi
sk19
91Ye
ast(
S.ce
revi
siae
)H
epat
itis
Bvi
rus
surf
ace
antig
en(H
bsA
g)
Enge
rix-
BG
laxo
Smith
-K
line
1987
HB
Vsu
rfac
ean
tigen
Prop
hyla
xis
ofH
BV
infe
ctio
n(a
ctiv
eim
mun
izat
ion)
Yeas
t(S.
cere
visi
ae)
2001
Gen
H-B
-Vax
Ave
ntis
-Pa
steu
r,C
hiro
n,B
ehri
ng
1986
Fact
orV
III
Kog
enat
eG
enen
tech
,B
ayer
1994
Oct
ocog
alfa
Hem
ophi
liaA
Mam
mal
ian
(BH
K)
2004
Hel
ixat
eG
enen
tech
,A
vent
is-
Beh
ring
1994
Oct
ocog
alfa
Rec
ombi
nate
Gen
etic
sIn
stitu
te,
Bax
ter
1992
Rur
ioct
ocog
alfa
Mam
mal
ian
(CH
O)
ReF
acto
Phar
mac
ia–
Upj
ohn,
Wye
th–
Aye
rst
1999
Mor
octo
cog
alfa
2005
(con
tinue
dov
erle
af)
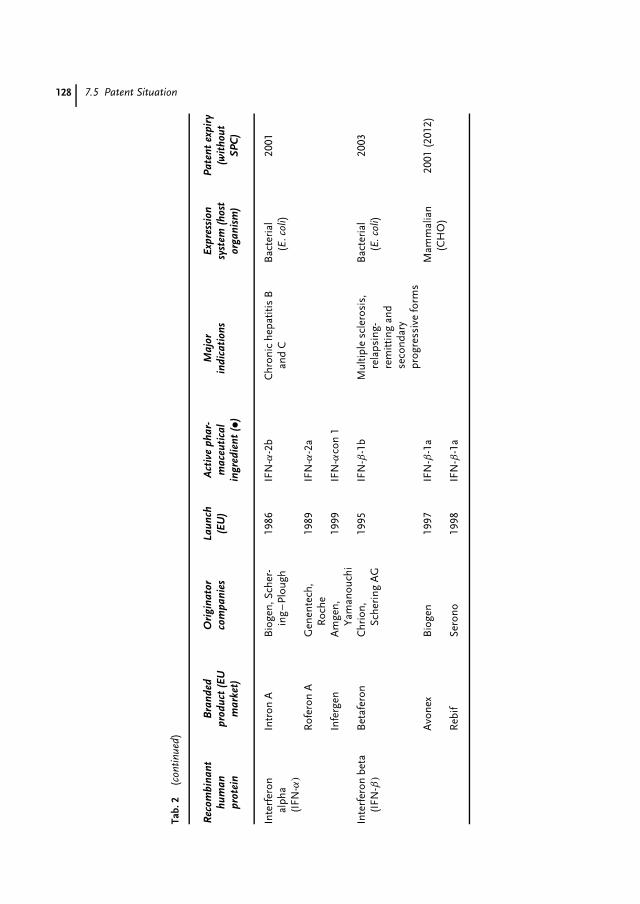
128 7.5 Patent Situation
Tab.
2(c
ontin
ued)
Rec
ombi
nant
hum
anpr
otei
n
Bra
nded
prod
uct
(EU
mar
ket)
Ori
gina
tor
com
pani
esLa
unch
(EU
)A
ctiv
eph
ar-
mac
euti
cal
ingr
edie
nt(•)
Maj
orin
dica
tion
sEx
pres
sion
syst
em(h
ost
orga
nism
)
Pate
ntex
piry
(wit
hout
SPC
)
Inte
rfer
onal
pha
(IFN
-α)
Intr
onA
Bio
gen,
Sche
r-in
g–
Plou
gh19
86IF
N-α
-2b
Chr
onic
hepa
titis
Ban
dC
Bac
teri
al(E
.col
i)20
01
Rof
eron
AG
enen
tech
,R
oche
1989
IFN
-α-2
a
Infe
rgen
Am
gen,
Yam
anou
chi
1999
IFN
-αco
n1
Inte
rfer
onbe
ta(I
FN-β
)
Bet
afer
onC
hrio
n,Sc
heri
ngA
G19
95IF
N-β
-1b
Mul
tiple
scle
rosi
s,re
laps
ing-
rem
ittin
gan
dse
cond
ary
prog
ress
ive
form
s
Bac
teri
al(E
.col
i)20
03
Avo
nex
Bio
gen
1997
IFN
-β-1
aM
amm
alia
n(C
HO
)20
01(2
012)
Reb
ifSe
rono
1998
IFN
-β-1
a

Biogeneric Drugs 129
certain actions using this protected matterare not regarded as infringement. Cur-rently, nearly all member states belongingto the World Intellectual Property Organ-isation (WIPO) include experimental-useexcemptions in their national patent laws.Typically, these exceptions are relatedto noncommercial research experiments.Several countries outside the EuropeanUnion have installed the aforementionedBolar provisions either in their patent lawor in the rulings of pharmaceutical regis-tration. What does Bolar provision mean?The term ‘‘Roche–Bolar’’ arose from a le-gal procedure held in 1984 in the UnitedStates, Roche Prods. Inc. versus BolarPharm Co. [14]. The court decided thattesting of generic versions of pharmaceu-ticals during patent life was infringementand added no weight to the experimental-use excemption of the US patent law.Later, this decision was overruled by theHatch–Waxman Act (section 505 of theFDA Act), which provided the legal ba-sis for generic drug development and thefiling of an ANDA (see section above)without infringing existing patents anylonger. Briefly, a Bolar provision allowsall developmental, testing, and experimen-tal work required for the registration ofa generic medicine to take place duringthe patent period of the original product.The purpose of such a provision is to en-sure that generic medicines are on themarket immediately after patent expiry.Bolar-type provision exists in the UnitedStates, as a part of the Hatch–Waxman Act,Canada, Japan, Australia, Israel, and someEastern European countries such as Hun-gary, Poland, and Slovenia [15]. Among theEuropean Union countries, with the excep-tion of Portugal, no Bolar provision existsso far. There are enormous efforts takenby the generic lobby to influence a har-monized European Regulation following
a Bolar-type exemption. It seems that theEuropean Commission is now in favorof Bolar provisions in the European Unionand has proposed an appropriate Bolar rul-ing under article 9 of the regulation for aEuropean community patent. In addition,a recent proposal for an amending Di-rective 2001/83/EC included Bolar rulingunder article 10. A valid Bolar provisionwould be a breakthrough for the genericindustry in Europe.
The generic manufacturers traditionallymake use of patent-free countries andcountries with Bolar provisions to developand manufacture their products and to per-form there clinical bioequivalence studies.After patent expiry, they are free to transferthe production to their facilities in Europe.This is much more difficult for a biogenericdevelopment, which requires a high tech-nical standard according to the guidelinesof the International Conference on Har-monisation (ICH). As already mentioned,biologics will be approved taking into con-sideration the production process and theproduction site. Thus, the European bio-generic companies are in a dilemma. Theyhave to perform the complete developmentand production within patent-free territo-ries or within the Bolar countries outsidethe European Union. They have to chargecontract research organisations (CROs)and manufacturers (CMOs) or enter intostrategic alliances with biopharmaceuticalcompanies, for example, in Canada or Asia.On the other hand, they deal with multi-source biotech of high complexity, whichmakes the transfer of the bioprocess toanother site, from the technical and regu-latory point of view, a time-consuming andexpensive step, bearing always the risk ofadditional bridging studies. Furthermore,in the Bolar countries, the biotech produc-tion capacities available for the biogenericcompanies, if any, are very limited. Strong

130 7.6 Biogeneric Targets: First Wave
competition between the biogeneric com-panies took place for these few resourcesand will continue further due to the in-creasing number of projects.
Considering everything, one can under-stand why the generic industry urgentlycraves for a Bolar-type ruling in the Euro-pean Union. This regulation unequivocallyshould also cover biosimilar products. Thisis currently not clear for many Bolar provi-sions, including the Hatch–Waxman Actin the United States. Such a policy wouldallow the biogeneric industry to invest indedicated multipurpose biotech facilitiesand would facilitate a market entry moreclose to the patent expiry.
7.6Biogeneric Targets: First Wave
Table 2 summarizes potential target pro-teins for a first wave of biogeneric prod-ucts. It must be emphasized that this isa personal selection and not a completeone. This table only includes those candi-dates that will have patent expirations upto the year 2008. Furthermore, the mar-ket volumes [16] have also been used forthis selection. Owing to the high costsfor the development of a biogeneric, onlytop-selling products are of interest to thecompanies. There are further exclusionsfor the selection of this panel. Recombi-nant proteins alone have been considered.Monoclonal antibodies are believed not toappear in a first wave. Although antibodiespresent a very important cluster of success-ful products, they are not discussed here.There are two reasons against a genericantibody development. Firstly, a copy ofa monoclonal antibody seems possible ona functional rather than on a molecularlevel, leading to extended preclinical andclinical development. Secondly, antibodies
require much higher volumes in produc-tion caused by relatively high dosages.Likewise, some other candidates are notconsidered because of difficult clinical de-velopment, for example the plasminogenactivators.
7.6.1Erythropoietin (EPO)
EPO exerts an exceptional attraction forthe generic industry. EPO has the high-est market volume among all therapeuticproteins and is a real blockbuster. It isnot speculation to assume that nearlyall of the biogeneric companies have anEPO in their pipeline. A recent reviewreported 16 companies developing thisproduct and at least 7 of them have plannedto enter the European market [8]. Threeproducts are currently present in Europe:Eprex (epoetin alfa) from Johnson &Johnson, NeoRecormon (epoetin beta)from Roche, and Aranesp (darbepoetinalfa) from Amgen. The former, Johnson& Johnson’s Eprex, is the best-sellinggenetically engineered drug ever, with$3.4 billion in sales in 2001 [17]. The latter,Amgen’s Aranesp, is a second-generationproduct, containing a mutein with pro-longed bioavailability. This was achievedby the introduction of additional glycosyla-tion sites. Aranesp is certainly not in thecurrent focus of biogeneric developments.A fourth product, Dynepo (epoetin delta)from Aventis and Transkaryotic Therapies(TKT), is already approved. The methodused by TKT was an activation of the hu-man chromosomal EPO gene instead ofcloning and expressing the gene in Chi-nese hamster ovary (CHO) cells. This lookslike a typical patent strategy leading at theend to a biosimilar product. However, itwill be very difficult for Aventis to main-tain noninfringement before court. It is

Biogeneric Drugs 131
assumed that Aventis will wait as longas Kirin–Amgen’s basic patent expiresin December 2004. The question whetherDynepo is a generic or not is justified. Ac-cording to the definition given in Sect. 7.3,Dynepo will likely not appear as a genericon the market because Aventis, a highlyreputed innovator company, will sell theproduct with scientific arguments ratherthan by price reduction. A fifth EPO, pro-duced with baby hamster kidney (BHK)cells, designated as epoetin omega andknown for a long time [18], is again underclinical investigation. This product proba-bly belongs now to Lek (Slovenia), whichwas the sponsor of the studies [19]. Lekwas recently acquired by Novartis.
The analytical and biological differencesbetween epoetin-alfa and -beta, two CHO-derived EPOs, do not seem to be of clinicalrelevance [20]. Nevertheless, a manufac-turer of an EPO can apply for a newINN, because slight differences in thegylcosylation pattern cannot be avoided.EPO products applied today contain a verycomplex mixture of many different iso-forms that just differ in their glycosylationstructure. The capability for sufficient gly-cosylation is provided by the host cell,for example, CHO; however, upstreamand downstream production procedureshave a considerable influence on the fi-nal isoform composition. The necessaryquality of a new EPO product is de-fined by a monograph of the EuropeanPharmacopoeia [21], but the specificationsmentioned are just minimum require-ments. The true measure is set by theoriginator products. Nevertheless, the ex-istence of a monograph is welcome for ageneric approach.
Recently, there was growing concernin France about case reports of purered-cell aplasia (PRCA) developing in13 patients with chronic renal anemia
treated with Eprex (epoetin alfa). Neutral-izing antibodies induced by EPO therapycross-neutralize natural endogenous EPO,leading to severe transfusion-dependentanemia [22]. One year later, the numberof reported PRCA cases, as of Novem-ber 2002, had already increased to morethan 175 patients. This reflects an inci-dence of 20 per 100 000 per year. The mostlikely explanation for this serious side ef-fect is a subtle change in the EPO molecule,probably introduced by the manufacturingand/or formulation changes in 1998 [23].There were two coincidences for the emer-gence of PRCA, (1) with the reformulationof Eprex, mainly the withdrawal of al-bumin under pressure of the CPMP, and(2) with the shift from intravenous to sub-cutaneous administration [17]. Procrit,the US version of Eprex, is still formu-lated with human albumin and seems notto be correlated with PRCA. As a conse-quence, the EMEA recently restricted theuse of Eprex in renal anemia to intra-venous administration.
It is likely that PRCA will have additionalimpact for generic versions of EPO. TheCPMP is particularly concerned with theproblem of immunogenicity and mayrequire postmarketing monitoring for atleast one year [17]. Furthermore, it cannotbe excluded that this may also influencethe requirements for approval of otherprotein drugs.
7.6.2Colony-stimulating Factors (CSFs)
Two important endogenous growth fac-tors, granulocyte-colony-stimulating fac-tor (G-CSF) and granulocyte/macrophage-colony-stimulating factor (GM-CSF) reg-ulate the proliferation and differentia-tion of progenitor cells within the bone

132 7.6 Biogeneric Targets: First Wave
marrow and the release of mature neu-trophils into the peripheral blood. Cancerchemotherapy, which affects rapidly di-viding cells, frequently leads to a sideeffect termed ‘‘neutropenia’’. Neutropeniais a decrease in counts of neutrophilicgranulocytes in the peripheral blood andaffects more than one in three patientsreceiving chemotherapy for cancer. Pa-tients driven into neutropenia can developfever and have an increased risk for in-fections. Life-threatening gastrointestinaland pulmonary infections occur, as doessepsis. A subsequent cycle of chemother-apy may have to be delayed until thepatient has recovered from neutropenia.Recombinant human G-CSF and GM-CSFare effective pharmaceutical substancesand have been successfully applied totreat chemotherapy-induced neutropenia.They restore the number of neutrophilsin the blood and keep it above the criticallevel [24].
From a marketing point of view, G-CSFis more successful than GM-CSF. Scien-tific reasons for the preferential applicationof G-CSF in prophylaxis of neutropeniaexist too. G-CSF is more specific for gran-ulocytes. It takes a position within theregulation cascade of hemopoiesis, onestep behind of GM-CSF and closer to-ward the differentiation of neutrophils.The dominant product on the marketis Amgen’s Neupogen containing fil-grastim, an Escherichia coli–expressed,recombinant human met-G-CSF. The sec-ond G-CSF product, Chugai’s Granocyte,is derived from recombinant CHO cellsand glycosylated. With the exception ofFrance, this product has only little marketshares. This is explained by the respectivemarketing power of the pharmaceuticalcompanies rather than by scientific rea-sons.
In 2002, Amgen additionally launchedNeulasta, a second-generation, PEG-ylated filgrastim. This product has a pro-longed pharmacokinetic half-life profileand requires less frequent application. Itis assumed that Neulasta will expand theCSF market and will replace Neupogen
to some extent before the patents expire.The most attractive generic target amongthe CSF products is certainly Neupogen.However, generic versions appearing af-ter patent expiry then have to competewith Neulasta. This will happen at theearliest in 2006. Nevertheless, the highcost of the therapy limits its widespreaduse [24], and a lower price for a genericG-CSF could justify the application of afirst-generation drug and would even favorbroader application.
Today, the market volume of GM-CSFseems too low to guarantee return of in-vestment for a generic version within areasonable time. Furthermore, GM-CSFand G-CSF compete in the indicationof neutropenia. However, GM-CSF hassome opportunities lying in new indi-cations. One promising development isthe successful treatment of active Crohn’sdisease [25]. The product of Immunex,Leukine (sargramostim), which was notlaunched in Europe so far, was sold in2002 to Schering AG, upon the acquisitionof Immunex by Amgen. Sargramostim isa mutein and differs from the naturalprotein. It has a substitution of leucinein position 23, and carries heterogeneousyeast-type glycosylation, due to the Saccha-romyces expression system applied. It canbe assumed that Schering AG will launchthis product in Europe and will further in-vest in the immunomodulatory potential ofGM-CSF for therapy of morbus Crohn. Incontrast, Leucomax (malgramostim; No-vartis) is expressed in E. coli and notglycosylated. Likewise, this product was

Biogeneric Drugs 133
sold in 2002. Novartis transferred theproduct rights to its comarketing partnerSchering–Plough. It remains to be seen ifunder new ownership the GM-CSF prod-ucts will gain in market volumes in thenear future.
7.6.3Human Growth Hormone (hGH)
Recombinant hGHs are well-establishedproducts, launched in Europe in 1988. Themarket is highly competitive, as five dif-ferent branded products are available for alimited number of patients (see Table 2).Some of the products, in certain patientgroups, still have orphan drug status. Theoriginal indication of hGH was growthdeficiency in children caused by the lackor insufficient endogenous production ofthe hormone. The goal of treatment inthese cases is to stimulate linear growth,hopefully reaching a final height withinthe normal range. Nowadays, hGH hasan expanded therapeutic spectrum in chil-dren [26] and is used additionally foradolescents and adult patients having sec-ondary weight losses or muscular atrophywith or without hGH deficiencies [27].All the five available recombinant growthhormones, four expressed in E. coli andone in murine cells, have identical aminoacid sequence and are not glycosylated.These products are indeed ‘‘comparable’’or ‘‘biosimilar’’; they have the same INN,somatropin, and the same clinical efficacy.This is a good example where indepen-dent developments lead to very similarproducts. In this background, one can ex-pect that the requirements for approval ofa generic hGH are much less than, forexample, for a glycoprotein like EPO. Con-sidering also some further growth of themarket, hGH is a promising candidate fora biogeneric product portfolio. With the
exception of some specific patents, whichblock certain methods of production andformulation, hGH is already patent-freeand additional products are awaited inthe next years. There are several life cy-cle extension strategies by the originatorsmoving in the direction of easier admin-istration, such as needle-pen, needle-freeinjection, slow release formulations, oreven oral and inhaled forms. This willdrive generic companies to innovate too.
7.6.4Insulins
Diabetes mellitus is a global epidemic af-fecting about 150 million people aroundthe world. These numbers are believedto grow rapidly along with the increas-ing problems of age and obesity. It wasestimated that the numbers will doublewithin 25 years. Disease management oftype 1 and type 2 diabetes is performedby insulin therapy. Three human-identicalrecombinant insulins are on the Europeanmarket, including Eli Lilly’s pioneer prod-uct Huminsulin being the first genetic-engineered polypeptide drug launched [5].This protein and Insuman of Aventis areboth synthesized in E. coli, whereas NovoNordisk produces its Insulin Actrapid inyeast. For the daily glycemic control in di-abetics, there are usually three differenttypes of insulin required: (1) a rapid-actingvariant, (2) an intermediate-acting vari-ant, and (3) a long-acting variant. Thesetypes differ in their pharmacokinetic pro-file accomplished by classical formulationstrategies [28]. Briefly, regular insulin actsrapidly, the long-acting (retard) insulinis achieved by neutral-protamin-Hagedorn(NPH) formulation, and the intermediatevariant is just a mixture of regular andNPH insulin.

134 7.6 Biogeneric Targets: First Wave
Three novel insulin analogues, allmuteins, are of importance. Regular in-sulin has the tendency to locally builddimers and hexamers upon injection,hence the release is not as rapid as de-sired. This prompted Eli Lilly and NovoNordisk to develop muteins with an al-tered carboxy-terminus in the B-chain. EliLilly’s solution for obtaining a more rapidinsulin was a switch of the two amino acidsin position 28 and 29, proline and lysine,thus preventing aggregation. The result-ing protein was named ‘‘insulin lispro’’and the corresponding product Huma-log was launched in 1996. Novo Nordiskreached the same effect by introducing as-partic acid instead of proline in position 28.The mutein was named ‘‘insulin aspart,’’and the corresponding product Novolog
was launched in 1999. In contrast, Aventiswent the opposite direction toward a long-acting insulin obtained by exchange of theC-terminal asparagine with glycine in theA-chain, together with prolonging the C-terminus of the B-chain by attaching twoarginine residues. The resulting productLantus, containing the mutein ‘‘insulinglargine,’’ was approved in 2000. Takingtogether, these three novel insulins presentexpanding options in diabetes manage-ment [29].
The huge and continuously growingmarket of diabetes is in the focus of genericcompanies. The patents for regular humaninsulin expire in 2003. Therefore, genericversions will likely appear within thenext years. Human insulin is a relativelysmall nonglycosylated protein, and therequirements of drug approval are believedto have a similar low extent as for hGH(see above). However, a disadvantage ofinsulin should be mentioned too. Becausethe dosages of insulin are very high,several tons of recombinant insulin haveto be produced per year to supply all
patients. Therefore, the manufacturersproduce their insulins in largest scales, andthe prices calculated per gram protein arerelatively low. This could be a significantmarket entry barrier for generic insulins,which may have a limited potential forprice reduction. It should be mentionedthat ratiopharm and B. Braun have alreadylaunched a generic, semisynthetic humaninsulin in 2000.
7.6.5Hepatitis B Vaccine
Vaccines are a special category of biolog-ical products, and only the recombinantvaccine for hepatitis B virus (HBV) is con-sidered in this chapter. The prophylaxis ofHBV infections is successfully performedusing a subunit vaccine, based on a singlerecombinant protein, hepatitis B surfaceantigen (HBsAg). This antigen is producedefficiently in yeast and shows excellentimmunogenicity. After seroconversion ofanti-HBsAg, immunity is maintained forat least 10 years [30]. There are two prod-ucts approved in Europe, Engerix-B andGen H-B-Vax from GlaxoSmithKline andAventis respectively. There are several rea-sons why these products are attractivegeneric targets. The patent protection forthese products has already expired. In ad-dition, the market volume is large and willfurther increase, especially in view of in-ternational HBV control programs alreadyrunning or entering many countries inthe next years. New trends for innovationshould be mentioned too. Firstly, there arenew generation vaccines combining HB-sAg and Hepatitis A (HAV) immunogens,for example Twinrix of GlaxoSmithKline,and, secondly, additional recombinant vi-ral antigens of HBV are combined withHBsAg to provide against low- or nonrep-sonder of HBsAg immunization.

Biogeneric Drugs 135
7.6.6Factor VIII (FVIII)
Hemophilia A is one of the most com-mon inherited bleeding disorders and iscaused by genetic deficiency of coagulationfactor VIII (FVIII) [31]. The replacementtherapy with plasma-derived FVIII and,later, recombinant FVIII has substan-tially improved the quality of life and thelife expectancy of hemophilia A patients.In the early 1980s, significant numbersof hemophilia A patients have been in-fected with hepatitis viruses and HIV.The virus transmissions were caused byFVIII batches isolated from contaminatedplasma pools [3]. Subsequently, this led toa dramatic change in the safety philosophyfor plasma-derived products and for bio-logics in general. Likewise, there were de-mands for the development of safe recom-binant FVIII, which was a scientific anda technical challenge. Activated naturalFVIII mainly consists of a heterodimericform of two different polypeptide chainslinked via calcium ions. The heterodimerconsists of 2351 amino acids and has asize of 127 kDa. Furthermore, there are 25potential glycosylation sites, and the totalmolecular weight of the native glycopro-tein is around 300 kDa. After all, in theearly 1990s the first recombinant productsappeared on the market. Today, four re-combinant FVIII products are registeredin Europe (Table 2), and indeed no trans-mission of hepatitis or HIV attributing torecombinant FVIII has been reported [32].The latest product, Refacto (moroctocogalfa), was launched in 1999 and is a muteinhaving a deletion of the B-domain, whichrenders the protein more stable. Refacto
(Wyeth) and Recombinate (Baxter) con-tain CHO-expressed FVIII, whereas Bayer(Kogenate) and Aventis (Helixate useBHK cells for the expression of FVIII. The
patents for FVIII will expire in 2004 and2005 and further products could emerge.The market opportunities are promising.However, biogeneric developers would en-ter a highly difficult field. Recombinantexpression, purification, and stabilizationof FVIII requires considerable experience,more than that required for the aforemen-tioned targets.
7.6.7Interferons (IFN)
Interferons (IFN) are a class of relatedcytokines with multiple activities. Theyare grouped in three major categories,namely IFN-α, IFN-β, and IFN-γ , ac-cording to their different cellular origin.Besides their historically described antivi-ral activities, they are also known to possessimmunomodulatory and cell-proliferativepotential [32]. The nomenclature of the var-ious IFNs in the past was chaotic and haschanged over time. It is very difficult torelate descriptions in old papers or patentsto the actual molecular definitions. Besidessome antitumour applications, two widelyused IFN therapies have been successfullyestablished. These therapies are (1) IFN-αfor the treatment of chronic hepatitis Cvirus (HCV) infections and (2) IFN-β forthe treatment of multiple sclerosis (MS).These two applications are highly attractivefor generic companies and the productsshould be discussed in more detail.
IFN-α, formerly ‘‘leukocyte IFN,’’ is amixture of many distinct protein homo-logues. At least 23 different IFN-α genesare known. For the recombinant pro-duction, Roche, respectively Genentech,decided to use IFN-α-2a, whereas Scher-ing–Plough’s product is related to IFN-α-2b. By contrast, Amgen has developed anartificial consensus IFN-α (IFN alfacon 1),probably following a patent strategy. All

136 7.6 Biogeneric Targets: First Wave
three recombinant proteins are producedin E. coli. The corresponding products, Ro-feron A, Intron A, and Infergen are allapproved for treatment of HBV and HCVinfections. Emphasis must be added ontwo recent developments in HCV therapy,which strongly influenced generic strate-gies. At first, Schering–Plough introduceda combination therapy with IFN-α andribavirin (Rebetol), a classical chemicalantiviral compound of ICN pharmaceuti-cals, until then only used for treatmentof pulmonary infections of respiratorysnycitium virus (RSV). This combinationtherapy was a breakthrough for treatmentof chronic HCV infections and set a newstandard within a short time. Secondly,Schering–Plough launched PEG-Intron
in 2000, a PEGylated IFN-α-2b basedon Enzon’s technology for PEGylation.This product offers an advantageous once-weekly administration, but more impor-tantly, the clinical efficacy has been im-proved. Again, within a short time, themarket switched nearly completely to thesecond-generation product. On the sameway is Roche, which developed a PEG-ylated IFN-α-2a, Pegasys, using the PEGtechnology of Shearwater. This productwill be marketed together with its own rib-avirin brand (Copegus) for combinationtherapy. Because of synergistic intellec-tual property issues between Roche andSchering–Plough, it is assumed that thesetwo companies will dominate the HCVmarket for a long time. This is a goodexample for successful life cycle man-agement at the end of patent protection.Schering–Plough, for instance, has fileda panel of patent applications claimingthe combination therapy with ribavirinand IFN-α. The introduction of PEGylatedIFN-α will make it difficult, if not impossi-ble, to sell nonPEGylated generic versions.Some generic companies, like Bioceuticals
(Stada) and BioGeneriX (ratiopharm) havegiven up on plans for the development ofIFN-α [33].
IFN-β, formerly ‘‘fibroblast IFN,’’ has along history of therapeutic applications.The latest and most important indica-tion entered by this cytokine was multiplesclerosis (MS). MS is a chronic, noncur-able, progressive, and neurological diseaseand is the prototype of an inflamma-tory autoimmune disorder of the centralnervous system (CNS). The main patholog-ical feature is demyelination of ganglions,which causes the various symptoms [34].The use of IFN in MS has been stud-ied for more than two decades; however,the mechanism of its action in MS isstill not understood. Likewise, this holdstrue for the etiology of MS. Since 1995,IFN-β products are approved for treat-ment of relapsing-remitting and secondaryprogressive MS. Three products are onthe market, which differ in several as-pects. Betaferon (Schering AG) containsa nonglycosylated mutein of IFN-β, ex-pressed in E. coli. The active substance istermed ‘‘IFN-β-1b’’ and differs from thenatural amino acid sequence by havingserine instead of cysteine in position 17.In contrast, Avonex (Biogen) and Rebif
(Serono) are both expressed in CHO cells.The proteins are N-glycosylated and havean identical sequence to the natural pro-tein. The INN (IFN-β-1a) is the same forboth glycoproteins, although slight differ-ences in glycosylation exist. It must beemphasized that Betaferon has a ratherlow specific activity of 32 million interna-tional units (IU) per milligram comparedto Avonex, which has 200 IU/mg. Thus,the dosages for these two products, byamount of protein, differ by nearly oneorder of magnitude. Avonex is indicatedto be administered at 30 µg once a week,while Betaferon is indicated at 250 µg to

Biogeneric Drugs 137
treat MS [35]. In contrast, the latest productRebif was administered with 44 µg threetimes a week. Moreover, whereas Avonex
is applied intramuscularly, Serono decidedon a subcutaneous application of Rebif.This high-dose strategy seems to be suc-cessful. Recent clinical trials showed thatthe therapy with Rebif was significantlymore effective than Avonex [36]. This wasof great importance for Serono, becausethey were able to overcome the existingorphan drug status in the United States.The question rose whether Rebif is ageneric or not. Basic patents have alreadyexpired, or have been rejected after oppo-sitions. Undoubtedly, Rebif is a copy ofAvonex and could claim for ‘‘biosimilar-ity.’’ Also, the generic feature of a lowerprice might be fulfilled. The therapies withRebif and Avonex have similar calcu-lated costs based on time and patients.Thus, because of the higher dosage ofRebif, the price for the substance IFN-β isindeed significantly lower for Rebif thanfor Avonex. However, in spite of these ar-guments, nobody, neither physicians, norpatients, nor Serono itself, would acceptany relationship between Rebif and a bio-generic product. Interestingly, Avonex
also has a biogeneric history. Paradoxi-cally, the FDA approval of Avonex wasbased along a comparability approach,just on biological, biochemical, and bio-physical data, without clinical trials [35].The background is as follows: Biogen andRentschler Arzneimittel (Laupheim, Ger-many) founded a joint venture company,called Bioferon, for the development ofIFN-β. The product received the name ofthe company, Bioferon, and has goneinto phase III trials. Later on, there wasa breach between Rentschler and Biogenand the joint venture went into receiver-ship during the trials. The rights for theclinical data stayed with Biogen and the
rights for clone, process, and substancestayed with Rentschler. Hence, Biogenwas obliged to develop a new cell bankand product. They used the clinical data ofBioferon and showed analytical compara-bility to Avonex. This was assumed to be atest case for biogeneric science [35]. Subse-quently, on the basis of additional phase IVclinical data, Avonex received the EMEAapproval. Interestingly, Rentschler in co-operation with BioPartners is now rede-veloping Bioferon, which in principle isthe reference product of the ‘‘biogeneric’’Avonex. They want to convince regula-tors once again about the comparability ofthese two products [37].
Generic developers of IFN-β should con-sider one drawback: although the patentsituation allows free operation in Europe,there is one patent of Rentschler [38],expiring in 2012, claiming IFN-β prepara-tions with improved glycosylation patterns.This patent, an inheritance of Bioferon,also covers the existing products. An op-position of Biogen was recently rejectedby the European Patent Office. There-fore, a pure generic strategy would requirelicense, if available, whereas a circumvent-ing strategy would lead to a noncompa-rable product. Further risks should bementioned too. In contrast to replacementtherapies, like for EPO, hGH, insulin,or FVIII, the therapy with immunomod-ulators bears the risk of substitution.Especially in MS, it seems possible that inthe near future IFN-β could be substitutedby more efficient drugs. Several alternativesubstances are in clinical development oralready approved. Alternatively and simi-lar to the change in HCV standard therapy(combination with ribavirin), IFN-β couldbe used as adjuvant therapy in combina-tion with chemical drugs. Furthermore,the benefits of IFN-β in MS, also in view

138 7.7 Biogeneric Developments and its Requirements
of the high costs, are controversially dis-cussed. At least the clinical effectivenessbeyond the first year of treatment is calledin question [39]. In conclusion, among theinterferons, IFN-β-1a, is the most promis-ing biogeneric candidate; however, the riskof substitution in the indication for MS issignificantly higher than for other targets.
7.7Biogeneric Developments and itsRequirements
In principle, the requirements for the de-velopment of a therapeutic recombinantprotein are independent of a generic strat-egy. The ICH guidelines provide a frame-work that is indispensable for an EMEAapproval. The ICH tripartite documentsdefine ‘‘good manufacturing practice’’(GMP), which specifies the requirementsand conditions for manufacturing the Ac-tive Pharmaceutical Ingredients API or fi-nal product. Furthermore, the ICH guide-lines instruct, for example, the analysis ofthe expression constructs, cell hosts andsubstrates, viral safety evaluations, analyt-ical procedures and their validation, andstability testing. In addition, the CPMPguidelines have been considered, too. Asalready outlined in Sect. 7.4, the regulatoryprerequisites for approval of a biosimilarrecombinant protein are far different fromthe abridged pathway applied for chemi-cal generics. Biogenerics are more close tonew drugs. The product development of abiotech product is characterized by threemain sections: (1) process development,(2) development of analytical methods, and(3) preclinical and clinical development.Table 3 gives an overview of the varioussteps, which at the end have to be puttogether to obtain the market authoriza-tion under the legalities of drug approval.
Noncompliance may have a severe impacton the time schedule and in the worstsituation might be even irreversible. It isobvious that much less difficulties occurfor biogeneric developers than for the orig-inators. Many problems have been solvedin advance and there is no need for a proofof principle. This results in abbreviatedtime lines. Figure 1 gives an example ofthe follow-up of a biogeneric project usingCHO expression and shows the sequenceand links of the various parts of develop-ment on a timely basis. This sequence isoptimized for maximum overlapping. Theproduct launch is realized seven to eightyears after starting the project with molec-ular biology. By using E. coli as expressionsystem, the project would be finished ap-proximately one year earlier. In the follow-ing sections, emphasis is added to specialfeatures of biogeneric development.
7.7.1Process Development
The required gene and its sequence are inthe public domain. Typically, the DNA willbe chemically synthesized. The selection ofthe host cell and the construction of usefulexpression systems is the standard tech-nology of molecular biology. Biogenericcompanies have to deal mainly with twoexpression systems, E. coli and CHO(see Table 2). Occasionally, Saccharomycesand BHK might be alternative expressionhosts. The first milestone for the project isthe finishing of the master cell bank (MCB)and the manufacturing working cell bank(WCB). Comprehensive analytical work isnecessary, especially for the mammaliancells, to obtain all the safety data requiredfor release of the cell banks into produc-tion. It should be mentioned that in theearliest step of development, for examplefor clone selection, it is indispensable to
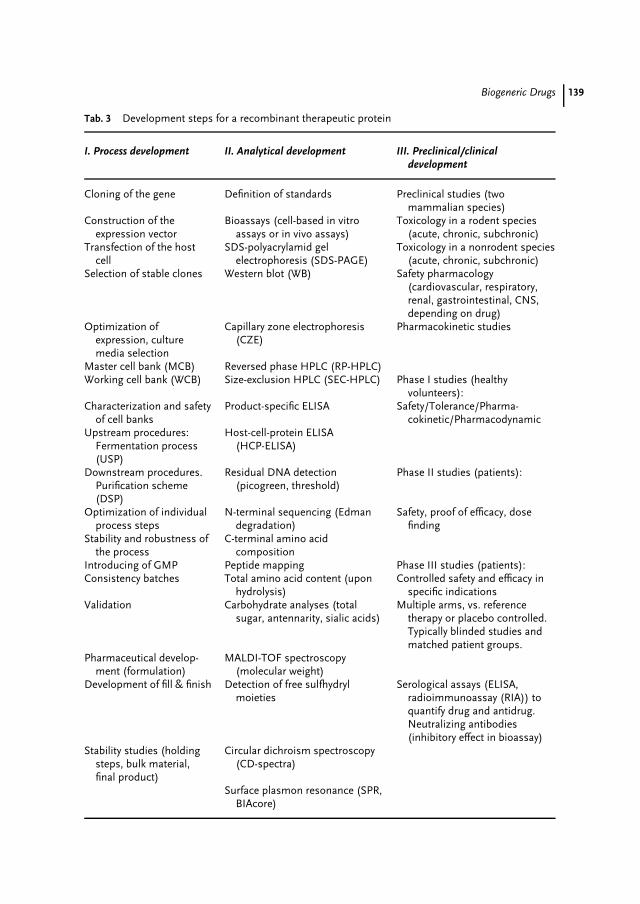
Biogeneric Drugs 139
Tab. 3 Development steps for a recombinant therapeutic protein
I. Process development II. Analytical development III. Preclinical/clinicaldevelopment
Cloning of the gene Definition of standards Preclinical studies (twomammalian species)
Construction of theexpression vector
Bioassays (cell-based in vitroassays or in vivo assays)
Toxicology in a rodent species(acute, chronic, subchronic)
Transfection of the hostcell
SDS-polyacrylamid gelelectrophoresis (SDS-PAGE)
Toxicology in a nonrodent species(acute, chronic, subchronic)
Selection of stable clones Western blot (WB) Safety pharmacology(cardiovascular, respiratory,renal, gastrointestinal, CNS,depending on drug)
Optimization ofexpression, culturemedia selection
Capillary zone electrophoresis(CZE)
Pharmacokinetic studies
Master cell bank (MCB) Reversed phase HPLC (RP-HPLC)Working cell bank (WCB) Size-exclusion HPLC (SEC-HPLC) Phase I studies (healthy
volunteers):Characterization and safety
of cell banksProduct-specific ELISA Safety/Tolerance/Pharma-
cokinetic/PharmacodynamicUpstream procedures:
Fermentation process(USP)
Host-cell-protein ELISA(HCP-ELISA)
Downstream procedures.Purification scheme(DSP)
Residual DNA detection(picogreen, threshold)
Phase II studies (patients):
Optimization of individualprocess steps
N-terminal sequencing (Edmandegradation)
Safety, proof of efficacy, dosefinding
Stability and robustness ofthe process
C-terminal amino acidcomposition
Introducing of GMP Peptide mapping Phase III studies (patients):Consistency batches Total amino acid content (upon
hydrolysis)Controlled safety and efficacy in
specific indicationsValidation Carbohydrate analyses (total
sugar, antennarity, sialic acids)Multiple arms, vs. reference
therapy or placebo controlled.Typically blinded studies andmatched patient groups.
Pharmaceutical develop-ment (formulation)
MALDI-TOF spectroscopy(molecular weight)
Development of fill & finish Detection of free sulfhydrylmoieties
Serological assays (ELISA,radioimmunoassay (RIA)) toquantify drug and antidrug.Neutralizing antibodies(inhibitory effect in bioassay)
Stability studies (holdingsteps, bulk material,final product)
Circular dichroism spectroscopy(CD-spectra)
Surface plasmon resonance (SPR,BIAcore)

140 7.7 Biogeneric Developments and its Requirements
Phases of development
Year
01 02 03 04 08070605
• Cloning, cellbiology
• Non-GMP process development
• Master and working cell bank
• Pilot scale (partially GMP)
• Process development
• Preclinical studies (toxicology)
• Development of formulation
• Scale-up (fully GMP), validation
• Clinical studies (Phase I, II/III)
• Work out of the dossier
• EMEA centralized procedure
• Product launch
Fig. 1 Development of a biogeneric project (example for time schedule).
quantify and to analyze the product. Thisis easier to establish for biogeneric devel-opments, because specific antibodies orcomplete ELISA products for clinical diag-nosis are available for most products. Also,reference material can be obtained in highpurity from the sales product.
In contrast, the development of the up-stream procedures (USP) does not differfor a biogeneric. This has to be done inde-pendently. There is no access to any experi-ence from the reference product. However,the fermentation techniques applied arestandardized and routine in the hands ofexperienced manufacturers. Nevertheless,this is an important and time-consumingsection, and difficulties may occur alongwith scale-up. The downstream process(DSP), which always contains a sequenceof different chromatographic steps, ac-companied by some filtration procedures,can be deduced from published litera-ture, supported by experience in proteinchemistry. It is very likely that biogeneric
developments will end up with their ownand unique DSP. There are many alter-native methods available. The necessity ofDSP is the removal of impurities, such ashost cell proteins (HCP), DNA, endotox-ins, pyrogens, and other process-relatedsubstances. The requirements on purityfor a therapeutic grade protein are high.As discussed in Sect. 7.5, DSP is a field forsecondary patent applications and the de-velopment often has to be performed alonga circumventing strategy. The same is truefor the development of the pharmaceuticalformulation (see Sect. 7.5).
Much work and high costs are relatedto the stage of production of substancefor use in clinical trials, especially forphase III. This requires introduction ofGMP and huge amount of validationwork later on. The phase III materialhas to be taken from the commercialscale. In contrast to originators, whichhave to walk more carefully step-by-step,considering feedbacks from preclinical or

Biogeneric Drugs 141
clinical phases, biogeneric companies havethe possibility to move on and to produceall the materials from the final process andscale. This would substantially lower therisk for noncomparability of the variousmaterials during different stages and thusavoids bridging studies. The developmentof the final product, which is a prefilledsyringe in the majority of cases, showsno biogeneric-specific aspects. Because ofpatents, it is assumed that biogenericproducts will appear with their ownformulations, which differ more or lessfrom the original product. The formulationof the reference product, in respect ofstability, serves as the gold standard.
7.7.2Development of Analytical Methods
Table 3 presents a selection of assays fre-quently taken for the characterization of atherapeutic protein. The field is complexand I can mention only few aspects. Theassays are applied for in-process controls(IPC), batch release, extended character-ization of the purified protein, processvalidation, or stability studies. Most of theassays are standard biochemical methods,which have to be adapted and validatedfor the specific protein. Biogeneric de-velopers will relate their standards to thetarget product, especially if they have de-cided to go for a comparability strategy.In such a case, it seems wise to use thereference product throughout the develop-ment. In some cases such as EPO or FVIII,there are defined reference standards fromthe European Pharmacopoeia (BiologicalReference Preparations, (BRP) available.Other sources for biological standards arethe World Health Organisation (WHO)and the British National Institute for Bi-ological Standards and Control (NIBSC).Of special importance is the potency of
the protein, analyzed by specific bioassays.These assays determine the biologic activ-ity, mostly in terms of international units(IU). Typical bioassays are (1) in vivo an-imal systems, for example mice (EPO),(2) cell-based proliferation assays (CSF)or antiviral assays (IFN). The potency tobind to the natural receptor can effectivelybe demonstrated with so-called ‘‘reportergene activation assays’’, which transducethe receptor-binding signal via gene acti-vation to an easily detectable marker.
For the characterization of the product,one has to consider purity, residual con-taminants, biological activity, and physico-chemical properties. Several sophisticatedmethods are required and have to pro-vide structural evidence for a biosimilarproduct. The N-terminus and C-terminushave to be intact. Free sulfhydryl groupsand disulfide bridges have to be in theright position. Altered versions of theproteins, which are termed as ‘‘product-related impurities’’, for instance causedby methionine oxidation or deamidation,truncated species, dimers, and aggregates,have to be characterized and quantifiedat very low levels. Additional analyticalmethods are utilized for glycoproteins. Theanalysis of the carbohydrate compositionis mandatory. The antennarity structureand the specific number of sialic acidshave a strong influence on potency and onthe pharmacokinetic behavior in vivo. Forthe immunological detection of HCP im-purities, a source- and process-specific testhas to be developed on the basis of mockmaterial used for immunization. Mockmaterial is a kind of control protein prepa-ration, without the product, just modelingthe impurities. Although it is tremendouswork to establish and validate all these as-says, the biogeneric developers can refer topublications, European Pharmacopoeia in

142 7.8 Conclusions
the best case, and charge specialized ser-vice laboratories that offer these methods.However, most of the methods requiredfor the routine quality control, IPC andbatch release, need to be installed at thesite of manufacturing.
7.7.3Preclinical and Clinical Development
This is the most significant part of the to-tal development costs of a pharmaceutical.Biogeneric developers, however, under-went low risk for failure in clinical studies.This is the major difference in the devel-opment of a new protein drug. Dependingon the kind of product and the degreeof analytical comparability, the extent ofclinical studies will vary considerably forbiogenerics. This has been discussed inSect. 7.4 in more detail. For the preclinicalphase, the study program for a biogenericdrug can be reduced to a certain extent.Nevertheless, there are some irreplaceableparts showing toxicology, safety, and phar-macokinetics in two mammalian species.This is also mandatory for entering in hu-man trials. The companies have to decidewhether they should design their preclin-ical studies in direct comparison with thereference product or perform a stand-aloneapproach. Generally, published data areavailable for preclinical results of the orig-inal drug substance. This can be used assupporting material.
Before entering clinical studies, the for-mulation has to be fixed and stability dataof at least three months are necessary.Most of the biogeneric products are liquid-formulated parenteralia presented as pre-filled syringes. There are some importantdifferences for the clinical trials of bio-generics. The studies are designed, at leastpartially, as comparator studies. Phase Iwill be performed with healthy volunteers
or occasionally even with selected patientgroups. The study has to show bioequiva-lence and comparable pharmacodynamicsto the comparator product. To achieve bioe-quivalence, precise dosing per kilogramof body weight is essential. In addition,the phase I study also covers safety as-pects. There is a residual risk for missingbioequivalence and thus showing non-comparability. This would have a severeimpact on further clinical development.The classical separation between phase IIand III studies (see Table 3) is not rea-sonable for biogenerics. Once proven anequivalent pharmacokinetic and pharma-codynamic response, a dose finding or ageneral proof of efficacy (phase II) doesnot seem necessary at all. A typical bio-generic phase III study is designed asa comparator study with two arms andcrossing-over. This should show a com-parable efficacy to the reference productand provide sufficient evidence that bothproducts are interchangeable. In addition,a second part of the phase III study hasto provide safety data based on a statisti-cally calculated number of patients. Thesafety matter is the most important aspectthroughout the different preclinical andclinical stages. The design of the clinicalstudies is one of the major questions tobe addressed to members of the CPMPduring a scientific advice procedure.
Finally, it should be mentioned thatfor both the animal studies and theclinical studies in humans, serologicalassays have to be developed in advance.These tests are required for the detectionof the protein drug in serum or plasma(pharmacokinetic) as well as for thedetection of specific antibodies evoked bythe drug. If antibodies appeared, they haveto be further analyzed for neutralizingactivity. This could be demonstrated byan inhibitory effect in the bioassay.

Biogeneric Drugs 143
7.8Conclusions
Recombinant therapeutic proteins are inthe sight of the generic industry sinceseveral years, and product developmentshave begun. The differences betweenchemical generics and biogenerics aredominated by the kind of approval, the longtime lines, and in the overall high costsfor a project. In these aspects, biogenericsresemble new drugs rather than classicalgenerics. Significant investment and lackof biotech know-how are the two majorbarriers for the generic industry to enterthis kind of business. The risks can beminimized by recruiting experienced staffand competent partners for development,production, and distribution. Key factorsfor success are excellence in biotech,management of alliances and partnering,legal competence, and marketing anddistribution power. The first wave ofrecombinant proteins will run off patentfrom now until 2008. The target productshave been presented in detail and many ofthem will have to compete with therapeuticequivalent biogenerics in the near future.
The common term ‘‘biogenerics’’ wasused throughout the text, although it is dis-liked by the regulators. Other suggestions,such as ‘‘multisource products,’’ ‘‘biosim-ilar products,’’ or ‘‘comparable products’’are used preferentially in relation to theirapproval. They all mean the same. It isof minor importance what kind of word-ing will be used at the end. To overcomeany misunderstanding, it was necessary todefine biogenerics unequivocally in directcomparison to chemical generics. Besidesthe regulatory conditions, market aspectsare also included for a definition. Other-wise, biopharmaceutical products, whichare accepted in the medical community as
innovations, for example Rebif (IFN-β-1a) or Dynepo (epoetin delta), would bepure biogenerics. Indeed, it will be diffi-cult in the future to classify products asbiogenerics. This is due to additional in-novations, own brands, multiple licensestrategies, and expected moderate pricereductions.
The biogeneric business urgently re-quires the development of a more favorablelegal framework. This situation, althoughevolving, has not yet become a reality. Twoindependent projections in the EuropeanUnion are in progress: a more precise guid-ance for the approval of biogenerics and theruling of a Bolar provision. The expectedrulings would solve many conflicts thebiogeneric developers actually deal with.Unfortunately, the recent concerns aboutthe immunogenicity of Eprex (epoetinalfa) leading to the life-threatening PRCAsyndrome will not make it easier to approvegeneric erythropoietins.
The complex patent situation for bio-logics favors future litigation. Very likely,biogeneric companies will receive aggres-sive legal opposition from the originators.One of the strategies they can use, called‘‘evergreening’’, means to file ‘‘secondarypatents’’ some years after the basic patent,which claim essential parts of the old prod-uct, and thus prolonging the exclusivemarket period. In some cases, especiallyin the United States, this was discussedas a kind of patent law abuse. Besidespatents, innovators have further methodsto protect their products, which are not dis-cussed in this overview. These are (1) dataexclusivity periods (not directly relevant forbiogenerics), (2) supplementary protectioncertificates (SPC), and (3) orphan drug sta-tus. Altogether, these legal instrumentsmaintain a reasonable period of marketexclusivity for the originator.

144 7.8 Conclusions
Another strategy of originators is lifecycle management. The introduction ofsecond-generation products at the endof patent expiry and possibly phasingout the predecessor product is a well-known strategy against generics. Thiswas successfully demonstrated for IFN-α and indicates the risks for biogenericcompanies.
Altogether, the competition betweenoriginators and generic companies putpressure on both sides to innovate. Thisresults in benefits for patients (improvedtherapy) and the health system (lowercosts).
References
1. K. Maleck, F. Pollano, Eur. Biopharm. Rev.2001, 3, 19–21.
2. P. Brown, M. Preece, J. P. Brandel et al.,Neurology 2000, 55(8), 1075–1081.
3. P. M. Mannucci, P. L. Giangrande, Hematol.J. 2000, 1(2), 72–76.
4. G. Schernthaner, Diabetes Care 1993, 16(Suppl. 3), 155S–165S.
5. R. E. Chance, B. H. Frank, Diabetes Care1993, 16 (Suppl. 3), 133S–142S.
6. H. Schellekens, Nat. Rev. Drug Discov. 2002,1(6), 457–462.
7. Note for guidance on comparability ofmedicinal products containing biotechnol-ogy derived proteins as drug substance,CPMP/BWP/3207/00 (September 20, 2001).
8. S. Usdin, BioCentury 2003, 11(20), A1–A6.9. SCRIP World Pharm. News, 2003, 6, 2855.
10. Draft Annex I of Directive 2001/83/EC onthe community code relating to medicinalproducts for human use.
11. K. Haan, S. Usdin, BioCentury 2002, 10(35),A1–A6.
12. T. L. Gerrard, BioProcess Int. 2003, 1(5),38–42.
13. A. Dove, Nat. Biotechnol. 2003, 21, 495–498.14. Roche Prods. Inc. v. Bolar Pharm Co, U.S.
Court of Appeals for the Federal Circuit, Fed.Cir., 1984, 733F.2d 858.
15. National Economic Research Association(NERA), Policy Relating to Generic Medi-cines in the OECD. Study Carried Out onBehalf of the European Commission, 1998.
16. K. Robinson, BioPharm Int. 2003, 14(1),24–26.
17. G. Kelley, BioPharm Int. 2002, 15(12), 44.18. J. S. Powell, K. L. Berkner, R. V. Lebo, Proc.
Natl. Acad. Sci. U. S. A. 1986, 83, 6465–6469.19. A. Sikole, G. Spasovski, D. Zafirov et al.,
Clin. Nephrol. 2002, 57(3), 237–245.20. P. L. Storring, R. J. Tiplady, R. E. Gaines Das
et al., Brit. J. Hematol. 1998, 100, 79–89.21. Council of Europe, Pharmacop 2002, 1316,
1123–1128.22. N. Casadevall, J. Naaf, B. Viron et al., N. Engl.
J. Med. 2002, 346(7), 469–475.23. H. Schellekens, Nephrol. Dial. Transplant.
2003, 18, 1257–1259.24. D. C. Dale, Drugs 2002, 62 (Suppl. 1),
1S–15S.25. B. K. Dieckgraefe, J. R. Korzenik, Lancet
2002, 360, 1478–1480.26. M. J. Henwood, A. Grimberg, T., Moshang
Jr, Curr. Opin. Pediatr. 2002, 14(4), 437–442.27. P. Iglesias, J. J. Diez, Expert Opin. Pharma-
cother. 1999, 1(1), 97–107.28. A. M. Gualandi-Signorini, G. Giorgi, Eur.
Rev. Med. Pharmacol. Sci. 2001, 5(3), 73–83.29. J. E. Gerich, Am. J. Med. 2002, 113(4),
308–316.30. G. M. Keating, S. Noble, Drugs 2003, 63(10),
1021–1051.31. J. Klinge, N. M. Ananyeva, C. A. Hauser
et al., Semin. Thromb. Hemost. 2002, 28(3),309–322.
32. M. De Andrea, R. Ravera, D. Gioia, et al.,Eur. J. Pediatr. Neurol. 2002, 6 (Suppl. A),A41S–A46S.
33. S. Usdin, BioCentury 2002, 10(17), A1–A13.34. H. Wiendl, B. C. Kieseier, Expert Opin.
Investig. Drugs 2003, 12(4), 689–712.35. K. Haan, BioCentury 2002, 10(35), A7–A8.36. F. Patti, A. Reggio, Int. J. Clin. Pract. Suppl.
2002 (Sep); (131), S23–S32.37. S. Usdin, BioCentury 2003, 11, A6.38. Bioferon Biochemische Substanzen GmbH
& Co, EP-529300-B1, 1992.39. G. Filippini, L. Munari, B. Incorvaia et al.,
Lancet 2003, 361(9357), 545–552.

Part IIITherapeutic Proteins – SpecialPharmaceutical Aspects
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

147
8Pharmacokinetics andPharmacodynamics of BiotechDrugs
Bernd MeibohmUniversity of Tennessee Health Science Center, Memphis, TN, USA
Hartmut DerendorfUniversity of Florida, Gainesville, FL, USA
8.1Introduction
In the last two decades, an increasingfraction of pharmaceutical R&D has beendevoted to biotechnology-derived drugs(biotech drugs) – large molecules such assoluble proteins, monoclonal antibodies,and antibody fragments, as well as smallerpeptides, antisense oligonucleotides, andDNA preparations for gene therapy [1].Biotech and genomic companies currentlyperform nearly one-fifth of all pharmaceu-tical R&D, a figure that is set to doublewithin the next 10 years [2]. These biotech-related drug development efforts have sofar been quite successful. Biotech prod-ucts accounted for more than 35% of the37 new active substances (NASs) that werelaunched in 2001, and it has been pre-dicted that half of all NASs developedin the next 10 to 15 years will resultfrom research into antibodies alone [1].Numerous approved biotech drug prod-ucts with blockbuster character underline
this success – erythropoietin (Epogen,Procrit), abciximab (Rheopro), andtrastuzumab (Herceptin) to name a few.Since the development of biotech drugsgenerally rests on a fundamental un-derstanding of the related disease, theirclinical development has also proven tobe more successful than for conventionalsmall molecules (new molecular entities:NCEs). Only 8% of the NCEs that enteredclinical drug development between 1996and 1998 reached the market comparedto 34% of biotech drugs. On the basis ofthese facts, it can be predicted that biotechdrugs will play a major, if not dominant,role in the drug development arena of thenext decades. Thus, biotech-based medica-tions might serve as the key for the aspired‘‘personalized medicine’’ in the health caresystems of the future [3].
The basis for the pharmacotherapeuticuse of biotech drugs is similar to thatof small molecules, a defined relationshipbetween the intensity of the therapeuticeffect and the amount of drug in the body,
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8
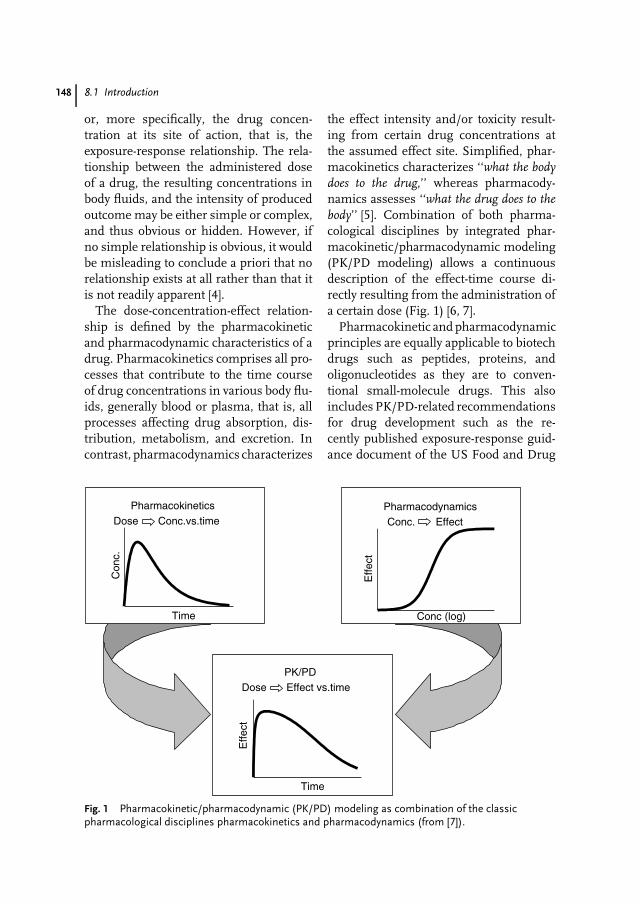
148 8.1 Introduction
or, more specifically, the drug concen-tration at its site of action, that is, theexposure-response relationship. The rela-tionship between the administered doseof a drug, the resulting concentrations inbody fluids, and the intensity of producedoutcome may be either simple or complex,and thus obvious or hidden. However, ifno simple relationship is obvious, it wouldbe misleading to conclude a priori that norelationship exists at all rather than that itis not readily apparent [4].
The dose-concentration-effect relation-ship is defined by the pharmacokineticand pharmacodynamic characteristics of adrug. Pharmacokinetics comprises all pro-cesses that contribute to the time courseof drug concentrations in various body flu-ids, generally blood or plasma, that is, allprocesses affecting drug absorption, dis-tribution, metabolism, and excretion. Incontrast, pharmacodynamics characterizes
the effect intensity and/or toxicity result-ing from certain drug concentrations atthe assumed effect site. Simplified, phar-macokinetics characterizes ‘‘what the bodydoes to the drug,’’ whereas pharmacody-namics assesses ‘‘what the drug does to thebody’’ [5]. Combination of both pharma-cological disciplines by integrated phar-macokinetic/pharmacodynamic modeling(PK/PD modeling) allows a continuousdescription of the effect-time course di-rectly resulting from the administration ofa certain dose (Fig. 1) [6, 7].
Pharmacokinetic and pharmacodynamicprinciples are equally applicable to biotechdrugs such as peptides, proteins, andoligonucleotides as they are to conven-tional small-molecule drugs. This alsoincludes PK/PD-related recommendationsfor drug development such as the re-cently published exposure-response guid-ance document of the US Food and Drug
PharmacokineticsDose Conc.vs.time
Con
c.
Time
PK/PDDose Effect vs.time
Time
Effe
ct
PharmacodynamicsConc. Effect
Conc (log)
Effe
ct
Fig. 1 Pharmacokinetic/pharmacodynamic (PK/PD) modeling as combination of the classicpharmacological disciplines pharmacokinetics and pharmacodynamics (from [7]).

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 149
Administration and the ICH E4 guidelineof the International Conference on Har-monization of Technical Requirements forRegistration of Pharmaceuticals for Hu-man Use [8, 9]. Since biotech drugs arefrequently identical or similar to endoge-nous substances, they oftentimes exhibitunique pharmacokinetic and pharmaco-dynamic properties. These may pose extrachallenges and questions during their pre-clinical and clinical drug development thatare different from small-molecule drugcandidates and may require additional re-sources and unique expertise. Some ofthese problems and challenges will be dis-cussed in the following.
8.2Bioanalytical Challenges
The availability of an accurate, precise,and specific bioanalytical technique for thequantification of active drug moieties inplasma, blood, or other biological fluids isan essential prerequisite for the evaluationof the relationship between dose, concen-tration, and effect of biotech drugs. In anal-ogy to small molecules, these analyticaltechniques have to be validated and haveto meet prespecified criteria regarding ac-curacy, precision, selectivity, sensitivity,reproducibility, and stability, for example,those recommended by the US Food andDrug Administration [10–12]. Additionalrequirements for bioanalytical method val-idation for macromolecules have recentlybeen published [11].
A further level of complexity is oftenadded to the bioanalytics of biotech drugsby the fact that numerous of these com-pounds are endogenous substances thatare already in the body before drug admin-istration. Thus, the analytical techniquewill detect a so-called baseline level prior
to drug exposure. This baseline level caneither be constant or undergo complexchanges, for example, circadian rhythms orirregular time courses. In order to charac-terize the clinical pharmacology of biotechdrugs naturally present in the body, base-line values have to either be accounted forin the pharmacokinetic and pharmacody-namic analysis or be suppressed beforeexogenous drug administration [13–15].The suppression of endogenous base-line levels is oftentimes facilitated viaphysiological regulation or feedback mech-anisms. This approach was, for example,used to suppress the endogenous releaseof insulin and somatotropin (growth hor-mone, GH) via infusions of glucose andsomatostatin respectively, prior to their ex-ogenous administration for evaluation oftheir pharmacokinetics [16].
In contrast to the bioanalytics of small-molecule drugs, immunoassays and bioas-says are frequently applied to quantify pep-tides and proteins in biological samples.Immunoassays are considered the analyt-ical method of choice for concentrationdeterminations of peptides and proteinsas they can relatively rapidly and easily beperformed [17]. These generally compriseenzyme immunoassays as well as radioim-munoassays with poly- and monoclonalantibodies. While both techniques have asufficient sensitivity and reproducibility,their specificity for the active drug com-pound to be quantified has to be carefullyevaluated during the assay validation pro-cess. Immunoassays may be insensitive torelatively minor changes in the primaryor secondary structure of proteins. Forrecombinant interferon-γ , for example,bioavailability was reported to be >100%for subcutaneous compared to intravenousadministration, which was produced byassay artifacts due to slightly modifieddegradation products [16]. Other potential

150 8.3 Pharmacokinetics of Peptides and Proteins
interferences with immunoassays includematrix effects, specific binding proteins,proteases, and cross-reactivity toward en-dogenous proteins [17].
Bioassays are frequently used as analternative or in addition to immunoas-say techniques. Bioassays, in contrast toimmunoassays, quantify not the phar-macologically active substance, but itsbiological activity, for example, in cell cul-ture models based on cell differentiation,cell proliferation, or cytotoxicity as well asgene expression assays or whole animalmodels. Frequent major problems withbioassays comprise a high variability in themeasured parameters, lack of precision,and their time- and labor-intensive per-formance. Furthermore, bioassays often-times also lack specificity for the measuredcompound, as they may also detect theresponse to bioactive metabolites [16, 17].
Because of some of the problems withbioassays and immunoassays, liquid chro-matography (LC)-based techniques are in-creasingly applied as an alternative. Whilemodern LC-based assays have a compa-rable sensitivity to immunoassays, theyoftentimes are characterized by a higherselectivity [18, 19]. Muller et al., for ex-ample, used LC/mass spectrometry withmatrix-assisted laser desorption ioniza-tion in ex vivo pharmacokinetic studiesin combination with enzyme inhibitionexperiments to investigate the complexmetabolism of dynorphin A1-13, a pep-tide with opioid activity, up to the fifthmetabolite generation [20, 21].
Biodistribution studies for peptides arefrequently performed with radioactivelylabeled compound. The radioactivity caneither be introduced by external labelingwith 125I or by internal labeling of alreadypresent atoms, for example, via additionof 3H, 14C, or 35S radioactively labeledamino acids to cell cultures producing
recombinant proteins. External labelingchemically modifies the protein, whichmay affect its activity and pharmacokinet-ics. Internal labeling circumvents thesepotential problems, but has the disad-vantage that radioactively labeled aminoacids might be reused in the endogenousamino acid pool. Thus, when using ra-dioactive labeling, it is generally necessaryto investigate whether the physicochemi-cal and biological properties of the proteinsare unchanged. In addition, it is cru-cial to differentiate whether the measuredradioactivity represents intact protein, la-beled metabolites, or the released labelitself. Radioactivity that can be precipitatedwith trichloroacetic acid, for example, canbe used to delineate active protein fromreleased label and metabolites of smallmolecular weight [16].
8.3Pharmacokinetics of Peptides and Proteins
Although traditional pharmacokinetic pri-nciples are also applicable for peptides andproteins, their in vivo disposition is to alarge degree affected by their physiologicalfunction. Peptides, for example, whichfrequently have hormone activity, usuallyhave short elimination half-lives, whichis desirable for a close regulation oftheir endogenous levels and thus function.Contrary to that, transport proteins likealbumin or antibodies have eliminationhalf-lives of several days, which enablesand ensures the continuous maintenanceof necessary concentrations in the bloodstream [16]. The reported terminal half-life for SB209763, a humanized respiratorysyncytial virus monoclonal antibody, forexample, was reported as 22 to 50days [22].

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 151
8.3.1Absorption
Traditionally, the largest obstacle for asuccessful pharmacotherapy with peptideand protein drugs is their delivery to thedesired site of action. A clinically usableabsorption of exogenously applied pep-tides and protein after oral applicationwith conventional dosage forms is usuallynot present [19, 23]. This lack of systemicbioavailability is mainly caused by two fac-tors, high gastrointestinal enzyme activityand the function of the gastrointestinalmucosa as absorption barrier. There issubstantial peptidase and protease activ-ity in the gastrointestinal tract, makingit to the most efficient body compart-ment for peptide and protein metabolism.Furthermore, the gastrointestinal mucosapresents a major absorption barrier forwater-soluble macromolecules like pep-tides and protein [13, 19]. This is at leastfor peptides complemented by the func-tional system of cytochrome P450 3A andp-glycoprotein activity [24–26].
The lack of activity after oral admin-istration for most peptides and proteinsresulted in the past besides parenteral ap-plication into the utilization of nonoral ad-ministration pathways, for example, nasal,buccal, rectal, vaginal, percutaneous, ocu-lar, or pulmonary drug delivery [27]. Drugdelivery via these administration routes,however, is also frequently accompaniedby presystemic degradation processes.Bioavailability of numerous peptides andproteins is, for example, markedly re-duced after subcutaneous or intramus-cular administration compared to theirintravenous administration. The pharma-cokinetically derived apparent absorptionrate constant is thus the combination of ab-sorption into the systemic circulation andpresystemic degradation at the absorption
site. The true absorption rate constant ka
can then be calculated as
ka = F · kapp
where F is the bioavailability comparedto intravenous administration. A rapidapparent absorption rate constant kapp canthus be the result of a slow absorption anda fast presystemic degradation, that is, alow systemic bioavailability [13].
8.3.2Distribution
Whole body distribution studies are es-sential for classical small-molecule drugsin order to exclude tissue accumulation ofpotentially toxic metabolites. This problemdoes not exist for protein drugs in whichcatabolic degradation products are aminoacids recycled in the endogenous aminoacid pool. Therefore, biodistribution stud-ies for peptides and proteins are primarilyperformed to assess targeting to specifictissues as well as to identify the majorelimination organs [28].
The volume of distribution of proteinsis usually small and limited to the vol-ume of the extracellular space because oftheir high molecular weight and the relatedlimited mobility because of impaired pas-sage through biomembranes [29, 30]. Afterintravenous application, peptides and pro-teins usually follow a biexponential plasmaconcentration–time profile that can bestbe described by a two-compartment phar-macokinetic model [13]. This has, for ex-ample, been described for leuprorelin,a synthetic agonist analog of luteiniz-ing hormone-releasing hormone (LH-RH) [31], for clenoliximab, a macaque-human chimeric monoclonal antibodyspecific to the CD4 molecule on the surfaceof T-lymphocytes [32], and for AJW200, ahumanized monoclonal antibody to von

152 8.3 Pharmacokinetics of Peptides and Proteins
Willebrand factor [33]. The central com-partment in this model represents primar-ily the vascular space and the interstitialspace of well-perfused organs with perme-able capillary walls, especially liver andkidneys, while the peripheral compart-ment comprises the interstitial space ofpoorly perfused tissues like skin and (in-active) muscle [19].
Thus, the volume of distribution ofthe central compartment in which pep-tides and proteins initially distribute afterintravenous administration is typically 3to 8 L, approximately equal to slightlyhigher than the plasma volume [19] (ap-proximate body water volumes for a 70-kg person: interstitial 12 L, intracellular27 L, intravascular 3 L) [34]. The total vol-ume of distribution (Vd) is frequently14 to 20 L, not more than twice theinitial volume of distribution (Vc) [13,28]. This distribution pattern has, forexample, been described for the somato-statin analog octreotid (Vc 5.2–10.2 L,Vd 18–30 L), the t-PA analog tenecteplase(Vc 4.2–6.3 L, Vd 6.1–9.9 L), and the gly-coprotein IIb//IIIa inhibitor eptifibatide(Vc 9.2 L) [35–37]. Active tissue uptake andbinding to intra- and extravascular pro-teins, however, can substantially increasethe volume of distribution of peptide andprotein drugs, as, for example, observedwith atrial natriuretic peptide (ANP) [38].
There is a tendency for Vd and Vc tocorrelate with each other, which impliesthat the volume of distribution is predom-inantly determined by distribution in thevascular and interstitial space as well asunspecific protein binding in these dis-tribution spaces. The distribution rate isinversely correlated with molecular sizeand is similar to that of inert polysac-charides, suggesting that passive diffusionthrough aqueous channels is the primarydistribution mechanism [19].
Distribution, elimination, and pharma-codynamics are, in contrast to conven-tional drugs, frequently interrelated forpeptides and proteins. The generally lowvolume of distribution should not neces-sarily be interpreted as low tissue pene-tration. Receptor-mediated specific uptakeinto the target organ, as one mechanism,can result in therapeutically effective tis-sue concentrations despite a relativelysmall volume of distribution [28]. Nar-togastrim, a recombinant derivative ofthe granulocyte-colony stimulating factor(G-CSF), for example, is characterizedby a specific, dose-dependent, and sat-urable tissue uptake into the target organbone marrow, presumably via receptor-mediated endocytosis [39].
8.3.3Protein Binding
It is a general pharmacokinetic principle,which is also applicable to peptides andproteins, that only the free, unboundfraction of a drug substance is accessibleto distribution and elimination processesas well as interactions with its targetstructure (e.g. receptor) at the site ofaction. Hence, the activity of a drug isbetter characterized by its free rather thantotal concentration if there is no constantrelationship between free and total drugconcentration.
Physiologically active endogenous pep-tides and proteins are frequently interact-ing with specific binding proteins that areinvolved in their transport and regulation.Furthermore, interaction with bindingproteins may enable or facilitate cellu-lar uptake processes and thus affect thedrug’s pharmacodynamics. Specific bind-ing proteins were, for example, describedfor IGF-1 (insulin-like growth factor), t-PA,interleukin-2, and somatotropin [16].

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 153
Six specific binding proteins were iden-tified for IGF-1, with one binding at least95% of IGF-1 in plasma. Since the bindingaffinity of IGF-1 to this binding proteinis substantially higher than IGF receptors,the binding protein is assumed to have areservoir function that protects the bodyfrom insulin-like hypoglycemia. Further-more, the elimination half-life for boundIGF-1 is significantly longer than for freeIGF-1, since only the unbound IGF-1 isaccessible to elimination via glomerularfiltration or peritubular extraction [28, 40].
Somatotropin, another example, has atleast two binding proteins in plasma [16].This protein binding substantially reducessomatotropin elimination with a tenfoldsmaller clearance of total compared to freesomatotropin, and also decreases its activ-ity via reduction of receptor interactions.
Apart from these specific bindings,peptides and proteins may also be non-specifically bound to plasma proteins. Forexample, metkephamid, a met-enkephalinanalog, was described to be 44 to 49%bound to albumin [41], and octreotid is upto 65% bound to lipoproteins [35].
8.3.4Elimination
Peptide and protein drugs are nearly exclu-sively metabolized via the same catabolicpathways as endogenous or dietetic pro-teins, leading to amino acids that arereutilized in the endogenous amino acidpool for the de novo biosynthesis of struc-tural or functional body proteins.
Nonmetabolic elimination pathwayssuch as renal or biliary excretion arenegligible for most peptides and proteins.Amino acids as well as some peptidesand proteins such as immuneglobulineA, however, are excreted into the bile [19].For octreotid, biliary excretion is, at least
in rat and dog, an important eliminationpathway [35]. If biliary excretion ofpeptides and proteins occurs, it generallyresults in subsequent metabolism of thesecompounds in the gastrointestinal tract(see Sect. 8.3.4.2) [13].
The elimination of peptides and proteinscan occur unspecifically nearly everywherein the body or can be limited to a specificorgan or tissue. Locations of intensivepeptide and protein metabolism are liver,kidneys, gastrointestinal tissue, and alsoblood and other body tissues. Molecularweight determines the major metabolismsite as well as the predominant degradationprocess [13, 42] (Table 1).
The metabolism rate generally increaseswith decreasing molecular weight fromlarge to small proteins to peptides, butis also dependent on other factors likesecondary and tertiary structure as well asglycosylation. The clearance of a peptide orprotein describes the irreversible removalof active substance from the extracellularspace, which also includes cellular uptakebesides metabolism. Because of the un-specific degradation of numerous peptidesand proteins in blood, clearance can exceedcardiac output, that is, >5 L/min for bloodclearance and >3 L/min for plasma clear-ance [13]. Investigations on the detailedmetabolism of peptides and proteins arerelatively difficult because of the myriad ofmolecule fragments that may be formed.
8.3.4.1 ProteolysisProteolytic enzymes such as proteasesand peptidases are ubiquitously availablethroughout the body, but are especiallylocalized in blood, in the vascular endothe-lium, and also on cell membranes andwithin cells. Thus, intracellular uptake isper se more an elimination rather than adistribution process [13]. While peptidasesand proteases in the gastrointestinal tract

154 8.3 Pharmacokinetics of Peptides and Proteins
Tab. 1 Molecular weight as major determinant of the elimination mechanisms of peptides andproteins. As indicated, mechanisms may overlap. Endocytosis may occur at any molecular weightrange (modified from [19, 28])
Molecularweight
Eliminationsite
Predominant eliminationmechanisms
Majordeterminant
<500 Blood, liver Extracellular hydrolysis Passivelipoid diffusion
Structure, lipophilicity
500–1000 Liver Carrier-mediated uptakePassive lipoid diffusion
Structure, lipophilicity
1000–50 000 Kidney Glomerular filtration andsubsequent degradationprocesses (see Fig. 2)
Molecular weight
50 000–200 000 Kidney, liver Receptor-mediated endocytosis Sugar, charge200 000–400 000 Opsonization α2-macroglobulin, IgG>400 000 Phagocytosis Particle aggregation
and in lysosomes are relatively unspecific,soluble peptidases in the interstitial spaceand exopeptidases on the cell surface havea higher selectivity and determine the spe-cific metabolism pattern of an organ [19].The proteolytic activity of subcutaneoustissue, for example, results in a partial lossof activity of subcutaneously compared tointravenously administrated interferon-γ .
8.3.4.2 Gastrointestinal EliminationFor orally administered peptides and pro-teins, the gastrointestinal tract is themajor site of metabolism. Presystemicmetabolism is the primary reason fortheir lack of oral bioavailability. Parenter-ally administered peptides and proteins,however, may also be metabolized in theintestinal mucosa following intestinal se-cretion. At least 20% of the degradationof endogenous albumin takes place in thegastrointestinal tract [13].
8.3.4.3 Renal EliminationFor parenterally administered and endoge-nous peptides and proteins, the kidneysare the major elimination organ if they are
smaller than the glomerular filtration limitof ∼60 kD, although the effective moleculeradius is probably the limiting factor. Theimportance of the kidneys as elimina-tion organ could, for example, be shownfor interleukin-2, M-CSF and interferon-α [16, 19].
Various renal processes are contributingto the elimination of peptides and proteins(Fig. 2). For most substances, glomeru-lar filtration is the dominant, rate-limitingstep as subsequent degradation processesare not saturable under physiologic condi-tions [13, 43]. Hence, the renal contribu-tion to the overall elimination of peptidesand proteins is reduced if the metabolicactivity for these proteins is high in otherbody regions, and it becomes negligiblein the presence of unspecific degrada-tion throughout the body. In contrast tothat, the contribution to total clearance ap-proaches 100% if the metabolic activityis low in other tissues or if distributionis limited. For recombinant IL-10, for in-stance, elimination correlates closely withglomerular filtration rate, making dosageadjustments necessary in patients with im-paired renal function [44].
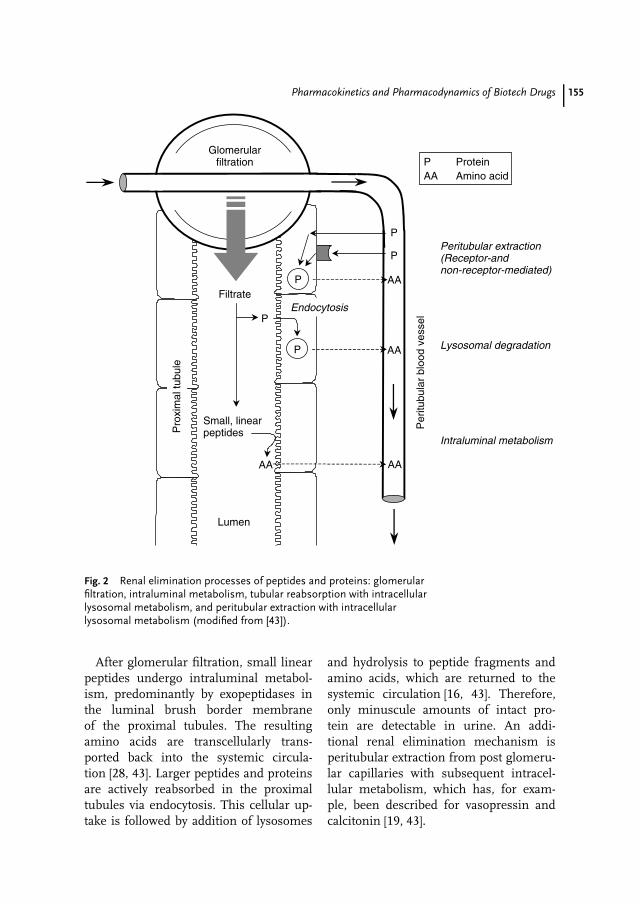
Pharmacokinetics and Pharmacodynamics of Biotech Drugs 155
Per
itubu
lar
bloo
d ve
ssel
Pro
xim
al tu
bule
Lumen
Glomerularfiltration
P AA
PEndocytosis
Lysosomal degradation
AAAA
P
Intraluminal metabolism
P AA
Peritubular extraction(Receptor-and non-receptor-mediated)
Small, linearpeptides
P ProteinAA Amino acid
P
Filtrate
Fig. 2 Renal elimination processes of peptides and proteins: glomerularfiltration, intraluminal metabolism, tubular reabsorption with intracellularlysosomal metabolism, and peritubular extraction with intracellularlysosomal metabolism (modified from [43]).
After glomerular filtration, small linearpeptides undergo intraluminal metabol-ism, predominantly by exopeptidases inthe luminal brush border membraneof the proximal tubules. The resultingamino acids are transcellularly trans-ported back into the systemic circula-tion [28, 43]. Larger peptides and proteinsare actively reabsorbed in the proximaltubules via endocytosis. This cellular up-take is followed by addition of lysosomes
and hydrolysis to peptide fragments andamino acids, which are returned to thesystemic circulation [16, 43]. Therefore,only minuscule amounts of intact pro-tein are detectable in urine. An addi-tional renal elimination mechanism isperitubular extraction from post glomeru-lar capillaries with subsequent intracel-lular metabolism, which has, for exam-ple, been described for vasopressin andcalcitonin [19, 43].

156 8.3 Pharmacokinetics of Peptides and Proteins
8.3.4.4 Hepatic EliminationApart from proteolysis and the kidneys,the liver substantially contributes to themetabolism of peptide and protein drugs.Proteolysis usually starts with endopepti-dases that attack in the middle part ofthe protein, and the resulting oligopep-tides are then further degraded by ex-opeptidases. The ultimate metabolites ofproteins, amino acids, and dipeptides arefinally reutilized in the endogenous aminoacid pool. The rate of hepatic metabolismis largely dependent on specific amino acidsequences in the protein [28].
An important first step in the hep-atic metabolism of proteins and pep-tides is the uptake into hepatocytes.Small peptides may cross the hepatocytemembrane via passive diffusion if theyhave sufficient hydrophobicity. Uptake oflarger peptides and proteins is facili-tated via various carrier-mediated, energy-dependent transport processes. Receptor-mediated endocytosis is an additionalmechanism for uptake into hepatocytes(see Sect. 8.3.4.5) [28]. In addition, pep-tides such as metkephamid can already bemetabolized on the surface of hepatocytesor endothelial cells [41].
8.3.4.5 Receptor-mediated EliminationReceptor binding is usually negligiblecompared to total amount of drug inthe body for conventional small-moleculedrugs and rarely affects their pharma-cokinetic profile. In contrast to that, asubstantial fraction of a peptide and pro-tein dose can be bound to receptors. Thisbinding can lead to elimination throughreceptor-mediated uptake and subsequentintracellular metabolism. The endocyto-sis process is not limited to hepatocytes,but can occur in other cells as well, in-cluding the therapeutic target cells. Sincethe number of receptors is limited, their
binding and the related drug uptake canusually be saturated within therapeuticconcentrations. Thus, receptor–mediatedelimination constitutes a major source fornonlinear pharmacokinetic behavior of nu-merous peptide and protein drugs, that is,a lack of dose proportionality.
M-CSF, for example, undergoes besideslinear renal elimination a nonlinear elim-ination pathway that follows Michaelis-Menten kinetics and is linked to a receptor-mediated uptake into macrophages. Atlow concentrations, M-CSF follows lin-ear pharmacokinetics, while at high con-centrations, nonrenal elimination path-ways are saturated, resulting in nonlin-ear pharmacokinetic behavior (Fig. 3) [45,46]. Other examples for receptor-mediatedelimination are insulin, t-PA, epidermalgrowth factor (EGF), ANP, and interleukin-10 [19, 28, 38, 44, 47].
Eppler et al., for example, had to developa mechanism-based, target-mediated drugdistribution model in order to accuratelydescribe the nonlinear pharmacokineticsof a recombinant human vascular en-dothelial growth factor (rhVEGF165) inpatients with coronary artery disease [48].Nonlinearity was caused by elimination ofrhVEGF165 by binding to specific and sat-urable high-affinity receptors followed byinternalization and degradation.
8.3.5Species Specificity and Allometry
Peptides and proteins exhibit distinctspecies specificity with regard to structureand activity. Peptides and proteins withidentical physiological function may havedifferent amino acid sequences in differ-ent species and may have no activity orbe even immunogenic if used in a differ-ent species. The extent of glycosylation isanother factor of species differences, for
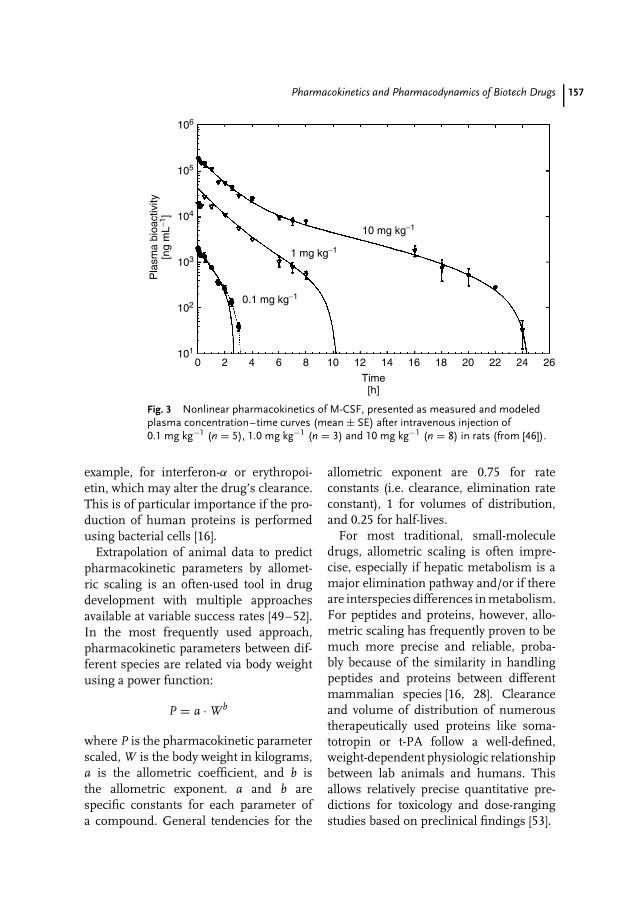
Pharmacokinetics and Pharmacodynamics of Biotech Drugs 157
106
105
104
103
102
101
Pla
sma
bioa
ctiv
ity[n
g m
L−1]
0 2 4 6 8 10 12 14 16 18 20 22 24 26
Time[h]
10 mg kg−1
0.1 mg kg−1
1 mg kg−1
Fig. 3 Nonlinear pharmacokinetics of M-CSF, presented as measured and modeledplasma concentration–time curves (mean ± SE) after intravenous injection of0.1 mg kg−1 (n = 5), 1.0 mg kg−1 (n = 3) and 10 mg kg−1 (n = 8) in rats (from [46]).
example, for interferon-α or erythropoi-etin, which may alter the drug’s clearance.This is of particular importance if the pro-duction of human proteins is performedusing bacterial cells [16].
Extrapolation of animal data to predictpharmacokinetic parameters by allomet-ric scaling is an often-used tool in drugdevelopment with multiple approachesavailable at variable success rates [49–52].In the most frequently used approach,pharmacokinetic parameters between dif-ferent species are related via body weightusing a power function:
P = a · Wb
where P is the pharmacokinetic parameterscaled, W is the body weight in kilograms,a is the allometric coefficient, and b isthe allometric exponent. a and b arespecific constants for each parameter ofa compound. General tendencies for the
allometric exponent are 0.75 for rateconstants (i.e. clearance, elimination rateconstant), 1 for volumes of distribution,and 0.25 for half-lives.
For most traditional, small-moleculedrugs, allometric scaling is often impre-cise, especially if hepatic metabolism is amajor elimination pathway and/or if thereare interspecies differences in metabolism.For peptides and proteins, however, allo-metric scaling has frequently proven to bemuch more precise and reliable, proba-bly because of the similarity in handlingpeptides and proteins between differentmammalian species [16, 28]. Clearanceand volume of distribution of numeroustherapeutically used proteins like soma-totropin or t-PA follow a well-defined,weight-dependent physiologic relationshipbetween lab animals and humans. Thisallows relatively precise quantitative pre-dictions for toxicology and dose-rangingstudies based on preclinical findings [53].
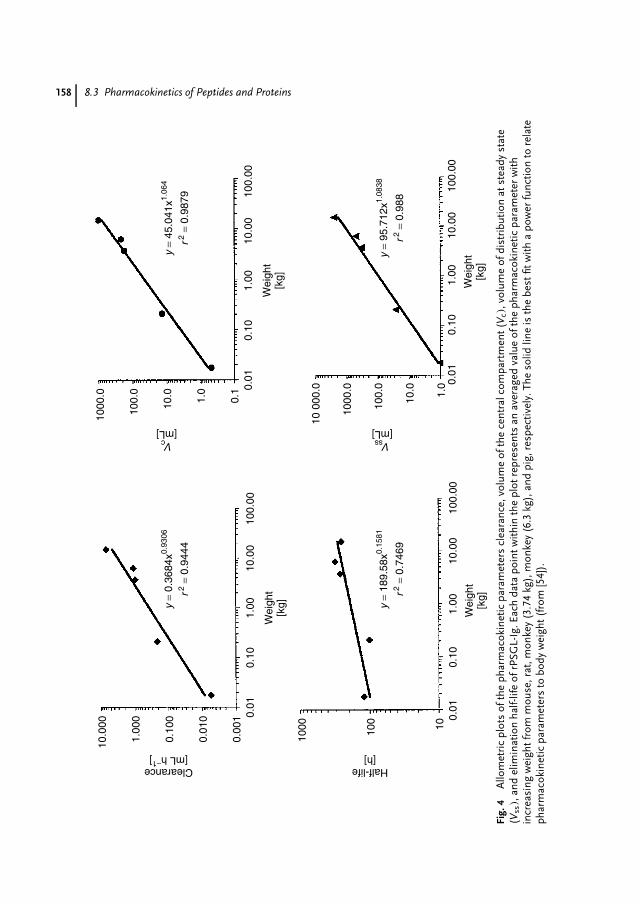
158 8.3 Pharmacokinetics of Peptides and Proteins10
.000
Clearance[mL h−1]
1.00
0
0.10
0
0.01
0
0.00
1 0.01
0.10
1.00
Wei
ght
[kg]
10.0
010
0.00
1000
.0
Vc[mL]
100.
0
10.0 1.0
0.1 0.
010.
101.
00
Wei
ght
[kg]
10.0
010
0.00
10 0
00.0
Vss[mL]
1000
.0
100.
0
10.0 1.0 0.
010.
101.
00
Wei
ght
[kg]
10.0
010
0.00
0.01
0.10
1.00
Wei
ght
[kg]
10.0
010
0.00
1000
Half-life[h]
100 10
y =
0.36
84x0.
9306
r2 =
0.94
44
y =
45.0
41x1.
064
r2 =
0.98
79
y =
189.
58x0.
1581
r2 =
0.74
69
y =
95.7
12x1.
0838
r2 =
0.98
8
Fig.
4A
llom
etri
cpl
ots
ofth
eph
arm
acok
inet
icpa
ram
eter
scl
eara
nce,
volu
me
ofth
ece
ntra
lcom
part
men
t(V
c),v
olum
eof
dist
ribu
tion
atst
eady
stat
e(V
ss),
and
elim
inat
ion
half-
life
ofrP
SGL-
Ig.E
ach
data
poin
twith
inth
epl
otre
pres
ents
anav
erag
edva
lue
ofth
eph
arm
acok
inet
icpa
ram
eter
with
incr
easi
ngw
eigh
tfro
mm
ouse
,rat
,mon
key
(3.7
4kg
),m
onke
y(6
.3kg
),an
dpi
g,re
spec
tivel
y.Th
eso
lidlin
eis
the
best
fitw
itha
pow
erfu
nctio
nto
rela
teph
arm
acok
inet
icpa
ram
eter
sto
body
wei
ght(
from
[54]
).

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 159
Figure 4 shows the allometric plots ofpharmacokinetic parameters for the re-combinant, soluble, and chimeric form ofP-selectin glycoprotein ligand-1 (rPSGL-Ig), an antagonist to P-selectin for thetreatment of P-selectin–mediated diseaseslike thrombosis, reperfusion injury, anddeep vein thrombosis. Human rPSGL-Igpharmacokinetic parameters could accu-rately be predicted on the basis of datafrom mouse, rat, monkey, and pig usingallometric power functions [54].
8.3.6Immunogenicity
Because of the antigenic potential ofproteins, formation of antibodies is afrequently observed phenomenon duringchronic therapy with protein drugs, espe-cially if human proteins are used in animalstudies or if animal-derived proteins areapplied in human clinical studies [16].
Most monoclonal antibodies are murinein nature and their systemic adminis-tration can lead to the development ofhuman antimouse immunoglobulin anti-body (HAMA) response, which is in mostcases directed against the constant regionsof the immunoglobulin. Genetically engi-neered mouse–human chimeric antibod-ies try to minimize this immunogenicityin man by joining variable domains ofthe mouse to the constant regions ofhuman immunoglobulins [55]. The anti-EGFR (epidermal growth factor receptor)IgG monoclonal antibody cetuximab is anexample of a murine–human chimericantibody currently under clinical investi-gation for various cancer indications [56].
Extravascular injection is known to stim-ulate antibody formation more than in-travenous application, most likely dueto the increased immunogenicity of pro-tein aggregates and precipitates formed
at the injection site [57]. The presenceof antibodies can obliterate the biologi-cal activity of a protein drug. In addition,protein–antibody complexation can alsomodify the distribution, metabolism, andexcretion, that is, the pharmacokinetic pro-file, of the protein drug. Elimination caneither be increased or decreased. Fasterelimination of the complex occurs if thereticuloendothelial system is stimulated.Elimination is slowed down if the anti-body–drug complex forms a depot for theprotein drug. This effect would prolong thedrug’s therapeutic activity that might bebeneficial if the complex formation doesnot decrease therapeutic activity [19, 28,57]. Furthermore, antibody binding mayalso interfere with bioanalytical methodslike immunoassays.
8.4Pharmacokinetics of Oligonucleotides
Antisense oligonucleotides hold greatpromise as novel therapeutic agents de-signed to specifically and selectively in-hibit the production of disease-relatedproducts, with fomivirsen being the firstapproved antisense oligonucleotide drugproduct [58]. So far, a significant body ofpreclinical and human pharmacokineticdata is only available for phosphothioateoligonucleotides (PONs).
Oral bioavailability is generally very low,ranging from 1 to 3%. Ongoing studies,however, indicate that oral bioavailabilitycan be increased by the appropriate re-lease of drug and permeability-enhancingexcipients [59]. PONs have also been ad-ministered via subcutaneous, intradermal,and pulmonary application routes.
After intravenous administration, PONsfollow generally two-compartment char-acteristics and are rapidly cleared from
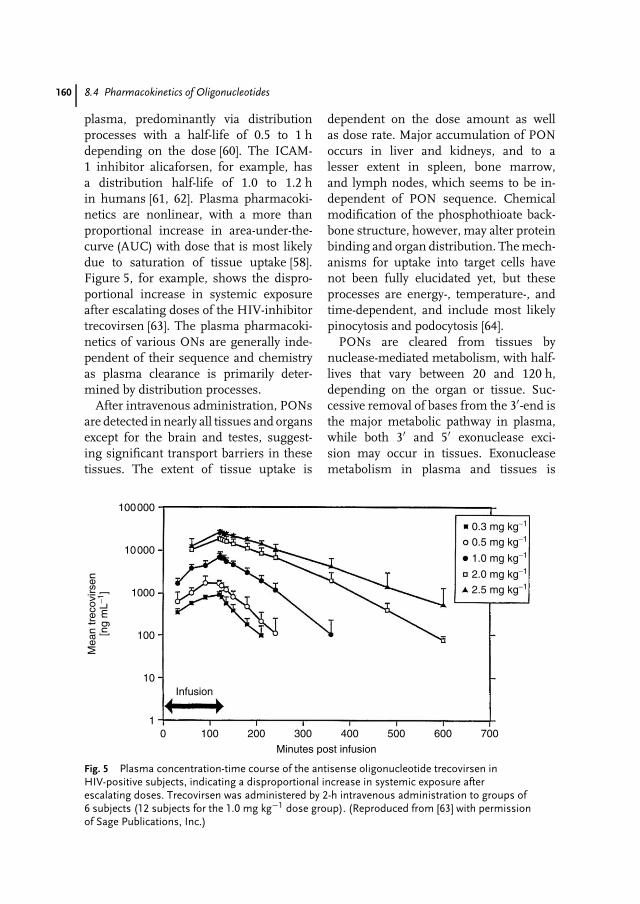
160 8.4 Pharmacokinetics of Oligonucleotides
plasma, predominantly via distributionprocesses with a half-life of 0.5 to 1 hdepending on the dose [60]. The ICAM-1 inhibitor alicaforsen, for example, hasa distribution half-life of 1.0 to 1.2 hin humans [61, 62]. Plasma pharmacoki-netics are nonlinear, with a more thanproportional increase in area-under-the-curve (AUC) with dose that is most likelydue to saturation of tissue uptake [58].Figure 5, for example, shows the dispro-portional increase in systemic exposureafter escalating doses of the HIV-inhibitortrecovirsen [63]. The plasma pharmacoki-netics of various ONs are generally inde-pendent of their sequence and chemistryas plasma clearance is primarily deter-mined by distribution processes.
After intravenous administration, PONsare detected in nearly all tissues and organsexcept for the brain and testes, suggest-ing significant transport barriers in thesetissues. The extent of tissue uptake is
dependent on the dose amount as wellas dose rate. Major accumulation of PONoccurs in liver and kidneys, and to alesser extent in spleen, bone marrow,and lymph nodes, which seems to be in-dependent of PON sequence. Chemicalmodification of the phosphothioate back-bone structure, however, may alter proteinbinding and organ distribution. The mech-anisms for uptake into target cells havenot been fully elucidated yet, but theseprocesses are energy-, temperature-, andtime-dependent, and include most likelypinocytosis and podocytosis [64].
PONs are cleared from tissues bynuclease-mediated metabolism, with half-lives that vary between 20 and 120 h,depending on the organ or tissue. Suc-cessive removal of bases from the 3′-end isthe major metabolic pathway in plasma,while both 3′ and 5′ exonuclease exci-sion may occur in tissues. Exonucleasemetabolism in plasma and tissues is
100000
10000
1000
100
10
10 100 200 300 400
Minutes post infusion
Mea
n tr
ecov
irsen
[ng
mL−1
]
Infusion
0.3 mg kg−1
0.5 mg kg−1
1.0 mg kg−1
2.0 mg kg−1
2.5 mg kg−1
500 600 700
Fig. 5 Plasma concentration-time course of the antisense oligonucleotide trecovirsen inHIV-positive subjects, indicating a disproportional increase in systemic exposure afterescalating doses. Trecovirsen was administered by 2-h intravenous administration to groups of6 subjects (12 subjects for the 1.0 mg kg−1 dose group). (Reproduced from [63] with permissionof Sage Publications, Inc.)

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 161
rapid, with 30 to 40% of PON havingat least one nucleotide removed after5 min in plasma. Endonuclease-mediateddegradation of PONs is generally notobserved [64].
PONs are highly bound to plasma pro-teins that protect them from renal fil-tration [65]. Plasma protein binding ofISIS2503, for example, ranged from 95to 97% in rats and monkeys, but was sat-urable at high concentrations [66]. Urinaryexcretion is a major route of excretion forPONs, regardless of sequence or chemicalstructure, with the majority being shorter-length metabolites rather than unchangedparent drug [60]. Urinary excretion is non-linear, with a greater fraction excreted athigher doses. Potential mechanisms in-clude saturation of plasma protein bindingas well as tubular reuptake mechanisms.Only a minor fraction of the dose is ex-creted into feces although enterohepaticrecirculation has been suggested [58].
ISIS 104838 is a tumor necrosis factor-α (TNF-α) inhibiting second-generationON containing five 2′-O-(2-methoxyethyl)modified (2′-MOE) nucleosides at the3′- and 5′-terminus, respectively. Thepharmacokinetic pattern for this second-generation PON was similar to first-generation PONs, except for a less pro-nounced nonlinearity in systemic expo-sure and a substantially prolonged termi-nal half-life of 27 ± 3.8 days, most likelydue to the complete blockade of exonu-clease digestion by the MOE modifica-tion [67].
8.5Pharmacokinetics of DNA
In comparison to peptides, proteins, andoligonucleotides, much less is known
about the pharmacokinetics of recombi-nant plasmid DNA used like a ‘‘drug’’in the novel treatment approach of genetherapy.
The in vivo disposition of plasmidDNA and its complexes depends largelyon its physicochemical characteristics, astrong negative charge and high molecu-lar weight [68]. After intravenous admin-istration in rats, pDNA is detected inall major organs including lungs, liver,kidney, and spleen. Low-level detectionin the brain is most likely an artifactfrom residual blood, given that pDNAis unlikely to cross the blood-brain bar-rier [69].
After intravenous administration inmice, pDNA is rapidly eliminated fromthe plasma due to extensive uptake intothe liver as well as rapid degradationby nucleases, with hepatic uptake clear-ance approaching liver plasma flow. pDNAis preferentially taken up by liver non-parenchymal cells, such as Kupffer andendothelial cells via receptor-mediated pro-cesses [70].
Analysis of the functional forms ofpDNA in rats revealed that supercoiledpDNA rapidly disappears from plasmawith a half-life of 0.15 min. Approxi-mately 60% of supercoiled pDNA is de-graded to open circular pDNA, whichis subsequently nearly completely con-verted to linear pDNA. Conversion ofopen circular to linear pDNA followedMichaelis–Menten kinetics, while linearpDNA was removed with a half-life of2.1 min. The slower elimination of opencircular and linear pDNA compared tosupercoiled pDNA was suggested to be re-lated to a stronger interaction with plasmamacromolecules that might offer someprotection from plasma nuclease degra-dation [69].

162 8.6 Exposure/Response Correlations for Biotech Drugs
8.6Exposure/Response Correlations forBiotech Drugs
Since biotech drugs are usually highlypotent compounds with steep dose-effectcurve, a careful characterization ofthe dose-concentration-effect relationshipshould receive particular emphasis duringthe preclinical and clinical drug devel-opment process. Integrated pharmacoki-netic/pharmacodynamic (PK/PD) model-ing approaches have widely been appliedfor the characterization of biotech drugs.PK/PD modeling does not only allow for acontinuous description of the time courseof effect as a function of the dosing regimeand comprehensive summary of availabledata but also enables testing of competinghypotheses regarding processes altered bythe drug, allows to make predictions ofdrug effects under new conditions, andfacilitates the estimation of inaccessiblesystem variables [6, 71].
The application of PK/PD modeling isbeneficial in all phases of preclinical andclinical drug development, with a focus ondosage optimization and identification ofcovariates that are causal for intra- and in-terindividual differences in drug responseand/or toxicity [72]. It has recently furtherbeen endorsed by the publication of theExposure-Response Guidance documentby the U.S. Food and Drug Administra-tion [8]. Mechanism-based PK/PD model-ing appreciating the physiological eventsinvolved in the elaboration of the observedeffect has been promoted as superior mod-eling approach as compared to empiricalmodeling, especially because it does notonly describe observations but also offerssome insight into the underlying biologicalprocesses involved and thus provides flex-ibility in extrapolating the model to other
clinical situations [7, 73]. Since the molec-ular mechanism of action of biotech drugsis generally well understood, it is oftenstraightforward to transform this avail-able knowledge into a mechanism-basedPK/PD modeling approach that appropri-ately characterizes the real physiologicalprocess leading to the drug’s therapeu-tic effect.
In the following, the application of thethree most common PK/PD modelingclasses, direct link models, indirect linkmodels, and indirect response models,will be discussed in more detail. In ad-dition, extensions of these concepts andmore complex approaches will be intro-duced in illustrative examples. However,it should be mentioned that PK/PD mod-els for biotech drugs are not only limitedto continuous responses as shown in thefollowing but are also used for binary orgraded responses. Lee et al., for example,used a logistic PK/PD modeling approachto link cumulative AUC of the anti-TNF-αprotein etanercept with the American Col-lege of Rheumatology response criterionof 20% improvement (ARC20) in patientswith rheumatoid arthritis [74].
8.6.1Direct Link PK/PD Models
While drug concentrations are usually an-alytically quantified in plasma, serum, orblood, the magnitude of the observed re-sponse is determined by the concentrationof the drug at its effect site, the site ofaction in the target tissue [6]. The relation-ship between the drug concentration inplasma and at the effect site may eitherbe constant or undergo time-dependentchanges. If equilibrium between both con-centrations is rapidly achieved or the siteof action is within plasma, serum or blood,there is practically a constant relationship

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 163
between both concentrations with no tem-poral delay between plasma and effect site.In this case, measured concentrations candirectly serve as input for a pharmacody-namic model. The most frequently useddirect link pharmacodynamic model is thesigmoid Emax-Model:
E = Emax · Cn
ECn50 + Cn
with Emax as maximum achievable effect,C as drug concentration at the effectsite, and EC50 the concentration of thedrug that produces half of the maximumeffect. The Hill-coefficient n is a shapefactor that allows for an improved fitof the relationship to the observed data.Thus, a direct link model directly connectsmeasured concentration to the observedeffect without any temporal delay [5, 6].
Racine-Poon et al. provided an exam-ple for a direct link model by relatingthe serum concentration of the antihu-man immunoglobulin E (IgE) antibodyCGP 51901 for the treatment of sea-sonal allergic rhinitis to the reductionof free IgE via an inhibitory Emax-model [75]. Radwanski et al. used a similarapproach to assess the effect of recom-binant interleukin-10 on the ex vivo re-lease of the proinflammatory cytokinesTNF-α and interleukin-1β (IL-1β) in LPS-stimulated leukocytes [76].
8.6.2Indirect Link PK/PD Models
The concentration-effect relationship ofmany biotech drugs, however, cannot bedescribed by direct link PK/PD models,but is characterized by a temporal dissoci-ation between the time courses of plasmaconcentration and effect. In this case,concentration maxima would occur be-fore effect maxima, effect intensity would
increase despite decreasing plasma con-centrations and would persist beyond thetime drug concentrations in plasma areno longer determinable. The relationshipbetween measured concentration and ob-served effect follows a counterclockwisehysteresis loop. This phenomenon can ei-ther be caused by an indirect responsemechanism (see Sect. 8.6.3) or by a distri-butional delay between the concentrationsin plasma and at the effect site. Thelatter can conceptually be described byan effect-compartment model, which at-taches a hypothetical effect-compartmentto a pharmacokinetic compartment model.The effect compartment does not accountfor mass balance and only defines thechanges in concentration at the effectsite via the time course of the effect it-self [5, 77].
An effect-compartment approach was,for example, applied by Gibbons et al.to quantify the reduction in mean arte-rial blood pressure by the antiadrener-gic peptoid CHIR 2279 [78]. The sameconcept was used by Pihoker et al. tocharacterize the relationship between theserum concentration of the somatotropin-releasing peptide GHRP-2 and soma-totropin (Fig. 6) [79].
8.6.3Indirect Response PK/PD Models
The effect of most biotech drugs, however,is not mediated via a direct interactionbetween drug concentration and responsesystems, but frequently involves severaltransduction processes that include at theirrate-limiting step the stimulation or inhibi-tion of a physiologic process, for example,the synthesis or degradation of a molec-ular response mediator like a hormoneor cytokine. In these cases, mechanism-based indirect response models should be
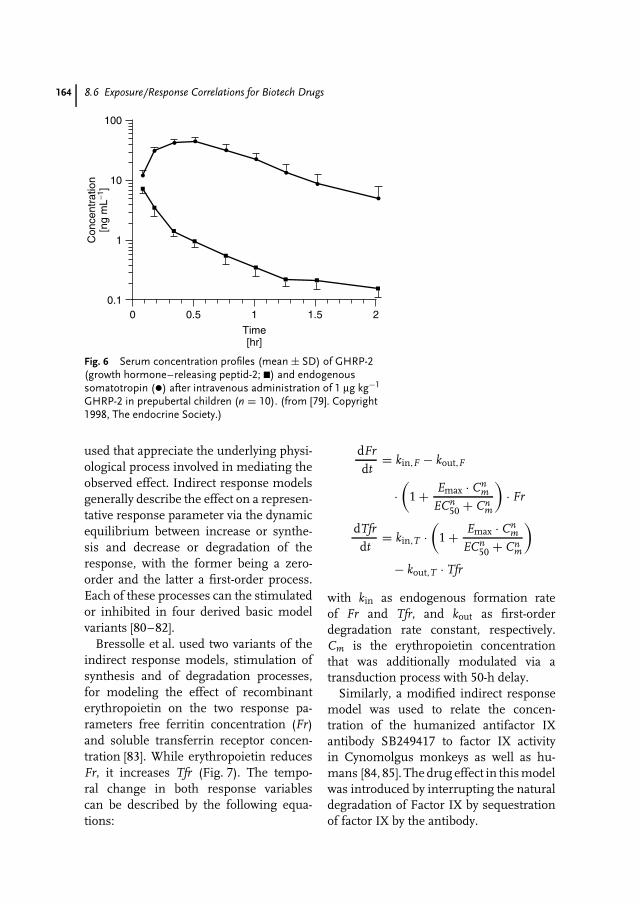
164 8.6 Exposure/Response Correlations for Biotech Drugs
0.10 0.5 1
Time[hr]
1.5 2
1
10
100
Con
cent
ratio
n[n
g m
L−1]
Fig. 6 Serum concentration profiles (mean ± SD) of GHRP-2(growth hormone–releasing peptid-2; �) and endogenoussomatotropin (•) after intravenous administration of 1 µg kg−1
GHRP-2 in prepubertal children (n = 10). (from [79]. Copyright1998, The endocrine Society.)
used that appreciate the underlying physi-ological process involved in mediating theobserved effect. Indirect response modelsgenerally describe the effect on a represen-tative response parameter via the dynamicequilibrium between increase or synthe-sis and decrease or degradation of theresponse, with the former being a zero-order and the latter a first-order process.Each of these processes can the stimulatedor inhibited in four derived basic modelvariants [80–82].
Bressolle et al. used two variants of theindirect response models, stimulation ofsynthesis and of degradation processes,for modeling the effect of recombinanterythropoietin on the two response pa-rameters free ferritin concentration (Fr)and soluble transferrin receptor concen-tration [83]. While erythropoietin reducesFr, it increases Tfr (Fig. 7). The tempo-ral change in both response variablescan be described by the following equa-tions:
dFr
dt= kin,F − kout,F
·(
1 + Emax · Cnm
ECn50 + Cn
m
)· Fr
dTfr
dt= kin,T ·
(1 + Emax · Cn
m
ECn50 + Cn
m
)
− kout,T · Tfr
with kin as endogenous formation rateof Fr and Tfr, and kout as first-orderdegradation rate constant, respectively.Cm is the erythropoietin concentrationthat was additionally modulated via atransduction process with 50-h delay.
Similarly, a modified indirect responsemodel was used to relate the concen-tration of the humanized antifactor IXantibody SB249417 to factor IX activityin Cynomolgus monkeys as well as hu-mans [84, 85]. The drug effect in this modelwas introduced by interrupting the naturaldegradation of Factor IX by sequestrationof factor IX by the antibody.
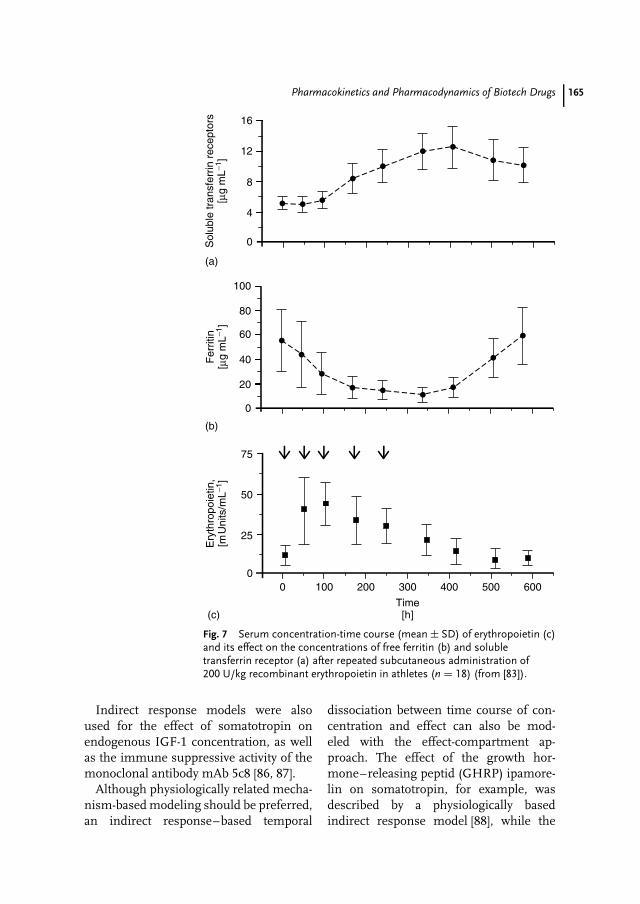
Pharmacokinetics and Pharmacodynamics of Biotech Drugs 165
(c)
(a)
(b)
Sol
uble
tran
sfer
rin r
ecep
tors
[µg
mL−1
]
16
12
4
8
0
Fer
ritin
[µg
mL−1
]
100
80
60
40
20
0
Ery
thro
poie
tin,
[mU
nits
/mL−1
]
75
50
25
00
Time[h]
300200100 400 500 600
Fig. 7 Serum concentration-time course (mean ± SD) of erythropoietin (c)and its effect on the concentrations of free ferritin (b) and solubletransferrin receptor (a) after repeated subcutaneous administration of200 U/kg recombinant erythropoietin in athletes (n = 18) (from [83]).
Indirect response models were alsoused for the effect of somatotropin onendogenous IGF-1 concentration, as wellas the immune suppressive activity of themonoclonal antibody mAb 5c8 [86, 87].
Although physiologically related mecha-nism-based modeling should be preferred,an indirect response–based temporal
dissociation between time course of con-centration and effect can also be mod-eled with the effect-compartment ap-proach. The effect of the growth hor-mone–releasing peptid (GHRP) ipamore-lin on somatotropin, for example, wasdescribed by a physiologically basedindirect response model [88], while the

166 8.6 Exposure/Response Correlations for Biotech Drugs
already mentioned GHRP-2 effect onsomatotropin was characterized with aneffect-compartment approach [79]. Simi-larly, effect compartment as well as indirectresponse models were applied for char-acterizing the effect of insulin on bloodglucose levels. A recent comparative study,however, suggests that a mechanism-based indirect response model is a moreappropriate approach for modeling thePK/PD of insulin [89].
8.6.4Precursor Pool PK/PD Models
An extension of indirect response mod-els are precursor pool-dependent indirectresponse models that include the libera-tion of an endogenous compound froma storage pool. These models possessthe unique ability to characterize bothtolerance and rebound phenomena [90].Such a model was, for example, usedto describe the effect of interferon-β1aon neopterin, an endogenous marker forcell-mediated immunity, in humans andmonkeys (Fig. 8) [91, 92]. The primaryelimination mechanism of interferon-β 1awas modeled as receptor-mediated endo-cytosis, and the pharmacodynamic modelwas driven by the amount of internalizeddrug-receptor complex DR∗:
dP
dt= k0 ·
(1 + Smax · DR∗
SC50 + DR∗
)− kp · P
where P is the concentration of neopterinprecursor (neopterin triphosphate), k0 isthe zero-order production rate of precursorP, and kp is the first-order rate constantcharacterizing the conversion of precursorP to neopterin. The amount of internalizeddrug-receptor complex DR∗ stimulatesprecursor production via a stimulationfunction governed by the maximum effectparameter Smax and a sensitivity parameterSC50, the concentration that results in halfof Smax. The rate of change in neopterinconcentration NP is then defined by thefollowing expression:
dNP
dt= kp · P − kout · NP
with kout as the first-order elimination rateconstant of NP in the body.
8.6.5Complex PK/PD Models
Since the effect of most biotech drugs ismediated via complex regulatory physio-logic processes including feedback mecha-nisms and/or tolerance phenomena, somePK/PD models that have been describedfor biotech drugs are much more sophis-ticated than the four classes of modelspreviously discussed. One example of sucha complex modeling approach has beendeveloped by Nagaraja et al. for the ther-apeutic effects of the LH-RH antagonistcetrorelix [93–95].
Pk0
−Smax, SC50
NPkoutkP
DR*
Fig. 8 Schematic representation of anindirect response model with precursorpool used to describe the effect ofinterferon-β 1a (represented by itsinternalized drug-receptor complex DR∗)on endogenous neopterin concentrations(NP) via stimulation of the synthesis ofits precursor neopterin triphosphate (P).See text for discussion of the details(from [91, 92]).
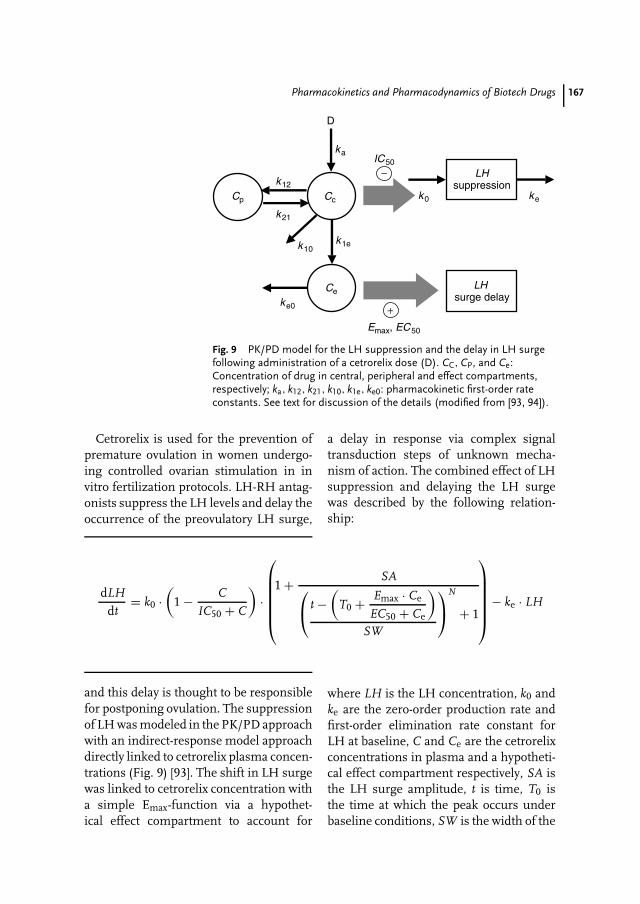
Pharmacokinetics and Pharmacodynamics of Biotech Drugs 167
−IC50
Ce
k1e
Cp
k12
k21
Cc
D
ka
ke0
k10
LH surge delay
+Emax, EC50
LHsuppression
k0 ke
Fig. 9 PK/PD model for the LH suppression and the delay in LH surgefollowing administration of a cetrorelix dose (D). CC, CP, and Ce:Concentration of drug in central, peripheral and effect compartments,respectively; ka, k12, k21, k10, k1e, ke0: pharmacokinetic first-order rateconstants. See text for discussion of the details (modified from [93, 94]).
Cetrorelix is used for the prevention ofpremature ovulation in women undergo-ing controlled ovarian stimulation in invitro fertilization protocols. LH-RH antag-onists suppress the LH levels and delay theoccurrence of the preovulatory LH surge,
dLH
dt= k0 ·
(1 − C
IC50 + C
)·
1 + SA t −
(T0 + Emax · Ce
EC50 + Ce
)
SW
N
+ 1
− ke · LH
and this delay is thought to be responsiblefor postponing ovulation. The suppressionof LH was modeled in the PK/PD approachwith an indirect-response model approachdirectly linked to cetrorelix plasma concen-trations (Fig. 9) [93]. The shift in LH surgewas linked to cetrorelix concentration witha simple Emax-function via a hypothet-ical effect compartment to account for
a delay in response via complex signaltransduction steps of unknown mecha-nism of action. The combined effect of LHsuppression and delaying the LH surgewas described by the following relation-ship:
where LH is the LH concentration, k0 andke are the zero-order production rate andfirst-order elimination rate constant forLH at baseline, C and Ce are the cetrorelixconcentrations in plasma and a hypotheti-cal effect compartment respectively, SA isthe LH surge amplitude, t is time, T0 isthe time at which the peak occurs underbaseline conditions, SW is the width of the
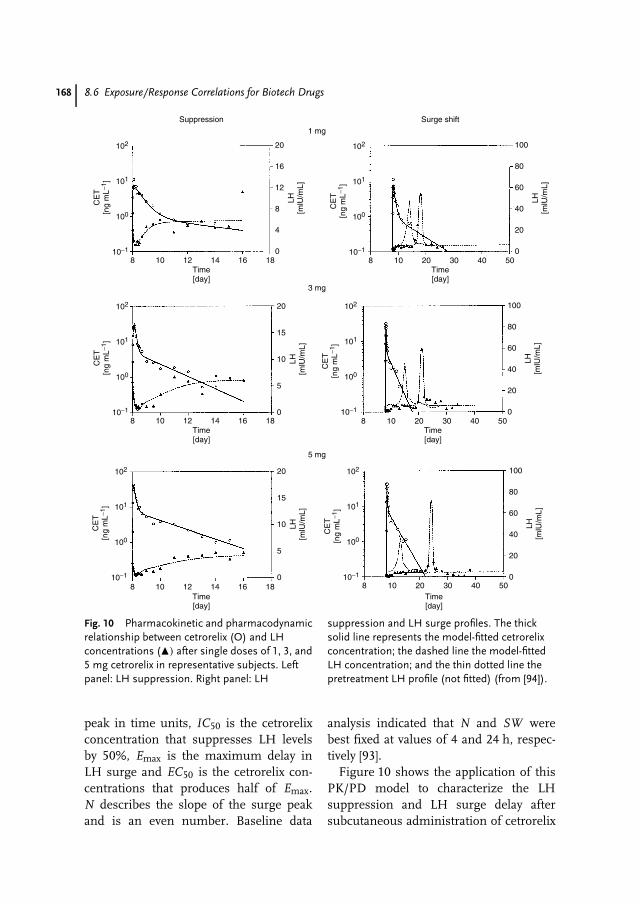
168 8.6 Exposure/Response Correlations for Biotech Drugs
102
101
100
10−1
CE
T[n
g m
L−1]
LH[m
lU/m
L]
LH[m
lU/m
L]
20
16
12
8
4
08 10 12
Time[day]
Time[day]
1 mgSuppression Surge shift
3 mg
5 mg
Time[day]
Time[day]
Time[day]
Time[day]
14 16 18
102
101
100
10−1
CE
T[n
g m
L−1]
100
80
60
40
20
08 10 20 30 40 50
102
101
100
10−1
CE
T[n
g m
L−1]
LH[m
lU/m
L]
LH[m
lU/m
L]
20
15
10
5
08 10 12 14 16 18
102
101
100
10−1
CE
T[n
g m
L−1]
100
80
60
40
20
08 10 20 30 40 50
102
101
100
10−1
CE
T[n
g m
L−1]
LH[m
lU/m
L]
LH[m
lU/m
L]
20
15
10
5
08 10 12 14 16 18
102
101
100
10−1
CE
T[n
g m
L−1]
100
80
60
40
20
08 10 20 30 40 50
Fig. 10 Pharmacokinetic and pharmacodynamicrelationship between cetrorelix (O) and LHconcentrations (�) after single doses of 1, 3, and5 mg cetrorelix in representative subjects. Leftpanel: LH suppression. Right panel: LH
suppression and LH surge profiles. The thicksolid line represents the model-fitted cetrorelixconcentration; the dashed line the model-fittedLH concentration; and the thin dotted line thepretreatment LH profile (not fitted) (from [94]).
peak in time units, IC50 is the cetrorelixconcentration that suppresses LH levelsby 50%, Emax is the maximum delay inLH surge and EC50 is the cetrorelix con-centrations that produces half of Emax.N describes the slope of the surge peakand is an even number. Baseline data
analysis indicated that N and SW werebest fixed at values of 4 and 24 h, respec-tively [93].
Figure 10 shows the application of thisPK/PD model to characterize the LHsuppression and LH surge delay aftersubcutaneous administration of cetrorelix

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 169
to groups of 12 women at different doselevels. The analysis revealed a markeddose-response relationship for the LHsurge and thus predictability of drugresponse to cetrorelix [94].
An even more complex mechanism-based modeling approach including tol-erance phenomena was used for the effectof antide, an LH-RH antagonist, on theendogenous regulatory mechanisms andplasma concentrations of LH and testos-terone [96].
8.7Summary
In general, biotech drugs underlie thesame pharmacokinetic and pharmacody-namic principles as traditional, small-molecule drugs. On the basis of theirsimilarity to endogenous compounds ornutrients, however, numerous caveats andpitfalls related to bioanalytics and phar-macokinetics have to be considered andaddressed during the development pro-cess and may require additional resources.Furthermore, pharmacodynamics is fre-quently complicated owing to close inter-action with endogenous substances andspecific feedback mechanisms.
Biotech drugs, including peptides, pro-teins and antibodies, oligonucleotides, andDNA, are projected to cover a substan-tial market share in the health caresystems of the future. It will be cru-cial for their widespread application inpharmacotherapy, however, that their re-spective drug development programs aresuccessfully completed in a rapid, cost-efficient, and goal-oriented manner. Amore widespread application of pharma-cokinetic and pharmacodynamic conceptsincluding exposure-response correlations
has repeatedly been promoted by indus-try, academia, and regulatory authoritiesfor all preclinical and clinical phasesof drug development and is believed toresult in a scientifically driven, evidence-based, more focused and accelerated drugproduct development process [72]. Thus,PK/PD concepts are likely to continue andexpand their role as a decisive factor in thesuccessful development of biotechnologi-cally derived drug products in the future.
References
1. S. Arlington, S. Barnett, S. Hughes et al.,Pharma 2010: The Threshold to Innovation,IBM Business Consulting Services, Somers,New York, 2002.
2. M. Hosseini, K. Nagels, Scr. Mag. 2001,December, 22–25.
3. T. Nagle, C. Berg, R. Nassr et al., Nat. Rev.Drug Discov. 2003, 2(1), 75–79.
4. G. Levy, J. Allergy Clin. Immunol. 1986, 78(4Pt 2), 754–761.
5. N. H. Holford, L. B. Sheiner, Pharmacol.Ther. 1982, 16(2), 143–166.
6. B. Meibohm, H. Derendorf, Int. J. Clin.Pharmacol. Ther. 1997, 35(10), 401–413.
7. H. Derendorf, B. Meibohm, Pharm. Res.1999, 16(2), 176–185.
8. CDER/FDA, Exposure Response Relationships:Guidance for Industry, US Department ofHealth and Human Services, Food and DrugAdministration, Center for Drug Evaluationand Research, Rockville, 2003.
9. International Conference on Harmoniza-tion, ICH E4 – Dose-Response Information toSupport Drug Registration, European Agencyfor the Evaluation of Medicinal Products,London, 1994.
10. CDER/FDA, Bioanalytical Method Validation:Guidance for Industry, US Department ofHealth and Human Services, Food and DrugAdministration, Center for Drug Evaluationand Research, Rockville, 2001.
11. K. J. Miller, R. R. Bowsher, A. Celniker et al.,Pharm. Res. 2001, 18(9), 1373–1383.
12. V. Shah, K. Midha, J. Finlay et al., Pharm.Res. 2000, 17(12), 1551–1557.

170 8.7 Summary
13. W. Colburn, Peptide, peptoid, and pro-tein pharmacokinetics/pharmacodynamicsin Peptides, Peptoids, and Proteins (Eds.:P. Garzone, W. Colburn, M. Mokotoff), Har-vey Whitney Books, Cincinnati, OH, 1991,pp. 94–115.
14. K. Nakao, A. Sugawara, N. Morii et al., Eur.J. Clin. Pharmacol. 1986, 31(1), 101–103.
15. F. S. Labella, J. D. Geiger, G. B. Glavin,Peptides 1985, 6(4), 645–660.
16. R. J. Wills, B. L. Ferraiolo, Clin. Pharma-cokinet. 1992, 23(6), 406–414.
17. A. Chen, D. Baker, B. Ferraiolo, Points toconsider in correlating bioassays andimmunoassays in the quantitation ofpeptides and proteins in Peptides, Peptoids,and Proteins (Eds.: P. Garzone, W. Colburn,M. Mokotoff), Harvey Whitney Books, Cin-cinnati, OH, 1991, pp. 54–71.
18. G. Rodes, V. Boppana, Novel derivatizationapproaches in the analysis of peptidesin biological fluids by high performanceliquid chromatography in Peptides, Peptoids,and Proteins (Eds.: P. Garzone, W. Colburn,M. Mokotoff), Harvey Whitney Books,Cincinnati, OH, 1991, pp. 73–79.
19. C. Mcmartin, Adv. Drug Res. 1992, 22,39–106.
20. S. Muller, G. Hochhaus, Pharm. Res. 1995,12(8), 1165–1170.
21. S. Muller, A. Hutson, V. Arya et al., J. Pharm.Sci. 1999, 88(9), 938–944.
22. H. C. Meissner, J. R. Groothuis, W. J. Rod-riguez et al., Antimicrob. Agents Chemother.1999, 43(5), 1183–1188.
23. A. Fasano, J. Pharm. Sci. 1998, 87(11),1351–1356.
24. V. J. Wacher, J. A. Silverman, Y. Zhanget al., J. Pharm. Sci. 1998, 87(11), 1322–1330.
25. B. Meibohm, I. Beierle, H. Derendorf, Clin.Pharmacokinet. 2002, 41(5), 329–342.
26. L. B. Lan, J. T. Dalton, E. G. Schuetz, Mol.Pharmacol. 2000, 58(4), 863–869.
27. V. Lee, Problems and solutions in peptideand protein drug delivery in Peptides,Peptoids, and Proteins (Eds.: P. Garzone,W. Colburn, M. Mokotoff), Harvey WhitneyBooks, Cincinnati, OH, 1991, pp. 81–92.
28. R. Braeckman, Pharmacokinetics and phar-macodynamics of peptide and proteindrugs in Pharmaceutical Biotechnology(Eds.: D. Crommelin, R. Sindelar), HarwoodAcademic Publishers, Amsterdam, 1997,pp. 101–122.
29. H. Berger, N. Heinrich, H. Schafer et al., LifeSci. 1988, 42(9), 985–991.
30. R. M. Reilly, J. Sandhu, T. M. Alvarez-Diezet al., Clin. Pharmacokinet. 1995, 28(2),126–142.
31. P. Periti, T. Mazzei, E. Mini, Clin. Pharma-cokinet. 2002, 41(7), 485–504.
32. D. R. Mould, C. B. Davis, E. A. Minthornet al., Clin. Pharmacol. Ther. 1999, 66(3),246–257.
33. S. Kageyama, H. Yamamoto, H. Nakazawaet al., Arterioscler. Thromb. Vasc. Biol. 2002,22(1): 187–192.
34. M. Rowland, T. Tozer, Clinical Pharmacoki-netics: Concepts and Applications, 3rd ed.,Williams & Wilkins, Media, PA, 1993.
35. P. Chanson, J. Timsit, A. G. Harris, Clin.Pharmacokinet. 1993, 25(5), 375–391.
36. P. Tanswell, N. Modi, D. Combs et al., Clin.Pharmacokinet. 2002, 41(15), 1229–1245.
37. K. B. Alton, T. Kosoglou, S. Baker et al., Clin.Ther. 1998, 20(2), 307–323.
38. A. C. Tan, F. G. Russel, T. Thien et al., Clin.Pharmacokinet. 1993, 24(1), 28–45.
39. T. Kuwabara, T. Uchimura, H. Kobayashiet al., Am. J. Physiol. 1995, 269(1 Pt 1), E1–E9.
40. D. R. Clemmons, M. L. Dehoff, W. H. Busbyet al., Endocrinology 1992, 131(2): 890–895.
41. Y. Taki, T. Sakane, T. Nadai et al., J. Pharm.Pharmacol. 1998, 50(9), 1013–1018.
42. R. Wills, A kinetic/dynamic perspectiveof a peptide and protein: GRF andrHuIFN-alpha-2a in Peptides, Peptoids, andProteins (Eds.: P. Garzone, W. Colburn,M. Mokotoff), Harvey Whitney Books,Cincinnati, OH, 1991, pp. 117–127.
43. T. Maack, C. Park, M. Camargo, Renalfiltration, transport and metabolism ofproteins in The Kidney (Eds.: D. Seldin,G. Giebisch), Raven Press, New York, 1985,pp. 1773–1803.
44. S. Andersen, L. Lambrecht, S. Swan et al., J.Clin. Pharmacol. 1999, 39(10), 1015–1020.
45. A. Bartocci, D. S. Mastrogiannis, G. Miglio-rati et al., Proc. Natl. Acad. Sci. U. S. A. 1987,84(17), 6179–6183.
46. R. J. Bauer, J. A. Gibbons, D. P. Bell et al., J.Pharmacol. Exp. Ther. 1994, 268(1), 152–158.
47. T. Murakami, M. Misaki, S. Masuda et al., J.Pharm. Sci. 1994, 83(10), 1400–1403.
48. S. M. Eppler, D. L. Combs, T. D. Henryet al., Clin. Pharmacol. Ther. 2002, 72(1),20–32.

Pharmacokinetics and Pharmacodynamics of Biotech Drugs 171
49. R. L. Dedrick, J. Pharmacokinet. Biopharm.1973, 1(5), 435–461.
50. H. Boxenbaum, J. Pharmacokinet. Biopharm.1982, 10(2), 201–227.
51. I. Mahmood, J. D. Balian, Clin. Pharma-cokinet. 1999, 36(1), 1–11.
52. I. Mahmood, Am. J. Ther. 2002, 9(1), 35–42.53. J. Mordenti, S. A. Chen, J. A. Moore et al.,
Pharm. Res. 1991, 8(11), 1351–1359.54. S. P. Khor, K. Mccarthy, M. Dupont et al., J.
Pharmacol. Exp. Ther. 2000, 293(2), 618–624.55. S. K. Batra, M. Jain, U. A. Wittel et al., Curr.
Opin. Biotechnol. 2002, 13(6), 603–608.56. R. S. Herbst, C. J. Langer, Semin. Oncol.
2002, 29(1 Suppl. 4), 27–36.57. P. Working, P. Cossum, Clinical and
preclinical studies with recombinant humanproteins: effect of antibody productionin Peptides, Peptoids, and Proteins (Eds.:P. Garzone, W. Colburn, M. Mokotoff),Harvey Whitney Books, Cincinnati, OH,1991, pp. 158–168.
58. B. H. Dvorchik, Curr. Opin. Mol. Ther. 2000,2(3), 253–257.
59. ISIS Pharmaceuticals, New data on CapsuleForm of ISIS 104838 Demonstrate AntisenseDrugs Can Be Administered Orally, PressRelease, Toronto, 2002.
60. S. Agrawal, J. Temsamani, W. Galbraithet al., Clin. Pharmacokinet. 1995, 28(1),7–16.
61. B. R. Yacyshyn, W. Y. Chey, J. Goff et al., Gut2002, 51(1), 30–36.
62. B. R. Yacyshyn, C. Barish, J. Goff et al.,Aliment Pharmacol. Ther. 2002, 16(10),1761–1770.
63. D. Sereni, R. Tubiana, C. Lascoux et al., J.Clin. Pharmacol. 1999, 39(1): 47–54.
64. A. A. Levin, Biochim. Biophys. Acta 1999,1489(1), 69–84.
65. S. T. Crooke, M. J. Graham, J. E. Zucker-man et al., J. Pharmacol. Exp. Ther. 1996,277(2), 923–937.
66. R. Z. Yu, R. S. Geary, J. M. Leeds et al., J.Pharm. Sci. 2001, 90(2), 182–193.
67. K. L. Sewell, R. S. Geary, B. F. Baker et al.,J. Pharmacol. Exp. Ther. 2002, 303(3),1334–1343.
68. M. Nishikawa, S. Takemura, Y. Takakuraet al., J. Pharmacol. Exp. Ther. 1998, 287(1),408–415.
69. B. E. Houk, R. Martin, G. Hochhaus et al.,Pharm. Res. 2001, 18(1), 67–74.
70. Y. Takakura, M. Nishikawa, F. Yamashitaet al., Eur. J. Pharm. Sci. 2001, 13(1), 71–76.
71. D. E. Mager, E. Wyska, W. J. Jusko, DrugMetab. Dispos. 2003, 31(5), 510–518.
72. B. Meibohm, H. Derendorf, J. Pharm. Sci.2002, 91(1), 18–31.
73. G. Levy, Clin. Pharmacol. Ther. 1994, 56(4),356–358.
74. H. Lee, H. C. Kimko, M. Rogge et al., Clin.Pharmacol. Ther. 2003, 73(4), 348–365.
75. A. Racine-Poon, L. Botta, T. W. Chang et al.,Clin. Pharmacol. Ther. 1997, 62(6),675–690.
76. E. Radwanski, A. Chakraborty, S. Van Wartet al., Pharm. Res. 1998, 15(12), 1895–1901.
77. L. B. Sheiner, D. R. Stanski, S. Vozeh et al.,Clin. Pharmacol. Ther. 1979, 25(3),358–371.
78. J. A. Gibbons, A. A. Hancock, C. R. Vittet al., J. Pharmacol. Exp. Ther. 1996, 277(2),885–899.
79. C. Pihoker, G. L. Kearns, D. French et al.,J. Clin. Endocrinol. Metab. 1998, 83(4),1168–1172.
80. N. L. Dayneka, V. Garg, W. J. Jusko, J.Pharmocokinet. Biopharm. 1993, 21(4),457–478.
81. A. Sharma, W. Jusko, Br. J. Clin. Pharmacol.1998, 45, 229–239.
82. Y. N. Sun, W. J. Jusko, J. Pharm. Sci. 1999,88(10), 987–990.
83. F. Bressolle, M. Audran, R. Gareau et al.,J. Pharmacokinet. Biopharm. 1997, 25(3),263–275.
84. L. J. Benincosa, F. S. Chow, L. P. Tobia et al.,J. Pharmacol. Exp. Ther. 2000, 292(2),810–816.
85. F. S. Chow, L. J. Benincosa, S. B. Shethet al., Clin. Pharmacol. Ther. 2002, 71(4),235–245.
86. J. V. Gobburu, C. Tenhoor, M. C. Roggeet al., J. Pharmacol. Exp. Ther. 1998, 286(2):925–930.
87. Y. N. Sun, H. J. Lee, R. R. Almon et al.,J. Pharmacol. Exp. Ther. 1999, 289(3),1523–1532.
88. J. V. Gobburu, H. Agerso, W. J. Jusko et al.,Pharm. Res. 1999, 16(9), 1412–1416.
89. S. Lin, Y. W. Chien, J. Pharm. Pharmacol.2002, 54(6), 791–800.
90. A. Sharma, W. F. Ebling, W. J. Jusko, J.Pharm. Sci. 1998, 87(12), 1577–1584.

172 8.7 Summary
91. D. E. Mager, B. Neuteboom, C. Efthymio-poulos et al., J. Pharmacol. Exp. Ther. 2003,26, 26.
92. D. E. Mager, W. J. Jusko, Pharm. Res. 2002,19(10), 1537–1543.
93. N. V. Nagaraja, B. Pechstein, K. Erb et al., J.Clin. Pharmacol. 2003, 43(3), 243–251.
94. N. V. Nagaraja, B. Pechstein, K. Erb et al.,Clin. Pharmacol. Ther. 2000, 68(6), 617–625.
95. B. Pechstein, N. V. Nagaraja, R. Hermannet al., J. Clin. Pharmacol. 2000, 40(3),266–274.
96. K. E. Fattinger, D. Verotta, H. C. Porchetet al., Am. J. Physiol. 1996, 271(4 Pt 1),E775–E787.

173
9Formulation of Biotech Products
Ralph LippSchering AG, Berlin, Germany
Erno PungorBerlex Biosciences, Richmond, CA, USA
9.1Introduction
Today erythropoietin, insulin, and inter-ferons belong to the 10 top drug sub-stances on a global scale. Use of furtherproteins like somatotropin, for example,is rapidly growing and in future therewill be an enhanced access to a grow-ing number of therapeutically relevantproteins. This will most likely lead to a fur-ther increased importance of protein- andpeptide-based drugs. Even today, drugsand drug candidates from the latter classesare being produced with high efficiencyby novel biotechnological methods, andadditional benefit is expected by the useof methods from the area of proteomicsin the near future [1]. Novel transgenicapproaches of protein and peptide pro-duction show significant advantages withrespect to lower costs of goods for largerpeptides in comparison to traditional re-combinant methods [2]. However, in theclass of small peptides that are based on
less than 20 amino acids, the classicalchemical synthesis still serves as a versatilemethod of cost-efficient drug production.In order to fully exploit the therapeuticpotential of proteins, highly specific for-mulations are required that need to meetchallenging targets from the areas of sta-bilization, specific application routes, andin some cases, drug-targeting aspects aswell. These formulation and applicationroute specifics will be highlighted in thesubsequent paragraphs.
9.2General Considerations on the Formulationof Proteins and Peptides
The stability, biological activity, and phar-macological activity of proteins and pep-tides are largely dependent on their intactprimary, secondary, tertiary, and quater-nary structure. Proteins and peptides canbe easily modified by physical or chemicalmeans [3]. Table 1 provides an overview of
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

174 9.2 General Considerations on the Formulation of Proteins and Peptides
Tab. 1 Main degradation pathways of proteins and peptides
Degradation pathways
Physical Chemical
Denaturation (�T, pH) Oxidation (O2, h · ν), e.g. Met, Cys, His, TrpNoncovalent aggregation Deamidation (�T, pH) e.g. Asn, GlnPrecipitation Peptide cleavage (�T) e.g. Asp-XAdsorption Disulfide interchange (pH) Cys
Beta-elimination (pH) e.g. Ser, Thr, Cys, LysDisulfide formation (pH, O2) CysCovalent aggregationCyclization (pH), e.g. Asp, Glu
the major degradation pathways that haveto be considered when dealing with thesemolecules.
The effects of degradations can bevery complex (losses in biological activity,changes in pharmacokinetics, pharmaco-dynamics, toxicity, biodistribution, elim-ination pathways, antigenecity, immuno-genecity, etc.). In reality, the number of po-tential degradation pathways is even largersince some of the proteins are chemicallymodified: glycosylated, phoshorylated, etc.These nonprotein modifications may alsohave significant impact on the drug per-formance [4] (for example, glycosylationoften controls circulating half-life, themechanism of drug elimination and im-munogenicity, phosphorylation in manycases contributes to biological activity andspecificity of action), therefore the formu-lation has to protect these modifications,too. Unfortunately, there are no generalrules to predict the effects of the indi-vidual degradation events; the effects ofsimilar changes are widely variable onthe pharmaceutical performances of dif-ferent protein drugs. From the regulatoryperspective, the degradation products aregenerally considered impurities, they needto be strongly controlled and listed in thespecifications. These specifications have
to reflect the clinically safe levels of theseimpurities. If a particular degradation isextensive, the presence of the degradationproduct on the drug safety may have to bespecifically evaluated (in model systems oreven in clinical studies).
As indicated in Table 1, there are severalcommon triggers for instability of pro-teins and peptides, like presence of oxygen,shifts to extreme low or high pH values,elevated temperature and so on. Althoughthere are some common approaches (e.g.using surfactants to prevent/reduce aggre-gation, or using antioxidants, like ascorbicacid to slow oxidation of the proteins),there is no general strategy for stabi-lization that will serve all drugs in anequivalent manner. In contrast, there is atendency to develop strongly tailored man-ufacturing processes and formulations toserve the stabilization need of the individ-ual compounds. Manufacturers also haveto demonstrate the ability of the manu-facturing processes to yield products ofreproducible quality by process validation.
The variety of protein degradation path-ways and the necessity to demonstratecontrol present a significant analyticalchallenge to the manufacturers of pro-tein drugs. Besides the traditional an-alytical tools of protein chemistry (gel

Formulation of Biotech Products 175
electrophoresis with protein staining orimmunoblots, N-terminal sequencing, UVspectral analysis, etc.), a variety of tech-niques are used. These include differ-ent chromatographic (e.g. ion exchange,reverse phase, isoelectric focusing) sep-arations of the intact proteins or enzy-matically digested proteins (peptide map),separations on capillary systems, massspectroscopic analysis of proteins, andpeptide maps to assess chemical modi-fications. Circular dichroism is used toassess secondary and tertiary structures;light scattering techniques (sometimes incombination with chromatographic sep-aration) and field flow fractionation areprimarily applied to assess aggregations.If the protein is glycosylated, specific car-bohydrate profile analysis and sequencingmethods (liquid and/or gas chromato-graphic) are used to assess the integrityof the carbohydrates.
9.3Application Routes for Proteins andPeptides
Directly from the beginning of the thera-peutic use of proteins and peptides, par-enteral formulations were of utmost im-portance, like sc formulations of insulin,for example. This was mainly triggered byspecial aspects from three different areas:
• Suboptimal physicochemical propertiesof the drugs for absorption throughbiological membranes.
• Instability of the drugs during nonpar-enteral administration.
• Specific therapeutic requirements withrespect to onset or duration of action.
The specific physicochemical character-istics of proteins, like high molecular mass
(often far beyond Mrel of 500), in manycases, high hydrophilicity, and the carriageof molecular charges largely cause the in-ability of these drugs to easily permeatebiological membranes and thus dimin-ish the bioavailability (BA) of the relatedcompounds. Stability aspects may play asignificant role during administration andabsorption as well. The sensitivity of manyproteins against extreme pH values has al-ready been highlighted (see Table 1), andit is obvious that acidic pH values inthe GI-tract are deleterious to the respec-tive compounds. Furthermore, enzymaticdegradation by proteases plays a signif-icant role after oral administration andsubsequent GI-passage.
In addition to the aforementioned ab-sorption hurdles, the sometimes highlyspecific therapeutic requirements ask for,for example, a very rapid onset of action,like in the case of oxitocin-mediated laborinduction or lactation, or the administra-tion of insulin in the context of food intake.Some indications, on the other hand, askfor a long duration of action or even a con-tinuous drug delivery like in the case ofbasal insulin administration or the appli-cation of β-interferon in the treatment ofmultiple sclerosis.
Today and in future, the demand fornonparenteral administration of proteinsand peptides will be significantly growing.This is especially triggered by the wishof the patient to receive a convenientnoninvasive treatment, rather than aninvasive one. In order to fulfill this wish,two main hurdles need to be overcome.Firstly, in order to enable an effective,reliable, and safe application of proteins,specialized formulations and drug deliverysystems for various application routesneed to be developed. Secondly, in orderto compensate for the significantly lowerdrug utilization, in many cases caused by

176 9.4 Parenteral Application
lower bioavailability after nonparenteralapplication, the manufacturing processesfor the proteins need to be optimized inorder to lower the costs of goods far enoughto allow for an economical overall therapy.
Owing to their overall importance, theparenteral application route and the threeapplication routes that are currently ofstrong academic interest (enteral, espe-cially oral), increasing clinical relevance(pulmonary), or therapeutic potential(nasal) are described in more detail in thesubsequent paragraphs. Further, noninva-sive routes of protein and peptide applica-tion like, for example, buccal [5], transder-mal [6], colonic [7], and rectal [8] applica-tion will not be discussed within this chap-ter because of their lower significance ver-sus the aforementioned ones, as of today.
9.4Parenteral Application
The most widely used parenteral admin-istration avenues are intravenous (iv), in-tramuscular (im), and subcutaneous (sc).In addition, there are several minor appli-cations (e.g. intraarterial). Application ofa protein drug by the different main par-enteral administration routes may haveprofound effects on the pharmacologicalperformances. When the drug is admin-istered iv, it is immediately available foraction in the circulation, while drugs ad-ministered im or sc need more time toreach the blood (depot effect), and conse-quently the pharmacokinetic (PK) profilescould be different. Besides the PK, theroute of administration may have influ-ence on the primary distribution of thedrug. For example, when administeredsc, smaller and hydrophillic proteins tendto enter the venous system, while largerand/or more hydrophobic proteins tend to
be absorbed through the lymphatic system.The different routes of parenteral admin-istration could also have effect on theantigenecity and immunogenecity of thedrugs [9]. Several other aspects may also betaken into account when deciding on theapplication route. If, for example, chronicdosing is required (like with insulin andinterferon therapies), sc and im admin-istrations may offer added benefits as thepatients can perform injections as opposedto the iv dosing, which is normally done inhospital settings.
Parenteral protein drugs are traditionallypresented in vials with liquid, frozen, orlyophilized protein formulations. Stabilityand economical considerations influencethe choice of formulation. Generally, theliquid formulations are less stable, manyof the chemical degradation reactions areslowed down or practically eliminated inthe frozen state or in the lyophilized cakewith low water activity. From the manu-facturing economy perspective, there aresome trade-offs. Liquid and frozen formu-lations are less expensive to produce thanthe freeze-dried formulations. At the sametime, many lyophilized formulations arestable at room temperature or higher, al-lowing for less costly shipping of the drug.Frozen formulations and some liquidformulations only stable under refriger-ation conditions require a cool chain forshipping to assure stability. Liquid formu-lations may present additional problems inshipping and handling the vials: proteinsare often highly surface active compoundscapable of forming stable foam.
The manufacturers also have to assurethe integrity of the container closure sys-tem used for the packaging of the drug(vials, syringes, various injector types, etc.)and the stability of the drug in the ap-proved presentation (syringe, iv bag, etc.).As the number of protein drugs that can

Formulation of Biotech Products 177
be self-administered by the patients is in-creasing, a variety of approaches have beentaken to improve convenience (packag-ing of liquid formulation in syringes andlyophilized formulation in double cham-ber syringes allowing an ‘‘in-line’’ recon-stitution to reduce manipulation requiredby the patients). Similarly, attempts weremade to reduce injection pain (for exampleby using needleless injectors delivering thedrug in a high-speed jet stream). The vari-ous delivery systems may create additionalchallenges in demonstrating drug integrity(syringes and even more so, the needlelessinjectors create high shear force duringinjection, which may cause proteins to ag-gregate or undergo partial denaturation).In all these cases, the manufacturers arerequired to demonstrate that the drugmaintains the specified properties all theway to the point when it is applied to thepatients in the specific systems.
9.5Oral Application
Amongst the different routes of enteralapplication, the oral route is clearly the
most attractive one from the patient’s pointof view. Upon oral application, the drugrapidly reaches the stomach and thereafterthe small intestine comprising duodenum,jejunum, and ileum, and, subsequently,colon and rectum.
Several factors are promoting the druguptake, like, for example, the high innersurface of approximately 100 m2, the longcontact time of ca. 16 h, and the presenceof Peyer’s patches. Factors that mightdecrease drug absorption on the otherhand are low pH values in the stomach,the presence of endogenous proteases andbacterial enzymes, physical barriers likethe mucus, and the glycocalix coveringthe microvilli, as well as the first liverpassage.
In order to further discuss the oral routeof administration, it is helpful to recall thebasic epithelial transport routes in the firstplace (see Fig. 1).
For large molecules, it is assumed thattranscytotic vesicular or – after openingof the tight junctions – paracellular trans-port may play a significant role, whereastranscellular transport is deemed to playa less significant role. Carrier-mediatedtransport so far is mainly discussed for
Apical
Basolateral
Efflux system
Paracellular Transcellular Carrier
Vesicular
Fig. 1 Epithelial transport routes.

178 9.5 Oral Application
dipeptides and in the context of efflux sys-tems like, for example, P-glycoprotein [10].
The uptake of proteins and peptidesafter enteral administration is largelyprohibited by physical and enzymaticbarriers [11]. The mucus and the glycocalyxthat cover the microvilli of the brush bordermembrane need to be permeated prior to acontact of the drug with the epithelial celland thus serve as physical barriers againstprotein uptake. The tight junctions thatform the very close connection betweenadjacent to epithelial cells also build astrong physical barrier (see Fig. 2).
Enzymatic barriers against protein up-take stem from various classes of pro-teases. For example, gastric proteases likepepsin, and intestinal pancreatic proteaseslike trypsin and alpha-chymotrypsin [11].In the microvilli, aminopeptidases as wellas carboxypeptidases are located, and uponthe passage of the cell membrane, thedrugs are confronted with the contact ofcytosolic petidases like di-tripeptidases. Inaddition to the aforementioned endoge-nous enzymes, enzymes of the intestinalbacteria have to be considered along withpotential catalysts of protein degradation.
Taking into account the efficiency of thephysical and biological barriers againstprotein uptake, it is obvious that the
absorption of proteins after peroral appli-cation is typically low. One example for lowbioavailability after peroral administrationis the one of sal-calcitonin in dogs [12].
In Fig. 3, the bioavailability of sal-calcitonin upon application via several ad-ministration routes is displayed. Whereasinfusion into the portal vein leads to anearly full bioavailability, thus demonstrat-ing that there is no relevant effect of thefirst liver passage in this specific case, andsubcutaneous administration still leads toa moderate bioavailability of 50%, whereasit is zero after peroral application. Regionaladministration of the drug directly into ei-ther the duodenum, the ileum, or the colonled to improved bioavailability in all casesversus peroral administration. However,the uptake was still negligible and variedin the range from 0.02 to 0.06%.
In order to overcome the barriers of pro-tein uptake, several attempts have beentried in the recent past, which will bediscussed subsequently. In the area offormulation approaches, gastric resistantcoatings of the protein containing dosageforms have proven to be a versatile stabi-lizer against the low pH value that prevailsin the stomach. Owing to the fact thatthe drug is only released after passage ofthe pylorus, the deleterious effect of gastric
Mucus
Glycocalyx
Microvilli
Tight junction
Epithelial cell
Capillary
Lamina propriaFig. 2 Physical barriers againstpermeation.

Formulation of Biotech Products 179
0 20 40 60 80 100 BA[%]
0 0.06 BA[%]
Portal vein (0.05 mg)
Subcutaneous (0.1 mg)
Perorally (25 mg)
Duodenally (25 mg)
Into the ileum (25 mg)
Into the colon (25 mg)
0.02 0.04 0.08 0.1
Fig. 3 Bioavailability of sal-calcitonin in dogs.
proteases like pepsin is also minimized viaa gastric resistant coating [13]. Inhibitionof catabolic proteases can be achieved indifferent ways, and is mainly performedin the large intestine. Firstly, protease in-hibitors like, for example, puromycin [14]may be added to the formulation. Secondly,a less specific approach may be followedvia shifting the pH to lower values wheresome proteases exhibit less activity. In thatrespect, compounds like, for example, cit-ric acid are useful formulation additives.A practical example of a formulation ap-proach using the combined effect of agastric resistant coating and the addition ofcitric acid to a capsule-based sal-calcitoninformulation is discussed below.
Microencapsulation is a further formu-lation tool that helps overcome the ab-sorption barriers against proteins. Firstly,encapsulation minimizes the susceptibil-ity of the drugs against proteolysis, andsecondly, particle uptake is discussedby several authors as well [15]. Math-iowitz et al. reported the in vivo effectof PLGA/FA-encapsulated insulin on fedrats after peroral administration. The lat-ter formulation of 20 I.U. insulin led tounchanged glucose levels upon feeding,whereas the same dose administered asa simple solution could not prevent an
increase in blood glucose by ca. 40 mgdL−1, 1.5 h upon feeding.
Bioadhesion, mediated either by meansof polymer particles or lectines, is dis-cussed to prolong the gastrointestinaltransit time and thereby enhance the ab-sorption potential of the drugs [16]. Pene-tration enhancers are derived from variouscompound classes such as bile salts andfatty acids [13], for example. Some of thesecompounds are deemed to interact withthe lipid bilayer of cell membranes, thus in-creasing their fluidity and decreasing theirresistance against drug permeation. A veryspecific permeation enhancer on the otherhand is the zona occludens toxin, whichopens the tight junctions, and thereby al-lows higher penetration rates [13].
Besides the means to increase bioavail-ability through optimization of formula-tions, some efforts concentrate on theoptimization of the proteins themselveswith respect to use of analogues, like, forexample, sal-calcitonin instead of humancalcitonin due to the long in vivo half-lifeof the nonhuman analogue. Pegylation is afurther means of creating analogues withoptimized pharmacokinetical characteris-tics. In the area of prodrug formation,vitamin B12 derivatives play an importantrole because of their susceptibility to the

180 9.6 Nasal Application
vitamin B12-carrier system, which allowsfor higher (pro)drug uptake [17].
In order to substantially increase thebioavailability of proteins, a combinationof elements from the aforementionedenhancement technologies is often ap-plied. Lee et al. [12] report a study wheresal-calcitonin in formulations containingup to 570 mg of citric acid in a hardgelatine capsule with a gastric-resistantcoat were administered, thus combiningthree approaches: stable analogue, gastricacid protection, and protease inhibition.Trypsin, for example, is known to exhibitits maximum activity at a pH of 5 to 6,whereas it displays only 15% activity atpH 3.5. Capsules loaded with 1.2-mg sal-calcitonin were administered perorally tobeagle dogs. The pH value in the vicin-ity of the drug-carrying formulation wasmonitored via a Heidelberger capsule. Theoutcome of this study was that in individ-ual dogs the area under the curve (AUC)of the drug was increased up to 70-fold.
An additional route of uptake uponperoral administration is the passage ofthe M-cells of the Peyer’s patches [18].Although this route in essence has only alow transport capacity, it is of importancefor mucosal (peroral) vaccination, forexample. The Peyer’s patches are locatedin the small intestine where M-cells, whichneither possess a mucus nor a glycocalixlayer in comparison to the adjacententerocytes, allow the drug uptake viatranscytosis. The drug is then transportedinto the lymphatic system and causesan immune response in the case of thevaccination approach. Absorption via M-cells in essence is possible due to thedecreased enzymatic activity and theirrather high permeability.
Although there is a rather high numberof research initiatives ongoing in the areaof peroral application of pharmacologically
active compounds, no breakthrough withrespect to reasonably high bioavailabilityrates has been made so far.
9.6Nasal Application
The nose is characterized by a four-chambered structure, with the ostries di-viding the front chamber from the lower,middle, and upper chamber [19]. One ofthe characteristics of the upper chamberis its coverage with cilliars. The cilliars’function is to clean the chambers fromparticles that stem from the air by a di-rected movement toward the throat. Theyare covered by a protective mucus that isfully renewed every 20 min owing to theaforementioned transport mechanism. Afactor that is accountable for increasedabsorption potential after nasal applica-tion is the availability of certain ‘‘pores’’that specifically increase the absorption ofsmall molecules of Mrel up to 300, espe-cially hydrophilic ones [20]. Furthermore,the nasal cavity is easily accessible by med-ications, for example, in form of dropletsof 20 to 30 µm in diameter. Furthermore,there will be obviously no deleterious ef-fects from liver first pass metabolism nordegradation from gastric or pancreatic en-zymes upon nasal application.
Factors limiting the drug uptake afternasal application are the limited absorp-tion area of 160 cm2 and the short contacttime of 20 min due to cilliaric transport(sa). Furthermore, there are proteases andpeptidases located in the mucus of thenasal tissue, however, at concentrationsthat are easily saturable in many cases.
Several peptide products are in themarketplace today, which achieve high ac-ceptance by the patients, predominantlydue to their ease of use. These drugsstem from the class of oligopeptides

Formulation of Biotech Products 181
such as Luteinizing Hormone ReleasingHormone (LHRH) and octreoid, for ex-ample [21]. Formulations for drugs withhigher molecular weight, such as calci-tonin or insulin, for example, are beingdeveloped as well.
In order to increase bioavailability af-ter nasal application, several approacheswere reported. Enzyme inhibition us-ing puromycine or bile salts is onemeans [22]. The use of middle-chain phos-pholipids as penetration enhancers isanother one. Powder formulations basedon dextran or chitosan, for example, aredescribed as well [23]. Besides the formu-lation approaches, the prodrug approachis followed, for example, by formingacyloxymethyl-derivatives of amines [24].Furthermore, studies on the absorptionof larger molecules like, for example, in-sulin have been reported as well. Hussainshows how the addition of the enzyme in-hibitor puromycine, for example, inhibitsthe cleavage of Leu-enkephalin to des-tyrosin Leu-enkephalin during its passagethrough nasal tissue in a concentration-dependent manner [22].
Figure 4 provides absolute bioavailabil-ity values of several proteins and peptidesafter nasal administration in man.
Although rather high bioavailability val-ues have been reported for drugs such asLeuprorelin and Insulin, for example, ithas to be pointed out that the variability ofthe drug uptake is sometimes pronounced.For calcitonin, for example, it has been re-ported to vary from 0.3 to 30% with a meanof 3% in one study. High variance, how-ever, will not be acceptable for drugs witha narrow therapeutic window.
Formulations for nasal application needto be sterile and free of cilliotoxic sub-stances. Solutions may be applied viapump dispensers. Nasal powders on theother side require special application sys-tems like, for example, the Jetilizer [25].This system follows a twin constructionpattern with two nozzles. The capsule con-taining the particle-based formulation isopened by needles. A pump-activated airstream aerosolizes the powder in an equi-librium chamber. After passing a conicaltube, the powder is applied with a high de-position rate to the absorption area withinthe nose.
Owing to the benefits mentioned above,the nasal route of peptide applicationalready plays a significant role in theclinical practice of today. Depending onthe outcome of the development activities
0 5 10 15 20 25 30
Bioavailibility[%]
Insulin (6 kD)
Calcitonin (4.5 kD)
Leuprorelin (1.2 kD)
Desmopressin (1.1 kD)
Octreoid (1.0 kD)
Market
Clinic
Varies from 0.3 to 30%
Fig. 4 Bioavailability of proteins and peptides after nasaladministration in man.

182 9.7 Pulmonary Application
in the areas of formulations and devices,this role will be growing in the near future,eventually even reaching importance forthe application of larger compounds.
9.7Pulmonary Application
With regard to the aim to achieve a high ab-solute bioavailability, the pulmonary routeof administration bears several beneficialaspects. First of all, the large absorptionsurface of approximately 100 m2 has tobe mentioned. This surface is providedby the alveoli of the deep lung, whichare characterized by a layer consistingpredominantly of very thin so-called typeI-cells, which are characterized by a heightof only ca. 0.2 µm. This layer forms, to-gether with the adjacent layer of capillaryendothelium cells, the main part of thebarrier between the air-filled space of thelung and the bloodstream in the capil-laries [26]. Furthermore, the existence ofpores within this barrier is discussed. Ob-viously, there will be no metabolic effectof a liver first pass effect after pulmonaryapplication. However, factors potentiallydecreasing the drug uptake after pul-monary administration are potentially lowalveolar deposition rates, which might bedue to the fact that large particles ordroplets will not reach the deep lung butmight be deposited in the segments ofthe upper lung, or, in case of very smallparticles or droplets, exhalation might takeplace even after arriving at the deep lung inthe first place. Furthermore, the so-calledsurfactant, which is a layer consisting ofamphiphilic substances [26] covering theapical side of the alveolar type I-cell layermight hamper the drug uptake as wellas macrophages, which are physiologically
cleaning the alveoli from exogenic parti-cles and germs. Furthermore, it has to bepointed out that the cell layer of the alveoliis rather tight, and does not easily allowfor paracellular transport.
Patton proposed [27] the existence ofdifferent transport routes for macro-molecules depending on their size. Com-pounds smaller than 40 kD are supposed toutilize paracellular and transcytotic routesin parallel, whereas larger compoundsshould utilize the latter only, leading toan reduced uptake into the bloodstreamof the capillaries, but to an relatively ele-vated uptake into the lymph system, andthe venoles as well.
The proteolytic activity of the alveolarepithelium has been studied by, for exam-ple, Yang et al. [28]. They showed that thehalf-life of LHRH in type-I cell cultures invitro was 5 h, and that the stability couldbe significantly increased by substitutingthe Gly6 by a D-Ala6, leading to an approx-imately threefold more stable analogue ofthe drug. Even larger proteins are reportedto exhibit good absorption rates upon pul-monary administration like, for example,parathyroid hormone (PTH) [29], and anoverview of the bioavailability of proteinsand peptides after pulmonary administra-tion is given in Fig. 5.
The high bioavailability values rangingin the area of up to 40% as well asthe fact that rather large compoundssuch as Somatotropin (22 kD) demonstratemoderate to good bioavailability are veryimpressive.
In order to apply proteins to thelung with the target to achieve highbioavailability, it is important to formulatethem in ways that allow for a high amountof deep lung deposition in the first place.Two main formulation approaches haveto be differentiated, the particle-based andthe droplet-based one.
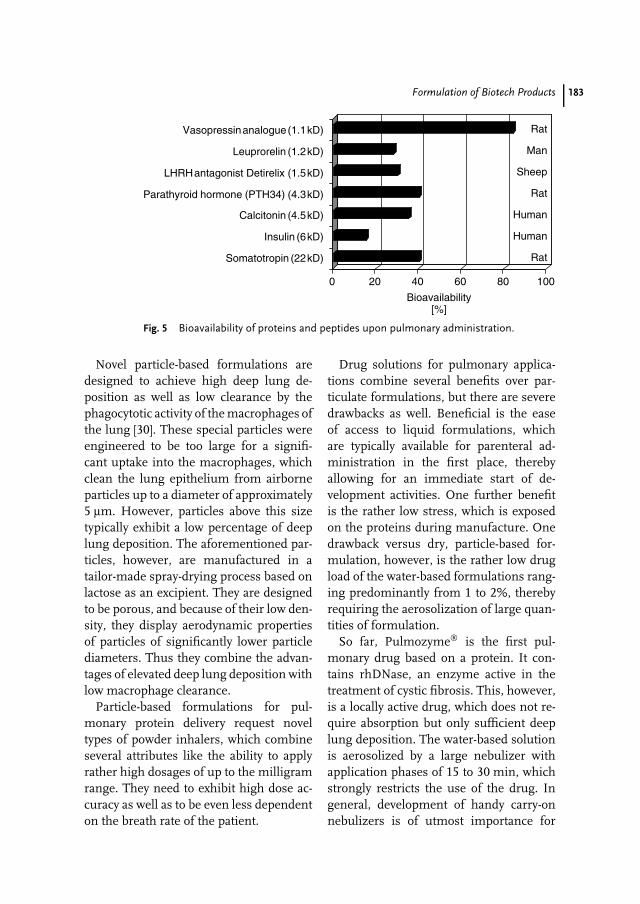
Formulation of Biotech Products 183
0 20 40 60 80 100
Bioavailability[%]
Somatotropin (22kD)
Insulin (6kD)
Calcitonin (4.5kD)
Parathyroid hormone (PTH34) (4.3kD)
LHRH antagonist Detirelix (1.5kD)
Leuprorelin (1.2kD)
Vasopressin analogue (1.1kD) Rat
Man
Sheep
Rat
Human
Human
Rat
Fig. 5 Bioavailability of proteins and peptides upon pulmonary administration.
Novel particle-based formulations aredesigned to achieve high deep lung de-position as well as low clearance by thephagocytotic activity of the macrophages ofthe lung [30]. These special particles wereengineered to be too large for a signifi-cant uptake into the macrophages, whichclean the lung epithelium from airborneparticles up to a diameter of approximately5 µm. However, particles above this sizetypically exhibit a low percentage of deeplung deposition. The aforementioned par-ticles, however, are manufactured in atailor-made spray-drying process based onlactose as an excipient. They are designedto be porous, and because of their low den-sity, they display aerodynamic propertiesof particles of significantly lower particlediameters. Thus they combine the advan-tages of elevated deep lung deposition withlow macrophage clearance.
Particle-based formulations for pul-monary protein delivery request noveltypes of powder inhalers, which combineseveral attributes like the ability to applyrather high dosages of up to the milligramrange. They need to exhibit high dose ac-curacy as well as to be even less dependenton the breath rate of the patient.
Drug solutions for pulmonary applica-tions combine several benefits over par-ticulate formulations, but there are severedrawbacks as well. Beneficial is the easeof access to liquid formulations, whichare typically available for parenteral ad-ministration in the first place, therebyallowing for an immediate start of de-velopment activities. One further benefitis the rather low stress, which is exposedon the proteins during manufacture. Onedrawback versus dry, particle-based for-mulation, however, is the rather low drugload of the water-based formulations rang-ing predominantly from 1 to 2%, therebyrequiring the aerosolization of large quan-tities of formulation.
So far, Pulmozyme is the first pul-monary drug based on a protein. It con-tains rhDNase, an enzyme active in thetreatment of cystic fibrosis. This, however,is a locally active drug, which does not re-quire absorption but only sufficient deeplung deposition. The water-based solutionis aerosolized by a large nebulizer withapplication phases of 15 to 30 min, whichstrongly restricts the use of the drug. Ingeneral, development of handy carry-onnebulizers is of utmost importance for

184 9.8 Conclusion
Tab. 2 Absolute bioavailibility of proteins and peptides afternoninvasive administration in comparison
Route of administration Class of drug
Proteins Peptides
Oral Up to ∼ 1%Nasal Up to ∼ 10% Up to ∼ 30%Pulmonary Up to ∼ 40%
the success and the future acceptance ofinhalative protein drugs formulated as so-lutions. An example of an effective, smalldevice, which provides effective nebuliza-tion by the passage of the drug solutionthrough micro nozzles is given in [30].
In summary, pulmonary application ofproteins and peptides seems to be apromising approach to enable rather highbioavailabilites of the drugs applied. Maintasks of the near future remain to clarifythe long-term safety of pulmonary proteinapplication on the lung physiology andfunction by means of, for example, forcedexpiratory volume measurement and chestX rays. This is especially important, sinceall drugs under current development aretargeting at chronic administration.
9.8Conclusion
Table 2 provides a condensed informationon the bioavailability ranges that havebeen reported so far for the oral, nasal,and pulmonary application of proteinsand peptides, clearly demonstrating theadvantage of pulmonary application overnasal and especially oral application in thisrespect.
In the area of oral delivery, a largenumber of preclinical and even a fewearly clinical trials are still being carried
out. In the area of nasal delivery, thereare various activities in research anddevelopment ongoing. Most important,some products based on oligo-peptidesare successful in the market for years.Pulmonary drug delivery is characterizedby various activities in preclinic andclinic. Especially, several insulin-baseddrug developments are in a late stage ofphase III trials [31], and with the locallyacting Pulmozyme one protein-basedpulmonary drug, although acting locally,is being marketed already.
References
1. A. Dove, Nat. Biotechnol. 1999, 17, 233–236.2. P. W. Latham, Nat. Biotechnol. 1999, 17,
755–757.3. Basic review of formulation development of
proteins for parenteral use. J. A. Bontempo,(Ed.), Development of BiopharmaceuticalParenteral Dosage Forms, Marcel Dekker, NewYork, Basel, Hong Kong, 1994.
4. Basic review of protein modificationsand their effects. D. J. Graves, B. L. Martin,J. H. Wang, Co- and Post-TranslationalModification of Proteins. Chemical Principlesand Biological Effects, Oxford UniversityPress, New York, Oxford, 1994.
5. H. E. Junginger, J. A. Hoogstrate, J. C. Ver-hoef, J. Control. Release 1999, 62, 149–159.
6. J. E. Riviere, M. C. Heit, Pharm. Res. 1997,14(6), 686–697.
7. H. Tozaki, J. Nishioka, J. Komoike et al., J.Pharm. Sci. 2001, 90(1), 89–97.

Formulation of Biotech Products 185
8. T. Uchida, S. Sakakibara, Y. Toida et al.,Pharm. Pharmacol. Commun. 1999, 5,523–527.
9. S. E. Grossberg, Human antibody develop-ment to therapeutic response modifiers inProgress in Oncology, Update on Cytokines(Ed.: E. C. Borden), Mediscript, London, UK,1990, 17–23.
10. V. H. L. Lee, C. Chu, E. D. Mahlin, J. Control.Release 1999, 62, 129–140.
11. A thorough review of oral protein delivery.W. Wang, J. Drug Target 1996, 4(4), 195–232.
12. Y. H. Lee, P. Sinko, Adv. Drug Deliv. Rev.2000, 36, 225–238.
13. Review on insulin delivery highlightingseveral enhancement strategies for oralbioavailability. G. P. Carino, E. Mathiowitz,Adv. Drug Deliv. Rev. 1999, 35, 249–257.
14. A. Bernkop-Schnurch, J. Control. Release1998, 1–16.
15. S. P. Baldwin, W. M. Saltzman, Adv. DrugDeliv. Rev. 1998, 33, 71–86.
16. G. Ponchel, J. M. Irache, Adv. Drug Deliv.Rev. 1998, 191–219.
17. J. Alsenz, G. J. Russel-Jones, S. Westwoodet al., Pharm. Res. 2000, 17(7), 825–832.
18. F. Niedergang, J. P. Kraehenbuhl, Trends CellBiol. 2000, 10, 137–141.
19. Basic reference on anatomy and physiologyof the nose. N. Mygind, R. Dahl, Adv. DrugDeliv. Rev. 1998, 29, 3–12.
20. V. Agarwal, B. Mishra, Ind. J. Exp. Biol. 1999,37, 6–16.
21. Thorough review on the nasal administrationof peptide hormones. A. E. Pontiroli, Adv.Drug Deliv. Rev. 1998, 29, 81–87.
22. Basic review on nasal drug delivery.A. A. Hussain, Adv. Drug Deliv. Rev. 1998,29, 39–49.
23. L. Illum, N. F. Farraj, S. S. Davis, Pharm.Res. 1994, 11(8), 1186–1189.
24. R. Krishnamoorthy, A. K. Mitra, Adv. DrugDeliv. Rev. 1998, 29, 135–146.
25. Detailed comparison of devices fornasal application. M. Nomura, A. Yanagawa,O. Tokomo et al., Pharm. Technol. Eur. 1998,10(10), 48–58.
26. Thorough review of absorption routes of thelung. J. S. Patton, Adv. Drug Deliv. Rev. 1996,19, 3–36.
27. J. S. Patton, Nat. Biotechnol. 1998, 16,141–143.
28. X. Yang, J. K. A. Ma, C. J. Malanga et al., Int.J. Pharm. 2000, 195, 93–101.
29. J. S. Patton, Adv. Drug Del. Rev. 2000, 42,239–248.
30. D. A. Edwards, A. Ben-Jeriba, R. Langer, J.Appl. Physiol. 1998, 85(2), 379–385.
31. Comparison of different pulmonary proteindelivery platforms. K. Haan, Biocentury 2002,10(51), A1–A6.

187
10Patents in the PharmaceuticalBiotechnology Industry: Legaland Ethical Issues
David B. ResnikEast Carolina University, Greenville, NC, USA
10.1Introduction
This chapter will provide the reader with anoverview of patenting in the pharmaceuti-cal biotechnology industry and summarizesome of the key legal and ethical issuesrelated to the patenting of biomedical prod-ucts and processes. It will examine thelegal aspects of patenting before consider-ing the ethical and policy issues. This essaywill focus primarily on the US patent laws,which are very similar to the Europeanpatent laws. The essay will note some dif-ferences between the US and Europeanlaws, and it will mention some relevantinternational intellectual property treaties.
10.2Patent Law
10.2.1What is a Patent?
A patent is a type of intellectual prop-erty. All properties can be understood asa collection of rights to control a partic-ular thing. Tangible properties give the
property holder rights to control tangiblethings, such as cars or land. Intellectualproperties, on the other hand, give theproperty holder rights to control intan-gible things, such as inventions, poems,or computer programs. Tangible thingshave a particular location in space andtime, whereas intangible things do not.The main types of intellectual property arepatents, copyrights, trademarks, and tradesecrets [1].
A patent is a private right granted bythe government to someone who inventsa new and useful product or process.The initial patent holder, the inventor,has the right to exclude others frommaking, using, or commercializing hisinvention. The patent holder may transferall or part of his rights to anotherparty, including another individual or acorporation. Researchers who work forbiotechnology companies usually assigntheir patent rights to the company inexchange for a salary, a fee, or a shareof royalties. Assignment of patent rightstransfers all the rights to the assignee, whobecomes the new patent holder. Patentholders may also grant licenses to otherparties in exchange for royalties or a fee.
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

188 10.2 Patent Law
For example, a biotechnology companywith a patent on a gene therapy techniquecould grant individuals or companieslicenses to use the technique [1].
In the United States, a patent holder hasthe right to refrain from making, using,or licensing his invention, if he or she sodesires. In the United States, a patent con-fers rights to make, use, or commercializea thing but implies no corresponding obli-gations. As a result, some companies in theUnited States use patents to block techno-logical development to gain an advantageover their competitors. Some Europeancountries, however, have compulsory li-censing, which requires the patent holderto make, use, or commercialize his orher invention or license others to doso [1].
The term of patent in the UnitedStates and most countries that belongto the European Union (EU) lasts for20 years from the time the inventorsubmits his application. A patent is notrenewable. Once the patent expires, theinvention becomes part of the publicdomain, and anyone can make, use,or commercialize the invention withoutthe permission from the inventor [1]. Inthe pharmaceutical industry, the averageinterval between the discovery of a newdrug and its final approval by the Foodand Drug Administration (F.D.A.) forhuman consumption is 10 years, whichincludes the time required to conductclinical research, product development,as well as an F.D.A. review. Thus, mostpharmaceutical companies can expect thatthey will have about 10 years to recoup themoney they have invested in a new drugbefore the patent expires. Once the patentexpires, the name of the drug may still havetrademark protection, but other companiescan manufacture and market a generic
version of the drug without obtainingpermission from the company [2].
The main policy rationale for patentlaws is that they promote the progressof science, technology, and industry byproviding financial incentives for inven-tors, entrepreneurs, and investors [1]. Bygranting property rights over inventions,the patent system gives inventors, privatecompanies, and other organizations theopportunity to profit from their invest-ments of time and money in research anddevelopment. Since most new scientificdiscoveries and technological innovationsbenefit the society, the public benefits fromgranting private rights over intellectualproperty. However, excessive private con-trol over intellectual property can impedeaccess to science and technology. Thus,patent laws attempt to strike an appropri-ate balance between the public and privatecontrol of inventions. A good example ofthis balancing is the length of a patent: ifthe term of a patent is too short, companiesand researchers will not have enough timeto obtain a fair return on their investment;if the term is too long, the public will nothave adequate access to the technology.
10.2.2How Does One Obtain a Patent?
To obtain a patent, one must submit apatent application to the patent office.In the United States, the Patent andTrademark Office (PTO) examines thepatent applications. The application mustprovide a description of the inventionthat would allow someone trained in therelevant practical art to make and usethe invention. One or more individualsmay be listed as inventors on the patentapplication. The application need notinclude a sample or model of the invention;a written description will suffice. The

Patents in the Pharmaceutical Biotechnology Industry: Legal and Ethical Issues 189
application will contain information aboutthe invention, background references,data, as well as one or more claimspertaining to the invention. The claimsstated on the patent application willdetermine the scope of the inventor’spatent rights.
If the PTO rejects a patent applica-tion, the inventor may submit a re-vised application. The process of submis-sion/revision/resubmission, otherwise kn-own as ‘‘prosecuting’’ a patent, may con-tinue for months or even years. If the PTOrejects the patent, the applicant may appealthe decision to a federal court [1].
The PTO will award a patent to aninventor only if he or she provides evidencethat his or her invention satisfies all of thefollowing conditions (EU countries havesimilar requirements [1, 3]):
1. Originality: The invention is new andoriginal; it has not been previouslydisclosed in the prior art. The rationalefor this condition is that the public doesnot benefit when the patent office grantsa patent on something that has alreadybeen invented. Thus, if someone elsehas already submitted an application forthe same invention, this would qualifyas a prior disclosure. Also, disclosurecould occur if a significant part ofthe invention has been published orused [1].
2. Nonobviousness: The invention is notobvious to someone who is trained inthe relevant practical art.
3. Usefulness: The invention has somedefinite, practical utility. The utility ofthe invention should not be merelyhypothetical, abstract, or contrived. Therationale for this condition is self-explanatory: the public does not benefitfrom useless patents. Recently, the U.S.PTO raised the bar for proving the
utility of patents on DNA in response toconcerns that it was granting patents onDNA sequences when the inventors didnot even know the biological functionsof those sequences [4].
In addition to satisfying these threeconditions, to obtain a patent in the UnitedStates, the inventor must exhibit duediligence in submitting an application anddeveloping the invention. In the UnitedStates, the person who is the first toconceive an invention will be awarded thepatent provided he exhibits due diligence.If the first inventor does not exhibit duediligence, the PTO may award the patent toa second inventor, if that inventor reducesthe invention to practice and submits anapplication before the first inventor [1].
Once the PTO awards the patent, theapplication becomes part of the public do-main, and other inventors and researchersmay use the knowledge contained in theapplication. Indeed, the ‘‘patent bargain’’is an agreement between the governmentand a private party in which the partyagrees to disclose the knowledge related tohis invention to the public in exchange fora limited monopoly on the invention [1].The public benefits from this bargain be-cause it encourages the inventor to avoidprotecting his knowledge through tradesecrecy. A great deal of the world’s scien-tific and technical information is disclosedin patent applications [5]. For example,the PTO has a large online, searchabledatabase of patent applications [6].
10.2.3What is the Proper Subject Matter for aPatent?
Under the US law, the PTO canaward patents on articles of manufacture,compositions of matter, machines, or

190 10.2 Patent Law
techniques or improvements thereof [7].The EU countries allow patents on simi-lar types of things [3]. Although differentpatent laws use different terms to describethe subject matter of patents, there arethree basic types of subjects for patents:(1) products (or materials), (2) processes(or methods), and (3) improvements. Forexample, one could patent a mousetrap (aproduct), a method for making a mouse-trap (a process), or a more efficient andhumane mousetrap (an improvement) [1].
One of the most important doctrinesin patent law is that patents only ap-ply to products or processes that resultfrom human ingenuity (or inventiveness).Thus, the US courts have held that onemay not patent laws of nature or nat-ural phenomena, since these would bepatents on a product of nature. Over twodecades ago, a landmark US SupremeCourt case, Diamond v. Chakrabarty, setthe legal precedent in the United Statesfor patents on life forms [8]. Chakrabartyhad used recombinant DNA techniques tocreate a type of bacteria that metabolizescrude oil. The PTO had rejected his patentapplication on the grounds that the bacte-ria did not result from human ingenuity,but the Supreme Court vacated this rulingand held that Chakrabarty could patenthis genetically engineered life forms [8, 9].This decision helped to establish the legalprecedent for other patents on life forms,such as patents on laboratory mice, celllines, and bioengineered tissues and or-gans [10]. The EU countries have followedthe United States in allowing patents onlife forms that result from human ingenu-ity [11].
Before Chakrabarty received his patent,the PTO had also granted inventors patentson DNA, proteins, and recombinant DNAtechniques [12–14]. In granting patentson organic compounds that occur in
living organisms, such as animals orplants, patent agencies have distinguishedbetween naturally occurring compoundsand isolated and purified compounds [15].For example, DNA in its natural stateoccurs in virtually all organisms and isunpatentable in its natural state. However,scientists can use various chemical andbiological techniques to create isolatedand purified samples of DNA, whichare patentable. The reason why patentagencies allow patents on isolated andpurified compounds is that they resultfrom human ingenuity [16].
Another important doctrine in patentlaw is that patents apply to applications,not to ideas. Ideas are part of the pub-lic domain. For example, courts in theUnited States have ruled that mathemati-cal algorithms are unpatentable ideas butthat the computer programs that use algo-rithms to perform practical functions arepatentable [17].
10.2.4Types of Patents in PharmaceuticalBiotechnology
There are many different types of patentsthat may be available to researchers andcompanies in the field of pharmaceuticalbiotechnology. Following the distinctionin Sect. 10.2.3 between products andprocesses, potential patents might includethe following:
1. Patents on pharmaceutical and biomed-ical products, such as bioengineereddrugs, proteins, receptors, neurotrans-mitters, oligonucleotides, hormones,genes, DNA, DNA microchips, RNA,cell lines, bioengineered tissues and or-gans, and genetically modified bacteria,viruses, animals, and plants.

Patents in the Pharmaceutical Biotechnology Industry: Legal and Ethical Issues 191
2. Patents on pharmaceutical and biotech-nological processes, such as methodsfor genetic testing, gene therapy proce-dures, DNA cloning techniques, meth-ods for culturing cells and tissues, DNAand RNA sequencing methods, andxenotransplantation procedures.
3. Patents on improvements of pharma-ceutical, biomedical, and biotechnolog-ical products and processes.
For any of these products or processesto be patentable, they would need to resultfrom human ingenuity.
10.2.5Patent Infringement
Patent infringement occurs when some-one uses, makes, or commercializes aninvention without the permission of thepatent holder. In the United States, thepatent holder has the responsibility ofbringing an infringement claim againsta potential infringer and proving that in-fringement occurred [1]. A court may issuean injunction to stop the infringementor award the patent holder damages forloss of income due to infringement. Thereare three types of infringement: directinfringement, indirect infringement, andcontributory infringement. Patent holdersmay also settle infringement claims outof court. Researchers, corporations, anduniversities usually try to avoid any involve-ment in an infringement lawsuit, sincepatent infringement litigation is expensiveand time consuming [16].
Many EU countries have a defense topatent infringement known as the researchexemption [3]. The United States also hasa research exemption (also known asthe experimental use exemption), whichhas been used very infrequently [18].Under this exemption, someone who
uses or makes a patented inventionfor pure research with no commercialintent can assert this defense in aninfringement lawsuit to avoid an adverselegal decision. The research exemptionis similar to the ‘‘fair use’’ exemptionin copyright law insofar as it permitssome unconsented uses of intellectualproperty [18]. There are some problemswith the exemption, however. First, theresearch exemption is not well publicized.Second, the research exemption is not welldefined [18]. Indeed, in the United Statesthe research exemption has no statutorybasis but is a creation of case law. Somecommentators have argued that countriesshould clarify and strengthen the researchexemption in order to promote researchand innovation in biotechnology and avoidexcessive private control of inventions [3].
10.2.6International Patent Law
Every country has the authority to makeand enforce its own patent laws and toaward its own patents. Thus, a patentholder must apply for a patent in everycountry where he wants patent protection.For example, a corporation that patentsa new drug in the United States mustalso apply for a patent in Germany, ifit desires patent protection in Germany.Furthermore, complex matters relatingto jurisdiction can arise when someoneinfringes a patent that is protected inone country but not in another. Forexample, if someone infringes a US patentin Germany, but the invention is notprotected by the German patent laws, thenthe patent holder will need to bring alawsuit in a court in the United States,which may or may not have jurisdiction.
To deal with international disputes aboutintellectual property and to harmonize

192 10.3 Ethical and Policy Issues in Biotechnology Patents
intellectual property laws, many countrieshave signed intellectual property treaties.Most of these treaties define minimumstandards for intellectual property protec-tion and obligate signatories to cooperatein the international enforcement of prop-erty rights. The most important treatyrelated to patents is the Trade Related As-pects of Intellectual Properties agreement(TRIPS), which has been developed andnegotiated by the World Trade Organiza-tion (WTO). The TRIPS agreement definesminimum standards for patent rights. Forexample, it requires that patents last for20 years. Countries that have signed theagreement agree to adopt patent laws thatprovide at least the minimum level of pro-tection under the agreement. Countriesmust also agree to cooperate in the en-forcement of patent rights. TRIPS allowscountries to override patents rights to dealwith national emergencies, such as publichealth crisis [1].
10.3Ethical and Policy Issues in BiotechnologyPatents
Having provided the reader with somebackground information on patenting inbiotechnology, this chapter will brieflyreview some important ethical and policyissues.
10.3.1No Patents on Nature
In the 1990s, a variety of writers, politicalactivists, theologians, ethicists, and profes-sional organizations opposed patents onbiotechnological products and processesfor a variety of reasons. Many of thesecritics argued that patents on living bod-ies, as well as patents on body parts, are
unethical because they are patents on nat-ural things [19]. They argued that it isimmoral and ought to be illegal to patentorganisms, tissues, DNA, proteins, andother biological materials. Some of thesecritics based their opposition to biotechnol-ogy patents on religious convictions [20],while others based their opposition on ageneral distrust of biotechnology and thebiotechnology industry [21, 22]. Some ofthe more thoughtful critics of biotechnol-ogy patents accepted some types of patentson biological materials, but objected topatents on other types of biological materi-als, such as patents on genes or cell lines,on the grounds that these types of patentsattempt to patent nature [23, 24].
As noted in Sect. 10.2.3, patents onproducts of nature are illegal; a prod-uct or process must have resulted fromhuman ingenuity to be patentable. Buthow much human ingenuity should berequired to transform something froman unpatentable product of nature to apatentable, human invention? Definingthe boundaries between the products ofnature and human inventions is a fun-damental issue in patent law and policythat parallels the tenuous distinction be-tween the natural and artificial [25]. Whilemost people can agree on paradigmaticcases of things that are natural, such asgold, and things that are artificial, such asgold jewelry, it difficult to reach an agree-ment on borderline cases, such as DNAsequences. On the one hand, DNA se-quences exist in nature and can thereforebe regarded as natural. On the other hand,isolated and purified DNA sequences donot exist in nature and are produced onlyunder laboratory conditions. They are, insome sense, human artifacts. However, thenucleotide sequences in isolated and pu-rified DNA are virtually identical to thesequences in naturally occurring DNA.

Patents in the Pharmaceutical Biotechnology Industry: Legal and Ethical Issues 193
There is probably no objective (i.e. sci-entific) basis for distinguishing betweennaturally occurring DNA and isolated andpurified DNA. Likewise, there is probablyno objective basis for distinctions betweennatural cell lines versus artificial cell lines,natural proteins versus artificial proteins,and natural organisms versus artificial or-ganisms.
If the distinction between a productof nature and a human invention is notobjective, then it depends, in large part,on human values and interests. It is likeother controversial distinctions in biomed-ical law and ethics, such as human versusnonhuman and alive versus dead. Thebest way to deal with these controver-sial distinctions is to carefully consider,negotiate, and balance competing valuesand interests in light of the particularfacts and circumstances. Laws and poli-cies that define patentable subject mattershould also attempt to promote an optimalbalance between competing interests andvalues and should carefully consider thefacts and circumstances relating to eachitem of technology [25]. Policies adoptedby the United States and the EuropeanUnion with respect to the patenting ofDNA appear to strike an optimal balancebetween competing interests and valuesbecause these policies disallow the patent-ing of DNA in its natural state but allowthe patenting of isolated and purifiedDNA [11, 15].
10.3.2Threats to Human Dignity
Critics of biotechnology patents have alsoclaimed that patents on human body parts,such as genes, cell lines, and DNA, areunethical because they treat people as mar-ketable commodities [19, 21, 22, 26]. Somehave even compared patents on human
genes to slavery [27]. The issues con-cerning the commercialization of humanbody parts are complex and emotionallycharged. They also have implications formany different social policies, includingorgan transplantation, surrogate parent-ing, and prenatal genetic testing. Thischapter will give only a brief overview ofthis debate.
According to several different ethicaltheories, including Kantianism and theJudeo–Christian tradition, human beingshave intrinsic moral value (or dignity) andshould not be treated as if they have onlyextrinsic value. An entity (or thing) hasintrinsic value if it is valuable for itsown sake and not merely for the sakeof some other thing. A commodity is athing that has a value – a market valueor price – which serves as a basis forexchanging it for some other thing. Forexample, one can exchange a barrel of oilfor $30 or exchange a visit to the dentistfor $50. Treating an entity as a commodityis treating it as if it has only extrinsic valueand not intrinsic value. Thus, it wouldbe unethical to treat a human being as acommodity because this would be treatingthat person as if they have only extrinsicvalue and no intrinsic value. Slavery istherefore unethical because it involves thebuying and selling of whole human beings.People are not property [28, 29].
Even though treating a whole humanbeing as a commodity violates humandignity, one might argue that treatinga human body part as a commoditydoes not violate human dignity. Humanbeings have billions of different body parts,ranging from DNA, RNA, proteins, andlipids to membranes, organelles, cells,tissues, and organs. Properties that weascribe to the parts of a thing do notnecessarily transfer to the whole thing;inferences from parts to wholes are

194 10.3 Ethical and Policy Issues in Biotechnology Patents
logically invalid. For example, the factthat a part of an automobile, such asthe front tire, is made of rubber doesnot imply that the whole car is made ofrubber [28]. Likewise, treatment of a partof human being, such as blood or hair, as acommodity does not imply treatment of thewhole human being as a commodity. It ispossible to commodify (or commercialize)a human body part without commodifyingthe whole human being.
This argument proves that buying andselling hair, blood, or even a kidneyis not equivalent to slavery. Even so,one might argue that treating humanbody parts as commodities constitutesincomplete commodification of humanbeings; partial commodification of humanbeings can threaten human dignity evenif it does not violate human dignity [26].Incomplete commodification can threatenhuman dignity because it can lead toexploitation, harm, and injustice, as wellas complete commodification of humanbeings. For example, in the now famouscase of Moore v. Regents of University ofCalifornia, the desire to patent a valuablecell line played an important role inthe exploitation of a cancer patient [24].The researchers took cells from Moore’sbody that overexpress cytokines. Theresearchers did not tell Moore what theyplanned to do with the tissue samplesthey took from him or that the samplescould be worth millions of dollars [30].One might argue that treating human bodyparts as commodities inevitably leads tothe abuse of human rights and dignity asin the Moore case. Although incompletecommodification of human beings is notintrinsically immoral, it can lead thesociety down a slippery slope towardvarious types of immorality and injustices.In order to stop the slide down this slipperyslope, society should forbid activities that
constitute incomplete commodification ofhuman beings, such as the patenting ofcell lines and DNA, a market in humanorgans, surrogate pregnancy contracts,cloning for reproduction, and sellinghuman gametes [31].
One could reply to this argument by ac-knowledging that the slippery slope posesa genuine threat to human dignity butmaintain that it may be possible to preventexploitation, injustice, and other abusesby developing clear and comprehensiveregulations on practices that commodifyhuman body parts. Regulations should re-quire informed consent to tissue donation,gamete donation, and organ donation, aswell as fair compensation for subjects thatcontribute biological materials to researchand product development activities. Regu-lations should also protect the welfare andprivacy of human research subjects andpatients [28, 32]. These regulations shouldalso state that some human biological ma-terials, such as embryos, should not betreated as commodities because they posean especially worrisome threat to humandignity. Although an embryo is not a hu-man being, it should be illegal to buy, sell,or patent a human embryo. However, itshould be legal to buy or patent embry-onic stem cells, provided that the societyhas appropriate regulations [33]. Althoughselling organs is illegal in many countries,including the United States and many Eu-ropean nations, some have argued thatorgans could be bought and sold, pro-vided that appropriate regulations are inplace [34, 35].
10.3.3Access to Technology
One of the most important ethical andpolicy concerns raised by the critics ofbiotechnology patenting is that patenting

Patents in the Pharmaceutical Biotechnology Industry: Legal and Ethical Issues 195
will have an adverse impact on access tomaterials and methods that are vital to re-search and innovation in biotechnology aswell as medical tests and treatments. Thenegative effects of patenting on science,industry, and medicine will constitute agreat social cost rather than a social ben-efit. In Sect. 10.2.1, we noted that theprimary rationale for the patent system isthat it benefits the society by encouragingprogress in science, technology, and indus-try. However, this argument loses its forcewhen patenting has the opposite effect.If patenting does more harm than good,then we should forbid or greatly restrictpatenting [24, 36]. The issue of access tomaterials and methods in biotechnology,like the issues discussed in Sects. 10.3.1and 10.3.2, is very complex and contro-versial. This chapter will not attempt toexplore these issues in great depth, but itwill attempt to provide the reader with anoutline of the arguments on both sides.
Concerns about access to materials andmethods stem from potential problemswith the licensing of patents on prod-ucts and processes that are useful in re-search and innovation of biomedicine andbiotechnology [37]. First, if a researcheror a company wants to develop a newproduct or process in biotechnology andbiomedicine, then he or she may needto negotiate and obtain dozens of differ-ent licenses from various patent holdersin order to avoid patent infringement.The researcher or company might needto fight through a ‘‘patent thicket’’ in orderto develop a new and useful invention. Forexample, DNA chip devices test for thou-sands of different genes in one assay. Ifdozens of companies hold patents on thesedifferent genes, then one may need to ob-tain dozens of different licenses to developthis new product. Although larger biotech-nology and pharmaceutical companies are
prepared to absorb the legal and admin-istrative transaction costs associated withlicensing, smaller companies and univer-sities may find it difficult to navigate the‘‘patent thicket’’ [38].
Second, ‘‘blocking patents’’ in biotech-nology could prevent the development ofdownstream products and processes [39].In industries with many different interde-pendent products and processes, someonewho holds a particular invention may beable to affect or control the development ofsubsequent inventions that depend on thatprior invention. These prior inventions arealso known as ‘‘upstream’’ inventions, andthe subsequent inventions are also knownas ‘‘downstream’’ inventions. Some com-panies may obtain patents for the solepurpose of preventing competitors fromdeveloping useful inventions in biotech-nology. In the United States, these com-panies would have no obligation to use,make, market, or license such inventions.They could use their inventions to blockthe development of downstream productsand processes. In countries that have com-pulsory licensing, companies would havea legal duty to make, use, commercialize,or license their inventions, but they couldstill use other means to prevent the devel-opment of downstream technologies, suchas setting very high licensing fees.
Third, high licensing fees could imposea heavy toll on research and innovationin biotechnology and biomedicine [37].Companies with patents on upstreaminventions might issue licenses on thecondition that they receive a percentageof profits from downstream inventions.While downstream patent holders haveno legal obligation to share their profitswith upstream patent holders, upstreampatent holders may try to acquire a portionof downstream profits by issuing these‘‘reach through’’ licenses. Even companies

196 10.3 Ethical and Policy Issues in Biotechnology Patents
that do not issue ‘‘reach through’’ licensesmay still set high licensing fees. For ex-ample, many commentators have claimedthat Myriad Genetics’ high licensing feesfor its tests for BRCA1 and BRCA2 mu-tations, which increase the risk of breastand ovarian cancer, have had a negativeimpact on research and innovation, anddiagnostic and predictive testing [40].
These aforementioned problems relatedto licensing – the patent thicket, blockingpatents, and high licensing fees – couldundermine not only research and innova-tion but could also have an adverse impacton health care by undermining the accessto new medical products and services, suchas genetic tests. For example, if a companyis unable to develop a genetic test, due tolicensing problems, then the patients willnot benefit from that test. If a companydevelops a genetic test but charges a highfee to conduct the test or charges a highfee to license the test, then many patientsmay not be able to afford the test. In ei-ther case, problems related to the licensingof biotechnology products and processescould prevent the public from benefitingfrom new developments in biomedicine.
On the other hand, many commentatorsand industry leaders have rebutted thesecriticisms of biotechnology patenting by ar-guing that the free market, patent offices,and the legal system will keep potential li-censing problems in check [41–43]. Com-panies will not have any major difficultiesin negotiating and obtaining licenses be-cause they will all understand the impor-tance of cooperation in the biotechnologyindustry. Few companies will developblocking patents because these patentswill usually prove to be unprofitable: onecan make much more money from mar-keting or licensing a new invention thanfrom keeping it on the shelf. Finally, highlicensing costs will decline in response
to lower consumer demands, especially ifcompetitors are able to enter the marketby developing new inventions that workaround existing ones. (A ‘‘work around’’ in-vention is an improvement on a patentedinvention or an alternative to a patentedinvention.) Industry leaders also pointout that the potential licensing problemsfaced by the biotechnology industry arenot new because many other industrieshave faced – and solved – similar prob-lems [41]. For example, many differentcompanies in the semiconductor industryhave worked together to develop licensingagreements [44]. There are many interde-pendent products and processes in thesemiconductor industry and many differ-ent patent holders, but companies havemanaged to avoid licensing problems andthe industry has thrived. Indeed, the semi-conductor industry is one of the mostsuccessful and innovative industries theworld has ever known.
Some commentators have argued thatsocieties should reform the patent systemto prevent licensing problems from occur-ring and to ensure that new biomedicaltechnologies are affordable and accessible.These proposed reforms, some of whichhave been mentioned above, include thefollowing:
1. Banning patents on particular kinds ofproducts or processes, such as patentson genes that are associated withdiseases or patents on genetic tests [23].
2. Expanding and clarifying the researchexemption in biotechnology [3, 16].
3. Raising the bar for the various con-ditions for awarding patents, such asnovelty and utility [3, 16].
4. Restricting the scope of biotechnologypatents in order to allow for ‘‘workaround’’ inventions and to promotecompetition [3, 16].

Patents in the Pharmaceutical Biotechnology Industry: Legal and Ethical Issues 197
5. Applying antitrust laws to the biotech-nology industry to promote fair compe-tition [16].
6. Conducting an ethical review ofpatent applications to address ethicaland policy issues before awardingpatents [3, 45].
7. Developing a patent pool in the biotech-nology industry to promote efficientlicensing [46].
Most of these proposed reforms, withthe exception of banning some typesof biotechnology patents, would proba-bly promote research and innovation inbiotechnology and biomedicine withoutundermining the financial incentives forresearchers and companies. Many of thesereforms could be enacted without any addi-tional legislation, since patent offices andthe courts already have a great deal ofauthority to shape patent law and policythrough their interpretation and applica-tion of existing statutes [47].
10.3.4Benefit Sharing
The final issue this chapter will considerinvolves the sharing of the benefits of re-search and innovation in biotechnology.Some critics of biotechnology patents haveclaimed that the distribution of the ben-efits of research and innovation is oftenunfair [22, 24, 48, 49]. According to thesecritics, pharmaceutical and biotechnologycompanies benefit greatly from researchand innovation by earning large profits,but individual patients or research sub-jects, populations, or communities benefitvery little. For example, to study a geneticdisease, researchers need to take tissuesamples from patients/subjects. Very of-ten, researchers do not offer to pay subjectsany money for their tissue samples or
promise them any royalties from the com-mercialization of their research or its appli-cations. If a company develops a profitablegenetic test from free genetic samples,patients/subjects could argue that the com-pany is not sharing the benefits fairly.Unequal distributions of benefits couldalso occur between companies and entirecommunities or countries. For example,some pharmaceutical and biotechnologycompanies are now developing drugs onthe basis of the knowledge obtained fromindigenous populations concerning theirmedicinal plants. If a company developsa profitable medication from this indige-nous knowledge and does not offer thepopulation any compensation, the popu-lation could argue that the company hasnot shared the benefits of research fairly.Unequal distributions of benefits couldalso take place between developed nationsand developing nations. For example, if re-searchers, patients, and companies fromthe developed world benefit a great dealfrom biotechnology, but people in the de-veloping world do not, one might arguethat the benefits of biotechnology havebeen distributed unfairly.
Several writers and organizations havecalled for the fair distribution of the ben-efits of research in biotechnology [50–53].Some writers appeal directly to theoriesof justices, such as utilitarianism, egal-itarianism, or social contract theory, toargue for a fair distribution of researchbenefits [47, 54]. Others appeal to theconcept of a common heritage relatingto human biological materials, such asDNA [52, 53]. Regardless of how one justi-fies a general principle of benefit sharing inbiotechnology, the most important practi-cal problems involve determining how thebenefits should be shared. What wouldbe a fair sharing of benefits between

198 10.4 Conclusion
researchers and companies and sub-jects/populations/communities? Shouldresearchers and companies offer to givesubjects/populations/communities finan-cial compensation for providing researchmaterials and methods, such as tissue sam-ples of indigenous knowledge? Should re-searchers and companies offer to pay royal-ties for the commercialization of researchto subjects/populations/communities? Al-though financial compensation might beuseful and appropriate in some situations,such as giving communities royalties forindigenous knowledge or providing somesubjects with compensation for their valu-able tissues (as in the Moore case, dis-cussed in Sect. 10.3.2), in other situations,direct financial compensation may not bevery useful or appropriate. For example,if a company collects thousands of tissuesamples from subjects and uses the knowl-edge gained from those samples to developa commercial product, the financial bene-fit offered to any particular subject mightbe miniscule, since the benefits wouldneed to be divided among thousands ofsubjects. Moreover, it may be impossi-ble to estimate the potential benefits tosubjects prior to the development of theproduct, since most new products are notprofitable. Furthermore, subjects in somecultures might not be interested in finan-cial rewards for participation. Perhaps thebest way to share the benefits in situationslike these would be to offer to providethe population or community with nonfi-nancial benefits, such as improvements inhealth care, education, or infrastructure.In any case, these are complex questionsthat cannot be addressed in depth in thischapter. To answer questions about thefair distribution of research benefits inany particular case, one needs to applythe theories and concepts of distributivejustice.
Even though there is little consensusabout how to distribute the benefits ofresearch and innovation in biotechnol-ogy, almost everyone with an interestin the issue agrees that subjects shouldbe informed about the plans for ben-efit sharing (if there are any) [55]. Forexample, the researchers in the Moorecase should have told Moore that theyplanned to develop a cell line from histissue and that they were not planningto offer him any financial compensa-tion. If researchers conduct a study thatinvolves an entire population or com-munity, they should discuss the benefit-sharing plans with the representativesof the community or population [56]. In-deed, respect for human dignity requiresnothing less than fully informing the sub-jects of the material facts related to theirresearch participation, including facts per-taining to the commercialization of re-search [57, 58].
10.4Conclusion
This essay has provided the reader with anoverview of the legal, ethical, and policyissues relating to the patenting of productsand processes used in pharmaceuticalbiotechnology. Although this essay hasattempted to provide the reader with up-to-date information, it is possible that someof this information may soon be out-of-date, due to the changes in technology,case law, legislation, and internationaltreaties. Since most of these issues arevery complex and constantly changing,those who are interested in learningmore about this topic should reviewthe relevant documents, guidelines, andpolicies relating to their particular areas ofresearch and development.

Patents in the Pharmaceutical Biotechnology Industry: Legal and Ethical Issues 199
References
1. A useful overview of intellectual propertylaw. A. Miller, M. Davis, Intellectual Property,West Publishing, St. Paul, MN, 2000.
2. A useful overview of legal and ethical issuesinvolving DNA patents. A. Goldhammer,Accountability Res. 2001, 8, 283–292.
3. The Nuffield Council on Bioethics, The Ethicsof Patenting DNA, Nuffield Council, London,2002.
4. U.S. Patent and Trademark Office, Fed.Register 1999, 64(244), 71440–71442.
5. Derwent Information, Frequently askedquestions, http://www.derwent.com/, Ac-cessed: December 31, 2002.
6. U.S. Patent and Trademark Office,www.uspto.gov, Accessed: December 31,2002.
7. U.S. Patent Act. 35 U.S.C. 101, 1995.8. A landmark court case relating to the
patenting of biological materials. Diamondv. Chakrabarty, 447 U.S. 303, 1980.
9. A. Chakrabarty, US 4259444, 1980.10. R. Eisenberg, Nat. Genet. 1997, 15(2),
125–130.11. A clear statement of the E.U.’s opinions
on some ethical issues in biotechnology.European Commission, Opinions of theGroup of Advisors on the Ethical Implicationsof Biotechnology of the European Commission,European Commission, Brussels, 1998.
12. A useful overview of legal issues relating tothe patenting of DNA. R. Eisenberg, EmoryLaw J. 1990, 39, 721–745.
13. D. Kevles, A. Berkowitz, Brooklyn Law Rev.2001, 67, 233–248.
14. S. Cohen, H. Boyer, US 4237224, 1980.15. Outlines the U.S. PTO’s DNA patent policy.
J. Doll, Science 1998, 280, 689–690.16. A useful overview of legal and ethical issues
involving DNA patents. D. Resnik, Sci. Eng.Ethics 2001, 7(1), 29–62.
17. Parker v. Flook, 437 U.S. 584, 1978.18. J. Karp, Yale Law J. 1991, 100, 2169–2188.19. A useful summary of religious objections to
patents on biological materials. M. Hanson,Hastings Cent. Rep. 1997, 27(6) (SpecialSupplement), 1–21.
20. R. Land, B. Mitchell, First Things 1996, 63(2),20–22.
21. A. Kimbrell, The Human Body Shop,Gateway, Washington, 1997.
22. J. . Rifkin, The Biotech Century, PenguinPutnam, New York, 1998.
23. J. Merz, M. Cho, Camb. Q. Healthc. Ethics1998, 7, 425–428.
24. L. Andrews, D. Nelkin, Body Bazaar, CrownPublishers, New York, 2000.
25. D. Resnik, Discoveries, inventions, and genepatents in Who Owns Life? (Eds.: D. Magnus,A. Caplan, G. McGee), Prometheus Press,New York, 2002, pp. 171–181.
26. M. Radin, Contested Commodities, HarvardUniversity Press, Cambridge, MA, 1996.
27. Joint Appeal Against Human and AnimalPatenting, Press Conference Text, May 17,1995. Board of Church and Society of theUnited Methodist Church, Washington, DC,1995.
28. A useful discussion of some ethical issuesin patenting human DNA. D. Resnik, J. Law,Med. Ethics 2001, 29(2), 152–162.
29. R. Green, Kennedy Inst. Ethics J. 2001, 11(3),247–261.
30. A key court case involving informed consentand the commercialization of research.Moore vs. Regents of the University ofCalifornia, 793 P.2d 479, Cal. 1990.
31. M. Hanson, J. Med. Philos. 1999, 24(3),267–287.
32. D. Resnik, Bioethics 2001, 15(1), 1–25.33. D. Resnik, Health Care Anal. 2002, 10,
127–54.34. S. Wilkinson, Health Care Anal. 2000, 8,
352–61.35. American Medical Association, Council on
Ethical and Judicial Affairs, Arch. Intern. Med.1995, 155, 581–89.
36. A. Caplan, J. Merz, Br. Med. J. 1996, 312, 926.37. An influential essay outlining some potential
problems with licensing in biotechnology.M. Heller, R. Eisenberg, Science 1998, 280,698–701.
38. C. Shapiro, Navigating the patent thicket:cross-licenses, patent pools, and standardsetting in Innovation Policy and the Economy(Eds.: A. Jaffe, J. Lerner, S. Stern), MIT Press,Cambridge, MA, 2000, pp. 119–150.
39. L. Guenin, Theor. Med. Bioethics 1996, 17,279–314.
40. J. Merz, M. Cho, M. Robertson, D. Leonard,Mol. Diagn. 1997, 2(4), 299–304.
41. R. Scott, Testimony before the HouseJudiciary Subcommittee on Courts andIntellectual Property (July 13, 2000).

200 10.4 Conclusion
42. J. Tribble, Camb. Q. Healthc. Ethics 1998, 7,429–432.
43. G. Woolett, O. Hammond, An industryperspective on the gene patenting debatein Perspectives on Gene Patenting (Ed.:A. Chapman), American Association for theAdvancement of Science, Washington, 1999,pp. 43–50.
44. H. Hall, R. Ziedonis, Rand J. Econ. 2001, 32,101–128.
45. T. Caulfield, R. Gold, Clin. Genet. 2000, 57,370–375.
46. Discuss patents pools in biotechnology. U.S.PTO, Patent Pools: A Solution to the Problemof Access in Biotechnology Patents? U.S. PTO,Washington, 2000.
47. Thorough discussion of legal, ethical, andpolicy issues related to DNA patenting.D. Resnik, Owning the Genome: A MoralAnalysis of DNA Patenting, S.U.N.Y. Press,Albany, New York, 2004.
48. M. Knoppers, M. Hirtle, K. Glass, Science1999, 286, 2277–2278.
49. V. Shiva, Biopiracy: The Plunder of Natureand Knowledge, South End Press, Boston,1996.
50. R. Crespi, Sci. Eng. Ethics 2000, 6(2),157–180.
51. Human Genome Organization (HUGO),Statement on Benefit Sharing, HUGO,Bethesda, MD, 2000.
52. M. Sturges, Am. Univ. Int. Rev. 1997, 13,219–261.
53. A useful discussion of the common heritageargument. P. Ossario, Common heritagearguments and the patenting of DNAin Perspectives on Gene Patenting (Ed.:A. Chapman), American Association for theAdvancement of Science, Washington, DC,1999, pp. 89–110.
54. D. Resnik, Health Policy 2003, 65, 181–197.55. E. Clayton, K. Steinberg, M. Khoury,
E. Thomson, L. Andrews, M. Kahn,L. Kopelman, J. Weiss, J. Am. Med. Assoc.1995, 274, 1786–1792.
56. R. Sharp, M. Foster, J. Law, Med. Ethics 2000,28(1), 41–49.
57. World Medical Association, J. Am. Med.Assoc. 2000, 284, 3043–3046.
58. A useful discussion of ethical and policyissues relating research involving humanbiological materials. National BioethicsAdvisory Commission (NBAC), ResearchInvolving Human Biological Materials:Ethical Issues and Policy Guidance, NBAC,Washington, DC, 1998.

201
11Drug Approval in the EuropeanUnion and the United States
Gary WalshUniversity of Limerick, Limerick City, Ireland
11.1Introduction
The pharmaceutical sector is arguablythe most highly regulated industry inexistence. Legislators in virtually all theregions of the world continue to en-act/update legislation, controlling everyaspect of pharmaceutical activity. Interpre-tation, implementation, and enforcementof these laws is generally delegated bythe lawmakers to dedicated agencies. Therelevant agencies within the EuropeanUnion (EU) and the United States (USA)are the European Medicines EvaluationAgency (EMEA) and the US Food andDrug Administration (FDA), respectively.This monograph focuses upon the struc-ture, remit, and operation of both theseorganizations, specifically in the context ofbiopharmaceutical products.
11.2Regulation within the European Union
11.2.1The EU Regulatory Framework
The founding principles of what we nowcall the European Union are enshrined in
the treaty of Rome, initially adopted bysix countries in 1957. While this treatycommitted its signatories to a range of co-operation and harmonization measures,it largely deferred health care–related is-sues to individual member states. As aconsequence, each member state draftedand adopted its own set of pharmaceu-tical laws, enforced by its own nationalregulatory authority. Although the mainprinciples underpinning elements of na-tional legislation were substantially similarthroughout all European countries, de-tails did differ from country to country.As a result, pharmaceutical companiesseeking product-marketing authorizationswere forced to apply separately to eachmember state. Uniformity of regulatoryresponse was not guaranteed and eachcountry enforced its own language require-ments, scale of fees, processing times, andso on. This approach created enormousduplication of effort, for companies andregulators alike.
In response, the European Commis-sion (EC, Brussels) began a determinedeffort to introduce European-wide phar-maceutical legislation in the mid-1980s.The commission represents the EU body
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

202 11.2 Regulation within the European Union
with responsibility for drafting (andsubsequently ensuring the implementa-tion) of EU law, including pharmaceuticallaw. In pursuing this objective, it has at itsdisposal two legal instruments, ‘‘regula-tions’’ and ‘‘directives’’. Upon approval, aregulation must be enforced immediatelyand without alteration by all EU memberstates. A directive, in contrast, is a ‘‘softer’’legal instrument, requiring member statesonly to introduce its ‘‘essence’’ or ‘‘spirit’’into national law.
By the early 1990s, some 8 regulationsand 18 directives had been introduced,which effectively harmonized pharmaceu-tical law throughout the European Union.In addition to making available the leg-islative text, the European Commissionhas also facilitated the preparation andpublication of several adjunct documentsdesigned to assist industry and other inter-ested parties to interpret and conform tothe legislative requirements. Collectivelythese documents are known as ‘‘the rulesgoverning medicinal products in the Eu-ropean union’’ and they make compulsoryreading for those involved in any aspectof pharmaceutical regulation. The nine-volume (Table 1) publication is regularlyupdated and hard copies may be pur-chased from the commission’s publication
office [1] or may be consulted/downloaded(for free) from the relevant EU website [2].
11.2.2The EMEA
Harmonization of pharmaceutical lawmade possible the implementation of anEU wide system for the authorizationand subsequent supervision of medicinalproducts. The EMEA was set up tocoordinate and manage the new system [3].Based in Canary Wharf, London, theagency became operational in 1995. TheEMEA mission statement is ‘‘to contributeto the protection and promotion of publicand animal health’’. It seeks to achievethis by
• providing high-quality evaluation ofmedicinal products;
• advising on relevant R & D programs;• providing a source of drug and other
relevant information to health careprofessionals/users;
• controlling the safety of medicines forhumans and animals.
An outline structure of the EMEA isprovided in Fig. 1. From a technical stand-point, the most significant organizational
Tab. 1 The volumes comprising ‘‘the rules governing medicinal products within the Europeanunion’’
Volume Title
1 Pharmaceutical legislation: Medicinal products for human use2 Notice to applicants: Medicinal products for human use3 Guidelines. Medicinal products for human use4 Good manufacturing practices. Medicinal products for human and veterinary use5 Pharmaceutical legislation: Veterinary medicinal products6 Notice to applicants. Veterinary medicinal products7 Guidelines. Veterinary medicinal products8 Maximum residue limits. Veterinary medicinal products9 Pharmacovigilance. Medicinal products for human and veterinary use

Drug Approval in the European Union and the United States 203
Management board
Executive director
Preauthorizationevaluation of medicinesfor human use
Postauthorizationevaluation of medicinesfor human use
Veterinary medicinesand inspections
Committee forOrphan MedicinalProducts (COMP)
Committee forProprietary MedicinalProducts (CPMP)
Committee forVeterinary MedicinalProducts (CVMP)
Fig. 1 Simplified structural overview of the EMEA. Refer to [3] for further details.
structures are the following:
• The unit for preauthorization evaluationof medicines for human use.
• The unit for post authorization evalua-tion of medicines for human use.
• The unit for veterinary medicines andinspections.
A more detailed description of these unitsand their responsibilities is available onthe EMEA home page [3]. Two additionalstructural units also exist: administration,and communications and networking.
From a drug approval perspective, at theheart of the functioning of the EMEA arethree key scientific committees:
• The Committee for Proprietary Medici-nal Products (CPMP)
• The Committee for Veterinary Medici-nal Products (CVMP)
• The Committee for Orphan MedicinalProducts (COMP).
Each committee is composed of a numberof (mainly technical) experts, the majority
of whom are drawn from the nationaldrug regulatory authorities of each EUmember state (Table 2). The functionof these committees in the context ofnew biotechnology drug approvals will bediscussed in the next section. In additionto these three committees, the EMEAhas at its disposal a bank of some 3000European technical experts (the majority ofwhom, again, are drawn from the nationalregulatory authorities). The EMEA drawsupon this expert advice as required.
11.2.3New Drug Approval Routes
The rules governing medicinal productsin the European Union provide for twoindependent routes by which new poten-tial medicines may be evaluated. Theseare termed the ‘‘centralized’’ and ‘‘decen-tralized’’ procedures, respectively, and theEMEA plays a role in both [4]. The central-ized procedure is compulsory for biotechmedicines and as such is described ingreatest detail below. This route may alsobe used to evaluate new chemical entities.

204 11.2 Regulation within the European Union
Tab. 2 National drug regulatory authorities of the 15 current EU member states. Internetaddresses given if listed in the EMEA annual report, 2002
Austria BelgiumFederal ministry for labour, health and
social affairs, Wien.Ministere des affaires socials de la sante
publique et de l’environment, Brussleshttp://www.afigp.fogv.be
Denmark FinlandDanish medicines agency, Bronshoj.
http://www.dkma.dkNational agency for medicines, Helsinki
France GermanyAgence Francaise de securite sanitaire des
produits de sante, Saint Denis, Cedexhttp://www.afssaps.sante.fr
BfArM, Bonn http://www.bfarm.de
Greece IrelandNational organization for medicines, Athens Irish medicines board, Dublin
http://www.imb.ieItaly LuxembourgDipartimento della valutazione dei
medicinali e della farmacovigilanza,Rome http://www.sanita.it/farmaci
Division de la pharmacie et desmedicaments, Luxembourg
The Netherlands PortugalMedicines evaluation board, Den Haag
http://www.cbg-meb.nlInfarmed, Lisbona http://www.infarmed.pt
Spain SwedenAgencia Espanola del medicamento, Madrid
http://www.agemed.esMedical products agency, Uppsala
http://www.mpa.seUKMedicines control agency, London.
http://www.gov.uk/mca
11.2.3.1 The Centralized ProcedureUnder the centralized route, marketingauthorization applications (dossiers) aresubmitted directly to the EMEA. Beforeevaluation begins, the EMEA staff first val-idate the application, by scanning throughit to ensure that all necessary informationis present and presented in the cor-rect format. This procedure usually takesone to two working weeks to complete.Biotech-based dossiers are termed ‘‘partA applications’’, whereas new chemicalentities are termed ‘‘part B applications’’.
The validated application is then pre-sented at the next meeting of the CPMP(human medicine applications) or CVMP(veterinary medicines). This committee
then appoints one of its members to act as‘‘rapporteur’’ for the application. The rap-porteur organizes technical evaluation ofthe application (product safety, quality, andefficacy), and this evaluation is often car-ried out in the rapporteur’s home nationalregulatory agency. Another member of thecommittee (a corapporteur) is often alsoappointed to assist in this process. Uponcompletion of the evaluation phase, therapporteurs draw up a report, which theypresent, along with a recommendation,at the next CPMP (or CVMP) meeting.After discussion, the committee issues ascientific opinion on the product, eitherrecommending acceptance or rejection ofthe marketing application. The EMEA then

Drug Approval in the European Union and the United States 205
transmits this scientific opinion to theEuropean Commission in Brussels (whorepresent the only body with the legalauthority to actually grant marketing au-thorizations). The Commission, in turn, is-sues a final decision on the product (Fig. 2).
Regulatory evaluation of marketing au-thorization applications must be com-pleted within strict time limits. The EMEAis given a 210-day window to evaluatean application and provide a scientificopinion. However, during the applica-tion process, if the EMEA officials seekfurther information/clarification on anyaspect of the application, this 210-day‘‘clock’’ stops until the sponsoring com-pany provides satisfactory answers. Theaverage duration of active EMEA evalua-tion of biotech-based product applicationsis in the region of 175 days, well withinthis 210-day time frame. The duration ofclock stops can vary widely – from 0 daysto well over 300 days. Most applications,however, incur clock stops of the orderof 30 to 80 days. Upon receipt of the
EMEA opinion, the commission is given amaximum of 90 days in which to translatethis opinion into a final decision. Overalltherefore, the centralized process shouldtake a maximum of 300 ‘‘active’’ evaluationdays. EMEA opinions provided in 2002 forboth human- and veterinary-based biotechdrugs are listed in Table 3.
11.2.3.2 Mutual RecognitionThe second route facilitating product au-thorization is termed ‘‘mutual recogni-tion’’ or the ‘‘decentralized procedure’’.This is open to non-biotechnology prod-ucts, and the procedure entails the initialsubmission of an authorization appli-cation to a single national regulatoryagency of an EU member state (Table 2).This agency then assesses the application(within 210 days), formulates an opinion,and either grants or rejects the application.If authorization is granted, the sponsor-ing company may then apply via ‘‘mutualrecognition’’ to extend the market au-thorization to the remaining EU states.
Fig. 2 Overview of the EUcentralized procedure. Refer totext for details.
Marketing authorizationapplication submitted
Final Commission decision
Validation & presentation atnext CPMP or CVMP meeting
CPMP (or CVMP)scientific opinion issued
Opinion transmitted toEuropean Commission
Evaluation,210 days, maximum
Commission evaluation,90 days, maximum
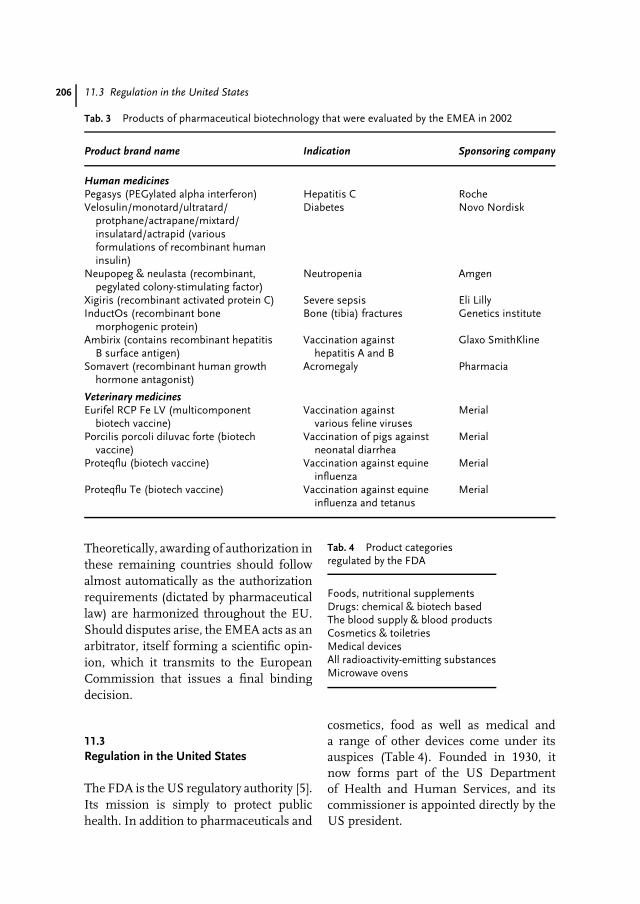
206 11.3 Regulation in the United States
Tab. 3 Products of pharmaceutical biotechnology that were evaluated by the EMEA in 2002
Product brand name Indication Sponsoring company
Human medicinesPegasys (PEGylated alpha interferon) Hepatitis C RocheVelosulin/monotard/ultratard/
protphane/actrapane/mixtard/insulatard/actrapid (variousformulations of recombinant humaninsulin)
Diabetes Novo Nordisk
Neupopeg & neulasta (recombinant,pegylated colony-stimulating factor)
Neutropenia Amgen
Xigiris (recombinant activated protein C) Severe sepsis Eli LillyInductOs (recombinant bone
morphogenic protein)Bone (tibia) fractures Genetics institute
Ambirix (contains recombinant hepatitisB surface antigen)
Vaccination againsthepatitis A and B
Glaxo SmithKline
Somavert (recombinant human growthhormone antagonist)
Acromegaly Pharmacia
Veterinary medicinesEurifel RCP Fe LV (multicomponent
biotech vaccine)Vaccination against
various feline virusesMerial
Porcilis porcoli diluvac forte (biotechvaccine)
Vaccination of pigs againstneonatal diarrhea
Merial
Proteqflu (biotech vaccine) Vaccination against equineinfluenza
Merial
Proteqflu Te (biotech vaccine) Vaccination against equineinfluenza and tetanus
Merial
Theoretically, awarding of authorization inthese remaining countries should followalmost automatically as the authorizationrequirements (dictated by pharmaceuticallaw) are harmonized throughout the EU.Should disputes arise, the EMEA acts as anarbitrator, itself forming a scientific opin-ion, which it transmits to the EuropeanCommission that issues a final bindingdecision.
11.3Regulation in the United States
The FDA is the US regulatory authority [5].Its mission is simply to protect publichealth. In addition to pharmaceuticals and
Tab. 4 Product categoriesregulated by the FDA
Foods, nutritional supplementsDrugs: chemical & biotech basedThe blood supply & blood productsCosmetics & toiletriesMedical devicesAll radioactivity-emitting substancesMicrowave ovens
cosmetics, food as well as medical anda range of other devices come under itsauspices (Table 4). Founded in 1930, itnow forms part of the US Departmentof Health and Human Services, and itscommissioner is appointed directly by theUS president.

Drug Approval in the European Union and the United States 207
The FDA derives its legal authorityfrom the federal food, drug, and cosmetic(FD&C) act. Originally passed into law in1930, the act has been updated/amendedseveral times since. The FDA interpretsand enforces these laws. Although thereare many parallels between the FDA andthe EMEA, its scope is far broader thanthat of the EMEA and its organizationalstructure is significantly different. Over-all, the FDA now directly employs some9000 people, has an annual budget in theregion of US$1 billion and regulates overUS$1 trillion worth of products annually(Table 4). A partial organizational struc-ture of the FDA is presented in Fig. 3. Inthe context of pharmaceutical biotechnol-ogy, the centre for Drug Evaluation and Re-search (CDER) and, in particular, the Cen-tre for Biologics Evaluation and Research(CBER) are the most relevant FDA bodies.
11.3.1CDER and CBER
A major activity of CDER is to eval-uate new drugs and decide if marketauthorization should be granted or not. Ad-ditionally, CDER also monitors the safety
and efficacy of drugs already approved(i.e. post marketing surveillance and re-lated activities). CDER predominantly reg-ulates ‘‘chemical’’-based drugs (i.e. drugswhich are usually of lower molecularweight and often manufactured by directchemical synthesis). Included are prescrip-tion, generic, and over-the-counter drugs.CDER also regulates some products ofpharmaceutical biotechnology, includingrecombinant hormones (e.g. recombinantinsulins and gonadotrophins) and certaincytokines (e.g. recombinant interferons).
The CBER undertakes many activitiessimilar to that of CDER, but it focusesupon biologics and related products. Theterm ‘‘biologic’’ historically has a specificmeaning, relating to ‘‘a virus, therapeu-tic serum, toxin, antitoxin, vaccine, blood,blood components or derivatives, or al-lergenic products that are used in theprevention, treatment, or cure of diseasesof human beings’’ [6]. CDER thereforeregulates products such as vaccines andblood factors, whether they are producedby traditional or modern biotechnologicalmeans (i.e. by nonrecombinant or re-combinant means). Additional ‘‘biologicalproducts’’, including cell, gene therapy,
FDA
Centre for Drug Evaluation& Research (CDER)
Centre for Biologics Evaluation& Research (CBER)
Centre for VeterinaryMedicine (CVM)
Centre for Devices& RadiologicalHealth (CDRH)
Centre for Food Safety &Applied Nutrition
Fig. 3 Partial organizational structure of the FDA.

208 11.3 Regulation in the United States
and tissue-based products also fall underthe auspices of CBER.
11.3.2The Approvals Procedure
The overall procedure by which biotech-nology and other drugs are evaluated andapproved by CDER or CBER are pre-dictably very similar, although some ofthe regulatory terminology used by thesetwo centers differ. A summary overview ofthe main points along the drug develop-ment/approval road where CDER/CBERplay key regulatory roles is provided inFig. 4.
Once a sponsor (company, research in-stitute, etc.) has completed the preclinicalevaluation of a proposed new drug, itmust gain FDA’s approval before institut-ing clinical trials. The sponsor seeks thisapproval by submitting an investigationalnew drug (IND) application to either CDERor CBER, as appropriate. The application,which is a multivolume work of severalthousand pages, contains information de-tailing preclinical findings, methods ofproduct manufacture, and proposed proto-cols for initial clinical trials. The regulatoryofficials then assess the data provided andmay seek more information/clarificationfrom the sponsor if necessary. Evaluationis followed by a decision to either permitor block clinical trials. Should clinical
trials commence, the sponsor and regu-latory officials hold regular meetings inorder to keep the FDA appraised of trialfindings. Upon successful completion ofclinical trials, the sponsor then usuallyapplies for marketing authorization. InCDER speak, this application is termeda new drug application (an NDA). NDAsusually consist of several hundred volumescontaining over 100 000 pages in total.The NDA contains all the preclinical aswell as clinical findings and other perti-nent data/information. Upon receipt of anNDA, the CDER officials check throughthe document ensuring completeness (aprocess similar to the EMEA’s validationphase). Once satisfied, they ‘‘file’’ the ap-plication and evaluation begins.
The NDA is reviewed by various reg-ulatory experts, generally under topicheadings such as ‘‘medical,’’ ‘‘pharmacol-ogy,’’ ‘‘chemistry,’’ ‘‘biopharmaceutical,’’‘‘statistical,’’ and ‘‘microbiology’’ reviews.Reviewers may seek additional informa-tion/clarification from the sponsor as theyfeel necessary. Upon review completion,the application is either approved or re-jected. If approved, the product may goon sale but regulatory officials continue tomonitor its performance (postmarketingsurveillance). Should unexpected/adverseevents be noted, the regulatory authorityhas the legal power (and responsibility) to
Preclinicaltesting
General medical useApprovalClinicaltrials
IND application
Regular regulatorymeetings
NDA/BLAapplication
Postmarketing surveillance
Fig. 4 Summary overview of the main points during a drug’s lifetime at which the FDA plays a keyregulatory role. Refer to text and Ref. [5] for further details.

Drug Approval in the European Union and the United States 209
suspend/revoke/modify the approval, asappropriate.
The review process undertaken by CBERofficials upon biologic and related prod-ucts is quite similar to that describedabove for CDER-regulated product. CBER-regulated investigational drugs may enterclinical trials subject to gaining IND sta-tus. The application process for marketingauthorization undertaken by the sponsorsubsequent to completion of successfulclinical trials is termed the licensure phasein CBER terminology. The actual productapplication is known as a biologics licenceapplication (BLA). Overall, the content andreview process for a BLA is not dissimi-lar to that of the analogous CDER NDAprocess, as discussed above. The bottomline is that the application must supportthe thesis that the product is both safeand effective and that it is manufacturedand tested to the highest quality standards.Overall, the median time between submis-sion and approval of product marketingapplication to CBER/CDER stands at ap-proximately 12 months.
While the majority of biotech-baseddrugs are regulated in the United Statesby either CBER or CDER, it is worth not-ing that some such products fall outsidetheir auspices. Bone morphogenic pro-teins (BMPs) function to stimulate boneformation. As such, several have been ap-proved for the treatment of slow-healingbone fractures. Product ‘‘administration’’requires surgical implantation of the BMPin the immediate vicinity of the fracture,usually as part of a supporting device. Assuch, in the United States, these productsare regulated by the FDA’s Centre for De-vices and Radiological Health (CDRH) [7].Drugs (both biotech and nonbiotech) des-tined for veterinary use also fall outsidethe regulation of CBER or CDER. Mostsuch veterinary products are regulated by
the FDA’s Centre for Veterinary Medicine(CVM), although veterinary vaccines (andrelated products) are regulated not by theFDA but by the Centre for Veterinary Bi-ologics (CVB), which is part of the USDepartment of Agriculture [8].
11.4International Regulatory Harmonization
Europe, the United States, and Japanrepresent the three main global pharma-ceutical markets. As such, pharmaceuticalcompanies usually aim to register mostnew drugs in these three key regions.Although the underlining principles aresimilar, detailed regulatory product au-thorization requirements differ in thesedifferent regions, making necessary someduplication of registration effort. The in-ternational conference on harmonizationof technical requirements for registra-tion of pharmaceuticals for human use(the ICH process) is an initiative aimedat harmonizing regulatory requirementsfor new drug approvals in these re-gions. The project was established in1990 and brings together both regula-tory and industry representatives fromEurope, the United States, and Japan.ICH is administered by a steering com-mittee consisting of representatives of theabove-mentioned groupings. The steer-ing committee in turn is supported byan ICH secretariat, based in Geneva,Switzerland [9]. The main technical work-ings of ICH are undertaken by expertworking groups charged with developingharmonizing guidelines. The guidelinesare grouped under one of the followingheadings:
• Efficacy (clinical testing and safetymonitoring–related issues)

210 11.4 International Regulatory Harmonization
Tab. 5 Finalized ICH guidelines that specifically focus upon products ofpharmaceutical biotechnology
Guideline number Guideline title
Q5A Viral safety evaluation of biotechnology productsQ5B Quality of biotechnology products: analysis of the
expression construct in cells used for theproduction of rDNA-derived products
Q5C Quality of biotechnological products: stability testingof biotechnological/biological products
Q5D Quality of biotechnological products: derivation andcharacterization of cell substrates used forproduction of biotechnological/biological products
Q6B Specifications: test procedures and acceptance criteriafor biotechnological/biological substances
S6 Preclinical safety evaluation of biotechnology-derivedpharmaceuticals
• Quality (pharmaceutical developmentand specifications)
• Safety (preclinical toxicity and relatedissues)
• Multidisciplinary (topics not fitting theabove descriptions).
Thus far, 37 guidelines aimed at both tra-ditional and biotechnology-based productshave been produced and are being imple-mented (Table 5). One of the ICHs mostambitious initiatives to date has been thedevelopment of the common technical doc-ument. This provides a harmonized formatand content for new product authorizationapplications within the European Union,the United States, and Japan. When thisand the other guidelines are fully im-plemented, considerable streamlining ofthe drug development and, in particular,registration process will be evident. This
will make more economical use of boththe company’s and regulatory authorities’time, will reduce the cost of drug develop-ment and speed up the drug developmentprocedure, ensuring faster public access tonew drugs.
References
1. Office for Official Publications ofthe European Communities, http://www.publications.eu.int.
2. http://www.pharmacos.eudra.org/.3. http://www.emea.eu.int/.4. G. Walsh, Nat. Biotechnol. 1999, 17, 237–240.5. http://www.fda.gov/.6. http://www.fda.gov/cber/index.html.7. http://www.fda.gov/cdrh/index.html.8. http://aphis.usda.gov/vs/cvb/.9. http://www.ich.org/.

Part IVBiotech 21 – Into the NextDecade
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

213
12Rituximab: Clinical Developmentof the First Therapeutic Antibodyfor Cancer
Antonio J. Grillo-LopezNeoplastic and Autoimmune Diseases Research Institute, Rancho Santa Fe, CA, USA
12.1Introduction
After many years of research, monoclonalantibodies (Mabs) were a source of dis-appointment to many but a small coregroup of investigators [1]. In about oneyear, between 1991 and 1992, rituximabwas engineered as a chimeric anti-CD20Mab. Then, in late 1992, the investiga-tional new drug (IND) application forrituximab (Rituxan, MabThera) was sub-mitted and clinical trials were initiated inFebruary 1993 (Table 1) [2]. In the year2002, rituximab became the number one,brand name, cancer therapeutic productin the world (approximately US$1.3 billionin sales). Over 300 000 patients have beentreated with rituximab. The events of theintervening 10 years changed the courseof history. The clinical development phasewas completed in record time for a lym-phoma agent (three years from the firstto the last patient enrolled) and includedcombination trials with chemotherapy, bi-ologicals, and radioimmunotherapy. TheInternational Workshop Response Crite-ria for non-Hodgkin’s lymphoma (NHL)had their origin in the criteria used forthe rituximab clinical trials and were
validated using the rituximab database.The dossier filed simultaneously with theFDA in the United States and with theEMEA in Europe was available in elec-tronic format. This was the first electronicBiologics License Application (BLA) filedunder the FDA’s developing electronicsubmission standards. For the first time,a Mab used as a single agent showedsufficient clinical activity to warrant world-wide approvals for a cancer indication.For the first time, a Mab was approvedspecifically for the treatment of patientswith NHLand an agent (combined withCyclophosphamide HydroxydaunorubieinOncovin Preonisone (CHOP) chemother-apy) was shown to be superior to CHOPalone in patients with aggressive NHL – anew ‘‘gold standard’’ was established.Rituximab + CHOP produced a signifi-cant increase in overall survival (OS)as compared to CHOP alone. The Ze-valin treatment regimen includes ritux-imab as the cold antibody. In February2001, Zevalin became the first radioim-munotherapy approved for the treatmentof NHL. Rituximab has surpassed its orig-inal indications, and today, it is beinginvestigated in clinical trials for a varietyof autoimmune disorders. Enthusiasm for
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8
214 12.2 Clinical Development and Regulatory Approvals
Tab. 1 Milestones in the history of rituximab
Date Milestone
January 1991 Mice immunized with human CD20 antigen. Anti-CD20 antibodies isolated. Parentantibody for rituximab (murine anti-CD20 antibody, IDEC-2B8) identified andcharacterized.
June 1991 Chimeric antibody IDEC-C2B8 engineered with murine variable and humanconstant regions (IgG1 kappa isotype).
August 1991 Vector engineered, introduced by electroporation into CHO cells, antibodyproduced by fermentation process.
December 1992 IND filed with the US FDA – 1st Phase I trial protocol submitted.February 1993 First patient treated with rituximab (single dose Phase I trial)April 1994 First patient treated on the 1st combination study – CHOP + rituximab in LG/NHL.March 1995 First patient treated on the Phase III (pivotal) trial that led to the approval of
rituximab by regulatory authorities in the United States and Europe.March 1996 Last patient entered on the Phase III (pivotal trial).February 1997 Biologics License Application (BLA) filed with the US FDA and simultaneous filing
of the European dossier with the EMEA.July 1997 Biologics Response Modifiers Advisory Committee of the US FDA meets in
Washington DC and recommends approval.November 1997 Rituximab approved in the United States by the FDA for patients with
LG/NHL – 1st Mab approved for the treatment of cancer, 1st Mab approved forthe treatment of NHL.
June 1998 Rituximab approved in Europe by the EMEA for LG/NHL.December 2000 Results of the Grupe Dej Etudes De Lymphome D’adultes (GELA) study presented
at the American Society of Hematology meeting showing statistically significantsuperiority of CHOP + rituximab over CHOP alone in patients with IntermediateGrade NHL.
October 2001 Committee for Proprietary Medicinal Products (CPMP) of the EMEA recommendsapproval of rituximab for patients with Intermediate Grade NHL.
December 2002 Rituximab named the number one selling, brand name, cancer therapeutic productin the world (approximately US$1.3 billion).
clinical and laboratory research in the areaof antibody therapeutics was renewed andgrew in an accelerated fashion yieldingnumerous new Mabs for cancer and au-toimmune diseases, some of which arealready approved [3]. This chapter providesinformation and insight as to how thesehistorical events came about.
12.2Clinical Development and RegulatoryApprovals
The IND for rituximab was submittedto the US FDA in December 1992 [4].
The anti-CD20 Mab had been purposelyengineered with human constant regions(IgG1 kappa isotype) to ensure that itwould effectively bind complement and,through Fc receptors, effector cells so thatit could effect complement-dependent cy-totoxicity (CDC) and antibody-dependentcellular cytotoxicity (ADCC) [5, 6] (Fig. 1).This was confirmed in vitro. Additionally,in vivo experiments in monkeys revealedimmediate, profound, and specific B-celldepletion with recovery within 100 days.The effects of the antibody on the humanimmune system were unknown. It wasexpected to produce B-cell depletion and

Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 215
B-cell
Rituximab
CD20
CDCComplement
Effector cells ADCC
− Variable regions−murine− Constant regions−human
Apoptosis
Fig. 1 Mechanism of action of rituximab. The chimeric (mouse/human)antibody, rituximab, binds to the CD20 antigen on B-cells and(a) activates complement to effect CDC, (b) attracts effector cells via Fcreceptors to effect ADCC, and (c) transmits a signal into the cell toinduce apoptosis. (See Color Plate p. xxii).
to decrease lymphomatous nodes, masses,and infiltrates. Its effects on immunoglob-ulin levels were unknown. The timing andduration of B-cell depletion could not beaccurately predicted from the animal stud-ies. Thus, the initiation of the first PhaseI, single dose, clinical trial was delayed for2 months (beyond the usual 30-day wait)because of the FDA’s safety concerns andspecifically their concerns regarding ef-fects on immunoglobulins and on B-cells.Eventually, an agreement was reached ona starting dose of 10 mg m−2. Today, weknow that this dose represents less than1% of the total dose that patients receiveover four infusions (about 3 gm for the av-erage patient). Clinical trials were initiatedin February 1993.
12.2.1Clinical Development
The clinical development of ritux-imab was conducted entirely by IDEC
Pharmaceuticals Corporation, San Diego,California. The Division of Medical Re-search and Regulatory Affairs (M&RA) atIDEC was responsible for all aspects ofclinical development and regulatory inter-actions worldwide including preparationand defence of the BLA and European sub-missions through the review process andto approvals. The clinical trials were alldesigned, implemented, conducted, ana-lyzed, interpreted, and reported by a smallgroup of professionals (staff of seven in1992) at IDEC’s M&RA division. Investiga-tors were chosen from important academicinstitutions in the United States andCanada. A Clinical Research and Develop-ment Agreement (CRADA) was initiatedwith the US NCI around 1996. However,the US NCI did not participate in the devel-opment studies. The first study conductedunder US NCI sponsorship was the CHOPversus R + CHOP intergroup study thatstarted in 1997.

216 12.2 Clinical Development and Regulatory Approvals
IDEC began collaborations with Genen-tech Inc., San Francisco, California, inMarch 1995, for the manufacturing, mar-keting, and sales of rituximab in theUnited States. Shortly thereafter, collabo-rations began with F. Hoffmann-La RocheLtd., Basel, Switzerland, for the develop-ment of rituximab in the European Union,and with Zenyaku Kogyo Co., Ltd, Tokyo,Japan, for the development of rituximab inJapan.
12.2.2The Clinical Development Plan
The clinical development plan was de-signed to achieve an early approval basedon single-agent efficacy in patients withrelapsed or refractory LG/F NHL. Re-sources were limited and it was notpossible to conduct large randomizedtrials or to pursue an additional indi-cation (such as aggressive NHL). Thus,the plan relied on single-arm studiesthat utilized surrogate endpoints (e.g. re-sponse rates) and qualified for approvalunder ‘‘accelerated approval’’ guidelines.However, three pilot studies of differentcombinations were carried out (combina-tion trials with chemotherapy, biologicals,and radioimmunotherapy). These were
considered important as it was clear thatthe Mab would eventually be used as partof a combination or multimodality therapyand not just as a single agent. If success-ful, these pilot studies could lead to largerrandomized trials. The single-agent stud-ies in patients with LG/F NHL includedPhase I single dose, Phase I/II multipledose, Phase II in patients with bulky dis-ease, Phase II re-treatment, Phase II 8infusion, and Phase III (pivotal) studies.The first patient was treated in Febru-ary 1993 and the last patient (includedin the regulatory dossiers) was enrolled inFebruary 1996. Completion of enrolmentin all of these studies in a three-year pe-riod established a record in NHL wheremost cooperative group studies take yearsto complete. An aggressive but realisticclinical development plan, including bothsingle-agent and combination studies, setthe pace that made these achievementspossible.
12.2.3Clinical Trials Methodology
The methodology utilized in the imple-mentation, conduct, analysis, and inter-pretation of the clinical development plan
Tab. 2 Key investigators in rituximab clinical trials
Investigator Site Study participation
David Maloney, MD Stanford U. Med. C. 102–01, 02, 05Thomas Davis, MD Stanford U. Med. C. 102–07, 08Peter McLaughlin, MD M.D. Anderson C.C. 102–05, PKMyron Czuczman, MD Roswell Park C.C. 102–03, 05, 06Neil Berinstein, MD Toronto-Sunnybrook C.C. 102–05, PKLarry Piro, MD Scripps Clinic 102–06, PK
Notes: U: University; C: Center; CC: Cancer Center;Many other investigators and staff at investigational sites made importantcontributions and are not listed because of space constraints.

Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 217
was critical to its success. A limited num-ber of academic institutions (about 30in the United States and Canada) werechosen to participate in the clinical tri-als and most enrolled patients in two ormore studies (Table 2). This served severalpurposes: (a) the staff at these sites were in-structed on clinical trials methodology onlyonce as it was consistent across all stud-ies, (b) the investigator’s meetings couldaddress several studies and the overallnumber of meetings was decreased, (c) theinvestigators and the company staff inter-acted more frequently and more efficiently,(d) the site staff became experts at study-ing drug administration as well as safetyand efficacy monitoring and reporting. Therequired bureaucracy (including clinicaltrials agreements, confidentiality agree-ments, adverse event reporting, queriesand audit trails, accounting for experi-mental drug, etc.) was consistent and thuscould be simplified and minimized. Proto-cols, Case Report forms (CRFs), and datacollection were standardized. Importantly,a peer-level relationship based on mutualprofessional respect was established be-tween the investigators and their staff andthe company clinician and staff.
Clinical trials conducted during develop-ment (as described below in Sects. 12.2.6and 12.2.7) were similar in a number ofways. Inclusion and exclusion criteria werealmost identical across studies. These stud-ies focused on the treatment of patientswith relapsed or refractory LG/F NHL.Patients were dosed at 375 mg m−2 of rit-uximab by intravenous infusion weekly fora total of four doses. This consistency re-sulted in a degree of homogeneity thatallowed for analyses across studies as wellas comparisons between studies. Any ex-ceptions to these general rules are notedin the individual study descriptions below.
12.2.4Response Criteria
Defining a set of clinical response criteriawas a difficult task. At the time the clinicaltrials started in 1993, the WHO and East-ern Cooperative Oncology Group (ECOG)criteria were being utilized [7, 8]. Histor-ically, these criteria had been developedfor the efficacy evaluation of patients withsolid tumors. There were no standard crite-ria for NHL. The WHO and ECOG criteriawere inadequate for the evaluation of NHLpatients as they were based on the dis-appearance of a tumor mass, whereas inlymphoma the ‘‘tumor mass’’ is in parta normal anatomical structure, a lymphnode, that may decrease in size but willnot disappear. We convened a panel ofNHL experts from the United States todraft lymphoma-specific criteria for therituximab clinical trials [9]. These criteriawere subsequently endorsed by a groupof European NHL experts [10]. In Octo-ber 1996, these rituximab NHL responsecriteria were reviewed and approved bythe Biologic Response Modifiers AdvisoryCommittee of the US FDA [11, 12]. Athird-party blinded panel of NHL experts(Lymphoma Experts Confirmation of Re-sponse, LEXCOR) evaluated patients in therituximab Phase III (pivotal) trial by apply-ing these criteria [13, 14, 15]. The criteriawere accepted by the US FDA in 1997 [16].In February 1998, we collaborated withthe US NCI to convene an internationalworking group in order to reach a con-sensus on new response criteria for NHLthat could be accepted and applied world-wide. We invited, in addition to the USNHL experts, a number of internationalexperts from Europe and other areas in-cluding: Coiffier B (France), Connors JM(Canada), Lister TA (United Kingdom),Hagenbeek A (Netherlands), Hiddemann

218 12.2 Clinical Development and Regulatory Approvals
W (Germany), and others. The committee,at a meeting in Washington DC, drafteda set of criteria. These criteria were testedby application to the rituximab clinical tri-als database. This database included rawdata from all patients treated with ritux-imab in the clinical trials conducted duringdevelopment. The tumor measurementsfor these patients had been collected ini-tially at the investigational sites by theprincipal investigators, radiologists, andtheir staff. All CT scans were collectedon an ongoing basis and were subse-quently subject to a centralized and blindedreview by an independent (third party)panel of NHL experts (oncologists andradiologists) termed the LEXCOR panel.The LEXCOR panel included the follow-ing: Hematologists/Oncologists – ChesonB (US NCI), Horning S (Stanford U),Just R (San Diego), Kossman C (SanDiego), Morrison V (U Minn.), Peter-son B (CALGB), and Rosen P (UCLA);and Radiologists – Carter W (San Diego),Klippenstein D (Roswell Park), and Ko-rtman K (San Diego). These experts mea-sured all the lesions on each CT scan for ev-ery patient. Some patients had more than50 measurable lesions. To our knowledge,the resulting database (with bidimensionalmeasurements of all lesions) is the onlyone of its kind as investigators usuallymeasure only 6 to 10 ‘‘sentinel’’ lesions.
These International Working Group Re-sponse Criteria for NHL (IWRC), pub-lished in 1999, have become the stan-dard criteria for response evaluation inNHL [17] and have been applied to therituximab studies [18, 19].
12.2.5The Medical Research and RegulatoryAffairs Staff
The company staff included a core groupof experienced professionals: clinical
scientists, clinical research associates (sitemonitors), statisticians, medical writers,regulatory specialists, and others (Table 3).This core staff had years of experiencein cancer drug development in pharma-ceutical industry as well as in academiccenters. In the biotech world of 1992, hav-ing an experienced core clinical staff wasthe exception rather than the rule. Manybiotech companies were relatively smallwith inadequate funds and resources andtheir staff had limited clinical trials expe-rience. Frequently that experience was atthe level of clinical trials of institutional orcooperative group type and not the highlyregimented studies required by regulatoryagencies. Those who have not had the ex-perience of conducting a clinical trial thatmust meet worldwide regulatory require-ments do not comprehend the degree ofrigor, detail, accuracy, specificity, and clar-ity demanded of such studies. Such studiesrepresent the best clinical science and arenot just designed to meet, the sometimesarbitrary, regulatory requirements. In theacademic world, ‘‘peer review’’ is consid-ered to be the highest-level test that amanuscript must undergo in order to bepublished. This usually entails review bytwo or three anonymous reviewers withvarying degrees of expertise who will nothave access to the raw data. ‘‘Peer review’’of a regulatory dossier, the clinical trials,results, and interpretation, is a much moredetailed and rigorous process. The review-ers have access to the raw data and willreview it in detail. A representative sam-ple of the sites participating in the clinicaltrials will be audited. When a manuscriptfails peer review, it can be rewritten andresubmitted. When a regulatory dossierfails peer review, the consequences havea more significant impact, as the work ofmany years may have to be repeated. Thus,

Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 219
Tab. 3 Key IDEC Pharmaceuticals staff in rituximab clinical trials
Name and title Responsibilities
Antonio J. Grillo-Lopez, MD, Chief Medical Officerand Senior VP, Medical and Regulatory AffairsDivision
Chief Medical Officer and Project Clinician1992–2001.
Brian K. Dallaire, Pharm D, Senior Director, ClinicalOperations
Clinical Scientist and Divisional operations,plans, and resources 1993–2001.
Christine White, MD, Senior Director, Hematologyand Oncology
Safety Officer and Clinical Scientist1995–2001.
Chester Varns, Director, Clinical Trials Monitoring Clinical trials monitoring, studyimplementation, data acquisition.
Anne McClure, MS, Director, Medical Writing Medical writing.David Shen, PhD, Senior Director, Biometrics Biostatistics, data entry and analysis.Jay Rosenberg, PhD, Clinical Immunology LaboratoryJohn Leonard, PhD, Senior Director, Project Planning
and Regulatory AffairsProject planning and regulatory affairs.
Alice Wei, Senior Director, Regulatory Affairs Regulatory filings and interactions.
Notes: Clinical trials with rituximab began in 1993. The BLA was filed with the US FDA in February1997 and the MAA was filed simultaneously with the EMEA in Europe. Approval was granted in theUnited States in November 1997 and in Europe in June 1998. Many others at IDEC Pharmaceuticalsmade important contributions and are not listed because of space constraints.
having an experienced, professional, anddedicated clinical staff is invaluable.
Some companies chose to conduct theirwork through the use of consultantsand contractors. The so-called virtualcompany has been justified by the expectedfiscal efficiency and the lower overhead.However, consultants and contractorscan be more expensive than in-housepersonnel and will never have the degreeof loyalty and dedication. Continuity isa major problem as the outside staff isusually subject to greater turnover andchanges of assignment. The critical issue isloss of control. Someone other than you ishaving daily contact with the investigatorsand sites. Someone else is interacting withthe FDA. Importantly, the database is notheld by the company and the consistencyand quality of the data is at risk. All of thesefactors constitute the real price of having avirtual company. Virtual companies many
times generate costly ‘‘virtual data’’. It isimportant for the small biotech companiesto have their own clinical developmentstaff and thus hold in their own hands thereigns to their ultimate success.
12.2.6Phase I and I/II Clinical Trials
FDA and EMEA approvals of rituximabwere based on five single-agent studiesconducted primarily in patients with re-lapsed or refractory, low-grade or follicular,CD20+, B-cell NHL. Clinical trial resultsare listed in Table 4. Two of these werePhase I (single dose) or I/II (multiple dose)studies.
The first Phase I study, single rituximabinfusions ranging from 10 to 500 mg m−2
in 15 patients, reached the highest dosewithout dose-limiting toxicity [20]. Themaximum tolerated dose (MTD) was not
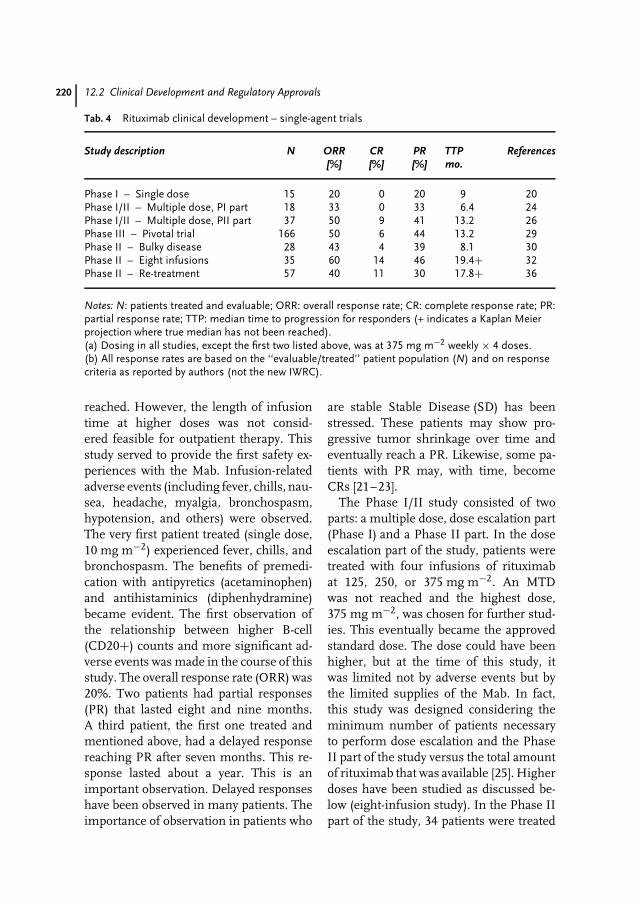
220 12.2 Clinical Development and Regulatory Approvals
Tab. 4 Rituximab clinical development – single-agent trials
Study description N ORR[%]
CR[%]
PR[%]
TTPmo.
References
Phase I – Single dose 15 20 0 20 9 20Phase I/II – Multiple dose, PI part 18 33 0 33 6.4 24Phase I/II – Multiple dose, PII part 37 50 9 41 13.2 26Phase III – Pivotal trial 166 50 6 44 13.2 29Phase II – Bulky disease 28 43 4 39 8.1 30Phase II – Eight infusions 35 60 14 46 19.4+ 32Phase II – Re-treatment 57 40 11 30 17.8+ 36
Notes: N: patients treated and evaluable; ORR: overall response rate; CR: complete response rate; PR:partial response rate; TTP: median time to progression for responders (+ indicates a Kaplan Meierprojection where true median has not been reached).(a) Dosing in all studies, except the first two listed above, was at 375 mg m−2 weekly × 4 doses.(b) All response rates are based on the ‘‘evaluable/treated’’ patient population (N) and on responsecriteria as reported by authors (not the new IWRC).
reached. However, the length of infusiontime at higher doses was not consid-ered feasible for outpatient therapy. Thisstudy served to provide the first safety ex-periences with the Mab. Infusion-relatedadverse events (including fever, chills, nau-sea, headache, myalgia, bronchospasm,hypotension, and others) were observed.The very first patient treated (single dose,10 mg m−2) experienced fever, chills, andbronchospasm. The benefits of premedi-cation with antipyretics (acetaminophen)and antihistaminics (diphenhydramine)became evident. The first observation ofthe relationship between higher B-cell(CD20+) counts and more significant ad-verse events was made in the course of thisstudy. The overall response rate (ORR) was20%. Two patients had partial responses(PR) that lasted eight and nine months.A third patient, the first one treated andmentioned above, had a delayed responsereaching PR after seven months. This re-sponse lasted about a year. This is animportant observation. Delayed responseshave been observed in many patients. Theimportance of observation in patients who
are stable Stable Disease (SD) has beenstressed. These patients may show pro-gressive tumor shrinkage over time andeventually reach a PR. Likewise, some pa-tients with PR may, with time, becomeCRs [21–23].
The Phase I/II study consisted of twoparts: a multiple dose, dose escalation part(Phase I) and a Phase II part. In the doseescalation part of the study, patients weretreated with four infusions of rituximabat 125, 250, or 375 mg m−2. An MTDwas not reached and the highest dose,375 mg m−2, was chosen for further stud-ies. This eventually became the approvedstandard dose. The dose could have beenhigher, but at the time of this study, itwas limited not by adverse events but bythe limited supplies of the Mab. In fact,this study was designed considering theminimum number of patients necessaryto perform dose escalation and the PhaseII part of the study versus the total amountof rituximab that was available [25]. Higherdoses have been studied as discussed be-low (eight-infusion study). In the Phase IIpart of the study, 34 patients were treated
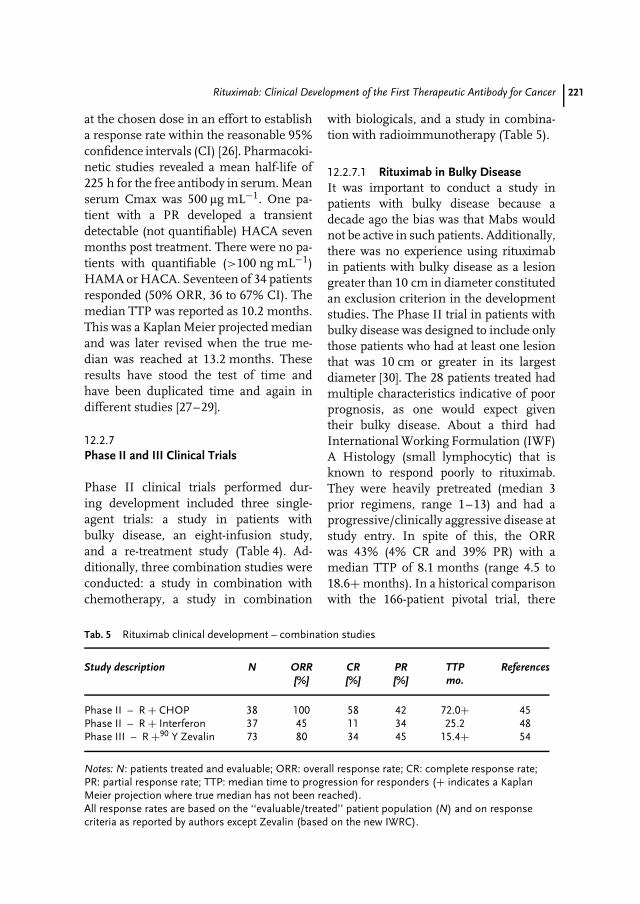
Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 221
at the chosen dose in an effort to establisha response rate within the reasonable 95%confidence intervals (CI) [26]. Pharmacoki-netic studies revealed a mean half-life of225 h for the free antibody in serum. Meanserum Cmax was 500 µg mL−1. One pa-tient with a PR developed a transientdetectable (not quantifiable) HACA sevenmonths post treatment. There were no pa-tients with quantifiable (>100 ng mL−1)HAMA or HACA. Seventeen of 34 patientsresponded (50% ORR, 36 to 67% CI). Themedian TTP was reported as 10.2 months.This was a Kaplan Meier projected medianand was later revised when the true me-dian was reached at 13.2 months. Theseresults have stood the test of time andhave been duplicated time and again indifferent studies [27–29].
12.2.7Phase II and III Clinical Trials
Phase II clinical trials performed dur-ing development included three single-agent trials: a study in patients withbulky disease, an eight-infusion study,and a re-treatment study (Table 4). Ad-ditionally, three combination studies wereconducted: a study in combination withchemotherapy, a study in combination
with biologicals, and a study in combina-tion with radioimmunotherapy (Table 5).
12.2.7.1 Rituximab in Bulky DiseaseIt was important to conduct a study inpatients with bulky disease because adecade ago the bias was that Mabs wouldnot be active in such patients. Additionally,there was no experience using rituximabin patients with bulky disease as a lesiongreater than 10 cm in diameter constitutedan exclusion criterion in the developmentstudies. The Phase II trial in patients withbulky disease was designed to include onlythose patients who had at least one lesionthat was 10 cm or greater in its largestdiameter [30]. The 28 patients treated hadmultiple characteristics indicative of poorprognosis, as one would expect giventheir bulky disease. About a third hadInternational Working Formulation (IWF)A Histology (small lymphocytic) that isknown to respond poorly to rituximab.They were heavily pretreated (median 3prior regimens, range 1–13) and had aprogressive/clinically aggressive disease atstudy entry. In spite of this, the ORRwas 43% (4% CR and 39% PR) with amedian TTP of 8.1 months (range 4.5 to18.6+ months). In a historical comparisonwith the 166-patient pivotal trial, there
Tab. 5 Rituximab clinical development – combination studies
Study description N ORR[%]
CR[%]
PR[%]
TTPmo.
References
Phase II – R + CHOP 38 100 58 42 72.0+ 45Phase II – R + Interferon 37 45 11 34 25.2 48Phase III – R +90 Y Zevalin 73 80 34 45 15.4+ 54
Notes: N: patients treated and evaluable; ORR: overall response rate; CR: complete response rate;PR: partial response rate; TTP: median time to progression for responders (+ indicates a KaplanMeier projection where true median has not been reached).All response rates are based on the ‘‘evaluable/treated’’ patient population (N) and on responsecriteria as reported by authors except Zevalin (based on the new IWRC).

222 12.2 Clinical Development and Regulatory Approvals
were no significant differences in ORRbetween patients with bulky disease andpatients with lesions of 5 to 7 cm, or withthe general pivotal trial population [30].Rituximab was shown to be active inpatients with bulky disease. This clinicaltrial remains the only reported study ofrituximab in patients with bulky disease.
12.2.7.2 Optimizing the Dose andScheduleThe eight-infusion study was necessary aspatients in the four-infusion dosing expe-rience (at 375 mg m−2) had not reachedeither MTD or steady state/plateau. Thedose and schedule of administration hadnot been optimized. Given the positivecorrelation between higher serum levelsof antibody and response, it was importantto explore a higher total dose adminis-tered over eight doses (375 mg m−2 weekly× 8 doses) [31]. The eight-infusion studywas significant in showing a numericallyhigher ORR and CR than the previousfour-infusion studies [32]. The ORR in35 treated patients was 60% (14% CRand 46% PR). The median TTP exceeded19.4 months and was also longer than his-torical controls using four infusions. Phar-macokinetic studies revealed a progressiveincrease in serum concentration levels ofrituximab beyond the fourth infusion witha possible plateau following the seventhand eighth infusions [32, 33]. Aviles et al.have reported on a six-infusion study inwhich they also achieved better ORR andlonger TTP than with four infusions [34].O’Brien et al., in a Phase I dose escalationtrial in patients with Chronic Lympho-cytic Leukemia (CLL), have also shown thebenefit of higher doses of rituximab. Acontrolled, randomized study will proba-bly never be carried out to formally settlethe issue of whether or not more rituximabis better as these pilot studies suggest.
Nevertheless, there is a pharmacokineticand biologic rationale and the results re-ported to date have sufficed for the USFDA to include the option of eight-infusiondosing in the package insert [35].
12.2.7.3 Repeated Treatment – asMaintenance or Following DiseaseProgressionA number of issues regarding repeatedtreatment with rituximab had to be ad-dressed. Can rituximab treatment be re-peated safely? What are the long-termeffects of sustained B-cell depletion? Dopatients continue to respond and for howlong? Should treatment be repeated uponrelapse or is maintenance therapy feasi-ble and preferable? The timing of relapsefor the individual patient cannot be pre-dicted. We know that the median TTP forresponders is about one year, but there isa wide range with some patients relapsingearly on and others having prolonged sus-tained remissions with no other therapy(Fig. 2) [37, 38]. It is also clear that B-cell re-covery in peripheral blood is not a markerfor disease progression (PD) as some pa-tients relapse before (during depletion)and many patients remain in remissionbeyond the point of B-cell recovery. Also, ithas been shown that the median tumor vol-ume for responding patients, as measuredby the sum of the products of the per-pendicular diameters (SPD), continues todecrease even after B-cells have recoveredin peripheral blood (Fig. 3). It would havebeen nice to have a simple marker, such asthe B-cell count, to indicate PD. However,it is clear that normal B-cell recovery can-not be equated with lymphomatous B-cellrecovery or with PD [39].
Patients enrolled on the re-treatmentstudy had been previously treated withrituximab, responded and later relapsed.They were required to have PD and
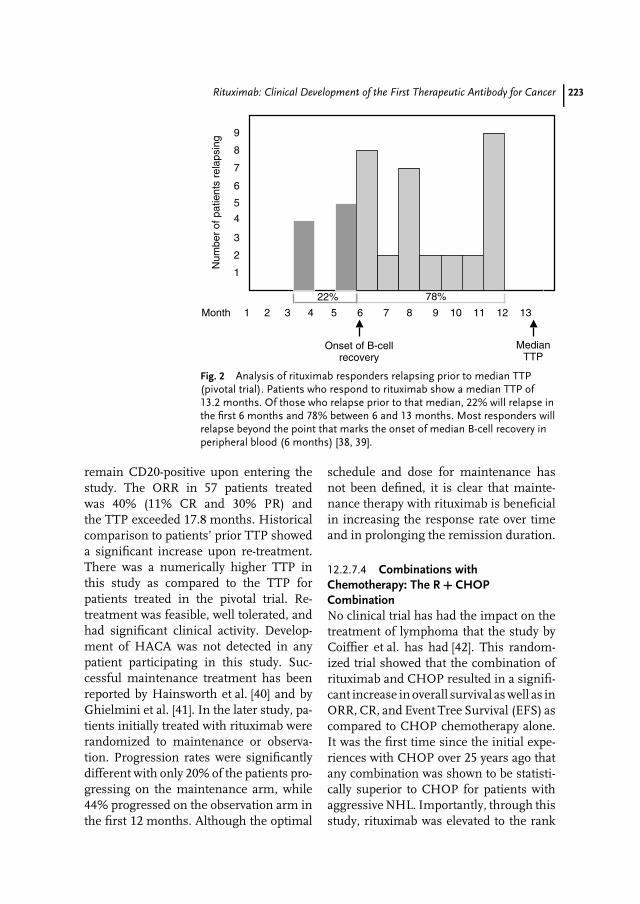
Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 223
Onset of B-cellrecovery
1 2 3 4 5 6 7 8 9 10 11 12 13
2
1
3
5
6
7
8
9
Month
Num
ber
of p
atie
nts
rela
psin
g
22% 78%
MedianTTP
4
Fig. 2 Analysis of rituximab responders relapsing prior to median TTP(pivotal trial). Patients who respond to rituximab show a median TTP of13.2 months. Of those who relapse prior to that median, 22% will relapse inthe first 6 months and 78% between 6 and 13 months. Most responders willrelapse beyond the point that marks the onset of median B-cell recovery inperipheral blood (6 months) [38, 39].
remain CD20-positive upon entering thestudy. The ORR in 57 patients treatedwas 40% (11% CR and 30% PR) andthe TTP exceeded 17.8 months. Historicalcomparison to patients’ prior TTP showeda significant increase upon re-treatment.There was a numerically higher TTP inthis study as compared to the TTP forpatients treated in the pivotal trial. Re-treatment was feasible, well tolerated, andhad significant clinical activity. Develop-ment of HACA was not detected in anypatient participating in this study. Suc-cessful maintenance treatment has beenreported by Hainsworth et al. [40] and byGhielmini et al. [41]. In the later study, pa-tients initially treated with rituximab wererandomized to maintenance or observa-tion. Progression rates were significantlydifferent with only 20% of the patients pro-gressing on the maintenance arm, while44% progressed on the observation arm inthe first 12 months. Although the optimal
schedule and dose for maintenance hasnot been defined, it is clear that mainte-nance therapy with rituximab is beneficialin increasing the response rate over timeand in prolonging the remission duration.
12.2.7.4 Combinations withChemotherapy: The R + CHOPCombinationNo clinical trial has had the impact on thetreatment of lymphoma that the study byCoiffier et al. has had [42]. This random-ized trial showed that the combination ofrituximab and CHOP resulted in a signifi-cant increase in overall survival as well as inORR, CR, and Event Tree Survival (EFS) ascompared to CHOP chemotherapy alone.It was the first time since the initial expe-riences with CHOP over 25 years ago thatany combination was shown to be statisti-cally superior to CHOP for patients withaggressive NHL. Importantly, through thisstudy, rituximab was elevated to the rank
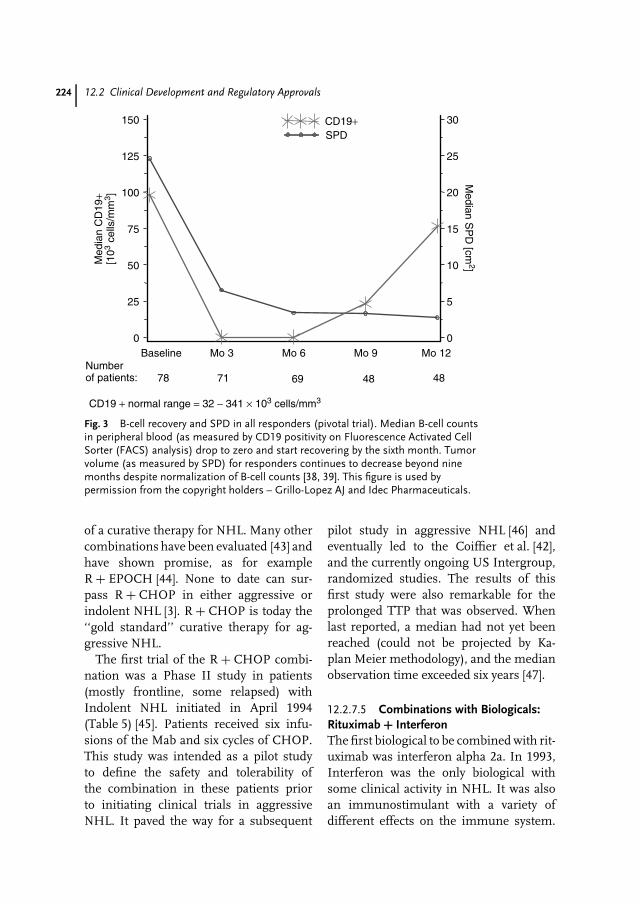
224 12.2 Clinical Development and Regulatory Approvals
CD19 + normal range = 32 − 341 × 103 cells/mm3
Med
ian
CD
19+
[103
cells
/mm
3 ]
Numberof patients: 78 71 69 48 48
0
25
50
75
100
125
150
Baseline Mo 3 Mo 6 Mo 9 Mo 120
5
10
15
20
25
30
Median S
PD
[cm2]
SPDCD19+
Fig. 3 B-cell recovery and SPD in all responders (pivotal trial). Median B-cell countsin peripheral blood (as measured by CD19 positivity on Fluorescence Activated CellSorter (FACS) analysis) drop to zero and start recovering by the sixth month. Tumorvolume (as measured by SPD) for responders continues to decrease beyond ninemonths despite normalization of B-cell counts [38, 39]. This figure is used bypermission from the copyright holders – Grillo-Lopez AJ and Idec Pharmaceuticals.
of a curative therapy for NHL. Many othercombinations have been evaluated [43] andhave shown promise, as for exampleR + EPOCH [44]. None to date can sur-pass R + CHOP in either aggressive orindolent NHL [3]. R + CHOP is today the‘‘gold standard’’ curative therapy for ag-gressive NHL.
The first trial of the R + CHOP combi-nation was a Phase II study in patients(mostly frontline, some relapsed) withIndolent NHL initiated in April 1994(Table 5) [45]. Patients received six infu-sions of the Mab and six cycles of CHOP.This study was intended as a pilot studyto define the safety and tolerability ofthe combination in these patients priorto initiating clinical trials in aggressiveNHL. It paved the way for a subsequent
pilot study in aggressive NHL [46] andeventually led to the Coiffier et al. [42],and the currently ongoing US Intergroup,randomized studies. The results of thisfirst study were also remarkable for theprolonged TTP that was observed. Whenlast reported, a median had not yet beenreached (could not be projected by Ka-plan Meier methodology), and the medianobservation time exceeded six years [47].
12.2.7.5 Combinations with Biologicals:Rituximab + InterferonThe first biological to be combined with rit-uximab was interferon alpha 2a. In 1993,Interferon was the only biological withsome clinical activity in NHL. It was alsoan immunostimulant with a variety ofdifferent effects on the immune system.

Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 225
Patients on the combination study received12 doses of interferon on a weekly basisand 4 infusions of rituximab on weeks5 through 8 [48]. The combination wassafe and well tolerated. The ORR was 45%(CR 11% and PR 34%) and was lowerthan expected. However, the TTP for re-sponders was 25.2 months. Interferon iscurrently approved for the prolongation ofchemotherapy-induced remissions. Eventhough it did not increase the response rateof rituximab, it appears to have prolongedthe TTP. Despite these encouraging results(confirmed by others [49]), investigatorshave not shown much interest in utiliz-ing interferon during maintenance ther-apy following rituximab. Other promisingrituximab combinations include inter-leukin 2, GM-CSF, G-CSF, alemtuzumab,epratuzumab, and so on.
12.2.7.6 Combinations withRadioimmunotherapy: Rituximab + ZevalinRituximab has been shown to synergisewith chemotherapeutic agents [50]. Thereis also some early data that anti-CD20Mabs may synergise with radiation. Al-though the clinical development of Zevalin(90Yttrium-labeled ibritumomab tiuxetan)began utilizing murine IDEC-2B8 (ibri-tumomab) as the cold antibody in thetreatment regimen, we had always plannedto switch to rituximab as soon as feasible[51 to 54]. The activity of the Zevalin treat-ment regimen, including rituximab, wasestablished during clinical development.In the Phase III randomized study, Ze-valin had an ORR of 80% (34% CR and45% PR) with a TTP of 15.4+ monthsin responders. In February 2001, Ze-valin became the first radioimmunother-apy approved for the treatment of NHL.Other radioimmunotherapies are underdevelopment (radiolabeled tositumomab,epratuzumab, etc.).
12.2.8Regulatory Dossiers, Review, and Approvals
Communication and coordination with theUS FDA was a very important factor in car-rying out the clinical development plan andproceeding on to the dossier submission,review, and approval processes. A profes-sional and peer-level relationship based onopenness and mutual trust was establishedbetween the clinical and regulatory staffat IDEC Pharmaceuticals and the FDA re-viewers. This served to preempt or expediteresolution of the many issues that alwaysarise during development. The IND forrituximab was submitted to the US FDAin December 1992 [4]. Clinical trials wereinitiated in February 1993 and enrolmentcompleted three years later in February1996. The dossier, filed simultaneouslywith the FDA in the United States and withthe EMEA in Europe in February 1997,was available in electronic format. Thiswas the first electronic BLA filed underthe FDA’s developing electronic submis-sion standards and IDEC Pharmaceuticals’first BLA. At the time, the US FDA hadpublished a draft guidance manual forComputer-Assisted Product License Ap-plications (CAPLAs); however, final stan-dards and requirements were still underdevelopment. The BLA was a new en-tity, and no guidelines had been publishedspecifically for electronic submission ofthis type of application. Therefore, IDECworked with the Center for Biologics Eval-uation and Research (CBER) to design auser-friendly e-BLA while simultaneouslylaying the groundwork for future stan-dards. On the basis of the success of thefirst e-BLA submission, IDEC geared upfor a second electronic filing three yearslater. This second product developed byIDEC Pharmaceuticals, Zevalin (90Y ib-ritumomab tiuxetan), for the treatment

226 12.4 Conclusions: Achievements, Current Role, and Future Applications of Rituximab
of non-Hodgkin’s lymphoma, ultimatelybecame the first radioimmunotherapy ap-proved by the US FDA (dossier underreview by EMEA in Europe).
The rituximab presentation to the Bio-logics Response Modifiers Advisory Com-mittee (BRMAC) of the FDA took placeon July 25, 1997 and the final approvalwas granted on November 26, 1997 (Fig. 4)[55–58]. The EMEA granted approval to rit-uximab in Europe in June 1998. Rituximabbecame the first monoclonal antibody ap-proved for the treatment of cancer andspecifically for patients with NHL.
12.3Rituximab: Other Indications/Applications
Rituximab has been approved in theUnited States for relapsed or refractoryLG/F NHL and in Europe for relapsedor refractory LG/F NHL and for aggres-sive NHL. Multiple other indications havebeen explored including hematologic ma-lignancies – CLL, multiple myeloma, andso on; autoimmune disorders – ImmuneThrombocytopenic Purpura (ITP), heuma-toid arthritis, Systemic Lupus Erythemato-sus (SLE), hemolytic anemias, and so on(Figs. 5 and 6) [4, 59, 60].
12.4Conclusions: Achievements, Current Role,and Future Applications of Rituximab
Rituximab represents the most importantscientific achievement of the past decade.It was the first therapeutic antibody ap-proved for the treatment of cancer andspecifically for NHL. The current IWRC forNHL had their origin with the rituximab re-sponse criteria. Clinical development wascompleted in record time. Dossiers werefiled simultaneously in the United Statesand in Europe. The US filing utilized anelectronic format (computer-aided productlicense application, CAPLA). These andmany other achievements during clinicaldevelopment served to provide tremen-dous impetus to the monoclonal antibodyresearch area. The renewed enthusiasm inthis area has yielded many new Mabs withactivity in both hematologic malignanciesand in autoimmune diseases. Several ofthese have been approved (e.g. herceptin,alemtuzumab, mylotarg, others) and manyothers are under active investigation.
Rituximab is approved for the treatmentof patients with relapsed or refractoryLG/F NHL. In Europe, it is also approvedfor aggressive NHL (R + CHOP). It isused as a single agent, in combinations
Augusta Cerny C. David Shen, Ph.D
Chet Varns Christine A. White, M.D.
Alice Wei Brian K. Dallaire, Pharm.D.
William Hauser Susan K. Langley
IDEC pharmaceuticals staff
Rituximab: BRMAC presentation25 July 1997
Antonio J. Grillo-López, M.D.
Fig. 4 Rituximab: BRMACpresentation – 25 July 1997. The IDECPharmaceuticals staff’s presentation and therituximab data were commended by theBRMAC advisory committee members forthe excellent organization, clarity, scientificquality, and methodological rigor. Thevideotaped presentation is now used bypharmaceutical companies as a teachingtool for advisory committee presentations[55–58]. Presenters: Alice Wei – regulatory,Antonio Grillo-Lopez – medical andscientific. (This figure is used by permissionfrom the copyright holders – Grillo-Lopez AJand IDEC Pharmaceuticals).

Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 227
Fig. 5 Rituximab: literature reports(neoplastic diseases). The indicationsand clinical applications for rituximabare expanding as research in the therapyof multiple neoplastic diseases isreported. This list is a sample of theliterature reports on the manyindications currently underinvestigation.
Rituximab: Literature reportsNeoplastic diseases
• Low-grade NHL• Intermediate−and high-grade NHL• Waldenstrom’s macroglobulinemia• Chronic lymphocytic leukemia• Acute prolymphocytic leukemia• Acute lymphoblastic leukemia• Hodgkin’s disease• Cutaneous B-cell lymphoma• Colon cancer and other solid tumors
Fig. 6 Rituximab: literature reports(autoimmune diseases). The indicationsand clinical applications for rituximabare expanding as research in the therapyof multiple autoimmune diseases isreported. This list is a sample of theliterature reports on the manyindications currently underinvestigation.
Rituximab: Literature reportsAutoimmune diseases
• Idiopathic thrombocytopenic purpura• Acquired Factor VIII inhibitors• Pure red cell aplasia• Systemic lupus erythematosus• Rheumatoid arthritis• Hemolytic anemias• Posttransplant lymphoproliferative disease• Paraneoplastic pemphigus• IgM polyneuropathies• Myasthenia gravis• Graft versus host disease
(with chemotherapy, biologicals, radioim-munotherapies), and as part of myeloabla-tive regimens. Its part (as cold antibody) inthe Zevalin treatment regimen led to theapproval of this new therapeutic as the firstradioimmunotherapy for the treatment ofcancer. A wide variety of other indicationsare being explored.
In 2002, rituximab became the numberone, brand name, cancer therapeutic inthe world. It is an important componentof the current curative combination foraggressive NHL (R + CHOP). In thefuture, rituximab may become a partof other curative treatment regimens forhematologic malignancies and will alsofind utility in the treatment of majordiseases such as rheumatoid arthritis andother autoimmune disorders.
Acknowledgments
None of the research described in thischapter could have been performed with-out the selfless, courageous, and generousparticipation of the numerous lymphomapatients who volunteered for the initialclinical trials. This is particularly true ofthose who participated in our Phase Itrials, at a time when the efficacy andsafety of the antibody were unknown.They had hope, faith, and courage. Theytrusted us with their lives. Without theseheroes, Rituxan would not exist today. We(the researchers), and the many patientswho will benefit from their participationin these studies, owe them a debt ofgratitude.

228 12.4 Conclusions: Achievements, Current Role, and Future Applications of Rituximab
References
1. A. J. Grillo-Lopez, Oncol. Spec. 2001, 2,700–705.
2. Review of the clinical data from theinitial development studies of rituximab.A. J. Grillo-Lopez, C. A. White, C. Varnset al., Semin. Oncol. 1999, 26 (Suppl. 5, 14),66–73.
3. Presents new therapeutic paradigms in thetreatment of NHL. A. J. Grillo-Lopez, ExpertRev. Anticancer Ther. 2002, 2(3), 323–329.
4. A candid review of historical events duringthe development of rituximab. A. J. Grillo-Lopez, Semin. Oncol. 2000, 27 (Suppl. 6, 12),9–16.
5. Provides an insight into the key laboratoryand in vivo studies that helped elucidatethe mechanisms of action of rituximab.D. R. Anderson, A. Grillo-Lopez, C. Varnset al., Biochem. Soc. Trans. 1997, 2, 705–708.
6. Review of the preclinical data on rituximab.M. E. Reff, K. Carner, K. S. Chambers et al.,Blood 1994, 83(2), 435–445.
7. World Health Organization, WHO Hand-book for Reporting Results of Cancer Treatment,Offset Publication, Geneva, 1979, p. 48.
8. M. Oken, R. Creech, D. Tormey et al., Am. J.Clin. Oncol. 1982, 5, 649–655.
9. S. Horning, B. Cheson, B. Peterson et al.,Proc. Am. Soc. Clin. Oncol. 1997, 16, 18a(#62).
10. A. J. Grillo-Lopez, S. Horning, B. D. Chesonet al., Exp. Hematol. 1997, 25(8), 732 (#17).
11. A. J. Grillo-Lopez, Response Evaluation inPatients with Low-Grade or Follicular NHL,Presentation to the Biological ResponseModifiers Advisory Committee of the FDA,Washington, DC, 1996.
12. A. J. Grillo-Lopez, Response criteria andQA auditing of responses in low-gradeor follicular lymphomas: experience in theIDEC-C2B8 single arm pivotal trial in CBER,FDA (Chair), Clinical Trials in Biotechnology(Ed.: K. Weiss), Workshop Sponsored by theDrug Information Association, Dana Point,CA, 1997.
13. A. J. Grillo-Lopez, B. Cheson, S. Horninget al., Blood 1998, 92(10), 412a (#1701).
14. Important to all who evaluate NHL patientsfor response and also to those whowould like to compare clinical results fromone publication in the medical literature
with others. A. J. Grillo-Lopez, B. D. Cheson,S. J. Horning et al., Ann. Oncol. 2000, 11,399–408.
15. A. J. Grillo-Lopez, B. D. Cheson, S. J. Horn-ing et al., Ann. Oncol. 2000, 11, 399–408.
16. A. J. Grillo-Lopez Scientific and Medical Sum-mary on IDEC-C2B8: Rituxan (rituximab),Biologics License Application Presentationto the Biological Response Modifiers Advi-sory Committee of the FDA, Washington,DC, 1997.
17. Current response criteria for lymphoma arediscussed and presented in this important pa-per. B. D. Cheson, S. J. Horning, B. Coiffieret al., J. Clin. Oncol. 1999, 17(4), 1244–1253.
18. A. J. Grillo-Lopez, P. Mclaughlin, B. Chesonet al., Proc. Am. Soc. Clin. Oncol. 1999, 18,13a (#44).
19. A. J. Grillo-Lopez, C. Varns, D. Shen et al.,Ann. Oncol. 1999, 10(3), 178. (#660).
20. Report on the first clinical trialwith rituximab. D. G. Maloney, T. M. Liles,D. K. Czerwinski et al., Blood 1994, 84(8),2457–2466.
21. A. J. Grillo-Lopez, D. Shen, D. Lee et al.,Blood 2000, 96(11), 238b (#4760).
22. M. S. Czuczman, A. J. Grillo-Lopez, C. A.White et al., Blood 2001, 98(11), 601a (#2519).
23. E. E. Hedrick, R. I. Fisher, B. K. Link et al.,Blood 98(11, Part 2), 229b (#4636).
24. D. G. Maloney, A. J. Grillo-Lopez, D. J. Bod-kin et al., J. Clin. Oncol. 1997, 15(10),3266–3274.
25. A. J. Grillo-Lopez, Semin. Oncol. 2000, 27(Suppl. 6, 12), 9–16.
26. D. G. Maloney, A. J. Grillo-Lopez, C. A. Whiteet al., Blood 1997, 90(6), 2188–2195.
27. J. M. Foran, R. K. Gupta, D. Cunninghamet al., Br. J. Haematol. 2000, 109(1), 81–88.
28. M. Ghielmini, S. F. Schmitz, K. Burki et al.,Ann. Oncol. 2000, 11 (Suppl. 1), S123–S126.
29. Report on the pivotal trial that lead tothe approval of rituximab. P. Mclaughlin,A. J. Grillo-Lopez, B. K. Link et al. J. Clin.Oncol. 1998, 16, 2825–2833.
30. T. A. Davis, C. A. White, A. J. Grillo-Lopezet al., J. Clin. Oncol. 1999, 17(6), 1851–1857.
31. Key article on the pharmacokineticsof rituximab. N. L. Berinstein, A. J. Grillo-Lopez, C. A. White et al., Ann. Oncol. 1998,9, 995–1001.
32. L. D. Piro, C. A. White, A. J. Grillo-Lopezet al., Ann. Oncol. 1999, 10, 655–661.

Rituximab: Clinical Development of the First Therapeutic Antibody for Cancer 229
33. A. Saven, A. J. Grillo-Lopez, N. Janakiramanet al., Blood 2000, 96(11), 730a (#3155).
34. A. Aviles, M. I. Leon, J. C. Diaz-Maqueoet al., J. Hematother. Stem Cell Res. 2001,10(2), 313–316.
35. Rituximab (Rituxan, MabThera) PackageInsert, FDA Website at http://www.fda.gov/.
36. T. A. Davis, A. J. Grillo-Lopez, C. A. Whiteet al., J. Clin. Oncol. 2000, 18(17), 3135–3143.
37. A. J. Grillo-Lopez, D. Shen, D. Lee et al.,Blood 2000, 96(11), 238b (#4760).
38. T. A. Davis, A. J. Grillo-Lopez, P. Mclaughlinet al., Blood 2000, 96(11), 733a (#3171).
39. A. J. Grillo-Lopez, P. Mclaughlin, K. Weyet al., Blood 2000, 96(11), 332a (#14342).
40. J. D. Hainsworth, Semin. Oncol. 2002, 29(1)(Suppl. 2), 25–29.
41. M. Ghielmini, S. -F. Hsu Schmitz, S. Cog-liatti et al., Blood 2002, 100, 161a. (abstract#604).
42. Key article on the new ‘‘gold standard’’ inthe therapy of aggressive NHL. B. Coiffier,E. Lepage, J. Briere et al., N. Engl. J. Med.2002, 346(4), 235–242.
43. J. Boye, T. Elter, A. Engert, Ann. Oncol. 2003,14, 520–535.
44. W. H. Wilson, S. R. Frankel, N. Drbohlavet al., Proc. Am. Soc. Clin. Oncol. 2000, 20(1),290a. (#1158).
45. First study of the rituximab + CHOPcombination. M. S. Czuczman, A. J. Grillo-Lopez, C. A. White et al., J. Clin. Oncol. 1999,17(1), 268–276.
46. J. M. Vose, B. K. Link, M. L. Grossboardet al., J. Clin. Oncol. 2001, 19, 389–397.
47. M. S. Czuczman, A. J. Grillo-Lopez, C. A.White et al., Blood 2001, 98(11), 601a.(#2519).
48. T. A. Davis, D. G. Maloney, A. J. Grillo-Lopez et al., Clin. Cancer Res. 2000, 6,2644–2652.
49. S. Sacchi, M. Frederico, U. Vitolo et al.,Haematologica 2001, 86, 951–958.
50. A. Demidem, T. Lam, S. Alas et al., CancerBiother. Radiopharm. 1997, 12(3), 177–185.
51. A. J. Grillo-Lopez, P. Chinn, R. Morena,et al., Blood 1995, 86(10), 55a (#207).
52. S. J. Knox, M. L. Goris, K. Trisler et al., Clin.Cancer Res. 1996, 2, 457–470.
53. T. E. Witzig, C. A. White, G. A. Wisemanet al., J. Clin. Oncol. 1999, 17(12), 3793–3803.
54. A. J. Grillo-Lopez, Expert Rev. AnticancerTher. 2002, 2(5), 485–493.
55. A. J. Grillo-Lopez, IDEC-C2B8: Rituxan
(rituximab), Oral/slide Presentation – Bio-logical Response Modifiers AdvisoryCommittee, Washington, DC, July 1997,(Data on file – IDEC Pharmaceuticals andUS FDA).
56. A. J. Grillo-Lopez, C. A. White, B. K. Dallaireet al., Curr. Pharm. Biotechnol. 2000, 1(1),1–9.
57. A. Grillo-Lopez, Presentation to BR-MAC, Scientific and Medical Sum-mary of IDEC-C2B8, July 25, 1997,www.fda.gov/OHRMS/dockets/ac/97/transcpt/3311t2.rtf.
58. A. Wei, A. J. Grillo-Lopez, Rituximab Pre-sentation to the US FDA Biologic ResponseModifiers Advisory Committee, FDA Advi-sory Committee Meetings on Video Tape,F-D-C Reports, Special Projects Depart-ment (Phone: 301-657-9830), July 25, 1997,http://www.fdaadvisorycommittee.com/FDC/advisorycommittee/toc.htm.
59. A. J. Grillo-Lopez, B. K. Dallaire, A. Mcclureet al., Curr. Pharm. Biotechnol. 2001, 2,301–311.
60. A. J. Grillo-Lopez, Rituxan: The First Decadeand the Future, Grand Rounds Presentationat Teacher’s Hospital, San Juan, Puerto Rico,February 2003.

231
13Somatic Gene Therapy –Advanced BiotechnologyProducts in Clinical Development
Matthias Schweizer, Egbert Flory, Carsten Muenk and Klaus CichutekPaul-Ehrlich-Institut, Langen, Germany
Uwe GottschalkPharma-Biotechnology, Wuppertal, Germany
13.1Introduction
Innovative biopharmaceuticals of the fu-ture include gene transfer medicinal prod-ucts [1, 2]. It can be assumed that bymid-2003, approximately 4000 patientsor healthy individuals have been treatedwithin a clinical gene therapy trial, ap-proximately 600 of those in Europe andapproximately 260 in Germany. Most ofthe clinical trials are currently in phaseI or II because, due to a great diversityof ongoing developments, clinical expe-rience must first be gained before target-orientated product development and phaseIII clinical trials can be initiated. In thisregard, investigator-driven gene therapystrategies developed by biomedical labora-tories together with special clinical teamsare very distinct from those developed bythe pharmaceutical industry. Investigator-driven gene therapy strategies are being in-vented by teams of biomedical researchersand physicians while developing a new
approach for the treatment of a specialdisease in a defined stage. This is used,for the first time, on a selected groupof patients in first clinical trials of phaseI/II and is aimed at proving the safetyof the medicinal product. In clinical trialssponsored by the pharmaceutical indus-try, this phase of orientation has oftenalready been completed and further de-velopment in phase II or III is aimedat dose finding or proving efficacy. Con-cerning product development, there areno standard approaches because, at thisstage of development, little experience hasbeen gained and the types of gene trans-fer medicinal products are very diverse [3].Therefore, in the following sections themain current clinical developments will bedescribed while a brief outline of a singleexample of a manufacturing process, alsodue to manifold diversity, is given.
Gene transfer medicinal products forhuman use are medicinal products usedfor in vivo diagnosis, prophylaxis, ortherapy (Fig. 1). They contain or consist of
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8
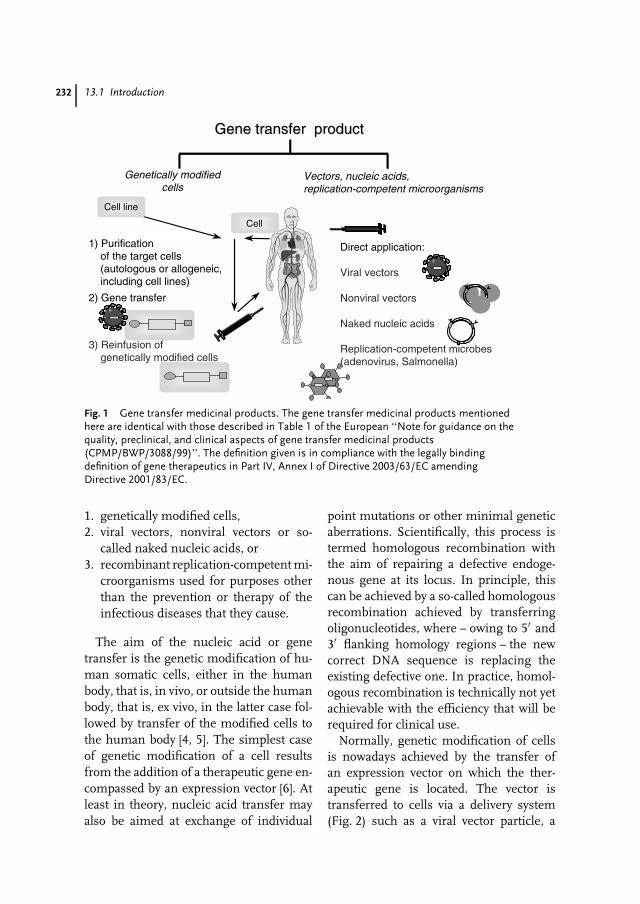
232 13.1 Introduction
Gene transfer productGene transfer product
Genetically modifiedcells
Vectors, nucleic acids,replication-competent microorganisms
1) Purification of the target cells (autologous or allogeneic, including cell lines)
2) Gene transfer
3) Reinfusion of genetically modified cells
Direct application:
Viral vectors
Nonviral vectors
Naked nucleic acids
Replication-competent microbes(adenovirus, Salmonella)
Cell line
Cell
Fig. 1 Gene transfer medicinal products. The gene transfer medicinal products mentionedhere are identical with those described in Table 1 of the European ‘‘Note for guidance on thequality, preclinical, and clinical aspects of gene transfer medicinal products(CPMP/BWP/3088/99)’’. The definition given is in compliance with the legally bindingdefinition of gene therapeutics in Part IV, Annex I of Directive 2003/63/EC amendingDirective 2001/83/EC.
1. genetically modified cells,2. viral vectors, nonviral vectors or so-
called naked nucleic acids, or3. recombinant replication-competent mi-
croorganisms used for purposes otherthan the prevention or therapy of theinfectious diseases that they cause.
The aim of the nucleic acid or genetransfer is the genetic modification of hu-man somatic cells, either in the humanbody, that is, in vivo, or outside the humanbody, that is, ex vivo, in the latter case fol-lowed by transfer of the modified cells tothe human body [4, 5]. The simplest caseof genetic modification of a cell resultsfrom the addition of a therapeutic gene en-compassed by an expression vector [6]. Atleast in theory, nucleic acid transfer mayalso be aimed at exchange of individual
point mutations or other minimal geneticaberrations. Scientifically, this process istermed homologous recombination withthe aim of repairing a defective endoge-nous gene at its locus. In principle, thiscan be achieved by a so-called homologousrecombination achieved by transferringoligonucleotides, where – owing to 5′ and3′ flanking homology regions – the newcorrect DNA sequence is replacing theexisting defective one. In practice, homol-ogous recombination is technically not yetachievable with the efficiency that will berequired for clinical use.
Normally, genetic modification of cellsis nowadays achieved by the transfer ofan expression vector on which the ther-apeutic gene is located. The vector istransferred to cells via a delivery system(Fig. 2) such as a viral vector particle, a

Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 233
Expression systemor vector
(encompasses thetherapeutic gene)
Delivery system:here, viralvector particle
Genetically modified cell
MessengerRNA
Therapeuticprotein
Therapeuticgene
Fig. 2 Delivery system and expression vector used as gene transfer medicinal products. Theterminology complies with the definition of gene therapeutics in Part IV, Annex I of Directive2003/63/EC amending Directive 2001/83/EC.
nonviral vector complex, or a plasmid. Inthe latter case, the expression vector isinserted into and is therefore part of abacterial plasmid, which allows its manu-facture and amplification in bacteria. Viralexpression vectors contain the sequencesignals (nucleic acid sequences) requiredfor transfer by a particular viral vectorparticle. For retroviral vectors, for exam-ple, such signals are encompassed by theflanking ‘‘Long Terminal Repeat’’ (LTR)sequences, the packaging signal Psi (�)required for incorporation of the expres-sion vector by the retroviral vector particle,and other sequence signals. For nonviralvector complexes and naked nucleic acid,the expression vector is part of a bacterialcarrier, the so-called plasmid DNA. Nonvi-ral vectors are, for example, plasmid DNAmixed with a transfection reagent, whereasnaked DNA does not contain a transfectionreagent.
Another example of a gene trans-fer medicinal product is recombinantmicroorganisms such as conditionallyreplication-competent adenoviruses for tu-mor therapy [7]. Here, neither an en-dogenous cellular gene is repaired byhomologous recombination nor is a non-adenoviral therapeutic gene transferred.The transfer of conditional replicating ade-noviruses to the malignant tumor cellsinduces cell lysis and local tumor abla-tion. The entire genome of the adenovirusis transferred without an additional thera-peutic gene. The adenoviral genome maytherefore be considered as the therapeu-tic gene.
Gene transfer efficiency plays a centralrole in gene transfer. It depends on a num-ber of factors, for example, target cell, typeof application (ex vivo or in vivo strategy),the tissue or organ containing the targetcells, the physiological situation, and the

234 13.2 Gene Transfer Methods
Tab. 1 Gene transfer methods (vectors/delivery systems)
Delivery system Description Chromosomal integration
Naked nucleic acid Plasmid DNA, in the absence oftransfection reagents
No (after im inoculation)
Nonviral vector Plasmid DNA/transfection reagentmixture
No (applicationdependent)
Viral vectorRetroviral vector Derived from murine leukemia virus
(MLV)Yes
Lentiviral vector Derived from HIV-1 YesAdenoviral vector Deletions in the virus genes E1, E3 or E4,
E2ts, combinations thereof, or ‘‘gutted’’(gene-depleted)
No
Conditionallyreplication-competentadenovirus
No therapeutic gene except for the virusgenome
No
Adeno-associated virus(AAV) vector
Wild-type AAV-derived Yes/no (applicationdependent)
Smallpox virus vector MVA (Modified Vaccinia Ancara) NoALVAC (Avian Vaccinia) NoVaccinia No
Alphavirus vector Semliki Forest virus (SFV) NoHerpes-viral vector Herpes simplex virus No
disease and disease stage. Table 1 showsthe most common viral vectors currentlyin clinical use. The vectors shown are repli-cation incompetent and only transfer theexpression vector void of any viral genesas much as possible. So-called integratingvectors mediate chromosomal integrationof the expression vector (e.g. retroviral vec-tors) [8], whereas nonintegrating vectorslead to an episomal status of the expres-sion vector in the cell (e.g. adenoviralvectors), or to its cytoplasmatic replication(e.g. alphavirus-derived vectors, vaccinia).Vectors derived from vaccinia, for exam-ple, used for tumor vaccination, may bereplication incompetent such as ModifiedVaccinia Ankara (MVA) or Avian Vaccinia(ALVAC), or replication competent, butattenuated like vaccinia.
After uptake by human somatic cells,the expression vector is transcribed like a
normal cell gene. The resulting messengerRNA (mRNA) is translated and the ther-apeutic protein is synthesized by the cel-lular machinery. When so-called ribozymegenes are used, the mRNA acts like acatalytic enzyme and is itself the therapeu-tic gene product. As already mentioned,when a recombinant microorganism suchas a conditionally replication-competentadenovirus (RCA) is used, the genome ofthe microorganism may be seen as thetherapeutic gene.
13.2Gene Transfer Methods
The objective of clinical gene transferis the transfer of nucleic acids for thepurpose of genetically modifying humancells (Fig. 3).
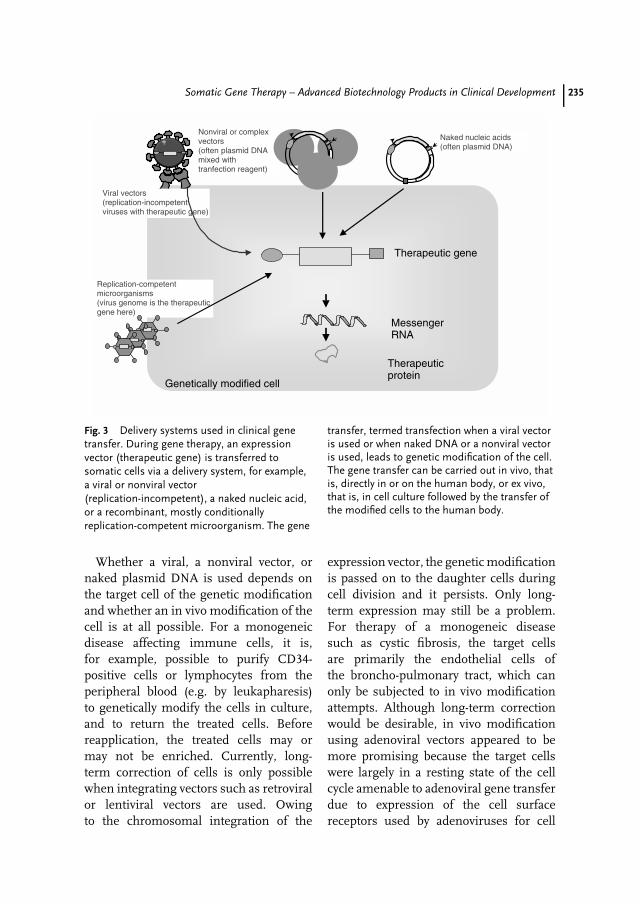
Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 235
Nonviral or complexvectors(often plasmid DNAmixed withtranfection reagent)
Viral vectors(replication-incompetentviruses with therapeutic gene)
Naked nucleic acids(often plasmid DNA)
Replication-competentmicroorganisms(virus genome is the therapeuticgene here)
Genetically modified cell
MessengerRNA
Therapeuticprotein
Therapeutic gene
Fig. 3 Delivery systems used in clinical genetransfer. During gene therapy, an expressionvector (therapeutic gene) is transferred tosomatic cells via a delivery system, for example,a viral or nonviral vector(replication-incompetent), a naked nucleic acid,or a recombinant, mostly conditionallyreplication-competent microorganism. The gene
transfer, termed transfection when a viral vectoris used or when naked DNA or a nonviral vectoris used, leads to genetic modification of the cell.The gene transfer can be carried out in vivo, thatis, directly in or on the human body, or ex vivo,that is, in cell culture followed by the transfer ofthe modified cells to the human body.
Whether a viral, a nonviral vector, ornaked plasmid DNA is used depends onthe target cell of the genetic modificationand whether an in vivo modification of thecell is at all possible. For a monogeneicdisease affecting immune cells, it is,for example, possible to purify CD34-positive cells or lymphocytes from theperipheral blood (e.g. by leukapharesis)to genetically modify the cells in culture,and to return the treated cells. Beforereapplication, the treated cells may ormay not be enriched. Currently, long-term correction of cells is only possiblewhen integrating vectors such as retroviralor lentiviral vectors are used. Owingto the chromosomal integration of the
expression vector, the genetic modificationis passed on to the daughter cells duringcell division and it persists. Only long-term expression may still be a problem.For therapy of a monogeneic diseasesuch as cystic fibrosis, the target cellsare primarily the endothelial cells ofthe broncho-pulmonary tract, which canonly be subjected to in vivo modificationattempts. Although long-term correctionwould be desirable, in vivo modificationusing adenoviral vectors appeared to bemore promising because the target cellswere largely in a resting state of the cellcycle amenable to adenoviral gene transferdue to expression of the cell surfacereceptors used by adenoviruses for cell

236 13.2 Gene Transfer Methods
entry. In addition, the amount and titers ofadenoviral vectors seemed suitable. Theseexamples illustrate that a number of factorscontribute to the choice of the treatmentstrategy, the vector, and the route ofadministration. No single ‘‘ideal’’ vectoris therefore suitable for a large variety ofgene therapies [9]. In the past 15 years,many novel gene transfer techniqueshave been developed and used in clinicalstudies [10]. In the following section andin Table 1, specific characteristics of thevectors most commonly used in the clinicare summarized.
13.2.1Nonviral Vectors and Naked Nucleic Acid
The advantage of nonviral gene transfersystems compared with viral gene trans-fer systems is the smaller size limitationsfor the genes to be transferred. The ex-pression vectors are nowadays usually partof a bacterial plasmid that can easily beamplified and grown in bacterial cultures.Plasmid DNA of up to 20 kb pairs encom-passing an expression vector of up to 17 kbpairs can easily be manufactured. Promis-ing methods for the in vivo administrationof plasmid DNA include intradermal orintramuscular injection for the so-callednaked nucleic acid transfer. Needle injec-tion or application by medical devices suchas gene guns can be used for this purpose.For so-called nonviral vectors, such assynthetic liposomes or other transfectionreagents mixed with plasmid DNA, theDNA-binding liposomes mediate contactwith the cellular plasma membrane, thusreleasing the DNA into the cytoplasmaof the cell where uptake by the nucleushas to occur subsequently [11]. Duringreceptor-mediated uptake of nonviral vec-tors, cell surface proteins (receptors), suchas asialoglycoprotein or the transferrin
receptor, mediate cellular uptake of theDNA complex containing a specific recep-tor ligand [12, 13].
13.2.2Viral Vectors
During evolution, viruses have been opti-mized to efficiently enter mammalian cellsand replicate. Infected mammalian cellstranscribe the viral genes and synthesizethe viral gene products with high effi-ciency, sometimes to the disadvantage ofendogenous protein production. Viral vec-tors are replication-incompetent particlesderived from viruses by genetic engineer-ing that no longer transfer to cells thecomplete set or any viral genes. Instead,an expression vector with one or moretherapeutic genes is transferred to cells.Since no complete viral genome is trans-ferred, virus replication is impossible or,in some cases, impaired as with first-or second-generation adenoviral vectors.The following section briefly describes theproperties of the currently frequently usedviral vectors.
13.2.2.1 Retroviral VectorsThe retroviral vectors in clinical usehave mainly been derived from murineleukemia virus (MLV) [14]. MLV causesleukemia in mice and replication-com-petent retrovirus (RCR) in a contaminatedvector preparation was shown to causeleukemia in severely immunosuppressedmonkeys. RCR absence therefore hasto be verified before human use ofretrovirally modified cells; MLV vectoruse in vivo has been very rare. Thegenome of the retroviral vectors consistsof two copies of single-stranded RNA,which contains one or more coding regionsflanked by the viral control elements, theso-called ‘‘long terminal repeat’’ (LTR)

Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 237
regions. In the infected cells, the RNA istranslated into double-stranded viral DNAand integrated into the cell. The integratedvector DNA is the expression vector. MLVvectors allow efficient genetic modificationof proliferating cells by chromosomallyintegrating the expression vector.
Advantages of retroviral vectors includehigh gene transfer (transduction) effi-ciency, and long-term modification of cellsdue to stable integration of the expressionvector into the chromosome of the cells.In addition, the MLV envelope proteinscan be exchanged against those from otherviruses (which is termed ‘‘vector pseudo-typing’’). This allows preparation of MLVvectors with improved transduction effi-ciency for certain cell types. Disadvantagesof retroviral vectors include the small sizeof the coding region (approximately 9 kbpairs or less), the restriction of transduc-tion to proliferating cells only, insertionalmutagenesis due to integration, and thelow titer of usually not more than 108
transducing units per milliliter of vec-tor preparation. Although chromosomalintegration occurs generally at random,it may lead to activation of cellular can-cer genes, so-called proto-oncogenes, or,theoretically, to inactivation of tumor sup-pressor cells. In conjunction with addi-tional genetic mutations, this may resultin very low frequency in malignant celltransformation. Hundreds of patients whohave been treated with retrovirally mod-ified hematopoietic cells years ago havenot shown any signs of cancer related tothe gene transfer except for two patientstreated during a SCID-X1 gene therapytrial in France. In the latter two leukemiacases, the vector-mediated overexpressionof the proto-oncogene LMO2, possibly inconjunction with the therapeutic γ c chaingene (which may influence cell prolifer-ation and signal transduction) and the
SCID-X1 disease, is the probable causeof leukemia.
13.2.2.2 Lentiviral VectorsLentiviral vectors have been derived fromhuman immunodeficiency virus type 1(HIV-1), simian immunodeficiency virus(SIV) isolated from various Old Worldmonkeys, feline lentivirus (FIV), andequine infectious aneamia virus (EIAIV)isolated from horses [15, 16]. Lentivirusescause an acquired immunodeficiency syn-drome and replication-competent virushas therefore to be excluded before humanuse by batch-to-batch analysis and verifi-cation of replication-competent lentivirus(RCL) absence. Lentiviral vectors maytransfer coding regions of up to 9 kb pairsand allow pseudotyping just like MLV vec-tors. Their advantage is the dual capacity totransfer therapeutic genes into nonprolif-erating cells in conjunction with persistentgenetic modification due to chromosomalintegration. This could be useful for ex vivomodification of stem cells and in vivo mod-ification of neuronal cells. Most lentiviralvectors have been pseudotyped with the Gprotein of vesicular stomatitis virus (VSV-G) or the envelope proteins of Gibbon apeleukemia virus. The first clinical study us-ing lentiviral vectors has started in 2003and involves the ex vivo modification ofautologous lymphocytes of HIV infectedpatients with a therapeutic ribozyme geneshown in vitro to inhibit HIV-1 replication.
13.2.2.3 Adenoviral VectorsThe adenoviral genome consists of double-stranded DNA that persists episomally,that is, inside the nucleus, but not inte-grated into the chromosome of the cell [17].Therefore, the genetic modification may belost during cell proliferation. Adenoviralvectors are the currently preferred vectors

238 13.3 Clinical Use
for the in vivo transduction of a varietyof human somatic cells including non-proliferating cells. In contrast to lentiviralvectors, they allow insertion of larger cod-ing regions of therapeutic genes above10 kb pairs and are not associated with adetectable risk of insertional oncogenesis.In addition, vector titers above 1011 trans-ducing units per milliliter can usually beachieved. The lack of long-term expressionis in part due to the fact that certain ade-novirus genes have been kept on first- orsecond-generation adenoviral expressionvectors, and because of the frequent gener-ation of RCA during production. So-calledgutless vectors are void of any adenoviralgenes, but have to be purified from RCAafter production.
Some wildtype (replicating) adenovirusstrains cause inflammations of the airwaysand the conjunctivae. Adenoviral vectorsmay therefore also be transferred by in-halation of aerosols, and inflammationsobserved following vector applications aremainly local, transient, and associatedwith very high titer applications. High-titeradenoviral vectors are no longer systemi-cally administrated because one patienthad died during systemic administrationof a maximum dose of approximately1013 vector particles during gene therapyof the monogeneic disease OTC (Or-nithine Transcarbamylase) deficiency, alife-threatening metabolic disorder.
13.2.2.4 AAV (Adeno-associated Viral)VectorsAdeno-associated viruses (AAVs) belongto the family of parvoviruses [18, 19].Their genome consists of single-strandedDNA. Wild-type AVV can only replicatein the presence of helper viruses likeadenovirus or herpesvirus and has notbeen associated with any disease. AAVcan infect hematopoietic cells including
nonproliferating cells. Integration in in-fected human somatic cells is oftenconfined to a distinct locus on humanchromosome 19. AAV-derived vectors areusually classified as integrating vectors, al-though vector integration is unfortunatelyno longer confined to chromosome 19, butabsence of integration may be observed,for example, following intramuscular ad-ministration The size of the coding regionis very limited (approximately 4 kb pairs).
13.2.2.5 Poxvirus VectorsPoxvirus vectors encompass vaccinia de-rived from the smallpox vaccine and moreattenuated variants like ALVAC or MVA(Modified Vaccinia Ankara). Their genomeconsists of single-stranded DNA of 130 to300 kb pairs. Replication is restricted tothe cytoplasm of cells and high amounts ofprotein are synthesized by the cell follow-ing transduction. Most applications there-fore involve intramuscular vaccination.
13.3Clinical Use
13.3.1Overview on Clinical Gene Therapy Trials
A number of clinical trials show promisingresults (see Table 2). In the past few years,it has become increasingly clear that foreach disease, the development of a partic-ular and specific gene transfer method inconnection with a particular treatment ap-proach will probably be necessary. The firststandard use of an approved gene transfermedicinal product is to be expected withinthe next seven years since approximately1% of the clinical gene therapy studies arein an advanced stage of phase II or phaseIII clinical trial.
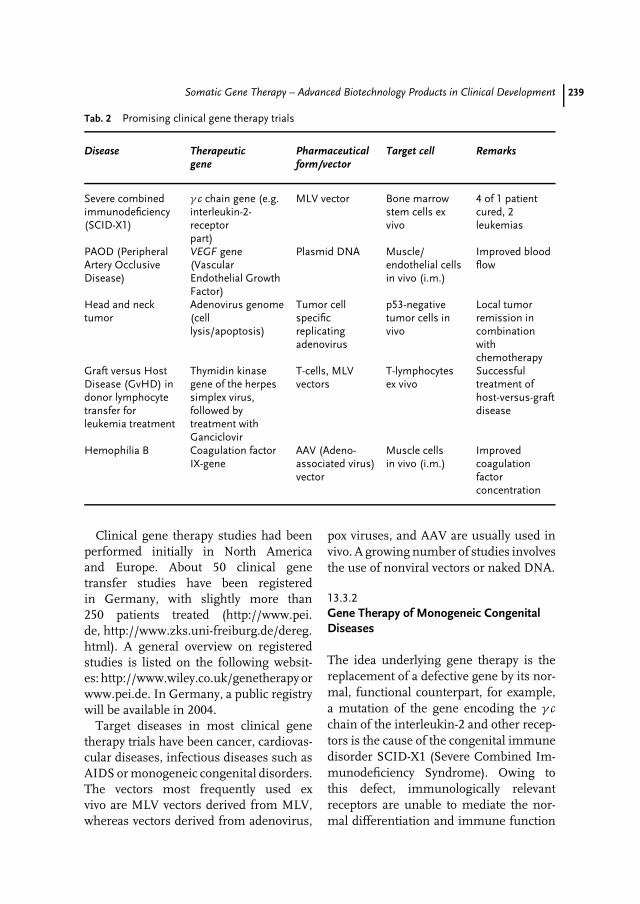
Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 239
Tab. 2 Promising clinical gene therapy trials
Disease Therapeuticgene
Pharmaceuticalform/vector
Target cell Remarks
Severe combinedimmunodeficiency(SCID-X1)
γ c chain gene (e.g.interleukin-2-receptorpart)
MLV vector Bone marrowstem cells exvivo
4 of 1 patientcured, 2leukemias
PAOD (PeripheralArtery OcclusiveDisease)
VEGF gene(VascularEndothelial GrowthFactor)
Plasmid DNA Muscle/endothelial cellsin vivo (i.m.)
Improved bloodflow
Head and necktumor
Adenovirus genome(celllysis/apoptosis)
Tumor cellspecificreplicatingadenovirus
p53-negativetumor cells invivo
Local tumorremission incombinationwithchemotherapy
Graft versus HostDisease (GvHD) indonor lymphocytetransfer forleukemia treatment
Thymidin kinasegene of the herpessimplex virus,followed bytreatment withGanciclovir
T-cells, MLVvectors
T-lymphocytesex vivo
Successfultreatment ofhost-versus-graftdisease
Hemophilia B Coagulation factorIX-gene
AAV (Adeno-associated virus)vector
Muscle cellsin vivo (i.m.)
Improvedcoagulationfactorconcentration
Clinical gene therapy studies had beenperformed initially in North Americaand Europe. About 50 clinical genetransfer studies have been registeredin Germany, with slightly more than250 patients treated (http://www.pei.de, http://www.zks.uni-freiburg.de/dereg.html). A general overview on registeredstudies is listed on the following websit-es: http://www.wiley.co.uk/genetherapy orwww.pei.de. In Germany, a public registrywill be available in 2004.
Target diseases in most clinical genetherapy trials have been cancer, cardiovas-cular diseases, infectious diseases such asAIDS or monogeneic congenital disorders.The vectors most frequently used exvivo are MLV vectors derived from MLV,whereas vectors derived from adenovirus,
pox viruses, and AAV are usually used invivo. A growing number of studies involvesthe use of nonviral vectors or naked DNA.
13.3.2Gene Therapy of Monogeneic CongenitalDiseases
The idea underlying gene therapy is thereplacement of a defective gene by its nor-mal, functional counterpart, for example,a mutation of the gene encoding the γ cchain of the interleukin-2 and other recep-tors is the cause of the congenital immunedisorder SCID-X1 (Severe Combined Im-munodeficiency Syndrome). Owing tothis defect, immunologically relevantreceptors are unable to mediate the nor-mal differentiation and immune function

240 13.3 Clinical Use
of lymphoid cells such as T-cells and nat-ural killer lymphocytes (NK). Therefore,newborn babies suffering from SCID-X1have a very limited immune system andmust live in a germ-free environment.Their life expectancy is strongly reduced.Conventional treatment, that is, bone mar-row transplantation, can provide a cure toa certain extent, but involves a high riskif no HLA haploidentical donor is avail-able. For the latter situation, gene therapywithin the framework of a clinical studywas considered in France [20].
In this study, autologous CD34-positivebone marrow stem cells were retrovirallymodified to express the functional γ c chaingene. T-cells and other hematopoietic cellsderived form corrected stem cells wereshown to repopulate the hematopoieticcell compartment, and over a periodof up to 3 years, 11 treated patients,mostly newborns, displayed a functionaland nearly normal immune system. Thisrepresents the first reproducible cure of adisease by gene therapy.
A leukemia-like lympho-proliferativedisease was diagnosed roughly three yearsafter treatment of two obviously cured pa-tients. Treatment had been started at theage of a few months. Subsequent analy-sis revealed that the leukemia-like diseasewas indeed caused by the MLV vector; thedisease mechanism is termed ‘‘insertionaloncogenesis’’ resulting from insertionalmutagenesis of the proto-oncogene LMO2.According to the current knowledge, upto 50 cells with an integration in LMO2may have been administered together withthe approximately 108 genetically mod-ified CD34-positive bone marrow cells.Owing to the expression vector integra-tion, the transcription of the LMO2 genewas deregulated and activated. Under nor-mal circumstances, the body can cope withindividual cells presenting preneoplastic
changes like the one described. In the twotreated children, however, further geneticchanges must have accumulated to finallyresult in leukemia. Contributing factorsdiscussed include the effect of the thera-peutic γ c chain gene, the product of whichinfluences cell proliferation and differen-tiation, and other so far unknown geneticchanges that may have occurred duringthe massive in vivo cell replication. InSCID-X1, the T-cell compartment is com-pletely depleted, and is replenished aftergene therapy by differentiation and repli-cation of a few genetically corrected bloodstem cells. During this process, geneticaberrations may occur with substantial fre-quency. However, further analysis will berequired to understand the exact cause ofleukemia development in SCID-X1 genetherapy. Since hundreds of patients treatedwith retrovirally modified cells in the past10 years have not developed leukemia up tonow, it is currently assumed that a practi-cal risk of leukemia exists only in SCID-X1gene therapy.
Gene therapy of hemophilia B alsoseems promising. Here, AAV vectorsencoding a smaller but functional versionof the human coagulation factor IX-gene were administered by intramuscularinjection. A detectable increase in factorIX plasma concentration was observed.Even repeated AAV injections were welltolerated.
13.3.3Tumor Gene Therapy
There are various gene therapy approachesthat are being developed for the treatmentof cancer. They are aimed at inhibitingmolecular pathways underlying malignantcell transformation. In other cases, tumorcell ablation by directly applying cell-killing

Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 241
mechanisms, or, more indirectly, by im-proving immunological defense mecha-nisms directed against tumor cells areattempted [21].
A number of gene therapy studies in-volving the adenoviral transfer of tumorsuppressor genes like p53 have alreadybeen performed. This is aimed at revertingmalignant cell transformation or at in-ducing apoptosis. However, transductionfollowing, for example, needle inoculationinto tumors has been shown to be limitedto a few cells close to the needle tracks.Direct tumor cell ablation by local injec-tion of conditionally replication-competentadenoviruses in head and neck tumorsled to detectable local tumor regressionby direct virus-mediated cell lysis, espe-cially if chemotherapy was used in parallel.Here, virus replication improved transduc-tion efficiency in vivo. For the treatmentof malignant brain tumors, variant her-pesviruses have been inoculated into thetumor in order to lyse the tumor cells invivo, especially if prodrugs have been ad-ministered that are converted by the viralthymidin kinase gene to a toxic drug.
In addition, a number of clinical ap-proaches have already been tested that ledto an improvement of immune recogni-tion of tumors [22]. They involved intra-tumoral injection of vectors that transferforeign MHC genes, such as B7.1 orB7.2, or cytokine genes, for example,interleukin-2 or granulocyte-macrophagecolony stimulating factor (GM-CSF). Here,vaccinia-derived vectors such as MVA orALVAC have often been used. Autolo-gous or allogeneic tumor cells were alsomodified ex vivo by transfer of immunos-timulating genes. Promising results havebeen reported from a phase I studyin which autologous tumor cells wereadenovirally modified with the GM-CSF
gene and rapidly reinoculated to stimulateantitumor immunity.
13.3.4Gene Therapy of Cardiovascular Diseases
Local intramuscular injection of plasmidDNA or adenoviral vectors encoding vas-cular epithelial growth factor or fibroblastgrowth factor, both able to induce the for-mation of new blood vessels, have beenused to improve microcirculation in is-chemic tissue. Needle injection of plasmidDNA has been used in leg muscle, catheterapplication or needle injection was alsotried in ischemic heart muscle. The for-mation of new blood vessels and animprovement in the microcirculation hasbeen observed.
A narrowing of the blood vessels(restenosis) often occurs after coronaryblood vessel dilatation by stent implan-tation. This is probably caused by the pro-liferation of smooth muscle cells followinginjuring of the blood vessel endotheliumby the stent. Here, the role of adenoviralor plasmid DNA–mediated transfer of thegene encoding inducible nitroxid synthase(iNOS) is thought to result in reduced cellproliferation.
13.3.5Preventive Vaccination and Gene Therapyof Infectious Diseases
During the past 5 to 10 years, effec-tive medicines have been developed forthe treatment of AIDS. Combinationsof effective chemotherapeutics are ableto inhibit various steps of the replica-tion cycle of HIV-1. This often resultsin reduction of the viral load in theperipheral blood, sometimes down toa level barely detectable with moderntechniques. Because of the requirement

242 13.4 Manufacture and Regulatory Aspects
for long-term treatment and the mas-sive adverse effects related to conven-tional treatment by chemotherapy, genetherapy of HIV infection could offer ad-ditional therapy options. Ex vivo retrovi-ral transfer of HIV-inhibiting genes intoperipheral blood lymphocytes or CD34-positive human cells has been attempted,so far with little success. The thera-peutic molecules used include (1) decoy-RNA specifying multiple copies of theRev- or the Tat-responsive element, so-called poly-TAR or poly-RRE sequences,(2) miniantibodies (single chain Fv, scFv)able to capture viral gene products withinthe cell, (3) transdominant negative mu-tants of viral proteins such as RevM10,or (4) ribozyme RNA that enzymaticallycleaves RNA. Other genes still under de-velopment are designed to prevent entryor chromosomal integration of HIV. Itremains to be shown whether such genetherapy approaches present a suitable ther-apeutic option compared with existingchemotherapy.
The best prevention of infectious dis-eases is achieved by prophylactic vac-cines [23]. Clinical trials using vectoredvaccines based on ALVAC or MVA havebeen initiated. Other clinical trials pursuethe goal of developing vaccines againstHIV-1, malaria, hepatitis B, tuberculo-sis, and influenza A virus infections [24].Vaccination regiments using poxvirus vec-tors such as ALVAC or MVA in com-bination with naked DNA as a primevaccine, sometimes followed by furtherbooster injections of recombinant viralantigens, are being tested in humans.Such regiments have been shown to pre-vent disease progression after lentivirusinfection of monkeys. This illustratesthe complexity of vaccination strategiesthat are currently pursued in vaccine re-search.
13.3.6Clinical Gene Therapy for the Treatment ofOther Diseases
Clinical gene therapy can also be usedfor the treatment of diseases not necessar-ily caused by single known gene defects,if promising therapeutic genes can bereasonably applied. Patients with chronicrheumatoid arthritis, for instance, shouldbenefit from a reduction of the inflamma-tions in joints. Such inflammations arecaused or at least maintained by a cascadeof events including the overexpression andincreased release of a number of inflam-matory cytokines. Monoclonal antibodiesthat are able to reduce the local concentra-tion of the tumor necrosis factor (TNF)have already been successfully used totreat disease. Here, clinical gene transferapproaches involve the transfer of autol-ogous synovial cells modified ex vivo bya therapeutic gene encoding interleukin-1receptor antagonist. Alternatively, adenovi-ral vectors with the same gene have beendirectly injected into the affected joint.
13.4Manufacture and Regulatory Aspects
The regulation of gene therapy is very com-plex and differs considerably in the Euro-pean Union and the United States [25]. InPart IV, Annex I of Directive 2003/63/EC(which replaces Annex I of Directive2001/83/EC), a definition of so-called genetherapeutics is given. As gene therapy notonly includes therapeutic but also preven-tive and diagnostic use of vectors, nucleicacids, certain microorganisms, and ge-netically modified cells, the term ‘‘genetransfer medicinal products’’ as used inthe relevant European guideline ‘‘Note forguidance on the quality, preclinical and

Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 243
clinical aspects of gene transfer medicinalproducts (CPMP/BWP/3088/99)’’ seemsmore exact. An accurate listing of themedicinal products that belong to thegroup of gene transfer medicinal prod-ucts can be found in the table containedin the guideline. The definition given inthe first chapter of this article is in ac-cordance with this guideline and is inagreement with the definition of gene ther-apeutic products of Directive 2003/63/EC.The annex of the latter directive containslegally binding requirements for qualityand safety specifications of gene transferproducts. Although targeted at product li-censing, these requirements may have abearing on their characterization beforeclinical use. Active ingredients of genetransfer medicinal products may include,for example, vectors, naked plasmid DNA,or certain microorganisms such as condi-tionally replicated adenovirus. For the exvivo strategy, the active ingredients are thegenetically modified cells.
Written approval by a competent author-ity in conjunction with positive appraisalby an ethics committee will in future benecessary for the initiation of clinical genetherapy trials. Respective regulatory pro-cesses are currently established in all EUmember states during transformation ofDirective 2001/20/EC. The manufactureof clinical samples in compliance withGood Manufacturing Practice (GMP) willbecome compulsory. Germ-line therapy isillegal in the European Union. The lawrelevant for clinical gene therapy trials andmanufacture of gene transfer medicinalproducts in Germany is the German DrugLaw (AMG) and respective decrees andoperation ordinances. The law governingthe physicians’ profession stipulates in the‘‘Guidelines on gene transfer into humansomatic cells’’ (‘‘Richtlinien zum Gen-transfer in menschlichen Korperzellen’’)
that the competent ethics committee mayseek advice from the central ‘‘Commis-sion of Somatic Gene Therapy’’ of theScientific Council of the German MedicalAssociation before coming to its vote. ThePaul–Ehrlich Institut is the competent au-thority in Germany and offers informationon current clinical trial regulations.
Gene transfer medicinal products willbe licensed via the centralized procedureby the European Commission. The licen-sing process is coordinated by the EMEA(European Agency for the Evaluation ofMedicinal Products) following submissionof a licensing application. The market-ing authorization is governed by CouncilRegulation (EC) No. 2309/93. The recom-mendation in favor or against marketingauthorization is made on the basis of Direc-tives 75/319/EEC and 91/507/EEC by ex-perts of the national competent authoritieswho are members of the ‘‘Committee forProprietary Medicinal Products’’ (CPMP).
In the United States, the Center forBiologics Evaluation and Research (CBER)of the ‘‘Food and Drug Administration’’(FDA) is responsible for clinical trialapproval and marketing authorization.
The assessment of the licensing applica-tion focuses on the quality, safety, efficacy,and environmental risk of a gene trans-fer medicinal product. The manufacturingprocess has to be designed and performedaccording to Good Manufacturing Process(GMP) regulations. Like other biologicals,gene therapy products have considerablylarger size and complexity compared tochemicals, and analysis of the finishedproduct is not sufficient to control theirquality and safety. A suitable process man-agement, in-process control of all criticalparameters identified within process vali-dation are decisive factors. Gene transfermedicinal products containing or consist-ing of genetically modified organisms are

244 13.4 Manufacture and Regulatory Aspects
also subject to contained-use regulationsbefore licensing and until these organismsare applied to humans.
From the economic point of view, proce-dures for the manufacture of therapeuticDNA must be scalable and efficient, and, atthe same time, simple and robust. Manu-facturing processes are as manifold as thegene transfer methods used in gene ther-apy. As an example, manufacture of plas-mid DNA for naked nucleic acid transfercan be briefly described as follows [26]. Themethods available for plasmid productiontoday largely originate from lab proceduresfor the production of DNA for analytical
purposes (mini preparations) and havebeen adapted to fit process scale [27]. Toxicsubstances and those that present a hazardto the environment, expensive ingredi-ents, and nonscalable methods must beavoided [28]. In this context, the experi-ence gained from industrial manufactureof raw materials with the aid of bacte-rial cultures and virus production for thepurpose of vaccine production are usefulfor fermentation [29]. Suitable methods fordownstream processing above all includechromatographic methods with high dy-namic capacity and selectivity as well ashigh throughput [30, 31].
Process step Purpose
E. coli batch fermentation
Cell separation Volume reduction
Alkaline lysis(NaOH/SDS)
K. acetate precipitation
Removal ofmembrane fragments
proteinsgenomic DNA
Clarification(filtration/centrifugation) Removal of
precipitate
Anion exchangechromatography(‘‘Capture step’’)
Removal ofRNA
host cell proteinsendotoxin
Isopropanolprecipitation Removal of
host cell proteins
Gel filtration/Reversed-phasechromatography(‘‘Polishing step’’)
Removal ofgenomic DNA
RNAendotoxins
‘‘open circle’’ plasmid
Formulation
Fig. 4 Therapeutic plasmid DNA: Typical manufacturing process.

Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 245
Test Specification (Method)Appearance Clear colourless solution (visual inspection)Size, restriction interfaces(identity)
Agreement with plasmid card (agarose gelelectrophoresis, restriction enzyme assay)
Circular plasmid DNA (ccc) > 95 % (Agarose gel electrophoreses, HPLC)E. coli DNA < 0.02 mg/mg plasmid DNA (southern blot)Protein Not detectable (BCA protein assay)RNA Not detectable (agarose gel electrophoresis)Endotoxin < 0.1 EU/mg plasmid DNA (LAL assay)Sterility No growth after 14 days (USP)Specific activity Conforms to reference standard (in vitro
transfection)
Fig. 5 Therapeutic plasmid DNA: Typical release specifications.
In a typical procedure for the man-ufacture of therapeutic plasmid DNA(cf. Figs. 4 and 5), the first step is batchfermentation of Escherichia coli cells froma comprehensively characterized ‘‘Mas-ter Working Cell Bank’’ (MWCB). Forthis purpose, modern methods use high-density fermentation with optimized andsafe E. coli K12 strains bearing a highnumber of copies of the required plas-mid. The bacterial cells are harvestedfor further processing, resuspended ina small buffer volume, and lysed in analkaline lysis procedure [32]. By neutraliza-tion, the plasmid DNA is renatured whilea large quantity of proteins, membranecomponents, and genomic DNA remaindenatured. After separation of the pre-cipitate by filtration, a chromatographicstep can be performed as ‘‘capture step’’.Because of the anionic character of thenucleic acid, anion exchange chromatog-raphy (AEX) is the method of choice. Infractionated gradient elutions, differencesin the charge enable the separation fromcontaminated RNA. Gel filtration (GF) orreversed phase (RP) steps can be usedfor fine purification. For final productanalysis, evidence must be provided batch-by-batch that besides the correct identityand homogeneity, critical impurities likemicroorganisms, host cell proteins, ge-nomic DNA, RNA, or endotoxins have
been reduced below the specified lim-its [33]. Removal of endotoxins is criticalfor in vivo gene transfer efficiency achievedwith naked DNA.
Some established methods from pro-tein chemistry can be used for process-ing therapeutic DNA. Parallels with theprocessing of proteins, however, cannotconceal the fact that nucleic acids havesome very specific properties. These in-clude the extremely high viscosity of DNAsolutions, the high sensitivity of nucleicacids to gravity, the low static and dy-namic capacity of their chromatographicadsorption, and the ability to penetratefiltration media with porosities well be-low their molecular weight (‘‘spaghettieffect’’).
After first experience, plasmid concen-trations of approximately 200 mg L−1 fer-mentation broth can be obtained in high-density fermentation (optical density >
50), corresponding to a yield of ap-proximately 800 mg plasmid DNA perkilogram of dry biomass. Thus, froma fermenter of 1000 L usable volume,approximately 100 g plasmid DNA canbe isolated per run in a batch fer-mentation at a purification yield of ap-proximately 50%. Consequently, capaci-ties for production of kilogram amountscan be built up with existing technolo-gies [34].

246 13.5 First Experience with the Clinical Use of Gene Transfer Medicinal Products
13.5First Experience with the Clinical Use ofGene Transfer Medicinal Products
The development of somatic gene ther-apy is still in its infancy. A number oftheoretical risks of gene therapy have beenlisted, and numerous approaches and genetransfer methods are being developed inthe clinic, even more in preclinical experi-ments.
Until today, SCID-X1 patients have ap-parently been cured by gene therapy usingretrovirally modified bone marrow stemcells. At the same time, the occurrenceof leukemia in 2 of the approximately10 successfully treated children showedthat, at this point of development, theoret-ical risks cannot be clearly distinguishedfrom clinically relevant risks due to the sofar insufficient clinical experience. Trends,however, show that each pathological situ-ation will require the development of acertain adapted gene therapy approach.Thus, in the long run, gene therapy willpresent real therapy or prevention op-tions, especially for a number of up-to-nowinsufficiently treatable or untreatable dis-eases.
References
1. F. Anderson, Sci. Am. 1995, 273, 96B–98B.2. K. W. Culver, Gene Therapy, Mary Ann
Liebert, New York, 1994.3. R. A. Morgan, W. F. Anderson, Annu. Rev.
Biochem. 1993, 62, 191–217.4. R. C. Mulligan, Science 1993, 260, 926–932.5. U. Gottschalk, S. Chan, Arzneim.-Forsch./
Drug Res. 1998, 48, 1111–1120.6. P. Tolstoshev, W. F. Anderson, Genome Res.
Mol. Med. Vir. 1993, 7, 35–47.7. D. Armentato, C. Sookdeo, G. White, J. Cell
Biochem. 1994, 18A, 102–107.8. R. G. Vile, S. J. Russell, Br. Med. Bull. 1995,
51, 12–15.
9. C. P. Hodgson, Biotechnology 1995, 13,222–229.
10. K. K. Jain, Vectors for Gene Therapy, ScripReports, PJB Publications Ltd, Surrey,1996.
11. R. J. Mannino, S. Gould-Fogerite, Biotech-niques 1988, 6, 682–688.
12. S. I. Michael, D. T. Curiel, Gene Ther. 1994,1, 223–232.
13. J. W. Wilson, M. Grossmann, J. A. Cabreraet al., J. Biol. Chem. 1992, 267,11483–11489.
14. C. J. Buchholz, J. Stitz, K. Cichutek, Curr.Opin. Mol. Ther. 1999, 5, 613–621.
15. J. Stitz, C. J. Buchholz, K. Cichutek et al.,Virology 2000, 273, 16–20.
16. J. Stitz, M. D. Muhlebach, K. Cichutek et al.,Virology 2001, 291, 191–197.
17. V. Randrianarison-Jewtoukoff, M. Perricau-det, Biologicals 1995, 23, 145–147.
18. R. M. Kotin, Hum. Gene Ther. 1994, 5,793–797.
19. S. Hacein-Bey-Abina, A. Fischer, M. Cavaz-zana-Calvo, Int. J. Hematol. 2002, 76,295–298.
20. V. Randrianarison-Jewtoukoff, M. Perricau-det, Biologicals 1995, 23, 145–147.
21. R. A. Spooner, M. P. Deonarian, A. A. Epe-netos, Gene Ther. 1995, 2, 1–11.
22. B. Gansbacher, K. Zier, B. Daniels, J. Exp.Med. 1990, 172, 1217–1222.
23. K. Cichutek, Intervirology 2000, 43, 331–338.24. R. S. Nussenzweig, C. A. Long, Science 1994,
265, 1381–1384.25. O. Cohen-Haguenauer, F. Rosenthal, B. Gan-
sbacher et al., Hum. Gene Ther. 2002, 13,2085–2110.
26. N. A. Horn, J. A. Meek, G. Budahazi et al.,Hum. Gene Ther. 1995, 6, 565–573.
27. M. Muller, Considerations for the scale-upof plasmid DNA purification in Nucleic AcidIsolation Methods (Eds.: B. Bowlen, P. Durre),American Scientific Publishers, New York,2003.
28. M. Marquet, N. A. Horn, J. A. Meek, Bio-pharm 1995, 10, 26–37.
29. C. Prior, P. Bay, B. Ebert, Pharm. Technol.1995, 19, 30–52.
30. A. P. Green, G. M. Prior, N. M. Helvestonet al., Biopharm 1997, 10, 52–62.
31. G. Chandra, P. Patel, T. A. Kost et al., Anal.Biochem. 1992, 203, 169–177.
32. H. C. Birnboim, Methods Enzymol. 1983, 100,243–249.

Somatic Gene Therapy – Advanced Biotechnology Products in Clinical Development 247
33. M. Marquet, N. A. Horn, J. A. Meek, Part 1:Biopharm 1997, 10(7), 42–50; Part 2:Biopharm 1997, 10(8), 40–45.
34. U. Gottschalk, The industrial perspective ofsomatic gene therapy in Interdisciplinary
Approaches to Gene Therapy (Eds.: S. Muller,J. W. Simon, J. Vesting), Springer, 1997.

249
14Nonviral Gene Transfer Systemsin Somatic Gene Therapy
Oliver KayserFreie Universit
..at Berlin, Berlin, Germany
Albrecht F. KiderlenRobert Koch-Institut, Berlin, Germany
14.1Introduction
Somatic gene therapy might develop intoone of the most important therapeuticstrategies of the near future. Innate oracquired genetic defects are held respon-sible for a number of diseases such ashemophilia, cystic fibrosis (mucoviscido-sis), adenosine-deaminase (ADA) deficit,and AIDS. Substituting or supplement-ing malfunctioning or missing geneticinformation by transiently or permanentlyinserting the appropriate gene appears tobe a plausible therapeutic strategy espe-cially from the patients’ point of view.Attempts in gene therapy began in theearly 1990s with great expectations thathave only partially been met. No diseasewith a defined genetic background has sofar been causally cured by gene therapy.Viral gene transfer systems have causedsevere problems that could not be broughtunder control to date. The death of the 18-year-old Jesse Gelsinger is a tragic evidenceof the basic deficits of viral transfection
systems. In consequence, attention is nowbeing focused on chemical and physicalgene transfer systems. This review cov-ers nonviral gene transfection strategies ofcurrent interest with special reference toexperimental results found in vivo and forclinical trials.
14.2What is Gene Therapy?
Gene therapy may be defined as theexpression of a gene that has been in-troduced into a target cell or a targettissue in order to alter an existing func-tion or to introduce a new function withthe aim of curing a patient from a spe-cific disease. In many countries this isrestricted by law to somatic cells. In Ger-many, for example, genetic manipulationof germ cells is forbidden according toSection 5 Embryonenschutz Gesetz (Es-chG) and attracts a penalty of up to fiveyears imprisonment [1]. German legisla-tion on gene transfection as a form of
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

250 14.3 Strategies in Gene Therapy
therapy or medicine is still incomplete. Anamendment specifically covering somaticgene therapy has not yet been passed.German Drug Regulation (Section 2 andSection 3 Deutsches Arzneimittelgesetz,AMG) defines DNA introduced for ther-apeutic purposes in a very general manneras a pharmaceutical product. GermanGene Technology Regulation (Section 2 (3)Deutsches Gentechnikgesetz, GenTG) ex-plicitly excludes the regulation of genetherapy [2]. Interestingly, gene therapymight be indirectly affected: theoretically(sensu strictu), a patient undergoing genetherapy becomes a genetically modifiedorganism. Consequently, his release fromhospital should be subject to authorization.When discussing DNA as a pharmaceuti-cal, a look at the European legislation, forexample, at the guidelines of the EuropeanAgency for the Evaluation of MedicinalProducts (EMEA) might be helpful. Ac-cording to Council Regulations (EEC) No.2309/93 – ANNEX (List A), DNA is con-sidered a pharmaceutical product for genetherapy [3]. The necessary vector is simplydefined as an additive.
Most likely, gene therapy will only workfor a limited number of ‘‘suitable’’ diseasesin a restricted commercial environment.It is very costly and affords a highlycomplex technology as well as an indi-vidually tailored strategy for gene delivery,depending on the relevant circumstancesand aims [4]. Monogenetic diseases suchas hemophilia [5], sickle cell anemia,adenosine-deaminase deficit [6], cystic fi-brosis [7], or Duchenne muscular dystro-phy [8] are likely first candidates, as theyrequire the replacement or substitution ofonly a single gene. Trisomy 21 (Down Syn-drome), on the other hand, which is alsofrequently mentioned in this context, is apoor candidate, as it is much more diffi-cult to silence the additional chromosome
21 than to replace nonfunctional genes.In different forms of cancer and heart-or circulatory failures or of acquired ge-netic dysfunctions such as hepatitis orAIDS, two or more genes must be sub-stituted or otherwise manipulated. Here,successful gene therapy seems unlikely atthe moment owing to technical limitationsin DNA transport into target cells and intissue-specific forms of application.
Among the known innate diseases, genetherapy has been most intensively investi-gated in ADA deficiency and cystic fibrosis.Both reduce mean life expectancy to below20 years, accompanied by severe symp-toms that strongly affect the quality oflife [6, 7].
14.3Strategies in Gene Therapy
Independent of the respective methodof gene transfer, two basic strategiesfor gene therapy may be discriminated(Fig. 1). Following the ex vivo strategy,cells or tissues are first removed fromthe patient, then exposed in vitro tothe therapeutic genetic construct. If thetransfection has been successful, thematerial is reimplanted in the patient [9,10]. In the alternative in vivo methods,the therapeutic gene or DNA sequence isintegrated into a vector and this constructis injected locally or systemically intothe patient. Among other deficits, thelatter method is characterized by poortissue selectivity, rapid extracellular DNAdegradation, and the danger of inducingoncogenes when using viral vectors.
Further differentiation is possibleat the molecular level (Fig. 2): Anintact (therapeutic) gene may simplybe added to the defect one (Fig. 2a),a missing gene may be substituted
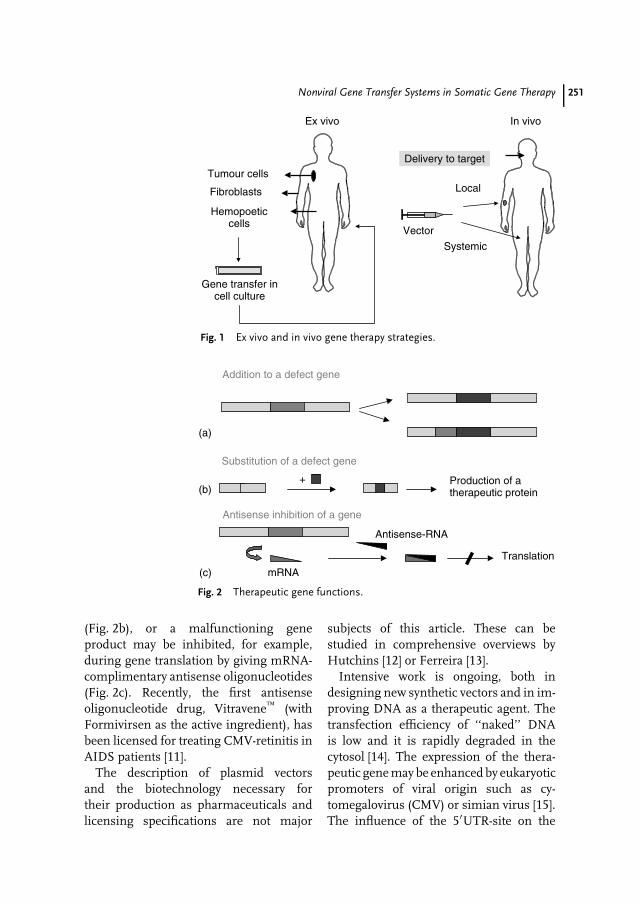
Nonviral Gene Transfer Systems in Somatic Gene Therapy 251
Tumour cells
Fibroblasts
Hemopoeticcells
Gene transfer incell culture
Delivery to target
Vector
Local
Systemic
In vivoEx vivo
Fig. 1 Ex vivo and in vivo gene therapy strategies.
Addition to a defect gene
Antisense inhibition of a gene
mRNA
Antisense-RNA
Translation
(a)
(b)
(c)
Substitution of a defect gene
+ Production of atherapeutic protein
Fig. 2 Therapeutic gene functions.
(Fig. 2b), or a malfunctioning geneproduct may be inhibited, for example,during gene translation by giving mRNA-complimentary antisense oligonucleotides(Fig. 2c). Recently, the first antisenseoligonucleotide drug, Vitravene (withFormivirsen as the active ingredient), hasbeen licensed for treating CMV-retinitis inAIDS patients [11].
The description of plasmid vectorsand the biotechnology necessary fortheir production as pharmaceuticals andlicensing specifications are not major
subjects of this article. These can bestudied in comprehensive overviews byHutchins [12] or Ferreira [13].
Intensive work is ongoing, both indesigning new synthetic vectors and in im-proving DNA as a therapeutic agent. Thetransfection efficiency of ‘‘naked’’ DNAis low and it is rapidly degraded in thecytosol [14]. The expression of the thera-peutic gene may be enhanced by eukaryoticpromoters of viral origin such as cy-tomegalovirus (CMV) or simian virus [15].The influence of the 5′UTR-site on the

252 14.4 Gene Transfer Systems
translation efficiency of mRNA is also be-ing studied [16, 17]. The insertion of atleast one intron into the cDNA may lead to100-fold enhancement of mRNA [18]. Theaddition of a suitable terminal poly(A)-signal (bovine growth factor) has similareffects [19]. Special attention is given to thedevelopment of tissue-specific promoters,restricting transfected gene expression tothe necessary therapeutic sites [20, 21]. Avery elegant strategy for controlling theexpression of the transfected gene is byturning it on or off with common oraldrugs such as tetracycline or progesterone-antagonist. These interact with a mutatedreceptor acting as a transgene, thus initi-ating a signal transduction cascade, whichfinally induces the transcription (or its in-hibition) of the respective gene for justas long as the drug is kept at a sufficientlevel [22, 23].
14.4Gene Transfer Systems
Owing to their rapid degradation inthe cytosol, genes or DNA-sequences
are only rarely transfected as ‘‘naked’’molecules [24]. The commonly used genetransfer methods may be segregated intobiological, physical, and chemical systems(Table 1). The biological systems involvingviral vectors (retro-, adeno-, or poxviruses)make up 77% of all gene transfectionstudies published to date [25]. However,physical and chemical methods have expe-rienced a relative increase in recent years(12% from 1998 to 2003). This tendencyreflects the often highly serious and hardlycontrollable side effects of viral systems.Despite recent modifications of wild-typevectors, the future of such systems forhuman application appears limited. Onemain problem of viral vectors is theirstrong immunogenicity. Already, the first(high dose) application may initiate an im-mune reaction against the given proteins,which may lead to all sorts of allergic reac-tions, even lethal anaphylactic shock, whenrepeated. Possible reversion of the virus towild-type is another danger. The poten-tial induction of oncogenes by retrovirusesmust also be mentioned [26, 27].
On this background, the development ofchemical or physical gene transfer systems
Tab. 1 Gene transfer methods in somatic gene therapy [10]
Method Application Tissueselectivity
Transfectionefficiency
Expressionduration
Chemical Liposomes,Ca-phosphate-precipitation
Ex vivo/(invivo)
No Low Transient/stabile
Physical Microinjection,electroporation,‘‘particle-bombardment’’
Ex vivo/(in vivo)
No Moderate Transient
Biological Nonviral:ligand/receptor
(Ex vivo)/invivo
Yes Low–moderate Transient
Viral: (e.g. retro-,adeno-, adeno-associated)
Ex vivo/invivo
Some Moderate–high Transient/stabile

Nonviral Gene Transfer Systems in Somatic Gene Therapy 253
appears especially interesting, combin-ing simple usage with maximum safety.Further requirements for an ideal genetransfer system are minimal infectivity andimmunogenicity, defined chemical andphysical characteristics, and the possibilityof multiple dosing [27].
When looking for suitable alternativesto viral vectors, the pharmaceutical in-dustry can offer a broad spectrum ofthoroughly investigated and readily avail-able medicinal carrier systems. As far asmolecular transport and organ- or cell-specificity are concerned, the demands onmodern drug delivery and gene transfersystems exhibit so much similarity, thatthe latter may profit significantly in thefields of biotechnology and pharmacol-ogy [28, 29]. Among the chemical vectors,liposomes, polymer nanoparticles, andpolylysine particles deserve special discus-sion. Physical transfection systems suchas electroporation and bioballistics arefurther examples of an efficient transferof existing know-how in pharmaceuticaltechnology to somatic gene therapy. Itmight be mentioned that gene transferin a nonviral system must correctly beaddressed as transfection, whereas in bi-ological systems it should be referred toas transduction. Table 1 suggests a sys-tematic arrangement of methods in genetransfer technology. In the following, themost important contemporary nonviral
gene transfer systems are described andassessed.
14.5Physical Gene Transfer Systems
14.5.1Electroporation
During electroporation, cells or tissues areexposed to an electric field with high volt-age (up to 1 kV). Short, rapid pulses causetransient membrane instability and the for-mation of pores with a mean lifetime ofminutes [30]. Soluble DNA constructs thathave been added to the culture medium orinjected into the tissue may thereby enterthe cell and ultimately reach the nucleus.The basic principle is also known to phar-macists as ‘‘iontophoresis’’ and is usedfor transdermal drug application [31]. Thisgene transfection method has so far beenwell accepted by patients and is safe, espe-cially due to the low risk of infection. Todate, mainly liver, muscle, and skin cellshave been transformed this way, mostlyvia the transdermal route [32].
14.5.2Bioballistics
Bioballistical methods are already widelyused in biotechnology, for example, as‘‘gene guns’’ for injecting DNA vaccines(Fig. 3). For this, linear or circular
Fig. 3 Accela gene gun(Powderject) [33]. Nozzle
Absorber
Cassette
Gas cylinder
Safety catch Trigger

254 14.6 Chemical Vectors
(plasmid) DNA is adsorbed to nanometer-sized gold or tungsten particles. These areshot from a cylinder with compressed ni-trogen or helium as propellant, therebyreaching speeds of up to 900 m s−1 [33].When applied to the skin, the DNA/metalparticles pass through dead tissue suchas the Stratum corneum, reaching livingcells. Statistically, only one of 10 000 par-ticles reach the interior of viable cells,thereby loosing their DNA. For the DNAto then enter the nucleus, active trans-port mechanisms are probably necessary.Bioballistical gene transfer methods areobviously not suitable for systemic appli-cation. Further drawbacks are the needfor very stable DNA and the high devel-opment costs. Their greatest advantage,on the other hand, is the fact that re-lated technologies are already approvedand commercially available such as theAccell (Powderject) (Fig. 3) or the He-lios (BioRad) systems that are used forvaccination [34].
14.6Chemical Vectors
A multitude of synthetic chemical vectorsare being developed. Of these, cationiclipids and cationic polymers are probablythe most thoroughly investigated. Alreadyin 1987, Felgner et al. could demonstratein vitro gene transfection using cationiclipids [35]. Transfer efficiency was system-atically improved [36, 37], leading to thefirst clinical studies on cystic fibrosis pa-tients [38, 39].
Irrespective of their highly variantmolecular composition and 3D struc-ture, chemical vectors such as liposomesand polymer particles have many biolog-ical and physical features in common.Positively charged amine functions areespecially important as these can be
loaded with the negatively charged DNAmolecules [40]. Optimal interaction be-tween plasmid-DNA and cationic additivesleads to the development of colloidal,positively charged particles that may ad-here to and be taken up by negativelycharged cells. This is a cornerstone ofchemical vector technology. Type andstructure of the amine functions deter-mine the stability of the complex, itscellular uptake in a phagosome, its re-lease from the phagosome into the cytosoland dissociation of the DNA molecule, andeven DNA-transport to the nucleus [41].
Nucleic acids as drugs are still rather un-usual in pharmaceutics. They are highlynegatively charged and range in sizes from103 kDa (oligonucleotides) to 106 kDa(genes). Their targets are invariably intra-cellular. In contrast to most conventionalpharmaceuticals, nucleic acids are toolarge and too strongly charged to passcell membranes by simple passive means.Furthermore, free, that is, ‘‘naked’’ nucleicacids are rapidly degraded by cellular nu-cleases. Chemical vectors therefore havethe additional job to protect the DNA theycarry from enzymatic degradation. In thecontext of gene transfer, liposomes arealso referred to as lipoplex, polymers aspolyplex, and combinations as lipopolyplexparticles.
14.6.1Cationic Liposomes (Lipoplex)
Cationic liposomes were developed and al-ready complexed with DNA over 20 yearsago [35]. However, it was in the Hu-man Genome Project that their po-tential as gene transfer vehicles re-ally became apparent to geneticists andpharmacists. Cationic liposomes consistof cationic phospholipids that may bedivided according to their number of

Nonviral Gene Transfer Systems in Somatic Gene Therapy 255
ONH
NH
O
DC-Chol
Multivalent cationic lipids
Monovalent cationic lipids
O
ON
+
N+
N+
O
O
O
NH3+
NH3+
NH2+
H2N
DOSPA
O
O
O
O
O
OHO
DMRIE
DOTAP
Fig. 4 Cationic lipids: DOSPA: 2,3-Dioleoyloxy-N-[2-(spermincarboxyamido)ethyl]-N,N-dimethyl-1-propanamminiumchloride; DOTAP: 1,2-Dioleoyloxypropyl-3-N,N,N-trimethylammoniumchloride; DC-chol: 3β-[N-(N′,N′-dimethyl-aminoethane)carbamoyl]-cholesterol; DMRIE: 1,2-Dimyristyloxypropyl-3-N,N-dimethylhydroxyammoniumbromide [42, 43].
tertiary amine functions into monova-lent and multivalent cationic lipids [42,43]. An overview of the most commonlyused cationic lipids is given in Fig. 4.DOTAP (1,2-Dioleoyloxypropyl-3-N,N,N-trimethylammoniumchloride) and DC-chol (3β-[N′,N′-dimethylaminoethanecar-bamoyl]-cholesterol) are possibly themost important. However, due tohigh toxicity, they must be mixedwith helper lipids such as Di-oleoylphosphatidylethanolamine (DOPE)before processing to liposomes [44].A well-known transfection agent isLipofectin, which consists of equalparts of N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride(DOTMA) and DOPE [45].
The high general toxicity of the firstcationic lipids initiated the synthesis ofa variety of derivatives [46]. The published
in vitro data allows a first structure/activityanalysis. The presence of a tertiary aminefunction and of 1 or 2 hydrophobicaliphatic chains are most important forproducing stable liposomes and for bind-ing negatively charged DNA. The tertiaryamine functions can be combined withthe hydrophobic lipid chains by either es-ter or ether bonds. Ester bonds are splitmore readily under physiological condi-tions than ether bonds and the lipoplexparticles are consequentially metabolizedmore rapidly. These different metaboliccharacteristics are exemplified by DOTMAand DOTAP; the former is synthesized asester, the latter as ether.
The aliphatic lipid chain may be sat-urated (DMRIE) or may contain doublebonds (DOTAP, DOSPA). In vivo, dialkyl-chains with a length of 12 to 18 carbonatoms each seem to promote the best

256 14.6 Chemical Vectors
gene transfection [38]. A substitution ofthe alkyl chains with cholesterol, as inDC-chol, shows further advantages in thetreatment of cystic fibrosis [38, 39]. Choles-terol structures have strong affinities tobroncho-epithelial cells and interact lesswith plasma proteins [39].
Positively charged liposomes loadedwith negatively charged DNA must main-tain an overall positive charge in order toapproach and contact target cells that arenormally negatively charged. Liposomesthat have been overloaded with DNA andthus receive a negative total charge ex-hibit low endocytosis rates and enhancedmetabolic degradation [38, 39]. Proper se-lection of the cationic lipids and of thelipid-to-DNA ratio are decisive for the rateof complexation and the colloidal structureof the product. Finally, conformations maychange in time, a process also referred toas ‘‘aging’’ of the DNA/lipid complex [47].
Recent studies show that intracellularstability of the lipid/DNA complex andthe necessary dissociation of the DNAmolecule from its vector depend both onthe pKs of the chosen lipids and on thepH of the product [48]. Safinya et al. de-scribe how not only the main cationiclipid but also the helper lipids influencethe physical characteristics of the result-ing lipid phase [49]. For instance, whenformulating DOTAP liposomes with Di-oleoylphosphatidylcholine (DOPC), lamel-lar structures are achieved. However, whenexchanging DOPC for DOPE as helperlipid, inverse hexagonal micelles are pro-duced. One may speculate whether dif-ferent lipid phase characteristics lead todefined biopharmaceutical variations [50].
Cationic liposomes have also been inten-sively investigated in vivo. In combinationwith neutral helper lipids, they appear to bewell tolerated even at high concentrations;they are neither immunogenic nor do they
induce toxic side effects [36]. This appliesto different routes of application such asintravenous, pulmonary, or nasal, none ofwhich have been described as significantlyunpleasant by the recipient patients. Nev-ertheless, cationic liposomes also pose anumber of problems such as high serumprotein binding and rapid metabolic degra-dation in the liver.
Liposomes are generally the most effi-cient at a diameter ranging from 400 to500 nm. Larger particles are almost com-pletely removed from the circulation bycells of the monocytic phagocytes system ofliver and lung, and therefore do not reachother target areas [51, 52]. Pegylation is onemeans of improving pharmacokinetics.However, the useful extent of pegylation isrestricted as further reduction of the zetapotential at the surface of the liposomescorrelates with their reduced cellular up-take [7, 53].
Low transfection rates are a further prob-lem. Reasons are, among others, insuffi-cient lysosomal release of the therapeuticDNA from its carrier and/or its rapidenzymatic degradation within the lyso-some or in the cytosol. The DNA thatwas integrated in the liposomes must bereleased to become effective (see Fig. 5).Studies on microinjections of lipid/DNAcomplexes directly into the nucleus clearlyshow reduced transfection rates [54]. Asthe nuclear pores have mean diametersof only 25 to 50 nm and passage formacromolecules is further controlled bythe nuclear pore complex, passive dif-fusion into the karyosol is limited toparticle sizes below 45 kDa [51, 55]. Con-ventional recombinant plasmids normallyhave a molecular weight ranging from 50to 100 kDa, and therefore require activetransport into the nucleus [55].
Admission of plasmid-DNA into thekaryosol is steered by nuclear localization

Nonviral Gene Transfer Systems in Somatic Gene Therapy 257
5. Transport toand in the nucleus
Proteins
Target cell
Gene transfer complex
4. Endosomal lysis
Plasmid DNA
−−
−−−
−
−
Cationic polymer/lipid
++
++
+
mRNA
1. Condensation of the DNA
2. Binding to cell surface3. Endocytosis
Fig. 5 Uptake, transport, and release of DNA in particulate gene transfer systems.
sequences that are assisted by importin-ß,guanin-nucleotide-binding protein (Ran),and nuclear transport factor (NTF). Inthe karyosol, the therapeutic DNA istranscribed by RNA polymerases intomRNA, which is transported back into thecytosol where it attaches to ribosomes fortranslation into protein [55, 56]. In order toimprove its transport into the nucleus, thetherapeutic DNA may be coupled to suchnucleus localizing sequences. The lattercan be found naturally in certain virusesthat have developed this strategy for a mostefficient nuclear invasion. For example,Rudolph et al. coupled DNA to shortTAT sequences taken from the arginine-rich motif of the HIV-1-TAT proteinand achieved significant enhancement oftransfection. Of the 101–amino acid–longHIV-TAT sequence they synthesized, a12–amino acid–long oligopeptide boundit to polyarginine and thus achieved a 390-fold enhanced transfection rate [57].
Unsatisfactory transfection rates and lowcell- or organ specificity also initiatedthe development of simple liposomes
to virosomes or immunoliposomes. Vi-rosomes are liposomes that contain vi-ral proteins or fusiogenic peptides forenhanced DNA release from the endo-some and for generally improved genetransfection rates [58]. Immunoliposomesare characterized by target-specific mono-clonal antibodies bound to their surface.These were first developed in the eightiesfor tumor-specific delivery of liposome-entrapped drugs and have since beenmodified for gene therapy [55, 58].
14.6.2Polymer Particles (Polyplex)
A further class of synthetic gene vectorsthat has received attention in past yearsis cationic polymers, which condenseand package DNA with high efficiency.Polymerized or oligomerized branched ornonbranched amino acid chains composedof lysine or arginine are common [59, 60].Polyethylenimine, however, developed in1995 by Boussif [61] and already used for

258 14.6 Chemical Vectors
NH2
O
*
NH2
NH2NH2
*NH
NH
NH
NH
NH
N
O
O
O
O
O
NH2
NH2
n
NH2NNNH2
OO
NHO
NH2
PLL
PEI
pAMEAMA dendrimer (1. Generation)
*NH
n* *
N+
n
PVP
OOO
OHNH2
OOH
O
NH2**
OH
OH n
Chitosan
Fig. 6 Chemical structures of cationic polymers.
gene transfection experiments in vitro aswell as in vivo, appears to be the mostpromising cationic polymer at the moment(see Fig. 6). Basically, cationic polymergene transfer systems reveal the samepharmaceutical problems and intracellularbarriers as described above for cationicliposomes.
14.6.3Poly-L-Lysine (PLL) and Poly-L-Arginine(PLA)
Poly-L-lysine (PLL) has already been inten-sively used as a polymeric gene transfersystem [60, 62]. It is synthesized by poly-merization of the N-carboxyanhydrid oflysine. Arginine is polymerized in a similarmanner. PLL/DNA complexes are pro-duced by dissolving both components inaqueous media and precipitating the par-ticulate complexes. These particles, whichnormally range from 400 to 500 nm,are capable of transporting nucleic acidsranging from short molecules to large
artificial yeast chromosomes [62, 63]. Thefirst in vivo studies, however, revealed sub-stantial toxicity combined with low DNAtransfection efficiency. Different chemicalmodifications and variations in particlesize were then tested. Toxicity is substan-tially reduced by coating the particles withPEG derivatives [64], and the transfectionrate is enhanced by attaching ligands suchas transferrin, folate, or target-specificmonoclonal antibodies [65]. Interestingly,pegylation also achieved significant reduc-tion in unwanted hepatic metabolizationof the particles.
Apart from lysine, polymers of othercationic amino acids such as arginine andhistidine were also investigated. Conjuga-tion of histidine to ε-L-histidine enabledthe development of highly interestingPLH/DNA complexes characterized byhigh transfection rates [66]. One explana-tion for the elevated transfection efficiencymay be that the highly protonated histidinestructure that develops in the generallyacidic (pH 6) endosomal environment may

Nonviral Gene Transfer Systems in Somatic Gene Therapy 259
cause rapid destruction of the endoso-mal membrane, and thus an enhancedrelease of the therapeutic DNA into thecytosol.
14.6.4Polyethyleneimine (PEI)
Linear or branched polyethyleneiminesgenerally range in size from 1.8 to800 kDa [60, 67, 68]. They are synthesizedby cationic polymerization. Starting from2-substituted-2-oxazoline-monomers, lin-ear PEI with a mean molecular weight of22 kDa, also known as ExGen 500 [69], areproduced by hydrolysis. PEI/DNA com-plexes have already been used in manyin vivo studies in animals following iv in-jection [70–74]; clinical studies, however,have not been reported to date. PEI/DNAcomplexes have repeatedly revealed veryhigh transfection efficacy. One advantageof PEI/DNA complexes over lipoplex- orPLL particles is their intrinsic buffer capac-ity at lysosomal pH, leading, as describedabove for PLL-particles, to rapid destruc-tion of the lysosomal membrane andDNA release [75]. This effect, also termed‘‘proton-sponge’’, is brought about by thechemical structure of PEI. Polymerizationproduces particles with primary amines atthe surface and secondary as well as ter-tiary amines in the interior. This causes ashift in pKa from approximately 6.9 to 3.9.The strong protonation at a pH below 6induces an osmotic gradient across the en-dosomal membrane, resulting in an influxof water, swelling, and finally disruption ofthe endosome with release of the PEI/DNAparticles into the cytosol [75].
Though the literature reveals some dis-crepancies, there seems to exist an inverserelationship between the molecular weightof the PEI particles and their transfection
efficiency. The interesting physicochem-ical characteristics of PEI-based genetransfer systems encouraged their furtherdevelopment, for example, to dendrimers(see below). In direct comparison, lin-ear PEI complexes (e.g. PEI 22) seem topossess better transfection characteristicsthan branched (e.g. PEI 25) [70]. One ex-planation may be a premature dissociationand subsequent degradation of the DNAmolecule.
Clinical trials with PEI complexes asgene transfer systems could so far notbe undertaken because of their frequentlyintolerable general toxicity. Depending onthe chemical structure, the lethal dosefor mice ranges from 40 to 100 mg kg−1
body weight [76]. The main problem liesin the strong interaction between PEIcomplexes and erythrocytes leading totheir aggregation and the danger of emboli.Pegylation of the PEI complexes may onlypartially help solve the problem, as a highdegree of pegylation generally reducesparticle uptake and thus transfectionefficacy [77].
14.6.5Dendrimers
The name ‘‘dendrimer’’ refers to thestar- or tree-shaped, branched structuresof this relatively new class of cationicgene transfer systems [8, 78–81]. Theyare frequently synthesized from polyami-doamines with special chemical or physicalfeatures. Probably best known are the‘‘starburst’’ dendrimers with particle sizesranging from 5 to 100 nm. These particlesreveal a highly regular branched ‘‘den-dritic’’ symmetry. Starburst dendrimersare three-dimensional oligomeric or poly-meric compounds, which, initiated fromsmall molecules as nuclei, are builtlayer-by-layer (‘‘generations’’) by repeated

260 14.6 Chemical Vectors
chemical reaction cycles. This allows anexquisite steering of the final size, three-dimensional form, and surface chemistryof a starburst polymer by the individualselection of components and binding pro-cedures for each generation [82].
The physical and chemical characteris-tics of dendrimers are mainly the result ofthe number and type of amine functionson the particle surface, but the secondaryand tertiary amines in the inside also af-fect their biological features. Despite theirhigh molecular weights, dendrimers aresoluble in water. They complex DNA withgreat efficiency, thus giving excellent vehi-cles for gene transfer. Their high transferefficiency, however, is probably less owingto high DNA adsorption rates but ratherto protonation of the amine functions af-ter endosomal uptake. As described abovefor the PEI complexes, this induces anosmotic gradient, leading to osmotic ly-sis of the organelle and enhanced releaseof the complex into the cytosol [82, 83].Dendrimer/DNA complexes have proventheir gene transfer efficacy in in vivo stud-ies [79–81]. However, as already exhibitedby the PEI complexes, they show strong,undesired interaction with erythrocytes,causing hemolysis. Again, the free primaryamine functions on the particle surface areheld responsible. Depending on the typeof dendrimer and the target cell, cationicdendrimers also reveal general cytotoxic-ity at concentrations ranging from 50 to300 µg mL−1 [84]. On the other hand, nodendrimer has to date been reported to in-duce tumors or to substantially affect theimmune system [85].
14.6.6Chitosan
Chitosan is a fiber produced by hydroly-zing chitin, mostly from crustaceans [86].
Owing to its free amine functions, chi-tosan may also be protonated (pKa = 5.6).In in vitro studies with Hela-cells, chi-tosan/DNA complexes showed a gene-transfection potency similar to that re-ported for PEI/DNA complexes. Plainchitosan, however, is almost insolublein water at neutral pH (but soluble atacidic pH). For this reason, trimethylated,quaternary chitosan derivatives have beenproduced that are sufficiently soluble un-der physiological conditions and easilycomplex DNA molecules. In in vitro exper-iments with COS-1 and CaCo-2 cells, theseinnovative chitosan derivatives proved su-perior to nontreated chitosan polymers,particularly as they showed no unspecificcytotoxicity [87].
14.6.7Poly(2-dimethylamino)ethylmethacrylate
Methacrylate polymers are used in phar-maceutical technology for microencapsu-lation. They are synthesized by polymeriza-tion of monomeric dimethylaminoacrylicacid to poly(2-dimethylamino)ethylmeth-acrylate (pDMAEMA), which is both sim-ple and cheap. Their low general toxicitymakes these polymers interesting alsoas gene transfer vehicles [88]. In in vitroexperiments with OVCAR-3 and COS-7 cells, some pDMAEMA/DNA particlesshowed high transfection rates [88, 89].This proved to be highly dependent ontheir size and charge. In HEPES-buffer(pH 7.4) pDMAEMA particles exhibit apositive zeta potential of around 25 mVand an average size of 100 to 200 nm. Fol-lowing endosomal uptake, again the outerprimary amine functions are protonated,leading to osmotic lysis of the endosomeand release of the pDMAEMA/DNA parti-cles into the cytosol.

Nonviral Gene Transfer Systems in Somatic Gene Therapy 261
Studies of DNA absorption ontopDMAEMA particles show that linearDNA (e.g. antisense oligonucleotides) isadsorbed more strongly than circularplasmid DNA [91]. However, this strongeradsorption has a negative influence onthe gene transfer efficiency, as the DNAmolecule is less likely to dissociate fromthe pDMAEMA particle in the cytosol. Forthis reason, circular DNA is preferred.As expected, DNA that is adsorbed topDMAEMA is protected from degradationby DNAse I [88].
14.7Outlook
The nonviral gene transfection systemsintroduced in this review bear significantadvantages over the viral systems, but haveserious drawbacks as well. One fact infavor is that many such systems are al-ready well established in classical areas ofpharmaceutical technology. Their produc-tion methods have already been optimizedand safety aspects have been investigatedin detail. Simple transfer of knowledgemay substantially reduce developing costs.Nonviral systems are noninfectious. Theyallow significantly higher DNA-loadingrates than viral systems, which reach theirlimits around 30 kb (Herpes virus). Nonvi-ral systems are only weakly immunogenicand therefore allow – in stark contrast toviral systems – multiple application.
The main drawback of nonviral systemsis that they normally only lead to thetransient expression of the therapeuticgene as it is not permanently integratedinto the host genome. In consequence,the therapeutic gene transfer must beregularly repeated, possibly over a longperiod of time. Further disadvantages areinsufficient cell- or tissue specificity and
low DNA transfer rates from the cytosolto the nucleus. Taken together, the gene-transfection performances of nonviralsystems are even weaker than those ofviral systems.
To date, it is still not possible to make aclear decision between viral and nonviralgene transfer systems. Mixed systems,hybrid vectors, which can be envisaged as‘‘denucleated’’ viruses, are in the pipeline.A combination of viral surface proteins andliposomes or the integration of therapeuticDNA into artificial cells or viruses arefurther innovative ideas for improvingsomatic gene therapy. Most importantis the rapid progress in three fields:in cell biology, unspecific and specificintracellular trafficking of macromoleculesstill raises questions; in biochemistry,further DNA carriers must be broughtforward for testing; and pharmaceuticaltechnology must supply improved andcell/tissue-specific drug delivery systems.Brought together in a rational form,such progress should make somatic genetherapy possible for selected disease formsin the near future.
References
1. German Regulation for the Protection ofHuman Embryos (Embryoschutz Gesetz,EschG) as proclaimed 13th December 1990,BGBl Part I, 1990, p. 2747.
2. German Regulation for Gene Technology(Gentechnik Gesetz, GenTG) as proclaimed16th December 1993, BGBl Part I, p. 2066.Last altered (2. GenTG-Anderung) 8thAugust 2002, BGBl Part I, 2002, 59,pp. 3220–3244.
3. EMEA Council Regulation 2309/93 asproclaimed 22nd July 1993, (OJ EC L 214).
4. G. M. Rubanyi, Mol. Aspects Med. 2001, 22,113–142.
5. H. S. Kingdon, R. L. Lundblad, Biotechnol.Appl. Biochem. 2002, 35(2), 141–148.

262 14.7 Outlook
6. R. M. Blaese, K. W. Culver, A. D. Miller,et al., Science 1995, 270, 475–480.
7. T. R. Flotte, Curr. Opin. Mol. Ther. 1999, 1,510–516.
8. W. D. Biggar, H. J. Klamut, P. C. Demacio,et al., Clin. Orthop. 2002, 401, 88–106.
9. C. Bordignon, L. D. Notarangelo, N. Nobili,et al., Science 1995, 270(5235), 470–475.
10. U. Gottschalk, S. Chan, Arzneimittelforschung1998, 48, 1111–1120.
11. R. M. Orr, Curr. Opin. Mol. Ther. 2001, 3,288–294.
12. B. Hutchins, Curr. Opin. Mol. Ther. 2000, 2,131–135.
13. G. N. M. Ferreira, G. A. Monteiro, M. F.Duarte, et al., Trends Biotechnol. 2000, 18,380–938.
14. D. M. Feltquate, J. Cell. Biochem. Suppl. 1998,30–31, 304–311.
15. L. Qin, Y. Ding, D. R. Pahud, et al., Hum.Gene Ther. 1997, 8, 2019–2029.
16. M. Kozak, J. Biol. Chem. 1991, 266,19867–19870.
17. M. Kozak, Annu. Rev. Cell. Biol. 1992, 8,197–225.
18. M. T. Huang, C. M. Gorman, Nucleic AcidsRes. 1990, 18, 937–947.
19. N. S. Yew, D. M. Wysokenski, K. X. Wang,et al., Hum. Gene Ther. 1997, 8, 575–584.
20. M. E. Coleman, F. DeMayo, K. C. Yin, et al.,J. Biol. Chem. 1995, 270, 12109–12116.
21. K. Anwer, M. Shi, M. F. French, et al., Hum.Gene Ther. 1998, 9, 659–670.
22. M. Gossen, S. Freundlieb, G. Bender, et al.,Science 1995, 268, 1766–1769.
23. Y. Wang, B. W. O’malley, S. Y. Tsai, MethodsMol. Biol. 1997, 63, 401–413.
24. J. A. Wolff, R. W. Malone, P. Williams, et al.,Science 1990, 247, 1465–1468.
25. URL: http://www.wiley.co.uk/genmed/clinical/ (1st June 2003).
26. G. Romano, P. P. Claudio, H. E. Kaiser,et al., In Vivo 1998, 12, 59–67.
27. A. Fischer, Cell Mol. Biol. (Noisy-le-grand)2001, 47, 1269–1275.
28. H. Ma, S. L. Diamond, Curr. Pharm.Biotechnol. 2001, 2, 1–17.
29. R. I. Mahato, L. C. Smith, A. Rolland, Adv.Gen. 1999, 41, 95–156.
30. J. Gehl, Acta Physiol. Scand. 2003, 177,437–447.
31. N. Kanikkannan, BioDrugs 2002, 16, 339–47.32. S. Satkauskas, M. F. Bureau, M. Puc, et al.,
Mol. Ther. 2002, 5, 133–140.
33. T. L. Burkoth, B. J. Bellhouse, G. Hewson,et al., Crit. Rev. Ther. Drug Carrier Syst. 1999,16, 331–384.
34. A. D. Barrett, Curr. Opin. Investig. Drugs2002, 3, 992–995.
35. P. L. Felgner, T. R. Gadek, M. Holm, et al.,Proc. Natl. Acad. Sci. U. S. A. 1987, 84,7413–7417.
36. N. S. Templeton, Expert Opin. Biol. Ther.2003, 3, 57–69.
37. D. Niculescu-duvaz, J. Heyes, C. J. Springer,Curr. Med. Chem. 2003, 10, 1233–1261.
38. E. R. Lee, J. Marshall, C. S. Siegel, et al.,Hum. Gene Ther. 1996, 7, 1701–1717.
39. C. Wheeler, P. L. Felgner, Y. J. Tsai, et al.,Proc. Natl. Acad. Sci. U. S. A. 1996, 93,11454–11459.
40. I. Koltover, K. Wagner, C. R. Safinya, Proc.Natl. Acad. Sci. U. S. A. 2000, 97,14046–14051.
41. A. J. Lin, N. L. Slack, A. Ahmad, et al., J.Drug Target. 2000, 8, 13–27.
42. Z. Li, Z. Ma, Curr. Gene Ther. 2001, 1,201–226.
43. S. Simoes, P. Pires, N. Duzgunes, et al.,Curr. Opin. Mol. Ther. 1999, 1, 147–157.
44. I. Koltover, T. Salditt, J. O. Radler, et al.,Science 1998, 281, 78–81.
45. P. L. Felgner, Y. J. Tsai, L. Sukhu, et al., Ann.N. Y. Acad. Sci. 1995, 772, 126–139.
46. B. J. Stevenson, A. D. Carothers, W. A.Wallace, et al., Gene Ther. 1997, 4, 210–218.
47. E. Tomlinson, A. P. Rolland, 1996, 39,357–372.
48. D. V. Schaffer, N. A. Fidelman, N. Dan,et al., Biotechnol. Bioeng. 2000, 67, 598–606.
49. C. R. Safinya, Curr. Opin. Struct. Biol. 2001,11, 440–448.
50. J. O. Radler, I. Koltover, T. Salditt, et al.,Science 1997, 275, 810–814.
51. R. Wattiaux, N. Laurent, S. Wattiaux-deConinck, et al., Adv. Drug Deliv. Rev. 2000,41, 201–208.
52. G. Osaka, K. Carey, A. Cuthbertson, et al., J.Pharm. Sci. 1996, 6, 612–618.
53. R. Z. Yu, R. S. Geary, J. M. Leeds, et al.,Pharm. Res. 1999, 16, 1309–1315.
54. J. Vacik, B. S. Dean, W. E. Zimmer, et al.,Gene Ther. 1998, 6, 1006–1014.
55. M. Johnson-Saliba, D. A. Jans, Curr. DrugTargets 2001, 2, 371–399.
56. C. K. Chan, D. A. Jans, Immunol. Cell Biol.2002, 80, 19–130.

Nonviral Gene Transfer Systems in Somatic Gene Therapy 263
57. C. Rudolph, C. Plank, J. Lausier, et al., J. Biol.Chem. 2003, 278, 11411–11418.
58. N. Shi, R. J. Boado, W. M. Pardridge, Pharm.Res. 2001, 18, 1091–1095.
59. W. Zauner, M. Ogris, E. Wagner, Adv. DrugDeliv. Rev. 1998, 30, 115–131.
60. C. W. Pouton, P. Lucas, B. J. Thomas, et al.,J. Control. Release 1998, 53, 289–299.
61. O. Boussif, F. Lezoualch, M. A. Zanta, et al.,Proc. Natl. Acad. Sci. U. S. A. 1995, 92,7297–301.
62. C. P. Lollo, M. G. Banaszczyk, P. M. Mullen,et al., Methods Mol. Med. 2002, 69, 1–13.
63. P. Marschall, N. Malik, Z. Larin, Gene Ther.1999, 6, 1634–1637.
64. M. Mannisto, S. Vanderkerken, V. Toncheva,et al., J. Control. Release 2002, 83, 169–182.
65. E. Wagner, M. Ogris, W. Zauner, Adv. DrugDeliv. Rev. 1998, 30, 97–113.
66. P. Midoux, E. Lecam, D. Coulaud, et al.,Somat. Cell. Mol. Genet. 2002, 27, 27–47.
67. B. Brissault, A. Kichler, C. Guis, et al.,Bioconjugate Chem. 2003, 14, 581–587.
68. D. A. Tomalia, G. R. Killat in Encyclopedia ofPolymer Science and Engineering Vol. 1 (Ed.:J. I. Kroschwitz), 2nd ed., Wiley, New York,1985, pp. 680–739.
69. J. L. Merlin, A. N’Doye, T. Bouriez, et al.,Drug News Perspect. 2002, 15, 445–451.
70. L. Wightman, R. Kircheis, V. Rossler, et al.,J. Gene Med. 2001, 3, 362–372.
71. C. Plank, M. X. Tang, A. R. Wolfe, et al.,Hum. Gene Ther. 1999, 10, 319–332.
72. A. Bragonzi, G. Dina, A. Villa, et al., GeneTher. 2000, 7, 1753–1760.
73. I. Chemin, D. Moradpour, S. Wieland, et al.,J. Viral. Hepat. 1998, 5, 369–375.
74. S. M. Zou, P. Erbacher, J. S. Remy, et al., J.Gene Med. 2000, 2, 128–134.
75. J. Behr, Chimia 1997, 51, 34–36.
76. A. G. Schatzlein, Anti-Cancer Drugs 2001, 12,275–304.
77. H. Petersen, P. M. Fechner, A. L. Martin,et al., Bioconjugate Chem. 2002, 13, 845–854.
78. M. J. Cloninger, Curr. Opin. Chem. Biol.2002, 6, 742–748.
79. S. E. Stiriba, H. Frey, R. Haag, Angew.Chem., Int. Ed. Engl. 2002, 41, 1329–1334.
80. C. L. Gebhart, A. V. Kabanov, J. Control.Release 2001, 73, 401–416.
81. G. De jong, A. Telenius, S. Vanderbyl, et al.,Chromosome Res. 2001, 9, 475–485.
82. G. M. Dykes, J. Chem. Technol. Biotechnol.2001, 76, 903–918.
83. G. R. Newkome, C. N. Moorefield, F. Vogtle,Dendrimers and Dendrons: Concepts, Syntheses,Applications, Wiley-VCH, New York, 2001.
84. N. Malik, R. Wiwattanapatepee, R. Klopsch,et al., J. Control. Release 2000, 65, 133–148.
85. J. F. Kukowska-Latallo, A. U. Bielinska,J. Johnson, et al., Proc. Natl. Acad. Sci. U.S. A. 1996, 93, 4897–4902.
86. L. Rompp in Biotechnologie und Gentechnik(Eds.: W. D. Decjwer, A. Puhler, E. Schmid),2nd ed., Georg Thieme Verlag, Stuttgart,New York, 1999.
87. M. Thanou, B. I. Florea, M. Geldof, et al.,Biomaterials 2002, 23, 153–159.
88. J. H. Van steenis, E. M. Van maarseveen,F. J. Verbaan, et al., J. Control. Release 2003,87, 167–176.
89. N. J. Zuidam, G. Posthuma, E. T. J. De vries,et al., J. Drug Target. 2000, 8, 51–66.
90. C. Arigita, N. J. Zuidam, D. J. Crommelin,et al., Pharm. Res. 1999, 16, 1534–1541.
91. F. Verbaan, Colloidal Gene DeliverySystems – In vivo Fate of poly(2-(dimethyl amino) ethyl methacrylate)-based Transfection Complexes, Ph.D.Thesis, Utrecht University, Holland, 2002,pp. 14–15.

265
15Xenotransplantation inPharmaceutical Biotechnology
Gregory J. BrunnTransplantation Biology and the Departments of Pharmacology and Experimental Therapeutics,Mayo Clinic, Rochester, MN, USA
Jeffrey L. PlattSurgery, Immunology, and Pediatrics, Mayo Clinic, Rochester, MN, USA
No area of medicine has generated moreexcitement or controversy than the field oftransplantation. Organ allotransplantationallows ‘‘curative’’ treatments for failureof the heart, kidney, liver, and lungs byreplacing these diseased organs with phys-iologically normal ones. Replacement ofbeta cells via pancreatic islet or whole pan-creas transplantation offers curative treat-ment to patients with diabetes. The mainlimitation to applying transplantation forthe treatment of diseases is a shortage ofhuman donors. This shortage limits theclinical application of organ transplanta-tion to approximately 5% of the numberof transplants that would be performedwere the supply of organs unlimited [1,2]. Possible solutions to this limitationhave garnered considerable interest andinclude the use of artificial organs, ‘‘engi-neered tissues,’’ stem cell transplants, andxenotransplants. Although some newertechnologies have excited interest, xeno-transplants of the heart, lung, kidney, and
pancreatic islets are known to functionwell enough to sustain life. Enthusiasmfor xenotransplantation also stems fromthe possibility that animal tissues and or-gans might be less susceptible to diseaserecurrence compared to allotransplants.Advances in cellular and molecular biol-ogy and in genetics open possibilities foruse of cells, tissues, and organs to addressthe complications of disease, not only byreplacement of abnormal cells and tissuesbut also by the use of transplanted tissuesto impart novel physiological functions.In this regard and for some purposes,xenografts may be an ideal vehicle forintroducing a novel gene or biochemicalprocess that could be of value to the trans-plant recipient.
If interest in xenotransplantation issubstantial, the hurdles to its applicationare equally so. For the past three decades,the first and preeminent obstacle totransplanting organs and tissues betweenspecies has been the immune reaction of
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

266 15.2 The Biologic and Immunologic Responses to Xenotransplantation
the host against the graft. A second, andstill theoretical, hurdle is the possibilitythat beyond the immune barrier, theremight be physiologic limitations to thesurvival or function of a xenograft andthe possibility that a xenotransplant mightengender medical complications for thexenogeneic host. A third hurdle is thepossibility that a xenograft might transferinfectious agents from the donor to thehost, and that from the host such agentsmight spread to other members of society.This communication will consider thecurrent state of efforts to overcome thevarious hurdles to xenotransplantation andwill evaluate how genetic engineeringmight be applied to this end.
15.1The Pig as a Source of Tissues and Organsfor Clinical Xenotransplantation
Although it might be intuitive that the bestsource of xenogeneic tissues for clinicaltransplantation is nonhuman primates, itis the pig that is the focus of most effortsin this field. The reasons for favoring thepig as a xenotransplant source includethe availability of pigs in large numbers,the ease with which the pig can bebred, the limited risk of zoonotic diseaseengendered by the use of pigs, and thepossibility of introducing new genes intothe germline of the pig.
Genetic engineering of pigs using trans-genic techniques and nuclear transfer hascertain advantages over conventional genetherapy (Table 1). Introducing genetic ma-terial directly into the porcine germ cellobviates the need for a vehicle, whichmay vary in reliability of gene deliveryand may introduce secondary unintendedconsequences due to the vector itself. Sec-ond, the genetic material introduced into
the germline can be expressed constitu-tively in all cells, especially in stem cells,and passed on to subsequent generations.Third, with the use of transgenic tech-niques, only the donor is manipulated; inconventional gene therapy, both the donorand the recipient may be affected.
Recent advances in cloning pigs [3–5]through nuclear transfer also allows‘‘knocking out’’ genes. Besides the advan-tage of gene knockouts, nuclear transfercan be done with cultured somatic cellsobviating the need for embryonic stemcells.
15.2The Biologic and Immunologic Responsesto Xenotransplantation
All xenografts elicit an immune response,including antibodies, cell-mediated immu-nity, natural killer (NK) cells, and inflam-mation [6]. However, the fate of xenograftsconfronted with these responses is dictatedin part by the way in which the graft re-ceives its vascular supply (Fig. 1). Isolatedcells, such as hepatocytes, and ‘‘free’’ tis-sues, such as pancreatic islets and skin,derive their vascular supply through theingrowth of host blood vessels. The pro-cess of neovascularization, as such, mightbe impaired in a xenograft by incompati-bility of donor growth factors with the hostmicrovasculature. To the extent that neo-vascularization or graft function dependsupon hormones and cytokines of host ori-gin, the function of the xenograft mightalso be impaired. As the host microcir-culation is established, however, a xeno-geneic tissue may be relatively protectedfrom attack by host immune elements.Whole-organ grafts provide their own mi-crocirculation and growth factors, and, as aresult, incompatibility between the donor

Xenotransplantation in Pharmaceutical Biotechnology 267
Tab. 1 Genetic engineering in xenotransplantation: conventional gene therapy versus transgenictherapy versus cloning
Conventionalgene therapy
Conventionaltransgenic techniques
Cloning
Delivery Vector or vehicle required Injection of genetic mat-erial directly into pro-nuclei of fertilized egg
Transfection of culturedsomatic cells
Expression Dependent on ability ofeach cell to take upgenetic material
Genetic materialintroduced into thegermline, leading toexpression in a line ofanimals
Genetic materialintroduced into thegermline, leading toexpression in a lineof animals
Requires treatment forevery transplant orrecipient
May require repeatedtreatment
One manipulation One manipulation
Immunogenicity Delivery vehicle ortransgene may beimmunogenic
The transgene may elicitimmune response
The transgene mayelicit immuneresponse
Target of geneticmanipulation
The recipient and the graftmay be transduced
Genetic manipulation ofthe donor only
Genetic manipulationof the donor only
Geneticmanipulation
Gene addition Dominantnegative
Gene addition Dominantnegative
Gene additionDominant negativeGene knockout
and the recipient is less likely to have animpact on cellular function. On the otherhand, because the circulation is of donororigin, the immune, inflammatory, andcoagulation systems of the recipient canact directly on donor cells, sometimes withdramatic and devastating consequences.
15.3Hyperacute Rejection
An organ transplanted from a pig into aprimate such as a human is subject to hy-peracute rejection. Hyperacute rejectionbegins immediately upon reperfusion ofthe graft and destroys the graft within min-utes to a few hours. Hyperacute rejection ischaracterized histologically by interstitialhemorrhage and thrombosis, the thrombi
consisting mainly of platelets [7]. Researchover the past decade has clarified themolecular basis for the hyperacute re-jection of pig organs by primates [8, 9],and this knowledge has led to the devel-opment of new and incisive therapeuticapproaches to averting this problem. Hy-peracute rejection was once consideredthe most daunting hurdle to clinical ap-plication of xenotransplantation; however,hyperacute rejection can now be preventedin nearly every case.
Hyperacute rejection of porcine organxenografts by primates is initiated bythe binding of xenoreactive natural an-tibodies to the graft [7, 10–12]. Xenore-active natural antibodies are presentin the circulation without a knownhistory of sensitization [13]. Contraryto expectations, xenoreactive antibodies

268 15.3 Hyperacute Rejection
Organ transplant
Recipientvessel
Donorvessel
Cell transplant
Tissue transplant
Donororgan
Donorcells
Donortissue
Fig. 1 Mechanisms of xenograft vascularization. Organ xenograftsreceive recipient blood exclusively through the donor blood vessels(top). Free tissue xenografts (e.g. pancreatic islets and skin) arevascularized partly by the ingrowth of recipient blood vessels andpartly by spontaneous anastomosis of donor and recipientcapillaries (middle). Cellular xenografts (e.g. hepatocytes and bonemarrow cells) are vascularized by the ingrowth of recipient bloodvessels (bottom).
are predominantly directed against onlyone antigen, a saccharide consisting ofterminal Galα1,3Gal [14–17]. The impor-tance of Galα1,3Gal as the primary anti-genic barrier to xenotransplantation wasdemonstrated recently by experiments inwhich anti-Galα1,3Gal antibodies werespecifically depleted from baboons usingimmunoaffinity columns before trans-plantation of pig organs [18]. Antibodybinding to the newly transplanted organswas largely curtailed, and hyperacute re-jection did not occur.
Although the identification of the rel-evant antigen for pig-to-primate xeno-transplantation allows specific depletionof the offending antibodies, more endur-ing and less intrusive forms of therapywould be preferred. One approach toovercoming the antibody–antigen reactionis to develop lines of pigs with low
levels of antigen expression [19]. Vari-ous genetic approaches aimed at ‘‘re-modeling’’ the antigenicity of donor tis-sues by reducing Galα1,3Gal expres-sion are under investigation. These ap-proaches can be separated into threecategories: (1) interference with the func-tion of α1,3galactosyltransferase (α1,3GT),the enzyme that catalyzes the synthesis ofthe Galα1,3Gal moiety; (2) expression ofα galactosidase, which cleaves αgalactosylresidues; and (3) deletion of the geneencoding α1,3GT from the pig genometo prevent synthesis of the saccharide.The utility of genetic modification ofpig tissues to reduce Galα1,3Gal ex-pression has recently been demonstratedby Sharma and colleagues [20], whogenerated transgenic pigs expressing theH-transferase. Transgenic pigs expressingH-transferase express H antigen at the

Xenotransplantation in Pharmaceutical Biotechnology 269
terminus of some sugar chains insteadof Galα1,3Gal. Another genetic approachto modify expression of Galα1-3Gal wasproposed by Osman et al. [21]. Expres-sion of α-galactosidase, which cleavesα-galactosyl residues [16], in conjunctionwith other galactosyltransferases, signif-icantly reduces expression of Galα1-3Gal [21]. A third strategy to modifyGalα1,3Gal was demonstrated by Miya-gawa and coworkers, who generatedtransgenic pigs expressing the humanβ-1,4-N-acetylglucosaminyltransferase IIIgene (GnT-III) [22]. This enzyme catalyzestransfer of N-acetylglucosamine to matur-ing mannose-modified proteins as theypass through the Golgi apparatus andleads to diminished αGalα1,3Gal expres-sion both by competing with α1,3GTand by preventing subsequent modifi-cations of α1,3GT by insertion of anN-acetylglucosamine onto the growingmannose chain. The net result was di-minished natural antibody reactivity ontransgenic pig tissue when transplantedinto primates. Although these advancesillustrate the utility of genetic modifi-cation of pig donor tissue, the mainlimitation of these genetic approaches isthat residual Galα1,3Gal may be suffi-cient to allow rejection reactions to oc-cur [23].
The most obvious approach to devel-oping xenograft donors with diminishedreactivity with host antibodies would be togenetically target or ‘‘knock out’’ the en-zyme α1,3-galactosyltransferase. Embry-onic stem cells were used to knock thisgene out in mice [24], demonstrating thatremoval of this enzyme is not lethal. Theintense interest in generating pigs thatlack the α1,3GT gene has fueled a race todelete this gene from the pig genome, andrecently several groups have succeeded inthis endeavor. Prather and colleagues [25]
and Ayares and coworkers [26] used sim-ilar strategies to disrupt one allele of theα1,3GT gene (GGTA1) in pigs by firsttargeting the gene for disruption in fetalporcine fibroblasts. Selected clones wereused as nuclear donors for enucleated pigoocytes and were the resulting embryosimplanted into surrogate gilts. Both ap-proaches yielded live, healthy piglet clonesin which one copy of GGTA1 had been dis-rupted. These achievements demonstratedthat nuclear transfer technology could beapplied to pig embryos, which are notori-ously fragile and difficult to manipulate.Recently, the generation of cloned pigsharboring a functional knockout of bothalleles of α1,3GT was reported [27]. Thisfirst success required some serendipityin that the α1,3GT-deficient pigs werefound not to be homozygous knockouts,but rather functional knockouts that arosefrom a process in which the knockout ofone allele paired with a spontaneous sin-gle base change in the remaining GGTA1gene resulted in an inactivating aminoacid substitution in α1,3GT. Irrespectiveof how the inactivation of α1,3GT wasachieved in these animals, it may now befeasible to make an incisive determina-tion as to the utility of these geneticallymodified pig tissues for avoiding hypera-cute rejection, and possibly other hurdlesto using xenotransplantation. A cautionarynote has recently come to light, however,indicating that pig cells that lack bothcopies of the GGTA1 gene may still syn-thesize the Galα1,3Gal antigen, albeit atvery low levels [28]. While the generation ofα1,3Gal knockout pigs may help overcomean important hurdle to xenotransplanta-tion by preventing hyperacute rejection,it may not avert other potent xenogeneicimmune responses, as will be discussedbelow.

270 15.4 Complement Activation
15.4Complement Activation
A second and essential step in the develop-ment of hyperacute rejection is activationof the complement system of the recipienton donor blood vessels [11]. Complementactivation is triggered by the binding ofcomplement-fixing xenoreactive antibod-ies to graft endothelium, and to a smallerextent perhaps, by reperfusion injury. Re-gardless of the mechanism leading to com-plement activation, a xenograft is extraor-dinarily sensitive to complement-mediatedinjury because of multiple defects in theregulation of complement (Fig. 2) [29–31].Under normal circumstances, the comple-ment cascade is regulated or inhibited byvarious proteins in the plasma and onthe surface of cells. These proteins protectnormal cells from suffering inadvertent
injury during the activation of comple-ment. The proteins that regulate thecomplement cascade function in a species-restricted fashion; that is, complementregulatory proteins inhibit homologouscomplement far more effectively than het-erologous complement [30, 32]. Accord-ingly, the complement regulatory proteinsexpressed in a xenograft are ineffective atcontrolling the complement cascade of therecipient, and the graft is subject to severecomplement-mediated injury [29].
To address this problem, lines of animalshave been developed that are transgenic forhuman complement regulatory proteinsand that are able to control activation ofcomplement in the xenograft (Fig. 2) [30,33, 34]. Animals transgenic for humandecay-accelerating factor (hDAF), whichregulates complement at the level ofC3, together with CD59, which regulates
C3 convertase
ClassicalC1, C4, C2
AlternativeC3b, B, D, P
AnaphylotoxinsC3a, C5a
LysisC5, C6, C7, C8, C9n
OpsonizationC3b
MCPDAF CD59
Fig. 2 Regulation of the complement system. The complementcascade, which can be activated via the classical or alternative pathway,is regulated under normal circumstances by various proteins in theplasma and on the cell surface. Three of the cell surface complementregulatory proteins are shown here. Decay-accelerating factor (DAF)and membrane cofactor protein (MCP) regulate complement activationby dissociating or promoting the degradation of C3 convertase. CD59,also known as protectin, prevents the functions of terminalcomplement complexes by inhibiting C8 and C9. An organ grafttransplanted into a xenogeneic recipient is especially sensitive tocomplement-mediated injury because DAF, MCP, and CD59 expressedon the xenograft endothelium cannot effectively regulate thecomplement system of the recipient.

Xenotransplantation in Pharmaceutical Biotechnology 271
complement at the level of C8 and C9 [34],or CD46, which controls complementactivation at the level of C3 and C4 [35],have demonstrated that the expressionof even low levels of hDAF and CD59or CD46 in porcine-to-primate xenograftsis sufficient to allow a xenograft toavoid hyperacute rejection [36, 37]. Theseresults, and the dramatic prolongation ofxenograft survival achieved by expressinghigher levels of hDAF factor in thepig [38], underscore the importance ofcomplement regulation as a determinantof xenograft outcomes.
One of the major obstacles in testingthe effects of transgenes in pig organs hasbeen the difficulty in generating transgenicpigs. Recent work by Lavitrano, et al. [39]may accelerate the rate at which transgenicpigs may be generated and tested intransplant models. These investigatorsused sperm-mediated gene transfer toincorporate the hDAF gene into pigs andobtained a high efficiency of transgenesis(80% of pigs incorporated hDAF into thegenome) and hDAF expression (43% ofthe transgenic pigs). The transgenic hDAFwas functional in vitro, and transmittedto progeny as expected. This method,in theory, could be used to introducemultiple genes at once, or a tailor-madeset of human genes that may be useful fortransplant-mediated genetic therapies, asmentioned earlier.
15.5Acute Vascular Rejection
If hyperacute rejection of a xenograft isaverted, a xenograft is subject to thedevelopment of acute vascular rejection,so named because of its resemblance toacute vascular rejection of allografts [40,41]. Acute vascular rejection (sometimes
called delayed xenograft rejection) may be-gin within 24 hours of reperfusion andlead to graft destruction over the followingdays and weeks [40, 42, 43]. Although thefactors important in the pathogenesis ofacute vascular rejection are incompletelyunderstood, there is growing evidence thatacute vascular rejection is triggered at leastin part by the binding of xenoreactive an-tibodies to the graft. The importance ofxenoreactive antibodies in triggering acutevascular rejection is suggested by threelines of evidence: (1) antidonor antibodiesare present in the circulation of recipientswhose grafts are subject to acute vascu-lar rejection [11, 40, 44, 45], (2) depletionof antidonor antibodies delays or pre-vents acute vascular rejection [46], and(3) administration of antidonor antibodiesleads to the development of acute vascu-lar rejection [47]. Recent studies suggestthat among the antibodies that provokeacute vascular rejection are those directedagainst Galα1-3Gal [48, 49]. This prob-lem thus constitutes further impetus forthe continued development of α1,3Gal-deficient pigs. Regardless of which ele-ments of the immune system trigger acutevascular rejection, it is commonly thoughtthat this type of rejection, and especiallythe intravascular coagulation characteris-tically associated with it, are caused bythe activation of endothelial cells in thetransplant [40, 50, 51]. Activated endothe-lial cells express procoagulant molecules,such as tissue factor (TF), and proinflam-matory molecules, such as E-selectin andcytokines [30]. The pathogenesis of acutevascular rejection is summarized in Fig. 3.
Although various therapeutic manipula-tions have proven successful in preventinghyperacute rejection, acute vascular rejec-tion poses a more difficult problem, inpart, because therapies are needed on anongoing basis. For this reason, genetic

272 15.6 Accommodation
Platelets E-selectionTF PAI-1
NKMO
Ab C
IL1a
Fig. 3 Pathogenesis of acute vascular rejection. Activation of endotheliumby xenoreactive antibodies (Ab), complement (C), platelets, and perhaps byinflammatory cells (natural killer (NK) cells and macrophages (M�) leadsto the expression of new pathophysiologic properties. These newproperties, such as the synthesis of tissue factor (TF) and plasminogenactivator inhibitor type 1 (PAI-1), promote coagulation; the synthesis ofE-selectin and cytokines such as IL1α promote inflammation. Thesechanges in turn cause thrombosis, ischemia, and endothelial injury, thehallmarks of acute vascular rejection. (Adapted from Nature 1998:392(Suppl.) 11–17, with permission.) (See Color Plate p. xxiii).
modification of the donor may provemore important for dealing with acutevascular rejection than with hyperacute re-jection. The various possible approachesfor combating acute vascular rejectionare listed in Table 2. Among these ap-proaches, the reduction of Galα1,3Gal inxenotransplant donors may be an im-portant part of the overall strategy, tothe extent that Galα1,3Gal proves to bean important antigenic target in acutevascular xenograft rejection. Preliminarystudies suggest that the level of antibodybinding needed to initiate acute vascu-lar rejection is considerably lower thanthe level needed to initiate hyperacuterejection [23]. Accordingly, the antigen ex-pression would have to be reduced verysignificantly to achieve therapeutic benefitfor acute vascular rejection. The availabilityof α1,3galactosyltransferase-deficient pigsshould prove to be an ideal model totest this concept. In addition to lower-ing antigen expression, it is likely thatexpression of human complement regu-latory proteins will be helpful in prevent-ing acute vascular rejection. Preliminarystudies suggest that interfering with theantigen–antibody reaction and controlling
the complement cascade may be suffi-cient to prevent acute vascular rejectionfor at least some period of time [46]. Thesegoals were accomplished by using animalstransgenic for hDAF factor and CD59 asa source of organs, and baboons depletedof immunoglobulin as recipients. Cozziand associates [38] achieved prolonged sur-vival of xenografts, presumably preventingacute vascular rejection, by using trans-genic pigs expressing high levels of DAFand cynomolgus monkeys treated withvery high doses of cyclophosphamide. Theimmunosuppression perhaps preventedthe synthesis of antidonor antibodies.
Work in rodents points to the potentialinvolvement of NK cells and macrophagesin mediating acute vascular rejection.However, the ability of immunoglobulinmanipulation to prevent acute vascular re-jection suggests that the involvement ofNK cells and macrophages might be lessimportant than in vitro studies and stud-ies in rodents have suggested [33, 51]. Onthe other hand, NK cells might exacer-bate the injury triggered by xenoreactiveantibodies, as human NK cells have beenshown to activate porcine endothelial cellsin vitro [52–54].

Xenotransplantation in Pharmaceutical Biotechnology 273
Tab. 2 Therapeutic strategies for acute vascular xenograft rejection
Possible mechanismtargeted
Manipulation of
Recipient Donor
Antibody–antigeninteraction
Specific depletion of xenoreactiveantibodies
Generating transgenic pigs withlow levels of antigen
Prevention of xenoreactiveantibody synthesis (e.g.cyclophosphamide,leflunomide)
Generation of pig clones lackingantigen
Complementactivation
Systemic anticomplement therapy(e.g. CVF, sCR1, gammaglobulin)
Generation of donor pigstransgenic for humancomplement regulatoryproteins
Endothelial cellactivation
Administration ofanti-inflammatory agents
Inhibition of NFκB functionIntroduction of protectivegenes
Molecularincompatibilities
Administration of inhibitors (e.g.inhibitors of complement orcoagulation)
Introduction of compatiblemolecules
15.6Accommodation
Fortunately, the presence of antidonorantibodies in the circulation of a graft re-cipient does not inevitably trigger acutevascular rejection. If antidonor antibodiesare temporarily depleted from a recipient,an organ transplant can be establishedso that rejection does not ensue whenthe antidonor antibodies are returned tothe circulation [55]. This phenomenon isreferred to as ‘‘accommodation’’ [30]. Ac-commodation may reflect a change inthe antibodies, in the antigen, or in thesusceptibility of the organ to rejection.If accommodation can be established, itmay be especially important in xenotrans-plantation because it would obviate theneed for ongoing interventions to inhibitantibody binding to the graft. One poten-tial approach to accommodation may bethe use of genetic engineering to reducethe susceptibility of an organ transplant to
acute vascular rejection and the endothe-lial cell activation associated with it [51].Unfortunately, successful intervention atthe level of such effector mechanismsis yet to be achieved. However, disrup-tion of antibody–antigen interaction hasbrought about accommodation in humansubjects [50, 55].
15.7Cellular Mediated Immune Responses
Organ transplants and cellular and freetissue transplants are subject to cellularrejection. In allotransplantation, cellularrejection is controlled by conventional im-munosuppressive therapy, but there isconcern that, for several reasons, cel-lular rejection may be especially severein xenotransplants. First, the great vari-ety of antigenic proteins in a xenograftmay lead to recruitment of a diverseset of ‘‘xenoreactive’’ T-cells. Second, the

274 15.8 Physiologic Hurdles to Xenotransplantation
binding of xenoreactive antibodies andactivation of the complement system maylead to amplification of elicited immuneresponses [56]. For example, deposition ofcomplement in a graft may cause activationof antigen-presenting cells, in turn stimu-lating T-cell responses. Still another factorthat might amplify the elicited immuneresponse to a xenotransplant involves ‘‘im-munoregulation,’’ which ordinarily wouldcircumscribe cellular immune responses,but may fail or be deficient across species.Such failure could reflect limitations in therecognition of xenogeneic cells or incom-patibility of relevant growth factors, as buttwo examples.
Induction of immunologic tolerance hasbeen an erstwhile goal of transplant sur-geons and physicians. Especially in thecase of xenotransplantation, if the cur-rent immunosuppressive regimens are notsufficient, induction of immunologic tol-erance may be required. At least three ap-proaches are being pursued: (1) the gener-ation of mixed hematopoietic chimerism,(2) the establishment of microchimerismby various means, and (3) thymic trans-plantation [57–59]. The development ofmixed hematopoietic chimerism throughthe introduction of donor bone mar-row [60] has worked very well across rodentspecies [61, 62], although success may belimited by xenoreactive antibodies and theengraftment impaired by incompatibilityof host growth factors or microenviron-ment [63]. Fortunately, there is evidencethat these problems can be overcome [58].Various approaches to peripheral toler-ance, such as the blockade of costimulationby administration of a fusion protein con-sisting of a soluble form of the CTLA-4molecule and immunoglobulin (CTLA-4-Ig), are being pursued.
Still another factor in the cellularresponse to a xenotransplant involves the
action of NK cells. Natural killer cell func-tions can be amplified by cell surface recep-tors that recognize Galα1,3Gal [64]. Natu-ral killer cell functions are downregulatedby receptors that recognize homologousmajor histocompatibility complex (MHC)class I [65, 66]. Human NK cells may beespecially active against xenogeneic cellsbecause of stimulation on the one handand failure of downregulation on the other.The possible involvement of NK cells inxenograft rejection might be addressed bygeneration of transgenic pigs expressingon the cell surface MHC-like moleculesthat will more effectively recognize corre-sponding receptors on NK cells and thatwill downregulate the function of NK cells.
How a xenogeneic donor could be mod-ified genetically to enhance the develop-ment of tolerance or to limit elicited im-mune responses is still uncertain. Clearly,efforts to control the natural immune barri-ers to xenotransplantation may contributeto limiting the elicited immune response.To the extent that recipient T-cells recog-nize donor cells directly, that is, the T-cellreceptors of the recipient recognizing na-tive MHC antigens on donor cells, a xeno-geneic donor might be engineered in sucha way to reduce corecognition (throughCD4 and CD8) or costimulation (throughCD28 or other T-cell surface molecules)or to express inhibitory molecules suchas CD59 or Fas ligand. These approachesand the expression of inhibitory molecules,which are being considered as approachesto gene therapy in allotransplantation, maywell prove more effective in xenotrans-plantation because inhibitory genes can beintroduced as transgenes and thereby ex-pressed in all relevant cells in the graft.Another useful and perhaps necessary ap-proach will involve genetic modificationsto allow the survival and function of donorbone marrow cells.

Xenotransplantation in Pharmaceutical Biotechnology 275
15.8Physiologic Hurdles to Xenotransplantation
Progress in addressing some of the im-munological obstacles to xenotransplanta-tion has brought into focus the question ofthe extent to which a xenotransplant wouldfunction optimally in a foreign host. A re-cent demonstration that the porcine kidneyand the porcine lung can replace the mostimportant functions of the primate kidneyand primate lung are encouraging [67, 68].Subtle defects in physiology across speciesmay nevertheless exist. Organs such as theliver, which secrete a variety of proteinsand which depend on complex enzymaticcascades, may prove incompatible with aprimate host. Accordingly, one importantapplication of genetic engineering in xeno-transplantation may be the amplificationor modulation of xenograft function to al-low for more complete establishment ofphysiologic function or to overcome crit-ical defects. For example, recent studiesby Akhter and associates [69] and Kypsonand coauthors [70] aimed at improving thefunction of cardiac allografts by manipu-lation of β-adrenergic signaling, and thistechnique might be adapted to the xeno-transplant to improve cardiac function. Onthe other hand, most cellular processesand biochemical cascades are intrinsicallyregulated to meet the overall physiologicneeds of the whole individual. The keyquestion then is, which of the many poten-tial defects actually need to be repaired.
Another potential hurdle to the clinicalapplication of xenotransplantation is thepossibility that the xenograft may disturbnormal metabolic and physiologic func-tions in the recipient. For example, Lawsonand coworkers [71, 72] have shown thatporcine thrombomodulin fails to inter-act adequately with human thrombin andprotein C to generate activated protein C.
This defect could lead to a prothromboticdiathesis because of failure of generationof activated protein C. Of even greaterconcern is the possibility that the trans-plantation of an organ, such as the liver,could add prothrombotic or proinflam-matory products into the blood of therecipient. Although perhaps a great manyphysiologic defects can be detected at themolecular level, the critical question willbe, which of these defects is important atthe whole-organ level or with respect to thewell being of the recipient, and which mustbe repaired by pharmaceutical or geneticmeans.
15.9Zoonosis
The increasing success of experimen-tal xenotransplants and therapeutic trialsbring to the fore the question of zoono-sis, that is, infectious disease introducedfrom the graft into the recipient. The trans-fer of infectious agents from the graft tothe recipient is a well-known complicationof allotransplantation. To the extent thatinfection of the recipient in this way in-creases the risks of transplantation, therisk can generally be estimated and adecision made on the basis of the riskversus the potential benefits conferred bythe transplant. The concern about zoono-sis in xenotransplantation is not so muchthe risk to the recipient of the trans-plant, but the risk that an infectious agentwill be transferred from the recipient tothe population at large. Fortunately, allof the microbial agents known to infectthe pig can be detected by screening andpotentially eliminated from a population ofxenotransplant donors. There is concern,however, that the pig may harbor endoge-nous retroviruses, which are inherited withgenomic DNA and which might become

276 15.10 A Scenario for the Clinical Application of Xenotransplantation
activated and transferred to the cells of therecipient. For example, Patience and coau-thors [73] recently reported that a C-typeretrovirus endogenous to the pig could beactivated in pig cells, leading to the re-lease of particles that can infect humancell lines. Whether this virus or other en-dogenous viruses can actually infect acrossspecies and whether such infection wouldlead to disease are unknown, but remain asubject of current epidemiologic investiga-tion. If cross-species infection does proveto be an important issue, genetic therapiesmight also be used to address this prob-lem. The simplest genetic therapy wouldinvolve breeding out the organism, butthis approach might fail if the organismwere widespread or integrated at multipleloci. Some genetic therapies have beendeveloped to potentially control humanimmunodeficiency viruses [74]. Althoughthese therapies have generally failed be-cause it has been difficult or impossible togain expression of the transferred genes instem cells and at levels sufficient to dealwith high viral loads, the application ofsuch therapies might be much easier inxenotransplantation because the therapeu-tic genes could be delivered through thegermline. Ultimately, if elimination of en-dogenous retroviruses were necessary, itcould potentially be accomplished by genetargeting and cloning, as discussed above.
15.10A Scenario for the Clinical Application ofXenotransplantation
Successful application of xenotransplanta-tion in the clinical arena requires insightsinto not only immunology but also phys-iology and infectious disease, all of whichhave been discussed briefly here in thecontext of genetic therapy. In recent years,
important advances have been made inelucidating the immunologic hurdles ofpig-to-primate transplantation. Althoughthis scientific progress is important andexciting, xenotransplantation will likelyenter the clinical arena through a step-by-step process. A first step, free tissuexenografting, is in limited clinical tri-als already [75–77], and preliminary ev-idence is encouraging as porcine freetissue xenografts appear to endure in ahuman recipient [77]. One immediate ap-plication of free tissue xenografting wouldbe treatment of cirrhosis caused by hep-atitis virus, using targeted infusion ofporcine hepatocytes [6]. The promise ofthis approach is enhanced because (1) pighepatocytes are resistant to viral rein-fection, (2) rat models of cirrhotic liverfailure indicate that porcine hepatocytexenotransplants may endure and func-tion well [78], and (3) predicted demandfor hepatic transplantation due to hepatitisC-induced cirrhotic liver disease is likely toworsen the already acute shortage of liversavailable for transplant. Another potentialextension of free tissue xenografting isthe transplantation of xenogeneic islets oflangerhans for the treatment of type 1 di-abetes. Xenogeneic islet transplants mayprove to be less liable to destruction bythe autoimmune processes that underliesthis disease. Temporary or ‘‘bridge’’ organtransplantation will probably follow freetissue xenografting. Bridge transplants willnot address the problem of the shortage ofhuman organs, but incisive analysis of theoutcomes of these transplants will provideimportant information about the remain-ing immunologic hurdles and the potentialphysiologic and infectious considerations.With this information, further therapiesincluding genetic engineering may allowthe use of porcine organs as permanentreplacements. Even then, one can envision

Xenotransplantation in Pharmaceutical Biotechnology 277
ongoing efforts to apply genetic therapiesthat will optimize graft function and limitthe complications of transplantation.
While it may be that the use of pigs as asource of organs and tissues for transplan-tation is not far off, exciting advances in tis-sue engineering, stem cell technology, andin vitro organogenesis may broaden theuse of animals in human medicine. Adultand embryonic stem cell culture has givenrise to organ-specific tissues with func-tional characteristics of the correspondingorgans [79]. Although these cultures areunlikely to yield fully developed functionalorgans for transplantation into humans,pigs or other animals could be used as re-cipients of these culture-initiated tissuesand allow completion of development.These organs, grown and maintained inanimals, may then be available for trans-plantation on an ‘‘as needed’’ basis. Pigsmay thus serve as xenograft ‘‘recipients’’prior to becoming organ ‘‘donors.’’ Therecent success in cloning of animals,including pigs, raises the possibility oftransferring nuclei from a human patient’scells into enucleated stem cells of an ani-mal, and then growing the cells in animalsto generate differentiated human tissuethat is autologous with the patient. Thelessons learned from genetic manipula-tion of animals in the quest to make animalorgans suitable for transplantation into hu-mans may find their best application ingenerating animals suitable for use as bi-ological reactors to grow human organssuitable for transplantation into humans.
Acknowledgments
Supported by grants from the Heart,Lung, and Blood Institute of the NationalInstitutes of Health.
References
1. 2001 Annual Report of the U.S. OrganProcurement and Transplantation Networkand the Scientific Registry for TransplantRecipients: Transplant Data 1991–2000, De-partment of Health and Human Services,Health Resources and Services Administra-tion, Office of Special Programs, Divisionof Transplantation, Rockville, MD; UnitedNetwork for Organ Sharing, Richmond, VA;University Renal Research and EducationAssociation, Ann Arbor, MI. Retrieved De-cember 9, 2002, from the World Wide Web:http://www.optn.org//data/annualReport.asp.
2. R. W. Evans in Xenotransplantation (Ed.:J. L. Platt), ASM Press, Washington, DC,2001, pp. 29–51.
3. A. Onishi, M. Iwamoto, T. Akita et al.,Science 2000, 289, 1188–1190.
4. I. A. Polejaeva, S. Chen, T. D. Vaught et al.,Nature 2000, 407, 86–90.
5. J. Betthauser, E. Forsberg, M. Augensteinet al., Nat. Biotechnol. 2000, 18, 1055–1059.
6. M. Cascalho, J. L. Platt, Immunity 2001, 14,437–446.
7. J. L. Platt, Hyperacute Xenograft Rejection,Austin: R.G. Landes, 1995.
8. J. L. Platt in Samter’s Immunologic Diseases(Eds.: K. F. Austen, M. M. Frank et al.),Lippincott Williams & Wilkins, Philadelphia,2001, pp. 1132–1146.
9. J. L. Platt, Transplantation 2000, 69,1034–1035.
10. D. K. C. Cooper, P. A. Human, G. Lexeret al., J. Heart Transplant. 1988, 7, 238–246.
11. J. L. Platt, R. J. Fischel, A. J. Matas et al.,Transplantation 1991, 52, 214–220.
12. J. L. Platt, M. A. Turman, H. J. Noreen et al.,Transplantation 1990, 49, 1000–1001.
13. R. Y. Calne, Transplant. Proc. 1970, 2,550–556.
14. M. S. Sandrin, H. A. Vaughan, P. L. Dabkow-ski et al., Proc. Natl. Acad. Sci. U. S. A. 1993,90, 11391–11395.
15. A. H. Good, D. K. C. Cooper, A. J. Malcolmet al., Transplant. Proc. 1992, 24, 559–562.
16. B. H. Collins, W. Parker, J. L. Platt, Xeno-transplantation 1994, 1, 36–46.
17. W. Parker, D. Bruno, Z. E. Holzknecht et al.,J. Immunol. 1994, 153, 3791–3803.

278 15.10 A Scenario for the Clinical Application of Xenotransplantation
18. S. S. Lin, D. L. Kooyman, L. J. Daniels et al.,Transplant. Immunol. 1997, 5, 212–218.
19. C. G. Alvarado, A. H. Cotterell, K. R.McCurry et al., Transplantation 1995, 59,1589–1596.
20. A. Sharma, J. F. Okabe, P. Birch et al., Proc.Natl. Acad. Sci U. S. A. 1996, 93, 7190–7195.
21. N. Osman, I. F. McKenzie, K. Ostenriedet al., Proc. Natl. Acad. Sci. U. S. A. 1997,94, 14677–14682.
22. S. Miyagawa, H. Murakami, Y. Takahagiet al., J. Biol. Chem. 2001, 276, 39310–39319.
23. W. Parker, S. S. Lin, J. L. Platt, Transplanta-tion 2001, 71, 313–319.
24. A. D. Thall, P. Maly, J. B. Lowe, J. Biol. Chem.1995, 270, 21437–21440.
25. L. Lai, D. Kolber-Simonds, K. W. Park et al.,Science 2002, 295, 1089–1092.
26. Y. Dai, T. D. Vaught, J. Boone et al., Nat.Biotechnol. 2002, 20, 251–255.
27. C. J. Phelps, C. Koike, T. D. Vaught et al.,Science 2003, 299, 411–414.
28. A. Sharma, B. Naziruddin, C. Cui et al.,Transplantation 2003, 75, 430–436.
29. S. Miyagawa, H. Hirose, R. Shirakura et al.,Transplantation 1988, 46, 825–830.
30. J. L. Platt, G. M. Vercellotti, A. P. Dalmassoet al., Immunol. Today 1990, 11, 450–456.
31. A. P. Dalmasso, Immunopharmacology 1992,24, 149–160.
32. A. P. Dalmasso, G. M. Vercellotti, J. L. Plattet al., Transplantation 1991, 52, 530–533.
33. D. White, J. Wallwork, Lancet 1993, 342,879–880.
34. J. L. Platt, J. S. Logan, Transplant. Rev. 1996,10, 69–77.
35. L. E. Diamond, C. M. Quinn, M. J. Martinet al., Transplantation 2001, 71, 132–142.
36. K. R. McCurry, D. L. Kooyman, C. G. Alva-rado et al., Nat. Med. 1995, 1, 423–427.
37. G. W. Byrne, K. R. McCurry, M. J. Martinet al., Transplantation 1997, 63, 149–155.
38. E. Cozzi, N. Yannoutsos, G. A. Langfordet al. in Xenotransplantation: The Transplan-tation of Organs and Tissues Between Species(Eds.: D. K. C. Cooper, E. Kemp, J. L. Plattet al.), Springer, Berlin, 1997, pp. 665–682.
39. M. Lavitrano, M. L. Bacci, M. Forni et al.,Proc. Natl. Acad. Sci. U. S. A. 2002, 99,14230–14235.
40. J. R. Leventhal, A. J. Matas, L. H. Sun et al.,Transplantation 1993, 56, 1–8.
41. K. A. Porter in Pathology of the Kidney (Ed.:R. H. Heptinstall), Little Brown & Company,Boston, 1992, pp. 1799–1933, Vol. 3.
42. J. C. Magee, B. H. Collins, R. C. Harlandet al., J. Clin. Investig. 1995, 96, 2404–2412.
43. S. S. Lin, J. L. Platt, J. Heart Lung Transplant.1996, 15, 547–555.
44. J. J. McPaul, P. Stastny, R. B. Freeman, J.Clin. Investig. 1981, 67, 1405–1414.
45. L. C. Paul, F. H. J. Claas, L. A. Van Es et al.,N. Engl. J. Med 1979, 300, 1258–1260.
46. S. S. Lin, B. C. Weidner, G. W. Byrne et al.,J. Clin. Investig. 1998, 101, 1745–1756.
47. R. J. Perper, J. S. Najarian, Transplantation1967, 5, 514–533.
48. A. H. Cotterell, B. H. Collins, W. Parkeret al., Transplantation 1995, 60, 861–868.
49. K. R. McCurry, W. Parker, A. H. Cotterellet al., Hum. Immunol. 1997, 58, 91–105.
50. W. Parker, S. Saadi, S. S. Lin et al., Immunol.Today 1996, 17, 373–378.
51. F. H. Bach, H. Winkler, C. Ferran et al.,Immunol. Today 1996, 17, 379–384.
52. D. J. Goodman, M. Von Albertini, A. Willsonet al., Transplantation 1996, 61, 763–771.
53. A. M. Malyguine, S. Saadi, J. L. Platt et al.,Transplantation 1996, 61, 161–164.
54. A. M. Malyguine, S. Saadi, R. A. Holzknechtet al., J. Immunol. 1997, 159, 4659–4664.
55. M. W. Chopek, R. L. Simmons, J. L. Platt,Transplant. Proc. 1987, 19, 4553–4557.
56. N. S. Ihrcke, L. E. Wrenshall, B. J. Lindmanet al., Immunol. Today 1993, 14, 500–505.
57. T. E. Starzl, A. J. Demetris, N. Murase et al.,Immunol. Today 1993, 14, 326–332.
58. D. H. Sachs, T. Sablinski, Xenotransplanta-tion 1995, 2, 234–239.
59. Y. Zhao, K. Swenson, J. J. Sergio et al., Nat.Med. 1996, 2, 1211–1216.
60. M. Sykes, L. A. Lee, D. H. Sachs, Immunol.Rev. 1994, 141, 245–276.
61. H. Li, C. Ricordi, A. J. Demetris et al.,Transplantation 1994, 57, 592–598.
62. I. Aksentijevich, D. H. Sachs, M. Sykes,Transplantation 1992, 53, 1108–1114.
63. H. A. Gritsch, R. M. Glaser, D. W. Emeryet al., Transplantation 1994, 57, 906–917.
64. L. Inverardi, B. Clissi, A. L. Stolzer et al.,Transplant. Proc. 1996, 28, 552.
65. H.-G. Ljunggren, K. Karre, Immunol. Today1990, 11, 237–244.
66. L. L. Lanier, J. H. Phillips, Immunol. Today1996, 17, 86–91.

Xenotransplantation in Pharmaceutical Biotechnology 279
67. J. H. Lawson, J. L. Platt in Dialysis andTransplantation: A Companion to Brennerand Rector’s The Kidney (Eds.: W. Owen,B. Pereira, M. Sayegh), W. B. Saunders Co,Philadelphia, 2000, pp. 653–660.
68. C. W. Daggett, M. Yeatman, A. J. Lodgeet al., J. Thorac. Cardiovasc. Surg. 1998, 115,19–27.
69. S. A. Akhter, C. A. Skaer, A. P. Kypson et al.,Proc. Natl. Acad. Sci. U. S. A. 1997, 94,12100–12105.
70. A. P. Kypson, K. Peppel, S. A. Akhter et al.,J. Thorac. Cardiovasc. Surg. 1998, 115,623–630.
71. J. H. Lawson, J. L. Platt, Transplantation1996, 62, 303–310.
72. J. H. Lawson, R. D. Sorrell, J. L. Platt, Circu-lation 1997, 96, 565.
73. C. Patience, Y. Takeuchi, R. A. Weiss, Nat.Med. 1997, 3, 282–286.
74. B. A. Sullenger, H. F. Gallardo, G. E. Un-gers et al., Cell 1990, 63, 601–608.
75. C. G. Groth, O. Korsgren, A. Tibell et al.,Lancet 1994, 344, 1402–1404.
76. R. S. Chari, B. H. Collins, J. C. Magee et al.,N. Engl. J. Med. 1994, 331, 234–237.
77. T. Deacon, J. Schumacher, J. Dinsmoreet al., Nat. Med. 1997, 3, 350–353.
78. H. Nagata, M. Ito, J. Cai et al., Gastroenterol-ogy 2003, 124, 422–431.
79. J. A. Thomson, J. Itskovitz-Eldor, S. S. Sha-piro et al., Science 1998, 282, 1145–1147.

281
16Sculpturing the Architecture ofMineralized Tissues: TissueEngineering of Bone fromSoluble Signals to SmartBiomimetic Matrices
Ugo Ripamonti, Lentsha Nathaniel Ramoshebi, Janet Patton, June Teare, Thato Matsaba andLouise RentonBone Research Unit of the Medical Research Council and the University of the Witwatersrand,2193 Parktown, Johannesburg, South Africa
16.1Introduction
The study of the molecular and cellu-lar biology of bone morphogenetic andosteogenic proteins (BMPs/OPs), mor-phogens endowed with the striking prerog-ative of initiating de novo bone formationby induction, has profoundly modifiedour understanding of cell differentiationand the induction of tissue morphogen-esis [1–14]. Indeed, in vivo studies overthe past 12 years have revealed how os-teoblastic differentiation and the inductionof bone formation are controlled via thedeployment of a set of specific solublesignals, the BMPs/OPs, members of thetransforming growth factor-β (TGF-β) su-pergene family. To induce bone formation,however, the soluble signals require to bereconstituted with an insoluble substra-tum that triggers the bone differentiationcascade, as shown in nonhuman and hu-man primates [15–19].
The capability of the pharmaceutical in-dustry to develop peptides and proteins or
morphogens, defined as form-generatingsubstances [20] capable of imparting spe-cific different pathways to responding cellsinitiating the cascade of pattern formationand the attainment of tissue form andfunction, has increased markedly in thetwenty-first century. Morphogens of thetransforming growth factor-β (TGF-β) su-perfamily have now become available incommercially viable quantities producedas rationally designed gene products inaddition to naturally occurring ones in pu-rified form and in large scale for use in clin-ical contexts [17, 21, 22]. However, even ifmorphogens’ availability by recombinantDNA technology produced by the pharma-cological industry is cost-effective, signif-icant challenges to morphogens’ deliverystill limit their utilization as therapeuticagents. Morphogens of the TGF-β super-family are autocrine- and paracrine-solublesignals that have widespread pleiotropicfunctions in vivo [3, 5, 7, 8]. Followingtheir direct administration into the bloodstream, it is highly improbable that tissue
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

282 16.2 Osteogenic Soluble Signals of the TGF-β Superfamily
engineers will be able to direct such mor-phogens to specific receptors on target cellsin sufficient quantities to evoke a desiredtherapeutic response without promotingpotentially deleterious side effects in sev-eral tissues and organs of the mammalianbody.
This monograph describes the solublesignals that initiate bone formation by in-duction and outlines novel concepts ofbone tissue engineering as evaluated inprimate models. The monograph furtherreports the apparent redundancy in endo-chondral bone formation by molecularlydifferent members of the TGF-β super-family. Our studies reported below indi-cate that in the adult primate – and in theadult primate only – recombinantly pro-duced or naturally derived TGF-β isoformsare powerful inducers of endochondralbone formation, with a specific activityequal to, if not higher than, identicaldoses of recombinant hBMPs/OPs. Wewill further highlight the use of novelbiomimetic matrices to deliver the bio-logical activity of the osteogenic membersof the TGF-β superfamily. Experimentalstudies in ovariectomized (OVX) primatesof the species Papio ursinus will describethe local delivery of naturally derived andrecombinantly produced hBMPs/OPs us-ing a reconstituted basement membranegel (Matrigel) to treat systemic bone lossin nonhuman primates and thus by ex-tension to human primates affected byosteoporosis. Lastly, we describe the devel-opment and use of biomimetic matriceswith site-specific geometric modificationsand endowed with the striking preroga-tive of initiating de novo bone formationby induction in heterotopic extraskeletalsites of primates even in the absenceof exogenously applied osteogenic geneproducts of the TGF-β superfamily [16,23–25].
16.2Osteogenic Soluble Signals of the TGF-βSuperfamily
Bone is in both a soluble and a solid state,and there is a continuum between thesoluble and insoluble states regulated bysignals in solution interacting with the in-soluble extracellular matrix [7, 10]. Naturerelies on common yet limited molecularmechanisms tailored to provide the emer-gence of specialized tissues and organs.The TGF-β superfamily is indeed an ele-gant example of nature’s parsimony in pro-gramming multiple specialized pleiotropicfunctions deploying molecular isoformswith minor variations in amino acid motifswithin highly conserved carboxy-terminalregions.
In preclinical and clinical contexts, tissueregeneration in postnatal life recapitulatesevents that occur in the normal course ofembryonic development and morphogene-sis [5, 8, 10]. Both embryonic developmentand tissue regeneration are equally regu-lated by a selected few, highly conservedfamilies of morphogens, the soluble sig-nals of the TGF-β superfamily. Amongthe many tissues in the body, bone hasconsiderable potential for repair and re-generation and could well be considereda prototype for tissue repair and regenera-tion in molecular terms [3–5, 14, 16, 25].
Bone morphogenetic proteins/osteo-genic proteins (BMPs/OPs), members ofthe TGF-β superfamily, are soluble media-tors of tissue morphogenesis and powerfulregulators of cartilage and bone differ-entiation in embryonic development andregeneration in postnatal life [1, 3–5, 14,16, 25]. A striking prerogative of the os-teogenic members of the TGF-β superfam-ily, whether naturally derived or producedby DNA recombinant technologies, is theirability to induce de novo endochondral

Sculpturing the Architecture of Mineralized Tissues 283
bone formation in extraskeletal heterotopicsites in postnatal life as a recapitulation ofevents that occur in the normal courseof embryonic development [3, 5, 12, 14,25]. To induce endochondral bone dif-ferentiation to be exploited in preclinicaland clinical contexts, the osteoinductivesoluble signals require the reconstitutionwith an insoluble signal or substratumthat triggers the bone differentiation cas-cade [7–9].
The menu for enunciating the rulesthat sculpt the architecture of cortico-cancellous structures of the bone andregulate bone regeneration and bonetissue engineering in clinical contexts listscomplex interactions between soluble andinsoluble signals [7]. Tissue engineeringin clinical contexts requires three keycomponents: an osteoinductive signal;an insoluble substratum that deliversthe signal and acts as a scaffold fornew bone formation; and host cellscapable of differentiation into bone cells inresponse to the osteoinductive signal. Thesignals responsible for osteoinduction areconferred by the osteogenic members ofthe TGF-β superfamily [10, 15, 25].
The reconstitution of doses of recombi-nant human osteogenic protein-1 (rhOP-1), with insoluble substrata such as theinactive insoluble collagenous bone ma-trix additionally prepared from xenogeneicsources, restores the biological activityand results in the long-term efficacy ofsingle applications of gamma-irradiatedhOP-1 delivered by xenogeneic bovine col-lagenous matrices in regenerating largedefects of membranous bone preparedin the calvarium of the adult primatePapio ursinus [15, 26]. The operational re-constitution of the soluble morphogeneticsignal (hOP-1) with an insoluble sub-stratum (the collagenous insoluble bonematrix) underscores the critical role of
the insoluble signal of the collagenousmatrix for the induction of tissue mor-phogenesis and regeneration [10, 15, 27].These findings obtained in the adult pri-mate indicate that a single application ofgamma-irradiated hOP-1 combined withthe gamma-irradiated xenogeneic bovinecollagenous bone matrix carrier is effectivein regenerating and maintaining the archi-tecture of the induced bone at doses of 0.5-and 2.5-mg hOP-1 per gram of carrier ma-trix (Fig. 1) [15]. Information concerningthe efficacy and safety of gamma-irradiatedosteogenic devices in nonhuman primatesis an important prerequisite for clinicalapplications.
The fact that a single BMP/OP initi-ates bone formation by induction doesnot preclude the requirement for inter-actions with other morphogens deployedsynchronously and synergistically duringthe cascade of bone formation by induc-tion, which may proceed via the com-bined action of several BMPs/OPs residentwithin the natural milieu of the extra-cellular matrix of bone [15, 25]. Partiallypurified preparations from bone matrixare known to contain, in addition to spe-cific BMPs/OPs, several other proteinsand some as yet poorly characterizedmitogens [28]. Indeed, 90 days after im-plantation, regenerated tissue induced by2.5 mg of partially purified BMPs/OPscombined with gamma-irradiated matrix,had mineralized bone and osteoid volumescomparable to specimens induced by 0.5-mg hOP-1 devices (Fig. 1) [15].
Partially purified preparations frombone matrix obtained using chroma-tographic procedures as described [15,29–31] are known to contain BMP-2,BMP-3, and OP-1 but not detectable TGF-βs [32]. In a clinical trial in humans,osteogenic devices prepared by partiallypurified BMPs/OPs reconstituted with
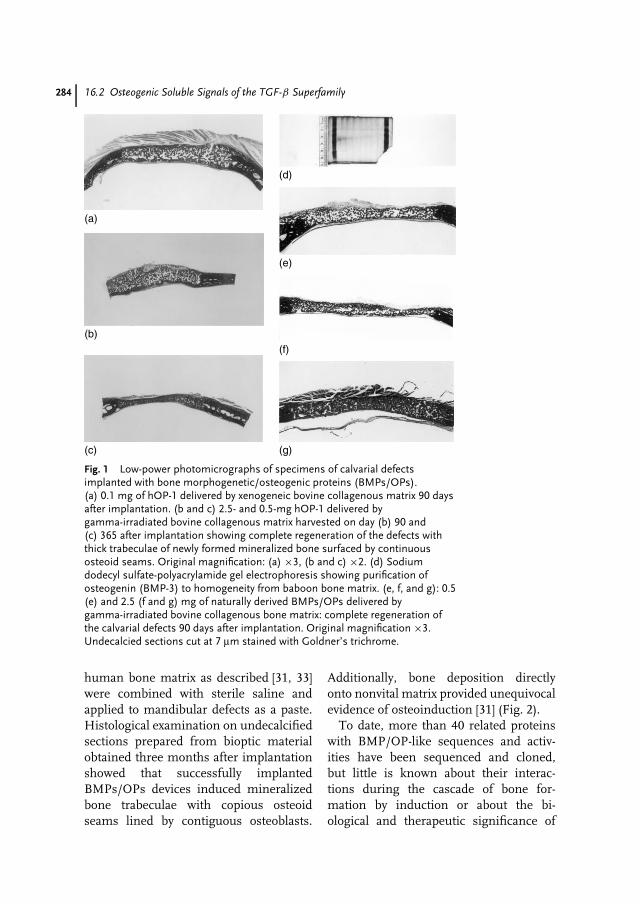
284 16.2 Osteogenic Soluble Signals of the TGF-β Superfamily
(a)
(b)
(c)
(d)
(e)
(f)
(g)
Fig. 1 Low-power photomicrographs of specimens of calvarial defectsimplanted with bone morphogenetic/osteogenic proteins (BMPs/OPs).(a) 0.1 mg of hOP-1 delivered by xenogeneic bovine collagenous matrix 90 daysafter implantation. (b and c) 2.5- and 0.5-mg hOP-1 delivered bygamma-irradiated bovine collagenous matrix harvested on day (b) 90 and(c) 365 after implantation showing complete regeneration of the defects withthick trabeculae of newly formed mineralized bone surfaced by continuousosteoid seams. Original magnification: (a) ×3, (b and c) ×2. (d) Sodiumdodecyl sulfate-polyacrylamide gel electrophoresis showing purification ofosteogenin (BMP-3) to homogeneity from baboon bone matrix. (e, f, and g): 0.5(e) and 2.5 (f and g) mg of naturally derived BMPs/OPs delivered bygamma-irradiated bovine collagenous bone matrix: complete regeneration ofthe calvarial defects 90 days after implantation. Original magnification ×3.Undecalcied sections cut at 7 µm stained with Goldner’s trichrome.
human bone matrix as described [31, 33]were combined with sterile saline andapplied to mandibular defects as a paste.Histological examination on undecalcifiedsections prepared from bioptic materialobtained three months after implantationshowed that successfully implantedBMPs/OPs devices induced mineralizedbone trabeculae with copious osteoidseams lined by contiguous osteoblasts.
Additionally, bone deposition directlyonto nonvital matrix provided unequivocalevidence of osteoinduction [31] (Fig. 2).
To date, more than 40 related proteinswith BMP/OP-like sequences and activ-ities have been sequenced and cloned,but little is known about their interac-tions during the cascade of bone for-mation by induction or about the bi-ological and therapeutic significance of

Sculpturing the Architecture of Mineralized Tissues 285
(a) (b)
(c)
Fig. 2 Photomicrographs of tissue inductionand morphogenesis in bioptic material 90 daysafter implantation of naturally derivedBMPs/OPs purified from bovine bone matrix inhuman mandibular defects. (a) Trabeculae ofnewly formed mineralized bone covered bycontinuous osteoid seams within highly vascularstroma. (b) and (c) High-power views showing
cellular mineralized bone surfaced by osteoidseams. Newly formed and mineralized bonedirectly opposing the implanted collagenousmatrix carrier (arrows) confirms bone formationby induction. Undecalcified sections at 7 µmstained with Goldner’s trichrome. Originalmagnification: (a) ×14; (b) ×40; and (c) ×50.(See Color Plate p. xxiii).
this apparent redundancy. Recombinantlyproduced hBMP-2, hBMP-4, and hOP-1singly initiate bone formation by induc-tion in vivo [1, 34–36]. It is likely that theendogenous mechanisms of bone repairand regeneration in postnatal life necessi-tates the deployment and concerted actionsof several of the BMPs/OPs present withinthe extracellular matrix of bone [15, 25].Whether the biological activity of partiallypurified BMPs/OPs is the result of thesum of a plurality of BMP/OP activitiesor of a truly synergistic interaction amongBMP/OP family members deserves appro-priate investigation [15, 25].
In addition to bone induction in post-fetal life, the BMPs/OPs are involved
in inductive events that control patternformation during morphogenesis andorganogenesis in such disparate tissuesand organs as the kidney, eye, nervous sys-tem, lung, teeth, skin, and heart [37]. Thesestrikingly pleiotropic effects of BMPs/OPsmay spring from minor amino acid se-quence variations in the carboxyl-terminalregion of the proteins [38] as well as inthe transduction of distinct signal path-ways by individual Smad proteins aftertransmembrane serine-threonine kinasereceptor activation [39, 40].
In vitro studies indicate that both hOP-1 and hBMP-2 modulate messenger RNA(mRNA) expression of related BMP/OPfamily members and in vivo studies are

286 16.2 Osteogenic Soluble Signals of the TGF-β Superfamily
now mandatory to identify therapeuticapproaches on the basis of the informationof gene regulation by hBMPs/OPs [41, 42].Ultimately, it will be necessary to gain in-sights into the distinct spatial and temporalpatterns of expression of BMPs/OPs andTGF-ß family members elicited by a singleapplication of recombinant hBMPs/OPs,including hOP-1.
To investigate expression patterns ofgene products following single applica-tions of a recombinant hBMP/OP, dosesof hOP-1 (0.1, 0.5, and 2.5 mg) were im-planted in heterotopic extraskeletal sites ofthe rectus abdominis muscle and orthotopi-cally in calvarial defects of adult primatePapio ursinus. mRNA expression of OP-1, TGF-ß, BMP-3, and collagens Type IIand IV showed upregulation of the genesin ossicles generated by the higher doseof the hOP-1 as evaluated on day 15 and30, though with significant differences be-tween tissues generated in orthotopic andin heterotopic sites [Thato Matsaba, un-published data]. In vivo studies in primatemodels should now be used to design ther-apeutic approaches on the basis of theinformation of gene regulation by hOP-1 [25].
The processes of tissue morphogen-esis and regeneration of the complextissue morphologies of the periodontaltissues rest on the sequential expressionof BMP/OP proteins during regenerativeevents and also on the expression andsynthesis of related morphogenetic geneproducts of the large TGF-ß superfam-ily [43]. The initiating events in periodontalregeneration are transitory and lead to se-quential molecular and cellular outcomesstimulating subsequent events such aschemotaxis, differentiation, proliferation,and angiogenesis, leading ultimately tothe morphogenesis and remodeling of theperiodontal tissues [43–45].
The pleiotropic functions of the BMPs/OPs have been further shown after theirimplantation in furcation defects preparedin adult primates Papio ursinus [43, 44, 46,47]. Undecalcified semi-thin sections offurcation defects of the chacma baboon Pa-pio ursinus treated with doses of highly pu-rified bone-derived BMPs/OPs showed notonly alveolar bone but also cementogenesisand periodontal ligament regeneration, theessential ingredients to engineer periodon-tal tissue regeneration [44, 46, 48] (Fig. 3).
Single applications of relatively lowdoses of hOP-1 (0.1- and 0.5-mg hOP-1per gram of collagenous matrix as carrier)preferentially induced cementogenesis asevaluated 60 days after implantation in sur-gically induced furcation defects in Papioursinus [47]. This seemingly specific ce-mentogenic function of hOP-1 suggestedthat a structure/activity profile could re-side within BMP/OP family members tocontrol tissue morphogenesis and regen-eration of disparate tissues and organs [43,44, 47, 48].
More challenging was the demon-stration of cementogenesis and alveolarbone regeneration in periodontally in-duced furcation defects with root sur-faces chronically exposed to periodon-tal pathogens [43]. A pathogenic humanstrain of Porphyromonas gingivalis was in-oculated into the furcation areas of thefirst and second mandibular molars offour adult chacma baboons twice a monthfor 12 months. Chronic periodontitis wasinduced in all four animals as assessedby probing periodontal pocket depths, in-traoral radiographs, and microbiologicalanalyses that confirmed the presence ofPorphyromonas gingivalis [43]. Two monthsafter scaling, root planing, and a plaque-control regimen with clinical resolutionof gingivitis, mucoperiosteal flaps wereelevated to expose Class II furcation
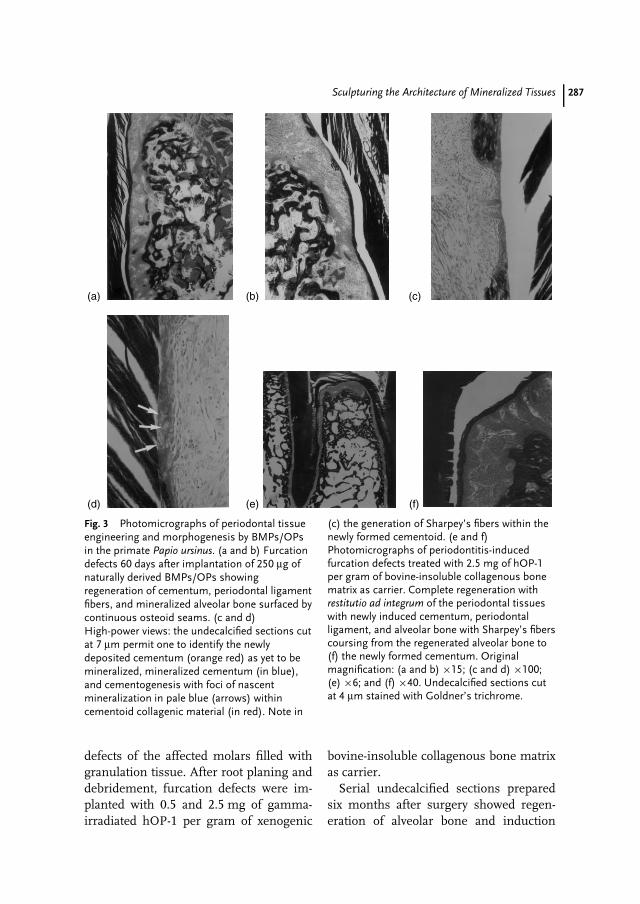
Sculpturing the Architecture of Mineralized Tissues 287
(a) (b) (c)
(d) (e) (f)
Fig. 3 Photomicrographs of periodontal tissueengineering and morphogenesis by BMPs/OPsin the primate Papio ursinus. (a and b) Furcationdefects 60 days after implantation of 250 µg ofnaturally derived BMPs/OPs showingregeneration of cementum, periodontal ligamentfibers, and mineralized alveolar bone surfaced bycontinuous osteoid seams. (c and d)High-power views: the undecalcified sections cutat 7 µm permit one to identify the newlydeposited cementum (orange red) as yet to bemineralized, mineralized cementum (in blue),and cementogenesis with foci of nascentmineralization in pale blue (arrows) withincementoid collagenic material (in red). Note in
(c) the generation of Sharpey’s fibers within thenewly formed cementoid. (e and f)Photomicrographs of periodontitis-inducedfurcation defects treated with 2.5 mg of hOP-1per gram of bovine-insoluble collagenous bonematrix as carrier. Complete regeneration withrestitutio ad integrum of the periodontal tissueswith newly induced cementum, periodontalligament, and alveolar bone with Sharpey’s fiberscoursing from the regenerated alveolar bone to(f) the newly formed cementum. Originalmagnification: (a and b) ×15; (c and d) ×100;(e) ×6; and (f) ×40. Undecalcified sections cutat 4 µm stained with Goldner’s trichrome.
defects of the affected molars filled withgranulation tissue. After root planing anddebridement, furcation defects were im-planted with 0.5 and 2.5 mg of gamma-irradiated hOP-1 per gram of xenogenic
bovine-insoluble collagenous bone matrixas carrier.
Serial undecalcified sections preparedsix months after surgery showed regen-eration of alveolar bone and induction
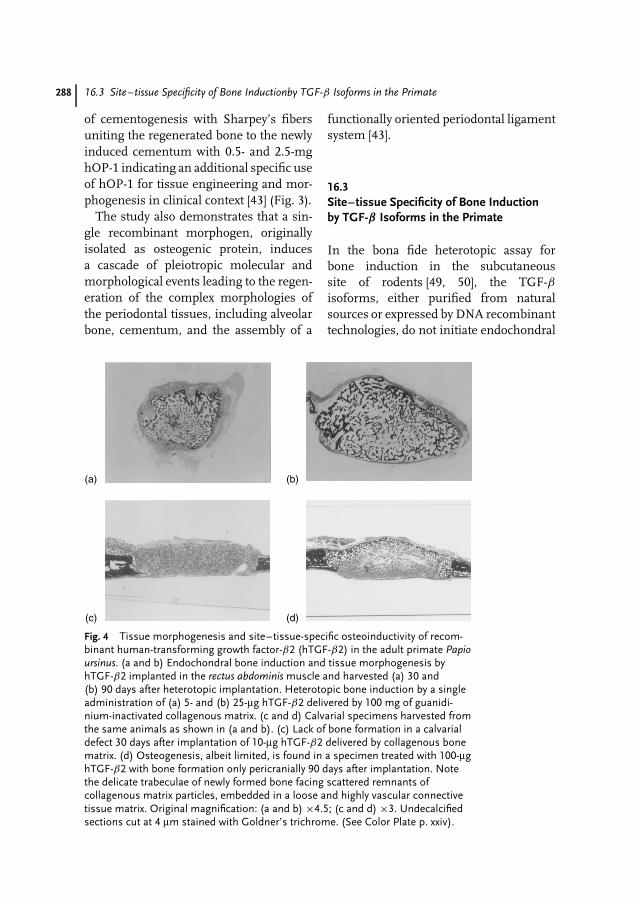
288 16.3 Site–tissue Specificity of Bone Inductionby TGF-β Isoforms in the Primate
of cementogenesis with Sharpey’s fibersuniting the regenerated bone to the newlyinduced cementum with 0.5- and 2.5-mghOP-1 indicating an additional specific useof hOP-1 for tissue engineering and mor-phogenesis in clinical context [43] (Fig. 3).
The study also demonstrates that a sin-gle recombinant morphogen, originallyisolated as osteogenic protein, inducesa cascade of pleiotropic molecular andmorphological events leading to the regen-eration of the complex morphologies ofthe periodontal tissues, including alveolarbone, cementum, and the assembly of a
functionally oriented periodontal ligamentsystem [43].
16.3Site–tissue Specificity of Bone Inductionby TGF-β Isoforms in the Primate
In the bona fide heterotopic assay forbone induction in the subcutaneoussite of rodents [49, 50], the TGF-βisoforms, either purified from naturalsources or expressed by DNA recombinanttechnologies, do not initiate endochondral
(a) (b)
(c) (d)
Fig. 4 Tissue morphogenesis and site–tissue-specific osteoinductivity of recom-binant human-transforming growth factor-β2 (hTGF-β2) in the adult primate Papioursinus. (a and b) Endochondral bone induction and tissue morphogenesis byhTGF-β2 implanted in the rectus abdominis muscle and harvested (a) 30 and(b) 90 days after heterotopic implantation. Heterotopic bone induction by a singleadministration of (a) 5- and (b) 25-µg hTGF-β2 delivered by 100 mg of guanidi-nium-inactivated collagenous matrix. (c and d) Calvarial specimens harvested fromthe same animals as shown in (a and b). (c) Lack of bone formation in a calvarialdefect 30 days after implantation of 10-µg hTGF-β2 delivered by collagenous bonematrix. (d) Osteogenesis, albeit limited, is found in a specimen treated with 100-µghTGF-β2 with bone formation only pericranially 90 days after implantation. Notethe delicate trabeculae of newly formed bone facing scattered remnants ofcollagenous matrix particles, embedded in a loose and highly vascular connectivetissue matrix. Original magnification: (a and b) ×4.5; (c and d) ×3. Undecalcifiedsections cut at 4 µm stained with Goldner’s trichrome. (See Color Plate p. xxiv).
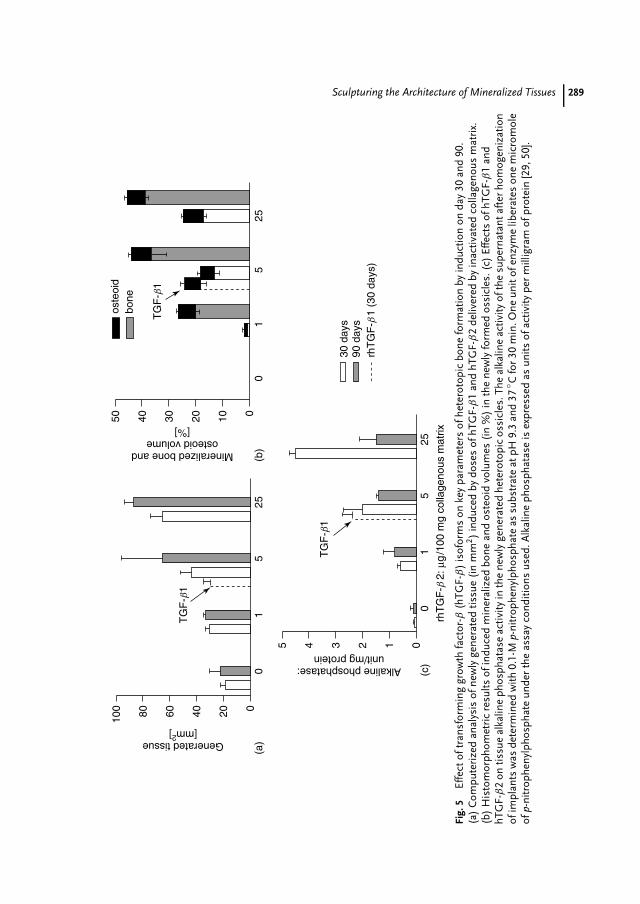
Sculpturing the Architecture of Mineralized Tissues 289
100
(a)Generated tissue
[mm2]T
GF
-b1
80 60 40 20 00
15
25(b
)
50os
teoi
dbo
ne
TG
F-b
1
Mineralized bone andosteoid volume
[%]
40 30 20 10 00
15
25
30 d
ays
90 d
ays
rhT
GF
-b1
(30
days
)
(c)
TG
F-b
1
5
Alkaline phosphatase:unit/mg protein
4 3 2 1 00
15
rhT
GF
-b 2
: µg
/100
mg
colla
geno
us m
atrix
25
Fig.
5Ef
fect
oftr
ansf
orm
ing
grow
thfa
ctor
-β(h
TGF-
β)
isof
orm
son
key
para
met
ers
ofhe
tero
topi
cbo
nefo
rmat
ion
byin
duct
ion
onda
y30
and
90.
(a)
Com
pute
rize
dan
alys
isof
new
lyge
nera
ted
tissu
e(i
nm
m2)
indu
ced
bydo
ses
ofhT
GF-
β1
and
hTG
F-β
2de
liver
edby
inac
tivat
edco
llage
nous
mat
rix.
(b)
His
tom
orph
omet
ric
resu
ltsof
indu
ced
min
eral
ized
bone
and
oste
oid
volu
mes
(in
%)
inth
ene
wly
form
edos
sicl
es.(
c)Ef
fect
sof
hTG
F-β
1an
dhT
GF-
β2
ontis
sue
alka
line
phos
phat
ase
activ
ityin
the
new
lyge
nera
ted
hete
roto
pic
ossi
cles
.The
alka
line
activ
ityof
the
supe
rnat
anta
fter
hom
ogen
izat
ion
ofim
plan
tsw
asde
term
ined
with
0.1-
Mp-
nitr
ophe
nylp
hosp
hate
assu
bstr
ate
atpH
9.3
and
37◦ C
for
30m
in.O
neun
itof
enzy
me
liber
ates
one
mic
rom
ole
ofp-
nitr
ophe
nylp
hosp
hate
unde
rth
eas
say
cond
ition
sus
ed.A
lkal
ine
phos
phat
ase
isex
pres
sed
asun
itsof
activ
itype
rm
illig
ram
ofpr
otei
n[2
9,50
].

290 16.3 Site–tissue Specificity of Bone Inductionby TGF-β Isoforms in the Primate
bone formation [51, 52]. Strikingly, and incontrast, the mammalian TGF-β isoformshave shown a marked site- and tissue-specific endochondral osteoinductivity, yetremarkably in the primate only [53–57](Figs. 4 and 5). In the higher vertebratessuch as the primate species, the presenceof several related but different molecularforms with osteogenic activity highlightsthe biological significance of this apparentredundancy and indicates multiple interac-tions during both embryonic developmentand bone regeneration in postnatal life [25,54]. Indeed, a potent and accelerated syn-ergistic interaction in endochondral boneformation has been shown with the bi-nary application of human recombinantor native TGF-β1 with hOP-1 in both het-erotopic and orthotopic sites of primates(Fig. 6) [25, 54, 55, 58].
The TGF-β isoforms are powerful induc-ers of endochondral bone when implantedin the rectus abdominis muscle of theprimate Papio ursinus at doses of 1, 5,and 25 µg per 100 mg of collagenous ma-trix as carrier, yielding large corticalizedossicles by day 90 (Figs. 4 and 5). Endo-chondral bone initiated by TGF-β isoformsexpresses mRNA of bone induction mark-ers including BMP-3 and OP-1 [55–57].
A significant striking result is that thebone-inductive activity of TGF-β isoformsin the primate is site- and tissue-specificwith rather substantial endochondral boneinduction in the rectus abdominis musclebut absent osteoinductivity in orthotopicsites on day 30 and limited osteogenesis inorthotopic sites on day 90 (Fig. 4).
The observed site and tissue specificity ofinduction in the nonhuman primate Papioursinus and thus by extension to homo sapi-ens may be due to the presence or absenceof multiple variable-responding cells, theexpression of inhibitory binding pro-teins or the influence of the downstream
antagonists of the TGF-β signaling, Smad6and Smad7 [59–61]. Indeed, current re-sults of mRNA studies on tissues gener-ated by TGF-β isoforms in heterotopic andorthotopic sites demonstrated robust ex-pression of the TGF-β self-regulatory pro-teins Smad6 and Smad7 orthotopically, butonly modest expression in tissue from het-erotopic sites (unpublished observation).These findings represent one possible ex-planation for the poor osteoinductivity ofTGF-β isoforms observed in nonhealingcalvarial defects and indicate that overex-pression of Smad6 and Smad7 downregu-late the osteoinductivity of the TGF-β iso-forms when deployed orthotopically [61].
Conceivably, the rapid induction of en-dochondral bone by hTGF-β isoformscould be utilized for the generation oflarge ossicles in the rectus abdominis mus-cle of human patients. Thirty days afterheterotopic implantation, generated ossi-cles could be harvested and morsellizedfragments transplanted into bony defectsaffecting the same patient in an autoge-nous fashion to treat defects either in theaxial or craniofacial skeleton including pe-riodontal osseous defects. The rapidity oftissue morphogenesis and induction ofbone formation complete with mineral-ization of the outer cortex of the ossiclesand bone marrow formation by day 30 is ofparticular importance for repair and regen-eration of bone in the elderly, where repairphenomena are temporally delayed andhealing progresses slower than in youngerpatients. Potentially, fragments of autoge-nously induced bone could be morsellizedfrom ossicles induced in the rectus abdo-minis after the binary application of hOP-1and relatively low doses of a TGF-β iso-form, a synergistic strategy known to yieldmassive mineralized ossicles with largeseams of osteoid populated by contiguous
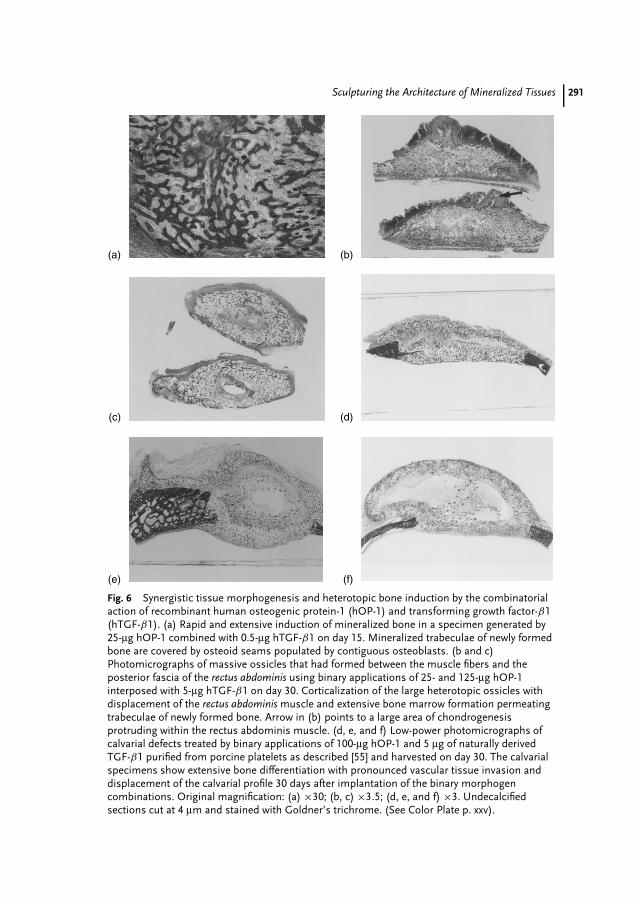
Sculpturing the Architecture of Mineralized Tissues 291
(a)
(d)
(b)
(e)
(c)
(f)
Fig. 6 Synergistic tissue morphogenesis and heterotopic bone induction by the combinatorialaction of recombinant human osteogenic protein-1 (hOP-1) and transforming growth factor-β1(hTGF-β1). (a) Rapid and extensive induction of mineralized bone in a specimen generated by25-µg hOP-1 combined with 0.5-µg hTGF-β1 on day 15. Mineralized trabeculae of newly formedbone are covered by osteoid seams populated by contiguous osteoblasts. (b and c)Photomicrographs of massive ossicles that had formed between the muscle fibers and theposterior fascia of the rectus abdominis using binary applications of 25- and 125-µg hOP-1interposed with 5-µg hTGF-β1 on day 30. Corticalization of the large heterotopic ossicles withdisplacement of the rectus abdominis muscle and extensive bone marrow formation permeatingtrabeculae of newly formed bone. Arrow in (b) points to a large area of chondrogenesisprotruding within the rectus abdominis muscle. (d, e, and f) Low-power photomicrographs ofcalvarial defects treated by binary applications of 100-µg hOP-1 and 5 µg of naturally derivedTGF-β1 purified from porcine platelets as described [55] and harvested on day 30. The calvarialspecimens show extensive bone differentiation with pronounced vascular tissue invasion anddisplacement of the calvarial profile 30 days after implantation of the binary morphogencombinations. Original magnification: (a) ×30; (b, c) ×3.5; (d, e, and f) ×3. Undecalcifiedsections cut at 4 µm and stained with Goldner’s trichrome. (See Color Plate p. xxv).

292 16.4 Treatment of Systemic Bone Loss by Local Induction of Bone
osteoblastic cells by day 15 and 30 afterheterotopic implantation [25, 54, 55, 58].
16.4Treatment of Systemic Bone Loss by LocalInduction of Bone
The biosynthesis and assembly of ex-tracellular matrix with angiogenesis andvascular invasion is a prerequisite to re-store the architecture of skeletal structureswith a constellation of extracellular ma-trix components [5]. Therefore, a criticalprovision for tissue engineering is thesculpturing of the optimal extracellularmatrix scaffolding [5] for the transforma-tion of responding cells into secretory bonecells and osteoblasts.
We have thus investigated extracellu-lar matrix components reconstituted asa biomimetic carrier matrix as deliverysystems for naturally derived, highlypurified BMPs/OPs and hOP-1 in the
SC
SC
ROI
A
A
B
35−40°
Fig. 7 Schematic representation of(a) the primate Papio ursinus lumbarvertebra and (b) the bone marrowbiopsy needle positioning during localadministration of naturally derived bonemorphogenetic proteins. Insertion ofthe needle was monitored on X-rayimage intensifier. Bottom panel: sagittalrepresentation of (a) the lumbarvertebral body and its region of interest(ROI); SC: spinal canal.
rodent bioassay [62]. Matrigel, a solubleextract of the Engelbreth-Holm-Swarm tu-mor [63] gels at room temperature to forma reconstituted basement membrane [63].It contains laminin, type IV collagen, en-tactin, nidogen, heparan sulphate proteo-glycan, and additional growth factors [63],and was tested to deliver the BMPs/OPsafter subcutaneous implantation in ro-dents [62]. The matrix proteins lamininand type IV collagen bind BMPs/OPs [64].The combination of the Matrigel carrierwith morphogens successfully inducedbone formation, indicating that Matrigel
is an effective carrier of osteogenic solublesignals [62].
We have used the Matrigel matrix re-constituted with BMPs/OPs for a novellocal treatment of systemic bone loss in ourcohort of OVX primates Papio ursinus [62].The bone mineral density of the lumbarvertebrae of the OVX primates was signif-icantly affected by estrogen depletion [62].Histomorphometric data on iliac crestbiopsies showed that bone was perma-nently lost 36 months after ovariectomy.Injections of 0.5 mg of naturally derivedBMPs/OPs into the lumbar vertebrae 3and 4 using an X-ray image intensifier forguidance was successfully performed forthe first time in primates [62] (Fig. 7). Thisnovel method may prove to be valuable forthe treatment of systemic bone loss by lo-calized injections of osteogenic proteins ofthe TGF-β superfamily in clinical contexts.
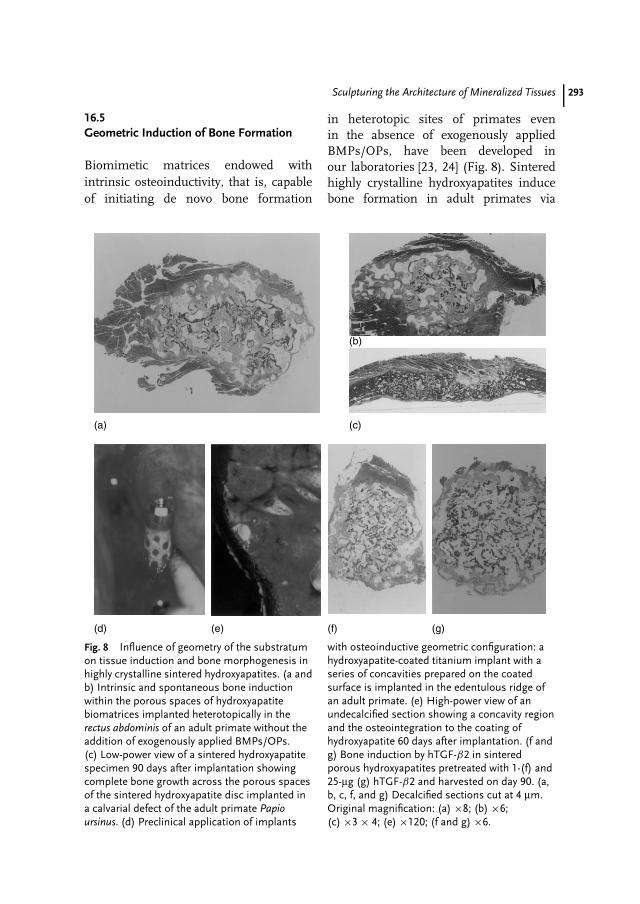
Sculpturing the Architecture of Mineralized Tissues 293
16.5Geometric Induction of Bone Formation
Biomimetic matrices endowed withintrinsic osteoinductivity, that is, capableof initiating de novo bone formation
in heterotopic sites of primates evenin the absence of exogenously appliedBMPs/OPs, have been developed inour laboratories [23, 24] (Fig. 8). Sinteredhighly crystalline hydroxyapatites inducebone formation in adult primates via
(a)
(d) (e) (f) (g)
(c)
(b)
Fig. 8 Influence of geometry of the substratumon tissue induction and bone morphogenesis inhighly crystalline sintered hydroxyapatites. (a andb) Intrinsic and spontaneous bone inductionwithin the porous spaces of hydroxyapatitebiomatrices implanted heterotopically in therectus abdominis of an adult primate without theaddition of exogenously applied BMPs/OPs.(c) Low-power view of a sintered hydroxyapatitespecimen 90 days after implantation showingcomplete bone growth across the porous spacesof the sintered hydroxyapatite disc implanted ina calvarial defect of the adult primate Papioursinus. (d) Preclinical application of implants
with osteoinductive geometric configuration: ahydroxyapatite-coated titanium implant with aseries of concavities prepared on the coatedsurface is implanted in the edentulous ridge ofan adult primate. (e) High-power view of anundecalcified section showing a concavity regionand the osteointegration to the coating ofhydroxyapatite 60 days after implantation. (f andg) Bone induction by hTGF-β2 in sinteredporous hydroxyapatites pretreated with 1-(f) and25-µg (g) hTGF-β2 and harvested on day 90. (a,b, c, f, and g) Decalcified sections cut at 4 µm.Original magnification: (a) ×8; (b) ×6;(c) ×3 × 4; (e) ×120; (f and g) ×6.

294 16.5 Geometric Induction of Bone Formation
intrinsic osteoinductivity regulated bythe geometry of the substratum [23, 24](Fig. 8).
Current experiments in our laborato-ries have confirmed that the geometry ofbiomimetic matrices is not the only drivingforce in osteoinduction since the struc-ture of the insoluble signal dramaticallyinfluences and regulates gene expression,with the induction of bone as a sec-ondary response [25, 65]. Soluble signalsinduce morphogenesis; physical forces im-parted by the geometric topography ofthe insoluble signal dictate biological pat-terns, constructing the induction of boneand regulating the expression of selectedmRNA of gene products of the TGF-βsuperfamily as a function of the struc-ture [66].
Our molecular, biochemical, and mor-phological data have indicated that the spe-cific geometric configuration in the formof concavities within highly crystalline hy-droxyapatite biomimetic matrices is thedriving molecular and morphogenetic mi-croenvironment, conducive and induciveto a specific sequence of events leadingto bone formation by induction [23, 24].The specific geometry of the biomimeticmatrices initiates a bone-inductive mi-croenvironment by providing geometricalstructures biologically and architecturallyconducive and inducive to optimal seques-tration and synthesis of osteogenic mem-bers of the TGF-β superfamily [23–25] andparticularly capable of stimulating angio-genesis, a prerequisite for osteogenesis.Angiogenesis may indeed provide a tem-porally regulated flow of cell populationscapable of expression of the osteogenicphenotype.
We have recently investigated whetherthe BMPs/OPs shown to be present byimmunolocalization in the concavities areadsorbed onto the sintered biomimetic
matrices from the circulation or ratherare locally produced after local expres-sion and synthesis by transformed cellularelements resident within the concavitymicroenvironment [23, 65]. We now pro-pose the following cascade of molecularand morphogenetic events culminatingin the induction of bone in heterotopicsites of primates and initiating withinconcavities of the smart biomimetic ma-trices:
1. Vascular invasion and capillary sprout-ing within the invading tissue with cap-illary elongation in close contact withthe implanted hydroxyapatite bioma-trix.
2. Attachment to and differentiation ofmesenchymal cells at the hydroxyap-atite/soft tissue interface of the con-cavities. Expression of TGF-β andBMPs/OPs family member genes indifferentiating osteoblast-like cells res-ident within the concavities of thesmart biomimetic matrices as shownby Northern blot analyses of tissueharvested from the concavities of thesubstratum [65].
3. Synthesis of specific TGF-β superfam-ily member proteins as markers of boneformation by induction from residenttransformed osteoblast-like cells ontothe sintered crystalline hydroxyapatiteas shown by immunolocalization ofOP-1 and BMP-3 within the cellularcytoplasm and at the interface of thehydroxyapatite biomatrix with the in-vading mesenchymal tissue [23, 24, 65].
4. Intrinsic osteoinduction with furtherdifferentiation of osteoblastic cell lines,which is dependent upon a criti-cal threshold of endogenously pro-duced BMPs/OPs initiating bone for-mation by induction as a secondaryresponse [65].

Sculpturing the Architecture of Mineralized Tissues 295
16.6Conclusion
Tissue regeneration in postnatal life reca-pitulates events that occur in the normalcourse of embryonic development andmorphogenesis. Both embryonic develop-ment and tissue regeneration are equallyregulated by a selected few and highlyconserved families of morphogens. Thisplurality of gene products are membersof the TGF-β superfamily. The initia-tion of bone formation during embryonicdevelopment and postnatal osteogenesisinvolves a complex cascade of molecularand morphogenetic processes that ulti-mately lead to the architectural sculptureof precisely organized multicellular struc-tures. In the primate only, heterotopicbone induction is initiated by naturallyderived BMPs/OPs and TGF-βs, recom-binant hBMPs/OPs and hTGF-βs, andsintered hydroxyapatites biomimetic ma-trices with a specific geometric configu-ration. Bone tissue develops as a mosaicstructure in which members of the TGF-β superfamily singly, synergistically, andsynchronously initiate and maintain thedeveloping morphological structures andplay different roles at different time pointsof the morphogenetic cascade. Osteogenicmembers of the TGF-β superfamily aresculpturing tissue constructs, helping toengineer skeletal tissue regeneration inmolecular terms: morphogens exploitedin embryonic development are reexploitedand redeployed in postnatal tissue regen-eration.
Biomimetic biomaterial matrices arenow designed to obtain specific biologi-cal responses so much so that the use ofbiomaterials capable of initiating bone for-mation via osteoinductivity even in the ab-sence of exogenously applied BMPs/OPsis fast altering the horizons of therapeutic
bone regeneration. Our results have indi-cated that the geometry of the substratumis not the only driving force since the struc-ture of the insoluble signal dramaticallyinfluences and regulates gene expressionand the induction of bone as a secondaryresponse. Soluble signals induce morpho-genesis; physical forces imparted by the ge-ometric topography of the insoluble signaldictate biological patterns, constructingthe induction of bone and regulating theexpression of osteogenic gene transcriptsand their translation products initiatingbone formation as a function of the struc-ture [66–68].
Acknowledgments
This work is supported by grants from theSouth African Medical Research Council,the University of the Witwatersrand, Jo-hannesburg, the National Research Foun-dation and by ad hoc grants from the BoneResearch Unit. We thank the Central An-imal Services of the University for thecontinuous help with primate experimen-tation.
References
1. J. M. Wozney, V. Rosen, A. J. Celeste et al.,Science 1988, 242, 1528–1534.
2. J. M. Wozney, Mol. Reprod. Dev. 1992, 32,160–167.
3. A. H. Reddi, Curr. Opin. Cell Biol. 1992, 4,850–855.
4. A. H. Reddi, Cytokine Growth Factor Rev.1997, 8, 11–20.
5. A. H. Reddi, J. Cell Biochem. 1994, 56,192–195.
6. A. H. Reddi, Curr. Opin. Cell Biol. 1994, 4,737–744.
7. A. H. Reddi, Cell 1997, 159–161.8. A. H. Reddi, Nat. Biotechnol. 1998, 16,
247–252.

296 16.6 Conclusion
9. A. H. Reddi, Biochem. Soc. Trans. 2000, 28,345–349.
10. A. H. Reddi, Tissue Eng. 2000, 6, 351–359.11. U. Ripamonti, A. H. Reddi, Adv. Plast.
Reconstr. Surg. 1995, 11, 47–65.12. U. Ripamonti, S. Vukicevic, South Afr. J. Sci.
1995, 91, 277–280.13. U. Ripamonti, South Afr. J. Bone Joint Surg.
1997, 7, 21–32.14. U. Ripamonti, N. Duneas, Plast. Reconstr.
Surg. 1998, 101, 227–239.15. U. Ripamonti, B. Van Den Heever, J. Crooks
et al., J. Bone Miner. Res. 2000, 15,1798–1809.
16. U. Ripamonti, L. N. Ramoshebi, T. Matsabaet al., J. Bone Joint Surg. Am. 2001, 83-A,S1-166–127.
17. G. E. Friedlaender, C. R. Perry, J. D. Cole, J.Bone Joint Surg. Am. 2001, 83-A, S151–S158.
18. E. H. Groeneveld, E. H. Burger, Eur. J.Endocrinol. 2000, 142, 9–21.
19. H. G. Moghadam, M. R. Urist, G. K. Sandoret al., J. Craniofac. Surg. 2001, 12, 119–127.
20. A. M. Turing, Philos. Trans. R. Soc. London1952, 237, 37–72.
21. P. J. Boyne, J. Bone Joint Surg. Am. 2001,83-A, S146–S150.
22. U. M. Wikesjo, R. G. Sorennsen, J. M.Wozney, J. Bone Joint Surg. Am. 2001, 83-A,S136–S145.
23. U. Ripamonti, J. Crooks, A. N. Kirkbride,South Afr. J. Sci. 1999, 95, 335–343.
24. U. Ripamonti, Smart biomaterials withintrinsic osteoinductivity: geometric controlof bone differentiation in Bone Engineering(Ed.: J. E. Davis), EM2 Corporation, Toronto,Canada, 2000, pp. 215–222.
25. U. Ripamonti, Osteogenic proteins of thetransforming growth factor-ß superfamily inEncyclopedia of Hormones (Eds.: H. L. Henry,A. W. Norman), Academic Press, 2003,pp. 80–86.
26. U. Ripamonti, B. Van Den Heever, T. K.Sampath et al., Growth Factors 1996, 13,273–289.
27. T. K. Sampath, A. H. Reddi, Proc. Natl. Acad.Sci. U. S. A. 1981, 78, 7599–7603.
28. P. V. Haushka, A. E. Mavrakos, M. D. Iafratiet al., J. Biol. Chem. 1996, 261, 12665–12674.
29. U. Ripamonti, A. Ma, N. Cunningham et al.,Matrix 1992, 12, 369–380.
30. U. Ripamonti, A. Ma, N. Cunningham et al.,Plast. Reconstr. Surg. 1993, 91, 27–36.
31. C. Ferretti, U. Ripamonti, J. Craniofac. Surg.2002, 13, 434–444.
32. A. H. Reddi, N. S. Cunningham, Biomateri-als 1990, 11, 33–34.
33. U. Ripamonti, C. Ferretti, Mandibularreconstruction using naturally derived bonemorphogenetic proteins: a clinical trialreport in Advances in Skeletal reconstructionUsing Bone Morphogenetic Proteins (Ed.:T. S. Lindholm), World Scientific PublishingCo., 2002, pp. 277–289.
34. E. A. Wang, V. Rosen, J. S. D’alessandroet al., Proc. Natl. Acad. Sci. U. S. A. 1990,87, 2220–2224.
35. R. G. Hammonds, R. Schwall, A. Dudleyet al., Mol. Endocrinol. 1991, 5, 149–155.
36. T. K. Sampath, J. C. Maliakal, P. V. Haushkaet al., J. Biol. Chem. 1992, 267, 20352–20362.
37. G. Thomadakis, L. N. Ramoshebi, J. Crookset al., Eur. J. Oral Sci. 1999, 107, 368–377.
38. K. Staehling-Hampton, P. D. Jackson, M. J.Clark et al., Cell Growth Differ. 1994, 5,585–593.
39. K. Miyazono, Bone 1999, 25, 91–93.40. K. Miyazono, P. Ten Dijke, C. H. Heldin,
Adv. Immunol. 2000, 75, 115–157.41. D. Chen, M. A. Harris, G. Rossini et al.,
Calcif. Tissue Int. 1997, 60, 238–290.42. Y. Honda, R. Kniutsen, D. D. Strong et al.,
Calcif. Tissue Int. 1997, 60, 297–301.43. U. Ripamonti, J. Crooks, J. Teare et al., South
Afr. J. Sci. 2002, 98, 361–368.44. U. Ripamonti, A. H. Reddi, Crit. Rev. Oral
Biol. Med. 1997, 8, 154–163.45. U. Ripamonti, J. R. Tasker, U. Ripamonti
et al., Curr. Pharm. Biotechnol. 2000, 1,47–55.
46. U. Ripamonti, M. Heliotis, van den Heeveret al., J. Periodontal Res. 1994, 29, 439–445.
47. U. Ripamonti, M. Heliotis, D. C. Ruegeret al., Arch. Oral Biol. 1996, 41, 121–126.
48. U. Ripamonti, Induction of cementogenesisand periodontal ligament regenerationby bone morphogenetic proteins inBone Morphogenetic Proteins: BiologicalCharacteristics and Reconstructive Repair,Chapter 17 (Ed.: T. S. Lindholm), R.G.Landes Company and Academic Press, 1996,pp. 189–198.
49. M. R. Urist, Science 1965, 150, 893–899.50. A. H. Reddi, C. B. Huggins, Proc. Natl. Acad.
Sci. 1972, 69, 1601–1605.

Sculpturing the Architecture of Mineralized Tissues 297
51. A. B. Roberts, M. B. Sporn, R. K. Assoianet al., Proc. Natl. Acad. Sci. U. S. A. 1986,83, 4167–4171.
52. T. Matsaba, L. N. Ramoshebi, J. Crookset al., Growth Factors 2001, 19, 73–86.
53. U. Ripamonti, C. Bosch, B. Van Den Heeveret al., J. Bone Miner. Res. 1996, 11, 938–945.
54. U. Ripamonti, N. Duneas, B. Van DenHeever et al., J. Bone Miner. Res. 1997, 12,1584–1595.
55. N. Duneas, J. Crooks, U. Ripamonti, GrowthFactors 1998, 15, 259–277.
56. U. Ripamonti, J. Crooks, T. Matsaba et al.,Growth Factors 2000, 17, 269–285.
57. U. Ripamonti, J. Teare, T. Matsaba et al.,Site, tissue and organ specificityof endochondral bone induction andmorphogenesis by TGF-beta isoforms in theprimate Papio ursinus, Proceedings of the 2001FASEB Summer Research Conference, Tucson,Arizona, USA, 2001.
58. U. Ripamonti, J. R. Tasker, Bone inductionby transforming growth factor-ß in theprimate and synergistic interaction withbone morphogenetic proteins in Advancesin Skeletal Reconstruction Using BoneMorphogenetic Proteins (Ed.: T. S. Lindholm),World Scientific Publishing Co., Singapore,2002, pp. 79–95.
59. T. Imamura, M. Takase, A. Nishihara et al.,Nature 1997, 389, 622–626.
60. A. Nakao, M. Afrakhte, A. Moren et al.,Nature 1997, 389, 631–635.
61. U. Ripamonti, J. Teare, T. Matsaba et al.,Site, tissue and organ specificityof endochondral bone induction and
morphogenesis by TGF-ß isoforms in theprimate Papio ursinus, FASEB SummerConference: The TGF-ß Superfamily: Signalingand Development, Tucson, Arizona, USA, July7–12, 2001.
62. U. Ripamonti, B. Van Den Heever, M. Helio-tis et al., South Afr. J. Sci. 2002, 98, 429–433.
63. H. K. Kleinman, M. L. McGarvey, J. R.Hassell et al., Biochemistry 1986, 25,312–318.
64. V. M. Paralkar, A. K. N. Nandekar, R. H.Pointer et al., J. Biol. Chem. 1990, 265,17281–17284.
65. U. Ripamonti, L. N. Ramoshebi, J. Pattonet al., Soluble signals and insolublesubstrata: Novel molecular cues instructingthe induction of bone in The Skeleton (Eds.:E. J. Massaro, J. M. Rogers), Humana Press,Totowa, NJ, 2003, in press.
66. U. Ripamonti, T. Matsaba, J. Teare et al., In-trinsic osteoinductivity by smart biomimeticmatrices, Abstract in the 4th InternationalConference on Bone Morphogenetic Proteins,Sacramento, USA, October 17–21, 2002.
67. U. Ripamonti, J. Teare, T. Matsaba et al.,Tissue engineering by bone morphogeneticproteins, Abstract in the 4th InternationalConference on Bone Morphogenetic Proteins,Sacramento, USA, October 17–21, 2002.
68. U. Ripamonti, J. Patton, T. Matsaba et al.,Endochondral bone induction by TGF-ß proteins: site and tissue specificityof induction in the primate, Abstract inthe 4th International Conference on BoneMorphogenetic Proteins, Sacramento, USA,October 17–21, 2002.

299
Subject Index
aA. coloradensis 13A. mediterranei 14A. orientalis 14A. teicomyceticus 14AAV (Adeno-associated viral) 238abciximab 147Accela 253‘‘accommodation’’ 273accuracy 110acetoin 24acetylation 39, 104N-acetylglucosamine 269β-1,4-N-acetylglucosaminyltransferase III 269acremonium chrysogenum
Cephalosporin C 26acromegaly 206actinomycete 9actinorhodin 11acute vascular rejection 271acyl carrier protein 18ADCC (antibody-dependent cell-mediated
cytotoxicity) 108adeno-associated virus 234adenoviral vector 237adenovirus 233adjuvant 69, 79, 84adsorption 174ADMA/tox 7aggregate 141aggregation 122agrobacterium 49Agrobacterium tumefaciens 28AIDS 5, 120, 239, 249AIDS vaccine 63airlift reactor 36alanin racemase 26albumin 39, 150alcohol fermentation 23alemtuzumab 225
algae 52alicaforsen 161allergic 120allometry 157allotransplantation 265alpha-chymotrypsin 178alphavirus 234
RNA replicon 81alveoli 182Alzheimer 3ambirix 206Amgen 120Amgen’s Aranesp 130amino- 22aminooxy- 22amphotericin 13Amycolatopsis (A.) orientalis 10analytical method 141animals 190anion exchange chromatography (AEX) 245anthocyanin 29anthraquinone 29anti-idiotype antibody 63anti-protein antibody 114antibacterial 13antibiotic resistance gene 80antibiotics 25antibody 108antidonor antibody 273antifoam agent 104antigen 59, 79antigen presentation 82antigen-presenting cells (APCs) 82antigenecity 174antiinfectives market 10antiparasitic 10antisense oligonucleotide 147antitrust laws 197antitumor 13application route 175
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H.Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

300 Subject Index
aps (amplification-promoting sequence) 47aquayamycin 18, 21Arabidopsis 53Aranesp 130arginine 258arteriosclerosis 28‘‘artificial’’ gene 20Arzneimittelgesetz 250ascomycin 13Asialoglycoprotein 236Aspergillus 25Aspergillus nidulans 26Aspergillus niger 24Aspergillus terreus 25assay calibration 110asthma 3atrial natriuretic peptide (ANP) 152Atropa belladonna 28, 29Attenuated vaccine 67, 95Aureofaciens 10autoimmune reaction 94avermectin 13, 15avian vaccinia 234avilamycin 13, 17avonex 128, 136avoparcin 13azido- 22
bB-cells 113, 116baby hamster kidney (BHK) 131Bacillus amyloliquefaciens 24Bacillus subtilis 24Bacillus thuringensis 37bacteria 13bacterial origin of replication 80banana 53banning patents 196benefit sharing 197berberine 29betaferon 128, 136BfArM 204bialaphos 13bicinchoninic 105binding assays 106bioadhesion 179bioanalytical 149
assay 103method 7
bioassay 105, 139, 141, 150, 292bioavailability 37, 151, 154, 175, 179bioballistics 253biochemical assay 108
biodistribution 92, 151, 174bioengineered tissues 190bioequivalence 37, 109, 142bioferon 137biogen 120biogenerics 119
definition 121patent 62, 124, 126, 128registration 71, 97, 122regulatory 122
BioGeneriX 119bioinformatics 7biological standards 141biomanufacturing 36biomedicine 195biomimetic matrix 281biopharmaceuticals 35, 119, 120BioRad 254bioreactors 36biosynthesis 11biotech production 9biotechnology 3, 195biotechnology industry
legal 187biotherapeutics 112biuret 105bleomycin 13blockbusters 76, 119, 147blocking patents 195bone 281booster vaccination 94borreliosis 65Boston consulting group 7Bradford 105brain tumors 241BRCA1 196BRCA2 196Bt corn 372,3-butanediol 24
cCa-phosphate-precipitation 252CaCo-2 260cadaverin 29calcitonin 156calibration 110cancer 3candicidin 13candidate 68capillary zone electrophoresis 139CAPLA 226capture step 244carbohydrate analysis 139carbon flux 15

Subject Index 301
N-carboxyanhydrid 258γ -carboxylation 104carboxypeptidases 178carcinogenicity 69caspases 83Catharanthus roseus 24cationic liposomes 254
lipoplex 254cauliflower 53CBER 207CD20+ 220CD34 235, 242CD4 64, 274CD59 270CD8 274CDER 207CDRH 207CDC (complement-dependent cytotoxicity)
108cell lines 190cell wall synthesis 13center for biologics evaluation and research
(CBER) 124centralized procedure 204, 205, 243centrifugation 244cetrorelix 169chemical vector 254, 256, 258, 260Chinese hamster ovary (CHO) cells 130
expression 35chitin 260chitin synthase 13chitosan 181, 260chloramphenicol 13chlortetracycline 13cholesterol 5, 25chromosomal integration 235circular dichroism 139cirrhotic liver failure 276clavulanic acid 13clinical development 138, 231cloning 5, 194CMV-retinitis 251coagulation 120coagulation factor 135codon usage 50Coleus blumei 29collagenic material 287colon 177colony-stimulating factor 131Committee for Orphan Medicinal Products
(COMP) 203Committee for Proprietary Medicinal Products
(CPMP) 123, 203, 204
Committee for Veterinary Medicinal Products(CVMP) 203, 204
complement activation 270, 273complex PK/PD model 169Contract Research Organisations (CROs) 129Coomassie 49copegus 136Coptis japonica 29copyright 187corapporteur 204corn 37COS-1 260cosmetics 206Creutzfeld–Jacob 120Crohn’s disease 132crop cultivation 35cross-presentation 82cross-reactivity 106CSF 225CVM 207cyclohexamide 13cyclosporin 25cydosporine A 12cystic fibrosis 183, 250cytochrome P450 17, 151cytokine genes 241cytokines 63, 92, 124cytomegalovirus 3, 80, 251cytotoxicity 150
ddactinomycin 13daptomycin 13database 7daunorubicin 13DC-chol 255deamidation 104, 113, 141deimmunisation 11512b-desrhodinosyl-urdamycin G 21delivery system 235demographics 6denaturation 174dendrimers 259, 260dendritic cells 82dengue 65denucleated 261deoxy- 22depot effect 176desmin 81dextran 181dextrin 23diabetes 206diabetes mellitus 133

302 Subject Index
diagnostics 3diarrhea 30dimers 141directives 123, 202distribution 148, 151
of benefits 197disulphide 104DMRIE 255DNA 85, 190, 233
genomic DNA 89naked nucleic acid 234oligonucleotides 147pharmacokinetics 162plasmid 233therapeutic 112
DNA binding 13DNA cloning 191DNA delivery 85DNA gyrase 13DNA intercalation 13DNA microchips 190DNA technology 3DNA transfer 11DNA vaccine 63, 79, 87, 253
construction 79, 80, 82, 84expression plasmids 80
DNase I 261DOPE 255doramectin 15dose-concentration-effect relationship 148DOSPA 255DOTAP 255double chamber syringe 177down syndrome 250downstream 131
processing 48, 67, 244doxorubicin 12, 13drug absorption 148drug delivery 39, 151, 175, 253, 261drug development 6drug discovery 9drug stability 106Duchenne muscular dystrophy 250duodenum 177dynepo 130
eE. coli
K12 245efficacy 96egalitarianism 197electroporation 83, 252, 253elimination 153
ELISA 108Embryonenschutz 249EMEA 41, 98, 123, 201, 243
annual report 204210-day window 205legislation 201organizational structure 207
endocytosis 155endogenous retrovirus 275endoplasmatic reticulum 45endothelial cell activation 273endotoxin 54, 104, 140, 244engerix 127engerix-B 134‘‘engineered tissues’’
stem cell transplant 265enkephalin 153enteral application 177entrepreneurs 188environment 244epitope mapping 106EPO see erythropoietinepoetin alfa 126epoetin beta 126epogen 147Epratuzumab 225Eprex 126, 130Erwinia uredovora 28Erypo 126erythromycin A 12, 13, 17erythronolide macrolactone 17erythropoietin (EPO) 35, 112, 126, 130, 147,
167, 173Escherichia coli 30, 35, 80, 245ethical 187, 192, 194, 196
issues 37review 197
eucaryotic cells 9euphorbia milli 29Eurifel 206European commission 205, 243European legislation 250European pharmacopoeia 131European technical experts 203ex vivo 233excretion 148ExGen 500 259exonuclease 162exopeptidase 155exposure-response relationship 148expression system 37expression vector 51, 139, 232extraction 54

Subject Index 303
ffab molecules 107factor IX 239factor VIII 120, 127factor VIII (FVIII) 135FD&C 207FDA 3, 41, 98, 123, 201, 206, 243
guideline 110fermentation 9, 30, 36, 51, 67, 140fermenter 245ferritin 27filgrastim 126filtration 244financial compensation 198FKBP12 13fluorescence 108fluoroimmunoassay 110fomivirsen 3foot-and-mouth-disease (FMD) 66formivirsen 251formulation 122, 125, 139, 173fowl pest 37free cell suspension 30functional food 28fungi 104
gα1,3-galactosyltransferase 268α1,3-galactosyltransferase-deficient pigs 272β-galactosidase 24β-glucanase 24G-CSF 131Ganciclovir 239gastrointestinal elimination 154gavibamycin 14gel filtration 244, 245Geldanamycin 13Gen 127Gen H-B-Vax 134gene clusters 14gene delivery 266gene guns 82, 86, 236, 253gene knockout 50gene pharming 8gene therapy 147gene transfer 231
methods 252Genentech 120generic pharmaceuticals 121
criteria 121generics 122gene 79genetic drift 43
genetic stability 51genetic therapy 5genetically modified bacteria 190genomic DNA 89genomics 6, 9gentamicin 13Gentechnikgesetz 250germ-line therapy 243GFP (green fluorescent protein) 47GI-passage 175GI-tract 175glomerular filtration 155glucocerebrosidase 39glycoalkaloid 27glycocalix 177glycoprotein 236glycorandomization 20glycosylation 39, 40, 45, 104, 105, 157
pattern 113GM-CSF 241GMP 35Goldner’s trichrome 284Golgi apparatus 45good manufacturing practice 88, 243graft 239host disease 239granocyte 126granulocyte colonystimulating factor (G-
CSF) 126granulocyte-macrophage colonystimulating
factor (GM-CSF) 126gravimetry 105greenhouses 35guanin-nucleotide-binding protein 257
hH 127ε-L-histidine 258H-transferase 268Hafnia alvei 29hairy roots 30half-life 174Hatch–Waxman act 129hDAF factor 271heavy metal ion 104Heidelberger capsule 180helios 254helixate 127, 135hemophilia A 135hemophilia B 239hemorrhage 267hepatic elimination 156hepatitis 30, 86, 97, 120

304 Subject Index
hepatitis A 134hepatitis B vaccine 134hepatitis C 206hepatocytes 156, 266heptapeptide 23herbicide-resistance 43herbicides 10herceptin 147, 226herpes 261herpes simplex virus 234Hevea brasiliensis 29hGH see human growth hormonehigh throughput 17, 244hill-coefficient 164histocompatibility complex class I 81HIV 242HIV-1-TAT 257HMG-CoA 25HMG-CoA-reductase 29HO 21homologous recombination 232horizontal transmission 43host cell proteins 244HPLC 139hTGF-β2 288HuCAL 113humalog 134human antimouse immunoglobulin antibody
(HAMA) 161human dignity 193human genome project 254human glycosylation 45human granulocyte macrophage-colony
stimulating factor (rhGM-CSF) 113human growth hormone (hGH) 120, 127,
133human leader sequence 50humatrope 127humidity 106Huminsulin 127, 133humira 113humulin 4, 120hydrodynamic pump 36hydrophilicity 115hydrophobicity 156(3S)-hydroxy-3-methylglutaryl-CoA 25hydroxyapatites 293hyoscyamin-6ß-hydroxylase 28, 29Hyoscyamus niger 28, 29
iICH 149
guidelines 138, 210
Q5A 210Q5B 210Q5C 210Q5D 210Q6B 210S6 210
identity 54, 69, 103, 105IFN alfacon 1 135IFN-α 135IFN-β 135IFN-γ 135IGF-1 (insulin-like growth factor) 153IgG1 214ileum 177immune reaction 120immune system 45, 224immunoassay 105, 149immunogenic epitope 115immunogenicity 69, 99, 107, 111, 112, 114,
116, 118, 120, 123, 160, 253immunoglobulins 120immunohistochemistry 106‘‘immunoregulation’’ 274immunostimulant 224immunosuppressive drug 25immunotoxicology 95importin-ß 257impurity 54, 69, 104
elimination 89process-related 104product-related 89
impurity standard 105in vivo 233in-study validation 111incidence 112IND 213inductos 206infectivity 253infergen 128, 136inflammatory cell invasion 94innovator 123insertional mutagenesis 98insulin 120, 127, 133, 173, 179insulin actrapid 127insulin glargine 134insulin lispro 134insulin-like hypoglycemia 153insuman 127intellectual property law 192interferons 124, 125
interferon alpha 128interferon beta 128interferons (IFN) 135
interleukin-2 153, 225

Subject Index 305
international conference on harmonisation(ICH) 129
international nonproprietary name (INN) 122international patent law 191international regulatory harmonization 209,
210intramuscular 113intron A 80, 128, 136investigational new drug (IND) 208investments 7, 72investors 188ion exchange 175iontophoresis 253ischemia 272isoelectric focusing 175isopenicillin 26isothermal titration calorimetry 107IX 120Ixodes scapularis 87
jjejunum 177Jesse Gelsinger 249jetilizer 181
kkanamycin 13ketoreductase 18Kjeldahl 105Klebsiella pneumoniae 24‘‘knocking out’’ genes 266kogenate 127, 135
llactoferrin 27β-lactams 26lactose 183laminin 292landomycin 18Langerhans cells 82lantus 134large-scale production 120LDL 5leachates 104lenograstim 126lentiviral vector 234, 237lettuce 27leucomax 126leukapharesis 235leukemia 236leukine 132leuprorelin 151
LEXCOR panel 218license 195licensing 71, 97
fees 195limit of quantitation 111Lincomycin 13linearity 110, 111lipid-to-DNA ratio 256liposomes 236, 252, 253
activity analysis 255general toxicity 255metabolic degradation 256structure analysis 255
Lithospermum erythrorhizon 29liver first pass effect 182long terminal repeat 233lovastatin 25Lowry 105LT-B 30luciferase 108luminescence 108lupus-prone 95lycopene 28lycopene ß-cyclase 28lymphocytes 84lymphokine 25lyophilization 67lyophilized 176lysin-decarboxylase 29lysine 258lysosomes 155
mMabThera 213maize 27major histocompatibility complex (MHC) 115malaria 5, 65MALDI (matrix assisted laser desorption
ionization) 105, 150MALDI-TOF (matrix assisted laser desorption
ionization time of flight) 51, 139manufacture 244manufacturers (CMOs) 129market authorization application (MAA) 123marketing authorization 205mass spectrometry (MS) 104mass spectroscopic analysis 175master 67master cell bank 139matrigel 282measles 63mechanical 36media components 104

306 Subject Index
medicinal plants 197medicines 6
control agency 204membrane 13metabolic engineering 15metabolism 148, 153metabolite 156metagenome 16methicillin 16methionine oxidation 141methoxy- 22mevastatin 25MHC 82
antigens 274ligand 115
Michaelis–Menten kinetics 156Michaelis–Menten equation 36microencapsulation 179microinjection 252Micromonospora purpurea 13microvilli 177milbemycin 13mithramycin 13, 18mitomycin C 13moenomycin 13molecular farming 30Monascus 25monensin 13monoclonal antibodies (Mabs) 35, 103, 213Morinda citrifolia 29moroctocog alfa 127, 135morphogen 282, 292moss 50
bioreactor 51mucoviscidosis 249
adenosine-deaminase (ADA) 249multiple sclerosis 3mumps 63mutual recognition 205mycoplasma 104mylotarg 226
nnanoparticle 253nanorobotic devices 8Narcissus pseudonarcissus 28nasal application 180natamycin 13natural antioxidant 28natural killer 266natural products 10NDA 208nebulizer 183
neomycin 13neopterin 169neorecormon 126neulasta 132neupogen 126, 132neupopeg 206neutral-protamin-Hagedorn (NPH) formula-
tion 133neutropenia 132, 206new drug application 208Nicotiana tabacum 29, 30, 39nicotin 29nikkomycin 13NK cells 272non-Hodgkin’s lymphoma 48, 213noninvasive routes 176nonspecific background (NSB) 114nonviral vector 235, 236norditropin 127novobiocin 13nuclear localization 257nuclease 162nucleic acid 232nucleotidyl-transferase 22nutraceutical 27nutritional supplement 206nystatin 13
ooctocog alfa 127octreotid 153oligonucleotide 3, 84, 161, 251, 254
pharmacokinetics 162oligopeptide (N-142-160-C) 66oncogene 88oncogene LMO2 237oncogenic sequence 95onkomouse 4open circle plasmid 244opsonization 154oral application 177, 178organs 190, 194ornithin-decarboxylase 29ornithine transcarbamylase 238‘‘orphan’’ gene cluster 17orthosomycins 17OSMAC 16osteoinduction 294osteoinductivity 290ovarian cancer 196OVCAR-3 260oxazolidinone antibiotic linezolid 16oxidation 104oxytetracycline 13

Subject Index 307
pP. stipitis 24p53 241packaging signal 233palitaxel 29pancreas 120pancreas transplantation 265pancreatic islet 265Papio ursinus 282paracellular transport 182parasitology 63parenteral application 176parenteral injection 85particle-bombardment 252patent 62, 187
Diamond v. Chakrabarty 190human ingenuity 190Moore v. Regents of University of Califor-
nia 194nonobviousness 189originality 189requirements 189subject matter 189supreme court 190types of patents 190usefulness 189
patent application 188patent infringement 125, 191patent law 187, 188, 190patent system 188patent thicket 195pathogenesis
acute vascular rejection 271PCR 4, 81pDMAEMA 260peanut 27PEG 258PEG-Intron 136pegasys 136, 206PEGylated filgrastim 132PEI/DNA 259penicillin 4, 16, 88Penicillium chrysogenum 26Penicillium citrinum 25peptidase 151, 154peptide mapping 104, 139peptides 173Perilla frutescens 29personalized 6
genetic profiling 5medicine 147
pertussis 39Peyer’s patches 177phagocytosis 154
pharmaceutical biotechnology 265pharmaceutical legislation 202pharmaceutical research and manufacturers of
America 3pharmaceuticals 3, 5pharmacodynamics 37, 91, 147pharmacogenetic 6pharmacokinetics 37, 109, 139, 147pharmacovigilance 98, 202phase I 71, 142phase II 71, 142phase III 70, 71, 142phase IV 71phosphorylation 39, 104photobioreactors 52, 53Physcomitrella patens 50physical gene transfer systems 253PK/PD modeling 148, 163plant expression 37, 38plants 190plasmids 80, 88, 256
consistency 90identity 90impurities 90potency assay 90purity 90stability 90
pneumatical 36point mutation 232policy issues 192, 194, 196poliomyelitis 63polishing 67polishing step 244poly(2-dimethylamino)ethylmethacrylate 260poly-L-arginine 258poly-L-lysine 258polyclonal antibody 104polyethyleneimine 259polyketides 10, 18polyketide synthases 20polymer particles (polyplex) 257polypeptides 120polysaccharides 152porcilis porcoli diluvac forte 206porcine endothelial cells 272porcine hepatocytes 276Porphyromonas gingivalis 286posttranslational modifications 45postmarketing clinical trials 71potatoes 27potency 54, 103
determination 107, 109powderject 253poxvirus vector 238

308 Subject Index
pravastatin 25pre-study validation 110preauthorization 203precipitation 174precision 110preclinical 138
safety 93precursor pool 168pregnancy contracts 194preservatives 104primary 15
patents 125prions 69, 104pristinamycins 13, 17procaryotic 9process development 138procrit 131, 147(pro)drug 180progenitor cells 131promoter 44, 80protease 151, 154, 178protein 3, 173
quantification 105recombinant 104, 106, 108
protein binding 152protein degradation 174protein drugs 119protein glycosylation 45protein C 275proteolysis 153, 179proteomics 173proteqflu 206proton-sponge 259protoplasts 51provitamin A 28pulmonary application 182pulmozyme 183purity 69, 103, 104
impurity 103pylorus 178pyrogens 140pyroglutamate 104
qquality 90quantity 54, 103
rR&D 6radioactivity 150radioimmunoassay (RIA) 108radioimmunotherapy 213radioisotopes 105
ramoplanin 14rapamycin 14, 20rapporteur 204rebif 128, 136‘‘reach through’’ licenses 195rebound phenomena 169receptor-mediated elimination 156recombinant 3recombinant DNA 79recombinant protein chemistry 3recombinant proteins 35, 103, 119recombinate 127, 135recombivax 4reconstitution 177rectum 177red-cell aplasia (PRCA) 131refacto 127, 135registration 97
dossier 70regulation of the complement system 270regulations 202regulatory authority 204renal elimination 154, 155replicase 81restenosis 241resveratrol 27, 29retroviral vector particle 233reverse-phase HPLC 104rhDNase 183rheopro 147Rhizopus oryzae lactate dehydrogenase 24rhizosecretion 48ribosome 13rice 27rifamycin 14rituxan 213rituximab 213
application 226approval 225B-cell recovery 222clinical development 215combination 223development 221EMEA 225first phase I study 219future applications 226, 228IDEC 219indications 226interferon 224mechanism of action 215optimizing the dose and schedule 222pharmacokinetic 222phase I and I/II clinical trials 219phase I/II 216

Subject Index 309
phase II 216, 221re-treatment study 221regulatory approval 214, 216, 218, 220,
222, 224regulatory dossiers 225
RNA 190, 234RNA polymerase II promoter 81RNA sequencing method 191Roche–Bolar 129roferon A 128, 136rosmarinic acid 29rotavirus 69royalties 197rubella 63rurioctocog alfa 127
sS. albus 14S. ambofaciens 14S. argillaceus 13, 18S. aureofaciens 13, 14S. avermitilis 13, 15S. avermitlis 15S. carbophilus 25S. cattleya 14S. cinnamonensis 13S. clavuligerus 13S. coelicolor 15S. cyanogenus 18S. fradiae 13, 14, 18S. ghanaensis 13S. griseus 13, 14S. hygroscopicus 13, 14S. kanamyceticus 13S. lavendulae 13S. lincolnensis 13S. nataensis 13S. niveus 13S. nodosus 13S. noursei 13S. parvulus 13S. peucetius 10, 13S. pristinaespiralis 13, 17S. rimosus 13S. roseosporus 13S. staurosporeus 14S. tendae 13S. venezuelae 13S. verticillus 13S. virginiae 14S. viridochromogenes 13Sac. spinosa 14saccharomyces 132, 138
saccharomyces cerevisiae 23, 24, 26, 29Saccharopolyspora (Sac.) erythraea 10, 13safety 96
pharmacology 139saizen 127salicin 37salinomycin 14salmonella 95Salmonella enterica 22sargramostim 126SCID-X1 237, 239
newborns 240scopolamin 29SDS-polyacrylamid gel 139
electrophoresis 104secondary metabolism 15secondary metabolite 10secondary patent 125selectivity 110semiconductor industry 196Semliki forest virus 81, 234serum concentration-time course 165serum half-life 107severe sepsis 206shelf-life specifications 106Shigella 95shikonin 29siderophores 15signal pathway 285Sindbis virus 81size-exclusion chromatography 104size-exclusion HPLC 139slavery 194smallpox virus 234soil-DNA 16somatic gene therapy 231, 249
DNA 232guideline 242manufacture 242, 244quality 242regulatory aspect 242, 244
somatotropin 153, 182somatropin 133somavert 206specificity 54, 103, 106spinosyn 14spiramycin 14spray-drying 183stability 54, 67, 106, 110, 111, 176
storage 106stabilization 174standardization 7, 104starburst 259starch-decomposing enzymes 23

310 Subject Index
statins 25staurosporin 14stem cells 5sterility 106sterols 29stilbene-synthase 29stratum corneum 254Streptomyces coelicolor 11, 15Streptomyces 10, 11, 14, 15streptomycin 14strictosidine 24strictosidine beta-glucosidase 24strictosidine synthase 24string-of-beads 81subcutaneous applications 113sulfation 104sulfoxidation 104superpathogens 43supplementary protection certificates (SPCs)
124surfactant 182SWOT 39syncytial virus 65synercid 18synthetic genes 82
tT-cell epitopes 81T-lymphocytes 79, 152tacrolimus 14Taxus 29teicoplanin 14, 23tenecteplase 152terminator 80tetanus 39tetracycline 12, 14, 252TGF-β 281TGF-β superfamily 282, 294thalictrum minor 29therapeutic 3
genes 235, 236, 250protein 104
thienamycin 14thiosugar phosphates 22thrombomodulin 275thrombopoietin 115thrombosis 28, 160, 267, 272thymic transplantation 274thymidin kinase 239tight junction 177tissue 281TMV-based vectors 48tobacco mosaic virus 43
toiletries 206Tolypocladium inflatum 25tomatoes 44toxic substances 244toxicity 7, 37, 94, 174toxicology 139toxin conjugates 95toxins 95, 105trade related aspects of intellectual properties
(TRIPS) 192trade secrets 187trademarks 187transcription 80, 104transcytosis 180transdermal route 253transduction 237, 253transfection 139, 233, 236, 253, 259, 267transferrin 236transferrin receptor 167transgene insertion 50transgenic organisms 8transgenic plants 28transplantation 83trastuzumab 147trecovirsen 161trisomy 250trypanosoma 65trypsin 178TSE (transmissible spongiform
encephalopathies) 40tubular reabsorption 155tumor gene therapy 240tumorigenicity 94twinrix 134tylosin 14type I-cell 182
uUDP derivatives 22upstream 131
process 67urdamycin 18, 21urokinase 39US Department of Health and Human
Services 206utilitarianism 197
vvaccination 206, 241vaccine 39, 59, 68, 79, 134
herpes simplex virus 61antigen candidate 60B-cell lymphoma 61

Subject Index 311
borrelia/Lyme disease 61CEA-tumors 61clinical development 70cytomegalovirus 61delivery of DNA vaccines 84economic aspects 72helicobacter pylori 61hepatitis C virus 61HIV/AIDS 61HRSV 61impurity 89infectious agents 61leishmania 61licensing 97life span 75malaria 61melanoma 61parainfluenza 61patent 62pharmacodynamic 91pharmacokinetic 91preclinical development 67preclinical safety 90, 92, 94predevelopment 60production facility 70prostata carcinoma 61recombinant technique 63registration 71, 97research concept 59, 60rotavirus 61salmonella 61schistosoma 61screening 85, 86shigella 61streptococcus 61success rates 62, 74toxicology 69toxoplasma 61trypanosoma 61
vaccine antigens 66protectivity 66
vaccine candidates 64, 81vaccine development 61, 72vaccine production 3vaccinia ancara 234validation 7, 104, 109, 110, 139vancomycin 12, 14, 23vascular invasion 294vasopressin 156vax 127
vector 232, 250, 266velosulin 206vertical transmission 43veterinary vaccine 62viral reinfection 276viral vector particle 232viral vectors 48, 236, 253virginiamycin 14virus replication 236virus-like particle 82viruses 104, 190viscosity 26vitamin C 28vitamin E 28Vitis sp. 29Vitis vinifera 29vitravene 3, 251
wwestern blot (WB) 139WHO 16Willebrand factor 152willow bark 37working cell banks 67, 139working seeds 67world trade organization (WTO) 192
xxenogeneic cells 274xenografts 266xenoreactive antibody 267‘‘xenoreactive’’ T-cells 273xenotransplantation 5, 191, 265, 267, 268
hyperacute rejection 267, 268immunologic responses 266pig 266
xigiris 206xylosyl transferase 51
yyeast genetics 23
zzevalin 225zomacton 127zona occludens toxin 179zoonosis 275

xv
Color Plates
Fig. 2.1 Photography of a sporulated Streptomyces strain growing on solid medium. The blue dropsindicate the production of an antibiotic (aromatic polyketide).
Pharmaceutical Biotechnology, Drug Discovery and Clinical Applications. Edited by O. Kayser and R.H. Muller.Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.ISBN: 3-527-30554-8

xvi Color Plates
Maj
or te
chno
logy
Mam
mal
ian
(CH
O)
cells
Tra
nsge
nic
mam
mal
milk
Tra
nsge
nic
chic
ken
eggs
Tra
nsge
nic
plan
ts
Cro
ptec
h(B
lack
sbur
g, V
A)
(San
Die
go, C
A)
(Ow
ensb
oro,
KY
)
(Cle
rmon
t-F
erra
nd, F
ranc
e)
(Col
lege
Sta
tion,
TX
)
Epi
cyte
Larg
e S
cale
Bio
logy
Mer
iste
m T
hera
peut
ics
Pro
dige
ne
Avi
geni
cs(A
then
s, G
A)
(Bur
linga
me,
CA
)
(Shr
ewsb
ury,
MA
)
(Pla
ntat
ion,
FL)
(Ann
Arb
or, M
I)
(Nan
tes,
Fra
nce)
Orig
en T
hera
peut
ics
Tra
nXen
oGen
Vira
gen
Gen
eWor
ks
Viv
alis
GT
C B
ioth
erap
eutic
s
PP
L T
hera
peut
ics
Bio
Pro
tein
(Par
is, F
ranc
e)
(Edi
nbur
gh, U
K)
(Fra
min
gham
, MA
)(T
hous
and
Oak
s, C
A)
(Lei
den,
Net
herla
nds)
uses
hum
an c
ells
(S. S
an F
ranc
isco
, CA
)ot
her
curr
ent b
iolo
gics
man
ufac
ture
rs;
Am
gen
Gen
ente
ch
Cru
cell
$150
*Com
pany
est
imat
es
$1−$
2$1
−$2
$0.0
5
Com
pani
es
Est
imat
ed c
ost
(cos
t/g r
aw m
ater
ial)*
Fig.
3.2
Com
pani
esan
dte
chno
logi
esin
biom
anuf
actu
ring
.Aco
mpa
riso
nof
diffe
rent
expr
essi
onsy
stem
ssh
ows
the
big
diffe
renc
esin
term
sof
cost
s,ra
ngin
gfr
omU
S$15
0pe
rgr
amfo
rC
HO
cells
toU
S$0.
05pe
rgr
amfo
rtr
ansg
enic
plan
ts[1
1].

Color Plates xvii
Strengths• Access new manufacturing facilities• High production rates/high protein yield• Relatively fast ‘gene-to-protein’ time• Safety benefits; no hum. pathogens/no TSE• Stable cell lines/high genetic stability• Simple medium (water, minerals & light)• Easy purification (ion exchange vs. prot A)
TRENDS in Biotechnology Vol.20 No.12, 2002
Minus Plus
Opportunities • Reduce projected COGS • Escape capacity limitations • Achieve human-like glycosylation
Weaknesses• No approved products yet (but Phase III)• No final guidelines yet (but drafts available)
Threats• Food chain contamination• Segregation risk
Fig. 3.3 SWOT analysis of plant expression systems. Plant expression systems have a lot ofadvantages (plus) over other systems and are therefore mostly shown on the right-hand side of thepicture (Raskin I et al., Plants and human health in the twenty-first century. Trends in Biotechnol. 200220, 522–531.). Herein different systems (transgenic animals, mammalian cell culture, plants, yeast,and bacteria) are compared in terms of speed (how quickly they can be developed), operating andcapital costs and so on, and plants are obviously advantageous. Even for glycosylation, assembly andfolding, where plants are not shown on the right-hand side (meaning other systems areadvantageous), some plant expression systems are moving in that direction (as will be shownexemplarily in the section for moss). Also, the weaknesses and threats can be dealt with, using theappropriate plant expression system [20].
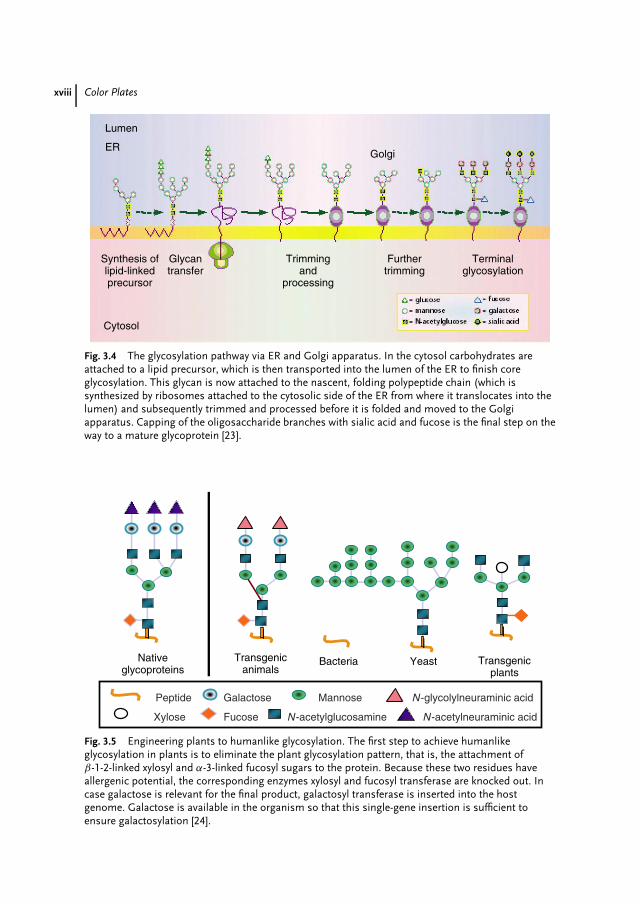
xviii Color Plates
Lumen
Golgi
Cytosol
Synthesis oflipid-linkedprecursor
Glycantransfer
Trimmingand
processing
Furthertrimming
Terminalglycosylation
ER
Fig. 3.4 The glycosylation pathway via ER and Golgi apparatus. In the cytosol carbohydrates areattached to a lipid precursor, which is then transported into the lumen of the ER to finish coreglycosylation. This glycan is now attached to the nascent, folding polypeptide chain (which issynthesized by ribosomes attached to the cytosolic side of the ER from where it translocates into thelumen) and subsequently trimmed and processed before it is folded and moved to the Golgiapparatus. Capping of the oligosaccharide branches with sialic acid and fucose is the final step on theway to a mature glycoprotein [23].
Bacteria Yeast Transgenicplants
Transgenicanimals
Nativeglycoproteins
N-glycolylneuraminic acid
N-acetylneuraminic acid
Mannose
Fucose
Galactose
Xylose
Peptide
N-acetylglucosamine
Fig. 3.5 Engineering plants to humanlike glycosylation. The first step to achieve humanlikeglycosylation in plants is to eliminate the plant glycosylation pattern, that is, the attachment ofβ-1-2-linked xylosyl and α-3-linked fucosyl sugars to the protein. Because these two residues haveallergenic potential, the corresponding enzymes xylosyl and fucosyl transferase are knocked out. Incase galactose is relevant for the final product, galactosyl transferase is inserted into the hostgenome. Galactose is available in the organism so that this single-gene insertion is sufficient toensure galactosylation [24].

Color Plates xix
• Root secretion, easy recovery• Greenhouse-contained tanks• High-density tissue• Salts and water only• Tobacco is well characterized• Stable genetic system
Phytomedics (tobacco):
Fig. 3.6 Secretion of the biopharmaceuticals via tobacco roots. The tobacco plants are geneticallymodified in such a way, that the protein is secreted via the roots into the medium (‘‘rhizosecretion’’).In this example, the tobacco plant takes up nutrients and water from the medium and releases GFP(green fluorescent protein). Examination of root-cultivation medium by its exposure tonear-ultraviolet illumination reveals the bright green-blue fluorescence characteristics of GFP in thehydroponic medium (left flask in panel lower left edge). The picture also shows a schematic drawingof the hydroponic tank, as well as tobacco plants at different growth stages, for example, callus,–fullygrown and greenhouse plantation [24].
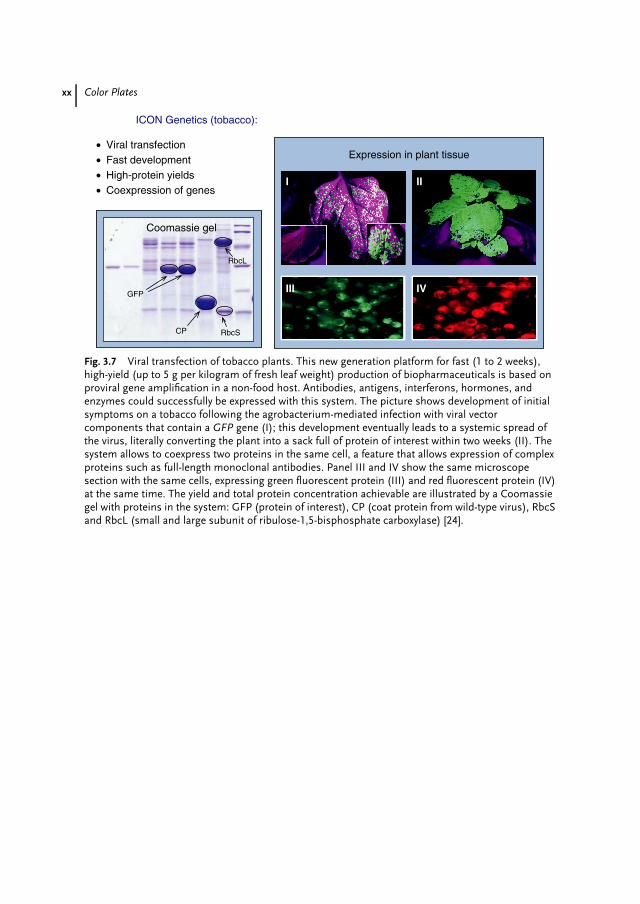
xx Color Plates
• Viral transfection• Fast development• High-protein yields• Coexpression of genes
ICON Genetics (tobacco):
RbcL
RbcSCP
GFP
Expression in plant tissue
Coomassie gel
I
IVIII
II
Fig. 3.7 Viral transfection of tobacco plants. This new generation platform for fast (1 to 2 weeks),high-yield (up to 5 g per kilogram of fresh leaf weight) production of biopharmaceuticals is based onproviral gene amplification in a non-food host. Antibodies, antigens, interferons, hormones, andenzymes could successfully be expressed with this system. The picture shows development of initialsymptoms on a tobacco following the agrobacterium-mediated infection with viral vectorcomponents that contain a GFP gene (I); this development eventually leads to a systemic spread ofthe virus, literally converting the plant into a sack full of protein of interest within two weeks (II). Thesystem allows to coexpress two proteins in the same cell, a feature that allows expression of complexproteins such as full-length monoclonal antibodies. Panel III and IV show the same microscopesection with the same cells, expressing green fluorescent protein (III) and red fluorescent protein (IV)at the same time. The yield and total protein concentration achievable are illustrated by a Coomassiegel with proteins in the system: GFP (protein of interest), CP (coat protein from wild-type virus), RbcSand RbcL (small and large subunit of ribulose-1,5-bisphosphate carboxylase) [24].

Color Plates xxi
• Simple medium (photoautotrophic plant needs only water and minerals)• Robust expression system (good expression levels from 15 to 25°C)
• Secretion into medium via human leader sequence (broad pH range: 4−8)• Easy purification from low-salt medium via ion exchange
• Easy genetic modifications to cell lines• Stable cell lines/high genetic stability
• Codon usage like human (no changes required)• Inexpensive bioreactors from the shelf
• Nonfood plant (no segregation risk)• Good progress on genetic modification of glycosylation
pathways (plant to human)
Greenovation (moss system):
Fig. 3.8 Greenovation use a fully contained moss bioreactor. This company has established aninnovative production system for human proteins. The system produces pharmacologically activeproteins in a bioreactor, utilizing a moss (Physcomitrella patens) cell culture system with uniqueproperties [24].
30 L pilot reactor for moss Two weeks after incubation
Fig. 3.11 Scaling of photobioreactors up to several 1000 L. The moss bioreactor is based on thecultivation of Physcomitrella patens in a fermenter. The moss protonema is grown underphotoautotrophic conditions in a medium that consists essentially of water and minerals. Light andcarbon dioxide serve as the only energy and carbon sources. Cultivation in suspension allows scalingof the photobioreactors up to several 1000 L. Adaptation of existing technology for large-scalecultivation of algae is done in cooperation with the Technical University of Karlsruhe. Courtesy ofgreenovation Biotech GmbH (Freiburg, Germany) and Professor C. Posten, Technical University(Karlsruhe, Germany).
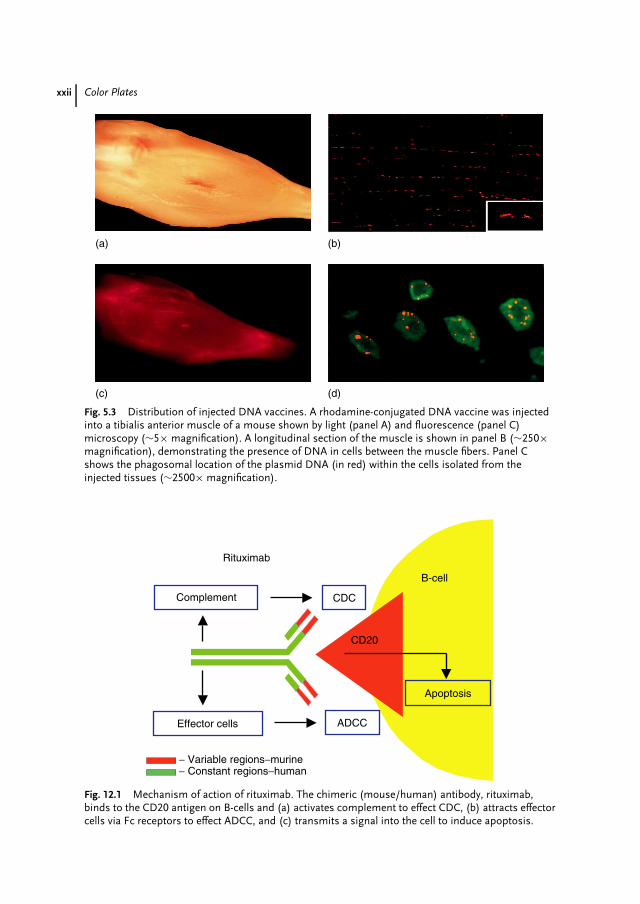
xxii Color Plates
(a) (b)
(c) (d)
Fig. 5.3 Distribution of injected DNA vaccines. A rhodamine-conjugated DNA vaccine was injectedinto a tibialis anterior muscle of a mouse shown by light (panel A) and fluorescence (panel C)microscopy (∼5× magnification). A longitudinal section of the muscle is shown in panel B (∼250×magnification), demonstrating the presence of DNA in cells between the muscle fibers. Panel Cshows the phagosomal location of the plasmid DNA (in red) within the cells isolated from theinjected tissues (∼2500× magnification).
B-cell
Rituximab
CD20
CDCComplement
Effector cells ADCC
− Variable regions−murine− Constant regions−human
Apoptosis
Fig. 12.1 Mechanism of action of rituximab. The chimeric (mouse/human) antibody, rituximab,binds to the CD20 antigen on B-cells and (a) activates complement to effect CDC, (b) attracts effectorcells via Fc receptors to effect ADCC, and (c) transmits a signal into the cell to induce apoptosis.
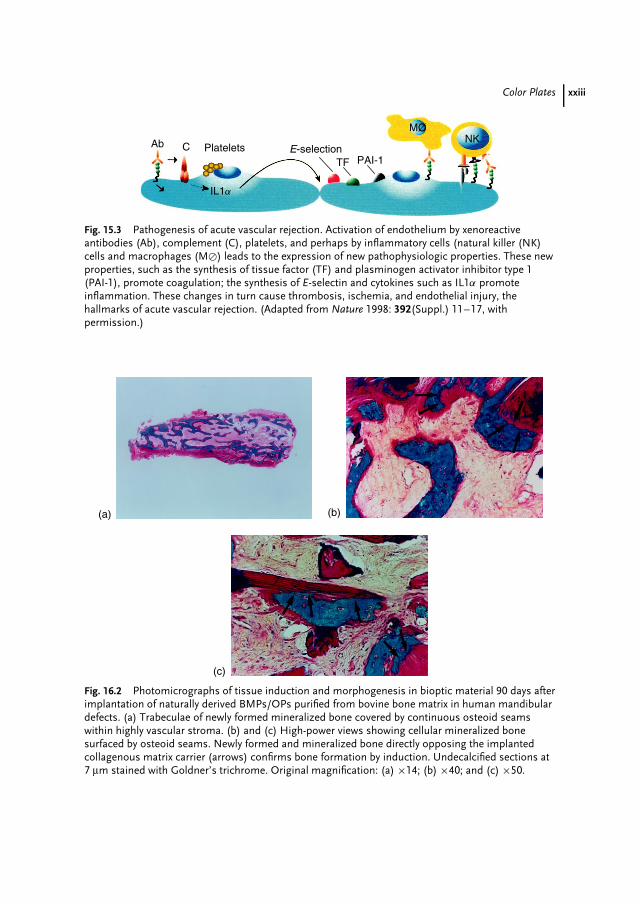
Color Plates xxiii
Platelets E-selectionTF PAI-1
NKMO
Ab C
IL1a
Fig. 15.3 Pathogenesis of acute vascular rejection. Activation of endothelium by xenoreactiveantibodies (Ab), complement (C), platelets, and perhaps by inflammatory cells (natural killer (NK)cells and macrophages (M�) leads to the expression of new pathophysiologic properties. These newproperties, such as the synthesis of tissue factor (TF) and plasminogen activator inhibitor type 1(PAI-1), promote coagulation; the synthesis of E-selectin and cytokines such as IL1α promoteinflammation. These changes in turn cause thrombosis, ischemia, and endothelial injury, thehallmarks of acute vascular rejection. (Adapted from Nature 1998: 392(Suppl.) 11–17, withpermission.)
(a) (b)
(c)
Fig. 16.2 Photomicrographs of tissue induction and morphogenesis in bioptic material 90 days afterimplantation of naturally derived BMPs/OPs purified from bovine bone matrix in human mandibulardefects. (a) Trabeculae of newly formed mineralized bone covered by continuous osteoid seamswithin highly vascular stroma. (b) and (c) High-power views showing cellular mineralized bonesurfaced by osteoid seams. Newly formed and mineralized bone directly opposing the implantedcollagenous matrix carrier (arrows) confirms bone formation by induction. Undecalcified sections at7 µm stained with Goldner’s trichrome. Original magnification: (a) ×14; (b) ×40; and (c) ×50.
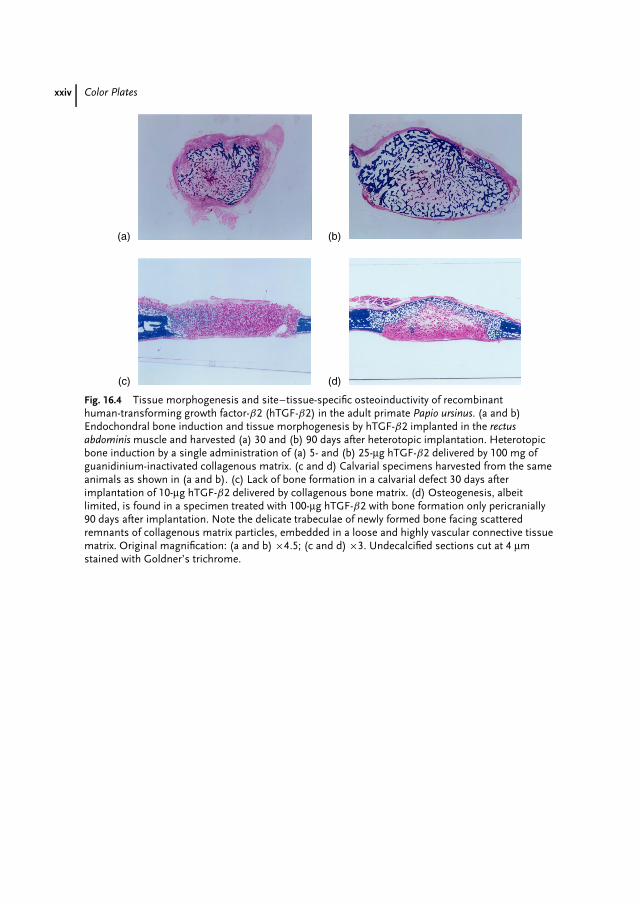
xxiv Color Plates
(a) (b)
(c) (d)
Fig. 16.4 Tissue morphogenesis and site–tissue-specific osteoinductivity of recombinanthuman-transforming growth factor-β2 (hTGF-β2) in the adult primate Papio ursinus. (a and b)Endochondral bone induction and tissue morphogenesis by hTGF-β2 implanted in the rectusabdominis muscle and harvested (a) 30 and (b) 90 days after heterotopic implantation. Heterotopicbone induction by a single administration of (a) 5- and (b) 25-µg hTGF-β2 delivered by 100 mg ofguanidinium-inactivated collagenous matrix. (c and d) Calvarial specimens harvested from the sameanimals as shown in (a and b). (c) Lack of bone formation in a calvarial defect 30 days afterimplantation of 10-µg hTGF-β2 delivered by collagenous bone matrix. (d) Osteogenesis, albeitlimited, is found in a specimen treated with 100-µg hTGF-β2 with bone formation only pericranially90 days after implantation. Note the delicate trabeculae of newly formed bone facing scatteredremnants of collagenous matrix particles, embedded in a loose and highly vascular connective tissuematrix. Original magnification: (a and b) ×4.5; (c and d) ×3. Undecalcified sections cut at 4 µmstained with Goldner’s trichrome.
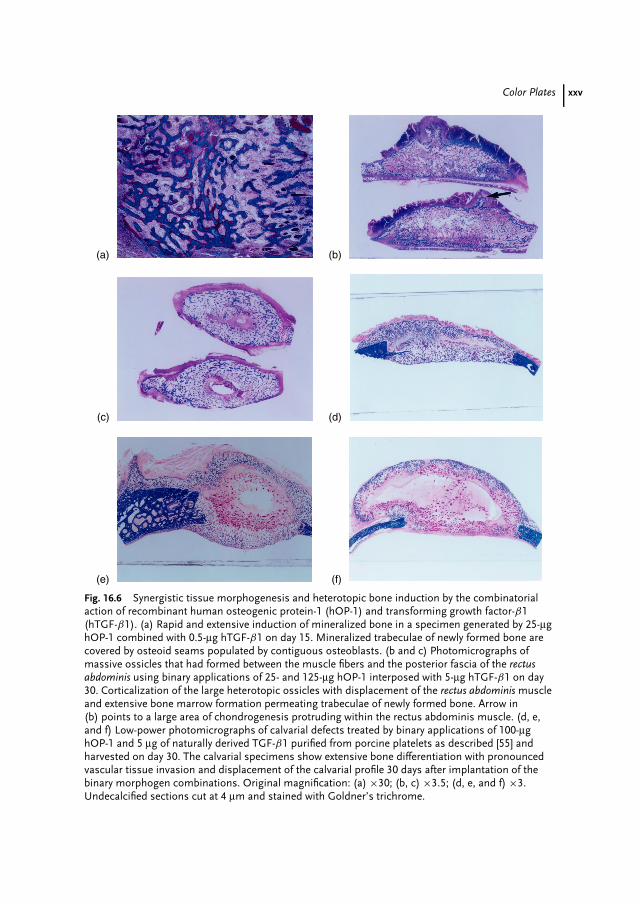
Color Plates xxv
(a)
(d)
(b)
(e)
(c)
(f)
Fig. 16.6 Synergistic tissue morphogenesis and heterotopic bone induction by the combinatorialaction of recombinant human osteogenic protein-1 (hOP-1) and transforming growth factor-β1(hTGF-β1). (a) Rapid and extensive induction of mineralized bone in a specimen generated by 25-µghOP-1 combined with 0.5-µg hTGF-β1 on day 15. Mineralized trabeculae of newly formed bone arecovered by osteoid seams populated by contiguous osteoblasts. (b and c) Photomicrographs ofmassive ossicles that had formed between the muscle fibers and the posterior fascia of the rectusabdominis using binary applications of 25- and 125-µg hOP-1 interposed with 5-µg hTGF-β1 on day30. Corticalization of the large heterotopic ossicles with displacement of the rectus abdominis muscleand extensive bone marrow formation permeating trabeculae of newly formed bone. Arrow in(b) points to a large area of chondrogenesis protruding within the rectus abdominis muscle. (d, e,and f) Low-power photomicrographs of calvarial defects treated by binary applications of 100-µghOP-1 and 5 µg of naturally derived TGF-β1 purified from porcine platelets as described [55] andharvested on day 30. The calvarial specimens show extensive bone differentiation with pronouncedvascular tissue invasion and displacement of the calvarial profile 30 days after implantation of thebinary morphogen combinations. Original magnification: (a) ×30; (b, c) ×3.5; (d, e, and f) ×3.Undecalcified sections cut at 4 µm and stained with Goldner’s trichrome.

















