Page 1
Phosphate uptake from water on a Surfactant-Modified Zeoliteand Ca-zeolites
Joachim Schick • Philippe Caullet •
Jean-Louis Paillaud • Joel Patarin •
Stephanie Freitag • Claire Mangold-Callarec
Published online: 15 July 2011
� Springer Science+Business Media, LLC 2011
Abstract The phosphate adsorption kinetics are deter-
mined in batch-wise (noted B) and fixed-bed column (noted
C) experiments on a Surfactant-Modified Zeolite (SMZ) and
various Ca-zeolites. The influence of phosphate concentra-
tion (0.08 or 0.8 mmol/L), presence of NO3-, HCO3
-,
SO42- and Cl- competing anions (individual concentra-
tion = 0.8 meq/L) and flow rate Q (1–30 mL/min) is stud-
ied. Preliminary experiments lead to the selection of the
most efficient Ca-LTA and SMZ samples for the subsequent
studies. In B experiments, the nature of the used system does
not influence the equilibrium removal rate R (&80%) but
affects the adsorption kinetics. The equilibrium times are
shorter on SMZ than on Ca-LTA, increasing with the
phosphate concentration and the presence of competing
anions, respectively in the *0.5–6 or *3–24 h ranges. In C
experiments, the phosphate uptake performances on SMZ
are higher than in the corresponding B experiments, with in
particular higher final q/qm values. The deterioration of the
performances on SMZ in presence of competing anions or
with increase of Q is due to the effect of the slow phosphate
ion-exchange kinetics and the short used contact time. For
similar reasons, sorption on Ca-LTA is lower than on SMZ.
For instance, with a 0.8 mmol/L phosphate concentration
and a 10 mL/min flow rate, the time-decreasing R values
become close to 50 and 10% after filtration of 10 bed-vol-
umes respectively in presence of SMZ and Ca-LTA. Glob-
ally, SMZ is clearly more efficient than Ca-LTA, being
furthermore a versatile and easily regenerable material.
Keywords Phosphate removal � Batch � Fixed-bed
column � SMZ � Ca-zeolites
1 Introduction
The presence of phosphate anions in waters, together with
nitrate anions, is mainly due to human activities, i.e. use of
industrial detergents or house-hold washing powders and
over application of fertilizers in agriculture, and is
responsible for a major environmental problem, the so-
called eutrophication phenomenon [1]. Although the
phosphate concentrations in rivers are in fact generally
lower than the ones of nitrate, approaching a few mg/L, the
average concentrations in wastewaters are much higher and
close to several tens of mg/L. The French regulation of
December 22, 1994 imposes a maximal phosphate con-
centration in the discharges of water-treatment plants in the
3–6 mg/L range (depending on the BOD5 value corre-
sponding to the amount of dissolved oxygen consumed in
5 days by bacteria that perform biological degradation of
organic matter) [2].
The elimination of phosphate from waters is possible
through various methods, i.e. biological processes [3],
physical processes (reverse osmosis [4], nanofiltration [5]
or electrodialysis [6]) and physico-chemical processes.
Among the latter, two main methods can be distinguished,
J. Schick � P. Caullet � J.-L. Paillaud (&) � J. Patarin
Equipe Materiaux a Porosite Controlee (MPC), Institut de
Science des Materiaux de Mulhouse (IS2M), LRC CNRS
7228-UHA, ENSCMu, 3, rue Alfred Werner, 68093 Mulhouse
Cedex, France
e-mail: [email protected]
S. Freitag
Groupe Securite et Ecologie Chimiques, Laboratoire Propre
Integre (LPI), ENSCMu, 3 rue Alfred Werner, 68093 Mulhouse
Cedex, France
C. Mangold-Callarec
FR Environnement Nautique, BP 97371, 29673 Morlaix, France
123
J Porous Mater (2012) 19:405–414
DOI 10.1007/s10934-011-9488-3
Page 2
either chemical precipitation [7, 8] by aluminum or iron
salts or by lime, or adsorption and/or ion exchange reac-
tions on a variety of materials. These materials are either
natural, for instance dolomite [9], soils [10], residues of
blast furnaces [11], coal fly ashes [12]… or synthetic,
namely activated carbons [13], iron or aluminum hydrox-
ides [14, 15], biopolymers [16]… Apart from these mate-
rials, compounds characterized by an organized porosity,
either zeolitic (natural [17–19] or synthetic [20–24]) or
mesostructured (functionalized) [25, 26] were also used.
With the notable exception of the mesostructured func-
tionalized materials, the preparation of which is rather
difficult and expensive, all the phosphate adsorption pro-
cesses, implying in particular zeolitic materials, are clearly
promising as they operate under economical and simple
conditions. The phosphate removal mechanism on zeolitic
compounds can correspond to a precipitation-induced
sorption process. Several related papers [10, 17, 21–24]
concluded to a strong correlation between the Al-, Fe- and
also Ca-contents in the materials and the extent of phos-
phate uptake. The phosphate removal on zeolites can also
be due to an ion-exchange process, in the case of the so-
called Surfactant-Modified-Zeolites (referred to as SMZ)
[27]. These SMZ are prepared in a simple way, typically
through a treatment by a cationic surfactant (typically
hexadecyltrimethylammonium, hereafter abbreviated as
HDTMA) solution from the very cheap natural clinoptilo-
lite. The sorbed surfactant species form bilayers on the
zeolite crystals surface, with the first layer retained by
cation exchange, the second layer being attached by
hydrophobic bonding (van der Waals forces) and stabilized
by counteranions. SMZ can thus be used as anionic
exchangers to adsorb a variety of anions, such as chromate,
antimonate, arsenate, selenate, phosphate and nitrate… [18,
19, 27–34]. Taking into account this ion-exchange sorption
mechanism, the regeneration of SMZ can be easily per-
formed, which is also very important in the perspective of
industrial applications.
To our knowledge, the number of papers specifically
devoted to the sorption of phosphate ions on SMZ is rather
low, and imply the use of clinoptilolite zeolite [18, 19] or
of LTA-type zeolite [33]. SMZ prepared from clinoptilolite
is only employed in batch-wise experiments, Hrenovic
et al. [19] dealing more precisely with the interaction of
SMZ and phosphate- accumulating bacteria in the phos-
phorus removal from wastewaters. Bansiwal et al. [33]
examine specifically the possibility to use SMZ as a carrier
for fertilizer and for slow release of phosphorus. No kinetic
data are given and the influence of competing anions is
only partly examined. Furthermore, no information is
available about fixed-bed column experiments, which are
obviously essential in order to provide a more realistic
simulation of dynamic field conditions. The present paper
focuses thus on the use of a SMZ sample prepared from
clinoptilolite in various batch-wise (hereafter noted B) and
fixed-bed column (hereafter noted C) experiments and also
on the comparison with the use of Ca-forms of several
zeolites, including clinoptilolite and two synthetic zeolites
(FAU and LTA structural types). The behavior of the latter
materials is indeed expected to differ greatly from the one
of SMZ, taking into account the completely different
sorption mechanisms, i.e. precipitation reaction instead of
anion-exchange. The parameters studied in this paper are,
beside the sorption B or C mode, the initial phosphate
concentration, the presence of the NO3-, HCO3
-, SO42-
and Cl- competing anions and the Q flow rate value (C).
The influence of the contact time (B mode) or the residence
time (C mode) is investigated. The chosen initial concen-
trations are equal to 0.08 or 0.8 mmol/L, this concentra-
tions range being approximately representative of the
real concentrations detected in rivers, groundwaters (a few
mg/L) and waste waters (several tens of mg/L). Higher
phosphate concentrations (up to 29 mmol/L) were only
used in specific experiments performed in order to deter-
mine the anionic adsorption capacity qm of the SMZ
sample. The questions of the regeneration of the SMZ and
of the removal of the leached surfactant from the effluents
are not discussed in this paper as they were examined in
previous studies relative to the nitrate sorption on SMZ in
B or C experiments [35, 36].
2 Experimental section
2.1 Characterization methods
The details relative to the methods of X-ray diffraction
(XRD), scanning electron microscopy (SEM), X-ray fluo-
rescence spectroscopy (XRF), Zeta potential measurement,
Cationic Exchange Capacity (CEC) and External Cationic
Exchange Capacity (ECEC) measurements (the latter are
only performed on the raw clinoptilolite sample, see
hereafter) are given in our previous paper [35].
The Si/Al framework molar ratio of the FAU-type
zeolite (Na-X type) was determined from the correspond-
ing XRD pattern using the following empirical relations
[37]:
Si
Al¼ NSi
NAl
¼ ð192� NAlÞNAl
ð1Þ
NAl ¼ 101:202a0
f� 24:2115
� �ð2Þ
where NSi and NAl correspond respectively to the number
of Si and Al atoms in the faujasite unit cell (192 T atoms),
a0 is the unit cell parameter and f is a correction factor
specific for each FAU-type zeolite, namely f = 0.9957 for
406 J Porous Mater (2012) 19:405–414
123
Page 3
NaX faujasite. The XRD pattern was collected using a
PANalytical MPD X’Pert Pro diffractometer operating
with Cu Ka radiation (k = 0.15418 nm) in the 2h range
0.5–50 and equipped with a X’Celerator real-time multiple
strip detector. The refinement of the unit cell parameter
(FAU- structure type, cubic symmetry) was performed by
using the Werner’s trial and error indexing program [38].
Some samples, after being outgassed (1 h at 90 �C then
300 �C overnight), were studied by N2 adsorption manom-
etry (77 K) using a Micromeritics ASAP 2420 analyzer.
The anion (phosphate, nitrate, sulphate, bicarbonate,
chloride and bromide) concentrations were determined
using P/ACE system MDQ Capillary Electrophoresis (CE)
instrument (Beckman Coulter Spectrometer). Experimental
details are displayed in Ref. [35].
2.2 Characterization of the used adsorbent samples
The natural raw zeolite sample used in this study is a
clinoptilolite-rich tuff from Bulgaria, with an estimated
clinoptilolite weight content close to 90%. Prior to the
preparation of the adsorbent samples, this raw material was
sieved to particles in the 0.8–2 mm range. Additional
information (nature of impurities, chemical composition,
CEC and ECEC values) can be found in Ref. [35]. In
particular, the CEC and ECEC values are close to 1,850
and 150 meq/Kg respectively. Three clinoptilolite phos-
phate adsorbents were used, namely the raw form, the
Ca-exchanged form and the Surfactant-Modified form
(SMZ sample). The Ca-form is prepared through a first full
ionic exchange with NH4? ions (L/S = 10 mL/g, room
temperature, 24 h, NH4Cl 1 M), followed by a subsequent
exchange with Ca2? ions (L/S = 10 mL/g, room temper-
ature, 24 h, CaCl2 1 M). This procedure is adapted
from the one proposed by Ji et al. [39]. Actually, the
Ca-exchange could not be fully achieved in accordance
with Ming [40], with a calcium content corresponding
respectively in the exchanged and raw sample close to
about 70 and 40% of the CEC. The SMZ sample was
prepared by treatment of the raw clinoptilolite sample by a
hexadecyltrimethylammonium solution according to the
procedure related in detail in Ref. [35] and is characterized
by a qm value close to 90 meq/Kg.
This is significantly lower to the value expected from
the ECEC value, i.e.150 meq/Kg. The observed discrep-
ancy was previously interpreted in terms of a restricted
accessibility of the large HDTMA cations to the clinop-
tilolite crystals surface, hindering thus the formation of a
continuous surfactant bilayer [35].
Besides, the Ca-forms of the LTA- and FAU-type zeo-
lites were prepared from commercial extrudates purchased
from Sigma-Aldrich. The initial Na-forms consist of
spherical beads (diameter close to 2 mm), made of small
micrometric (&1–2 lm) zeolite crystals and a binder.
According to XRD, SEM and XRF characterizations, the
binder is amorphous, with a composition and a morpho-
logical appearance consistent with the ones of a clay-like
compound. The Si/Al framework ratio of the FAU-type
zeolite deduced from the XRD pattern (see Sect. 2.1) is
close to 1.2. The Ca-forms were prepared directly from the
Na-forms through two successive exchanges with Ca2?
ions (L/S = 100 mL/g, room temperature, 24 h, CaCl21 M) [41]. Under these experimental conditions, a com-
plete exchange is achieved. N2 adsorption manometry
measurements were performed on Na-FAU and Ca-LTA
samples and led, by comparison with the results obtained
on pure zeolitic samples (Sigma–Aldrich) to an estimated
zeolite weight content close to 80% in the extrudates.
2.3 Sorption experiments
Batch kinetic B experiments were performed at room
temperature up to 24 h by introducing 5 g of adsorbent
sample in a polypropylene flask containing 50 mL of
aqueous solutions of potassium salts (Fluka). The mixtures
were stirred with a reciprocating platform shaker and the
pH values were systematically in the 5–6 range. The effect
of the initial phosphate concentration ([H2PO4-]i = 0.08
and 0.8 mmol/L) was first investigated in the presence of
one of the five adsorbents described in the preceding Sect.
2.2.
The most effective adsorbents, i.e. the Ca-LTA and SMZ
samples, were then used for the competitive adsorption of
phosphate in the simultaneous presence of the NO3-,
HCO3-, SO4
2- and Cl- anions, each individual initial con-
centration being equal to 0.8 meq/L. 0.5 mL of samples was
taken at various times, filtered through 0.2 lm syringe filters
and analyzed by capillary electrophoresis. Total taking of
samples does not exceed 5 mL, i.e. 10% of the initial solution
and does thus not disturb significantly the course of the
experiment. The adsorption isotherm of the SMZ sample was
determined ([H2PO4-]i = 0.08; 0.8; 2.42; 4.83; 14.5;
29 mmol/L, L/S = 10 mL/g, room temperature, 24 h con-
tact time) in order to determine the qm experimental value,
the Langmuir isotherm model [42] being used in order to fit
the sorption equilibrium data.
Fixed-bed column C sorption experiments were carried
out in a 120 cm length and 0.9 cm internal diameter vertical
glass column, the zeolite bed depth being equal to 90 cm and
corresponding to a Bed Volume (BV) equal to 57 mL (zeo-
lite weight = 40 g). Only the most effective adsorbents,
Ca-LTA and SMZ, were used in these flow mode experi-
ments. The influent was introduced downward as a contin-
uous flow and at a given constant flow rate using a peristaltic
pump (Cole Parmer MasterFlex L/S), glass wool being set at
the bottom of the column to avoid any loss of the filtering
J Porous Mater (2012) 19:405–414 407
123
Page 4
medium particles. Some precautions were taken in order to
avoid the presence of air in the column. First, the column is
filled with distilled water before the introduction of the
clinoptilolite particles. Afterwards, the aqueous solution
level is kept constant in the column in order that the particles
remain continuously immersed, this being simply obtained
thanks to the presence of a siphon between the column and
the recovery container. The macroporosity of the zeolite bed
was estimated by measuring the residence time tR (see Ref.
[36]). For this purpose, a 100 mmol/L potassium nitrate
aqueous solution was percolated down flow with a 3 mL/min
flow rate through the zeolite column containing 40 g of raw
clinoptilolite. As raw zeolites are obviously unable to
remove nitrate ions from aqueous solutions, tR corresponds
to the time when NO3- begins to be detected in the effluent.
In C experiments, the effects of the initial phosphate
concentration (0.08 and 0.8 mmol/L KH2PO4 solutions) at
a constant 10 mL/min Q flow rate and of the Q values (1,
10 or 30 mL/min) at the 0.8 mmol/L initial phosphate
concentration were examined. Competitive adsorption
between H2PO4- and NO3
-, HCO3-, SO4
2- and Cl-
anions was also studied, with initial individual concentra-
tions equal to 0.8 meq/L and a flow rate of 10 mL/min.
Each filtration experiment was continued until a complete
breakthrough (removal efficiency equal to 0%) for phos-
phate ions was observed. The totality of the effluent vol-
ume is recovered during the filtration in a 25 L
polypropylene vessel. Samples (&20 mL) were withdrawn
at various times simultaneously at the exit of the column
and in the 25 L vessel and further filtered and analyzed by
CE.
The percentage removal rate (R) of phosphate (or more
generally of any anion) was calculated as
R ¼ C0 � Ct
C0
� 100% ð3Þ
where C0 and Ct are the concentrations in solution (mmol/L)
at the beginning of the experiment and after a contact or
filtration time t.The exchange ratio was calculated for the
SMZ sample and is defined as:
q
qm
� 100% ð4Þ
where q corresponds to the amount of anionic exchanged
sites (meq/Kg). In the fixed bed column experiments, the q
values were determined directly from the H2PO4- ion
concentration (or more generally from the individual con-
centration of the various anions present) measured in the
totality of the effluent volume recovered during the filtra-
tion. Global or individual q/qm values can thus be calcu-
lated. The samplings are considered not to disturb this
estimation of q, as the totality of the withdrawn volumes
remains negligible (\10%) with regard to the global fil-
tered volume.
3 Results and discussion
In the following text and figures, raw clinoptilolite,
Ca-exchanged clinoptilolite, Ca-exchanged zeolite A and
Ca-exchanged zeolite X are respectively referred to as
HEU, Ca-HEU, Ca-LTA and Ca-FAU. The results will be
presented in two successive parts, corresponding to B
(Sect. 3.1) and C (Sect. 3.2) sorption experiments.
3.1 B sorption experiments
The results are displayed in two consecutive parts, corre-
sponding to preliminary experiments relative to solutions
of H2PO4- ions alone and then to competing anions-con-
taining H2PO4- solutions.
3.1.1 Preliminary experiments: solutions of H2PO4- ions
alone
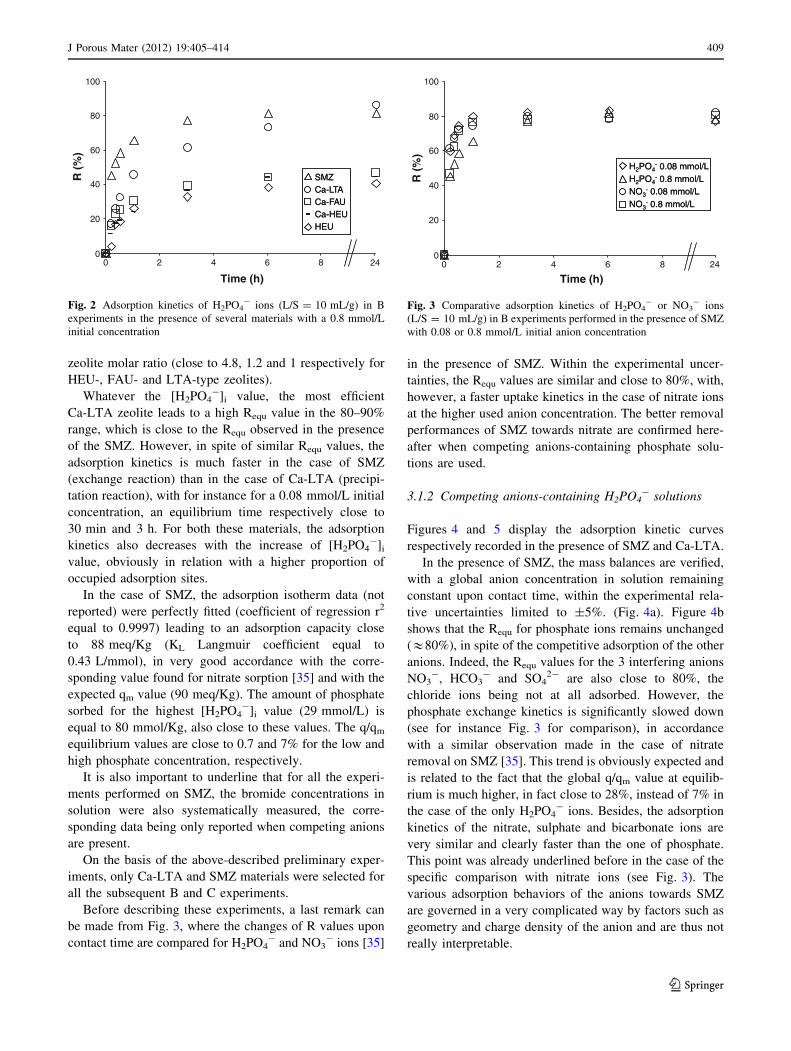
Figures 1 and 2 correspond respectively to a 0.08 and
0.8 mmol/L initial concentration and describe the behavior
of the 5 adsorbents used.
For both concentrations, the increasing efficiency order
in terms of equilibrium R values (Requ) is rather similar,
namely HEU ? Ca-HEU ? Ca-FAU ? Ca-LTA ? SMZ,
even if there are some slight differences according to the
initial concentration value.
With respect to the Ca-zeolites, the phosphate removal
is due to a calcium phosphate precipitation reaction. The
affinity for H2PO4- increases obviously with the growing
concentration of Ca2? ions, i.e. with the increase of the
calcium-exchange rate and/or the decrease of the Si/Al
0
20
40
60
100
0 2 4 6 8
Time (h)
R (
%)
24
80
SMZCa-LTACa-FAUCa-HEUHEU
Fig. 1 Adsorption kinetics of H2PO4- ions (L/S = 10 mL/g) in B
experiments in the presence of several materials with a 0.08 mmol/L
initial concentration
408 J Porous Mater (2012) 19:405–414
123
Page 5
zeolite molar ratio (close to 4.8, 1.2 and 1 respectively for
HEU-, FAU- and LTA-type zeolites).
Whatever the [H2PO4-]i value, the most efficient
Ca-LTA zeolite leads to a high Requ value in the 80–90%
range, which is close to the Requ observed in the presence
of the SMZ. However, in spite of similar Requ values, the
adsorption kinetics is much faster in the case of SMZ
(exchange reaction) than in the case of Ca-LTA (precipi-
tation reaction), with for instance for a 0.08 mmol/L initial
concentration, an equilibrium time respectively close to
30 min and 3 h. For both these materials, the adsorption
kinetics also decreases with the increase of [H2PO4-]i
value, obviously in relation with a higher proportion of
occupied adsorption sites.
In the case of SMZ, the adsorption isotherm data (not
reported) were perfectly fitted (coefficient of regression r2
equal to 0.9997) leading to an adsorption capacity close
to 88 meq/Kg (KL Langmuir coefficient equal to
0.43 L/mmol), in very good accordance with the corre-
sponding value found for nitrate sorption [35] and with the
expected qm value (90 meq/Kg). The amount of phosphate
sorbed for the highest [H2PO4-]i value (29 mmol/L) is
equal to 80 mmol/Kg, also close to these values. The q/qm
equilibrium values are close to 0.7 and 7% for the low and
high phosphate concentration, respectively.
It is also important to underline that for all the experi-
ments performed on SMZ, the bromide concentrations in
solution were also systematically measured, the corre-
sponding data being only reported when competing anions
are present.
On the basis of the above-described preliminary exper-
iments, only Ca-LTA and SMZ materials were selected for
all the subsequent B and C experiments.
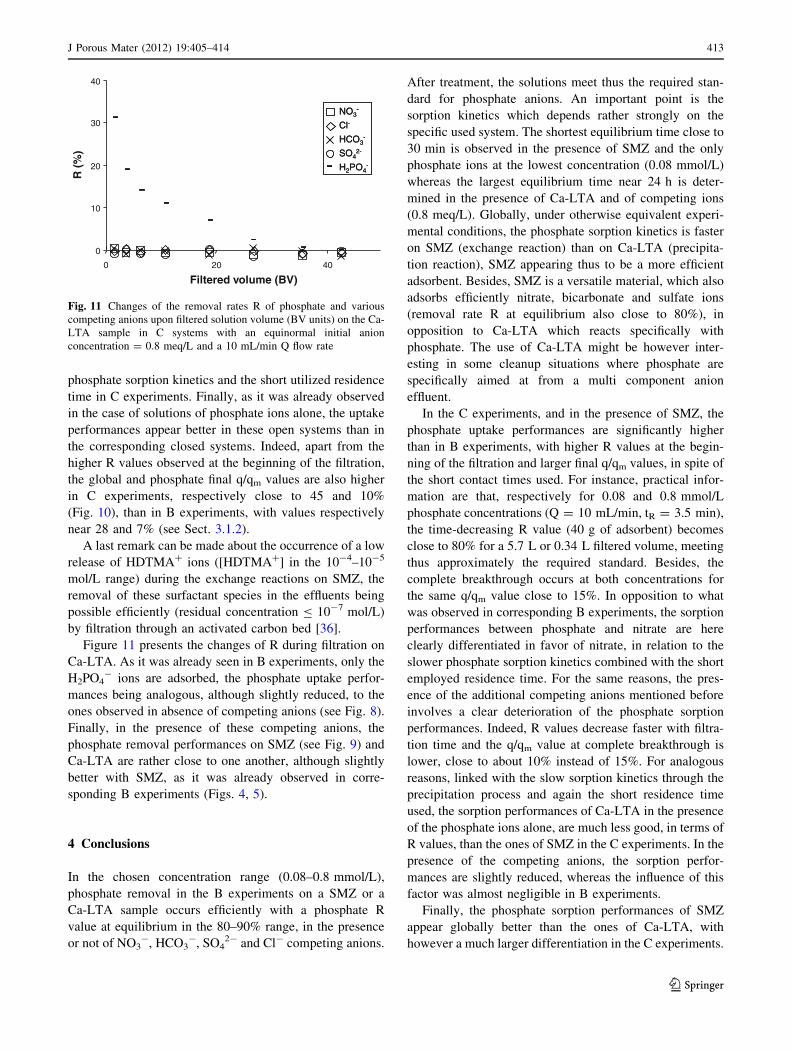
Before describing these experiments, a last remark can
be made from Fig. 3, where the changes of R values upon
contact time are compared for H2PO4- and NO3
- ions [35]
in the presence of SMZ. Within the experimental uncer-
tainties, the Requ values are similar and close to 80%, with,
however, a faster uptake kinetics in the case of nitrate ions
at the higher used anion concentration. The better removal
performances of SMZ towards nitrate are confirmed here-
after when competing anions-containing phosphate solu-
tions are used.
3.1.2 Competing anions-containing H2PO4- solutions
Figures 4 and 5 display the adsorption kinetic curves
respectively recorded in the presence of SMZ and Ca-LTA.
In the presence of SMZ, the mass balances are verified,
with a global anion concentration in solution remaining
constant upon contact time, within the experimental rela-
tive uncertainties limited to ±5%. (Fig. 4a). Figure 4b
shows that the Requ for phosphate ions remains unchanged
(&80%), in spite of the competitive adsorption of the other
anions. Indeed, the Requ values for the 3 interfering anions
NO3-, HCO3
- and SO42- are also close to 80%, the
chloride ions being not at all adsorbed. However, the
phosphate exchange kinetics is significantly slowed down
(see for instance Fig. 3 for comparison), in accordance
with a similar observation made in the case of nitrate
removal on SMZ [35]. This trend is obviously expected and
is related to the fact that the global q/qm value at equilib-
rium is much higher, in fact close to 28%, instead of 7% in
the case of the only H2PO4- ions. Besides, the adsorption
kinetics of the nitrate, sulphate and bicarbonate ions are
very similar and clearly faster than the one of phosphate.
This point was already underlined before in the case of the
specific comparison with nitrate ions (see Fig. 3). The
various adsorption behaviors of the anions towards SMZ
are governed in a very complicated way by factors such as
geometry and charge density of the anion and are thus not
really interpretable.
SMZCa-LTACa-FAUCa-HEUHEU
0
20
40
80
100
0 2 4 6 8
Time (h)
R (
%)
24
60
SMZCa-LTACa-FAUCa-HEUHEU
Fig. 2 Adsorption kinetics of H2PO4- ions (L/S = 10 mL/g) in B
experiments in the presence of several materials with a 0.8 mmol/L
initial concentration
H2PO4- 0.08 mmol/L
H2PO4- 0.8 mmol/L
NO3- 0.08 mmol/L
NO3- 0.8 mmol/L
0
20
40
60
100
0 2 4 6 8
Time (h)
R (
%)
H2PO4- 0.08 mmol/L
H2PO4- 0.8 mmol/L
NO3- 0.08 mmol/L
NO3- 0.8 mmol/L
24
80
Fig. 3 Comparative adsorption kinetics of H2PO4- or NO3
- ions
(L/S = 10 mL/g) in B experiments performed in the presence of SMZ
with 0.08 or 0.8 mmol/L initial anion concentration
J Porous Mater (2012) 19:405–414 409
123
Page 6
In the presence of Ca-LTA (Fig. 5), only the phosphate
anions are removed, the adsorption being practically not
influenced (very close Requ and uptake kinetics) by the
presence of competing anions (see Fig. 3 for comparison).
This result illustrates naturally a calcium salts precipitation
sorption process [43, 44]. Indeed, in the used 5–6 pH range,
the formation of Ca(HPO4) with pKs & 6.6 is possible,
phosphate anions being mainly present as H2PO4- and
HPO42- species, whereas the precipitations of calcium
nitrate (soluble), of CaSO4 (pKs & 4.2) and of CaCO3
(pKs & 8.2, but the main species in solution are H2CO3
and HCO3-) are highly improbable.
Finally, in the presence of the mentioned competing
anions, the performances of both Ca-LTA and SMZ become
very analogous, as far as the Requ value and the adsorption
kinetics are considered (see for comparison Figs. 4, 5).
3.2 C sorption experiments
In this section, in addition to the influence of the parameters
already examined in Sect. 3.1, the influence of a specific
parameter, i.e. the flow rate Q, is also investigated. The
experimental data are again displayed in two consecutive
parts, corresponding to solutions of H2PO4- ions alone and
to competing anions-containing H2PO4- solutions.
The macroporosity of the zeolite bed (BV = 57 mL) was
estimated according to the method described previously
in Sect. 2.3 by measuring the residence time tR (see Ref. [36])
of a 100 mmol/L potassium nitrate aqueous solution
(Q = 3 mL/min) in the column. The tR values are almost the
same for both Ca-LTA and SMZ materials, i.e. close to
12 min, which leads to a macroporosity value of about
60%. Residence times tR can thus be estimated at about 1.1, 3.5
and 34 min respectively for the flow rate of 30, 10 and 1 mL/
min used in the following phosphate sorption experiments.
3.2.1 Solutions of H2PO4- ions alone
The changes of R and q/qm upon filtration time on SMZ are
respectively shown in Figs. 6 and 7. As mentioned before,
the mass balances are verified (Br- concentrations not
reported here), with a global anion concentration in solu-
tion remaining constant (±5%) upon filtration time.
Whereas the initial R values are higher than about 95%
at the beginning of the filtration, whatever the phosphate
concentration and flow rate values, they decrease then
progressively with increasing time, the more rapidly the
initial phosphate concentration is higher (Q flow
rate = 10 mL/min). This expected trend, previously evi-
denced during adsorption of nitrate ions on the same
material [36], is obviously related to a faster increase of the
q/qm value. For the same reasons proposed in Ref. [36], the
uptake performances appear better in these open systems
than in corresponding closed systems, in spite of the short
used residence time close to 3.5 min. Indeed, beside the
very high R values evidenced at the beginning of the fil-
tration, the final q/qm values are also much higher, close to
15% and independent of the [H2PO4-]i, instead of 0.7 or
7% depending on the phosphate concentration in B
experiments (see Sect. 3.1, preliminary experiments).
Whereas in B experiments the Requ values for nitrate and
phosphate anions were the same (&80%), with only a
NO3-
Cl-
Br-
HCO3-
SO42-
H2PO4-
NO3-
Cl-
Br-
HCO3-
SO42-
H2PO4-
NO3-
Cl-
Br-
HCO3-
SO42-
H2PO4-
0,0
0,8
1,6
2,4
Time (h)C
on
c. (
meq
/L)
(a)
00
20
40
60
100
Time (h)
R (
%)
80
(b)
2 4 6 240 2 4 6 24
Fig. 4 Changes upon time of
the concentrations (a) and the
removal rates R (b) of
phosphate and various
competing anions in B
experiments on SMZ with an
equinormal initial anion
concentration = 0.8 meq/L and
L/S = 10 mL/g
NO3-
Cl-
HCO3-
SO42-
H2PO4-
0
20
40
80
100
0 2 4 6 8 24
60NO3
-
Cl-
HCO3-
SO42-
H2PO4-
NO3-
Cl-
HCO3-
SO42-
H2PO4-
0
20
40
80
100
Time (h)
R (
%) 60
Fig. 5 Changes upon time of the removal rates R of phosphate and
various competing anions in B experiments on Ca-LTA with an
equinormal initial anion concentration = 0.8 meq/L and L/S =
10 mL/g
410 J Porous Mater (2012) 19:405–414
123
Page 7
difference in the sorption kinetics (faster for nitrate ions,
see Figs. 3, 4), the sorption performances are here clearly
differentiated in favor of NO3-. For instance, with an ini-
tial anion concentration of 0.8 mmol/L (Q = 10 mL/min),
and respectively for nitrate and phosphate, the filtered
volume corresponding to a 50% R value is close to 50 or 10
BV, the complete breakthrough occurs at 100 or 30 BV and
ultimately the final q/qm value is close to 55 and 15% (see
for comparison data corresponding to a 0.8 mmol/L initial
nitrate concentration in Figures 2 and 3 of Ref. [36]). The
observed differentiation between phosphate and nitrate
behaviors in C experiments is related to the lower sorption
kinetics of the phosphate ions (see Figs. 3, 4) in combi-
nation with the short employed residence time.
The influence of the Q flow rate value ([H2PO4-]i =
0.8 mmol/L) is not really significant. This seems to be at
variance with the behavior evidenced in the case of NO3-
ions ([NO3-] = 1.6 mmol/L), where the increase of the
flow rate led to a clear deterioration of the sorption per-
formances (see Figure 4 in Ref. [36]). This discrepancy is
probably only apparent, the influence of the Q value being
not visible here, due to the relatively low amounts of sor-
bed phosphate and the corresponding larger resulting
uncertainties.
The changes of R during filtration time on Ca-LTA are
displayed in Fig. 8. Whereas the Requ in B experiments
were similar and close to 80% for both Ca-LTA and SMZ
adsorbents, with however a slower adsorption kinetics in
the case of Ca-LTA (see Fig. 2), the performances of
Ca-LTA in C experiments become less good than the ones
of SMZ (with the exception of the experiment with
[H2PO4-] = 0.8 mmol/L and Q = 1 mL/min, where the
performances are rather close). The general performances
decrease is probably correlated to the slower phosphate
sorption kinetics by Ca-LTA (see Fig. 2) and the short used
residence time. For the same reasons, the increase of Q has
a marked negative effect on the sorption efficiencies,
whereas the influence of this parameter was negligible in
the case of SMZ (Fig. 6). Finally, one can observe that the
increase of [H2PO4-]i has practically no influence on the
observed R values, whereas it led to significantly lower R
values in the case of SMZ (Fig. 6).
3.2.2 Competing anions-containing H2PO4- solutions
Figures 9 and 10 display respectively the changes of R and
q/qm values during filtration in the presence of SMZ. The
anion concentrations displayed in Fig. 9a allow to check
0.08 mmol/L - 10 mL/min0.8 mmol/L - 10 mL/min0.8 mmol/L - 1 mL/min0.8 mmol/L - 30 mL/min
00
0
20
40
60
80
100
Filtered volume (BV)R
(%
)
(a)
50 100 150 200 10 20 30 40 50
Filtered volume (BV)
(b)Fig. 6 a Changes of the R
removal rates of phosphate upon
filtered solution volume (BV
units) on SMZ in C experiments
for 0.08 and 0.8 mmol/L
phosphate solution with
different Q flow rates (b gives a
zoom of the 0–50 BV range)
0
5
10
15
20
Filtered volume (BV)
q/q
m
0.08 mmol/L - 10 mL/min
0.8 mmol/L - 10 mL/min
0.8 mmol/L - 1 mL/min
0.8 mmol/L - 30 mL/min
0 50 100 150 200 0 10 20 30 40 50
Filtered volume (BV)
(a) (b)Fig. 7 a Changes of the q/qm
values upon filtered solution
volume (BV units) on SMZ in C
experiments for 0.08 and
0.8 mmol/L phosphate solution
with different Q flow rates
(b gives a zoom of the 0–50 BV
range)
J Porous Mater (2012) 19:405–414 411
123
Page 8
that the global anion concentration in solution remains
constant (±5%) upon filtration time.
The increasing order of anion selectivity towards
SMZ is similar to the one observed in B experiments, i.e.
Cl- (null affinity) � H2PO4- \HCO3
- \SO42- \NO3
-.
Whereas in B experiments, the presence of competing
anions did not change the phosphate Requ value, with
however a decrease in the adsorption kinetics (see Fig. 4),
it involves in these C experiments a marked decrease of the
phosphate R value. For instance, the filtered volume cor-
responding to a 50% R value are respectively close to 3 and
8 BV, depending on whether competing anions are present
or not (see Figs. 6, 9b). Besides, the final q/qm value also
decreased to about 10% instead of 15% (see Figs. 7, 10).
Again, the behavior difference observed between these two
experimental systems is probably related to the low relative
00
20
40
60
80
Filtered volume (BV)R
(%
)20 40 0 10 20
Filtered volume (BV)
0.08 mmol/L - 10 mL/min0.8 mmol/L - 10 mL/min0.8 mmol/L - 1 mL/min0.8 mmol/L - 30 mL/min
(a) (b) 0.08 mmol/L - 10 mL/min0.8 mmol/L - 10 mL/min0.8 mmol/L - 1 mL/min0.8 mmol/L - 30 mL/min
Fig. 8 Changes of the R
removal rates of phosphate upon
filtered solution volume (BV
units) on Ca-LTA in C
experiments for 0.08 and
0.8 mmol/L phosphate solution
with different Q flow rates
(b gives a zoom of the 0–20 BV
range)
0
1
2
3
Filtered volume (BV)
Co
nc.
(m
eq/L
)
NO3-
Cl-
Br-
HCO3-
SO42-
H2PO4-
0
20
40
60
100
0 20 40 0 20 40
Filtered volume (BV)
R (
%)
80 NO3-
Cl-
Br-
HCO3-
SO42-
H2PO4-
NO3-
Cl-
Br-
HCO3-
SO42-
H2PO4-
(a) (b)Fig. 9 Changes of
concentrations (a) and of the
removal rates R (b) of
phosphate and various
competing anions upon filtered
solution volume (BV units) on a
SMZ sample in C systems with
an equinormal initial anion
concentration = 0.8 meq/L and
a 10 mL/min Q flow rate
0
20
40
60
0 20 40
Filtered volume (BV)
q/q
m
NO3-
Cl-
HCO3-
SO42-
totalH2PO4
-
0 10 20
Filtered volume (BV)
(a) (b)
NO3-
Cl-
HCO3-
SO42-
totalH2PO4
-
3-
Cl-
HCO3-
SO42-
totalH2PO4
-
Fig. 10 Changes of the
individual and global q/qm
values of phosphate and various
competing anions upon filtered
solution volume (BV units) on a
SMZ sample in C systems with
an equinormal initial anion
concentration = 0.8 meq/L and
a 10 mL/min Q flow rate
(b gives a zoom of the 0–20 BV
range)
412 J Porous Mater (2012) 19:405–414
123
Page 9
phosphate sorption kinetics and the short utilized residence
time in C experiments. Finally, as it was already observed
in the case of solutions of phosphate ions alone, the uptake
performances appear better in these open systems than in
the corresponding closed systems. Indeed, apart from the
higher R values observed at the beginning of the filtration,
the global and phosphate final q/qm values are also higher
in C experiments, respectively close to 45 and 10%
(Fig. 10), than in B experiments, with values respectively
near 28 and 7% (see Sect. 3.1.2).
A last remark can be made about the occurrence of a low
release of HDTMA? ions ([HDTMA?] in the 10-4–10-5
mol/L range) during the exchange reactions on SMZ, the
removal of these surfactant species in the effluents being
possible efficiently (residual concentration B 10-7 mol/L)
by filtration through an activated carbon bed [36].
Figure 11 presents the changes of R during filtration on
Ca-LTA. As it was already seen in B experiments, only the
H2PO4- ions are adsorbed, the phosphate uptake perfor-
mances being analogous, although slightly reduced, to the
ones observed in absence of competing anions (see Fig. 8).
Finally, in the presence of these competing anions, the
phosphate removal performances on SMZ (see Fig. 9) and
Ca-LTA are rather close to one another, although slightly
better with SMZ, as it was already observed in corre-
sponding B experiments (Figs. 4, 5).
4 Conclusions
In the chosen concentration range (0.08–0.8 mmol/L),
phosphate removal in the B experiments on a SMZ or a
Ca-LTA sample occurs efficiently with a phosphate R
value at equilibrium in the 80–90% range, in the presence
or not of NO3-, HCO3
-, SO42- and Cl- competing anions.
After treatment, the solutions meet thus the required stan-
dard for phosphate anions. An important point is the
sorption kinetics which depends rather strongly on the
specific used system. The shortest equilibrium time close to
30 min is observed in the presence of SMZ and the only
phosphate ions at the lowest concentration (0.08 mmol/L)
whereas the largest equilibrium time near 24 h is deter-
mined in the presence of Ca-LTA and of competing ions
(0.8 meq/L). Globally, under otherwise equivalent experi-
mental conditions, the phosphate sorption kinetics is faster
on SMZ (exchange reaction) than on Ca-LTA (precipita-
tion reaction), SMZ appearing thus to be a more efficient
adsorbent. Besides, SMZ is a versatile material, which also
adsorbs efficiently nitrate, bicarbonate and sulfate ions
(removal rate R at equilibrium also close to 80%), in
opposition to Ca-LTA which reacts specifically with
phosphate. The use of Ca-LTA might be however inter-
esting in some cleanup situations where phosphate are
specifically aimed at from a multi component anion
effluent.
In the C experiments, and in the presence of SMZ, the
phosphate uptake performances are significantly higher
than in B experiments, with higher R values at the begin-
ning of the filtration and larger final q/qm values, in spite of
the short contact times used. For instance, practical infor-
mation are that, respectively for 0.08 and 0.8 mmol/L
phosphate concentrations (Q = 10 mL/min, tR = 3.5 min),
the time-decreasing R value (40 g of adsorbent) becomes
close to 80% for a 5.7 L or 0.34 L filtered volume, meeting
thus approximately the required standard. Besides, the
complete breakthrough occurs at both concentrations for
the same q/qm value close to 15%. In opposition to what
was observed in corresponding B experiments, the sorption
performances between phosphate and nitrate are here
clearly differentiated in favor of nitrate, in relation to the
slower phosphate sorption kinetics combined with the short
employed residence time. For the same reasons, the pres-
ence of the additional competing anions mentioned before
involves a clear deterioration of the phosphate sorption
performances. Indeed, R values decrease faster with filtra-
tion time and the q/qm value at complete breakthrough is
lower, close to about 10% instead of 15%. For analogous
reasons, linked with the slow sorption kinetics through the
precipitation process and again the short residence time
used, the sorption performances of Ca-LTA in the presence
of the phosphate ions alone, are much less good, in terms of
R values, than the ones of SMZ in the C experiments. In the
presence of the competing anions, the sorption perfor-
mances are slightly reduced, whereas the influence of this
factor was almost negligible in B experiments.
Finally, the phosphate sorption performances of SMZ
appear globally better than the ones of Ca-LTA, with
however a much larger differentiation in the C experiments.
NO3-
Cl-
HCO3-
SO42-
H2PO4-
0
20
40
30
10
0 4020
Filtered volume (BV)
R (
%)
NO3-
Cl-
HCO3-
SO42-
H2PO4-
-
Fig. 11 Changes of the removal rates R of phosphate and various
competing anions upon filtered solution volume (BV units) on the Ca-
LTA sample in C systems with an equinormal initial anion
concentration = 0.8 meq/L and a 10 mL/min Q flow rate
J Porous Mater (2012) 19:405–414 413
123
Page 10
The SMZ displays furthermore two other important
advantages, being easily regenerable and, besides, able to
adsorb other pollutants, anionic such as nitrate ions, but also
cationic species and even organic molecules.
Acknowledgments The authors would like to thank FR Environn-
ement Nautique for financial support, A.N.R.T. for a CIFRE doctoral
grant to J.S. (no. 183/ 2007) and Pr. A. Louati (ENSCMu, UHA) for
his assistance in the anion analysis by capillary electrophoresis.
References
1. S.N. Levine, D.W. Schindler, Can. J. Fish. Aquat. Sci. 46, 2–10
(1989)
2. http://www.legifrance.gouv.fr/
3. S. Srivastava, A.K. Srivastava, Biochem. Eng. J. 40, 227–232
(2008)
4. S. Yeoman, T. Stephenson, J.N. Lester, R. Perry, Environ. Pollut.
49, 183–189 (1988)
5. C. Visvanathan, P. Roy, Environ.Technol. 18, 551–556 (1997)
6. T. Helfgott, J.V. Hunter, Chem. Eng. Prog. 65, 97–103 (1969)
7. R.G. Penetra, M.A.P. Reali, E. Foresti, J.R. Campos, Water Sci.
Technol. 40, 137–143 (1999)
8. L.L. Ruixia, G. Jinlong, T.J. Hongxiao, J. Colloid Interface Sci.
248, 268–274 (2002)
9. H. Roques, L. Nugroho-Judy, A. Lbugle, Water Res. 25, 959–965
(1991)
10. K. Sakadevan, H.J. Bavor, Water Res. 32, 393–399 (1998)
11. E. Oguz, J. Hazard. Mater. 114, 131–137 (2004)
12. N.M. Agyei, C.A. Strydom, J.H. Potgieter, Cement Concrete Res.
30, 823–826 (2000)
13. C. Namasivayam, D. Sangeetha, J. Colloid Interface Sci. 280,
359–365 (2004)
14. Y. Seida, Y. Nakano, Water Res. 36, 1306–1312 (2002)
15. D.A. Georgantas, H.P. Grigoropoulou, J. Colloid Interface Sci.
315, 70–79 (2007)
16. R. Wu, K.H. Lam, J. Lee, T.C. Lau, Chemosphere 69, 289–294
(2007)
17. S.H. Gharaibeh, I.M. Dwairi, Chem. Tech.-Leipzig 48, 215–218
(1996)
18. A.D. Vujakovic, M.R. Tomasevic-Canovic, A.S. Dakovic, V.T.
Dondur, Appl. Clay Sci. 17, 265–277 (2000)
19. J. Hrenovic, M. Rozic, L. Sekanovic, A. Anic-Vucinic, J. Hazard.
Mater. 156, 576–582 (2008)
20. M.S. Onyango, D. Kuchar, M. Kubota, H. Matsuda, Ind. Eng.
Chem. Res. 46, 894–900 (2007)
21. J. Chen, H. Kong, D. Wu, Z. Hu, Z. Wang, Y. Wang, J. Colloid,
Interface Sci. 300, 491–497 (2006)
22. B. Zhang, D. Wu, C. Wang, S. He, Z. Zhang, J. Environ. Sci. 19,
540–545 (2007)
23. D. Wu, B. Zhang, C. Li, Z. Zhang, H. Kong, J. Colloid Interface
Sci. 304, 300–306 (2006)
24. N. Murayama, S. Yoshida, Y. Takami, H. Yamamoto, J. Shibata,
Separ. Sci. Technol. 38, 113–130 (2003)
25. R. Saad, S. Hamoudi, K. Belkacemi, J. Porous Mat. 15, 315–323
(2008)
26. E.W. Shin, J.S. Han, M. Jang, S. Min, J.K. Park, R.M. Rowel,
Environ. Sci. Technol. 38, 912–917 (2004)
27. G.M. Haggerty, R.S. Bowman, Environ. Sci. Technol. 28,
452–458 (1994)
28. E.J. Sullivan, R.S. Bowman, I.A. Legiec, J. Environ. Qual. 32,
2387–2391 (2003)
29. Z. Li, Microp. Mesopor. Mater. 61, 181–188 (2003)
30. Z. Li, I. Anghel, R. Bowman, J. Disper. Sci. Technol. 19,
843–857 (1998)
31. Z. Li, R.S. Bowman, Environ. Sci. Technol. 31, 2407–2412
(1997)
32. Z. Li, R. Beachner, Z. McManama, H. Hanlie, Microp. Mesopor.
Mater. 105, 291–297 (2007)
33. N.K. Bansiwal, S.S. Rayalu, N.K. Labhasetwar, A.A. Juwarkar,
S. Devotta, J. Agric. Food Chem. 54, 4773–4779 (2006)
34. A.I. Perez Cordoves, M. Granda Valdes, J.C. Torres Fernandez,
G. Pina Luis, J.A. Garcıa-Calzon, M.E. Dıaz Garcıa, Microp.
Mesopor. Mater. 109, 38–48 (2008)
35. J. Schick, P. Caullet, J.L. Paillaud, J. Patarin, C. Mangold-
Callarec, Microp. Mesopor. Mater. 132, 395–400 (2010)
36. J. Schick, P. Caullet, J.-L. Paillaud, J. Patarin, C. Mangold-
Callarec, Microp. Mesopor. Mater. 142, 549–556 (2011)
37. V. Jorik, Zeolites 13, 187 (1993)
38. P.E. Werner, L. Eriksson, M. Stendhal, J. Appl. Crystallogr. 18,
367–370 (1985)
39. Z.Y. Ji, J.S. Yuan, X.G. Li, J. Hazard. Mater. 141, 483–488
(2007)
40. D. Ming, J. Dixon, Clay. Clay Miner. 35, 463–468 (1987)
41. T. Kyotani, An. Sci. 22, 961–964 (2006)
42. I. Langmuir, J. Am. Chem. Soc. 38, 2221–2295 (1916)
43. D.R. Lide, Handbook of Chemistry and Physics, 76th edn. (CRC
Press, Boca Raton, 1995)
44. G. Charlot, Les Reactions Chimiques en Solution Aqueuse, 7th
edn. (Masson, Paris, 1983)
414 J Porous Mater (2012) 19:405–414
123