Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1236 Pharmacotherapy for Parkinson’s Disease – Observations and Innovations BY DAG NYHOLM ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2003
Transcript
Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1236
Pharmacotherapy for Parkinson’s Disease– Observations and Innovations
BY
DAG NYHOLM
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2003
SE-75185
Solen förgyller dagen.
Månan försilfrar natten. Men sökes visdomsskatten,
lys Dig sjelf, så är lagen!
Erik Gustaf Geijer
The sun gilds the day. The moon silvers the night.
But if you seek the treasure of insight, enlighten yourself, that is the rule, I say!
Erik Gustaf Geijer (translation by Dag Nyholm)
Front cover: Etching signed by Höfer, available at the University Library. Uppsala University, founded in 1477, was given the University Hall as a present from the Swedish state in 1877. The building was constructed by Herman Teodor Holmgren and was inaugurated on May 17, 1887. The statue in memory of Erik Gustaf Geijer was sculptured by John Börjeson and unveiled on October 30, 1888.
Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals:
I Nyholm D, Lennernäs H, Gomes-Trolin C, Aquilonius SM.
Levodopa pharmacokinetics and motor performance during activities of daily living in parkinsonian patients on individual drug combinations. Clin Neuropharmacol 2002; 25: 89-96.
II Nyholm D, Askmark H, Gomes-Trolin C, Knutson T,
Lennernäs H, Nyström C, Aquilonius SM. Optimizing levodopa pharmacokinetics – intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol 2003; in press.
III Nilsson D, Nyholm D, Aquilonius SM. Duodenal levodopa
Motor complications ................................................................................26 Fluctuations .........................................................................................26 Dyskinesias..........................................................................................29
The management of motor complications................................................31 Oral drug-delivery ...............................................................................31 Intravenous infusion ............................................................................32 Intestinal infusion ................................................................................34 Subcutaneous infusion.........................................................................36 Infusion systems ..................................................................................37
Methods ........................................................................................................41 Patients .....................................................................................................41 Study designs and medications ................................................................41 Measuring levodopa and 3-O-methyldopa in blood.................................44
System for intestinal infusion of levodopa/carbidopa..............................48 Automatic dose dispenser for oral levodopa/carbidopa ...........................49
Statistics and data management................................................................51
Results...........................................................................................................53 Levodopa pharmacokinetics (papers I and II)..........................................53
Oral SR formulation versus intestinal infusion ...................................54 Influence of food .................................................................................57 Gastric infusion ...................................................................................59
Levodopa and motor function (papers I, II, III and V).............................60 Experience with levodopa infusion (papers II and III).............................63 Usability of an automatic dose dispenser (paper IV) ...............................64 Compliance and usability of patient diaries (paper V).............................65
Discussion.....................................................................................................67 Impact of fluctuating levodopa pharmacokinetics ...................................67 The demand for individualisation of therapy ...........................................72 Frequent observations and true compliance with patient diaries .............73 Future prospects .......................................................................................74
3-O-MD 3-O-methyldopa 5-HT 5-hydroxytryptamine AADC aromatic amino acid decarboxylase AUC area under the plasma concentration-time curve BBB blood brain barrier CADD continuous ambulatory drug delivery CDS continuous dopaminergic stimulation Cmax maximum concentration in blood after a dose Cmin minimum concentration in blood after a dose CNS central nervous system COMT catechol-O-methyltransferase CR controlled-release Css concentration at steady-state CV coefficient of variation DAT dopamine transporter DBS deep-brain stimulation DOPA dihydroxyphenylalanine EBM evidence-based medicine EMG electromyography GABA gamma-amino butyric acid GPi globus pallidus interna HBS hydrodynamically balanced system HPLC high-performance liquid chromatography IR immediate-release LDR long-duration response LNAA large neutral amino acid MAO monoamine oxidase MT movement time NMDA N-methyl-D-aspartate NUDS Northwestern University disability scale PD Parkinson’s disease PDQ-39 Parkinson’s disease questionnaire (39 items) PEG percutaneous endoscopic gastrostomy PET positron emission tomography PLM posturo-locomotor-manual (test) SD standard deviation
SDR short-duration response SNr substantia nigra pars reticulata SPECT single-photon emission computed tomography SR sustained-release, slow release STN subthalamic nucleus t ½ elimination half-life of a substance Tmax time to reach Cmax UPDRS unified Parkinson’s disease rating scale VAS visual analogue scale VDS verbal descriptive statistics VT volume transmission WRS Webster rating scale WT wiring transmission
9
Introduction
Background Parkinson’s disease (PD) is a progressive, neurodegenerative disorder affecting primarily dopaminergic neuronal systems with impaired motor function as a consequence. The diagnosis might conceal a wide pattern of similar disorders with different causes and courses. However, a few criteria and a clear response to dopaminomimetic therapy constitute the idiopathic PD. The classical PD criteria are bradykinesia, tremor and rigidity. Postural instability has lately joined to form a tetrad. The disease is diagnosed on a clinical basis and there are no diagnostic tests. However, a positive response to levodopa administration is obvious and separates idiopathic PD from other disorders, which present with parkinsonism. The clinical characteristics were first described as the shaking palsy by James Parkinson in 1817 (Parkinson, 1817).
Up to now, the causes of PD are unknown and genetic research has not solved the problem, although a few hereditary forms of PD have been found (Spacey and Wood, 1999 for review). Since the first PD gene locus was discovered within an Italian family (Polymeropoulos et al., 1996), a number of PARK genes (1-8) have recently been identified to be involved in development of parkinsonism (Foltynie et al., 2002). Some known mutations result in dysfunctional properties of α-synuclein, a protein suggested to be involved in the formation of synaptic vesicles for dopamine storage. A reduced number of vesicles, related to mutations in α-synuclein, might lead to increased oxidative stress by the unstored dopamine, a possible explanation to the pathogenesis of PD (Lotharius and Brundin, 2002).
Motor symptoms are the predominant features of PD, however other symptoms may occur, e.g. neuropsychiatric problems, autonomic dysfunction and sleep disorders. Some of these complications might be induced by dopaminomimetic therapy whereas some features are only linked to disease progression. Since levodopa was introduced as the first effective treatment as late as 1967 there are some, but incomplete, data available on the natural course of the untreated disease.
10
The introduction of levodopa was revolutionary in that it became an excellent treatment for the earlier essentially untreatable disorder. Mortality has been nearly normalised and levodopa is still considered the most effective agent for treatment of PD. However, disabling motor complications occur in the late stages of PD and the need for a better, perhaps more physiologically based therapy for this complex disorder remains.
Extensive research is ongoing and has provided many new treatment alternatives in the recent years. Much progress has been made and there are promising novelties for the future, but it remains the case that the advanced stages of PD constitute a sometimes frustrating challenge.
Epidemiology Studies of epidemiology of PD are few, often restricted in scope and not always reliable. One of the difficulties is that there is no ante-mortem diagnostic test for PD. The diagnostic methods and accuracy differ between different diagnosticians. This means that some cases of PD are in fact essential tremor or neurodegenerative disorders, such as progressive supranuclear palsy or multiple system atrophy, which are not always easily distinguishable. Some instances, especially of elderly patients, who in fact have PD are misdiagnosed as cases of depression or “normal” aging (Tanner and Goldman, 1996).
Prevalence (the total number of persons with the disease at a fixed point in time) in most European and US studies is 0.1–0.2 %, but up to 0.3 % has occasionally been reported (Aquilonius and Hartvig, 1986). The prevalence is highest in Europe and North America, lower in Asia and about 0.05% in Africa (Li et al., 1985; Schoenberg et al., 1988; Okada et al., 1990). Several studies in Europe and the US have suggested that about 1 % of people above 65 years of age are affected by PD (Marttila and Rinne, 1976; Sutcliffe and Meara, 1995).
The age at onset of PD is about 55–60 years. In 1967, Hoehn and Yahr published that 2/3 of the patients’ onset were between 50 and 69 years of age (Hoehn and Yahr, 1967). The results are still relevant according to new studies of distribution of age at onset. The duration of PD in the population was investigated in a British study from 1992 (Sutcliffe and Meara, 1995). The distribution of disease duration in the 374 patients ranged from 0 to 42 years and the median was 6 years. Eight percent of the patients had been affected by PD for more than 20 years.
One of the first rating scales for disease severity was the Hoehn & Yahr scale (Hoehn and Yahr, 1967). It comprises five stages, from unilateral disease (stage I) to confinement to bed or wheelchair unless aided (stage V).
11
The median duration of illness since onset, in patients before levodopa therapy was introduced, being 3 years in stage I and 14 years in stage V. Twenty years later, Hoehn suggested that the duration of each stage was prolonged by 3–5 years, when patients were treated with levodopa (Hoehn, 1987).
Pathophysiology The basal ganglia are involved in normal motor control via a number of internal circuits, finally projecting to the thalamus and cerebral cortex. Dopaminergic projections from the substantia nigra pars compacta terminate on dendrites of the medium-sized spiny neurons in the striatum (caudate nucleus and putamen) (Kotter, 1994). In turn, these GABAergic efferents project, directly and indirectly, to the internal segment of globus pallidus (GPi) and substantia nigra pars reticulata (SNr), which are the major output nuclei for the basal ganglia (Chase et al., 1998). Basal ganglia output is then directed to several thalamic and brainstem nuclei. The direct pathway from the striatum to GPi/SNr is monosynaptic, while the indirect pathway includes synapses in the external segment of globus pallidus and the subthalamic nucleus (STN).
Dopamine receptors are divided into two families, D1 and D2. The D3 and D4 subtypes of receptors are members of the D2 family and D5 is D1-like (Sibley et al., 1993). Striatal D1 receptors are mainly located on neurons in the direct pathway and D2 receptors in the indirect projection (Gerfen et al., 1990). Dopamine acts via excitatory transmission in the direct D1-mediated pathway and inhibitory in the indirect D2-mediated pathway (Clark and White, 1987).
The firing pattern of the nigrostriatal dopaminergic neurons is regular and with a frequency of 3–6 Hz. Bursts of 15–20 Hz occur in response to salient visual or auditory stimuli (Schultz, 1994). The constant striatal dopaminergic stimulation regulates glutamatergic input from the cortex to the direct and indirect pathways (Levy et al., 1997).
PD is mainly characterised by denervation of nigrostriatal dopaminergic neurons, although other neuronal systems also are involved (Agid, 1989). Striatal dopamine receptors, at least D2, are upregulated in untreated PD as a compensatory response to dopamine deficiency (Guttman, 1992). However, the loss of dopamine leads to an imbalance in the basal ganglia, directing neurotransmission to the indirect pathway. This results in increased inhibitory output from the GPi causing a decreased excitatory cortical projection from the thalamus.
12
The decreased facilitation of cortical motor areas is manifested as bradykinesia (Wichmann and DeLong, 1993). Symptom onset appears when about 80% of the nigrostriatal neurons have degenerated and, when motor response complications are experienced, more than 90% have been lost (Hornykiewicz and Kish, 1987). The attrition of pigmented neurons in the substantia nigra pars compacta occurs in normal ageing at a rate of 4.7% per decade (Fearnley and Lees, 1991). Recent studies of progression have used the PET technique to study 6-[18F]fluorodopa uptake and dopamine transporter (DAT) imaging (Nurmi et al., 2001; Ma et al., 2002a). The relative reduction in fluorodopa uptake and DAT binding in PD was lower compared to the post-mortem studies.
In the research on intercellular communication in the CNS other systems than classical synaptic transmission must be identified. The terms wiring and volume transmission (WT and VT) have been introduced for this purpose (Agnati et al., 1995). WT is defined as a fast one-to-one transmission via classical synapses, and VT is defined as as a slow one-to-many intercellular communication within the extracellular space and cerebrospinal fluid. In PD, the therapeutic efficacy of levodopa is thought to depend on the VT of dopamine released in striatal extracellular space more than on the WT of the few remaining dopaminergic neurons (Zoli et al., 1999). It is suggested that potentiation of VT, switching from short-distance to long-distance VT is a compensatory mechanism for the loss of dopaminergic neurons to delay onset of motor symptoms (Agnati et al., 1995; Zoli et al., 1998). In the progressing disease this compensation is not enough to cover the degeneration, resulting in shorter response duration of each levodopa tablet. When the drug-induced dopaminergic stimulation ends and PD symptoms reappear, the patient goes from “on” to “off” state.
Extrasynaptic diffusion of transmitters is thought to be common in central synapses (Zoli et al., 1999). This could explain why dopamine reuptake sites are located outside the synapses (Nirenberg et al., 1997) and dopamine receptors are concentrated in extrasynaptic membranes of dendrites (Yung et al., 1995). Inhibition of reuptake seems not to influence dopamine concentration at the synapse (Garris et al., 1994).
Internalisation, from plasma membrane to cytoplasm, of striatal D1 receptors occurs in levodopa treated rodents whether unlesioned or lesioned with 6-hydroxydopamine (Muriel et al., 2002). This desensitisation of D1 receptors may have consequences for reduced efficacy of therapy but further studies of this phenomenon is required.
Repeated pharmacological stimulation of dopaminergic receptors results in a progressive enhancement of responsiveness; the behavioural sensitisation phenomenon. It has been suggested that this sensitisation is
13
increased by pulsatile and decreased by continuous dopaminergic stimulation (Blanchet et al., 1995).
Sleep benefit is a curious phenomenon in PD. More than half of patients report improved motor function after sleep and more than one third consider awakening the best time of the day, which allows them to postpone the morning anti-PD medication. The phenomenon has been found to be more common in elderly patients and in patients with long disease duration and is thought to result from increased presynaptic dopamine storage during sleep (Parkes, 1983; Merello et al., 1997a).
Pharmacotherapy
History The first treatment of PD, or paralysis agitans, was anticholinergics in the 19th century (Ordenstein, 1867), probably based on a misinterpretation of drooling as hypersalivation. The anticholinergic alkaloid scopolamine was known to block salivation. Effects on the parkinsonian symptoms were very moderate. The first steps in the direction towards a treatment for the motor symptoms were taken by Arvid Carlsson in 1957 when he identified dihydroxyphenylalanine (DOPA) as a reserpine antagonist (Carlsson et al., 1957). The sedation and hypokinetic syndrome induced by the antipsychotic agent reserpine in experimental animals could be reversed. Soon thereafter the group published a theory with dopamine as a neurotransmitter and a possible involvement in motor control (Carlsson et al., 1958). Dopamine was up to this point considered to be a physiologically inactive precursor to noradrenaline and it was not until a few years later, when Carlsson’s research group could demonstrate the cellular localization of dopamine, noradrenaline and serotonin, that their claim was generally accepted. Meanwhile, supportive studies were presented, e.g. dopamine deficit in brains from PD patients (Ehringer and Hornykiewicz, 1960) and spectacular therapeutic effect of intravenous DOPA in PD patients (Birkmayer and Hornykiewicz, 1961). The doses of levodopa were about 100 mg and other authors were unable to confirm any major effect as compared to placebo in double-blind studies (Fehling, 1966; Rinne and Sonninen, 1968). In 1967, Cotzias and coworkers presented the dramatic effects of much larger oral doses of DOPA, 1.6–12.6 g, on PD patients (Cotzias et al., 1967) and two years later L-DOPA was introduced as a clinically applicable therapy (Barbeau, 1969; Cotzias et al., 1969). The high doses of levodopa brought many side effects. Nausea was very common but vanished almost totally
14
when peripheral decarboxylation of levodopa was inhibited (Pletscher and Bartholini, 1971). Sustained-release formulations of levodopa and its decarboxylase inhibitor were developed in the 1980s as an attempt to improve the pharmacokinetic profile after oral delivery (Cedarbaum et al., 1987). The increasing knowledge of monoamines led to trials with monoamine oxidase inhibitors in the 1960's, and in 1978 the facilitation of dopamine by selegiline was presented (Knoll, 1978). During the 1990s inhibitors of another enzyme, catechol-O-metyltransferase (COMT), was developed to further increase the utilisation of levodopa (Mannisto and Kaakkola, 1999). Bromocriptine, the oldest dopamine receptor agonist was accompanied by several new agonists in the late 1990s. The introduction of levodopa led to a markedly reduced mortality and it has been demonstrated that early levodopa treatment is superior to late, regarding life expectancy (Diamond et al., 1987).
Treatment strategies Dopaminomimetic therapy is one of the greatest successes of clinical neuropharmacology in the 20th century and current therapeutic research focuses mainly on refinement of this therapy, although other transmitter systems are studied as well.
Dopamine binds more avidly to D2 receptors, especially the D3 subtype. Therefore most dopamine agonists are designed for D2 stimulation. However, neurophysiological and behavioural data from animal models of PD suggest that D1 stimulation, alone or in combination with D2 stimulation, may be preferable (Walters et al., 1987).
A controversy on whether treatment of PD should start with levodopa or a dopamine agonist is currently ongoing (Montastruc et al., 1999; Weiner, 1999). Long-term studies of bromocriptine as initial therapy with later add-on levodopa versus levodopa as initial and only medication showed no favourable effect of an early use of the dopamine agonist on mortality (Lees et al., 2001; Montastruc et al., 2001). After some years of high expectations from new dopamine agonists the current opinion seems to be shifted in favour of levodopa (Djaldetti et al., 2003; Wooten, 2003).
Continuous dopaminergic stimulation (CDS) has been in focus ever since it was shown that a stable constant-rate intravenous infusion of levodopa produced a complete removal of motor fluctuations in patients with fluctuating response to oral levodopa (Shoulson et al., 1975).
Attempts have been made to compile standardised algorithms for the therapeutic management of PD (Olanow et al., 2001). However, criticism has been raised against such algorithms if sponsored by the pharmaceutical industry rather than related to evidence-based medicine (EBM). A literature
15
review performed in accordance with criteria of EBM was recently presented (Movement Disorder Society, 2002). Creation of standardised algorithms is almost impossible considering the extreme variability in patients’ clinical presentations, drug responses, etc. (Lees, 2002). Each patient needs an individually tailored therapy. Pharmacogenetics, or pharmacogenomics, deals with genetic polymorphisms in drug-metabolising enzymes, transporters and receptors, which may explain inter-individual differences in efficacy and toxicity of, for example, dopaminomimetic medication. The difference in dose requirement between patients is obvious in PD and will probably be a major issue in future refinement of therapy.
Drugs
Levodopa
Pharmacokinetics and pharmacodynamics The dopamine precursor levodopa (Levo-3,4-dihydroxyphenylalanine) is a large neutral amino acid (LNAA), like tyrosine, phenylalanine, tryptophan, leucine, isoleucine and valine. Levodopa is transformed into dopamine in cells containing the enzyme aromatic L-amino acid decarboxylase (AADC), i.e. dopamine-containing neurons, but also 5-HT neurons and pericytes (Cooper et al., 1991). Orally administered levodopa reaches the maximum blood concentration (Cmax) after 45–120 minutes, partly depending on the formulation of the tablet. The pharmacokinetics of levodopa is also dependent on gastric emptying as absorption occurs in the proximal one-third of the small intestine (duodenum/jejunum), not in the stomach (Rivera-Calimlim et al., 1971a; Sasahara et al., 1981). Gastric emptying is therefore a determining factor for the access to orally administered levodopa. The motility of the stomach varies with fed and fasted state (Smith and Feldman, 1986) and erratic gastric emptying gives a fluctuating levodopa concentration-time curve (Kurlan et al., 1988b). Moreover, gastric emptying time is delayed in some PD patients compared with a control population (Hardoff et al., 2001). Levodopa itself may slow gastric emptying (Robertson et al., 1990) although accelerated levodopa absorption has been suggested in patients exposed to long-term levodopa therapy (Murata et al., 1996). The longer levodopa remains in the stomach or small intestine, the more extensively it is metabolised and made unavailable for absorption (Rivera-Calimlim et al., 1971b). Levodopa absorption is fast and complete but the bioavailability is only 30% due to decarboxylation to dopamine by the enzyme AADC in the gastrointestinal mucosa. The AADC inhibitors benserazide and carbidopa inhibit extracerebral decarboxylation efficiently,
16
thereby enhancing the bioavailability of levodopa in the combined drugs Madopar and Sinemet. All commercially available levodopa formulations now contain an AADC inhibitor. Therefore, when levodopa is mentioned in the rest of this thesis, it is always in presence of benserazide or carbidopa. Some of the administered levodopa is metabolised by COMT into 3-O-methyldopa. Therefore a COMT inhibitor may be added to levodopa and the AADC inhibitor. Levodopa is completely metabolised into dopamine, noradrenaline or homovanillic acid, which are mainly eliminated in urine.
Levodopa is transported across the intestinal mucosa and the blood-brain barrier (BBB) by the large neutral amino acid (LNAA) transport system, which means that some amino acids competitively inhibit levodopa membrane transport (Wade et al., 1973; Mearrick et al., 1974; Lennernas et al., 1993). A PET study of intravenous Levo-[18F]fluorodopa and amino acid loading demonstrated a significant reduction in tracer uptake into the brain, thus proving the competition between levodopa and amino acids for uptake across the BBB (Leenders et al., 1986). Doses of levodopa taken with a meal, particularly with high protein content, will be less efficacious than doses taken on an empty stomach (Nutt et al., 1984). This has led to the use of a protein redistribution diet for patients with motor fluctuations (Pincus and Barry, 1988; Karstaedt and Pincus, 1992). By restricting most of the daily protein intake to evening meals, daytime plasma levodopa levels are more predictable and motor performance is improved, including so called dopa-resistant “off” periods.
Oral levodopa therapy thus gives a somewhat unpredictable onset of response as the gastric emptying rate varies during the day and as amino acids in meals can inhibit the uptake of levodopa from intestine into blood and subsequent transport into the brain. However, under controlled conditions regarding diet and medication, the levodopa response was found to be predictable also in patients who reported diurnal response fluctuations (Frankel et al., 1990). The authors concluded that plasma levodopa levels predict motor response to a great extent.
The therapeutic effect of levodopa is very good early in the course of the disease and normal mobility can be achieved by the replacement therapy. A single dose of levodopa gives a fast motor response, which can be maintained several hours due to a preserved dopamine buffering capacity in the neurons. The fluctuating pharmacokinetics is no problem in early PD. As the disease progresses within a few years the response duration gets shorter (“wearing-off” phenomenon) and the therapeutic window is narrowed (Mouradian et al., 1988). The dopaminergic medication must be increased for onset of response while the threshold for involuntary movements, dyskinesias, decreases.
17
As PD progresses the effect-time of each levodopa dose becomes shorter, but the half-life of levodopa is not thought to vary with PD duration or treatment (Fabbrini et al., 1988; Mouradian et al., 1988). Peripheral pharmacokinetics of levodopa should remain unchanged during the course of the disease, according to studies of plasma clearance (Chase et al., 1987). However, a comparison of stable and fluctuating patients showed increased area under the plasma concentration time curve (AUC) and Cmax and decreased Tmax and T ½ in fluctuating patients (Murata et al., 1996). AUC is significantly greater in elderly patients because of lower systemic clearance (Robertson et al., 1989; Contin et al., 1991). There is also a gender difference where women have significantly greater levodopa bioavailability than men in terms of weight-corrected AUC and Cmax (Kompoliti et al., 2002). Levodopa pharmacokinetics does not seem to be altered by any of the dopamine agonists bromocriptine, cabergoline or pramipexole although inconsistent results of the interaction between levodopa and bromocriptine have been published (Contin et al., 1992; Del Dotto et al., 1997; Kompoliti et al., 2002).
In the advancing stages of the disease patients take small and frequent doses of levodopa, as continuously as possible, to reduce fluctuations in plasma levodopa levels and motor performance (Obeso et al., 1994). The gastric emptying then becomes a determinant for onset of effect.
Different formulations To overcome the problem of the slow onset of action of oral levodopa, soluble formulations have been studied. Levodopa solutions offer the opportunity for individualised dose adjustment and are possibly less dependent on gastric emptying. In a double-blind crossover comparison of such a solution and standard tablets, patients responded to liquid levodopa with significantly improved “on” time, without an increase in the severity of dyskinesia (Pappert et al., 1996). However, another study did not find any significant difference between tablets and the solution regarding plasma levodopa oscillations and motor response fluctuations (Metman et al., 1994). Several studies have shown that the levodopa solution results in shorter time to peak plasma levodopa concentrations. This includes a dispersible formulation of benserazide/levodopa (Contin et al., 1999). Failure of liquid formulations to affect plasma levodopa oscillations is attributed to erratic gastric emptying, as determined in a study where even continuous gastric levodopa infusion failed to significantly reduce plasma fluctuations as compared to oral therapy (Kurlan et al., 1988a).
The concept of small and frequent dosage has led to development of microtablets of 1.25/5 mg carbidopa/levodopa for individual drug dispensing
18
by counting (Aquilonius et al., 1998). The microtablet concept has not yet reached clinical trials.
Two sustained-release (SR) formulations have been designed to achieve smooth plasma concentration levels (Cedarbaum, 1989 for review). Madopar HBS (hydrodynamically balanced system) is a gelatin capsule, which is transformed into a mucous body floating on the surface of gastric fluid, releasing its benserazide/levodopa by slow diffusion. In Sinemet CR (controlled-release), the final product of five prototypes, carbidopa/levodopa is dispersed within an erodible polymer matrix. Plasma levodopa peaks were delayed until 2–3 hours after a dose and “on” time without choreic dyskinesia was significantly increased as compared to the immediate-release formulation (Cedarbaum et al., 1987; Goetz et al., 1987; LeWitt et al., 1989). Madopar HBS showed similar properties with more stable plasma levodopa levels in most patients, as compared to standard tablets (Poewe et al., 1986) but failed to reduce motor fluctuations and dyskinesias compared with the standard formulation in a 5-year trial (Dupont et al., 1996). The bioavailability of both sustained-release formulations was lower than for standard formulations. Long-term treatment with these formulations showed to be difficult in terms of controlling motor fluctuations over time, partly because of unpredictable plasma levels and long Tmax (Pezzoli et al., 1988; Cedarbaum, 1989). Plasma levodopa variability was the same for IR and SR formulations in a group of ten patients. Three individuals, however, experienced benefit with the SR formulation, because they were able to maintain levodopa levels above the threshold for “on” state (McHale et al., 1990a). In a 5-year comparison of IR and SR levodopa there were no large differences in outcome between the two formulations, except for a significant improvement in UPDRS part II (activities of daily living) in favour of the SR formulation (Block et al., 1997). After gastric emptying the transit time in the small bowel is limited to a few hours with rapid absorption or constant enzymatic metabolisation. Thus the perfect SR tablet cannot be produced. Still, SR formulations produce less variation in plasma levodopa fluctuations as compared to immediate release tablets. Studies comparing standard Sinemet 25/100 and Sinemet CR (50/200) have shown significantly reduced variability of plasma levodopa levels with the CR formulation. Cedarbaum et al (1989) found a lower coefficient of variation for Sinemet CR, 55.5% versus 68.8%, while the range (maximum to minimum) of plasma levodopa concentrations did not differ between the two formulations. Bypassing the stomach by intravenous or intestinal infusion is the only way to markedly reduce fluctuations in plasma levodopa concentrations.
19
Non-motor response The action of levodopa is not solely related to motor function. Mood response to a 2-hour infusion has been studied in patients during the first year of levodopa therapy (Maricle et al., 1998). After a 2-day levodopa holiday mood elevation was evident after an acute infusion dose. The authors concluded that the mood response is more pronounced in advanced PD. Levodopa is used for mood-enhancing purposes in patients who have undergone surgery for deep-brain stimulation (Krack et al., 1998).
Toxicity? It has long been discussed whether levodopa is toxic and induces motor complications or not (Agid, 1998 for review). There is growing evidence that levodopa is only toxic in vitro, in the absence of glia, and that this has no physiological relevance. In animals (Hefti and Melamed, 1981; Lyras et al., 2002) and humans (Quinn et al., 1986) levodopa has not been found to damage dopaminergic neurons, but instead there are indications that levodopa therapy may promote functional recovery of dopaminergic nigrostriatal neurons (Murer et al., 1998; Datla et al., 2001). Further, there is a consensus that levodopa is the most effective substance in ameliorating parkinsonian symptoms, implying that prescription of levodopa should be based only on consideration of its efficacy and side-effect profile for the individual patients’ needs (Agid et al., 1999; Katzenschlager and Lees, 2002).
AADC inhibitors Benserazide and carbidopa are peripheral AADC inhibitors that reduce the amount of levodopa required to produce a given response by about 75% (Calne et al., 1971). When levodopa and an AADC inhibitor are administered together, levodopa plasma levels are increased up to 5-fold and the plasma half-life of levodopa is slightly increased (Reid et al., 1972). The elimination half-life of a single-dose of oral levodopa is about 1.5 hours in the presence of the inhibitor and about 1 hour without it (Cedarbaum, 1987 for review). Following oral administration of carbidopa to an intravenous infusion of levodopa it was estimated that the the bioavailability of levodopa was doubled (Nutt et al., 1985). A blinded crossover trial revealed no difference in therapeutic effects or adverse reactions between the two levodopa/AADC inhibitor preparations (Greenacre et al., 1976).
COMT inhibitors In the 1990s inhibitors of catechol-O-methyltransferase (COMT), namely entacapone and tolcapone were introduced. COMT is an enzyme which O-methylates levodopa and dopamine in the brain, but is also present in the gut
20
and the liver. The O-methylated derivative of levodopa, 3-O-methyldopa (3-O-MD), cannot be converted to dopamine in the brain and has been proposed to compete with levodopa for the transport across the blood-brain barrier (BBB) (Reches et al., 1982). However, a PET study in cynomolgus monkeys that received 3-O-MD infusion and 6-[18F]fluorodopa did not support this hypothesis (Guttman et al., 1992). Tolcapone is able to cross the BBB itself, whereas entacapone only acts peripherally. These drugs ingested together with levodopa/AADC inhibitor may provide up to 75% longer elimination half-life and up to 90% increase of the AUC of levodopa (Nutt et al., 1994; Jorga et al., 1998). The formation of 3-O-MD from levodopa is substantially decreased when a COMT inhibitor is used (Mannisto and Kaakkola, 1999 for review). Thus, the levodopa dose can be reduced with the same effect achieved.
In single-dose studies in rodents, striatal levels of levodopa and dopamine were significantly increased with the addition of entacapone or tolcapone (Nissinen et al., 1992; Kaakkola and Wurtman, 1993).
The clinical benefits observed with COMT inhibitors can probably be attributed to an increase in minimum plasma concentrations (Cmin) of levodopa related to a prolonged elimination half-life (Heikkinen et al., 2001). Maximum plasma concentrations have been reported to be unaffected, at least after single doses of levodopa and COMT inhibitor (Nutt et al., 1994). If this is true also for repeated dosage of levodopa together with COMT inhibitor, increased Cmin and unchanged Cmax would imply decreased fluctuations in plasma. It has been reported that plasma levodopa levels are kept more sustained with, as compared to without, a COMT inhibitor (Nutt et al., 1994; Nutt, 2000). However, only very little data on variability of levodopa concentrations in plasma during repeated dosage have been published. The increased levodopa bioavailability with repeated doses of levodopa and tolcapone may increase both Cmax and Cmin, elevating the concentration-time curve, seemingly without affecting levodopa variability (Baas et al., 2001). Regarding variability it was found that the coefficient of variation was 38% with entacapone and a lower levodopa dose compared with 49% with levodopa as monotherapy (Nutt et al., 1994). Mean plasma levodopa concentrations as well as trough and peak concentrations were higher during entacapone therapy. For mildly and moderately affected patients with not so narrow therapeutic windows, COMT inhibition may involve improvement in raising the plasma troughs and keeping peaks not too far above the dyskinesia threshold. However, in advanced patients there might be a risk of inducing hyperkinesia unless the levodopa dose is decreased. The severity of dyskinesias was not increased but the duration of dyskinesias was prolonged in a study of entacapone (Ruottinen and Rinne, 1996).
21
The COMT genotype in relation to the efficacy of entacapone has been investigated, but the genotype was considered to have only minor clinical relevance (Lee et al., 2002).
MAO-B inhibitors The selective monoamine oxidase-B (MAO-B) inhibitor selegiline has been used for many years with the inhibition of dopamine metabolism as the objective. Selegiline has also been proposed to slow the progress of the disease by a neuroprotective action (Heinonen and Lammintausta, 1991; Jenner and Olanow, 1996). The DATATOP study showed that initiation of levodopa therapy could be delayed after the use of selegiline (Shoulson, 1998). Some palliative benefit has been found but selegiline tends to increase peak levodopa concentrations with a subsequent risk of adverse events (Birkmayer et al., 1975; Poewe et al., 1987). The present view is that there is no evidence for neuroprotective benefit from selegiline (Miyasaki et al., 2002).
Dopamine agonists The first clinically used dopamine agonist was bromocriptine, which was introduced in the 1970s as add-on therapy to levodopa (Calne et al., 1974). Recently, a number of new agonists have entered the market, i.e. cabergoline, pramipexole and ropinirole. They differ in dopamine receptor selectivity and pharmacokinetics. All dopamine agonists except apomorphine have longer half-lives than levodopa, thus they may provide a somewhat more continuous dopaminergic stimulation than levodopa.
In a parallel-group trial of the COMT inhibitor tolcapone versus the dopamine agonist pergolide as add-on to levodopa it was found that levodopa in combination with tolcapone was better tolerated, showed a better adverse-event profile and provided greater improvements in quality of life (Koller et al., 2001).
Alternative drug-delivery systems for continuous use of dopamine agonists are currently being investigated. One such promising example is the rotigotine patch which offers continuous transdermal delivery (Metman et al., 2001).
Brain imaging techniques (PET and SPECT) will likely be used for future studies of neuroprotective strategies. Recent data indicate that dopamine agonists may slow the degenerative progression compared with levodopa, but it has been proposed that the results should be cautiously interpreted and compared with clinical signs of disease progression (Marek et al., 2002).
22
Apomorphine Apomorphine is a potent D1 and D2 receptor agonist which is administered subcutaneously, either as injection or as continuous infusion (Pietz et al., 1998). The elimination half-life is short, about 30 minutes (Gancher et al., 1989), onset of effect is rapid and apomorphine is therefore most commonly used in advanced PD as injections in the severe “off” state. Such injections result in significantly faster onset of action as compared to liquid levodopa according to a double-blind single-dose study (Merello et al., 1997b). Its potency in anti-PD response is comparable to that of levodopa, which makes apomorphine unique among the dopamine agonists (Kempster et al., 1990). Usually levodopa is used concomitantly with apomorphine, but a recent study showed that apomorphine infusion can be used as long-term monotherapy (Manson et al., 2002). Trials with intranasal (Kapoor et al., 1990) and sublingual (Lees et al., 1989) apomorphine have been performed with good effects but with some practical problems and poor local tolerability. Intravenous infusion via port-a-cath has been carried out for up to 13 months without complications (Stocchi et al., 1999). However this method resulted in severe thrombotic complications in another patient material (Manson et al., 2001).
Bromocriptine Bromocriptine has been used as add-on to levodopa therapy for decades with good effects (Lieberman and Goldstein, 1985). Early use of bromocriptine is not linked to reduced mortality compared to treatment with levodopa (Montastruc et al., 2001). In a randomised trial comparing levodopa and bromocriptine as initial treatment it was found that patients in the bromocriptine arm returned to baseline disability levels one year earlier than patients in the levodopa arm (Lees et al., 2001).
Cabergoline Cabergoline has the longest t ½ of all dopamine agonists, more than 60 hours. As compared to levodopa, cabergoline as initial treatment significantly delays development of motor complications, but the risk of serious adverse events is slightly higher (Rinne et al., 1998). Motor improvement after a dose is significant 24 hours after dose intake (Ahlskog et al., 1994). In animal studies, cabergoline reduces the intensity of levodopa-induced dyskinesia, suggesting that continuous stimulation may reverse dyskinesia (Hadj Tahar et al., 2000).
Pramipexole Pramipexole has been compared to levodopa as initial treatment (Parkinson Study Group, 2000). Development of dyskinesias was less common in the
23
group randomised to pramipexole, but, despite supplemental open-label levodopa, this group did not reach the same level of motor improvement as the levodopa group according to UPDRS.
Ropinirole A five-year study of ropinirole versus levodopa showed that ropinirole treated patients had a lower rate of dyskinesias, but more hallucinations and a significantly smaller improvement in motor scores as compared with levodopa treated patients (Rascol et al., 2000).
Other treatments
Levodopa esters The methyl and ethyl esters of levodopa are more water-soluble than levodopa itself and have therefore been tested for different routes of administration. The esters are readily hydrolysed to levodopa in plasma. The methyl ester showed to be equivalent to levodopa in reversing reserpine-induced akinesia in mice (Cooper et al., 1984) and intravenous and jejunal levodopa methyl ester infusions (100–250 mg/ml) in humans have shown significant motor improvement and close to constant levodopa levels in plasma (Juncos et al., 1987; Ruggieri et al., 1989). However, sublingual levodopa methyl ester was ineffective and subcutaneous injections provided an unpredictable response, which suggested that the methyl ester would be unlikely to become a practical treatment option (Kleedorfer et al., 1991). Subcutaneous and intramuscular injections of the ethyl ester have been suggested as a new rescue therapy for disabling “off” situations (Djaldetti and Melamed, 1996). In a rodent study, subcutaneously and intraperitoneally administered ethyl ester was found to produce striatal levodopa and dopamine concentrations similar to those obtained after intraperitoneal levodopa injection (Djaldetti et al., 1996). The accumulation of methanol/ethanol might be a potential problem for these approaches.
The butyl ester of levodopa has recently showed to produce adequate plasma levodopa concentrations after transdermal administration in rats (Sudo et al., 2002).
Glutamate antagonists Glutamatergic receptor blockade may improve motor response complications. Amantadine is a noncompetitive NMDA receptor antagonist, which has been shown to inhibit drug-indiced dyskinesias, but it also has other effects, such as enhanced dopamine release, inhibition of dopamine reuptake and anticholinergic effects. It was originally introduced as prophylaxis for influenza but was also found to have mild antiparkinsonian
24
effects (Parkes et al., 1974). In a double-blind placebo-controlled study it was found that a 2-hour intravenous infusion of amantadine improved dyskinesias by 50% without any loss of antiparkinsonian benefit from levodopa (Del Dotto et al., 2001). Amantadine is administered orally in clinical practice.
Memantine is another NMDA antagonist which is structurally similar to amantadine. It may improve parkinsonian symptoms but has no effect on drug-induced dyskinesia (Merello et al., 1999).
Anticholinergics Anticholinergic drugs were used in the pre-levodopa era but have no place in modern pharmacotherapy of PD. However, low doses may be efficacious against dystonia (Poewe et al., 1988).
Non-pharmacological treatment
Physiotherapy The phenomenon of cueing is a puzzling feature of PD. A patient who cannot voluntarily initiate gait could easily climb a flight of stairs or start walking if a floor marker was placed in front of the feet (Azulay et al., 1999). These paradoxical abilities seem to have no relation to pharmacological stimulation. Various types of sensory cueing (auditory, visual) have been investigated (Rubinstein et al., 2002 for review) and it has been proposed that cueing is an important complement to conventional therapy in the daily management of gait disturbances associated with advanced PD. Cueing also shows up in studies where patients are obliged to perform motor tasks like finger tapping or walking. A patient in an “off” situation can suddenly perform the task on demand, immediately falling back into the “off” state after the required performance, which in a way can bias the motor outcome in a clinical study. Frequent and lengthy observations may overcome this problem to reflect the motor function as correctly as possible.
Physiotherapy is generally considered an important complement to pharmacotherapy in PD, for increased motor control (Nieuwboer et al., 2001). There are numerous physiotherapy techniques and only a small number of patients studied. Therefore, in a Cochrane review, it was concluded that there was insufficient evidence to support or refute the efficacy of physiotherapy in PD (Deane et al., 2001).
25
Neurosurgery The interest in neurosurgery for the management of PD has been growing in the recent years. The lesioning techniques pallidotomy and thalamotomy are the oldest interventions on the menu, while deep brain stimulation (DBS) is a recent alternative. Stimulation of the thalamus is a successful treatment for tremor-dominant PD (Koller et al., 2000), whereas high-frequency stimulation of the subthalamic nucleus (STN) affects all motor symptoms of PD and drug-induced dyskinesia (Bejjani et al., 2000). DBS of the internal globus pallidus (GPi) is more effective in ameliorating drug-induced dyskinesia, but with successive reduction in dopaminergic pharmacotherapy after STN stimulation a similar reduction in dyskinesia can be achieved (Krack et al., 1998). Therefore, considering that STN DBS is more effective against akinesia, bilateral STN stimulation has become the method of choice. The features of DBS opens up new insights in the function of the cortical-basal ganglia-cortical circuits, and STN stimulation is proposed to involve not only motor, but also psychomotor regulation (Krack et al., 2001). Acute high-frequency STN stimulation mimics an acute levodopa challenge with a similar decrease in UPDRS motor scores. “Off” period dystonia is then suppressed but an increased voltage can induce dystonic dyskinesias, resembling diphasic dyskinesias seen with levodopa treatment. A further increase in voltage leads to choreic peak-dose pattern dyskinesias (Krack et al., 1999). This implies that different levels of STN stimulation result in different motor response, which could be translated to dose dependent motor outcome with different levels of levodopa administration.
The motor response to DBS has been compared to the long-duration response of levodopa in early PD, because of the reduced interdose trough disability. Short-term intravenous levodopa infusion produces the same benefit as DBS whether stimulation is on or not, but when the infusion is stopped the patient turns akinetic off stimulation and only moderately bradykinetic on stimulation (Nutt et al., 2001). Some patients have been able to completely withdraw levodopa therapy following STN DBS. These patients experience no motor fluctuations or dyskinesias, but patients still on medication do (Vingerhoets et al., 2002). Patients who require medication therefore seem to be dependent of continuous dopaminergic stimulation.
The pattern of DBS-related adverse events includes severe and irreversible complications, such as intracranial hemorrhage, and even the rate of mortality is alarmingly high according to some of the recent literature (Hariz, 2002 for review). Postoperative changes in mood and in personality have also been reported (Houeto et al., 2002). Therefore, a careful patient selection for DBS surgery is important to avoid side effects. Young, non-demented patients with no psychopathology, who have failed to control symptoms with conventional therapy, are ideal candidates.
26
Transplantation Experimental research with cell transplantation, e.g. embryonic stem cells, has shown promising results (Piccini et al., 1999), but this is not a clinically useful strategy for different reasons. Instead, grafting of animal cells (Larsson et al., 2001) or stem cells cultured in vitro are alternatives for the future and hopefully a cure of PD. Regulation of the growth of the graft is one of the challenges, to prevent tumours and “off” phase dyskinesia (Hagell et al., 2002). Dyskinesia in the absence of dopaminergic medication has in fact complicated the outcome of neuronal transplantation (Ma et al., 2002b).
The discovery of neurogenesis in adult brains (Eriksson et al., 1998) has encouraged further research of cell transplantation in PD.
Motor complications
Fluctuations The anti-PD medication with levodopa is very effective and uncomplicated in the early stages of the disease. When patients are “on”, symptoms are virtually completely eliminated. This early stage of PD is often called “the honeymoon period” or “the good years”. After a few years, however, patients feel the effect duration of each dose of levodopa and they experience predictable “wearing-off” episodes, also called end-of-dose deterioration (Djaldetti and Melamed, 1998). Wearing-off has been described to occur when the plasma levodopa level has fallen to approximately 50% of the peak concentration (Kempster et al., 1989).
Early in PD, the response of a levodopa dose is long, but it decays slowly, proportional to the progression of the disease. This seems related to reduced dopamine storage capacity as the amount of remaining nerve terminals is reduced (Nutt et al., 1995).
A clearly defined therapeutic window then manifests itself, with a lower threshold for response and an upper threshold for involuntary movements, hyperkinesias (Chase, 1998a). This window narrows with the progression of the disease. In very advanced stages the “on-off” phenomenon appears. The ”on-off phenomenon” describes a clinical pattern of mobility/immobility, in which a patient who is mobile or “on” suddenly changes to immobility or “off”. This state is very difficult to manage with conventional oral medication. In 1974, the features of the “on-off” phenomenon were well described (Sweet and McDowell):
27
The functional changes for the patient are very distressing. A patient who is quite mobile, although bothered by dyskinesia, suddenly becomes very akinetic over a period of minutes. Tremor also may re-emerge while the dyskinesia has stopped. These changes may be reversed within a few minutes or a few hours. The effect is so sudden that it has been likened to a light switch action and termed the ‘on-off-response’. Since it is unpredictable and severe, this response is quite incapacitating. A patient who goes out shopping or walking never knows when he may become immobile in a store or riveted to a park bench.
Most patients treated with levodopa develop fluctuations in motor performance. After 3–5 years of treatment one third, after 5–7 years about half, and after 10-12 years nearly all patients suffer from the fluctuations (Rinne, 1983; Markham and Diamond, 1986). These figures are in line with a recent literature review, where the authors suggest a 40% likelihood of developing motor fluctuations within 4–6 years of levodopa treatment (Ahlskog and Muenter, 2001).
In a study of the evolution of motor fluctuations in PD it was found that patients who develop a fluctuating response to levodopa have better long-term functional ability because of a greater levodopa response (McColl et al., 2002). Non-fluctuating patients were more prone to develop mid-line, axial, disability, such as dysarthria and balance problems.
Non-motor symptoms, such as pain, fatigue, anxiety and depression may also vary during a day and are often of greater impact for the patient than are the classical motor symptoms (Marr, 1991; Shulman et al., 2002). Some symptoms are always linked to “off” episodes and relieved when the patient turns “on”, thus non-motor fluctuations are important to evaluate along with pure motor fluctuations (Hillen and Sage, 1996). Mood elevation and anxiety reduction was related to improved motor performance after levodopa infusion in a double-blind placebo-controlled study (Maricle et al., 1995).
Mechanisms of motor fluctuations The typical motor response produced by a single dose of levodopa, lasting for minutes to hours and then rapidly declining, is called the short-duration response (SDR) (Muenter and Tyce, 1971). The long-duration response (LDR) is instead developing over days to weeks of chronic levodopa therapy and would decline at the same slow rate with cessation of levodopa (Cotzias et al., 1967; Muenter and Tyce, 1971). LDR might be explained by silent binding sites, i.e. buffering molecules which protect their ligands (e.g. dopamine) from fast degradation, thus assuring physiologically relevant concentrations of dopamine in the extracellular space for a prolonged period (Zoli et al., 1999). In early PD, the response of a levodopa dose is long, partly because storage of dopamine is possible, and the SDR is obscured by the LDR (Nutt and Holford, 1996). The LDR disappears slowly with time,
28
proportional to the progression of the disease (Nutt et al., 1995). A study of the responses in 18 levodopa naïve patients before and 4 years after introduction of levodopa revealed a progressive increase in SDR magnitude after levodopa withdrawal (Nutt et al., 2002). The magnitude of the SDR was defined as difference between peak and baseline finger tapping speeds. A shortening of the SDR was not seen over the 4 years, implying that changes in the LDR is more important to the development of motor fluctuations.
The mechanisms behind the development of motor complications are still unknown but it was shown in the early stages that there is a clear relationship between levodopa concentration in plasma and the degree of parkinsonism (Tolosa et al., 1973). A simultaneous monitoring of levodopa in plasma and clinical response has brought some understanding of the unexpected fluctuations in motor performance. For example the impaired effect of levodopa after meals could objectively be explained by plasma curves showing a lower amount of absorbed levodopa (Nutt et al., 1984).
When PD has reached the wearing-off state, fluctuations in response to levodopa reflect the fluctuations in plasma levels (Kempster et al., 1989; Riley and Lang, 1993). Motor response latency from orally ingested levodopa decreases from about 60 minutes in early PD to 20 minutes in late PD (Sohn et al., 1994). The close time-effect relationship between levodopa in plasma, striatal uptake and release of dopamine in advanced PD has been demonstrated using PET methodology (Tedroff et al., 1992). The results, showing a very short time to effect in advanced PD, are probably due to decreasing capacity of dopamine storage as a consequence of the dopaminergic denervation. Displacement of the dopamine antagonist [11C]raclopride has been reported to correlate with disability of PD. A large striatal dopaminergic nerve-terminal deficiency correlates to a high capacity for levodopa to increase synaptic dopamine and displace [11C]raclopride binding (Tedroff et al., 1996). These presynaptic mechansisms are likely to play a key role in the pathogenesis of motor complications.
Postsynaptic changes contribute to the pathogenesis of motor fluctuations as well. It has been suggested that intermittent stimulation of postsynaptic dopaminergic receptors leads to alterations in synaptic transmission within the basal ganglia, favouring the appearance of motor fluctuations and dyskinesias (Chase et al., 1996). It has been hypothesized that pulsatile dopaminergic stimulation leads to enhanced NMDA receptor subunit phosphorylation in the striatum. These receptors then become more sensitive to glutamatergic corticostriatal input, resulting in motor fluctuations and dyskinesias (Metman et al., 2000).
29
Dyskinesias The most common side effect of dopaminergic medication is dyskinesia. The nomenclature is somewhat confusing and often the term dyskinesia means choreic involuntary movements, but also dystonia is in fact included in the expression. Dyskinesia involves three categories: “off” period dystonia, biphasic dyskinesia and peak-dose dyskinesia (Marconi et al., 1994). “Off” period dystonia occurs in the “off” state and disappears with dopaminergic therapy (Poewe et al., 1988). Biphasic dyskinesias are also referred to as diphasic or onset- and end-of-dose dyskinesias or dystonia-improvement-dystonia sequences. The relation to high or low levels of levodopa is unclear and the phenomenon may possibly rather be induced by changes in concentration. A rise in the concentration produces dyskinesia, often dystonia, followed by a period of PD symptom relief, sometimes associated with peak-dose dyskinesia, and when the concentration falls, more dystonia or violent hyperkinesia appears. The third category of dyskinesia, peak-dose dyskinesia, is monophasic and predominantly choreic. This type of dyskinesia can also be called hyperkinesia, or interdose dyskinesia and is typically associated with a plasma levodopa peak and complete relief of PD symptoms. The term “on” with dyskinesia mostly refers to the mobile state only disturbed by peak-dose hyperkinesia.
A video-electromyographic study of dyskinesias, with assessments every 5 minutes, revealed that there was no strict dichotomy between biphasic and monophasic dyskinesias (Marconi et al., 1994). Instead, there seemed to be a continuum between dystonia as a trough-dose marker and hyperkinesia as a peak-dose consequence.
Dyskinesia does not appear immediately when medication is first started, but develops after a few years. In the pre-levodopa era, patients that were exposed to levodopa late in the course of PD developed dyskinesias within a few months (Yahr et al., 1969). A present-time review of the literature showed that the risk of experiencing dyskinesias is approximately 40% after 4–6 years of levodopa therapy and 0% within the first year (Ahlskog and Muenter, 2001). The severity is progressively increasing (Nutt et al., 2002).
Dystonia occurred in the pre-levodopa era and foot dystonia was described as a common phenomenon of the disease (Stewart, 1898). However, long-term dopaminomimetic therapy has been suggested to worsen “off”-period dystonia and one-third of patients treated with levodopa for five years experience painful foot dystonia (Poewe and Lees, 1987 for review).
Drug-induced dyskinesia may occur in other conditions than idiopathic PD, for example in cortical-basal degeneration and progressive supranuclear palsy, which are differential diagnoses to PD (Frucht et al., 2000; Kim et al., 2002), but not in healthy subjects.
30
Mechanisms of dyskinesias There are studies claiming that levodopa is toxic in vitro (Jenner and Brin, 1998 for review). This has led to restrictions in levodopa treatment and a maximised amount of add-on therapies for the patient population. There is an ongoing debate on whether treatment of PD should start with levodopa or a dopamine agonist (Montastruc et al., 1999; Weiner, 1999). Comparisons of the new dopamine agonists and levodopa show that development of dyskinesia is delayed in the agonist treated groups, whereas the motor scores are generally more improved in the levodopa groups (Rascol et al., 2000). This might be a consequence of the superiority of levodopa regarding efficacy – the motor performance gets better but dyskinesia develops faster. Severity and the time-period to the development of motor complications during levodopa therapy have been suggested to be approximately the same, no matter if levodopa is prescribed early or late in the course of PD (Blin et al., 1988; Caraceni et al., 1991).
Short-acting dopaminomimetic agents tend to induce dyskinesia to a greater extent than long-acting drugs in monkeys (Bedard et al., 1986). A short-acting dopamine agonist known to induce dyskinesia was given as a continuous infusion to monkeys without inducing dyskinesia (Morissette et al., 1997). In this study it was found that the different modes of administration differently affected gene expression in the striatum, with possible dyskinetic/anti-dyskinetic consequences.
Dyskinesia may occur as a response to salient visual or auditory stimuli, regardless of pharmacological stimulation. This is explained by a burst firing of dopamine (Schultz, 1994).
Pattern of side effects should be taken into consideration when choosing therapy, e.g. patients with late onset of PD run a greater risk of developing hallucinations and are therefore often recommended to start with levodopa only. The development of dyskinesia is of relatively minor importance in these patients.
The severity of PD is probably the most important co-factor when it comes to development of dyskinesia. In a study from 1969 of 60 patients, previously untreated, who were treated with levodopa 3–8 g daily for 4–12 months, 37 developed involuntary movements within this short period of time. Only nausea was a more common side effect in this study where no AADC inhibitor was available (Yahr et al., 1969). The fact that dyskinesia develops fast in severely denervated patients implies that the mechanisms are more related to the disease itself than to the therapeutic agents.
In summary, the development of dyskinesia is probably related to disease severity and the mode of administration of dopaminergic therapy. Each dyskinetic episode is explained by a peak concentration of the anti-PD drug, or a sudden dopamine-releasing stimulus.
31
The management of motor complications
Oral drug-delivery A detailed history taking by the physician and education of the patient is of great importance for optimal therapeutic management (Verhagen Metman, 2002). Documentation of symptoms during normal activities of daily living should be encouraged. Monitoring of plasma levodopa concentrations might be useful in fluctuating patients, to determine if fluctuations are predicted by pharmacokinetic factors, which then can be modified by individualisation of the treatment (Frankel et al., 1990; Okereke, 2002).
The dosage of oral levodopa is difficult to establish when the therapeutic window is narrow and patients experience “on-off” fluctuations in motor performance. The strategy to optimise the dose is to decrease the doses and increase the number of dosage occasions (Calne et al., 1974). This improves the pharmacokinetic profile in plasma and the dopamine production is thereby smoother with several moderately high peaks during the day instead of few high peaks. This mimics the physiological tonic dopaminergic stimulation better than when levodopa is given only 3–4 times a day. Most patients in the severely fluctuating stage of PD take levodopa tablets once per hour or once per 2 hours.
Round-the-clock dopaminergic stimulation by means of cabergoline showed a reduction in the intensity of levodopa-induced dyskinesia in an animal study. The results indicate that continuous stimulation may reverse motor complications (Hadj Tahar et al., 2000). The severity of dyskinesias after a levodopa challenge was reduced after long-term DBS of the STN, suggesting that motor complications are at least partially reversible (Bejjani et al., 2000).
Blockade of glutamatergic transmission at the NMDA receptor, with for example amantadine, may ameliorate motor response complications, especially dyskinesias (Verhagen Metman et al., 1998).
Sustaine-release levodopa formulations were designed to produce stable plasma levodopa levels, but, as described above, are unpredictable and have a long Tmax. SR medication is to be taken infrequently, which is convenient but may prolong both “off” and hyperkinetic states in fluctuating patients. Therefore, a strategy to combine IR and SR formulations has been suggested, for immediate onset of action and prolonged response (LeWitt, 1992). This strategy has resulted in the development of a dual-release formulation of levodopa and benserazide, which provides a significantly shorter time to “on” compared with SR formulation, but with similar t ½ (Ghika et al., 1997; Descombes et al., 2001).
32
Inhibition of COMT increases AUC of levodopa and is also thought to reduce plasma levodopa fluctuations (Nutt, 2000) and provide more “on” time without dyskinesias (Heikkinen et al., 2001). However, increased Cmax has been shown in repeated dosing, and thus no large reduction in plasma levodopa variability (Baas et al., 2001).
“Time to on”, i.e. waiting for a dose to induce “on”, stands for 70% of the daily “off” periods (Merims et al., 2002), implying that doses should be taken more frequently. Frequent dosing requires lower individualised doses.
Levodopa solutions, with ascorbic acid added for chemical stability, allows for frequent dosage of small volumes of liquid. Another advantage is the rapid onset of liquid levodopa as compared to solid form for fast relief of “off” situations.
Attempts have been made to create computed algorithms for individualised therapy (Albani et al., 1987) but such techniques are probably difficult to use because of large inter-individual differences and temporal intra-individual alterations in dose requirements.
Frequent dose intakes are cumbersome and the effect is unreliable, mainly because of the erratic gastric emptying. When oral medication fails, infusion becomes an alternative for delivery of levodopa.
The principle behind constant levodopa infusion is to achieve continuous dopaminergic stimulation with an optimised dose that can be kept stable within the patient’s therapeutic window. To make this possible, the gastric emptying must be by-passed. Levodopa cannot be administered by transdermal or rectal routes. The alternatives remaining are parenteral and enteral routes of administration.
Intravenous infusion The first human experiments with intravenous levodopa infusion showed favourable effects in five severely “on-off” fluctuating patients, as compared to oral administration of levodopa (Shoulson et al., 1975). It was found that constant intravenous infusion of levodopa resulted in stable plasma levodopa concentrations and virtual disappearance of motor fluctuations when patients were supine. However, when rising from the bed, parkinsonian symptoms reappeared.
In a study of levodopa absorption and transport, continuous intravenous infusion was used (Nutt et al., 1984). The authors claimed that bypassing absorption by constant infusion produced a stable clinical state lasting for up to 36 hours. It has been shown, by means of intravenous infusion, that levodopa concentrations in ventricular cerebrospinal fluid mirror, but lag behind, concentrations in plasma (Woodward et al., 1993).
33
In studies of patients with motor response fluctuations described as chaotic and unpredictable, oral levodopa was replaced with continuous intravenous infusion (Quinn et al., 1982; Quinn et al., 1984). All patients remained continuously mobile and ambulant on infusion.
In twenty patients with predictable or unpredictable motor fluctuations in relation to oral levodopa, intravenous constant-rate infusion brought about a dramatic extension in the duration of motor response. Based on these results, development of SR levodopa was proposed for improved control of response fluctuations (Hardie et al., 1984). Further studies in seven “on-off” fluctuating patients who were given intravenous levodopa infusion at rates of 32-80 mg/h produced a smooth clinical response following plasma levodopa concentrations within narrow limits (Hardie et al., 1986). Considerable inter-individual differences in levodopa levels were found and plasma concentrations related to optimum response were between 0.3 and 1.6 mg/l.
Twelve patients with wearing-off or “on-off” responses were maintained in a fully ambulatory state during 1–4 weeks on intravenous infusion by means of a portable pump (Juncos et al., 1985; Chase et al., 1987). All patients had stable plasma levodopa levels and in the wearing-off patients response variations were virtually abolished. However, the “on-off” fluctuating patients continued to manifest significant fluctuations. In further studies of patients with “on-off” fluctuations constant-rate infusion actually improved motor outcome over time, although at a substantially slower improvement rate than in the wearing-off group of patients (Mouradian et al., 1987). Chase and co-workers later postulated that since levodopa infusion brought palliative benefits, it could also have prophylactic benefits in the earlier stages of PD (Chase et al., 1994).
The duration of the antiparkinsonian action of levodopa after sudden withdrawal of a constant, optimal-dose intravenous infusion was studied in 48 patients (Fabbrini et al., 1988). The duration of effect after withdrawal was significantly shorter in patients with “on-off” fluctuations compared to stable patients.
Round-the-clock intravenous infusion of levodopa in 12 patients for 7 to 12 days showed a gradual decrease of fluctuations in motor performance (Mouradian et al., 1990). The decreased motor variability persisted for several days after return to conventional oral therapy, but then started to increase again, back to baseline levels. It was concluded that virtually all patients who have developed motor complications would respond to steady-state infusions of levodopa. The authors suggested development of practical means for providing stable dopaminergic replacement chronically for ambulatory parkinsonian patients.
A study of intravenous infusions of levodopa and the dopamine agonist lisuride once again confirmed that continuous dopaminergic stimulation
34
could strikingly reduce “off” episodes in complicated PD (Stocchi et al., 1986). The authors further discussed the impossibility of referring to a standard treatment schedule, as the dosage of L-dopa was found to be highly variable from patient to patient. They concluded that intravenous infusion could be a valuable form of estimating the single individual’s drug requirements.
It is suggested in several studies that continuous levodopa infusion not only improves motor fluctuations but also widens the therapeutic window (Dizdar et al., 1996; Stocchi et al., 1996). The dyskinesia dose-response curve can be shifted to the right by continuous intravenous infusion and the maximum intensity of dyskinesias can be reduced, while the dose-response for relief of parkinsonism is not altered (Schuh and Bennett, 1993).
Since visual hallucinations may occur in advanced PD, probably as a consequence of hyperdopaminergic stimulation, high-dose intravenous levodopa infusion in five non-demented patients with daily visual hallucinations was tested (Goetz et al., 1998). The infusion doses were 1.5 mg/kg/h, administered as steady infusion or pulse infusion for 4 hours. Plasma levodopa levels with steady infusion provided a smooth curve whereas the curve produced with pulse infusion showed peaks and troughs. Although some patients became prominently dyskinetic, none developed visual hallucinations during the infusions. It was concluded that visual hallucinations do not relate simply to high plasma levodopa levels.
Short-term intravenous infusions may result in sclerosis of peripheral veins, necessitating central venous access, and since levodopa is too acidic and too insoluble it is not convenient for long-term parenteral administration (LeWitt, 1993; Chase, 1998b).
Intravenous levodopa infusion is sometimes used for standardised dosage in study settings. Often a 2-hour infusion of levodopa 1 mg/ml is used with concomitant oral AADC inhibitor (Nutt et al., 2002). These studies are not contributing to the experience with the outcome of infusions, because of the short time of infusion and the fact that steady-state is only reached, not sustained.
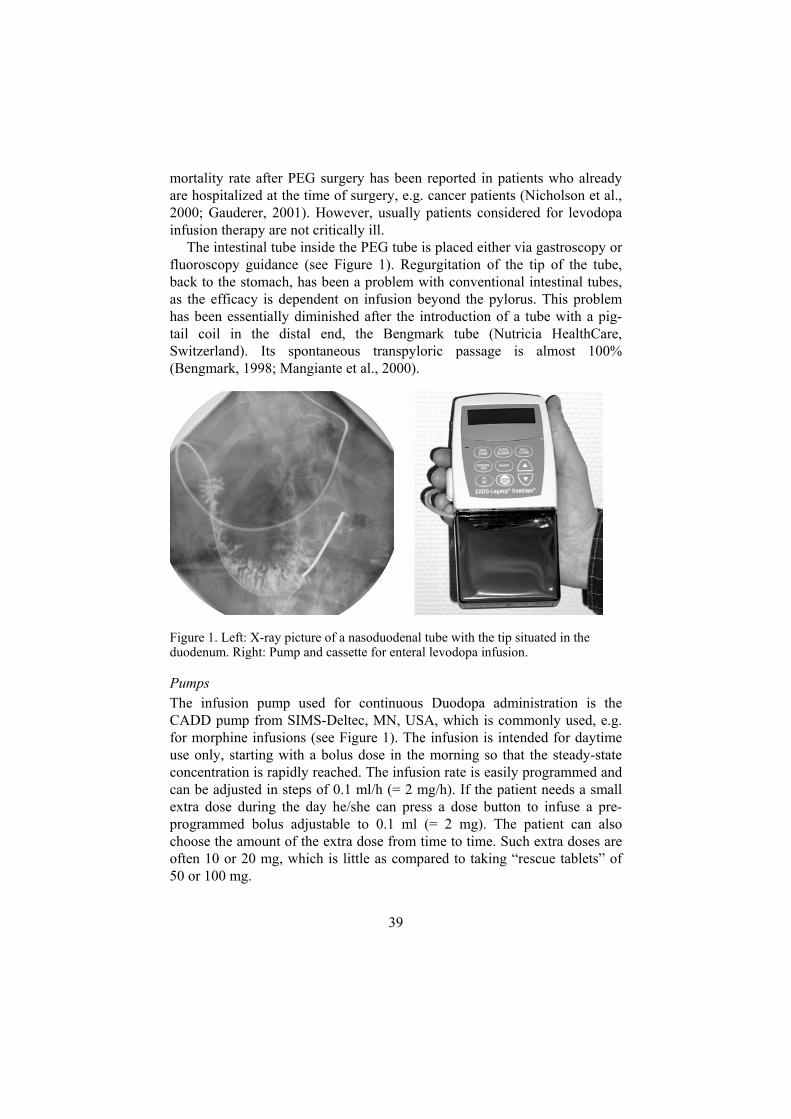
Intestinal infusion The term “enteral infusion” may sometimes incorrectly involve gastric infusion, besides duodenal/jejunal. The word “intestinal” is used to emphasize that the medication is infused into the small intestine and not into the stomach. The difference is of great importance in levodopa administration in PD.
In 1986, the first intraduodenal infusions were presented (Kurlan et al., 1986). The motor response in three patients with so-called resistant “on-off”
35
fluctuations was dramatically improved, well comparable to the effects of intravenous infusions. The infusion study was performed by means of nasoduodenal tubes. The authors concluded that the mode of delivery is suitable for chronic therapy, particularly with the advent of improved ambulatory infusion-pump technology. The group later presented results from 10 patients who were given nasoduodenal, nasogastric and oral levodopa (Kurlan et al., 1988a). The same reduction in motor fluctuations as in the previous study was found with duodenal infusion, but also gastric infusion produced improved mobility compared to standard oral therapy.
Intraduodenal levodopa infusion (50–70 mg/h) for up to 9 hours in four fluctuating patients resulted in a sustained “on” state with stable plasma levodopa levels (Frankel et al., 1989). Plasma levels were unaffected by oral protein loads but motor performance deteriorated anyway, probably because of a transport competition over the BBB.
In a patient with incapacitating motor fluctuations continuous daytime enteral infusion of levodopa/carbidopa increased the proportion of “on”-time to 100% (Cedarbaum et al., 1990). It was also found that the patient could progressively reduce levodopa intake while remaining continuously “on”.
Experience of long-term duodenal levodopa infusions has been reported by a group in New Brunswick, NJ, USA. They have presented several publications on reduced motor fluctuations with continuous intraduodenal levodopa infusions. The first reports were on four patients with severe “on-off” phenomena who used infusion of levodopa for at least 4 months (Sage et al., 1988a; Sage et al., 1988b). On infusion, motor fluctuations virtually disappeared. Further experience was presented (Sage et al., 1989b; Sage et al., 1990), indicating that continuous long-term levodopa infusion is a practical but complex form of therapy for patients failing on conventional treatment. Long-term experience from 22 patients was reported in 1998 (Syed et al., 1998). Patients chosen for infusion therapy had severe motor fluctuations unresponsive to conventional medical management. They had intractable and frequent “off” periods, intolerable dyskinesias, or both. Nearly all patients continued to have dramatically increased “on” time for the duration of follow up with daytime continuous levodopa infusion. Night-time-only infusion was carried out in one patient with sleep disturbance (Sage and Mark, 1991). Sleep was immediately improved but also daytime motor performance was gradually improved both regarding “off” episodes and dyskinesia, suggesting a carry-over benefit from the infusion. Intraduodenal infusion was also successfully used for treatment of complex dystonia in patients who had very narrow dystonia-free therapeutic windows (Sage et al., 1989a; McHale et al., 1990b).
A short-term, double-blind, placebo-controlled, crossover study of continuous duodenal and intermittent oral levodopa/carbidopa administration
36
in 10 patients (Kurth et al., 1993) showed that all patients had significantly decreased variability in plasma levodopa levels permitting better titration of levodopa dosage to individual requirements. Regarding motor function, seven patients experienced increased functional “on” hours and decreased number of “off” episodes. Because of the perceived benefit noted during the open-label phase of the trial, all patients elected to continue nasoduodenal infusion therapy after completion of the study. The authors concluded that duodenal infusion might be useful and beneficial for younger, motivated patients who experienced severe motor fluctuations in spite of optimal tablet therapy.
The occurrence of motor fluctuations despite a constant-rate levodopa infusion was studied in eleven fluctuating patients (Nutt et al., 1997). Some of the motor variability was explained by LNAA competition, but also pharmacodynamic factors were suggested to be responsible for the fluctuations.
The development of a new formulation intended for enteral infusion (Duodopa) was introduced in 1991 (Bredberg et al., 1993; Nilsson et al., 1998) and is described in detail below.
The issue of tolerance development to levodopa has been investigated by means of 2-hour and 21-hour intravenous infusions (Nutt et al., 1993). The duration of response after discontinuing infusion was shorter after the 21-hour infusion and it was suggested that long-term infusion might induce tolerance. The same was proposed in one patient on intravenous round-the-clock infusion, where the dose had to be increased with declining motor performance (Cedarbaum et al., 1990). However, the function was restored when the patient used daytime infusion instead. A shift in the dose-response curve to the right was also found in patients on 24-hour intravenous infusion, but motor response was still improved compared to oral therapy (Mouradian et al., 1990). Daytime intraduodenal infusion showed an increased sensitivity to levodopa with time, i.e. the opposite of tolerance, as average Css could be decreased by 50% after 1 month of infusion (Sjalander-Brynne and Paalzow, 1998). Also jejunal delivery of levodopa methyl ester showed decreased dose requirement with time (Ruggieri et al., 1989) and subcutaneous apomorphine infusion did not demonstrate any increase in dosage over time, even in patients on 24-hour infusions (Manson et al., 2002).
Subcutaneous infusion Continuous subcutaneous infusions of dopamine agonists such as apomorphine (Obeso et al., 1987; Colzi et al., 1998) and lisuride (Ruggieri et al., 1988; Stocchi et al., 2002) have shown similar stable effects as levodopa infusion. The pattern of side effects is different and long-term subcutaneous
37
infusion often produces skin reactions. Such infusions are intended as add-on therapy to oral levodopa, which means that motor fluctuations caused by fluctuating concentrations of levodopa still remain. However, apomorphine infusion has recently shown promising results as monotherapy (Manson et al., 2002). These patients demonstrated a gradual decrease in dyskinesia severity. Dyskinesias gradually returned after 2-3 months in patients who discontinued infusion monotherapy. There was a trend that patients who had been treated with apomorphine monotherapy for a longer period took longer time to deteriorate after discontinuation of monotherapy.
Infusion systems It has been known for a considerable period of time that advanced PD patients can benefit from continuous levodopa infusion. However, the experimental infusion systems were impractical. Intravenous long-term therapy is not desirable and the limited solubility of levodopa demanded large volumes of infusion fluid, making pumps and solutions laborious to handle. In one of the studies of intravenous levodopa infusion described above (Stocchi et al., 1986) it was stated that the low solubility, requiring large quantities of solvent and large infusion pumps, rules out an application in standard chronic therapy. Further, the chemical stability of levodopa and carbidopa in water solution is poor, necessitating frequent change of supply of the infusion fluid.
The gel suspension concept The problems with the early infusion systems led to the development of a gel suspension of levodopa and carbidopa (Duodopa). In studies preceding this new formulation both intravenous and intraduodenal infusions were used for pharmacokinetic calculations (Bredberg et al., 1990). The first study of the levodopa suspension was published in 1993 (Bredberg et al.). Five patients’ conditions were improved regarding motor fluctuations and plasma levodopa concentrations varied at most 2 fold as compared to 3-10 fold during oral therapy. A long-term follow-up was presented in 1998 (Nilsson et al.), showing sustained efficacy and decreased levodopa consumption (Sjalander-Brynne and Paalzow, 1998) after 2 ½ years.
Duodopa is an aqueous suspension of levodopa (20 mg/ml) and carbidopa (5 mg/ml) in a methylcellulose gel. The suspension is dispensed into 100 ml cassettes, connectable to an ambulatory pump for infusion via an intestinal tube. The chemical and physical stability of the active substances is high as compared to a water solution and one cassette is intended for one day’s use, approximately 16 hours of infusion.
38
The principle of the method is to continuously infuse levodopa and carbidopa directly into the duodenum. Absorption problems with erratic gastric emptying, early metabolization and LNAA competitive inhibition will be markedly reduced as levodopa can be quickly absorbed into the bloodstream.
Dosage of levodopa is very individualised and predefined dosage or dose schedules are impossible to use in the advanced stage of the disease. The possibility in individualizing the doses is one of the greatest advantages of infusion therapy with the new formulation. In clinical practice, the patient collaborates with the doctors and nurses responsible for adjustments of dose. For example, a patient can sense when an “off”-period is approaching, before it is visible for the observer. In that case, an extra dose of 10 mg might be taken in order to stabilize the condition. If a “rescue tablet” of 50 or 100 mg would be taken, a marked peak would show in the concentration-time curve, disturbing the smooth steady-state concentration, and the patient would become dyskinetic for a while. Such a peak can actually have consequences for the rest of the day, as the levodopa administration is constant, and the plasma levodopa concentration is shifted to a higher level.
The pharmacokinetic data show that a constant plasma levodopa level is held during infusion of the levodopa gel formulation. The concept implies an easier dosage regimen where very small adjustments in the infusion rate (0.1 ml/h, i.e. 2 mg/h) can be made. When oral levodopa dosage is adjusted the change is generally 50 or 100 mg on one dose intake at a time accompanied by large changes in plasma concentrations. Levodopa infusion offers a lower risk for over consumption.
In clinical practice the levodopa suspension is mostly used as monotherapy, which utilizes the continuous dopaminergic stimulation as far as possible without interference from other drugs with uncontrolled pharmacokinetics.
Tubing In short-term studies of enteral infusion, nasoduodenal tubes can be used, but in order to get long-term access to the small bowel, surgery is needed. The surgical techniques used are simple and well-known methods (Wollman et al., 1995 for review), e.g. percutaneous endoscopic gastrostomy (PEG) (Gauderer et al., 1980) with intestinal tube or radiologic gastrojejunostomy (Preshaw, 1981). Only local anaesthesia is needed. After the operation the infusion system can be used immediately and the patient can be mobilised. All patients feel pain from the wound but usually for not more than two days. The pain is seldom severe. The post-operative risk of complications is very small as no general anaesthesia is used and as the incision is very small. Transient local infection may appear but is uncomplicated. An increased
39
mortality rate after PEG surgery has been reported in patients who already are hospitalized at the time of surgery, e.g. cancer patients (Nicholson et al., 2000; Gauderer, 2001). However, usually patients considered for levodopa infusion therapy are not critically ill.
The intestinal tube inside the PEG tube is placed either via gastroscopy or fluoroscopy guidance (see Figure 1). Regurgitation of the tip of the tube, back to the stomach, has been a problem with conventional intestinal tubes, as the efficacy is dependent on infusion beyond the pylorus. This problem has been essentially diminished after the introduction of a tube with a pig-tail coil in the distal end, the Bengmark tube (Nutricia HealthCare, Switzerland). Its spontaneous transpyloric passage is almost 100% (Bengmark, 1998; Mangiante et al., 2000).
Figure 1. Left: X-ray picture of a nasoduodenal tube with the tip situated in the duodenum. Right: Pump and cassette for enteral levodopa infusion.
Pumps The infusion pump used for continuous Duodopa administration is the CADD pump from SIMS-Deltec, MN, USA, which is commonly used, e.g. for morphine infusions (see Figure 1). The infusion is intended for daytime use only, starting with a bolus dose in the morning so that the steady-state concentration is rapidly reached. The infusion rate is easily programmed and can be adjusted in steps of 0.1 ml/h (= 2 mg/h). If the patient needs a small extra dose during the day he/she can press a dose button to infuse a pre-programmed bolus adjustable to 0.1 ml (= 2 mg). The patient can also choose the amount of the extra dose from time to time. Such extra doses are often 10 or 20 mg, which is little as compared to taking “rescue tablets” of 50 or 100 mg.
40
Objective
The general objective for the present investigation was to develop new techniques for assessment and management of motor complications in PD.
An observational study of levodopa pharmacokinetics and motor function was performed in an unselected population of patients during activities of daily living. Levodopa pharmacokinetics was further studied in fluctuating patients on sustained-release tablets as monotherapy compared to continuous intestinal infusion of levodopa. In these two studies, variability in concentration-time curves and in motor performance was investigated to test the hypothesis that fluctuating pharmacokinetics is responsible for motor fluctuations.
Patients with long-term intestinal levodopa infusion were followed for assessment of effects, side effects and dose adjustments over time to investigate any signs of tolerance development or sensitisation to this new route of administration of levodopa.
In the development of techniques for individualisation of levodopa dosage an automatic dose dispenser was tested for usability purposes. Patients’ thoughts on such an instrument are valuable for future applications.
Self-assessment of symptoms is important in the individual management of PD and enables investigations of patients in normal daily activities. Paper diaries are not reliable and therefore an electronic diary has been studied to evaluate the rationale for its use in clinical trials or routine treatment of motor complications in PD.
41
Methods
Patients Eighty-two patients (27 women and 55 men) with PD participated in the five studies. In total, the results from 94 patients are reported, with some participating in more than one study. Age ranged from 39–85 years and all stages of PD were represented. The research ethics committee of Uppsala University approved all studies before any interventions were made and all patients gave their written informed consent to participate.
In study II there were four early drop-outs because of withdrawn consents and their data are not presented. In study III six patients stopped infusion therapy during the long-term observation, but their data are included as far as possible. In the detailed long-term follow-up of nine patients within study III, four dropped out. Two of them had stopped infusion treatment because of cognitive impairment and two were restrained from participation because of severe orthostatism and severe kyphosis, respectively.
Study designs and medications
Study I An observational study in 10 patients at different stages of the disease (Hoehn and Yahr range 2–4) and on individual drug combinations. The aim was to define both symptomatology and levodopa pharmacokinetics after oral administration in an unselected group of PD patients on different drug combinations during daily activities at home and at work. A physician and a nurse visited the patients’ homes during one day, starting with the first morning dose of levodopa and for the following 10 hours. Patients were on their ordinary anti-PD medication and spent the day following normal routines. To follow the intra-individual variations in plasma concentrations of levodopa and 3-O-MD during the day at a high resolution level, blood samples were taken every 20 minutes. Concurrently, motor function was
42
rated by the patient and by the investigators. With regard to the time schedule and study design, evaluation procedures such as the tapping test, walking time and UPDRS were not applicable as they would disturb normal activities. Investigators’ estimations were done by observation only, using a global “on-off”-scale graded from -3 (severely “off”) to +3 (severely hyperkinetic) where 0 is considered as a state of “normality”. The patients themselves evaluated mobility on a 10-graded VAS including the categories “tremor”, “bradykinesia”, “hyperkinesia” and “pain”. All medications during the 10 hour period were recorded regarding the active drug substances, the dose given, and exact dosing times. All food and fluid intake were monitored with the exact times given. Consequently, a dietician classified the intake of animal proteins and fat over the study period into four classes: none, low, moderate, and high. A quantitative assessment was not possible since the food was not weighed or analysed, which would have disturbed the daily routines.
Study II A randomised non-blinded crossover trial in 12 patients with motor fluctuations. The aim was to compare pharmacokinetics of levodopa between oral sustained-release tablets and intraduodenal infusion of carbidopa/levodopa. During a baseline week, all patients were converted from earlier medication regimens to monotherapy with SR carbidopa/levodopa 50/200 mg (Sinemet CR) with IR carbidopa/levodopa (Sinemet 12.5/50 mg) taken when needed. This “wash-in” week allowed for the elimination of prior long-acting anti-PD medication. Patients were randomised to continue either SR tablets (group 1) or to start nasoduodenal infusion of levodopa (group 2) during weeks 1–3. After week 3 patients were crossed over to infusion (group 1) and SR tablets (group 2), for the next 3 weeks. On the last day of the baseline week, plasma levodopa concentrations were determined every 30 minutes from 8 a.m. to 5 p.m. Medication was individually titrated for optimal effect, “near normality”, during the first week of each treatment period but could be further adjusted throughout the study if needed. One test day was run during the second week and two non-consecutive test days during the third week. Test days involved collections of blood samples every 30 minutes from 8 a.m. to 5 p.m., standardized video recordings hourly from 8 a.m. and opto-electronic movement analysis (PLM-test) hourly from 8:30 a.m. A comparative study between two drug preparations ideally should be double-blind and placebo-controlled. In this study, however, we found such a design impossible to perform in practice. Patients who have felt the benefits of smooth levodopa delivery can easily recognize it and distinguish it from oral medication. Patients have proved to sense when the duodenal tube has been dislocated to the stomach, as
43
levodopa is then only absorbed when the stomach empties. A double-blind study design where a dose for the levodopa infusion is set during a baseline week and used several weeks later is difficult to follow if it is not allowed to adjust the infusion rate. Pharmacodynamic changes with up- and down-regulation of striatal dopamine receptors may alter the dose requirement over a few days with any antiparkinsonian therapy. Further, the disease continuously progresses, which might have impact on the dose needed. Thus, small dose adjustments must always be allowed, if needed, for ethical reasons. Very small bolus doses (5–20 mg of levodopa) are possible with the infusion pump, whereas such small amounts cannot be provided with tablets. The use of “rescue tablets” to compensate for a slightly too low infusion dose disturbs the pharmacokinetics and subsequently the effects. And if the preset infusion dose would be too high, the patient would be dyskinetic all the time, which could be very distressing. Such a design is far from clinical practice, where small adjustments of the infusion dose are needed quite frequently in the beginning and must be allowed for best possible treatment. Patients were at the hospital during adjustments of therapy and on test days but could choose to stay at home the rest of the time. During test days ordinary meals were served at fixed times. The location of the nasoduodenal tube was examined on the last day of infusion for all patients to confirm that all tubes were situated in the duodenum.
Study III A prospective and retrospective long-term follow-up of 28 very advanced PD patients started on continuous levodopa infusion treatment at Uppsala University Hospital during the period 1991–1998. Nine of the patients were evaluated in detail with repeated test occasions while applicability and complications were reported for the whole group. The nine patients were tested when administered optimal oral therapy on two non-consecutive days and then tested on several occasions, over periods of two non-consecutive days, while on infusion therapy during a period of 4–7 years. Preliminary results of follow-up after 2 ½ years have been published previously (Nilsson et al., 1998). On each test day patients were studied every 15–20 minutes from 8 a.m. to 5 or 7 p.m. by means of the PLM-test. Further, the patients were video-filmed every 30 minutes when performing a standardised sequence of motor tasks, “piano playing”, alternating hand movements, rising from a chair and walking. In addition, different disability scales were applied after overnight drug holidays to follow the progress of the disease. On each test occasion the catheter tip was checked by fluoroscopy to be situated in the duodenum. Change in levodopa consumption and concomitant medication was monitored in order to assess the long-term effects of the
44
infusion therapy. Safety aspects and reasons for discontinuation of infusion therapy were retrospectively investigated.
Study IV The aim of this study was to present and evaluate a new drug administration concept, which includes an electronic automatic dose dispenser for adjustable individualised delivery of microtablets. Twenty patients with PD tested the usability of this dispensing device and offered their opinion of the concept. Microtablets containing 1.25/5 mg of carbidopa/levodopa were produced and their properties were tested regarding dimensions, porosity, friability and tensile strength. The content of levodopa and carbidopa in twenty microtablets was analysed using HPLC. Only placebo microtablets were used for the usability tests and patients did not ingest any microtablets.
Study V A randomised usability test of a new electronic patient diary for real-time data capture in comparison with a conventional paper diary. The aim was to determine whether the electronic diary produced at least as good compliance as the paper diary, or better, with respect to answers within a determined time-frame. Twenty patients with PD diagnosed at least five years before the screening visit were enrolled. Ten patients were randomised to the paper diary and ten were randomised to the electronic diary. Patients were asked to fill in the daily questionnaire every two hours on two predefined non-consecutive days per week over four weeks. On each occasion, 10–11 questions were to be answered within 15 minutes of the scheduled time. An alarm was set on the electronic diary for each scheduled time. No reminders were provided for patients using the paper diary, but a predefined time schedule was enclosed and the patients were asked to enter the time at which they really filled in the questionnaire. The questionnaires were constructed to collect subjective symptoms and quality-of-life data.
Measuring levodopa and 3-O-methyldopa in blood
High-performance liquid chromatography Plasma levels of levodopa and 3-O-MD were assayed by reversed phase high-performance liquid chromatography (HPLC) with electrochemical detection, using a modification of the procedure described by Blandini et al. (1997).
45
Venous blood samples were collected into vacuum tubes containing 0.1 ml of sodium heparin (1000 U/ml), chilled on ice, and immediately centrifuged at 1200 g for 10 minutes. The supernatants were stored at -72°C prior to the analysis. Samples were thawed at room temperature and an aliquot of 100 µl plasma was deproteinized by adding 100 µl of 4.0 N HClO4. This was followed by dilution 1:5 with water and centrifugation at 12 000 g for 10 min at 4°C. Twenty µl of the supernatant were injected onto the HPLC system.
The HPLC system consisted of a pump (SunFlow 100, Spark Deutschland), equipped with a C18 reversed-phase column (150 x 4 mm, 5 µm). The system was connected to an autosampler (Marathon, Spark Holland). The coulochem II electrochemical detector had a 5011 high sensitivity analytical cell (ESA, Chelmsford, MA, USA). The potential of the work electrode was set at +0.35 V versus the reference electrode. Chromatograms were analyzed by a computing integrator (Turbochrom 4.1, Perkin Elmer NCI 900). The mobile phase (pH 3.1) consisted of 100 mM NaH2PO4, 0.5 mM OSA and 1.0 mM EDTA, mixed with 10 % (v/v) methanol. The elution was carried out isocratically at room temperature, at a flow-rate of 0.8 ml/min.
Calibration curves were constructed for levodopa and 3-O-MD (range 0.005 - 4.0 µg/ml) and linear regression was used to fit a straight line through the calibration points. The concentrations of levodopa and 3-O-MD in the samples (µg/ml plasma) were derived from the respective calibration curve by comparing peak heights of the two compounds in the samples with the peak heights of the standard mixture of both compounds. The detection limits for plasma levels of levodopa and 3-O-MD, at a signal to noise ratio of 3, were 2.0 and 3.0 ng/ml respectively. The recovery of the method was 98 % for levodopa and 99 % for 3-O-MD.
Pharmacokinetic calculations The area under the concentration-time curve (AUC 0-t) was calculated by using the linear and the logarithmic trapezoidal rule for ascending and descending blood concentrations, respectively, up to the last time-point.
The observed peak plasma concentrations (Cmax) and the times when they occurred (Tmax) for levodopa were derived directly from the plasma concentration-data for each subject. The steady state plasma concentration (Css) was calculated for both levodopa and 3-O-MD. Intra-individual variability is given as the coefficient of variation (CV = standard deviation/mean).
Oral clearance was calculated as dose divided by AUC.
46
Assessments of symptoms
Computerised assessments For objective assessment of PD symptoms in studies II and III we have used the PLM-test (Qualisys AB, Gothenburg, Sweden), which measures postural (P), locomotor (L) and manual (M) movements (Johnels et al., 1989). The technique is a development from computer assisted optoelectronic movement analysis, used in gait physiology and elite sports training (Steg et al., 1989). The test involves picking up an object from the floor, walking 1.5 meters forwards and placing the object on a shelf at the height of the patient’s chin. These movements should be repeated as many times as possible within 30 seconds at maximal possible speed. The patient wears reflective markers and the object is also reflective, allowing an infrared light-emitting camera to detect the movements. Only the time when the object was moved forward from the floor to the shelf is evaluated. The data are processed in a computer, resulting in a figure of movement time (MT), the mean of all completed movements in a 30 seconds session. Calculations of each of the three phases P, L and M are also available from the computer software. The PLM-test is considered strictly objective and has shown to provide fully reproducible evaluation of motor response after a single-dose of levodopa (Johnels et al., 1993).
Clinical assessments The need to describe PD symptoms in a standardised and trustworthy manner has led to development of a great number of clinical rating scales, which are often modified and later rejected because of the great difficulties in assessing PD. The Hoehn and Yahr scale is one of the oldest and describes the progression of PD in five stages (Hoehn and Yahr, 1967). The original scale did not include axial involvement or differentiation in stages of postural instability and it was therefore modified when it became a part of the Unified PD Rating Scale (UPDRS) (Fahn et al., 1987). The Hoehn and Yahr scale does not include dyskinesia as a marker for disease severity.
UPDRS is the most widely used scale of today (Mitchell et al., 2000) and it is also required by authorities for clinical drug trials. The six parts of the UPDRS are I. Mentation, behaviour and mood, II. Activities of daily living, III. Motor examination, IV. Complications of therapy, V. Modified Hoehn and Yahr staging, VI. Schwab and England activities of daily living scale. When a total UPDRS score is mentioned, it is normally the score of parts I-IV. Part II is historical information for both “on” and “off”. Part III, the
47
motor examination should be made in “off” state, preferably off drug, to evaluate the severity of PD. It may also be used for repeated characterisation of patients in clinical trials, but it is too time-consuming and cumbersome to use for frequent examinations.
We have also used Webster Rating Scale (WRS) and Northwestern University Disability Scale (NUDS). The WRS is scored from 0–30 where 0 is normal. NUDS is scored from 0–50 where 50 is normal.
“On-off” rating of a patient’s motor performance on scales for parkinsonism and dyskinesias have been used both in real-time and in video recordings for subsequent evaluations. In studies II and III, a standardised sequence of motor tasks was used; “piano playing”, alternating hand movements, rising from a chair and walking. This examination includes items 25, 27 and 29 of the UPDRS. The parkinsonism scale, derived from UPDRS item 31, was graded from –3 (marked bradykinesia) to 0 (normal) and the dyskinesia scale, derived from Goetz’ Dyskinesia rating scale (Goetz et al., 1994) was graded from 0 (normal) to 3 (severe choreic dyskinesia). To estimate the change in time the patients spent in different motor states on the video ratings, a “near normal” state was defined to correspond to the function between -1, 0 or +1, an “off” state to -3 and -2 and a “hyperkinetic” state to +2 and +3. “Near normality” represents a state where patients are practically undisturbed by symptoms, such as mild slowness or mild dyskinesias. The video recordings could not be assessed in blinded fashion since patients wore tubes and pumps during infusion therapy.
Patient self-assessments Rating scales for self-assessment can be an important support for clinical evaluation, and can be used in the home environment.
The visual analogue scale (VAS) is widely used in symptom evaluation of pain and could also be used for PD symptoms (Favre et al., 2000). VAS consists of a line, commonly 10 cm in length, which is anchored at either end by the extremes (“no disability” and “maximal disability”) of the variable to be measured, and the assessment is made by a mark on this line at an appropriate place between the anchor points.
The PD questionnaire-39 (PDQ-39) is considered the most appropriate instrument with which to measure health-related quality of life in PD (Marinus et al., 2002). PDQ-39 consists of 39 questions using verbal descriptive scales (VDS), with an ordering of the different outcomes on a categorical scale (Jenkinson et al., 1997).
In clinical trials of new antiparkinsonian drugs, entries in patient diaries should be made frequently, preferably several times a day, to record any fluctuations in motor performance. Although the accuracy and reliability of
48
self-assessment is high in PD patients (Brown et al., 1989; Louis et al., 1996), studies of compliance with patient diaries have shown that they are not filled in at the appropriate times, implying low compliance and non-representative outcomes (Stone et al., 2002). The use of electronic diaries could possibly improve the reliability of data. The device used to collect data for the electronic patient diary of study V was an Ericsson MC218. The electronic diary was connected to a Siemens S35 cellular phone. The equipment was placed in a briefcase measuring 32 × 25 × 5 cm and weighed about 1.5 kg. Data were transferred to the database without wires. The device was battery-sensitive and the battery level could be measured remotely. All diary data, patient compliance data, and technical performance data could be monitored through a secured website. The three questionnaires used were PDQ-39, daily questions and usability questions. Patients were asked to fill in the daily questionnaire every two hours on two predefined non-consecutive days per week over four weeks. On each occasion, 10–11 questions were to be answered within 15 minutes of the scheduled time. The PDQ-39 questionnaire was applied at the inception of the study and on the last day of the study. The patients using the electronic diary were asked to fill in usability questions regarding their experience of the method. Precautions were taken to minimize the influence of bias introduced by technical problems and any misunderstanding of the questions. The clinician demonstrated the questionnaire to each patient on the first visit, and on any following visit upon request. The demonstration of the hand-held computer was repeatable an unlimited number of times and these data were not recorded in the database. The time spent for initial training varied between 10 and 20 minutes per patient. The only operation required of the patient was to respond to the questionnaire. The electronic diary was automatically turned on by the alarm signal and data were automatically sent to the database.
System for intestinal infusion of levodopa/carbidopa Duodopa (US patent number 5,635,213, manufactured by NeoPharma Production AB, Uppsala, Sweden) is a stable gel suspension of levodopa and carbidopa. The suspension was administered through a nasoduodenal tube (Kangaroo, 8 FR, Sherwood Medical, UK) or through a permanent PEG with intestinal tube (Freka PEG Universal-Intestinal, Fresenius, Germany) by a portable pump (CADD-PCA, SIMS Deltec, MN, USA) (see Figure 1). A plastic cassette is attached to the pump. The cassette contains a plastic bag with a short tube, which is to be connected to a duodenal tube. Each cassette contains 100 ml of carbidopa/levodopa in concentration 5/20 mg/ml and is
49
intended for one day's use. The cassettes are disposable. The gel is infused into the duodenum at a rate of usually 1–6 ml/h (20–120 mg levodopa per hour). The pump and cassette are carried in a bag around the waist or over the shoulder.
The nasoduodenal tubes in study II were placed with the assistance of fluoroscopy (see Figure 1). If clinical signs of deterioration in motor performance appeared, the location of the tube was checked by means of fluoroscopy. All tubes were checked on the last day of infusion treatment. One cassette (2,000 mg levodopa) per day was sufficient for most patients though some required two cassettes. Infusion was given during the daytime only, from approximately 6 a.m. to 10 p.m.
Automatic dose dispenser for oral levodopa/carbidopa
The concept The concept of individual drug dispensing “by counting” is presented, as a potential solution to the shortcomings of standard oral formulations. This new concept is based on the use of standardised dosage units, each containing a sub-therapeutic amount of the active ingredient. These units are referred to as microtablets (diameter: 3 mm, thickness: 1.3 mm).
For the application in PD, the dispenser would contain microtablets, each containing 5 mg levodopa and 1.25 mg carbidopa. The automatic dispenser delivers the correct dose for each patient who then is able to swallow them either undissolved or dissolved in liquid.
Because of the limited size of the tablets, some kind of counting device is required for patient assistance. In this study, an automatic dose dispenser is described and evaluated (Figure 2). This is an improved and upgraded version of a prototype presented previously (Aquilonius et al., 1998).
The cassette portion of the device, which can be manually refilled but is also planned to be available as a new prefilled cassette for convenience, contains >2000 microtablets. The microtablets are transported within the dispenser by the plastic components which are moved by a battery driven motor. A photocell monitors the number of microtablets transported from the cassette to the receiving compartment and the electronic motor stops when the pre-set number of microtablets has passed the photocell. The actuator then causes the microtablets to be emptied from the receiving compartment in the device into an external collector or a glass of water. A digital display guides the patient through the process, i.e. using the buttons, the patient starts the dispenser device and sets the correct dose; instructions then appear to empty the microtablets from the device into the collector. The weight of
50
the device, without microtablets, is 232 grams and the dimensions are 132 mm (height), 63 mm (width) and 32 mm (thickness).
Figure 2. The dispenser comprises a cassette filled with microtablets, buttons operated by the patient (with an associated digital display) for dose adjustment, a battery-driven electronic motor, a photocell monitoring the number of microtablets dispensed from the cassette to a receiving compartment, and an actuator, by which the microtablets are emptied from the receiving compartment into a collector or a glass of water.
The patient study The usefulness of the automatic dose dispenser and patient acceptance of the device were evaluated in 20 patients with PD.
Patients were characterised according to medication, concomitant diseases and severity of PD. The UPDRS, parts I, II and IV, and the modified Hoehn and Yahr scale were applied.
All patients were instructed once on how to use the dispensing device. After the demonstration, the patients were asked to operate the dispenser themselves. Any additional instructions were recorded. Patients were asked to start the dispenser, to set the dose to 65 mg (from a default dose of 20 mg), to confirm the dose and to release the 13 microtablets into a glass. The patients were also asked to pick up five microtablets from the table. These tests were observed by the investigators, who recorded the outcomes. After the tests, all patients answered 15 usability questions relating to their impressions of the method. Only placebo microtablets were used for the tests and patients did not ingest any microtablets.
51
Statistics and data management
Studies I and IV Only descriptive statistics were used together with calculations of mean values and SD. Pharmacokinetic calculations are described earlier.
Study II Pharmacokinetic calculations (Css, SD and CV) were performed as described earlier. Statistical significance was calculated at 95% CI.
During a test day on infusion for one patient, the cassette was accidentally disconnected from the pump for a few hours leading to decline in both plasma levodopa concentrations and motor performance. The test day continued with the cassette reconnected and the plasma levodopa concentrations were used in the analysis of statistical significance but excluded in terms of mean Css and mean CV because of the period of absence of levodopa administration.
Study III In the nine patients tested, movement time (MT, from the PLM-test) on infusion was compared to corresponding MT during oral therapy (the 1st and the 2nd day separately). The non-parametric Kruskal-Wallis test was applied for statistical analysis. The Squared Ranks test for variance was used to obtain a comparison of MT variability. The statistical calculations have been adjusted to the expected increase of MT with age in a healthy population (Johnels et al., 1989). Analyses were performed by means of StatView 4.02 (Abacus Concepts, Inc., Berkeley, CA).
Study V An independent clinical research organisation (IRW Consulting, Täby, Sweden) performed all data entry for data collected in the paper diaries. All data management activities were measured to estimate the time taken.
In the comparison of the two methods of data collection, patient compliance was defined as the number of time points at which records were completed in the electronic diaries against the number of time points at which records were completed in the paper diaries. Because the patients were instructed to fill in the time points at which each questionnaire was completed in the paper diaries, compliance with the predefined 15-minute time intervals was also checked (the same interval was used for the electronic diary). The primary objective was to compare the electronic diary with the paper diary with respect to compliance, measured as the number of completed data records and the number of dropouts. Secondary outcomes
52
were PDQ-39 score, self-assessment of symptoms, data validity, and security, and the experience of the method as assessed by the patients. Data were summarized by descriptive statistics, with compliance presented as percentages, PDQ-39 data presented as the total sums of the two groups, and daily questionnaires presented by the medians and ranges of intra-individual differences. Statistical hypothesis testing was performed using a Mann-Whitney U test to compare the paper and electronic diaries with respect to the proportion of completed records. The differences between PDQ-39 scores at the start and the end of the study were tested using a Wilcoxon matched pairs test (non-parametric test). Statistical tests were two-tailed with a significance level of p < 0.05. Statistica 5.5 (Statsoft Inc, Tulsa, USA) was used for statistical calculations.
53
Results
Levodopa pharmacokinetics (papers I and II) The levodopa dose was correlated to plasma Css and AUC for all 22 patients but one. This patient, in study I, who was the only one treated with tolcapone, diverged from the correlation coefficient of dose and AUC as compared to the other patients. A clear peak in the plasma concentration-time curve for levodopa was obviously related to the oral intake of levodopa in 44 out of 58 dosing events in the ten patients of study I. Seven of the patients responded with plasma peaks to more than 75% of the doses taken.
Oral levodopa therapy was associated with fluctuations in plasma levodopa levels in all patients to an extent of 25–70% (CV) in the 10 patients of study I and 19–74% in the 12 patients on SR levodopa in study II. On infusion, the latter patients showed a significantly decreased variability range in plasma levodopa; 5–36% (see Table 1). There were large inter-individual differences in dosage in both studies.
Figure 3. Patient E took 50 mg levodopa on 10 occasions and patient F took 100 mg levodopa on 5 occasions during the 10 hours.
0
1
2
3
00:00 05:00 10:00Time (hours)
Plas
ma
levo
dopa
(µg/
ml)
EF
54
The strategy to manage motor fluctuations with frequent small doses was illustrated in study I, where two patients had a total dose of 500 mg of levodopa distributed on 10 and 5 occasions respectively. Their plasma concentration-time profiles are shown in Figure 3. Frequent small doses resulted in lower peaks and higher troughs, whereas Css was the same for both patients. The decreased variability was described by the CV; 52% for the hourly regime versus 70% for dosage every two hours.
There was a considerable variation in the plasma concentration-time curves of levodopa in the four patients that used COMT inhibitors, mean CV was 41% (range 34–56%). As expected, the ratio of Css 3-O-MD to Css levodopa was significantly lower in these patients.
CV in plasma levodopa levels in the use of SR tablets of levodopa/carbidopa and SR capsules of levodopa/benserazide was similar to CV in IR formulations. Two patients in study I combined IR and SR formulations to get quick onset and long duration of the levodopa dose. CV was 56% and 25% respectively in these individuals. CV was 69% in a patient who used SR levodopa every 5 ½ hours. In this patient, levodopa plasma concentration went down to zero in between the doses, but no symptoms appeared.
Table 1. Data on plasma levodopa Css and CV from studies I and II.
Study Css (µg/ml) CV (%) I oral range min-max 0.5 – 7.1 25 – 70 I oral mean (± SD) 2.0 (± 1.9) 48 I oral median 1.3 49 II oral SR range min-max 1.0 – 4.3 19 – 74 II oral SR mean (± SD) 2.7 (± 0.8) 34 II oral SR median 2.5 31 II infusion range min-max 1.4 – 4.8 5 – 36 II infusion mean (± SD) 2.7 (± 0.9) 14 II infusion median 2.7 13 I oral, n = 10. II oral, n = 36. II infusion, n = 34. In study II n represents number of test days in the 12 patients.
Oral SR formulation versus intestinal infusion Variation in plasma levodopa concentrations expressed as mean Cmax divided by mean Cmin per patient ranged 2.4–9.2 fold during oral SR tablet therapy, and 1.3–2.5 fold during intestinal infusion. Mean intra-individual CV for the plasma concentrations of levodopa following oral medication was significantly decreased during continuous infusion (see Table 1 & Figure 4).
55
Figure 4. Mean values of intra-individual CV for plasma levodopa in the 12 patients on oral SR levodopa tablets and intestinal levodopa infusion.
Figure 5. Distribution of CV for plasma levodopa from all test days in the 12 patients; 36 test days on oral SR tablets and 34 test days on intestinal infusion.
Figure 6. The typical pharmacokinetic profiles of SR levodopa tablets and intestinal levodopa infusion. All test days from one patient (no. 14) are presented. The upper panel shows concentration-time curves on SR tablets and the lower panel shows the curves on infusion. CV for plasma levodopa are 25%, 27% and 22% on the three test days on tablets and 6%, 6% and 10% on infusion. Oral SR medication was 500 mg at 7:00 and 400 mg at 10:00, 13:00 and 16:00. Extra IR doses were taken as 50 mg at 14:00 on test day 1, 50 mg at 9:20 on test day 2, and 50 mg at 8:50 on test day 3 of oral therapy. Infusion rates were 116 mg/h on the first two test days and 112 mg/h on test day 3 of infusion therapy. No extra doses were taken during this period.
0
1
2
3
4
5
6
7
07:00 10:00 13:00 16:00Clock time
Plas
ma
levo
dopa
(µg/
ml)
Test day 1 Test day 2 Test day 3
0
1
2
3
4
5
6
7
07:00 10:00 13:00 16:00Clock time
Plas
ma
levo
dopa
(µg/
ml)
Test day 1 Test day 2 Test day 3
57
The significant difference in CV between the treatments was 19% (p < 0.01). Median ranges between lowest and highest daily CV were 27–37% for tablets and 9–18% for infusion, further data are presented in Figure 5. The mean of all patients’ mean variances was 0.9 (range 0.4–2.0) [µg/ml]2 on CR tablets and 0.2 (range 0.0–0.6) [µg/ml]2 on infusion. This difference was also significant (p < 0.01). The plasma concentration-time profiles are exemplified in Figure 6.
Mean Css was 2.7 µg/ml for both methods of levodopa administration. Whereas the CV of the overall mean curve was 16% with oral therapy, it was 3% with infusion. The correlation coefficient was 78% when mean Cssoral was plotted against mean Cssinfusion of all patients. All patients had lower Cmax of levodopa given by infusion as compared to tablets. Mean (± SD) Cmax of levodopa was 4.6 ± 1.4 µg/ml on SR tablets and 3.4 ± 1.2 µg/ml on infusion. The decrease in Cmax ranged from 8–40% (mean 24%). All but two had higher mean Cmin of levodopa given by infusion as compared to tablets. Mean (± SD) Cmin of levodopa was 1.4 ± 0.8 µg/ml on SR tablets and 2.0 ± 0.7 µg/ml on infusion. The increase in Cmin ranged from -17–542% (mean 85%). The mean variability of plasma levodopa levels, expressed as difference between Cmax and Cmin, was 3.2 ± 0.9 µg/ml with SR tablets and 1.4 ± 0.7 µg/ml with infusion. This difference between treatments is statistically significant (p < 0.0001).
Mean (± SD) oral clearance for levodopa was 609 ± 94 ml/min on SR tablets and 575 ± 119 ml/min on infusion.
Average intra-individual CV of 3-O-MD concentrations was 6% for both treatments. Mean Css was 11.0 µg/ml on SR tablets and 10.5 µg/ml on infusion. Mean ratio Css3-O-MD/Csslevodopa across all patients was 3.9. No correlation was seen between this ratio and dose or duration of PD.
Influence of food No restricted diets were followed in the studies. In study I, as assessed by a dietician, intake of animal protein was low or moderate for all patients and none for a patient who was a vegetarian. In two patients the peaks in the concentration-time profile of levodopa did not occur after lunch with increased symptomatology as a consequence, the “no-on” phenomenon (Melamed et al., 1986). This food related decrease in the clinical effect of levodopa is exemplified in Figure 7. Another regular levodopa dose was required to normalize motor function in both cases, and as expected a clear peak in plasma levodopa concentration was observed.
58
Figure 7. The impact of a meal containing low fat and low animal proteins on levodopa pharmacokinetics. The patient took 50 mg levodopa immediately after lunch (13:30) and an extra dose of 25 mg, 40 minutes later. Plasma levels of levodopa were virtually unaffected until the next regular dose, taken at 15:30, was absorbed. The greatest value of either bradykinesia, tremor or pain is given as symptoms on VAS.
Figure 8. The patient took 200 mg of SR levodopa/carbidopa at 7:00, 10:00, 13:00 and 16:00. He was moderately hyperkinetic at 12:00, declining to parkinsonism at 15:00. Lunch was ingested at about noon.
The “no-on” phenomenon was also found in study II, when expected peaks from the SR tablets were missing. The plasma levodopa concentration-time curve of a patient who was very sensitive to amino acid competition from proteins in food is exemplified in Figure 8.
Gastric infusion After the second test day of infusion treatment in patient 4 in study II, fluoroscopy revealed that the tube had been dislocated from the duodenum to the stomach. The tube was re-positioned and the patient’s motor abilities were much improved during the last test day, with 90% of video observations in near normal state as compared to 50% and 44% on the first two test days, when the tube was probably dislocated. The plasma levodopa CV is markedly higher during the two first test days, 28% and 36% as compared to 9% on fluoroscopy-verified duodenal infusion on the third test day (mean CV on tablet therapy was 29% for this patient). The plasma concentration-time curves of test days 2 and 3 are shown in Figure 9, together with a curve from one of the test days with oral SR levodopa, to depict the difference in levodopa absorption when administered orally and as infusion into the stomach or directly into the duodenum.
Figure 9. On oral therapy this patient received 300 mg SR levodopa at 7:00, followed by 100 mg at 10:00 and 200 mg at 13:00 and 16:00. On infusion, a morning bolus of 50–70 mg was followed by infusion at a rate of 46–50 mg/hr.
Levodopa and motor function (papers I, II, III and V) In study I, patients’ VAS-based self-rated motor function was often closely related to plasma levodopa levels, regardless of concomitant antiparkinson medication. In several cases the patients’ ability to assess their mobility was more sensitive when compared to ratings by the investigators. This fact was striking in a patient who was rated by the investigators as mildly bradykinetic at a stable level for the first nine hours. However, the patient himself reported fluctuating changes in his symptoms that later were found to be related to fluctuating plasma concentrations of levodopa.
In study V, motor function was not investigated in relation to medication, but similar fluctuations as seen in study I were reported. Intra-individual variability in motor performance was high, both within a day and between days. Fluctuations in motor and non-motor symptoms over a day were co-varying in some patients. For example, in one case, the pattern of pain and depression clearly followed the motor function. In this case the curves describing parkinsonism were very similar on the two different scales used for data collection, VDS and VAS.
Video scoring, study II Video evaluations of the fluctuating patients in study II showed a significantly increased number of observations in “near normal” state during levodopa infusion as compared to SR tablets (p < 0.01). Further, a significant decrease in both “off”-state (p < 0.01) and hyperkinesia (p = 0.03) was found (see Table 2). The mean variance of scores in video recordings was 1.1 during infusion therapy and 2.0 during oral therapy.
Table 2. Percentage of video observations in different motor states, studies II and III.
Study “off” “near normal” “hyperkinesia” II oral SR 25 61 14 II infusion 12 80 8 III oral 25 29 46 III infusion 3–8 months 18 49 33 III infusion 4–7 years 32 42 25 n=12 patients in study II and n=6 patients in study III
Video scoring, study III Ratings from video tapes of the very advanced patients in study III indicated an improvement with more time spent in the “near normal” state after 4–7 years of infusion as compared to oral therapy, however less than after 3–8
61
months of infusion (Table 2). The mean time spent in “off” state after 4–7 years had increased as compared to the two earlier occasions, mainly because patient G had difficulties walking and therefore was scored as “off” at every recording. The hyperkinesia was decreased after 3–8 months of duodenal therapy as compared to oral and even further decreased after 4–7 years. If patient G is excluded from the calculations, the amount of “off” time after 4–7 years of treatment decreases to 18%, “hyperkinesia” would be 31% and the time spent in “near normal” state would be 51% (n=5).
PLM-test, study II Intra-individual mean MT from the PLM-test was lower in crossover period 2 in all patients but one, whereas 9 out of 12 patients had less variation in MT on infusion treatment compared to oral treatment. In the analysis of median MT and median MT variance the variability of data was high, since the MT is designated to 30 seconds when an akinetic patient is unable to walk. No significant difference between treatments was found in the two groups because of the skewed data distribution.
PLM-test, study III Median movement time, oral therapy versus 4–7 years of continuous infusion therapy – When comparing the first test day MT was significantly decreased in two cases and unchanged in the other three patients after 4–7 years. When comparing the second test day MT was significantly decreased in three cases.
Median movement time, 3–8 months versus 4–7 years of continuous infusion therapy – Two patients showed significant MT improvement on both test days, and one patient significantly increased his MT both test days after 7 years.
Movement time variance – The MT variance at 4–7 years of infusion as compared to oral therapy was significantly decreased both days in one patient and one day in another patient. The variance was significantly increased both days in one patient. The MT variance at 4–7 years as compared to 3–8 months of infusion was significantly decreased on one of the days in one patient, significantly increased both days in another patient.
Rating scales The total score of UPDRS did not change much during the short-term study II. The motor examination (part III) was not performed on any predefined occasions in relation to dose intake. However, difference in total UPDRS score was statistically significant in favour of infusion therapy (p < 0.01). In part IV all patients had fewer complications with infusion therapy. Median
62
score was 7.5 at baseline and after oral therapy, and 4.5 after infusion therapy. The difference was statistically significant (p < 0.01).
Before infusion therapy began patients A and B of study III scored 21 and 22 respectively on the WRS and 20 and 22, 7 years later. The NUDS scores in the same patients were 37 and 32 before infusion and 40 and 24 respectively after 7 years. Among the other five patients remaining on infusion therapy after 4–5 years three still had better scores in UPDRS part I-II as compared to before infusion treatment, and four had lesser complications (part IV).
“Accidental double-blind withdrawal” During the last test day on infusion for patient 2 in study II, motor performance became gradually and severely impaired in the afternoon after a stable motor performance with mild hyperkinesia in the morning. After about 3 hours, the cassette was discovered to have been accidentally disconnected from the pump. The test day continued with the cassette reconnected, and some small boluses to reach steady-state again. The plasma levodopa concentration-time curve of this sudden withdrawal of therapy is shown in Figure 10.
Figure 10. Accidentally interrupted infusion. The patient was scored as severely “off” at 14:00, 15:00 and 16:00 and MT of the PLM-test increased from 1.86 ± 0.05 seconds in the first six tests (from 8:30 to 13:30) to 9.70 and 8.78 seconds at 14:30 and 15:30. At 16:30 the patient was completely akinetic and could not perform the test.
Experience with levodopa infusion (papers II and III)
Dosage and concomitant medication Mean levodopa consumption during test periods (up to 5:00 p.m.) in study II was 1075 ± 383 mg with SR tablets and 1003 ± 330 mg with infusion. Mean morning doses of levodopa were 308 mg of SR tablets and 131 mg of morning bolus with the infusion therapy. Comparing intra-individual doses from the first test day with the last test day on each treatment arm showed that, with respect to oral tablets, four patients had the same dose, six had higher and two had lower dose on the last test day, whereas on infusion four had a higher and eight had a lower dose on the last test day. Extra tablets were used in total a mean of 3.3 times per patient per 24 hours during the three test days and extra boluses of enteral administration were used a mean of 4.3 times. The “rescue” levodopa tablets were 50 mg each and the fine-tuned bolus doses were 14 mg, on average. Night-time oral levodopa intake was frequently needed in six cases, but no differences between treatments could be detected in this short-term study. Patients returned to their earlier medication after the study. However some chose to continue with the optimised SR tablet medication. Nine of the 12 patients later chose to continue levodopa infusion therapy by a permanent infusion system with PEG and duodenal tube.
The mean daily levodopa consumption in the long-term follow-up in study III was gradually reduced in most of the very advanced patients after the introduction of infusion therapy. At the time of last follow-up half of patients had decreased their levodopa doses as compared to before infusion therapy. The median value of the percentage change in daily dose from oral baseline to last follow-up of infusion therapy was -1% (range -71–163%). Twenty-one out of the 28 patients used other antiparkinsonian drugs concomitantly to oral levodopa, before infusion but at the last test occasion on infusion only 12 patients were still treated with oral antiparkinsonian drugs. Five of these patients used lower doses of concomitant medication as compared to oral treatment. Stereotactic surgery (pallidotomy) was performed in one patient in addition to infusion therapy.
Treatment time and safety The total infusion time in the 28 cases of study III was 1045 months, i.e. 87 patient-years. Six of the patients had returned to oral therapy over the years. Two patients curtailed their treatment due to deterioration of their condition, both had developed symptoms of multiple system atrophy. Three patients terminated treatment due to problems in handling the system as they
64
developed dementia and one patient had stopped treatment due to lack of improvement.
There were no major complications or serious adverse events obviously related to the drugs in the studies. One patient, diagnosed with atrial fibrillations during study II continued the study protocol as planned. One patient in study III developed EMG-verified polyneuropathy in his feet, but continued infusion therapy and the symptoms gradually disappeared.
Technical matters In study III, technical problems with the pumps infrequently resulted in exchange and repair necessitating a supply of spare pumps at the ward. The gastrostomy was infected transiently post-operatively in six cases and minor discharge was common during infusion but at an acceptable tolerance-level. Problems occurred with the infusion system leading to exchange or adjustment of the intestinal tube by gastroscopy (35 times) or by fluoroscopic guidance. X-ray was also used for planned inspections of the tube position. In total, fluoroscopy/X-ray was used 162 times during the 87 patient-years, i.e. approximately once every 6 months per patient. The most common reasons for adjustment have been firstly, difficulties in placing the catheter tip in the duodenum during the initial operation and, secondly, the tip has sometimes regurgitated from the duodenum into the stomach during infusion. In some cases, often after several years of treatment, the tube in use was exchanged due to increased resistance to infusion.
Usability of an automatic dose dispenser (paper IV) All patients were able to handle the dispensing device independently and were generally satisfied with their own management of the automat. One patient had rheumatoid arthritis but this did not affect her ability to handle the device. One left-handed patient used his right hand for operating the device without difficulty. This is an important finding, suggesting that the device can be used by hemiparkinsonian patients whose dominant hand is affected.
Since the microtablets were very small (diameter: 3 mm), it was anticipated that patients with tremor and/or dyskinesia might find it troublesome to handle them. However, all patients managed to pick up five tablets from a flat table surface.
Most patients were positive about future use, but many mentioned modifications of the current prototype. Most patients found the device too large and some thought it too heavy. Many patients suggested changing the size to more closely resemble that of cellular telephones. The buttons were
65
too small for seven patients and the text of the display was considered too small by eleven patients; however, all patients were able to read the text. All patients but one felt comfortable with the automatic counting of the correct dose, but some claimed they would check the number of tablets for the first few times of use. All but one were positive about the concept of dose administration in general and 17 patients were interested in using the device for their own medication, thus replacing the conventional levodopa tablets. One patient who used levodopa infusion said that he would not exchange his infusion for microtablets, but would prefer microtablets before standard tablets if he had to choose. One patient was newly diagnosed with PD and preferred ordinary tablets, but he stated he would possibly change his mind in the future. One patient claimed that conventional tablets were easier and another did not want to use the device since he already took a great number of other medications. The outcome of the tests was not clearly correlated with age or PD severity.
Compliance and usability of patient diaries (paper V) Median compliance for the daily questionnaire was 88% (range 61–100%) for the electronic diary and 98% (range 75–100%) for the paper diary. Compliance strictly linked to the scheduled times, as recorded in the paper diaries, was 78% (range 57–98%) and is defined as “true compliance”. This is significantly lower than both compliance with the electronic diary and raw compliance with the paper diary (p < 0.05). The difference between compliance with the electronic diary and raw compliance with the paper diary was not significant (p = 0.15). The distribution of the timing of patients responding to the diary shows that some paper-diary answers were completed more than an hour after the scheduled time. PDQ-39 compliance was 100% for the electronic diary and 94% for the paper diary. Four individuals using the electronic diary completely omitted a total of six days of entries in the daily questionnaire, and one individual using the paper diary missed one day’s entries.
Of the 10 patients using the electronic diary, five had no earlier experience with computers. There is no evidence that a lack of earlier experience with computers reduced the patient’s ability to use the electronic diary. Each patient using the electronic diary responded to the usability questions. There was variation between patients but very few responded with extreme answers such as “Strongly agree” or “Strongly disagree”.
Patient data, patient compliance, and technical performance were successfully monitored by the web application. Battery problems with electronic diaries were observed in this study, both by patients and by the
66
study monitor. Those problems were limited to non-critical issues arising from the battery alarms used in the electronic diaries and remotely measured battery levels at the monitoring web site. A patient who did not complete a diary question at a particular time (non-compliant patient) was alerted by the study monitor and the site staff. Site staff could contact patients and ensure the continuation of the daily diary report.
67
Discussion
Impact of fluctuating levodopa pharmacokinetics
Study designs and variability measures When evaluating the well-known interaction between pharmacokinetics and pharmacodynamics it is of great importance to acquire a true profile of plasma concentrations. In the case of levodopa, with a short half-life and unpredictable absorption because of erratic gastric emptying, resolution in observations is crucial. Plasma samples were obtained every 20 minutes in study I and every 30 minutes in study II. Hourly measurements, as used in some other studies, cannot properly reflect levodopa variability in plasma. To evaluate the difference between 20-minute and 60-minute sampling in a patient in study I the original curve was compared to a plot where two out of three values per hour were deleted. With hourly sampling Cmax was decreased from 2.09 to 1.71 µg/ml, AUC was decreased from 595 to 550 µg/ml⋅min and CV was increased from 47 to 55%.
CV of frequent plasma levodopa concentrations describes the actual variability well, as all samples are used in the calculation of mean value and SD, in contrast to describing the difference between Cmax and Cmin, which are only two values per test period. Thus, CV is mainly used in the following discussion on fluctuations in levodopa pharmacokinetics.
Oral levodopa and fluctuating plasma levels In the two pharmacokinetic studies there were large inter-individual differences in dosage and plasma concentrations. A comparison of mean values of the two studies therefore needs cautious interpretation. However, fluctuations in plasma levodopa levels after oral administration were similar in the two studies, but slightly smaller in study II when only SR tablets were used, as expected. Interestingly, the lowest CV (25%) of plasma levodopa in study I was seen in a patient who combined IR and SR formulations and the second highest CV (69%) was found in a patient who took only two SR capsules during the 10-hour study period. The high CV in this patient can be
68
explained by the infrequent dosing, which allowed each dose to be completely eliminated from the plasma compartment. Even though plasma levodopa levels dropped to zero, the patient had continuous effect from the first dose. This long-duration response is probably explained by preserved dopamine storage capacity in the remaining terminals, but could also be referred to concomitant medication with ropinirole. All patients in study I used dopamine agonists, but the motor fluctuations observed were mostly attributed to changes in plasma levodopa levels. This is likely related to the short t ½ of levodopa but also to the great potency of the drug. Levodopa is always needed in the advanced stages for symptom relief, but the fluctuations in pharmacokinetics are responsible for the disabling fluctuations in motor performance. This suggests that levodopa therapy, rather than dopamine agonists, must be a target for individualisation and refinement. In this area COMT inhibitors have been developed with great expectations. In study I, COMT inhibition showed a significant decrease in formation of 3-O-MD. However, fluctuations in plasma levodopa concentrations did not seem to differ from the data of non-COMT-inhibited levodopa administration. Intra-individual comparisons were not performed with and without COMT inhibitor, so finer conclusions on COMT inhibitors and plasma levodopa fluctuations cannot be drawn.
The rationale for small and frequent levodopa doses was supported by the lower CV of the patient who took hourly doses of 50 mg, as compared to the one who took 100 mg every two hours (Figure 3). The first patient’s lower peaks and higher troughs encourage further development of individualised therapy, like the concept with microtablets (study IV). The technique with levodopa infusion (studies II and III) is the concept driven to extremes, with very small doses delivered continuously.
The problem with interactions between food and levodopa was elucidated in two cases in study I (see Figure 7), when expected plasma levodopa peaks did not appear after intake of meals with low content of both fat and proteins. This resulted in prolonged “off” periods. The reason for a missing plasma peak is probably the delayed gastric emptying after a meal containing fat or the competitive inhibition from LNAAs in the food, both in uptake from the intestine and in transport across the BBB. The food interaction is obviously a problem for some patients and it might be important to recognize.
Motor fluctuations in PD are well known and easy to detect for an observer. Non-motor fluctuations, on the contrary, have not been well characterised and are not as obvious for the observer as they are for the patient. There is a growing interest in non-motor fluctuations and new therapies aiming at decreasing motor fluctuations should also take non-motor symptoms into account (Witjas et al., 2002). In study I, a patient reported
69
subjective fluctuations although the observers rated him at a quite constant level of motor performance. When later analysing the plasma levodopa concentrations it was found that the self-reported symptoms were related to fluctuations in plasma levodopa. Thus, it seems that some patients can sense the pulsatile dopaminergic stimulation before motor fluctuations are visible. An example of non-motor fluctuations related to motor fluctuations was found in study V, where pain and depression were clearly linked to impaired motor performance.
Intestinal levodopa infusion Intra-individual variation in plasma levodopa was significantly decreased with infusion as compared to oral administration of SR tablets (see Figures 4, 5 & 6). Several interesting issues of pharmacokinetics were observed in study II apart from this main finding.
The formulation of SR levodopa tablets is an attempt to smooth out plasma levodopa levels, but for a number of reasons, as discussed above, a perfectly sustained release of levodopa can never be accomplished with oral formulations. Although extended action is wanted SR tablets are sometimes difficult to use because of the temporal unpredictability, e.g. long Tmax and the risk of overdose when adding extra doses of IR levodopa. The unpredictability of plasma profiles from day to day was seen in some patients in the study. For example, Cmax after the morning dose (at 7:00) of SR levodopa appeared at 8:00 one day and at 10:30 another day in one patient (Figure 11). On the day of short Tmax after the first dose, the Cmax of the second dose was very low. This problem was probably not attributed to SR tablets as such, but rather to erratic gastric emptying. Adding an IR tablet to the morning SR dose could possibly solve the problem with slow onset of effect. IR tablets might be generally preferable in such patients as the dosage intervals could be decreased. As seen in Figure 11, plasma levodopa levels were substantially smoother with infusion, and the variation in Cmax and Cmin was likewise, predictably stabilized from day to day. In this patient mean Cmin from the three test days increased from 0.31 µg/ml to 1.99 µg/ml with infusion as compared to oral medication.
70
Figure 11. Unpredictability of SR tablets. The patient took 200 mg SR levodopa at 7:00 and 13:00, and 100 mg at 10:00 and 16:00 on both days presented. On infusion, a morning bolus of 90 mg was followed by an infusion rate of 52 mg/hr.
Gastric levodopa infusion was accidentally studied in a patient whose duodenal tube was dislocated to the stomach (Figure 9). The plasma profile indicates a slow leakage of the levodopa suspension by the pylorus and a peak as the stomach empties all its content into the duodenum. This levodopa portion is then absorbed and the subsequent decrease in plasma levodopa indicates a closed pylorus. Then again, the stomach empties the accumulated amount of levodopa suspension, possibly more frequently than in the morning, as levodopa concentration is kept surprisingly stable. The experience from the long-term study is that, although plasma levodopa levels may be relatively stable with gastric infusion, patients immediately notice if the tube is dislocated from the intestine to the stomach.
Another accidental example of the impact of pharmacokinetics in the therapeutic window is the patient who met with sudden withdrawal of the infusion (Figure 10). This result could in a way be regarded as a double-blind, single-subject, withdrawal study. Levodopa infusion was monotherapy and both patient and investigators thought that active drug was given. The decline in plasma levodopa is presented and the fast deterioration in motor performance is typical for patients with advanced PD and a virtually non-existing long-duration response.
The smoothened pharmacokinetic profile achieved with levodopa infusion also brought a smoothened motor performance in studies II and III. Motor outcome in study II with hourly ratings from video tapes showed significant
decrease in both hyperkinesia and in the “off” state. The ratings were not blinded because patients wore nasoduodenal tubes when on infusion. In a forthcoming study dummy tubes will be used to enable blinded evaluations and the frequency of recordings will be increased to obtain a more valid picture of motor performance. The PLM test was also performed hourly, and only once hourly, which is not optimal. Even if the non-blinded motor ratings in study II could be criticized, the impact of smooth plasma levodopa levels in these fluctuating patients was significant.
The long-term experience of levodopa infusion revealed that the improvement in motor performance could be sustained for several years, also in patients with very severe PD at the start of infusion therapy. The fact that patients on levodopa infusion performed as good, or better, as compared to optimised oral therapy in the PLM-test (both speed and variability) and in the video scoring after up to 7 years must be regarded as a major breakthrough for long-term use of infusion therapy. For these very advanced patients it was surprising that very little or no progress of the disease was seen in five out of the seven patients after 4–7 years of continuous infusion, according to the rating scales. This might, speculatively, represent a slowing of the disease progress following long-term stable levodopa/dopamine levels.
A common clinical observation is that patients tend to develop hyperkinesia after some weeks on the same dose that initially produced the “on”-state without hyperkinesia. The infusion dose is then decreased to a level where hyperkinesia disappears but patients are kept in the “on”-state. This phenomenon has not been shown in any controlled study, but infusion doses were decreased for 8 of the 12 patients in study II between test day 1 and 3 (duration 1–2 weeks) and for half of the 28 patients in study III when comparing the dose at last follow-up with the oral dose before initiation of infusion therapy. Moreover, most of them discontinued or decreased concomitant anti-PD treatments. It seems, for these patients, that the anti-parkinsonian threshold gets lower with infusion therapy. The fact that they become hyperkinetic on the initial dose suggests that the therapeutic window is not widened but moved down the y-axis of the concentration-time curve. These findings are in line with earlier reports that daytime levodopa infusion seems to restore and enhance sensitivity of striatal dopamine receptors (Cedarbaum et al., 1990). Thus, there would be no risk of infusion-induced tolerance to levodopa.
Levodopa infusion reduces the core symptoms of PD and the drug-induced dyskinesias, but there are also indications that symptoms like freezing of gait and dystonia may be improved, although these symptoms are previously considered difficult to treat with dopaminergic therapy. Further, a
72
more stable mood and reduction of anxiety may be expected from infusion therapy (Maricle et al., 1995).
Adverse events were recorded in the studies and judging by these data levodopa infusion is a safe treatment. No unexpected or serious adverse events related to infusion therapy have been found.
Most patients or caregivers easily handled the infusion system. The greatest technical problem was dislocation of the intestinal tubes from the duodenum into the stomach, probably by either retrograde peristalsis or pulling of the tube by PEGs directed upwards and to the left in the stomach. Even if 162 X-ray examinations seem numerous it should be considered that the total duration of therapy was 1045 months and that some patients were over-represented. Whether frequent tube dislocations can be explained by individual anatomical factors or technical PEG placement needs further evaluation. The shape of the tip of the tube seems to have a great impact on long-term intestinal placement. The coiled Bengmark tube has reduced the number of dislocations according to our experience. The infusion system is not very laborious to handle and patient compliance is good, as no interventions should be needed during daytime.
As seen in the early studies of intravenous and intraduodenal levodopa infusion, wearing-off fluctuations can easily be controlled by infusion. Also “on-off” fluctuations can be decreased with infusion therapy, probably because the mechanisms are in part explained by unpredictable pharmacokinetics. However, there are central pharmacodynamic aspects besides pharmacokinetics and both “off” episodes and hyperkinesia do occur in patients treated with infusion. Those fluctuations are probably caused by psychological stimuli, for example stressful situations, and cannot be treated with pharmacotherapy. Still, in these patients, fluctuations that are dependent of pharmacokinetics can be abolished, thus increasing the total time of the “on”-state without dyskinesia, although a 100% response rate may not be accomplished. Patients with wearing-off fluctuations may become 100% “on” without dyskinesia and patients with severe “on-off” fluctuations can be helped in the same direction.
The demand for individualisation of therapy There was a wide range of intra-individual dosage requirements of antiparkinsonian medication in all studies. Generally, low dosage was used in early PD and higher dosage in late PD, but the severity of the disease is not clearly reflected in the dosage. The total daily dosage should be individualised, involving two parameters; the size of each dose and the frequency of dose intakes. Examples were observed in study I, where
73
patients took levodopa doses of 50–250 mg in intervals of 1–5.5 hours. The dosage is individually titrated in clinical practice, however, in early PD stereotyped infrequent standard doses are often used. The observation that 50 mg per hour results in reduced plasma variability as compared to 100 mg per two hours indicates that an approach with small and frequent doses could be valuable also in early PD, although the immediate clinical outcome would not be different from standard treatment when LDR is still achievable. There is an awareness among patients that such therapeutic management might be beneficial in the long term. This was found in study IV, where some patients divided their levodopa tablets into pieces smaller than half a tablet. In this study together with a previous study with an early prototype of the automatic dose dispenser 30 out of 34 patients were interested in using the system with microtablets in the future.
The demand for individualised therapy is further shown in the levodopa infusion studies II and III. An extreme fine-tuning of the infusion rates in steps of 2 mg/h was possible. In these advanced patients the therapeutic window is extremely narrow, thus oral levodopa doses cannot find this window for sufficiently long periods, as the fluctuations are so steep. A non-fluctuating steady-state concentration is required in these cases for sustained SDR and this can only be achieved with infusion, either intravenous or intraduodenal.
Frequent observations and true compliance with patient diaries High-resolution assessments are important in patients with fluctuating symptoms, especially when studying patients with cognitive impairment. However, high-frequency assessment may be stressful, so the number of questions to be answered should be kept to a minimum and the study should preferably be performed in the home environment, as experienced in study I. Disabling non-motor symptoms, such as pain, fatigue, and depression, may be caused by factors other than PD itself, and are difficult but important to evaluate. With high-resolution observations, non-motor fluctuations in relation to mobility can be identified, giving a clue to the mechanisms involved.
Study V was the first to use electronic patient diaries for PD patients. The new wireless electronic data-capture device was well tolerated, with a high compliance rate. Compliance was also unexpectedly high for the paper diary. This can be explained by the highly motivated patients, who considered the study important both for PD patients in general, and for themselves in particular, in providing their physicians with a detailed description of their
74
daily functions. It may also be the result of invalidly completed (prospectively or retrospectively) diaries. Stone et al. (2002) reported that only 10% of diary data on paper are entered at the appropriate time-points. There are two ways of collecting self-assessed patient data in a completely reliable sense. Either, the investigator is present when the patient fills in the diary, or the diary must be electronic with a time-stamp for each answered question.
Differences in patient compliance between the two methods used in study V were not statistically significant with respect to raw data. However, true compliance (when the time factor was considered) was significantly reduced in patients using paper diaries. Furthermore, true compliance with the paper diary may still be overestimated because of falsely reported time-points, as reported by Stone et al. (2002). The reliability of the data collected with the electronic diary is absolute, as the timing of answers is recorded and is therefore secured.
Future prospects All five studies of this thesis naturally lead to future prospective research. Pharmacokinetic studies in the home environment are possible to undertake, intestinal levodopa infusion offers a number of interesting approaches of study, the automatic dose dispenser with levodopa microtablets should be further developed, and more evaluation and practical use of electronic diaries lie in the near future.
The neurodegenerative progress of PD with constant-rate infusion of levodopa in comparison with oral levodopa should be studied, preferably with PET or SPECT. It would be very interesting to study long-term levodopa infusion in a de novo patient to compare progression and development of complications with controls treated with conventional oral therapy. It would be difficult to find volunteers for such a trial, but the idea to study effects of CDS in early PD is presently used in the development of long-acting dopamine agonists and inhibitors of COMT.
Safety with continuous daytime levodopa infusion is presently studied in a retrospective study with individual treatment times of up to 11 years and total treatment exposure of more than 200 patient-years. These data will direct the further development of the infusion therapy.
Further, levodopa infusion will be studied in a multi-centre crossover study comparing infusion as monotherapy with any individually optimised conventional treatment. This study will provide a comparison of the efficacy of infusion to the “best method”, which is a more valid method than placebo-controlled studies in this specific case.
75
A new gastrostomy port for enteral infusion is presently studied. The device consists of a titanium port with a jejunal catheter (T-port, Transcutan AB, Sweden). The system is installed under fluoroscopy without gastroscope, using local anaesthesia. The port is implanted under the skin and is fixated by ingrowth of subcutaneous tissue, through specially designed holes in the device. A small central part (diameter: 12 mm) is protruding through the skin for connection to the infusion pump system (Figure 12). The prospects with the port are less local complications, such as hypergranulation tissue and leakage, and a greater success rate of long-term duodenal/jejunal tube placement when compared with conventional PEG. In addition, the cosmetic result and the convenience for the patients are expected to be improved. The outcome of the T-port as intestinal access for levodopa infusion is promising so far, although only three patients have been operated during a period of two months.
Figure 12. The transcutaneous titanium port for enteral levodopa infusion may solve the problems earlier encountered with PEG tubes. The picture was taken three weeks postoperatively and the patient had not experienced any complications. The tip of the tube was located in the proximal jejunum.
Technical progress is foreseen in most of these prospects. Infusion therapy will likely be improved with development of better and safer tube systems, lighter pumps, and possibly further refined formulations of levodopa suspensions, maybe combining AADC- and COMT inhibitors. The automatic dose dispenser should be user-friendlier and completely safe in counting microtablets, containing 1.25/5 mg of carbidopa/levodopa. The pharmacokinetics and pharmacodynamics of these microtablets in relation to standard tablets will have to be studied.
76
The telecom development is rapid, producing more convenient solutions in the field of electronic data capture. Future approaches in designing studies of PD will probably involve electronic patient diaries of different kinds. Real-time communication between patient and investigator will allow, for example, pharmacological interventions in the home environment. This could reduce the need for hospitalisation to some extent, and have health-economical benefits.
Health-economy will probably be increasingly important in a time where advanced and expensive treatments can easily be offered. Evaluation of economical long-term benefits must be put in relation to the costs. And most important of all, patients’ health-related quality of life must always be in focus.
77
Conclusions
It is concluded that fluctuation in delivery of levodopa to the CNS is the main reason for motor fluctuations in PD. The pharmacokinetics of levodopa is fluctuating already in the early stages of PD and it appears that refined oral treatment can only partly reduce these problems.
The need for refined levodopa formulations for individualised dosage is evident and patients have appeared to be well aware of this. Most patients are interested in future use of an automatic dose dispenser for microtablets of levodopa/carbidopa.
Administering levodopa as an intestinal infusion significantly reduces plasma levodopa fluctuations and can markedly reduce motor complications like wearing-off, “on-off” fluctuations and dyskinesia in both short- and long-term perspectives. There are indications that levodopa infusion may also ameliorate problems with dystonia and freezing, at least if these motor complications are dose-dependent. Probably, pharmacokinetics can explain more of unpredictable “off” periods and between-day variations than previously believed. Enteral levodopa infusion is safe and efficacious as long-term therapy. There have been few complications with the infusion system and refined systems are presently studied.
Electronic patient diary systems, with real-time and time-stamped data capture, are suitable for studies where frequent symptom fluctuations are investigated. This involves clinical trials of drugs for continuous dopaminergic stimulation, but also clinical evaluations of new therapies, preferably during normal activities of daily living.
The more I learn, the less I know
Lloyd Cole
79
Acknowledgments
The work involved in submitting this thesis has been substantially facilitated by the help of the following people, to whom I express my gratitude:
Sten-Magnus Aquilonius, my tutor, for his continuous enthusiasm and never-ending flow of innovative ideas, for his skilful advice in clinical neurology and for friendly and committed supervision at any hour of the day and any day of the year,
Hans Lennernäs, my co-tutor, for his inspiring devotion to the studies of bowels and pharmacokinetics, and for taking the time for frequent meetings when needed,
Dag Nilsson, “Dag the first”, co-author, for both the scientific and the clinical introduction to levodopa infusion, and for his excellent advice and training in fluoroscopy-guided tube relocation,
Cecilia Gomes-Trolin, co-author, for analysing thousands of plasma samples, for always being in a cheerful mood and for her impressive skills in laboratory methodology,
Håkan Askmark, co-author, for support, mainly in the clinical part of the work with the patients at the ward,
Christer Nyström, co-author, for his impressive feeling for formulations of pharmaceuticals and his exactitude in the preparation of manuscripts,
Susanne Bredenberg, co-author, for excellent and extremely effective collaboration in the last study of both our theses,
Jan Kowalski, co-author, for his enthusiasm and scientific interest in electronic data capture and the statistics therein,
80
Tina Knutson, co-author, for her assistance in relation to nasoduodenal tube placement, sometimes at short notice and with little time, but in a spirit of calmness and sympathy,
Katarina Johansson for sharing her deep knowledge in the appearance of Parkinson’s disease, for her sense of humour, for hard work, and for not letting work disturb leisure time too much,
Mikael Sättler for his enthusiastic demonstrations of tubes, for “sharing everything” with me in study I, for skilful vein punctures, a good sense of humour and some thrilling games of table tennis,
the 82 persons with Parkinson’s disease in studies I–V, who I must not mention by their names, but who have educated me so well, some of them have struggled through tough study protocols, hopefully gaining at least some personal benefit in the end,
all my colleagues at the department of Neuroscience and at the clinic of neurology, especially Ingela Nygren for a good introduction into my very first real clinical work, Anders Johansson for introductions into emergency cases and for pleasant conversations on topics of mutual music interest, Dan Ribom for sharing his recent knowledge in the art of preparing a dissertation, Torsten Danfors for providing a pleasant research atmosphere in our room, Titti Ekegren for discussions and laboratory help, and Gun Schönnings for help in all matters, and for always having everything under control,
all other personnel at the neurology clinic, i.e. ward 85 D and outpatient clinic, for hard work day and night, good humour, interest in innovations and excellent patient care,
Carina Bodin for her positive attitude, for co-driving in study I and keeping the patients happy during study II,
Gitte Johansson for being the experienced research nurse in study II,
Henry Engler and Bo Johnels for introducing me to movement analysis,
Tommy Lewander for sharing his great knowledge in clinical trial methodology and for interesting discussions on neuropsychopharmacology,
81
Peter LeWitt for hospitality during my stay in Detroit and for valuable scientific discussions,
Christina Persson for dietary assessments in study I,
Berit Scott for support and useful suggestions in the performance of study I,
Susanna Lindvall and colleagues at the Swedish Parkinson's disease Association for help in recruiting patients to study II,
Margareta Löfholm and colleagues at the Hospital Pharmacy for correct deliveries of drugs in both clinical trials and daily use of the levodopa suspension,
Curt Nilsson and Tommy Forsgren for enthusiasm in medical innovations,
Lars Knutson, Håkan Liljeholm and others at Enköping Hospital, and the surgeons of the “upper gastro team” at Uppsala University Hospital for fruitful discussions on PEG tubes, stomas and gastrointestinal surgery,
Rickard Nyman and Anders Sundin at the radiology department, and Stig Bengmark in London, for research progress in the challenges of long-term tube placement in the small intestine,
The companies Clinitrac, NeoPharma, Sensidose and Transcutan for good collaboration in implementing new solutions for the problems of management of PD,
my parents-in-law, Britt-Marie and Lars Johansson, for contributing to study I with accommodation at Barkarö,
my parents, Stina and Alf, and my sister Karolina for their support and love throughout my entire life
and finally, but primarily, my beloved wife Lena and my adorable son Gustav for all their love.
The work was performed with financial support from the Swedish Medical Research Council (grant no. 4373), Clinitrac AB, NeoPharma Production AB, the Faculty of Medicine at Uppsala University and its foundation for research in psychiatry and neurology.
82
References
Agid Y. Dopaminergic systems in Parkinson's disease. In: Quinn N and Jenner P, editors. Disorders of movement: Clinical, pharmacological and physiological aspects. London: Academic Press, 1989: 85-107.
Agid Y. Levodopa: is toxicity a myth? Neurology 1998; 50: 858-863. Agid Y, Ahlskog E, Albanese A, Calne D, Chase T, De Yebenes J, et al. Levodopa
in the treatment of Parkinson's disease: a consensus meeting. Mov Disord 1999; 14: 911-913.
Agnati LF, Zoli M, Stromberg I, Fuxe K. Intercellular communication in the brain: wiring versus volume transmission. Neuroscience 1995; 69: 711-726.
Ahlskog JE, Muenter MD, Maraganore DM, Matsumoto JY, Lieberman A, Wright KF, et al. Fluctuating Parkinson's disease. Treatment with the long-acting dopamine agonist cabergoline. Arch Neurol 1994; 51: 1236-1241.
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16: 448-458.
Albani C, Asper R, Hacisalihzade SS, Baumgartner G. Individual levodopa therapy in Parkinson's disease. Adv Neurol 1987; 45: 497-501.
Aquilonius SM, Hartvig P. A Swedish county with unexpectedly high utilization of anti-parkinsonian drugs. Acta Neurol Scand 1986; 74: 379-382.
Aquilonius SM, Sandell A, Sundell S, Nyström C. A handy dose-automat for adjustable delivery of oral levodopa/carbidopa. Mov Disord 1998; 13 (Suppl 2): 79.
Azulay JP, Mesure S, Amblard B, Blin O, Sangla I, Pouget J. Visual control of locomotion in Parkinson's disease. Brain 1999; 122 ( Pt 1): 111-120.
Baas H, Zehrden F, Selzer R, Kohnen R, Loetsch J, Harder S. Pharmacokinetic-pharmacodynamic relationship of levodopa with and without tolcapone in patients with Parkinson's disease. Clin Pharmacokinet 2001; 40: 383-393.
Barbeau A. L-dopa therapy in Parkinson's disease: a critical review of nine years' experience. Can Med Assoc J 1969; 101: 59-68.
Bedard PJ, Di Paolo T, Falardeau P, Boucher R. Chronic treatment with L-DOPA, but not bromocriptine induces dyskinesia in MPTP-parkinsonian monkeys. Correlation with [3H]spiperone binding. Brain Res 1986; 379: 294-299.
Bejjani BP, Arnulf I, Demeret S, Damier P, Bonnet AM, Houeto JL, et al. Levodopa-induced dyskinesias in Parkinson's disease: is sensitization reversible? Ann Neurol 2000; 47: 655-658.
Bengmark S. Progress in perioperative enteral tube feeding. Clin Nutr 1998; 17: 145-152.
83
Birkmayer W, Hornykiewicz O. Der L-Dioxyphenylalanin (= DOPA)-Effekt bei der Parkinson-Akinesie. Wien Klin Wschr 1961; 73: 787-788.
Birkmayer W, Riederer P, Youdim MB, Linauer W. The potentiation of the anti akinetic effect after L-dopa treatment by an inhibitor of MAO-B, Deprenil. J Neural Transm 1975; 36: 303-326.
Blanchet PJ, Calon F, Martel JC, Bedard PJ, Di Paolo T, Walters RR, et al. Continuous administration decreases and pulsatile administration increases behavioral sensitivity to a novel dopamine D2 agonist (U-91356A) in MPTP-exposed monkeys. J Pharmacol Exp Ther 1995; 272: 854-859.
Blandini F, Martignoni E, Pacchetti C, Desideri S, Rivellini D, Nappi G. Simultaneous determination of L-dopa and 3-O-methyldopa in human platelets and plasma using high-performance liquid chromatography with electrochemical detection. J Chromatogr B Biomed Sci Appl 1997; 700: 278-282.
Blin J, Bonnet AM, Agid Y. Does levodopa aggravate Parkinson's disease? Neurology 1988; 38: 1410-1416.
Block G, Liss C, Reines S, Irr J, Nibbelink D. Comparison of immediate-release and controlled release carbidopa/levodopa in Parkinson's disease. A multicenter 5-year study. The CR First Study Group. Eur Neurol 1997; 37: 23-27.
Bredberg E, Tedroff J, Aquilonius SM, Paalzow L. Pharmacokinetics and effects of levodopa in advanced Parkinson's disease. Eur J Clin Pharmacol 1990; 39: 385-389.
Bredberg E, Nilsson D, Johansson K, Aquilonius SM, Johnels B, Nystrom C, et al. Intraduodenal infusion of a water-based levodopa dispersion for optimisation of the therapeutic effect in severe Parkinson's disease. Eur J Clin Pharmacol 1993; 45: 117-122.
Brown RG, MacCarthy B, Jahanshahi M, Marsden CD. Accuracy of self-reported disability in patients with parkinsonism. Arch Neurol 1989; 46: 955-959.
Calne DB, Reid JL, Vakil SD, Rao S, Petrie A, Pallis CA, et al. Idiopathic Parkinsonism treated with an extracerebral decarboxylase inhibitor in combination with levodopa. Br Med J 1971; 3: 729-732.
Calne DB, Teychenne PF, Leigh PN, Bamji AN, Greenacre JK. Treatment of parkinsonism with bromocriptine. Lancet 1974; 2: 1355-1356.
Caraceni T, Scigliano G, Musicco M. The occurrence of motor fluctuations in parkinsonian patients treated long term with levodopa: role of early treatment and disease progression. Neurology 1991; 41: 380-384.
Carlsson A, Lindquist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957; 180: 1200-1201.
Carlsson A, Lindquist M, Magnusson T, Waldeck B. On the presence of 3-hydroxytyramine in brain. Science 1958; 127: 471.
Cedarbaum JM. Clinical pharmacokinetics of anti-parkinsonian drugs. Clin Pharmacokinet 1987; 13: 141-178.
Cedarbaum JM, Breck L, Kutt H, McDowell FH. Controlled-release levodopa/carbidopa. II. Sinemet CR4 treatment of response fluctuations in Parkinson's disease. Neurology 1987; 37: 1607-1612.
84
Cedarbaum JM. The promise and limitations of controlled-release oral levodopa administration. Clin Neuropharmacol 1989; 12: 147-166.
Cedarbaum JM, Kutt H, McDowell FH. A pharmacokinetic and pharmacodynamic comparison of Sinemet CR (50/200) and standard Sinemet (25/100). Neurology 1989; 39: 38-44; discussion 59.
Cedarbaum JM, Silvestri M, Kutt H. Sustained enteral administration of levodopa increases and interrupted infusion decreases levodopa dose requirements. Neurology 1990; 40: 995-997.
Chase TN, Juncos J, Serrati C, Fabbrini G, Bruno G. Fluctuation in response to chronic levodopa therapy: pathogenetic and therapeutic considerations. Adv Neurol 1987; 45: 477-480.
Chase TN, Engber TM, Mouradian MM. Palliative and prophylactic benefits of continuously administered dopaminomimetics in Parkinson's disease. Neurology 1994; 44: S15-18.
Chase TN, Engber TM, Mouradian MM. Contribution of dopaminergic and glutamatergic mechanisms to the pathogenesis of motor response complications in Parkinson's disease. Adv Neurol 1996; 69: 497-501.
Chase TN. The significance of continuous dopaminergic stimulation in the treatment of Parkinson's disease. Drugs 1998a; 55 Suppl 1: 1-9.
Chase TN. Levodopa therapy: consequences of the nonphysiologic replacement of dopamine. Neurology 1998b; 50: S17-25.
Clark D, White FJ. D1 dopamine receptor--the search for a function: a critical evaluation of the D1/D2 dopamine receptor classification and its functional implications. Synapse 1987; 1: 347-388.
Colzi A, Turner K, Lees AJ. Continuous subcutaneous waking day apomorphine in the long term treatment of levodopa induced interdose dyskinesias in Parkinson's disease. J Neurol Neurosurg Psychiatry 1998; 64: 573-576.
Contin M, Riva R, Martinelli P, Albani F, Baruzzi A. Effect of age on the pharmacokinetics of oral levodopa in patients with Parkinson's disease. Eur J Clin Pharmacol 1991; 41: 463-466.
Contin M, Riva R, Martinelli P, Albani F, Baruzzi A. No effect of chronic bromocriptine therapy on levodopa pharmacokinetics in patients with Parkinson's disease. Clin Neuropharmacol 1992; 15: 505-508.
Contin M, Riva R, Martinelli P, Cortelli P, Albani F, Baruzzi A. Concentration-effect relationship of levodopa-benserazide dispersible formulation versus standard form in the treatment of complicated motor response fluctuations in Parkinson's disease. Clin Neuropharmacol 1999; 22: 351-355.
Cooper DR, Marrel C, Testa B, van de Waterbeemd H, Quinn N, Jenner P, et al. L-Dopa methyl ester--a candidate for chronic systemic delivery of L-Dopa in Parkinson's disease. Clin Neuropharmacol 1984; 7: 89-98.
Cooper J, Bloom F, Roth R. The biochemical basis of neuropharmacology: Oxford University Press, 1991.
Cotzias GC, Van Woert MH, Schiffer LM. Aromatic amino acids and modification of parkinsonism. N Engl J Med 1967; 276: 374-379.
85
Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism--chronic treatment with L-dopa. N Engl J Med 1969; 280: 337-345.
Datla KP, Blunt SB, Dexter DT. Chronic L-DOPA administration is not toxic to the remaining dopaminergic nigrostriatal neurons, but instead may promote their functional recovery, in rats with partial 6-OHDA or FeCl(3) nigrostriatal lesions. Mov Disord 2001; 16: 424-434.
Deane KH, Jones D, Playford ED, Ben-Shlomo Y, Clarke CE. Physiotherapy for patients with Parkinson's Disease: a comparison of techniques. Cochrane Database Syst Rev 2001: CD002817.
Del Dotto P, Colzi A, Musatti E, Strolin Benedetti M, Persiani S, Fariello R, et al. Clinical and pharmacokinetic evaluation of L-dopa and cabergoline cotreatment in Parkinson's disease. Clin Neuropharmacol 1997; 20: 455-465.
Del Dotto P, Pavese N, Gambaccini G, Bernardini S, Metman LV, Chase TN, et al. Intravenous amantadine improves levadopa-induced dyskinesias: an acute double-blind placebo-controlled study. Mov Disord 2001; 16: 515-520.
Descombes S, Bonnet AM, Gasser UE, Thalamas C, Dingemanse J, Arnulf I, et al. Dual-release formulation, a novel principle in L-dopa treatment of Parkinson's disease. Neurology 2001; 56: 1239-1242.
Diamond SG, Markham CH, Hoehn MM, McDowell FH, Muenter MD. Multi-center study of Parkinson mortality with early versus later dopa treatment. Ann Neurol 1987; 22: 8-12.
Dizdar N, Kagedal B, Lindvall B, Olsson JE. Clinical and biochemical studies of intravenous L-dopa in treatment of Parkinson's disease. 31st Scandinavian Congress of Neurology. Acta Neurol Scand 1996; 94 (Suppl 167): 17.
Djaldetti R, Atlas D, Melamed E. Effect of subcutaneous administration of levodopa ethyl ester, a soluble prodrug of levodopa, on dopamine metabolism in rodent striatum: implication for treatment of Parkinson's disease. Clin Neuropharmacol 1996; 19: 65-71.
Djaldetti R, Melamed E. Levodopa ethylester: a novel rescue therapy for response fluctuations in Parkinson's disease. Ann Neurol 1996; 39: 400-404.
Djaldetti R, Melamed E. Management of response fluctuations: practical guidelines. Neurology 1998; 51: S36-40.
Djaldetti R, Treves TA, Merims D, Sroka H, Melamed E. Effect of late initiation of levodopa treatment in patients with long-standing Parkinson's disease. Clin Neuropharmacol 2003; 26: 24-27.
Dupont E, Andersen A, Boas J, Boisen E, Borgmann R, Helgetveit AC, et al. Sustained-release Madopar HBS compared with standard Madopar in the long-term treatment of de novo parkinsonian patients. Acta Neurol Scand 1996; 93: 14-20.
Ehringer H, Hornykiewicz O. Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramine) im Gehirn des Menschen und ihr Verhalten bei Erkrankungen des extrapyramidalen Systems. Klin Wochenschr 1960; 38: 1236-1239.
86
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nat Med 1998; 4: 1313-1317.
Fabbrini G, Mouradian MM, Juncos JL, Schlegel J, Mohr E, Chase TN. Motor fluctuations in Parkinson's disease: central pathophysiological mechanisms, Part I. Ann Neurol 1988; 24: 366-371.
Fahn S, Elton RL, Committee UPDRS. Unified Parkinson's Disease Rating Scale. In: Fahn S, Marsden CD, Calne DB and Goldstein M, editors. Recent developments in Parkinson's disease. Vol 2. Florham Park, New Jersey: Macmillan Healthcare Information, 1987: 153-163.
Favre J, Burchiel KJ, Taha JM, Hammerstad J. Outcome of unilateral and bilateral pallidotomy for Parkinson's disease: patient assessment. Neurosurgery 2000; 46: 344-353; discussion 353-345.
Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 1991; 114 ( Pt 5): 2283-2301.
Fehling C. Treatment of Parkinson's syndrome with L-dopa. A double blind study. Acta Neurol Scand 1966; 42: 367-372.
Frankel JP, Kempster PA, Bovingdon M, Webster R, Lees AJ, Stern GM. The effects of oral protein on the absorption of intraduodenal levodopa and motor performance. J Neurol Neurosurg Psychiatry 1989; 52: 1063-1067.
Frankel JP, Pirtosek Z, Kempster PA, Bovingdon M, Webster R, Lees AJ, et al. Diurnal differences in response to oral levodopa. J Neurol Neurosurg Psychiatry 1990; 53: 948-950.
Gancher ST, Woodward WR, Boucher B, Nutt JG. Peripheral pharmacokinetics of apomorphine in humans. Ann Neurol 1989; 26: 232-238.
Garris PA, Ciolkowski EL, Pastore P, Wightman RM. Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci 1994; 14: 6084-6093.
Gauderer MW, Ponsky JL, Izant RJ, Jr. Gastrostomy without laparotomy: a percutaneous endoscopic technique. J Pediatr Surg 1980; 15: 872-875.
Gauderer MW. Percutaneous endoscopic gastrostomy-20 years later: a historical perspective. J Pediatr Surg 2001; 36: 217-219.
Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr., et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990; 250: 1429-1432.
Ghika J, Gachoud JP, Gasser U. Clinical efficacy and tolerability of a new levodopa/benserazide dual-release formulation in parkinsonian patients. L-Dopa Dual-Release Study Group. Clin Neuropharmacol 1997; 20: 130-139.
Goetz CG, Tanner CM, Klawans HL, Shannon KM, Carroll VS. Parkinson's disease and motor fluctuations: long-acting carbidopa/levodopa (CR-4-Sinemet). Neurology 1987; 37: 875-878.
87
Goetz CG, Stebbins GT, Shale HM, Lang AE, Chernik DA, Chmura TA, et al. Utility of an objective dyskinesia rating scale for Parkinson's disease: inter- and intrarater reliability assessment. Mov Disord 1994; 9: 390-394.
Goetz CG, Pappert EJ, Blasucci LM, Stebbins GT, Ling ZD, Nora MV, et al. Intravenous levodopa in hallucinating Parkinson's disease patients: high-dose challenge does not precipitate hallucinations. Neurology 1998; 50: 515-517.
Greenacre JK, Coxon A, Petrie A, Reid JL. Comparison of levodopa with carbidopa or benserazide in parkinsonism. Lancet 1976; 2: 381-384.
Guttman M. Dopamine receptors in Parkinson's disease. Neurol Clin 1992; 10: 377-386.
Guttman M, Leger G, Cedarbaum JM, Reches A, Woodward W, Evans A, et al. 3-O-methyldopa administration does not alter fluorodopa transport into the brain. Ann Neurol 1992; 31: 638-643.
Hadj Tahar A, Gregoire L, Bangassoro E, Bedard PJ. Sustained cabergoline treatment reverses levodopa-induced dyskinesias in parkinsonian monkeys. Clin Neuropharmacol 2000; 23: 195-202.
Hagell P, Piccini P, Bjorklund A, Brundin P, Rehncrona S, Widner H, et al. Dyskinesias following neural transplantation in Parkinson's disease. Nat Neurosci 2002; 5: 627-628.
Hardie RJ, Lees AJ, Stern GM. On-off fluctuations in Parkinson's disease. A clinical and neuropharmacological study. Brain 1984; 107 ( Pt 2): 487-506.
Hardie RJ, Malcolm SL, Lees AJ, Stern GM, Allen JG. The pharmacokinetics of intravenous and oral levodopa in patients with Parkinson's disease who exhibit on-off fluctuations. Br J Clin Pharmacol 1986; 22: 429-436.
Hardoff R, Sula M, Tamir A, Soil A, Front A, Badarna S, et al. Gastric emptying time and gastric motility in patients with Parkinson's disease. Mov Disord 2001; 16: 1041-1047.
Hariz MI. Complications of deep brain stimulation surgery. Mov Disord 2002; 17 Suppl 3: S162-166.
Hefti F, Melamed E. Dopamine release in rat striatum after administration of L-dope as studied with in vivo electrochemistry. Brain Res 1981; 225: 333-346.
Heikkinen H, Nutt JG, LeWitt PA, Koller WC, Gordin A. The effects of different repeated doses of entacapone on the pharmacokinetics of L-Dopa and on the clinical response to L-Dopa in Parkinson's disease. Clin Neuropharmacol 2001; 24: 150-157.
Heinonen EH, Lammintausta R. A review of the pharmacology of selegiline. Acta Neurol Scand Suppl 1991; 136: 44-59.
Hillen ME, Sage JI. Nonmotor fluctuations in patients with Parkinson's disease. Neurology 1996; 47: 1180-1183.
Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967; 17: 427-442.
Hoehn MM. Parkinson's disease: progression and mortality. Adv Neurol 1987; 45: 457-461.
Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. Adv Neurol 1987; 45: 19-34.
88
Houeto JL, Mesnage V, Mallet L, Pillon B, Gargiulo M, du Moncel ST, et al. Behavioural disorders, Parkinson's disease and subthalamic stimulation. J Neurol Neurosurg Psychiatry 2002; 72: 701-707.
Jenkinson C, Fitzpatrick R, Peto V, Greenhall R, Hyman N. The Parkinson's Disease Questionnaire (PDQ-39): development and validation of a Parkinson's disease summary index score. Age Ageing 1997; 26: 353-357.
Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 1996; 47: S161-170.
Johnels B, Ingvarsson PE, Thorselius M, Valls M, Steg G. Disability profiles and objective quantitative assessment in Parkinson's disease. Acta Neurol Scand 1989; 79: 227-238.
Johnels B, Ingvarsson PE, Holmberg B, Matousek M, Steg G. Single-dose L-dopa response in early Parkinson's disease: measurements with optoelectronic recording technique. Mov Disord 1993; 8: 56-62.
Jorga KM, Fotteler B, Heizmann P, Zurcher G. Pharmacokinetics and pharmacodynamics after oral and intravenous administration of tolcapone, a novel adjunct to Parkinson's disease therapy. Eur J Clin Pharmacol 1998; 54: 443-447.
Kaakkola S, Wurtman RJ. Effects of catechol-O-methyltransferase inhibitors and L-3,4-dihydroxyphenylalanine with or without carbidopa on extracellular dopamine in rat striatum. J Neurochem 1993; 60: 137-144.
Kapoor R, Turjanski N, Frankel J, Kleedorfer B, Lees A, Stern G, et al. Intranasal apomorphine: a new treatment in Parkinson's disease. J Neurol Neurosurg Psychiatry 1990; 53: 1015.
Karstaedt PJ, Pincus JH. Protein redistribution diet remains effective in patients with fluctuating parkinsonism. Arch Neurol 1992; 49: 149-151.
Katzenschlager R, Lees AJ. Treatment of Parkinson's disease: levodopa as the first choice. J Neurol 2002; 249 Suppl 2: II19-24.
Kempster PA, Frankel JP, Bovingdon M, Webster R, Lees AJ, Stern GM. Levodopa peripheral pharmacokinetics and duration of motor response in Parkinson's disease. J Neurol Neurosurg Psychiatry 1989; 52: 718-723.
Kempster PA, Frankel JP, Stern GM, Lees AJ. Comparison of motor response to apomorphine and levodopa in Parkinson's disease. J Neurol Neurosurg Psychiatry 1990; 53: 1004-1007.
Kim JM, Lee KH, Choi YL, Choe GY, Jeon BS. Levodopa-induced dyskinesia in an autopsy-proven case of progressive supranuclear palsy. Mov Disord 2002; 17: 1089-1090.
Knoll J. The possible mechanisms of action of (-)deprenyl in Parkinson's disease. J Neural Transm 1978; 43: 177-198.
Koller W, Lees A, Doder M, Hely M. Randomized trial of tolcapone versus pergolide as add-on to levodopa therapy in Parkinson's disease patients with motor fluctuations. Mov Disord 2001; 16: 858-866.
Koller WC, Pahwa PR, Lyons KE, Wilkinson SB. Deep brain stimulation of the Vim nucleus of the thalamus for the treatment of tremor. Neurology 2000; 55: S29-33.
Kompoliti K, Adler CH, Raman R, Pincus JH, Leibowitz MT, Ferry JJ, et al. Gender and pramipexole effects on levodopa pharmacokinetics and pharmacodynamics. Neurology 2002; 58: 1418-1422.
Kotter R. Postsynaptic integration of glutamatergic and dopaminergic signals in the striatum. Prog Neurobiol 1994; 44: 163-196.
Krack P, Pollak P, Limousin P, Hoffmann D, Xie J, Benazzouz A, et al. Subthalamic nucleus or internal pallidal stimulation in young onset Parkinson's disease. Brain 1998; 121 ( Pt 3): 451-457.
Krack P, Pollak P, Limousin P, Benazzouz A, Deuschl G, Benabid AL. From off-period dystonia to peak-dose chorea. The clinical spectrum of varying subthalamic nucleus activity. Brain 1999; 122 ( Pt 6): 1133-1146.
Kurlan R, Rubin AJ, Miller C, Rivera-Calimlim L, Clarke A, Shoulson I. Duodenal delivery of levodopa for on-off fluctuations in parkinsonism: preliminary observations. Ann Neurol 1986; 20: 262-265.
Kurlan R, Nutt JG, Woodward WR, Rothfield K, Lichter D, Miller C, et al. Duodenal and gastric delivery of levodopa in parkinsonism. Ann Neurol 1988a; 23: 589-595.
Kurlan R, Rothfield KP, Woodward WR, Nutt JG, Miller C, Lichter D, et al. Erratic gastric emptying of levodopa may cause "random" fluctuations of parkinsonian mobility. Neurology 1988b; 38: 419-421.
Kurth MC, Tetrud JW, Tanner CM, Irwin I, Stebbins GT, Goetz CG, et al. Double-blind, placebo-controlled, crossover study of duodenal infusion of levodopa/carbidopa in Parkinson's disease patients with 'on-off' fluctuations. Neurology 1993; 43: 1698-1703.
Larsson LC, Frielingsdorf H, Mirza B, Hansson SJ, Anderson P, Czech KA, et al. Porcine neural xenografts in rats and mice: donor tissue development and characteristics of rejection. Exp Neurol 2001; 172: 100-114.
Lee MS, Kim HS, Cho EK, Lim JH, Rinne JO. COMT genotype and effectiveness of entacapone in patients with fluctuating Parkinson's disease. Neurology 2002; 58: 564-567.
Leenders KL, Poewe WH, Palmer AJ, Brenton DP, Frackowiak RS. Inhibition of L-[18F]fluorodopa uptake into human brain by amino acids demonstrated by positron emission tomography. Ann Neurol 1986; 20: 258-262.
90
Lees AJ, Montastruc JL, Turjanski N, Rascol O, Kleedorfer B, Peyro Saint-Paul H, et al. Sublingual apomorphine and Parkinson's disease. J Neurol Neurosurg Psychiatry 1989; 52: 1440.
Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57: 1687-1694.
Lees AJ. Drugs for Parkinson's disease. J Neurol Neurosurg Psychiatry 2002; 73: 607-610.
Lennernas H, Nilsson D, Aquilonius SM, Ahrenstedt O, Knutson L, Paalzow LK. The effect of L-leucine on the absorption of levodopa, studied by regional jejunal perfusion in man. Br J Clin Pharmacol 1993; 35: 243-250.
LeWitt PA, Nelson MV, Berchou RC, Galloway MP, Kesaree N, Kareti D, et al. Controlled-release carbidopa/levodopa (Sinemet 50/200 CR4): clinical and pharmacokinetic studies. Neurology 1989; 39: 45-53; discussion 59.
LeWitt PA. Clinical studies with and pharmacokinetic considerations of sustained-release levodopa. Neurology 1992; 42: 29-32; discussion 57-60.
Levy R, Hazrati LN, Herrero MT, Vila M, Hassani OK, Mouroux M, et al. Re-evaluation of the functional anatomy of the basal ganglia in normal and Parkinsonian states. Neuroscience 1997; 76: 335-343.
Li SC, Schoenberg BS, Wang CC, Cheng XM, Rui DY, Bolis CL, et al. A prevalence survey of Parkinson's disease and other movement disorders in the People's Republic of China. Arch Neurol 1985; 42: 655-657.
Lieberman AN, Goldstein M. Bromocriptine in Parkinson disease. Pharmacol Rev 1985; 37: 217-227.
Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 2002; 3: 932-942.
Louis ED, Lynch T, Marder K, Fahn S. Reliability of patient completion of the historical section of the Unified Parkinson's Disease Rating Scale. Mov Disord 1996; 11: 185-192.
Lyras L, Zeng BY, McKenzie G, Pearce RK, Halliwell B, Jenner P. Chronic high dose L-DOPA alone or in combination with the COMT inhibitor entacapone does not increase oxidative damage or impair the function of the nigro-striatal pathway in normal cynomologus monkeys. J Neural Transm 2002; 109: 53-67.
Ma Y, Dhawan V, Mentis M, Chaly T, Spetsieris PG, Eidelberg D. Parametric mapping of [18F]FPCIT binding in early stage Parkinson's disease: a PET study. Synapse 2002a; 45: 125-133.
Ma Y, Feigin A, Dhawan V, Fukuda M, Shi Q, Greene P, et al. Dyskinesia after fetal cell transplantation for parkinsonism: a PET study. Ann Neurol 2002b; 52: 628-634.
Mangiante G, Marini P, Fratucello GB, Casaril A, Ciola M, Facci E, et al. [The Bengmark tube in surgical practice and in the critically ill patient]. Chir Ital 2000; 52: 573-578.
91
Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev 1999; 51: 593-628.
Manson AJ, Hanagasi H, Turner K, Patsalos PN, Carey P, Ratnaraj N, et al. Intravenous apomorphine therapy in Parkinson's disease: clinical and pharmacokinetic observations. Brain 2001; 124: 331-340.
Manson AJ, Turner K, Lees AJ. Apomorphine monotherapy in the treatment of refractory motor complications of Parkinson's disease: Long-term follow-up study of 64 patients. Mov Disord 2002; 17: 1235-1241.
Marconi R, Lefebvre-Caparros D, Bonnet AM, Vidailhet M, Dubois B, Agid Y. Levodopa-induced dyskinesias in Parkinson's disease phenomenology and pathophysiology. Mov Disord 1994; 9: 2-12.
Marek K, Jennings D, Seibyl J. Do dopamine agonists or levodopa modify Parkinson's disease progression? Eur J Neurol 2002; 9 Suppl 3: 15-22.
Maricle RA, Nutt JG, Valentine RJ, Carter JH. Dose-response relationship of levodopa with mood and anxiety in fluctuating Parkinson's disease: a double-blind, placebo-controlled study. Neurology 1995; 45: 1757-1760.
Maricle RA, Valentine RJ, Carter J, Nutt JG. Mood response to levodopa infusion in early Parkinson's disease. Neurology 1998; 50: 1890-1892.
Marinus J, Ramaker C, van Hilten JJ, Stiggelbout AM. Health related quality of life in Parkinson's disease: a systematic review of disease specific instruments. J Neurol Neurosurg Psychiatry 2002; 72: 241-248.
Markham CH, Diamond SG. Long-term follow-up of early dopa treatment in Parkinson's disease. Ann Neurol 1986; 19: 365-372.
Marr JA. The experience of living with Parkinson's disease. J Neurosci Nurs 1991; 23: 325-329.
Marttila RJ, Rinne UK. Epidemiology of Parkinson's disease in Finland. Acta Neurol Scand 1976; 53: 81-102.
McColl CD, Reardon KA, Shiff M, Kempster PA. Motor response to levodopa and the evolution of motor fluctuations in the first decade of treatment of Parkinson's disease. Mov Disord 2002; 17: 1227-1234.
McHale DM, Sage JI, Sonsalla PK, Heikkila RE, Duvoisin RC. Steady plasma levodopa concentrations required for good clinical response to CR-4 in patients with 'on-off'. Eur Neurol 1990a; 30: 90-92.
McHale DM, Sage JI, Sonsalla PK, Vitagliano D. Complex dystonia of Parkinson's disease: clinical features and relation to plasma levodopa profile. Clin Neuropharmacol 1990b; 13: 164-170.
Mearrick PT, Wade DN, Birkett DJ, Morris J. Metoclopramide, gastric emptying and L-dopa absorption. Aust N Z J Med 1974; 4: 144-148.
Melamed E, Bitton V, Zelig O. Episodic unresponsiveness to single doses of L-dopa in parkinsonian fluctuators. Neurology 1986; 36: 100-103.
Merello M, Hughes A, Colosimo C, Hoffman M, Starkstein S, Leiguarda R. Sleep benefit in Parkinson's disease. Mov Disord 1997a; 12: 506-508.
Merello M, Pikielny R, Cammarota A, Leiguarda R. Comparison of subcutaneous apomorphine versus dispersible madopar latency and effect duration in
Merello M, Nouzeilles MI, Cammarota A, Leiguarda R. Effect of memantine (NMDA antagonist) on Parkinson's disease: a double-blind crossover randomized study. Clin Neuropharmacol 1999; 22: 273-276.
Merims D, Djaldetti R, Melamed E. Waiting for on: A major problem in patients with Parkinson's disease and on-off motor fluctuations. Mov Disord 2002; 17 (Suppl 5): S81.
Metman LV, Hoff J, Mouradian MM, Chase TN. Fluctuations in plasma levodopa and motor responses with liquid and tablet levodopa/carbidopa. Mov Disord 1994; 9: 463-465.
Metman LV, Konitsiotis S, Chase TN. Pathophysiology of motor response complications in Parkinson's disease: hypotheses on the why, where, and what. Mov Disord 2000; 15: 3-8.
Metman LV, Gillespie M, Farmer C, Bibbiani F, Konitsiotis S, Morris M, et al. Continuous transdermal dopaminergic stimulation in advanced Parkinson's disease. Clin Neuropharmacol 2001; 24: 163-169.
Mitchell SL, Harper DW, Lau A, Bhalla R. Patterns of outcome measurement in Parkinson's disease clinical trials. Neuroepidemiology 2000; 19: 100-108.
Miyasaki JM, Martin W, Suchowersky O, Weiner WJ, Lang AE. Practice parameter: initiation of treatment for Parkinson's disease: an evidence-based review: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2002; 58: 11-17.
Montastruc JL, Rascol O, Senard JM. Treatment of Parkinson's disease should begin with a dopamine agonist. Mov Disord 1999; 14: 725-730.
Montastruc JL, Desboeuf K, Lapeyre-Mestre M, Senard JM, Rascol O, Brefel-Courbon C. Long-term mortality results of the randomized controlled study comparing bromocriptine to which levodopa was later added with levodopa alone in previously untreated patients with Parkinson's disease. Mov Disord 2001; 16: 511-514.
Morissette M, Goulet M, Soghomonian JJ, Blanchet PJ, Calon F, Bedard PJ, et al. Preproenkephalin mRNA expression in the caudate-putamen of MPTP monkeys after chronic treatment with the D2 agonist U91356A in continuous or intermittent mode of administration: comparison with L-DOPA therapy. Brain Res Mol Brain Res 1997; 49: 55-62.
Mouradian MM, Juncos JL, Fabbrini G, Chase TN. Motor fluctuations in Parkinson's disease: pathogenetic and therapeutic studies. Ann Neurol 1987; 22: 475-479.
Mouradian MM, Juncos JL, Fabbrini G, Schlegel J, Bartko JJ, Chase TN. Motor fluctuations in Parkinson's disease: central pathophysiological mechanisms, Part II. Ann Neurol 1988; 24: 372-378.
Mouradian MM, Heuser IJ, Baronti F, Chase TN. Modification of central dopaminergic mechanisms by continuous levodopa therapy for advanced Parkinson's disease. Ann Neurol 1990; 27: 18-23.
Movement Disorder Society. Management of Parkinson's disease: an evidence-based review. Mov Disord 2002; 17 Suppl 4: S1-166.
93
Muenter MD, Tyce GM. L-dopa therapy of Parkinson's disease: plasma L-dopa concentration, therapeutic response, and side effects. Mayo Clin Proc 1971; 46: 231-239.
Murata M, Mizusawa H, Yamanouchi H, Kanazawa I. Chronic levodopa therapy enhances dopa absorption: contribution to wearing-off. J Neural Transm 1996; 103: 1177-1185.
Murer MG, Dziewczapolski G, Menalled LB, Garcia MC, Agid Y, Gershanik O, et al. Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery, in rats with moderate nigrostriatal lesions. Ann Neurol 1998; 43: 561-575.
Muriel MP, Orieux G, Hirsch EC. Levodopa but not ropinirole induces an internalization of D1 dopamine receptors in parkinsonian rats. Mov Disord 2002; 17: 1174-1179.
Nicholson FB, Korman MG, Richardson MA. Percutaneous endoscopic gastrostomy: a review of indications, complications and outcome. J Gastroenterol Hepatol 2000; 15: 21-25.
Nieuwboer A, De Weerdt W, Dom R, Truyen M, Janssens L, Kamsma Y. The effect of a home physiotherapy program for persons with Parkinson's disease. J Rehabil Med 2001; 33: 266-272.
Nilsson D, Hansson LE, Johansson K, Nystrom C, Paalzow L, Aquilonius SM. Long-term intraduodenal infusion of a water based levodopa-carbidopa dispersion in very advanced Parkinson's disease. Acta Neurol Scand 1998; 97: 175-183.
Nirenberg MJ, Chan J, Pohorille A, Vaughan RA, Uhl GR, Kuhar MJ, et al. The dopamine transporter: comparative ultrastructure of dopaminergic axons in limbic and motor compartments of the nucleus accumbens. J Neurosci 1997; 17: 6899-6907.
Nissinen E, Linden IB, Schultz E, Pohto P. Biochemical and pharmacological properties of a peripherally acting catechol-O-methyltransferase inhibitor entacapone. Naunyn Schmiedebergs Arch Pharmacol 1992; 346: 262-266.
Nurmi E, Ruottinen HM, Bergman J, Haaparanta M, Solin O, Sonninen P, et al. Rate of progression in Parkinson's disease: a 6-[18F]fluoro-L-dopa PET study. Mov Disord 2001; 16: 608-615.
Nutt JG, Woodward WR, Hammerstad JP, Carter JH, Anderson JL. The "on-off" phenomenon in Parkinson's disease. Relation to levodopa absorption and transport. N Engl J Med 1984; 310: 483-488.
Nutt JG, Woodward WR, Anderson JL. The effect of carbidopa on the pharmacokinetics of intravenously administered levodopa: the mechanism of action in the treatment of parkinsonism. Ann Neurol 1985; 18: 537-543.
Nutt JG, Carter JH, Woodward W, Hammerstad JP, Gancher ST. Does tolerance develop to levodopa? Comparison of 2- and 21-H levodopa infusions. Mov Disord 1993; 8: 139-143.
Nutt JG, Woodward WR, Beckner RM, Stone CK, Berggren K, Carter JH, et al. Effect of peripheral catechol-O-methyltransferase inhibition on the pharmacokinetics and pharmacodynamics of levodopa in parkinsonian patients. Neurology 1994; 44: 913-919.
94
Nutt JG, Carter JH, Woodward WR. Long-duration response to levodopa. Neurology 1995; 45: 1613-1616.
Nutt JG, Holford NH. The response to levodopa in Parkinson's disease: imposing pharmacological law and order. Ann Neurol 1996; 39: 561-573.
Nutt JG, Carter JH, Lea ES, Woodward WR. Motor fluctuations during continuous levodopa infusions in patients with Parkinson's disease. Mov Disord 1997; 12: 285-292.
Nutt JG. Effect of COMT inhibition on the pharmacokinetics and pharmacodynamics of levodopa in parkinsonian patients. Neurology 2000; 55: S33-37; discussion S38-41.
Nutt JG, Rufener SL, Carter JH, Anderson VC, Pahwa R, Hammerstad JP, et al. Interactions between deep brain stimulation and levodopa in Parkinson's disease. Neurology 2001; 57: 1835-1842.
Nutt JG, Carter JH, Lea ES, Sexton GJ. Evolution of the response to levodopa during the first 4 years of therapy. Ann Neurol 2002; 51: 686-693.
Obeso JA, Grandas F, Vaamonde J, Rosario Luguin M, Martinez-Lage JM. Apomorphine infusion for motor fluctuations in Parkinson's disease. Lancet 1987; 1: 1376-1377.
Obeso JA, Grandas F, Herrero MT, Horowski R. The role of pulsatile versus continuous dopamine receptor stimulation for functional recovery in Parkinson's disease. Eur J Neurosci 1994; 6: 889-897.
Okada K, Kobayashi S, Tsunematsu T. Prevalence of Parkinson's disease in Izumo City, Japan. Gerontology 1990; 36: 340-344.
Okereke CS. Role of integrative pharmacokinetic and pharmacodynamic optimization strategy in the management of Parkinson"s disease patients experiencing motor fluctuations with levodopa. J Pharm Pharm Sci 2002; 5: 146-161.
Olanow CW, Watts RL, Koller WC. An algorithm (decision tree) for the management of Parkinson's disease (2001): treatment guidelines. Neurology 2001; 56: S1-S88.
Ordenstein L. Sur la paralysie agitante et la sclérose en plaque generalisée. Paris: Martinel, 1867.
Pappert EJ, Goetz CG, Niederman F, Ling ZD, Stebbins GT, Carvey PM. Liquid levodopa/carbidopa produces significant improvement in motor function without dyskinesia exacerbation. Neurology 1996; 47: 1493-1495.
Parkes JD, Baxter RC, Marsden CD, Rees JE. Comparative trial of benzhexol, amantadine, and levodopa in the treatment of Parkinson's disease. J Neurol Neurosurg Psychiatry 1974; 37: 422-426.
Parkes JD. Variability in Parkinson's disease; clinical aspects, causes and treatment. Acta Neurol Scand Suppl 1983; 95: 27-35.
Parkinson J. An essay on the shaking palsy. London: Whittingham and Rowland for Sherwood, Neely and Jones, 1817.
Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. Parkinson Study Group. Jama 2000; 284: 1931-1938.
95
Pezzoli G, Tesei S, Ferrante C, Cossutta E, Zecchinelli A, Scarlato G. Madopar HBS in fluctuating parkinsonian patients: two-year treatment. Mov Disord 1988; 3: 37-45.
Piccini P, Brooks DJ, Bjorklund A, Gunn RN, Grasby PM, Rimoldi O, et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson's patient. Nat Neurosci 1999; 2: 1137-1140.
Pietz K, Hagell P, Odin P. Subcutaneous apomorphine in late stage Parkinson's disease: a long term follow up. J Neurol Neurosurg Psychiatry 1998; 65: 709-716.
Pincus JH, Barry K. Protein redistribution diet restores motor function in patients with dopa-resistant "off" periods. Neurology 1988; 38: 481-483.
Pletscher A, Bartholini G. Selective rise in brain dopamine by inhibition of extracerebral levodopa decarboxylation. Clin Pharmacol Ther 1971; 12: 344-352.
Poewe W, Gerstenbrand F, Ransmayr G. Experience with selegiline in the treatment of Parkinson's disease. J Neural Transm Suppl 1987; 25: 131-135.
Poewe WH, Lees AJ, Stern GM. Treatment of motor fluctuations in Parkinson's disease with an oral sustained-release preparation of L-dopa: clinical and pharmacokinetic observations. Clin Neuropharmacol 1986; 9: 430-439.
Poewe WH, Lees AJ. The pharmacology of foot dystonia in parkinsonism. Clin Neuropharmacol 1987; 10: 47-56.
Poewe WH, Lees AJ, Stern GM. Dystonia in Parkinson's disease: clinical and pharmacological features. Ann Neurol 1988; 23: 73-78.
Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science 1996; 274: 1197-1199.
Preshaw RM. A percutaneous method for inserting a feeding gastrostomy tube. Surg Gynecol Obstet 1981; 152: 658-660.
Quinn N, Marsden CD, Parkes JD. Complicated response fluctuations in Parkinson's disease: response to intravenous infusion of levodopa. Lancet 1982; 2: 412-415.
Quinn N, Parkes JD, Marsden CD. Control of on/off phenomenon by continuous intravenous infusion of levodopa. Neurology 1984; 34: 1131-1136.
Quinn N, Parkes D, Janota I, Marsden CD. Preservation of the substantia nigra and locus coeruleus in a patient receiving levodopa (2 kg) plus decarboxylase inhibitor over a four-year period. Mov Disord 1986; 1: 65-68.
Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342: 1484-1491.
Reches A, Mielke LR, Fahn S. 3-o-methyldopa inhibits rotations induced by levodopa in rats after unilateral destruction of the nigrostriatal pathway. Neurology 1982; 32: 887-888.
Reid JL, Calne DB, Vakil SD, Allen JG, Davies CA. Plasma concentration of levodopa in Parkinsonism before and after inhibition of peripheral decarboxylase. J Neurol Sci 1972; 17: 45-51.
96
Riley DE, Lang AE. The spectrum of levodopa-related fluctuations in Parkinson's disease. Neurology 1993; 43: 1459-1464.
Rinne UK, Sonninen V. A double blind study of 1-dopa treatment in Parkinson's disease. Eur Neurol 1968; 1: 180-191.
Rinne UK. Problems associated with long-term levodopa treatment of Parkinson's disease. Acta Neurol Scand Suppl 1983; 95: 19-26.
Rinne UK, Bracco F, Chouza C, Dupont E, Gershanik O, Marti Masso JF, et al. Early treatment of Parkinson's disease with cabergoline delays the onset of motor complications. Results of a double-blind levodopa controlled trial. The PKDS009 Study Group. Drugs 1998; 55 Suppl 1: 23-30.
Rivera-Calimlim L, Dujovne CA, Morgan JP, Lasagna L, Bianchine JR. Absorption and metabolism of L-dopa by the human stomach. Eur J Clin Invest 1971a; 1: 313-320.
Rivera-Calimlim L, Morgan JP, Dujovne CA, Bianchine JR, Lasagna L. L-3,4-dihydroxyphenylalanine metabolism by the gut in vitro. Biochem Pharmacol 1971b; 20: 3051-3057.
Robertson DR, Wood ND, Everest H, Monks K, Waller DG, Renwick AG, et al. The effect of age on the pharmacokinetics of levodopa administered alone and in the presence of carbidopa. Br J Clin Pharmacol 1989; 28: 61-69.
Robertson DR, Renwick AG, Wood ND, Cross N, Macklin BS, Fleming JS, et al. The influence of levodopa on gastric emptying in man. Br J Clin Pharmacol 1990; 29: 47-53.
Rubinstein TC, Giladi N, Hausdorff JM. The power of cueing to circumvent dopamine deficits: A review of physical therapy treatment of gait disturbances in Parkinson's disease. Mov Disord 2002; 17: 1148-1160.
Ruggieri S, Stocchi F, Carta A, Bravi D, Bragoni M, Giorgi L, et al. Comparison between L-dopa and lisuride intravenous infusions: a clinical study. Mov Disord 1988; 3: 313-319.
Ruggieri S, Stocchi F, Carta A, Catarci M, Agnoli A. Jejunal delivery of levodopa methyl ester. Lancet 1989; 2: 45-46.
Ruottinen HM, Rinne UK. Entacapone prolongs levodopa response in a one month double blind study in parkinsonian patients with levodopa related fluctuations. J Neurol Neurosurg Psychiatry 1996; 60: 36-40.
Sage JI, Schuh L, Heikkila RE, Duvoisin RC. Continuous duodenal infusions of levodopa: plasma concentrations and motor fluctuations in Parkinson's disease. Clin Neuropharmacol 1988a; 11: 36-44.
Sage JI, Trooskin S, Sonsalla PK, Heikkila R, Duvoisin RC. Long-term duodenal infusion of levodopa for motor fluctuations in parkinsonism. Ann Neurol 1988b; 24: 87-89.
Sage JI, McHale DM, Sonsalla P, Vitagliano D, Heikkila RE. Continuous levodopa infusions to treat complex dystonia in Parkinson's disease. Neurology 1989a; 39: 888-891.
Sage JI, Trooskin S, Sonsalla PK, Heikkila RE. Experience with continuous enteral levodopa infusions in the treatment of 9 patients with advanced Parkinson's disease. Neurology 1989b; 39: 60-63; discussion 72-63.
97
Sage JI, Sonsalla PK, McHale DM, Heikkila RE, Duvoisin RC. Clinical experience with duodenal infusions of levodopa for the treatment of motor fluctuations in Parkinson's disease. Adv Neurol 1990; 53: 383-386.
Sage JI, Mark MH. Nighttime levodopa infusions to treat motor fluctuations in advanced Parkinson's disease: preliminary observations. Ann Neurol 1991; 30: 616-617.
Sasahara K, Nitanai T, Habara T, Morioka T, Nakajima E. Dosage form design for improvement of bioavailability of levodopa V: Absorption and metabolism of levodopa in intestinal segments of dogs. J Pharm Sci 1981; 70: 1157-1160.
Schoenberg BS, Osuntokun BO, Adeuja AO, Bademosi O, Nottidge V, Anderson DW, et al. Comparison of the prevalence of Parkinson's disease in black populations in the rural United States and in rural Nigeria: door-to-door community studies. Neurology 1988; 38: 645-646.
Schuh LA, Bennett JP, Jr. Suppression of dyskinesias in advanced Parkinson's disease. I. Continuous intravenous levodopa shifts dose response for production of dyskinesias but not for relief of parkinsonism in patients with advanced Parkinson's disease. Neurology 1993; 43: 1545-1550.
Schultz W. Behavior-related activity of primate dopamine neurons. Rev Neurol (Paris) 1994; 150: 634-639.
Shoulson I, Glaubiger GA, Chase TN. On-off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology 1975; 25: 1144-1148.
Shoulson I. DATATOP: a decade of neuroprotective inquiry. Parkinson Study Group. Deprenyl And Tocopherol Antioxidative Therapy Of Parkinsonism. Ann Neurol 1998; 44: S160-166.
Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord 2002; 8: 193-197.
Sibley DR, Monsma FJ, Jr., Shen Y. Molecular neurobiology of dopaminergic receptors. Int Rev Neurobiol 1993; 35: 391-415.
Sjalander-Brynne L, Paalzow LK. Pharmacodynamic aspects of drug administration. Tolerance development. Adv Drug Deliv Rev 1998; 33: 235-240.
Smith HJ, Feldman M. Influence of food and marker length on gastric emptying of indigestible radiopaque markers in healthy humans. Gastroenterology 1986; 91: 1452-1455.
Sohn YH, Metman LV, Bravi D, Linfante I, Aotsuka A, Mouradian MM, et al. Levodopa peak response time reflects severity of dopamine neuron loss in Parkinson's disease. Neurology 1994; 44: 755-757.
Spacey SD, Wood NW. The genetics of Parkinson's disease. Curr Opin Neurol 1999; 12: 427-432.
Stewart P. Paralysis agitans with an account of a new symptom. Lancet 1898; 2: 1258-1260.
Stocchi F, Ruggieri S, Brughitta G, Agnoli A. Problems in daily motor performances in Parkinson's disease: the continuous dopaminergic stimulation. J Neural Transm Suppl 1986; 22: 209-218.
98
Stocchi F, Bonamartini A, Vacca L, Ruggieri S. Motor fluctuations in levodopa treatment: clinical pharmacology. Eur Neurol 1996; 36 Suppl 1: 38-42.
Stocchi F, Farina C, Nordera G, Ruggieri S. Implantable venous access system for apomorphine infusion in complicated Parkinson's disease. Mov Disord 1999; 14: 358.
Stocchi F, Ruggieri S, Vacca L, Olanow CW. Prospective randomized trial of lisuride infusion versus oral levodopa in patients with Parkinson's disease. Brain 2002; 125: 2058-2066.
Stone AA, Shiffman S, Schwartz JE, Broderick JE, Hufford MR. Patient non-compliance with paper diaries. Bmj 2002; 324: 1193-1194.
Sudo J, Iwase H, Higashiyama K, Kakuno K, Miyasaka F, Meguro T, et al. Elevation of plasma levels of L-dopa in transdermal administration of L-dopa-butylester in rats. Drug Dev Ind Pharm 2002; 28: 59-65.
Sutcliffe RL, Meara JR. Parkinson's disease epidemiology in the Northampton District, England, 1992. Acta Neurol Scand 1995; 92: 443-450.
Sweet RD, McDowell FH. Plasma dopa concentrations and the "on-off" effect after chronic treatment of Parkinson's disease. Neurology 1974; 24: 953-956.
Syed N, Murphy J, Zimmerman T, Jr., Mark MH, Sage JI. Ten years' experience with enteral levodopa infusions for motor fluctuations in Parkinson's disease. Mov Disord 1998; 13: 336-338.
Tanner CM, Goldman SM. Epidemiology of Parkinson's disease. Neurol Clin 1996; 14: 317-335.
Tedroff J, Aquilonius SM, Hartvig P, Bredberg E, Bjurling P, Langstrom B. Cerebral uptake and utilization of therapeutic [beta-11C]-L-DOPA in Parkinson's disease measured by positron emission tomography. Relations to motor response. Acta Neurol Scand 1992; 85: 95-102.
Tedroff J, Pedersen M, Aquilonius SM, Hartvig P, Jacobsson G, Langstrom B. Levodopa-induced changes in synaptic dopamine in patients with Parkinson's disease as measured by [11C]raclopride displacement and PET. Neurology 1996; 46: 1430-1436.
Tolosa ES, Martin WE, Cohen HP. Value of plasma-dopa determinations in the management of Parkinson's disease. Lancet 1973; 1: 942.
Wade DN, Mearrick PT, Morris JL. Active transport of L-dopa in the intestine. Nature 1973; 242: 463-465.
Walters JR, Bergstrom DA, Carlson JH, Chase TN, Braun AR. D1 dopamine receptor activation required for postsynaptic expression of D2 agonist effects. Science 1987; 236: 719-722.
Weiner WJ. The initial treatment of Parkinson's disease should begin with levodopa. Mov Disord 1999; 14: 716-724.
Verhagen Metman L, Del Dotto P, Blanchet PJ, van den Munckhof P, Chase TN. Blockade of glutamatergic transmission as treatment for dyskinesias and motor fluctuations in Parkinson's disease. Amino Acids 1998; 14: 75-82.
Verhagen Metman L. Recognition and treatment of response fluctuations in Parkinson's disease: Review article. Amino Acids 2002; 23: 141-145.
Wichmann T, DeLong MR. Pathophysiology of parkinsonian motor abnormalities. Adv Neurol 1993; 60: 53-61.
99
Vingerhoets FJ, Villemure JG, Temperli P, Pollo C, Pralong E, Ghika J. Subthalamic DBS replaces levodopa in Parkinson's disease: two-year follow-up. Neurology 2002; 58: 396-401.
Witjas T, Kaphan E, Azulay JP, Blin O, Ceccaldi M, Pouget J, et al. Nonmotor fluctuations in Parkinson's disease: frequent and disabling. Neurology 2002; 59: 408-413.
Wollman B, D'Agostino HB, Walus-Wigle JR, Easter DW, Beale A. Radiologic, endoscopic, and surgical gastrostomy: an institutional evaluation and meta-analysis of the literature. Radiology 1995; 197: 699-704.
Woodward WR, Olanow CW, Beckner RM, Hauser RA, Gauger LL, Cedarbaum JM, et al. The effect of L-dopa infusions with and without phenylalanine challenges in parkinsonian patients: plasma and ventricular CSF L-dopa levels and clinical responses. Neurology 1993; 43: 1704-1708.
Wooten GF. Agonists vs levodopa in PD: the thrilla of whitha. Neurology 2003; 60: 360-362.
Yahr MD, Duvoisin RC, Schear MJ, Barrett RE, Hoehn MM. Treatment of parkinsonism with levodopa. Arch Neurol 1969; 21: 343-354.
Yung KK, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience 1995; 65: 709-730.
Zoli M, Torri C, Ferrari R, Jansson A, Zini I, Fuxe K, et al. The emergence of the volume transmission concept. Brain Res Brain Res Rev 1998; 26: 136-147.
Zoli M, Jansson A, Sykova E, Agnati LF, Fuxe K. Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol Sci 1999; 20: 142-150.