UNIVERSITY OF COPENHAGEN DEPARTMENT OF BIOLOGY PhD thesis Rasmus Nielsen Klitgaard, M.Sc. Antibiotic Drug Discovery Potentiation of the quinolones and targeting the initiation of DNA replication This thesis has been submitted to the PhD School of The Faculty of Science, University of Copenhagen, Denmark, 28. February 2018.
Transcript
U N I V E R S I T Y O F C O P E N H A G E N D E P A R T M E N T O F B I O L O G Y
PhD thesis Rasmus Nielsen Klitgaard, M.Sc.
Antibiotic Drug Discovery Potentiation of the quinolones and targeting the initiation of DNA replication
This thesis has been submitted to the PhD School of The Faculty of Science, University of Copenhagen,
Denmark, 28. February 2018.
Dan Andersson
Department of Medical Biochemistry and Microbiology
University of Uppsala, Sweden.
Mogens Kilstrup
Department of Biochemistry and Biomedicine
Metabolic Signaling and Regulation
Danish Technical University, Denmark.
Signe Lo Svenningsen
Department of Biology
Biomolecular Sciences
University of Copenhagen, Denmark.
Submitted: 28.02.2018
Academic advisor Anders Løbner-Olesen
Department of Biology
Functional Genomics
University of Copenhagen, Denmark.
Assessment committee
1
Acknowledgements
First, I would like to thank my supervisor Anders Løbner-Olesen for his excellent support and
guidance throughout my PhD. I have highly appreciated that Anders has been available more or
less every day and gladly discussed any questions I might have had.
I would also like to thank Godefroid Charbon, not only for our collaboration on paper II
presented in this thesis, but also for always taking time to discuss and give advice on my other
projects. It has been greatly cherished.
Furthermore, I would like to thank the staff at Naicons srl. and all of the people who have been
part of the ALO lab: Thomas T. Thomsen, Jakob Frimodt-Møller, Maria S. Haugan, Christoffer
Campion, Anna E. Ebbensgard, Michaela Lederer, Henrik Jakobsen, Leise Riber and Belén M.
Chamizo.
A thanks, should also be given to my bachelor student, Anne Kristine Schack, who contributed
to the construction of the screening system presented in paper III.
Finally, I would like to thank my girlfriend, Marie, my family and my friends for their great
support and interest in my work.
2
Abstract – English
Antibiotic resistance has been deemed as one of the biggest threats to the global public health by the
World health Organization. In 2050, an estimated 10 million deaths per year will be attributed to
antimicrobial resistance, thus proper action needs to be taken to stop this negative development. An
important mean in the arms race against antibiotic resistance is the discovery and development of novel
antibiotics, but also preserving the efficacy of the antibiotics that are already in clinical use.
In paper I, we search for ciprofloxacin helper drug targets in an effort to preserve the use
of this widely applied antibiotic. Using a combined genetic and transcriptomic approach, the AcrAB-TolC
efflux pump and the SOS response genes, RecA and RecC, are identified as potential targets for helper
drugs in Escherichia coli strains with low-level ciprofloxacin resistance. In addition, our results also
indicate that reversing high-level ciprofloxacin resistance is likely not plausible.
In paper II, we present two novel cell based screens for identifying inhibitors of the
chromosomal DNA replication initiation in bacteria. The screens are based on growth rescue of cells that
rigorously over-initiate the DNA replication, due to either increased regeneration of the active ATP
bound form of the replication initiator protein DnaA, or by being deficient in the process known as
regulatory inactivation of DnaA (RIDA). Screening a library of 400 microbial extracts, revealed the iron
chelator deferoxamine as a compound that rescues the growth of over-initiating cells. Albeit not by
decreasing the replication initiation frequency, but by reducing the production of reactive oxygen
species. Substantiating the model that oxidative DNA damage and its repair promotes the lethal action
of hyper-replication.
In paper III, we constructed and verified a novel high throughput, cell based, fluorescence
screen for inhibitors of chromosome replication initiation in bacteria. The screen utilizes an E. coli
mutant that is resistant to replication initiation inhibitors and holds a fluorescence reporter system for
DNA replication inhibitors. This screen was also subjected to the above-mentioned library of microbial
extracts, though it did not lead to any positive hits.
3
Abstract – Danish
Verdens Sundheds organisationen, WHO, har udnævnt antibiotika resistens til at være en af de største
trusler mod det globale sundhedssystem. Det er blevet estimeret at i 2050 vil ca. 10 millioner dødsfald
årligt være associeret med antibiotika resistens. Det er derfor yderst vigtigt at der allerede nu tages de
nødvendige initiativer til at begrænse denne negative udvikling. En af de væsentligste faktorer i kampen
mod antibiotika resistens er udviklingen af nye antibiotika, samt at præservere virkningen af de antibiotika
som allerede bruges i klinikken.
I et forsøg på at præservere den kliniske anvendelighed af det ofte benyttede antibiotika
ciprofloxacin. Søger vi i artikel I efter gener i Escherichia coli hvis deletion reverserer ciprofloxacin resistens
og dermed kan bruges som mål for ciprofloxacin hjælpestoffer. Ved hjælp af genetisk deletions analyse
identificerede vi efflux pumpen, AcrAB-tolC, samt SOS-respons proteinerne, RecA og RecC som mulige mål
for ciprofloxacin hjælpestoffer i lav-resistente stammer af E. coli. Ydermere viste vores resultater også at
det formentlig ikke er muligt at reverserer ciprofloxacin resistens i høj-resistente stammer af E. coli.
I artikel II præsenterer vi to nye screeningssystemer til at identificere inhibitorer af
initieringen af kromosomal DNA replikation i bakterier. Disse to screeningssystemer er baseret på celler der
over-initierer DNA replikationen, via henholdsvis forhøjet regenerering af den ATP bundne form af
initieringsproteinet DnaA eller mangel på processen kendt som regulativ inaktivering af DnaA (RIDA).
Denne over-initiering er lethal for cellerne. Under screening af et bibliotek bestående af 400 mikrobielle
ekstrakter, identificerede vi jern chelatoren deferoxamine, som et stof der kan redde væksten af celler der
over-initierer replikationen. Dog ikke ved at nedsætte initierings frekvensen, men ved at reducere
produktionen af reaktive oxygen radikaler. Hvilket ydermere fast slår modellen, at oxidativ DNA skade og
dets reparation medierer celledød i bakterier det over-initierer DNA replikationen.
I artikel III konstruerede og verificerede vi endnu et nyt screeningssystem til inhibitorer af
DNA replikations initieringsprocessen. Denne screen består af en E. coli mutant der er resistent over for
stoffer der blokerer replikations initierings processen og samtidig indeholder et fluorescens baseret
reporter system der aktiveres af replikations initierings inhibitorer. Denne screen blev også testet mod det
ovennævnte bibliotek af mikrobielle ekstrakter, men gav ingen positive hits.
4
List of papers
Paper I Can Ciprofloxacin Resistance be Reversed by Helper Drugs? Rasmus N. Klitgaard, Bimal Jana, Luca Guardabassi, Karen Leth Nielsen and Anders Løbner-Olesen.
Paper II A strategy for finding DNA replication inhibitors in E. coli identifies iron chelators as molecules that promote survival of hyper-replicating cells. Godefroid Charbon, Rasmus Nielsen Klitgaard, Charlotte Dahlmann Liboriussen, Peter Waaben
Thulstrup, Sonia Ilaria Maffioli, Stefano Donadio and Anders Løbner-Olesen.
Paper III A Novel Fluorescence Based Screen for Inhibitors of the Initiation of DNA Replication in Bacteria. Rasmus N. Klitgaard and Anders Løbner-Olesen.
Papers not included in the thesis Ciprofloxacin intercalated in fluorohectorite clay: Identical pure drug activity and toxicity with higher adsorption and controlled release rate. E. C. dos Santos, Z. Rozynek, E. L. Hansen, R. Hartmann-Petersen, R. N. Klitgaard, A. Løbner-Olesen, d L.
Michels, A. Mikkelsen, T. S. Plivelic, H. N. Bordallo and J. O. Fossum.
Mutations in the Bacterial Ribosomal Protein L3 and Their Association with Antibiotic Resistance. Rasmus N. Klitgaard, Eleni Ntokou, Katrine Nørgaard, Daniel Biltoft, Lykke H. Hansen, Nicolai M.
LIST OF PAPERS ............................................................................................................... 5
TABLE OF CONTENTS ...................................................................................................... 6
A BRIEF HISTORY OF ANTIBIOTICS ................................................................................ 9
The early days ................................................................................................................................................................... 9
The golden age of antibiotics ........................................................................................................................................... 9
The present and future of antibiotics ............................................................................................................................ 10
PART I: POTENTIATION OF THE QUINOLONES ........................................................... 11
Discovery and development of the quinolones ............................................................................................................. 11
The quinolone targets ..................................................................................................................................................... 12
Mechanism of action ....................................................................................................................................................... 14 Fragmentation of the bacterial chromosome ................................................................................................................ 14
Reactive oxygen species and quinolone lethality .......................................................................................................... 15 Are ROS involved in quinolone lethality? ................................................................................................................... 15
The SOS response, an endogenous defense against quinolones .................................................................................. 16 Regulation and induction of the SOS response ............................................................................................................ 17 Repair of quinolone mediated double stranded DNA breaks by the SOS response ..................................................... 17
Quinolone resistance ....................................................................................................................................................... 18 Target site mutations .................................................................................................................................................... 18 Non-target site mutations involved in quinolone resistance ........................................................................................ 19 Plasmid mediated quinolone resistance ....................................................................................................................... 19
Reversing antibiotic resistance by helper drugs .......................................................................................................... 22 Potential targets for potentiation of quinolones ........................................................................................................... 23
6
PART II: TARGETING THE INITIATION OF CHROMOSOMAL DNA REPLICATION IN BACTERIA ........................................................................................................................ 23
Initiation of chromosomal DNA replication in E. coli ................................................................................................. 24 DNA replication and the cell cycle. ............................................................................................................................. 24 Initiation of replication ................................................................................................................................................ 25 The origin of replication .............................................................................................................................................. 26 The initiator protein DnaA ........................................................................................................................................... 27
Replication initiation by DnaAATP ................................................................................................................................. 29 Formation of the DnaAATP initiation complex ............................................................................................................. 29 DUE unwinding ........................................................................................................................................................... 30 DnaB helicase loading ................................................................................................................................................. 31
Regulation of the replication initiation ......................................................................................................................... 31 The dual role of DiaA in regulating replication initiation ............................................................................................ 32 Regulatory inactivation of DnaAATP (RIDA) ............................................................................................................... 33 datA-dependent DnaAATP-hydrolysis (DDAH) ............................................................................................................ 33 Regulation of DDAH activity ...................................................................................................................................... 34 SeqA, a negative regulator of the replication initiation ............................................................................................... 35 Rejuvenation of the cellular DnaAATP pool .................................................................................................................. 36
The lethal action of severe over-initiation of the DNA replication ............................................................................. 39
Targeting the Initiation of replication .......................................................................................................................... 40
PAPER I: CAN CIPROFLOXACIN RESISTANCE BE REVERSED BY HELPER DRUGS?............................................................................................................................ 42
PAPER II: A STRATEGY FOR FINDING DNA REPLICATION INHIBITORS IN E. COLI IDENTIFIES IRON CHELATORS AS MOLECULES THAT PROMOTE SURVIVAL OF HYPER-REPLICATING CELLS. ....................................................................................... 57
PAPER III: A NOVEL FLUORESCENCE BASED SCREEN FOR INHIBITORS OF THE INITIATION OF DNA REPLICATION IN BACTERIA. ....................................................... 94
Potentiation of the quinolones ..................................................................................................................................... 102
Targeting the commencement of DNA replication in bacteria ................................................................................. 104
Why is severe over-initiation of the DNA replication lethal? ................................................................................... 106
The authors declare that they have no competing interests. 224
Funding 225
Study was funded with financial support from the University of Copenhagen Centre for Control of 226
Antibiotic Resistance (UC-Care) and by the Center for Bacterial Stress Response and Persistence 227
(BASP) funded by a grant from the Danish National Research Foundation (DNRF120). 228
Authors´ contributions 229
RNK carried out all experimental work, designed the study, analysed the data and prepared the 230
final manuscript. ALO supervised all aspects of the study and helped prepare the final manuscript. 231
BJ assisted and supervised the experimental part of the RNAseq. LG supervised and delivered the 232
ST131 UR40 strain. KLN performed genomic analyses and delivered the EC38 strain carrying the qnrS 233
gene. All authors read and approved the final manuscript 234
Acknowledgments 235
53
We acknowledge the financial support from the University of Copenhagen Centre for Control of 236
Antibiotic Resistance (UC-Care) and by the Center for Bacterial Stress Response and Persistence 237
(BASP) funded by a grant from the Danish National Research Foundation (DNRF120). 238
References 239
1. Mitscher LA: Bacterial topoisomerase inhibitors: quinolone and pyridone 240 antibacterial agents. Chemical reviews 2005, 105(2):559-592. 241
2. Linder JA, Huang ES, Steinman MA, Gonzales R, Stafford RS: Fluoroquinolone 242 prescribing in the United States: 1995 to 2002. The American Journal of Medicine, 243 118(3):259-268. 244
3. Emmerson AM, Jones AM: The quinolones: decades of development and use. The 245 Journal of antimicrobial chemotherapy 2003, 51 Suppl 1:13-20. 246
4. Aldred KJ, Kerns RJ, Osheroff N: Mechanism of quinolone action and resistance. 247 Biochemistry 2014, 53(10):1565-1574. 248
5. Werner NL, Hecker MT, Sethi AK, Donskey CJ: Unnecessary use of 249 fluoroquinolone antibiotics in hospitalized patients. BMC Infectious Diseases 250 2011, 11:187-187. 251
6. Dalhoff A: Global Fluoroquinolone Resistance Epidemiology and Implictions for 252 Clinical Use. Interdisciplinary Perspectives on Infectious Diseases 2012, 2012:37. 253
7. White AR, Kaye C, Poupard J, Pypstra R, Woodnutt G, Wynne B: Augmentin 254 (amoxicillin/clavulanate) in the treatment of community-acquired respiratory 255 tract infection: a review of the continuing development of an innovative 256 antimicrobial agent. The Journal of antimicrobial chemotherapy 2004, 53 Suppl 257 1:i3-20. 258
8. Rodríguez-Martínez JM, Machuca J, Cano ME, Calvo J, Martínez-Martínez L, 259 Pascual A: Plasmid-mediated quinolone resistance: Two decades on. Drug 260 Resistance Updates 2016, 29:13-29. 261
9. Cerquetti M, Giufrè M, García-Fernández A, Accogli M, Fortini D, Luzzi I, Carattoli 262 A: Ciprofloxacin-resistant, CTX-M-15-producing Escherichia coli ST131 clone 263 in extraintestinal infections in Italy. Clinical Microbiology and Infection, 264 16(10):1555-1558. 265
10. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita 266 M, Wanner BL, Mori H: Construction of Escherichia coli K‐12 in‐frame, single‐267 gene knockout mutants: the Keio collection. Molecular systems biology 2006, 2(1). 268
11. McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, 269 Vanderpool CK, Tjaden B: Computational analysis of bacterial RNA-Seq data. 270 Nucleic Acids Res 2013, 41(14):e140. 271
12. Avasthi TS, Kumar N, Baddam R, Hussain A, Nandanwar N, Jadhav S, Ahmed N: 272
Genome of Multidrug-Resistant Uropathogenic Escherichia coli Strain NA114 273 from India. Journal of bacteriology 2011, 193(16):4272-4273. 274
54
13. Cirz RT, Chin JK, Andes DR, de Crécy-Lagard V, Craig WA, Romesberg FE: 275 Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. 276 PLoS Biol 2005, 3(6):e176. 277
14. Tamae C, Liu A, Kim K, Sitz D, Hong J, Becket E, Bui A, Solaimani P, Tran KP, 278 Yang H et al: Determination of Antibiotic Hypersensitivity among 4,000 Single-279 Gene-Knockout Mutants of Escherichia coli. Journal of bacteriology 2008, 280 190(17):5981-5988. 281
15. Yamada J, Yamasaki S, Hirakawa H, Hayashi-Nishino M, Yamaguchi A, Nishino K: 282 Impact of the RNA chaperone Hfq on multidrug resistance in Escherichia coli. 283 The Journal of antimicrobial chemotherapy 2010, 65(5):853-858. 284
16. Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH: Antibiotic 285
sensitivity profiles determined with an Escherichia coli gene knockout 286 collection: generating an antibiotic bar code. Antimicrobial agents and 287 chemotherapy 2010, 54(4):1393-1403. 288
17. Marcusson LL, Frimodt-Møller N, Hughes D: Interplay in the Selection of 289 Fluoroquinolone Resistance and Bacterial Fitness. PLoS Pathog 2009, 290 5(8):e1000541. 291
18. Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Bush K, 292 Hooper DC: Fluoroquinolone-modifying enzyme: a new adaptation of a common 293 aminoglycoside acetyltransferase. Nature medicine 2006, 12(1):83-88. 294
19. Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR: RecA Inhibitors 295 Potentiate Antibiotic Activity and Block Evolution of Antibiotic Resistance. Cell 296 chemical biology 2016, 23(3):381-391. 297
20. Brisse S, Diancourt L, Laouénan C, Vigan M, Caro V, Arlet G, Drieux L, Leflon-298 Guibout V, Mentré F, Jarlier V et al: Phylogenetic Distribution of CTX-M- and 299
Non-Extended-Spectrum-β-Lactamase-Producing Escherichia coli Isolates: 300 Group B2 Isolates, Except Clone ST131, Rarely Produce CTX-M Enzymes. 301 Journal of Clinical Microbiology 2012, 50(9):2974-2981. 302
21. López-Cerero L, Navarro MD, Bellido M, Martín-Peña A, Viñas L, Cisneros JM, 303 Gómez-Langley SL, Sánchez-Monteseirín H, Morales I, Pascual A et al: Escherichia 304 coli belonging to the worldwide emerging epidemic clonal group O25b/ST131: 305 risk factors and clinical implications. Journal of Antimicrobial Chemotherapy 306 2014, 69(3):809-814. 307
22. Tran T, Ran Q, Ostrer L, Khodursky A: De Novo Characterization of Genes That 308 Contribute to High-Level Ciprofloxacin Resistance in Escherichia coli. 309 Antimicrobial agents and chemotherapy 2016, 60(10):6353-6355. 310
23. Drlica K, Malik M, Kerns RJ, Zhao X: Quinolone-mediated bacterial death. 311 Antimicrobial agents and chemotherapy 2008, 52(2):385-392. 312
24. Michel B: After 30 Years of Study, the Bacterial SOS Response Still Surprises 313 Us. PLoS Biology 2005, 3(7):e255. 314
25. Kuzminov A: RuvA, RuvB and RuvC proteins: cleaning-up after 315 recombinational repairs in E. coli. BioEssays : news and reviews in molecular, 316 cellular and developmental biology 1993, 15(5):355-358. 317
55
26. Chase JW, Richardson CC: Exonuclease VII of Escherichia coli : MECHANISM 318 OF ACTION. Journal of Biological Chemistry 1974, 249(14):4553-4561. 319
27. Schneider R, Travers A, Kutateladze T, Muskhelishvili G: A DNA architectural 320 protein couples cellular physiology and DNA topology in Escherichia coli. 321 Molecular microbiology 1999, 34(5):953-964. 322
28. Mazzariol A, Tokue Y, Kanegawa TM, Cornaglia G, Nikaido H: High-Level 323 Fluoroquinolone-Resistant Clinical Isolates of Escherichia coli Overproduce 324 Multidrug Efflux Protein AcrA. Antimicrobial agents and chemotherapy 2000, 325 44(12):3441-3443. 326
29. Yilmaz S, Altinkanat-Gelmez G, Bolelli K, Guneser-Merdan D, Ufuk Over-Hasdemir 327 M, Aki-Yalcin E, Yalcin I: Binding site feature description of 2-substituted 328 benzothiazoles as potential AcrAB-TolC efflux pump inhibitors in E. coli. SAR 329 and QSAR in Environmental Research 2015, 26(10):853-871. 330
30. Opperman TJ, Kwasny SM, Kim HS, Nguyen ST, Houseweart C, D'Souza S, Walker 331 GC, Peet NP, Nikaido H, Bowlin TL: Characterization of a novel pyranopyridine 332 inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrobial agents and 333 chemotherapy 2014, 58(2):722-733. 334
31. Aparna V, Dineshkumar K, Mohanalakshmi N, Velmurugan D, Hopper W: 335 Identification of Natural Compound Inhibitors for Multidrug Efflux Pumps of 336 Escherichia coli and Pseudomonas aeruginosa Using In Silico High-Throughput 337 Virtual Screening and In Vitro Validation. PloS one 2014, 9(7):e101840. 338
32. Bohnert JA, Schuster S, Kern WV: Pimozide Inhibits the AcrAB-TolC Efflux 339 Pump in Escherichia coli. The open microbiology journal 2013, 7:83-86. 340
33. Chevalier J, Bredin J, Mahamoud A, Malléa M, Barbe J, Pagès J-M: Inhibitors of 341 Antibiotic Efflux in Resistant Enterobacter aerogenes and Klebsiella 342 pneumoniae Strains. Antimicrobial agents and chemotherapy 2004, 48(3):1043-343 1046. 344
34. Culyba MJ, Mo CY, Kohli RM: Targets for Combating the Evolution of Acquired 345 Antibiotic Resistance. Biochemistry 2015, 54(23):3573-3582. 346
35. Blázquez J, Couce A, Rodríguez-Beltrán J, Rodríguez-Rojas A: Antimicrobials as 347 promoters of genetic variation. Current Opinion in Microbiology 2012, 15(5):561-348 569. 349
36. Recacha E, Machuca J, Díaz de Alba P, Ramos-Güelfo M, Docobo-Pérez F, 350 Rodriguez-Beltrán J, Blázquez J, Pascual A, Rodríguez-Martínez JM: Quinolone 351 Resistance Reversion by Targeting the SOS Response. mBio 2017, 8(5). 352
353
354
56
Paper II: A strategy for finding DNA replication
inhibitors in E. coli identifies iron chelators as
molecules that promote survival of hyper-replicating
cells.
Currently in review at: Molecular Microbiology.
57
A strategy for finding DNA replication inhibitors in E. coli identifies iron chelators as molecules 1
that promote survival of hyper-replicating cells 2
3
Godefroid Charbon1†, Rasmus Nielsen Klitgaard1†, Charlotte Dahlmann Liboriussen1, Peter Waaben 4
Thulstrup2, Sonia Ilaria Maffioli3, Stefano Donadio3 and Anders Løbner-Olesen1* 5
6
From the 1University of Copenhagen, Dept. of Biology, Ole Maaløes Vej 5, 2200 Copenhagen N, 7
Denmark. 2University of Copenhagen, Dept. of Chemistry, Universitetsparken 5, 2100 Copenhagen 8
MeCN, flow: 1 ml min-1 at 50°C, UV detection: 230 nm. Linear gradient of phase B: 10 to 95% in 428
18 minutes followed by 5 minutes at 95%. 24 fractions (1 ml each) were collected. 100 l of each 429
fraction were stored for LC/MS analysis while the remaining was dried in a speedvac at 40°C 430
overnight and re-dissolved in 100 µl 10% dmso for the screening. 431
432
Identification of Deferoxamine from extract 18C9 by LC-MS. 433
LC-MS analyses was carried out using a Dionex UltiMate 3000 coupled with an LCQ Fleet mass 434
spectrometer equipped with an electrospray interface (ESI) and a tridimensional ion trap. The 435
following settings were used for liquid chromatography: 1 minute of pre-concentration at 10%, a 7 436
minutes linear gradient from 10 to 95%, followed by an isocratic step at 95% of 2 minutes and 1 437
minute of re-equilibration at 10% of CH3CN with an aqueous phase of 0.05% formic acid. The 438
column was an Atlantis T3 C18 5 μm x 4.6 mm x 50 mm at a flow rate of 0.8 ml min -1. The m/z 439
range (120-2000) and the ESI conditions were as follows: spray voltage of 3500 V, capillary 440
temperature of 275 °C, sheat gas flow rate at 35 and auxiliary gas flow rate at 15. The mass data 441
(.RAW files) from Xcalibur were converted to .mzXML file format, followed by submission to the 442
Global Natural Products Social Molecular Networking (Wang et al., 2016) database for de-443
replication. 444
445
Marker frequency analysis by qPCR 446
76
Cells centrifuged 5 minute 8000x g the supernatant discarded and the cells resuspended in 100 l of 447
cold 10 mM Tris pH7.5. The cells were then fixed by adding 1 ml of 77% ethanol and stored at 4 °C 448
until use. For the qPCR analysis, 100 l of ethanol fixed cells were centrifuged 7 minutes at 17000 449
x g, the supernatant discarded and the samples centrifuged again for 30 seconds at 17000 x g, 450
followed by removal of the remaining ethanol. The cell pellet was resuspended in 1ml cold water 451
and 2 µl was used as template for qPCR analysis. The Quantitative-PCR was performed using a 452
Takara SYBR Premix Ex Taq II (RR820A) in a BioRAD CFX96. All ori/ter ratios were 453
normalized to the ori/ter ratio of MG1655 treated with rifampicin for 2h. The origin and terminus 454
was quantified using primers 5′-TTCGATCACCCCTGCGTACA-3′ and 5′-455
CGCAACAGCATGGCGATAAC-3′ for the origin and 5′-TTGAGCTGCGCCTCATCAAG-3′ and 456
5′-TCAACGTGCGAGCGATGAAT-3′ for the terminus. 457
458
Flow cytometry 459
Preparation of samples for determination of number of origin per cell: 1 ml of cell culture was 460
incubated at 37°C for 2 to 4 hours with 300 µg ml-1 rifampicin and 36 µg ml-1 cephalexin. Cells 461
were fixed in 70% ethanol and stored at 4°C, as described for the marker frequency analysis by 462
qPCR. 463
Preparation of samples for determination of cell size: 1 ml of cell culture was placed on ice and 464
fixed as described for the marker frequency analysis by qPCR. 465
DNA Staining; 100-300 µl of fixed cells were centrifugated at 15,000 x g for 15 min. The 466
supernatant was discarded and the pellet resuspended in 130 µl “Staining solution” (90 µg ml-1 467
mithramycin, 20 µg ml-1 ethidium bromide, 10 mM MgCl2, 10 mM Tris pH 7.5). Samples were 468
then kept on ice for a minimum of 10 min. prior to flow cytometric analysis. Flow cytometry was 469
77
performed using an Apogee A10 Bryte instrument. For each sample, 30 000 to 200 000 cells were 470
analyzed. 471
472
Minimal inhibitory concentration 473
The MIC of DFO was determined by micro-dilution in a 96-well plate. MG1655 was grown to an 474
OD600 of 0.5 in minimal rich medium. The culture was diluted to an OD600 of 0.001 in minimal rich 475
medium. 100 µl diluted culture was added to each well of a 96-well plate containing a dilution 476
series of DFO in minimal rich medium, giving a final concentration range of 512 to 0.5 µg ml-1 of 477
DFO. The 96-well plate was incubated at 37 °C for 24 hours and inspected for visible growth 478
inhibition. 479
480
Minimal rescuing concentration 481
MG1655 hda::cat was grown overnight in minimal poor medium at 37°C. The overnight culture 482
was diluted 100x in minimal rich medium and grown for four hours at 37°C. The culture was 483
diluted to an OD600 of 0.001 in minimal rich medium and 100 µl culture was added to each well of a 484
96-well plate containing a dilution series of DFO in minimal rich medium, giving a final 485
concentration range of 512 to 0.5 µg ml-1 of DFO. The growth at 37 °C during continuous shaking 486
was monitored for sixteen hours, using a Biotek Synergy H1 plate reader. 487
Acknowledgments 488
The authors were funded by grants from the Danish National Research Foundation (DNRF120) 489
through the Center for Bacterial Stress Response and Persistence (BASP) and by the University of 490
Copenhagen Centre for Control of Antibiotic Resistance (UC-Care). 491
78
References 492
Babu, V.M.P., M. Itsko, J.C. Baxter, R.M. Schaaper & M.D. Sutton, (2017) Insufficient levels of the nrdAB-493 encoded ribonucleotide reductase underlie the severe growth defect of the Δhda E. coli strain. 494 Molecular microbiology 104: 377-399. 495
Barata, J.D., P.C. D'Haese, C. Pires, L.V. Lamberts, J. Simoes & M.E. De Broe, (1996) Low-dose (5 mg/kg) 496 desferrioxamine treatment in acutely aluminium-intoxicated haemodialysis patients using two drug 497 administration schedules. Nephrol Dial Transplant 11: 125-132. 498
Barona-Gomez, F., U. Wong, A.E. Giannakopulos, P.J. Derrick & G.L. Challis, (2004) Identification of a cluster 499 of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J Am Chem 500 Soc 126: 16282-16283. 501
Bolivar, F., R.L. Rodriguez, P.J. Greene, M.C. Betlach, H.L. Heyneker, H.W. Boyer, J.H. Crosa & S. Falkow, 502 (1977) Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. 503 Gene 2: 95-113. 504
Campbell, J.L. & N. Kleckner, (1990) E. coli oriC and the dnaA gene promoter are sequestered from dam 505 methyltransferase following the passage of the chromosomal replication fork. Cell 62: 967-979. 506
Chan, G.C., S. Chan, P.L. Ho & S.Y. Ha, (2009) Effects of chelators (deferoxamine, deferiprone and 507 deferasirox) on the growth of Klebsiella pneumoniae and Aeromonas hydrophila isolated from 508 transfusion-dependent thalassemia patients. Hemoglobin 33: 352-360. 509
Charbon, G., L. Bjorn, B. Mendoza-Chamizo, J. Frimodt-Moller & A. Lobner-Olesen, (2014) Oxidative DNA 510 damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic 511 Acids Res 42: 13228-13241. 512
Charbon, G., C. Campion, S.H. Chan, L. Bjorn, A. Weimann, L.C. da Silva, P.R. Jensen & A. Lobner-Olesen, 513 (2017a) Re-wiring of energy metabolism promotes viability during hyperreplication stress in E. coli. 514 PLoS Genet 13: e1006590. 515
Charbon, G., L. Riber, M. Cohen, O. Skovgaard, K. Fujimitsu, T. Katayama & A. Lobner-Olesen, (2011) 516 Suppressors of DnaA(ATP) imposed overinitiation in Escherichia coli. Mol Microbiol 79: 914-928. 517
Charbon, G., L. Riber & A. Lobner-Olesen, (2017b) Countermeasures to survive excessive chromosome 518 replication in Escherichia coli. Curr Genet. 519
Clark, D.J. & O. Maaløe, (1967) DNA replication and the division cycle in Escherichia coli. J.Mol.Biol. 23: 99-520 112. 521
Cooper, S. & C.E. Helmstetter, (1968) Chromosome replication and the division cycle of Escherichia coli B/r. 522 J Mol Biol 31: 519-540. 523
D'Onofrio, A., J.M. Crawford, E.J. Stewart, K. Witt, E. Gavrish, S. Epstein, J. Clardy & K. Lewis, (2010) 524 Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem Biol 525 17: 254-264. 526
Donachie, W.D., (1968) Relationship between cell size and time of initiation of DNA replication. Nature 219: 527 1077-1079. 528
Fossum, S., G. De Pascale, C. Weigel, W. Messer, S. Donadio & K. Skarstad, (2008) A robust screen for novel 531 antibiotics: specific knockout of the initiator of bacterial DNA replication. FEMS Microbiol Lett 281: 532 210-214. 533
Foti, J.J., B. Devadoss, J.A. Winkler, J.J. Collins & G.C. Walker, (2012) Oxidation of the guanine nucleotide 534 pool underlies cell death by bactericidal antibiotics. Science 336: 315-319. 535
Frimodt-Moller, J., G. Charbon, K.A. Krogfelt & A. Lobner-Olesen, (2015) Control regions for chromosome 536 replication are conserved with respect to sequence and location among Escherichia coli strains. 537 Front Microbiol 6: 1011. 538
79
Fujimitsu, K., T. Senriuchi & T. Katayama, (2009) Specific genomic sequences of E. coli promote replicational 539 initiation by directly reactivating ADP-DnaA. Genes Dev 23: 1221-1233. 540
Gero, D., K. Modis, N. Nagy, P. Szoleczky, Z.D. Toth, G. Dorman & C. Szabo, (2007) Oxidant-induced 541 cardiomyocyte injury: identification of the cytoprotective effect of a dopamine 1 receptor agonist 542 using a cell-based high-throughput assay. Int J Mol Med 20: 749-761. 543
Guyer, M.S., R.R. Reed, J.A. Steitz & K.B. Low, (1981) Identification of a sex-factor-affinity site in E. coli as 544 gamma delta. Cold Spring Harb.Symp.Quant.Biol. 45: 135-140. 545
Hider, R.C. & X. Kong, (2010) Chemistry and biology of siderophores. Natural product reports 27: 637-657. 546 Hirota, Y., J. Mordoh & F. Jacob, (1970) On the process of cellular division in Escherichia coli. 3. 547
Thermosensitive mutants of Escherichia coli altered in the process of DNA initiation. J Mol Biol 53: 548 369-387. 549
Imlay, J.A., S.M. Chin & S. Linn, (1988) Toxic DNA damage by hydrogen peroxide through the Fenton 550 reaction in vivo and in vitro. Science 240: 640-642. 551
Johnsen, L., C. Weigel, J. von Kries, M. Moller & K. Skarstad, (2010) A novel DNA gyrase inhibitor rescues 552 Escherichia coli dnaAcos mutant cells from lethal hyperinitiation. J Antimicrob Chemother 65: 924-553 930. 554
Kasho, K. & T. Katayama, (2013) DnaA binding locus datA promotes DnaA-ATP hydrolysis to enable cell 555 cycle-coordinated replication initiation. Proc Natl Acad Sci U S A 110: 936-941. 556
Kato, J. & T. Katayama, (2001) Hda, a novel DnaA-related protein, regulates the replication cycle in 557 Escherichia coli. EMBO J 20: 4253-4262. 558
Kellenberger-Gujer, G., A.J. Podhajska & L. Caro, (1978) A cold sensitive dnaA mutant of E. coli which 559 overinitiates chromosome replication at low temperature. Mol Gen Genet 162: 9-16. 560
Kjelstrup, S., P.M.P. Hansen, L.E. Thomsen, P.R. Hansen & A. Løbner-Olesen, (2013) Cyclic Peptide Inhibitors 561 of the β-Sliding Clamp in <italic>Staphylococcus aureus</italic>. PloS one 8: e72273. 562
Kling, A., P. Lukat, D.V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S.C. Wenzel, 563 C. Konig, N.C. Ammerman, M.B. Barrio, K. Borchers, F. Bordon-Pallier, M. Bronstrup, G. 564 Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D.W. Heinz, H. Hoffmann, S. Klieber, M. 565 Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J.H. Grosset, S. Lagrange 566 & R. Muller, (2015) Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. 567 Science 348: 1106-1112. 568
Kurokawa, K., S. Nishida, A. Emoto, K. Sekimizu & T. Katayama, (1999) Replication cycle-coordinated change 569 of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. EMBO J 18: 6642-6652. 570
Leonard, A.C. & J.E. Grimwade, (2015) The orisome: structure and function. Front Microbiol 6: 545. 571 Liu, Y., S.C. Bauer & J.A. Imlay, (2011) The YaaA protein of the Escherichia coli OxyR regulon lessens 572
hydrogen peroxide toxicity by diminishing the amount of intracellular unincorporated iron. J 573 Bacteriol 193: 2186-2196. 574
Lu, M., J.L. Campbell, E. Boye & N. Kleckner, (1994) SeqA: a negative modulator of replication initiation in E. 575 coli. Cell 77: 413-426. 576
Messer, W., (2002) The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate 577 DNA replication. FEMS Microbiol Rev 26: 355-374. 578
Messer, W., F. Blaesing, J. Majka, J. Nardmann, S. Schaper, A. Schmidt, H. Seitz, C. Speck, D. Tungler, G. 579 Wegrzyn, C. Weigel, M. Welzeck & J. Zakrzewska-Czerwinska, (1999) Functional domains of DnaA 580 proteins. Biochimie 81: 819-825. 581
Miller, J.H., (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, 582 NY. 583
Raymond, K.N., E.A. Dertz & S.S. Kim, (2003) Enterobactin: an archetype for microbial iron transport. Proc 584 Natl Acad Sci U S A 100: 3584-3588. 585
Riber, L., J. Frimodt-Moller, G. Charbon & A. Lobner-Olesen, (2016) Multiple DNA Binding Proteins 586 Contribute to Timing of Chromosome Replication in E. coli. Front Mol Biosci 3: 29. 587
80
Riber, L., J.A. Olsson, R.B. Jensen, O. Skovgaard, S. Dasgupta, M.G. Marinus & A. Lobner-Olesen, (2006) Hda-588 mediated inactivation of the DnaA protein and dnaA gene autoregulation act in concert to ensure 589 homeostatic maintenance of the Escherichia coli chromosome. Genes Dev 20: 2121-2134. 590
Robins-Browne, R.M. & J.K. Prpic, (1985) Effects of iron and desferrioxamine on infections with Yersinia 591 enterocolitica. Infection and Immunity 47: 774-779. 592
Robinson, A., R.J. Causer & N.E. Dixon, (2012) Architecture and conservation of the bacterial DNA 593 replication machinery, an underexploited drug target. Curr Drug Targets 13: 352-372. 594
Roosenberg, J.M., 2nd, Y.M. Lin, Y. Lu & M.J. Miller, (2000) Studies and syntheses of siderophores, microbial 595 iron chelators, and analogs as potential drug delivery agents. Current medicinal chemistry 7: 159-596 197. 597
Saha, M., S. Sarkar, B. Sarkar, B.K. Sharma, S. Bhattacharjee & P. Tribedi, (2016) Microbial siderophores and 598 their potential applications: a review. Environ Sci Pollut Res Int 23: 3984-3999. 599
Sekimizu, K., D. Bramhill & A. Kornberg, (1987) ATP activates dnaA protein in initiating replication of 600 plasmids bearing the origin of the E. coli chromosome. Cell 50: 259-265. 601
Sever, M.J. & J.J. Wilker, (2004) Visible absorption spectra of metal-catecholate and metal-tironate 602 complexes. Dalton Trans: 1061-1072. 603
Simmons, L.A., A.M. Breier, N.R. Cozzarelli & J.M. Kaguni, (2004) Hyperinitiation of DNA replication in 604 Escherichia coli leads to replication fork collapse and inviability. Mol Microbiol 51: 349-358. 605
Skarstad, K. & T. Katayama, (2013) Regulating DNA replication in bacteria. Cold Spring Harb Perspect Biol 5: 606 a012922. 607
Takahashi, N., C.C. Gruber, J.H. Yang, X. Liu, D. Braff, C.N. Yashaswini, S. Bhubhanil, Y. Furuta, S. Andreescu, 608 J.J. Collins & G.C. Walker, (2017) Lethality of MalE-LacZ hybrid protein shares mechanistic attributes 609 with oxidative component of antibiotic lethality. Proc Natl Acad Sci U S A. 610
Terlain, B. & J.P. Thomas, (1971) [Structure of griselimycin, polypeptide antibiotic extracted Streptomyces 611 cultures. I. Identification of the products liberated by hydrolysis]. Bull Soc Chim Fr 6: 2349-2356. 612
Thompson, M.G., B.W. Corey, Y. Si, D.W. Craft & D.V. Zurawski, (2012) Antibacterial Activities of Iron 613 Chelators against Common Nosocomial Pathogens. Antimicrobial agents and chemotherapy 56: 614 5419-5421. 615
von Freiesleben, U., M.A. Krekling, F.G. Hansen & A. Lobner-Olesen, (2000) The eclipse period of Escherichia 616 coli. EMBO J 19: 6240-6248. 617
Wang, M., J.J. Carver, V.V. Phelan, L.M. Sanchez, N. Garg, Y. Peng, D.D. Nguyen, J. Watrous, C.A. Kapono, T. 618 Luzzatto-Knaan, C. Porto, A. Bouslimani, A.V. Melnik, M.J. Meehan, W.T. Liu, M. Crusemann, P.D. 619 Boudreau, E. Esquenazi, M. Sandoval-Calderon, R.D. Kersten, L.A. Pace, R.A. Quinn, K.R. Duncan, 620 C.C. Hsu, D.J. Floros, R.G. Gavilan, K. Kleigrewe, T. Northen, R.J. Dutton, D. Parrot, E.E. Carlson, B. 621 Aigle, C.F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B.T. 622 Murphy, L. Gerwick, C.C. Liaw, Y.L. Yang, H.U. Humpf, M. Maansson, R.A. Keyzers, A.C. Sims, A.R. 623 Johnson, A.M. Sidebottom, B.E. Sedio, A. Klitgaard, C.B. Larson, C.A.B. P, D. Torres-Mendoza, D.J. 624 Gonzalez, D.B. Silva, L.M. Marques, D.P. Demarque, E. Pociute, E.C. O'Neill, E. Briand, E.J.N. Helfrich, 625 E.A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J.J. Kharbush, Y. Zeng, J.A. Vorholt, 626 K.L. Kurita, P. Charusanti, K.L. McPhail, K.F. Nielsen, L. Vuong, M. Elfeki, M.F. Traxler, N. Engene, N. 627 Koyama, O.B. Vining, R. Baric, R.R. Silva, S.J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. 628 Hoffman, V. Agarwal, P.G. Williams, J. Dai, R. Neupane, J. Gurr, A.M.C. Rodriguez, A. Lamsa, C. 629 Zhang, K. Dorrestein, B.M. Duggan, J. Almaliti, P.M. Allard, P. Phapale, et al., (2016) Sharing and 630 community curation of mass spectrometry data with Global Natural Products Social Molecular 631 Networking. Nature biotechnology 34: 828-837. 632
Weed, M.R., K.E. Vanover & W.L. Woolverton, (1993) Reinforcing effect of the D1 dopamine agonist SKF 633 81297 in rhesus monkeys. Psychopharmacology (Berl) 113: 51-52. 634
81
Fig. 1. Concept of the screen.A) Principle of the screen. In absence of Hda or in presence of multiple copies of DARS2, DNA replication commences too soon and/ or too often resulting in inviability. An anti-DnaA molecule that reduces DnaAactivity reestablishes the initiation frequency to a level that restores viability.Such an anti-DnaA molecule reduces DnaA activity in wild-type cells to a level that no longer sustains viability. B) Schematic representation of the screening method. Hda deficient cells or wild-type cells containing a pBR322-DARS2 plasmid are propagated under permissive growth conditions, i.e. either anaerobic or in minimal poor medium. An estimated twenty thousand cells are spread on two types of agar plates: minimal poor (permissive conditions) and minimal rich (non-permissive conditions) medium. A diffusion assay is performed by punching holes in the agar and introducing 5 ml bioactive extract into each. The plates are incubated aerobically at 37oC for 16h and visually inspected. On the non-permissive conditions plates, positive “hits” are depicted by a small clearing area separating a zone of growth encircling the hole from which the specific extract has been diffusing. The same extract on permissive conditions is depicted by a small clearing area encircling the hole from which the extract has been diffusing.
82
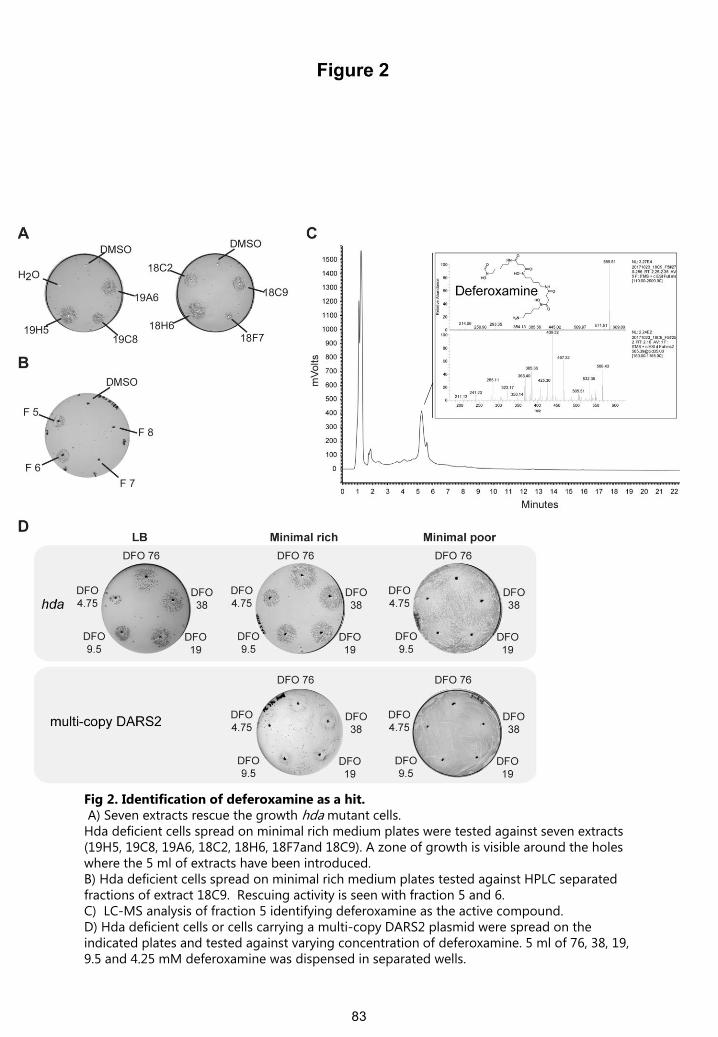
Fig 2. Identification of deferoxamine as a hit.A) Seven extracts rescue the growth hda mutant cells.
Hda deficient cells spread on minimal rich medium plates were tested against seven extracts (19H5, 19C8, 19A6, 18C2, 18H6, 18F7and 18C9). A zone of growth is visible around the holes where the 5 ml of extracts have been introduced.B) Hda deficient cells spread on minimal rich medium plates tested against HPLC separated fractions of extract 18C9. Rescuing activity is seen with fraction 5 and 6.C) LC-MS analysis of fraction 5 identifying deferoxamine as the active compound.D) Hda deficient cells or cells carrying a multi-copy DARS2 plasmid were spread on the indicated plates and tested against varying concentration of deferoxamine. 5 ml of 76, 38, 19, 9.5 and 4.25 mM deferoxamine was dispensed in separated wells.
83
Fig. 3. Deferoxamine does not affect initiation of DNA replication.The indicated cells were grown exponentially at 37 °C in minimal medium supplemented with minimal poor medium (blue) and then diluted into minimal rich medium and incubated for 4 hours at 37 °C in absence of DFO (green) or presence of DFO at a final concentration of 150mM (orange). Cells were treated with rifampicin and cephalexin prior to flow cytometricanalysis. Each panel represents a minimum of 30000 cells. When relevant, the average ori/cell (O/C), ori/mass (O/M) relative to the wild-type (wt) and mass doubling time (t) are shown in the histograms. Inserts show growth of culture where no meaningful doubling time could be obtained. N.D. – Not Determined, N.R. – Not Relevant.
84
Fig. 4. Deferoxamine restores growth hda mutant cells during fast growth.A. Wild-type and Hda deficient cells were grown in LB supplemented with 150 mM DFO. Cells were diluted 5 times in LB without DFO and maintained by dilution in fresh medium for two hours. When indicated Iron II perchlorate was added at a final concentration of 200 mM to titrate DFO. Insert: Growth of hda mutant cells was followed by measuring OD600. Cells were treated with rifampicin and cephalexin prior to flow cytometric analysis. Each panel represents a minimum of 30000 cells. When relevant, the average ori/cell (O/C), ori/mass (O/M) relative to the wild-type (wt) and mass doubling time (t) are shown in the histograms. Orange histograms represent cells grown in the presence of DFO whereas green histograms were derived from cells grown/incubated without DFO for the indicated time. N.D. – Not Determined, N.R. – Not Relevant.B. DFO promotes replication fork progression in Hda deficient cells or wild-type cells containing a pBR322-DARS2 plasmid. The indicated cells were grown exponentially at 37 °C in minimal poor medium (blue), shifted to minimal rich medium and incubated for 4 hours at 37 °C in absence of DFO or presence of DFO at a final concentration of 150 mM. The ori/ter ratios were determined by qPCR analysis. Shown is the mean ± s.d. (n=3).
85
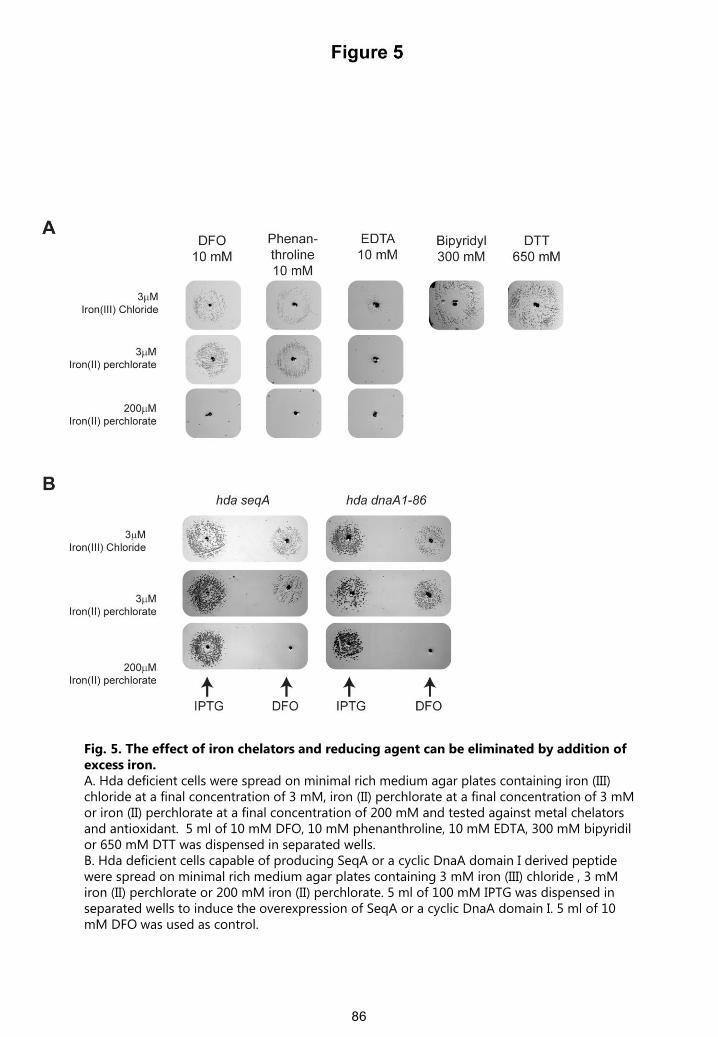
Fig. 5. The effect of iron chelators and reducing agent can be eliminated by addition of excess iron. A. Hda deficient cells were spread on minimal rich medium agar plates containing iron (III) chloride at a final concentration of 3 mM, iron (II) perchlorate at a final concentration of 3 mMor iron (II) perchlorate at a final concentration of 200 mM and tested against metal chelatorsand antioxidant. 5 ml of 10 mM DFO, 10 mM phenanthroline, 10 mM EDTA, 300 mM bipyridilor 650 mM DTT was dispensed in separated wells. B. Hda deficient cells capable of producing SeqA or a cyclic DnaA domain I derived peptide were spread on minimal rich medium agar plates containing 3 mM iron (III) chloride , 3 mMiron (II) perchlorate or 200 mM iron (II) perchlorate. 5 ml of 100 mM IPTG was dispensed in separated wells to induce the overexpression of SeqA or a cyclic DnaA domain I. 5 ml of 10 mM DFO was used as control.
86
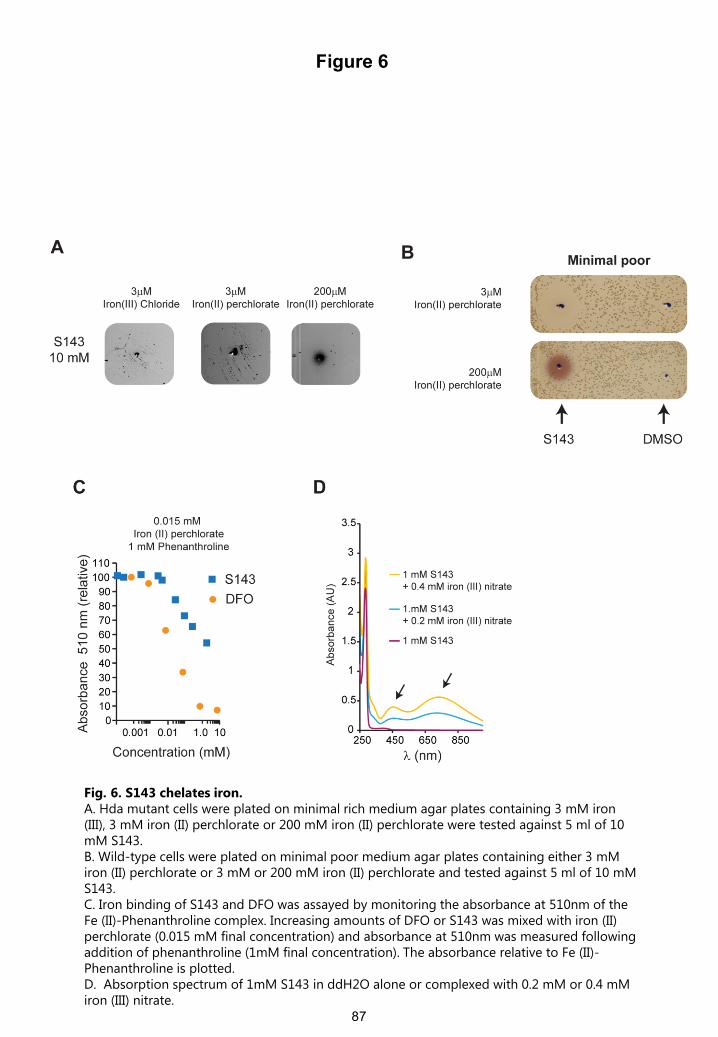
Fig. 6. S143 chelates iron.A. Hda mutant cells were plated on minimal rich medium agar plates containing 3 mM iron (III), 3 mM iron (II) perchlorate or 200 mM iron (II) perchlorate were tested against 5 ml of 10 mM S143. B. Wild-type cells were plated on minimal poor medium agar plates containing either 3 mMiron (II) perchlorate or 3 mM or 200 mM iron (II) perchlorate and tested against 5 ml of 10 mMS143. C. Iron binding of S143 and DFO was assayed by monitoring the absorbance at 510nm of the Fe (II)-Phenanthroline complex. Increasing amounts of DFO or S143 was mixed with iron (II) perchlorate (0.015 mM final concentration) and absorbance at 510nm was measured following addition of phenanthroline (1mM final concentration). The absorbance relative to Fe (II)-Phenanthroline is plotted.D. Absorption spectrum of 1mM S143 in ddH2O alone or complexed with 0.2 mM or 0.4 mMiron (III) nitrate.
87
Fig. 7. Model structure for DFO and S143 chelating ironA single DFO molecule forms six bonds with iron (III) while up to three S143 molecules can interact with one iron (III) through chelation by their catechol moieties.
88
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.0 2.0 4.0 6.0 8.0 10.0 12.0 14.0 16.0
32 g DFO ml-1
16 g DFO ml-1
8 g DFO ml-1
4 g DFO ml-1
2 g DFO ml-1
1 g DFO ml-1
0.5 g DFO ml-1
no DFO
Time (hours)
O.D
. 600
nm
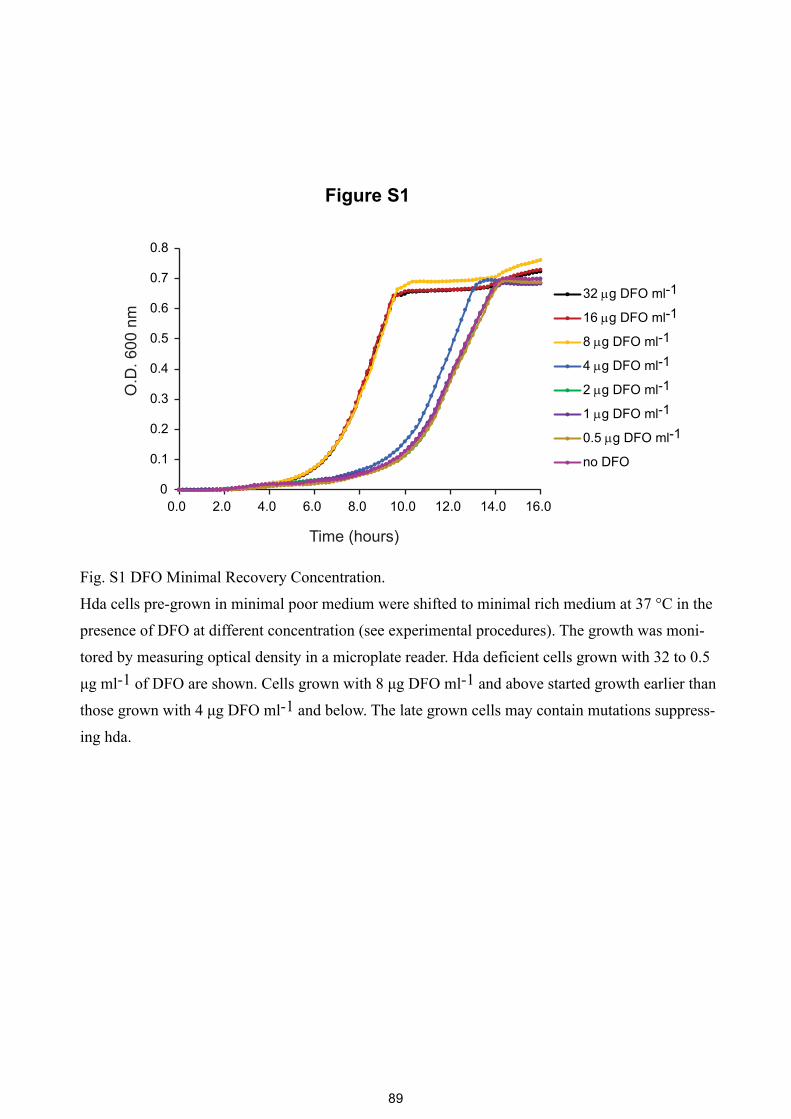
Figure S1
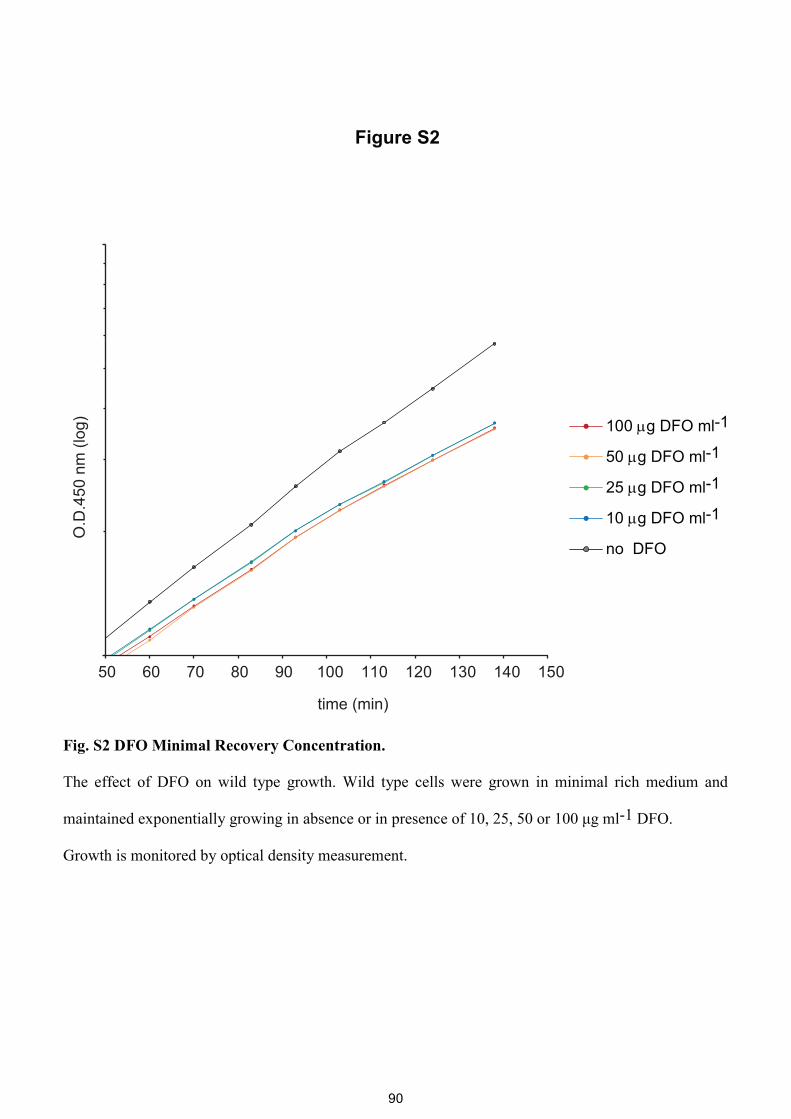
Fig. S1 DFO Minimal Recovery Concentration.
Hda cells pre-grown in minimal poor medium were shifted to minimal rich medium at 37 °C in the
presence of DFO at different concentration (see experimental procedures). The growth was moni-
tored by measuring optical density in a microplate reader. Hda deficient cells grown with 32 to 0.5
μg ml-1 of DFO are shown. Cells grown with 8 μg DFO ml-1 and above started growth earlier than
those grown with 4 μg DFO ml-1 and below. The late grown cells may contain mutations suppress-
ing hda.
89
50 60 70 80 90 100 110 120 130 140 150
100 g DFO ml-1
50 g DFO ml-1
25 g DFO ml-1
10 g DFO ml-1
no DFO
O.D
.450
nm
(log
)
time (min)
Figure S2
Fig. S2 DFO Minimal Recovery Concentration.
The effect of DFO on wild type growth. Wild type cells were grown in minimal rich medium and
maintained exponentially growing in absence or in presence of 10, 25, 50 or 100 μg ml-1 DFO.
Growth is monitored by optical density measurement.
90
DFO DMSO
200M Iron(II) perchlorate
glu +casa
3M Iron(III) Chloride
Figure S3
Fig. S3 Excess iron counteracts the effect of DFO in the pBR322-DARS2 screen.
Cells carrying a multi-copy DARS2 plasmid were spread on minimal rich medium agar plates
containing iron (III) chloride at a final concentration of 3 μM or iron (II) perchlorate at a final
concentration of 200 μM and tested against DFO. 5 μl of 10 mM DFO or DMSO was dispensed in
separated wells.
91
0 10 30 50
O.D
. 600
nm
(log
)
time (min)
wt
wt 150M DFO
wt 150M DFO 200 M iron (II) perchlorate
Figure S4
Fig. S4 The effect of DFO on wild type growth is counteracted by iron.
Wild type cells were grown in minimal rich medium and maintained exponentially growing in absence
of DFO, in presence of 150μM DFO or 150μM DFO and excess iron (II). Growth was monitored by
optical density measurement.
92
Abs
orba
nce
(AU
)
10 mM S1431.2 mM S1430.3 mM S143
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0.16
0.18
0.2
350 450 550 650
0.05 mM S143No S143
(nm)
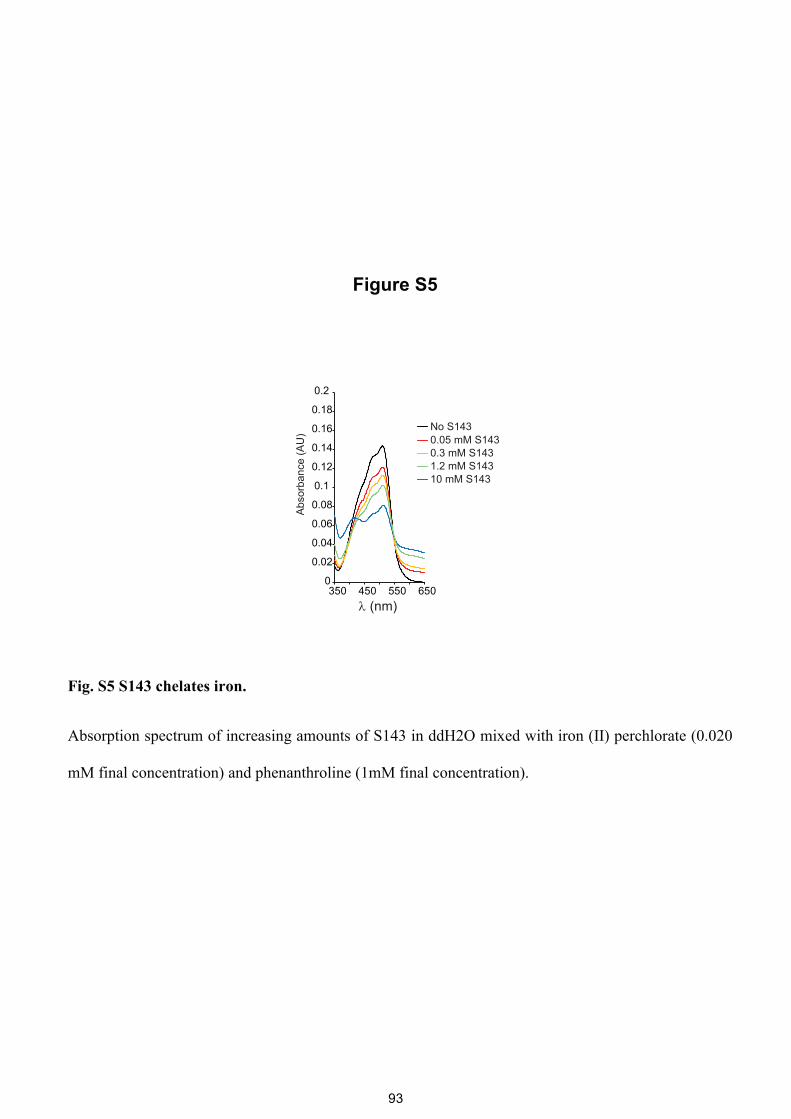
Figure S5
Fig. S5 S143 chelates iron.
Absorption spectrum of increasing amounts of S143 in ddH2O mixed with iron (II) perchlorate (0.020
mM final concentration) and phenanthroline (1mM final concentration).
93
Paper III: A Novel Fluorescence Based Screen for
Inhibitors of the Initiation of DNA Replication in
Bacteria.
Currently in review at: Current Drug Discovery Technologies.
A Novel Fluorescence Based Screen for Inhibitors of the Initiation of DNA
Replication in Bacteria
Rasmus N. Klitgaarda and Anders Løbner-Olesena*
aDepartment of Biology, Faculty of SCIENCE, University of Copenhagen, Copenhagen, Denmark.
Abstract: Background: One of many strategies to overcome antibiotic resistance is the discovery of
compounds targeting cellular processes, which have not yet been exploited. Methods and materials:
Using various genetic tools, we constructed a novel high throughput, cell based, fluorescence screen for
inhibitors of chromosome replication initiation in bacteria. Results: The screen was validated by
expression of an intra-cellular cyclic peptide interfering with the initiator protein DnaA and by over-
expression of the negative initiation regulator SeqA. We also demonstrate that neither tetracycline nor
ciprofloxacin triggers a false positive result. Finally, 400 extracts isolated mainly from filamentous
actinomycetes were subjected to the screen. Conclusion: We conclude that the presented screen is
applicable for identifying putative inhibitors of DNA replication initiation in a high throughput setup.
Keywords: DNA replication initiation, inhibitors, DnaA, high throughput screen, fluorescence, microbial
extracts.
1. INTRODUCTION
Antibiotic resistance is one of the major health care
problems in the world; therefore, it is important to discover
compounds targeting unexploited processes essential to the
growth or viability of bacteria. One such process is
replication of the bacterial chromosome. Targeting the DNA
replication is attractive for a number of reasons; i) The
replisome number per cell is low, ii) The replication complex
is a multi-protein machinery and therefore contains a large
number of potential targets, iii) Key components of the
replication machinery is well conserved and has low
sequence homology with human replication proteins and iiii)
The DNA replication is an under exploited target, this far
only type-II topoisomerase inhibitors are used in the
clinic[1]. A number of different compounds have been
identified targeting components of the replication machinery
including; DNA ligase (LigA)[2, 3], DNA polymerase III[4,
5], the sliding clamp[6, 7] and single-stranded DNA-binding
proteins[8]. In contrast, only a few efforts have been made to
discover inhibitors of the initiation process of the bacterial
DNA replication [9-11].
In most bacteria, replication of the chromosomal
DNA is initiated from a single origin of replication, termed
oriC. The initiation process is best characterized in
Escherichia coli, where DNA replication is initiated by
binding of the initiator protein DnaA, in its active ATP-
bound form, to the oriC. When sufficient DnaAATP
molecules are bound to the oriC it forms a nucleoprotein
complex responsible for separation of the DNA double
strand[12]. Following duplex opening the nucleoprotein
complex loads the DnaB helicase, with help from the
helicase loader protein DnaC [13, 14]. Loading of DnaB then
triggers the assembly of the remaining parts of the
replication machinery[12]. The initiation process, including
the DnaC assisted loading of the DnaB helicase, is highly
conserved across bacterial species [15], and is therefore an
interesting target for novel antibiotics.
E. coli rnhA mutants, lacking RNase HI activity, are
able to initiate the DNA replication by a protein synthesis
and DnaA/oriC independent pathway called constitutive
Stable DNA Replication (cSDR)[16, 17]. cSDR is initiated
at a number of alternative sites on the chromosome, termed
oriK[18]. It is believed that lack of RNase HI activity leads
to stabilization of nascent RNA transcripts, which anneals to
the DNA template behind the moving RNA polymerase,
creating an R-loop. The RNA is thought to act as a primer
for extension by DNA polymerase I, creating a D-loop like
structure. This structure is then bound by PriA, initiating
assembly of the PriA dependent primosome, loading of the
DnaB helicase and assembly of the replisome [19, 20]. The
DnaA/oriC independency of cSDR makes it a valuable tool
when searching for inhibitors targeting the initiation of DNA
replication.
We here present a high throughput fluorescence based screen, which can be used to specifically screen for inhibitors that targets DNA replication initiation.
*Address correspondence to this author at the Department of Biology, Faculty of SCIENCE, University of Copenhagen, Copenhagen, Denmark; E-mail: [email protected]
Short Running Title of the Article Journal Name, 2014, Vol. 0, No. 0 7
[40] Langer U, Richter S, Roth A, Weigel C, Messer W.
A comprehensive set of DnaA-box mutations in the
replication origin, oriC, of Escherichia coli. Molecular
microbiology. 1996;21(2):301-11.
Received: March 20, 2014 Revised: April 16, 2014 Accepted: April 20, 2014
101
Discussion
Antibiotic resistance has become an urgent problem, which not only threatens the future of health care,
as we know it today, but also might turnout out to be a heavy burden for the global economy (12, 13).
An effective strategy to overcome antibiotics resistance will need to be multidimensional. Thus,
facilitate a sustainable use of antibiotics in the clinic as well as in the industry, diminish the spread of
infectious disease, and encourage development of novel drugs and preservation of existing antibiotics.
In the last 10 years over 50 national and international initiatives have been founded with the goal of
encouraging research and development of antibiotics (258). Though action has already been taken;
evidence and experience show that the current market for antibiotics does not foster major investments
from the large pharmaceutical companies, who have set their course for more profitable markets (12).
We have contributed to the fight against antibiotic resistance, by searching for potential helper drug
targets to reverse quinolone resistance and thereby preserve the efficacy of one of the most important
classes of antibiotics. Furthermore, we have developed and verified two distinct strategies for the
discovery of novel classes of antibiotics targeting the initiation of chromosomal DNA replication in
bacteria.
Potentiation of the quinolones
In paper I, we sought to identify targets for potentiation of ciprofloxacin by introduction of more than
twenty separate single gene deletions in a high-level ciprofloxacin resistant strain. None of the tested
gene deletions rendered the high-level resistant strain clinically susceptible. However, deletion of acrA,
tolC, recA or recC decreased the MIC of a low-level ciprofloxacin resistant strain beneath the clinical
break point. Indicating that inhibition of the AcrAB-tolC efflux-pump or HR repair of DNA DSBs, via RecA
or RecC inhibition, is a plausible strategy for reversal of low-level ciprofloxacin resistance. These findings
are in agreement with the observations made by Tran et al. and Recacha et al.(129, 130).
The discovery and development of putative inhibitors of RecA, and thereby the SOS
response, is attractive for several reasons. Most bactericidal antibiotics are inducers of the SOS response
(55), thus inhibition of RecA might not only potentiate the quinolones but also other classes of
bactericidal antibiotics. The SOS response also plays an important role in the evolution of antibiotic
resistance, by inducing horizontal gene transfer (HGT) of antibiotic resistance genes (259) and by
promoting mutagenesis via the error prone DNA polymerases IV and V (86). In addition, induction of the
SOS response has been shown to promote HGT of pathogenicity associated genes in E. coli and S.
102
aureus. Thus, RecA inhibitors could potentially exert a dual mode of action in increasing antibiotic
susceptibility and decreasing evolution of antibiotic resistance and pathogenicity.
As described earlier, multiple efforts have been made to identify putative inhibitors of
RecA, leading to the discovery of several compounds that blocks the ATPase activity of RecA in vitro
(121, 122, 124). It is unknown if any these compounds are still under development. Copper
phtalocyanine-3, 4´, 4´´,4´´´-tetrasulfonic acid (CuPTA) and suramin are the only compounds that
evidently inhibits RecA in vivo (119, 125). In paper I, we report that CuPTA does not change the
ciprofloxacin MIC for a high- or low-level ciprofloxacin resistant strain. Indicating that either CuPTA is a
weak RecA inhibitor or that a given inhibitor needs to completely inactivate RecA for potentiating
ciprofloxacin. Hence, development of an effective RecA inhibitor in regards to potentiation of the
quinolones and other bactericidal antibiotics might prove difficult. In contrast to RecA, there only exist a
single report of identification of putative inhibitors of the RecBCD complex, however the potential of the
identified compounds to potentiate the quinolones has not been assessed (260).
Inhibition of efflux pumps, like AcrAB-TolC, is a well investigated mean of potentiating
antibiotics. Similar to inhibition of RecA, efflux pump inhibition generates some desirable side effects in
addition to decreasing the MIC. In E. coli, deletion of acrAB delays the emergence of levofloxacin
resistance (261), while the virulence of Salmonella enterica to some extend relies on the functionality of
its drug efflux systems (262). A described earlier several EPIs that targets the AcrAB-TolC efflux pump
have been identified. However, not a single EPI has made it through clinical trials and into the clinic
(263, 264). One of the major challenges in EPI development is the broad compound specificity exerted
by most efflux pumps. Consequently, it is challenging to setup guidelines for discriminating between
efflux pump substrates and inhibitors, making it difficult to pick suitable compound libraries for
screening (264). Current challenges in clinical development of EPIs are; toxicity, pharmacokinetics,
potency and spectrum of activity (265). Hence, further insight into the structure and function of efflux
pumps is essential for successful discovery and development of EPIs in the future.
Tran et al. showed that combinatorial disruption of the AcrAB-TolC efflux pump and the
SOS response rendered a high-level ciprofloxacin resistant strain clinically susceptible (129). However,
deployment of this observation in the clinic would require a three drug combinatorial treatment.
Experience from combinatorial drug therapy of cancer, has shown that treatment with multiple drugs is
challenging due to overlapping toxicities and differences in pharmacological profiles (266). Indicating,
that it would be difficult to develop such a treatment, though there is no doubt that combinatorial
inhibition of drug efflux and the SOS response, would be a powerful weapon in overcoming antibiotic
resistance.
103
Based on the findings presented in paper I, it seems that reversing high-level ciprofloxacin
resistance with a single helper drug is not possible. However, during the gene deletion analysis, we were
not able to delete priA in the high-level resistant strain, LM693. A similar observation was reported by
Cirz et al., who were unable to construct an E. coli gyrA(S83L) ΔpriA mutant (72). PriA is the initiator
protein of replication restart, a housekeeping process that facilitates the restart of stalled replication
forks during normal growth (267). E. coli priA- strains suffers from severe growth retardation and are
hyper-susceptible to ciprofloxacin with a MIC below 1 ng/ml (72). The above observations indicates that
PriA inhibition is possibly lethal to bacterial strains with gyrA mutations conferring ciprofloxacin
resistance. Thus, combinatorial treatment of a PriA inhibitor with ciprofloxacin, or cycling between the
two, could be an effective mean of treating infections caused by ciprofloxacin resistant bacteria.
Targeting the commencement of DNA replication in bacteria
Duplication of the chromosome is an essential part of the bacterial cell cycle and its initiation could
potentially serve as a novel antibiotic target. We have presented two novel cell based strategies for
identifying DNA replication initiation inhibitors. The replication initiation process and its regulation is
complex and can therefore, potentially be inhibited via multiple different targets, of which directly
targeting DnaA is the most apparent one. However, the negative or positive regulation of the initiation,
might also serve as potential targets. Emphasized by the fact that deletion of either datA, DARS1 or
DARS2 in E. coli, results in a reduced ability to colonize the large intestine in mice (183).
DnaA binding to the DnaA boxes in the oriC is a plausible target for putative inhibitors of
the replication initiation. Specifically, targeting the HTH motif of DnaA domain IV could in principal block
the binding of DnaA to both the high and low affinity DnaA boxes in the oriC. Interference with the
assembly of the DnaAATP-OriC nucleoprotein complex is attainable in multiple ways. Binding of an
inhibitor in the nucleotide-binding pocket of the AAA+ module of domain III, could block ATP binding
and lock the structural conformation of DnaA in an apo-DnaA or DnaAADP-like state that is inactive in
DnaA oligmerization. In addition, the cooperative binding of DnaAATP could also be inhibited by
interfering with the DnaA-DnaA interactions mediated by specific residues of DnaA domain I and III.
However, DnaA boxes in the oriC are not arranged similarly across bacterial species, indicating that
there are many different ways of assembling the nucleoprotein complex. Thus, it is not necessarily the
same residues that mediates the DnaA-DnaA interactions in different bacterial species (188). Due to
their stimulatory role in the replication initiation process, factors like; IHF, DiaA, HU, and H-NS, might
also serve as targets for inhibiting replication initiation. Although none of these factors are essential for
replication initiation, their deletion does lead to under-initiation (163, 268-270), though it remains to be
assessed if it has a lethal effect in a hostile environment like the human body.
104
Loading of the DnaB helicase by DnaA is an essential step in initiating the replication
process and is therefore an obvious target. Inhibiting the binding of DnaC would likely block the loading
of DnaB onto the ssDUE. As DnaB would not be locked into to the open conformation that is essential
for its loading onto the ssDUE (193). In addition, hindering the interaction between DnaB and DnaA
domain I should also block the loading of the DnaB helicase.
As mentioned above, the processes that regulates the DnaAATP/DnaAADP ratio is also a
potential target for interfering with the replication initiation. Interestingly, over-initiation seems to be
more lethal than decreasing the initiation frequency (237). Thus, inhibiting the conversion of DnaAATP to
DnaAADP by RIDA or DDAH, or the sequestration of the oriC by SeqA, is likely more favorable than
targeting the rejuvenation of DnaAATP. Obstructing SeqA binding to the GATC in the oriC and the DnaA
promoter region, would likely lead to over-initiation and increased levels of DnaAATP. However, the
increase in initiation observed when seqA is deleted is not detrimental to the cell (271). DDAH is less
efficient in stimulating DnaAATP hydrolysis than RIDA (170), indicating that RIDA is the favorable target.
RIDA inactivation could be achieved by; i) hindering binding of ADP to Hda, ii) blocking the ATPase
stimulatory interaction of the Hda Arg-finger with the DnaAATP ATPase in domain III, iii) inhibiting the
stabilizing interactions between the C-terminal of Hda and DnaAATP domain I. Though the idea of
inducing over-initiation is intriguing, the fact that secondary mutations arise quickly in cells that are
over-initiating (240), designates that resistance would quickly develop. Furthermore, the ROS
dependency of the lethal action of hyper-replication suggests, that the strategy of inducing over-
initiation is not plausible at low ROS conditions i.e. anaerobsis or when free iron availability is limited.
Inhibition of the regeneration of DnaAATP via DARS1 or DARS2, is likely achievable by
blocking the DnaA-DnaA interactions in the DnaA box core region, or by hindering the binding of IHF or
Fis to the regulatory region of DARS2. The mechanisms that lead to DnaAATP regeneration mediated by
phospholipids are unknown. Consequently, inhibition of the phospholipid synthesis is the only known
mean by which this process could be inhibited. Targeting the regulatory mechanisms of the replication
initiation has a downside, as it currently not known how many bacterial species that uses the DnaAATP
level to regulate replication initiation. Hda homologs have been identified in Caulobacter and most
enterobacteria, but not in Bacillus, Staphylococcus and H. pylori (131, 272). DARS1 and DARS2 are likely
conserved in proteobacteria that are closely related to E. coli, while more distantly related
proteobacteria have DARS-like DnaA box clusters in other intergenic regions. Non-proteobacterial
species like S. aureus, Mycobacterium tuberculosis and Bacillus subtilis have DnaA box clusters near
dnaA, though with a different arrangement of the DnaA boxes than the one in E. coli (148).
Consequently, compounds that target the regulation of replication initiation will likely not have a broad
bacterial spectrum.
105
Despite all of the above mentioned potential targets, it is curios that not a single
compound that targets the replication initiation process has been identified. This fact could indicate that
the replication initiation is not an ideal target for development of novel antibiotics. Furthermore, the
complexity of its regulation may enhance the occurrence of secondary compensatory mutations that
counteract the inhibitory action of a given compound. Yet, it is important to keep in mind that direct
efforts at identifying replication initiation inhibiters has so far been scarce.
The screens presented in paper II and III have some distinct differences concerning their
specificity and practical applicability. The fluorescence-based screen relies on replication by cSDR and is
therefore not able to identify putative inhibitors of the DnaB helicase loading; as such compounds would
kill the cells. Conversely, the screens based on over-initiating cells cannot be used to identify inhibitors
that induce over-initiation. Whereas, over-initiation of the mini-chromosome in the fluorescence based
screen, could potentially lead to instability and loss of the mini-chromosome and thereby expression of
the green fluorescent protein (GFP). Concerning the practical applicability, the fluorescence based
screen is performed in 96-well plates in a high throughput manner. In contrast, the agar plate based
platform of the hda and DARS2 screens needs to undergo further optimization for use in a high
throughput setup. The agar plate based platform has a significant advantage, as a concentration
gradient is created when the extract or compound diffuse from the well into the agar. Thus, several drug
concentrations are tested at once, in contrast to the fluorescence based screen, where only a single
concentration is tested.
Why is severe over-initiation of the DNA replication lethal?
In paper II, we show that the iron chelator deferoxamine rescues the growth of over-initiating cells by
promoting the processivity of replication forks. Deferoxamine has been shown to inhibit the Fenton
reaction and thereby the production of ROS both in vivo and in vitro. Our findings therefore support the
proposed model that the lethal action of over-initiating the chromosomal DNA replication is caused by
formation of DSBs in the DNA, when replication forks encounters 8-oxo-dGTP lesions that are under
repair by the GO system (235). Since overproduction of ribonucleotide reductase (RNR), restores the
growth of over-initiating cells, it has been suggested that during replication over-initiation the cells are
starved for dNTPs because of the increased number of replication forks (238, 239, 241). The dNTP
starvation is proposedly responsible for the severe growth retardation of cells deficient in Hda (239).
However, several observations contradict this model; i) the reduction in the dNTP pool of the hda
mutant is not significant, ii) the inviability of an hda mutant does not resemble the inviability observed
for cells starved in dNTPs, as dNTP starvation leads to obliteration of the oriC, in contrast to the
observed increase in oriC copy number for hda mutants, iii) an increase in all four dNTPs is not observed
106
when RNR is overexpressed, specifically the dGTP level remains more or less unchanged, thus a hda
mutant overexpressing RNR is still starved in dGTP (271). Conversely, the observed suppression of the
hda phenotype by overproduction of RNR is more likely caused by a reduction in replication initiation, as
hda mutants overproducing RNR has an origin concentration resembling that of a wild-type (241, 271).
Hence, over-expression of RNR in an hda mutant lowers the initiation frequency, giving time for efficient
repair of 8-oxo-dGTP lesions.
Conclusions
Utilizing genetic screens and differential gene-expression analysis, we have shown that reversing
ciprofloxacin resistance in a high-level ciprofloxacin resistant E. coli strain is likely not possible. However,
our genetic screen revealed the AcrAB-tolC efflux pump, and the SOS response proteins RecA and RecC,
as plausible targets for ciprofloxacin helper drugs in E. coli strains with a MIC just above clinical
breakpoint.
We have constructed and verified three screens for identifying inhibitors of the initiation
of chromosomal DNA replication in bacteria. One screen relies on replication inhibition of an OriC
dependent mini-chromosome, leading to expression of GFP and thereby a detectable increase in
fluorescence. The two other screens are based on growth rescue of cells that exerts lethal over-initiation
of the DNA replication, by either harboring multiple copies of DARS2 or being deficient in Hda.
As a pilot screen for inhibitors of the replication initiation process, we subjected our novel
screens to a library of 400 actinomycetes extracts. Even though we did not identify any initiation
inhibitors, the iron chelator deferoxamine, a known inhibitor of the Fenton reaction (245), was
identified as a compound that rescues the growth of over-initiating cells. Corroborating the model that
the lethality of over-initiating the chromosomal DNA replication is caused by formation of DSBs in the
DNA, when replication forks encounters 8-oxo-dGTP lesions that are under repair by the GO system
(235). In addition, we also showed that the growth rescue of over-initiating cells exerted by the
suggested gyrase inhibitor (±)-6-Chloro-PB hydrobromide (S143) is, at least in part, due to its ability to
chelate iron.
Future perspectives
Antibiotic resistance will be a remaining threat to global health care. However, by heavily investing in
antibiotic drug development and regulating the use of antibiotics globally. It may be possible to halt or
slow the current negative development. Based on our findings that the AcrAB-TolC efflux pump and the
107
SOS response genes RecA and RecC, might serve as ciprofloxacin helper drug targets, in treating low-
level resistant strains. In addition to the fact that such helper drugs have the potential to potentiate
other known antibiotics and decrease the evolution of antibiotic resistance, it would be interesting to
setup screens for identifying inhibitors of these three targets. Moreover, it would be attractive to assess
if PriA deletion is truly lethal for bacterial strains carrying gyrA mutations that confer ciprofloxacin
resistance.
As we now have two distinct strategies for identifying replication inhibitors, the next
logical step is to obtain several chemical or natural extract libraries that could be subjected to the
screens. The 400 extracts that were screened in paper II and III, are part of a large library of more than
4000 microbial extracts, owned by our collaborator Naicons srl., which has proven to be a source of
novel antibiotic compounds (273, 274). Hopefully, future funding will give us the opportunity to screen
the remainder of this vast library of bioactive natural extracts.
108
Bibliography
1. Fleming A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. British journal of experimental pathology. 1929;10(3):226-36. 2. Ehrlich P, and Hata, S. Die Experimentelle Chemotherapie der Spirilosen1910. 3. Aminov R. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Frontiers in Microbiology. 2010;1(134). 4. Achari A, Somers D, Champness JN, Bryant PK, Rosemond J, Stammers DK. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nature structural biology. 1997;4:490. 5. Gould K. Antibiotics: from prehistory to the present day. Journal of Antimicrobial Chemotherapy. 2016;71(3):572-5. 6. Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nature Chemical Biology. 2007;3:541. 7. Fleming A. Penicillin, Nobel lecture,1945. 8. Leach KL, Brickner SJ, Noe MC, Miller PF. Linezolid, the first oxazolidinone antibacterial agent. Annals of the New York Academy of Sciences. 2011;1222:49-54. 9. Long KS, Vester B. Resistance to Linezolid Caused by Modifications at Its Binding Site on the Ribosome. Antimicrobial agents and chemotherapy. 2012;56(2):603-12. 10. Eliopoulos GM, Meka VG, Gold HS. Antimicrobial Resistance to Linezolid. Clinical Infectious Diseases. 2004;39(7):1010-5. 11. The PEW Charitable Trusts. Antibiotics currently in global clinical development 2017 [Available from: http://www.pewtrusts.org/~/media/assets/2017/12/antibiotics_currently_in_clinical_development_09_2017.pdf?la=en. 12. Simpkin VL, Renwick MJ, Kelly R, Mossialos E. Incentivising innovation in antibiotic drug discovery and development: progress, challenges and next steps. The Journal of antibiotics. 2017;70:1087. 13. Review on Antimicrobial Resistance. Resistance: Tackling a Crisis for the Health and Wealth of Nations 2014 [Available from: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf. 14. Mitscher LA. Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chemical reviews. 2005;105(2):559-92. 15. Linder JA, Huang ES, Steinman MA, Gonzales R, Stafford RS. Fluoroquinolone prescribing in the United States: 1995 to 2002. The American Journal of Medicine.118(3):259-68. 16. Emmerson AM, Jones AM. The quinolones: decades of development and use. The Journal of antimicrobial chemotherapy. 2003;51 Suppl 1:13-20. 17. Werner NL, Hecker MT, Sethi AK, Donskey CJ. Unnecessary use of fluoroquinolone antibiotics in hospitalized patients. BMC Infectious Diseases. 2011;11:187-. 18. Dalhoff A. Global Fluoroquinolone Resistance Epidemiology and Implictions for Clinical Use. Interdisciplinary Perspectives on Infectious Diseases. 2012;2012:37. 19. Bisacchi GS. Origins of the Quinolone Class of Antibacterials: An Expanded “Discovery Story”. Journal of Medicinal Chemistry. 2015;58(12):4874-82. 20. Lesher GY, Froelich EJ, Gruett MD, Bailey JH, Brundage RP. 1,8-Naphthyridine Derivatives. A New Class of Chemotherapeutic Agents. Journal of Medicinal and Pharmaceutical Chemistry. 1962;5(5):1063-5. 21. Aldred KJ, Kerns RJ, Osheroff N. Mechanism of quinolone action and resistance. Biochemistry. 2014;53(10):1565-74. 22. Owens RC, Ambrose, Paul G. CLINICAL USE OF THE FLUOROQUINOLONES. Medical Clinics of North America. 2000;84(6):1447-69.
109
23. Goss WA, Deitz WH, Cook TM. Mechanism of Action of Nalidixic Acid on Escherichia coli II. Inhibition of Deoxyribonucleic Acid Synthesis. Journal of bacteriology. 1965;89(4):1068-74. 24. Hane MW, Wood TH. Escherichia coli K-12 mutants resistant to nalidixic acid: genetic mapping and dominance studies. Journal of bacteriology. 1969;99(1):238-41. 25. Gellert M, Mizuuchi K, O'Dea MH, Itoh T, Tomizawa JI. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(11):4772-6. 26. Sugino A, Peebles CL, Kreuzer KN, Cozzarelli NR. Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(11):4767-71. 27. Kato J, Nishimura Y, Imamura R, Niki H, Hiraga S, Suzuki H. New topoisomerase essential for chromosome segregation in E. coli. Cell. 1990;63(2):393-404. 28. Hooper DC. Mode of action of fluoroquinolones. Drugs. 1999;58 Suppl 2:6-10. 29. Aldred KJ, McPherson SA, Turnbough CL, Jr., Kerns RJ, Osheroff N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013;41(8):4628-39. 30. Aldred KJ, McPherson SA, Wang P, Kerns RJ, Graves DE, Turnbough CL, Jr., et al. Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry. 2012;51(1):370-81. 31. Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol. 2010;17(99):1152-3. 32. Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiology and molecular biology reviews : MMBR. 1997;61(3):377-92. 33. Khodursky AB, Zechiedrich EL, Cozzarelli NR. Topoisomerase IV is a target of quinolones in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(25):11801-5. 34. Ferrero L, Cameron B, Crouzet J. Analysis of gyrA and grlA mutations in stepwise-selected ciprofloxacin-resistant mutants of Staphylococcus aureus. Antimicrobial agents and chemotherapy. 1995;39(7):1554-8. 35. Gillespie SH, Voelker LL, Ambler JE, Traini C, Dickens A. Fluoroquinolone resistance in Streptococcus pneumoniae: evidence that gyrA mutations arise at a lower rate and that mutation in gyrA or parC predisposes to further mutation. Microbial drug resistance (Larchmont, NY). 2003;9(1):17-24. 36. Fournier B, Zhao X, Lu T, Drlica K, Hooper DC. Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrobial agents and chemotherapy. 2000;44(8):2160-5. 37. Kreuzer KN, Cozzarelli NR. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. Journal of bacteriology. 1979;140(2):424-35. 38. D'Arpa P, Beardmore C, Liu LF. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer research. 1990;50(21):6919-24. 39. Malik M, Zhao X, Drlica K. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Molecular microbiology. 2006;61(3):810-25. 40. Hiasa H, Yousef DO, Marians KJ. DNA strand cleavage is required for replication fork arrest by a frozen topoisomerase-quinolone-DNA ternary complex. The Journal of biological chemistry. 1996;271(42):26424-9. 41. Shea ME, Hiasa H. Interactions between DNA helicases and frozen topoisomerase IV-quinolone-DNA ternary complexes. The Journal of biological chemistry. 1999;274(32):22747-54.
110
42. Shea ME, Hiasa H. Distinct effects of the UvrD helicase on topoisomerase-quinolone-DNA ternary complexes. The Journal of biological chemistry. 2000;275(19):14649-58. 43. Shea ME, Hiasa H. The RuvAB branch migration complex can displace topoisomerase IV.quinolone.DNA ternary complexes. The Journal of biological chemistry. 2003;278(48):48485-90. 44. Pohlhaus JR, Kreuzer KN. Norfloxacin-induced DNA gyrase cleavage complexes block Escherichia coli replication forks, causing double-stranded breaks in vivo. Molecular microbiology. 2005;56(6):1416-29. 45. Zhao X, Malik M, Chan N, Drlica-Wagner A, Wang JY, Li X, et al. Lethal action of quinolones against a temperature-sensitive dnaB replication mutant of Escherichia coli. Antimicrobial agents and chemotherapy. 2006;50(1):362-4. 46. Deitz WH, Cook TM, Goss WA. Mechanism of action of nalidixic acid on Escherichia coli. 3. Conditions required for lethality. Journal of bacteriology. 1966;91(2):768-73. 47. Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X. Quinolones: action and resistance updated. Current topics in medicinal chemistry. 2009;9(11):981-98. 48. Imlay JA, Fridovich I. Superoxide Production by Respiring Membranes of Escherichia Coli. Free Radical Research Communications. 1991;12(1):59-66. 49. Imlay JA. Diagnosing oxidative stress in bacteria: not as easy as you might think. Current opinion in microbiology. 2015;24:124-31. 50. Belenky P, Ye JD, Porter CB, Cohen NR, Lobritz MA, Ferrante T, et al. Bactericidal Antibiotics Induce Toxic Metabolic Perturbations that Lead to Cellular Damage. Cell Rep. 2015;13(5):968-80. 51. Becerra MC, Albesa I. Oxidative stress induced by ciprofloxacin in Staphylococcus aureus. Biochemical and Biophysical Research Communications. 2002;297(4):1003-7. 52. Albesa I, Becerra MC, Battan PC, Paez PL. Oxidative stress involved in the antibacterial action of different antibiotics. Biochem Biophys Res Commun. 2004;317(2):605-9. 53. Becerra MC, Páez PL, Laróvere LE, Albesa I. Lipids and DNA oxidation in Staphylococcus aureus as a consequence of oxidative stress generated by ciprofloxacin. Molecular and Cellular Biochemistry. 2006;285(1):29-34. 54. Goswami M, Mangoli SH, Jawali N. Involvement of Reactive Oxygen Species in the Action of Ciprofloxacin against Escherichia coli. Antimicrobial agents and chemotherapy. 2006;50(3):949-54. 55. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell. 2007;130(5):797-810. 56. Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Molecular systems biology. 2007;3:91. 57. Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the Guanine Nucleotide Pool Underlies Cell Death by Bactericidal Antibiotics. Science. 2012;336(6079):315-9. 58. Kottur J, Nair DT. Reactive Oxygen Species Play an Important Role in the Bactericidal Activity of Quinolone Antibiotics. Angewandte Chemie (International ed in English). 2016;55(7):2397-400. 59. Hassan HM, Moody CS. Induction of the manganese-containing superoxide dismutase in Escherichia coli by nalidixic acid and by iron chelators. FEMS Microbiology Letters. 1984;25(2-3):233-6. 60. Greenberg JT, Monach P, Chou JH, Josephy PD, Demple B. Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in Escherichia coli. Proceedings of the National Academy of Sciences. 1990;87(16):6181-5. 61. Tsaneva IR, Weiss, B. soxR, a locus governing a superoxide response regulon in Escherichia coli K-12. Journal of bacteriology. 1990;172(8):4197-205. 62. Seo Sang W, Kim D, Szubin R, Palsson Bernhard O. Genome-wide Reconstruction of OxyR and SoxRS Transcriptional Regulatory Networks under Oxidative Stress in <em>Escherichia coli</em> K-12 MG1655. Cell Reports.12(8):1289-99. 63. Nunoshiba T, Hidalgo E, Amábile Cuevas CF, Demple B. Two-stage control of an oxidative stress regulon: the Escherichia coli SoxR protein triggers redox-inducible expression of the soxS regulatory gene. Journal of bacteriology. 1992;174(19):6054-60.
111
64. Chou JH, Greenberg JT, Demple B. Posttranscriptional repression of Escherichia coli OmpF protein in response to redox stress: positive control of the micF antisense RNA by the soxRS locus. Journal of bacteriology. 1993;175(4):1026-31. 65. Li X-Z, Nikaido H. Efflux-Mediated Drug Resistance in Bacteria. Drugs. 2004;64(2):159-204. 66. Wang X, Zhao X, Malik M, Drlica K. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. The Journal of antimicrobial chemotherapy. 2010;65(3):520-4. 67. Malik M, Hussain S, Drlica K. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrobial agents and chemotherapy. 2007;51(1):28-34. 68. Liu Y, Imlay JA. Cell Death from Antibiotics Without the Involvement of Reactive Oxygen Species. Science. 2013;339(6124):1210-3. 69. Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. Killing by Bactericidal Antibiotics Does Not Depend on Reactive Oxygen Species. Science. 2013;339(6124):1213. 70. Radman M. SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic life sciences. 1975;5a:355-67. 71. Lewin CS, Howard BM, Ratcliffe NT, Smith JT. 4-quinolones and the SOS response. Journal of medical microbiology. 1989;29(2):139-44. 72. Cirz RT, Chin JK, Andes DR, de Crécy-Lagard V, Craig WA, Romesberg FE. Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. PLoS Biol. 2005;3(6):e176. 73. Little JW, Edmiston SH, Pacelli LZ, Mount DW. Cleavage of the Escherichia coli lexA protein by the recA protease. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(6):3225-9. 74. Schlacher K, Goodman MF. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat Rev Mol Cell Biol. 2007;8(7):587-94. 75. Little JW. Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie. 1991;73(4):411-21. 76. Hiom K. DNA Repair: Common Approaches to Fixing Double-Strand Breaks. Current Biology. 2009;19(13):R523-R5. 77. Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DB. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature. 2004;432(7014):187-93. 78. Wyman C, Ristic D, Kanaar R. Homologous recombination-mediated double-strand break repair. DNA repair. 2004;3(8–9):827-33. 79. Morgan-Linnell SK, Becnel Boyd L, Steffen D, Zechiedrich L. Mechanisms Accounting for Fluoroquinolone Resistance in Escherichia coli Clinical Isolates. Antimicrobial agents and chemotherapy. 2009;53(1):235-41. 80. Lindgren PK, Marcusson LL, Sandvang D, Frimodt-Møller N, Hughes D. Biological Cost of Single and Multiple Norfloxacin Resistance Mutations in Escherichia coli Implicated in Urinary Tract Infections. Antimicrobial agents and chemotherapy. 2005;49(6):2343-51. 81. Marcusson LL, Frimodt-Møller N, Hughes D. Interplay in the Selection of Fluoroquinolone Resistance and Bacterial Fitness. PLoS Pathog. 2009;5(8):e1000541. 82. CLSI. Performance Standards for Antimicrobial Susceptibility Testing. 27th ed. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute; 2017. 83. Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions? Current Opinion in Microbiology. 2006;9(5):461-5. 84. Andersson DI, Hughes D. Effects of Antibiotic Resistance on Bacterial Fitness, Virulence, and Transmission. Evolutionary Biology of Bacterial and Fungal Pathogens: American Society of Microbiology; 2008. 85. Komp Lindgren P, Karlsson Å, Hughes D. Mutation Rate and Evolution of Fluoroquinolone Resistance in Escherichia coli Isolates from Patients with Urinary Tract Infections. Antimicrobial agents and chemotherapy. 2003;47(10):3222-32.
112
86. Fuchs RP, Fujii S, Wagner J. Properties and functions of Escherichia coli: Pol IV and Pol V. Advances in protein chemistry. 2004;69:229-64. 87. Grkovic S, Brown MH, Skurray RA. Regulation of Bacterial Drug Export Systems. Microbiology and Molecular Biology Reviews. 2002;66(4):671-701. 88. Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Molecular microbiology. 1995;16(1):45-55. 89. Delihas N, Forst S. MicF: an antisense RNA gene involved in response of Escherichia coli to global stress factors11Edited by D. Draper. Journal of molecular biology. 2001;313(1):1-12. 90. Cama J, Bajaj H, Pagliara S, Maier T, Braun Y, Winterhalter M, et al. Quantification of Fluoroquinolone Uptake through the Outer Membrane Channel OmpF of Escherichia coli. J Am Chem Soc. 2015;137(43):13836-43. 91. Chapman JS, Georgopapadakou NH. Routes of quinolone permeation in Escherichia coli. Antimicrobial agents and chemotherapy. 1988;32(4):438-42. 92. Poole K, Tetro K, Zhao Q, Neshat S, Heinrichs DE, Bianco N. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrobial agents and chemotherapy. 1996;40(9):2021-8. 93. Ziha-Zarifi I, Llanes C, Köhler T, Pechere J-C, Plesiat P. In Vivo Emergence of Multidrug-Resistant Mutants of Pseudomonas aeruginosa Overexpressing the Active Efflux System MexA-MexB-OprM. Antimicrobial agents and chemotherapy. 1999;43(2):287-91. 94. Llanes C, Hocquet D, Vogne C, Benali-Baitich D, Neuwirth C, Plésiat P. Clinical Strains of Pseudomonas aeruginosa Overproducing MexAB-OprM and MexXY Efflux Pumps Simultaneously. Antimicrobial agents and chemotherapy. 2004;48(5):1797-802. 95. Hooper DC, Jacoby GA. Mechanisms of drug resistance: quinolone resistance. Annals of the New York Academy of Sciences. 2015;1354(1):12-31. 96. Martinez-Martinez L, Pascual A, Jacoby GA. Quinolone resistance from a transferable plasmid. Lancet (London, England). 1998;351(9105):797-9. 97. Jacoby GA. Mechanisms of Resistance to Quinolones. Clinical Infectious Diseases. 2005;41(Supplement_2):S120-S6. 98. Robicsek A, Jacoby GA, Hooper DC. The worldwide emergence of plasmid-mediated quinolone resistance. The Lancet Infectious diseases. 2006;6(10):629-40. 99. Munshi MH, Sack DA, Haider K, Ahmed ZU, Rahaman MM, Morshed MG. Plasmid-mediated resistance to nalidixic acid in Shigella dysenteriae type 1. Lancet (London, England). 1987;2(8556):419-21. 100. Jacoby GA, Strahilevitz J, Hooper DC. Plasmid-mediated quinolone resistance. Microbiology spectrum. 2014;2(2):10.1128/microbiolspec.PLAS-0006-2013. 101. Vetting MW, Hegde SS, Fajardo JE, Fiser A, Roderick SL, Takiff HE, et al. Pentapeptide repeat proteins. Biochemistry. 2006;45(1):1-10. 102. Tran JH, Jacoby GA. Mechanism of plasmid-mediated quinolone resistance. Proceedings of the National Academy of Sciences. 2002;99(8):5638-42. 103. Hata M, Suzuki M, Matsumoto M, Takahashi M, Sato K, Ibe S, et al. Cloning of a Novel Gene for Quinolone Resistance from a Transferable Plasmid in Shigella flexneri 2b. Antimicrobial agents and chemotherapy. 2005;49(2):801-3. 104. Jacoby GA, Walsh KE, Mills DM, Walker VJ, Oh H, Robicsek A, et al. qnrB, another plasmid-mediated gene for quinolone resistance. Antimicrobial agents and chemotherapy. 2006;50(4):1178-82. 105. Wang M, Guo Q, Xu X, Wang X, Ye X, Wu S, et al. New plasmid-mediated quinolone resistance gene, qnrC, found in a clinical isolate of Proteus mirabilis. Antimicrobial agents and chemotherapy. 2009;53(5):1892-7. 106. Cavaco LM, Hasman H, Xia S, Aarestrup FM. qnrD, a Novel Gene Conferring Transferable Quinolone Resistance in Salmonella enterica Serovar Kentucky and Bovismorbificans Strains of Human Origin. Antimicrobial agents and chemotherapy. 2009;53(2):603-8.
113
107. Albornoz E, Tijet N, De Belder D, Gomez S, Martino F, Corso A, et al. qnrE1, a Member of a New Family of Plasmid-Located Quinolone Resistance Genes, Originated from the Chromosome of Enterobacter Species. Antimicrobial agents and chemotherapy. 2017;61(5). 108. Pons MJ, Gomes C, Ruiz J. QnrVC, a new transferable Qnr-like family. Enfermedades Infecciosas y Microbiología Clínica. 2013;31(3):191-2. 109. Xiong X, Bromley EH, Oelschlaeger P, Woolfson DN, Spencer J. Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: conserved surface loops direct the activity of a Qnr protein from a gram-negative bacterium. Nucleic Acids Res. 2011;39(9):3917-27. 110. Vetting MW, Hegde SS, Wang M, Jacoby GA, Hooper DC, Blanchard JS. Structure of QnrB1, a plasmid-mediated fluoroquinolone resistance factor. The Journal of biological chemistry. 2011;286(28):25265-73. 111. Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, et al. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nature medicine. 2006;12(1):83-8. 112. Rodríguez-Martínez JM, Machuca J, Cano ME, Calvo J, Martínez-Martínez L, Pascual A. Plasmid-mediated quinolone resistance: Two decades on. Drug Resistance Updates. 2016;29:13-29. 113. Yamane K, Wachino J-i, Suzuki S, Kimura K, Shibata N, Kato H, et al. New Plasmid-Mediated Fluoroquinolone Efflux Pump, QepA, Found in an Escherichia coli Clinical Isolate. Antimicrobial agents and chemotherapy. 2007;51(9):3354-60. 114. Reading C, Cole M. Clavulanic Acid: a Beta-Lactamase-Inhibiting Beta-Lactam from Streptomyces clavuligerus. Antimicrobial agents and chemotherapy. 1977;11(5):852-7. 115. White AR, Kaye C, Poupard J, Pypstra R, Woodnutt G, Wynne B. Augmentin (amoxicillin/clavulanate) in the treatment of community-acquired respiratory tract infection: a review of the continuing development of an innovative antimicrobial agent. The Journal of antimicrobial chemotherapy. 2004;53 Suppl 1:i3-20. 116. Worthington RJ, Melander C. Combination Approaches to Combat Multi-Drug Resistant Bacteria. Trends in biotechnology. 2013;31(3):177-84. 117. Van Bambeke F, Pages JM, Lee VJ. Inhibitors of bacterial efflux pumps as adjuvants in antibiotic treatments and diagnostic tools for detection of resistance by efflux. Recent patents on anti-infective drug discovery. 2006;1(2):157-75. 118. Sabatini S, Gosetto F, Serritella S, Manfroni G, Tabarrini O, Iraci N, et al. Pyrazolo[4,3-c][1,2]benzothiazines 5,5-dioxide: a promising new class of Staphylococcus aureus NorA efflux pump inhibitors. J Med Chem. 2012;55(7):3568-72. 119. Nautiyal A, Patil KN, Muniyappa K. Suramin is a potent and selective inhibitor of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. The Journal of antimicrobial chemotherapy. 2014;69(7):1834-43. 120. Bellio P, Brisdelli F, Perilli M, Sabatini A, Bottoni C, Segatore B, et al. Curcumin inhibits the SOS response induced by levofloxacin in Escherichia coli. Phytomedicine. 2014;21(4):430-4. 121. Sexton JZ, Wigle TJ, He Q, Hughes MA, Smith GR, Singleton SF, et al. Novel Inhibitors of E. coli RecA ATPase Activity. Current Chemical Genomics. 2010;4:34-42. 122. Lee AM, Ross CT, Zeng B-B, Singleton SF. A Molecular Target for Suppression of the Evolution of Antibiotic Resistance: Inhibition of the Escherichia coli RecA Protein by N6-(1-Naphthyl)-ADP. Journal of Medicinal Chemistry. 2005;48(17):5408-11. 123. Lee AM, Singleton SF. Inhibition of the Escherichia coli RecA protein: zinc(II), copper(II) and mercury(II) trap RecA as inactive aggregates. Journal of inorganic biochemistry. 2004;98(11):1981-6. 124. Cline DJ, Holt SL, Singleton SF. Inhibition of Escherichia coli RecA by rationally redesigned N-terminal helix. Organic & biomolecular chemistry. 2007;5(10):1525-8. 125. Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR. RecA Inhibitors Potentiate Antibiotic Activity and Block Evolution of Antibiotic Resistance. Cell chemical biology. 2016;23(3):381-91.
114
126. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular systems biology. 2006;2(1). 127. Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, et al. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrobial agents and chemotherapy. 2010;54(4):1393-403. 128. Tamae C, Liu A, Kim K, Sitz D, Hong J, Becket E, et al. Determination of Antibiotic Hypersensitivity among 4,000 Single-Gene-Knockout Mutants of Escherichia coli. Journal of bacteriology. 2008;190(17):5981-8. 129. Tran T, Ran Q, Ostrer L, Khodursky A. De Novo Characterization of Genes That Contribute to High-Level Ciprofloxacin Resistance in Escherichia coli. Antimicrobial agents and chemotherapy. 2016;60(10):6353-5. 130. Recacha E, Machuca J, Díaz de Alba P, Ramos-Güelfo M, Docobo-Pérez F, Rodriguez-Beltrán J, et al. Quinolone Resistance Reversion by Targeting the SOS Response. mBio. 2017;8(5). 131. Grimwade JE, Leonard AC. Targeting the Bacterial Orisome in the Search for New Antibiotics. Frontiers in Microbiology. 2017;8:2352. 132. van Eijk E, Wittekoek B, Kuijper EJ, Smits WK. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. Journal of Antimicrobial Chemotherapy. 2017;72(5):1275-84. 133. Schaechter M, Maaloe O, Kjeldgaard NO. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. Journal of general microbiology. 1958;19(3):592-606. 134. Cooper S, Helmstetter CE. Chromosome replication and the division cycle of Escherichia coli B/r. Journal of molecular biology. 1968;31(3):519-40. 135. Helmstetter C, Cooper S, Pierucci O, Revelas E. On the bacterial life sequence. Cold Spring Harbor symposia on quantitative biology. 1968;33:809-22. 136. Masters M, Broda P. Evidence for the bidirectional replications of the Escherichia coli chromosome. Nature: New biology. 1971;232(31):137-40. 137. Fossum S, Crooke E, Skarstad K. Organization of sister origins and replisomes during multifork DNA replication in <em>Escherichia coli</em>. The EMBO journal. 2007;26(21):4514. 138. Skarstad K, Boye E, Steen HB. Timing of initiation of chromosome replication in individual Escherichia coli cells. The EMBO journal. 1986;5(7):1711-7. 139. Katayama T, Kasho K, Kawakami H. The DnaA Cycle in Escherichia coli: Activation, Function and Inactivation of the Initiator Protein. Frontiers in Microbiology. 2017;8(2496). 140. Riber L, Frimodt-Møller J, Charbon G, Løbner-Olesen A. Multiple DNA Binding Proteins Contribute to Timing of Chromosome Replication in E. coli. Frontiers in Molecular Biosciences. 2016;3:29. 141. Davey MJ, Fang L, McInerney P, Georgescu RE, O’Donnell M. The DnaC helicase loader is a dual ATP/ADP switch protein. The EMBO journal. 2002;21(12):3148-59. 142. Fang L, Davey MJ, O'Donnell M. Replisome Assembly at oriC, the Replication Origin of E. coli, Reveals an Explanation for Initiation Sites outside an Origin. Molecular Cell. 1999;4(4):541-53. 143. Robinson A, van Oijen AM. Bacterial replication, transcription and translation: mechanistic insights from single-molecule biochemical studies. Nat Rev Micro. 2013;11(5):303-15. 144. Katayama T, Kubota T, Kurokawa K, Crooke E, Sekimizu K. The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosomal replicase. Cell. 1998;94(1):61-71. 145. Kitagawa R, Ozaki T, Moriya S, Ogawa T. Negative control of replication initiation by a novel chromosomal locus exhibiting exceptional affinity for Escherichia coli DnaA protein. Genes & development. 1998;12(19):3032-43. 146. Lu M, Campbell JL, Boye E, Kleckner N. SeqA: a negative modulator of replication initiation in E. coli. Cell. 1994;77(3):413-26.
115
147. von Freiesleben U, Rasmussen KV, Schaechter M. SeqA limits DnaA activity in replication from oriC in Escherichia coli. Molecular microbiology. 1994;14(4):763-72. 148. Fujimitsu K, Senriuchi T, Katayama T. Specific genomic sequences of E. coli promote replicational initiation by directly reactivating ADP-DnaA. Genes & development. 2009;23(10):1221-33. 149. Bramhill D, Kornberg A. A model for initiation at origins of DNA replication. Cell. 1988;54(7):915-8. 150. Schaper S, Messer W. Interaction of the initiator protein DnaA of Escherichia coli with its DNA target. The Journal of biological chemistry. 1995;270(29):17622-6. 151. McGarry KC, Ryan VT, Grimwade JE, Leonard AC. Two discriminatory binding sites in the Escherichia coli replication origin are required for DNA strand opening by initiator DnaA-ATP. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):2811-6. 152. Rozgaja TA, Grimwade JE, Iqbal M, Czerwonka C, Vora M, Leonard AC. Two oppositely-oriented arrays of low affinity recognition sites in oriC guide progressive binding of DnaA during E. coli pre-RC assembly. Molecular microbiology. 2011;82(2):475-88. 153. Sakiyama Y, Kasho K, Noguchi Y, Kawakami H, Katayama T. Regulatory dynamics in the ternary DnaA complex for initiation of chromosomal replication in Escherichia coli. Nucleic Acids Research. 2017;45(21):12354-73. 154. Samitt CE, Hansen FG, Miller JF, Schaechter M. In vivo studies of DnaA binding to the origin of replication of Escherichia coli. The EMBO journal. 1989;8(3):989-93. 155. Cassler MR, Grimwade JE, Leonard AC. Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. The EMBO journal. 1995;14(23):5833-41. 156. Fujita MQ, Yoshikawa H, Ogasawara N. Structure of the dnaA region of Micrococcus luteus: conservation and variations among eubacteria. Gene. 1990;93(1):73-8. 157. Messer W, Blaesing F, Majka J, Nardmann J, Schaper S, Schmidt A, et al. Functional domains of DnaA proteins. Biochimie. 1999;81(8):819-25. 158. Nozaki S, Ogawa T. Determination of the minimum domain II size of Escherichia coli DnaA protein essential for cell viability. Microbiology (Reading, England). 2008;154(Pt 11):3379-84. 159. Felczak MM, Simmons LA, Kaguni JM. An essential tryptophan of Escherichia coli DnaA protein functions in oligomerization at the E. coli replication origin. The Journal of biological chemistry. 2005;280(26):24627-33. 160. Simmons LA, Felczak M, Kaguni JM. DnaA Protein of Escherichia coli: oligomerization at the E. coli chromosomal origin is required for initiation and involves specific N-terminal amino acids. Molecular microbiology. 2003;49(3):849-58. 161. Abe Y, Jo T, Matsuda Y, Matsunaga C, Katayama T, Ueda T. Structure and function of DnaA N-terminal domains: specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. The Journal of biological chemistry. 2007;282(24):17816-27. 162. Keyamura K, Abe Y, Higashi M, Ueda T, Katayama T. DiaA dynamics are coupled with changes in initial origin complexes leading to helicase loading. The Journal of biological chemistry. 2009;284(37):25038-50. 163. Ishida T, Akimitsu N, Kashioka T, Hatano M, Kubota T, Ogata Y, et al. DiaA, a Novel DnaA-binding Protein, Ensures the Timely Initiation of Escherichia coli Chromosome Replication. Journal of Biological Chemistry. 2004;279(44):45546-55. 164. Su'etsugu M, Harada Y, Keyamura K, Matsunaga C, Kasho K, Abe Y, et al. The DnaA N-terminal domain interacts with Hda to facilitate replicase clamp-mediated inactivation of DnaA. Environmental microbiology. 2013;15(12):3183-95. 165. Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nature reviews Microbiology. 2007;5(5):343-54. 166. Zhang Q, Zhou A, Li S, Ni J, Tao J, Lu J, et al. Reversible lysine acetylation is involved in DNA replication initiation by regulating activities of initiator DnaA in Escherichia coli. Scientific reports. 2016;6:30837.
116
167. Kawakami H, Ozaki S, Suzuki S, Nakamura K, Senriuchi T, Su'etsugu M, et al. The exceptionally tight affinity of DnaA for ATP/ADP requires a unique aspartic acid residue in the AAA+ sensor 1 motif. Molecular microbiology. 2006;62(5):1310-24. 168. Nishida S, Fujimitsu K, Sekimizu K, Ohmura T, Ueda T, Katayama T. A Nucleotide Switch in the Escherichia coli DnaA Protein Initiates Chromosomal Replication: EVIDENCE FROM A MUTANT DnaA PROTEIN DEFECTIVE IN REGULATORY ATP HYDROLYSIS IN VITRO AND IN VIVO. Journal of Biological Chemistry. 2002;277(17):14986-95. 169. Su'Etsugu M, Kawakami H, Kurokawa K, Kubota T, Takata M, Katayama T. DNA replication-coupled inactivation of DnaA protein in vitro: a role for DnaA arginine-334 of the AAA+ Box VIII motif in ATP hydrolysis. Molecular microbiology. 2001;40(2):376-86. 170. Kasho K, Katayama T. DnaA binding locus datA promotes DnaA-ATP hydrolysis to enable cell cycle-coordinated replication initiation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(3):936-41. 171. Kawakami H, Keyamura K, Katayama T. Formation of an ATP-DnaA-specific initiation complex requires DnaA Arginine 285, a conserved motif in the AAA+ protein family. The Journal of biological chemistry. 2005;280(29):27420-30. 172. Felczak MM, Kaguni JM. The box VII motif of Escherichia coli DnaA protein is required for DnaA oligomerization at the E. coli replication origin. The Journal of biological chemistry. 2004;279(49):51156-62. 173. Ozaki S, Noguchi Y, Hayashi Y, Miyazaki E, Katayama T. Differentiation of the DnaA-oriC subcomplex for DNA unwinding in a replication initiation complex. The Journal of biological chemistry. 2012;287(44):37458-71. 174. Ozaki S, Kawakami H, Nakamura K, Fujikawa N, Kagawa W, Park SY, et al. A common mechanism for the ATP-DnaA-dependent formation of open complexes at the replication origin. The Journal of biological chemistry. 2008;283(13):8351-62. 175. Li S, Zhang Q, Xu Z, Yao Y-F. Acetylation of Lysine 243 Inhibits the oriC Binding Ability of DnaA in Escherichia coli. Frontiers in Microbiology. 2017;8(699). 176. Fujikawa N, Kurumizaka H, Nureki O, Terada T, Shirouzu M, Katayama T, et al. Structural basis of replication origin recognition by the DnaA protein. Nucleic Acids Res. 2003;31(8):2077-86. 177. Blaesing F, Weigel C, Welzeck M, Messer W. Analysis of the DNA-binding domain of Escherichia coli DnaA protein. Molecular microbiology. 2000;36(3):557-69. 178. Erzberger JP, Pirruccello MM, Berger JM. The structure of bacterial DnaA: implications for general mechanisms underlying DNA replication initiation. The EMBO journal. 2002;21(18):4763-73. 179. Shimizu M, Noguchi Y, Sakiyama Y, Kawakami H, Katayama T, Takada S. Near-atomic structural model for bacterial DNA replication initiation complex and its functional insights. Proceedings of the National Academy of Sciences. 2016;113(50):E8021-E30. 180. Keyamura K, Katayama T. DnaA protein DNA-binding domain binds to Hda protein to promote inter-AAA+ domain interaction involved in regulatory inactivation of DnaA. The Journal of biological chemistry. 2011;286(33):29336-46. 181. Noguchi Y, Sakiyama Y, Kawakami H, Katayama T. The Arg Fingers of Key DnaA Protomers Are Oriented Inward within the Replication Origin oriC and Stimulate DnaA Subcomplexes in the Initiation Complex. The Journal of biological chemistry. 2015;290(33):20295-312. 182. Ozaki S, Katayama T. Highly organized DnaA-oriC complexes recruit the single-stranded DNA for replication initiation. Nucleic Acids Res. 2012;40(4):1648-65. 183. Frimodt-Møller J, Charbon G, Krogfelt KA, Løbner-Olesen A. Control regions for chromosome replication are conserved with respect to sequence and location among Escherichia coli strains. Frontiers in Microbiology. 2015;6:1011. 184. Kaur G, Vora MP, Czerwonka CA, Rozgaja TA, Grimwade JE, Leonard AC. Building the bacterial orisome: high-affinity DnaA recognition plays a role in setting the conformation of oriC DNA. Molecular microbiology. 2014;91(6):1148-63.
117
185. Duderstadt KE, Chuang K, Berger JM. DNA stretching by bacterial initiators promotes replication origin opening. Nature. 2011;478:209. 186. Duderstadt KE, Mott ML, Crisona NJ, Chuang K, Yang H, Berger JM. Origin remodeling and opening in bacteria rely on distinct assembly states of the DnaA initiator. The Journal of biological chemistry. 2010;285(36):28229-39. 187. Costa A, Hood IV, Berger JM. Mechanisms for initiating cellular DNA replication. Annual review of biochemistry. 2013;82:25-54. 188. Leonard AC, Grimwade JE. The orisome: structure and function. Frontiers in Microbiology. 2015;6:545. 189. Arai K, Yasuda S, Kornberg A. Mechanism of dnaB protein action. I. Crystallization and properties of dnaB protein, an essential replication protein in Escherichia coli. The Journal of biological chemistry. 1981;256(10):5247-52. 190. Reha-Krantz LJ, Hurwitz J. The dnaB gene product of Escherichia coli. II. Single stranded DNA-dependent ribonucleoside triphosphatase activity. The Journal of biological chemistry. 1978;253(11):4051-7. 191. Bujalowski W, Klonowska MM, Jezewska MJ. Oligomeric structure of Escherichia coli primary replicative helicase DnaB protein. The Journal of biological chemistry. 1994;269(50):31350-8. 192. Donate L-E, Llorca Ó, Bárcena M, Brown SE, Dixon NE, Carazo J-Ma. pH-controlled quaternary states of hexameric DnaB helicase. Journal of molecular biology. 2000;303(3):383-93. 193. Chodavarapu S, Jones AD, Feig M, Kaguni JM. DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Research. 2016;44(1):210-20. 194. Seitz H, Weigel C, Messer W. The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Molecular microbiology. 2000;37(5):1270-9. 195. Soultanas P. Loading mechanisms of ring helicases at replication origins. Molecular microbiology. 2012;84(1):6-16. 196. Makowska-Grzyska M, Kaguni JM. Primase directs the release of DnaC from DnaB. Mol Cell. 2010;37(1):90-101. 197. Keyamura K, Fujikawa N, Ishida T, Ozaki S, Su'etsugu M, Fujimitsu K, et al. The interaction of DiaA and DnaA regulates the replication cycle in E. coli by directly promoting ATP DnaA-specific initiation complexes. Genes & development. 2007;21(16):2083-99. 198. Zhou H-X. The Affinity-Enhancing Roles of Flexible Linkers in Two-Domain DNA-Binding Proteins. Biochemistry. 2001;40(50):15069-73. 199. Camara JE, Breier AM, Brendler T, Austin S, Cozzarelli NR, Crooke E. Hda inactivation of DnaA is the predominant mechanism preventing hyperinitiation of Escherichia coli DNA replication. EMBO Rep. 2005;6(8):736-41. 200. Kato J-i, Katayama T. Hda, a novel DnaA-related protein, regulates the replication cycle in Escherichia coli. The EMBO journal. 2001;20(15):4253-62. 201. Su'etsugu M, Nakamura K, Keyamura K, Kudo Y, Katayama T. Hda monomerization by ADP binding promotes replicase clamp-mediated DnaA-ATP hydrolysis. The Journal of biological chemistry. 2008;283(52):36118-31. 202. Su'etsugu M, Shimuta TR, Ishida T, Kawakami H, Katayama T. Protein associations in DnaA-ATP hydrolysis mediated by the Hda-replicase clamp complex. The Journal of biological chemistry. 2005;280(8):6528-36. 203. Kim JS, Nanfara MT, Chodavarapu S, Jin KS, Babu VMP, Ghazy MA, et al. Dynamic assembly of Hda and the sliding clamp in the regulation of replication licensing. Nucleic Acids Res. 2017;45(7):3888-905. 204. Katayama T, Ozaki S, Keyamura K, Fujimitsu K. Regulation of the replication cycle: conserved and diverse regulatory systems for DnaA and oriC. Nature reviews Microbiology. 2010;8(3):163-70. 205. Kitagawa R, Mitsuki H, Okazaki T, Ogawa T. A novel DnaA protein-binding site at 94.7 min on the Escherichia coli chromosome. Molecular microbiology. 1996;19(5):1137-47.
118
206. Nozaki S, Yamada Y, Ogawa T. Initiator titration complex formed at datA with the aid of IHF regulates replication timing in Escherichia coli. Genes to cells : devoted to molecular & cellular mechanisms. 2009;14(3):329-41. 207. Ogawa T, Yamada Y, Kuroda T, Kishi T, Moriya S. The datA locus predominantly contributes to the initiator titration mechanism in the control of replication initiation in Escherichia coli. Molecular microbiology. 2002;44(5):1367-75. 208. Kasho K, Tanaka H, Sakai R, Katayama T. Cooperative DnaA Binding to the Negatively Supercoiled datA Locus Stimulates DnaA-ATP Hydrolysis. Journal of Biological Chemistry. 2017;292(4):1251-66. 209. Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. Journal of bacteriology. 1999;181(20):6361-70. 210. Frimodt-Møller J, Charbon G, Krogfelt KA, Løbner-Olesen A. DNA Replication Control Is Linked to Genomic Positioning of Control Regions in Escherichia coli. PLoS genetics. 2016;12(9):e1006286. 211. Morigen, Lobner-Olesen A, Skarstad K. Titration of the Escherichia coli DnaA protein to excess datA sites causes destabilization of replication forks, delayed replication initiation and delayed cell division. Molecular microbiology. 2003;50(1):349-62. 212. Cagliero C, Grand RS, Jones MB, Jin DJ, O'Sullivan JM. Genome conformation capture reveals that the Escherichia coli chromosome is organized by replication and transcription. Nucleic Acids Res. 2013;41(12):6058-71. 213. Messer W, Bellekes U, Lother H. Effect of dam methylation on the activity of the E. coli replication origin, oriC. The EMBO journal. 1985;4(5):1327-32. 214. Russell DW, Zinder ND. Hemimethylation prevents DNA replication in E. coli. Cell. 1987;50(7):1071-9. 215. Campbell JL, Kleckner N. E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell. 1990;62(5):967-79. 216. Boye E. The hemimethylated replication origin of Escherichia coli can be initiated in vitro. Journal of bacteriology. 1991;173(14):4537-9. 217. Waldminghaus T, Skarstad K. The Escherichia coli SeqA protein. Plasmid. 2009;61(3):141-50. 218. Kurokawa K, Nishida S, Emoto A, Sekimizu K, Katayama T. Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. The EMBO journal. 1999;18(23):6642-52. 219. Skarstad K, Lobner-Olesen A. Stable co-existence of separate replicons in Escherichia coli is dependent on once-per-cell-cycle initiation. The EMBO journal. 2003;22(1):140-50. 220. Skarstad K, Katayama T. Regulating DNA Replication in Bacteria. Cold Spring Harbor Perspectives in Biology. 2013;5(4):a012922. 221. Taghbalout A, Landoulsi A, Kern R, Yamazoe M, Hiraga S, Holland B, et al. Competition between the replication initiator DnaA and the sequestration factor SeqA for binding to the hemimethylated chromosomal origin of E. coli in vitro. Genes to cells : devoted to molecular & cellular mechanisms. 2000;5(11):873-84. 222. Nievera C, Torgue JJ, Grimwade JE, Leonard AC. SeqA blocking of DnaA-oriC interactions ensures staged assembly of the E. coli pre-RC. Mol Cell. 2006;24(4):581-92. 223. Bach T, Morigen, Skarstad K. The initiator protein DnaA contributes to keeping new origins inactivated by promoting the presence of hemimethylated DNA. Journal of molecular biology. 2008;384(5):1076-85. 224. Torheim NK, Skarstad K. Escherichia coli SeqA protein affects DNA topology and inhibits open complex formation at oriC. The EMBO journal. 1999;18(17):4882-8. 225. Bogan JA, Helmstetter CE. DNA sequestration and transcription in the oriC region of Escherichia coli. Molecular microbiology. 1997;26(5):889-96.
119
226. Riber L, Løbner-Olesen A. Coordinated Replication and Sequestration of oriC and dnaA Are Required for Maintaining Controlled Once-per-Cell-Cycle Initiation in Escherichia coli. Journal of bacteriology. 2005;187(16):5605-13. 227. Kasho K, Fujimitsu K, Matoba T, Oshima T, Katayama T. Timely binding of IHF and Fis to DARS2 regulates ATP–DnaA production and replication initiation. Nucleic Acids Research. 2014;42(21):13134-49. 228. Sekimizu K, Kornberg A. Cardiolipin activation of dnaA protein, the initiation protein of replication in Escherichia coli. The Journal of biological chemistry. 1988;263(15):7131-5. 229. Crooke E, Castuma CE, Kornberg A. The chromosome origin of Escherichia coli stabilizes DnaA protein during rejuvenation by phospholipids. The Journal of biological chemistry. 1992;267(24):16779-82. 230. Castuma CE, Crooke E, Kornberg A. Fluid membranes with acidic domains activate DnaA, the initiator protein of replication in Escherichia coli. The Journal of biological chemistry. 1993;268(33):24665-8. 231. Xia W, Dowhan W. In vivo evidence for the involvement of anionic phospholipids in initiation of DNA replication in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(3):783-7. 232. Zheng W, Li Z, Skarstad K, Crooke E. Mutations in DnaA protein suppress the growth arrest of acidic phospholipid-deficient Escherichia coli cells. The EMBO journal. 2001;20(5):1164-72. 233. Fingland N, Flåtten I, Downey CD, Fossum-Raunehaug S, Skarstad K, Crooke E. Depletion of acidic phospholipids influences chromosomal replication in Escherichia coli. MicrobiologyOpen. 2012;1(4):450-66. 234. Hansen FG, Christensen BB, Atlung T. The initiator titration model: computer simulation of chromosome and minichromosome control. Research in microbiology. 1991;142(2):161-7. 235. Charbon G, Bjørn L, Mendoza-Chamizo B, Frimodt-Møller J, Løbner-Olesen A. Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic Acids Research. 2014;42(21):13228-41. 236. Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). Journal of bacteriology. 1992;174(20):6321-5. 237. Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Molecular microbiology. 2004;51(2):349-58. 238. Gon S, Camara JE, Klungsoyr HK, Crooke E, Skarstad K, Beckwith J. A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. The EMBO journal. 2006;25(5):1137-47. 239. Babu VMP, Itsko M, Baxter JC, Schaaper RM, Sutton MD. Insufficient levels of the nrdAB-encoded ribonucleotide reductase underlie the severe growth defect of the Δhda E. coli strain. Molecular microbiology. 2017;104(3):377-99. 240. Riber L, Olsson JA, Jensen RB, Skovgaard O, Dasgupta S, Marinus MG, et al. Hda-mediated inactivation of the DnaA protein and dnaA gene autoregulation act in concert to ensure homeostatic maintenance of the Escherichia coli chromosome. Genes & development. 2006;20(15):2121-34. 241. Fujimitsu K, Su'etsugu M, Yamaguchi Y, Mazda K, Fu N, Kawakami H, et al. Modes of Overinitiation, dnaA Gene Expression, and Inhibition of Cell Division in a Novel Cold-Sensitive hda Mutant of Escherichia coli. Journal of bacteriology. 2008;190(15):5368-81. 242. Riber L, Fujimitsu K, Katayama T, Lobner-Olesen A. Loss of Hda activity stimulates replication initiation from I-box, but not R4 mutant origins in Escherichia coli. Molecular microbiology. 2009;71(1):107-22. 243. Charbon G, Campion C, Chan SHJ, Bjørn L, Weimann A, da Silva LCN, et al. Re-wiring of energy metabolism promotes viability during hyperreplication stress in E. coli. PLoS genetics. 2017;13(1):e1006590.
120
244. Charbon G, Riber L, Cohen M, Skovgaard O, Fujimitsu K, Katayama T, et al. Suppressors of DnaA(ATP) imposed overinitiation in Escherichia coli. Molecular microbiology. 2011;79(4):914-28. 245. Imlay J, Chin S, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240(4852):640-2. 246. Liu Y, Bauer SC, Imlay JA. The YaaA protein of the Escherichia coli OxyR regulon lessens hydrogen peroxide toxicity by diminishing the amount of intracellular unincorporated iron. Journal of bacteriology. 2011;193(9):2186-96. 247. Surivet JP, Lange R, Hubschwerlen C, Keck W, Specklin JL, Ritz D, et al. Structure-guided design, synthesis and biological evaluation of novel DNA ligase inhibitors with in vitro and in vivo anti-staphylococcal activity. Bioorganic & medicinal chemistry letters. 2012;22(21):6705-11. 248. Mills SD, Eakin AE, Buurman ET, Newman JV, Gao N, Huynh H, et al. Novel bacterial NAD+-dependent DNA ligase inhibitors with broad-spectrum activity and antibacterial efficacy in vivo. Antimicrobial agents and chemotherapy. 2011;55(3):1088-96. 249. Rose Y, Ciblat S, Reddy R, Belley AC, Dietrich E, Lehoux D, et al. Novel non-nucleobase inhibitors of Staphylococcus aureus DNA polymerase IIIC. Bioorganic & medicinal chemistry letters. 2006;16(4):891-6. 250. Tarantino PM, Jr., Zhi C, Wright GE, Brown NC. Inhibitors of DNA polymerase III as novel antimicrobial agents against gram-positive eubacteria. Antimicrobial agents and chemotherapy. 1999;43(8):1982-7. 251. Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, et al. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science. 2015;348(6239):1106-12. 252. Kjelstrup S, Hansen PMP, Thomsen LE, Hansen PR, Løbner-Olesen A. Cyclic Peptide Inhibitors of the β-Sliding Clamp in <italic>Staphylococcus aureus</italic>. PloS one. 2013;8(9):e72273. 253. Marceau AH, Bernstein DA, Walsh BW, Shapiro W, Simmons LA, Keck JL. Protein interactions in genome maintenance as novel antibacterial targets. PloS one. 2013;8(3):e58765. 254. Fossum S, De Pascale G, Weigel C, Messer W, Donadio S, Skarstad K. A robust screen for novel antibiotics: specific knockout of the initiator of bacterial DNA replication. FEMS Microbiol Lett. 2008;281(2):210-4. 255. Johnsen L, Weigel C, von Kries J, Moller M, Skarstad K. A novel DNA gyrase inhibitor rescues Escherichia coli dnaAcos mutant cells from lethal hyperinitiation. The Journal of antimicrobial chemotherapy. 2010;65(5):924-30. 256. Yamaichi Y, Duigou S, Shakhnovich EA, Waldor MK. Targeting the Replication Initiator of the Second Vibrio Chromosome: Towards Generation of Vibrionaceae-Specific Antimicrobial Agents. PLOS Pathogens. 2009;5(11):e1000663. 257. Weigel C, Schmidt A, Seitz H, Tungler D, Welzeck M, Messer W. The N-terminus promotes oligomerization of the Escherichia coli initiator protein DnaA. Molecular microbiology. 1999;34(1):53-66. 258. Renwick MJ, Simpkin V, Mossialos E. European Observatory Health Policy Series. Targeting innovation in antibiotic drug discovery and development: The need for a One Health - One Europe - One World Framework. Copenhagen (Denmark): European Observatory on Health Systems and Policies
(c) World Health Organization 2016 (acting as the host organization for, and secretariat of, the European Observatory on Health Systems and Policies). 2016. 259. Beaber JW, Hochhut B, Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2003;427:72. 260. Amundsen SK, Spicer T, Karabulut AC, Londoño LM, Eberhart C, Fernandez Vega V, et al. Small-Molecule Inhibitors of Bacterial AddAB and RecBCD Helicase-Nuclease DNA Repair Enzymes. ACS chemical biology. 2012;7(5):879-91. 261. Singh R, Swick MC, Ledesma KR, Yang Z, Hu M, Zechiedrich L, et al. Temporal Interplay between Efflux Pumps and Target Mutations in Development of Antibiotic Resistance in Escherichia coli. Antimicrobial agents and chemotherapy. 2012;56(4):1680-5.
121
262. Nishino K, Latifi T, Groisman EA. Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Molecular microbiology. 2006;59(1):126-41. 263. Mahamoud A, Chevalier J, Alibert-Franco S, Kern WV, Pages JM. Antibiotic efflux pumps in Gram-negative bacteria: the inhibitor response strategy. The Journal of antimicrobial chemotherapy. 2007;59(6):1223-9. 264. Hannah YM, Shirin J, Sutton JM, Khondaker MR. Current Advances in Developing Inhibitors of Bacterial Multidrug Efflux Pumps. Current medicinal chemistry. 2016;23(10):1062-81. 265. Opperman TJ, Nguyen ST. Recent advances toward a molecular mechanism of efflux pump inhibition. Frontiers in Microbiology. 2015;6:421. 266. Lopez JS, Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nature Reviews Clinical Oncology. 2016;14:57. 267. Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404(6773):37-41. 268. Filutowicz M, Roll J. The requirement of IHF protein for extrachromosomal replication of the Escherichia coli oriC in a mutant deficient in DNA polymerase I activity. The New biologist. 1990;2(9):818-27. 269. Ogawa T, Wada M, Kano Y, Imamoto F, Okazaki T. DNA replication in Escherichia coli mutants that lack protein HU. Journal of bacteriology. 1989;171(10):5672-9. 270. Katayama T, Takata M, Sekimizu K. The nucleoid protein H-NS facilitates chromosome DNA replication in Escherichia coli dnaA mutants. Journal of bacteriology. 1996;178(19):5790-2. 271. Charbon G, Riber L, Løbner-Olesen A. Countermeasures to survive excessive chromosome replication in Escherichia coli. Current Genetics. 2018;64(1):71-9. 272. Wargachuk R, Marczynski GT. The Caulobacter crescentus Homolog of DnaA (HdaA) Also Regulates the Proteolysis of the Replication Initiator Protein DnaA. Journal of bacteriology. 2015;197(22):3521-32. 273. Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, et al. Antibacterial Nucleoside-Analog Inhibitor of Bacterial RNA Polymerase. Cell. 2017;169(7):1240-8.e23. 274. Jabés D, Brunati C, Candiani G, Riva S, Romanó G, Donadio S. Efficacy of the New Lantibiotic NAI-107 in Experimental Infections Induced by Multidrug-Resistant Gram-Positive Pathogens. Antimicrobial agents and chemotherapy. 2011;55(4):1671-6.